valence bond description of the co ligand
TRANSCRIPT

• In the CO molecule both the C and the O atoms are sp hybridized.
• The singly occupied sp and pz orbitals on each atom form a σ and a π bond,
respectively.
• This leaves the C py orbital empty, and the O py orbital doubly occupied, and so the
second π bond is formed only after we have formed a dative bond by transfer of the
lone pair of O py electrons into the empty C py orbital.
Valence Bond description of the CO ligand
• This transfer leads to a Cδ−−−−−Oδ+• This transfer leads to a Cδ−−−−−Oδ+
polarization of the molecule, which is
almost exactly canceled out by a partial
Cδ+−−−−Oδ− polarization of all three
bonding orbitals because of the higher
electronegativity of oxygen.
• The free CO molecule therefore has a
net dipole moment very close to zero.
C Osp sp spsp
p
p p
pdative
1

MO correlation diagram for CO
2p
C
HOMO
LUMO
OC
2p
O
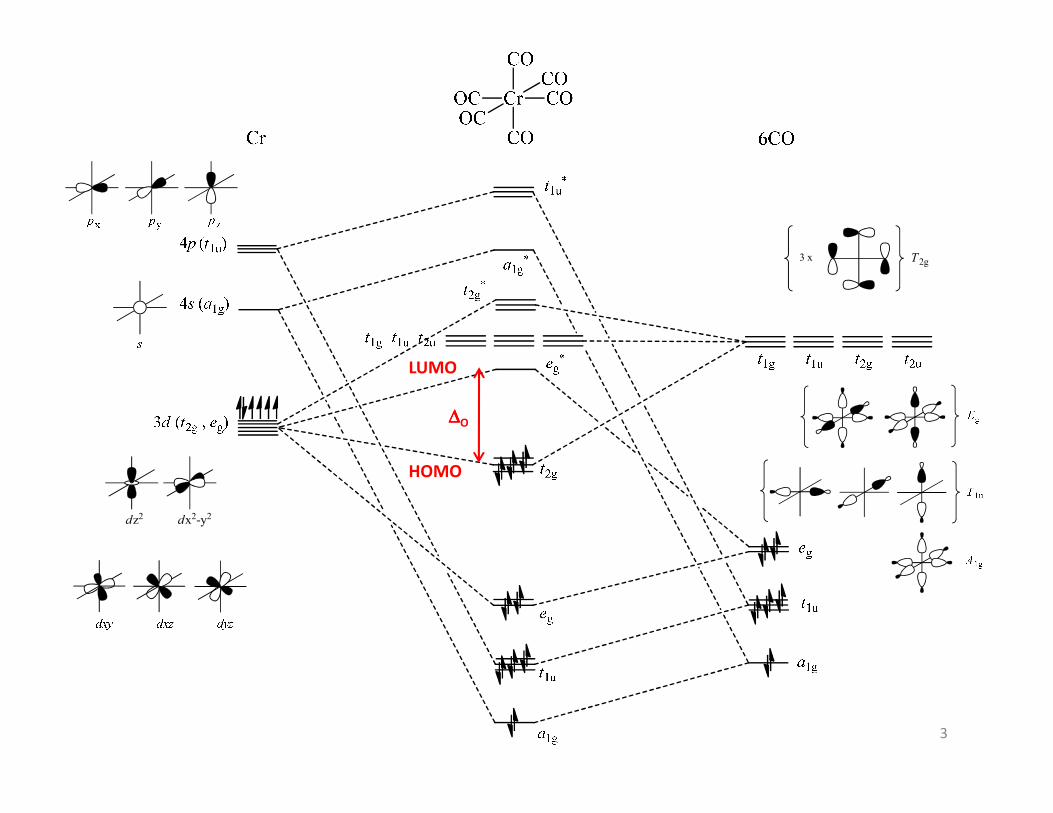
The CO LUMO orbitals are anti bonding (π*). These
are empty orbitals and accept electron density
from the metal centre via π-backbonding with the
metal dxy, dxz and dyz orbitals.
2s
2s
2p
bond order = (8 - 2)/2 = 3
The CO HOMO orbital is a bonding orbital of σsymmetry with significant electron density on the
carbon. This orbital forms a σ bond with metal p , dz2
and dx2–y2 orbitals. This is a filled orbital and
donates electron density to the metal centre.
2
LUMO
∆∆∆∆
T2g3 x
dx2-y2dz2
HOMO
∆∆∆∆O
3

4

• The net result is that C becomes more positive on coordination, and O becomes more negative.
This translates into a polarization of the CO on binding.
• This metal-induced polarization chemically activates the CO ligand making C more sensitive to
nucleophilic attack and O more sensitive to electrophilic attack.
• The polarization will be modulated by the effect of the other ligands on the metal and by the
net charge on the complex.
� In LnM(CO), the CO carbon becomes particularly δ+ in character if the L groups are good π
acids or if the complex is cationic, e.g. Mo(CO)6 or [Mn(CO)6]+, because the CO-to-metal σ-
donor electron transfer will be enhanced at the expense of the metal to CO back donation.
� If the L groups are good donors or the complex is anionic, e.g. Cp2W(CO) or [W(CO)5]2−,
back donation will be encouraged, the CO carbon will lose its pronounced δ+ charge, but
the CO oxygen will become significantly δ−.
• The range can be represented in valence bond terms the extreme in which CO acts as a pure σdonor, through to the extreme in which both the π∗x and π∗y are both fully engaged in back
bonding.
5

CO's render the electron rich Cr metal electrophilic via strong π-backbonding. Complexation of benzene with the
electrophilic Cr(CO)3 fragment withdraws electon density from the aromatic ring activating it towards nucleophilic
Semmelhack JACS 1980 (102) 5926
π-acceptor effects on reactivity
electrophilic Cr(CO)3 fragment withdraws electon density from the aromatic ring activating it towards nucleophilic
attack.
Yamamoto JACS 1971 (93)3350.
Acrolein is thought to act as a π-acid, withdrawing electron density from the Ni(II) complex via π-backbonding and
promoting elimination of the diethyl fragment to reduce the metal.6

Dewar-Chatt-Duncanson model
Explain the trend in CO vibrations for the following series of compoundsExplain the trend in CO vibrations for the following series of compounds
v(CO) cm-1
[Ti(CO)6]2- 1748
[V(CO)6]- 1859
Cr(CO)6 2000
[Mn(CO)6 ]+ 2100
[Fe(CO)6 ]2+ 2204
7

The increase in electron density at the nickel from PR3 σ-
donation is dispersed through the M-L π system via π-
backbonding. Much of the electron density is passed onto
M-CO ππππ-Backbonding and the trans-effect
CO stretching frequencies
measured for Ni(CO)3L where L are
PR3 ligands of different σ-donor
abilities. [v(CO) =2143 cm-1]
backbonding. Much of the electron density is passed onto
the CO π* and is reflected in decreased v(CO) stretching
frequencies which corresponds to weaker CO bonds.
Recall: Band position in IR is governed by :
1. force constant of the bond (f) and
2. individual masses of the atoms (Mx and My).
Stronger bonds have larger force constants than weaker bonds.
Tolman Chem. Rev. 1977 (77) 313 8

• In the L→M π-donor system filled p ligand orbitals exist slightly lower in energy
than the metal t2g set with which overlap occurs.
• The bonding t2g MO’s formed are again lower in energy than the initial metal t2g
set, however, the corresponding t2g* anti-bonding MO’s are lower in energy than
the eg* σ anti-bonding orbitals.
• Additionally, as the ligand t2g p orbitals are typically of lower energy than the
metal t2g set, the bonding t2g MO’s formed are predominantly ligand based, with
the corresponding t * anti-bonding MO’s being predominantly metal based.
MO description for L→M ππππ-donor system in an Oh complex
the corresponding t2g* anti-bonding MO’s being predominantly metal based.
• Thus the LFSE ∆o is decreased relative to the σ only system. The t2g bonding MO’s
are stabilized which is countered by occupation of the t2g* anti-bonding orbitals.
• Overall this combined σ and π donation from ligand to metal results in an
increased M-L bond order and a stronger bond, however, the metal now becomes
more electron rich which can decrease the bond strengths of the remaining ligand
set making ligand-metal π bonding a less favorable interaction.
9

4s (a1g)
4p (t1u)
CrIII 6Cl-
eg*
t1u*
a1g*
Cl
CrCl ClCl
Cl
Cl
t2*
3-
T2g3 x
a1g
3d (t2g , eg)
t1u
eg
a1g
t1u
eg
t2g
t2g
t2g
dx2-y2dz2
10

MO description for L→M ππππ-donor system in an Oh complex
The energy of the HOMO is directly affected by M-L π-bonding. Ligand to metal π-
donation increases the energy of the HOMO making the metal more basic. π-donor
ligands stabilize electron poor, high oxidation state metals. Very prevalent for early TM
complexes (low d electron count) and less so for late TM (high d electron count).
11

• Alkoxy, amido and halido ligands are
sp2 hybridized so only one set of p
electrons are available for π bonding
12

Summary of ππππ-bonding in a Oh complexes
ππππ donor ligands result in L→M ππππbonding, a smaller ∆∆∆∆o favoring high spin
configurations and a decreased stability.
ππππ acceptor ligands result in M→L ππππ bonding, a larger ∆∆∆∆o
favoring low spin configurations with an increased
stability.13

Types of bonding
σ π δ σ π δ σ π δ σ π δ
• Bond polarity is evaluated by the difference in electronegativities of neighboring atoms
14

Ionic bonding
• Greater when elements of high and opposite charge interact.
• Differences in charge are paralleled in differences in
electronegativities.
• Large differences in electronegativity favor strong ionic bonding.
• M-L σ-bonding in early metals has significant ionic character.
M-L σ bond polarization
Covalent bonding
• Greater when orbitals of similar energies interact.
• The energy of atomic orbitals is inversely proportional to the
element's electronegativity (i.e. the orbital energy of an
electronegative element is lower than that of a electropositive
element).
• Small differences in electronegativity favor strong covalent bonding.
• M-L σ-bonding in late metals has a high degree of covalent bonding.
15

• Both the covalent model and the ionic model differ only in the way the electrons
are considered as coming from the metal or from the ligands
- emphasize model…not a true representation of metal charge!!!
• Each model is often invoked without any warning in the literature therefore it is
important to be able to identify their use.
• The ionic model is most commonly used for traditional M−−−−L inorganic
coordination compounds therefore coordinating ligands are treated equally in
both models.
Ionic vs. covalent model
both models.
• The ionic model is more appropriate for high-valent metals with N, O or Cl ligands.
• In the ionic model the M−X bond is considered as arising from a cationic M+ and
an anionic X− (heterolytic)
• The covalent model is sometimes preferred for organometallic species with low-
valent metals where the metal and ligand oxidation states cannot be
unambiguously defined.
• In the covalent model the M−X bond is considered as arising from a neutral metal
and ligand radical X• (homolytic).16

Hydride
Alkyl (e.g. methyl)
Alkenyl
Alkynyl
Allyl
-1
-1
-1
-1
-1
-1
2
2
2
2
2
2
0
0
0
0
0
0
1
1
1
1
1
1
ηηηη1 (hapticity = 1)
Ionic model covalent model
# of e- # of e-
charge donated charge donated
Aryl
Cyclopentadienyl
Carbene
Carbyne
Acyl
Carbon monoxide
Nitrile
Fulminate
-1
-1
-2
-3
-1
0
-1
-1
2
2
4
6
2
2
2
2
0
0
0
0
0
0
0
0
1
1
2
3
1
1
1
1
17

Aqua
Hydroxyl
Oxo
Alkoxide
Ether
(Sulfide/Tioether O = S)
ηηηη1 (hapticity = 1)
Ionic model covalent model
# of e- # of e-
charge donated charge donated
0
-1
-2
-1
0
-1
2
2
4
2
2
2
+1
0
0
0
+1
0
1
1
2
1
1
1
Carboxylate
Carbonate
Cyanate(Thiocyanate O = S)
Isocyanate(Isothiocyanate O = S)
Sulfoxide
-1
-1
-1
0
0
2
2
2
2
2
0
0
0
+1
+1
1
1
1
1
1
18

Isocyanate
Nitrogen
Amine
Pyridyl
Imine
Amide
Nitrile
ηηηη1 (hapticity = 1)
Ionic model covalent model
# of e- # of e-
charge donated charge donated
-1
0
0
0
0
-1
0
2
2
2
2
2
2
2
0
+1
+1
+1
+1
0
+1
1
1
1
1
1
1
1Nitrile
Isonitrile
Nitrosyls
Nitro
Nitrito
Phosphine
Phosphide
Halide (e.g. Cl)
0
0
-1
0
-1
-1
0
-1
-1
2
2
2
2
2
2
2
2
2
+1
+1
0
+1
0
0
+1
0
0
1
1
1
1
1
1
1
1
1 19

µµµµ-hydride
µµµµ-oxo
µµµµ-alkoxide
µµµµ-halide
µµµµ-CO
HM M
O
M M
O
M M
R
X
M M
M
C
M
O
H2N M'
Bridging ligands (non-chelating)
Ionic model covalent model
# of e- # of e-
charge donated charge donated
n/a
-2
-1
-1
-2
0
n/a
4
4
4
4
2x2
-1
0
+1
-1
0
+2
2
2
2
2
2
2x1
µµµµ-en (ethylene diamine)
µµµµ-pyrazine
µµµµ-4,4’-bipyridine
µµµµ-dppe
[1,2-bis(diphenyl phosphino)ethane]
NH2M
NM N M
NM N M'
0
0
0
0
2x2
2x2
2x2
2x2
+2
+2
+2
+2
2x1
2x1
2x1
2x1
20

• Common unsaturated ππππ donating ligands encountered in organotransition-metal chemistry
together with the respective numbers of electrons relevant to the application of the 18 VE rule.21

• When applying the 18VE rule the following should be considered
1. The intramolecular partitioning of the electrons has to ensure that the total
charge of the complex remains unchanged (ionic or covalent model).
The 18 VE rule
2. A M−M bond contributes one electron to the total electron count of a single
metal atom.
3. The electron pair of a bridging ligand donates one electron to each of the
bridged metal atoms.
22

1. The intramolecular partitioning of the electrons has to ensure that the total
charge of the complex remains unchanged.
Electron counting - revision
23

2. A M−−−−M bond contributes one electron to the total electron count of a single
metal atom.
What is the d electron count of Mn in the unstable (CO)5Mn monomer?
24

3. The electron pair of a bridging ligand donates one electron to each of the
bridged metal atoms.
25

• Transition metal complexes can be divided into 3 classes:
Class # valence electrons 18 VE rule
I . . . 16 17 18 19 . . . not obeyed
II . . . 16 17 18 not exceeded
III 18 obeyed
The 18 VE rule
• How does the nature of the central atom and of the ligands determine whether a
complex belongs to class I, II, or III?
• Guiding principles to construction of a transition metal complex:
� antibonding orbitals should not be occupied
� nonbonding orbitals may be occupied
� bonding orbitals should be occupied
26

Class I
� The splitting of ∆∆∆∆0 is relatively small for 3d metals as well as for σ ligands at
the lower end of the spectrochemical series.
� t2g is non-bonding and can be occupied by 0 − 6 electrons.
� eg* is weakly anti-bonding and can be occupied by 0 − 4 electrons.� eg* is weakly anti-bonding and can be occupied by 0 − 4 electrons.
� Thus 12 −−−− 22 valence electrons can be accommodated, i.e. the 18VE rule is not
obeyed.
� Owing to their inherently small ∆0 splitting tetrahedral complexes also belong
to this class.
27

28

Class II
� ∆∆∆∆0 is larger for 4d and 5d metals (especially in the higher oxidation states) as
well as for strong σ donating ligands.
� t2g is essentially non-bonding and can be occupied by 0 − 6 electrons.
� eg* is more strongly anti-bonding and is no longer available for occupancy.� eg* is more strongly anti-bonding and is no longer available for occupancy.
� Consequently the valence shell contains 18 VE or less.
� A similar splitting ∆0 is also observed for complexes of 3d metals with ligands
that exhibit extremely high ligand-field strength (e.g. CN−)
29

30

Class III
� ∆0 is largest for ligands at the upper end of the spectrochemical series (good π
acceptors, such as CO , PF3, olefins, arenes).
� t2g is now bonding due to π interactions with orbitals of the ligands and
should be occupied by 6 electrons.
� eg* is strongly anti-bonding and remains unoccupied.
� The valence shell contains 18 VE obeying the 18VE rule.
� Exceptions can occur usually for steric reasons, e.g. V(CO)6 contains 17VE but
is readily reduced to [V(CO)6]−.
Organometallic complexes of transition metals almost exclusively belong to Class III
31

32

T2g3 x
dx2-y2dz2
33

• For M(d8) late transition metals 16 VE configurations are favored
Examples: M (d8)
[Ni(CN)4]2− 16 VE
[Rh(CO)2Cl2]− 16 VE square planar, CN 4
[AuCl4]− 16 VE
Deviations from the 18VE rule
• For M(d10) late transition metals 14 VE configurations are favored
Examples: M (d10)
[Ag(CN)]− 14 VE
Ph3PAuCl 14 VE linear, CN 2
• d8 (16e−−−−) complexes prefer square-planar
• d0, d5, d10 complexes usually tetrahedral34

• According to the 18VE rule, a coordination number (CN) of 5 is expected for d8
late transition metals.
• This is found for early transition metal complexes with a d8 configuration.
Examples: M (d8)
Fe(CO)5 18 VE
Mn(CO) −−−− 18 VE trigonal bipyramidal, C.N. 5Mn(CO)5−−−− 18 VE trigonal bipyramidal, C.N. 5
[(ηηηη4−−−−C4H8)(ηηηη6−−−−C6H6 )Ru] 18 VE
35

• A qualitative explanation for deviations from the 18VE rule takes into a/c the
following:
� electroneutrality principle (Pauling, 1948)
� ππππ−−−−donating character of the ligands
� change in energy separation between (n – 1)d, ns, and np orbitals within a � change in energy separation between (n – 1)d, ns, and np orbitals within a
transition metal series.
36

• Increase in atomic number is accompanied by a decrease in orbital energies
across the periodic table as additional valence electrons only partially screen the
higher nuclear charge.
� For (n-1)d electrons, the decrease in energy is more pronounced than for ns
and np electrons
(d orbitals are more diffuse and experience a lower nuclear shielding relative (d orbitals are more diffuse and experience a lower nuclear shielding relative
to s and p orbitals of the same principle quantum number)
� Successive addition of d electrons across a transition series therefore
increases the effective nuclear charge and stabilizes the d orbitals.
� The energy separation between the (n-1)d, ns, np atomic orbitals also
increases with an increasing positive charge of the atom.
37

Electroneutrality Principle
“Stable complexes are those with structures such that each atom has only a small
electric charge. Stable M-L bond formation generally reduces the positive charge
on the metal as well as the negative charge and/or e- density on the ligand. The
result is that the actual charge on the metal is not accurately reflected in its
formal oxidation state”
- Pauling; The Nature of the Chemical Bond, 3rd Ed.;1960, pg. 172.- Pauling; The Nature of the Chemical Bond, 3rd Ed.;1960, pg. 172.
• The 18VE rule often fails for early transition metals.
• Formal oxidation state is not an accurate description of electron density at the
metal.
• High oxidation state, early TM complexes are stabilized via π-donation
i.e. a shifting of electron density from π-donor ligands to the metal.
• This in part accounts for the extreme oxophilicity of early TM.
38

1) Determine the oxidation state of the metal
(Balance ligand charges with overall charge
of the complex)
2) Determine the number of valence electrons
d electron count = (Group #) – (metal
oxidation state)
Revision: Electron Counting
3) Count electrons donated by neutral
or anionic ligands
(bridging ligands donate half electrons to
either metal per coordination site)
4) Metal – Metal bonds
(one electron per bond)
39

40

Homework: Construct a ππππ MO diagrom for Titanium
Tetraisopropoxide, Ti(OiPr)4
41

General properties of organometallic complexes
• There are several differences between organometallic complexes and coordination
compounds:
� The metals are more electron rich, i.e. the metal bears a greater negative charge in the
organometallic complex.
� The M−L bonds are much more covalent and often have a substantial π component.
� The metal d orbitals are higher in energy and by back donation perturb the electronic
structure of the ligands much more than is the case for coordination compounds.
� The organometallic ligands can be polarized and therefore activated toward chemical
reactions, σ and π bonds in the ligands can be weakened or broken, and chemical bonds
can be made or broken within and between different ligands.
� Readiness to change the coordination number and the lability of the M−C σ bond are
essential for organometallic catalysis
• This rich pattern of reactions is characteristic of organometallic chemistry.
42