usp• - university of the south...
TRANSCRIPT

THE UNIVERSITY OF THE SOUTH PACIFICLIBRARY
Author Statement of Accessibility
Name of Candidate
Degree
Department/School
Institution/University
Thesis Title
Date of completion ofrequirements for award
, SPAS
/
1 This thesis may be consulted in the Library without the author's permission
2. This thesis may be cited without the author's permission providing it is suitablyacknowledged.
3. This thesis may be photocopied in whole without the author's written permission
4. This thesis may be photocopied in proportion without the author's written permissionPart that may be copied:
Under 10%---------- 40---60%
10- 2 0 % - - - - - - - - - - - - - - - - - - - 6 0 - - - 8 0 % •
20- 40% Over 80%
5. I autliorise the University to produce a microfilm or microfiche copy for retention and usein the Library according to rules 1-4 above (for security and preservation purposes mainly)
6. I authorise the Library to retain a copy of this thesis in e-format for archival andpreservation purposes.
7. After a period of 5 years from the date of publication, the USP Library may issue the thesisin whole or in part, in photostat or microfilm or e-format or other copying medium,without first seeking the author's written permission,
8. I authorise the Universitytoauthorised users,
Signed:
Date: 2/11/2005
Contact Address SPAS
Permanent Address
make this thesis av
USP•
YesNo

STUDIES ON SOME NATURALLY OCCURRING
QUINONES FROM FIJIAN PLANTS
by
Sadaquat Ali, B.Sc.
A thesis submitted to the University
of the South Pacific in part:
fulfilment of the. requirements
for the degree of Master of Science
Chemistry DepartmentSeptember 1978
University of the South Pacific '

PREFACE
This thesis which is subMitted for coNsideration of an
award of degree of Master of Science has not previously been
presented for any degree. I t is a record of research work
carried out in the Chemistry Department of the University of
the South Pacific (Part A) between January 1976 and
September 1977, and in the Chemistry Department of the
University of Aberdeen (Part B) between October 1977 and
August 19 78. The work described is believed to be original
except whore due reference is made.

ACKNOWLEDGEMENTS
I wish to thank my supervisors Professor R.H. Thomson of the
University of Aberdeen (UA) , and Dr. R.M, Smith and Dr. A. Singh of
the University of the South Pacific (US?), for their help interest,
valuable advice and inspiring guidance which have been the motive force
in bringing forth this thesis to the present shape. Only imagination,
not words., can reflect my true feelings towards my mentors. I also wish
to record my thanks to the Vice Chancellor, Dr. J,A. Maraj (USP), and to
the Head of the Chemistry Department, Professor A.J. Dandy (USP), for
their immeasurable support and encouragement during the in i t i a l stages.
Thanks are due to Dr. D.C.C. Smith of the University of Manchester,
for a generous gift of 8-hydroxy~l-naphthaldehyde, and to Dr. R.A. Howie
(UA) for supplying samples of Fremy's s a l t . Thanks are also due to
the members of the technical staff (UA and US?) for their assistance,
and to my colleagues (too many to mention) for useful discussions,
I should like to express my particular appreciation to the
University of the South Pacific for a postgraduate studentship and study
leave. The support of funds granted through the Research Committee (USP)
is gratefully acknowledged,
It is a pleasure to express my thanks to the Executive Committee of
the Association of Commonwealth Universities (ACU) for the award of a \
studentship at the University of Aberdeen.
Last, but by no means least , I would like to thank Mrs. M. Riddoch \
who typed this thesis with exceptional s k i l l .

CONTENTS
SUMMARY CD
PART A
Studies on C. alata
CHAPTER 1 Introduction
CHAPTER 2 Isolation and Identificationof Anthraquinones
CHAPTER 3 Isolation and Identificationof Glycosides
CHAPTER 4 Experimental
REFERENCES
17
34
PART B
Studies on H, tiliaceous
CHAPTER 1 Introduction
CHAPTER 2 Isolation and Identification,of Sesquiterpenoids
CHAPTER 3 Synthesis of o- and p-NaphthoquinonGS-perl-aldehydes
CHAPTER 4 Experimental
REFERENCES
37
57
75
90

E R R A T A
In the whole thesis H. tiliaceoue should read H.. tiliaceus,
p.3, 1. 5p.4, 1. 7p.7, 1. 4P . 3 , 1.7p.14,1.15p.14, 1.26p.22, 1. 8p.25, 1.13p.28, 1.21,p.32, 1. 8
from
25
p.32, headingp.34, ref.
ref.ref.ref.
p.35. ref.p.44, 1. 1p.44, 1. 21p.58, 1. 1p.58, 1. 3p.61, 1. 4p.76, 1. 10p.77, 1.13p.78, 1 . 2p.81, 1 . 6p.81, 1. 9
310121226
from
bottom
bottomfrom bottom
from bottom
p . 8 1 , 1 . 2 from bottomp.83, 1. 6 from bottomp.84, 1 . 2p.85, 1 . 2
p.87, 1 . 1
p.88, 1. 2
p.88, 1 . 2 from bottom
p 8 9 , 1. 5 from bottom
p.89
p.92, ref. 44p.92, ref. 46, 47
diureticPhilippinesultra-visibletriacetylfraction227descendingBuckhv.rst Parktriacetatedescendinghydrolysis toHesenauer
T.A. Neubern de ToledoPauloBirkinshawdelete - (furan ring)anhydridemechanism ..,. wasMaruyamaXXXIXC Hedelete -(See also Fig.12.82 (2H, JL, J = 8Ka ...)230, of line 5 froM bottoM 278. 406.(14 H, eacH 2 X OCH3 )
4.36 and 4.68 (2H . . . . )Lactone (XXXIX)1730cm-
H, 3.06; M+, 202.0260. C1
requires C, 65.4; H, 3%diazoraethane (5.3 m Mole)337 ran
224 M+(81Br), 100%....
238 (M+(81Br), 95%) . . . .
II for naphthoquinone- a t m/e 158;
M+ - -2C0 for 2-methoxy-1 , 4-naphthoquinoneat m/e132.
: Magnussons lavit •

( i )
This thesis is divided into two par ts , Included in the first
part is a discussion on the uses of Cassia plants in folkmedicine,
and a review of the anthraquinones and their derivatives known in
ia alata (Leguminosae) . The presence of aloe-emodin and rhein
in the leaves of C. alata is confirmed, and the isolation and
identification of two new constituents, isochrysophanol and the 1-
glucoside of physcion is reported.
The second part is primarily concerned with, the chemistry of
Hibiscus (Malvaceae) , and reported herein is the isolation and
characterization of hibisconea A-C, lapachol and a new ketone, named
hibiscone D from the stems of Hibiscus tiliaceous (Fiji) . The isolat
and structural elucidation of gossypol, mansonone D and F from the roo
of the same plant from Brazil is also reported. The occurrence of
terpenoid o-and p-quinones in Malvaceae i s discussed, and the synthes
of o- and p_~naphthoquinone-peri-alde.hydes, structurally-related to the
terpenoids in Malvaceae is described.

PART A
Studies on Cassia alata
(Leguminosae)

-1-
Chapter 1
Introduction
Quinones are naturally occurring coloured compounds and arc found to
occur mainly in plants. They also occur in microorganisms, lichens and
insects. A very large number of quinones are known and they are sub-
divided into the following groups!
1. Benzoquinones
2. Naphthoquinones
3. Anthraquinones
4. Extended quinones
Anthraquinones constitute the greatest group of the quinones
and to date approximately 300 are known to occur in higher
plants. The are generally found in the following plant
families :
1. Rubiaeceae
2.Rhamnaceae
3.Leguminosae
4.Polygonnaceae
5.Bignoniaceae
6.Verbenaceae
7.Serophulariaceae
8.Liliaceae
Rubiaceae accounts for half the known number and the rest are
fairly well distributed among the remaining plant families.
Anthraquinones are generally present in the entire plant
system but higher concentrations

-2 -
are sometimes found in young leaves, sends, roots and bark.
Anthraquinones may be found in free, combined (glycosides), dimeric
and reduced forms (anthrones and dianthrones, both free and combined).
The distribution of anthraquinones in the family Leguminosae appears
to be scattered while the distribution of the major classes of secondary
constituents such as flavonoids, oligosaccharides, alkaloids, amino acids
show significant correlations with the systematics of the family
(chemotaxonomy) . Anthraquinones characteristically occur in the plants
that belong to the genus Cassia (family Leguminosae) . They are found
regularly in at least three of the subgenera. Early surveys" indicated
that they were confined to the section Chainaesenna of the subgenus
Senna. Within this section they were found only in three series
(Pachycarpae 15 spp,, Pictae 9 spp., Brachycarpre 8 spp.) and were
reported absent in the other five series. However, later detailed
surveys3 showed that they had a much broader distribution being present
within the section Chamaesenna in two other series and also occurring in
the subgenera Fistula (4 spp.) and Lasiorhegma (8 spp.). How far
anthraquinones occur in the neighbouring genera remains for future
investigation. It is of chemotaxonomic interest to note that anthra-
quinones and their derivatives have bean found in all the Cassia species
(about 40). Other groups of compounds isolated from the genus Cassia
are triterpenoids, sterols, alkaloids, flavonoids and xanthones.
Many plants that belong to the genus Cassia have been known since
the beginning of the nineteenth century because of their importance in
folklore medicine and also for their bioaesthetic (beauty) properties,
Senna* was noted for its cathartic properties" and was used mainly for
curing patients suffering from chronic constipation by great Arabian
* Senna is the accepted common name for a l l the plants that "belong togenus Cassia.

-3—
physicians. Today one will find many shrubs (all belonging to the
genus Cassia that could be used as cathartic drugs but two are quite
outstanding for their effects and are worth noting. One native in
Egypt is usually called Alexandriaii senna (Cassia acutifolia) and the
other is an Arabian shrub which was taken to Southern India and
cultivated near Tinnevelly and thus called the Tinnevelly senna
(Cassia angustifolia). Today the leaves and pods of C. acutifolia
C. angustifolia are interchangeably used in cases of chronic
constipation (laxative). They act chiefly in evacuating the contents of
the colon in six to eight hours after taking the drug. They are used
mostly as the dry powder of the leaflets after they have been cleaned
and crushed,
The other members of the genus which are medicinally important includ
Cassia fistula5 (Indian laburnum, cassia stick tree) and Cassia reticulata
The aqueous extracts of these plants show some antibacterial activity due
to the presence of an anthraquinone. Aqueous extracts from various
Cassia species called "bush tear" in the West Indies are also noted in
the folklore medicine. These extracts were used to remedy kidney
disorders, indigestion, ringworm, constipation, gout, rheumatism and
general pains of undefined origin. The active constituents in these
extracts are s t i l l to be identified but there are strong possibilities
that anthraquinones may be responsible for the remedies.'1 Extracts from
Cassia_ appendiculata have been noted for antibiotic activity and Cassia
silberiana is said to be a good duretic agent.8 Cassia glanca is used
to treat diabetes and the search for the active component is s t i l l in
progress,'! Recently an antifungal component believed to bo an anthra-
quinone was isolated from Cassia tora.9
Cassia alata commonly known in Fiji and neighbouring islands as

"ringworm bush" or "candle flower" is a pantropical shrub and a
widely used folk medicine. 10 I t is found growing wild in Fi j i 1 1
especially in the wet areas. I t grows about 2 to 4 metres high and
the stems show a tendency to creep along the ground. The flowers are
yellow and surrounded by thin yellow oblong-concave brads , One
peculiar characteristic of the Fijian C. alata which distinguishes
from plants found in other countries (India, Phi l lpines , etc.) is that
it does not produce seeds.
The leaves of C alatahave been shown to possess some laxative
action,12 antitumour activity13 and insecticidal properties.14 The
leaflets are popularly used against ringworm (a skin disease of fungal
origin which is very common in tropical countries), scabies, ulcer,
impetigo and eczema.15 The juice obtained from the leaflets after
vigorous grinding is rubbed against the body infected with the disease,
The leaves are also used as an abortifacient (strong infusion) and when
combined with flowers as an expectorant in bronchitis, dyspnea and
asthma,15 Other uses of the plant are in the treatment of Herpes
circine, darts and epidermomycoses . 1 6
Haupttnann and Nazario17 isolated from the leaves of this plant rhein
(l,8-dihydroxy-9,10-anthraquinone-3-carboxylic acid, I) and glucose
after hydrolysis of the crude glycosidic extract and also a "yellow
crystalline compound" later named18 "cassiaxanthone" (VIII) (this
compound was suspected of being an artefact resulting from alkaline
treatment of some anthraquinone present in Cassia species, however ai
later study119 suggested the possibility that i t may be not an artefact;
but a true metabolite).
*personal observation
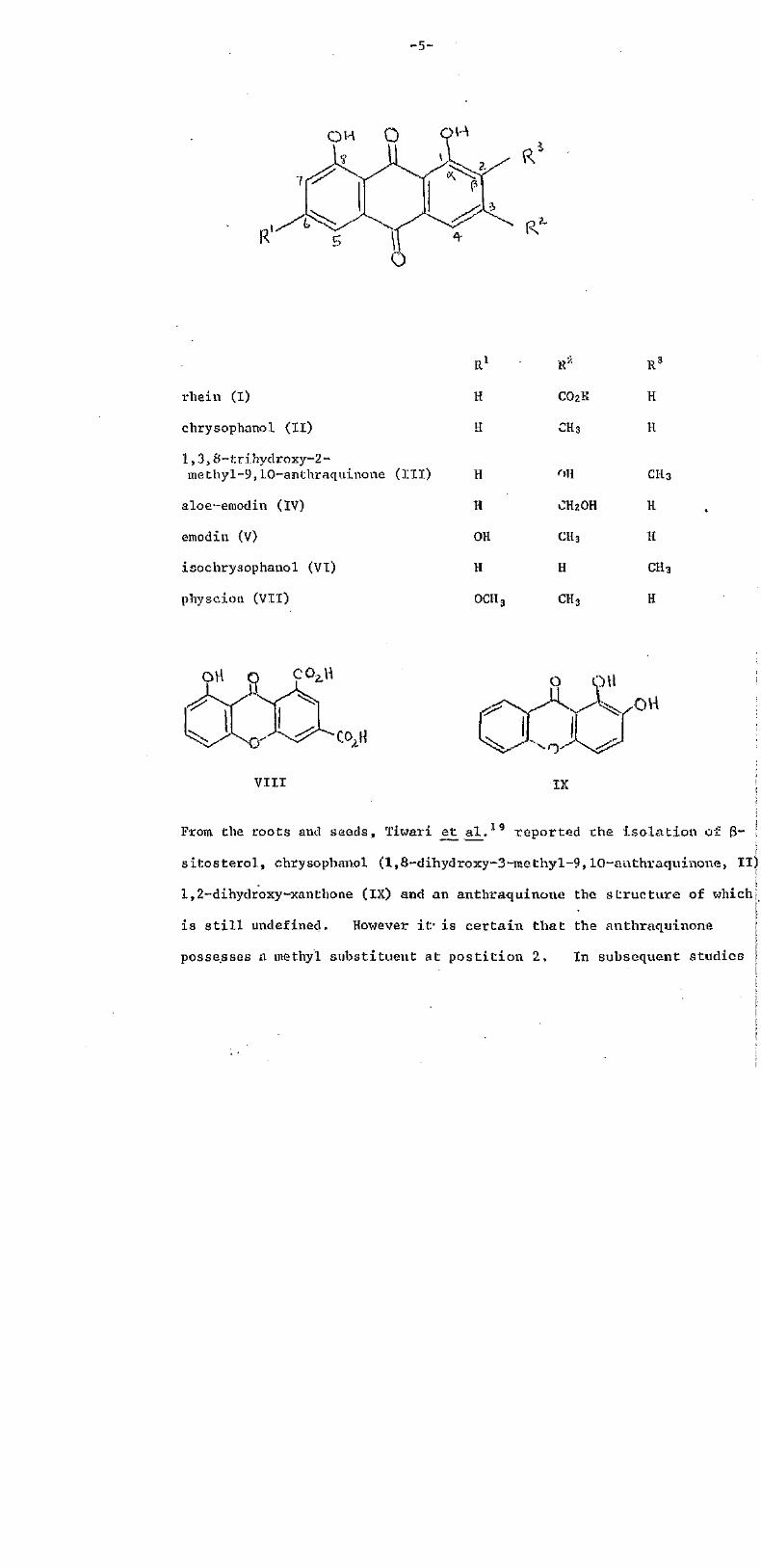
-5-
rbein (I)
chrysophanol (11)
1,3,8-triHydrosy-2-methyl-9,10-anthraquinone (III)
aloe-emodin (IV)
emodin (V)
isochrysophanol (VI)
pHyscion (VII)
R1
H
H
H
H
OH
H
OCH
CO2H
CH3
CH2OH
CH3
H
CH3
H
H
CH3
H
H
CH3
H
From, the roots and seeds, Tiwari et al. 19 reported the isolation of 3~
sitosterol, chrysophanol (1,8-dihydroxy-3-methyl-9,10-authraqquinone, II)
1,2-dihydroxy-xanthone (IX) and an anthraquinotte the structure of which
is s t i l l undefined. However i t is certain that the anthraquinone
possesses a methyl substituent at postition 2, In subsequent studies

-6-
Twari and co-workers20 showed the presence of 1,3,8-trihydroxy-2-
methyl-9,10-anthraquinone (III) and a glucoside. . (see Chapter 3).
Rao and co-workers reported two further compounds from the leaves of
C . alata, alo-emodin (1,8-dihydroxy-3-hydroxyinethyl-9 ,10-anthraquinone,
IV) and kaempferol (a flavonoid) , Aloc-ernodin (IV) was recently noted
to possess some antitumour activity. Recently Villaroya and
Bernal-Santos15 reported the isolation of emodin (1, 6,8-trihydroxy~3-
methy,1-9,10-anthraquinone, V) together with rhein ( I ) , chrysophanol (II)
and aloe-emodin (IV). To date no dianthraquinonyls have been isolated
from C. alata. The only an throne isolated i s that of rhein ( I ) . 17
**
In 197 3 Hodges and his co-workers at Massey "University carried out
in vitro studies on extracts from Fijian C. alata. They found activity
against Trichophyton mentagraphztes (ringworm) and Candida albians
mainly in the ethanolicextract. However on further fractionations
the activity was lost. In an earlier similar study on extracts from
C. excelsea, 'de Lima and co-workers 23 showed the antibiotic property
against Candida was due to rhein (1) .
In the light of these many medicinal uses attributed to the
presence of anthraquinones and their derivatives and in view of
uncertainties in the exact chemical composition, a systematic investigate
of the leaves of the Fijian C, alata was undertaken.
See page. 16
personal communication

Isolations and Identifications
Anthrquinones from C. alata
The oven dried powdered leaves of C. alata from 'Fiji were
repeatedly extracted with ethanol, and the residue obtained by
evaporation of the solvent was suspended in a minimum volume of water.
This was then exhaustively extracted with petroleum ether followed by
ether. The petroleum extract which consisted mainly of lipids and
chlorophyll was discarded and the ether fraction was partitioned into
"neutral" and bicarbonate-soluble (acidic) fractions, The neutral
fraction on column chromatography yielded two anthraquinones in
crystalline form designated as A and B, A yellow compound in high
yield was also obtained from this fraction which was identified as
kaempferol (a flavonoid). From the acidic fraction an anhraquinone
designated as C was obtained.
A. Identification of Anthraquinone A as Isochrysophanol (VI)
Anthraquinone A was recrystall ised from ethanol as yellow needles,
m.p. 175-176deg.. The purity of the compound was tested in several
chromatographic solvent systems using chromatoplates whereupon a single
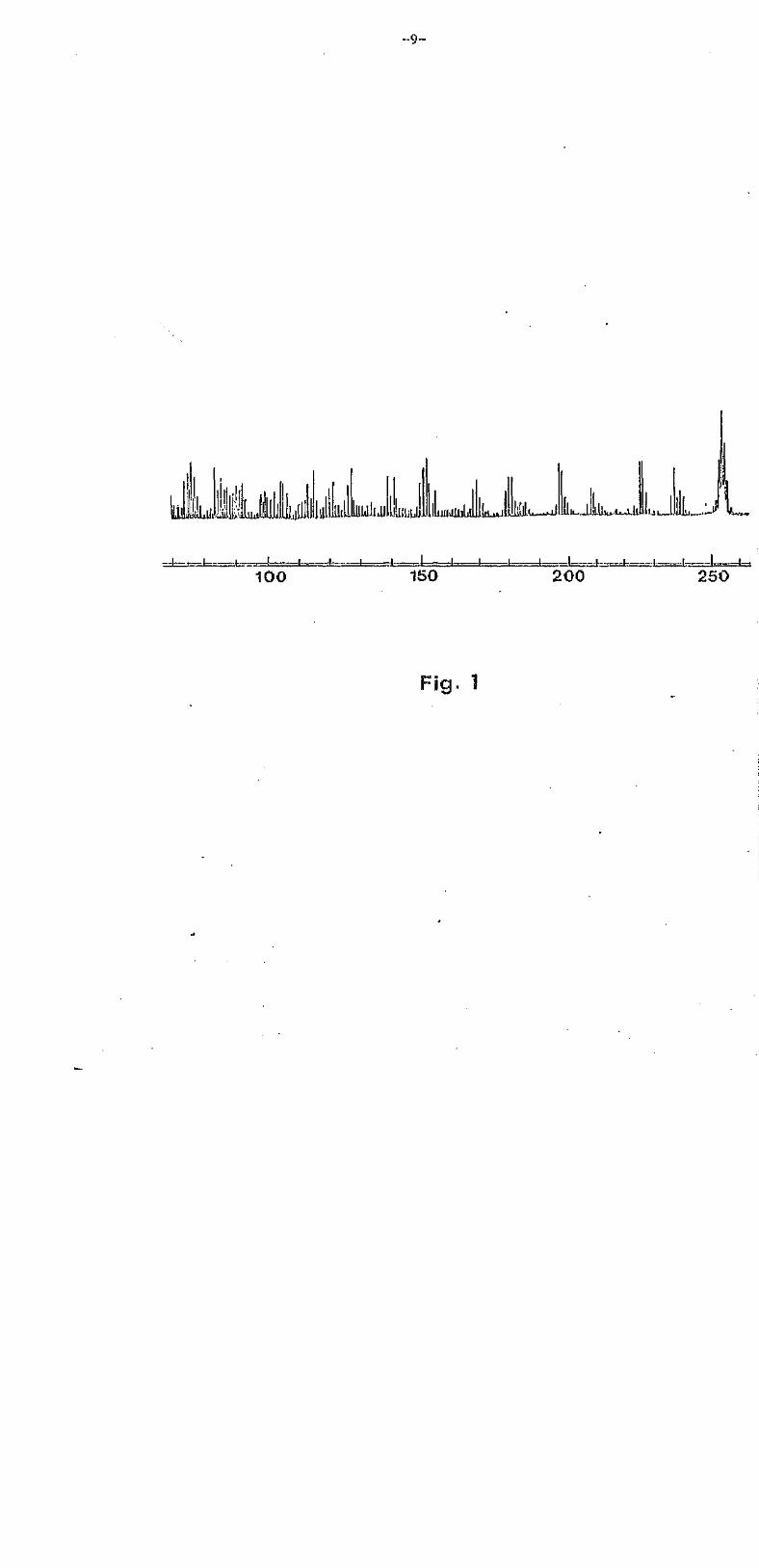
spot was obtained in each case. The M.S. spectrum i s given in Fig. 1.
High resolution measurement indicated a molecular composition of C
(Found: 254.2432, 100%. Required: 254.2426). Other ion fragments that
should be noted appeared at m/e 237 (M+ -OH,2%), 226 (M+-CO, 3.5%), 225
(M+-CH0, 3.5%), 198 (M+-2C0, 2%) and 197 (M+-CO-CHo, 3.5%). The success
elimination of two molecules of carbon monoxide from the molecular ion

-8-
is typical of anthraquinones.
The colour of A (in chloroform) changed from yellow to red on
addition of 1% inethanollc sodium hydroxide solution.1 Also a yellow
spot of this solution on a piece of filter pnper changed to orange
after being sprayed with 0.5% methanolic magnesium acetate solution
and heated at 90° for 5 min.24 These colour reactions clearly indicutt
the existence of anthraqtiinone hydroxyl groups.
On acetylation with pyridine and acetyl chloride, a diacetyl
derivative was obtained demonstrating the presence of two hydroxyl grou
The absence of 3-hydroxyl groups was Indicated by its insolubility in
aqueous sodium carbonate25 and its solubility in aqueous sodium hydroxl
solution,1 Based on this information the two a-hydroxyl groups might fa
expected to be present at one of the relationships: 1:4, 1:5 or 1:8.
The U.V. .spectrum [A (EtOII) 228, 256, 287 and 432 nm] resemblemax .
that of 1,8-dihydroxyanthraquinones 26 and in particular chrysophanol (t
[A (EtOH) 228, 257, 287 and 429 nm] very closely. From the work
Briggs and collaborators27 and of Birkinshaw26 it is known chat the ;
anthraquinone absorption band in the region 320-480 nm is strongly
influenced by the number of a- hydroxyl groups, while B-hydroxyl groups
have little influence. In unsubstituted anthraquinone this band is at
360 nm and the presence of two a-hydroxyl groups shifts it to the
420-450 ran region. Also a band in the 480-520 nm region indicates two
hydroxyl groups in 1:4 relationship. Since anthraquinone A had Amax
at 432 nm and none at longer wavelength it was taken that the two
hydroxyl groups were either at positions 1:8 or 1:5. The possibility !
of 1:5 relationship of the two a-hydroxyl groups was finally eliminated
after a thorough study of the I.R. spectrum of the pigment. It is kno
from the work of Bellamy28 that singly chelated anthraquinone carbonylsi
- 9 -
!!!!!!!!!!!!
!! !!!!!
100J I _ I ! I
150!I. _! ! . _ . ! , , .| !
200 250
Fig. 1

-10-
absorb in the region 1631-1637 cm"1 in which anthracquinone A had no
absorption indicating the absence of 1:5 and also 1:4 relationships.
In the I.R. spectrum carbonyl absorptions were seen at 1670 and 1620 cm"1
representing unchelated and chelated carbonyl groups," respectively.
Also in the spectrum there was no definite hydroxyl band which in fact
further supported the total absence of any 3-hydroxyl group. Briggs
and Nicholls30 and Bloom et al.29 have shown that the anthraquinones
having hydroxyl groups in a-positions do not exhibit the characteristic
hydroxyl frequency in the region 3600-3250 cm-1 due to some form of
association of those hydroxyl groups with the carbonyl groups,
Moreover according to Bloom et al.29 the presence of of 8~hydroxyl groups
in an anthraquinone can be ascertained by r.hs appearance of a peak in
the region of 3600-3250 cm"1.
The P.M.P. spectrum of the compound in deuterochJ.orof orm is given in
Fig. 2. The two signals in the downfield region 12.02 and 12.. 426 are
ascribable to chelated and free hydroxyl groups and a three proton
singlet at 2.2.78 is due to a methyl group. Complex signals from
benzenoid protons arc seen in the aromatic region (7.48 to 7.80 tS) as
expected.
In the light of the above experimental information the obvious
candidate was chrysophanol (II) which is fair ly widely distributed in
Cassia species. However the melting point of anthraquinone A (175-176°)
was different from that of chrysophanol (196°).1
The possibility of the anthraquinone being chrysophanol was
eliminated on a more cogent evidence since a marked depression was
observed in the melting point of the anthraquinone when mixed in equal
proportion with an authentic samp,le of chrysophanol ( I I ) , However, no
such depression was observed in the case with an authentic sample of

-11"
15
\
10 8
* CHCl3
6 4
F i g . 2
2 0

-12-
isochrysophanol (VI). The melting point (175-176°) of isochryso-
phanol reported31 corresponds well with A. Therefore the anthraquinone
was identified as isochrysophanol (l,8'-dihydroxy--2-methylanthraquinone,
VI) and this is the f i rs t report of i t s isolat ion from C. alata.
The only other Cassia species in which isochrysophanol i s reported to
occur is C.occidentalis.32
B. Identification of Anthraquinone B as Aloe-emodin (IV)
The neutral fraction when subjected to column chromatography and
eluted with chloroform afforded a second anthraquinone (B ) . This
anthraquinone was recrystallised from acetone into orange-yellow needles,
m.p. 222-223°. The purity of the compound was checked on thin-layer
chromatoplates whereby a single spot was obtained in each chromatographic
solvent system. The high resolution M.S. measurement showed the
molecular formula to he C15H10O5 [Found: 270.2452, 100%, Required
270.2420]. Diagnostically significant ions occurred at m/e 253 (M+ -OH,
1.5%), 242 (M+-CO, 56%), 241 (M+-CHO, 63%), 239 (M+-CH2OH, 2%), 214
(M+-2C0, 2%), and 213 (M+-CO-CHO, 5.5%). The molecular ion peak
occurred 16 mass units higher than that of isochrysophanol (VI). This
indicated the possibility of the anthraquinone being one stage further
oxidised than isochrysophaol, since this type of pattern is frequently
encountered in nature. I t is also worth noting the similari ty of the
fragmentation pattern with those of the previous anthraquinone. For
example successive loss of two molecules of carbon monoxide from the
molecular ion.
The colour reactions were very similar with those of isochrysophanol,
suggesting a polyhydrosyanthraquinone. I t gave a pink colouration with

-13-
0.5% methanolic solution of magnesium acetate 21 and a red colouration
with 10% methanolic potassium hydroxide solution. The anthraquinone
was insoluble in 5% aqueous sodium carbonate solution and soluble in
aqueous sodium hydroxide solution, thereby showing the absence of any
B-hydroxyl groups. The compound was also insoluble in 5% aqueous
sodium bicarbonate solution indicating the absence of a carboxyl group.
A diacetyl derivative was obtained on acetylation with dried pyridine
and acetyl chloride at room temperature.
I t was certain that the pigment was a dihydroxyanthraquinone and
had one of the three possible relationships: 1:4, 1:5 and 1:8. The
possibility of the compound having either 1:4 or 1:5 relationship was
eliminated with the help of U.V. and I.E.. data. The electronic
spectrum [A (EtOH) 225, 255, 277, 288, 432 and 457(sh) ran] resembled
that of aloe-emodin (IV) [A (EtOH) 225, 254, 276.5, 287, 430 and
457(sh) nm] very closely. The bathochromic shift observed (430 to
510 nm) by addition of sodium hydroxide solution indicated that the
hydroxyl group was peri to the carbonyl function. This observation
in fact goes further in confirming the existence of only two cx-hydroxyl
groups. The absence of absorption in the 480-520 nm region eliminated
the possibility of a 1:4 relationship.2 6 , '2 7 The I.R. spectrum had
significant bands at 3400 (free hydroxyl), 1675 (free carbonyl) and
1631 cm-1 (chelated carbonyl). Since the band at 1675 (chelated carbony)
is absent in the I.R. spectra of 1:4 and 1:5 dihydroxyanthraquinones
(all phenolic hydroxyls are involved in hydrogen bonding with the two per:
carbonyls), the presence of this band in the spectrum of anthraquinone B
confirmed the 1:8 relationship (one carbonyl hydrogen bonded to one
hydroxyl and the other carbonyl bexng free) .28-30
Examination of the P.M.R. spectrum revealed a l l the peaks expected of

-14-
aloe-emodin in CD3OD, Signals for the benzenoid protons appeared at
7.22-7.82 6 as a 5 proton multiplet while the methlene protons of the
hydroxy methyl appeared as a two proton singlet at 4.608
The above spectroscopic data together with the chemical information
suggested that compound B was aloe-emodin (IV). The identity was
confirmed by mixed melting point and co-chromatography (in several solvenl
solvents) with an authentic sample of aloe-emodin.
It should be noted that as speculated earlier aloe-emodin is indeed
one stage further oxidized than isochrysophanol but the position of the
substituent in this case is at 3 whereas in isochrysophanol it is at 2,
Therefore it is assumed that the biogenetic pathway of isochrysophanol
is different from that of aloe-emodin in C. alata.
C. Identification of Anthraquinone C as Rhein (I)
This was the last of the free anthraquinones and was isolated from
the acidic fration of the crude extract, The anthraquinone was re-
crystallised from methanol affording dark yellow crystals, m.p. 315-316°.:
Again the purity of the pigment was tested on thin-layer chromato-
plates in various different chromatographic systems whereby a single
anthraquinone spot was obtained in each case. The compound readily
dissolved with effervescence in 1% aqueous sodium bicarbonate solution
which was indicative of the presence of a carboxyl group. This was
confirmed by the appearance of peaks at 3100 and 1680 cm"1 in the I.E..
spectrum.
High resolution M.S. of the anthraquinone indicated a molecular form
of C15H8O6 [Found: 284.2301, 100%, Required: 284.2254], Prominent
peaks appeared at m/e 267 (M+-0H 3.5%), 256 (M+-C0, 5%), 277 (M+-CO.-CHO,

-15-
2.5%), 228 (M+-ZCO, 3%), and 239 <M+-CO2H 4.5%). After comparing this
fragmentation pattern with those of isochrysophanol and aloe-emodin i t
was quite conclusive of this anthraquinone being also a dihydroxyquinone.
As with the previous two anthraquinones the colour reactions were positive
(suggesting a polyhydroxyanthraquinone) . On acetylation with pyridine
and acetyl chloride a diacetyl derivative was obtained demonstrating the
presence of two hydroxyl groups, As usual, the position of the two
hydroxyl groups was then solved with the help of U.V. and I.R. data.
Briggs and Nichols"29 and Bloom et al29 have shown that the anthra-
quinones having hydroxyl groups in a-position do not exhibit the
characteristic hydroxyl frequency in the region 3600-3250 cm"1 of I.R.
Moreover according to Bloom et al.29 the presence of g-hydroxyl groups
in an anthraquinone nucleus can be ascertained by the appearance of a
peak in the said region. Anthraquinone C did not show this band
indicating the abssnce of any free f3-hydroxyl group. The band at
3100 cm"1 was indicative of the hydroxyl of a carboxylic group. 33
It was fairly certain like the other anthraquinones that the G had
also one of the following combinations: 1:4, 1:5 or 1:8. The U.V.
absorption A (EtOH) 431 nm indicated the presence of two a-hydroxyl
groups (Briggs et al. 27 and Berkinshaw26 ) . According to Bloom et a l . 2 9
if an anthraquinone contains two a-hydroxyl groups at positions 1:4 or
1:5 there appears a single peak in the carbonyl stretching frequency
region in the I.R. for the chelated carbonyl groups only which l ies between
1645 and 1608 cm -1. The I.R. spectrum of anthraquinone C did not show
this band in the carbonyl stretching frequency region and therefore was
not according to the aforesaid observations. This clearly indicated the
absence of two a-hydroxyl groups at positions 1:4 and 1:5. Thus the only
possibility left was 1:8 for the two hydroxyl groups (confirmed by

-16-
magnesium acetate test 24) .
Because of the small amount of material available and also its
relative insolubility in a suitable solvent, the P.M.R spectrum was
not obtained. However together with the above spectroscopic
information and chemical data i t was possible to identify the anthra-
quinone as rhein (I) . The identity was confirmed by mixed melting
point and co-chromatography (in several solvent systems) with an
authentic sample of rhein.
Thus the present work confirms the presence of rhein (1) and
aloe-emodin (IV) in C. alata and reports a new constituent isochrysophanol
(VI). Although the separations were monitored by T.L.C. no further
free anchraquinones appeared to be present despite the earlier reports
of chrysophanol (II) 5 l.3,8-trihydroxy-2.-methyl-9,l0-anthraquinone
(III) and emodin (V) .
Villaroya and Bernal-Santos1 from their work on C. alata obtained
five anthraquinones and assigned one of them (a liquid) to chrysophanol
(II) . The author finds no justification in their assignment since the
sample was non-crystalline and thus grossly impure, Their reported I,R.
and U.V. spectra do not agree and even the RF, value is wrong. The I.R.
spectrum suggests a fatty acid ester or glyceride which would be a liquid,
If the sample did contain an anthraquinone i t could have equally been
isochrysophanol (VI) - the new constituent isolated in the present work.
-17-
Chapter 3
Isolation arid 'Identification
of Glycosides
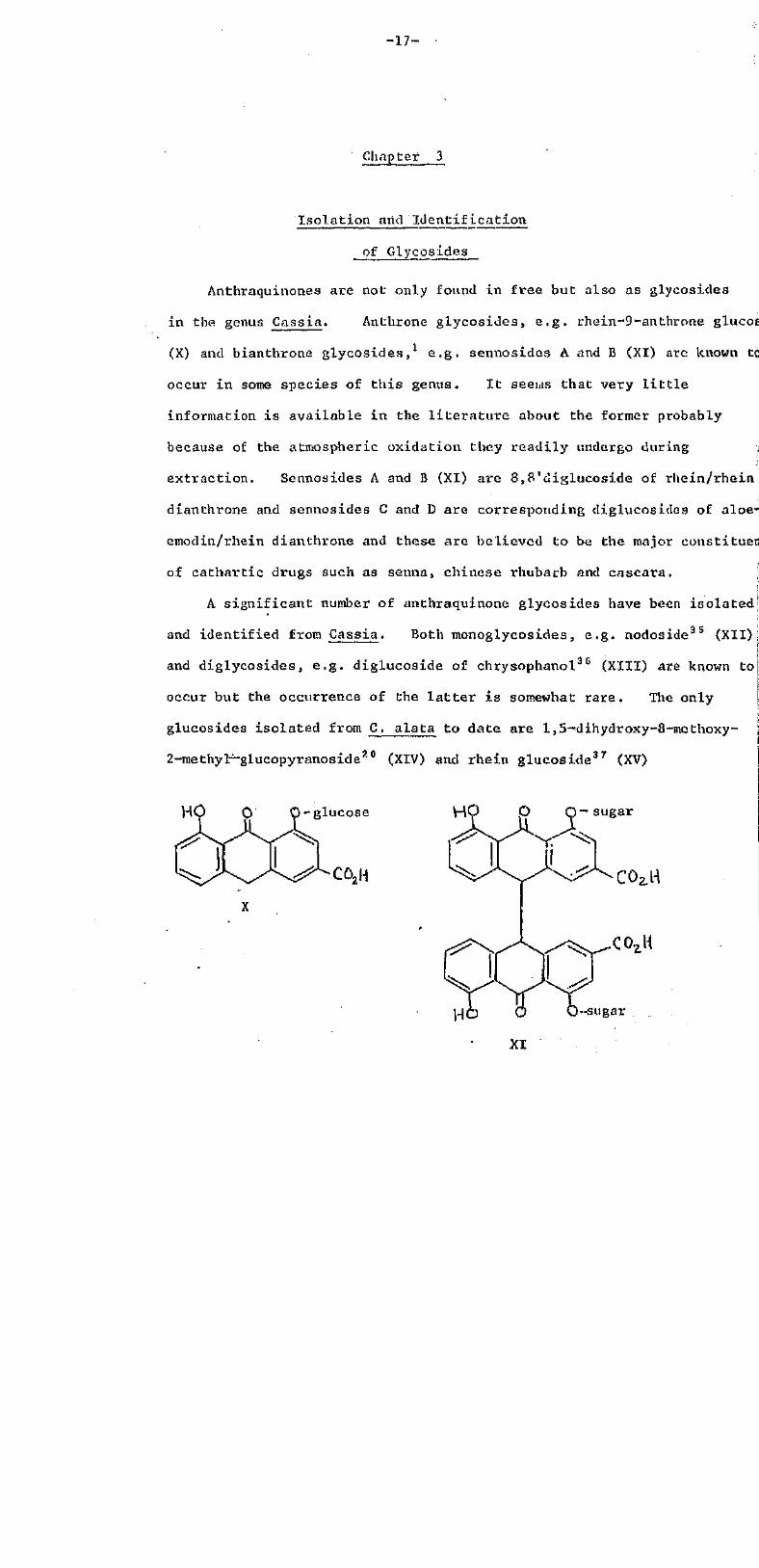
Anthraquinones are not only found in free but also as glycosides
in the genus Cassia. Anthrone glycosides, e.g. rhein-9-an throne glucose34
(X) and bianthrone glycosides,1 e.g. sennosides A and B (XI) arc known tc
occur in some species of this genus. It seems that very little
information is available in the literature about the former probably
because of the atmospheric oxidation they readily undergo during ,
extraction. Sennosides A and B (XI) are 8,8'diglucoside of rhein/rhein
dianthrone and sennosides C and D are corresponding diglucosiddes of aloe-
emodin/rhein dianthrone and these are believed to be the major constituents
of cathartic drugs such as senna, Chinese rhubarb and cascara,
A significant number of anthraquinone glycosides have been isolated
and identified from Cassia. Both monoglycosides, e.g. nodoside35 (XII)
and diglycosides, e.g. diglucoside of chrysophanol36 (XIII) are known to
occur but the occurrence of the la t ter is somewhat rare. The only
glucosides isolated from C. alata to date are l,5~dihydroxy-8-methoxy-
2-methy-l-glucopyranoside20 (XIV) and rhein glucoside37 (XV)
H -glucose
CO2H
H ~ sugar

-18-
glucose-o O
XII
glucose- O- glucose
H3CO Q OK
HO-D-(+)-glucopyranosyl
XIV
HO O glucose
-glucose
XVI

-19-
In the present work a new glucoside was obtained from C. alata
and structurally characterised as physcion-l~glucoside (XVI).
The oven dried powdered leaves were successively extracted with
chloroform and ethanol, The chloroform extract was discarded since
it consisted mostly of chlorophyll, lipids, free anthraquinones and
flavonoids. The crude ethanol extract was suspended in water and
extracted many times with ether to remove last traces of free anthra-
quinones and flavonoids followed by extraction with n-butanol. The
n-butanol extract was concentrated and then redissolved in methanol.
The methanolic solution was then added to a large volume of ether to
give an immediate precipitation. The brown precipitate was filtered
off and dried under vacuum. The precipitate was found to be very
-a
hygroscopic. The dried precipitate was subjected to column
chromatography using polyamide30 as the adsorbent. On elution with
methanol, the fast running band was collected and on crystallization
from methanol gave as yellow powdery material. The powder gave
positive flavonoid test and on acid hydrolysis afforded kaempferol and
glucose (identified by paper chromatography). No attempt was made to
determine the position of the sugar moiety on the flavonoid nucleus, A
slower moving band was also eluted and this gave positive results when
tested for anthraqttinone and sugar (Molisch's test). This fraction when
crystallized from methanol-chloroform (1:1) gave orange yellow needles
which on mild acid hydrolysis yielded an anthraquinone (aglycone) and a
sugar.

-20-
Identification of aglycone as Physcion (VII)
The aglycone obtained from acid hydrolysis was recrystal l ised from
hexane-toluene (1:1) as yellow needles, m.p. 205-206°. As usual the
purity of the compound was tested in various solvent systems whereby a
single spot was been in each case. The mass spectrum of the compound
is given in Fig. 3. High resolution mass measurement gave the molecular
formula as C16H12O5 [Found: 284.3029, 100%. Required: 284.2690]
The rest of the mass spectrum had a fragmentation pattern very
similar to those oil anthraquinones isolated e a r l i e r . The colour
reactions were characteristic of dihydroxyanthraquinones .1,24 The
dihydroxy nature of the pigment was demonstrated by preparing the
diacetyl derivative. The dye like others was insoluble in 5% aqueous
sodium carbonate solution but soluble in 10% aqueous sodium hydroxide
solution,1 thus clearly indicating the absence of any 3-hydroxyl substituents
The I.R. spectrum of the anthraquinone did not show any peak in the region I
3600-3250 cm"1 which farther supported the absence ofg-hydroxyl groups.29,30
Significant peaks were only seen at 1670 and 1620 cm"1 thus showing the
chelated nature of one of the carbonyl groups.28 In other words for this
to be true the two hydroxyl groups can only be at positions 1 and 8. i
The U.V. spectrum had an absorption maximum at 434 ran, again a clear
indication of two a-hydroxyl groups.26'27 The P.M.R. spectrum in DMSO-d6
contained signals for the benzenoid protons in the 6.60 to 7.60 6 region, |
and signals for the two phenolic protons appeared as singlets at 12,15
and 12.26 6. A three proton, singlet at 2,45 6 was almost identical in
chemical shift to that of the methyl protons in isochrysophanol (Fig. 2).
Also an additional three proton singlet appeared at a much lower field
(3.92 6).

- 2 1 -
100 150 2001 1 1 1 11—..
250 284
Fig .3

-22-
The above spectrOscopic data together with chemical informatioN
were in agreement with those of physcion (VII), The identity was
confirmed by use of an authentic sample of physcion (mixed melting point
and co-chromatography) .
OVA
CH3
VII
o OH
XVII
Identification of glycoside sugar
The aqueous hydrolysate which consisted mostly of sugar from the
glycoside was neutralised and the volume reduced. With the aid of
standard sugars and using the decending paper chromatography technique
the sugar was identified as glucose,

-23-
Determination of the position of glucose on the aglycone (physcion)
The glucoside was permethylated using dimethyl sulphate /potassium
carbonate and then hydrolysed under mild acid conditions. On reduction
of the volume of the reaction mixture, a residue was obtained which on
recrystallisation from methanol furnished reddish brown needles, m.p,
177-179°. This was identified as l~hydroxy-3-methyl-6 ,8-dimethoxy-
9,10-anthraquinone (XVII) (lit./0 178°).
The isolation of (XVII) clearly eliminated the possibility of the
sugar moiety being attached at the 8-hydroxyl in the glucoside, Thus
the glucoside was characterised as 1glucoside of 1,8-dihydroxy-6-
methoxy-3-methyl-9,10-anthraquinone (XVI). This glucoside is known to
occur in only one species of the genus Cassia, i.e. C. occidentalis.-1
The present work thus reports a new constituent 1-glucoside of physcion
(XVI) in C. alata. The presence of l,5-dihydroxy-8-methoxy-2-methyl-
glucopyranoside (XIV) and rhein glucoside (XV) as reported earlier could
not be confirmed using the above techniques.

- 2 4 -
Chapter 4
Experimental
Melting points were measured on a Koflcr hot stage microscope
and are all corrected.
Electronic spectra (U.V.-visible) were recorded on a Perkin Elmer
402 Ultra-visible spectrophotometer in ethanol (95%) . Shoulder peaks
are indicated as (sh) and log e values are given in parenthesis.
Infrared spectra (I.R.) were recorded on a Perkin-Elmer 177 Grating
spectrophotometer in potassium bromide discs. Only the main absorptions
of interest are reported, Intensities are reported as strong (s) ,
medium (m) or weak (w) , In some cases important assignments are also
made in parenthesis.
Mass spectra (M.S.) were recorded on an AET MS 30 high resolution mass
spectrometer and peak intensities are raported as a percentage of the
base peak.
The Proton Magnetic Resonance (P.M.R..) spectra were recorded on a
Varian HA 100D model. The solvent is specified in each case. Chemical
shifts are reported in ppm on the g scale using tetramethyslilane as the
internal reference. The proton integral and spin multiplicity (s =
singlet, d = doublet, t = t r iplet , q = quartet and m = multiplet) are
given in parenthesis immediately after the chemical shift. Where
possible assignments are also made.
All solvents used were analytical grade. Analytical thin-layer
chromatography (T.L.C.) was carried out on 20x20 cm plates using 0.25 mm
layer of Merck Silica Gel GF254.

- 2 5 -
All known compounds were identified (U.V.-visible and IR. spectra;
co-t.l .c. by direct comparison with authentic specimens. Spots of the
anthraquinones dissolved in ether (or acetone) were placed on si l ica
gel plates and developed with
(i) chloroform (neat; solvent system 1)
(ii) chloroform:methanol (9:1; solvent system 2)
(ii i) ethyl acetate:methanol:H2O (100:16:14; upper phase;
solvent system 3)
and (iv) ethyl acetate:benzene (1:1; solvent system 4)
All extractions were carried out at room temperature (24-26°) .
Extraction of Free Anthraquinones
The oven dried powdered leaves (collected from the fields opposite
Buchurst Park, Laucala Bay, Suva, Fiji Islands) of C. alata (2 kg) were
extracted many times with 95% ethanol (4 £) in flasks of 5 l i t r e capacity
until the extract was almost colourless. The dark greenish brown
viscous residue (65 g) obtained by evaporation of the solvent in vacuo
was suspended in distilled water (300 ml) and then extracted repeatedly
with light petroleum (b.p, 40-60°; 6x200ml) followed by diethyl ether
(6x200 ml) .
* the authenticity was checked by submission of samples of plant material
to the Department of Botany, Ministry of Agriculture, Fisheries and
Forests (MAFF), Rodwell Road, Suva, F i j i Islands.

1. Light petroleum extract
The dark green solution was reduced in volume using a rotary
evaporator and a spot of the crude extract was placed on a t . l . c . plate
and developed in solvent system 1. The chromatogram was dried and
sprayed with 10% inethanolic potassium hydroxide solution whereby negative
results were obtained for both anthraquinones and flavonoids. This
fraction consisted mainly of chlorophyll and lipids and so was discarded.
II . Diethyl ether extract
The solvent was removed in vacuo and similarly tested for anthra-
quinones and flavonoids. The dried chromatogram on spraying with NaOH
gave three spots that immediately changed to red and one spot that-
changed to yellow. The solvent was disti l led off and the residue. (5.4 g)
was thoroughly extracted with benzene )6x100 ml). The remaining
(insoluble) material (3.2 g) was shown to contain a significant proportion
of kaempferol (U.V., I.E.. with an authentic sample), and so was not
examined any further.
The benzene extract virtually contained a l l the free anthraquinones.
The residue (2.2 g) obtained by evaporation of the solvent in vacuo was
redissolved in ether (100 ml) and fractionated into "neutral" and
bicarbonate-soluble (acidic) fractions by repeatedly extracting with 1%
aqueous sodium bicarbonate solution (6x100 ml).

-27-
A. Diethyl_ ether (neural)fraction
(i) Fractionation
The solvent was distilled off and the residue (1,2 g) was
chromatographed on silica gel (150 g). Elution with toluene gave a
solid which was crystallised from ethanol to give isochryaophanol (VI)
(154 mg) aa yellow needles, in.p. 175-176° (lit.,31 176°); Rf: 0.90,
0.76, 0.51 and 0,47 in solvent systems 1, 2, 3 and 4, respectively; X
(EtOH) 228 (log e 4.30), 256 (4.18), 277 (4.01), 287 (4.01), and 432 nm
(4.14) (bathochromic shift to 512 nm with NaOH); V (KBr) 1670 (m,max.
unchelated C=0), 1620 (s, chclated C-0), 1580(sh) (C=C aromatic skeleton),
1670 and 1420 (m, phenolic OH), 1380 (broad, C-H of CH3), 1160 and 1070
(m, C-0 of phenolic OH), and 580 and 750 cm"1 (s, aromatic substitution);
see Figs. 1 and 2 for M.S, and P.M.R., respectively; (Found: M+
254.2432. C15H10O4 requires M, 254.2426).
The yellow anthraquinone was soluble in many organic solvents such
as chloroform, methanol, acetone, etc. and insoluble in 5% aqueous sodium
carbonate solution. It gave a deep red colour when dissolved in methanol
with a few drops of 2M aqueous sodium hydroxide solution. Spots of the
solution in acetone on a piece of filter paper turned orange on spraying
with 0.5% tnethanolic magnesium acetate solution and heating at 90° for
5 min.
Further elution of the column with chloroform gave a solid which was
crystallised twice from acetone to give aloe-emodin (IV) (215 mg) as
yellow orange needles, in.p. 222-223° (lit.,1 223°); Rf: 0.83, 0.76, 0.62
and 0.54 in solvent systems 1, 2, 3 and 4, respectively; X (EtOH)
, max,225.5 (log e 4 .56) , 255 (4.31), 277 (3 .98) , 288 ( 4 . 0 0 ) , 432 (4 .01) , and457 nm (sh) (3.86) (bathochromic s h i f t to 510 nm with NaOH); V (KBr)
max.

-28-
3400 (s, OH of hydroxymethyl) , 1675 (w, free 0 0 ) , and 1631 cm"1 (s,
chelated C=0); g (CD30D) 4..60 (2H, s, 3-CH2OH) and 7.22-7.82 (511, m,
benzenoid protons) (Found: M+, 270.2452. C15H10O5 requires M, 270.2420).
The dark orange yellow anthraquinone was soluble in many organic
solvents such as chloroform, methanol, acetone, etc., and insoluble in
5% aqueous sodium carbonate solution. The yellow chloroform solution of
the anthraquinone immediately changed to red on addition of a few drops of
sodium hydroxide solution (2M) . The solution was also shotted on a piece
of filter paper and sprayed with 0.5% methyl alcoholic magnesium acetate
solution. The colour of the spot changed to orange after being heated
at 90° in the oven for 1.5 min.
(ii) Diacetate of isochrysophanol (VI)
To the ice-cold solution of (VI) (100 mg) in distilled pyridine
(5 ml) acetyl chloride (5 ml) was gradually added. The reaction
mixture was kept in the cold room for approximately 14 h. and then poured
into ice-cold distilled water (100 ml) whereby immediate precipitation
took place. The precipitate was collected, washed with cold and hot
water, dried and crystallised twice from chloroform-methanol (4:1) giving
yellowish green needles, m.p. 145-146° (63.5 mg); mixed m.p. with the
diacetate prepared from authentic isochrysophanol, 144-146°,
(iii) Diacetate of aloe-emodin (IV)
Aloe-emodin (100 mg) was acetylated with acetyl chloride (4 ml.) in
distilled pyridine (5 ml) . After work up the diacetate derivative was
crystallised from acetone as yellow plates (74 mg), m.p. 172-173°
(lit.,21 mp. 1.72°).

-29-
B Sodium bicarbonate (acidic) fraction
The aqueous sodium bicarbonate solution was treated with 2M HCl
(50 ml) and extracted exhaustively with ethyl acetate (4x100 ml).
The extracts were combined and dried over anhydrous sodium sulphate.
The residue obtained after removal of solvent was recrystallised from
methanol to give rhein (1) as orange yellow needles (32.5 ing), m.p.
316-318° (decomp.) (lit.,21 315-317° (decomp.); Rf: 0.45 and 0.38 in
solvent systems 3 and 4, respectively; X (EtoH) 228 (log e 4.56),
258 (4.35), 288 (4.01), and 431 ran (4.04) (Bathochromic shift to 512 nm
with NaOH); V (KBr) 3100 (s, OH of carboxyl), 1680 (s, free C=0),
and 1620 (s, chelated 00) (Found: M+, 284,2301. C15H8O6 requires
M, 284.2254).
The dark brown anthraquinone was found insoluble in most organic
solvents and only partially soluble in methanol. It was soluble in
DMSO. It dissolved with effervescence in 5% aqueous sodium carbonate
solution and also in 1% sodium bicarbonate solution. It gave a red
colour with godium hydroxide solution and an orange spot was revealed
after spraying a spotted piece of filter paper and heating for 5 min.
Rhein (70mg) was acetylated as above with acetyl chloride (2 ml)
in distilled pyridine (5 ml). After work-up, the acetyl derivative
was crystallised from methanol (9.2 mg), in.p 243-245° (decomp,) (lit,21
242-244° (decomp.)). Identified as the diacetyl derivative.

-30-
Isolation of Glycosides
For this extraction a slightly different procedure was chosen.
The powdered leaves (1.5 kg) were extracted exhaustively with
chloroform followed by 95% cthanol (4 l) . The chloroform extract was
left aside since this consisted mostly of lipids, chlorophyll, free
flavonoids and anthraquinones.
The ethanolic extract was concentrated to a dark brown syrup
(-100 g) using a rotary evaporator. The syrup was suspended in
distilled water (500 ml) and extracted repeatedly with ether (3x50 ml)
to remove the last traces of free anthraquinones and other compounds.
This was followed by n-butanol (3x500 ml) . The residue obtained after
removal of n-butanol was redissolved in methane1. (250 ml) and added to a
large volume of ether (2 l) , The precipitate which was immediately
formed was collected quickly by vacuum (very hygroscopic) and subjected
to column chroma tography using polyaraide33 (150 g) as the absorbent
and methanol as the only eluting solvent. Two relatively clear bands
were obtained. The fast moving band (yellowish) after necessary
purification ( t . l .c . ) gave positive results for both flavonoid (dark
yellow with NaOH) and sugar (Molisch's t es t ) . The aglycone obtained on
mild acid hydrolysis had identical U.V., I.R. anu chrotnatographic
behaviour with that of kaempferol. The slow moving band (reddish brown)
was collected and the residue obtained after the removal of solvent was
crystallised from chloroformmethanol (9:1) to give 1-glucoside of
physcion (XVI) as orange-yellow needles (very hygroscopic) (250 mg),
m.p, 228-234°. I t was insoluble in most non-polar organic solvents
(chloroform, ether, petrol, etc.) whereas i t was very soluble in methanol
and water. It gave the colour reactions of anthraquinone (red with

— 3 1 —
sodium hydroxide solution and a colour with .5% methyl alcoholic
magnesium acetate solution after being heated for 5 min., which was hard
to distinguish between orange and purple) and gave positive Mool isch ' s
t e s t .
Physcion-l~glucoside (140 mg) was dissolved in methanol {100 ml)
and 7% aqueous sulphuric acid (50 ml was added to i t and hydrolysed
by refluxing for 2 h. over a steam bath. The reaction mixture wasa
then cooled, diluted with excess of water and extracted with ether
(6x100ml) . The ether extract was washed with d i s t i l l e d water to
remove traces of mineral acids and dried over anhydrous sodium
sulphate. The residue obtained after the removal of solvent was
crystallised from hexane-Toluene (1:1) to give physcion (VII) (94 mg) as
yellow needles, M.p. 205-206° ( l i t , 1 207°); RE0,51 and 0.34 in
solvent systems 3 and 4 respectively; \ (EtOH 255(sh) (log G 4.20),
265(sh) (4.19), 289(4.20), and 434 nM (4.05) (bathochromic shift to
512 nM with NaOH v (KBr) 3200 (w, OH), 1670 (w, unchelatedi C=0),
and 1620 cm"1 (s, chelated 0 0 ) ; 6 (CDCl3) 2.45 <3H, 3, 3-Me), 3.92 (3H,
s, 6-0Me), 6.60 (IH, d, 7-H), 7.04 (1H, bs, 2-H), 7.32 (1H, d, 5H) ,
7.57 (lH, brs, 4-H), 12.-15 and 12.26 (lHeach, s, 8 and1-OH) (Found: M+,
284.3029. C16H12 O5 requires M, 284.2690).
Physcion was found to be soluble in methanol, acetone and chloroform.
I t was soluble in 5% aqueous sodium hydroxide solut ion giving a red
coloured solution and insoluble in 5% aqueous sodium bicarbonate solution,
An orange spot was seen after spraying with 0.5% methanolic magnesium
acetate solution and heating at 900 for 5 rain.
Physcion (40 mg) was acetylated as above with acetyl chloride (2.5
in dis t i l led pyridine (5 ml) , After work up the acetyl derivative was
crystallised from methanol as yellow-green needles, (16.5 mg), m.p.

-32-
186-187°, mixed m.p. with diacetate prepared from authentic physcion,
185-186°.
The aqueous sugar solution of the hydrolysate a l ter neutralizing
with finely powdered barium hydroxide 0 2.5 g) was fil tered and
concentrated to 1 mi using a rotary evaporator. The f i l t r a t e along
with standard sugars ((+)- glucose, fractose, galactose, and sucrose)
were spotted on Whatman No, 1 paper and the chromatogram was run in
pyridine:ethyl acetate:water (20:50:70) for 24 h. using the decending
paper chromatography technique.''2 The chromatogram was dried in the
fume hood and then dipped into a plate containing saturated aqueous
silver nitrate solution (0.3ml) in acetone (1.00 mi) . The chromatogram
was dried, sprayed with aqueous sodium hydroxide solution (2M), washed
with water to remove remaining silver ions and then dried in the oven.
at 110-120°. Examination of the chroma to grain after 10 min. revealed
only a single dark spot from the f i l t ra te which had the same Rf value
as that of glucose.
Methylaton of glucoside (XVI) and hydrolysis of (XVII)
Physcion-l-glucoside (XVI) was dissolved in dry acetone (20 ml)
and refluxed with dimethyl sulphate (2.5 ml) and anhydrous potass ium
carbonate (5 g) for 24 h. The reaction mixture was cooled, fi l tered
and solvent removed In vacuo. The residue was taken up in water (50 ml)
and concentrated ammonia solution (10 ml) was added and warmed over a
steam bath to destroy the unreacted dimethyl sulphate. The aqueous
suspension was then extracted repeatedly with chlorcforni (3x25ml).
The extracts were combined, dried'over anhydrous sodium sulphate and
solvent removed in vacuo. The residue was redissoived in methanol

(25 ml) and refluxed with 7% aqueous sulphuric acid (10 ml) for 2 h.
over a steam bath. The reaction mixture was allowed to cool, diluted
with distilled water (25 ml) and extracted with ether (.3x25 ml) .
The ether extract waswashed many times with water to remove traces of
mineral acids, dried over anhydrous sodium sulphate and solvent
distilled off. The residue was crystallized from methanol as yellow
needles, m.p. 177-179° (lit.,40 m.p. of l-methyl ether of physcion 205°;
lit.,40 m.p. of 8-methyl ether of physcion 178°). Therefore the
glucoside was identified as the 1-glucoside of l,8-dihydroxy-6-
methoxy-3-methyl-9-10-anthraquinone or 1-glucoside of physcion.

-34-
References
1. R.H. Thomson, Naturally Occurring Quinones, 2nd edit.,
Academic Press, London, 1971.
2. E. Gilg and H. Heinemann, Festschriftr A. Tschirch-Tauchnitz,
1926, 52.
3. R. Hegnaver, Planta Med.1959,7, 344 and references therein.
A. C.E. Seaforth, Trop.Sci., 1962, 4, 159.
5. R.P. Fatel
and K.C. Patel, Indian J. Pharm. , 1956, 18, 107,
6. M. Anchel, J. Biol.Chem., 1949, 177, 169.
7. O.G. de Lima, I.L.D. Albiquerque, M.A.P. Barka, G. M. Maciel,and
J.F. de Mello, Ref. Inst. Antibiotic, Univ. Recife, 1966, 6, 3.8. P. Duquenois and R. Anton, Planta Med., 1968, 16, 184.9. T.K. Acharya and I.B. Chatterje, Sci. and Cult. , 1974, 40, '316.
10. J. Sterly, Heilflanzen der Finwohner Melanesiens, Komissions
Verlag Klaus Renner, Munich, 1970, p.341.
11. J.W. Parham, Plants of the Fiji Islandss, Fiji Government Press,
Suva, 1964.12. A. Tharcillo and J. de Newbern, Anais faculdade farmc. odontol,
Univ. Sao Pulo, 1949, 7_, 105; through C.A., 1950, 44, 11028h.
13. M. Belkin, B.D. Fitzgerald, and W. Cogan, J. Nat. Cancer Inst.,
1952, 13, 139.
14. A.F. Sieves et al. , J. Econ. Entomol., 1949, 42, 549.
15. M.L.E. Villaroya and R. Bernal-Santos, Asian J. Pharm., 1976, 3(1), 10|
16. J. Rageau, Les Plantes Medicinals de la Nouvelle-Caledono,
O.R.S.T.O.M., Paris, 1973.
17. H. Hauptmatin and L.L. Nazario, J . Amer. Chem. Soc. , 1950, 7_2_, 1492.
18. M.S.R. Nair, T.C. McMorrisland M, Anchel, Phytochemistry,1970, 9, 1153. .
19. R.D. Tiwari and T Joshi, Proc. Nat. Acad. Sci. (India), 1965,
35A, 448.

-35-
20. R.D. Tiwari and C.P. Yadava, Planta Med. , 1.971, 19, 299.
21. J.V.L.N. Seshagiri Rao, P.S.R. Sastry, R.V. Krishna Rao,and
M. Vimaladevi, Curr. Sci. ,1975, 44 , 736.
22. S.M. Kupchan and A . Karim, LLoydia , 1976 , 39 , 223 .
23. O.G. de Lima and G.P. Pinto, Rev. inst. antileroticos, Univ. Recife,1958, 1, 23 ; through C.A. , 1959, 53, 22212c.24. Shibata, M. Takido, and O. Tanaka, J. Amer. Chem. Soc. ,1950, 72, 2789.
25. L.H. Briggs and G.A. Nicholls, J. Chem. Soc., 1949, 1241.
1958, 1_, 23; through j^A. , 1959, 5_3_, 22212c...
24. S. Shibata, M. Takido^and 0. Tanaka, J. Amer. Chem. Soc.,
1950, _72_, 2789.25. L.H, Briggs and G.A. Nicholls, J. Chem. Sac, 1949, 1241.
26. J.H. Berkinshaw, Biochem. J. , 1955, 59, 485.
27. L.H. Briggs, G.A. Nicholls, and R.M.L. Paterson, J. Amer. Chem. Soc,
1952, 1718,
28. L.J. Bellamy, The Infra-Red Spectra of Complex Molecules,
Methven, London, 1956.
29. H Bloom, L.H. Briggs,and B. Cleverley, J. Chem. Soc., 1959, 178.
30. L.H. Briggs and G.A. Nicholls, J. Chem. Soc, 1953, 3068.
31. E.J.C. Brew and R.H. Thomsons, J.Chem. Soc.,(c), 1971, 2007
32. J. Lai and P.C. Gupta, Experientia, 1974, 30(8), 850.33. J.C.D. Brand and G. Eglinton, Applications of Spectroscopy to
Organic Chemistry, Oldbourne Chemistry Series, London, 1965, p. 149.34. G.J. Kapadia and M.L. Khorana, J. Chroma tog. , 1961, 6, 537.35. S.A.I. Rizvi, P.C. Gupta and R.K Kaul, Planta Med. , 1971, 19, 222.
36. W Poethe, D.A. Rao and D. Loescher, Phann, Zentralh.,
1966, 107, 57.37. R. Anton and P. Duquenois, Ann. Pharm. Fr., 1968, 26(11), 118.38. H. Wagner and G, Demuth, Z. Naturforsch., , 1974, 29c, 204,
39. L.F. Fieser and M. Fieser, Reagents for Organic Synthesis,
John Wiley and Sons, Inc., New York, 1967.
40. "Beilsteins" Handbuch der Organischen Chetnie, (Verlag Julius Springer,
Berlin-Heidelberg, 1925, 8, p.523.

-36-
41. J. Lal and P.O. Gupta, Experientia, 1973, ,(2) , 141.
42. R.J. Henry, Clinical Chemistry, Principles and Technics,
2nd edit,, Harper and Row, New York, 1974.

PART B
Studies on Hibiscus tiliacebua
(Malvaceae)
--37--
Chapter 1
Introduction
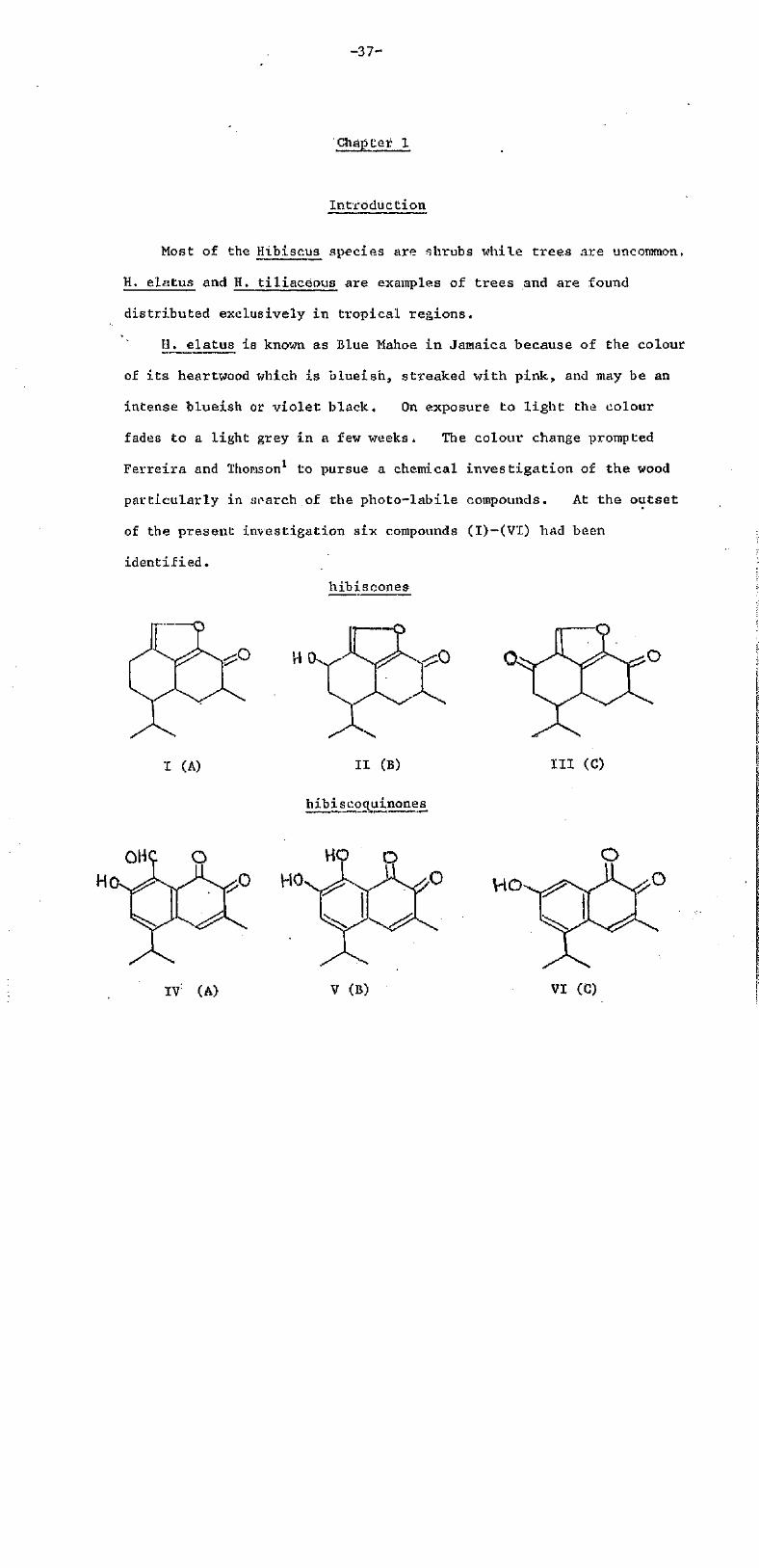
Most of the Hibiscus species are shrubs while trees are uncommon,
H. elatus and H. tiliaceous are examples of trees and are found
distributed exclusively in tropical regions.
H._ elatus is known as Blue Mahoe in Jamaica because of the colour
of i t s heartwood which is blueish, streaked with pink, and may be an
intense blueish or violet black. On exposure to l ight the colour
fades to a light grey in a few weeks. The colour change prompted
Ferreira and Thomson1 to pursue a chemical investigation of the wood
particularly in search of the photo-labile compounds. At the outset
of the present investigation six compounds (I)-(VI) had been
identified.
hibiscones
I (A) II (B)
hibiscoquinones
III (C)
IT (A) V (B) VI (C)

-38-
These are all sesquiterpenoids at various levels of oxidation. The
quinones fade on exposure to light, two of them [(V) and (VI)] slowly,
but the aldehydo-quinone (IV) very rapidly (see Chapter 3 for synthesis
of model compounds),
Since cv-quinones are usually dominant among terpenoid quinones
the occurrence of those quinones in Hibiscus is not very surprising.
However no terpenoid o-quinones are known from other Malvaceae genera.
For example, Bombax malabaricum and cotton plants (Gossypium hirsutun
and G. barbadense) contain only terpenoid freqtiinones and related
phenols and lactones (Table 1).
Table 1
Sesquiterpenoid p~quinones and selected
phenols present in Bombax malabaricum
arid Gossypium spp.
Plant Terpenoid quinones Terpenoid phenols
Bombax malabaricum VII,3 VIII3 IX3
Gossypium hirsutum VII4 . XI,5 XII,8 X6
and G. Barbadense XV,10 XVI11 + XIII,7 XIV9
t XV and XVI are diketones (2,3-d±hydro-1,4-quinones)
*Mansonones have been found recently in the wood of Azanza (=Thespesia)
spp.2 (Malvaceae),

-39-
o
0
VII (R=H, p-heirdgossypolone) IX
VIII (R=CH3 , 6-O-methyl-p-hemigossypolone)
X (R=R'-R"=H, 6-deoxyhemigossypol)
XI (R=R'=H, R"=OH, hemigossypol)
XII (R=OMe, R'=R"=H, gossyvertin)
OHC OH HO CHO
OH
XIII (gossypol)
XIV (3,4-dihydroxy-5-isopropyl-7-methyl-2H-naphthol [1,8-bc] f uran)

-40-
XV (RT=H, R=-CH2-CH2-CH=C(CH3)2, heliocide H2)
XVI (R=H, R1=-CH2-CH2-CH=C(CH3)2, heliocide H3
The terpenoid phenol, hemigossypol (XI) is probably the
penultimate compound in the biogenesis of the terpenoid p-quinones and
phenols in Bombax and Gossypium spp. In Gossypium it undergoes both
p-hydroxylatian leading to the formation of p-hemigossypolone (VII)
and oidative coupling leading to the formation of gossypol (XIII)
whereas in Bombax only the former oxidation is known,
These terpenoid phenols, for example, hemigossypol (XI) and
gossypol (XIII) are phytoalexins (i.e. they are produced in response
to fungal attack and have antifungal properties12) and the quinones
(XV) and (XVI) are toxic, or at least deterrent to tobacco budworm
(Heliothis virescens) and bollworm (Heliothis zea) which are important
pests on cotton.
In continuation of the work on Hibiscus, stems of H. tiliaceous
collected in Fiji, and roots of K. tiliaceous collected in Brazil
were investigated for terpenoids, H. tiliaceous, known as Vau in Fiji,
is one of the trees found growing wild, in coastal areas.1 3 The
fibres obtained from the bark of the tree were once used for making
women's dresses ("liku"), and the leaves were employed in folk medicine
as a painkiller in cases of ankle sprain.
* personal observation.

--41--
Chapter 2
Isolation and Identification of Sesguiterperioide
from II, tiliaceous (Malvaceae)
Dried wood of Fijian H. tiliaceous was extracted (soxhlet)
successively with petrol and chloroform. The petrol extract was
found to consist mostly of steroids and was not examined further-.
The chloroform extract showed five major bands on T.L.C. (detected
by U.V. and phloroglucinol spray reagent). by a combination of
column chromatography and P.L.C. these five bands were eventually
separated to give compounds A to E (in order of decreasing RF in
chloroform),
The petrol extract from the dried roots of Brazilian H. tiliaceous
on concentration deposited in relat ively high yield a yellow compound F,
The chloroform extract, after repeated P .L .C , furnished two further
coloured compounds, G and H (in order of decreasing 1RF in CHCl3 .
Identification, of compound A as lapachol (XVII)
Compound A was obtained as yellow, shiny, crystals, m.p. 140°,
It was shown to be lapachol (XVII) by direct comparison with an
authentic sample.

Identificatlon of compound B as hibiscone A (I)
Compound B was isolated in low yield as crystals, m.p. 94-95°.
It did not respond to the phloroglucinol spray reagent and gave a
positive test with Ehrlich's reagant only after prolonged heating,
It had the molecular formulaC15H20O2, showed intense U.V. absorption
at 280 ran and a strong conjugated carbonyl band at 1650 cm"1 in the
I.R. There were significant peaks in the M.S. at m/e 217 (M+-Me,
16%), 203 (M+-CHO, 20%), and 189 (M+-CHMe2, 50%). These data
suggested a sesquiterpenoid ketone and direct comparison showed that
they were identical with those of hibiscone. A (I) isolated earlier
from H, elatus .
Identification of compound C as hibiscone G (III)
This compound was obtained as shiny crysta ls , m.p. 125°, in
relatively high yield. No distinctive colour was observed on
heating with Ehrlich's reagent but an orange colour developed on
spraying with the phloroglucinol reagent. This compound, C15H18O3
contained one atom of oxygen more than compound B. It showed h
230 and 265 ran (on addition of base shifting to 412 nm), V 1670 cm"1,
and both P.M.R.. and M.S. provided evidence for the presence of a methyl
and isopropyl groups. These data suggested structure (III) for the
sesquiterpenoid and direct comparison (I.R., U.V., P.M.R., M.S. and
T.L.C) with an authentic specimen of hibiscone C (isolated previously
from H. elatus) confirmed the identity.
-43-
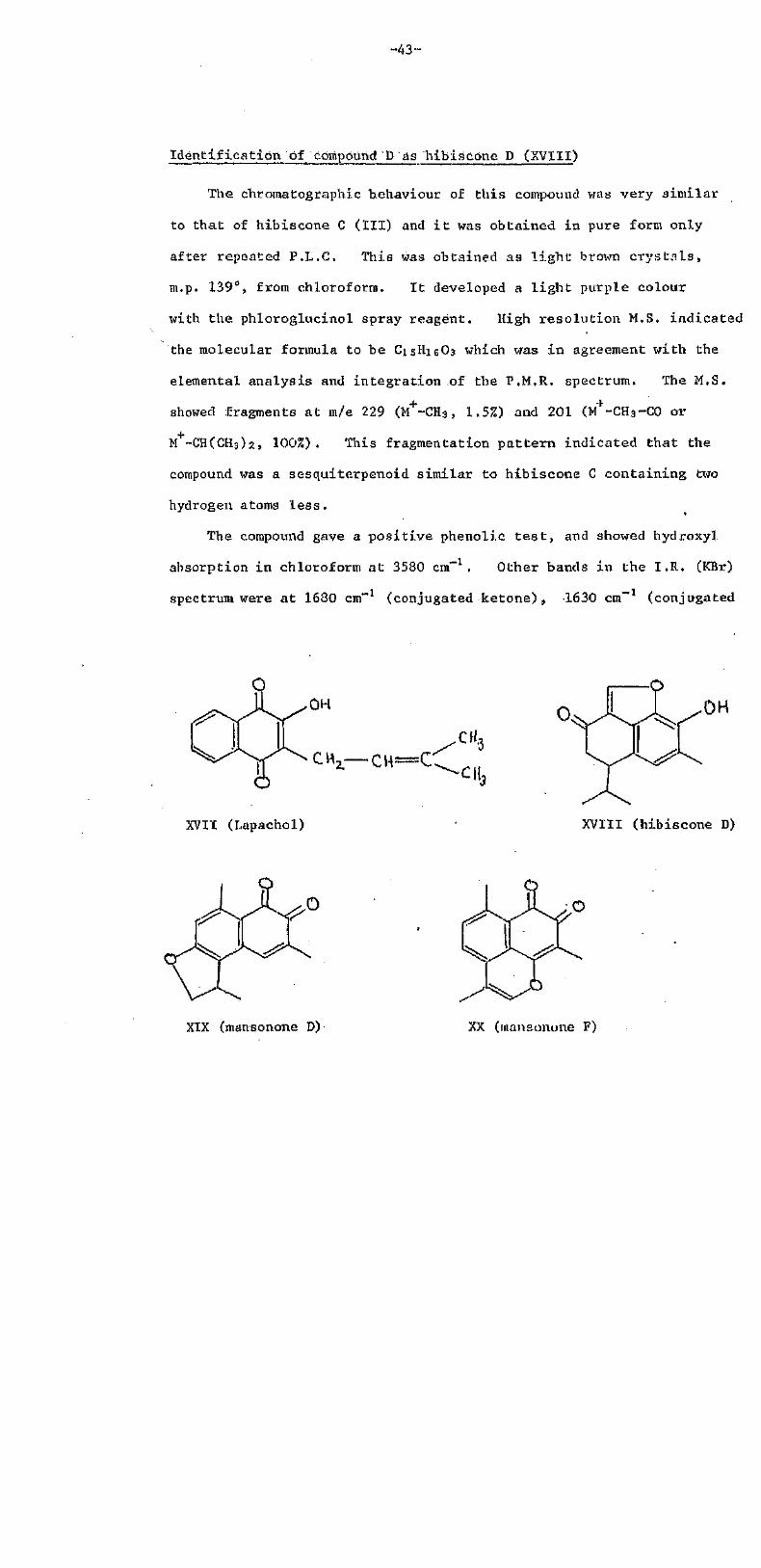
Identification of compound D as hibiscone D (XVIII)
The chroinatographic behaviour of this compound was very similar
to that of hibiscone C (III) and i t was obtained in pure form only
after repeated P.L.C. This was obtained as l ight brown crys ta ls ,
m.p. 139°, from chloroform. I t developed a l ight purple colour
with the phloroglucinol spray reagent. High resolution M.S. indicated
the molecular formula to be C15H16O3 which was in agreement with the
elemental analysis and integration of the P.M.R. spectrum. The M.S.
showed fragments at <m/e 229 (M+-CH3, 1.5%) and 201 (M+-CH3-C0 or
M+-CH(CH3)2, 100%). This fragmentation pattern indicated that the
compound was a sesquiterpenoid similar to hibiscone C containing two
hydrogen atoms less .
The compound gave a positive phenolic t e s t , and showed hydroxyl
absorption in chloroform at 3580 cm-1. Other bands in the I.R (KBr)
spectrum were at 1680 cm"1 (conjugated ketone), 1630 cm"1 (conjugated
XVIT (Lapachol) XVIII (hibiscone D)
XIX (mansonone D) XX (mansonone F)

olefin), and 850 cm-1 (furan ring). The U.V. spectrum exhibited
a broad maximum centred at 335 nm which underwent a bathochromic
shift of 70 nm on addition of base, thus confirming the phenolic
nature of the compound,
The P.M.R. (CDC5.3) spectrum is shown in Fig. 1. Irradiation
of the 1-H multiplet at 62,04 collapsed the. doublets at 60.82 and 0.90
to a broad singlet, and also the benzylic 1-H multiplet was simplified.
In turn, irradiation of this benzylic 1-H multiplet (63.26) simplified
the complex at 62.04, and in addition collapsed the 2-H doublet at 62.82
to a singlet. cased on these spin decoupling experiments, it was
concluded that the compound,like the previous two hibiscones, possessed.
an isopropyl group, the methine proton of which was coupled to a
benzylic proton, and this in turn was coupled to two equivalent
Tnethylene protons. In other words, an A2BX system (X being the
isopropyl methine) was apparent. Other features of the P.M.R.
spectrum included a broad 1-H singlet centred at 65.60 (disappearing
on addition of T2.O) , and a 3~H singlet at 52.40 assignable to an
aromatic methyl. At low field two 1-H singlets (66.94 and 7.98) were
observed, and were assigned to protons of the benzenoid and furan rings,;
respectively,
Compound D on acetylation (pyridine/acetic anhydrides) afforded
a monoacetyl derivative which even on repeated attempts failed to
crystallize, but was found to be homogenous by T.L.C, The P.M.R.
spectrum was almost identical to the parent compound except for the
appearance of a 3-H singlet at 62.33 assignable to the acetate methyl
and the absence of the singlet at 55.60.
On the basis of the above data and the co-occurrence of compound D

-45-
8-0
* CHCI3
4 - 0
Fig. 1
2 -0 PPM 0

-46-
with other hibiscones, structure (XVIII) is suggested for the
sesquiterpenoid ketone which is named hibiscone D. Further support
for the structure comes from the observation that aerial oxidation of
hibiscone D yields hibiscoquinone B (V)14 Hibiscone C (III) behaves
in the same way.
Identification of compound E as hibiscone "B (II)
This compound obtained as crystals, tn.p. 122-123°, was the most
polar hibiscone. Like the others i t failed to give a positive test
with Ehrlich's reagent and also no colour developed upon treatment with
the phlorogluciiiol spray reagent. It had the molecular formula C15H20O3
and thus had two hydrogen atoms more than hibiscone C. I.R. absorption
at 3450 cm-1 (OH) and 1650 cm-1 (C-0) suggested that compound E might be
hibiscone B (II) which was confirmed by direct comparison with an
authentic sample obtained from H. elatus, and by oxidation with 2,3™
dichloro-5,6-dicyano-l,4-benzoquinone (DDQ)15 to form hibiscone C,
Identification of compound F as gossypol (XIII)
This compound was isolated as small, yellow, shiny, crystals, m.p.
192-194°. I t gave a positive phenol tes t , and developed a dark purple
colour with the phloroglucinol spray reagent, I ts M.S. had a weak
parent peak at m/e 518 (M+ , 1.5%) and the base peak at m/e 482, correspo
to the loss of two molecules of water (M+ -2H2O) . I t had the molecular |
formula, C30H30O8which suggested that the compound was a dimer. The |
P.M.R. (CDCl3) spectrum indicated a symmetrical aromatic system with six
hydroxyl, two methyl, two isopropyl and two strongly chelated aldehyde
(confirmed by I.R.. Vc=o 16.10 cm"1) groups as substituents The overall

- - 4 7 -
data agreed well with that of gossypol (XIII) . • This was confirmed
by a comparison of the spectral and chemical properties (including
colour tests) with those of authentic gossypol.
Methylation of the dimer with dimethyl sulphate/potassium carbonate
yielded at least three methyl ethers but due to similarity in RF value
in many solvent systems i t was possible to isolate only one in pure
crystalline form. This was the hexamethyl ether, m.p. 214o ( l i t . , 1 6
216-218°), Unlike the parent compound, the M.S. of the ether showed
the molecular ion as the base peak at m/e 602, The P.M.R. spectrum
was very similar to that of gossypol except for the appearance of three
new 3~H singlets at tr ibutable to six methoxyl groups, and the disappeara
of the three l-H s inglets .
Identification of compound G as mansonone D (XIX)
Compound G crystall ised as dark, orange, prisms, m.p. 174°. I t was
a quinone, C15H24O3 readily decolourized by di th ioni te .
The presence of a significant M + 2 (28% ) peak in the M.S. indicated that
the compound was an o-quinone, The M.S. also included peaks at 227
(M+-15, 2%), 214 (M+-C0, 100%), 199 (M+-CH(CH9)2, 74%), 198 (M+-CH3-CI10,
94%), and 186 (M -2C0, 100%). The I.R. spectrum contained absorptions
at 1660, 1645, and 1600 cm"1.
The P.M.R. (CDCl3 spectrum revealed the presence of an aromatic
methyl (62.60), an aromatic proton (66.54), and a quinone methyl (52.04:
d, J=3Hz) coupled to an olefinic proton (q, J-3HZ) resonating at 67.18.
The spectrum also included a benzylic proton signal (53.60, m) coupled
to a methyl group (61.36, d) and two non-equivalent methylene protons
(64.36 and 4.68, m) .

-48 -
The above data indicated that compound G was mansonone D (XIX).
Direct comparison by T.R., M.M.P., P.M.R. and cb-chromatography with
an authentic specimen iso la ted from Mansonia alt isaitna establsihed
i t s identity.
Identification of compound H as mansononeF (XX)
This was obtained as dark, purple , c r y s t a l s , m.p. 213°. I t had
the molecular formula, C15H12O3 i . e . 2H l e s s than mansonone D, and a
strong M +2 peak - thus the M.S. again indicated an o-quinone. Other
important fragments in the M.S. appeared a t m/e 225 (M -CH3, 25%), 212
(M+-C0, 100%), 197 (M+-CH(CH3)2, 60%), 196 (M+-CH3-CHO, 80%), and 184
(M+-2C0, 75%). The I.R.. spectrum showed strong peaks a t 1680 and
1625 cm-1.
The PM.R.. (CDCl3) spectrum showed s ignals for 12 protons: one
aromatic methyl (62.70), one quinone methyl (51.96), one a l l y l i c methyl
(62.10, d, J=2Hz), two aromatic protons (67.40, bs ) , and one enol ic
proton (57,06, m, J=2Hz).
In view of the above data and the co-occurrence of compound H with
mansonone D (XIX) i t appeared that the quinone was mansonone F (XX) .
This was confirmed by undepressed M.M.P., superimposable P.M.R. and I.E..
spectra, and correspondences in other physical proper t ies with an
authentic sample of mansonone F (XX) i so la ted from Mansonia altissima17

-49-
Discussion
Thus the present work, reports the isolation of seven sesquiterpenoids
and one p-quinone (lapachol (XVII)). The seven terpenoids include two
sesquiterpenoid o-naphthoquinones (tnansonone D (XIX) and F (XX)), one
sesquiterpenoid phenol (gossypol (XIII)), three ketones (hibiscone A (I) ,
B (II) and C (III)) and a new ketone named hibiscone D (XVIII) .
Although lapachol (XVII) contains an isoprenoid fragment i t is not
a wholly terpenoid p--quinone. It is known to occur in at least three
families (Bignoniaceae, Verbenaceae and Proteaceae)16 but this is the
first report of i ts isolation from Malvaceae. This quinone has
significant anti-tumour properties in mice but unfortunately is not
suitable for use in man19 i
Mansonone D (XIX) and P (XX) belong to the cadinane group of
bicyclic sesquiterpenoids and are representative of a number of highly
oxidized quinonoid compounds found until recently, mainly in the heartwoodi
of Mansonia a l t i s s ima 1 7 (Sterculiaceae) . The only other family where
maosonones are known to occur is Ulmaceae. For example, mansonone C
(XXI) has been found along with naphthaldehydes in the heartwood of
U. procera (= U. glabra) .20 This finding of mansonone D and F in
Hibiscus (Malvaceae) is thus the f i r s t report of mansonones occurring
elsewhere than in Sterculiaceae and Ulmaceae. The occurrence of
gossypol (XIII) in Hibiscus is hardly surprising since i t also belongs to
the cadinane group and the co-occurrence of naphthaldehydes (compounds
structurally similar Co gossypol) with o-naphthoquinones (e.g. mansonone C
(XXI)) has been observed in Ulmaceae.20
Due to the restrict ion in rotation of the internuclear (7,7T) bond,
gossypol (XIII) is normally found in two optical forms, active.(+) and
* Mansonones have been found recently in the wood of Azanza (=Thespesia)spp.2 (Malvaceae)
-50-
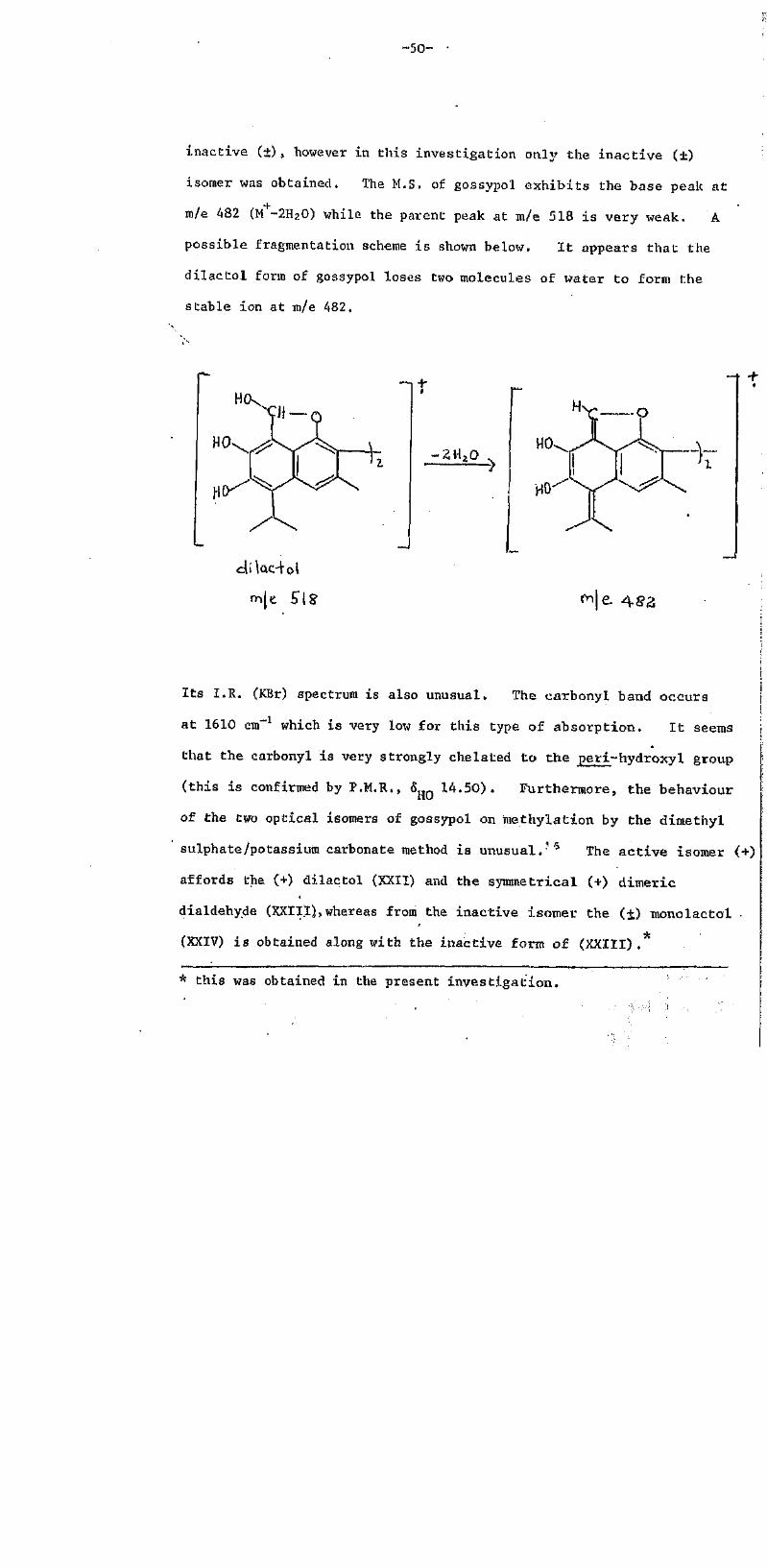
inactive (+) , however in this investigation only the inactive (±)
isomer was obtained. The M.S. of gossypol exhibits the base peak at
m/e 482 (M -2H2O) while the parent peak at m/e 518 is very weak. A
possible fragmentation scheme is shown below. It appears that the
dilactol form of gossypol loses two molecules of water to form the
stable ion at m/e 482,
m/e.518
t
m/e. 482
Its I.R. (KBr) spectrum is also unusual. The carbonyl band occurs
at 1610 cm-1 which is very low for this type of absorption. It seems
that the carbonyl is very strongly chelated to the peri-hydroxyl group
(this is confirmed by P.M.R., hHO 14.50). Furthermore, the behaviour
of the two optical isomers of gossypol on methylation by the dimethyl
sulphate/potassium carbonate method is unusual.16 The active isomer (+)
affords the (+) dilactol (XXII) and the symmetrical (+) dimeric
dialdehy.de (XXIII), whereas from the inactive isomer the (±) monolactol
(XXIV) is obtained along with the inactive form of (XXIII) .*
* this was obtained in the present investigation.

XXI (mansonone C)
OH
H3CO
XXIV
The hibiscones like the foregoing terpenoids belong to the cadinan
group. A common feature of the hibiscones but unusual in the cadinane
group is the oxygenation at C-8 which takes the form of a furan ring.
Hibiscone D (XVIII) is the most highly oxidized hibiscone and most
probably the biogenetic precursor of the hibiscoquinones. Support for
this comes, in part, from in vitro studies. Using DDQ hibiscone B (II;
was oxidized to hibiscone C (III), and it has been observed14' that on
aerial oxidation in the presence of base, hibiscone C (III) and D (XVIII)
are converted into hibiscoquinone B (V) . The mechanism believed to be]
operative in this alkaline oxidation is indicated in Scheme 1. The
attack by the base at the furan ring activated by the carbonyl group
leads to the formation of (XXV) which has two allylic (benzylic)
hydrogens easily lost (oxidized) to give (XXVI) , Opening of the lacto
ring (arrows) would give (XXVII) which by 3~cleavage would lead to
hibiscoquinone C (VI) or to hibiscoquinone A (IV) through oxidation. .
The formation of hibiscoquinone E (V) can be explained as follows. The

-52-
XVIII XXV
oOH
OH
XXVII / 3-cleavage
PvOOHC o O
VI
XXX
Scheme 1

-53-
mesomeric anion of (XXVII) reacts stepwise with oxygen (see below)
to give the hydroperoxide anion (XXVIII) which undergoes an internal
nucleophilic carbonyl addition forming the cyclic peroxide (XXIX).
This four membersd peroxide loses a formate ion via C-C cleavage and
forms (XXX) which is a tautomer of hibiscoquinone B (V).
This conversion of hibiscone D (XVIII) to the tautomer of
hibiscoquinone B (V) is analogous to the autoxidation of ketones under
alkaline conditions.21 ,22 For example, autoxidation of diphenylpyruvic
acid in aqueous sodium hydroxide affords benzophenone and oxalic
acid21 (Scheme 2) .
Ph Ph OH - Ph
Ph Ph Ph
diphenylpyruvic acid
Ph OoO o2 Ph 0<......... C-C -CO2H
Ph
OrO
c c
Ph o
iPh2CO + C02H
CO(..)
Scheme 2

-54-
2-Methyl-3-diraethylallyl or 3-ben2yl-l,4-naphthoquinones are also
known to behave similarly when autoxidized under similar conditions,
t t
e.g. in 'BuOH containing BuOK3-benzyl-2-methyl-l,4-naphthoquinone
undergoes autoxidation at the benzyl carbon atom to give phthiocol and
benzaldehyde2 3 (Scheme 3).
H— Ph
PhCHO • +CH-Ph
phthiocol anion
Scheme 3
The mechanism for the formation of a hydroperoxide anion from the
reaction of a carbanion with oxygen is unclear. A vast majority of
data supports an ionic mechanism (one two-electron step, R +02 -> RO2 )
whereas results obtained mainly by Russell and his co-workers24 suggest
that the possibility of a free radical reaction (two one-electron step

-55-
as shown below) cannot be eliminated, It appears that an ionic
R' + 02
R' + 02
R- initiation
"RO2- + R"—> R02 + R- propagation
mechanism is followed to some extern: but a radical reaction also takes
place.25
On the whole, the in vitro conversion of hibiscone D (XVIII) to
hibiscoquinone B (V) is similar to the Scheiffele and. Shirley's
alkaline autoxidation of gossypol (XIII) to (XXXI) ,26 It is interesting
to note that these authors suggest a Dakin type reaction ' but give
no mechanism. Obviously the mechanism for this oxidation is similar
to Scheme 1 and is indicated in Scheme 4
OHC
H C O 2
XXXI
Scheme 4

-56-
In view of the above in vitro conversions, the most likely
biogenetic pathway of the hibiscosesquiterpenoids is as follows:
hibiscoquinone A(IV)
hibiscone A -> hibiscone B -> hibiscone C ->hibiscone D->hibiscoquinone I
(I) (II) (III) (XVIII) \ (v)
hibiscoquinone C(VI)
Is,.

-57-
Chapter 3
Synthesis of o- and p-Naphthoquinone-peri-aldehydes
-Ferreira and Thomson found that one of the compounds responsible
for the striking colour change of the heartwood of H. elatus on
exposure to light was hibiscoquinone A (IV) . They found that a red
solution of (IV) in chloroform when left on a bench in daylight became
completely colourless in a couple of hours and the photo-product
obtained on work up was the isomeric lactone (XXXII) .
The mechanism involved in the formation of this lactone (XXXII)
is thought to be similar to the well-known intermolecular photochemical
quinone-aldehyde reactions. In these reactions, p-quinones react
with aromatic or aliphatic aldehydes to give esters (O-acylation) and
ketones (C-acylation) whereas o-quinones give exclusively esters (0-
acylation) . The mechanism for these reactions is shown in Scheme 5.
The formation of ketone involves the addition of acyl radical followed
by enolization of the adduct.
RCHO
)H
RCO
p-quinonesminor
o-quinonesScheme 5
+RCO
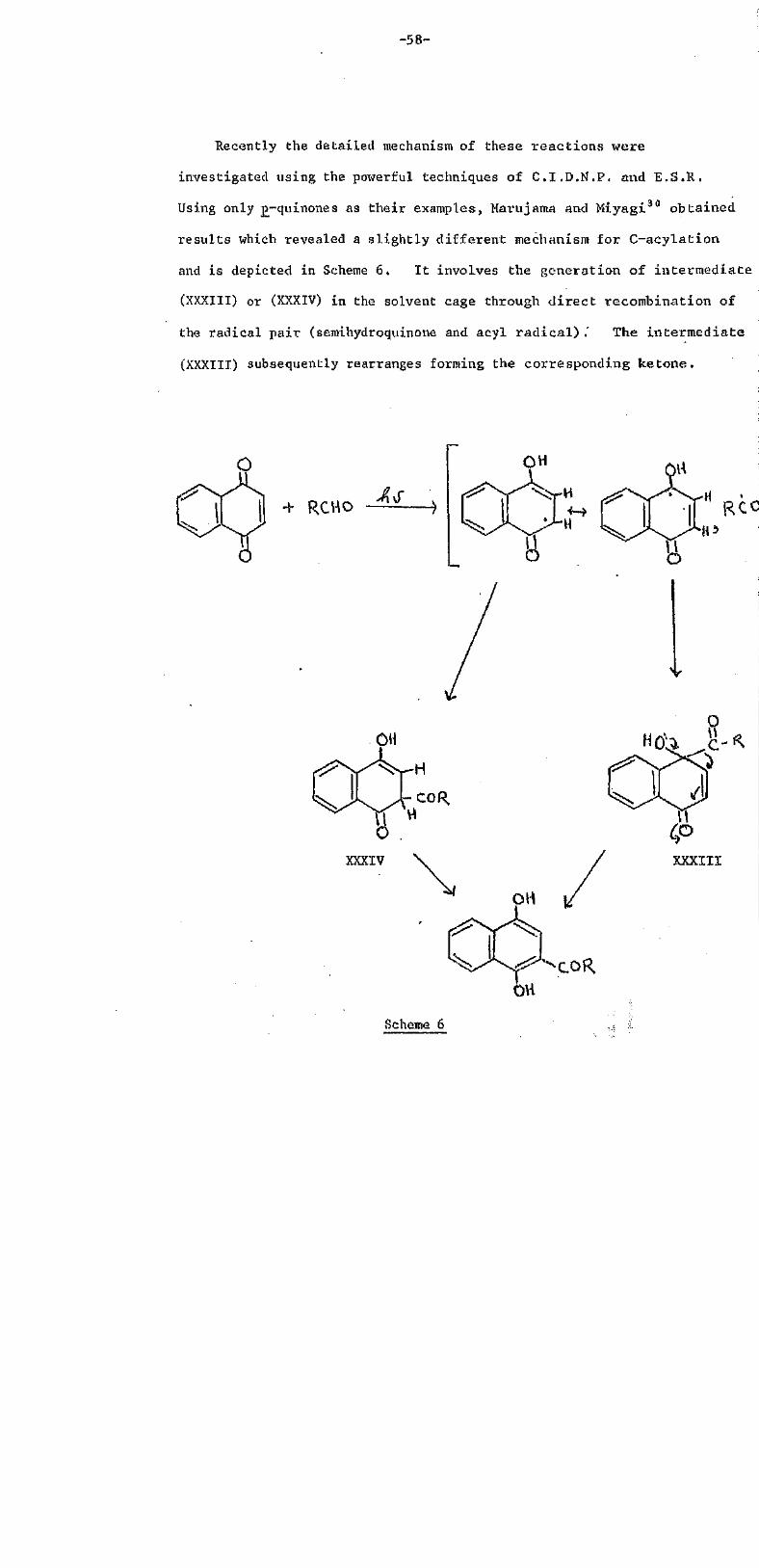
-58-
Recently the detailed mechanism of these r eac t ions were
investigated using the powerful techniques of C.I.D.N.P. and E.S.R.
Using only p-quinonesas their examples, Marujama and Miyagi 3 0 obtained,
resu l t s which revealed a s l igh t ly di f ferent mechanism for C-acylat ion
and is depicted in Scheme 6. I t involves the generat ion of in te rmedia te
(XXXIII) or (XXXIV) in the solvent cage through d i r e c t recombination of
the radical pai r (semihydroquinone and acyl r a d i c a l ) .' The in te rmedia te
(XXXIII) subsequently rearranges forming the corresponding ketone.
RCHO hs
-COR
XXXIII
Scheme 6
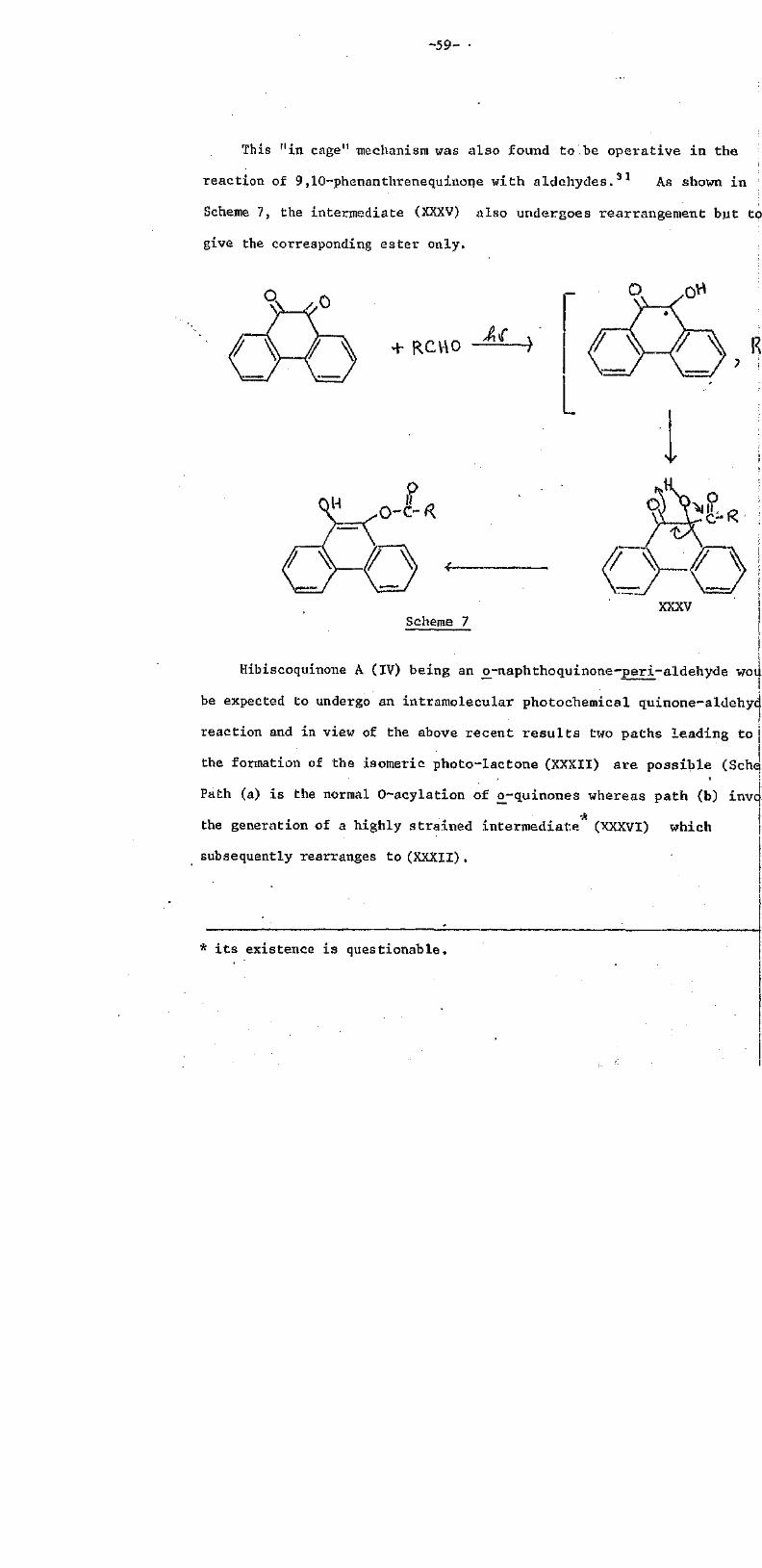
-59-
This "in cage" mechanism was also found to be operative in the
reaction of 9,10-phenanthrenequinone with aldehydes.31 As shown in
Scheme 7, the intermediate (XXXV) also undergoes rearrangement but to
give the corresponding ester only.
q OH
R
XXXVScheme 7
Hibiscoquinone A (IV) being an o-naphthoquinone-peri-aldehydei
be expected to undergo an intramolecular photochemical quinone-aldehyde
reaction and in view of the above recent results two paths leading toi
the formation of the isomeric photo-lactone (XXXII) are possible(Scheme 8)
Path (a) is the normal O-acylation of 0_-quinones whereas path (b) invc
the generation of a highly strained intermediate (XXXVI) which
subsequently rearranges to (XXXII) .
* i t s existence is questionable.

- 6 0 -
XXXVI
Scheme 8
Since t h i s in t ramolecu la r r e a c t i o n shown i n Scheme 8 i s new,
some simple model compounds, o - a n d p - q u i n o n e - p e r i - a l d e h y d e s were
synthesized to study t h e i r behaviour towards l i g h t .

Synthesis of 8-formyl-7-hyclroxy-l ,2-naphthoquinone (XXX.VIII)
and the lactone (XXXIX)
2,7-Dihydroxynaphthalene was formylated by the procedure of
Cramer and Windel3 2 to the formyl de r iva t ive (XXXVII) m . p . 157-158°
( l i t . 3 3 156.5-158.5°). This was oxidized wi th Fremy's s a l t 3 4 , 3 5 -
to give dark purple c rys ta l s of (XXXVIII). High r e s o l u t i o n M.S. and
OHC O
XXXVII XXXVIII
elemental analysis of (XXXVIII) indicated the molecular formula, C11H6O4
which was confirmed by in tegra t ion of the P.M.R. spectrum. The M.S.
exhibited the cha rac t e r i s t i c intense o--quinone M +2 peak and the I .R,
spectrum showed a very strong carbonyl absorpt ion a t 1650 cm~ . The
P.M.R. spectrum showed 1H doublets a t 6 6.34, 7.26, 7,42 and 7 .50, a l l of
equal coupling constants (8Hz) a t t r i b u t a b l e to the benzenoid and
quinonoid protons. The signal of the hydroxyl proton appeared as a
deuterium-exchangeable s ingle t a t b 12.34, and the aldehyde proton
resonated at 6 10.82. .
The isomeric a lka l i - so lub le product (XXIX) obtained from (XXXVIII)
on i r r ad i a t i on , showed in the I.R. spectrum a s t rong carbonyl absorpt ion
a t 1730 cm"1 which suggested that the compound was a c y c l i c l a c t o n e .
This lactone was found to be insoluble in. chloroform as opposed t o the

-62-
0
XXXVIII
XXIX
parent compound, and thus the P.M.R. spectrum was obtained in DMSO-dg.
The spectrum when compared with the P.21.R. spectrum of the quinonev
showed the absence of the aldehyde proton signal and the emergence of a
6H complex signal in the aromatic region which on "D2O shake" was
reduced to.a 4H m u l t i p l e t .
Synthesis of 8-formyl-hydroxy-1,-naphthoquinone (XLII)
1,7-Dihydroxynaphthalene was formylated as before to give a mixture
of formyl derivatives. The 2,8-dihydroxynaphthaldehyde (XL), m.p.
202-204° (lit.33203-204* ) was extracted from the isomeric 4,6-dihydroxy-
OH
dry HCl
XL XLI
naphthaldehyde (XLI) with boiling benzene, and was oxidized with Fremy's
sa l t to give a mixture of o- and o-quinones. The mixture was separated
OHCFremy's s a l t 'H
XXXVIII

-63-
on a silica gel column to give the title compound (XLII)and the o-
isomer (XXXVIII). The M,S. of (XL1I) was similar to that of (XXXVIII)
except for the absence of the intense peak at M +2. Its I.R. spectrum
displayed a strong carbonyl absorption at 1650 c m - 1 T h e P.M.R.
spectrum of this quinone when compared with that of (XXXVIII) showed a
significant difference in the aromatic region. The quinonoid protons
-appeared as a 2H singlet at 6 6.94 and the benzenoid protons as 1H
doublets at 6 7.30 and 8.24. The signal of the hydroxyl proton appeared
as a deuterium-exchangeable singlet at <S 12.76, and the aldehyde proton
resonated at 5 10.88. The b-isomer was identical (M,P., U.V.., I.R. ,
and P.M.R.) to the sample obtained from (XXXVII).
*
Attempted synthesis of 8-formyl-5-hydroxy-l,2-naphthoquinone (XLIV)
4,7-Dihydroxynaphthaldehyde (XLIII),m.p. 217° (dec) (lit,33
218° (dec.) was obtained from 1,6-dihydroxynaphthalene by the usual
formylation procedure.32 This aldehyde on Fremy's salt oxidation gave
a mixture of at least five products (T.L.G.) in approximately equal
proportions. After repeated P.L.C. and column chromatography one of the
products was isolated and identified as 6-hydroxy-l,4~naphthoquinone (XLV)

-64-
o 0!
Fremy'ssalt
XLIII
Dimethyl sulphate/potassium carbonate
XLVI
by comparison with an authentic specimen.
Failing to synthesize (XLIV), an attempt was made to partially
methylate (XLIII) to (XLVI) prior to oxidation. This partial methylation.
was not successful. Various attempts, using different methylating
agents (dimethyl sulphate and diazomethane) and different techniques,
gave only the dimethyl ether (M+ 216) .
Attempted synthesis of 8-formyl-5-hydroxy-l,4-naphthoquinone (XLVIII)
4,8-Dihydroxynaphthaldehyde. (XLVII), m.p. 278°(dec.) (lit.33
280o(dec.)) was obtained from 1,5-dihydroxynaphthalene by the usual
formylation procedure. This aldehyde was found to be only partially
soluble in aqueous methanol and similar media suitable for carrying out
Fremy's salt, oxidation. Therefore to overcome the solubility problem,
an attempt was made to reduce the polarity of the formyl derivative (XLVII)
by methylating the hydroxyl at position-4 (XLIX). This partial
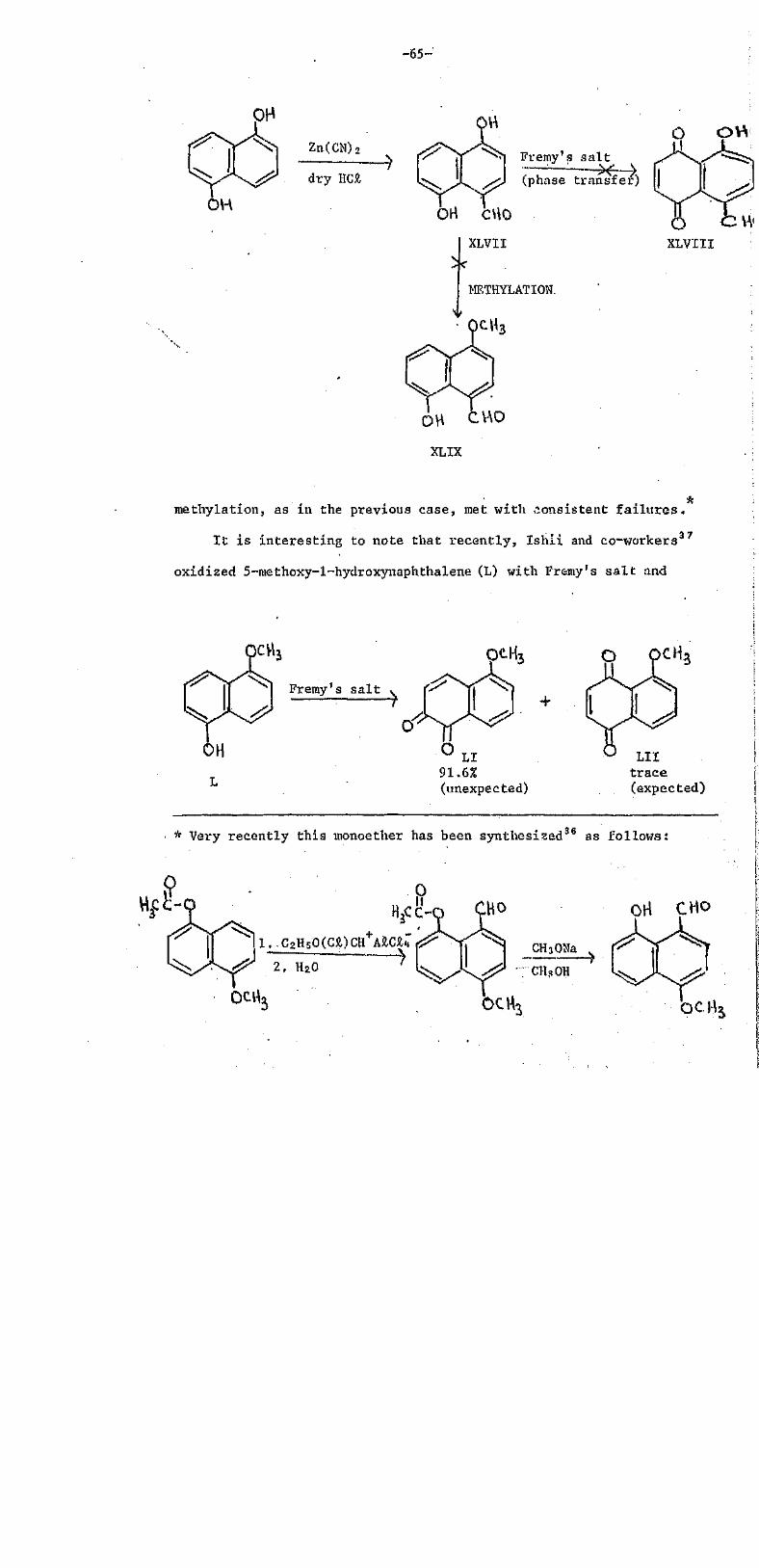
- 6 5 -
Zn(CN)
dry HCl
OH
Fremy's salt
(phase transfer)
OH CHOXLVII
METHYLATION.
Methylation, as in the previous case, met with consistent failures.
It is interesting to note that recently, Ishii and co-workers37
oxidized 5-methoxy-1-hydroxynaphthalene (L) with Fremy's salt and
OCH3
Premy's salt
o
LI91.6%(unexpected)
LIItrace(expected)
Very recently this monoether has been synthesized36 as follows:
OH CHO
1 . C2H5O(Cl)CH AlCl4
2 . H20

-66-1
obtained exclusively the o-uinone (LI) instead of the expected p-
isomer (LII).35 The question now arises, had the partial methylation
of (XLVII) been successful, which quinone isomer would have dominated
on Fremy's salt oxidation, (LIII) or (LI?)?
Fremy's salt or
oLIII LIV
Failing to achieve partial methylation, oxidation of the dihydroxy-
naphthaldehyde (XLV1I) in a two-phase system (ethyl acetate and water)
using Adogen 464 as the phase transfer agent38 was then attempted.
Although Fremy's salt was conveniently transferred into the ethyl
acetate phase ("new purple ethyl acetate") it was disappointing to find
on work up only intractable tars.
Synthesis of 8-formyl-1,4-naphthoquinone (LVI)
Using Fremy's salt, 8-hydroxynaphthaldehyde (LV) (kindly supplied
by Dr. D.C.C. Smith39) was conveniently oxidised to the title compound
OHC OHOHC
Fremy's salt
LV LVI

-67-
in 67% yield. This quinone had the molecular formula, C11H6O3 and
showed strong carbonyl absorptions (1655 and 1690 cm-1 ) in its I.R.
spectrum. TheP.M.R.. spectrum showed a 2H singlet at 6 7.04 assigned
to the quinonoid protons, a 1H singlet at 6 10.72 due to the aldehyde
proton, and a 3H multiplet in the aromatic region (6 7,80-8.40)
attributed to the benzenoid protons.
Attempted synthesis of 4-brom6-8-formyl-l,2-naphthoquinone (LVIII)
Having obtained the p-quinone without much difficulty from.8-
hydroxynaphthaldehyde (LV), the synthesis of the corresponding o-quinonc
was then the next choice. Generally the synthesis of 0-quinones is
difficult and even more difficult in the present situation where the
para position to the hydroxyl is unsubstituted. To overcome this
problem it was decided to block this position by introducing bromine and
then to subject the brorainated derivative to Fremy's salt oxidation,
which in principle (see notes on Fremy's salt on page 69) should mainly
lead to the formation of the bromo-substituted o-quinone (structurally
similar to hibiscoquinone A (IV)).
The general approach which was conceived for the synthesis of the
title compound (LVIII) is outlined in Scheme 9, Due to the limited
LV
LVII
Fremy's salt
Scheme 9
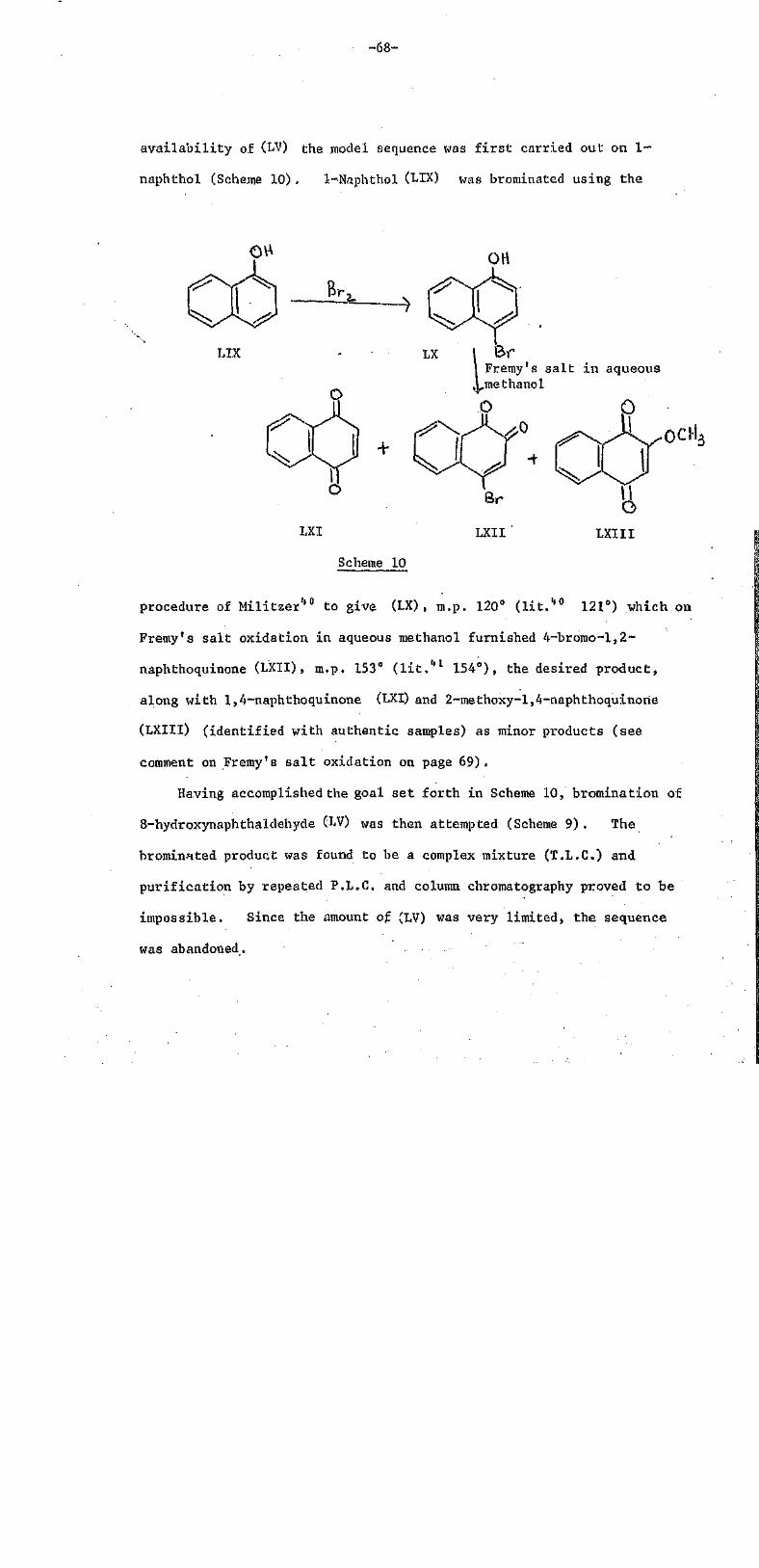
-68-
availability of (LV) the model sequence was first carried out on 1-
naphthol (Scheme 10). 1-Naphthol (LIX) was brominated using the
LIX LX
0
X X +
Fremy's sa l t in aqueousmethanol
LXI LXII
Scheme 10
procedure of Militzer40 to give (LX) , m.p. 120° (lit.40 121) which on
Fremy's salt oxidation in aqueous methanol furnished 4-bromo--l,2~
naphthoquinone (LXII), m.p. 153° (lit.41 154°), the desired product,
along with 1,4-naphthoquinone (LXI) and 2~methoxy-l,4-naphthoquinorie
(LXIII) (identified with authentic samples) as minor products (see
comment on Fremy's salt oxidation on page 69).
Having accomplished the goal set forth in Scheme 10, bromination of
8-hydroxynaphthaldehyde (LV) was then attempted (Scheme 9). The
brominated product was found to be a complex mixture (T.L.C.) and
purification by repeated P.L.C. and column chromatography proved to be
impossible. Since the amount of (LV) was very limited, the sequence
was abandoned.

-69-
Photochemistry
The behaviour of 8-formyl-7-hydroxy-l ,2-naphthoquinone (XXXVIII) •
towards light is found to be identical to that of hibiscoquinone A
(IV), i.e. a chloroform solution when left on a bench in daylight
becomes colourless in a couple of hours. On the other hand, solutions
of 8-formyl-7-hydroxy"l,4-naphthoquinone (XLII)and 87formyl-l,4-
naphthoquinone (LVI) are found to be light stable.,
A detailed study into the mechanism of this new intramolecular
quinone-aldehyde photochemical reaction using C.I.D.N.P. and E.S.R.
is being planned.
Notes on Premy's salt oxidation •
FreMy's salt [ION(SO3K)2]is an excellent selective free radical
oxidizing agent35 and is widely used in the synthesis of O~ and O-quinones
from phenols and anilines. Under mild conditions it oxidizes phenols
(unaubstituted at the para position) to the corresponding p-quinones
(Scheme 11) while o-quinones are obtained from para-substituted phenols.
Scheme 11

-70-
In 1953, Teuber42 proposed a mechanism which involves three steps
as shown in Scheme. 12, In the first step a radical of Fremy's salt
abstracts the hydrogen atom from the phenolic group resulting in the
formation of radical (LXIV) which is resonance stabilized. in the
second step, another radical of Fremy's salt couples with (LXIV)
HN(SO3K)
• •!;
Scheme 12
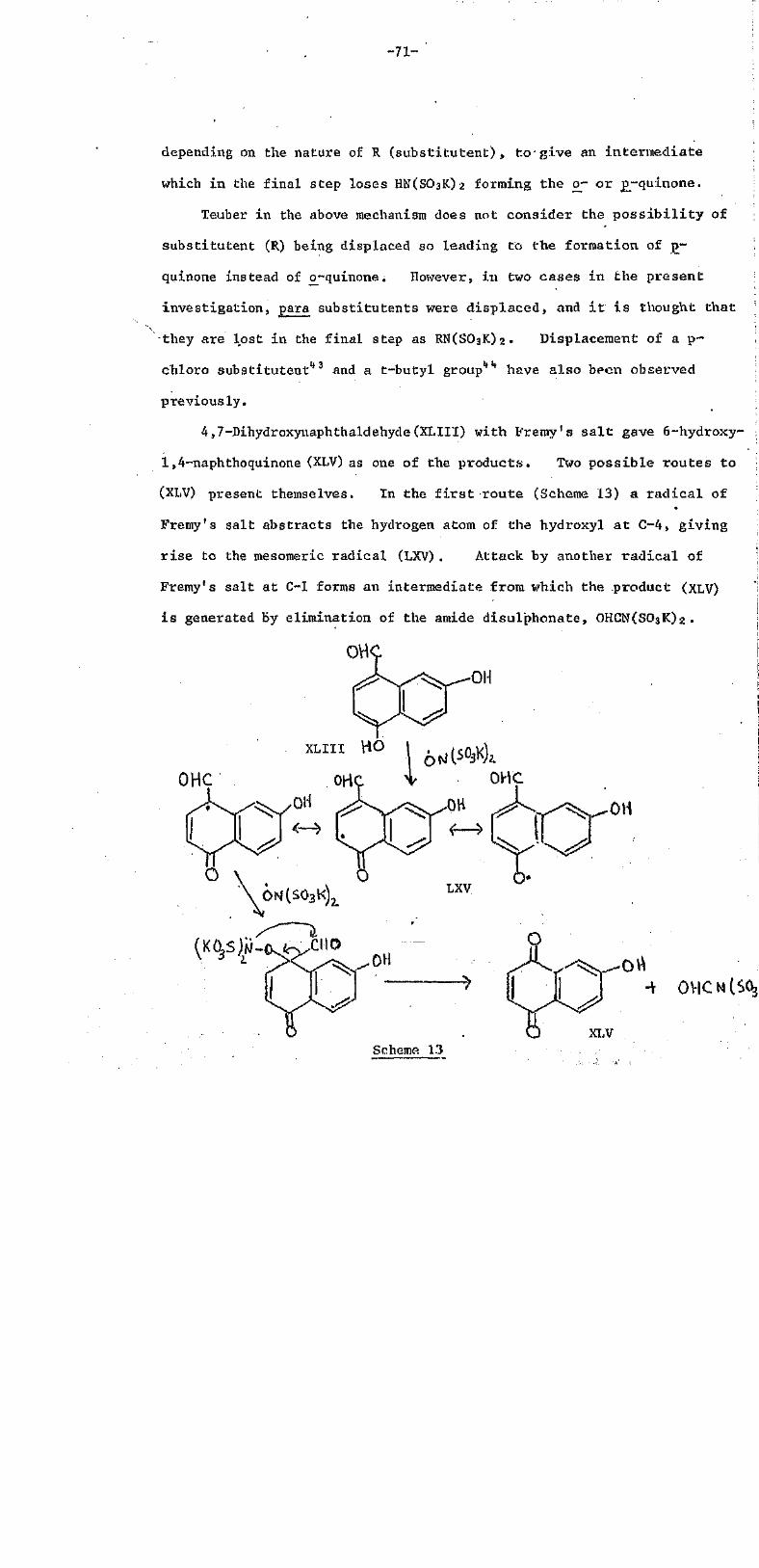
-71-
depending on the nature of R (substitutent), to give an intermediate
which in the final step loses HN(SO3K)2 forming the o- or p-quinone.
Teuber in the above mechanism does not consider the possibi l i ty of
substitutent (R) being displaced so leading to the formation of p-
quinone instead of o-quinone. However, in two cases in the present
investigation, para substitutents were displaced, and it i s thought that
•they are lost in the final step as RN(SO3K)2. Displacement of a p-
chloro substitutent43 and a t-butyl group44 have also been observed
previously.
4,7-Dihydroxynaphthaldehyde (XLIII) with Fremy's sal t gave 6-hydroxy-
1,4-naphthoquinone (XLV) as one of the products. Two possible routes to
(XLV) present themselves. In the f i r s t route (Scheme 13) a radical of
Fremy's salt abstracts the hydrogen atom of the hydroxyl at C-4, giving
r ise to the mesomeric radical (LXV). Attack by another radical of
Fremy's salt at C-1 forms an intermediate from which the product (XLV)
is generated by elimination of the amide disulphonate, OHCN(SO3K)2 .
OHC
XLVScheme 13

-72-
In the second route (Scheme 14) the hydrogen atom of the hydroxyl
at C-7 is abstracted and the resulting mesomeric radical reacts as in
Scheme 13 leading to the formation of (LXVI) which then aromatizes to
give the product (XLV) .
OHC
.0+ OHC
Cm• LXVI
Scheme 14

-73-
The second example where the substitutent was displaced was in
the oxidation of 4-bromo-l-naphthol (LX) with Fremy's salt to give (LXII)
along with two bromine-free products ((LXI) and (LXIII)) in small
amounts (Scheme 10), The mechanism by which these minor products
are formed is suggested in Scheme 15. It is similar to Scheme 13 and
O
HOCH3 (Solvent)
LXVIIILXIII
Scheme 15

-74-
14, except for the intermediate (LXVII) which in addition, is
capable of undergoing a nucleophilic attack at C-2 by a molecule of
solvent (CH3OH), This attack results in the removal of Br and
formation of another intermediate(LXVIII) from which (LXIII) is
generated by loss of HN(S03K)2.
The formation of 2-methoxy~l,4-naphthoquinone (LXIII)can also be
explained by a different route. The l,4~naphthoquinone(LXI) formed
by the loss of BrN(SO3K)2 shares a pair of its electrons with Lewis
acid,H+ from the medium (slightly acidic phosphate buffer) and froms
a conjugate acid which undergoes a nucleophilic attack at C-2 by a
molecule of solvent more readily than the parent compound. The attack
results in the formation of (LXIX) which is subsequently oxidized to
the product (LXIII). This route is similar to the reaction of quinones
with alcohols where ZnCl2 is the Lewis acid45 instead of H+ (Scheme 16).
+ ROH
R=Me, Et, Pr
Scheme 16

-75-
-Chapter 4
Experimental
See page 24 for explanatory notes. Solvent systems used in
this part of the investigation were:
(i) CHCl3 (solvent system 1)
(ii) CHCl3:MeOH (solvent system 2)
(iii) ethyl acetate:bcnzene (1:1, solvent system 3)
(iv) ethyl acetate;toluene:formic acid (10:9:1, solvent system 4)
Column chromatography was carried out on silica gel M.F.C.
(60-120 mesh) as supplied by Hopkin and Williams,
Elemental analyses were performed by the Micro-analysis Laboratories
of Aberdeen University.
NATURAL PRODUCTS WORK
Hibiscus tiliaceous
The stems of H. tiliaceous usad in this study were collected in
Suva, Fiji Islands during May 1977 and were identified by
Mrs. S. Siwatibau of the Department of Biology, University of the
South Pacific.
Extraction and chromatography
The air dried, powdered, stems (1,2 kg) were extracted in a Soxhlet
apparatus in 200 g portions with chloroform over 24 h. The extracts were
combined and evaporated in vacuo to leave a dark brownish gum (20.8 g)
which was chromatographed on acid-washed silica gel (1,5 kg) in chloroform

-76-
with progressively increasing proportions of methanol, and finally
20% methanol in chloroform. Twenty eight fractions (each approximately
250ml) were collected and screened for various components by T.L.C.
(solvent systems 2 and 3),
Isolation of lapachol CXVII) and hibiscone A(I)
Fractions 10-15 were pooled and evaporated in vacuo to give a
light brown residue (1.5 g) which showed two major spots (RF: 0.80 and
0.70, respectively, in solvent system 1) by T.L.C. These compounds were
then separated by P.L.C., developing first with CHCl3 (two developments)
followed by CHCl3 :Me0H (9:1). The bands were scraped off and extracted
with CHCh3 to give lapachol (XVII) and hibiscon?. A (I).
Lapachol was recrystallised from CHCl:petrol (9:1) as yellow
shiny crystals (42.5 ing) m.p. 140° (lit.18 140°); A (EtOH) 252.
(log E 4.06), 278 (4.16), and 330 nm (3.14); X (EtOH-NaOH) 281 and
486 nm; V (KBr) 3360 (s, OH), 1660 (s, C=0), 163.5 (s, chelated C=0),
and 1590 cm-1 (s, aromatic); 6 (CDCl3) 1.68 (3H, s, =CMe ), 1.78 (3H, s,
=CMe2), 3.30 (2H, d, J=8Hz,-CH2CH=C-) , 5.20 (lH, t, J=8Hz, -CH2-CH-i-),
7.30 (1H, s, OH, exchanged with D2O),7.66 (2H, M, ArH), and 8.06 (2H,
m, ArH); (Found:M+, 242.0946. C15H14O3 requires M, 242.0942);
identical (CO-T.L.C.;P.M.R., I.R., M.S., and U.V.) with authentic
lapachol.
Hibiscone A was crystallized from CHCl3 :petrol (1:1) as needles,
(20.5 mg), m.p. 94-95°; hmax (EtOH) 280 nm (log e 4.09); vmax (KBr)
1650 (s, conjugated C=0), and 850 cm-1 (m, furan ring);(Found: C, 77.6;
H, 8.8%; M+, 232.1460, C15H20O2 requires C, 77.6; H, 8.6%; H,
232,1463); identical (CO-T.L.C, I.R., U.V., and M.S.) with an authentic

-77-
sample of hibiscone A isolated previously from H. elatus.
y
Isolation of hibiscories C (III) arid D (XVIII)
Fractions 16-21 were pooled and evaporated to yield agvm (4,1 g) ,
T.L.C. examination of the gum by U.V. and with the phloroglucinol spray
reagent revealed the presence of two main spots whose RF values were almost
identical in several different solvent systems (1, 2 and 3), These were
eventually separated by repeated P.L.C, (developing five times in
solvent system 1, and then three times in solvent system 2) to give
hibiscones C (III) and D (XVIII).
Hibiscone C separated from CHCl3 :petroi (1:1) as crystals (560 rag),
m.p. 124-125°, gave an orange colour with the phlorogLucinoL spray
reagent; A (EtOH) 230 (log £ 4.16), and 265 nM (4.05); A
•(EtOH-NaOH) 230(sh) , 264 and 412 nm; V (KBr) 1670 (s, conjugated C O ) ,
1600 (s, conjugated OC) , and 1530 and 850 cm"1 (m, furan ring) (see
also Fig. 1); 6 (CDCl3) 0.90-1.10 (6H, two overlapping pairs of
doublets, J=8Hz, -CH(CH3)2), 1.36 (3H, d, J=8Kz, CH3-CH-) , 1.80-3.20
(8H, complex multiplets), and 8,08 (1H, s, furan-H); (Found: C, 73.2;
H, 7.0%; M+, 246.1254. C15H18e03 requires C, 73.2; H, 7.3%; M,
246.1255); identical (M.P., C0-TL.C, I.R., H.Y.., and P.M.R.) with
the specimen isolated earlier from H. elatus.
Hibiscone D was obtained from chloroform as light brown crystals
(19.6 mg), m.p. 139°; it gave a light purple colour with the phloroglucinol
spray reagent; A (EtOH) 229(sh) (log e 3.98), 243(sh) (3.93), andmax
335 nm (3.12); A (BtOH-NaOH) 230(s.h), 268, and 405 nm; V (KBr)max. max.
1680 (s, conjugated C=0), 1630 (m, conjugated C-C), 1500 (s, aromatic)
and 850 cm"1 (m, furan); V (CHCl3) 3580 cm-1 (phenolic OH); 6

-78-
(see also Fig. 1) 0,82 and 0.90 (each 3H, two pairs of doublets, J=8Hz,
-CH(CH3)2), 2.04 (1H, m, -CHMez), 2.40 (3H, s, ArCH3), 2.82 (2H, J=8H2,
equivalent methylene protons), 3.26 (1H, m, -CH -CH-CH-), 5.60 (1H, bs,
OH, exchanged with D20), 6.94 (1H, s, ArH), and 7.98 (1H, s, furan-H);
m/e (rel. int.) 244 (M+, 50%), 229 (M+-CH3, 1.5%), and 201 (M+-CH3~CO
or M+ -CH(CH3)2, 1.00%);(Found: C, 73.6; H, 6.5%; M+, 244.1101,
C15H16O3requires 6, 73.8; H, 6.6; M, 244..1099). 'The acetate was
obtained by treating hibiscone D (10 mg) with acetic anhydride (1 ml)
and dry pyridine (1 drop) overnight at room temperature. The reaction
mixture was poured into water (10 mil) and extracted with ether
(3x10 ml) . Combined ether fractions were dried over anhydrous MgSO4
and evaporated in vacuo to give a brown residue which after P.L.G.
(solvent system 1) afforded an oil (8 mg). This oil failed to
crystallize but was homogenous on T.L.C.; 6 (CDCl3) 0.82 and 0,90
(each 3H, two pairs of doublets, J=8Hz, -CH(CH3)2)= 2,02 (1H, m, -CHMe2) ,
2.33 (3H, s, GH3CO2-), 2.40 (3H, s, Ar-CH3), 2.80 (2H, d, J=8Hz,
equivalent methylene protons), 3.30 (1H, M, -CH2-CH-CH~), 7,04 (1H, s,
ArH), and 7.96 (1H, s, FUran-H); Vmax (CHCl3) 1750 (s, acetate),
Isolation of hibiscone B (II)
Fractions 22-26 were combined and evaporated in vacuo. The
residue (3.2 g) was transferred to a silica gel column (150 g) andl3:Me0H (5:1, 2:1 and 1:1 successively). The 50 ml |
fractions obtained from chloroform:inethanol (1:1) eluant were combined
and distilled off to give a residue (700 mg) which on crystallization from
CHCl3:Me0H (2:1) gave hibiscone B (II) as needles (260 mg), m.p. 122-123°
X (EtOH) 276 (log e 4.18); V (KBr) 3450 (s, OH), and 1650 cm"1

-79-
(conjugated C=O); S (CDCL3) 0,8
and 1.02 (each.3H, d, J=8Hz, -CH(CCH3)2),
1.32 (3H, d, .J=8Hz, -CH-CH3), 1,50-2.90 (9H, M), 5,01 (1H, s, OH,
exchanged with D20), and 7.64 (1H, s, furan-H);(Found: C, 72.6; H,
7.8%; M+, 248.1408. C15H20O3 requires C, 72,6; H, 8.1%; M,
248.1412); identical (CO-T.L.C. I.R, P.M.R. and M.S.) with an authentic
sample isolated, previously from H. elatus.
Oxidation of hibiscone B'to C using DDQ15
To a solution of hibiscone B (50.7mg) in CHCl3 (20 ml) was added
recrystallized DDQ (45,6 mg) and a few drops of 2M HCl. The reaction
mixture was heated under reflux and the reaction was monitored by T.L.C.
After 30 h the solvent was removed in vacuo and the residue chromatographed
on a silica gel column (20 g) with chloroform as eluant. The first band
was concentrated and the residue was subjected to P.L.C. to give
hibiscone C (40.1 mg), m.p. 123° (from CHCl3:petrol, 1:1); identical
in all respects (M.M.P., I.R., U.V., and P.M.R.) with the specimen
isolated from H. tiliaceouS.
Hibiscus tiliaceous (Brazil)
The roots of H. tiliaceous used in this investigation were collected
in Brazil.

-80-
ExtractioN and chromATography
Air-dried, powdered roots (300 g) were extracted in a soxhleT
apparatus in 150 g portions with petrol and chloroform, successively,
over 24 h.
Isolation of Gossypol (XIII)
Gradual concentration of the petrol extract gave a yellow powder
which was crystallized from CHCl3:petrol (1:2) to give small yellow
prisms (425 mg), m.p. 192-194° (dec.) (lIt.,16 195-197°); [a]0 0°
(CHCL3; C 2.10); A (MeOH) 238 (log e 4.86), 279 (4.45), and 381 ranmax
(4.23); X (MeOH-NaOH) 299 and 400 nM; V (KBr) 3520-3200' max max.
(br, phenolic OHs), 2900 (s, CH3), 1610 (chelated C=0), and 1570 cm"1
(s, aromatic); 6 (CDCl3) 1.45 (12H,. d, J=7Hz, 2HC(CH3)2), 2.18 (6H,
8, 2-Ar-CH2), 3.90 (2H, M, 2 HC(CH3)2), 5.96 (2H, s, 2 phenolic OHs,
disappeared on D20 shake), 6.38 (2H, s, 2 phenolic OHs,disappeared on
D20 shake), 7.78 (2H, s, 2Ar-H), 11.08 (2H, s, 2CH0) and 14.50 (2H, bs,
2 chelated OHs, disappeared on good D2O shake); (Found; C, 70.2; H,
5.9%; M+, 518.2101. C30H30O8 requires C, 70.0; H, 5.8%; M, 518.2107);
identical with an authentic sample of gossypol by M.M.P, and comparison
of I.R. (KBr) and P.M.R. (CDCl3).
Methylation of gossypol (XIII)- A solution of (XIII) (200 mg) in
acetone (30 ml) was heated under reflux with dimethyl sulphate (2 ml)
and anhydrous potassium carbonate (3 g) for 24 h. The reaction mixture
was cooled, filtered, and water (30 ml) added with a few drops of ammonia
solution. This was then warmed over" a steam bath for 1/2 h, and extracted
with chloroform. The chloroform extracts were combined, dried

-81-
and the solvent distilled off. T.L.C, analysis of the methylated
product revealed the. presence of at least two bands (R : 0.36 and 0.34
in solvent system 1) . The band with the lower RF value was
subsequently purified by P.L.C. (three developments in solvent system 1)
to give the hexamethyl derivative (90 mg). This was crystallized as
needles,from CHCl3-, m.p. 214° (lit.,16 216-218°); Xmax(MeOH) 2.30
(log e 4.70) and 254 tun (4.95); Vmax (CHCl3) 1700 (s, 00) , and 1245 cm-1
(C-O-C); 6 (CDCl3) 1.56 (12H, d, J=7HZ, 2 CH(CH3)O, 2.20 (6H, s, 2Ar-CH3),
3.24 (6H, s, 2 OCH3), 3.92 and 3.96 (14H, 2 OCH3 singlets overlapping
2H, m, 2 HC(CH3)2), 7.86 (2H, 2, 2Ar-H),and 10.56 (2H, s, 2CH0):(Fouud:
M+, 602.2879. C36H4208 requires M, 602.2878).
Isolation of mansonone P (XIX) and F (XX)
Analysis of the chloroform extract by T.L.C. indicated the presence
of two major coloured bands (RF: 0.40 and 0.28 in solvent system 1) and
also a trace of gossypol (XIII). The extract was concentrated under
reduced pressure to leave a viscous mass ( 55 g ) which, after
chromatography on a silica gel column (250 g) (eluent CHCl3) twice and
final purification by P.L.C. (three developments in solvent system 1)
afforded (in decreasing order of RF values in solvent system 1)
mansonone D and F.
Mansonone D (XIX) separated from chloroform:petrol (4:1) as dark
orange prisms (145 mg), tn.p. 174° (lit. ,'18*. 173-175°); h (EtOH) 243
(log e 4.14), 2.78 (4.14), and 4,06 nm (3.98); V (KBr) 2900 (s, CH3),
1660 and 1645 (s, C=O, and 1600 cm-1 (aromatic); 6 (CDCl3) 1.36 (3H, d,
J=8Hz, -CH-CH3), 2.04 (3H, d, J=3Hz, CH3-C=C-chH~), 2.60 (3H, s, Ar-CH3),
3.60 (1H, m, 0CH2-CH-), 4.36 and 4.68 (1H, m, OH2-(lH-), 6.54 (1H, s,
Ar-H) and 7.18 (1H, q, J=3HK, CH3-C=CH-) (Found: M+, 242.0944,

-82-
C15H14O3 requires M, 242.0942); identical (M.M.P., I.R., and P.M.R.)
with an authentic sample of mansonone D isolated from Mansonia altissima27
Marisorione F (XX) separated from CHCl3 as long purple shiny crystals
(40mg), m.p. 213° (lit.,18 214-21.5°)hmax 23?. (log F. 4.61), 285(sh)
(4,01), 338 (3,82), and 550 nm (3.70); V (KBr) 1680 and 1625 (s,axmax •
C=0), and 1600 cm"1 (s, aromatic); 6 (CDCl3) 1.96 (3H, s, quinone-CHs) ,
'2.10 (3H, d, J=2Hz, CH3-C=), 2.70 (3H, s, Ar-CHs), 7.06 (1H, m, J=2Hz,
7.40 (2H, bs, Ar-H); (Found: M+, 240.1094. C15H12O3
requires 240.1093); identical (M.M.P., I.R, and P.M.R.) with mansonone
F isolated from Mansonia altissima.17
SYNTHETIC WORK
General procedure for formylation of dihydroxynaphthalenes32
Into a solution of the dihydroxynaphthalene (13 g, 81.25 mmol) in
sodium-dried ether (150 ml) containing anhydrous zinc cyanide (10 g,
85.17 mmol), dry HCl gas was passed for 6 h. The yellow precipitate
was washed many times with sodium dried ether, and finally taken up in
distilled water (450 ml). On warming over a steam bath for 30 min.
the formyl derivative precipitated.
Fremy's salt solution34,35 -the solution used for oxidation was prepared
by dissolving freshly made Fremy's salt (10,0 g) in a mixture of
1/6M KH2PO4 buffer solution (170 ml) and distilled water (570 mi) .

-83-
8-Formy 1-7-hydroxy-l, 2-naphthoguinone (XXXVIII) .
2,7-Dihydroxynaphthaldehyde (XXXVII) was obtained from 2,7-
dihydroxynaphthalene, see above as yellow, fluffy, crystals (12.3 g,
80%), m.p. 157-158o (lit.33 156.5-158.5°); X (CH3OOH) 240, 277{sh),
289(sh), and 362 nm; X (MeOH-NaOH) 250, 300, and 392 nm; V
max. max.(KBr) 3400 (s, OH), and 1620 cm-1 (s, C=0).
\
2,7-Dihydroxynaphthaldehyde (XXXVII) (2,75 g, 14,64 mmol) in methanol
(150 mil) was treated at room temperature in the dark with Fremy's salt
solution (640 mil) to give immediately a purple precipitate. This was
filtered off and dissolved in acetone (20 ml). On addition of petrol
with cooling 8-formyl-7-hydroxy-l,2-naphthoquinone (XXXVIII) separated
as dark purple crystals (2.1 g, 71.2%), m.p. 149-150o; X (CHCl3)
250 (log e 3.93), 296 (4.16), 340(sh) (3.52), and 478 nm (3.27); V
(KBr) 3600-3350 (s,0H), and 1650 cm-1 (s, C=0); 6 (CDCl3) 6,34, 7,26,
7.42 and 7.50 (each 1H, d, J 8Hz, benzenoid and quinonoid protons),
10.82 (1H, s, CHO) and 12.34 (1H, s, OH, exchangeable with D2O); m/e
(Eel. int.) 202(M+, 94%), 174(M+-CO, 94%), and 146(M+-2CO, 100%);
(Found: C, 65.1; H, 3.2%; M+, 202.0264. -C11H16O4 requires C, 65.3;
H, 3.0%; M, 202.0266).
Lactone (XXIX)
8-Formyl-7-hydroxy-l,2-naphthoquinone (XXXVIII) (1-0 g, 5,0 mmol)
dissolved in chloroform (100 ml) was irradiated with U,V. for 45 min. '
at room temperature, Removal of chloroform by distillation under
reduced pressure and then crystallization from chloroform:methanol (4;1)
gave the lactone (XXXIX) as dark brown needles (0.76 g, 76%), m.p. 228-230°;

- 8 4 -
V (EtOH) 254 (log e 4.00) and 357 nm ( 3 , 9 8 ) ; V 3100-3500max• max
(br, OH), and 1730 (s, C=O); 6 (DMS0-d6) a set of multiplet in the
aromatic region (7.00 to 8.30) which integrated for 6H; m/e (rel. int.)
202(M+, 100%), 174(M+-C0, 1.5%), and 146(M+-2CO 56%);(Pound: C, 65.2;
H, 3.2%;M+ 202.0264. C11H6O4 requires C, 65.3; H, 3.0%; M,
202.0266).
8~Formyl7-hydroxy-1,4-naphthoquinone (XLII)
1,7-Dihydroxynaphthalene was formylated to give a mixture of
isomers (T.L.C.). The 2,8-dihydroxynaphthaldehyde (XL) was extracted
with boiling benzene (50 ml) and crystallized from methanol as small
yellow needles (5.81 g, 38.0%), m.p. 202-204° (lit.33 203-204o); V
(KBr) 3200 (br, OH) and 1650 cm"1 (s, 00) .
The aldehyde (2.0 g, 10.64 mmol) in methanol (110 ml) was treated
with Fremy's salt solution (470 ml) in the dark at ambient temperature,
and allowed to stand for 15min. The reaction mixture was then extracted
with chloroform (2x100 ml); the extracts were combined, dried (MgSO4),
and the solvent distilled off. The residual gum was chromatographed
on a silica gel column (100 g) in chloroform to give: (i) a solid which
was crystallized from chloroform to give 8-formyl~7-hydroxy-l,4-
naphthoquinone (XLII)(0.88 g, 41.0%), m.p. 135°; h (EtOH) 248 (logs
4 . 2 2 ) , 278(sh) ( 3 . 9 3 ) , 315(sh) ( 3 . 5 1 ) , and 380 nm (3 .39) ; Xmax
(EtOH-SaOH 234, 315, and 530 ran-, V (KBr) 3400-3300 ( s , OH), 1650
( s , C=0) , and 1600 cm"1 (w, a romat i c ) ; 6 (CDCl3) 6.94 (211, s , quinonoid
p r o t o n s ) , 7.30 (1H, d J 8Hz, Ar-H), 8.24 (1H, d, J 8Hz, Ar-H), 10.88
(1H, s , GHO), and 12.76 (1H, s, OH, exchangeable wi th D2O); m/e ( r e l . i n t . )

-85-
202{M+, 100%), 174(M+-CO, 10%), and 146(M+-2CO, 56%);(Found: C, 65.6;
H, 4.0%; M+, 202.0260. C11H6O4 requires C, 65.4; H, 4.0%; M,
202,0266) and (ii) the £-isomer (0.53 g, 24.7%) - identical in all
respects with 8~formyl-7-hydroxy-l,2-naphthoquinone (XXXVIII).
Attempted synthesis of 8-fornyl-5-hydr6xy-l,2-naphthoquinone (XLIV)
1,6-Dihydroxynaphthalene was formylated to give a mixture of
isomers. 4,7-Dihydroxynaphthaldehyde (XLIII) was obtained by washing
the product mixture exhaustively with chloroform, and then crystallizing
from methanol as silvery needles, (9.0 g, 59.0%), m.p. 217° (dec.) (lit.33
218° (dec.)); V (KBr) 3300-3200 (br, OH), and 1640 cm"1 (s, C=0).
A solution of this aldehyde (1.0 g, 5.3 mmol) in methanol (55 ml)
was treated at room temperature in the dark with Fremy's salt solution
(235 mil) . After 30 min. the reaction mixture was extracted with
chloroform (4x25 mil) and the extracts were combined, dried (MgSO4)., and
evaporated. The residue was transferred to a silica gel column (150 g)
and eluted with chloroform to give a product which on repeated P.L.C.
(solvent system 1) afforded yellow prisms of 6-hydroxy-l,4-naphthoquinone
(from benzene) (XLV, 0.4 g, 43%), m.p. 170° (lit.41 170°), X (MeOH)
262 and 396; X (MeOH-NaOH), 250, 300 and 500 nm; V (KBr) 3300
(s, OH), 1655 (s, C=0), and 1580 (s, aromatic)", identical (M.P., M.M.P.,
U.V., and I.R.) with an authentic specimen.
Attempted synthesis of 7-hydroxy-4-methoxynaphthaldehyde(XLV1)
A solution of dimethyl sulphate (670.2tng) in acetone (2 mil) was
added dropwise to a stirred refluxing solution of 4,7-dihydroxy-
naphthaldehyde (XLIII) (1.0 g, 5.3 mmol) in acetone (25 mi) containing
anhydrous potassium carbonate (5.0 g) over 2 h. The cooled filtered

-86-
solution was diluted with water (25 ml), warmed over a steam bath for
1 h. and extracted with chloroform (3x25 mi). The combined extract
was dried Na2SO4 and evaporated to give a yellow residue which on
crystallization from chloroform:methanol (9:1) gave needles of 4,7-
dimethoxynaphthaldehyde (0,52 g, 45.3%), m.p. 76° (dec.) (lit.48 76-77°);
V 1655 (s, C=0) and 1270 cm-1 (C-O-C); 6 (CD3COCD3) 4.06 and 4.10 (each
3H, s OCH3), 7.12. (2H, m, Ar-H), 7.44 (1H, m,Ar-11), 7.92 (2H, m, Ar-H)
and 10.94 (1H, s. CHO).
Attempted synthesis of 8-hydroxy-4-methoxy-naphthaldehyde (XLIX)
1,5-Dihydroxynaphthalene was formylated to give 4,8-dihydroxy-
naphthaldehyde (XLVI1) which separated from methanol: wter (9:1)-as
yellow crystals (10.1 g, 66.1%), m.p. 278° (dec.) (lit.33 280° (dec.));
Vmax (KBr) 3450-3380 (s, OH), and 1650 cm"1 (s, C=0)..
(i) Using dimethyl sulphate as methylating agent
A solution of 4,8-dihydroxynaphthaldehyde (XLVII) (1 g, 5.3 mmol)
in acetone (25 ml) containing potassium carbonate (5.0 g) was methylated
with a solution of dimethyl sulphate (670.2 mg) in acetone (2 ml) as
above. The mixture, after usual work up, gave the corresponding dimethyl
derivative (0.59 g, 47.0%), m.p. 126° (lit.1*7 126°); V (KBr) 1660 (s,max.
C=0) and 1270 cm-1 (s, C-O-C); 6 (CDCl3) 3.94 and 3.98 (each 3H, s, OCH3),
6.88 (211, m, Ar-H), 7.36 (1H, m, Ar-H), 7.96 (211, m, Ar-H), and 10.94
(1H, s, CHO).

- 8 7 -
(ii) Using diazomethane as the methylating agent
A solution of diazomethane (223 .4 mg) in ether (45 mi), prepared
from diazald (p_~tolylsulphenylmethylnitrosoamide) by the procedure
given in Vogel, 48 was added dropwise to a solution of 4,8-dihydroxy-
naphthaldehyde (XLVII (1,0 g, 5.3 mmol) in ether (25 ml) and the
reaction was monitored by T. L.C. The solvent was removed over a
steam bath and the resulting crude product was crystall l ized from
chloroform to give the dimethyl derivative identical to Chat obtained
in ( i ) .
Attempted synthesis of 8-fomyl-5-hydroxy-l,4-naphthoquinone (XLVIII)
by phase transfer
A solution of 4,8-dihydroxynaphthaldehyde (XLVII) (1.0 g, 5.3 mmol)
in ethyl acetate (50 ml) was added slowly over 10 min, to a stirred
solution of Fremy's salt (235 ml) containing Adogen 464 ( 8 ml).
Stirring vas continued for 30 min. at room temperature in the dark.
The reaction mixture was diluted with ethyl acetate (50 ml) , washed with
water and then evaporated to give a brown viscous mass, which could not be
purified either by column chromatography or P.L.C.
8-Formy1-1, 4-naphthoquinone (LVI)
8-Hydroxynaphthaldehyde (0.16 g, 0.93 mmol) in methanol (10 ml) was
treated with Fremy's salt solution (4 ml) and allowed to stand for 30min.
in the dark. The reaction mixture was extracted with chloroform
(3x25 ml). and the combined extract was dried (Na2SO4 Concentration of
the chloroform solution gave a gum which on crysta l l isa t ion from
chloroformipetrol (1:1) gave shiny brown prisms of 8-formyl-l,4-naphtho-

-88-
quinone (LV1) (0.116 g, 66.8%), m.p. 115-116°; "Xmax, (EtOH) 246
(log eed C=0) , and 1600 cm-1 (s, aromatic); <5 (CDCl3) 7.04 (2H, s,
quinonoid-H), 7.80-8.40 (3H, m, Ar-H), and 10.72 (IH, s, CHO); m/e
(rel. int.) 186(M+, 100%), 158(M+-CO, 50%), and 13.0(M+-2C0, 45%)}
(Found: C, 71.1; H, 3.4%; M+, 186.0320. C11H6O3 requires G, 71.0;
H, 3.2%; M, 186.0316).
4-Bromo-l-naphthol (LX)
A solution of iodine monobromide was prepared by addition of
powdered iodine (26 g) to a stirred solution of bromine (5 ml) in glacial
acetic acid (60 ml). This solution was warmed over a steam bath for 1 h.
and added within 10 min, into a solution of 1-naphthol(U.X) (14 g,
97.2 mmol) in glacial acetic acid (45 ml) . The reaction mixture was
left in the cold room and after 1 h. was poured into water (700 ml)
containing sodium bisulphite (16 g). The solution was then neutralized
with sodium bicarbonate (160 g) and the resulting white solid collected
and dried. Recrystallxzation from methanol gave crystals of the title
compound (LX) (12 g, 55.3%), m.p. 120° (lit.40 121-122*) ; h. (MeOH)
240, 309, and 330sh) nm; h (MeOH-NaOH) 253 and 343 nm; V (KBr)max• max•
3300-3100 (br, OH) and 1590cm"1 (s, aromatic), m/e (rel. int.) 224(Mt +2,
100%) and 222(M+, 100%).

-89-
4-BrommO-l,2-naphthoquinone (LXI1), 1,4-naphthoquinone (LXI) and
2-methoxy-1 ,4-naphthoquinbne (LXII1)
4-Bromo-l-naphthol (LX) (3.0 g, 13.45 iranol) in methanol (135 ml)
was treated at room temperature in the dark with Fremy's salt solution
(592 ml) and allowed to stand for 15 min. The reaction mixture was
then extracted with chloroform (2x100 ml) ; the extracts were combined,
dried (MgSO4), and evaporated to a pale yellow gum. P.L.C. of the gum
(solvent system 1) gave (i) 1,4-naphthoquinone (LXI) as yellow crystals
from chloroform (0.17 g, 8.0%), m.p. 127°(lit.41 126°); X (MeOH)
250 and 330 nmV max (Nujol) 1670 cm'1 (s, CO); m/e (rel. int.)
159(M+, 100%), 130(M+-C0, 55%), and 102(M+-2CO-, 60%); identical (I.R.,
U.V., and M.M..P.) with an authentic sample; (ii) 2-methoxy-l,4-naphtho-
quinone (LXIII) as pale yellow needles from methanol;chloroform (4: 1)
(0.38 g, 15.0%), m.p.186-187° (lit.143 185°); A (MeOH) 250, 275 and
335 ran; V (Nujol) 1670 cm-1 (s, C O ) ; m/e (rel. int.) 188(M+, 100%),max
160(M+C0, 80%), and .152(M+-2CO 50%); identical (I.R., M.M.P., and M.S.)
with an authentic specimen; and (iii) 4~bromo-l,2-naphthoquinone (LXII)
as light red needles from toluene (2.0 g, 62.7%), m.p. 153° (lit.''1 154°);
A (MeOH) 252 and 345 nm; V 1660 (s, C=0) and 1645 (w, C=0) ;max. max
m/e (rel. int.) 238(M++2, 95%), 236(M+, 100%), 208(M+C0, 89%), and 180
(M+-2C0, 45%).
Attempted synthesis of 4-bromo-8-formyl-l~hydroxynaphthalene (LVII)
8-Hydroxynaphthaldehyde (LV) (0.2 g, 1.2 mmol) was brominated as
above to give a complex mixture which could not be purified.

90-
References
1. M,A. Ferreira and R.H, Thomson, unpublished results.
2. Personal communication from Dr. R. Letcher to Professor R.H. Thomson.
3. V. Seshadri, A.K. Batl:a, and S. Rangaswami, Curr. Sci.,
1971, 40, 630.
4. J.R. Gray, T,J, Mabry, A,A. Bell, and R.D. Stipanovic,
J.C.S. Chem.Comm., 1976, 109.
5. V. Seshadri, A.K. Batta, and S. Rangaswami, Indian J. Ghent. ,
1976, 14(B). 616.
6. A.A. Bell, R.D, Stipanovic, C.R. Howell, and P.A Fryxell,
Phytochemistry, 1975, 14, 225.
7. R. Adams, T.A. Geissman, and J.D, Edwards, Chem Rev., 1960; 60, 555.
8. A.K. Karimdzhanov, A.I. Israailov, and Z.S. Abdulaev,
Khim. Prirod. Soedin., 1976, 238.
9. R.D. Stipanovic, A.A. Bell, and G.R. Howell, Phytochemistry,
1975, 14, 1809.
10. R.D. Stipanovic, A.A. Bell, D.H. O'Brien, and MJ, Lukefahr,
Tet. Lett., 1977, 567.
11. R.D, Stipanovic, A,A. Bell, D.H. O'Brien, and M.J. Lukefahr,
Phytochemistry, 1978, 17, 151.
12. A.A, Bell, 'Biologiual Control of Plant Insects and Disease1,
(F.G, Maxwell, ed.), Mississippi State University Press, Jackson,
1974, p.430.
13. J.W. Parham, 'Plants of the Fiji Islands', Fiji Government Press,
Suva, 1964
14. P. Singh, unpublished observation; Singh has since found hibiscone D
in H. tIliaceous from Sri Lanka.
15. L.F. Fieser and H. Fieser, 'Reagents for Organic Synthesis'
Wiley, New York, 1967.

-91-
16. S.C. Dtta, V.V.S. Murti, and T.R. Seshadri, Indian J, Chem.,
1972, 10, 263; 691.
17. G.B. Marini-Bettolo, C.G. Casinovi, and C. Galeffi,
Tet. Lett., 1965, 4857
18. R,H. Thomson, 'Naturally Occurring Quinones', 2nd ed., Academic Press,
London, 1971
19. K.V. Rao, T.J. McBride, and J.J. Olsson, Cancer Research,
1968, 28, 1952.
20. V. Krishnamoorthy and R.H. Thomson, Phytochemistry, 1970, 10_, 1669;
J.W. Rowe and M.K. Seikel, Phyto chemistry, 1972, 11, 2513.
21. J. Rigandy and C. Dufraisse, Compt. rend., 1949, 228, 253.
22. W.E. Doering and R.M. Haines, J. Amer. Chem. Soc, 1954, 76, 482
23. K. Chandrasenan and R.H. Thomson, Tetrahedron, 1971,27 2529.
24. G.A. Russell, Abs 17th Mat, Org. Chem. Symposium, Amer. Cheiti. Soc,
1961, 71.
25. A.C. Baillie, Ph.D. thesis, University of Aberdeen, 1966.
26. E.W. Scheiffele and D.A. Shirley, J. Org. Chem., 1964, 29;, 3617.
27. H.D. Dakin, Am. Chero.J., 1909, 42, 477.
28. D.H. Rosenblatt and R. Rosenthal, J. Amer. Chem. 'Soc, 1953, 75, 4607.
29. J.M. Bruce, Quart. Revs., 1967,21, 405.
30. K. Maruyama and Y. Mrai, and T. Otsuki, ibid, 1977, 50, 2777.
32. F, Cramer and H. Windel, Chem. Ber., 1956, 89, 354.
33. G.T. Morgan and V.C. Vining, J. Chem. Soc, 1921, 119_, 177.
34. H. J. Teuber and N. G8tz, Chem. Ber., 1954, 87, 1236.
35. H Zimmer, D.C. Larkin, and S.W. Horgan, Chem. Rev., 1971,71, 229.
36. O.N. 2hukovskaya, V.V. Mezheritxky, V.V. Zkachenko, and G.N. Borofeyenko,
Zhur. Organ. Khim. (USSR), 1978, 868.
,37. H. Ishii, T. Hanaoka, T. Asalca,-Y-. Harada, and N. Ikeda,
Tetrahedron, 1976, 32, 2693.

-92-
38, R.O. Hutchins, N.R. Natale, and W.J, Cook, Tet. Lett.,
1977, 4167 and references therein,
39. D. Berry and D.C.C. Smith, J. Chem. Soc. Perkin I, 1972, 699,
40, W. militZEr, J. Amer. Chem. Soc. , 1938, 60_, 256.
41. Elsevier's Encyclopaedia of Organic Chemistry., ed. F. Fadt,
Ser. Ill, Vol, 12, Elsevier Publishing Company, Amsterdam, 1952
42. H.-J. Teuber and W. Rau, Chem. Ber., 1953, 86, 1036,
43. H.-J. Teuber and 0. Glosauer, Chem. Ber., 1965, 98, 2643.
44. R. Magnussion, Acta Chem. Scand,, 1964, 18, 759.
45. E. Knoevenagel and C. Buckel, Ber., 1901, 34, 3993;
J.M. Singh and A.B. Turner, J Chem. Soc. Perlcin I, 1974, 2556.
46. N.P. Buu-Hoi' and D. Levit, Bull. Soc. chim. France, 1955, 1419.
47. N.P. Buu-Hoi' and D. Levit, J. Org. Chem., 1955, 20, 1191.
48. A,I. Vogel, ''Practical Organic Chemistry', 3rd ed., Longmans,
London, 1957.