urine screening of metabolic disorders
DESCRIPTION
Urinalysis presentation on urine screening of metabolic disorders like PKU, MSUD and cystinuriaTRANSCRIPT

Urine Screening of Metabolic Disorders

Overflow vs Renal DisordersOverflow
Overproduction of a normal or abnormal substance to the point that the tubules are not able to prevent it from escaping into the urine.
• Increased levels in blood & urine
• Inherited defect • Acquired defect
Renal Faulty tubular
reabsorption mechanism Increased levels seen in
urine only

Value of Urinalysis

Value of Urinalysis
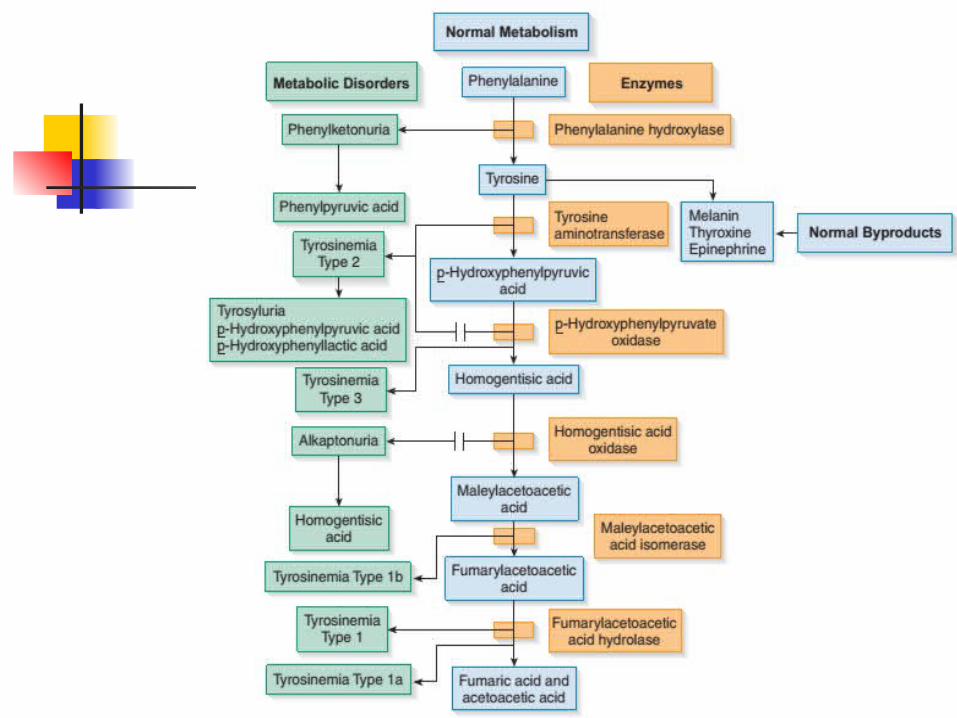

Phenylketonuria (PKU) “Mousy odor urine” 1/10,000 - 20,000 births Autosomal recessive trait Absence of phenylalanine
hydroxylase Urinalysis
Follow-up procedure in doubtful Dx cases
Screening test for proper dietary control in previously Dx cases
Monitoring the dietary intake of pregnant women known to lack phenylalanine hydroxylase

Phenylketonuria Diagnostic Tests
Microbial inhibition test (Guthrie)
Uses Bacillus subtilis (+) Phenylalanine
counteracts the action of β-2-thienylalanine,an inhibitor of B. subtilis (present in the media) growth is observed
Urinalysis1. Place 1 mL of urine in a
tube.2. Slowly add 5 qtts.of
10% ferric chloride.3. Observe colorpermanent blue green color
(+)

Tyrosyluria urine may contain excess tyrosine or its
degradation products, p-hydroxyphenylpyruvic acid and p-hydroxyphenyllactic acid.
Transitory tyrosinemia in premature infants
Acquired severe liver diseases
Enzyme deficiency
Type I - no fumarylacetoacetate hydrolase (FAH)
Type II – no tyrosine aminotransferase
Type III - p-hydroxyphenylpyruvic acid dioxygenase
Tested with nitroso-naphthol test and MS/MS
Autosomal Recessive
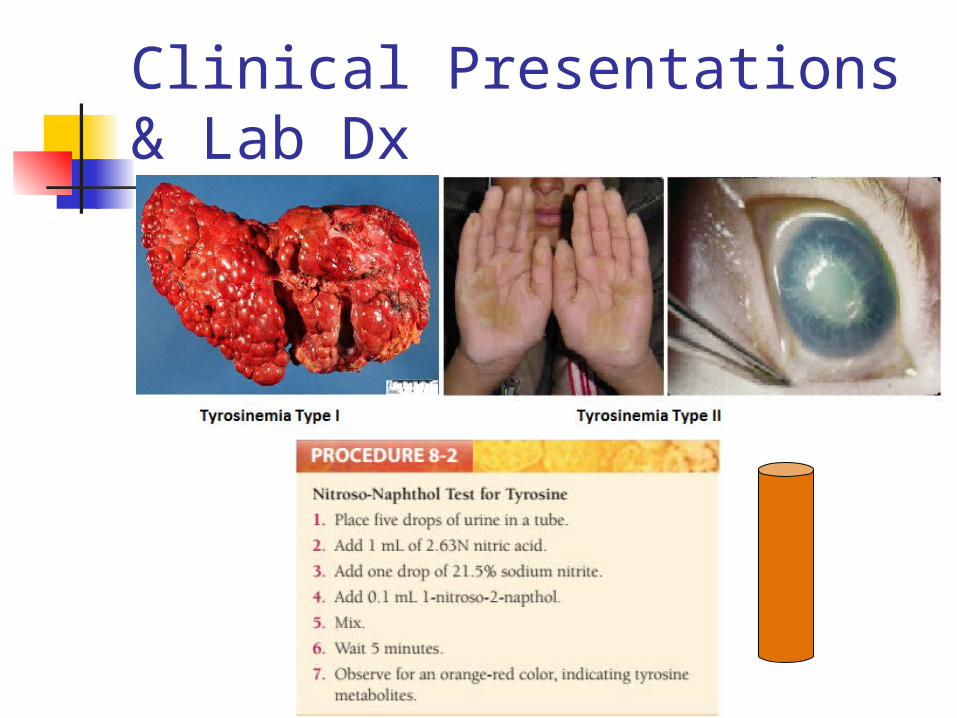
Clinical Presentations & Lab Dx

Tyrosine metabolized further to melanin, thyroxine, epinephrine, protein and tyrosine sulfate
Deficiency albinism Increased in urine urine darkening (after
exposure to air) Proliferation of melanocytes
malignant melanoma Tumors secrete 5,6- dihydroxyindole
melanogen melanin Melanin may react with Na nitroferricyanide
(acetone rgt. Strip) may producer ed color Ferric chloride tube test, a gray or black
precipitate forms in the presence
Melanuria

(“Alkali lover”) One of the 6 IEM described by Garrod
(1902) No Homogentisic acid oxidase HA
accumulation Brown or black-stained cloth or reddish-
stained disposable diapers May progress to arthritis, liver and
cardiac disorders Ferric chloride test deep blue color Clinitest yellow ppt. Addition of alkali in urine urine
darkening AgNO3 test, paper and TLC
Alkaptonuria

Amino Acid Disorders (Branched-Chain Amino Acid Disorders)
• Having a methyl group that branches from the main aliphatic C chain

Inherited as autosomal recessive trait
Failure to inherit gene for enzyme capable of oxidative decarboxylation of α-ketoisovaleric, α-ketoisocaproic, α-keto-β-methylvaleric
Exhibit failure to thrive w/in 1 wk
Maple Syrup odor is due to rapid accumulation of keto-acids
2,4-dinitrophenylhydrazine urine screening test
Maple Syrup Urine Disease

Sx: Early severe illness, vomiting, metabolic acidosis, hypoglycemia, ketonuria and increased serum NH3
Commonly: Isovaleric acidemia Propionic acidemia Methylmalonic acidemia
Isovaleric acidemia sweaty feet urine due to accumulation of isovaleryl glycine w/o isovaleryl CoA
Organic Acidemias
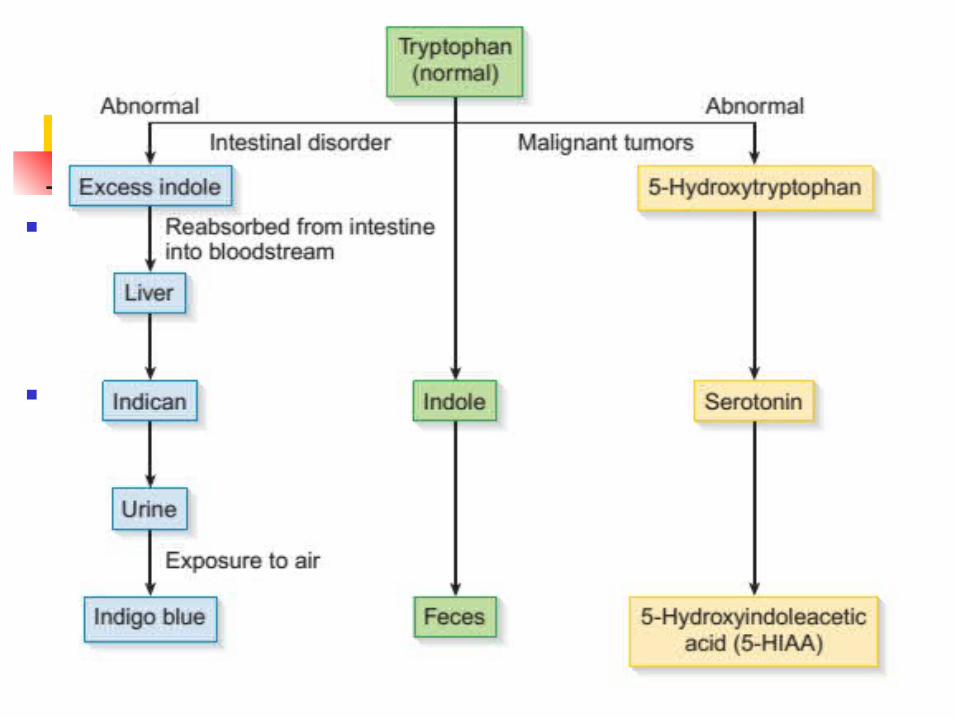
Indicanuria Due to obstruction, abnormal bacteria present,
malabsorption syndromes, Hartnup disease “Blue diaper syndrome”
5-hydroxyindoleacetic acid (5-HIAA) Carcinoid tumors involving argentaffin excess amounts
of serotonin increased urinary 5-HIAA levels Addition of nitrous acid and 1-nitroso-2-naphthrol purple
to black urine >25 mg/24h indicates tumor
Amino Acid Disorders (Tryptophan Disorders)

Cystine Disorders (Sulfur-like odor urine)
Cystinuria Inability of renal tubules
to reabsorb cystine Only amino acid found
during analysis of calculi Has two modes of
inheritance Poor reabsorption of all
four amino acids—cystine, lysine, arginine, and ornithine
cystine and lysine are not reabsorbed
Tested by cyanide-nitroprusside test red-purple color
Cystinosis Genuine IEM, can be
nepropathic/non-nepropathic Subdivided as infantile
and late-onset cystinosis
Defect in lysosomal membranes
Leads to crystalline deposits in cornea, bone marrow, lymph nodes, and internal organs

Fanconi syndrome Renal transplants and cystine-
depleting meds – extend lives from renal failure
Nonnephropathic – benign but may cause ocular disorders
Infantile nephropathic cystinosis Lab dx: poluria, generalized
aminoaciduria, (+) Clinitest for reducing substances, lack of urinary concentration
Cystinosis (cont’d)

Defect in metabolism of methionine Leads to failure to thrive, cataracts and mental
retardation, thromboembolic problems, and death (+) with cyanide-nitroprusside test Silver nitroprusside test – confirms presence Thromboembolism is related with:
Endothelial cell damage Smooth muscle cell proliferation Lipid peroxidation Upregulation of prothrombotic factors (XII and V) Downregulation of antithrombotic factors or endothelial-
derived nitric oxide
Homocystinuria

Porphyrin – heme intermediate Free erythrocyte protoporphyrin CDC
screening test for Pb poisoning Pophyrias commonly due to lead
poisoning, excessive alcohol intake, iron deficiency, chronic liver disease and renal disease
Rarity: Inherited porphyrias > Acquired
Related with vampirism
Porphyrin Disorders


Laboratory Diagnosis Ehrlich reaction - can be
used only for the detection of ALA and porphobilinogen
Watson-Schwartz reaction - differentiation between the presence of urobilinogen and porphobilinogen
Hoesch Test Fluorescence tests – does
not distinugish porphyrins

Mucopolysaccharide Disorders Mucopolysaccharides aka glycosaminoglycans
Incomplete breakdown of polysacchride portion in lysosomes of CT
Usually found in urine as dermatan, keratan and heparan sulfates.
Hurler syndrome – skeletal structure and mental retardation; cornea
Hunter syndrome – rarely seen in females, Sanfilippo syndrome – only mental retardation
Bone marrow transplants and gene replacement therapy U/A: Acid-Albumin, Cetyltrimethylammonium bromide
(CTAB) turbdity tests and metachromatic staining spot tests
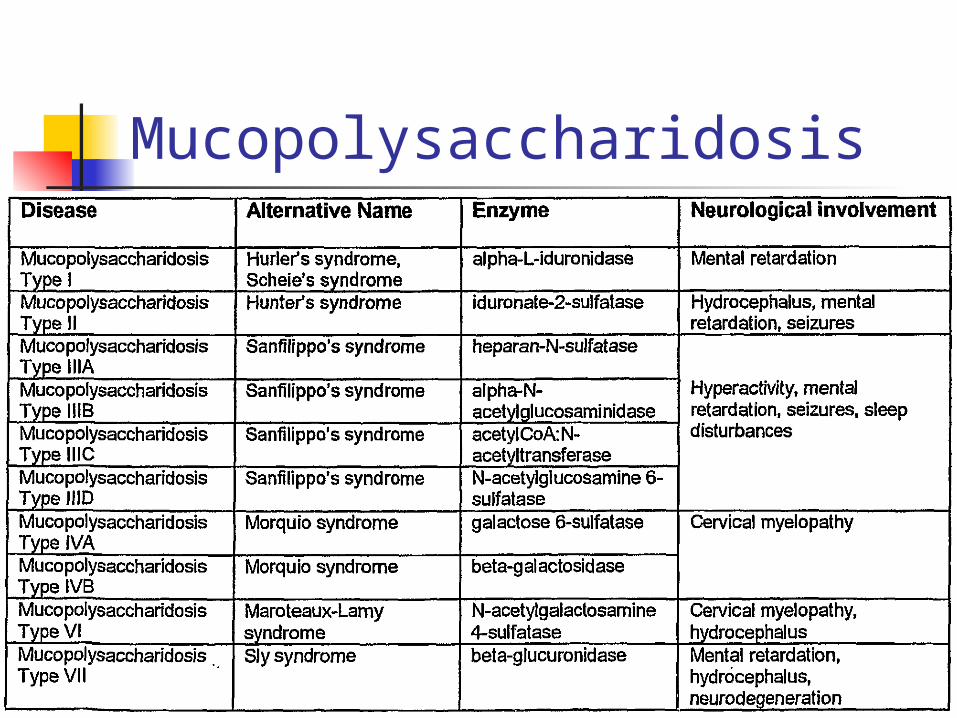
Mucopolysaccharidosis

Purine and CHO Disorders Lesch-Nyhan Disease
Inherited, sex-linked recessive Without enzyme hypoxanthine guanine
phosphoribosyltransferase Sx: severe motor defects, mental retardation, tendency
towards self destruction, gout and renal calculi. Uric acid like orange sand in diapers
Melituria Pentosuria Galactosuria – failure to thrive, liver disorders, cataracts and
severe mental retardation Deficiency in GALT, galactokinase, UDP-galactose-4-epimerase
Lactosuria Fructosuria