universidade federal do triÂngulo...
TRANSCRIPT

UNIVERSIDADE FEDERAL DO TRIÂNGULO MINEIRO
PRÓ-REITORIA DE PESQUISA E PÓS-GRADUAÇÃO
CURSO DE PÓS-GRADUAÇÃO EM CIÊNCIAS FISIOLÓGICAS
Thiago Salem Pançonato Teixeira
Estudo de modelagem molecular e de inibição enzimática das calicreínas
teciduais humanas 5 e 7 por compostos isocumarínicos e derivados de
isomanídeos.
Uberaba-MG
2011

Thiago Salem Pançonato Teixeira
Estudo de modelagem molecular e de inibição enzimática das calicreínas
teciduais humanas 5 e 7 por compostos isocumarínicos e derivados de
isomanídeos.
Dissertação apresentada ao Curso de Pós-
Graduação em Ciências Fisiológicas da
Universidade Federal do Triângulo Mineiro,
como requisito para a obtenção do título de
Mestre em Ciências Fisiológicas. Área de
Concentração I: Bioquímica, Fisiologia e
Farmacologia.
Orientador: Prof. Dr. Odonírio Abrahão Júnior
Co-orientador: Prof. Dr. Luciano Puzer
Uberaba – MG
2011


Dedico esse trabalho ao meu orientador Prof. Dr. Odonírio Abrahão Júnior e co-orientador Prof. Dr.
Luciano Puzer pelo apoio e ensino, bem como a todos envolvidos neste trabalho, direta ou
indiretamente, conferindo ao trabalho o caráter de “nosso”.

AGRADECIMENTOS
Agradeço a Deus pela oportunidade de vivenciar a realidade do pesquisador, concedendo-me
força, atenção e paciência, principalmente quando o cansaço e a saudade de casa
sobrepunham meus ânimos.
À minha mãe Maria Vitória por ser o sopro que me move, o coração a compartilhar a dor e a
alegria. A amiga que esteve presente com todo amor, mesmo a quilômetros de distância.
Ao meu pai Antônio Pançonato que me ensinou a resignar e ser paciente diante às
dificuldades que às vezes o destino impõe.
A minha tia Gabriela, minha segunda mãe, que com seu amor acompanhou meus primeiros
passos me ensinando a rir das dificuldades.
Aos meus irmãos Henrique e Hamilton que são exemplos e reflexões que sempre me
surpreendem. Mais que irmãos e amigos, são um destino a ser acompanhado.
À minha grande amiga Bruna que se confunde com irmã, por ter sido a força da risada entre
tantas adversidades fora de casa, o amor que sempre pulsa com intenso ardor.
Aos amigos de faculdade, Bárbara, Ilsione e Thiago Aparecido por terem sido o melhor
aprendizado, cada qual cativando com sua personalidade e humor característico.
Ao meu professor e orientador Dr. Odonírio Abrahão Júnior que desde a graduação me
ensinou a ser visionário e ter esperanças e forças ao visualizar o final e não o meio.
Ao meu professor e co-orientador Dr. Luciano Puzer por acreditar em mim para o
desenvolvimento do trabalho, e pelas oportunidades oferecidas.
Ao ex-aluno Renato Ferreira Freitas por ter sido amigo e professor, auxiliando-me no meu
trabalho e, motivando-me a seguir na vida acadêmica.
Aos funcionários do Laboratório de Biofísica e Bioquímica, Flávio, Geraldo e Marco Túlio e,
os alunos e colegas Fernando, Joyce, Leonardo Almeida, Leonardo Baesse, Lucas,
Luíza, Milton e Weder, não só pelo apoio no trabalho, mas pela amizade e risadas na
hora do café.
À professora Dra. Adriana K. Carmona e sua aluna Pollyana M. S. Melo pela ajuda e
paciência ao me concederem espaço para o desenvolvimento do trabalho.
E a todos que nesses seis anos em Uberaba foram amigos, colegas, conhecidos e inimigos, por
me mostrar meus erros e acertos, ajudando-me a tentar ser cada vez melhor.

Agradeço todas as dificuldades que enfrentei; não fosse
por elas, eu não teria saído do lugar. As facilidades
nos impedem de caminhar. Mesmo as críticas nos
auxiliam muito.
(Chico Xavier)

Resumo
A família das calicreínas teciduais humanas é composta de quinze serino proteases, sendo o
gene denotado por “KLK” e a proteína por “KLK” seguida do número correspondente. Elas
apresentam atividade semelhante à tripsina, com exceção das KLK3, 7 e 9 que apresentam
atividade semelhante à quimotripsina. São transcritas como pré-pró-enzimas, sendo que após
atingirem o local de ação perdem o segmento pré-peptídico. O segmento pró-peptídico
mantém a latência e, uma vez removido esse segmento, outros fatores controlam suas
atividades como inibição alostérica, clivagem interna e/ou inibição endógena. São expressas
em quase todos os tecidos, estando envolvidas em várias funções como regulação de tônus
vascular, liquefação do sêmen, remodelamento da matriz extracelular, remodelamento
neuronal, ativação de pró-hormônios, entre outros. Muitos estudos vêm sendo feitos sobre a
atuação dessas enzimas no desenvolvimento de câncer, uma vez que elas podem dificultar ou
facilitar o seu desenvolvimento através do seu envolvimento na metástase e angiogênese,
dependendo do tecido e tipo de fator presente no meio. Dentre essas enzimas, o enfoque desse
trabalho são as KLK5 e KLK7 que apresentam expressão em tecidos semelhantes, tendo
atuação sinérgica. Sua função mais bem descrita é a participação na descamação da pele
através de proteólise de proteínas da adesão celular. Quando seu inibidor natural está ausente,
o indivíduo apresenta um intenso processo descamativo (ictiose), causando infecções e
inflamações. Elas também atuam na degradação de componentes da matriz extracelular,
ativam peptídeos antimicrobianos e receptores pró-inflamatórios. Devido à ausência de
fármacos que agem sobre as KLK5 e KLK7, estudou-se o comportamento de uma classe de
compostos peptídicos miméticos (isomanídeos) e outra de compostos isocumarínicos. Dos
quinze isomanídeos, cinco foram solúveis em meio aquoso e, foram testados com a KLK5,
sendo que três apresentaram inibição reversível e competitiva na faixa de micromolar e duas
foram reversíveis não competitivas, porém com potência baixa. Os três compostos
isocumarínicos foram testados com ambas as enzimas, e foram inibidores reversíveis e
competitivos na faixa de micromolar, sendo as formas dímeras mais potentes. O estudo com
modelagem molecular mostrou que as interações hidrofóbicas e ligações de hidrogênio são as
principais forças envolvidas na estabilização enzima-inibidor. Os três inibidores peptídicos
miméticos que apresentaram inibição competitiva apresentaram modo de ligação semelhante,
posicionando seu substituinte R2 no subsítio S1, e o substituinte R1 em S2, sendo que o anel
de isomanídeo e o grupo O-benzil posicionou sobre S1’/S2’. Esses compostos fizeram quatro
ligações de hidrogênio, na qual duas foram com os resíduos da fenda de oxiânion. Já os
compostos de isocumarinas, para ambas as enzimas, se encaixam em S1, mas na KLK5 a
forma dímera se estende para S3/S4 e na KLK7 se estende para S1’. Na KLK5 esses
compostos formam interações de hidrogênio com os resíduos da fenda de oxiânion, porém o
mesmo não ocorre com a KLK7, indicando o porquê esses compostos são mais potentes para
a KLK5 do que para a KLK7, apesar desta ter uma superfície mais hidrofóbica e cavidade S1
maior. Propõe-se que compostos hidrofóbicos com cadeias semelhantes a aminoácidos
básicos ou hidrofóbicos potencializem a inibição para a KLK5 e KLK7, respectivamente.
Palavras chave: Calicreína, Isomanídeo, Isocumarina, Inibição, Docking Molecular.

Abstract
The human tissue kallikrein family is composed of fifteen serine proteases, being the
nomenclature for gene and protein denoted “KLK” and “KLK”, respectively, followed by its
corresponding number. Twelve are expected to have trypsin-like activity and three, KLK3, 7
and 9, to have chymotrypsin-like activity. All of them are expressed as pre-pro-enzyme,
which after reaching its main location the pre-peptide fragment is removed. The pro-peptide
keeps its latency and, once it is removed, other factors control their activity as allosteric
inhibition, internal cleavage and/or endogenous inhibition. They are expressed in almost every
tissue, being involved in many functions as vascular tonus regulation, semen liquefaction,
extracellular matrix remodeling, neuronal remodeling, pro-hormones activation, and so on.
Many studies are being made over the action of these enzymes on cancer development, which
depends on the tissue and factors involved in the medium, they can difficult or facilitate the
development through its involvement on metastases and angiogenesis. Among those enzymes,
we focused on KLK5 and KLK7, which have expression on the same tissues, with synergic
action. Their most well described function is on the skin desquamation through proteolysis of
the adhesion proteins. When their natural inhibitor is lacking, the patient has an intense skin
detachment process (ictiosys), causing infection and inflammation. They can act in the
extracellular matrix degradation, activating antimicrobial peptides and pro-inflammatory
receptors. Because of the lacking of drugs that act over KLK5 and KLK7, we studied the
behavior of a class of peptide mimetic compounds derived from isomannide, and
isocoumarine compounds. From the fifteen peptide mimetic compounds, five were soluble on
aqueous medium and were tested against KLK5, being three reversible and competitive
inhibitors on micromolar range and two reversible and noncompetitive inhibitors, but with
low potency. The three isocoumarine compounds were tested with both enzymes, and were
reversible and competitive on the micromolar range, being the dimer form more potent. The
molecular modeling study shows that the hydrophobic interaction and hydrogen bonds are the
main force involved in the enzyme-inhibitor interaction. The three competitive peptide
mimetic compounds have a similar binding mode, with its substituent R2 at S1 pocket, the R1
substituent at S2 pocket, and the isomannide ring and the O-benzyl group over S1’/S2’. These
compounds made four hydrogen bonds, being two with the residues of the oxyanion hole. The
isocumarine compounds, for both enzymes, fits on S1 pocket, and for KLK5 the dimer form
goes over S3/S4 pocket and for KLK7 over S1’. For the KLK5, they make hydrogen bonds
with the residues of the oxyanion hole, but the same do not happens with KLK7, which
indicates why they are more potent for KLK5 than for KLK7, even though KLK7 having a
more hydrophobic surface and a larger S1 pocket. We believe that hydrophobic compounds
with lateral chain similar to basic amino acid and similar to hydrophobic amino acid may give
better inhibition for KLK5 and KLK7, respectively.
Keywords: Kallikrein, Isomannide, Isocoumarine, Inhibition, Molecular Docking.

LISTA DE FIGURAS
FIGURA 1: LÓCUS, GENE E CARACTERÍSTICA DA PROTEÍNA CALICREÍNA.. ............................................................ 12
FIGURA 2: ALINHAMENTO DA SEQUÊNCIA PRIMÁRIA DAS CALICREÍNAS TECIDUAIS COM A QUIMOTRIPSINA
BOVINA. ......................................................................................................................................................... 13
FIGURA 3: MECANISMO GERAL DE REAÇÃO DE UMA SERINO PROTEASE. .............................................................. 15
FIGURA 4: ESTRUTURAS TRIDIMENSIONAIS DA KLK5 (A – CÓDIGO PDB: 2PSX) E DA KLK7 (B – CÓDIGO PDB:
2QXH).. ........................................................................................................................................................ 17
FIGURA 5: ESQUEMATIZAÇÃO DA DESCAMAÇÃO DA PELE. .................................................................................. 21
FIGURA 6: MECANISMO DO DOCKING MOLECULAR. ............................................................................................. 23
FIGURA 7: DEFINIÇÃO DE CORE E GRUPO ROTÂMERO. .......................................................................................... 24
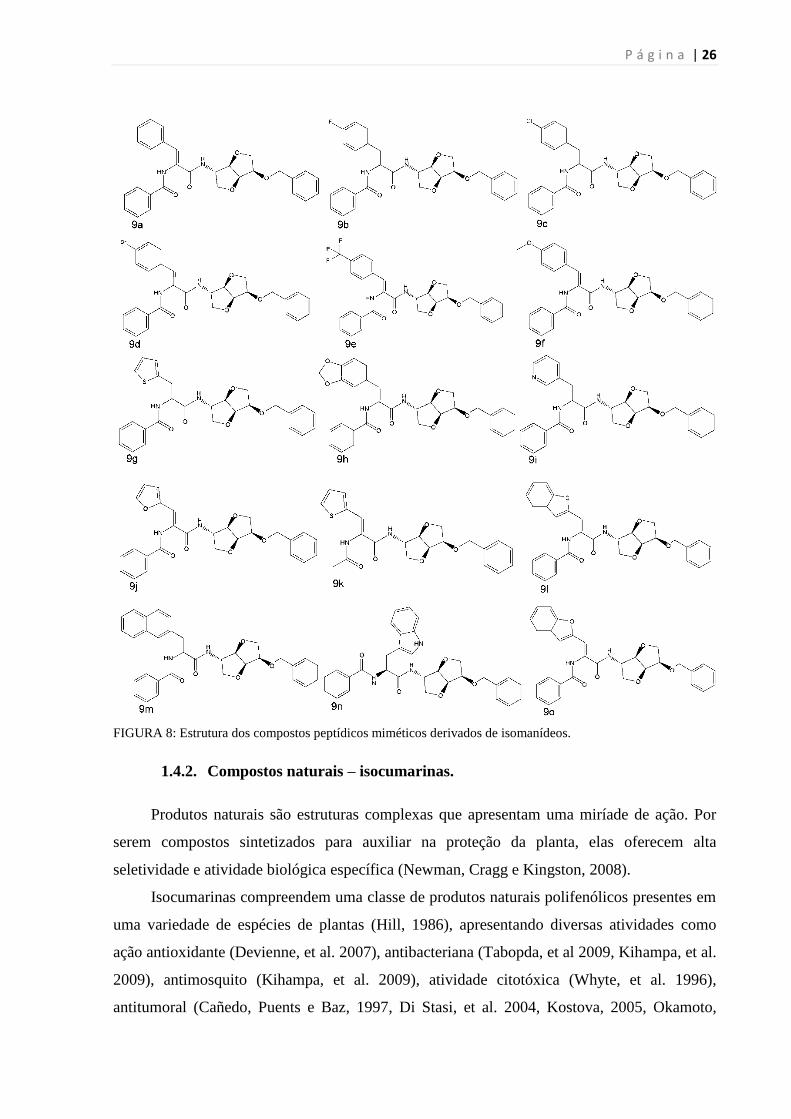
FIGURA 8: ESTRUTURA DOS COMPOSTOS PEPTÍDICOS MIMÉTICOS DERIVADOS DE ISOMANÍDEOS. ........................ 26
FIGURA 9: ESTRUTUA DOS COMPOSTOS ISOCUMARÍNICOS.. ................................................................................. 27
FIGURA 10: VALIDAÇÃO DO ESTUDO DE DOCKING. .............................................................................................. 32
FIGURA 11: GRÁFICO DE LINEWEAVER-BURK DA ENZIMA KLK5 COM OS COMPOSTOS PEPTÍDECOS MIMÉTICOS
DERIVADOS DE ISOMANÍDEOS. ....................................................................................................................... 36
FIGURA 12: GRÁFICO DE LINEWEAVER-BURK DA ENZIMA KLK5 COM OS COMPOSTOS PEPTÍDECOS MIMÉTICOS
DERIVADOS DE ISOMANÍDEOS.. ...................................................................................................................... 36
FIGURA 13: GRÁFICO DE LINEWEAVER-BURK DA ENZIMA KLK5 COM OS COMPOSTOS ISOCUMARÍNICOS. ......... 39
FIGURA 14: SOBREPOSIÇÃO DA LEUPEPTINA GERADA PELO REDOCKING.. ............................................................ 40
FIGURA 15: SOBREPOSIÇÃO DO LIGANTE K7J NA GERADA PELO REDOCKING. ...................................................... 41
FIGURA 16: POSIÇÃO DOS INIBIDORES PEPTÍDICOS MIMÉTICOS DERIVADOS DE ISOMANÍDEOS NA SUPERFÍCIE DA
KLK5. ........................................................................................................................................................... 42
FIGURA 17: DESTAQUE DOS PRINCIPAIS RESÍDUOS QUE FORMAM AS INTERAÇÕES HIDROFÓBICAS COM OS
INIBIDORES PEPTÍDICOS MIMÉTICOS. .............................................................................................................. 43
FIGURA 18: INTERAÇÃO DE HIDROGÊNIO DOS INIBIDORES PEPTÍDICOS MIMÉTICOS DERIVADOS DE ISOMANÍDEOS
COM A KLK5. ................................................................................................................................................ 45
FIGURA 19: VARIAÇÃO ESTRUTURAL DA KLK5 DETERMINADO POR DINÂMICA MOLECULAR. ............................. 46
FIGURA 20: POSIÇÃO DOS INIBIDORES DE ISOCUMARINAS SUPERFÍCIE DA KLK5.. .............................................. 48
FIGURA 21: DESTAQUE DOS PRINCIPAIS RESÍDUOS DA KLK5 QUE FORMAM AS INTERAÇÕES HIDROFÓBICAS COM
OS INIBIDORES DE ISOCUMARINAS. ................................................................................................................ 49
FIGURA 22: INTERAÇÃO DE HIDROGÊNIO DOS INIBIDORES ISOCUMARÍNICOS COM A KLK5. ................................ 51
FIGURA 23: POSIÇÃO DOS INIBIDORES DE ISOCUMARINAS SUPERFÍCIE DA KLK7. ............................................... 53
FIGURA 24: DESTAQUE DOS PRINCIPAIS RESÍDUOS DA KLK7 QUE FORMAM AS INTERAÇÕES HIDROFÓBICAS COM
OS INIBIDORES DE ISOCUMARINAS. ................................................................................................................ 54
FIGURA 25: INTERAÇÃO DE HIDROGÊNIO DOS INIBIDORES ISOCUMARÍNICOS COM A KLK7. ................................ 56

LISTA DE TABELAS
TABELA 1: MEMBROS DAS CALICREÍNA TECIDUAIS HUMANAS E PRINCIPAIS TECIDOS DE EXPRESSÃO. ................ 20
TABELA 2: VALORES DOS TESTES DE INIBIÇÃO (IC50 E KI EM µM) DOS COMPOSTOS PEPTÍDICOS MIMÉTICOS COM A
PROTEÍNA KLK5. .......................................................................................................................................... 34
TABELA 3: VALORES DE IC50 E KI (EM µM) DOS INIBIDORES DERIVADOS DE ISOCUMARINAS MEDIANTE A
ATIVIDADE DA KLK5. ................................................................................................................................... 37
TABELA 4: VALORES DE IC50 E KI (EM µM) DOS INIBIDORES DERIVADOS DE ISOCUMARINAS MEDIANTE A
ATIVIDADE DA KLK7. ................................................................................................................................... 37
TABELA 5: DISTÂNCIA DOS ÁTOMOS DA LEUPEPTINA EM RELAÇÃO À PROTEÍNA 2PSX NO ESTUDO DE REDOCKING.
...................................................................................................................................................................... 40
TABELA 6: DISTÂNCIA DOS ÁTOMOS DA K7J EM RELAÇÃO À PROTEÍNA 2QXH NO ESTUDO DE REDOCKING ........ 41
TABELA 7: DISTÂNCIA (EM Å) DOS ÁTOMOS DOS INIBIDORES PEPTÍDICOS MIMÉTICOS E DA KLK5 ENVOLVIDOS
NAS INTERAÇÕES DE HIDROGÊNIO. ................................................................................................................ 44
TABELA 8: DISTÂNCIA (EM Å) DOS ÁTOMOS DOS INIBIDORES DE ISOCUMARINAS E DA KLK5 ENVOLVIDOS NAS
INTERAÇÕES DE HIDROGÊNIO. ........................................................................................................................ 50
TABELA 9: DISTÂNCIA (EM Å) DOS ÁTOMOS DOS INIBIDORES DE ISOCUMARINAS E DA KLK7 ENVOLVIDOS NAS
INTERAÇÕES DE HIDROGÊNIO. ........................................................................................................................ 53

LISTA DE ABREVIATURAS
Abz Ácido ortoaminobenzóico
Ala, A Alanina
Arg, R Arginina
Asn, N Asparagina
Asp, D Aspartato
DMSO Dimetilsulfóxido
EDDNp Etilenodiaminodinitrofenol
Gln, Q Glutamina
Glu, E Glutamato
His, H Histidina
IC50 Concentração de inibição da metade da atividade máxima
Ile, I Isoleucina
Ki Constante de dissociação do inibidor
Km Constante de Michaelis
Lys, K Lisina
Phe, F Fenilalanina
Pro, P Prolina
RMSD Desvio médio quadrático padrão (Root mean square deviation)
Ser, S Serina
Thr, T Treonina
Tris Tris(hidroximetil)aminometano
Tyr, Y Tirosina

SUMÁRIO 1. INTRODUÇÃO. ........................................................................................................................................... 12
1.1. CLASSIFICAÇÃO, SEQUÊNCIA GÊNICA E ESTRUTURA DAS CALICREÍNAS TECIDUAIS
HUMANAS. ..................................................................................................................................................... 12
1.1.1. Estrutura das calicreínas teciduais humanas 5 e 7. ...................................................................... 16
1.2. ATIVAÇÃO E REGULAÇÃO DAS CALICREÍNAS TECIDUAIS HUMANAS............................. 18
1.3. FUNÇÃO FISIOLÓGICA DAS CALICREÍNAS TECIDUAIS HUMANAS. ................................... 19
1.3.1. Descamação da pele. ................................................................................................................... 20
1.3.2. Atuação das calicreínas na matriz extracelular, na resposta inflamatória e sobre peptídeos
antimicrobianos. ........................................................................................................................................... 21
1.4. FERRAMENTAS DE DOCKING MOLECULAR E O DESENVOLVIMENTO DE FÁRMACOS. 22
1.4.1. Moléculas peptídicos miméticas derivadas de isomanídeos. ....................................................... 25
1.4.2. Compostos naturais – isocumarinas. ........................................................................................... 26
2. OBJETIVOS. ................................................................................................................................................ 28
2.1. OBJETIVOS GERAIS. ........................................................................................................................ 28
2.2. OBJETIVOS ESPECÍFICOS. .............................................................................................................. 28
3. MATERIAIS E MÉTODOS. ........................................................................................................................ 29
3.1. MOLÉCULAS PARA ESTUDO DE INIBIÇÃO. ............................................................................... 29
3.1.1. Moléculas peptídico miméticas derivadas de isomanídeos. ........................................................ 29
3.1.2. Isocumarinas. .............................................................................................................................. 29
3.2. ENZIMAS, SUBSTRATOS E TAMPÕES. ........................................................................................ 29
3.2.1. Expressão, purificação e ativação da KLK5 e KLK7 recombinante. .......................................... 29
3.2.2. Substrato. ..................................................................................................................................... 29
3.2.3. Tampão. ....................................................................................................................................... 30
3.3. ENSAIOS DE INIBIÇÃO ENZIMÁTICA. ......................................................................................... 30
3.4. ESTUDO DE MECÂNICA MOLECULAR........................................................................................ 31
3.4.1. Estudo de docking molecular. ..................................................................................................... 31
3.4.2. Estudo de dinâmica molecular (DM). ......................................................................................... 33
4. RESULTADO E DISCUSSÃO. ................................................................................................................... 34
4.1. INIBIÇÃO DA KLK5 PELOS ISOMANÍDEOS. ............................................................................... 34
4.2. INIBIÇÃO DA KLK5 E KLK7 PELAS ISOCUMARINAS. .............................................................. 37
4.3. ESTUDO DE DOCKING MOLECULAR COMS OS INIBIDORES. ................................................ 40
4.3.1. Validação do método de docking molecular. .............................................................................. 40
4.3.2. Estudo de docking molecular dos isomanídeos na KLK5. .......................................................... 41
4.3.3. Docking das isocumarinas. .......................................................................................................... 47
5. CONCLUSÕES E PERSPECTIVAS. .......................................................................................................... 57
6. REFERÊNCIAS. .......................................................................................................................................... 60
7. ANEXOS ...................................................................................................................................................... 66

| 12 P á g i n a
INTRODUÇÃO.
1.1. CLASSIFICAÇÃO, SEQUÊNCIA GÊNICA E ESTRUTURA DAS CALICREÍNAS
TECIDUAIS HUMANAS.
As enzimas proteolíticas conhecidas como calicreínas são divididas em duas categorias:
calicreína plasmática e calicreína tecidual. Essas duas categorias diferem significativamente
quanto à massa molecular, especificidade pelo substrato, características imunológicas,
estrutura gênica e tipo de cinina liberada. De acordo com a nomenclatura oficial, o símbolo do
gene da calicreína e da proteína são denotados “KLK” e “KLK”, respectivamente, seguido do
número correspondente (Diamandis, et al. 2000).
FIGURA 1: Lócus, gene e característica da proteína calicreína. (A) O lócus KLK está localizado no braço
longo do cromossomo 19 na região citogenética 13.3 a 13.4. Os 15 genes das KLK, com exceção dos genes
KLK2 e KLK3, são transcritos do telômero para o centrômero. Os genes das KLKs clássicas (KLK1, KLK2 e
KLK3) são representados por cabeça de setas vermelhas, KLK4-KLK15 por cabeça de setas roxas e o pseudogene
ΨKLK1 por cabeça de seta amarela. (B) Gene da KLK com 5 éxons codificantes (caixa amarela), dois ou mais
éxons não codificantes (caixa vermelha) e 4 introns. A posição dos códons para os resíduos da tríade catalítica
são indicados pelas letras H (histidina), D (aspartato) e S (serina) nos seus respectivos éxons. (C) A proteína é
sintetizada como pré-pró-enzima. (Adaptado de Borgoño e Diamandis, 2004).
Em humanos, o lócus da calicreína tecidual se encontra no braço longo do cromossomo
19 na região citogenética 13.3 a 13.4 (FIG. 1 A). Os genes estão alinhados dentro de um lócus
de 300 Kb sem nenhuma intervenção por outro gene, com exceção das KLK1 e KLK3 na qual
o gene da KLK15 se localiza entre elas (FIG. 1 A). A direção da transcrição é do telômero ao
centrômero, com exceção da KLK2 e KLK3.

| 13 P á g i n a
Todos os genes consistem de cinco éxons codificantes e a organização deles é muito
similar com o primeiro éxon tendo uma região 5’ não traduzível. Os códons dos resíduos da
tríade catalítica histidina (H), ácido aspártico (D) e serina (S) se encontram, respectivamente,
no segundo, no terceiro e no quinto éxon (FIG. 1 B). Além do códon de parada, há uma região
3’ não traduzível com comprimento variável. Apesar das variações nas regiões não traduzíveis
originarem RNAm de comprimentos diferentes, a codificação origina as mesmas proteínas.
Embora o comprimento dos íntrons tenha variação considerável, os comprimentos dos éxons
são quase semelhantes ou idênticos.
FIGURA 2: Alinhamento da sequência primária das calicreínas teciduais com a quimotripsina bovina. Os
resíduos conservados são indicados pela caixa cinza. O resíduo N-terminal está destacado em azul escuro e os
resíduos da tríade catalítica em vermelho. Os resíduos que compõem as alças (loops) estão destacados em verde.
A numeração é de acordo com a numeração da quimotripsina. Os resíduos que compõem a α-hélice e folha-β
estão destacado com cilindro vermelho e seta amarela, respectivamente, abaixo (Adaptado de Goettig, Magdolen
e Brandstetter, 2010).
O alinhamento da sequência primária das calicreínas humanas mostra uma alta
similaridade entre elas (FIG. 2). O máximo de identidade nessas proteínas é encontrado na
região catalítica em torno dos aminoácidos histidina (WVLTAAHC), ácido aspártico

| 14 P á g i n a
(DLMLL) e serina (GDSGGPL). Em geral, a identidade dos aminoácidos nessas proteínas
está em torno de 40-80%, sendo que a cisteína é altamente conservada entre as 15 enzimas.
Todos os membros dessa família possuem de 10-12 resíduos de cisteína que são responsáveis
pelas pontes dissulfídicas (Yousef e Diamandis, 2001).
Todas as calicreínas são cadeias únicas e são sintetizadas como pré-pró-enzimas que são
então proteoliticamente processadas para formar pró-enzimas pela remoção do peptídeo sinal,
seguido pela ativação (também por proteólise) para formar a enzima ativa (FIG. 1 C). A
maioria das pró-formas é ativada pela quebra do final N-terminal tanto dos resíduos de lisina
como arginina (resíduos de maior preferência para clivagem pela tripsina). Como a maioria
das calicreínas humanas tem atividade semelhante à tripsina, elas podem atuar ativando elas
mesmas (autoativação) ou outras pró-formas de calicreínas (Yousef e Diamandis, 2001).
A massa molecular da fração pró-enzima varia de aproximadamente 23 a 26 kDa
(Yousef e Diamandis, 2001). No entanto, devido a glicosilação, maiores massas foram
observadas para várias calicreínas incluindo a KLK1 (Lu, et al. 1989), KLK3 (Belanger, et al.
1995), KLK5 (Brattsand e Egelrud, 1999) e KLK7 (Liu, et al. 1996) bem como as formas
recombinantes da KLK2 (Mikolajczyk, et al. 1997), KLK5 (Brattsand e Egelrud, 1999,
Yousef, et al. 2003), KLK6 (Bernett, et al. 2002), KLK7 (Liu, et al. 1996, Egelrud, et al.
1993) e KLK13 (Kapadia, et al. 2003).
Com exceção da KLK1, na qual foi observada a ocorrência de O-glicosilação (Lu, et al.
1989), todas as outras glicosilações relatadas envolvem a ligação N-carboidrato. A maioria
das KLKs possuem sítios ou sequências Asn-X-Ser/Thr (na qual X é qualquer aminoácido
exceto Pro) para N-glicosilação (Marshall, 1974).
As calicreínas teciduais humanas são serino proteases por possuírem a tríade catalítica
His-Asp-Ser, sendo a serina o resíduo que promove o ataque nucleofílico na ligação peptídica
do substrato (Blow, 1997). A fenda de oxiânion é formada pelos grupos aminos da cadeia
principal da Gly193 e Ser195 que formam um bolso com carga positiva responsável pela
ativação do carboxil da ligação peptídica a ser clivada e estabiliza a carga negativa do
oxiânion do intermediário tetraédrico.

| 15 P á g i n a
FIGURA 3: Mecanismo geral de reação de uma serino protease. A Ser195 promove o ataque nucleofílico
no carboxil da ligação peptídica do substrato, formando um intermediário tetraédrico que é estabilizado pelos
resíduos da fenda de oxiânion (Gly193 e Ser195). O intermediário tetraédrico é desfeito com a liberação do
segmento N-terminal. Na etapa seguinte uma molécula de água é desprotonada e promove o ataque nucleofílico,
formando um intermediário tetraédrico que se desfaz com a liberação do segmento C-terminal e a regeneração da
hidroxila da Ser195. (Hedstron, 2002).
Na primeira etapa da reação enzimática (acilação), a Ser195 ataca o carboxil peptídico
do substrato, assistido pela His57 (que age como uma base geral), para formar um tetraedro
intermediário (FIG. 3). A His57-H+ é estabilizada pela ligação de hidrogênio com Asp102. O
oxiânion do intermediário tetraédrico é estabilizado pela interação com os grupos aminos da
cadeia principal da fenda de oxiânion (Gly193 e Ser195). O intermediário tetraédrico se
desfaz com a liberação da sequência N-terminal, assistido pela His57-H+ (que age como um
ácido geral), para formar o intermediário acil-enzima. Na segunda etapa da reação
(desacilação), uma molécula de água é desprotonada pela His57 e promove o ataque
nucleofílico da acil-enzima, formando um segundo intermediário tetraédrico. Novamente a
His57-H+ desmancha o intermediário tetraédrico, resultando na liberação da sequência C-
terminal e a regeneração da hidroxila da Ser195.
Das quinze KLKs, é previsto que doze possuam atividade semelhante à tripsina,
enquanto três (KLK3, KLK7 e KLK9) possuam atividade semelhante à quimotripsina. O
aminoácido do subsítio S1, primariamente responsável pela especificidade da enzima pelo
substrato (Perona, et al. 1995), é encontrado em todas as calicreínas na posição 189, de acordo
com numeração da quimotripsina (Hartley, 1964). Múltiplos alinhamentos das sequências
proteicas indicam que doze calicreínas possuem um resíduo aspartato ou glutamato nessa
posição e, espera-se que clivem no lado carboxil de aminoácido básicos como arginina ou
lisina, semelhante à tripsina. Por outro lado, a KLK3, KLK7 e KLK9 têm o resíduo serina,
asparagina e glicina, respectivamente, na posição 189, conferindo uma especificidade
semelhante à quimotripsina (Yousef e Diamandis, 2001).

| 16 P á g i n a
1.1.1. Estrutura das calicreínas teciduais humanas 5 e 7.
Para ambas calicreínas (KLK5 e KLK7), suas estruturas cristalinas foram resolvidas
(Debela, et al. 2007a, 2007b) mostrando que são semelhantes com pequenas diferenças (FIG.
4).
Ambas se assemelham a uma estrutura oval com diâmetros de 35Å e 50Å,
respectivamente (Debela, et al. 2007a, 2007b). São formadas por dois barris-β, cada um
formado por seis fitas trans, duas α-hélices, uma 310-hélice e várias alças de superfície. A
tríade catalítica (His57, Asp102 e Ser195) está localizada ao longo da junção dos dois barris,
na qual o sítio catalítico junto com os subsítios de reconhecimento S4 a S3’ correm
perpendicularmente a essa junção (FIG. 4).
Como outras serino proteases, elas são sintetizadas na forma de pré-pró-enzima, sendo o
segmento pré-peptídico composto de 29 e 22 aminoácidos e, o segmento pró-peptídico
formado por 37 e 7 aminoácidos na KLK5 e KLK7, respectivamente. A enzima madura da
KLK5 contém 227 aminoácidos e a KLK7, 226 aminoácidos (Debela, et al. 2007a, 2007b).
De acordo com a numeração da quimotripsina, ambas as enzimas começam com o
resíduo Ile16, na qual seu grupo α-amônio forma uma ligação salina com o grupo carboxil da
cadeia peptídica do Asp194. Essa ligação (formado após a remoção do segmento pró-
peptídico) leva a formação da fenda de oxiânion que remodela o sítio S1. Como em outras
calicreínas, suas cadeias são conectadas por seis ligações dissulfídicas (Debela, et al. 2007a,
2007b).
A KLK5 apresenta quatro possíveis sítios de N-glicosilação (Asn18-Gly19-Ser20,
Asn122-Val123-Ser124, Asn159-Ile160-Ser161 e Asn252-Gly253-Ser254) e a KLK7 possui
um sítio em Asn239-Asp240-Thr241. Na estrutura cristalina da KLK5 há uma N-glicosilação
em Asn159 formada por duas moléculas de 2-acetamido-2-deoxi-β-D-glicopiranose
(GlucNAc) ligadas por ligação β-1,4 (FIG. 4 A). Já a estrutura cristalina da KLK7 não
apresenta glicosilação (o que pode ser devido à sua expressão em célula de inseto) (FIG. 4 B)
(Debela, et al. 2007b).

| 17 P á g i n a
FIGURA 4: Estruturas tridimensionais da KLK5 (A – código PDB: 2PSX) e da KLK7 (B – código PDB:
2QXH). Os resíduos da tríade catalítica (H57-D102-S195) estão destacados, bem como os resíduos 189, 190 e
218 do subsítio S1 que conferem atividade tipo-tripsina e tipo-quimotripsina para a KLK5 e KLK7,
respectivamente. A estrutura cristalina da KLK5 apresenta uma glicosilação.
Das alças que circundam a proteína, de grande interesse são a alça-37 e a alça-99. A
maioria das KLKs, com exceção das KLK5 e KLK14, tem o resíduo 37 deletado. O resíduo
37 na KLK5 é uma prolina, e a sua presença permite a alça se projetar no solvente de maneira
mais estendida que nas outras enzimas (Debela, et al. 2007a).
Na alça-99 ocorre a ligação do Zn2+
, que é um inibidor não competitivo. No caso da
KLK5, essa coordenação está bem definida pela estrutura cristalina, na qual o átomo Nδ1 da
His96, o átomo Nε2 da His99, e duas moléculas de água, formam uma esfera de coordenação
tetraédrica com o Zn2+
. Essa ligação desloca a cadeia lateral da His96 e His99 que é
acompanhada de alteração da cadeia principal que faz com que o átomo Nε2 da His57 forme
uma coordenação com o Zn2+
, e essa ligação altera orientação dos resíduos da tríade catalítica,
diminuindo sua atividade (Debela, et al. 2007a). No caso da KLK7, o resíduo His96 é mutado
por um Thr. Como a constante de inibição da KLK7 pelo Zn2+
é igual a da KLK5, acredita-se
que a força da coordenação do Thr96 seja semelhante da His96 (Debela, et al. 2007b).
O subsítio S1 da KLK5 é formado pelos resíduos Gly226-Val227-Tyr228 (formando o
fundo), os resíduos Ser214-Trp215-Gly216-Asp217-Tyr218-Pro219-Cys220 formam parte da
lateral e a borda superior e, os resíduos Asp189-Ser190-Gln192-Gly193-Asp194-Ser195
formando parte da lateral e a borda inferior. A preferência da KLK5 por arginina em P1 pode
ser explicada pelos resíduos que formam o bolso S1. A presença da Tyr218 provê uma
barreira hidrofóbica e a presença da Gly216 e Gly226 numa das bordas, permite a

| 18 P á g i n a
acomodação de grandes cadeias laterais. Juntamente com a presença do grupo carboxílico da
Asp189, o Oγ da Ser190 e o grupo carboxil da Asp217 contribuem para a formação de
ligações de hidrogênio que estabilizam a carga da arginina (Debela, et al. 2007a).
No caso da KLK7, o subsítio S1 é formado pelos resíduos Ala190-Ser195, Val213-
Cys220 e Pro225-Tyr228. Ela difere das outras KLKs pela mutação do resíduo Asp189 por
Asn189 e, do resíduo Ser190 por Ala190, tornando a parte central do bolso mais hidrofóbica.
A entrada é formada pela cadeia lateral hidrofóbica e polar da Phe218 e Asn192,
respectivamente. Assim, o bolso S1 possui um fundo polar, largo, mas não profundo como da
quimotripsina. Essas características tornam o subsítio S1 da KLK7 propício para acomodar
cadeias de médio a grande tamanho com pontas polares como resíduos de tirosina (Debela, et
al. 2007b).
O subsítio S2 da KLK5 é primariamente uma cavidade triangular polar, lateralmente
confinada pelas cadeias laterais imidazólicas da His57 e His99, no topo é revestido pelo grupo
fenólico da Tyr94, com o Trp215 fechando o fundo. O tamanho relativamente pequeno e as
pontas polares no subsítio S2 são consistentes com a preferência da KLK5 por resíduos de
pequeno a médio comprimento em P2 (Ser, Thr, Asn e Ala) (Debela, et al. 2006). A cadeia
lateral His99 da KLK5 é móvel, resultando numa cavidade S2 adaptável (Debela, et al.
2007a).
A cavidade S2 da KLK7 é estreita, formada pela cadeia lateral dos resíduos His57,
His99, Ser214 e Trp215. Essa pequena cavidade formada por resíduos polares, explica a
preferência por resíduos Tyr, seguido por resíduos pequenos e hidrofóbicos como Leu, Thr,
Met e Phe (Debela, et al. 2007b).
Em relação aos demais subsítios, há pouca descrição. Mas baseado na estrutura de
outras serino proteases, pode-se inferir que os subsítios S3-Sn podem ser determinantes na
especificidade do substrato (Bode, Turk e Karshikov, 1992), como exemplificado pela
elastase na qual a interação P3/S3 é dominante (Bode, Meyer e Powers, 1989).
1.2. ATIVAÇÃO E REGULAÇÃO DAS CALICREÍNAS TECIDUAIS HUMANAS
O modulador mais estudado da estimulação da transcrição das KLKs são os hormônios
sexuais. A KLK2 e KLK3 são modelos de expressão gênica regulada por hormônio
testosterona quase que exclusivamente na próstata (Cleutjens, et al. 1996). Por outro lado,
outros genes das KLKs, incluindo KLK5 e KLK6, são mais sensíveis ao estrógeno. A
expressão de quase todos os outros genes é regulada pela testosterona e/ou estrogênio.

| 19 P á g i n a
Como a ativação de protease é sempre irreversível, quatro mecanismos pós-traducionais
são envolvidos para prevenir proteólise indesejada da KLK e manter a integridade do tecido:
ativação de zimogênio, inibição endógena, inibição através de clivagem interna e regulação
alostérica.
A ativação do zimogênio é realizada por proteólise limitada do pró-peptídeo, que induz
uma mudança conformacional no sítio catalítico e no sítio de reconhecimento específico do
substrato, criando a enzima ativa. Estudos in vitro indicam que algumas KLKs podem se
autoativar (Mikolajczyk, et al. 1997, Magklara, et al. 2003) e podem ser ativadas por outras
KLKs (Lovgren, et al. 1997, Takayama, et al. 2001a, Takayama, et al. 2001b, Caubet, et al.
2004), bem como outras serino e metaloprotease (Takayama, Fujikawa e Davie, 1997,
Takada, Skidgel e Erdos, 1985).
Uma vez que ocorre a ativação, as KLKs são fortemente controladas por inibidores
endógenos principalmente α2-macroglobulina (α2M) e serpinas em fluidos e tecidos. Isto não
leva a inibição da atividade enzimática, mas previne as KLKs de interagirem com grandes
substratos ou inibidores por impedimento estérico (Sottrup-Jensen, et al. 1989). As KLKs
podem também perder atividade por clivagem interna. Cátions divalentes, especialmente o
zinco, mostraram ser inibidores reversíveis por se ligarem em sítios alostéricos (Debela,
2007c).
1.3. FUNÇÃO FISIOLÓGICA DAS CALICREÍNAS TECIDUAIS HUMANAS.
As calicreínas teciduais são expressas em vários tecidos e estão envolvidas em vários
processos fisiológicos. Devido à descoberta primária das calicreínas “clássicas”, suas funções
são mais bem descritas. A KLK1 é expressa em vários tecidos como rim, vasos sanguíneos,
sistema nervoso central, pâncreas, glândulas salivares e sudoríparas, baço e adrenal, sugerindo
uma natureza parácrina da enzima (Moreau et al. 2005). Sua função primária é a liberação de
lisil-bradicinina e, funções adicionais incluem processamento de proteínas, aumento da
formação de óxido nítrico e redução do estresse oxidativo (Borgoño, Michael e Diamandis,
2004).
A análise das demais calicreínas clássicas (KLK2 e KLK3) é mais restrita
principalmente à próstata e ao plasma seminal (TABELA 1). A análise de especificidade de
substrato mostra que a KLK2 ativa a KLK3 e ambas contribuem com a liquefação do coágulo
seminal através da hidrólise de proteínas da vesícula seminal, como seminogelina I e II e
fibronectina (Malm, et al. 2000).

| 20 P á g i n a
Sugere-se que a KLK4 participe na formação do esmalte, já que ela mostrou ser capaz
de clivar enamelina, uma das proteínas do esmalte dentário (Wright, et al. 2006, Yamakoshi,
et al. 2006). Vários trabalhos sugerem a participação de várias KLKs no processamento de
hormônios. Um grande número de KLKs (KLK5-8, 13 e 14) mostraram ser capazes de clivar
o hormônio de crescimento humano (hGH) in vitro (Komatsu, et al. 2007). Estudos imuno-
histoquímicos sugerem que KLK1, 6, 10 e 13 são fortemente expressas nas ilhotas de
Langerhans e talvez regulem a ativação das pró-formas da insulina, glucagon e somatostatina
(Petraki, et al. 2003a).
Achados mostram a atuação das KLKs no sistema nervoso central (SNC),
principalmente a KLK6 e KLK8 (TABELA 1), que apresentam expressão distinta no SNC de
adultos (Yousef, et al. 1999). Postula-se que a KLK6 esteja envolvida na doença de Parkinson
já que esta é capaz de clivar α-sinucleína, além de colocalizar em inclusões patológicas como
corpos de Lewy (Iwata, et al. 2003). Acredita-se que a KLK8 auxilie no remodelamento da
matriz extracelular neuronal (Tamura, et al. 2006).
TABELA 1: Membros das calicreína teciduais humanas e principais tecidos de expressão.
Enzima Localização tecidual
KLK1 Rim, Pâncreas, glândula salivar
KLK2 Próstata
KLK3 Próstata
KLK4 Próstata, dente
KLK5 Testículo, seios, pele
KLK6 Cérebro, pâncreas, rim
KLK7 Pâncreas, pele
KLK8 Pâncreas, cérebro, pele
KLK9 Pâncreas
KLK10 Pâncreas, seios, ovário, cólon, intestino, testículo, pulmão
KLK11 Pâncreas, pele, próstata, coração, testículo, cérebro
KLK12 Pâncreas
KLK13 Pâncreas, esôfago, apêndice
KLK14 Cérebro, medula óssea, fígado fetal
KLK15 Tireoide, próstata
Fonte: Debela, et al. 2006.
1.3.1. Descamação da pele.
Entre as células do estrato córneo (camada mais superficial de epiderme), uma forte
coesão é feita pelos corneodesmossomos, junções celulares derivadas dos desmossomos
(Chapman, et al. 1990). Estudos estruturais demonstraram que a degradação dos
corneodesmossomos é concomitante com a descamação (Chapman e Walsh, 1991, Fartasch,
et al. 1993).

| 21 P á g i n a
FIGURA 5: Esquematização da descamação da pele. As células da derme são unidas pro proteínas do
complexo corneodesmossomos. Nas camadas mais profundas da pele as enzimas calicreínas são inativas devido
por estarem na forma de zimogênio e também devido à alta concentração de zinco e LEKTI. Conforme as células
migram para as camadas mais superficiais, a concentração de zinco diminui, o pH diminui e assim a LEKTI se
desassocia das calicreínas e elas passam a ser ativas (por autólise ou por proteólise feita por outras enzimas)
promovendo a degradação dos corneodesmossomos e, consequentemente a descamação (Adaptado de Lundwall
e Brattsand, 2008).
Nas camadas mais profundas do EC, as KLK5 e KLK7 coexistem na forma de
zimogênio e forma ativa (FIG. 5). No entanto, a atividade da forma ativa é mínima porque
dois fatores estão presentes na regulação de suas atividades: o zinco, que é um inibidor
alostérico e, a LEKTI, que é um inibidor endógeno natural derivado da SPINK5 (Magert, et
al. 2002), que se associa à enzima em pH neutro (Descargues, et al. 2005). Conforme as
células migram para as camadas mais superficiais, o pH do meio se acidifica resultando na
dissociação da KLK com a LEKTI (Deraison, et al. 2007) e, a concentração de zinco vai
diminuindo. Então, as enzimas ativas podem ativar elas mesmas ou outras KLKs (Deraison, et
al. 2007), resultando na descamação da pele devido à clivagem dos corneodesmossomos.
Mutações em SPINK5 causa a síndrome de Netherton (Komatsu, et al. 2002), uma
síndrome multissistêmica caracterizada por ictiose, eritroderma, defeito no crescimento de
cabelo e características atópicas (Sun e Linden, 2006).
1.3.2. Atuação das calicreínas na matriz extracelular, na resposta inflamatória e
sobre peptídeos antimicrobianos.
Michael e colaboradores (2005) mostraram que a KLK5 cliva rapidamente componentes
da matriz extracelular como colágeno tipo I, II e III e, plasminogênio (Michael, et al.. 2005).

| 22 P á g i n a
Observou-se que a KLK7 cliva a E-caderina, uma das principais proteínas responsáveis pela
adesão célula a célula (Johnson, et al. 2007).
Queratinócitos epidérmicos sintetizam e secretam peptídeos antimicrobianos que
colaboram com a resposta imune inata da pele contra bactérias, fungos e infecções virais. As
catelicidinas são induzidas e depositadas em sítios inflamatórios após infecção (Braff, et al.
2005).
As KLK5 e KLK7 regulam a atividade da catelicidina, peptídeo antimicrobiano
secretado por queratinócitos. Diante de uma infecção este peptídeo é processado pela KLK5
para ativar a forma peptídica antimicrobiana, LL-37, que age como um agente quimiotático de
neutrófilos, monócitos, mastócitos e células-T (Yamasaki et al. 2006). Posteriormente, o
segmento LL-37 é clivado pelas KLK5 e KLK7 para gerar peptídeos menores (KS-22, KS30,
LL-29, RK-31 e KR30) que possuem atividade antimicrobiana. Subsequente a resolução da
infecção, as KLK5 e KLK7 processam o peptídeo LL-37 e demais peptídeos (Yamasaki et al.
2006) diminuindo a resposta imune.
Na inflamação, a ativação dos receptores 1 – 4 ativados por proteinases (proteinase
activated receptor 1-4 – PAR1-4) resulta na liberação de prostaglandinas (PG) (Kubota, et al.
2003, Asokananthan, et al. 2002, Frungieri, et al. 2002, Kong, et al. 1997). Estudos atestam o
papel das calicreínas como moduladores da sinalização de PAR, já que foi demonstrado que
elas podem ativar ou inativar esses receptores (Oikonomopoulou, et al. 2006).
Ekholm e Egelrud (1999) mostraram que na inflamação na psoríase, os níveis de KLK7
estão elevados e que ela pode ativar a IL-1 β por observarem que IL-1β ativa é formada
quando incubada com KLK7 (Nylander e Egelrud, 1997).
Embora as calicreínas apresentem um amplo espectro de ação (fisiológica e patológica),
não há no mercado moléculas disponíveis com potencial regulatório de suas atividades.
Existem moléculas inibidoras inespecíficas de serino proteases como PMSF (Pubchem CID
4784, http://pubchem.ncbi. nlm.nih.gov/) e DIFP (Pubchem CID 5936), porém elas são muito
tóxicas, inviabilizando seu uso clínico. Apesar do enfoque principal do desenvolvimento de
moléculas inibitórias seja para o uso clínico, essas moléculas também são úteis na pesquisa
para auxiliar na elucidação da ação dessas enzimas.
1.4. FERRAMENTAS DE DOCKING MOLECULAR E O DESENVOLVIMENTO DE
FÁRMACOS.
Tradicionalmente, fármacos eram descobertos após extensivos testes que envolviam a
síntese de compostos e, após testes in vitro e in vivo, eram modificados e novamente testados,

| 23 P á g i n a
tudo isso num processo que demandava tempo e dinheiro (van de Waterbeemd e Gifford,
2003). A seleção desses compostos pode ser mais eficiente com testes in silico (analogia aos
termos in vitro e in vivo por ser realizado por ferramentas computacionais). Um grande
número de programas é utilizado hoje pelas indústrias farmacêuticas e de biotecnologia,
principalmente os programas de docking molecular (FIG. 6), que a partir da estrutura
cristalográfica do receptor colocam a molécula em estudo no sítio de interesse, indicando se
pode ser um possível fármaco (Bleicher, et al. 2003).
FIGURA 6: Mecanismo do docking molecular. Após obtenção da estrutura do receptor e do ligante, são
aplicados algoritmos de docking para o cálculo da interação do ligante com o receptor. O cálculo considera a
complementação estrutural entre ligante e receptor e a complementação energética obtida por ligações não
covalente. Para cada pose gerada há um score.
O estudo de docking molecular consiste na predição da conformação e orientação (pose)
do ligante no sítio de ligação de um alvo, e do cálculo de sua energia de interação (score). O
processo de docking inicia com o posicionamento do ligante no receptor, e através de funções
de scoring, busca-se uma conformação do ligante que seja complementar ao receptor de
forma estrutural e energética. As características estruturais determinam os arranjos espaciais
moleculares e, as características energéticas são as ligações não covalentes (Sinnokrot e
Sherrill, 2003).
O programa Glide (grid-based ligand docking with energetics) (Friesner, et al. 2004) foi
desenvolvido para fazer uma busca exaustiva da posição, orientação e conformação espacial
do ligante no sítio do receptor (através de uma série de filtros). Inicialmente, é construída uma
“rede” (GRID) na região de interação do receptor. Em cada ponto dessa rede é feita a análise

| 24 P á g i n a
dos tipos de interação com cada tipo de átomo, fornecendo informações de diferentes campos
de forças (como interação eletrostática, van der Waals, hidrofóbica/hidrofílica, etc.) que provê
progressivamente scores mais acurados para a pose do ligante.
FIGURA 7: Definição de core e grupo rotâmero. O core é a parte mais rígida da molécula e o grupo
rotâmero consiste nas cadeias laterais que tem liberdade conformacional (Schrödinger, 2005).
O ligante é dividido em parte rígida (core) e “grupos rotâmeros” que são partes da
molécula ligada ao core por uma ligação que pode girar (FIG. 7). Então o programa procura
possíveis posições e orientações para o core no receptor. Uma vez encontrado posições
favoráveis, os grupos rotâmero são adicionados e minimizados. De três a seis estruturas com
menor energia são submetidas à análise conformacional de Monte Carlo que provoca
alterações nos ângulos e diedros do ligante em busca de conformações de mínima energia
global.
O diferencial do algoritmo Extra Precision Mode (XP) usado no programa Glide é a
aplicação de penalidades para poses que violam princípios físico-químicos, diminuindo a
formação de poses “falsos positivos”. O algoritmo de score é a soma das forças eletrostática,
van der Waals e demais forças que facilitam e dificultam a interação. O termo de penalidade
corresponde à tensão da ligação, perda de entropia e dessolvatação tanto do ligante como do
receptor. A comparação de vários programas de docking mostrou que o Glide é um dos
melhores atualmente (Cross, et al. 2009).
A seleção de moléculas para o estudo de docking pode ser feito por busca em bancos
de dados, ou quando de conhecimento do pesquisador, ser desenhada usando os programas
gráficos de modelagem molecular. Esses compostos podem ser de origem natural
(proveniente de plantas, bactérias, fungos ou animais) ou sintética (síntese química ou
modificação direta de moléculas naturais).

| 25 P á g i n a
1.4.1. Moléculas peptídicos miméticas derivadas de isomanídeos.
Uma das estratégias no desenvolvimento de fármacos é o uso de moléculas peptídico
miméticas, que consiste na modificação da cadeia principal ou cadeia lateral dos resíduos para
aumentar sua estabilidade contra proteases.
Anéis de isomanídeos são moléculas formadas pela fusão de dois anéis furano. O
enfoque no seu uso se deve à sua analogia estrutural com anéis cíclicos de dipeptídeos
(Bencsik, et al. 2003; Dietrich e Lubell, 2003), que são fixados em uma conformação bioativa,
mas são isentos das desvantagens que drogas peptídicas apresentam como baixa
permeabilidade e absorção, metabolismo rápido, alto clearance e baixa disponibilidade oral
(Hruby, 2002).
Uma série de compostos peptídicos miméticos derivados de isomanídeos (FIG. 8) foi
inicialmente designada como potentes inibidores da enzima NS3 do vírus da hepatite C
(HCV). A infecção por HCV pode levar ao desenvolvimento de cirrose, falência hepática ou
carcinoma hepatocelular (Burke e Cox, 2010). Não existe vacina para prevenir a infecção por
HCV e seu tratamento é caro e demorado (Grebely, Thomas e Dore, 2009).
HCV NS3 é uma serino protease semelhante à tripsina com a tríade catalítica His51-
Asp75-Ser135 (Bazan e Fletterick, 1989a, 1989b, Gorbalenya, et al. 1989). Como essa enzima
é essencial para a replicação viral, NS3 constitui um importante alvo para drogas antivirais.
Como a enzima NS3 pertence a mesma classe das calicreínas (serino proteases), esse
grupo de compostos peptídico mimético foi escolhido para o teste de inibição já que ele foi
desenhado para inibir essa classe enzimática.
| 26 P á g i n a
FIGURA 8: Estrutura dos compostos peptídicos miméticos derivados de isomanídeos.
1.4.2. Compostos naturais – isocumarinas.
Produtos naturais são estruturas complexas que apresentam uma miríade de ação. Por
serem compostos sintetizados para auxiliar na proteção da planta, elas oferecem alta
seletividade e atividade biológica específica (Newman, Cragg e Kingston, 2008).
Isocumarinas compreendem uma classe de produtos naturais polifenólicos presentes em
uma variedade de espécies de plantas (Hill, 1986), apresentando diversas atividades como
ação antioxidante (Devienne, et al. 2007), antibacteriana (Tabopda, et al 2009, Kihampa, et al.
2009), antimosquito (Kihampa, et al. 2009), atividade citotóxica (Whyte, et al. 1996),
antitumoral (Cañedo, Puents e Baz, 1997, Di Stasi, et al. 2004, Kostova, 2005, Okamoto,

| 27 P á g i n a
Kobayashi e Yoshida, 2005, Devienne, et al. 2005) e, potente inibição contra enzimas serino
proteases, incluindo quimotripsina, tripsina e elastase (Harper, Hemmi e Power, 1985) e
também contra cisteíno proteases (Severino, et al. 2011).
FIGURA 9: Estrutura dos compostos isocumarínicos. (A) paepalantina, (B) 8,8’-paepalantina dímero e
(C) vioxantina.
As isocumarinas paepalantina, 8,8’-paepalantina dímero e vioxantina (FIG. 9) foram
isoladas de Paepalanthus bromelioides, uma planta brasileira comumente encontrada na
região da Serra do Cipó, Minas Gerais, Brasil (Vilegas, et al. 1990, Coelho, et al. 2000,
Devienne, et al. 2007). Esses compostos são poderosos agentes para a proteção contra o
estresse oxidante imposto pela mitocôndria (Devienne, et al. 2007) e também, apresentam
atividade antimicrobiana (Devienne, et al. 2002, 2005, 2007, Varanda, et al. 2004).
Como há descrição de inibição de proteases pelas isocumarinas, as três moléculas de
isocumarinas (paepalantina, 8,8’-paepalantina dímero e vioxantina) foram utilizadas para
compor nossa biblioteca de compostos e observar se elas inibem as calicreínas teciduais
humanas 5 e 7.

| 28 P á g i n a
2. OBJETIVOS.
2.1. OBJETIVOS GERAIS.
Estudar o mecanismo de inibição das calicreínas teciduais humanas 5 e 7 por
compostos sintéticos (peptídico miméticos derivados de isomanídeos) e produtos
naturais (isocumarinas) através de testes de inibição enzimática e docking molecular.
2.2. OBJETIVOS ESPECÍFICOS.
Determinar a concentração necessária dos compostos sintéticos e naturais para a
inibição de 50% da atividade enzimática (IC50) da KLK5;
Determinar a concentração necessária dos compostos naturais para a inibição de 50%
da atividade enzimática (IC50) da KLK7;
Determinar a constante de inibição (Ki) da KLK5 pelos compostos sintéticos e
naturais;
Determinar a constante de inibição (Ki) da KLK7 compostos naturais;
Determinar o tipo de inibição da KLK5 pelos compostos sintéticos e naturais;
Determinar o tipo de inibição da KLK7 pelos compostos naturais;
Estudar o modo de interação da KLK5 com as moléculas sintéticas e naturais por
ferramentas de modelagem molecular;
Estudar o modo de interação da KLK7 com as moléculas naturais por ferramentas de
modelagem molecular.

| 29 P á g i n a
3. MATERIAIS E MÉTODOS.
3.1. MOLÉCULAS PARA ESTUDO DE INIBIÇÃO.
3.1.1. Moléculas peptídico miméticas derivadas de isomanídeos.
Um grupo de quinze compostos peptídicos miméticos derivados de isomanídeo foi
gentilmente cedido pela Profa. Dra. Estela Maris Freitas Muri da Universidade Federal
Fluminense (UFF). A síntese desses compostos foi feita pelo Prof. Dr. Octavio Augusto Ceva
Antunes (UFF) e se encontra descrito em Barros, et al. (2009).
As amostras foram diluídas em DMSO para uma concentração final de 100 mM.
3.1.2. Isocumarinas.
A segunda classe utilizada compreende três moléculas de isocumarinas, gentilmente
cedidas pela Profa. Dr. Karina Ferrazzoli Devienne Vincentine da Universidade Federal do
Triângulo Mineiro (UFTM). A extração e purificação estão descritas em Vilegas, et al. (1990)
e Coelho, et al. (2000).
As amostras foram diluídas em DMSO até não se observar a presença de precipitado. A
concentração final da paepalantina foi de 30,63 mM, a 8,8’-paepalantina dímero foi de 12,84
mM e da vioxantina foi de 7,4 mM.
3.2. ENZIMAS, SUBSTRATOS E TAMPÕES.
3.2.1. Expressão, purificação e ativação da KLK5 e KLK7 recombinante.
As enzimas maduras da KLK5 e da KLK7 foram clonadas, expressadas, purificadas e
ativadas pelo prof. Dr. Michael Blaber da Florida State Univesity (FSU), que gentilmente nos
cedeu amostras das duas enzimas.
3.2.2. Substrato.
Para os ensaios enzimáticos com as KLK5 e KLK7, foram utilizados substratos
específicos com transferência de energia fluorescente ressonante (FRET), sendo o substrato
Abz-KLRSSKQ-EDDnp para a KLK5 e Abz-KLYSSKQ-EDDnp para a KLK7,
respectivamente e, são excitados num comprimento de onde de 320nm e emitem fluorescência

| 30 P á g i n a
em 420nm. Esses substratos foram gentilmente cedidos pelo prof. Dr Luiz Juliano Neto da
Universidade Federal Paulista (UNIFESP).
3.2.3. Tampão.
Utilizou-se em todos os testes enzimáticos tampão Tris-HCl (Sigma-Aldrich) 50 mM,
pH 7,5, 0,01% v/v de Tween-20.
3.3. ENSAIOS DE INIBIÇÃO ENZIMÁTICA.
Para o Teste de IC50, na cubeta de ensaio foram adicionados 400 µL de tampão, 0,015
µM da enzima e incubada por 5 minutos à 37°C. Então foi adicionado o substrato numa
concentração de 24 µM e adicionado pelo menos cinco concentrações de inibidores em uma
faixa de 24 µM a 21,5 mM. As medidas foram conduzidas num fluorímetro Hitachi F2500
(comprimento de onda de excitação 320nm e emissão de 420nm). Os valores representam
uma média (±DP) de pelo menos três experimentos diferentes. Os valores de IC50 foram
obtidos de uma curva de regressão não linear usando o programa GraFit 7 (Leatherbarrow,
2009).
Para o teste de Ki, na cubeta de ensaio foram adicionados 400 µL de tampão, 0,015 µM
da enzima e, as moléculas numa concentração de 0 a 150 µM incubada por 5 minutos à 37°C.
A reação foi iniciada com a adição de seis concentrações diferentes de substrato (0,25 µM,
0,44 µM, 0,63 µM, 0,93 µM, 1,23 µM e 1,81 µM) e, as medidas foram conduzidas num
fluorímetro Hitachi F2500 (comprimento de onda de excitação 320nm e emissão de 420nm).
Os parâmetros obtidos foram plotados no gráfico duplo recíproco de Lineweaver-Burk, na
qual o eixo x corresponde ao inverso da constante de Michaelis-Menten (-Km) e o eixo y o
inverso da velocidade máxima (1/V). O valor de Ki foi determinado pela plotagem de Dixon
que consiste na plotagem do recíproco da velocidade inicial (1/V0) versus uma série de
concentrações diferentes do inibidor com concentração constante do substrato, na qual o
intersecto com o eixo x representa o valor de Ki.

| 31 P á g i n a
3.4. ESTUDO DE MECÂNICA MOLECULAR.
3.4.1. Estudo de docking molecular.
3.4.1.1. Preparo do receptor.
As coordenadas estruturais das enzimas utilizadas neste trabalho foram coletadas no site
Protein Data Bank (Berman, et al. 2000) (http://www.pdb.org/pdb/home/home.do). Foram
considerados as proteínas calicreína tecidual humana 5 (código 2PSX) e calicreína tecidual
humana 7 (código 2QXH).
A estrutura cristalina da KLK5, complexada com o inibidor leupeptina (Debela, et al.
2007a), apresenta resolução de 2,30Å, enquanto a KLK7, complexada com Suc-Ala-Ala-Pro-
Phe-clorometilcetona (K7J) (Debela, et al. 2007b), apresenta resolução de 2,00Å.
Como as estruturas cristalográficas não possuem átomos de hidrogênio (devido à
resolução do Raio-X) e várias moléculas e íons estão presentes junto ao cristal, a estrutura do
complexo foi preparado usando o programa Protein Preparation Wizard (Schröndinger, 2005).
Todas as moléculas que não fazem parte da estrutura da proteína (água, íons e outros solutos)
foram removidos, seguido da adição dos hidrogênios explícitos à proteína e a cuidada
investigação do estado de protonação dos resíduos ionizáveis. A estrutura foi minimizada
utilizando o campo de força OPLS-AA 2005 (Jorgensen, et al. 2005), sendo que o limite de
diferença entre a estrutura minimizada e a estrutura inicial não deveria ser maior do que 0,3Å
de RMSD.
O Grid foi gerado como uma caixa cúbica de bordas de comprimento de 25Å sendo que
seu centro se localizava no inibidor. Com isso garantimos que todos os resíduos do sítio de
clivagem se encontravam dentro da caixa. O GRID foi preparado para permitir a rotação do
grupo hidroxila da Ser195.
3.4.1.2. Preparo do ligante.
No estudo de docking, consideraram-se somente os ligantes que foram solúveis em meio
aquoso e apresentaram boa inibição, sendo os compostos 9g, 9j e 9k (peptídico mimético
derivados de isomanídeo) e, paepalantina, 8,8’-paepalantina dímero e vioxantina (compostos
isocumarínicos).

| 32 P á g i n a
Utilizou-se as ferramentas de desenho molecular implementado no programa Maestro
(Schrödinger, 2005) e a minimização das estruturas foi feita com o programa MacroModel
(Schrödinger, 2005).
3.4.1.3. Estudos de docking.
Antes de iniciar os estudos de docking, efetuou-se a validação da metodologia de
docking molecular pelo estudo de redocking dos ligantes presentes na estrutura cristalina das
proteínas KLK5 e KLK7. Este estudo consiste na retirada do ligante de sua coordenada
original e reposicionando-o novamente pelos cálculos de docking (FIG. 10).
√∑
(2)
As estruturas geradas pelo redocking devem ter um desvio médio quadrático (RMSD)
menor que 2,00 Å. Essa função matemática diz o quanto uma estrutura difere da outra por
comparar a disposição espacial (xyz) dos átomos, como visto na equação 2, na qual N é o
número de átomos sobre o qual o RMSD é medido e di é a distância (xyz) entre as
coordenadas espaciais do átomo i em duas conformações (Leach, 2006). Após validar a
metodologia, iniciou-se os estudos de docking com os compostos de isocumarinas e os
peptídico miméticos derivados de isomanídeos.
FIGURA 10: Validação do estudo de docking. As coordenadas do ligante leupeptina foram alteradas, e
após o docking, retorna à posição cristalográfica original (código PDB: 2PSX).
Todos os cálculos de docking foram efetuados usando o método Glide XP (Friesner, et
al. 2004, Schröndinger, 2005). Foram selecionados parâmetros para permitir o docking
flexível dos inibidores, bem como variar as conformações dos anéis (axial ou equatorial) e

| 33 P á g i n a
penalizar ligações peptídicas não planares. Para a geração das poses, foi especificado para
manter nas fases iniciais do cálculo de docking 5000 poses por ligante, sendo a janela de
energia para manter as poses iniciais de 100 Kcal/mol. Isso garante que as estruturas geradas
estejam dentro duma faixa de 100 Kcal/mol. Dessas conformações geradas, as 800 melhores
foram mantidos para minimização usando o parâmetro de amostragem expandida. O número
máximo de passos de minimização foi 100. Os demais parâmetros padrões do Glide foram
usados para as demais funções.
Os resultados obtidos foram analisados usando a própria interface gráfica do Glide
(Glide Visualizer) implementada no programa Maestro (Schrödinger, 2005). Para a geração
das figuras foi utilizado o programa Chimera (Pettersen, et al. 2004).
3.4.2. Estudo de dinâmica molecular (DM).
A dinâmica molecular (DM) é uma poderosa ferramenta utilizada no estudo de modelos
em escala microscópica. Esta técnica determina o movimento temporal de uma molécula
baseada nas interações com as partículas que fazem parte de seu sistema. Este efeito é
baseado na segunda lei de Newton que afirma que o movimento de uma partícula é
proporcional à força aplicada sobre ela, como descreve a fórmula 3:
(3)
na qual Fi é a força agindo na partícula i, mi é a massa da partícula i e, ai é a aceleração da
partícula i. O estudo da DM é feito sob pressão e temperatura constante.
Para o estudo de DM, utilizou-se o programa GROMACS (Berendsen, van der Spoel e
van Drunen, 1995, Lindahl, Hess e van der Spoel, 2001). A proteína KLK5 foi solvatada com
solvente explicito modo SPC, em uma caixa cúbica octaédrica com espaço de 1,2 nm em volta
do soluto e, 12 íons Cl- foram adicionados para solvatar o sistema. A energia de minimização
foi feita duas vezes usando o método steepest descent e método de gradiente conjugado. A
temperatura de referência foi fixada em 300 K. O campo de força GROMACS96 53a6 foi
usado para o cálculo da energia em um tempo de 20 ns. As coordenadas da trajetória foram
salvas a cada 2 ps. As ferramentas implementadas no GROMACS foram usadas para o
cálculo do RMSD e o gráfico foi visualizado usando o programa Xmgrace
(http://www.plasma-gate.weizmann.ac.il/Grace/).

| 34 P á g i n a
4. RESULTADO E DISCUSSÃO.
A velocidade máxima de clivagem do substrato Abz-QLRSSLK-EDDnp pela KLK5 é de
aproximadamente 300 µM/min, sendo o Km desta reação de aproximadamente 0,65 µM. Já a
clivagem do substrato Abz-QLYSSLK-EDDnp pela KLK7 foi de aproximadamente 325
µM/min, sendo o Km desta reação de aproximadamente 0,60 µM.
4.1. INIBIÇÃO DA KLK5 PELOS ISOMANÍDEOS.
Dos 15 isomanídeos testados, somente cinco (9g, 9h, 9i, 9j e 9k) foram solúveis na
solução aquosa de teste. Dos cinco compostos testados, três tiveram IC50 abaixo de 100 µM,
na qual o composto 9g foi o mais potente, com um IC50 de 57,9 µM, seguido do composto 9j
(IC50 = 85,3 µM) e 9k (IC50 = 95,7 µM), respectivamente (TABELA 2).
TABELA 2: Valores dos testes de inibição (IC50 e Ki em µM) dos compostos peptídicos miméticos com a
proteína KLK5.
Composto IC50 (µM) Ki (µM)
9a ns1
-
9b ns -
9c ns -
9d ns -
9e ns -
9f ns -
9g 57,9 (±9,6) 15,5
9h >1000 357,1
9i 323,5 200,0
9j 85,3 (±8,6) 25,0
9k 95,7 (±8,3) 111,1
9l ns -
9m ns -
9n ns -
9o ns - 1ns = não solúvel na solução aquosa de teste.
Para entender melhor o mecanismo de inibição desses compostos, determinou-se o
perfil de inibição com relação o substrato Abz-QLRSSLK-EDDnp. Os testes de atividade
foram feitos na ausência e na presença de três concentrações diferentes de inibidores (FIG. 11
e 12).
Para os compostos 9g, 9j e 9k, a plotagem do gráfico de duplo recíproco de Lineweaver-
Burk mostra o intercepto de todas as linhas (obtidos em quatro concentrações diferentes de
inibidores) convergindo para o eixo y (1/Vmax) e, o intercepto do eixo x (-1/Km) variando com
a concentração do inibidor (FIG. 11). Consequentemente, o valor de Vmax permaneceu
constante, enquanto o valor aparente de Km (Kmapp
, definido como Km (1 + [I]/Ki))

| 35 P á g i n a
aumentou com o aumento da concentração do inibidor. Esta análise estabeleceu que eles se
comportam como inibidores reversíveis e competitivos.
Já para os inibidores 9h e 9i o gráfico de Lineweaver-Burk mostra o intercepto de todas
as linhas convergindo para o eixo x (-1/Km) e, o intercepto do eixo y (1/Vmax) variando com a
concentração do inibidor (FIG. 12). Consequentemente, o valor de Kmapp
permaneceu
constante, enquanto o valor aparente de Vmáx aumentou com o aumento da concentração do
inibidor. Esta análise estabeleceu que eles se comportam como inibidores reversíveis e não-
competitivos.
A determinação da constante de inibição (Ki) desses inibidores foi feita pela plotagem de
Dixon. Observou-se que os valores de Ki seguem a mesma tendência como observado para o
IC50 (TABELA 2), na qual o composto 9g permanece o mais potente (Ki = 15,5 µM) seguido
do composto 9j (Ki = 25,0 µM) e 9k (Ki = 111,1 µM). Os compostos não competitivos9h e 9i
obtiveram um alto valor de Ki igual a 357,1 µM e 200,0 µM, respectivamente.

| 36 P á g i n a
FIGURA 11: Gráfico de Lineweaver-Burk da enzima KLK5 com os compostos peptídicos miméticos derivados de isomanídeos. Os compostos 9g (A), 9j (B) e 9k (C)
competem com o substrato pelo sítio catalítico, como pode ser visto pela intersecção no eixo y (1/Vmax). A concentração dos compostos variou numa faixa de 0µM a 150µM.
Os valores da coordenada x (1/[S]) estão em µM-1
e, os da coordenada y (1/Vmax) em µM-1
min-1
.
FIGURA 12: Gráfico de Lineweaver-Burk da enzima KLK5 com os compostos peptídicos miméticos derivados de isomanídeos. Os compostos 9h (A) e 9i (B) não competem
com o substrato pelo sítio catalítico, como pode ser visto pela intersecção no eixo x (-1/Km). A concentração dos compostos variou numa faixa de 0µM a 150µM. Os valores
da coordenada x (1/[S]) estão em µM-1
e, os da coordenada y (1/Vmax) em µM-1
min-1
.

| 37 P á g i n a
4.2. INIBIÇÃO DA KLK5 E KLK7 PELAS ISOCUMARINAS.
Foram realizados testes cinéticos de inibição da KLK5 e KLK7 com as isocumarinas.
Para a KLK5 (TABELA 3), a vioxantina foi o composto mais potente com IC50 de 53,7 μM,
seguido de 8,8’-paepalantina dímero (IC50 = 72,9 μM) e paepalantina (IC50 = 367,4μM). Já
para a KLK7 (TABELA 4), a 8,8’-paepalantine dímero obteve o menor valor de IC50
(112,4μM), seguido da vioxantina (IC50 = 135,3 μM) e paepalantina (IC50 = 165,8 μM).
Para melhor entender o mecanismo de ação das três isocumarinas naturais, determinou-
se o mecanismo de inibição com relação ao substrato Abz-KLRSSKQ-EDDnp para a KLK5 e
Abz-KLYSSKQ-EDDnp para a KLK7, empregando as três isocumarinas em quatro
concentrações diferentes (FIG.13).
TABELA 3: Valores de IC50 e Ki (em µM) dos inibidores derivados de isocumarinas mediante a atividade da
KLK5.
Composto KLK5
IC50(µM) Ki(µM)
Paepalantina 367,4 (±10,2) 47,5
8,8’-paepalantina dímero 72,9 (±3,0) 24,4
Vioxantina 53,7 (±3,7) 22,9
TABELA 4: Valores de IC50 e Ki (em µM) dos inibidores derivados de isocumarinas mediante a atividade da
KLK7.
Composto KLK7
IC50(µM) Ki(µM)
Paepalantina 165,8 (±15,3) 70,7
8,8’-paepalantina dímero 112,4 (±9,2) 12,2
Vioxantina 135,3 (±11,5) 37,2
O gráfico do duplo-recíproco de Lineweaver-Burk mostra o intercepto de todas as linhas
(obtidos em quatro concentrações diferentes de inibidores) convergindo para o eixo y (1/Vmax)
enquanto o intercepto no eixo x (-1/Km) varia com a concentração do inibidor.
Consequentemente, os valores remanescente de Vmáx permanecem constantes, enquanto o
valor aparente de Km (Kmapp
definido como Km (1 + [I]/Ki)) aumenta com o aumento da
concentração de inibidor. Este comportamento é consistente com ligação mútua entre inibidor
e substrato, na qual a isocumarina compete com o substrato pela enzima livre.
Usando os dados cinéticos, determinaram-se os valores de Ki dos compostos
isocumarínicos pela plotagem de Dixon (TABELA 3 e 4). Para a KLK5, a vioxantina
permanece o inibidor mais potente (Ki= 22,9 µM), seguido da 8,8’-paepalantine (Ki = 24,4
µM) e paepalantina (Ki = 47,5 µM). Para a KLK7, a 8,8’-paepalantina (Ki = 12,2 µM) é o mais

| 38 P á g i n a
potente, seguido pela vioxantina (Ki = 37,2 µM) e paepalantina (Ki = 70,7 µM). A análise dos
resultados cinéticos mostrou que para ambas as enzimas, a forma dímero é mais potente. Para
ambas as enzimas, os valores de Ki seguem a mesma tendência dos valores de IC50,
mostrando que a forma dímero é mais potente.
A comparação dos valores de inibição da KLK5 com as duas classes de compostos
(peptídico mimético e isocumarina) mostra que a isocumarina mais potentes apresenta um
valor de IC50 menor que o valor da molécula peptídico mimético mais potente (vioxantina:
IC50 = 53,7 µM; 9g: IC50 = 57,9 µM). Talvez essa melhor inibição ocorra devido à maior
presença de grupos polares na vioxantina que no composto 9g, já que no inibidor leupeptina
(presente na estrutura cristalina da KLK5 (código PDB: 2PSX)) a presença do grupo aldeído é
responsável pela interação com o resíduo Ser195 levando a uma inibição de 1,7 µM (Debela,
et al. 2007a).

| 39 P á g i n a
FIGURA 13: Gráfico de Lineweaver-Burk da enzima KLK5 com os compostos isocumarínicos. Os compostos paepalantina (A), 8,8’-paepalantina dímero (B) e
vioxantina (C) competem com o substrato pelo sítio catalítico da KLK5, como pode ser visto pela intersecção no eixo y (1/Vmax). A concentração dos compostos variou
numa faixa de 0µM a 30µM. Os valores da coordenada x (1/[S]) estão em µM-1
e, os da coordenada y (1/Vmax) em µM-1
min-1
.

| 40 P á g i n a
4.3. ESTUDO DE DOCKING MOLECULAR COMS OS INIBIDORES.
4.3.1. Validação do método de docking molecular.
Com o objetivo de avaliar se a metodologia do programa Glide apresenta bons
parâmetros para efetuar o docking, avaliou-se o redocking, que consiste em observar se o
programa é capaz de reproduzir a estrutura cristalina da proteína KLK5 e KLK7 com os seus
ligantes cristalizados.
FIGURA 14: Sobreposição da leupeptina gerada pelo redocking. A estrutura gerada pelo redocking
(bastão cor verde) apresenta disposição estrutural semelhante à estrutura cristalina (bastão cor cinza) com RMSD
de 0,65 Å.
Para a KLK5, o redocking posicionou o ligante leupeptina corretamente no sítio
catalítico. A diferença da estrutura gerada com a estrutura cristalina (RMSD) foi de 0,65 Å
(FIG. 14), sendo que o valor máximo aceito para considerar uma estrutura semelhante à outra
é de 2,00 Å. As distâncias de alguns átomos do ligante em relação à proteína foram
semelhantes, como visto na TABELA 5.
TABELA 5: Distância dos átomos da leupeptina em relação à proteína 2PSX no estudo de redocking.
Átomos da leupeptina Átomos da proteína Distância (Å)
PDB (2PSX) Redocking
NH1 Arg Oδ2 Asp189 2,70 2,75
NH2 Arg Oδ2 Asp189 3,01 2,65
NH(cadeia principal) Arg Carboxil Ser214 2,18 1,97
Carboxil Arg NH(cadeia principal) Gly193 2,06 2,37
NH(cadeia principal) Leu Carboxil Gly216 1,87 1,72

| 41 P á g i n a
O redocking do ligante K7J na KLK7 apresentou uma conformação semelhante com a
estrutura cristalina (FIG. 15), sendo a variação média da estrutura (RMSD) de 1,51 Å.
(TABELA 6). A principal variação estrutura ocorreu no grupo ACE e Suc.
FIGURA 15: Sobreposição do ligante K7J na gerada pelo redocking. A estrutura gerada pelo redocking
(bastão cor verde) apresenta disposição estrutural semelhante à estrutura cristalina (bastão cor cinza) com RMSD
de 1,51 Å.
TABELA 6: Distância dos átomos da K7J em relação à proteína 2QXH no estudo de redocking
Átomos da K7J Átomos da proteína Distância (Å)
PDB (2QXH) Redocking
Cα Phe Cβ Ala190 5,30 4,54
O ACE NH(cadeia principal) Gly193 2,37 1,92
Cα Pro Cε1 His57 4,11 4,36
NH(cadeia principal) Phe Carboxil Ser214 2,23 3,29
NH(cadeia principal) Ala Carboxil Gly216 2,28 2,00
4.3.2. Estudo de docking molecular dos isomanídeos na KLK5.
Para entender o modo de ligação e interação dos novos inibidores da KLK5, efetuou-se
o docking molecular dos compostos 9g, 9j e 9k no sítio de ligação da enzima, definido pelos
aminoácidos próximos da leupeptina na estrutura cristalina do complexo enzima-inibidor
(Debela, et al. 2007). Como não há descrição de sítio(s) alostérico(s) na KLK5, não se
realizou o docking dos compostos 9h e 9i.
Os valores de energia do docking correlacionaram com os resultados experimentais,
ranqueando o composto 9g com uma energia levemente menor (-7,70 Kcal/mol) do que 9j (-
7,27 Kcal/mol), enquanto 9k teve a terceira menor energia (-6,85 Kcal/mol). A análise dos
resultados de docking sugere fortemente que os compostos 9g, 9j e 9k compartilham modos

| 42 P á g i n a
de ligação semelhantes com a KLK5. A conformação do anel de isomanídeo é o mesmo no
composto 9g e 9j, mas difere em 9k (FIG. 16 D). O grupo O-benzil de todos inibidores ocupa
o bolso hidrofóbico S2’(FIG. 16). No outro lado do anel de isomanídeo, o substituinte R2 dos
três inibidores está posicionado no bolso S1 da KLK5. Como a única diferença entre o
composto 9g e 9k é o grupo R1, provavelmente esse grupo seja responsável pela diminuição
de 0,85 kcal/mol na interação do composto 9k com a enzima.
FIGURA 16: Posição dos inibidores peptídicos miméticos derivados de isomanídeos na superfície da KLK5. Os
compostos 9g (A), 9j (B) e 9k (C) têm o substituinte R2 no subsítio S1 e o substituinte R1 no subsítio S2. O anel
de isomanídeo ocupa o subsítio S1’ e o grupo O-benzil o subsítio S2’. A comparação das três moléculas (D)
mostra que eles possuem a mesma conformação com exceção do composto 9k que tem o anel de isomanídeo e o
grupo O-benzil levemente perpendicular ao dos outros compostos. A superfície hidrofóbica da proteína da
proteína é mostrada com cores variando de azul (hidrofílico) a vermelho (hidrofóbico). Os átomos de hidrogênio
não polar foram omitidos para melhor visualização.

| 43 P á g i n a
FIGURA 17: Destaque dos principais resíduos que formam as interações hidrofóbicas com os inibidores peptídicos miméticos. Os inibidores 9g (A) e 9j (B) formam
interação hidrofóbica principalmente com a Cys191 e o Trp215. Já o composto 9k (C) perde a interação com Trp215 devido à substituição em R1 do grupo fenil pelo grupo
metil.

| 44 P á g i n a
O substituinte R2, sendo o anel tiofeno para 9g e 9k e, furano para 9j, mostra uma
interação C-H∙∙∙π com o Cα da Cys191 (FIG. 17). Por outro lado, a maior diferença entre os
inibidores é em relação ao tamanho do substituinte R1. Os compostos mais potentes (9g e 9j)
têm um anel fenil localizado na entrada do bolso S2 (FIG. 17 A e B). Além das interações
hidrofóbicas que o anel fenil faz em S2, ele também faz interação C-H∙∙∙π com o Cβ do
Trp215. No composto 9k o anel fenil é substituído por um grupo metil e, as interações
mencionadas anteriormente são perdidas.
A habilidade de um inibidor se ligar à fenda de oxiânion pode ser considerada um fator
importante para uma boa inibição já que ela é responsável por estabilizar o substrato, como
exemplificado pelo inibidor benzamidina, na qual seu grupo amidino interage com o resíduo
Asp189, mas não com os resíduos da fenda do oxiânion e os resíduos dos subsítios catalítico,
apresentando uma constante de inibição alta que varia na faixa de µM a mM (Goetting,
Magdolen, Brandstetter, 2010).
Juntamente com as interações hidrofóbicas, a ligação desses inibidores é estabilizada
com quatro ligações de hidrogênio (TABELA 7). Há duas ligações de hidrogênio entre o
carboxil próximo ao anel de isomanídeo e os grupos amino da cadeia principal dos resíduos
catalíticos Ser195 e Gly193 da fenda oxônio (FIG. 18). O carboxil próximo à R1 forma uma
ligação de hidrogênio com o grupo amino da cadeia lateral da GLN192. Outra ligação de
hidrogênio é observada entre o grupo amino próximo à R1 com o carboxil da cadeia principal
da Ser214 (FIG. 18).
TABELA 7: Distância (em Å) dos átomos dos inibidores peptídicos miméticos e da KLK5 envolvidos nas
interações de hidrogênio.
Resíduos da KLK5 Átomos dos isomanídeos Distância (Å)
9g 9j 9k
NHε Gln192 Carboxil R1 1,83 1,84 1,97
NH(cadeia principal) Gly193 Carboxil anel isomanídeo 1,84 1,80 2,11
NH(cadeia principal) Ser195 Carboxil anel isomanídeo 2,29 2,30 1,91
Carboxil(cadeia principal) Ser214 Amino R1 2,18 2,24 2,12

| 45 P á g i n a
FIGURA 18: Interação de hidrogênio dos inibidores peptídicos miméticos derivados de isomanídeos com a KLK5. Os inibidores 9g (A), 9j (B) e 9k (C) formam
interações de hidrogênio (linha preta) com os mesmo resíduos, sendo os resíduos Gln192, Gly193, Ser195 e Ser214. Para melhor entendimento, somente os hidrogênios
polares (do ligante e do receptor) são mostrados.

| 46 P á g i n a
Devido ao desconhecimento de sítios alostéricos na qual os compostos 9h e 9i poderiam
estar se ligando, efetuou-se um estudo de dinâmica molecular da enzima para observar se a
estrutura geral da proteína modifica com o tempo (FIG. 19). Analisou-se a variação estrutural
de toda a proteína bem como dos resíduos que fazem parte dos subsítios S2’, S1’, S1 e S2. A
variação geral (RMSD) da proteína foi aproximadamente de 2,3 Å. Os subsítios S2’ e S1’
tiveram uma variação média de 1,3 Å e, o subsítio S1 de 1,4 Å, mostrando que esses sítios
possuem uma conformação estável, indicando que provavelmente esses sítios não se alteram
com a interação de um ligante noutra região da proteína.
Já o subsítio S2 teve uma variação média de 3,5 Å mostrando que é um sítio com
conformação altamente variável. Esse subsítio é caracterizado pela alça que compreende os
resíduos His96 e His99 que fazem parte da coordenação do Zn2+
na inibição alostérica nos
tecidos (Debela, et al. 2007a).
A partir desses dados, postulou-se que os compostos 9h e 9i não se liguem a uma região
afastada do sítio catalítico (como fazem inibidores não competitivo que, quando se ligam,
promovem uma alteração conformacional no sítio catalítico), mas que se ligam à S2 sem
bloquear os resíduos da região catalítica e, ao se ligarem a esse domínio provocam uma leve
alteração conformacional dos resíduos da tríade catalítica (semelhante ao efeito do Zn2+
),
levando a uma discreta redução da atividade proteolítica da enzima.
FIGURA 19: Variação estrutural da KLK5 determinado por dinâmica molecular. A variação (RMSD) da
proteína KLK5(linha preta) foi de 2,3Å, enquanto os subsítio S2’ (linha amarela) e S1’ (linha verde) tiveram
uma variação de 1,3Å, o subsítio S1 (linha vermelha) de 1,4Å e o subsítio S2 (linha azul) de 3,5Å, mostrando
que a conformação dos subsítios S2’, S1’ e S’ é bem conservada e, a do subsítio S2 é bem variável. O estudo foi
feito num tempo de 20 ns, e as variações foram salvas a cada 2ps.

| 47 P á g i n a
4.3.3. Docking das isocumarinas.
4.3.3.1. Calicreína tecidual humana 5.
De acordo com os valores de score, a 8,8’-paepalntina dímero foi ranqueada em
primeiro com o menor valor (-7,68 kcal/mol) do que a vioxantina (-7,49 kcal/mol), enquanto
paepalantina obteve a maior energia (-6,73 kcal/mol). Essa discordância dos valores de score
em relação aos valores de inibição pode ser explicada pelo fato que os programas de docking
são bons para prever a pose das estruturas, mas devido ao fator entrópico na interação
receptor-ligante, eles não são muito precisos em prever a afinidade. Uma diferença de 10
vezes na afinidade representa uma variação energética de ligação de 1,4 Kcal/mol (Stroud e
Finer-Moore, 2008).
Os resultados de docagem sugerem que o inibidor vioxantina e 8,8’-paepalantina
dímero partilham um modo de ligação semelhante com KLK5. Metade do dímero é
posicionada na entrada do subsítio S1 da KLK5 (FIG. 20) e a outra metade do dímero é
posicionada nos subsítios S3 e S4. Já o inibidor paepalantina ocupa o bolso S1 e parte do
subsítio S1’ (FIG. 20 A).
Junto à interação hidrofóbica com esses aminoácidos, vioxantina mostra uma interação
do tipo C-H∙∙∙π com Cδ e Cα da Gln192 e Asp217, respectivamente (FIG. 21). Esse inibidor
também apresenta uma interação N-H∙∙∙π com o Nε da Gln192. A 8,8’-papalantina dímero
também tem uma interação C-H∙∙∙π com o Cδ e Cβ da Gln192 e Trp215, respectivamente. Por
outro lado, paepalantina apresenta uma posição de ligação diferente, que é aproximadamente
perpendicular aos outros dois inibidores (FIG. 20A). Como observado previamente para os
outros inibidores, paepalantina também apresenta uma interação C-H∙∙∙π com Cα e Cδ da
Gln192 (FIG. 21 A).

| 48 P á g i n a
FIGURA 20: Posição dos inibidores de isocumarinas superfície da KLK5. Os inibidores paepalantina (A),
8,8’-paepalantina dímero (B) e vioxantina (C) ocupam a entrada do subsítio S1 da proteína e no caso da 8,8’-
paepalantina dímero e vioxantina, eles se estendem sobre os subsítios S3 e S4. A comparação das três moléculas
(D) mostra que as formas dímero possuem posição semelhante na proteína. A superfície hidrofóbica da proteína
da proteína é mostrada com cores variando de azul (hidrofílico) a vermelho (hidrofóbico). Os átomos de
hidrogênio não polar foram omitidos para melhor visualização.

| 49 P á g i n a
FIGURA 21: Destaque dos principais resíduos da KLK5 que formam as interações hidrofóbicas com os inibidores de isocumarinas. Os inibidores paepalantina (A), 8,8’-
paepalantina dímero (B) e vioxantina (C) formam interação hidrofóbica principalmente com Gln192, Trp215 e Asp217.
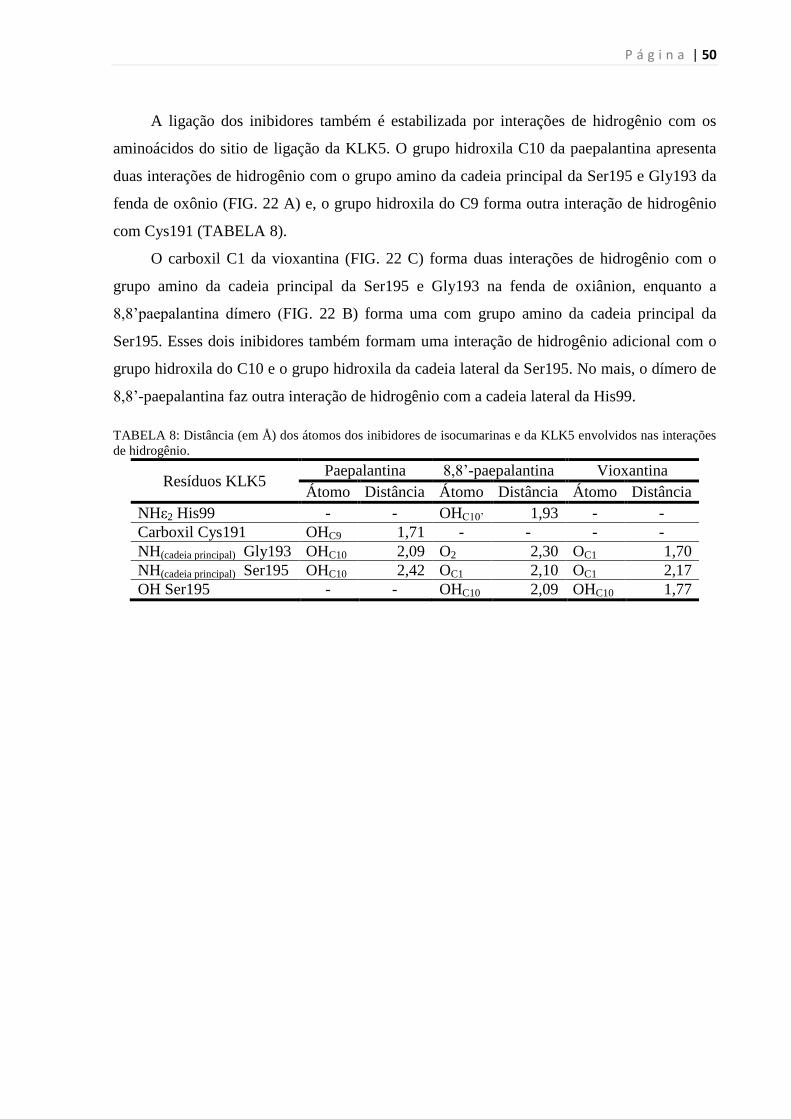
| 50 P á g i n a
A ligação dos inibidores também é estabilizada por interações de hidrogênio com os
aminoácidos do sitio de ligação da KLK5. O grupo hidroxila C10 da paepalantina apresenta
duas interações de hidrogênio com o grupo amino da cadeia principal da Ser195 e Gly193 da
fenda de oxônio (FIG. 22 A) e, o grupo hidroxila do C9 forma outra interação de hidrogênio
com Cys191 (TABELA 8).
O carboxil C1 da vioxantina (FIG. 22 C) forma duas interações de hidrogênio com o
grupo amino da cadeia principal da Ser195 e Gly193 na fenda de oxiânion, enquanto a
8,8’paepalantina dímero (FIG. 22 B) forma uma com grupo amino da cadeia principal da
Ser195. Esses dois inibidores também formam uma interação de hidrogênio adicional com o
grupo hidroxila do C10 e o grupo hidroxila da cadeia lateral da Ser195. No mais, o dímero de
8,8’-paepalantina faz outra interação de hidrogênio com a cadeia lateral da His99.
TABELA 8: Distância (em Å) dos átomos dos inibidores de isocumarinas e da KLK5 envolvidos nas interações
de hidrogênio.
Resíduos KLK5 Paepalantina 8,8’-paepalantina Vioxantina
Átomo Distância Átomo Distância Átomo Distância
NHε2 His99 - - OHC10’ 1,93 - -
Carboxil Cys191 OHC9 1,71 - - - -
NH(cadeia principal) Gly193 OHC10 2,09 O2 2,30 OC1 1,70
NH(cadeia principal) Ser195 OHC10 2,42 OC1 2,10 OC1 2,17
OH Ser195 - - OHC10 2,09 OHC10 1,77

| 51 P á g i n a
FIGURA 22: Interação de hidrogênio dos inibidores isocumarínicos com a KLK5. Os inibidores paepalantina (A), 8,8’-paepalantina dímero (B) e vioxantina (C)
formam interações de hidrogênio (linha preta) principalmente com os resíduos da fenda de oxiânion (grupo amino da cadeia principal da Gly193 e Ser195) além de outras
interações com outros resíduos que auxilia na estabilização do composto no sítio catalítico da enzima. Para melhor entendimento, somente os hidrogênios polares (do ligante e
do receptor) são mostrados.

| 52 P á g i n a
4.3.3.2. Calicreína tecidual humana 7.
Os estudos de docagem mostraram uma boa correlação com os resultados experimentais
dos inibidores com a KLK7, ranqueando em primeiro a 8,8’-paepalantina dímero com a
menor energia (-9,47kcal/mol), em segundo a vioxantina (-7,59 kcal/mol), enquanto a
paepalantina obteve a maior energia (-7,49kcal/mol).
Os resultados sugerem que o modo de ligação dos inibidores na KLK7 é distinto do
observado para a KLK5. Na KLK7 a ligação dos três inibidores adota uma conformação
semelhante e se ligam em dois subsítios, essencialmente através de interação hidrofóbica
(FIG. 23). Os inibidores maiores (8,8’-paepalantina dímero e vioxantina) se ligam dentro do
subsítio S1 bem como no subsítio S1’. Por outro lado, paepalantina, por ser menor, se encaixa
quase que completamente dentro do bolso S1.
O inibidor 8,8’-paepalantina mostra uma interação C-H···π com o Cε da His41,
enquanto vioxantina e paepalantina mostram o mesmo tipo de interação com Cα da Cys191 e
Trp215 (FIG. 24). Ainda, vioxantina também apresenta duas interações C-H···π com o Cβ e
Cδ da His57, respectivamente.

| 53 P á g i n a
FIGURA 23: Posição dos inibidores de isocumarinas superfície da KLK7. Os inibidores paepalantina (A),
8,8’-paepalantina dímero (B) e vioxantina (C) ocupam a entrada do subsítio S1 da proteína e no caso da 8,8’-
paepalantina dímero e vioxantina, eles se estendem sobre os subsítios S1’. A comparação das três moléculas (D)
mostra que as formas dímero possuem posição semelhante na proteína. A superfície hidrofóbica da proteína da
proteína é mostrada com cores variando de azul (hidrofílico) a vermelho (hidrofóbico). Os átomos de hidrogênio
não polar foram omitidos para melhor.
TABELA 9: Distância (em Å) dos átomos dos inibidores de isocumarinas e da KLK7 envolvidos nas interações
de hidrogênio.
Resíduos KLK5 Paepalantina 8,8’-paepalantina Vioxantina
Átomo Distância Átomo Distância Átomo Distância
NHε2 His57 - - OHC7’ 1,67
OHC7 2,20 OHC10’ 1,73
NH(cadeia principal) Gly193 - - OC1 2,12
- - OHC10 2,36
NH(cadeia principal) Ser195 - - OC1 2,15 - -
OH Ser195 OHC9 1,89
- - OHC10 2,07 OHC10 1,97

| 54 P á g i n a
FIGURA 24: Destaque dos principais resíduos da KLK7 que formam as interações hidrofóbicas com os inibidores de isocumarinas. Os inibidores paepalantina (A), 8,8’-
paepalantina dímero (B) e vioxantina (C) formam interação hidrofóbica principalmente com His41, His57, Cys192 e Trp215.

| 55 P á g i n a
As ligações de hidrogênio são importantes para ajudar a estabilizar a ligação dos
inibidores. A paepalantina forma duas interações de hidrogênio entre os grupos hidroxila C9 e
C10 e a cadeia lateral da Ser195 (FIG. 25 A).
O carboxil C1 da 8,8’-paepalantina forma duas interações de hidrogênio com o grupo
amino da Ser195 e Gly193 na fenda de oxônio (FIG. 25 B) e, o grupo hidroxila C10 forma
uma ligação de hidrogênio também com a Gly193 (TABELA 9). Também se observa duas
interações de hidrogênio com o carboxil da cadeia principal da His57 através dos grupos
hidroxila C7’ e C10’.
A vioxantina não forma interações de hidrogênio na fenda de oxônio. Ao contrário, seus
grupos hidroxila C7 e C10 fazem duas interações de hidrogênio com a cadeia lateral da His57
e Ser195, respectivamente (FIG. 25 C). Já que na vioxantina o grupo metila em C5 é ausente,
esse inibidor se liga mais profundamente no bolso S1 do que 8,8’-paepalantia. Por causa
disso, o carboxil C1 da vioxantina muda pra fora da fenda de oxônio. Além disso, os três
anéis da vioxantina que estão no bolso S1 são um pouco retorcido, comparado com a 8,8-
paepalantina. Isso evita o grupo hidroxila C10 de formar ligações de hidrogênio com a fenda
de oxônio.
Por ser mais hidrofóbico o subsítio S1 da KLK7, principalmente pela presença dos
resíduos Ala190 e Phe218, esperava-se que os inibidores isocumarínicos fossem mais
potentes em inibir a KLK7 do que a KLK5. Porém, observou-se o contrário. Duas
propriedades moleculares podem ajudar a justificar essa observação. Primeiro, embora o
subsítio S1 seja mais hidrofóbico, ele é menos fundo que o da KLK5, acomodando de
maneira menos favorável esses inibidores. Segundo, somente a 8,8’-paepalantina teve
interação de hidrogênio com os grupos aminos da cadeia principal dos resíduos da fenda de
oxiânion, enquanto que na KLK5 as três moléculas interagiram com esses resíduos.

| 56 P á g i n a
FIGURA 25: Interação de hidrogênio dos inibidores isocumarínicos com a KLK7. Os inibidores paepalantina (A), interage somente com a SER195, a 8,8’-paepalantina
dímero (B) interage com os resíduos HIS57, GLY193 e SER195 e, a vioxantina (C) interage com a His57 e Ser195. Para melhor entendimento, somente os hidrogênios
polares (do ligante e do receptor) são mostrados.

| 57 P á g i n a
5. CONCLUSÕES E PERSPECTIVAS.
Neste trabalho efetuou-se o estudo de inibição das calicreínas teciduais humanas 5 e 7
por duas classes de compostos, sendo três moléculas de isocumarinas naturais e cinco
compostos peptídicos miméticos derivados de um esqueleto molecular estereoespecífico
comum conhecido como isomanídeo. Todas as isocumarinas e os compostos peptídicos
miméticos 9g, 9j e 9k mostraram ser inibidores competitivos e reversíveis, na concentração de
micromolar. Os compostos 9h e 9i apresentaram comportamento de inibidor não competitivo,
porém com baixa potência.
Os inibidores isocumarínicos são mais potentes na inibição da atividade da KLK5. Dos
três compostos testados, a forma dímero (8,8’-paepalantina e vioxantina) foi mais potente.
Vioxantina foi o composto mais potente (IC50=53,7µM e Ki=22,9µM) seguido da 8,8’-
paepalantina (IC50=72,9µM e Ki=24,4µM). Esses dois compostos foram também os mais
potentes para a inibição da atividade da KLK7, sendo a 8,8’-paepalantina dímero o mais
potente (IC50 = 112,4 µM e Ki=12,2µM) e vioxantina o segundo mais potente (IC50 = 135,3
µM e Ki=37,2µM).
Esperava-se que os inibidores isocumarínicos fossem mais potentes para a KLK7 do
que para a KLK5 já que a primeira apresenta uma cavidade S1 maior e menos polar. A análise
do docking molecular mostra que esses compostos se posicionam de modo semelhante em
ambas as enzimas se ligando no bolso S1 e estendendo sobre os bolsos S3/S4 na KLK5 e
sobre S1’ na KLK7, com exceção da paepalantina que se liga somente à S1. As principais
forças envolvidas na estabilização dessas moléculas são interações hidrofóbicas e de
hidrogênio. Porém, os resíduos das proteínas envolvidos nas interações são diferentes, sendo
que na KLK5 os inibidores formam ligações de hidrogênio com os resíduos da fenda de
oxiânion (grupo amino da cadeia principal da Gly193 e Ser195). O mesmo não ocorre na
KLK7. Isso pode ser uma explicação do por que esses inibidores são menos potentes com ela.
Os ensaios de inibição dos isomanídeos mostraram que o composto 9g foi o mais
potente (IC50 = 57,9 µM e Ki=15.5µM) seguido de 9j (IC50 = 85,3 µM e Ki=25,0 µM) e 9k
(IC50 = 95,7 µM e Ki= 111,1 µM). A principal diferença entre os compostos 9g e 9k é a
substituição em R1 do grupo fenil na 9g por metil na 9k, o que pode ter contribuído para a
menor potência. Comparando com as isocumarinas, podemos observar que os valores dos dois
inibidores mais potentes são semelhantes, sugerindo que interações de hidrogênio e
hidrofóbicas são as principais forças envolvidas na inibição.
A análise do docking molecular dos isomanídeos mostrou que eles têm o mesmo modo
de ligação, com o substituinte R2 no bolso S1 e a cadeia se estendendo até S2’. A interação é

| 58 P á g i n a
estabilizada por interações hidrofóbicas e interações de hidrogênio e, semelhante às
isocumarinas, os inibidores peptídico miméticos interagem com os resíduos da fenda de
oxiânion.
A principal diferença dos compostos 9h e 9i em relação aos demais se deve ao
substituinte R2, sendo o um anel de benzodioxole e piridil (FIG. 8), respectivamente. Essas
estruturas são grupos hidrofóbicos e maiores que os demais substituintes R2. Como o subsítio
S1 da KLK5 é polar, isso pode caracterizar a inabilidade desses compostos se ligarem à
enzima.
O estudo por dinâmica molecular da KLK5 mostrou que os subsítios da enzima
apresentam conformação estável, com exceção do subsítio S2 que apresentam uma grande
mobilidade. Como esse subsítio é caracterizado por participar na inibição alostérica com o
Zn2+
, postulou-se que os inibidores não competitivos (9h e 9i) se ligam a esse sítio e assim
alteram a disposição conformacional dos resíduos da tríade catalítica e assim, diminuindo a
atividade enzimática.
Todo trabalho de desenvolvimento de fármacos visa obter moléculas com elevada
eficácia, potência na faixa de nanomolar e com efeito citotóxico mínimo. Este estudo foi o
primeiro a testar essas moléculas como prováveis inibidores de KLK5 e KLK7. Os resultados
foram promissores, apresentando inibição na faixa de micromolar. Por ser um estudo de
primeira linha, consideramos algumas sugestões para nortear o desenho de moléculas mais
potentes:
Pela análise de docking molecular, observou-se que as principais interações são
hidrofóbicas e as ligações de hidrogênio. Postulamos que moléculas com um
esqueleto hidrofóbico, com liberdade conformacional e substituintes polares
aumentarão a afinidade dessas moléculas.
O subsítio S1 da KLK5 é profundo e devido à presença do resíduo de aspartato
no seu fundo, tem preferência por resíduos básicos (como arginina e lisina).
Postulamos que a afinidade dos inibidores peptídico miméticos pela KLK5
aumentará com a substituição do anel hidrofóbico tiofeno (ou furano), na
posição R2, por um substituinte com cadeia hidrofóbica longa com ponta polar
(semelhante à arginina ou lisina).
O subsítio S1 da KLK7 apresenta uma cavidade menor com borda larga e
hidrofóbica e, devido à presença do aminoácido asparagina no seu fundo, tem
preferência por tirosina. Pode-se propor que a afinidade dos inibidores peptídico
miméticos pela KLK7 aumentará com a substituição em R2 por grupos
hidrofóbicos pequenos com pontas polares (semelhante à tirosina).

| 59 P á g i n a
Como parte de um projeto de pesquisa desenvolvido no Laboratório de Química
Computacional Medicinal da UFTM, estudos de inibição das calicreínas 5 e 7 continuam
sendo efetuados pelos alunos do laboratório. Efetuar-se-á o estudo de interação e inibição de
mais duas classes de compostos peptídicos miméticos derivados de isomanídeos, na qual se
observará a relação estrutura-atividade (QSAR) e o mecanismo de interação com as enzimas.
As moléculas de isocumarinas apresentadas neste trabalho e outras variações dessas
moléculas, como a presença de glicosilação, serão objetos de estudo quântico para observar o
potencial antioxidante, pKa, orbitais moleculares e diversas outras propriedades dessas
moléculas, para subsidiar a construção de um modelo farmacofórico de suas possíveis
atividades biológicas.

| 60 P á g i n a
6. REFERÊNCIAS.
Asokananthan N, Graham PT, Fink J, Knight DA, Bakker AJ, McWilliam AS, Thompson PJ, Stewart
GA. Activation of protease-activated receptor (PAR)-1, PAR-2, and PAR-4 stimulates IL-6, IL-8,
and prostaglandin E2 release from human respiratory epithelial cells. J Immunol 2002;168:3577–
3585.
Barros TG, Pinheiro S, Williamson JS, Tanuri A, Pereira HS, R. Brindeiro M, Neto JBA, Antunes
OAC, Muri EMF. Novel Peptide Mimetic Inhibitors of Hepatitis C Serine Protease Derived from
Isomannide. Synthesis. 2009;4:620-626.
Bazan JF, Fletterick RJ. Detection of a trypsin-like serine protease domain in flaviviruses and
pestiviruses, Virology 1989a;171:637-639.
Bazan JF, Fletterick RJ. Viral cysteine proteases are homologous to the trypsin-like family of serine
proteases: Structural and functional implications, PNAS 1989b;85:7872-7876.
Belanger A, Van Halbeek H, Graves HCB, Grandbois K, Stamey TA, Huang L, Poppe I, Labrie F.
Molecular mass and carbohydrate structure of prostate specific antigen: Studies for establishment
of an international PSA standard. Prostate 1995;27:187-197.
Bencsik JR, Kercher T, O’Sullivan M, Josey JA. Efficient, stereoselective synthesis of oxazolo[3,2-
a]pyrazin-5-ones: Novel bicyclic lactam scaffolds from the bicyclocondensation of 3-aza-1,5-
ketoacids and amino alcohols, Org. Lett. 2003;5:2727-2730.
Berendsen HJC, van der Spoel D, van Drunen R. GROMACS: a message-passing parallel molecular
dynamics implementation. Comput. Phys. Commun. 1995;91:43–56.
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The
Protein Data Bank, Nucleic Acids Res. 2000;28:235– 242.
Bernett MJ, Blaber SI, Scarisbrick IA, Dhanarajan P, Thompson SM, Blaber M. Crystal structure and
biochemical characterizarion of human kallikrein 6 reveals that a trypsin-like kallikrein is
expressed in the central nervous system. J. Biol. Chem. 2002;277:24562-24570
Bleicher KH, Böhm HJ, Müller K, Alanine AI. Hit and lead generation: Beyond high-throughput
screening. Nat. Rev. Drug Disc. 2003;2:369–378.
Blow DM. The tortuous story of Asp...His...Ser: Structural analysis of α-chymotrypsin. Trends
Biochem. Sci. 1997;22:405-408.
Bode W, Meyer EJr, Powers JC. Human leukocyte and porcine pancreatic elastase: X-ray crystal
structures, mechanism, substrate specificity, and mechanism-based inhibitors. Biochem.
1989;28:1951-1963.
Bode W, Turk D, Karshikov A. The refined 1.9-Å -ray crystal structure of D-Phe-Pro-Arg
chloromethylketone-inhibited human α-thrombin: Structure analysis, overall structure, electrostatic
properties, detailed active-site geometry, and structure-function relationships Protein Sci.
1992;1:426- 471.
Borgoño CA, Michael IP, Diamandis EP. Human Tissue Kallikreins: Physiologic Roles and
Applications in Cancer. Mol. Cancer Res. 2004;2:257-280.
Braff MH, Zaiou M, Fierer J, Nizet V, Gallo RL. Keratinocyte production of cathelicidin provides
direct activity against bacterial skin pathogens. Infect. Immun. 2005;73:6771–6781.
Brattsand M, Egelrud T. Purification, molecular cloning, and expression of a human stratum corneum
trypsin-like serine protease with possible function in desquamation. J. Biol. Chem.
1999;274:30033-30040.
Burke KP, Cox AL Hepatitis C virus evasion of adaptive immune responses: A model for viral
persistence. Immunol Res 2010;47:216-27
Cañedo LM, Puents JLF, Baz JP. A new isocoumarin antitumor agent produced by a marine bacteriu.
J. Antibiot. 1997;50:175-176.

| 61 P á g i n a
Caubet C, Jonca N, Brattsand M, Guerrin M, Bernard D, Schmidt R, Egelrud T, Simon M, Serre G.
Degradation of Corneodesmosome Proteins by Two Serine Proteases of the Kallikrein Family,
SCTE/KLK5/hK5 and SCCE/KLK7/hK7. J. Invest. Dermatol. 2004;122:1235–1244.
Chapman SJ, Walsh A, Jackson SM, Friedmann PS. Lipids, proteins and corneocyte adhesion. Arch
Dermatol Res. 1991;283:167–173.
Chapman SJ, Walsh A. Desmosomes, corneosomes and desquamation. An ultrastructural study of
adult pig epidermis. Arch Dermatol Res. 1990;282:304–310.
Coelho RG, Vilegas W, Devienne KF, Raddi MSG. A new cytotoxic naphthopyrone dimer from
Paepalanthus bromelioides, Fitoterapia 2000;71:497-500.
Cross JB, Thompson DC, Rai BK, Baber JC, Fan KY, Hu Y, Humblet C. Comparison of Several
Molecular Docking Programs: Pose Prediction and Virtual Screening Accuracy. J. Chem. Inf.
Model. 2009;49:1455–1474
Debela M, Goettig P, Magdolen V, Huber R, Schechter NM, Bode W. Structural Basis of the Zinc
Inhibition of Human Tissue Kallikrein 5. J. Mol. Biol. 2007a;373:1017–1031.
Debela M, Hess P, Magdolen V, Schechter NM, Steiner T, Huber R, Bode W, Goettig P.
Chymotryptic specificity determinants in the 1.0 Å structure of the zinc-inhibited human tissue
kallikrein 7. PNAS. 2007b;104:16086-16091.
Debela M, Magdolen V, Schechter N, Valachova M, Lottspeich F, Craik CS, Choe Y, Bode W,
Goettig P. Specificity Profiling of Seven Human Tissue Kallikreins Reveals Individual Subsite
Preferences, J. Biol. Chem. 2006;281:25678–25688.
Debela M. Crystal Structures of the Human Tissue Kallikreins 4, 5, 7, 10, Characterisation of Their
Substrate Specificity and Analysis of Their Various Zinc Inhibition Mechanisms. Max-Planck-
Institute of Biochemistry, TU Munich, Munich, 2007c, p. 191.
Deraison C, Bonnart C, Lopez F, Besson C, Robinson R, Jayakumar A, Wagberg F, Brattsand M,
Hachem JP, Leonardsson G, Hovnanian A. LEKTI fragments specifically inhibit KLK5, KLK7,
and KLK14 and control desquamation through a pH-dependent interaction. Mol. Biol.Cell.
2007;18:3607–3619.
Descargues P, Deraison C, Bonnart C, Kreft M, Kishibe M, Ishida-Yamamoto A, Elias P, Barrandon
Y, Zambruno G, Sonnenberg A, Hovnanian A. Spink5-deficient mice mimic Netherton syndrome
through degradation of desmoglein 1 by epidermal protease hyperactivity. Nat. Genet. 2005;37:56–
65.
Devienne KF, Cálgaro-Helena AF, Dorta DJ, Prado IMR, Raddi MSG, Vilegas W, Uyemura SA,
Santos AC, Curti C. Antioxidant activity of isocoumarins isolated from Paepalanthus bromelioides
on mitochondria. Phytochemistry. 2007;68:1075-1080.
Devienne KF, Raddi MSG, Coelho RG, Vilegas W. Structure–antimicrobial activity of some natural
isocoumarins and their analogues. Phytomedicine. 2005;12:378-381.
Devienne KF, Raddi MSG, Varanda EA, Vilegas W. In vitro cytotoxicity of some natural and semi-
synthetic isocoumarins from Paepalanthus bromelioides. Zeitschrift für Naturforschung.
2002;57c:85-88.
Di Stasi LC, Camuesco D, Nieto A, Vilegas W, Zarzuelo A, Galvez J. Intestinal anti-inflammatory
activity of paepalantine, an isocoumarin isolated from the capitula of Paepalanthus bromelioides in
the trinitrobenzenesulphonic acid model of rat colitis. Planta Med. 2004;70:315-320.
Diamandis EP, Yousef GM, Clements J, Ashworth LK, Yoshida S, Egelrud T, Nelson PS, Shiosaka S,
Little S, Lilja H, Stenman U-H, Rittenhouse HG, Wain H. New nomenclature for the human tissue
kallikrein gene family. Clinical Chem. 2000b;46:1855-1858.
Dietrich E, Lubell WD. Efficient synthesis of enantiopure pyrrolizidinone amino acid, J. Org. Chem.
2003;68:6988-6996.
Egelrud T. Purification and preliminary characterization of stratum corneum chymotryptic enzyme: a
proteinase that may be involved in desquamation. J. Invest. Dermatol. 1993;101:200-4.

| 62 P á g i n a
Ekholm E, Egelrud T. Stratum corneum chymotryptic enzyme in psoriasis. Arch Dermatol Res 1999;
291:195–200.
Fartasch M, Bassukas ID, Diepgen TL. Structural relationship between epidermal lipid lamellae,
lamellar bodies and desmosomes in human epidermis: An ultrastructural study. Br J Dermatol.
1993;128:1–9.
Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH,
Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. Glide: A new approach for rapid, accurate
docking and scoring. 1. Method and assessment of docking accuracy, J. Med. Chem.
2004;47:1739– 1749.
Frungieri MB, Weidinger S, Meineke V, Kohn FM, Mayerhofer A. Proliferative action of mast-cell
tryptase is mediated by PAR2, COX2, prostaglandins, and PPARgamma: Possible relevance to
human fibrotic disorders. Proc Natl Acad Sci USA 2002;99:15072–15077.
Goettig P, Magdolen V, Brandstetter H. Natural and synthetic inhibitors of kallikrein-related
peptidades (KLKs). Biochimie. 2010;92:1546-1567.
Gorbalenya AE, Donchenko AP, Koonin EV, Blinov VM. N-terminal domains of putative helicases of
flavi- and pestiviruses may be serine proteases, Nucleic Acids Res. 1989;17;3889-3897.
Grebely J, Thomas DL, Dore GJ. HCV reinfection studies and the door to vaccine development. J
Hepatol 2009;51:628-31
Harper JW, Hemmi K, Powers JC. Reaction of serine proteases with substituted isocoumarins –
discovery of 3,4-dichloroisocoumarin, a new general mechanism serine protease inhibitor,
Biochemistry 1985;24:1831-1841.
Hartley BS, Amino-acid sequence of bovine chymotrypsinogen-A. Nature. 1964;201:1284-1287.
Hedstron L. Serine Protease Mechanism and Specificity. Chem. Rev. 2002;102:4501 4523
Hill RA. Naturally occurring isocoumarins, Fortschritte der Chemie Organischer 1986;49:1–78.
Hruby VJ. Designing peptide receptor agonists and antagonists. Nature Rev. Drug Discov. 2002;1:
847-858.
Iwata A, Maruyama M, Akagi T, Hashikawa T, Kanazawa I, Tsuji S, Nukina N. Alpha-synuclein
degradation by serine protease neurosin: Implication for pathogenesis of synucleinopathies. Hum.
Mol. Genet.2003;12:2625–2635
Johnson SK, Ramani VC, Hennings L, Haun RS. Kallikrein 7 Enhances Pancreatic Cancer Cell
Invasion by Shedding E-cadherin. CANCER. 2007;109:1811-1820.
Jorgensen WL, Maxwell DS, Tirado-Rives J. Developmentand testing of the OPLS all-atom force
field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc.
1996;118:11225-11236.
Kapadia C, Chang A, Sotiropoulou G, Yousef GM, Grass L, Soosaipillai A, Xing X, Howarth DHC,
Diamandis EP. Human kallikrein 13: production and purification of recombinant protein and
monoclonal and polyclonal antibodies, and development of a sensitive and specific
immunofluorometric assay. Clin Chem 2003;49:77-86.
Kihampa C, Nkunya MH, Joseph CC, Magesa SM, Hassanali A, Heydenreich M, Kleinpeter E. Anti-
mosquito and antimicrobial nor-halimanoids, isocoumarins and an anilinoid from Tessmannia
densiflora. Phytochemistry 2009;70:1233–1238.
Komatsu N, Saijoh K, Otsuki N, Kishi T, Micheal IP, Obiezu CV, Borgono CA, Takehara K,
Jayakumar A, Wu HK, Clayman GL, Diamandis EP. Proteolytic processing of human growth
hormone by multiple tissue kallikreins and regulation by the serine protease inhibitor Kazal-Type5
(SPINK5) protein. Clin. Chim. Acta. 2007;377:228–236.
Komatsu N, Takata M, Otsuki N, Ohka R, Amano O, Takehara K, Saijoh K. Elevated stratum
corneum hydrolytic activity in Netherton syndrome suggests an inhibitory regulation of
desquamation by SPINK5-derived peptides. J. Invest. Dermatol.2002; 118: 436–443.

| 63 P á g i n a
Kong W, McConalogue K, Khitin LM, Hollenberg MD, Payan DG, Bohm SK, Bunnett NW. Luminal
trypsin may regulate enterocytes through proteinase- activated receptor 2. Proc Natl Acad Sci USA
1997;94:8884–8889.
Kostova I. Synthetic and natural coumarins as cytotoxic agents. Curr. Med. Chem. 2005;5:29-46.
Kubota Y, Nakahara T, Mitani A, Maruko T, Saito M, Sakamoto K, Ishii K. Possible involvement of
Ca(2+)-independent phospholipase A(2) in proteaseactivated receptor-2-mediated contraction of rat
urinary bladder. Naunyn Schmiedebergs Arch Pharmacol 2003;367:588–591.
Leach RA. Ligand-Based Approaches: Core Molecular Modeling. In:Taylor JB e Triggle DJ.
Comprehensive Medicinal Chmistry II. Volume 4: Computer assisted Drug Design.
Elsevier;2006. 88-118.
Leatherbarrow RJ. (2009) GraFit Version 7, Erithacus Software Ltd., Horley, U.K.
Lindahl E, Hess B, van der Spoel D. GROMACS 3.0: a package for molecular simulation and
trajectory analysis. J. Mol. Model.2001;7:306–317.
Liu K, Li QZ, Delgado-Vega AM, Abelson AK, Sánchez E, Kelly JA, Li L, Liu Y, Zhou J, Yan M, Ye
Q, Liu S, Xie C, Zhou XJ, Liu XL, Wazer DE, Watanabe K, Band V. Identification of a novel
serine protease-like gene, the expression of which is down-regulated during breast cancer
progression. Cancer Res. 1996;56:3371–3379.
Lovgren J, Rajakoski K, Karp M, Lundwall A, Lilja H. Activation of the zymogen form of prostate-
specific antigen by human glandular kallikrein 2. Biochem. Biophys. Res. Commun.
1997;238:549–555.
Lu HS, Lin F-K, Chao L, Chao J, Human urinary kallikrein. Complete amino acid sequence and sites
of glycosylation. International J. of Peptide and Protein Res. 1989;33:237-249.
Lundwall Å, Brattsand M, Kallikrein-related peptidases, Review, Cell. Mol. Life Sci. 2008;65:2019-
2038
Magert HJ, Kreutzmann P, Standker L, Walden M, Drogemuller K, Forssmann WG. LEKTI: a
multidomain serine proteinase inhibitor with pathophysiological relevance. Int. J. Biochem. Cell
Biol. 2002;34:573–576.
Magklara A, Mellati AA, Wasney GA, Little SP, Sotiropoulou G, Becker GW, Diamandis EP.
Characterization of the enzymatic activity of human kallikrein 6: autoactivation, substrate
specificity, and regulation by inhibitors. Biochem. Biophys. Res. Commun. 2003;307:948–955.
Malm J, Hellman J, Hogg P, Lilja H. Enzymatic action of prostate-specific antigen (PSA or hK3):
substrate specificity and regulation by Zn2+
, a tight-binding inhibitor. Prostate. 2000;45:132–139.
Marshall RD. The nature and metabolism of the carbohydrate-peptide linkages of glycoproteins.
Biochem. Soc. Symposia. 1974;40:17-26.
Michael IP, Sotiropoulou G, Pampalakis G, Magklara A, Ghosh M, Wasney G, Diamandis EP.
Biochemical and Enzymatic Characterization of Human Kallikrein 5 (hK5), a Novel Serine
Protease Potentially Involved in Cancer Progression. J. Biol. Chen. 2005;280:14628–14635.
Mikolajczyk SD, Millar LS, Marker KM, Grauer LS, Goel A, Cass MMJ, Kumar A, Saedi MS.
Ala217 is important for the catalytic function and autoactivation of prostate-specific human
kallikrein 2. Eur. J. Biochem. 1997;246:440–446.
Moreau ME, Garbacki N, Molinaro G, Brown NJ, Marceau F, Adam A. The Kallikrein-Kinin System:
Current and Future Pharmacological Targets. J. Pharmacol. Sci. 2005;99:6 – 38.
Newman DJ, Cragg GM, Kingston DGI. Natural Products as Pharmaceuticals and Soucers for Lead
Structures. In: Wermuth CG. The Practice of Medicinal Chemistry. Elsevier;2008.159-186.
Nylander LE, Egelrud T. Formation of active IL-1b from pro- IL-1b catalyzed by stratum corneum
chymotryptic enzyme in vitro. Acta Derm Venereol 1997;77:203–206
Okamoto T, Kobayashi T, Yoshida S. Chemical aspects of coumarin compounds for the prevention of
hepatocellular carcinomas, Curr. Mel. Chem. 2005;5:47-51.

| 64 P á g i n a
Oikonomopoulou K, Hansen KK, Saifeddine M, Tea I, Blaber M, Blaber SI, Scarisbrick I, Andrade-
Gordon P, Cottrell GS, Bunnett NW, Diamandis EP, Hollenberg MD. Proteinase-activated
receptors, targets for kallikrein signaling. J. Biol. Chem. 2006;281: 32095–32112.
Perona JJ, Craik CS. Structural basis of substrate specificity in the serine proteases. Protein
Sci.1995;4:337-60.
Petraki CD, Karavana VN, Diamandis EP. Human kallikrein 13 expression in normal tissues: an
immunohistochemical study. J. Histochem. Cytochem. 2003a;51:493–501.
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF
Chimera - a visualization system for exploratory research and analysis. J Comput
Chem. 2004;25:1605-12.
Schrödinger; Schrdinger LLC: New York, 2005; http://www.schrodinger.com
Severino RP, Guido RCV, Brömme D, Andricopulo AD, Vieira PC. Acridone alkaloids as potent
inhibitors of cathepsin V. Bioorg. Med. Chem. 2011;19:1477-1481.
Sinnokrot MO, Sherrill CD. Unexpected substituent effects in face-to-face π-stacking interactions. J
Chem Phys Chem A. 2003;107:8377-8379.
Sottrup-Jensen L, Sand O, Kristensen L, Fey GH. The α-macroglobulin bait region. Sequence
diversity and localization of cleavage sites for proteinases in five mammalian α-macroglobulins) J.
Biol.Chem. 1989;264:15781–15789.
Stroud RM, Finer-Moore J, Computational and Structural approach to drug discovery. Ligand-Protein
Interactions. University of California in San Francisco, San Francisco, CA, USA, 2008, p. 13.
Sun JD, Linden KG. Netherton syndrome: A case report and review of the literature. International J.
Dermatol. 2006; 45: 693-697.
Tabopda TK, Fotso GW, Ngoupayo J, Mitaine-Offer AC, Ngadjui BT, Lacaille-Dubois MA.
Antimicrobial dihydroisocoumarins from Crassocephalum biafrae. Planta Med. 2009;75:1258–
1261.
Takada Y, Skidgel RA, Erdos EG. Purification of human urinary prokallikrein. Identification of the
site of activation by the metalloproteinase thermolysin. Biochem. 1985;232:851–858.
Takayama TK, Fujikawa K, Davie EW. Characterization of the precursor of prostate-specific antigen.
Activation by trypsin and by human glandular kallikrein. J. Biol. Chem. 1997;272,21582–21588.
Takayama TK, McMullen BA, Nelson PS, Matsumura M, Fujikawa K. Characterization of hK4
(prostase), a prostate-specific serine protease: activation of the precursor of prostate specific
antigen (pro-PSA) and single-chain urokinase-type plasminogen activator and degradation of
prostatic acid phosphatase. Biochem. 2001;40:15341–15348.
Tamura H, Ishikawa Y, Hino N, Maeda M, Yoshida S, Kaku S, Shiosaka S. Neuropsin is essential for
early processes of memory acquisition and Schaffer collateral longterm potentiation in adult mouse
hippocampus in vivo. J. Physiol.2006;570:541–551.
van de Waterbeemd H, Gifford E. ADMET in silico modelling: Towards prediction paradise? Nat.
Rev. Drug Disc. 2003;2:192–204.
Varanda EA, Devienne KF, Raddi MS, Furuya EM, Vilegas W. Mutagenicity of paepalantine dimer
and glycoside derivatives from Paepalanthus bromelioides, Toxicol. In Vitro 2004;18:109-114.
Vilegas W, Roque NF, Salatino A, Giesbrecht AM, Davino S. Isocoumarin from Paepalanthus
bromelioides. Phytochemistry. 1990;29:2299-2301.
Whyte AC, Gloer JB, Scott JA, Malloch D. Cercophorins A-C: novel antifungal and cytotoxic
metabolites from the coprophilous fungus Cercophora areolata. J. Nat. Prod. 1996; 59:765–769.
Wright JT, Daly B, Simmons D, Hong S, Hart SP, Hart TC, Atsawasuwan P, Yamauchi M. Human
enamel phenotype associated with amelogenesis imperfecta and a kallikrein-4 (g.2142G > A)
proteinase mutation. Eur. J. Oral Sci. 2006;114:13–17.

| 65 P á g i n a
Yamasaki K, Schauber J, Coda A, Lin H, Dorschner RA, Schechter NM, Bonnart C, Descargues P,
Hovnanian A, Gallo RL. Kallikrein-mediated proteolysis regulates the antimicrobial effects of
cathelicidins in skin. FASEB J. 2006;20;2068–2080.
Yousef GM, Diamandis EP. The new human tissue kallikrein gene family: structure, function, and
association to disease. Endocrine Rev. 2001;22:184-204.
Yousef GM, Kapadia C, Polymeris ME, Borgoño C, Hutchinson S, Wasney GA, Soosaipillai A,
Diamandis EP. The human kallikrein protein 5 (hK5) is enzymatically active, glycosylated and
forms complexes with two protease inhibitors in ovarian cancer fluids. Biochimica et Biophysica
Acta - Gene Structure and Expression. 2003;1628:88-96
Yousef GM, Luo LY, Scherer SW, Sotiropoulou G, Diamandis EP. Molecular characterization of
Zyme/Protease M/Neurosin (PRSS9), a hormonally regulated kallikrein-like serine protease.
Genomics, v. 62, 1999, p. 251-259.

| 66 P á g i n a
7. ANEXOS

| 67 P á g i n a

| 68 P á g i n a

| 69 P á g i n a