universidad de chilerepositorio.uchile.cl/tesis/uchile/2010/qf-cataldo_lr/... · 2012-01-06 · en...
TRANSCRIPT

UNIVERSIDAD DE CHILE FACULTAD DE CIENCIAS QUÍMICAS Y FARMACÉUTICAS PROGRAMA DE MAGISTER EN BIOQUÍMICA
PARTICIPACIÓN DE LA VÍA WNT/-CATENINA EN LA REGULACIÓN DE LA EXPRESIÓN DE LA ISOFORMA C DE LA
ENZIMA CONVERTIDORA DE ENDOTELINA-1 (ECE-1c) Y SU ROL EN EL FENOTIPO TUMORAL INVASIVO EN CÁNCER DE COLON
Tesis presentada a la Universidad de Chile en cumplimiento de los
requisitos para optar al grado de Magister en Bioquímica
Área de especialización: Bioquímica Clínica Aplicada
y
Memoria para optar al Título de Bioquímico
por
LUIS RODRIGO CATALDO BASCUÑAN
Director de Tesis: Dr. Julio Tapia Pineda
Santiago-CHILE
Agosto-2010

2
UNIVERSIDAD DE CHILE FACULTAD DE CIENCIAS QUIMICAS Y FARMACEUTICAS
INFORME DE APROBACION
TESIS DE MAGISTER
Se informa a la Comisión de Postgrado de la Facultad de Ciencias Químicas y Farmacéuticas de la Universidad de Chile que la Tesis de Magíster presentada por el candidato
LUIS RODRIGO CATALDO BASCUÑAN
ha sido aprobada por la Comisión Informante de Tesis como requisito de Tesis para el Grado de Magíster en Bioquímica, en el examen de defensa de Tesis rendido el ………………………………………………………………… Director de Tesis: - Dr. Julio Tapia Pineda ..................................................... Comisión Informante de Tesis: - Dra. Carmen Romero Osses (Presidente) ............................................ - Dr. Sergio Lavandero Gonzalez ............................................. - Dr. Miguel Allende Connelly ............................................

3
FINANCIAMIENTO
Esta tesis fue realizada en el Laboratorio de Transformación Celular, Programa de
Biología Celular y Molecular, Instituto de Ciencias Biomédicas (ICBM), Facultad de Medicina,
Universidad de Chile.
Esta investigación fue financiada completamente con recursos de un proyecto
FONDECYT DE INICIACION N° 11070116.

4
AGRADECIMIENTOS
Quiero agradecer a todos los que de alguna forma han permitido que hoy llegue al fin
de una etapa importante en mi vida. A mi Director de Tesis, Dr. Julio Tapia, quien me abrió
las puertas de su laboratorio, me dio la confianza y me orientó durante el desafío de generar
ciencia en su laboratorio. Agradezco el tiempo que me dio para compartir experiencias de
vida, discutir, entregar consejos y comprender los errores. Agradezco a todos mis
compañeros de laboratorio: Catalina, Daniela, Eduardo, Roger, José Luis, Pablo e Ignacio, con
los cuales compartí la mayor cantidad de tiempo durante el periodo de Tesis, agradezco de
ellos su colaboración, su tolerancia y su sencillez. Agradezco a todos los jefes de laboratorios
vecinos y sus estudiantes que me apoyaron permanentemente con sus equipos, reactivos y
buena disposición. Especialmente los laboratorios de los doctores: Mario Galindo, Marcelo
Antonelli, José Suazo, Julieta González, Luisa Herrera y Gonzalo Cabrera. Quiero agradecer y
destacar la colaboración de la Dra. Cristina Fernández, Jefa Técnica del Servicio de Anatomía
Patológica del Hospital Clínico de la Universidad de Chile, quien amablemente dispuso la
entrega de las biopsias de pacientes con cáncer de colon. También quiero agradecer a
personas muy valiosas y que prestan una ayuda esencial y tienen siempre una palabra de
apoyo y empuje, Don Osvaldo y especialmente Don Javier.
No hubiese llegado a la instancia del desarrollo de mi Tesis si no hubiese cursado el
ciclo académico completo durante los últimos 5 años y medio, en los cuales recibí en todo
momento el apoyo monetario para pagar en parte lo que restaba del crédito universitario,
este apoyo se lo debo muy agradecidamente a la Sra. Dorothy Hochschild, una persona con
mucha sensibilidad social.
Por supuesto que quiero agradecer a los seres más queridos que siempre se
encuentran alrededor de uno apoyándolo y dando buenos consejos y amor; estas personas
para mí son mi familia, los cuales siempre e incondicionalmente han estado cerca sobretodo
en los peores momentos para tomarme y tirarme hacia arriba, demostrando el amor genuino
y sincero, y siendo duros si es necesario para hacerme superar los obstáculos que la vida nos
hace pasar, para sin duda hacernos más grandes, para demostrarnos que las experiencias

5
dolorosas nos hacen crecer. Esta linda familia está conformada, primero, por mis padres Luis
y Marianela, de quienes admiro su tolerancia, inteligencia, paciencia y esfuerzo; mis
hermanas Elibeth, Edith y Diana, agradezco de ellas la constante preocupación, la confianza y
el maravilloso cariño que me han dado, quienes de distinta forma, y con sus diversas
personalidades, han enriquecido mi vida. Finalmente a Macarena Palominos, una mujer que
como un regalo, sorpresivamente llegó a mi vida durante la Tesis, quien me ha hecho crecer
y recuperar la alegría, la tranquilidad y la propia confianza con mucho amor. Agradezco de
todos ellos el inmutable amor, el agente carburador que me da la razón para superar
cualquier problema y me hace ir siempre adelante, buscando la verdad y el destino que me
haga feliz.

6
RESUMEN
En el cáncer colorrectal es fundamental la participación de la vía de señalización Wnt-
canónica donde se encuentra aberrantemente activada en más del 80% de los casos. Esta vía
es transversal en los seres vivos y participa en el desarrollo, morfogénesis, proliferación y
regeneración celular. También está relacionada con numerosas patologías, incluyendo la
poliposis adenomatosa familiar (FAP), en la cual pacientes portadores de una mutación
heredada en APC (Adenomatous poliposis coli), desarrollan cientos de pólipos en el colon e
inevitablemente terminan desarrollando un cáncer.
-Catenina es la proteína central en la vía de señalización Wnt-canónica, ésta es
finamente regulada mediante el complejo de degradación conformado por las proteínas de
andamiaje axina y APC y las kinasas CK1 y GSK3las que en conjunto actúan marcando a -
catenina para enviarla a degradación proteosomal. Sin embargo, en forma aberrante alguna
de las proteínas del complejo pueden alterarse, activarse o expresarse otras proteínas que
promueven la estabilidad de -catenina, como la proteína kinasa CK2 o AKT/PKB, que en
consecuencia disminuyen o evitan la degradación proteosomal de -catenina, promoviendo
que ésta se acumule en el citosol, transloque al núcleo y transactive la expresión de genes
junto con los factores de transcripción TCF/LEF.
Uno de los genes blanco de la ruta Wnt-canónica es el péptido vasoactivo endotelina-
1 (ET-1). Este ha sido estudiado por sus efectos protumorales emergiendo recientemente
como un componente importante en el origen, desarrollo y progresión del cáncer debido a su
rol en la regulación de procesos como sobrevida, angiogénesis, transición epitelio
mesenquimática (TEM), invasión y metástasis.
La activación de ET-1 es llevada a cabo por la Enzima Convertidora de Endotelina-1
(ECE-1), la que está sobreexpresada en muchos cánceres, incluyendo el de colon, y presenta
4 isoformas, siendo la mayormente expresada en cáncer la isoforma c. En particular, esta
isoforma se ha reportado que tiene un rol proinvasivo en algunos cánceres, como ovárico,
prostático y mamario. No obstante, existe escasa información acerca de la regulación de su
expresión en estos tejidos.

7
En esta tesis se ha propuesto como hipótesis que “la vía Wnt-canónica regula
positivamente la expresión de ECE-1(c) promoviendo la invasividad de células de cáncer
colorectal”.
Para responder esta hipótesis se analizó mediante western blot, los niveles de
expresión de ECE-1 en células de cáncer colorectal, observándose un aumento relacionado
con el grado de malignidad de cada línea celular. También se activó la vía Wnt-canónica con
inhibidores farmacológicos, encontrándose que los niveles de la enzima aumentaron,
mientras que éstos disminuyeron cuando la ruta fue regulada negativamente. Asimismo, se
expresó ectópicamente en células embrionarias no tumorales HEK-293T proteínas que
participan activando la ruta Wnt-canónica, producto de lo cual los niveles de expresión de
ECE-1(c) también aumentaron. Por otro lado, se determinó la capacidad invasiva de distintas
líneas celulares de CCR y se observó una relación directa con los niveles de expresión de ECE-
1. Para conocer el rol específico de ECE-1c en la invasividad, ésta se expresó ectópicamente
en células CHO-K1, observándose un notable aumento en el potencial invasivo de las células
que la sobreexpresaron. Finalmente, para darle una proyección clínica a nuestros resultados,
se determinó los niveles de mRNA de ECE-1 y ECE-1c en biopsias de cáncer colorectal de
pacientes chilenos, encontrándose niveles significativamente más altos en la porción tumoral
de éstas respecto de la no tumoral.
En consecuencia, los resultados obtenidos sugieren fuertemente que ECE-1c es un
nuevo blanco de la vía Wnt-canónica y que además se comporta como un marcador de
progresión y de mal pronóstico en el CCR.

8
ABSTRACT
Canonical Wnt signaling pathway is essential in colorectal cancer (CRC) where is
activated over 80% of cases. This pathway participates in development, morphogenesis,
proliferation and cell regeneration. It is also been implicated in many diseases, including
familial adenomatous polyposis (FAP), where patients with an inherited mutation in APC
(Adenomatous Polyposis Coli) protein develop hundred of polyps in the colon which
consequently develop cancer.
-Catenin is the key protein in the canonical Wnt signaling pathway. It is regulated by
a negative multiprotein complex formed by Axin, APC and the kinases CK1 and GSK3
which targets -catenin to proteasomal degradation. However, abnormally expressed or
aberrantly activated proteins that promote -catenin stability, including protein kinase CK2
or AKT/PKB, can reduce or even avoid proteasomal degradation of -catenin, promoting its
cytosolic accumulation, nuclear migration and transactivation of target genes together with
TCF/LEF transcription factors.
One of such target genes encodes for endothelin-1 (ET-1) peptide. This has recently
emerged as an important component in the cancer progression because its role in regulating
cellular processes such as survival, angiogenesis, epithelial-mesenchymal transition (EMT),
invasion and metastasis. Activation of ET-1 is performed by the Endothelin-Converting
Enzyme-1 (ECE-1), which is overexpressed in many cancers, including colon. This is expressed
as four isoforms (a,b,c and d), being the “c” mostly expressed in cancers, including ovary,
prostate and breast, where promotes invasiveness. However, the regulatory mechanism of
ECE-1c expression in the above-mentioned cells as well as in colon cancer is unknown.
This thesis project proposed the following hypothesis: "The canonical Wnt signaling pathway
positively regulates the ECE-1 (c) expression, thereby promoting colon cancer cell
invasiveness".
In order to answer this hypothesis, up-regulation of -catenin activity was performed
by ectopically expressing GFP--catenin, GFP-CK2CA (a constitutively active mutant), or
incubating with a GSK3 inhibitor (LiCl) in human non-tumor, normal (HEK-293T) or colon

9
cancer (HT29-ATCC, DLD-1, HT29-US) cells of increasing malignity. Alternatively, down-
regulation of -catenin activity in the same cells was achieved by expressing HA-AKT-DN (a
dominant negative mutant), silencing with a siRNA specific for CK2, or using a specific CK2
inhibitor (TBB). Levels of mRNA and protein of ECE-1(c), survivin (as an endogen control for -
catenin activity) and actin were analyzed by RT-PCR and western-blot, respectively. To
evaluate cell invasion capability, a matrigel-based invasion chamber was used in CHO-K1 cells
transiently expressing ECE-1c. Finally, in order to demonstrate the clinical relevance of the
project, we used human samples to evaluate the alterations in mRNA levels of ECE-1(c) in
biopsies from Chilean colon cancer patients.
Our results showed that protein levels of ECE-1 positively correlated with the
malignant potential of colon cancer cell lines. Using HEK-293T cells, regulation of the Wnt/-
catenin signaling pathway significantly altered the mRNA and protein levels of ECE-1(c).
Additionally, ectopic expression of ECE-1c in CHO-K1 cells led to an increased cell invasion.
Finally, mRNA levels of ECE-1 and ECE-1c in biopsies of Chilean colon cancer patients showed
significantly higher levels in the tumor portion than the non-tumor control.
Therefore, the results obtained in this thesis strongly suggest that ECE-1c is a novel canonical
Wnt signaling pathway-dependent target as well as a tumor progression and poor-prognosis
marker in colon cancer.

10
LISTADO DE ABREVIATURAS
APC Adenomatous Poliposis Coli
Big-ET-1 Big Endotelina 1
CCR Cáncer Colorectal
CK1 Caseína Kinasa 1 subunidad alfa
CK2 Caseína Kinasa 2 subunidad alfa
COX-2 Ciclooxigenasa-2
DAG Diacilglicerol
ECA Enzima Convertidora de Angiotensina
ECE-1(c) Enzima Convertidora de Endotelina 1 isoforma c
ET-1 Endotelina 1
ETaR Receptor de Endotelina tipo a
ETbR Receptor de Endotelina tipo b
FAP Poliposis Adenomatosa Familiar
GSK3 Glicogeno sintetasa kinasa 3 beta
HNPCC Cáncer colorectal hereditario no polipósico
IP3 Inositol Trifosfato
MLS D-MEM Libre de Suero
MMPs Matriz-MetaloProteinasas
NEP Neprylisina
NES Secuencia de Exportación Nuclear
NLS Secuencia de localización Nuclear
SFB Suero Fetal Bovino
TBB 4,5,6,7-tetrabromobenzotriazole
TEM Transición Epitelio Mesenquimática
VEGF Factor de Crecimiento de Endotelio Vascular

11
TABLA DE CONTENIDOS
Financiamiento 3
Agradecimientos 4
Resumen 6
Abstract 8
Listado de Abreviaturas 10
Tabla de Contenidos 11
Introducción 12
Epidemiologia del cáncer colorectal
La ruta de señalización Wnt/-catenina y el cáncer colorectal
El Eje Endotelina y su relación con cáncer colorectal
La Enzima Convertidora de Endotelina-1 (ECE-1) y su relación con cáncer colorectal
Hipótesis 28
Objetivos 28
Objetivo General
Objetivos Específicos
Materiales y Métodos 29
Resultados 35
Discusión 50
Conclusiones 58
Modelo Propuesto 59
Referencias 60

12
INTRODUCCIÓN
Epidemiología del cáncer colorectal
El cáncer colorectal (CCR) es una enfermedad de creciente importancia debido a su
incidencia y alta mortalidad. Este cáncer es una enfermedad de países desarrollados y, de
acuerdo con esto, la mayoría de los registros con las más altas tasas de incidencia
corresponden a países de Europa, Norteamérica y Oceanía. Por otro lado, las tasas más bajas
son obtenidas en países de Asia, África y Sudamérica. Sin embargo, en los países que tienen
un nivel de desarrollo elevado, las tasas de incidencia y mortalidad han tendido a
estabilizarse o disminuir. En contraste, las naciones que están logrando estándares de países
desarrollados, lo que va de la mano de una occidentalización, han generado un aumento
sostenido en las tasas de incidencia y mortalidad en las últimas décadas [1].
En el mundo, cada año se diagnostica un millón de nuevos casos [2]. De acuerdo a los
datos más recientemente disponibles por la agencia internacional para la investigación en
cáncer (IARC), el CCR en el hombre es el cuarto cáncer más común a nivel mundial, mientras
que el tercero en la mujer. Las tasas de incidencia por cada 100.000 habitantes varían
marcadamente alrededor del mundo, con valores en hombre que van desde 4,1 en India a
59,1 en República Checa, y en la mujer desde 3,6 en India a 39,5 en Nueva Zelandia [1]. En
Chile, esta patología aumentó sostenidamente con una tasa de un 1.18% anual entre 1985 y
2005, siendo actualmente igual a 13 por cada 100.000 habitantes al año [1].
En cuanto a las tasas de muerte, éstas han estado disminuyendo lentamente en
aquellos países desarrollados que durante mucho tiempo han tenido las más altas tasas de
incidencia [1]. Algunos de estos países son EE.UU., Australia, Nueva Zelandia e Inglaterra.
Estas disminuciones en las tasas de mortalidad pueden ser reflejo de mejores tratamientos
que aumentan la sobrevida o posiblemente una detección más temprana debida a un
seguimiento oportuno o buen reconocimiento de los síntomas. Por el contrario, existen
países que muestran una tendencia al alza en las tasas de mortalidad. Lamentablemente
entre estos países se encuentran la mayoría de los países latinoamericanos, y entre ellos
Chile, donde el CCR representa la tercera causa de muerte por cáncer [1].

13
Existen también síndromes asociados a una mayor prevalencia en la formación del
CCR, los cuales producen en edades tempranas una gran cantidad de pólipos en el intestino.
Estos síndromes son los conocidos como poliposis adenomatosa familiar (FAP, por sus siglas
en inglés) y cáncer colorrectal hereditario no polipósico (HNPCC, o también llamado
Síndrome de Lynch) [3]. También se conocen ciertos grupos expuestos a una prevalencia
aumentada de este cáncer, como aquellos que tienen la enfermedad celíaca, cáncer de
mama, fumadores y obesos [4].
Diagnóstico, pronóstico y tratamiento del cáncer colorectal
Se ha observado que el adenocarcinoma de colon progresa lentamente y los síntomas
se vuelven más evidentes recién sobre los 5 años de aparición. Estos pueden incluir
sangramiento rectal, fatiga, anemia, falta de aliento, cambio en los hábitos intestinales,
molestias abdominales y obstrucción intestinal [5, 6]. La edad promedio de diagnóstico de
CCR es de 70 años en hombre y 71 años en mujer [7]. En el caso de pacientes con HNPCC o
FAP, en la mayoría de los casos se diagnostica antes de los 45 años y en estados avanzados
de la tumorigénesis [3, 6].
Por mucho tiempo se ha usado el examen de sangre oculta en heces, para el
seguimiento del CCR, pero el análisis clásico es la colonoscopía y, de hecho, actualmente se
recomienda hacer este análisis sobre los 50 años en pacientes con ausencia de factores de
riesgo, tales como algún familiar directo, ya sea con CCR o alguno de los síndromes de
poliposis familiar (FAP o HNPCC), o poseer alguna enfermedad ulcerativa o colitis crónica
durante un período mayor de 8 años. En caso negativo, se debe repetir la colonoscopía cada
10 años [4].
En cuanto al pronóstico, los dos factores más importantes en CCR son la profundidad
en la pared del intestino y la cantidad de linfonodos invadidos, con una mortalidad dentro de
los 5 años postcirugía entre un 60% y un 70% en pacientes con linfonodo positivo, aunque
casi el 30% de los pacientes nodo-negativo recaen y mueren de enfermedad diseminada [8].
El estadío del CCR entrega información pronóstica de estos factores. La clasificación
de Duke es simple, fácil de interpretar y ha sido ampliamente usada como uno de los más

14
valiosos indicadores pronósticos, además de que muestra una clara correlación entre la
etapa tumoral y la sobrevida. Se clasifica en categorías de acuerdo a su diseminación: Duke A,
en los cuales el carcinoma se limita a la pared del colon y/o recto, no extendiendose a ningún
tejido extrarrectal ni presentando metástasis en linfonodos; Duke B, donde el carcinoma se
ha diseminado por continuidad directa al tejido extrarectal pero aún no ha invadido los
linfonodos regionales; Duke C, aquellos en los que además se ha invadido los linfonodos
regionales; y Duke D, en los cuales el CCR ha diseminado, generando metástasis en sitios
distantes como hígado, pulmón o hueso [9].
El tratamiento principal para el CCR sigue siendo la cirugía, la que limpia la carga
tumoral, evita complicaciones del intestino (obstrucción, perforación, etc.) y al mismo tiempo
provee de información patológica. Asimismo, el principal fármaco antineoplásico usado en el
CCR es el 5-fluorouracilo (5-FU), el cual produce respuestas positivas que van entre un 8% y
un 85%, mientras que los tratamientos quimioterapéuticos habitualmente usados en esta
patología son FOLFOX-4 (oxaliplatino, fluorouracilo y acido folínico) ó LV5FU2 (fluorouracilo y
acido folínico) [7, 10].
Se ha encontrado que al momento del diagnóstico el 25% de los pacientes presentan
invasividad o metástasis en otros tejidos distantes. Peor aún, de aquellos pacientes
diagnosticados en etapas de CCR localizado, el 50% presentará posteriormente la
metastización del tumor [11]. Adicionalmente, el 50% de aquellos pacientes sometidos a
resección quirúrgica muere dentro de los 5 años por metástasis [8]. Por estos negativos
resultados en el manejo del CCR, la prevención y detección precoz se hacen esenciales. Lo
anterior lleva a plantear la necesidad de desarrollar nuevos y mejores marcadores
moleculares que permitan el diagnóstico, pronóstico y, eventualmente, el tratamiento del
CCR, de preferencia en etapas tempranas de la neoplasia.
La ruta de señalización Wnt/-catenina y el cáncer colorectal
Las células pueden regular a sus vecinas a través de la acción paracrina de factores de
crecimiento y de moléculas de adhesión. Existen muchos ejemplos de estas interacciones,
siendo una de las más conocidas la que involucra a los morfógenos Wnt [12]. Originalmente

15
descubiertos en Drosophila, estas proteínas han sido objeto de intenso estudio por su
relación con muchas enfermedades y especialmente el cáncer [12]. La función de estos
ligandos Wnt se lleva a cabo a través de dos tipos de ruta, no-canónicas y canónica. Las rutas
no-canónicas incluyen a su vez dos variantes, la de “polaridad celular planar”, que es
importante en el desarrollo del eje embrionario a través del citoesqueleto, y la “ruta del
calcio”, la cual regula la adhesión celular [12]. En cambio, la ruta Wnt-canónica regula la
adhesión celular, morfología, proliferación, migración y remodelamiento celular, es decir,
eventos claves para el desarrollo de un cáncer [12]. Los ligandos que activan la vía Wnt-
canónica incluyen wnt1, wnt3A, wnt8 y wnt8B [12]. Todas estas proteínas se unen a un
receptor de transmembrana acoplado a una proteína G, llamado Frizzled, cuya función
además está asociada a un correceptor denominado LRP5/6 (proteína relacionada con el
receptor de LDL-5/6) [13].
La vía de señalización Wnt-canónica es altamente conservada a lo largo de todo el
reino animal. El efector clave de esta vía es la proteína -catenina puesto que cumple un rol
fundamental como transactivador de la transcripción de genes relacionados con progresión y
viabilidad celular [14]. Es por lo anterior que a esta ruta se le conoce también como ruta de
señalización Wnt/-catenina (figura 1).
-Catenina fue inicialmente descubierta como un componente de las uniones
adherentes, promoviendo la asociación entre células, interactuando con el dominio
intracelular de la proteína de transmembrana E-cadherina (la que forma uniones homotípicas
dependientes de calcio) y haciendo de puente con el citoesqueleto de actina mediante la
unión con -catenina [15]. Por otro lado, la función señalizadora de -catenina es conferida
por una fracción citoplasmática la cual es altamente inestable en ausencia de una señal Wnt,
debido a que es fosforilada en múltiples sitios en su extremo N-terminal y como
consecuencia degradada proteosomalmente [16]. La eliminación de -catenina depende de
la acción combinada del supresor de tumores APC (Adenomatous Poliposis Coli, en inglés), la
proteína de anclaje Axina y dos serina/treonina kinasas, la glicógeno sintetasa kinasa 3
(GSK3) y la caseína kinasa 1 (CK1). Cuando aparece una señal Wnt, la fosforilación por las
kinasas CK1 y GSK3es inhibida y como consecuencia -catenina no es degradada,

16
estabilizándose y migrando al núcleo, donde promueve la transcripción de genes blanco tras
unirse a los factores de transcripción de la familia Tcf/Lef [16]. Existe un extenso listado de
genes que poseen sitios de unión para Tcf/Lef en sus promotores y, en consecuencia, son
regulados por la vía Wnt-canónica y además responsables de modular el desarrollo y
progresión del CCR. Entre ellos se puede mencionar a c-myc, mmp7, survivina, vegf y cox-2
[17-22].
Fig. 1. Componentes de la ruta Wnt-canónica. A la izquierda se muestra la ruta inactiva, mientras que a la
derecha la unión de un ligando Wnt gatilla la estabilización y migración de -catenina al núcleo. Obtenido de referencia [23].
El supresor de tumores APC tiene un rol crítico en promover la rápida degradación de
-catenina y su fosforilación por GSK3 [24]. Además se sabe que -catenina no posee
secuencias de localización nuclear (NLS) ni de exportación nuclear (NES), mientras que APC sí
las posee y se ha mostrado que puede inhibir la función transactivadora de -catenina a
través de su unión con ésta en el núcleo y posterior exportación hacia el citoplasma [25].
Asimismo, se ha demostrado que una región de APC típicamente deletada en tumores es
importante en la degradación de -catenina [26].

17
Sin embargo, el sistema es mucho más complejo porque, luego de estabilizarse, -
catenina requiere entrar al núcleo y mantenerse en este compartimiento a través de la
interacción con otras proteínas, siendo de esta manera su función transactivadora
comandada por interacciones específicas, algunas de las cuales la activan mientras que otras
la reprimen [27].
Existen a la fecha muchos antecedentes apoyando la idea de que la desregulación de
la vía Wnt-canónica puede llevar, a través de la expresión de numerosos genes, al descontrol
celular y que es un factor determinante en varias patologías, especialmente en cáncer, donde
se encuentra sobreactivada. Además, un indicador de la activación de la vía Wnt-canónica es
la localización nuclear de -catenina, lo cual ha sido incluso utilizado para estudiar el origen,
desarrollo y progresión de varios tumores. De hecho, en cáncer de mamas -catenina
funciona como un marcador de mal pronóstico [28], mientras que en el epitelio intestinal la
activación inapropiada de -catenina lleva frecuentemente al desarrollo de CCR,
encontrándose que en un 80% de los casos ocurre por alteraciones de la vía Wnt-canónica
[29].
El Eje Endotelina y su relación con cáncer colorectal
Uno de los genes blanco de la ruta de señalización Wnt-canónica y que se ha
estudiado mucho por su relación con CCR es endotelina-1 [21]. Las endotelinas (ETs) son una
familia de péptidos constituida por tres miembros: endotelina-1 (ET-1), endotelina-2 (ET-2) y
endotelina-3 (ET-3) [30]. Las ETs son producidas como polipéptidos preproendotelinas de 200
residuos que, luego de la remoción del péptido señal, constituyen la big-endotelina (big-ET)
formada por 38-41 residuos [31]. El péptido ET activo se forma cuando una endopeptidasa
conocida como Enzima Convertidora de Endotelina (ECE) corta a big-ET entre los residuos
Trp-21 y Val-22 [31]. No obstante, como cualquier péptido secretado, las concentraciones
extracelulares de ET son determinadas no sólo por su producción sino también por su
degradación. En este caso, la enzima endopeptidasa neutral (NEP), también denominada
neprylisina, inactiva una variedad de péptidos reguladores sobre la superficie celular, entre
ellos las ETs [32].

18
Las ETs actúan a través de dos subtipos de receptores acoplados a proteína G, ETaR y
ETbR. La afinidad de ETaR es para ET-1 ≥ ET-2 >> ET-3 y, de hecho, ET-1 y ET-2 pueden unirse
a ETaR a concentraciones fisiológicas, mientras que ET-3 no [33]. En cambio, ETbR tiene una
afinidad similar para los tres isopéptidos [33]. Así, se conoce como “eje ET-1” al conjunto de
componentes que permiten activar la vía, es decir, la preproET-1, ECE-1, y sus dos receptores
tipo A (ETaR) y tipo B (ETbR) [30] (figura 2).
Figura 2. Componentes del eje endotelina-1. Secuencia de activación del péptido ET-1 donde actúan los componentes del eje ET-1; la preproET1, ECE-1, ET-1 y sus dos receptores ETaR y ETbR.
ET-1, la principal isoforma de la familia, es un péptido pleiotrópico que está
involucrado en numerosos procesos fisiológicos y patológicos. ET-1 es un potente péptido
vasoactivo que regula el flujo sanguíneo y se ha sugerido su participación en numerosas
enfermedades, incluyendo hipertensión arterial, hipertensión pulmonar, falla renal aguda y
hemorragia subaracnoídea. Además, se ha involucrado a ET-1 en enfermedades relacionadas
con estados dolorosos, como la enfermedad isquémica de arterias coronarias, anemia
drepanocítica, enfermedades inflamatorias como la artritis e, incluso, el cáncer [30].
Las funciones pleiotrópicas de ET-1 pueden explicarse porque la activación de los
receptores de ET-1 estaría acoplada a una multiplicidad de eventos de señalización, entre
ellos: la activación de las fosfolipasas C y D [34]; aumento del nivel de Ca+2 y activación de la
PKC [35]; activación de proteínas kinasas de distinto tipo como MAPKs, ERKs, JNK, p38 [36-
39] y PI3K [40]; proteínas y receptores asociados a la actividad tirosina kinasa como, c-src,
PreProET1
200 aa
Big - ET1
38 - 41 aa
ECE - 1 ET1
21 aa NEP
ETaR ETbR

19
EGFR, y la tyr-kinasa dependiente de Ca+2 rica en prolina, Pyk2 [39, 41, 42]; y más
recientemente la generación de especies reactivas de oxígeno [43, 44]. (figura 3)
Figura 3. Múltiples vías de señalización activadas por endotelina-1 mediado por su unión al receptor ETaR y las respuestas celulares observadas. Obtenido de referencia [45].
El eje ET-1 ha emergido recientemente como un componente importante en el
origen, desarrollo y progresión del cáncer debido a su rol en la regulación de procesos como
mitogénesis, sobrevida celular, angiogénesis, regulación de infiltración de células inmune,
transición epitelio mesenquimática (TEM), invasión y diseminación de la metástasis [45].
Para investigar la propiedad mitogénica de ET-1 y su capacidad para inducir la
proliferación en CCR en respuesta a ET-1, Grant y colaboradores estudiaron sus efectos en 3
líneas celulares, con y sin inhibidores de ETaR, ETbR, subtipos de proteína G, PI3K ó PKC, y se
evaluó los niveles de proliferación y apoptosis. Se mostró que ET-1, mediado por el receptor
ETaR, es capaz de estimular el crecimiento a través de la proliferación y no de la disminución

20
de la apoptosis en todas las líneas celulares de CCR y que la estimulación del crecimiento
inducida por ET-1 es dependiente de la proteína G (Go/Gi), PI3K y PKC [46].
En otros tipos celulares, como en células de músculo liso vascular, ET-1 induce la
expresión de c-fos y c-myc aumentando así la proliferación [47]. Un efecto similar es visto en
fibroblastos y osteoblastos, entre otros tipos celulares, con la producción de inositol
trifosfato (IP3) y diacilglicerol (DAG) y la estimulación de la fosfolipasa C [48, 49].
Otros han demostrado que ET-1 suprime la apoptosis, perpetuando el crecimiento del
cáncer. En células de CCR que expresan normalmente Fas y Fas ligando y son resistentes a la
apoptosis mediada por Fas, fueron expuestas a Bosentan, un antagonista de ambos
receptores ETaR y ETbR, viéndose aumentada la respuesta apoptótica [50]. Más tarde, se
mostró que un antagonista específico de ETaR en células de CCR HT29 bloqueó la
proliferación e indujo apoptosis [51]. En cáncer ovárico se ha demostrado que ET-1 es capaz
de inhibir el efecto apoptótico que ejerce Paclitaxel, la droga más comúnmente usada para el
tratamiento de este cáncer, y además se observó que este efecto de sobrevida sería mediado
por ETaR y no por ETbR, ya que sólo el inhibidor específico de este receptor BQ-123 logró
inhibir la inducción de sobrevida por ET-1 en las líneas celulares de cáncer ovárico OVCA433 y
HEY [52].
Se sabe que un marcador de progresión de los cánceres de epitelio, como el CCR, es la
transición epitelio-mesenquimática (TEM) durante la cual las células tumorales sufren la
pérdida de polaridad celular y unión intercelular, adquiriendo la habilidad para invadir la
matriz extracelular y migrar a sitios distantes [45]. Similarmente a la ruta Wnt-canónica [53],
la activación del eje ET-1 regula negativamente la expresión de E-cadherina, aumentando la
expresión de N-cadherina de manera que promueve la TEM debido a que contribuye a
disminuir la interacción entre células tumorales y normales [54].
Por otro lado, el desarrollo tumoral depende de la neovascularización, lo que conlleva
a un reclutamiento y proliferación de células endoteliales y de la pared vascular (pericitos,
músculo liso, miofibroblastos) así como al remodelamiento celular [55]. En este contexto, ET-
1 también juega un papel importante, porque su potente efecto mitogénico estimula la
proliferación de estos tipos celulares [45]. Al igual que lo mostrado para la ruta Wnt-canónica

21
[20], en líneas celulares de cáncer ovárico la expresión de ET-1 se correlaciona con la
expresión de VEGF y neovascularización, reportándose el aumento de VEGF a nivel de mRNA
y proteína [56]. También, en células de melanoma primario y metastásico la activación de
ETbR mediante ET-1 y ET-3 en condiciones normóxicas aumenta la expresión de VEGF, COX-1
y COX-2, aumenta la actividad reportera de COX-2 y además aumenta la producción de
prostaglandina E2, lo que es aún más intenso en condiciones de hipoxia [57].
Interesantemente, tanto E-cadherina, VEGF y COX-2 han sido descritos como blancos
regulados por -catenina en la ruta Wnt-canónica [20, 53, 58].
Además de los efectos sobre crecimiento y angiogénesis, ET-1 también promovería la
invasión celular y metástasis. En líneas celulares de cáncer de ovario, ET-1 induce la secreción
y activación de metaloproteinasas (MMPs), reduce la expresión de conexina-43 (Cx43) y
reduce la comunicación intercelular a través de las gap junction tipo IC, promoviendo de esta
manera la invasión [45, 59]. En un ensayo in vivo en CCR, se evaluó el efecto de la infusión
intraportal del inhibidor específico del receptor ETaR, BQ123, en un modelo de ratón con CCR
y metástasis a hígado, encontrándose que ejerce una reducción significativa del peso del
tumor localizado en este órgano [60]. Estos datos sugieren fuertemente que la ET-1
producida por las células de CCR juega un rol importante en la metástasis a otros tejidos.
Al respecto, en un estudio clínico donde se midió la concentración plasmática de big-
ET-1 de muestras obtenidas previo a la cirugía en pacientes con CCR, se encontró valores
significativamente más altos en los pacientes con cáncer respecto a los controles. Además,
en muestras de plasma obtenido de vena porta se vieron aumentos de big-ET-1 en los
pacientes con grado D de Duke comparados con los pacientes con grado A, B y C. Estos
autores hallaron también una correlación entre la invasión microvascular y los aumentos de
big-ET-1 en las biopsias de los pacientes con CCR [61]. Asimismo, cuando se estudió los
niveles plasmáticos de ET-1 en pacientes con CCR se encontró que todos presentaban niveles
plasmáticos elevados del péptido ET-1 respecto a pacientes controles. Además, las células
tumorales metastásicas en hígado pero no las células normales expresan ET-1 [62]. Así, el
nivel de big-ET-1 y el de ET-1 aumenta significativamente en los grupos de pacientes con CCR
primario y metástasis hepática.

22
Se ha encontrado niveles plasmáticos elevados de ET-1 en el 80% de los casos de CCR
y en el mismo porcentaje de casos se ve una activación aberrante de la vía Wnt-canónica
[21]. De hecho, el gen de ET-1 es un blanco de esta ruta de señalización y es regulado
directamente por -catenina en células de CCR. Kim y colaboradores mostraron mediante
ensayos en cultivos de células de CCR, HT29-APC, que cuando se bloquea la proliferación y se
induce la apoptosis por la expresión de APC, lo cual desestabiliza a la -catenina
citoplasmática y por tanto se apaga la señalización de la ruta Wnt-canónica, el fenotipo
tumoral puede ser rescatado al suplementar el medio con el péptido activo ET-1 [21]. Estos
resultados destacan el rol esencial que juega ET-1 entre los genes regulados por la ruta Wnt-
canónica para ejercer el efecto protumoral.
Finalmente, una interacción más directa entre el eje ET-1 y la ruta Wnt-canónica se
encuentra en el cáncer de ovario, donde se ha reportado que ET-1 la estimula a través de la
inhibición de GSK3 dependiente de PI3K [63]. Así, ET-1 participa en un loop positivo con la
vía Wnt-canónica ya que, además de ser un blanco río abajo de ésta, es también capaz de
regularla positivamente a través de la activación de una vía dependiente de PI3K.
La Enzima Convertidora de Endotelina-1 (ECE-1) y su relación con cáncer colorectal
Los efectos de ET-1 relacionados con la carcinogénesis son ejercidos por la ET-1 activa,
cuya vida media es de sólo 1 minuto, a diferencia de big-ET-1 secretada que es de 23
minutos [45, 64]. Sin embargo, aunque la big-ET-1 tiene una vida media más prolongada, no
puede ser señal de los efectos protumorales por sí misma, sino que lo es la expresión y
actividad de la enzima encargada de su activación mediante corte proteolítico específico, la
Enzima Convertidora de Endotelina-1 (ECE-1).
Las ECEs juegan un rol clave en la biosíntesis de las endotelinas, debido a su habilidad
para cortar proteolíticamente los precursores inactivos big-ET-1, big-ET2 y big-ET3 [31] (ver
figura 2). Las ECEs pertenecen a la subfamilia de endopeptidasas neutras M13 que contiene 8
miembros, algunos de los cuales son NEP, KELL y PEX [65]. Estas enzimas son proteínas
integrales de membrana tipo II con un corto dominio citoplasmático, un único dominio
transmembrana y un gran dominio C-terminal conteniendo el sitio activo (figura 4) [65].

23
Figura 4. Estructura de la enzima convertidora de ET-1 (ECE-1). Arriba, esquema representando los dominios de la enzima ECE-1. En achurado, dominio intracelular variable para cada isoforma, en rojo, región intracelular común en todas las isoformas, en TM, dominio único de transmembrana, y HEXXH, dominio catalítico, de unión a zinc. Obtenido de referencia [66] . Abajo, estructura del dominio extracelular de la ECE-1, Obtenido de referencia [67].
A la fecha se han descrito 3 distintas enzimas, ECE-1, ECE-2 y ECE-3, y todas
constituyen metaloproteasas unidas a membrana [31, 68, 69]. ECE-1 y ECE-2 han sido
clonadas y caracterizadas, con una identidad total del 59%, difiriendo en la especificidad de
sustrato, pH óptimo, localización subcelular y distribución en tejidos [31, 68]. ECE-1 es la más
abundante y ha sido estudiada en detalle debido a que es la enzima favorita para la
conversión de big-ET-1 [65]. ECE-1 forma un dímero estabilizado por un puente disúlfuro que
une los dos grandes dominios extracitoplasmáticos de la proteína [70]. ECE-1 se expresa no
sólo en el endotelio, su principal sitio de expresión constitutiva, sino también en ciertos tipos
de células epiteliales y mesenquimáticas, células de ductos y glándulas en tejidos normales
humanos, incluyendo colon, estómago, hígado, endometrio, mamas, próstata, pulmón y
riñón, entre otros, lo que se relaciona con las conocidas acciones pleiotrópicas de las
endotelinas [71]. No obstante, ECE-1 parece desempeñar un rol importante en el desarrollo,
puesto que ratones KO para ECE-1 (ECE-1-/-) mueren in útero 30 minutos antes de nacer [72].

24
La ECE-1 activa proteolíticamente el péptido ET-1, lo que implica que la actividad de
ECE-1 tendría un rol crítico en los efectos locales de éste por cuanto serían ejercidos en las
células que poseen una mayor actividad de ECE-1 y no necesariamente en células que
secretan un mayor nivel de big-ET-1. Por ejemplo, cuando se evaluaron los niveles de
expresión de ET-1 en cuatro líneas celulares de adenocarcinoma endometrial, se encontró
altos niveles de mensajero de ET-1, sin embargo en dos de ellas no se encontraron niveles
detectables de ET-1 secretados al medio de cultivo [73]. Por ende, el estudio de la regulación
de la expresión de ECE-1 es un aspecto importante de considerar en la biología del cáncer y
especialmente en CCR.
Al igual que ET-1, la ECE-1 es sobreexpresada en varias líneas celulares tumorales y en
biopsias de distintos tejidos como mamas, pulmón, próstata, tiroides y colon [74-79], lo que
podría relacionarse con un rol importante de ECE-1 en la génesis y desarrollo del CCR.
Al comparar la expresión de los componentes del eje ET-1 es decir, prepro-ET-1, ECE-
1, ETaR y ETbR en CCR respecto al tejido normal, se observó una sobreexpresión de prepro-
ET-1 y de ECE-1 en las células de carcinoma y en los vasos del estroma [55]. También,
recientemente se usó la estrategia de siRNA contra ECE-1 en líneas celulares de cáncer
ovárico metastásico, encontrándose efectos dependientes de esta enzima, tales como:
menor fosforilación de p42/44MAPK dependiente de ET-1, una disminución de la invasividad
y actividad de la metaloproteinasa MMP2, una mayor adhesión a proteínas de la lamina basal
(laminina-1 y colágeno tipo IV) y aumento de la expresión de E-cadherina y disminución de N-
cadherina [80]. En resumen, estos datos sugieren que el silenciamiento de la ECE-1 permitiría
revertir varias de las características tumorigénicas normalmente observadas en células de
CCR.
Por otro lado, cuando se evaluó la inhibición de todas las isoformas de ECE-1, u otras
metaloproteasas similares como NEP o la enzima convertidora de angiotensina (ACE), sobre
el crecimiento de células de glioblastoma humano se encontró que sólo el inhibidor de ECE-1
afectó la síntesis de DNA [81]. Pero más importante aún, cuando en las mismas células se
agregó al medio de cultivo ET-1 o big-ET-1, se observó que éstas no revirtieron la disminución
del crecimiento generada por el inhibidor de ECE-1, sugiriendo que el inhibidor de ECE-1

25
bloqueó la proliferación de las células de glioblastoma humano muy probablemente por un
mecanismo independiente de la producción de ET-1.
ECE-1 se expresa como 4 isoformas distintas, las cuales han sido clonadas y
denominadas ECE-1a, ECE-1b, ECE-1c y ECE-1d, todas codificadas por el mismo gen pero
reguladas por promotores distintos que se encuentran localizados inmediatamente río arriba
de cada exón isoforma específico (ver figura 5) [82-84]. Todas las isoformas comparten un
dominio C-terminal similar donde radica su sitio activo, lo que explica que sus eficiencias
catalíticas (kcat) no presenten variaciones significativas [85], pero se diferencian en su
extremo N-terminal que es el responsable de la distribución subcelular específica de cada
isoforma [66].
Figura 5. Estructura del gen y alineamiento aminoacídico de las isoformas de la enzima convertidora de ET-1 (ECE-1). Arriba, esquema representando los promotores alternativos para cada isoforma de la ECE-1. Al medio, integración de exones específicos y comunes para cada isoforma de ECE-1. Abajo, Alineamiento de aminoácidos que componen la región intracelular variable de cada isoforma de ECE-1. (Obtenido de referencia [66]) .
EXON1c
EXON3
EXON2
EXON1a
EXON1b
EXON1d
EXON4-19
5’ 3’
P- 1c P- 1b P- 1d P- 1a

26
La expresión de las isoformas varía dependiendo del periodo del desarrollo así como
también es distinta en varias patologías [85]. Del mismo modo, la expresión relativa de cada
isoforma varía en los distintos tejidos, aunque ECE-1c es la mayormente expresada en varios,
incluso en cáncer [75, 83]. Particularmente, en el caso de las isoformas ECE-1a y ECE-1c,
ambas se expresan en la membrana celular [83] aunque se ha demostrado que tendrían roles
opuestos en células de cáncer de próstata PC3, siendo ECE-1c la que tendría la función
específica de promover la invasividad [86].
Actualmente no existen evidencias acerca de la regulación transcripcional de esta
enzima o de sus isoformas. Sin embargo, cuando se reportó la caracterización del promotor
de la isoforma ECE-1c se realizaron ensayos funcionales con el promotor completo (-968 bp
respecto al sitio de inicio de traducción) y con distintas deleciones del mismo clonados en un
vector reportero. Los resultados identificaron que la región comprendida entre 217 y 490 río
arriba a partir del sitio de inicio de la traducción en el promotor de la isoforma c de la ECE-1
abarca prácticamente la totalidad de la activación de la función promotora. Entre los
potenciales sitios de activación dentro de esta región promotora se encontró que entre -217
y -240 hay un aumento de unas 5,5 veces en la actividad reportera lo que podría estar
asociado a la unión de un factor de transcripción GATA. Luego, el aumento de 1,6 veces en la
actividad transcripcional cuando el tamaño de la secuencia promotora se extiende desde -
409 a -490 se sugirió a la unión de los factores de transcripción GATA y/o ETS (figura 6).
Por otra parte, un análisis más detallado de los resultados muestra que existió un
aumento de 2 veces en la actividad promotora de esta isoforma debido a algún factor de
transcripción no identificado que se une a la secuencia comprendida entre -240 y -409 (figura
6) [87]. A pesar de que los autores del trabajo no continuaron un estudio más detallado al
respecto, en nuestro laboratorio estudiamos in sílico esta región y encontramos un sitio
consenso de unión para el factor de transcripción TCF4, el cual podría ser responsable de
mediar a través de -catenina la regulación positiva de ECE-1c por la vía Wnt-canónica
(figura 6).

27
Figura 6. Secuencia del promotor especifico para la ECE-1c. A la Izquierda. Secuencia promotora de ECE-1c que
muestra encasilladas las regiones que comprenden los sitios consenso de unión para los factores de
transcripción putativos reportados que tendrían un efecto en la actividad transcripcional de ECE-1c, además en
rojo, el sitio consenso de unión para el factor de transcripción de la vía Wnt-canónica TCF. A la derecha.
Encasillado en verde, aumento en la actividad transcripcional cuando el promotor incorpora la secuencia
consenso de unión para un factor GATA. En azul, aumento en la actividad transcripcional cuando el promotor
incorpora la secuencia consenso de unión para los factores de transcripción GATA Y ETS. En rojo, aumento en la
actividad transcripcional cuando el promotor incorpora la secuencia consenso de unión para un factor TCF-4 (no
reportado). Obtenido de referencia [87].

28
HIPOTESIS
Existe mucha evidencia indicando que en el cáncer de colon se produce una activación
aberrante de la vía Wnt-canónica, lo que conlleva a una mayor expresión de varios genes
blancos, siendo uno de ellos ET-1. Este péptido para ser activo depende de la acción de ECE-
1, cuya isoforma c es sobreexpresada en muchos cánceres y especialmente el de colon.
Además datos in sílico obtenidos por nuestro laboratorio indican la presencia de un sitio de
unión a factores TCF/LEF en el promotor de ECE-1c. Finalmente, en cáncer de ovario y
próstata se ha descrito que esta isoforma tendría una función importante en invasividad
tumoral.
En consecuencia, la hipótesis del presente proyecto es:
“La vía Wnt-canónica regula positivamente la expresión de la isoforma c de la
Enzima Covertidora de Endotelina-1 [ECE-1(c)], lo que favorece la invasividad en
células de cáncer de colon”
OBJETIVOS OBJETIVO GENERAL
Estudiar si la expresión de ECE-1(c) depende de la vía Wnt-canónica y relacionarla con
el proceso invasivo en células de cáncer de colon.
OBJETIVOS ESPECÍFICOS
1) Determinar en líneas celulares normales y de CCR la expresión endógena de ECE-1 y
algún blanco conocido de la vía Wnt-canónica.
2) Estudiar en líneas celulares normales y de CCR si la activación o inhibición de la vía
Wnt-canónica aumenta o disminuye la expresión de ECE-1(c), respectivamente.
3) Analizar el grado de invasividad in vitro de las líneas celulares normales y de CCR
luego de la sobreexpresión o eliminación de ECE-1c.
4) Evaluar si existe correlación entre la expresión endógena de ECE-1(c) con el grado de
malignidad anatomopatológico en biopsias de pacientes con CCR.

29
MATERIALES Y METODOS Materiales y Reactivos. Las células (HEK-293T, HT29-ATCC, DLD-1 y HT29-US) se cultivaron en
medio de cultivo celular D-MEM high glucose (Hyclone), conteniendo suero fetal bovino (SFB,
Biological Industries, Israel). Para los experimentos de transfección se usó el reactivo de
transfección Superfect (SuperFect Transfection Reagent, Qiagen). Para la extracción de RNA
total se usó el reactivo TRIZOL (invitrogen, Life Technologies, Carlsbad, CA). Random Primer
(invitrogen), transcriptasa reversa M-MLV (invitrogen), inhibidor de RNAsa RNAsa OUT
(invitrogen). Para la extracción de proteínas a partir de células cultivadas, se usó el tampón
RIPA (25 mM Tris•HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS) y
la mezcla de inhibidores de proteasa (complete protease inhibitor cocktail tablets, Roche).
Para la detección de proteínas por WB se usó solución de reacción para quimioluminiscencia
de peroxidasa EZ-ECL (Biological Industries, Israel); anticuerpo monoclonal anti-ECE-1
humana (RyD systems), anticuerpo policlonal de conejo anti-survivina humana (RyD systems),
anticuerpo policlonal de cabra anti-actina (Santa Cruz Biotechnology) y anticuerpos
secundarios anti-IgG de ratón, conejo y cabra (Santa Cruz Biotechnology).
Cultivo celular. Se trabajó con 3 líneas celulares de cáncer de colon humano (HT29-ATCC,
DLD-1, y HT29-US), las cuales provienen de tumores que se encuentran en estadios B, C y D,
respectivamente, según la clasificación de Duke para la progresión tumoral de CCR. También
se usó dos líneas celulares normales (no tumorales), las células embrionarias de riñon
humano (HEK-293T), y las células epiteliales de ovario de hámster Chino (CHO-K1). Estas
últimas no presentan actividad ECE-1. Las 4 primeras fueron cultivadas y mantenidas con
medio D-MEM, mientras que la última con medio F12 (Gibco), ambos medios suplementados
con 10% de SFB, L-Glutamina (2 mM, Gibco), antibióticos (100 U/ml penicilina y 100 µg/ml de
estreptomicina, ambas Gibco) y antifúngico (0,5 µg/ml de fungizona anfotericina, Gibco),
mantenidas a 37ºC en ambiente humidificado con 5% CO2. Cuando se realizaron
tratamientos con inhibidores de CK2 (TBB, 10 mM) ó de GSK3LiCl, 1 M), se sembraron el
día previo al tratamiento 800.000 células en placas de 60 mm y en el momento del
tratamiento con la droga. El inhibidor se resuspendió en el volumen adecuado de D-MEM

30
suplementado para dejarlo a la concentración correspondiente ( TBB a 100 uM, LiCl a 20
mM), se lavaron las placas con PBS 1X dos veces y luego se agregó 2 mL del medio
conteniendo el inhibidor, mientras que a la placa control se agregó 2 ml de medio fresco, y
luego de 24 horas de tratamiento se volvió a lavar las placas 2 veces con PBS 1X y se extrajo
el RNA total o proteínas para el correspondiente estudio.
Transfección de células. Se realizaron transfecciones en dos escalas, en placas de 6 pocillos
(p6poc.) o en placas de 60 mm de diámetro (p60) dependiendo si el análisis posterior de las
células se haría mediante RT-PCR o Western Blot. El día previo a la transfección, se
sembraron 400.000 o 800.000 células en placas p6poc. o p60, respectivamente, con la idea
de que al momento de la transfección no sobrepasaran una confluencia del 60%. Luego se
mezclaron en tubos de 1,6 ml, 2 ó 5 µg (según se indica) de DNA con 100 ó 150 µl de D-MEM
sin suplementar (libre de suero y antibióticos) para placas p6poc ó p60, respectivamente.
Luego se le agregó el reactivo superfect (SF.) para que quedara en una relación de 1/5 de
DNA/SF. La mezcla se peleteó y se agitó para obtener una solución homogénea y luego se
dejó incubando durante 15 minutos a temperatura ambiente. Pasado el tiempo de formación
del complejo DNA/SF, se procedió a agregar 600 o 1000 µl de D-MEM suplementado a cada
tubo, se mezcló el contenido y se agregó suavemente a las placas inmediatamente después
de haber aspirado el medio antiguo. Las células se incubaron durante 3 horas y
posteriormente se procedió a aspirar la mezcla de transfección, reemplazándose por medio
D-MEM suplementado fresco para dejar expresar las respectivas proteínas durante 24 ó 48
hrs.
Extracción de RNA total. Para la obtención de RNA total desde biopsias, se realizó cortes de
tejido congelado con un tamaño de entre 100 y 150 mg y se depositaron en un criotubo con
1 ml del reactivo Trizol. Para la obtención de RNA total desde las placas de cultivo, se agregó
1 ml del reactivo Trizol, luego de homogenización se pasó el volumen total a tubos de 1,5 ml.
Tanto para el tejido de biopsias como para células en cultivo, el protocolo continuó de la
misma forma. Al homogenizado con Trizol, se le agregó 200 µL de cloroformo frío, se agitó en

31
forma suave por inversión. Se centrifugó durante 15 min a 12.000 g y 4° C y luego se
extrajeron 300 µL de fase acuosa (superior), se pasaron a un tubo de 1,5 ml nuevo y se
agregaron 500 µL de isopropanol, se mezclaron por inversión y se dejaron precipitar durante
1 hora a -20°C. Luego se volvió a centrifugar en las mismas condiciones previas, se eliminó el
sobrenadante y se lavó el pellet de RNA con 1 ml de solución de etanol 75% en agua/DEPC.
Por último, se obtuvo el pellet de RNA con una centrifugación extra durante 5 minutos a
8000 g, luego de finalmente resuspenderlo en 25 µL de agua/DEPC.
Análisis de expresión mediante RT-PCR. El RNA total, se cuantificó utilizando un método
fluorimétrico (Qubit, Invitrogen). El DNA complementario (cDNA) se sintetizó en una mezcla
de reacción de 25 µl. Inicialmente, se agregó 3 µg de RNA total por cada muestra, 0,5 µl de
random primers (3 µg/µl) y H2O/DEPC suficiente para 16,5 µL, esta mezcla se calentó
durante 5 minutos a 70°C y se enfrió otros 5 minutos en hielo. Luego se agregó buffer, dNTPs,
RNAsa OUT y la enzima transcriptasa reversa MMLV, para quedar a concentraciones finales
de 0,5 mM dNTPs, 2 U/µl RNAsa OUT y 8 U/µl M-MLV. Finalmente, el cDNA fue usado para
amplificar en una mezcla de reacción de PCR de 20 µL, conteniendo buffer 1X, 0,6 µl de
MgCl2 (50mM), 0,4 µl de dNTPs (10mM), 0,24 µl de Taq DNA polimerasa (Invitrogen, 5U/μl), 1
µl de partidor forward y 1 µl de reverse (10 uM). El PCR fue corrido bajo las siguientes
condiciones: una etapa de preincubación durante 3 min a 94°C; 20-28 ciclos de separación a
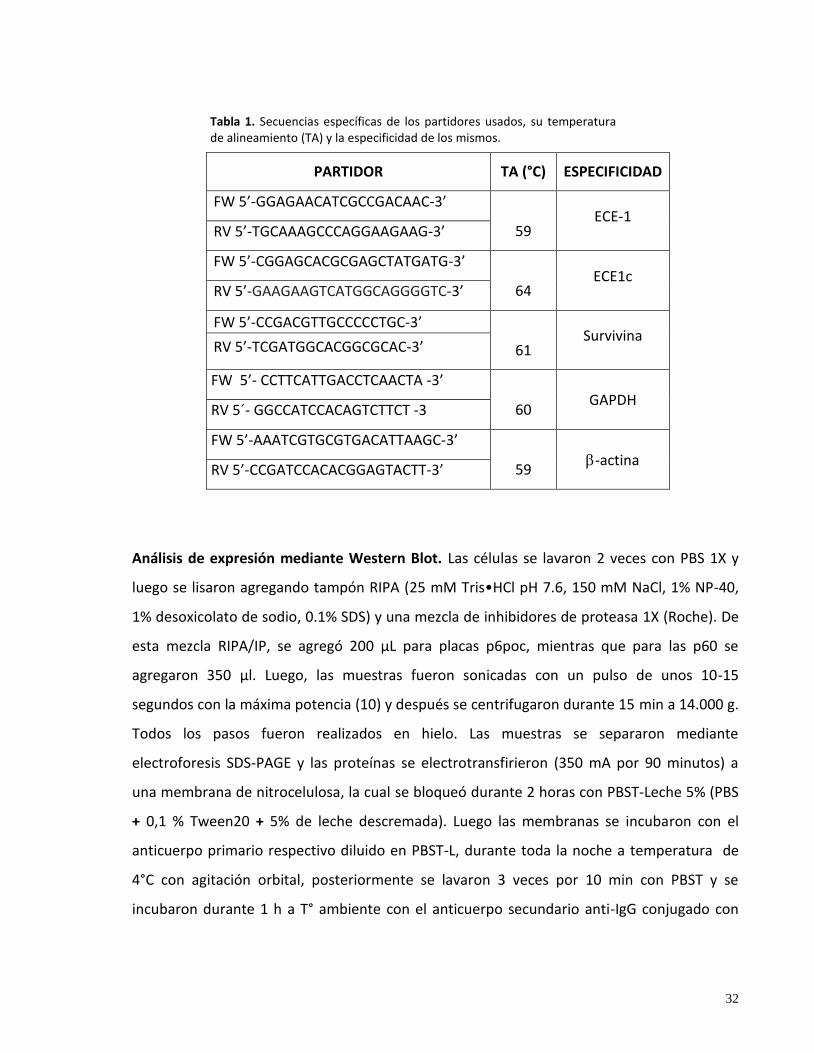
94°C durante 30 seg; alineamiento durante 45 seg a la temperatura específica (ver Tabla 1) y
extensión durante 45 seg a 72°C; finalmente, una etapa de extensión durante 5 minutos a
72°C. La secuencia de los partidores específicos y la T° de alineamiento (TA) para amplificar
ECE-1, ECE1c, survivina, -actina y GAPDH, se muestran en la Tabla 1. Los productos de PCR
fueron separados en geles de agarosa al 1,5% en TBE 1X y visualizados por tinción con
bromuro de etidio 0.06 μg/ml. La intensidad de las bandas fue cuantificada por
densitometría utilizando el programa Image J y todas fueron normalizadas por la expresión
de -actina ó GAPDH, como se indica.
32
PARTIDOR TA (°C) ESPECIFICIDAD
FW 5’-GGAGAACATCGCCGACAAC-3’
59 ECE-1
RV 5’-TGCAAAGCCCAGGAAGAAG-3’
FW 5’-CGGAGCACGCGAGCTATGATG-3’
64 ECE1c
RV 5’-GAAGAAGTCATGGCAGGGGTC-3’
FW 5’-CCGACGTTGCCCCCTGC-3’
61 Survivina
RV 5’-TCGATGGCACGGCGCAC-3’
FW 5’- CCTTCATTGACCTCAACTA -3’
60 GAPDH
RV 5´- GGCCATCCACAGTCTTCT -3
FW 5’-AAATCGTGCGTGACATTAAGC-3’
59 -actina
RV 5’-CCGATCCACACGGAGTACTT-3’
Análisis de expresión mediante Western Blot. Las células se lavaron 2 veces con PBS 1X y
luego se lisaron agregando tampón RIPA (25 mM Tris•HCl pH 7.6, 150 mM NaCl, 1% NP-40,
1% desoxicolato de sodio, 0.1% SDS) y una mezcla de inhibidores de proteasa 1X (Roche). De
esta mezcla RIPA/IP, se agregó 200 µL para placas p6poc, mientras que para las p60 se
agregaron 350 µl. Luego, las muestras fueron sonicadas con un pulso de unos 10-15
segundos con la máxima potencia (10) y después se centrifugaron durante 15 min a 14.000 g.
Todos los pasos fueron realizados en hielo. Las muestras se separaron mediante
electroforesis SDS-PAGE y las proteínas se electrotransfirieron (350 mA por 90 minutos) a
una membrana de nitrocelulosa, la cual se bloqueó durante 2 horas con PBST-Leche 5% (PBS
+ 0,1 % Tween20 + 5% de leche descremada). Luego las membranas se incubaron con el
anticuerpo primario respectivo diluido en PBST-L, durante toda la noche a temperatura de
4°C con agitación orbital, posteriormente se lavaron 3 veces por 10 min con PBST y se
incubaron durante 1 h a T° ambiente con el anticuerpo secundario anti-IgG conjugado con
Tabla 1. Secuencias específicas de los partidores usados, su temperatura de alineamiento (TA) y la especificidad de los mismos.

33
peroxidasa de rábano para, finalmente, detectar la quimioluminiscencia con el kit EZ-ECL
(Biological Industries, Israel).
Ensayo de invasividad in vitro. Se usaron cámaras de invasividad Matrigel BD Biocoat (BD
Biosciences, Bedford, MA, USA). Dos horas previas al experimento, se lava las células con PBS
1x y se deja con medio D-MEM libre de suero (MLS). En paralelo se rehidratan los insertos y
los pocillos de la placa con 500 µl de MLS. Pasado el tiempo de rehidratación, se tripsiniza las
células, se detuvo con D-MEM 10% SFB, se peletearon y luego se resuspendieron en MLS,
después se contaron sólo las células vivas (Tripan Blue) y se dejaron a una densidad de
100.000 cels/ml. Finalmente, las células se dejaron incubando en el inserto superior (500 µl,
50.000 cels) de las cámaras de matrigel, mientras al compartimiento inferior se le agregó 750
µl de D-MEM 10% (el que actúa como un quimioatractante para las células que se
encuentran en el inserto superior). Las células se incubaron durante 22 horas en condiciones
normales de crecimiento y posteriormente las que no atraviesan la membrana con matrigel
fueron removidas. En cambio, las que fueron capaces de atravesar la membrana y quedar
adheridas a ésta, fueron fijadas durante 2 min con 100% metanol, teñidas también durante 2
min con azul de toluidina 1% (disuelto en 1% tetraborato de sodio) y finalmente lavadas 2
veces durante 2 min con agua destilada. Después, se fotografiaron 4 campos distintos y se
contaron las células fijadas. Se expresaron los resultados calculando el promedio de los 4
campos.
Obtención de biopsias. Las biopsias de pacientes con diagnóstico de cáncer colorrectal
provinieron del Banco de Tejidos del Servicio de Anatomía Patológica del Hospital Clínico de
la Universidad de Chile, bajo la asesoría de la patóloga Dra. Cristina Fernández F., Jefe
Técnica del Servicio. Estas biopsias se obtuvieron siguiendo los protocolos bioéticos y de
preservación de tejidos apegados a las normas internacionales, que garantizan la
preservación de tejidos neoplásicos y no neoplásicos con integridad celular (DNA y proteínas,
con posibilidad de recuperar RNA) para estudios moleculares y genéticos. Se obtuvieron 14
muestras, que corresponden a biopsias de CCR, que incluyen tejido tumoral (TT) y tejido
adyacente sano (TA), de las etapas B y C de acuerdo a la clasificación de Duke. Los tejidos se

34
conservaron congelados a -80 °C, previa asignación de un número interno que garantiza la
confidencialidad de la información. En una base de datos se archiva además los
consentimientos informados de los pacientes y la información clínica, incluyendo el estadío
tumoral (Duke y TNM).
Análisis estadístico. Para análisis de varios grupos de datos, primero se analizaron para
conocer si poseían una distribución normal mediante el test de Kolmogorov-Smirnov. Para
saber si existió diferencias significativas entre los grupos de datos estudiados, se usó
posteriormente un test de ANOVA o un test de Kruskal-Wallis dependiendo de si existió una
distribución paramétrica o no paramétrica de los grupos de datos analizados
respectivamente. Luego, para saber entre cuales de ellos específicamente hubo o no
diferencia significativa, se usó un post-test de Tukey, en el caso de los datos distribuidos
paramétricamente, o un post-test de Dunn en el caso de los datos con distribución no
paramétrica. Estos post-test nos definen exactamente entre qué grupos de datos hubo
diferencia significativa. Cuando se analizó dos grupos de datos se aplicó t test de datos no
pareados. La significancia estadística fue indicada como sigue; * cuando se aplico como
norma un valor de P<0.05, ** para un valor de P<0.01 y *** para un valor de P<0.001. Los
análisis fueron desarrollados con el programa estadístico GraphPad Prism versión 5.0, el que
fue provisto amablemente por la Dra. Carmen Romero Osses, Jefa del Laboratorio de
Endocrinología y Biología de la Reproducción, Hospital Clínico de la Universidad de Chile.

35
RESULTADOS
El nivel endógeno de proteína ECE-1 es similar al blanco control de la vía Wnt-canónica,
survivina, en líneas celulares normal y de CCR.
Nuestro primer objetivo consistió en conocer si existía una asociación entre los
niveles de expresión de ECE-1c y el grado de malignidad tumoral del cual provienen las líneas
celulares de CCR humano en estudio, HT29-ATCC, DLD-1 y HT29-US, que según la clasificación
anatomopatológica de Duke se podrían asociar con las etapas B, C y D, respectivamente. Otro
propósito fue conocer si existía una relación entre la expresión de ECE-1c y el nivel de
actividad de la vía Wnt-canónica en las mismas células, lo que fue determinado
indirectamente por el nivel de expresión endógena de survivina, un blanco conocido de esta
ruta de señalización.
Para desarrollar este primer objetivo se analizó en células embrionarias humanas
HEK-293T y en las células de CCR humano la expresión proteica mediante western blot con
un anticuerpo específico para survivina como control de actividad de la vía Wnt-canónica [88]
y otro anticuerpo que reconoce todas las isoformas de ECE-1, puesto que no existe uno
específico para ECE-1c en el comercio. Como resultado, en los ensayos de western blot
encontramos que el nivel de expresión relativa de ECE-1 fue alto en la línea celular normal
HEK-293T, de igual forma que lo fue survivina (figura 7B). Esto es esperable ya que estas
células son extraídas de tejido embrionario, lo que implica que conservan un alto grado de
indiferenciación así como una alta capacidad de proliferación [89] y, por tanto, presentan
una alta actividad de la vía Wnt-canónica, lo cual se evidencia por el elevado nivel de
expresión de survivina (figura 7B).
Por otro lado, la línea celular HT29-ATCC presentó los niveles más bajos de expresión
de ECE-1 los cuales corresponden con lo esperado debido a que representan a un tumor
colorectal primario localizado (Duke B), con un alto potencial proliferativo pero sin potencial
metastásico. Similarmente, éstas células presentaron los niveles de survivina más bajos
(figura 7B).
La línea celular de CCR humano DLD-1 fue la que presentó los niveles de expresión de
ECE-1 más altos de todas las líneas celulares estudiadas, con un nivel significativamente

36
mayor respecto a las células de tumor localizado HT29-ATCC, lo que se explica porque estas
células corresponden a un cáncer de tipo invasivo capaz de infiltrar nódulos linfáticos,
correspondiendo a la clasificación Duke C (figura 7B).
La línea celular HT29-US proviene de las células HT29-ATCC y han sido generadas por
pasajes sucesivos de inoculación y recuperación de las metástasis de pulmón en ratones
inmunodeficientes desnudos [88], por lo tanto, tienen probablemente una capacidad
invasiva aumentada, representando así a células de un CCR humano de tipo Duke D. Como se
observa en la figura 7B, esta línea celular presentó niveles de expresión proteica de ECE-1
muy superiores a los de la línea celular parental (HT29-ATCC), lo que sugiere que el
fenómeno de transformación a una forma más maligna involucra el aumento de la expresión
de ECE-1 y, posiblemente, de la isoforma ECE-1c.

37
Finalmente, siendo survivina un blanco conocido de la vía Wnt-canónica, ésta mostró
niveles de expresión bastante similares a los observados por ECE-1 en todas las líneas
celulares (figura 7B). Estos resultados sugieren que, como sucede con el gen survivina, el
promotor de ece-1c podría ser también regulado por la vía de señalización Wnt-canónica, lo
que de ser cierto estaría de acuerdo con nuestra hipótesis.
FIGURA 7: Niveles proteicos endógenos de ECE-1 y survivina en distintas líneas celulares. A. Gel representativo de análisis de Western Blot donde se muestran los niveles de expresión de ECE-1 y survivina a partir de lisados de células normales HEK-293T y de CCR, HT29-ATCC, DLD-1 y HT29-US, estas últimas correspondiendo a las clasificaciones B, C y D de Duke, respectivamente. B. Gráfica que indica los niveles endógenos hallados de ECE-1 (barras negras) y de survivina (barras blancas) luego del
análisis densitométrico relativo a la expresión del gen constitutivo de -actina de los ensayos de western blot. Se utilizó el test estadístico Kruskal Wallis para comparación de valores de expresión de ECE-1 con distribución no paramétrica. Se halló un aumento significativo de ECE-1 entre las células HT29-ATCC y DLD-1 con un valor de P<0.001 (***). Las barras de error representan el error estándar de un total de 6 experimentos independientes.

38
La expresión de ECE-1c y survivina aumentan cuando la vía Wnt-canónica es activada y
disminuyen cuando ésta es inhibida.
Decidimos modular la actividad de la ruta Wnt-canónica en los distintos tipos
celulares anteriores mediante el tratamiento con drogas que inhiben a enzimas que
participan en ésta, regulándola positiva o negativamente.
La serina/treonina kinasa GSK3 es una enzima clave en la vía de señalización Wnt-
canónica ya que fosforila el extremo N-terminal de -catenina para marcarla y enviarla a su
degradación proteosomal y, consecuentemente, mantiene apagada la señalización Wnt-
canónica [90]. Por lo tanto, procedimos a aumentar la actividad de esta vía en las células
embrionarias no tumorales HEK-293T y en las células de cáncer HT29-ATCC y DLD-1 usando
LiCl, un conocido inhibidor de GSK3[22]. Células HEK-293T, HT29-ATCC y DLD-1 se
incubaron en presencia o ausencia de LiCl (20 mM) durante 24 h y luego se obtuvieron el
RNA y proteínas totales para realizar los ensayos de RT-PCR o western blot, respectivamente.
Como se observa en la figura 8A, el tratamiento con LiCl provocó un aumento en los
niveles de expresión de transcrito tanto de survivina como de la isoforma específica ECE-1c
en las células HEK-293T y DLD-1. Asimismo, a nivel proteico también provocó un aumento en
el nivel de expresión de ECE-1 en las células HEK-293T y HT-29ATCC (figura 8B y C). La
regulación pareció ser levemente más sensible en las células normales HEK-293T, las cuales
presentaron en promedio un 50% de aumento en el nivel proteico de ECE-1 respecto a las
células no tratadas, mientras que las células de CCR humano HT29-ATCC sólo mostraron un
aumento cercano al 36%, el cual fue estadísticamente significativo. El tratamiento con LiCl
también provocó aumentos en el nivel de expresión de survivina, lo que sirvió como control
del efecto de este compuesto en la actividad de la ruta Wnt-canónica (figura 8B).

39
FIGURA 8: Los niveles de mRNA de ECE-1c y proteína de ECE-1 aumentaron en células normales HEK-
293T y de CCR (DLD-1 y HT29-ATCC) debido al tratamiento con el inhibidor de la enzima GSK3LiCl. A. Análisis de RT-PCR mostrando la amplificación de la isoforma específica ECE-1c en células normales HEK-293T y las células DLD-1 tratadas durante 24 horas en ausencia (-) o presencia (+) de 20 mM de LiCl. B. Gel representativo de western Blot de ECE-1 y survivina desde lisados de células HEK-293T y HT29-ATCC, tratadas durante 24 horas en ausencia (-) o presencia (+) de 20 mM de LiCl. C. Gráfico de
análisis densitométrico de la expresión proteica, normalizando los niveles con -actina. Las barras negras indican nivel de expresión en células sin tratar y las barras blancas tratadas con LiCl (20 mM). Se muestran los valores promedio con sus errores estándar de un total de 3 experimentos independientes, indicando significancia estadística cuando correspondió luego de aplicar un t test de datos no pareados (*, P< 0,05).

40
Luego, analizamos si la inhibición de la vía Wnt-canónica producía algún efecto en los
niveles de mRNA y proteína de ECE-1. Para esto nuevamente usamos a survivina como un
gen blanco que es controlado por esta vía. Las 4 líneas celulares utilizadas en los
experimentos anteriores se trataron durante 24 h con un inhibidor específico de CK2 (TBB).
Resultados anteriores de nuestro laboratorio avalan el uso de este inhibidor como
modulador negativo de esta ruta puesto que la expresión y actividad de esta kinasa está
relacionada positivamente con la activación de la vía Wnt-canónica [91].
Se obtuvieron los extractos proteicos y el RNA total, para posteriormente realizar los
ensayos de WB y RT-PCR y medir, respectivamente, los niveles de proteína y mRNA de ECE-1,
ECE-1c y survivina. Como se observa en la figura 9A, los niveles de transcrito de ECE-1, ECE-1c
y survivina disminuyeron en todas las líneas celulares después del tratamiento con el
inhibidor TBB. Mientras que los niveles proteicos de ECE-1 cambiaron marcadamente,
mostrando una disminución de al menos un 50% en todas las líneas celulares tratadas con
TBB, siendo estos cambios significativos en las células de CCR DLD-1 y HT29-US, las que
poseen los mayores niveles de ECE-1 endógenos (figura 9B). Además, los niveles proteicos de
survivina también disminuyeron cuando estas células fueron tratadas con TBB, confirmando
que la actividad de la ruta Wnt-canónica fue disminuida (figura 9B).

41
FIGURA 9: La expresión de ECE-1 y survivina en células normales y de CCR disminuye al ser tratadas con un inhibidor de CK2. A. Análisis de RT-PCR de ECE-1, ECE-1c y survivina desde células no tumorales HEK-293T y tumorales HT29-ATCC, DLD-1 y HT29-US (Duke B, C y D, respectivamente), tratadas durante 24 horas en ausencia (-) o presencia (+) de 100 µM de TBB (arriba). Gel representativo de western Blot de ECE-1 y survivina desde lisados de células no tumorales HEK-293T y tumorales HT29-ATCC, DLD-1 y HT29-US, tratadas durante 24 horas en ausencia (-) o presencia (+) de 100 µM de TBB (abajo). B.
Gráfico de análisis densitométrico de los niveles proteicos, normalizando con -actina. Las barras negras indican el nivel de expresión en células sin tratar con TBB (100 µM) y las barras blancas tratadas. Se etiquetaron los valores promedio (sobre cada barra) de un total de 3 experimentos independientes, y se analizó estadísticamente los cambios mediante un t test de datos no pareados (*, P< 0,05 - **, P<0,01 - ***, P<0,001).

42
Debido a que está descrito que los inhibidores farmacológicos tienen la desventaja de
mostrar a veces efectos no específicos, se desarrolló una estrategia diferente para regular la
actividad de la vía Wnt-canónica. De este modo, células HEK-293T se transfectaron
transitoriamente con vectores de expresión que codifican para proteínas que participan
positivamente en la ruta, como son -catenina, CK2 o Akt-1 CA (constitutivamente activa).
Por el contrario, para regular negativamente la vía Wnt-canónica se transfectó con un RNA
interferente para CK2 (siRNA CK2) o un vector de expresión que codifica una mutante de
AKT que actúa como dominante negativa (AKT-1 DN).
Se analizó mediante western blot los niveles proteicos de ECE-1 y survivina en estas
células para conocer si estos tratamientos, luego de activar o inhibir la vía Wnt-canónica,
consecuentemente aumentaban o disminuían la expresión de ECE-1 y survivina. Como se
observa en la figura 10, la sobreexpresión de -catenina, CK2 y Akt-CA aumentó la
expresión de survivina y, muy interesantemente también, la de ECE-1, siendo este aumento
significativo al sobreexpresar -catenina (figura 10B).
Contrariamente, la transfección con un siRNA específico para CK2 y con el vector de
expresión de AKT-DN tuvo como efecto que los niveles de expresión de ECE-1 y survivina
fueran significativamente menores que aquellos mostrados por las células control
transfectadas con el vector vacío (mock). Tal como se esperaba, se observaron efectos
justamente opuestos en la expresión de ECE-1 al aumentar o inhibir la actividad de CK2 o
AKT, ambas kinasas regulando la ruta Wnt-canónica positivamente. Por lo tanto, estos
resultados, sumados a los mostrados en las figuras 8 y 9, apoyan fuertemente la idea de que
la vía Wnt-canónica participa positivamente en la regulación de la expresión de la ECE-1,
probablemente a través de la isoforma ECE-1c, en células normales y de CCR.

43
FIGURA 10: Los niveles proteicos de ECE-1 y survivina aumentan o disminuyen cuando se activa o inhibe la vía Wnt-canónica en células normales HEK-293. A. Gel representativo de Western Blot con células HEK-293T que fueron transfectadas con 5 µg de cada vector indicado arriba, mientras que en el caso del siRNA CK2α se utilizó una concentración de 200 nM. Se detectó ECE-1 y survivina con anticuerpos específicos. B. Gráfico de análisis densitométrico de la expresión proteica, normalizando los
niveles de ECE-1 (barras negras) y survivina (barras blancas) con -actina y presentando los resultados como porcentajes relativos al control con vector vacío arbitrariamente fijado como el 100%. Se compararon los valores promedio de expresión de cada proteína en las condiciones de transfección con respecto al vector vacío (mock) de un total de 3 experimentos independientes analizados estadísticamente mediante el t test de datos no pareados. (*, P< 0,05 - **, P<0,01 - ***, P<0,001).

44
La capacidad invasiva in vitro de las células de CCR está relacionada con la expresión
endógena de ECE-1 y es promovida por la isoforma específica ECE-1c.
Como se ha descrito anteriormente, la proteína ECE-1c está relacionada con un mayor
potencial invasivo en células de cáncer de mama, ovario y próstata [76, 80, 86]. Además,
nuestros resultados indican que los niveles de ECE-1 se encuentran aumentados en las
células que provienen de tipos de CCR con mayor grado Duke. Por consiguiente, estos datos
sugieren que ECE-1, o más específicamente ECE-1c, podría estar promoviendo un mayor
potencial invasivo en las líneas celulares de CCR. Para estudiarlo decidimos evaluar los
niveles de invasividad in vitro que presentan las tres líneas de CCR humano, HT29-ATCC, DLD-
1 y HT29-US, y la línea epitelial de ovario de hámster chino, CHO-K1. Escogimos esta última
línea celular no tumoral ya que está ampliamente descrito que presenta niveles de ECE-1 casi
despreciables, por lo que ha sido usada para estudiar y caracterizar bioquímicamente esta
enzima [92].
La células CHO-K1 son de origen epitelial y, por lo tanto, altamente diferenciadas y,
como era esperable, presentan bajos niveles de invasividad, siendo su capacidad invasiva
unas 6 veces menor que la mostrada por las células tumorales DLD-1 (figura 11A y B).
Por su parte, las líneas celulares de CCR presentaron niveles de invasividad que se
relacionaron con el grado Duke del tumor que representan. Así, las células HT29-ATCC
poseen una capacidad invasiva significativamente más baja que las células DLD-1 (figura 11A
y B), resultado esperable ya que estas células provienen de un tipo de cáncer localizado
(Duke B) y en consecuencia sin potencial invasivo.
La línea celular HT29-US, como se mencionó anteriormente, es una línea celular
obtenida artificialmente para aumentar su potencial metastásico y, como tal, presenta
niveles de invasividad 20 veces mayor que las células parentales HT29-ATCC (figura 11B).
Por último, la línea celular DLD-1 representa células de un CCR tipo Duke C, que se
caracteriza por ser una etapa avanzada con metástasis a tejido linfoide.

45
Además, fue muy interesante la correlación positiva hallada al comparar los niveles
endógenos de ECE-1 (figura 7B), con la capacidad invasiva que poseen las células de CCR
(figura 11B). Por ejemplo, las células DLD-1 presentaron los más altos niveles de invasividad
respecto a las otras dos líneas celulares de CCR y, de acuerdo con los resultados de la figura
11C, fue además la que presentó los más altos niveles de expresión de ECE-1. Así, los niveles
de expresión de ECE-1 se correlacionaron directamente con la capacidad invasiva mostrada
FIGURA 11: Los niveles basales de invasión en las líneas celulares de CCR se correlacionan con su grado de malignidad anatomopatológico. Ensayos de matrigel para determinar invasividad en líneas celulares de CCR y no tumorales CHO-K1. A. Fotografías representativas de microscopía de luz con aumento 40X indicando con flechas negras las células teñidas para la línea celular epitelial CHO-K1, y las tres líneas de CCR HT29-ATCC, DLD-1 y HT29-US. B. Gráfico del número promedio de células que invaden la matrigel en 3 experimentos distintos para cada línea celular. Se aplicó test Kruskal-Wallis para determinar si existió diferencia significativa en la capacidad invasiva entre las distintas líneas celulares, y mediante el post-test de Dunn, se halló significancia estadística entre las células HT29-ATCC y DLD-1 (*, P≤ 0,05). C. Correlación entre los niveles de expresión endógena de ECE-1 y el número de células que invaden la matrigel en células de CCR HT29-ATCC, HT29-US y DLD-1, mostrando un buen índice de correlación directa (R
2 = 0,9942).

46
por las células de CCR (figura 11C). En consecuencia, estos datos sugieren que la expresión de
ECE-1, y potencialmente la isoforma ECE-1c [86], tendría un rol importante en la capacidad
de las células de cáncer de colon para invadir y luego metastizar a otros tejidos.
Para estudiar más específicamente el rol que tendría la isoforma ECE-1c en promover
la metástasis en líneas celulares de CCR, realizamos el mismo ensayo de invasividad in vitro
pero esta vez utilizando la línea celular no tumoral de epitelio ovárico CHO-K1, las cuales son
ampliamente usadas para estudiar la ECE-1 puesto que no poseen actividad de ECE-1 y son
fácilmente transfectables. Es así como, realizamos una transfección transitoria de estas
células con el vector de expresión eucarionte pcDNA3-ECE1c, luego de lo cual se desarrolló
un ensayo de invasividad en matrigel y un análisis de Western Blot para comprobar la
expresión aumentada de la proteína. Como puede observarse en la figura 12, cuando se
sobreexpresó la isoforma especifica ECE-1c en casi 2 veces se produjo un significativo
aumento (4,5 veces) en la capacidad invasiva de estas células. Estos resultados confirmaron
el papel que tiene la isoforma ECE-1c en promover el potencial invasivo y sugiere un rol de la
misma en promover la metástasis en pacientes con cáncer de colon.
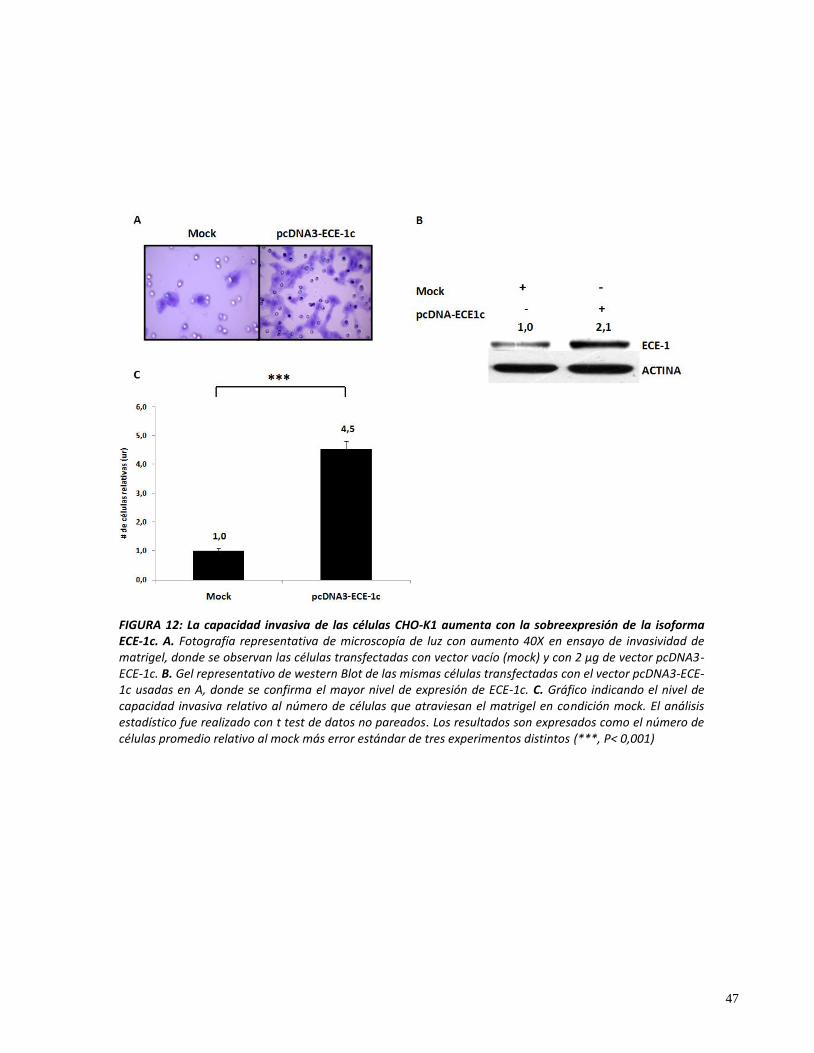
47
FIGURA 12: La capacidad invasiva de las células CHO-K1 aumenta con la sobreexpresión de la isoforma ECE-1c. A. Fotografía representativa de microscopía de luz con aumento 40X en ensayo de invasividad de matrigel, donde se observan las células transfectadas con vector vacío (mock) y con 2 µg de vector pcDNA3-ECE-1c. B. Gel representativo de western Blot de las mismas células transfectadas con el vector pcDNA3-ECE-1c usadas en A, donde se confirma el mayor nivel de expresión de ECE-1c. C. Gráfico indicando el nivel de capacidad invasiva relativo al número de células que atraviesan el matrigel en condición mock. El análisis estadístico fue realizado con t test de datos no pareados. Los resultados son expresados como el número de células promedio relativo al mock más error estándar de tres experimentos distintos (***, P< 0,001)

48
Los niveles de mRNA de ECE-1 y de la isoforma específica ECE-1c son mayores en tejido
tumoral que en tejido normal de pacientes con CCR.
Con el propósito de evaluar la existencia de una correlación entre la expresión
endógena de la isoforma ECE-1c con una mayor probabilidad de metástasis en el CCR,
cuantificamos los niveles de los mRNAs de ECE-1 total y de la isoforma específica ECE-1c
mediante RT-PCR desde biopsias de pacientes con CCR de distintos estadíos de acuerdo a la
clasificación de Duke. Se analizó 7 muestras de tejido tumoral (TT) y la misma cantidad a
partir del tejido no tumoral adyacente al tumor (TA).
Como se observa en la figura 13, el nivel promedio relativo de ECE-1 en las muestras
de tejido adyacente fue de 0,17 UR (unidades relativas), mientras que para las muestras
tumorales fue de 0,32 UR, lo que indica que existiría un 87% de aumento significativo en los
niveles de expresión de ECE-1 en pacientes con CCR con diverso grado de Duke. Asimismo, el
nivel del mensajero de ECE-1c en las muestras de tejido adyacente fue de 0,24 UR, mientras
que para las muestras tumorales fue de 0,41 UR, representando también un aumento
significativo del 70% en el nivel de expresión de ECE-1c en las muestras tumorales.
Aunque el bajo número de muestras analizadas no permite hacer un mejor estudio de
correlación entre el nivel de expresión de ECE-1c y el grado de malignidad según Duke, estos
resultados sugieren fuertemente que la isoforma ECE-1c jugaría un rol importante en la
progresión tumoral, y posiblemente la metástasis, del cáncer de colon en la población
chilena.

49
DISCUSIÓN
FIGURA 13: Niveles de mRNAs de ECE-1 y la isoforma especifica ECE-1c en biopsias de colon de pacientes chilenos. A. Gel de agarosa con bandas fluorescentes de productos de RT-PCR de ECE-1 y ECE-1c para analizar diferencias de expresión en tejidos adyacente (TA) y tumoral (TT) desde biopsias de CCR. B. Se representa la expresión del mRNA de ECE-1 y ECE-1c en tejidos adyacente (TA) y tumoral (TT), donde se grafica la normalización densitométrica de la banda de RT-PCR de 7 pacientes diagnosticados con CCR. Se muestra con línea roja el nivel promedio de expresión y se indica las diferencias significativas de expresión tanto de ECE-1 como de ECE-1c luego del análisis estadístico mediante un t test de datos no pareados (*, P< 0,05).

50
DISCUSION
La vía Wnt-canónica se encuentra activada en cáncer colorectal (CCR). De hecho, se
sabe que mutaciones en APC o -catenina se encuentran en más del 80% de los pacientes
con este tipo de cáncer. Esta ruta regula la expresión del gen de la preproET-1 [21], lo que
dependiendo de la expresión de ECE-1 podría producir también aumentos en la síntesis del
péptido protumoral ET-1. De esta forma, el aumento en la expresión de ECE-1 es una
modalidad importante de activación del péptido activo ET-1, de manera que los niveles de
expresión endógenos aumentados en las células de CCR pueden ser una señal importante de
su transformación maligna.
Por esta razón, nos interesó estudiar la expresión endógena de ECE-1 en 3 líneas
celulares de CCR humano, HT29-ATCC, DLD-1 y HT29-US, y compararla con la observada en
una línea no tumoral HEK-293T. Encontramos que los niveles de expresión proteica de ECE-1
son detectables en los 3 tipos celulares tumorales y que muestran una relación con el nivel
de malignidad anatomopatológico (grado Duke) al cual representan. Así, las células HT29-
ATCC, que corresponden a un cáncer tipo Duke B, mostraron los menores niveles de
expresión, mientras que las células HT29-US que corresponden a un tumor metastásico tipo
D de Duke, presentaron un nivel de expresión un 25% mayor que las parentales HT29-ATCC.
Por su parte, la línea celular DLD-1 se caracteriza por poseer una deleción en el gen de
APC que elimina la región de unión para -catenina, siendo incapaz de regular su
degradación proteosomal, lo que tiene como consecuencia que la vía Wnt-canónica sea
constitutivamente activa [93]. Esto podría explicar por qué estas células, que son una línea
celular de malignidad tumoral intermedia (Duke C), mostraron los más altos niveles de
expresión proteica de ECE-1, con un valor significativamente superior (38%) respecto de los
niveles expresados por la línea celular HT29-ATCC.
La línea celular no tumoral HEK-293T presentó también altos niveles de expresión de
ECE-1, lo que se explica porque estas células son de tipo embrionario y poseen varias vías de
señalización activadas, lo que las hace similar a una célula neoplásica [89]. De hecho, estas
células son ampliamente utilizadas como un modelo celular que posee la ruta Wnt-canónica
activada [88]. También la elevada expresión de ECE-1 que presentan estas células se puede

51
entender considerando esta enzima juega un rol importante en promover mitosis y sobrevida
a través de la activación del péptido ET-1 [50, 94].
Es importante destacar que los niveles de expresión endógena de survivina variaron
de una forma muy similar a los de ECE-1 en las cuatro líneas celulares estudiadas, lo que
sugirió que la ruta Wnt-canónica regulara también la expresión de ECE-1.
Como se mencionó anteriormente ECE-1 es crucial en la activación del péptido ET-1.
Nuestros resultados apoyan esta idea y otros autores lo han reportado también en cáncer
ovárico [80]. Este grupo cuantificó los niveles de péptido activo ET-1 secretado en dos líneas
celulares malignas, ES2 y OVCAR3, y observaron elevados niveles en ambas, sin embargo,
cuando se midieron los niveles de expresión del gen de la ET-1, sólo la línea celular ES2
mostraba niveles aumentados. Así, los altos niveles de ET-1 en células OVCAR3 se explicaron
porque esta línea celular expresa altos niveles de ECE-1 [80].
Por otro lado, está descrito que CK2 puede fosforilar a -catenina en treonina 393,
mejorando su estabilización y promoviendo su acumulación en citoplasma y núcleo y, en
consecuencia, aumentando la activación transcripcional de la vía Wnt-canónica [95] . Incluso
existe mayor evidencia que indicaría un rol esencial de CK2 en la activación de la vía Wnt-
canónica, ya que cuando se trata con el inhibidor de CK2, apigenina, previo a la estimulación
del receptor Frizzled, no aumenta la actividad promotora TCF, sugiriendo un rol de CK2 río
arriba de TCF y esencial en la activación de la vía Wnt-canónica [96]. Adicionalmente, en
nuestro laboratorio se ha descrito que la expresión de survivina es inhibida por el
tratamiento de las células con el inhibidor de CK2 TBB o DMAT [91]. Por esta razón, el uso de
inhibidores específicos de CK2 constituye una herramienta útil para inhibir la vía de
señalización Wnt-canónica.
Es así como, utilizando el inhibidor específico de CK2, TBB, se pudo comprobar que
existe una disminución en la expresión de la ECE-1 en las 4 líneas celulares empleadas, siendo
significativa en las células con mayor potencial maligno, DLD-1 y HT29-US. Además, no sólo
ECE-1 disminuyó sino que también lo hizo la isoforma especifica ECE-1c a nivel de mRNA (ver
figura 9).

52
En particular, los cambios en la expresión de la isoforma ECE-1c sólo pudieron ser
evaluados a nivel de mRNA, ya que a nivel proteico aún no existen anticuerpos comerciales
específicos para las isoformas de ECE-1. Los niveles de expresión proteica y de mRNA de
survivina presentaron también una disminución luego del tratamiento con el inhibidor de
CK2, confirmándose de esta manera que TBB está ejerciendo el efecto esperado, es decir,
inhibe la ruta Wnt-canónica y en consecuencia la expresión de ECE-1 (ver figura 9).
De igual modo, el tratamiento con el inhibidor de GSK3LiCl, apoyó la idea de que la
expresión de ECE-1, y específicamente ECE-1c, es regulada por la ruta Wnt-canónica. Es así
como las células no tumorales HEK-293T y tumorales HT29-ATCC y DLD-1, al ser tratadas con
el inhibidor de GSK3 y por lo tanto estimular la ruta Wnt-canónica, aumentaron los niveles
de expresión de ECE-1 y survivina a nivel proteico y específicamente de ECE-1c a nivel de
mensajero (ver figura 8).
Debido a que está descrito que el tratamiento con inhibidores puede a veces
presentar efectos inespecíficos, utilizamos la metodología de transfección transitoria de
células HEK-293T con vectores de expresión para proteínas participantes de la ruta Wnt-
canónica para regular así su activación o inhibición. Ya se ha establecido en nuestro
laboratorio que la expresión de -catenina de manera ectópica, así como CK2, promueve la
activación de la vía Wnt-canónica [91]. Asimismo, se ha descrito que la enzima AKT/PKB es
capaz de inactivar a GSK-3, permitiendo un aumento en la estabilización de -catenina [97].
También se ha descrito que AKT/PKB fosforila a -catenina, aumentando de este modo
igualmente la estabilización de -catenina cuando células HEK-293T son transfectadas con un
vector que codifica para esta enzima y por ende incrementando su actividad reportera [98].
Recientemente se ha aclarado la idea anterior, ya que se identificó que AKT/PKB
fosforila a -catenina exactamente en serina 552, lo que lleva a la disociación de su complejo
con E-cadherina, disminuyendo los contactos célula-célula y aumentando su acumulación en
el citosol y núcleo, en consecuencia mejorando la interacción con 14-3-3 y aumentando la
actividad de la ruta Wnt-canónica [99]. Debido a estos antecedentes, se valida el uso de la
expresión ectópica de AKT/PKB como una forma de regular positivamente la activación de la
ruta Wnt-canónica.

53
Los resultados de los ensayos de transfección transitoria apoyaron fuertemente
nuestra hipótesis, ya que cuando se sobreexpresó -catenina, CK2 o AKT-CA
(constitutivamente activa), las células presentaron niveles proteicos de ECE-1 bastante
superiores a los de las células control transfectadas con vector vacío, observándose
aumentos aproximados de un 80%, 120% y 160%, respectivamente (ver figura 10).
Por otra parte, cuando se transfectó con un siRNA específico para CK2α (siRNA CK2) o
con el vector que codifica para una AKT dominante negativa (la cual no tiene actividad
catalítica y compite con la enzima WT por sus sustratos), los efectos en la expresión de ECE-1
fueron también los esperados, encontrándose niveles promedio de expresión 40% y 45% más
bajos con respecto a las células control transfectadas con el vector vacío, respectivamente
(ver figura 10).
Importantemente, los incrementos en los niveles de ECE-1 generados por la
sobreexpresión de los componentes de la vía Wnt-canónica fueron congruentes con el
aumento de los niveles proteicos de survivina, mientras que se observó lo inverso cuando se
inhibió la ruta, comprobándose de esta forma que la ruta Wnt-canónica fue regulada como
se requería.
Cabe mencionar que estos datos no demuestran aún que el promotor de ECE-1c esté
siendo regulado positivamente por la vía Wnt-canónica. Para esto es necesario realizar
experimentos de actividad transcripcional mediante un vector reportero con la secuencia
promotora del gen y además con la secuencia promotora mutada en el putativo sitio de
unión al factor de transcripción TCF, el cual se une a -catenina para transactivar los genes
rio abajo de la vía Wnt-canónica. Estos experimentos fueron inicialmente considerados en
esta tesis, pero lamentablemente no se pudo diseñar el vector reportero dentro del plazo
que se disponía.
No existen actualmente reportes previos que demuestren nuestro modelo de
regulación para ECE-1. Sólo unos pocos mecanismos que regulan la expresión de ECE-1 han
sido descritos. Por ejemplo, en células endoteliales de cordón umbilical humano (HUVECs)
sometidas a condiciones de hipoxia se ha reportado un aumento en la expresión proteica de
ECE-1 y en los niveles de ET-1 [100]. También en estas mismas células tratadas en forma

54
dosis y tiempo dependiente con altas concentraciones de glucosa, se ha detectado mayores
niveles de expresión de ECE-1 a través del aumento en la expresión de PKC[101] Otros
estudios también sugieren que la sobreexpresión de ECE-1 podría ser mediada por especies
reactivas de oxígeno (ROS) en un mecanismo dependiente de la proteína activadora 1, donde
algunas kinasas como PI3K, Akt, Erk1/2 y Jun podrían estar involucradas [102].
Todos estos mecanismos de regulación convergen en un resultado similar, que la
expresión de ECE-1, y quizá más específicamente ECE-1c, son reguladas positivamente por
vías que se ven potenciadas o favorecidas en células de CCR. Una explicación razonable es
que estas células están sometidas a condiciones de hipoxia y estrés y, por lo tanto, se espera
una mayor producción de ROS [103]. Un aspecto importante es que en la mayoría de los
casos de CCR se encuentra sobreactivada la vía Wnt-canónica, de modo que tanto los
mecanismos de regulación de la ECE-1 anteriormente mencionados como el ahora propuesto
en esta tesis probablemente estarían actuando paralelamente en estas células.
El crecimiento autónomo de las células es una característica que se observa tanto en
un adenoma benigno como en un adenocarcinoma colorectal. Sin embargo, el paso esencial
en la progresión desde adenoma a carcinoma es la invasividad a otros tejidos, proceso que
requiere que la matriz extracelular sea degradada [104]. Así, las células neoplásicas que se
encuentran en un tumor localizado y tienen una alta tasa de proliferación, con poca irrigación
sanguínea y alto nivel de estrés, debieran aumentar la expresión de ECE-1/ECE-1c,
aumentando el potencial invasivo y favoreciendo el escape de las células para sobrevivir al
ambiente hostil generado [76, 80, 86].
De esta manera, nuestros resultados indican que las células de CCR que presentaron
mayores niveles de expresión de ECE-1 poseen un mayor potencial invasivo (ver figura 11C),
indicando una correlación directa, y apoyando fuertemente el rol invasivo que tendría ECE-1
en las células de CCR. Más interesante aún, nuestros resultados mostraron que la
sobreexpresión de la isoforma específica ECE-1c en células epiteliales CHO-K1 (las que
expresan niveles de ECE-1 despreciables) aumenta en forma significativa más de cuatro veces
la capacidad invasiva de estas células (ver figura 12). Esto es similar a lo mostrado por
Lambert y colaboradores en células de cáncer de próstata [86].

55
Así, los resultados anteriores sugirieron fuertemente un rol de la isoforma ECE-1c
promover la capacidad de las células de cáncer de colon para metastizar a sitios distantes.
Siendo muy probable observar que pacientes con CCR en etapas más agresivas expresen
mayores niveles de ECE-1 y ECE-1c. Por esta razón, quisimos evaluar los niveles de mRNA de
ECE-1 y de ECE-1c en tejidos de pacientes con cáncer de colon de distinto grado de
malignidad, definido por la clasificación de Duke. Esperábamos observar que aquellos
pacientes con CCR de mayor grado de Duke y, por lo tanto, con un cáncer de mayor potencial
invasivo, presentaran mayores niveles de expresión de mRNA de ECE-1c. Lamentablemente
no logramos obtener la cantidad de muestras necesarias para este estudio, aunque si
pudimos determinar que ECE-1c se encuentra aumentada en tejido tumoral respecto del
tejido sano, lo que sirvió de como un primer acercamiento para proyectar nuestros
resultados a un modelo in vivo (ver figura 13).
Es así como observamos un aumento significativo en la expresión tanto de ECE-1
como de la isoforma especifica ECE-1c en tejido tumoral comparado al tejido adyacente, en
un total de catorce muestras de biopsias de CCR. El aumento en los niveles de mRNA de ECE-
1 total, es decir las 4 isoformas, fue de aproximadamente un 86%, mientras que el mRNA de
la isoforma específica ECE-1c fue levemente menor, mostrando un aumento de
aproximadamente un 70% en las muestras provenientes desde tejido tumoral respecto del
tejido adyacente.
La diferencia observada en la expresión de tejido tumoral versus no tumoral podría
indicarnos que la isoforma que mayormente se regula positivamente en CCR es la isoforma c,
sin descartar que alguna de las otras 3 isoformas pudiera también aumentar su expresión en
menor proporción. Además, aunque no pudimos hacer la asociación entre el nivel de
expresión de ECE-1c con el grado de malignidad tumoral, se ha reportado previamente un
aumento en la expresión de esta isoforma en muestras que representan el progreso desde
un colon normal a un adenocarcinoma de colon, apoyando la idea del rol de ECE-1c en la
progresión maligna del CCR [55].
En cuanto al trabajo pendiente para responder efectivamente a la hipótesis
planteada, es necesario realizar algunos experimentos para dilucidar si el aumento en la

56
expresión de ECE-1 total luego de la regulación positiva por la vía Wnt-canónica es provocado
exclusivamente por el promotor de la isoforma ECE-1c, o si también incluye la regulación
positiva de alguna de las otras isoformas de ECE-1.
Otro punto interesante que debe continuarse indagando es aquel relacionado con el
rol que juega la ECE-1c en potenciar la capacidad invasiva de las células de CCR. En este
aspecto, faltó hacer un experimento en el cual silenciáramos la expresión de la isoforma ECE-
1c en las células de CCR, para luego determinar los niveles de invasividad in vitro mediante
ensayos de matrigel y así confirmar la participación de ECE-1c en aumentar la capacidad
invasiva en estas células.
Este trabajo puede ser el comienzo de otras investigaciones donde se estudie a esta
enzima como un nuevo blanco farmacológico para el tratamiento del CCR y, probablemente,
de otros tipos de cáncer. De este modo, generar un fármaco que permita lidiar contra un
blanco involucrado en el proceso metastásico adquiere mayor relevancia en el CCR donde se
conoce que la sobrevida a 5 años disminuye de un 90% a sólo un 10% en pacientes con un
cáncer localizado a pacientes con metástasis [5].
Actualmente ya existen antagonistas del receptor tipo A de ET-1 disponibles en forma
comercial para el tratamiento de enfermedades, como es el caso de Bosentan para la
hipertensión pulmonar, o el Atrasentan para el tratamiento del cáncer prostático refractario
al tratamiento hormonal [105]. Sin embargo, en algunos ensayos en falla cardíaca congestiva
los resultados han sido negativos, lo que ha llevado a la necesidad de desarrollar nuevos
enfoques que ataquen la formación del péptido activo ET-1, por ejemplo buscando
inhibidores de la ECE-1 [105].
Por lo tanto, el bloqueo farmacológico de la vía biosintética de ET-1 podría
representar una estrategia alternativa y/o complementaria al uso de antagonistas de los
receptores de este péptido, permitiendo un mejor bloqueo del eje endotelina. Algo similar ha
sido desarrollado para el sistema renina-angiotensina, donde el bloqueo simultáneo de la
enzima convertidora de angiotensina (ECA) y del receptor de angiotensina ha resultado ser
una estrategia eficaz en el control de la hipertensión arterial [85].

57
A pesar de los problemas propios de la realización de una tesis, los resultados
obtenidos nos permiten concluir que ECE-1c seria un nuevo blanco de la vía Wnt-canónica y
que además se comportaría como un marcador de progresión y de mal pronóstico en el CCR.
Al respecto, sería interesante continuar con estudios de esta enzima en ensayos in vivo para
llegar a controlar el crecimiento y diseminación del CCR. También dilucidar cómo ECE-1c
puede favorecer la capacidad invasiva de las células de CCR, es decir, si sólo es mediado por
el aumento en la producción del péptido activo ET-1 o si además la enzima por sí misma es
capaz de activar otros blancos con potencial tumorigénico o metastásico a través de su
función como una metaloproteasa.

58
CONCLUSIONES
• El nivel de expresión de ECE-1 es mayor en células con un nivel de actividad más alto
de la vía Wnt-canónica.
• La vía Wnt-canónica regula positivamente la expresión de ECE-1/ECE-1c en líneas
celulares de CCR.
• El nivel de expresión de ECE-1 se correlaciona con el grado de invasividad in vitro de
líneas celulares de CCR.
• La sobreexpresión de ECE-1c aumenta la capacidad invasiva de células epiteliales no
tumorales.
• Los niveles de mRNA de ECE-1c se encuentran significativamente aumentados en
muestras de pacientes chilenos con CCR.

59
MODELO PROPUESTO
Consistente con el origen epigenético del cáncer, las células que conforman el epitelio
del colon están sometidas a un ambiente rico en carcinógenos, lo que puede generar
mutaciones en algunas de ellas, otorgándoles ventaja en cuanto a proliferación acelerada
respecto a las células normales del entorno. El crecimiento acelerado hace que este pequeño
grupo de células esté sometido a una mayor probabilidad de generar sucesivas mutaciones,
desarrollándose finalmente un tumor primario. Por otro lado, consistente con el origen
genético del cáncer, en el CCR más del 80% de los casos se originan por mutaciones en
componentes de la vía Wnt-canónica, tales como APC y -catenina, que provocan la
estabilización de ésta última en el citosol y luego su migración al núcleo. En esta tesis
sugerimos que la activación de esta vía puede promover la expresión de la ECE-1c.
Probablemente otras mutaciones o cambios epigenéticos ocurridos durante el desarrollo
tumoral activaría a otras kinasas, como CK2 o AKT/PKB, que a su vez permitirían la
sobreactivación de la vía Wnt-canónica, regulando positivamente la expresión de ECE-1c. Por
último, ECE-1c anclada en la membrana de las células del tumor primario, aumentaría la
activación del péptido inmaduro big-ET-1. De esta forma, el péptido ET-1 a través de la unión
a sus receptores en células tumorales promovería la invasividad de éstas y la llegada a un
nuevo sitio de colonización o metástasis.

60
REFERENCIAS
1. Center, M.M., et al., Worldwide variations in colorectal cancer. CA Cancer J Clin, 2009. 59(6): p.
366-78.
2. Parkin, D.M., et al., Global cancer statistics, 2002. CA Cancer J Clin, 2005. 55(2): p. 74-108.
3. Burt, R.W., J.A. DiSario, and L. Cannon-Albright, Genetics of colon cancer: impact of inheritance
on colon cancer risk. Annu Rev Med, 1995. 46: p. 371-9.
4. Levine, J.S., Screening and surveillance for colorectal neoplasia: uncertainties of colonoscopic
management. Pol Arch Med Wewn, 2008. 118(5): p. 302-6.
5. Levin, B., et al., Screening and surveillance for the early detection of colorectal cancer and
adenomatous polyps, 2008: a joint guideline from the American Cancer Society, the US Multi-Society
Task Force on Colorectal Cancer, and the American College of Radiology. CA Cancer J Clin, 2008.
58(3): p. 130-60.
6. Colon Cancer. College of American Pathologists, 2006.
7. Sze, systemic treatment for colorectal cancer. The Hong Kong Medical Diary, 2007. 12(8).
8. Elahi, M.M. and N.W. Everson, Prognosis of colorectal cancer patients with elevated endothelin-1
concentrations. Asian J Surg, 2004. 27(1): p. 4-9.
9. Corman, M.L., Classic articles in colonic and rectal surgery. The classification of cancer of the
rectum. Dis Colon Rectum, 1980. 23(8): p. 605-11.
10. Larach, A., Novedades en cáncer colorectal. Rev. Med. Clin. Condes, 2006. 17(2): p. 66-9.
11. Reyes, J.M., Manejo actual del cáncer de colon y recto metastásico. Rev. Med. Clin. Condes, 2006.
17(2): p. 70-5.
12. Widelitz, R., Wnt signaling through canonical and non-canonical pathways: recent progress.
Growth Factors, 2005. 23(2): p. 111-6.
13. Tamai, K., et al., A mechanism for Wnt coreceptor activation. Mol Cell, 2004. 13(1): p. 149-56.
14. Bienz, M., beta-Catenin: a pivot between cell adhesion and Wnt signalling. Curr Biol, 2005. 15(2): p.
R64-7.
15. Kemler, R., From cadherins to catenins: cytoplasmic protein interactions and regulation of cell
adhesion. Trends Genet, 1993. 9(9): p. 317-21.
16. Polakis, P., Wnt signaling and cancer. Genes Dev, 2000. 14(15): p. 1837-51.
17. He, T.C., et al., Identification of c-MYC as a target of the APC pathway. Science, 1998. 281(5382): p.
1509-12.
18. Crawford, H.C., et al., The metalloproteinase matrilysin is a target of beta-catenin transactivation in
intestinal tumors. Oncogene, 1999. 18(18): p. 2883-91.
19. Zhang, T., et al., Evidence that APC regulates survivin expression: a possible mechanism
contributing to the stem cell origin of colon cancer. Cancer Res, 2001. 61(24): p. 8664-7.
20. Zhang, X., J.P. Gaspard, and D.C. Chung, Regulation of vascular endothelial growth factor by the
Wnt and K-ras pathways in colonic neoplasia. Cancer Res, 2001. 61(16): p. 6050-4.
21. Kim, T.H., et al., beta-Catenin activates the growth factor endothelin-1 in colon cancer cells.
Oncogene, 2005. 24(4): p. 597-604.
22. Torres, V.A., et al., Caveolin-1 controls cell proliferation and cell death by suppressing expression of
the inhibitor of apoptosis protein survivin. J Cell Sci, 2006. 119(Pt 9): p. 1812-23.
23. MacDonald, B.T., K. Tamai, and X. He, Wnt/beta-catenin signaling: components, mechanisms, and
diseases. Dev Cell, 2009. 17(1): p. 9-26.
24. Rubinfeld, B., et al., Binding of GSK3beta to the APC-beta-catenin complex and regulation of
complex assembly. Science, 1996. 272(5264): p. 1023-6.
25. Henderson, B.R. and F. Fagotto, The ins and outs of APC and beta-catenin nuclear transport.
EMBO Rep, 2002. 3(9): p. 834-9.
26. Munemitsu, S., et al., Regulation of intracellular beta-catenin levels by the adenomatous polyposis
coli (APC) tumor-suppressor protein. Proc Natl Acad Sci U S A, 1995. 92(7): p. 3046-50.
27. Harris, T.J. and M. Peifer, Decisions, decisions: beta-catenin chooses between adhesion and
transcription. Trends Cell Biol, 2005. 15(5): p. 234-7.

61
28. Lin, S.Y., et al., Beta-catenin, a novel prognostic marker for breast cancer: its roles in cyclin D1
expression and cancer progression. Proc Natl Acad Sci U S A, 2000. 97(8): p. 4262-6.
29. Kinzler, K.W. and B. Vogelstein, Lessons from hereditary colorectal cancer. Cell, 1996. 87(2): p.
159-70.
30. Nelson, J., et al., The endothelin axis: emerging role in cancer. Nat Rev Cancer, 2003. 3(2): p. 110-
6.
31. Xu, D., et al., ECE-1: a membrane-bound metalloprotease that catalyzes the proteolytic activation of
big endothelin-1. Cell, 1994. 78(3): p. 473-85.
32. Dawson, L.A., et al., Stromal-epithelial interactions influence prostate cancer cell invasion by
altering the balance of metallopeptidase expression. Br J Cancer, 2004. 90(8): p. 1577-82.
33. Rubanyi, G.M. and M.A. Polokoff, Endothelins: molecular biology, biochemistry, pharmacology,
physiology, and pathophysiology. Pharmacol Rev, 1994. 46(3): p. 325-415.
34. Clerk, A. and P.H. Sugden, Regulation of phospholipases C and D in rat ventricular myocytes:
stimulation by endothelin-1, bradykinin and phenylephrine. J Mol Cell Cardiol, 1997. 29(6): p. 1593-
604.
35. Pollock, D.M., T.L. Keith, and R.F. Highsmith, Endothelin receptors and calcium signaling. Faseb
J, 1995. 9(12): p. 1196-204.
36. Bogoyevitch, M.A., et al., Endothelin-1 and fibroblast growth factors stimulate the mitogen-activated
protein kinase signaling cascade in cardiac myocytes. The potential role of the cascade in the
integration of two signaling pathways leading to myocyte hypertrophy. J Biol Chem, 1994. 269(2): p.
1110-9.
37. Yamboliev, I.A., A. Hruby, and W.T. Gerthoffer, Endothelin-1 activates MAP kinases and c-Jun in
pulmonary artery smooth muscle. Pulm Pharmacol Ther, 1998. 11(2-3): p. 205-8.
38. Yoshizumi, M., et al., Effect of endothelin-1 (1-31) on extracellular signal-regulated kinase and
proliferation of human coronary artery smooth muscle cells. Br J Pharmacol, 1998. 125(5): p. 1019-
27.
39. Sorokin, A., et al., Protein-tyrosine kinase Pyk2 mediates endothelin-induced p38 MAPK activation
in glomerular mesangial cells. J Biol Chem, 2001. 276(24): p. 21521-8.
40. Foschi, M., et al., Biphasic activation of p21ras by endothelin-1 sequentially activates the ERK
cascade and phosphatidylinositol 3-kinase. Embo J, 1997. 16(21): p. 6439-51.
41. Flamant, M., et al., Epidermal growth factor receptor trans-activation mediates the tonic and
fibrogenic effects of endothelin in the aortic wall of transgenic mice. Faseb J, 2003. 17(2): p. 327-9.
42. Kodama, H., et al., Role of EGF Receptor and Pyk2 in endothelin-1-induced ERK activation in rat
cardiomyocytes. J Mol Cell Cardiol, 2002. 34(2): p. 139-50.
43. Cheng, T.H., et al., Reactive oxygen species modulate endothelin-I-induced c-fos gene expression in
cardiomyocytes. Cardiovasc Res, 1999. 41(3): p. 654-62.
44. Wedgwood, S., R.W. Dettman, and S.M. Black, ET-1 stimulates pulmonary arterial smooth muscle
cell proliferation via induction of reactive oxygen species. Am J Physiol Lung Cell Mol Physiol,
2001. 281(5): p. L1058-67.
45. Bagnato, A., F. Spinella, and L. Rosano, The endothelin axis in cancer: the promise and the
challenges of molecularly targeted therapy. Can J Physiol Pharmacol, 2008. 86(8): p. 473-84.
46. Grant, K., et al., Mechanisms of endothelin 1-stimulated proliferation in colorectal cancer cell lines.
Br J Surg, 2007. 94(1): p. 106-12.
47. Komuro, I., et al., Endothelin stimulates c-fos and c-myc expression and proliferation of vascular
smooth muscle cells. FEBS Lett, 1988. 238(2): p. 249-52.
48. Takuwa, N., et al., A novel vasoactive peptide endothelin stimulates mitogenesis through inositol lipid
turnover in Swiss 3T3 fibroblasts. J Biol Chem, 1989. 264(14): p. 7856-61.
49. Takuwa, Y., T. Masaki, and K. Yamashita, The effects of the endothelin family peptides on cultured
osteoblastic cells from rat calvariae. Biochem Biophys Res Commun, 1990. 170(3): p. 998-1005.
50. Eberl, L.P., et al., Endothelin receptor blockade potentiates FasL-induced apoptosis in colon
carcinoma cells via the protein kinase C-pathway. J Cardiovasc Pharmacol, 2000. 36(5 Suppl 1): p.
S354-6.
51. Peduto Eberl, L., R. Bovey, and L. Juillerat-Jeanneret, Endothelin-receptor antagonists are
proapoptotic and antiproliferative in human colon cancer cells. Br J Cancer, 2003. 88(5): p. 788-95.
52. Del Bufalo, D., et al., Endothelin-1 protects ovarian carcinoma cells against paclitaxel-induced
apoptosis: requirement for Akt activation. Mol Pharmacol, 2002. 61(3): p. 524-32.

62
53. Jamora, C., et al., Links between signal transduction, transcription and adhesion in epithelial bud
development. Nature, 2003. 422(6929): p. 317-22.
54. Jamal, S. and R.J. Schneider, UV-induction of keratinocyte endothelin-1 downregulates E-cadherin
in melanocytes and melanoma cells. J Clin Invest, 2002. 110(4): p. 443-52.
55. Egidy, G., et al., Modulation of human colon tumor-stromal interactions by the endothelin system.
Am J Pathol, 2000. 157(6): p. 1863-74.
56. Spinella, F., et al., Endothelin-1 induces vascular endothelial growth factor by increasing hypoxia-
inducible factor-1alpha in ovarian carcinoma cells. J Biol Chem, 2002. 277(31): p. 27850-5.
57. Spinella, F., et al., Endothelin-1 and endothelin-3 promote invasive behavior via hypoxia-inducible
factor-1alpha in human melanoma cells. Cancer Res, 2007. 67(4): p. 1725-34.
58. Howe, L.R., et al., Transcriptional activation of cyclooxygenase-2 in Wnt-1-transformed mouse
mammary epithelial cells. Cancer Res, 1999. 59(7): p. 1572-7.
59. Rosano, L., et al., Endothelin-1 induces tumor proteinase activation and invasiveness of ovarian
carcinoma cells. Cancer Res, 2001. 61(22): p. 8340-6.
60. Asham, E., et al., Increased endothelin-1 in colorectal cancer and reduction of tumour growth by
ET(A) receptor antagonism. Br J Cancer, 2001. 85(11): p. 1759-63.
61. Simpson, R.A., et al., Raised levels of plasma big endothelin 1 in patients with colorectal cancer. Br J
Surg, 2000. 87(10): p. 1409-13.
62. Shankar, A., et al., Raised endothelin 1 levels in patients with colorectal liver metastases. Br J Surg,
1998. 85(4): p. 502-6.
63. Sun, P., et al., Positive inter-regulation between beta-catenin/T cell factor-4 signaling and
endothelin-1 signaling potentiates proliferation and survival of prostate cancer cells. Mol Pharmacol,
2006. 69(2): p. 520-31.
64. Yildirim, Y., et al., Serum big endothelin-1 levels in female patients with breast cancer. Int
Immunopharmacol, 2008. 8(8): p. 1119-23.
65. Turner, A.J. and K. Tanzawa, Mammalian membrane metallopeptidases: NEP, ECE, KELL, and
PEX. Faseb J, 1997. 11(5): p. 355-64.
66. MacLeod, K.J., et al., Constitutive phosphorylation of human endothelin-converting enzyme-1
isoforms. J Biol Chem, 2002. 277(48): p. 46355-63.
67. Schulz, H., et al., Structure of human endothelin-converting enzyme I complexed with
phosphoramidon. J Mol Biol, 2009. 385(1): p. 178-87.
68. Emoto, N. and M. Yanagisawa, Endothelin-converting enzyme-2 is a membrane-bound,
phosphoramidon-sensitive metalloprotease with acidic pH optimum. J Biol Chem, 1995. 270(25): p.
15262-8.
69. Hasegawa, H., et al., Purification of a novel endothelin-converting enzyme specific for big
endothelin-3. FEBS Lett, 1998. 428(3): p. 304-8.
70. Shimada, K., et al., Rat endothelin-converting enzyme-1 forms a dimer through Cys412 with a
similar catalytic mechanism and a distinct substrate binding mechanism compared with neutral
endopeptidase-24.11. Biochem J, 1996. 315 ( Pt 3): p. 863-7.
71. Korth, P., et al., Cellular distribution of endothelin-converting enzyme-1 in human tissues. J
Histochem Cytochem, 1999. 47(4): p. 447-62.
72. Yanagisawa, H., et al., Dual genetic pathways of endothelin-mediated intercellular signaling revealed
by targeted disruption of endothelin converting enzyme-1 gene. Development, 1998. 125(5): p. 825-
36.
73. Pekonen, F., et al., Human endometrial adenocarcinoma cells express endothelin-1. Mol Cell
Endocrinol, 1992. 84(3): p. 203-7.
74. Gu, H., et al., Association of endothelin-converting enzyme-1b C-338A polymorphism with gastric
cancer risk: a case-control study. Eur J Cancer, 2008. 44(9): p. 1253-8.
75. Dawson, L.A., et al., Expression and localization of endothelin-converting enzyme-1 in human
prostate cancer. Exp Biol Med (Maywood), 2006. 231(6): p. 1106-10.
76. Smollich, M., et al., On the role of endothelin-converting enzyme-1 (ECE-1) and neprilysin in human
breast cancer. Breast Cancer Res Treat, 2007. 106(3): p. 361-9.
77. Ahmed, S.I., et al., Studies on the expression of endothelin, its receptor subtypes, and converting
enzymes in lung cancer and in human bronchial epithelium. Am J Respir Cell Mol Biol, 2000. 22(4):
p. 422-31.

63
78. Naidoo, V., et al., Localization of the endothelin system in human diffuse astrocytomas. Cancer,
2005. 104(5): p. 1049-57.
79. Van Beneden, R., et al., Increased expression of endothelin-1 converting enzyme in human thyroid
carcinoma. Clin Endocrinol (Oxf), 2004. 60(1): p. 146-7.
80. Rayhman, O., et al., Small interfering RNA molecules targeting endothelin-converting enzyme-1
inhibit endothelin-1 synthesis and the invasive phenotype of ovarian carcinoma cells. Cancer Res,
2008. 68(22): p. 9265-73.
81. Berger, Y., et al., Endothelin-converting enzyme-1 inhibition and growth of human glioblastoma
cells. J Med Chem, 2005. 48(2): p. 483-98.
82. Orzechowski, H.D., et al., Evidence of alternative promoters directing isoform-specific expression of
human endothelin-converting enzyme-1 mRNA in cultured endothelial cells. J Mol Med, 1997. 75(7):
p. 512-21.
83. Schweizer, A., et al., Human endothelin-converting enzyme (ECE-1): three isoforms with distinct
subcellular localizations. Biochem J, 1997. 328 ( Pt 3): p. 871-7.
84. Valdenaire, O., et al., A fourth isoform of endothelin-converting enzyme (ECE-1) is generated from
an additional promoter molecular cloning and characterization. Eur J Biochem, 1999. 264(2): p.
341-9.
85. Turner, A.J., et al., Isoforms of endothelin-converting enzyme: why and where? Trends Pharmacol
Sci, 1998. 19(12): p. 483-6.
86. Lambert, L.A., et al., Isoforms of endothelin-converting enzyme-1 (ECE-1) have opposing effects on
prostate cancer cell invasion. Br J Cancer, 2008. 99(7): p. 1114-20.
87. Funke-Kaiser, H., et al., Characterization of the c-specific promoter of the gene encoding human
endothelin-converting enzyme-1 (ECE-1). FEBS Lett, 2000. 466(2-3): p. 310-6.
88. Torres, V.A., et al., E-cadherin is required for caveolin-1-mediated down-regulation of the inhibitor
of apoptosis protein survivin via reduced beta-catenin-Tcf/Lef-dependent transcription. Mol Cell Biol,
2007. 27(21): p. 7703-17.
89. Al-Hajj, M. and M.F. Clarke, Self-renewal and solid tumor stem cells. Oncogene, 2004. 23(43): p.
7274-82.
90. Bienz, M. and H. Clevers, Linking colorectal cancer to Wnt signaling. Cell, 2000. 103(2): p. 311-20.
91. Tapia, J.C., et al., Casein kinase 2 (CK2) increases survivin expression via enhanced beta-catenin-T
cell factor/lymphoid enhancer binding factor-dependent transcription. Proc Natl Acad Sci U S A,
2006. 103(41): p. 15079-84.
92. Schmidt, M., et al., Molecular characterization of human and bovine endothelin converting enzyme
(ECE-1). FEBS Lett, 1994. 356(2-3): p. 238-43.
93. Morin, P.J., et al., Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-
catenin or APC. Science, 1997. 275(5307): p. 1787-90.
94. Del Bufalo, D., et al., Endothelin-1 acts as a survival factor in ovarian carcinoma cells. Clin Sci
(Lond), 2002. 103 Suppl 48: p. 302S-305S.
95. Song, D.H., et al., CK2 phosphorylation of the armadillo repeat region of beta-catenin potentiates
Wnt signaling. J Biol Chem, 2003. 278(26): p. 24018-25.
96. Gao, Y. and H.Y. Wang, Casein kinase 2 is activated and essential for wnt/beta -catenin signaling. J
Biol Chem, 2006.
97. Fukumoto, S., et al., Akt participation in the Wnt signaling pathway through Dishevelled. J Biol
Chem, 2001. 276(20): p. 17479-83.
98. Tian, Q., et al., Proteomic analysis identifies that 14-3-3zeta interacts with beta-catenin and
facilitates its activation by Akt. Proc Natl Acad Sci U S A, 2004. 101(43): p. 15370-5.
99. Fang, D., et al., Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional
activity. J Biol Chem, 2007. 282(15): p. 11221-9.
100. Khamaisi, M., et al., Diabetes and radiocontrast media increase endothelin converting enzyme-1 in
the kidney. Kidney Int, 2008. 74(1): p. 91-100.
101. Khamaisi, M., et al., Role of protein kinase C in the expression of endothelin converting enzyme-1.
Endocrinology, 2009. 150(3): p. 1440-9.
102. Soh, J.W., et al., Novel roles of specific isoforms of protein kinase C in activation of the c-fos serum
response element. Mol Cell Biol, 1999. 19(2): p. 1313-24.
103. Dang, C.V. and G.L. Semenza, Oncogenic alterations of metabolism. Trends Biochem Sci, 1999.
24(2): p. 68-72.

64
104. Brabletz, T., et al., beta-catenin regulates the expression of the matrix metalloproteinase-7 in human
colorectal cancer. Am J Pathol, 1999. 155(4): p. 1033-8.
105. Brands, M., et al., Selective indole-based ECE inhibitors: synthesis and pharmacological evaluation.
ChemMedChem, 2006. 1(1): p. 96-105.