try not to become a man of success but rather try to...
TRANSCRIPT

I
General introduction
"Try not to become a man of success but rather try to become a man of
value.”
~Albert Einstein

1
Chapter _ I
1.1. KINETIC STUDIES
iving organisms are all chemically reacting systems depending on the
continuance of oxidations and reductions for plants proliferate by using radiant
energy to reduce carbon dioxide whilst animals obtain their vital energy by the
oxidation of carbon compounds. Thus, the comprehension of mechanisms of
oxidation and reduction processes is essential to the understanding of the nature
of life and it is a task for the chemists to provide a clear picture of way in
which these chemical changes may take place with simple molecules, so that
biologists have a sure basis for their understanding of these behavior of
complex organisms.
Man knew chemical reactions long before chemistry had attained the
status of science and sensed that substances changed their properties under
certain external conditions, which is a characteristic of chemical reactions.
Kinetic studies are receiving much importance in the recent years. Since, it is
such a part of science that deals with the rate of chemical reaction, with all
factors, which influence the rate of reaction, and with the explanation of the
rate in terms of the reaction mechanism. To many chemists the real heart of
chemistry is the study of mechanisms. Thus, chemical kinetics can be defined
as that branch of chemistry concerned with the study and prediction of time
dependent systems.
Chemical kinetics with its dynamic viewpoint may be contrasted with
thermodynamics with its static viewpoint. Thermodynamics, a powerful tool
L
K

2
Chapter _ I
for investigating the conditions at equilibrium concerns mainly on the state of
equilibrium. Kinetics is concerned fundamentally with the details of the
process in which a system changes from state to other in a definite transition
state. The kinetic data will be the source of a great deal of detailed insight into
the mechanism of a reaction. Although, other types of experimental evidences
are also sought for purpose of formulating a reaction mechanism, the study of
reaction kinetics generally forms the backbone of a thorough mechanistic
investigation. Finally, as an area of pure science in itself, the study of rates and
mechanism is one of rich varieties, concerned with the chemistry of every
element and full of experimental challenges.
To understand the mechanism of any reaction we must know a reaction
as a function of time. The order of a reaction can be used to interpret the
reaction on molecular level. Chemists propose reaction mechanism by
predicting the sequence in which bonds break and atoms rearrange during the
reaction by considering the order of a reaction with respect to different reactive
species. Virtually all information regarding reaction mechanism comes by
inference of indirect evidence. Hence, it is the important job of chemists to
device the proper experiments to generate most conclusive confirmation. The
crucial steps in any kinetic investigation1 are: (1) product and intermediate
detection, (2) concentration determination of all species present, (3) deciding
on a method of following the rate, (4) the kinetic analysis and (5) determination
of the mechanism.

3
Chapter _ I
The award of Nobel Prize for the year 1992 to Prof. R. A. Marcus on the
“Electron Transfer Reactions”, 1999 to Prof. Ahmed Zewail for discovery in
“Femtochemistry”, Nobel Prize 2001 to Profs. William Knowles, K. Barry
Sharpless and Royji Noyori for their work on “Chirally Catalyzed
Hydrogenation Reactions” and Nobel Prize for the year 2005 to Profs. Robert
Grubbs, Richard Schrock, and Yves Chauvin on their research achievements on
“Metathesis Catalyst Technology” emphasize the importance of field of
reaction kinetics. Electron transfer reactions play a significant role in physical,
chemical and biological processes. Because of the ubiquity of electron transfer
processes, the study of electron transfer reactions, perhaps more so than that of
any other area of chemistry is characterized by a strong interplay of theory and
experiment2. The importance of electron transfer in transition metal redox
chemistry and in organic chemistry is well documented.3
The work of Henry Taube4 in redox systems unequivocally
demonstrated the transport of electron from reductant to oxidant. This
discovery certainly added many important features in the syntheses of
coordination complexes and organometallics. An oxidation process is always
accompanied by a reduction process;5 such reactions are called redox reactions.
Therefore, oxidation-reduction reaction needs at least two reactants, one
capable of gaining electrons (oxidant) and the other capable of losing electrons
(reductant), i.e., a reducing agent (reductant) by losing electrons, gets oxidized
and an oxidizing agent (oxidant) by gaining the electrons gets reduced. Redox

4
Chapter _ I
reactions are the basis for numerous biochemical pathways and cellular
chemistry, biosynthesis, and regulation.6
1.1.1. Oxidation-reduction in inorganic reactions
Two general classes of transition states emerge for redox reactions
involving metal complexes, the so called “outer-sphere and inner-sphere”
types.7 In outer sphere, electron transfer event occurs between chemical species
that remain separate intact before, during, and after the electron transfer
occurrence, whereas, in the inner sphere electron transfer the participating
redox sites undergoing electron transfer become connected by a chemical
bridge. Since in outer sphere electron transfer occurs between two non-
connected species, the electron is forced to move through space from
one redox center to the other. From Franck-Condon principle, it follows that
before electron transfer between two ions is possible, the energy of the electron
must be the same in the two sites. There must also be sufficient orbital overlap
between the two sites to provide for a reasonable probability of a transfer.
1.1.2. Oxidation-reduction in organic reactions
The oxidation-reduction concepts, however, are not so clearly applicable
in organic chemistry, when carbon compounds are oxidized their component

5
Chapter _ I
atoms are very seldom deprived of their surrounding complete electron shells.
Covalent bond fission is an essential feature of organic reactions and it can be
affected by two different pathways,7 viz., “Homolytic reactions” in which
electron pairs are symmetrically disrupted and “Heterolytic reactions” in
which electron pairs are transferred from one molecule to another as an
undivided entity. Electron removal by these two pathways has clearly
distinguishable characteristics.
1.1.3. Probable ways of electron transfer reactions
Oxidation-reduction reaction may involve one or more electron transfer.
Depending upon the number of electrons transferred between oxidant and
reductant, the reaction may proceed in one or more steps. Transition metals
such as iron and cobalt and several others usually exhibit stable oxidation states
differing by one electron and react with each other through one equivalent
steps. However, the stable oxidation states in post transition elements such as
arsenic, antimony etc., differ by two electrons. Thus, on the basis of their
pattern of reactivity, the reactions of these elements are classified into two
main categories,5,8
Two main types of electron-transfer reactions9,10
are
“Complementary reactions”11
in which the oxidant and reductant change their
oxidation state by an equal number of units, and “Non-complementary
reactions”12
in which the oxidant and the reductant change their oxidation state
by a different number of units.

6
Chapter _ I
Electron transfer reactions are governed by two classical principles:
(a) Michaelis principle of compulsory univalent oxidation steps13
(b) Shaffer’s principle of equivalent change14
Michaelis hypothesis states that an oxidation-reduction reaction takes
place in one or more successive single electron transfer steps, apart from the
reactions involving metal ions, many two equivalent redox reactions are now
known which proceed in one step through the transfer of hydride ion or an
oxygen atom.15
Example:
The second principle15
refers to the observation that non-complementary
reactions are often slow compared with complementary ones. Examples are the
slow reduction of thallium(III) by iron(II) or cerium(IV) by thallium(I) as
compared to the rapid reduction of thallium(III) by tin(II) and cerium(IV) by
iron(II).
1.1.4. Unstable oxidation states
The formation of unstable oxidation state during the course of non-
complementary reactions has been now anticipated in a number of such
reactions with sufficient proofs. For example, the reductions of thallium(III) by
iron(II),16
vanadium(III) or vanadium(IV)17,18
and chromium(VI) by
thallium(I)19
can only be explained through the formation of unstable
thallium(II) species. Similar unstable oxidation states have been observed in
NO3- + Cl
- NO2
- + OCl
-

7
Chapter _ I
other studies.20,21
The inter conversions between chromium(III) and
chromium(VI) always appear to involve the unstable states, chromium(IV) and
chromium(V). A number of studies of the catalysis by platinum metals of
oxidation reactions have been made.22
The catalysis by Ag(I),23
Cu(II),24
Mn(III)25
and Cr(III)26,27
in oxidation–reduction reactions are also found to
occur through formation of unstable oxidation states.
1.1.5. Active species
If a particular substance (oxidant, reductant or catalyst) is capable of
existence in several forms in aqueous solution, all the species existing may not
be active. Those species, which are involved in a slow step, will influence the
reaction. The reaction conditions will determine the nature of the active
species.
1.1.6. Catalysis
Any substance, other than reactants which influences the rate of
chemical reaction and itself remains unchanged chemically at the end, is called
a catalyst. The phenomenon of rate alteration is designated as catalysis.
Catalysts influence the reactions by changing the reaction path. Such catalytic
influences arise as consequences of lowering of the energy of activation.
In solution involving inorganic oxidations, the catalysts are ions having
unstable oxidation states. This case is a particular example of homogeneous

8
Chapter _ I
catalysis where the catalysts present in the same phase as a part of reactants.
Homogeneous catalysts are readily amenable to kinetic and mechanistic
studies, in particular using in situ spectroscopic studies. Homogeneous
catalysts are uniform, reproducible and easily modified in a controlled manner.
Though, the mechanism of catalysis depends on the nature of the substrate,
oxidant and other experimental conditions, it has been shown that metal ion
acts as catalyst by different pathways.28
Catalysis plays a vital role in the production of fuels, commodity
chemicals, fine chemicals, and pharmaceuticals, as well as providing the means
for strengthening environmental safeguards all over the world. More than 60%
of all chemical products and 90% of chemical processes are based on catalysis.
Catalysis is the most effective and most rational of all means for accelerating
the chemical reactions. Most of the technological processes introduced recently
in chemical industry incorporate catalytic reactions. In presence of catalyst the
rates of reactions are accelerated thousands and millions of times and they take
place at low temperature, which is an economical advantage. Catalysis is
utilized in manufacturing some of the most important inorganic products such

9
Chapter _ I
as hydrogen, ammonia etc. Often, very small quantities of catalysts like
Os(VIII),29
Pd(II),30
Cr(III),31
Ru(III),32
V(V)33
etc., causes appreciable rates
accelerations of particular reactions.
1.1.7. Applications of kinetics
Kinetics has a wide variety of applications in the world of medicine. In
addition to the ways in which the human body undergoes processes such as
respiration and metabolism, kinetics also play a large part in the administration
of drugs. For example, the mechanisms for the sustained release of drugs are
based on the half-life of the substances used and sometimes the pH of the body
as well. Practically, this affects the way in which dosages are determined and
prescribed. The rate of reactions and the various conditions under which they
occur are crucial for determining certain aspects of environmental protection.
For example, the destructive effect of chloroflourocarbons (CFCs) on the ozone
layer is best understood through an analysis of catalyzed chemical reactions.
The mathematical models that describe chemical reaction kinetics provide
chemists and chemical engineers with tools to better understand and describe
chemical processes such as food decomposition, microorganism growth,
stratospheric ozone decomposition, and the complex chemistry of biological
systems. These models can also be used in the design or modification of
chemical reactors to optimize product yield, more efficiently separate products,
and eliminate environmentally harmful by-products.

10
Chapter _ I
1.2. VOLTAMMETRIC STUDIES
Electrochemistry is a powerful and sensitive analytical tool used for
both qualitative and quantitative analysis over a wide range of concentrations.
The efficacy of electrochemical methods stems not only from their sensitivity
to trace amounts and the simplicity of the instrumentation, but also because
these methods can be used for separation of ionic species in addition to their
detection. Voltammetric technique is concerned mainly with the measurement
of electrical quantities, such as current, potential, or charge, and their
relationship to the chemical parameters.
The beginning of voltammetry was facilitated by the discovery
of polarography in 1922 by the Nobel Prize winning Chemist Jaroslav
Heyrovsky (Nobel Prize in 1959). Early voltammetric techniques had many
problems, limiting their viability for everyday use in analytical chemistry. In
1942, Hickling built the first three electrodes potentiostat.34
The 1960s and
1970s saw many advances in the theory, instrumentation, and the introduction
of computer added and controlled systems. These advancements improved
sensitivity and created new analytical methods. The coincidence of these
advances with the advent of low cost operational amplifiers also facilitated the
rapid commercial development of relatively inexpensive instrumentation.
Electron transfer plays a fundamental role in governing the pathways of
chemical reactions. Measurement of speed of the electron transfer process and
the number of electrons involved are difficult in traditional experimental

11
Chapter _ I
methods like Spectroscopy. Consequently our knowledge of the driving force
for many reactions remains elusive. Electrochemical methods offer the
potential to investigate these processes directly by the determination of the
number of electrons involved.35
1.2.1. Instrumentation
Electrochemistry is the study of reactions in which charged particles
(ions or electrons) cross the interface between two phases of matter, typically a
metallic phase (the electrode) and a conductive solution, or electrolyte. A
voltammetric system consists of an electrolysis cell, a potentiostat, a current-to-
voltage converter, and a data acquisition system. The electrolysis cell consists
of a working (indicator) electrode, counter (auxiliary) electrode, reference
electrode, and electrolytic solution. The working electrode’s potential is varied
linearly with time, while the reference electrode maintains a constant potential.
The counter electrode conducts electricity from the signal source to the
working electrode. The purpose of the electrolytic solution is to provide ions to
the electrodes during oxidation and reduction. A potentiostat is an electronic
device which uses a dc power source to produce a potential which can be
maintained and accurately determined, while allowing small currents to be
drawn into the system without changing the voltage. The current-to-voltage
converter measures the resulting current, and the data acquisition system
produces the resulting voltammogram.

12
Chapter _ I
1.2.2. Voltammetric techniques and their theoretical aspects
The different voltammetric techniques that are used are distinguished
from each other primarily by the potential function that is applied to the
working electrode to drive the reaction and by the material used as the working
electrode. These can be described as follows;
Cyclic voltammetry (CV): is popular for its relative simplicity and its
high information content. It is used most often as a diagnostic tool for
elucidating electrode mechanisms. Although it was first practiced at a hanging
mercury drop electrode,36
it gained widespread use when solid electrodes like
Pt, Au and carbonaceous electrodes were used, particularly to study anodic
oxidations.37
It is a potential sweep technique. The potential of the working electrode
is measured against a reference electrode which maintains a constant potential,
and the resulting applied potential produces an excitation signal such as that of
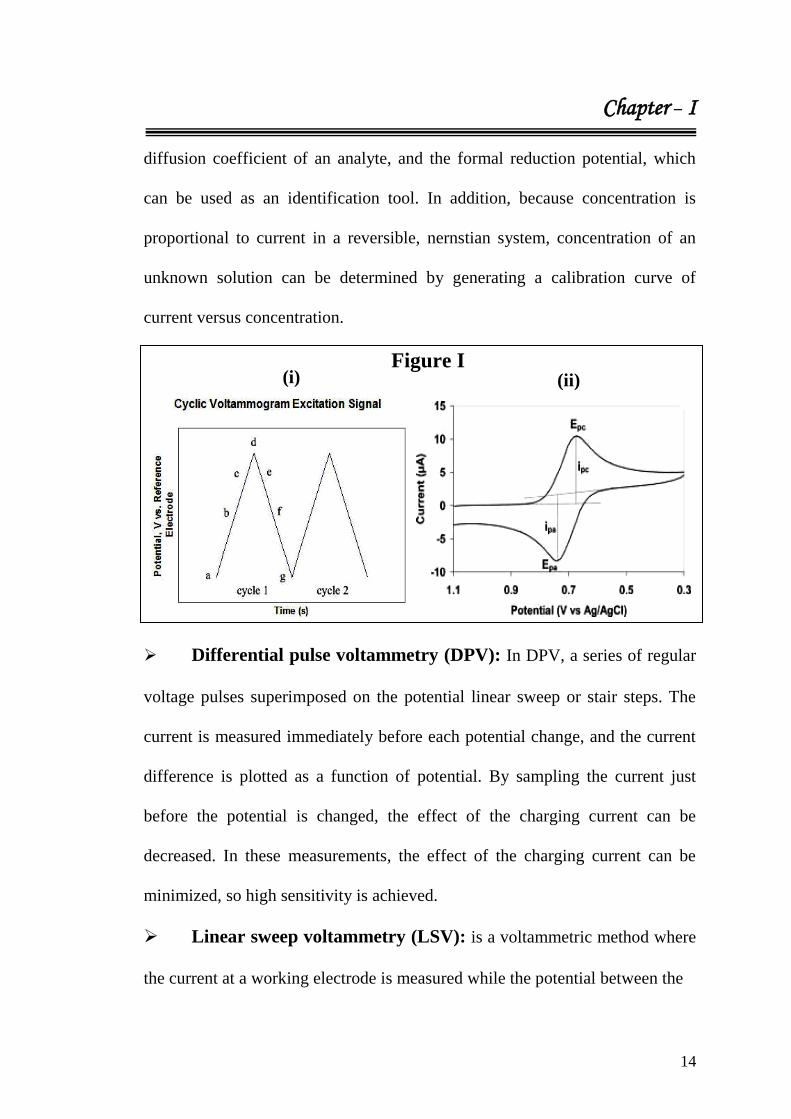
Figure I (i) (p. 14).38
In the forward scan, the potential first scans negatively,
starting from a greater potential (a) and ending at a lower potential (d). The
potential extrema (d) is called the switching potential, and is the point where
the voltage is sufficient enough to have caused an oxidation or reduction of an
analyte. The reverse scan occurs from (d) to (g), and is where the potential
scans positively. It shows a typical reduction occurring from (a) to (d) and an
oxidation occurring from (d) to (g). It is important to note that some analytes
undergo oxidation first, in which case the potential would first scan positively.

13
Chapter _ I
This cycle can be repeated, and the scan rate can be varied. The slope of the
excitation signal gives the scan rate used. Cyclic voltammetry is generally used
to study the electrochemical properties of an analyte in solution. A cyclic
voltammogram is obtained by measuring the current at the working electrode
during the potential scans. Figure I (ii) (p. 14) shows a cyclic voltammogram
resulting from a single electron reduction and oxidation. Consider the
following reversible reaction:
M+ + e
- <=> M
In Figure I (ii) (p. 14), the reduction process occurs from (a) the initial
potential to (d) the switching potential. In this region the potential is scanned
negatively to cause a reduction. The resulting current is called cathodic current
(ipc). The corresponding peak potential occurs at (c), and is called the cathodic
peak potential (Epc). The Epc is reached when all of the substrate at the surface
of the electrode has been reduced. After the switching potential has been
reached (d), the potential scans positively from (d) to (g). This results in anodic
current (Ipa) and oxidation to occur. The peak potential at (f) is called the
anodic peak potential (Epa), and is reached when all of the substrate at the
surface of the electrode has been oxidized.
Cyclic voltammetry can be used to study qualitative information about
electrochemical processes under various conditions, such as the presence of
intermediates in oxidation-reduction reactions, the reversibility of a reaction.
CV can also be used to determine the electron stoichiometry of a system, the
14
Figure I (ii) (i)
Chapter _ I
diffusion coefficient of an analyte, and the formal reduction potential, which
can be used as an identification tool. In addition, because concentration is
proportional to current in a reversible, nernstian system, concentration of an
unknown solution can be determined by generating a calibration curve of
current versus concentration.
Differential pulse voltammetry (DPV): In DPV, a series of regular
voltage pulses superimposed on the potential linear sweep or stair steps. The
current is measured immediately before each potential change, and the current
difference is plotted as a function of potential. By sampling the current just
before the potential is changed, the effect of the charging current can be
decreased. In these measurements, the effect of the charging current can be
minimized, so high sensitivity is achieved.
Linear sweep voltammetry (LSV): is a voltammetric method where
the current at a working electrode is measured while the potential between the

15
Chapter _ I
working electrode and a reference electrode is swept linearly in time. Oxidation
or reduction of species is registered as a peak or trough in the current signal at
the potential at which the species begins to be oxidized or reduced.
Square wave voltammetry (SWV): In this technique, a square
wave of constant amplitude is superimposed on the potential staircase sweep.
In this voltammetry the potential sweep is a series of stair steps. Due to the
lesser contribution of capacitative charging current the detection limits for
SWV are on the order of nanomolar concentrations.
Normal pulse voltammetry (NPV): In NPV, each potential step
begins at the same value and the amplitude of each subsequent steps increases
in small increments. The resulting voltammogram displays the sampled current
on the vertical axis and the potential to which the pulse is stepped on the
horizontal axis.
1.2.3. Electron transfer or charge transfer process
The electron transfer at the interface between the electrode and
electrolyte is central to an electrode reaction. Electro active species having
moved from the bulk of the solution by either diffusion or under forced
convection enters in the electrical double layer, which is under direct influence
of the electrode. On entering the double layer the species undergoes a structural
orientation so that it can take up or give up electrons from or to the electrode

16
Chapter _ I
surface respectively with the least activation energy when a suitable potential is
applied and macroscopically, we observe a current. This state of the reactant
species is known as transition state. Being unstable the species in transition
state converts itself to the final product by release of activation energy and gets
reduced or oxidized. This final product after undergoing suitable re-orientation
either gets deposited on the electrode surface or moves away from the electrode
surface into the bulk solution. The transfer of an electron to or from the
substrate is an activated process. The electron transfer process can be
“Reversible process” in which the electron transfer is much faster than the
mass transfer, “Irreversible process” in which electron transfer is much
slower than the mass transfer and “Quasi-reversible process” in which
electron transfer and mass transport has comparable rates.
1.2.4. Electrodes
The advent of modern electrochemistry created the need for new
electrodes and electrode set-ups. The most common arrangement today is the
electrochemical cell with three electrodes:
Working electrode (WE)
Reference electrode (RE)
Counter electrode (CE)
immersed in a solvent containing the analyte and also an excess of supporting
electrolyte. The choice of the solvent is dictated by the solubility of the analyte

17
Chapter _ I
and its redox activity and by solvent properties.39
Supporting electrolytes are
required in controlled-potential experiments to decrease the resistance of the
solution and also to maintain constant ionic strength.
1.2.4.1. Working electrode
The working electrode of an electrode system is the electrode, on the
surface of which, the analyte is oxidized or reduced (i.e. the electrode at which
the electrochemical reaction takes place). The active potential of the working
electrode is measured against a standard reference electrode. The working
electrode, where the reactions are studied, is usually made of noble metals,
carbon, or mercury. The most widely used metals are mercury, platinum, gold
and various forms of carbon. Numerous characteristics are required for this
kind of electrode: high signal-to-noise and reproducible response, no
interfering reactions in a certain range of potential, high electrical conductivity,
low surface capacitance, mechanical and chemical stability, low cost,
availability, low toxicity and long term stability.
1.2.4.2. Reference electrode
A reference electrode is an electrode which has a stable and well-
known electrode potential. The reference electrode is used as a half cell to build
an electrochemical cell. This allows the potential of the other half cell to be

18
Chapter _ I
determined. The calomel electrode and the silver/silver chloride electrode are
most commonly used reference electrodes.
1.2.4.3. Counter electrode
In voltammetric studies, the current flows between the working
electrode and counter electrode. Although the main interest is on the working
electrode, it must be ensured that the counter electrode does not complicate the
matters. It must not dissolve in the medium. The reaction product at the CE
must reach or react at the working electrode.
The counter electrode passes all the current needed to balance the
current observed at the working electrode. To achieve this current, the auxiliary
will often swing to extreme potentials at the edges of the solvent window,
where it oxidizes or reduces the solvent or supporting electrolyte. Platinum
electrodes fortunately meet most of these requirements and this is the most
widely used counter electrode in aqueous, non-aqueous as well as molten salt
media.
1.2.5. Applications of voltammetry
Voltammetry is the most effective and versatile electroanalytical
technique available for the mechanistic study of redox systems.40
It enables the
electrode potential to be rapidly scanned in search of redox couples once

19
Chapter _ I
located, a couple can then be characterized from the potential of peaks on the
cyclic voltammogram and from changes caused by variation of the scan rate.
The capability together with its variable time scale and good sensitivity
make this the most versatile electroanalytical technique. The determinations of
organic compounds in pharmaceuticals include many classes of drugs:
antibiotics, diuretics, antineoplastics, antidepressants, muscle relaxants,
neuroleptics, analgesics, vitamins, hormones, and others. Differential pulse
voltammetry/polarography (DPV/DPP) has been extremely useful for
determination of low amounts of electro-active compounds in pharmaceuticals.
A number of excellent voltammetric techniques are well described in the
literature.41,42
Recent advances in instrumental techniques, however, promise access to
molecular-level information about electrochemical systems that has been
unavailable. This exciting development opens up important new opportunities
in fundamental and applied science. In the future, it may become possible to
produce organized networks of molecules resembling, in their controlled
structure, biological systems, yet having properties different from those of any
material known today.

20
Chapter _ I
1.3. REFERENCES
1. M. R. Wright,
“An Introduction to Chemical Kinetics”, John Wiley and Sons, Inc.,
New York, (2004)
2. J. J. Zuckerman,
“Inorganic Reactions and Methods”, Vol. 15, VCH Publishers, Florida,
(1986)
3. Sir. G. Wilkinson,
“Comprehensive Coordination Chemistry”, Vol. 1, Pergamon Press,
(1987) p.332
4. H. Taube,
“Electron Transfer Reactions of Metal Complexes in Solution”,
Academic Press, New York, (1967)
5. J. Hrbac and R. Kohen,
Drugs Dev. Res., 50, 516 (2000)
6. M. Shapiro,
J. Surg. Res., 3, 138 (1972)
7. W. A. Waters,
“Mechanisms of Oxidation of Organic Compounds”, Methuen and
Co. Ltd., London, (1964) p.35
8. D. Benson,
“Mechanisms of Inorganic Reactions in Solution”, McGraw-Hill,
London, (1968)
9. F. Basolo and R. G. Pearson,
“Mechanisms of Inorganic Reactions”, 2nd Ed., John Wiley and Sons,
Inc., New York, (1967) p.454, 474 and 490
10. N. Sharma, R. Varadarajan, S. K. Mishra and P. D. Sharma,
Chem. Edu., 4, 38 (1988)
11. W. U. Malik, G. D. Tuli and R. D. Madan,
“Selected Topics in Inorganic Chemistry”, 6th Ed., S. Chand and
Company Ltd., New Delhi, (1996) p. 549

21
Chapter _ I
12. K. B. Wiberg,
“Oxidation in Organic Chemistry”, Academic Press, New York, (1965)
p.80
13. L. Michaelis,
Trans. Electrochem. Soc., 71, 107 (1937)
14. P. A. Shaffer,
J. Am. Chem. Soc., 55, 2169 (1933)
15. M. Anabar and H. Taube,
J. Am. Chem. Soc., 80, 1073 (1958);
R. Stewart,
Experimentia, 15, 401 (1959)
16. C. E. Johnson, Jr.,
J. Am. Chem. Soc., 74, 959 (1952);
K. J. Ashurst and W. C. E. Higginson,
J. Chem. Soc., 3044 (1953)
17. W. C. E. Higginson, D. R. Rosseinsky, J. B. Stead and A. G. Sykes,
Disc. Faraday Soc., 29, 49 (1960)
18. G. A. Hiremath, P. L. Timmanagoudar and S. T. Nandibewoor,
Pol. J. Chem., 70, 364 (1996)
19. S. A. Chimatadar, S. B. Koujalagi and S. T. Nandibewoor,
Transition Met. Chem., 27, 704 (2002)
20. S. A. Chimatadar, T. Basavaraj and S. T. Nandibewoor,
Inorg. React. Mech., 4, 209 (2002)
21. K. A. Thabaj, D. S. Munavalli, S. A. Chimatadar and S. T.
Nandibewoor,
Transition Met. Chem., 32, 902 (2007)
22. K. T. Sirsalmath, C.V. Hiremath and S. T. Nandibewoor,
Appl. Catal., A, 305, 79 (2006)
23. A. T. Gemeay, A. M. Habib and M. A. B. El-Din,
Dyes Pigments, 74, 458 (2007)

22
Chapter _ I
24. V. Sippola and O. Krause,
Catal.Today, 100, 237 (2005)
25. T. K. M. Shing, Y. Y. Yeung and P. L. Su,
Org. Lett., 8, 3149 (2006)
26. D. C. Bilehal, R. M. Kulkarni and S. T. Nnadibewoor,
J. Mol. Catal. A: Chem., 232, 21 (2005)
27. A. R. Supale and G. S. Gokavi, Catal. Lett., 124, 284 (2008)
28. P. Veersomaiah, K. Bal Reddy, B. Sethuram and T. Navaneeth Rao,
Indian J. Chem., 26A, 462 (1987)
29. S. J. Malode, N. P. Shetti and S. T. Nandibewoor,
Catal. Lett., 141, 1526, (2011)
30. V. C. Seregar, C. V. Hiremath and S. T. Nandibewoor,
Transition Met. Chem., 31, 541 (2006)
31. P. D. Pol, R. T. Mahesh and S. T. Nandibewoor,
React. Kinet. Catal. Lett., 81, 113 (2004)
32. S. J. Malode, J. C. Abbar and S. T. Nandibewoor,
Inorg. Chim. Acta, 363, 2430 (2010)
33. S. A. Farokhi, A. K. Kini and S. T. Nandibewoor,
Oxid. Commun., 30, 141 (2007)
34. A. Hickling,
Trans. Faraday Soc., 38, 27 (1942)
35. J. Heyrovsky,
Chem. Listy, 16, 256 (1922)
36. W. Kemula and Z. Kublik,
Nature, 182, 793 (1958)
37. R. N. Adams,
“Electrochemistry at Solid Electrodes”, Published by Marcel Dekker,
Inc., (1968)

23
Chapter _ I
38. P. T. Kissinger and W. R. Heineman,
J. Chem. Edu., 60, 702 (1983)
39. M. Noel and K. I. Vasu,
“Cyclic Voltammetry and the Frontiers of Electrochemistry”, 1st Ed.,
Oxford & IBH Publishing Co. Pvt. Ltd., New Delhi, (1990)
40. B. Uslu, S. A. Ozkan and Z. Senturk,
Anal. Chim. Acta, 555, 341 (2006);
R. N. Goyal and A. Dhawan, J. Electroanal. Chem., 591, 159 (2006)
41. J. Wang,
“Analytical Electrochemistry”, 3rd Ed., John Wiley & Sons, Inc.,
Hoboken, New Jersey, (2006)
42. C. Amatore, E. Maisonhaute and G. Siminneau,
J. Electroanal. Chem., 486, 141 (2000)