the virome of peony and the population structure of its
TRANSCRIPT

University of Arkansas, Fayetteville University of Arkansas, Fayetteville
ScholarWorks@UARK ScholarWorks@UARK
Graduate Theses and Dissertations
12-2020
The Virome of Peony and the Population Structure of Its Most The Virome of Peony and the Population Structure of Its Most
Prominent Viruses Prominent Viruses
Cullen Shaffer University of Arkansas, Fayetteville
Follow this and additional works at: https://scholarworks.uark.edu/etd
Part of the Botany Commons, Environmental Policy Commons, Plant Breeding and Genetics
Commons, and the Plant Pathology Commons
Citation Citation Shaffer, C. (2020). The Virome of Peony and the Population Structure of Its Most Prominent Viruses. Graduate Theses and Dissertations Retrieved from https://scholarworks.uark.edu/etd/3927
This Thesis is brought to you for free and open access by ScholarWorks@UARK. It has been accepted for inclusion in Graduate Theses and Dissertations by an authorized administrator of ScholarWorks@UARK. For more information, please contact [email protected].

The Virome of Peony and the Population Structure of Its Most Prominent Viruses
A thesis submitted in partial fulfillment
of the requirements for the degree of
Master of Science in Plant Pathology
by
Cullen Shaffer
Southern Arkansas University
Bachelor of Science in Biology, 2016
December 2020
University of Arkansas
This thesis is approved for recommendation to the Graduate Council.
Ioannis E. Tzanetakis, Ph.D Terry Kirkpatrick, Ph.D
Thesis Director Committee Member
Garry McDonald, Ph.D
Committee Member

Abstract
Peony (Peonia lactiflora, Pall.) is a popular ornamental that has been cultivated for
millennia. Due to its popularity, plant material is frequently moved across international borders
allowing for the spread of viruses. The virome of several peony plants was investigated and four
viruses; namely Amazon lily mild mottle virus (ALiMMV), Cycas necrotic stunt
virus (CNSV), Gentian Kobu-sho associated virus (GKaV) and Lychnis mottle virus (LycMoV)
were detected for the first time in the Western Hemisphere. Incidence ranged from a few plants
for ALiMMV to near universal infection for CNSV. GKaV was found in individuals that were
infected with Lemoine’s disease of peonies, a disorder causing root galls, and was absent from
asymptomatic individuals. Yet more plants affected must be assessed to determine association
between virus and disease. CNSV and LycMoV were the most prevalent viruses detected, the
majority of the times in asymptomatic infections. High throughput sequencing was employed to
examine the population structure of LycMoV and CNSV. Both viruses have homogenous
populations in peony and phylogenetic analyses indicate that those isolates form distinct clades.
The main evolutionary force identified was negative selection although there were few amino
acid positions in CNSV that undergo positive selection. An accurate, multiplex-diagnostic
method was developed for CNSV that can detect all published isolates, a valuable tool given that
CNSV is known to infect and cause disease in many agricultural and horticultural crops.

©2020 by Cullen Shaffer
All Rights Reserved

Acknowledgements
This work was partially funded by the Arkansas Agricultural Experimental Station and USDA-
NIFA Hatch project 1002361.
Committee members: Ioannis E. Tzanetakis Ph.D, Terry Kirkpatrick Ph.D, Garry McDonald
Ph.D.
Plant samples: David Michener Ph.D, Nastassia Vlasava Ph.D, Henry Chotkowski
Next Generation Sequencing: Kurt Lamour Ph.D
Tzanetakis Lab post-doctorate fellows: Tobiasz Druciarek, Asminia Katsiani, Dan E. V.
Villamor, Daisy B. Stainton
Tzanetakis Lab students: Jing Zhou Ph.D, Terea Bentley, Ava Wait, Andrea Sierra, Genesis
Espinoza, Tatiana Castillo
Devany Crippen
Baronger Bieger
I would also like to thank all my friends and family for being supportive throughout the years.

Dedication
I dedicate this research to my parents who have been the rocks of my life and have always helped
me through the many moves and trials of graduate school. For Laura Ortega, the girl with many
names, who quite honestly is the main reason I finished.

Table of Contents
Chapter 1.
Introduction…………………………………….…………………………………….….………...1
References………..……………………………………………….……………………………12
Figures…………..……………………………………………………………………………...17
Chapter 2. The population structure of Lychnis mottle virus ………......…………….……....... 18
References……………………………………………………………...…….............................28
Figures…………………………………………………………….…………………………...31
Chapter 3. Population structure of the emerging Cycas necrotic stunt virus and the development
of data-driven diagnostics………………………………..................................................48
References……….………………………………………………….…………………………59
Figures…………………………………………………………….…………………………...63
Conclusion……………………………………………………………………………………….74

1
Chapter 1: Introduction
Peony Cultivation and History
Paeonia lactiflora (syn. P. albiflora, P. sinensis) (Pall.), also known as Chinese peony
was created through hybridization between P. pergrina, P arietina, and P. officinalis. Legend has
it that the plant was named for the physician of the Greek gods, Paeon who was training under
Asclepius, the god of medicine. Paeon was given the peony on Mount Olympus by Leto and used
it to heal Hades from a wound. Ascelpius grew jealous of Paeon’s heroic deed and plotted to
murder him. As a reward for Paeon’s help, Hades decided to turn him into a peony to avoid death
(Harding 1917).
A member of the genus Paeonia, was first described in ‘Natural History’ around 77
A.D. by the naturalist Pliny. Pliny describes the plant and the seeds, but not the flowers
(Harding 1917). Jashemski and Meyer (2002) indicate that in ancient times there were four
species native to Italy, although it is not known what the species were. By 586 A.D. P.
lactiflora had become widespread across China where it was grown primarily for the medicinal
value of its roots. Additionally, flowers and seeds were also consumed. However, by the end of
the 11th century, growers were focused primarily on the ornamental aspects of the plant and by
the end of the 16th century there were more than thirty commercial varieties (Harding 1917)). P.
lactiflora was first described by the Prussian/German botanist Peter Simon Pallas in 1776 when
it was introduced to Europe from China where most breeding occurred. New varieties were
developed and brought to the New World by European immigrants in the early 1900s (Harding
1917). As the number of people who cultivated peonies grew, so did the number of cultivars.
Due to lackluster record keeping and growers actively misrepresenting their product, there were
thousands of varieties with no standard for verification of identity or approval of new cultivars.

2
The American Peony Society (APS) was formed in 1903 and sought to tackle the nomenclature
task. APS asked for plant donations for evaluation from across the US, and they received roots
representing over 2000 varieties from 22 American growers. Additionally, they received plants
from four other countries. One cultivar, ‘Edulis Superba’, was submitted under 23 different
names. The cultivar evaluation process ended in 1908 with 750 completed descriptions, but by
1911 some 2000 commercial cultivars were correctly named. Primarily due to this work, the APS
was chosen by the International Society for Horticulture Science as the worldwide registrar for
the genus Paeonia. Today there are more than 6900 cultivars currently registered (The
American Peony Society, 2020).
Peonies are commercially propagated via root cuttings both to ensure that the progeny
will have the same characteristics as the mother plant, and because seeds overcome double
dormancy to germinate. Typically, the crown is split after two or more years with each division
having 3-5 eyes and enough root system to support the new plant. Both the epicotyl and radicle
require chilling, but the epicotyl is only released from dormancy after the radicle has grown. A
chilling period is required for the radicle to break dormancy, a warm period is then needed to
allow radicle growth, and finally another cold period is needed to relieve the epicotyl from
dormancy (Yu et al. 2007).
Diseases of Peony
Peonies are relatively disease resistant, but they are affected by a few significant
pathogens. Botrytis blight, a fungal disease caused by Botrytis cinerea (Pers.) and B. paeoniae
(Oudem.) is the most common disease of garden peonies. Signs of disease include fungal growth
(gray mold) on new shoots. The fungi can also grow on developing flower parts causing bud

3
blast and flower blight, preventing the buds from opening (Plant Disease Diagnostic Clinic
Cornell University 2018). Powdery mildew also affects peonies. With this disease, plants exhibit
withered yellow leaves and premature senescence. The causal agent is Erysiphe paeoniae that
also is known to infect Paeonia. suffruticosa (Andr.). The signs of the fungus include the
appearance of circular white colonies that spread over the whole plant (Qian et al. 2016; Amano
1986).
The most prevalent bacterial disease is bacterial blight, caused by Xanthomonas hortorum
(Vauterin). Symptoms begin with spotting of leaves with spots progressing to severe blight
making plants unmarketable. Signs of the disease include bacterial streaming from lesions
(Oliver et al. 2012).
Peonies can also succumb to parasitism by plant-parasitic nematodes that feed on the
roots and, less commonly, on leaves. The root-knot nematode Meliodgyne hapla (Chitwood)
induces galls to form along the lateral roots (Vovlas et al. 2010). The foliar nematode,
Aphelenchoides fragariae (Christie), has been reported to feed on the leaves, and severe
infestation can lead to the inability of peonies to flower (Goodey et al. 1965; Lisa Kohl 2011).
The most mysterious ailment is Lemoine’s Disease of peony (LDP) (Figure 1).
Symptoms include galls of varying sizes along the roots. When galls are bisected, they exhibit
yellow inclusions (Garfinkel and Chastanager 2016). The earliest report of LDP appears to be by
Berkeley in England in the mid-19th century (Harding 1917). At that time, LDP was thought to
be distributed worldwide and most prevalent in the United States (Harding 1923). It is likely,
however, that LDP was mistakenly attributed to root-knot nematode disease since at the time, the
names were commonly used interchangeably (Harding 1923; Bessey 1911). Later work showed

4
that LDP was not associated with bacteria, fungi, or nematodes (Brown 1940) even though it was
originally hypothesized that the galls formed due to infection by the root-knot nematode. After
the dismissal of a nematode causality, is was hypothesized that LDP might be caused by a virus.
It is unclear how or why there was a paradigm shift, but it was probably due to the suspected
transmission of symptoms through pruning (Harding 1923).
Peony viruses
Tobacco rattle virus
Tobraviruses are rod-shaped viruses, with a bipartite, positive-sense, ssRNA genome.
The type member of the genus Tobravirus, Tobacco rattle virus (TRV), is the causal agent of
peony ringspot disease. The typical symptom is yellow mosaic on the foliage. Mechanical
inoculation of TRV onto P. lactiflora causes similar symptoms. Electron microscopy indicates
that long virus particles aggregate in the mitochondria whereas the shorter particles are dispersed
in the cytoplasm (Chang et al. 1976). TRV is transmitted by species of plant-parasitic nematodes
in the genera Trichodorus and Paratrichodorus in serotype-species-specific manner (Brown et
al. 1989). TRV has been detected in peony in Japan, Alaska, Michigan, and Ohio in the United
States (Chang et al. 1976: Robertson et al. 2009; Fisher 2012)
Alfalfa mosaic virus
Alfalfa mosaic virus (AMV) AMV has pleomorphic particles, with a multipartite, three
RNA genome (Spitsin et al. 1999). It is the only member of the genus Alfamovirus in the family
Bromoviridae. Aphids are known to transmit AMV in other hosts (Hill et al. 2001). AMV was
first identified in peony in 2003 in Italy in plants that exhibited yellowing, mosaic, oak-like
arabesques, and line-patterns in the older leaves (Bellardi et al. 2003). Electron micrographs

5
revealed an absence of TRV and other rod-shaped viruses, but mechanical inoculations onto
indicator plants caused virus-like symptoms (Bellardi et al. 2003). The presence of AMV was
confirmed by ELISA in peony and Vinca minor (L.). Graft transmission of AMV onto AMV-free
peonies in an aphid-proof cage, led to mild mosaic with ELISA confirming the presence of AMV
in symptomatic material (Bellardi et al. 2003).
Raspberry ringspot and Strawberry latent ringspot viruses
Raspberry ringspot virus (RRSV) and Strawberry latent ringspot virus (SLRSV) are
members of the family Secoviridae. They have a bipartite, positive sense RNA genome
encapsidated in spherical particles (Everett et al. 1994; Harrison et al. 1973). RRSV belongs to
the genus Nepovirus. RRSV and SLRSV are both transmitted by nematodes of the Longidorus
and Xiphinema genera respectively (Taylor and Robertson 1969; Harrison 1967) SLRSV was
originally classified as a nepovirus due to its transmission mode, but after phylogenetic analysis
it was moved to an unclassified status. The viruses were first detected in peony in Finland
(Bremer 1985). In their study, 37 P. officinalis (L.) plants were tested with ELISA and 19 plants
were found infected by at least one of the viruses, with RRSV being more common. No
symptoms were observed in the infected material.
Lychnis mottle virus
Lychnis mottle virus (LycMoV) (syn. Cnidium vein yellowing virus) is an unclassified
member of the family Secoviridae. This virus was first discovered in Lychnis cognata (L.) (Yoo
et al. 2015a) Cnidium (Cusson) (Yoo et al. 2015b) as well as Vincetoxicum acuminatum (Decne.)
plants displaying leaf mottling (Fujimoto et al. 2018) from Japan). LycMoV has two positive
strand RNA molecules encapsidated in spherical particles (Yoo et al. 2015a).

6
LycMoV was recently discovered in peony (Shaffer et al. 2019). It is important to note
that in Yoo et al. 2015b, there were two isolates of Cnidium vein yellowing virus (CnVYV)
deposited in GenBank that should also be classified as the same species as LycMoV following
the taxonomic criteria established by the International Committee on Taxonomy of Viruses.
Citrus leaf blotch virus
Citrus leaf blotch virus (CLBV) is a filamentous, monopartite, single-strand, positive-
sense RNA virus in genus Citrivirus. CLBV causes chlorotic spotting on leaves (Roistacher &
Blue 1968) and bud union incompatibility in susceptible citrus material (Galipienso et al. 2001).
CLBV is of great importance to the citrus industry and all new introductions of citrus germplasm
are grafted onto the known susceptible cultivar, ‘Dweet tangor’ ( Citrus reticulata Blanco X
Citrus sinensis L.) to identify the presence of CLBV (Krueger et al. 2005, 2012).
CLBV was first detected in peonies in the United States using next generation sequencing
and plants showing typical Lemoine’s disease symptoms. RT-PCR and a digoxigenin-labeled
probe were used to confirm the presence of CLBV in plants from Arkansas and Oregon.
CLBV was detected in both symptomatic and asymptomatic material so it is not believed to be
associated with LDP (Gress et al. 2016).
Cycas necrotic stunt virus
Cycas necrotic stunt virus (CNSV), a member of the genus Nepovirus was discovered in
Cycas revoluta (Thunb.) in Japan (Kusunoki et al. 1986). CNSV belongs to subgroup B of the
nepoviruses and has a bipartite positive sense RNA genome encapsidated in spherical particles
(Kusonoki et al. 1986). Mechanical transmission of CNSV from C. revoluta into several

7
indicator species was successful. In Chenopodium amaranticolor (Coste & Reyn.) and C.
serotinum (L.) seed transmission reached 30% demonstrating vertical transmission of the virus.
Viral particles were also observed in the cytoplasm of infected cells (Kusunoki et al. 1986).
CNSV was first reported in P. lactiflora in samples from Arkansas, Michigan, Alaska, Oregon,
and New York (Shaffer et al. 2019). The virus was present in both Lemoine’s Disease-affected
and asymptomatic material.
Amazon lily mild mottle virus
Amazon lily mild mottle virus (ALiMMV) is a member of the genus Anuluavirus making
it the second member of the genus (Fuji et al. 2012). The quasi-spherical, tripartite virus has a
positive sense RNA genome and was first isolated from cowpea (Vigna unguiculata L. Walp.),
after inoculation from infected Amazon lily (Eucharis grandiflora Planch. & Linden).
Researchers inoculated the virus in cowpea onto E. grandiflora plants and some plants showed
mild mottling or mosaic, fulfilling Koch’s postulates (Fuji et al. 2012). In Shaffer et al. 2020,
ALiMMV was reported in P. lactiflora in a few primarily asymptomatic plants out of more than
200 tested.
Gentian Kobu-sho-associated virus
Gentian Kobu-sho-associated virus (GKaV) is dsRNA virus with a monopartite genome
of 23 kbp. The closest relatives of GKaV to-date are members of the genus Pestivirus. Pestivirid
members infect non-human vertebrates and none of the current members is a plant pathogen
(Kobayashi et al. 2012).

8
GKaV was discovered in Japan in Gentiana triflora (Pall.) and G. scabra (Bunge)plants.
The virus is found in plants displaying tumors on the stems, nodes, and roots (Iwadate et al.
2006; Kodama et al. 2004; Kobayashi et al. 2012). Symptoms become apparent in the second or
third year of the planting and eventually lead to plant death a few seasons after the onset of
symptoms. In the field, the disease spreads inwards from the edges (Kodama et al. 2004).
Symptoms have never been seen after mechanical transmission, but the disease has spread
through grafting and tissue culture propagation (Iwadate et al. 2006; Chiba et al. 2008). Shoot
apical meristem culture was effective in eliminating symptoms (Takesawa et al. 2006). Although
Koch’s postulates could not be fulfilled with this suspected causal agent, symptoms were present
in more than 99% of individuals infected with GKaV indicating causality of the disease
(Kobayashi et al. 2012).
GKaV was found while sequencing peony material from 6 peony samples from the
cultivars: Bing Qing, Chun Xiao, Tryphan Park, Alice Harding, Alice Crousse, and Kirinmaru;
two of which (Alice Harding and Alice Crousse) had symptoms typical of LDP. Sequencing
gave 87% of the 23 KB genome whereas the rest was obtained using Sanger sequencing. PCR
primers designed against the peony isolate (GKaVF 5’-TTAGTGATGAGTGCCTTTTCC-3’ and
GKaVR 5’-CTGCCAGTCTTCTTGTGAACC-3’) were used to screen for the presence of GKaV
in plants from Arkansas and Michigan. Out of 144 Michigan samples 32 were labeled as stunted
whereas the rest were not. The presence of the virus was found in 18 of 32 (56%) of stunted
plants, which was significantly higher than the 6.5% (8/112) in plants that were not stunted.
GKaV was also found in 20% (34 of 166 plants) from Arkansas, but other than the cultivars
‘Alice Harding’ and ‘Alice Crousse’ the health status of the plants is unknown. Without
correlation of the disease in a large number of symptomatic plants and the absence of

9
asymptomatic plants and fulfillment of Koch’s postulates, the role of GKaV in LDP development
remains unclear. The peony isolate of GKaV was 88-89% identical at the nucleotide level
identity to the three Japanese isolates in GenBank (AB698917.1, AB698918.1, and
LC383792.1).
High Throughput Sequencing for Virus Detection
High throughput sequencing (HTS) is the use of modern sequencing technologies that
allow for the sequencing of millions of reads from both RNA and DNA templates, delivering
sequencing depth and breadth of nucleic acid targets. Used in tandem with bioinformatics
pipelines and software capable of handling megabytes of sequence data, HTS can be a powerful
tool to investigate the etiological causes of plant diseases and to describe the population structure
of plant pathogens. In plant virology HTS has been a boon in helping to detect and discover new
plant viruses, particularly viruses that are plant pathogenic.
The causal agents of many diseases that have been known for decades without
identification of a pathogenic agent, have been successfully solved with the use of HTS. For
example, Apple rubbery wood disease (ARWD) was first observed in 1935 (Wallace et al. 1944).
Its causal agents, Apple rubbery wood virus 1 and 2, were elucidated with the help of HTS
sequencing and the use of virus specific NGS analysis pipelines. Rott et al. (2018) were able to
determine that the cause of ARWD was two newly sequenced RNA viruses, both members of the
newly proposed genus Rubdoviridae in the order Bunyavirales.
A similar narrative involves citrus concave gum-associated virus (CCGaV), another
negative sense RNA infecting fruit trees (Navarro et al. 2018). CCGaV was found associated
with concave gum-blind pocket (CG) that was first described in the 1930s (Fawcett 1936).

10
Navarro and colleagues used a sRNA template to generate their NGS data using Illumina
platforms. Contig generated from de novo assembly of the sRNA libraries pointed to phlebo or
phlebo-like viruses in infected plants but their absence in CG-free material. The association of
CCGaV (84%) of CG-affected trees with multiplex RT-PCR testing and fulfillment of Koch’s
postulates ultimately led to the validation of CCGaV as the causal agent of the disease (Navarro
et al. 2018).
Lastly, Rose Rosette (RR) has been known to plague roses since the 1940s (Conners
1941). The virus was initially identified using dsRNA from a RR-afflicted individual subjected
to degenerate oligonucleotide-primed reverse transcriptase-PCR (DOP RT-PCR) and sequenced
using an Illumina platform (Laney et al., 2011). RRV was identified in the NGS data after de
novo assembly of contigs. Discovery of RRV ultimately led to the fulfillment of Koch’s
postulates by Di Bello et al. (2015) and the revelation that Phyllocoptes fructiphilus was the
eriophyid mite vector.
The viruses detected recently in peony (ALiMMV, CLBV, CNSV, LycMoV, and GKaV)
were all found using HTS technologies. HTS has been instrumental in numerous cases to help
elucidate the etiological relationship between viruses and diseases (Laney et al. 2011; Hassan et
al. 2017) and for virus discovery (Al Rwahnih et al. 2009; Villamor et al. 2016; 2017). HTS
allowed for the detection of these previously unknown peony viruses that could only have been
found otherwise through exhausting testing of known viruses by use of PCR or ELISA assays.
These viruses, found in the quest for the causal agent of LDP, highlight the unknowns of the
peony virome. All were previously described viruses that were detected in eight peony plants
subjected to HTS and give insight into a potentially larger peony virome.

11
Perennials, particularly ornamentals like peonies, may harbor these viruses
asymptomatically. In the cases of ALiMMV, CLBV, CNSV and LycMoV infection in peony in
the U.S. all are asymptomatic, whereas possible symptoms for GKaV are under investigation.
Peonies could easily allow movement of viruses across international borders. CLBV is a known
quarantine pest in citrus and LycMoV is a close relative of SLRSV, an important pathogen in the
berry industry, specifically in strawberry (Fragaria X ananassa Duchesne), raspberry (Rubus
idaeus L. ), and blackberry (Rubus fruticosus L.) (Murant 1974; Martin et al. 2013). These
viruses appear to cause no problems in peonies, but as the popularity and availability of peonies
is high, they could lead to introductions into new areas and crops.
As illustrated above, HTS can be a powerful tool for both virus discovery and detection.
However, HTS is not without its limitations and is just one of many tools that can be used to
elucidate virus presence. HTS in many cases can produce virus sequences in data-sets that are
unverifiable by other PCR and ELISA detection protocols. This means that they could be
artifacts generated by the sequencing/de novo assembly, or the concentration of the virus could
be too low to be detected by the aforementioned methods. HTS generates trillions of nucleotide
sequences in a single run and sorting and analyzing such large datasets presents a true challenge.
Most plant pathologists do not have the coding knowledge to design programs and algorithms to
sort the datasets that require specialized expertise or access to high performance computing
centers. Fortunately, there are publicly available pipelines available such as VirFind
(htpp://virfind.org/j/) and VirusDetect (http://virusdetect.feilab.net) that scientists can submit
datasets to in order to identify virus and virus-like sequences in their samples, ameliorating the
process and advancing the understanding of the plant virome.

12
References
Al Rwahnih, M., Daubert, S., Golino, D., Rowhani, A. 2009. Deep sequencing analysis of RNAs
from a grapevine showing Syrah decline symptoms reveals a multiple virus infection that
includes a novel virus. Virology. 387:395–401.
Amano, K. 1986. Host range and geographical distribution of the powdery mildew fungi. Japan
Scientific Societies Press. p. 741.
Bellardi, M. G., and Rubies-Autonell, C. 2003. First report of a Disease of Peony Caused by
Alfalfa mosaic virus. Plant Dis. 87:99.3-99.3
Bessey, E. A. 1911. Root-knot and Its Control. U.S. Government Printing Office. p. 18
Bremer, K. 1985. Strawberry latent ringspot virus in ornamental plants in Finland. Ann. Agric.
Fenn. 24:101-102
Brown, N. A. 1940. Nematodes and Lemoine disease. The role of nematodes and a chemical
stimulus in the Lemoine disease of peonies. American Peony Society Bulletin.
Brown, D. J. F., Ploeg, T. A., and Robinson, D. J. 1989. The association between serotypes of
tobraviruses and Trichodorus and Paratrichodorus species. EPPO Bulletin.
Chang, M. U., Doi Y., Yora, K. 1976. A Rod-Shaped Virus Found in the Peony Ringspot. Ann.
Phytophath. Sc. Japan 42:325-328.
Chiba, K-I., Onoda, K., Abe, J., Iwadate, Y., and Takesawa, T. 2008. Graft transmission of
gentian tumorous symptoms ‘Kobu-sho’ (in Japanese). Ann Rept Plant Prot North Japan 59:74–
76.
Conners, I. L. 1941. Twentieth Annual Report of the Canadian Plant Disease Survey, 1940. p.
98.
Cornell University College of Agriculture and Life Sciences: Plant Disease Diagnostic Clinic.
2018. Botrytis Blight of Peony: Botrytis paeoniae. Retrieved from
http://plantclinic.cornell.edu/factsheets/botrytisblightpeony.pdf
Di Bello, P. L., Ho, T., Tzanetakis, I. E. 2015. The evolution of emaraviruses is becoming more
complex: seven segments identified in the causal agent of Rose rosette disease. Virus Res.
210:241–244.
Everett, K. R., Milne, K. S., and Forster, R. L. S. 1994. Nucleotide sequence of the coat protein
genes of strawberry latent ringspot virus: lack of homology to the nepoviruses and comoviruses.
J. Gen. Virol. 75:1821–1825.
Fawcett, H. S. 1936. Citrus Diseases and Their Control. McGraw-Hill book Company,
Incorporated.

13
Fisher, J. R. 2012. First Report of Tobacco rattle virus Associated with Ring Spot and Line
Pattern Disease of Peony in Ohio. Plant Health Prog. 13:40.
Fuji, S., Kikuchi, M., Ueda, S., Toda, T., Furuya, H., Fukumoto, F., et al. 2012. Characterization
of a new Anulavirus isolated from Amazon lily plants. Arch. Virol. 158:201–206.
Fujimoto, Y., Nijo, T., Hosoe, N., Watanabe, K., Maejima, K., Yamaji, Y., and Namba, S. 2018.
Complete Genome Sequence of Lychnis Mottle Virus Isolated in Japan. Genome Announc. 6.
Galipienso, L., Vives, M. C., Moreno, P., Milne, R. G., Navarro, L., and Guerri, J. 2001. Partial
characterisation of citrus leaf blotch virus, a new virus from Nagami kumquat. Arch. Virol.
146:357–368.
Garfinkel, A. R., and Chastagner, G. A. 2016. Diseases of Peonies. In Handbook of Florists’
Crops Diseases, Handbook of Plant Disease Management, eds. Robert J. McGovern and Wade
H. Elmer. Cham: Springer International Publishing, p. 1–31.
Goodey, J. B., Franklin, M. T., and Hooper, D. J. 1965. T. Goodey’s “The nematode parasites of
plants catalogued under their hosts. Farmham Royal, Bucks, England, UK.
Gress, J. C., Smith. S., Tzanetakis, I. E. 2016. First Report of Citrus leaf blotch virus in Peony in
the U.S.A. Plant Dis. 101:637.
Harrison, B. D. 1967. The transmission of strawberry latent ringspot virus by Xiphinema
diversicaudatum (Nematoda). Ann. Appl. Biol. 60:405–409.
Harrison, B. D., Murant, A. F., Mayo, M. A., and Roberts, I. M. 1973. Distribution of
Determinants for Symptom Production, Host Range and Nematode Transmissibility between the
two RNA Components of Raspberry Ringspot Virus. J. Gen. Virol. 22:233–247.
Hassan, M., Di Bello, P. L., Keller, K. E., Martin, R. R., Sabanadzovic, S., and Tzanetakis, I. E.
2017. A new, widespread emaravirus discovered in blackberry. Virus Res. 235:1–5.
Harding, A. H. 1917. The Book of the Peony. J.B. Lippincott. p. 25-68.
Harding, A. H. 1923. Peonies in the Little Garden. Atlantic Monthly Press. p. 86-94.
Hill, J. H., Alleman, R., Hogg, D. B., Grau, C. R. 2001. First Report of Transmission of Soybean
mosaic virus and Alfalfa mosaic virus by Aphis glycines in the New World. Plant Dis. 85:561.
Iwadate, Y., Takesawa, T., Chiba, K-I., and Tada, K. 2006. ‘‘Kobu-sho’’ syndrome of gentian
(in Japanese). Plant Prot (Shokubutu boueki) 60:518–522.
Jashemski, W. F., and Meyer, F. G. 2002. The Natural History of Pompeii. Cambridge
University Press. p. 136.

14
Kodama, K., Naito, Y., Chiba, K-I., Katsube, K., Fujiwara, K., and Umesawa, M. 2004. On the
incidence of gentian Kobu-sho in Iwate Prefecture (Abstract in Japanese). Proceedings of
Tohoku Meeting of Japanese Soc. Hortic. Sci. 51–52.
Kohl, L. 2011. Astronauts of the Nematode World: An Aerial View of Foliar Nematode Biology,
Epidemiology, and Host Range. Astronauts of the Nematode World: An Aerial View of Foliar
Nematode Biology, Epidemiology, and Host Range. Retrieved from
https://www.apsnet.org/edcenter/apsnetfeatures/Pages/foliarnematodes.aspx
Kobayashi, K., Atsumi, G., Iwadate, Y., Tomita, R., Chiba, K., Akasaka, S., Nishihara, M.,
Takahashi, H., Yamaoka, N., Nishiguchi, M., and Sekine, K. 2012. Gentian Kobu-sho-associated
virus: a tentative, novel double-stranded RNA virus that is relevant to gentian Kobu-sho
syndrome. J. Gen. Plant Pathol. 79:56–63.
Krueger, R. R., J. A. Bash and R. F. Lee. 2005. Phytosanitary status of California
citrus. International Organization of Citrus Virologists Conference Proceedings (1957-20). 16:
468.
Krueger, R. R., J. A. Bash and R. F. Lee. 2012. Dweet mottle virus and Citrus leaf blotch virus.
Kusunoki, M., Hanada K., Iwaki M., Cho SU., Doi, Y., and Yora, K., 1986. Cycas necrotic stunt
virus, a new member of nepoviruses found in Cycas revoluta; host range, purification, serology,
and some properties. Jpn J Phytopathol. 52:302-311.
Laney, A. G., Keller, K. E., Martin, R. R., and Tzanetakis, I. E. 2011. A discovery 70 years in
the making: characterization of the Rose rosette virus. J. Gen. Virol. 92:1727–1732.
Martin, R. R., S, M., Sabanadzovic, S., Quito-Avila, D., Tzanetakis, I. E., Poudel, B. 2013.
Viruses and Virus Diseases of Rubus. Plant Dis. 97:168–182.
Murant, A. F. 1974. Strawberry latent ringspot virus. CMI/AAB Description of Plant Viruses.
126 p. 4.
Navarro, B., Minutolo, M., Stradis, A. D., Palmisano, F., Alioto, D., and Serio, F. D. 2018. The
first phlebo-like virus infecting plants: a case study on the adaptation of negative-stranded RNA
viruses to new hosts. Mol. Plant Pathol. 19:1075–1089.
Oliver, C. L., Cai, R., Vinatzer, B. A., Bush, E. A., and Hansen, M. A. 2012. First Report of
Bacterial Spot of Peony Caused by a Xanthomonas sp. in the United States. Plant Dis. 96:581.
Qian, H. W., Jing, J. C., Liang, C., Liang, W. X., and Huang, J. G. 2016. First Report of Powdery
Mildew Caused by Erysiphe paeoniae on Paeonia suffruticosa in China. Plant Dis. 100:1015.
Robertson, N. L., Brown, K. L., Winton, L. M., and Holloway, P. S. 2009. First Report of
Tobacco rattle virus in Peony in Alaska. Plant Dis. 93:675–675.

15
Roistacher, C. N., and R. L. Blue. 1968. A psorosis-like virus causing symptoms only on
‘Dweet’ tangor. International Organization of Citrus Virologists Conference Proceedings. 4:13-
18.
Rott, M. E., Kesanakurti, P., Berwarth, C., Rast, H., Boyes, I., Phelan, J. and Jelkmann, W. 2018.
Discovery of Negative-Sense RNA Viruses in Trees Infected with Apple Rubbery Wood Disease
by Next-Generation Sequencing. Plant Dis. 102:1254–1263.
Shaffer, C., J. C. Gress., Tzanetakis, I. E. 2019. First report of Cycas necrotic stunt virus and
Lychnis mottle virus in peony in the United States. Plant Dis. 103:5
Shaffer, C., Vakic, M., and Tzanetakis, I. E. 2020. First Report of Amazon lily mild mottle virus
in Peony in the USA. Plant Dis. (In press).
Spitsin, S., Steplewski, K., Fleysh, N., Belanger, H., Mikheeva, T., Shivprasad, S., Dawson, W.,
Koprowski, H., and Yubisov, V. 1999. Expression of alfalfa mosaic virus coat protein in tobacco
mosaic virus (TMV) deficient in the production of its native coat protein supports long-distance
movement of a chimeric TMV. PNAS. 96:2549–2553.
Takesawa, T., Chiba, K-I., Iwadate, Y., and Abe, J. 2006. Gentian tumorous symptoms are
transmitted by grafting and are not observed by shoot apex culture (in Japanese). Ann. Rept.
Plant Prot. N. J. 57:68–71.
Taylor, C. E., and Robertson, W. M. 1969. The location of raspberry ringspot and tomato black
ring viruses in the nematode vector, Longidorus elongatus (de Man). Ann. Appl. Biol. 64:233–
237.
The American Peony Society. 2020. The American Peony Society Story: International gate
keeper of the peony. Retrieved from https://americanpeonysociety.org/about/story/
Villamor, D. E. V., Mekuria, T. A., Pillai, S. S., and Eastwell, K. C. 2016. High-Throughput
Sequencing Identifies Novel Viruses in Nectarine: Insights to the Etiology of Stem-Pitting
Disease. Phytopathology. 106:519–527.
Villamor, D. E. V., Pillai, S. S., and Eastwell, K. C. 2017. High throughput sequencing reveals a
novel fabavirus infecting sweet cherry. Arch Virol. 162:811–816.
Vovlas, N., Troccoli, A., Minuto, A., Bruzzone, C., and Castillo, P. 2010. Parasitism of the root-
knot nematode Meloidogyne hapla on peony in northern Italy. Nematologia Mediterranea. 3:219-
221.
Wallace, T., Swarbrick, T., and Ogilvie, L. 1944. Some new troubles in apples; with special
reference to variety Lord Lambourne. Fruit Grower. 98.
Yoo, R. H., Zhao, F., Lim, S., Igori. D., Lee, S., and Moon, J. S., 2015a. The complete
nucleotide sequence and genome organization of lychnis mottle virus. Arch. Virol. 160:2891-
2894.

16
Yoo, R. H., Zhao, F., Lim, S., Igori, D., Kim, S., An, T., Lee, S., and Moon, J. S., 2015b. The
complete genome sequences of two isolates of cnidium vein yellowing virus, a tentative new
member of the family Secoviridiae. Arch Virol. 2015. 160:2911-4
Yu, X., Zhao, R., and Cheng, F. 2007. Seed Germination of Tree and Herbaceous Peonies: A
Mini-Review. Seed Sci. Biotech. 4:11-14.

17
Figures
Figure 1. Roots of ‘Coral Charm’ with galls typical of Lemoine’s disease of peony (LDP).

18
Chapter 2: The population structure of lychnis mottle virus
Abstract
The secovirid, Lychnis mottle virus (LycMoV), is one of several viruses recently detected
in peony. Given the high prevalence of the virus in the more than 300 samples tested, the
population structure of the virus was studied in-depth using 48 isolates representing at least 20
cultivars and collected from major producing and propagating states in the United States. The
studied population is homogeneous with proteins undergoing purifying selection. The
homogeneity of the United States population along with phylogenetic analyses of all publicly
available isolates point to the dissemination of the virus through propagation material rather that
active transmission. The role of peony in the spread of LycMoV and other viruses and its
possible effects on the health status of ornamental and other crops is discussed in depth.
Introduction
Peony (Paeonia lactiflora Pall.) is a vegetatively propagated ornamental grown around
the globe with an estimated market value of $440 million (Anonymous 2020). Peonies, as many
other ornamentals, cross borders without the stringent regulatory procedures that govern the
movement of agricultural crops (Gergerich et al., 2015). Upon passing visual inspection, plants
are cleared for propagation. If that material harbors viruses that are asymptomatic in peony, but
cause disease in other hosts, there is the potential for epidemics (Gergerich et al. 2015; Martin et
al. 2017). Citrus leaf blotch virus presents an excellent example, being asymptomatic in peony
(Gress et al. 2016), yet causing detrimental diseases in other crops (Galipienso et al. 2001; Wang
et al. 2016, Cao et al. 2017). Peonies are commercially propagated through root divisions. Given

19
the practice, the peony virome is of particular interest because of the worldwide distribution and
clonal propagation of the host; allowing for the undetected movement of viruses to new regions.
Lychnis mottle virus (LycMoV) is a virus that has been associated with mottling
symptoms in Lychnis cognata (Orange catchfly; Yoo et al. 2015a). At the same time another
isolate of the virus was published under the name Cnidium vein yellowing virus (CnVYV; Yoo et
al. 2015b) Given the 76-86% nt/ 88-89 % aa identities of the two viruses CnVYV should be
considered an isolate of LycMoV according to ICTV taxonomic criteria for members of the
Secoviridae (Thompson et al. 2017). In addition to lychnis and cnidium the virus was recently
detected in asymptomatic peonies in the United States (Shaffer et al. 2019). LycMoV along with
its close and well-studied relative, Strawberry latent ringspot virus (SLRSV), form a distinct
group and potentially a new genus within the Secoviridae (Dullemans et al. 2020). The two
viruses share genomic organization in the two RNA segments with RNA 1 coding for a
polyprotein with protease cofactor, helicase, genome-linked viral protein, cysteine protease and
RNA-dependent RNA polymerase (RdRp) motifs. RNA 2 codes for movement protein (MP), and
the two virus coat proteins (CPs) (Yoo et al. 2015a). Based on the knowledge of SLRSV biology
we can hypothesize that LycMoV could be transmitted by mechanical means (Tzanetakis et al.
2006), seed (Walkey and Whittingham-Jones 1970) and nematodes (Murant et al. 1974).
High-throughput sequencing (HTS) is a powerful tool for virus detection and discovery
(Adams et al. 2009; Al Rwahnih et al. 2009; Villamor et al. 2019) and it has successfully been
used for the detection of viruses in peony (Gress et al. 2016; Shaffer et al. 2019; 2020). HTS
usually involves the sequencing of total RNA or DNA that limits the amount of viral sequence
output substantially. However, HTS of PCR amplicons allows for the in-depth analysis of
hundreds of targeted virus sequences in a single sequencing reaction allowing for the study of the

20
population structure of a virus (Katsiani et al. 2020). The nature of the technique increases
sequencing depth of viral sequences and captures the viral quasi-species diversity.
In this study an amplicon-based Hi-Plex high-throughput sequencing approach was used
to characterize 48 LycMoV peony isolates allowing for the study of the virus population
structure using both phylogenetic and comparative analyses methods.
Materials and Methods
Plant Material and Screening
Three hundred and eleven (311) peony samples from Arkansas, Michigan, New York,
Oregon, and Alaska (Supplemental Table 1) were collected or mailed-in by peony growers.
Approximately 50 mg of leaf tissue was homogenized using the FastPrepⓇ-96 Instrument (MP
Biomedicals, USA). Nucleic acids were extracted according to the Poudel et al. (2013) or
Katsiani et al. (2020) protocols. Screening for LycMoV was done using primers (F 5′-
GGAGTCATGGCAAAGCTACG-3′/R 5′-CAAGCACCTCAATTATTTGCTCATC-3′)
performed as described in Shaffer et al. (2019). LycMoV was detected in 143 samples and 48
were selected for further analysis (Supplemental Table 2), with heavy emphasis on Arkansas
material as state growers hold a significant number and variety of cultivars that are relevant to
the commercial peony trade in the United States.
Multiplex Amplicon Sequencing
RNA was reverse-transcribed using virus-specific primers as well as a dT primer that
binds to the A tail of the virus (Supplemental Table 3) essentially as described in Katsiani et al.

21
(2020). The RT was carried out according to Poudel et al. (2012) whereas PCR amplification of
LycMoV RNA 1 confirmed efficient cDNA synthesis (Shaffer et al. 2019).
The population structure of the virus was assessed based on combined outputs of two Hi-
Plex sequencing reactions. Initially both RNA 1 and RNA 2 were targeted, but RNA 1 outputs
were low in the first reaction and what was retrieved shared 70-80% identities to the LycMoV-JP
isolate (LC382242.1), which was used as the reference sequence for primer design. The high
divergence of RNA 1 outputs did not allow design of effective sequencing primers. Fragments of
RNA 1 for all peony isolates were deposited in GenBank (Supplemental Table 4). DNA
amplicon size was set at 180-190 bases with at least 50 base overlap between fragments. One
hundred and thirty-six primer pairs, split into four files (93 for the first and 43 for the second
reaction that targeted only RNA 2; Supplemental Table 5), were designed and eight multiplex
PCRs were performed at Floodlight Genomics (Knoxville, TN) using proprietary technology
(Nguyen-Dumont et al. 2013a;b). Dual-index Illumina libraries were generated and sequenced
bidirectionally using a Hi-SeqX device to create 2X150 paired-end reads following the
manufacturer’s protocols (San Diego, California). The raw reads were trimmed to remove all
primer sequences and delivered as sample specific FASTQ files.
Annotation and Phylogenetic Analyses
Reads from all samples were mapped to the LycMoV-JP isolate (Yoo et al, 2015a) with
an amendment given that the 3’ UTR of this isolate had a repetitive region, possibly because of
false assembly. Reads were assembled into a contig when 90% of their length shared at least
80% identities to the reference sequence using CLC Genomics Workbench v20.3 (QIAGEN

22
Hilden, Germany). Isolate AR 44 had sequence gaps that were filled by Sanger sequencing of
two independent PCR amplicons.
All available LycMoV sequences were block-aligned using MUSCLE (Edgar et al. 2004)
in MEGA 7 (Kumar et al. 2016). The unaligned datasets for the individually coded proteins
were uploaded into Species Demarcation Tool version 1.2 (SDT) to calculate the nucleotide (nt)
and amino acid (aa) percentage pairwise identities (Muhire et al. 2014). SDT aligns each
sequence with every other, one at a time. The 48 isolates in this study along with the 12 fully
sequenced RNA 2 segments available in GenBank (Yoo et al. 2015a; 2015b; Fujimoto et al.
2018; Jiang et al. 2019; Dullemans et al. 2020) and the reference sequence for SLRSV
(NC006965.1), were also aligned using MUSCLE in MEGA 7 (Supplemental Table 6). The best-
fit nucleotide substitution model for all sequence datasets was determined by MEGA 7 using
default parameters. A neighbor-joining tree was made using the amino acid substitutions type
with a complete deletion strategy for any gaps/missing data treatment with a no branch swap
filter. Maximum-likelihood trees were constructed with the specified model (JTT+G for both the
MP and CPs) using 1000 bootstrap pseudoreplicates. The two coat proteins were treated as one
unit based on ICTV taxonomic criteria for secovirids (Thompson et al. 2017; Dullemans et al.
2020). SLRSV was used to root each tree. All branches with bootstrap support of <70% were
collapsed using TreeGraph 2 (Stöver and Müller 2010).
The proteins coded in LycMoV RNA2 were codon-aligned to compare homologous
codon sites. The LycMoV-JP sequence had degenerate bases and was manually edited using the
other isolates as a guide to determine the most probable amino acid. Sequences were analyzed
individually to determine the selection pressure using FUBAR (Murrell et al. 2013) within
SelectionMap version1.0 (Stenzel et al. 2014).

23
Results
Sequencing
The total number of reads and the number of reads mapped to the reference sequence are
provided in Supplemental Table 2. LycMoV RNA 2 (> 10X coverage for each site) was
assembled for 48 isolates whereas the low coverage areas of isolate AR 44 were PCR-amplified
and Sanger sequenced. The length of the molecule ranged from 3642-3659 nucleotides (nt),
primarily because of insertions/deletions in the UTRs. The complete sequence for the 48 isolates
for RNA 2 were deposited in GenBank (Accession numbers MW035143-90)
Isolates NY 7 and AR 62 both had nine nt deletions in the large coat protein coding
region (Supplemental Figure 1) which did not alter the reading frame. Both deletions were
confirmed with Sanger sequencing. The consensus sequences for NY 7 and AR 62 isolates were
deposited to GenBank under accession numbers MW035144 and MW035167, respectively.
Population structure
Percentage pairwise identities of the coding regions reveal that the peony isolates form a
distinct and homogenous population. The MP for the peony isolates shared percentage pairwise
identities of 95-100% and 96-100% at the nt and amino acid (aa) levels respectively
(Supplemental Figure 2). Within the peony isolates, the two coat proteins had very similar
percent identities ranging from 95-99%/95-100% (large/small CP) and 97-100% at the nt and aa
levels respectively (Supplemental Figure 2).
When all LycMoV isolates were analyzed the MP identities ranged from 69-100% and
61-100% for nt and aa respectively (Fig. 1). The lower identity in amino acids compared to

24
nucleotides is because three isolates (CnVYV1-SK, KR011029.1; CnVYV2-SK, KR011031.1;
and Lilium 14-00-NL 1 MF796980.1) have large deletions in the MP, reducing their pairwise
identity to other isolates. The pairwise identities of the large CP ranged from 70-99% and 82-
100%, whereas the small CP ranged from 69-100% and 80-100% for nt and aa respectively
(Supplemental Figure 2).
Maximum-likelihood phylogenetic trees constructed using the MP and CPs aa alignments
show that the majority of isolates are grouped by both host and country of origin in both the MP
and combined CPs trees (Fig. 1). All peony isolates fall into the same clade, but there is limited
geographical clustering within the clade. The peony isolates form a separate clade that includes
the Japanese Vincetoxicum isolate in both trees and the CnVYV1-SK (KR011029.1) isolate from
Cnidium in South Korea in the CPs tree (Fig. 1). The rest of the isolates cluster together based
on the host. Evolutionary analyses clearly demonstrate that all proteins studied are under
negative selection given the overabundance of synonymous over non-synonymous substitutions
in the proteins. The selection was strongest for the large CP (.1030), followed by the small CP
(.115) and the MP (.13) (Fig. 2).
Discussion
HTS has become a vital tool for studying the population structure of a virus. This is an
invaluable tool in understanding the genetic structure and evolutionary forces at play in the event
of a pathogen epidemic (Katsiani et al. 2020; Tamukong et al. 2020) and the Hi-Plex sequencing
approach was applied in the study of the population structure of LycMoV. This is the first such
study for LycMoV, an emerging virus.

25
The nucleotide identities for the three proteins for the virus population range from 66-
100% with the peony isolates sharing identities >95%. Isolates from a given host are highly
similar to each other, with the exception of Lilium 17-007-N (MG062674.1). The peony isolates
therefore represent a homogenous population with almost all of the variation in the LycMoV
population coming from isolates from other species and countries. Like peony, other LycMoV
isolates tend to cluster in host specific clades, however for many host species only one or a few
isolates are currently known.
In the MP tree all isolates cluster together by host, except for LycMoV-JP from
Vincetoxicum, which groups with the peony isolates. In the CPs trees, the same pattern is
exhibited with exception to C. officianale and host V. acuminatum (CnVYV1-SK and LycMoV-
JP respectively), which are present in the clade containing the peony isolates. Like peony, other
LycMoV tend to cluster in host specific clades, however for many host species only one or a few
isolates are currently known. Isolates cluster based on geographic origin other than CnVYV1-SK
and LycMoV-JP, but this may be because of the depth of data associated with LycMoV isolates
when we exclude the present study. Within the peony clade, there is little grouping of isolates by
state, which could be due to the movement of peonies across the country. However, as stated
earlier the number of peony isolates (n=48) compared to the rest available in GenBank (n=12)
does impact grouping. Nearly all of the nucleotide changes in the LycMoV population resulted in
synonymous mutations indicating that MP and CPs are under strong purifying selection as seen
in the MP and CPs of another secovirid, Grapevine fanleaf virus (Oliver et al. 2010).
CnVYV1-SK was present in the peony clade in the MP tree, but absent in the CPs which
could suggest a recombination event (Fig. 1). Yet, recombination was not investigated given that
the approach used in the present study assembles the consensus sequences from thousands of

26
short HTS reads mapped simultaneously, and the reads could be generated from any quasi-
species present in a plant. Forming a consensus creates an artificial isolate and not a true
biological entity, thus potentially leading to false assumptions when it comes to recombination
events (Katsiani et al. 2020).
The clustering of all US peony isolates is most probably because of the clonal
propagation of the host. LycMoV has been found in other hosts since it was first reported in
peony, but all other reports are from Asian countries, indicating a possible origin of the virus
(Yoo et al. 2015a; Uehara-Ichiki et al. 2016; Fujimoto et al. 2018). Peonies may have played an
important role in the introduction of LycMoV and potentially other viruses from Asia to the
United States. Sequencing of LycMoV from peonies in other locations and additional hosts could
confirm this hypothesis.
On the other hand the diversity observed in the US peony population could be a
‘founder’s effect’ where a single, recent introduction created the present population and the virus
did not have enough time to diversify in the new location (Hassan et al. 2019; Thekke-Veetil et
al. 2015). This hypothesis is highly unlikely given that peonies came to the United States largely
in the early 1900s as immigrants brought the plant from Europe after World War I (Harding
1917) with the viruses infecting them. In addition, the myriad of different peony cultivars, some
of which have been propagated for an eon or more, should have allowed for some diversification.
Peonies, and other ornamentals propagated through root divisions, present a particular
problem as daughter plants will carry all viruses harbored in the mother plants. Due to the
propagation regime and given that none of the modern virus elimination technologies (thermo-,
chemo-, cryotherapy) are applied in any commercial nurseries in the United States, it is

27
important to develop sensitive and efficient virus detection protocols. Those tools could be used
to identify infected material and eventually generate clean nursery stock, initiated from plants
that tested negative for the virus. LycMoV has been shown to infect a wide and varied range of
host plants, from ornamentals such as peony and lily to agricultural plants like alfalfa. This is of
concern as the host range indicates that the virus has the potential to cause significant economic
losses.

28
References
Adams, I. P., Glover, R. H., Monger, W. A., Mumford, R., Jackeviciene, E., Navalin-skiene, M.,
Samuitiene, M., Boonham, N. 2009. Next-generation sequencing and metagenomic analysis: A
universal diagnostic tool in plant virology. Mol. Plant Pathol. 10:537-545.
Al Rwahnih, M., Daubert, S., Golino, D., Rowhani, A. 2009. Deep sequencing analysis of RNAs
from a grapevine showing Syrah decline symptoms reveals a multiple virus infection that
includes a novel virus. Virol. J. 387:395–401.
Anonymous. 2020. https://www.marketwatch.com/press-release/peony-market-size-share-2020-
by-development-trend-key-manufacturers-price-supply-demand-growth-factor-and-end-user-
analysis-outlook-till-2026-2020-11-09
Cao, M. J., Yu, Y.-Q., Tian, X., Yang, F. Y., Li, R. H., Zhou, C. Y. 2017. First Report of Citrus
leaf blotch virus in Lemon in China. Plant Dis. 101:1561–1561.
Dullemans, A. M., Botermans, M., de Kock, M. J. D., de Krom, C. E., van der Lee, T. A. J.,
Roenhorst, J. W., Stulemeijer, I. J. E., Verbeek, M., Westenberg, M., van der Vlugt, R. A. A.
2020. Creation of a new genus in the family Secoviridae substantiated by sequence variation of
newly identified strawberry latent ringspot virus isolates. Arch. Virol. 165:21–31.
Edgar, R. C. 2004. MUSCLE: multiple sequence alignment with high accuracy and high
throughput. Nucleic Acids Res. 32:1792–1797.
Fujimoto, Y., Nijo, T., Hosoe, N., Watanabe, K., Maejima, K., Yamaji, Y., Namba, S. 2018.
Complete Genome Sequence of Lychnis Mottle Virus Isolated in Japan. Genome Announc. 6:25.
Galipienso, L., Vives, M. C., Moreno, P., Milne, R. G., Navarro, L., Guerri, J. 2001. Partial
characterisation of citrus leaf blotch virus, a new virus from Nagami kumquat. Arch. Virol.
146:357–368.
Gergerich, R. C., Welliver, R. A., Gettys, S., Osterbauer, N. K., Kamenidou, S., Martin, R. R.,
Golino D. A., Eastwell, K., Fuchs, M., Vidalakis G., I. E. Tzanetakis. 2015. Safeguarding Fruit
Crops in the Age of Agricultural Globalization. Plant Dis. 99:176–187.
Gress, J. C., Smith. S., Tzanetakis, I. E. 2016. First Report of Citrus leaf blotch virus in Peony in
the U.S.A. Plant Dis. 101:637.
Harding, A. H. 1917. The Mythology, and Ancient and Modern History of the Peony. In The
Book of the Peony. Philadelphia, Pennsylvania. J.B. Lippincott. p. 23-69.
Hassan, M., Tzanetakis, I. E. 2019. Population structure, evolution and detection of blackberry
leaf mottle-associated virus, an emerging emaravirus. Plant Path. 68:775–782.
Jiang, P., Shao, J., Nemchinov, L. G. 2019. Identification of emerging viral genomes in
transcriptomic datasets of alfalfa (Medicago sativa L.) Virol. J. 16:153.

29
Katsiani, A., Stainton, D., Lamour, K., Tzanetakis, I. E. 2020. The population structure of Rose
rosette virus in the USA. J. Gen. Virol. 101:676–684.
Kumar, S., Stecher, G., Tamura, K. 2016. MEGA7: Molecular Evolutionary Genetics Analysis
Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 33:1870–1874.
Martin, R.R., Constable, F., Tzanetakis, I.E. 2017. Quarantine regulations and the impact of
modern detection methods. Annu. Rev. Phytopathol. 54:189–205.
Muhire, B. M., Varsani, A., Martin, D. P. 2014. SDT: A Virus Classification Tool Based on
Pairwise Sequence Alignment and Identity Calculation. PLoS ONE. 9:e108277.
Murant, A. F. 1974. Strawberry latent ringspot virus. CMI/AAB Description of Plant Viruses. p.
126:4.
Murrell, B., Moola, S., Mabona, A., Weighill, T., Sheward, D., Kosakovsky Pond, S. L.,
Scheffler, K. 2013. FUBAR: a fast, unconstrained bayesian approximation for inferring
selection. Mol. Biol. Evol. 30:1196–1205.
Nguyen-Dumont, T., Pope, B. J., Hammet, F., Southey, M. C., Park, D. J. 2013a. A high-plex
PCR approach for massively parallel sequencing. BioTechniques. 55:69–74.
Nguyen-Dumont, T., Teo, Z. L., Pope, B. J., Hammet, F., Mahmoodi, M., Tsimiklis, H.,
Sabbaghian, N., Tischkowitz, M., Foulkes, W. D. 2013b. Hi-Plex for high-throughput mutation
screening: application to the breast cancer susceptibility gene PALB2. BMC Medical Genom.
6:48.
Oliver, J. E., Vigne, E., Fuchs, M. 2010. Genetic structure and molecular variability of
Grapevine fanleaf virus populations. Virus Res. 152:30–40.
Poudel, B., Sabanadzovic, S., Bujarski, J., Tzanetakis, I. E. 2012. Population structure of
Blackberry yellow vein associated virus, an emerging crinivirus. Virus Res. 169:272-275.
Poudel, B., Wintermantel, W.M., Cortez, A. A., Ho, T., Khadgi, A., Tzanetakis, I. E. 2013.
Epidemiology of Blackberry yellow vein associated virus. Plant Dis. 97: 1352-1357.
Shaffer, C., Gress, J. C., and Tzanetakis, I. E. 2019. First Report of Cycas Necrotic Stunt Virus
and Lychnis Mottle Virus in Peony in the United States. Plant Dis. 103:506.
Shaffer, C., Vakic, M., and Tzanetakis, I. E. 2020. First Report of Amazon lily mild mottle virus
in Peony in the USA. Plant Dis. 104.
Stenzel, T., Piasecki, T., Chrząstek, K., Julian, L., Muhire, B. M., Golden, M., Martin, D. P., and
Varsani, A. 2014. Pigeon circoviruses display patterns of recombination, genomic secondary
structure and selection similar to those of beak and feather disease viruses. J. Gen. Virol.
95:1338–1351.

30
Stöver, B. C., Müller, K. F. 2010. TreeGraph 2: Combining and visualizing evidence from
different phylogenetic analyses. BMC Bioinformatics. 11:7.
Tamukong, Y. B., Collum, T. D., Stone, A. L., Kappagantu, M., Sherman, D. J., Rogers, E. E.,
Dardick, C., Culver, J. N. 2020. Dynamic changes impact the plum pox virus population
structure during leaf and bud development. Virol. J. 548:192–199.
Thekke-Veetil, T., Polashock, J. J., Marn, M. V., Plesko, I. M., Schilder, A. C., Keller, K. E.,
Martin, R. R., Tzanetakis, I. E. 2015. Population structure of blueberry mosaic associated virus:
Evidence of reassortment in geographically distinct isolates. Virus Res. 201:79–84.
Thompson, J. R., Dasgupta, I., Fuchs, M., Iwanami, T., Karasev, A. V., Petrzik, K. Sanfaçon H.,
Tzanetakis, I. E., van der Vlugt, R. A. A., Wetzel, T., Yoshikawa, N. 2017. ICTV Virus
Taxonomy Profile: Secoviridae. J. Gen. Virol. 98:529–531.
Tzanetakis, I. E., Postman, J. D., Gergerich, R. C., Martin, R. R. 2006. A virus between families:
nucleotide sequence and evolution of Strawberry latent ringspot virus. Virus Res. 121:199–204
.
Uehara-Ichiki, T., Nakazono-Nagaoka, E., Ohashi, M., Sato, T., Hanada, K., Tatsuo, Y., Takao,
Y., Murakami, Y., Kurosaki, F., Aoki, T., Fujikawa, T. 2016. Detection of several plant viruses
from medical herbs, Rehmannia glutinosa varieties, and their nucleotide sequences. Jpn J
Phytopathol. 82:256.
Villamor, D. E. V., Ho, T., Al Rwahnih, M., Martin R. R., Tzanetakis, I. E. 2019. High
Throughput Sequencing for Plant Virus Detection and Discovery. Phytopathol. 109:5.
Walkey, D.G.A., Whittingham-Jones S.G. 1970. Seed transmission of Strawberry latent ringspot
virus in Celery (Apium graveolens var. Dulce). Plant Dis Rep. 54:802–803.
Wang, J., Zhu, D., Tan, Y., Zong, X., Wei, H., Liu, Q. 2016. First Report of Citrus leaf blotch
virus in Sweet Cherry. Plant Dis. 100:1027–1027.
Yoo, R. H., Zhao, F., Lim, S., Igori. D., Lee, S., Moon, J. S. 2015a. The complete nucleotide
sequence and genome organization of lychnis mottle virus. Arch. Virol. 160-2891-2894.
Yoo, R. H., Zhao, F., Lim, S., Igori, D., Kim, S.-M., An, T.-J., Lee, S.-H., Moon, J. S. 2015b.
The complete genome sequences of two isolates of cnidium vein yellowing virus, a tentative new
member of the family Secoviridae. Arch. Virol. 160:2911–2914.

31
Figures
Figure 1. Maximum-Likelihood trees of Lychnis mottle virus proteins coded in RNA 2. The trees
were drawn with 1000 replicates using the JTT+ G model. Both the MP (A) and CPs (B) trees
were drawn using amino acid muscle blocked alignments. All branches with less than 70%
support were collapsed using Tree Graph 2. All peony isolates are reported by the postal code
abbreviation of each state they were collected from, the isolate number and host genus. Each tree
is rooted with the SLRSV type isolate from Mentha. Isolates are color coded according to host:
Paeonia (red), Lilium (purple), Medicago (blue), Cnidium (green), Vincetoxicum (orange),
Lychnis (brown), and Mentha (black).
A

32
Figure 1 Cont.
B

33
A
B
C
Figure 2. A plot of natural selection along the length of the movement (A), large (B) and small
coat proteins (C) of lychnis mottle virus. The diagrams are visual representations of nt aligned
protein codon sites. At each codon the absolute (Abs) values of inferred synonymous substitution
rates subtracted from inferred non-synonymous substitution rates (dN-dS) are mapped
(determined by the FUBAR program). Sites with significant positive selection values appear
with a green bar while, blue indicates sites with significant values for negative selection. The
absence of a bar in a particular codon site indicates a gap in the CPs and MP gene alignments.
34
A
B
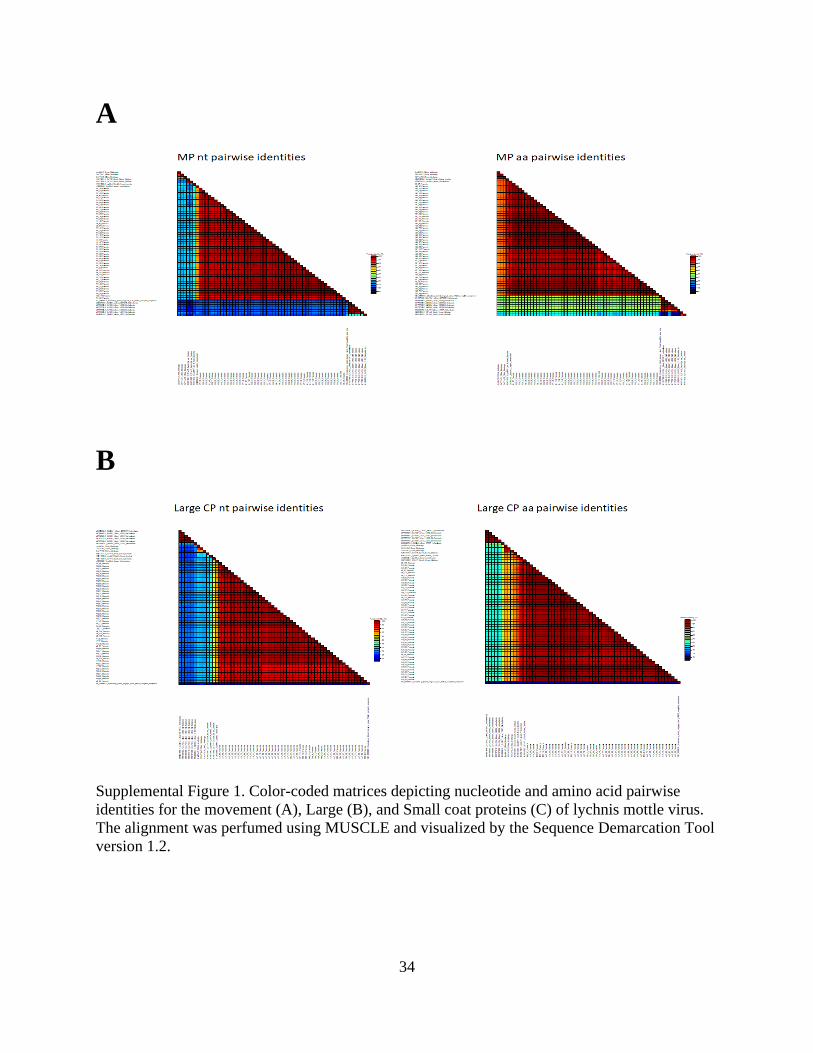
Supplemental Figure 1. Color-coded matrices depicting nucleotide and amino acid pairwise
identities for the movement (A), Large (B), and Small coat proteins (C) of lychnis mottle virus.
The alignment was perfumed using MUSCLE and visualized by the Sequence Demarcation Tool
version 1.2.

35
C
Supplemental Figure 1 Cont.
Supplemental Fig 2. Deletions present in the large coat protein of lychnis mottle virus isolates
NY 7 and AR 62 as confirmed by Sanger sequencing of PCR amplicons. The nine base deletions
in both isolates do not alter the reading frame.

36
Supplemental Table 1. Geographic distribution and number of samples tested positive for
lychnis mottle virus
State LycMoV Positive Tests/ Samples Tested
Michigan 49/144
Arkansas 38/87
New York 30/48
Alaska 22/24
Oregon 4/8
Total 143/311
Supplemental Table 2. Isolate origins and cultivar information of the lychnis mottle virus
isolates used in the study. Number of reads mapped and total reads for each isolate is also
provided.
Isolate GenBank Accession No Cultivar Reads Mapped Total Reads
NY6 MW035145 NA 929,579 1,323,598
NY7 MW035144 NA 1,134,412 1,166,271
NY28 MW035149 NA 4,472,214 5,871,706
NY46 MW035147 NA 881,827 1,884,974
NY 30 MW035148 NA 2,743,984 2,682,887
NY 47 MW035146 NA 1,645,171 2,114,132
OR 117 MW035143 NA 1,055,384 1,334,022
AK 66 MW035190 NA 1,970,372 2,029,251
AK 69 MW035189 NA 4,258,083 6,920,171
AK 77 MW035188 NA 83,642 3,199,999
MI 48 MW035151 La Perle 1,559,667 2,418,767
MI 87 MW035150 Lady Emily 500,698 1,098,871
MI 107 MW035155 Red Charm 2,185,601 3,713,936
MI 113 MW035154 Illini Belle 4,551,063 8,930,424
MI 114 MW035153 Illini Belle 2,931,749 4,699,002
MI 115 MW035152 Flame 257,812 2,399,147
AR 12 MW035187 Mrs. Franklin D. Roosevelt 2,432,950 4,448,754
AR 26 MW035186 White Cap 3,758,489 6,966,818
AR 27 MW035185 Hope 2,831,585 4,282,315
AR 28 MW035184 Christmas Velvet 2,017,359 2,831,901
37
Supplemental Table 2 Cont.
Isolate GenBank Accession No Cultivar Reads Mapped Total Reads
AR 31
MW035183
Mrs. Franklin D.
Roosevelt
1,565,342 2,654,507
AR 32
MW035182
Mrs. Franklin D.
Roosevelt
3,875,427 6,520,037
AR 33 MW035181 Flame 1,855,109 2,431,035
AR 38 MW035180 Joker 5,238,790 8,692,959
AR 39 MW035179 Joker Croots 2,416,616 3,724,600
AR 41 MW035178 Coral N’ Gold 3,100,997 5,260,796
AR 43 MW035177 Coral N’ Gold 4,471,555 6,655,217
AR 44 MW035176 Coral N’ Gold 2,047,462 2,357,717
AR 45 MW035175 White Cap 2,642,704 5,362,160
AR 46 MW035174 White Cap 3,757,074 8,180,333
AR 47 MW035173 White Cap 5,202,371 9,505,852
AR 49 MW035172 Henery Bockstoce 2,880,509 4,074,221
AR 50 MW035171 Henery Bockstoce 3,530,942 5,786,888
AR 53 MW035170 Mother’s Day 3,300,492 4,487,658
AR 55 MW035169 White Cap 2,753,379 4,751,932
AR 56 MW035168 White Cap 3,731,906 6,896,234
AR 62 MW035167 White Cap 3,197,713 6,798,981
AR 66 MW035166 Chippewa 3,388,204 5,332,601
AR 68 MW035165 Coral Charm 4,050,439 5,714,737
AR 69 MW035164 Coral Charm 3,287,633 4,865,933
AR 71 MW035163 Coral Charm 3,807,794 5,437,066
AR 73 MW035162 Ann Cousins 2,085,245 3,462,683
AR 74 MW035161 Hi-Mabel 2,545,341 4,339,004
AR 77 MW035160 Cora Stubbs 4,933,645 8,474,634
AR 78 MW035159 Cora Stubbs 4,927,364 9,329,122
AR 86 MW035158 Avalon 1,980,107 2,178,859
AR 88 MW035157 Dauntless 1,533,053 1,747,606
AR 90 MW035157 West Elkton 4,086,200 8,517,109
38
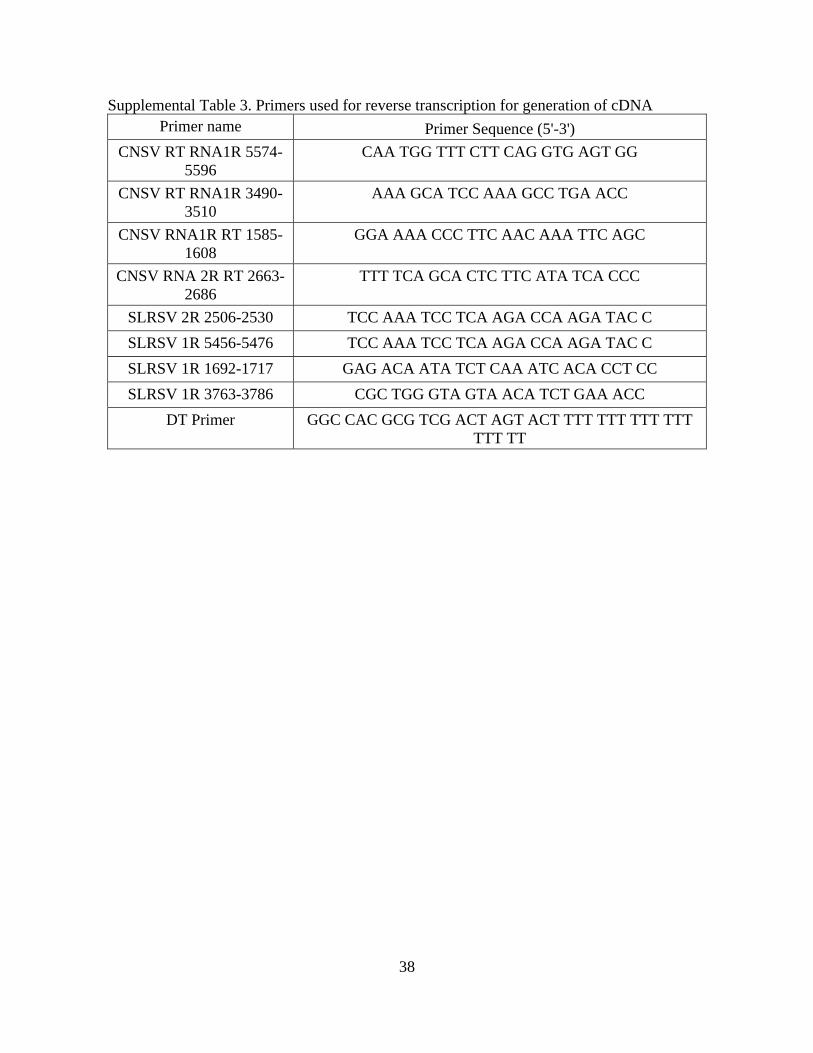
Supplemental Table 3. Primers used for reverse transcription for generation of cDNA
Primer name Primer Sequence (5'-3')
CNSV RT RNA1R 5574-
5596
CAA TGG TTT CTT CAG GTG AGT GG
CNSV RT RNA1R 3490-
3510
AAA GCA TCC AAA GCC TGA ACC
CNSV RNA1R RT 1585-
1608
GGA AAA CCC TTC AAC AAA TTC AGC
CNSV RNA 2R RT 2663-
2686
TTT TCA GCA CTC TTC ATA TCA CCC
SLRSV 2R 2506-2530 TCC AAA TCC TCA AGA CCA AGA TAC C
SLRSV 1R 5456-5476 TCC AAA TCC TCA AGA CCA AGA TAC C
SLRSV 1R 1692-1717 GAG ACA ATA TCT CAA ATC ACA CCT CC
SLRSV 1R 3763-3786 CGC TGG GTA GTA ACA TCT GAA ACC
DT Primer GGC CAC GCG TCG ACT AGT ACT TTT TTT TTT TTT
TTT TT

39
Supplemental Table 4. Lychnis mottle virus RNA 1 isolate sequence accessions used in this
study.
Isolate Designation GenBank Designation Mapped Reads Total Reads
NY 6 MW035097 240,997 991,093
NY 7 MW035096 626 21,478
NY 28 MW035102 4255777 7,599,307
NY 46 MW035101 338662 1,748,541
NY 30 MW035098 424823 2,937,761
NY 47 MW035100 205439 1,259,023
OR 117 MW035095 119,715 633,077
AK 66 MW035099 51490 1,036,853
AK 69 MW035142 992390 5,065,672
AK 77 MW035141 435861 3,120,111
MI 48 MW035104 382,773 1,540,066
MI 87 MW035103 186,064 982,913
MI 107 MW035108 635838 3,186,384
MI 113 MW035107 1,105,539 8,242,100
MI 114 MW035106 797,826 3,454,965
MI 115 MW035105 271,896 2,155,786
AR 12 MW035140 676,638 4,000,757
AR 26 MW035139 1,129,470 6,467,136
AR 27 MW035138 455,682 3,111,863
AR 28 MW035137 260,951 2,029,102
AR 31 MW035136 467,914 2,170,018
AR 32 MW035134 852,222 5,556,073
AR 33 MW035135 232,321 1,301,560
AR 38 MW035133 1,079,500 7,078,836
AR 39 MW035132 330,978 3,163,979
AR 41 MW035131 881,005 5,161,872
AR 43 MW035130 758,850 5,412,497
AR 44 MW035129 105,460 1,035,571
AR 45 MW035128 1,111,811 4,893,112
AR 46 MW035127 1,626,101 7,712,266
AR 47 MW035126 1,856,112 8,188,113
40
Supplemental Table 4 Cont.
Isolate
Designation Isolate Designation GenBank Designation Mapped Reads Total Reads
AR 49 AR 49 MW035125 246,306 2,756,597
AR 50 AR 50 MW035124 543,881 5,219,670
AR 53 AR 53 MW035123 578,601 3,641,240
AR 55 AR 55 MW035122 731,519 3,575,475
AR 56 AR 56 MW035121 1,179,093 5,642,321
AR 62 AR 62 MW035120 1,517,747 5,817,386
AR 66 AR 66 MW035119 790,165 4,076,770
AR 68 AR 68 MW035118 436,823 4,369,085
AR 69 AR 69 MW035117 280,877 4,297,594
AR 71 AR 71 MW035116 366,772 4,500,677
AR 73 AR 73 MW035115 355,616 2,759,125
AR 74 AR 74 MW035114 372,782 4,339,044
AR 77 AR 77 MW035113 1,900,361 8,474,634
AR 78 AR 78 MW035112 1,779,641 7,921,653
AR 86 AR 86 MW035111 64,989 850,356
AR 88 AR 88 MW035110 65,799 694,025
AR 90 AR 90 MW035109 703,969 7,951,965

41
Supplemental Table 5. Primers used for Hi-Plex PCR; F/R for forward/reverse, and the run in
which they were used and the primer sequence.
Primer Name HTS Run Primer Sequence (5'-3')
RNA 1 SLRSV F 1-24_1F Run 1 TTG AAA AGC AAT CTG CGA ACT TTG
RNA 1 SLRV F 97-118_2F Run 1 GCT TAT CCG GAT TCT CTC TTT G
RNA 1 SRLSV F 229-249_3F Run 1 TTC TAT GGG TTA CTC AAG AGG
RNA 1 SLRSV F 372-389_4F Run 1 GTG GGC TGG TTA GCA CAG
SLRSV F 496-520_1F Run 1 GTT CAT GTC AGA GTT GAT GGA CTT C
SLRSV F 624-644_2F Run 1 GGC TTG ATG TTG AGG RGT ACR
SLRSV F 764-789_3F Run 1 CCC TCT TTT GAT TAT ACA GGT CTT GG
SLRSV F 850-869_4F Run 1 TTT GGC TCG CTT TGT TTC CG
SLRSVF F 896-907_1F Run 1 GCT CCA GAT CTA ACG CTT TGG ATG
SLRSV F 1107-1128_2F Run 1 TTT CTG ACG AAT GGG TTG GGT G
SLRSV F 1226-1246_3F Run 1 GGT TCT GAA TCT TTG AGT CCC
SLRSV F 1348-1370_4F Run 1 CCA CAA TAA GAA ATT CAA TTT GG
SLRSV F 1476-1497_1F Run 1 GAG GAA CTC CAA TTG ATG AGG C
SLRSV F 1614-1635_2F Run 1 ACT ATG AGA AAG AGG CTC TTC C
SLSRV F 1756-1776_3F Run 1 GAT GGT GGT TAT TGA TGC CCC
SLRSV F 1883-1902_4F Run 1 TTT CTT GAC GGA GTC ATG GC
SLRSV F 2009-2030_1F Run 1 GAA AGA AAA GGA GAA TTG CTG C
SLRSV F 2138-2157_2F Run 1 CTG ACG GGT GCT GCT ATT GG
SLRSV F 2264-2285_3F Run 1 GAA ATC AAG CAT GTT GAT GAG C
SLRSV F 2384-2405_4F Run 1 ATC ACA CAC ATG AAA GAC TTG C
SLRSV F 2499-2523_1F Run 1 GGA AAA TAC TTT TGC TCT TAT GTC C
SLRSV F 2743-2764_3F Run 1 GAC AAC TGC AAC TCC AAC TTC C
SLRSV F 2869-2894_4F Run 1 CAT TAC ACA CTT TAA CAA TCT GTT GC
SLRSV F 2993-3013_1F Run 1 GTT GGT AAG TCA GTC TGC TCC
SLRSV F 3124-3146_2F Run 1 GCT GTC TTG TAT GAT GAT TTT GG
SLRSV F 3230-3253_3F Run 1 CTA GAA GCA AAG GGG AAT ACG TGC
SLRSV F 3360-3378_4F Run 1 AGG TCA CGG CAG TTG ATG G
SLRSV F 3478-3501_1F Run 1 GCA GTA TGC TGT CAA TCA GTC ACG
SLRSV F 3601-3621_2F Run 1 AAC AGC TGG AAT TGA GAT TCC
SLRSV F 3722-3745_3F Run 1 TAT AAT TCG AGC GCG TTT GGA TCC
SLRSV F 3842-3866_4F Run 1 CCA AAT CCC TAT TTT AAT TGC TCG C
SLRSV F 3955-3975_1F Run 1 AAA GGA GCT CGC TCA GGG TGC
SLRSV F 4062-4087_2F Run 1 GTT GGT TGA GAA ATC TTT ACA CGG CG
SLRSV F 4177-4198_3F Run 1 ATC TGG AAG GGA AAG GAG ACG C
SLRSV F 4307-4327_4F Run 1 TAT CCC GAT CAC CAC TAT CGG
SLRSV F 4436-4456_1F Run 1 GTC AAT AGG AAG CAT TGC AGG
SLRSV F 4564-4585_2F Run 1 TTT TAT GGC TGG GTC TGG TTG G
42
Supplemental Table 5 cont.
Primer Name HTS Run Primer Sequence (5'-3')
SLRSV F 4679-4701_3F Run 1 GCA GCA TTT TTG AAA GGA GTA GG
SLRSV F 4814-4834_4F Run 1 GGA AAA GGT CTG AAG ATT GCC
SLRSV F 4943-4964_1F Run 1 GGC GAT ATT AAG CAC CCT TTG G
SLRSV F 5194-5215_3F Run 1 TCT CGA CAT TTT TAA GAA GGG C
SLRSV F 5319-5341_4F Run 1 GAA TTT CCC AAA GGT GTC ACC TG
SLRSV F 5442-5462_1F Run 1 CAA ATG GGG AAG CTG GCA AGG
SLRSV F 5566-5587_2F Run 1 TTG TAT TGT GGG CTT GGA CAC C
SLRSV F 5689-5713_3F Run 1 CTT TGG TTC TTT TGT TGC TGA GTT G
SLRSV F 5809-5835_4F Run 1 TAA TTG GTT CAA TGG TGA TTA TTC CCG
SLRSV F 5937-5959_1F Run 1 AGA GCA ACA TCG AAT CAC ATA GG
SLRSV F 6065-6084_2F Run 1 GCT GGG AGC ATA TGA TGG CC
SLRSV F 6195-6215_3F Run 1 ACT GTT TCT AGC AGG CTT TGG
SLRSV F 6324-6345_4F Run 1 CTA TCT TGC TCC GTT GGC ATG G
SLRSV F 6451-6477_1F Run 1 GGA GAT AAG CAG TTG TAC TGT AAA TGG
SLRSV F 6640-6661_2F Run 1 ATC GCA CCA GGT GTT CAC ATC G
SLRSV F 6767-6788_3F Run 1 AGA TTC AAG CTC CTC AGG GTT C
SLRSV F 6893-6914_4F Run 1 GGG TTG CAG CCT TTT ATG CTT C
SLRSV F 7030-7052_1F Run 1 CTT TTT AGG CAT TTC TCT TGG AG
SLRSV F 7158-7182_2F Run 1 GTC AAA GAT CTT TGA ATC GAA GCC C
SLRSV F 7232-7256_3F Run 1 TTC GTG TGT TAG CTC TTT CTT GAG C
SLRSV F 7263-7283_4F Run 1 CGC TTA GTT ATG CTA CCC TGC
SLRSV RNA 2 F 1-29_1F Run 1 TTG AAA AGC AAT CTG CGA ACT TTG TTA CG
SLRSV RNA 2 F 118-139_2F Run 1 GGT TCT CTT CGT TAC TCG TCT G
SLRSV RNA 2 F 211-232_3F Run 1 TTC CCC GAT TTG TCA AGT CAG G
SLRSV RNA 2 F 339-360_4F Run 1 CTT TCT TTG GTA GGC GCT CCA C
SLRSV RNA 2 F 462-484_1F Run 1 CAG CAC AGA CTG AAA GTG GAA AG
SLRSV RNA 2 F 580-601_2F Run 1 CAA AGC TGT CGT CCA TGT TTC G
SLRSV RNA 2 F 827-849_4F Run 1 CTT CTG GAT GGT AAT CGA TCC AC
SLRSV RNA 2 F 947-971_1F Run 1 CCC AAT TTT GTA AAT CGA CTT CAG
SLRSV RNA 2 F 1076-1096_2F Run 1 CCG GAA CCT CTT CCT TAT TCC
SLRSV RNA 2 F 1198-1221_3F Run 1 TTA TAT GCC AAT ACA GCG TGC TGG
SLRSV RNA 2 F 1319-1339_4F Run 1 TAT GGT CAG CAG GAT AGA GCG
SLRSV RNA 2 F 1439-1462_1F Run 1 CAT GAA GAG TTG GTT CCT GCT TCG
SLRSV RNA 2 F 1555-1577_2F Run 1 AAT AAA TTC CAA CGT TGG TGC CG
SLRSV RNA 2 F 1677-1700_3F Run 1 GTT TGA TTG GTT CAG GGC CTA TAC
SLRSV RNA 2 F 1797-1819_4F Run 1 TTA TGC CAG GCA GGC ATG ATT CC
SLRSV RNA 2 F 1928-1949_1F Run 1 GAT GTT TCT GAC TAT GGG ATG G
SLRSV RNA 2 F 2056-2079_2F Run 1 GCT TTC AAC AAC AAC TTC CAA AGG

43
Supplemental Table 5 Cont.
Primer Name HTS Run Primer Sequence (5'-3')
SLRSV RNA 2 F 2181-2201_3F Run 1 GGA AGG GTG TTC TCT CAT TGG
SLRSV RNA 2 F 2308-2328_4F Run 1 CTC AGT GGT GGA TTT GAG AGC
SLRSV RNA 2 F 2433-2455_1F Run 1 ATA TCC TCA AAT TGG TGG TGC TG
SLRSV RNA 2 F 2564-2583_2F Run 1 CGG CTC AAT CCC AAG ACA AC
SLRSV RNA 2 F 2684-2707_3F Run 1 CAA AAG ACCT TCC GTC TGA TGA CG
SLRSV RNA 2 F 2808-2830_4F Run 1 AAT GCC CTT TGA TGC ATA GGA GC
SLRSV RNA 2 F 2920-2942_1F Run 1 CTC TCT CAC TTT TTC TGC TGT CC
SLRSV RNA 2 F 3038-3059_2F Run 1 GAG CAG GAT CAT CCT TTT GTG G
SLRSV RNA 2 F 3163-3182_3F Run 1 CAA AAC AAT GGC ACA GCT CC
SLRSV RNA 2 F 3251-3273_4F Run 1 TTA ACC ATC CCT GAT CCT TCA CC
SLRSV RNA 2 F 3367-3386_1F Run 1 TGG GCA TTA ACC AGG CAA GC
SLRSV RNA 2 F 3463-3486_2F Run 1 CTT TGA ATC GAA GCC CAA GTA TGG
SLRSV RNA 2 F 3580-3602_3F Run 1 GCT TTG TGT GTT TTC TTT AGA GC
SLRSV RNA 2 F 3682-3706_4F Run 1 TCT TTG AAT CGA AGC CCA AGT ATG G
SLRAV RNA 2F 3760-3782_1F Run 1 GAC TCT TTA TTG AGT TGT ACG CC
RNA 1 SLRSV R 153-180_1R Run 1 GTT CAA GTT AAG ACT TGA ATT CCA AAA G
RNA 1 SLRSV R 264-283_2R Run 1 GCA CAG CTT GGC TTT GTC AG
RNA 1 SLRSV R 398-419_3R Run 1 GAT TCT TCT CCT TCA TCC CAG C
RNA 1 SLRSV R 534-562_4R Run 1 AAC AGT ACC AAA GGA GAT ACC TGG
RNA 1 SLRSV R 664-686_1R Run 1 GAA GAA AGT TCC GTA CAC CAG C
RNA 1 SLRSV R 810-832_2R Run 1 AAA TAG GCC ATC CTT GCC AAA GC
RNA 1 SLRSV R 928-954_3R Run 1 GTG CCA AAA GAG ATA ACA TCA AGA TCG
RNA 1 SLRSV R 1168-1189_4R Run 1 GGG AAA ATA GAG TGC AAA CTC C
RNA 1 SLRSV R 1273-1293_2R Run 1 CAT TTG GTT CCA GGG GGA TAG
RNA 1 SLRSV R 1393-1416_3R Run 1 TCA GCA GGT TGC TCA ACA CAT TCC
RNA 1 SLRV R 1517-1537_4R Run 1 GGG ACT CTT ACC AAT TAG TGG
RNA 1 SLRSV R 1652-1668_1R Run 1 GGC ACC CCA TTA AGA TC
RNA 1 SLRSV R 1781-1804_2R Run 1 TGG AAA ATC TAA AAG CCA ACC AGG
RNA 1 SLRSV R 1922-1946_3R Run 1 ATG ATG CAG CAT TTA TAA CAG GAC C
RNA 1SLRSV R 2047-2071_4R Run 1 GAG AAA GCC ACA AAC ATA AAG CAG C
RNA 1 SLRSV R 2176-2197_1R Run 1 GGC TTG CCC TAA GAA CTG GAC C
RNA 1 SLRSV R 2308-2328_2R Run 1 TCC TCC TTT TCA GGA CTT TCC
RNA 1 SLRSV R 2435-2454_3R Run 1 GTC ACT CCT ACC CTT GTA GC
RNA 1 SLRSV R 2551-2574_4R Run 1 ATG AGA TCT TTT GCG CAG GAA AGG
RNA 1 SLRSV R 2661-2687_1R Run 1 TGT AAT CCA GAA ACT CAC TCT TAC TCC
RNA 1 SLRSV R 2785-2809_2R Run 1 ACA TAC TCT CAG TCG CTT TAA ACG C
RNA 1 SLRSV R 2910-2930_3R Run 1 CCT GTT GAC ACT CAA TTA GGG
RNA 1 SLRSV R 3035-3055_4R Run 1 CGC ATA ATC CAG TGA ATC AAG
44
Supplemental Table 5 Cont.
Primer Name
HTS
Run Primer Sequence (5'-3')
RNA 1 SLRSV R 3160-3183_1R Run 1 TTC CGC TTC ATC AAA ATG ACC ACC
RNA 1 SLRSV R 3293-3314_2R Run 1 CAC AGC ACT TGG TGT CAA ACC
RNA 1 SLRSV R 3400-3420_3R Run 1 TTT TGG TGG AGA GTA AAT CGG
RNA 1 SLRSV R 3521-3541_4R Run 1 GGG TGC ATC ACG GAG ATC AAC
RNA 1 SLRSV R 3648-3668_1R Run 1 GAA GTA AGT TAA CTG GTT GCC
RNA 1 SLRSV R 3772-3791_2R Run 1 AGG CAG CTT TTG GAA GTA GC
RNA 1 SLRSV R 3887-3910_3R Run 1 TGT CAC CTT GGG TTT CAA GAA ACG
RNA 1 SLRSV R 4006-4030_4R Run 1 TCC AAA ACC CAG CAA CAA CTT AAC C
RNA 1 SLRSV R 4118-4139_1R Run 1 AAG AAC CAG AAG CTT TCA TGG C
RNA 1 SLRSV R 4231-4252_2R Run 1 TAC ATC CTG GGC TTT TGC CTC C
RNA 1 SLRSV R 4347-4367_3R Run 1 GGT AAA CCA TTA GGA CAC GGG
RNA 1 SLRSV R 4469-4496_4R Run 1 AAT CTA TGA TAA CCA GGT CCT TGA GAC C
RNA 1 SLRSV R 4603-4626_1R Run 1 ACT TCT AGA ACA CCA TTT CTC TGG
RNA 1 SLRSV R 4725-4750_2R Run 1 ACC AGT TCC CGT GTC ATC GCA GAT GG
RNA 1 SLRSV R 4849-4866_3R Run 1 GTG GCT GAG AAG TGA TGC
RNA 1 SLRSV R 4983-5004_4R Run 1 TCG CCA CTC AGA CAG CCA GAC G
RNA 1 SLRSV R 5110-5133_1R Run 1 TCC CCT GTC AGA AGT TCC AAG TCC
RNA 1 SLRSV R 5237-5259_2R Run 1 GTG ACT GGG CTC ATA TCA GCA GC
RNA 1 SLRSV R 5486-5509_4R Run 1 TTC CCT GTT CAA CGT GTA ACA ACC
RNA 1 SLRSV R 5606-5632_1R Run 1 CAC CTC TTG ATA TAT TTT GGA GAG AGG
RNA 1 SLRSV R 5734-5756_2R Run 1 GAT TTA TCC CAA CCT TTG TAG GG
RNA 1 SLRSV R 5852-5879_3R Run 1 TAG CTA TTT CAA TGA GGA GAC ATC TTG G
RNA 1 SLRSV R 5978-5999_4R Run 1 ATT CCT CCA GAA ACA CGG TAC C
RNA 1 SLRSV R 6104-6127_1R Run 1 CAA CAG CCA ACT CCA CAC AAT CAG
RNA 1 SLRSV R 6232-6255_2R Run 1 GAT TTC TTT ATT TTT GTC CGA GCC
RNA 1 SLRSV R 6364-6385_3R Run 1 TCG TTT CCC TAA CCC AGT GAA G
RNA 1 SLRSV R 6493-6514_4R Run 1 CTC GGT TTC GGG AAC AAA GAT C
RNA 1 SLRSV R 6679-6704_1R Run 1 GGA ATT TCA TCA GGT TTC CAC TTA AG
RNA 1 SLRSV R 6809-6830_2R Run 1 TTT TTC AGA AAG CTC CGG ACC G
RNA 1 SLRSV R 6934-6957_3R Run 1 TGC TTT CAT ATA GGC TGA ATG ATG
RNA 1 SLRSV R 7063-7083_4R Run 1 CTA ATT AGT GCC CAT CTG GGC
RNA 1 SLRSV R 7202-7229_1R Run 1 CCC TAA CCT AAA TGC ATA AAT TAA ACA C
RNA 1 SLRSV R 7326-7348_2R Run 1 GAG CAT AAC AAC CAC GTA CAT CC
RNA 1 SLRSV R 7397-7422_3R Run 1 GCG CCA GAA CTA AAC CCG GTG AAA CC
RNA 1 SLRSV R 7418-7452_4R Run 1 TTT TTT TTT TTT TTT TAA AGC TTT TTC TAA GCG CC
SLRSV RNA 2 R 163-190_1R Run 1 CGT GGT GAC TGC CTA TAA TAA ACA CAG G
SLRSV RNA 2 R 280-305_2R Run 1 GAT AAC AAA GCA ACA AAA AGG AGA GG
SLRSV RNA 2 R 380-401_3R Run 1 TCT CCT TCG CAG AAT AGG AAG G

45
Supplemental Table 5. Cont.
Primer Name HTS Run Primer Sequence (5'-3')
SLRSV RNA 2 R 504-529_4R Run 1 TAA ACT CTT GAG CTG ACT AGT GTT GG
SLRSV RNA 2 R 619-647_1R Run 1 GAT GAG TTA GTT TAT CAT CAC ATA GAA CC
SLRSV RNA 2 R 747-770_2R Run 1 GTA TAG TGC AAA CAT CAG CAG ACC
SLRSV RNA 2 R 869-895_3R Run 1 CTC ATA AAG AGG TAG AAC ACC AAT ACC
SLRSV RNA 2 R 992-1013_4R Run 1 GAG AAC CTC CTC CGA GGA CAT C
SLRSV RNA 2 R 1115-1136_1R Run 1 TCA AGC CTC GCA CCC CAC ATG G
SLRSV RNA 2 R 1244-1268_2R Run 1 CCT TAA TTT GCG CCA CAT TAT CTT C
SLRSV RNA 2 R 1368-1389_3R Run 1 CCA ACT TTG TTG TGA AAT TGG C
SLRSV RNA 2 R 1483-1506_4R Run 1 ACA ACA GGC TGG GGG GAA AAG AAC
SLRSV RNA 2 R 1602-1626_1R Run 1 CAC TCA GTG TCT CTC AAT GAA GTC C
SLRSV RNA 2 R 1720-1745_2R Run 1 AAG TCT CAG AAG GAT CAT AAC ACA GC
SLRSV RNA 2 R 1844-1867_3R Run 1 ATC ACA AAA ACC TGC AGG GGT AGC
SLRSV RNA 2 R 1967-1989_4R Run 1 CCA AGT CCT ATT TTT TCC ACC TC
SLRSV RNA 2 R 2096-2115 _1R Run 1 GGA AGC AGG GGG TTA AGA GG
SLRSV RNA 2 R 2222-2244_2R Run 1 TCA ACA ATA CCT CCT GCC ATT GC
SLRSV RNA 2 R 2348-2368_3R Run 1 ATA GCC AGA CAA TGG CAC ACG
SLRSV RNA 2 R 2603-2623_1R Run 1 TAC AAT TGG TCC GCC AGA AGC
SLRSV RNA 2 R 2729-2754_2R Run 1 GGC GTA ACT GGA AAG TAG AGA AAA CC
SLRSV RNA 2 R 2852-2874_3R Run 1 GTC AGG TGA TAT TTG AGA GTA CC
SLRSV RNA 2 R 2965-2988_4R Run 1 GTA CTA TTA TCG ATG AAG CAC GGG
SLRSV RNA 2 R 3084-3110_1R Run 1 CAG TGG CAC CAA AAT GTG TAT TAG TCC
SLRSV RNA 2 R 3205-3228_2R Run 1 CTG TAC TCA ATA TCT GGC TCA ACC
SLRSV RNA 2 R 3324-3348_3R Run 1 TCC AAG AGA AAT GCC TAA GAG AAG G
SLRSV RNA 2 R 3409-3432_4R Run 1 TAA AAC AAC CAT GTA GAT CCA TGG
SLRSV RNA 2 R 3527-3552_1R Run 1 TCA ATA AAG AGT CAA CAC ATC AAG GC
SLRSV RNA 2 R 3620-3643_2R Run 1 AAG AAA CAA CTA CAT GCA TCC AGC
SLRSV RNA 2 R 3746-3770_3R Run 1 AAT AAA GAG TCA ACA CAT CAA GGC C
SLRSV RNA 2 R 3757-3782_4R Run 1 GGC GTA CAA CTC AAT AAA GAG TCA AC
SLRSV RNA 2 R 3923-3953_1R Run 1 AAA GCT TTA TCT AAG TGC CAG AAC TAA ACC C
1-28_1F Run 2 TTGAAAAGCAATCTGCGAACTTTGTTAC
379-400_1F Run 2 TCC TTC CTA YTC TGC RRA GGA G
582-605_1F Run 2 TGA AAG AGA AAC CCA AAG CTG TCG
974-996_1F Run 2 AAT CGA CTT CAG CTT TTG ACA AC
1190-1209_1F Run 2 GCT CGG CTC ACC AGR AGY GC
1617-1641_1F Run 2 GGACTTCATTGAGAGACACTGAGTG
2150-2173_1F Run 2 GTTCCTCTTCCATTTTGGAGAATC
2479-2503_1F Run 2 AGTGACACCTCAAAGAAAGGTTCTG
3054-3074_1F Run 2 AGC AGG ATC ATC CTT TTG TGG

46
Supplemental Table 5 Cont. Primer Name HTS Run Primer Sequence (5'-3')
1370-1388_1R Run 2 ATT GGC ACT TYT GGG GTG G
1875-1805_1R Run 2 TCC CTG CAG GGY TAR GGC TC
2319-2340_1R Run 2 AAT GCG GTG TGC TCT CAR ATC C -
2646-2669_1R Run 2 ACC TTT GAR ATA AAG GGC TCY TCC
2884-2901_1R Run 2 TCC AAG CGA GCT GTC AGG
3219-3240_1R Run 2 TAC TCA ATA TCT GGC TCA ACC C
3521-3540_1R Run 2 GMT AAC ACT CTW RCC TAA GC
261-281_2R Run 2 GRM AAA AGA TTG TTG TGA RAC
602-624_2R Run 2 CCT CCG AAA CAT GRA CGA C
893-915_2R Run 2 TGT GCC TCA TAA AGA GGT ARA AC
1175-1194_2R Run 2 CGA GCA GGT TGG AGA GGY GC
1617-1641_2R Run 2 CAC TCA GTG TCT CTC AAT GAA GTC C
2108-2126_2R Run 2 TCA TGG GCA GGR AGY ARG G
2336-2354_2R Run 2 ACA CGA GCA CGC AGA ATG C
2691-2713_2R Run 2 TAA YCG TCA TCA GAC GRA AGG TC
3039-3065_2R Run 2 AAT TCC ACA AAA GGA TGA TCC TGC TCC
3269-3291_2R Run 2 GAA ATA CTT AGG TGC AGG TGA AG
3594-3617_2R Run 2 CCT RGG AGG ATG CCT GGW TAA TCC
342-367_3R Run 2 CTA CCA AAG AAA GAA GTC ATC GTC C
669-689_3R Run 2 GTT CTG GAG TGT GGA CYA CAG
899-924_3R Run 2 CAT GTG AAT GTG CCT CAT AAA GAG G
1235-1262_3R Run 2 CTT CAA AAG TCA AAA CAG TTT TTG TGC C
1663-1685_3R Run 2 TTG GCT TAC ATC TCA AGT AGC GG
2147-2172_3R Run 2 GAA GAG GAA CAA CTY GGA TAA AAY TG
2350-2374_3R Run 2 CCC CRT AGC CAG AYA ATG GYA CAC G
2720-2741_3R Run 2 TAG AGA AAA CCR GAR GGA GCC C
3076-3096_3R Run 2 TGT RTT GGT CCA TCT RCC AGG
3351-3368_3R Run 2 TCY GGG CCA GGG ATA TTC
477-497_4R Run 2 TAA GAA TAG CYA TCT TYC CAC
728-745_4R Run 2 CCC TCC TGT GTC CTT GCC
910-930_4R Run 2 AAA AGG GCA TGT GAA TGT GCC
1337-1355_4R Run 2 CCG CTC TAT CCT GCT GRC C
1704-1727_4R Run 2 CTT CGC TTG TAT AGG CCC TG
2179-2207_4R Run 2 GAA CAC CYT TCC AAT ACC TAT GAT TCT CC
2493-2514_4R Run 2 CCT CAC AAG MAC AGA ACC TTT C
2858-2879_4R Run 2 GCT GTC AGG TGA TAY YTG AGA G
3196-3218_4R Run 2 AAA GTG AGA CAT CTC TCA CAT GG
3379-3397_4R Run 2 GCC TGA TTA GTG CCC ATC C
3540-3566_4R Run 2 GGA AAG AGC GTA CAA CTC AAY RAA GAG
47
Supplemental Table 6. Isolates from GenBank used for the evolutionary analyses of lychnis
mottle virus. GenBank
Designation
Isolate Designation Plant Species Country of
Origin
Publication
LC382243.1 Lychnis mottle virus J genomic RNA Vincetoxicum
acuminatum
Japan Fujimoto
et al. 2018
KR011033.1 Lychnis mottle virus isolate Andong Lychnis cognata South
Korea
Yoo et al.
2015a
KR011031.1 Cnidium vein yellowing virus 2 isolate Yeongyang2 Cnidium officinale South
Korea
Yoo et al.
2015b
KR011029.1 Cnidium vein yellowing virus 1 isolate Yeongyang1 Cnidium officinale South
Korea
Yoo et al.
2015b
MH237606.1 Lilium_5875017 Lilium sp. type
OrientalTrumpet
hybrid
Netherlands Dullemans
et al. 2020
MG062674.1 Lilium_17-007 Lilium sp. type:
Asiatic
Netherlands Dullemans
et al. 2020
MF797010.1 Lilium_14-035 Lilium sp. type
OrientalTrumpet
hybrid
Netherlands Dullemans
et al. 2020
MF797008.1 Lilium_14-034 Lilium sp. type
OrientalTrumpet
hybrid
Netherlands Dullemans
et al. 2020
MF796982.1 Lilium_14-002 Lilium sp. type:
Oriental-Trumpet
hybrid
Netherlands Dullemans
et al. 2020
MF796980.1 Lilium_14-001 Lilium sp. type
OrientalTrumpet
hybrid
Netherlands Dullemans
et al. 2020
LycMoV-A_China_Medicago Medicago sativa
L.
China Jiang et al.
2019
CnVYV-A2_China_Medicago Medicago sativa
L.
China Jiang et al.
2019
CnVYV-A1_China_Medicago Medicago sativa
L.
China Jiang et al.
2019
NC_006965.1 Strawberry_latent_ringspot_virus_RNA2_complete_sequence Mentha x gracilis,
'Variegata'
USA Tzanetakis
et al. 2006

48
Chapter 3: Population structure of the emerging cycas necrotic stunt virus and the
development of data-driven diagnostics
Abstract
Cycas necrotic stunt virus (CNSV) has an extensive host range and is emerging around
the globe, as a, pathogen of concern for several agricultural industries. One of the plant species
affected by CNSV is peony (Paeonia lactiflora Pall.). The virus is asymptomatic in most peony
cultivars, but there have been reports of leaf abnormalities and reddening in others. It is thus
important to study CNSV and its population structure to gain insights into its evolution and
epidemiology. The outputs of this study, in addition to the in-depth analysis of the virus
population structure, includes the development of a multiplex detection protocol that can
potentially amplify all available CNSV isolates, thus allowing for accurate, reliable detection of
the virus and safeguarding its susceptible, clonally propagated hosts.
Introduction
Cycas necrotic stunt virus (CNSV) is an emerging virus given the several reports on new
hosts and geographic areas in the past few years (Shaffer et al. 2019; Jiang et al. 2019; Lim et al.
2019; Igori et al. 2020; Tang et. al 2020). Symptoms depend on the host and the synergistic
effects of mixed infections of CNSV with other viruses (Shaffer et al. 2019; Igori et al. 2020;
Tang et. al 2020). Peony (P. lactiflora), a host of CNSV, is commercially propagated via root
cuttings and the virus is of great concern to the industry because it is asymptomatic in most
infections (Shaffer et al. 2019) and so it could be moved undetected through propagation
material. As CNSV appears to be increasing in incidence around the world, it is important to
investigate its population structure to better understand its epidemiology.

49
A member of the genus Nepovirus, CNSV is a bipartite, positive sense, single stranded
RNA virus. RNA 1 codes for the helicase, viral genome-linked protein (VPg), cysteine protease,
and RNA-dependent RNA polymerase (RdRp). RNA 2 codes for the homing (HP), coat (CP),
and movement (MP) proteins (Han et al. 2002). CNSV is only known to be transmitted by
mechanical means and by seed in Chenopodium amaranticolor and C. serotinum (Kusunoki al.
1986; Hanada et al. 2006) but given its taxonomy to the virus is likely also transmitted by pollen
and possibly by nematodes.
The first step in controlling the virus and minimizing its spread is the development of
accurate, reliable detection methods. A component of diagnostic accuracy is specificity which
can only be achieved with the prior knowledge of the pathogen diversity. Without a good
understanding of the virus population structure a diagnostic method could only detect a handful
of variants.
High-throughput sequencing (HTS) overcomes this obstacle by allowing for the unbiased
sequencing of all virus variants (Katsiani et al. 2020). The streamlined sequencing of diverse
isolates as provided by HTS allows for in-depth study of the virus population structure data used
for development of detection assays that would be accurate, and both sensitive and specific
(Rubio et al., 2020).
In this communication 17 CNSV isolates from peony alongside all available completely
sequenced isolates available in GenBank (Table S1) were analyzed and the population structure
of the virus was reconstructed using both phylogenetic and comparative analyses. Data were
used in the development of a diagnostic protocol targeting the 3’ UTR of the viral RNAs; a test
that could potentially detect all known isolates of the virus.

50
Methods and Materials
Plant material and high throughput sequencing
Three hundred and twenty-three (323) P. lactiflora leaf samples were obtained from from
Arkansas, Oregon, New York, Michigan, and Alaska (Table S2). Total RNA was extracted
following the Poudel et al. (2013) protocol and material was tested using the primers of Shaffer
et al. (2019) (F 5′-GAAGAGCATAGAAAGCATCTTTGA-3′/ R 5′-
CTTGGCACCAAGTCCAGTTC-3′). The PCR program consisted of two-minute denaturation at
94 ℃, followed by 40 cycles at 94 ℃ for 30 seconds, annealing for 30 seconds at 55 ℃ and
extension of 60 seconds at 72 ℃. The program terminated with a 10-minute elongation step at 72
℃. CNSV was present in at least one sample in every state sampled and in 85% of the material
tested (Table S1).
In order to understand the population structure of the virus, 17 isolates were selected
from Alaska, Arkansas, New York and Oregon and sequenced using both HTS and Sanger
sequencing. HTS was performed in two sets: for the first group (AR K, AR CX, and AR AHF)
total RNA was extracted according to Poudel et al. 2013 in two repeats. Both RNA extractions
were DNase treated, combined, and purified and used as the template for Illumina libraries.
Libraries were prepared using the Illumina Truseq Stranded RNA with Plant Ribo-zero kit and
sequenced using the Illumina NextSeq500/550 platform at 75 bp single end reads (Huntsman
Cancer Institute, University of Utah). Samples were pooled together for sequencing and were
delivered as sample-specific Illumina files. The second group of 14 isolates, dsRNA extractions
were completed essentially as described in Ho et al. (2018). For reverse transcription the dsRNA
template was denatured using an equal volume of 40mM methyl mercury in the presence of an
A-tail binding and virus specific primers (Table S3) with the reaction performed as described in

51
Shaffer et al. (2020; under review). The library was prepared using the Swift Biosciences Acell-
NGS 1S Plus DNA Library Kit with a selection size of 350 bp and pooled together for
sequencing using the HiSeq 2500 platform to generate 75 bp single end reads (Genomics &
Proteomics Center, Oklahoma State University). The reads for these samples were delivered as
sample-specific files with adapters trimmed off. Reads from all samples were additionally
analyzed using the default parameters in CLC Genomics Workbench v. 20.3 (QIAGEN Hilden,
Germany).
Annotation and Phylogenetic Analyses
Raw reads from AK 16, were mapped to contigs for RNA 1 and RNA 2 generated from
isolates from the peony population, Sequencing reads from AK 16 were then used as the
reference sequenced for downstream analyses. Sequences that were ≥90% of the length and also
≥80% similar to AK16 were mapped using CLC Genomics Workbench v20.3. The consensus
sequence from each isolate used in the analyses represent a ≥10X coverage per nucleotide. There
were sequence gaps in isolates MW, OR 117 and OR Itoh 3 that were PCR amplified and Sanger
sequenced; yet small gaps remained for eight isolates (0.1-5.2% of the coding regions; Table S4).
Phylogenetic and Comparative Analysis
Full length sequences of the virus available in GenBank alongside the peony isolates
comprised the 22 isolate dataset (Table S1). The full nucleotide (nt) and amino acid (aa)
polyprotein sequences for RNA 1 and RNA 2 were uploaded into Species Demarcation Tool
v1.2 (SDT; Muhire et al. 2014) and aligned using MUSCLE (Edgar et al. 2004) to determine
pairwise percent identities. After alignment, the best-fit nucleotide substitution model (JTT+G+I)
was determined for the complete RdRp amino acid (aa) dataset. An unrooted neighbor-joining
tree was constructed similar to Shaffer et al. (2020; under review) with 1000 bootstrap

52
pesudoreplicates. Any branches with less than 70% support were collapsed using TreeGraph 2
(Stöver and Müller 2010). Known nepoviral motifs were examined including the NTPase
binding domains (Gorbalenya et al. 1990), the protease GC and eight RdRp motifs (Sanfaçon et
al. 2009; Koonin 1991), the LPL of the movement protein (Koonin et al. 1991) and the FYGR in
the C terminus of the CP (Le Gall et al. 1995). The cleavage site between the MP and CP has
been experimentally verified (Demangeat et al. 1991) in tomato black ring virus, a member of
subgroup B nepoviruses alike CNSV, unlike the cleavage site between the HP and MP (Digiaro
et al. 2015). For this reason, the concatemer of the latter two proteins was used in the analysis.
Sequences for each protein were codon-aligned in MEGA 7 (Kumar et al. 2016) to
measure the selection pressure ω = dN/dS where dN depicts the non-synonymous substitutions
per nonsynonymous site and dS depicts synonymous substitutions per synonymous site (dS). An
amino acid is under negative selection when dN/dS is <1, neutral when dN/dS = 1 and positive
when dN/dS is >1. Analyses were conducted using the FUBAR method (Murrell et al. 2013) in
SelectionMap v.1.0 (Stenzel et al. 2014; Shaffer et al. 2020 un).
Detection
The information generated was used to design a multiplex-PCR detection test. The RT
was performed using an A-tail binding primer that adds a nucleotide scaffold for use in the
amplification step (OligoDT 5’-GGCCACGCGTCGACTAGTACTTTTTTTTTTTTTTTTTT-
3’). The cDNA was diluted 3:1 with nuclease-free water and used as a template for PCR
amplification. The CNSV specific primer (CNSV-F 5’AATGATGCGCGTTTATTGCACACG-
3’) shares 100% nt identities to all published virus isolates and used in PCR with reverse primer
(Adapter-R 5’-GGCCACGCGTCGACTAGTAC-3’) which binds to nucleotide scaffold
introduced during cDNA synthesis to yield a 209bp amplicon. The internal control primers

53
targeted a region of the NADH- β subunit gene (Tzanetakis et al. 2007). A 25uL reaction mixture
contained equal amounts of each primer (.4uM), 400 uM dNTPs, 1U of Taq DNA polymerase
(Genscript NJ), 10X Taq buffer (proprietary), 5% acetamide (Thermo Fisher Scientific MA) and
RNase/DNase-free water. The amplification program initiated with incubation at 94 ℃ for 2
minutes followed by 35 cycles at 94 ℃ for 30 seconds, annealing at 55 ℃ for 15 seconds and
extension at 72 ℃ for 45 seconds. The program concluded with a one-minute incubation at 72
℃.
Results
HTS Sequencing Outputs
The total number of reads ranged from 41-57M for the three samples using total RNA as
template and ~672K-12.5M reads for the cDNA generated from dsRNA. Virus specific reads
ranged from ~596-2.179K for the samples generated from total RNA template and ~7.8-290K for
the samples generated from dsRNA (Table S4). The complete RNA 1 coding sequence of was
obtained for 11 isolates, while the remaining six had 98.5-99.99% coverage. For RNA 2, the
complete coding sequence was attained for 14 isolates, with the remaining three having 94.8-
97.3% coverage. Even though the three samples using total RNA as a matrix had much higher
amounts of reads mapped specifically to CNSV, there was little difference in the amount of
coding sequenced attained (94.8-97.3%) when compared to the dsRNA samples with coverage of
98.5-99.99% for the isolates with gaps.
Evolutionary Analysis
Phylogenetic analysis of the RdRp indicate two major clades; one with all peony isolates
including the one from P. suffruticosa Andrews (MK512741.1) from South Korea (Lim et al.
2019) and a second with all other isolates (Figure 1).

54
The protein motifs studied were mostly conserved with minimal changes (Table 1). The
cleavage sites between proteins were conserved in the population.
Selection pressure was analyzed using Selection Map and the visual representations of
the selection are shown in Figure 2. The selection values ranged from ω=0.142 for the protease
to ω= 0.408 for the protease cofactor. All protease, CP, and VPg sites undergo negative selection
whereas there was a small number of sites that undergo positive selection in the protease
cofactor, helicase, RdRp and HP/MP (Table 2). All the positive selection sites of the polymerase
were concentrated at the C’ terminus pointing to an evolutionary hotspot whereas in the HP/MP
concatemer positive selection concentrates in the HP region and the N-terminus of the MP.
The nucleotide pairwise percent identity between the entire CNSV population is ~ 80-
99% with the US peony isolates sharing the highest % identities when compared to the rest of the
population (Figure 3).
Diagnostics
Infected samples have a dominant virus-specific band whereas the CNSV-free samples
have a dominant internal control band (Figure 4). This indicates that during PCR virus amplicons
are favored over the internal control. This is a preferred situation since the virus target will be
preferentially amplified over the internal control in a low virus titer situation. The internal
control amplicon validates the protocol by ensuring the quality of nucleic acids and the
successful enzymatic reactions (Thekke-Veetil et al. 2016). More than 25 samples were analyzed
with Sanger sequencing and they were all virus specific.
Discussion
Investigating the virus population structure will help better understand the epidemiology
and movement of CNSV. HTS was used to examine the population structure of CNSV and for

55
the development of accurate diagnostics. The selection pressures acting on the CNSV proteins
were also determined. Analyses indicate that the virus is under negative selection pressure with
few sites undergoing positive selection; especially at the C-terminus of the RdRp. These changes
might affect virus fitness in different hosts, environments or geographicr locations. All nepovirid
motifs were highly conserved in the entire population, as is the peony population, as all isolates
cluster together, including the P. suffruticosa isolate from South Korea (MK512741.1).
There is no universal control to slow the spread of plant viruses, outside vector control. In
the case of CNSV, for which is there is no known vector, control will be difficult. Yet, the best
way to control a virus infecting clonally propagated crops is to rely on accurate diagnostics to
eliminate infected material, thereby minimizing virus incidence to prevent its further spread and
potential widespread losses in crops (Gergerich et al., 2015; Martin and Tzanetakis, 2015; Martin
et al., 2016). Nepoviruses are primarily transmitted by nematodes, although there are exceptions
like Black currant reversion virus,that is transmitted by the eriophyid mite, Cecidophyopsis ribis
(Latvala et al. 1998) or Cherry leafroll and Blueberry leaf mottle viruses that are without known
vectors, but with confirmed high pollen and seed transmission rates (Childress and Ramsdell
1987; Rebenstorf et al. 2006). Pollen is a very effective way for viruses to be disseminated by
wind currents and human movement of pollinating insects that can sometime exceeds thousands
of miles (Melicher et al. 2019). CNSV could potentially be transmitted by any of the
aforementioned methods however there needs to be thorough investigations of alternative modes
of transmission of CNSV other than mechanical means (Kusonoki et al. 1986).
Due to the increase in reported cases (Shaffer et al. 2019; Jiang et al. 2019; Lim et al.
2019; Igori et al. 2020; Tang et. al 2020) and increasingly far-reaching distribution of CNSV
there may be some form of active spread, perhaps pollen/seed transmission of CNSV in peony.

56
CNSV has already been shown to be seed-transmissible in C. amaranticolor and C. serotinum
with vertical transmission rates as high as 89% (Kusunoki et al. 1986). Pollen-borne and seed-
borne viruses can also cross-infect plant species such as in the case of Blueberry shock virus
(BlShV) (Thomas-Sharma et al. 2017) that infects both cranberry and blueberry and CLRV that
naturally infects a variety of herbaceous and woody hosts (Rebenstorf et al. 2006). In BlShV
affecting blueberries, the virus moves horizontally to nearby plants by bee mediated
transmission, and seed transmission was documented in 1.2% of seedlings (Bristow and Martin
1999). On the other hand, in cranberries, BlShV had a vertical transmission rate up to 91%
whereas horizontal transmission via pollen was not reported (Thomas-Sharma
2017). Interspecies transmission has also been shown to occur horizontally for Raspberry bushy
dwarf virus (RBDV) when infected raspberry pollen was able to infect the stigma of Torenia
fournieri (torenia) resulting in horizontal transmission (Isogai et al. 2014). RBDV was then able
to move from infected torenia into the seeds demonstrating vertical transmission (Isogai et al.
2015). Thus RBDV, is able to infect torenia first through horizontal transmission and then
vertical transmission which could allow RBDV to enter a new population and spread quickly.
Due to the extremely high incidence of CNSV in peony and its global distribution
(Shaffer et al. 2019; Jiang et al. 2019; Lim et al. 2019; Igori et al. 2020; Tang et. al 2020) it is
likely that the virus is transmitting either horizontally through pollen or vertically through seed.
The probability of seed transmission is lower, however, because of the unique attributes of peony
seed germination (Yu et al. 2007). The nucleotide identity between the entire CNSV population
is ~ 80-99%, a wide range for isolates of the same virus, potentially allowing for virus plasticity
that facilitates different transmission strategies (da Silva et al. 2020).

57
There is only one clustering within the peony clade consisting of OR Itoh 3, AR K, and
NY 33 which could be due to the small dataset size. CNSV has been detected in both P.
suffruticosa, P. lactiflora, and Itoh hybrids, which point to active transmission of CNSV in
peony and potentially other hosts. The high rate of incidence of the virus and multiple reports in
peony could be a result of effective means of virus transmission, understudied till present. It is
also possible that CNSV could have been introduced into peony via bottleneck as discussed in
Shaffer et al. 2020 (under review), although given the low sequence diversity and the length of
time peonies have been cultivated this is not probable. The CNSV isolate from P. suffruticosa
from South Korea also groups with P. lactiflora from the US even though these are plants are
different species and have been separated by both spatially and temporally for potentially
decades or even centuries. The similarity between these two isolates gives more credence to a
possible pollen/seed transmission strategy for CNSV in peony. Mechanical transmission was
attempted repeatedly and though successful with LycMoV in Shaffer et al. 2019, it was never
achieved for CNSV (data not shown). Considering the number of peonies that have been in the
Nichols Arboretum collection since 1937 and have tested positive for the virus CNSV must be
moving actively via some means to be so ubiquitous, but with such a limited diversity in peony.
Regardless of the modes of transmission, the prevalence of CNSV in peony and its
increasing incidence worldwide merits further investigation. Having a wide distribution, and an
incompletely investigated host range, the virus biology needs to be studied in-depth to prevent
epidemics in agricultural and horticultural settings. As CNSV becomes more prevalent, the
development of accurate diagnostics will be a valuable tool to help safeguard plant species,
agriculture, and ecosystems from CNSV and to help establish the true incidence of CNSV in
different plant hosts and geographic regions. A diagnostic protocol was developed using a primer

58
that is 100% identical to both 3’ UTRs of all sequenced isolates, allows for detection of both
RNAs, increasing the sensitivity of the assay. This assay will help track virus spread and identify
its extended host range. The findings from this study indicate that CNSV is moving, in addition
to its clonal propagation through other modes, and that negative selection is a major factor in
shaping the population structure of the virus.

59
References
Bristow, P. R., and Martin, R. R. 1999. Transmission and the role of honeybees in field spread of
blueberry shock ilarvirus, a pollen-borne virus of highbush blueberry. Phytopathology. 89:124–
130.
Childress, A. M., and Ramsdell, D. C. 1987. Bee-mediated transmission of blueberry leaf mottle
virus via infected pollen in highbush blueberry. Phytopathology. 77: 167-172
da Silva, W., Kutnjak, D., Xu, Yi, Xu, Yimin, Giovannoni, J., Elena, S. F., and Gray, S., 2020.
Transmission modes affect the population structure of potato virus Y in potato. PLoS Pathog. 16
Demangeat, G., Hemmer, O., Fritsch, C., Gall, O. L., and Candresse, T. 1991. In vitro processing
of the RNA-2-encoded polyprotein of two nepoviruses: tomato black ring virus and grapevine
chrome mosaic virus. J. Gen. Virol. 72:247–252.
Digiaro, M., Yahyaoui, E., Martelli, G. P., and Elbeaino, T. 2015. The sequencing of the
complete genome of a Tomato black ring virus (TBRV) and of the RNA2 of three Grapevine
chrome mosaic virus (GCMV) isolates from grapevine reveals the possible recombinant origin of
GCMV. Virus Genes. 50:165–171.
Edgar, R. C. 2004. MUSCLE: multiple sequence alignment with high accuracy and high
throughput. Nucleic Acids Res. 32:1792–1797.
Gergerich, R. C., Welliver, R. A., Gettys, S., Osterbauer, N. K., Kamenidou, S., Martin, R. R.,
Golino, D. A., Eastwell, K., Fuchs, M., Vidalakis, G., and Tzanetakis, I. E. 2015. Safeguarding
Fruit Crops in the Age of Agricultural Globalization. Plant Dis. 99:176–187.
Gorbalenya, A. E., Koonin, E. V., and Wolf, Y. I. 1990. A new superfamily of putative NTP-
binding domains encoded by genomes of small DNA and RNA viruses. FEBS Lett. 262:145–
148.
Han, S. S., Karasev, A. V., Ieki, H., and Iwanami, T. 2002. Nucleotide sequence and taxonomy
of Cycas necrotic stunt virus. Arch. Virol. 147:2207–2214.
Hanada, K., Fukumoto, F., Kusunoki, M., Kameya-Iwaki, M., Tanaka, Y., and Iwanami, T.
2006. Cycas necrotic stunt virus isolated from gladiolus plants in Japan. J. Gen. Plant Pathol.
72:383–386.
Ho, T., Harris, A., Katsiani, A., Khadgi, A., Schilder, A., and Tzanetakis, I. E. 2018. Genome
sequence and detection of peach rosette mosaic virus. J. Virol. Methods. 254:8–12.
Igori, D., Lee, H.-K., Yang, H.-J., Lee, D.-S., Kim, S.-Y., Kwon, B., Oh, J., Kim, T.-D., An, C.,
Moon, J.-S., Lee, S. H. 2020. First report of the Cycas necrotic stunt virus infecting Cnidium
officinale in South Korea. Plant Dis. In press.

60
Isogai, M., Yoshida, T., Nakanowatari, C., and Yoshikawa, N. 2014. Penetration of pollen tubes
with accumulated Raspberry bushy dwarf virus into stigmas is involved in initial infection of
maternal tissue and horizontal transmission. Virology. 452–453:247–253.
Isogai, M., Yoshida, T., Shimura, T., and Yoshikawa, N. 2015. Pollen tubes introduce Raspberry
bushy dwarf virus into embryo sacs during fertilization processes. Virology. 484:341–345.
Jiang, P., Shao, J., and Nemchinov, L. G. 2019. Identification of emerging viral genomes in
transcriptomic datasets of alfalfa (Medicago sativa L.). Virol. J. 16:153.
Katsiani, A., Stainton, D., Lamour, K., and Tzanetakis, I. E. 2020. The population structure of
Rose rosette virus in the USA. J. Gen. Virol. 101:676–684.
Koonin, E. V. 1991. The phylogeny of RNA-dependent RNA polymerases of positive-strand
RNA viruses. J. Gen. Virol. 72 :2197–2206.
Koonin, E. V., Mushegian, A. R., Ryabov, E. V., and Dolja, V. V. 1991. Diverse groups of plant
RNA and DNA viruses share related movement proteins that may possess chaperone-like
activity. J. Gen. Virol. 72:2895- 2903.
Kumar, S., Stecher, G., and Tamura, K. 2016. MEGA7: Molecular Evolutionary Genetics
Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 33:1870–1874.
Kusunoki, M., Hanada, K., Iwaki, M., Chang, M. U., Doi, Y., and Yora, K. 1986. Cycas Necrotic
Stunt Virus, a New Member of Nepoviruses Found in Cycas revoluta Host Range, Purification,
Serology and Some Other Properties. Jpn. J. Phytopathol. 52:302–311.
Latvala, S., Susi, P., Kalkkinen, N., and Lehto, K. 1998. Characterization of the coat protein
gene of mite-transmitted blackcurrant reversion associated nepovirus. Virus Res. 53:1–11.
Le Gall, O., Candresse, T., and Dunez, J. 1995. A multiple alignment of the capsid protein
sequences of nepoviruses and comoviruses suggests a common structure. Arch. Virol. 140: 2041-
2053
Lim, S., Kwon, S.-Y., Lee, J.-H., Cho, H. S., Kim, H.-S., Park, J. M., Lee. S.-H., Moon, J.-S.
2019. Genomic detection and molecular characterization of two distinct isolates of cycas necrotic
stunt virus from Paeonia suffruticosa and Daphne odora. Virus Genes. 55:734–737.
Martin, R. R., and Tzanetakis, I. E. 2015. Control of virus diseases of berry crops. Adv. Virus
Res. 91:271–309.
Martin, R. R., Constable, F., and Tzanetakis, I. E. 2016. Quarantine Regulations and the Impact
of Modern Detection Methods. Annu. Rev. Phytopathol. 54:189–205.

61
Melicher, D., Wilson, E. S., Bowsher, J. H., Peterson, S. S., Yocum, G. D., and Rinehart, J. P.
2019. Long-Distance Transportation Causes Temperature Stress in the Honey Bee, Apis
mellifera (Hymenoptera: Apidae). Environ. Entomol. 48:691–701.
Muhire, B. M., Varsani, A., and Martin, D. P. 2014. SDT: A Virus Classification Tool Based on
Pairwise Sequence Alignment and Identity Calculation ed. Jens H. Kuhn. PLoS ONE.
9:e108277.
Murrell, B., Moola, S., Mabona, A., Weighill, T., Sheward, D., Kosakovsky Pond, S. L., and
Scheffler K. 2013. FUBAR: a fast, unconstrained bayesian approximation for inferring selection.
Mol. Biol. Evol. 30:1196–1205.
Poudel, B., Wintermantel, W. M., Cortez, A. A., Ho, T., Khadgi, A., and Tzanetakis, I. E. 2013.
Epidemiology of Blackberry yellow vein associated virus. Plant Dis. 97:1352–1357.
Rebenstorf, K., Candresse, T., Dulucq, M. J., Büttner, C., and Obermeier, C. 2006. Host Species-
Dependent Population Structure of a Pollen-Borne Plant Virus, Cherry Leaf Roll Virus. J. Virol.
80:2453–2462.
Rubio, L., Galipienso, L., and Ferriol, I. 2020. Detection of Plant Viruses and Disease
Management: Relevance of Genetic Diversity and Evolution. Front. Plant Sci. 11:1092
Sanfaçon, H., Wellink, J., Le Gall, O., Karasev, A., van der Vlugt, R., and Wetzel, T. 2009.
Secoviridae: a proposed family of plant viruses within the order Picornavirales that combines the
families Sequiviridae and Comoviridae, the unassigned genera Cheravirus and Sadwavirus, and
the proposed genus Torradovirus. Arch. Virol. 154:899–907.
Shaffer, C., Gress, J. C., and Tzanetakis, I. E. 2019. First Report of Cycas Necrotic Stunt Virus
and Lychnis Mottle Virus in Peony in the United States. Plant Dis. 103:1048–1048.
Stenzel, T., Piasecki, T., Chrząstek, K., Julian, L., Muhire, B. M., Golden, M., Martin, D. P., and
Varsani, A. 2014. Pigeon circoviruses display patterns of recombination, genomic secondary
structure and selection similar to those of beak and feather disease viruses. J. Gen. Virol.
95:1338–1351.
Stöver, B. C., and Müller, K. F. 2010. TreeGraph 2: Combining and visualizing evidence from
different phylogenetic analyses. BMC Bioinformatics. 11:7.
Tang, J., Ward, L., and Delmiglio, C. 2020. Occurrence of Cycas Necrotic Stunt Virus in
Paeonia lactiflora in New Zealand. Plant Dis. 104:1567–1567.
Thekke-Veetil, T., Ho, T., Moyer, C., Whitaker, V. M., and Tzanetakis, I. E. 2016. Detection of
Strawberry necrotic shock virus using conventional and TaqMan® quantitative RT-PCR. J.
Virol. Methods. 235:176–181.

62
Thomas-Sharma, S., Wells-Hansen, L., Page, R., Kartanos, V., Saalau-Rojas, E., Lockhart,
Benham. E. L., and McManus, P. S. 2017. Characterization of Blueberry shock virus, an
Emerging Ilarvirus in Cranberry. Plant Dis. 102:91–97.
Tzanetakis, I. E., Postman, J. D., and Martin, R. R. 2007. Identification, detection and
transmission of a new vitivirus from Mentha. Arch. Virol. 152:2027–2033.
Yu, X., Zhao, R., and Cheng, F. 2007. Seed Germination of Tree and Herbaceous Peonies: A
Mini-Review. Seed Sci. Biotech. 4:11-14

63
Figures
Figure 1. Unrooted maximum-likelihood phylogenetic tree using the aligned amino acid
alignment of the RNA-dependent RNA polymerase of Cycas necrotic stunt virus. The tree was
generated using the JTT+G+I model with 1000 bootstrap replicates on MEGA 7 (Kumar et al.
2016). The bar represents the amino acid changes per site.
64
(A) Protease Cofactor
(C) Viral protein genome-linked
Figure 2. SelectionMap v1.0 (Stenzel et al. 2014) generated visual profiles for each of the Cycas
necrotic stunt virus proteins indicating both negative (blue) and positive (green) selection as
calculated by FUBAR (Murrell et al. 2013). Average dN/dS is depicted above each graph.
1 10 20 30 40 50 60 70 80 90 100 110 120 130 140 150 160 170 180 190 200 210 220 230 240 250 260 270 280 290 300 310 320 330 340 350 360 370 380 390 400 410 420 430 440 450 460 470 480 490 500 510 520 530 540 550 560
Ab
s (
dN
- d
S)
0
2.5
5
7.5
10
Average dN/dS = .407016
09-07-20 Protease cofactor nt aa algined Selection map incomplete codon fixed
Negative selection
Positive selection
1 10 20
Ab
s (
dN
- d
S)
0
2.5
5
7.5
10
Average dN/dS = .262616
09-07-20 VPg nt aa algined Selection map no incomplete codons
Negative selection
Positive selection
Average dN/dS =.142373
Average dN/dS =.408255
(B) Protease

65
(D)Helicase
(E) RNA-dependent RNA polymerase
(F) Homing Protein and MP
(G) CP
Figure 2 Cont.
1 10 20 30 40 50 60 70 80 90 100 110 120 130 140 150 160 170 180 190 200 210 220 230 240 250 260 270 280 290 300 310 320 330 340 350 360 370 380 390 400 410 420 430 440 450 460 470 480 490 500 510 520 530 540 550 560 570 580 590 600 610 620 630 640 650 660 670 680 690
Ab
s (
dN
- d
S)
0
2.5
5
7.5
10
Average dN/dS = .171647
09-07-20 Helicase nt aa algined Selection map incomplete codon fixed
Negative selection
Positive selection
1 10 20 30 40 50 60 70 80 90 100 110 120 130 140 150 160 170 180 190 200 210 220 230 240 250 260 270 280 290 300 310 320 330 340 350 360 370 380 390 400 410 420 430 440 450 460 470 480 490 500 510 520 530 540 550 560 570 580 590 600 610 620 630 640 650 660 670 680 690 700 710 720 730 740 750 760 770 780 790 800 810 820 830 840 850
Ab
s (
dN
- d
S)
0
2.5
5
7.5
10
Average dN/dS = .190653
09-07-20 RdRP nt aa algined Selection map incomplete codons fixed and stops deleted
Negative selection
Positive selection
1 10 20 30 40 50 60 70 80 90 100 110 120 130 140 150 160 170 180 190 200 210 220 230 240 250 260 270 280 290 300 310 320 330 340 350 360 370 380 390 400 410 420 430 440 450 460 470 480 490 500 510 520 530 540 550 560 570 580 590 600 610 620 630 640 650 660 670 680 690 700 710 720 730 740 750 760 770 780 790
Ab
s (
dN
- d
S)
0
2.5
5
7.5
10
Average dN/dS = .32705
09-07-20 MP nt aa algined Selection map incomplete codon fixed
Negative selection
Positive selection
1 10 20 30 40 50 60 70 80 90 100 110 120 130 140 150 160 170 180 190 200 210 220 230 240 250 260 270 280 290 300 310 320 330 340 350 360 370 380 390 400 410 420 430 440 450 460 470 480 490 500 510 520 530 540 550
Ab
s (
dN
- d
S)
0
2.5
5
7.5
10
Average dN/dS = .174149
09-07-20 CP nt aa algined stop deleted for Selection map and incomplete codon fixed
Negative selection
Positive selection
Average dN/dS =.171647
Average dN/dS =.190653
Average dN/dS =.32705
Average dN/dS =.174149

66
Figure 3. Percent pairwise identities mapped in color-coded matrices for the Cycas necrotic stunt
virus RNA 1 and 2 polyproteins for nt and aa alignments. The visuals were created by Species
Demarcation Tool version 1.2 (Muhire et al. 2014) using the MUSCLE (Edgar et al. 2004)
program to create the individual alignments.
RNA 1 polyprotein aa identities
RNA 2 polyprotein aa identities RNA 2 polyprotein nt identities
RNA 1 polyprotein nt identities
RNA 1 polyprotein aa identities RNA 1 polyprotein nt identities
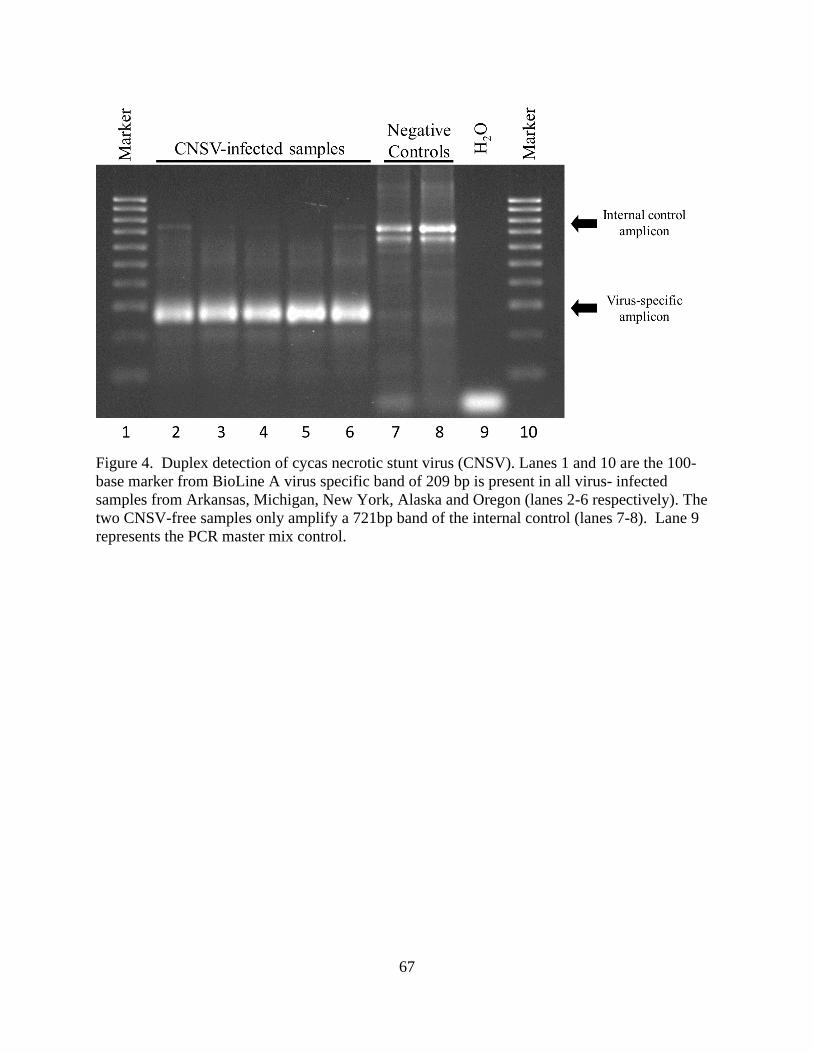
67
Figure 4. Duplex detection of cycas necrotic stunt virus (CNSV). Lanes 1 and 10 are the 100-
base marker from BioLine A virus specific band of 209 bp is present in all virus- infected
samples from Arkansas, Michigan, New York, Alaska and Oregon (lanes 2-6 respectively). The
two CNSV-free samples only amplify a 721bp band of the internal control (lanes 7-8). Lane 9
represents the PCR master mix control.

68
Table 1. Amino acid changes in each Cycas necrotic stunt virus protein motif examined
presented by isolate.
CNSV Isolate Viral motif AA change
JN127336.1 RdRp 1 T218I
MK521837.1 RdRp 1 T218I
BK010916.1 RdRp 1 T218V
NC_003791.1 RdRp 2 L228P
MW RdRp 3 H269R
AK 50 RdRp 3 H269R
AR K RdRp 3 H269R
AR CX RdRp 4 S291R
NC_003791.1 RdRp 4 G292S
NC_003791.1 RdRp 5 I379V
BK010916.1 RdRp 5 I379V
MK521837.1 RdRp 6 V398A
JN127336.1 RdRp 8 S472T
BK010916.1 RdRp 8 S472T
MK528137.1 RdRp 8 S472T
NC_003791.1 RdRp 8 S472T
NY 33 NTPase Binding Domain A F790W
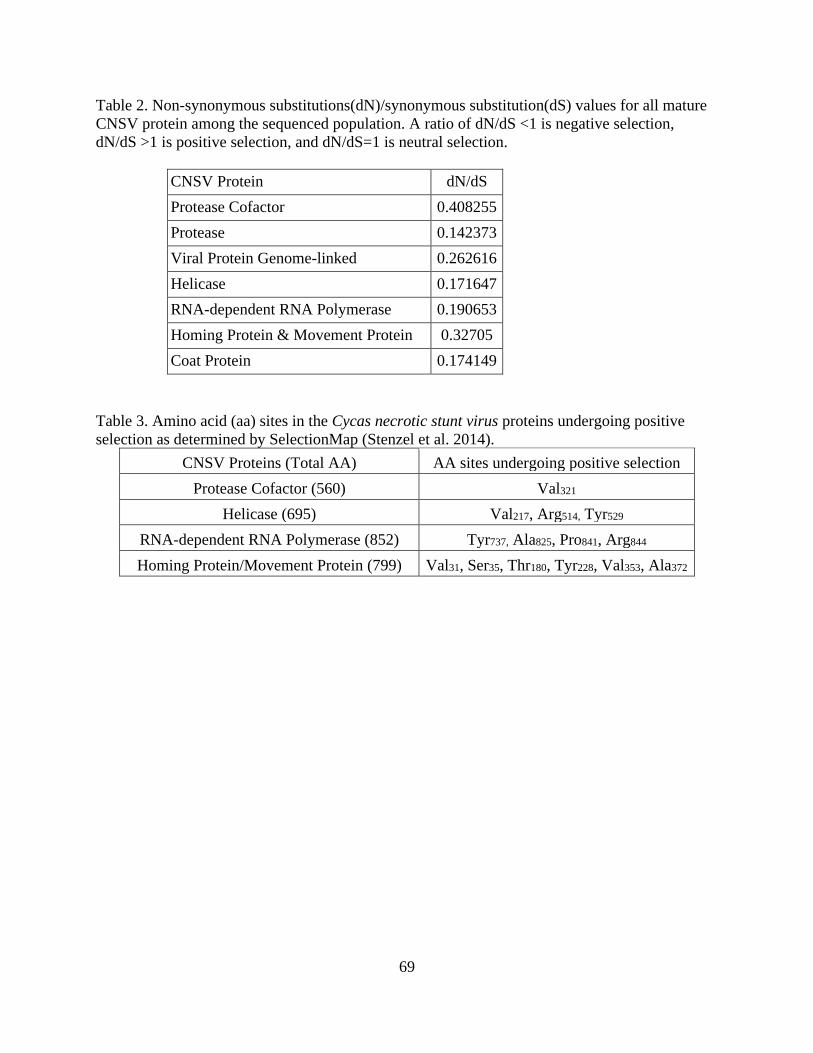
69
Table 2. Non-synonymous substitutions(dN)/synonymous substitution(dS) values for all mature
CNSV protein among the sequenced population. A ratio of dN/dS <1 is negative selection,
dN/dS >1 is positive selection, and dN/dS=1 is neutral selection.
CNSV Protein dN/dS
Protease Cofactor 0.408255
Protease 0.142373
Viral Protein Genome-linked 0.262616
Helicase 0.171647
RNA-dependent RNA Polymerase 0.190653
Homing Protein & Movement Protein 0.32705
Coat Protein 0.174149
Table 3. Amino acid (aa) sites in the Cycas necrotic stunt virus proteins undergoing positive
selection as determined by SelectionMap (Stenzel et al. 2014).
CNSV Proteins (Total AA) AA sites undergoing positive selection
Protease Cofactor (560) Val321
Helicase (695) Val217, Arg514, Tyr529
RNA-dependent RNA Polymerase (852) Tyr737, Ala825, Pro841, Arg844
Homing Protein/Movement Protein (799) Val31, Ser35, Thr180, Tyr228, Val353, Ala372
70
Supplementary Table 1. Cycas necrotic stunt virus sequences used in this study. Accession
numbers, isolate name, host, country of origin, and publication are shown.
GenBank
Designation
Isolate Designation Plant Species Country of
Origin
Publication
MK521837.1
isolate DJ segment RNA1,
complete sequence Daphne odora South Korea
Lim et al.
2019
MK521838.1
isolate DJ segment RNA2,
complete sequence Daphne odora South Korea
Lim et al.
2019
MK512741.1
isolate AD segment
RNA1, complete sequence Paeonia suffruticosa South Korea
Lim et al.
2019
MK512742.1
isolate AD segment
RNA2, complete sequence Paeonia suffruticosa South Korea
Lim et al.
2019
BK010916.1
TPA_asm: segment RNA1,
complete sequence
Medicago sativa
cultivar Dryland China
Jiang et al.
2019
BK010917.1
TPA_asm: segment RNA2,
complete sequence
Medicago sativa
cultivar Dryland China
Jiang et al.
2019
NC_003791.1 RNA 1, complete sequence Cycas revoluta Japan
Han et al.
2002
NC_003792.2 RNA 2, complete sequence Cycas revoluta Japan
Han et al.
2002
JN127336.1
isolate Lily1 polyprotein 1
gene, partial cds Lilium longiflorum Australia
Wylie et al.
2012
JN127337.2
isolate Lily1 segment
RNA2, complete sequence Lilium longiflorum Australia
Wylie et al.
2012
Supplementary Table 2. Number of samples tested positive for Cycas necrotic stunt virus over
the total number of samples tested.
State CNSV Positive Tests/Samples Tested
Alaska 22/24
Arkansas 67/87
Michigan 125/144
New York 51/58
Oregon 9/10
Total 274/323

71
Supplementary Table 3. Primers used in the generation of cDNA from samples that used dsRNA
as template.
Primer name Sequence (5’-3’)
CNSV RT RNA1R 5574-5596 CAA TGG TTT CTT CAG GTG AGT GG
CNSV RT RNA1R 3490-3510 AAA GCA TCC AAA GCC TGA ACC
CNSV RNA1R RT 1585-1608 GGA AAA CCC TTC AAC AAA TTC AGC
CNSV RNA 2R RT 2663-2686 TTT TCA GCA CTC TTC ATA TCA CCC
5’ RNA 1 R 425-450 CAT ATG AAG CAT TAT AGG TGG
3’ RNA 1 F 6907-6930 CTT GAT GGT ACC AGA GAT CTG ATC
5’ RNA 2 R 905-927 TGC AGC CAA GCG TCA AAA GTT GC
3’ RNA 2 F 3946-3967 GAG GGT TTT GGT CCT GAA GAG G
72
Supplementary Table 4. Amount of reads mapped from each sample to Cycas necrotic stunt
virus RNAs with percent of the coding sequence complete stated; in parentheses when
incomplete. Total reads/sample are also presented.
Isolate
Reads Mapped to RNA1 (% of
coding sequence completed)
Reads Mapped to RNA 2 (% of
coding sequence completed)
Total Reads
Generated
AK 16 104,055 180,927 3,231,905
AK 323 185,465 289,819 2,719,989
AK 50 92,723 35,216 (95.2%) 2,036,850
AK 81 22,886 (99.5%) 36,393 950,328
AR AHF 922,449 1,079,542 57,502,821
AR CX 1,927,567 2,178,613 45,522,587
AR K 595,682 (99.9%) 455,411 (94.8%) 41.419,357
MW 48,306 (99.7%) 55,712 1,674,507
NY 33 7,654 (98.5%) 9,417 1,318,811
NY 41 29,368 34,883 671,640
NY 51 26,945 38,271 3,185,361
NY 6 23,254 26,900 992,510
OR 117 76,481 (98.9%) 96,565 860,144
OR 122 71,350 60,529 3,026,986
OR Itoh 1 52,241 79,690 (97.3%) 1,837,562
OR Itoh 3 47,839 86,101 1,654,036
OR Itoh 4 57,984 (98.7%) 59,468 12,486,352

73
Conclusions
This research investigated the virome of peony in association with the causal agent(s) of
Lemoine’s disease (LDP). High-throughput sequencing (HTS) was used on seven P. lactiflora
plants, each representing a different cultivar. This led to the discovery of four viruses infecting
peony: Cycas necrotic stunt virus (CNSV), Lychnis mottle virus (LycMoV), Amazon lily mild
mottle virus (ALiMMV), and Gentian kobu-sho associated virus (GKaV). These viruses were
each surveyed for their incidence in the peony population with a wide range of incidence from a
few plants for ALiMMV to a near universal infection for CNSV. Due to the widespread nature
and number of viruses found in peony by exploratory HTS it would be prudent to screen more
peony plants from different geographic localities, cultivars, and species to better understand the
peony virome.
Although it was not the focus of this Thesis because of its late discovery, GKaV is
perhaps the most interesting. PCR screening detected GKaV in a high proportion of stunted
peony samples. This finding, in conjunction with the ability of the virus to cause tumors similar
to those observed on LDP material in ornamental gentian plants, makes GKaV a prime suspect as
the causal agent of LDP. However, given the limited material assayed more research must be
done to confirm causality.
LycMoV and CNSV were frequently found in peony plants (45% and 85% respectively),
but interestingly, caused no visual symptoms. Both viruses have homogenous populations in
peony with phylogenetic analyses showing peony isolates forming distinct clades from other
isolates. Isolates from other hosts and geographic locations exhibited more genetic diversity in
both cases. The transmission modes and the biology of both viruses are poorly understood and
should be investigated further. The high incidence and worldwide distribution of CNSV and

74
LycMoV make them candidates to cause epidemics under the right conditions if they were to
move to susceptible hosts. Reliable diagnostics for all published isolates of CNSV were
developed, and the information generated in our study for LycMoV could be used to develop
diagnostics in the future, crucial to identifying infected material and preventing virus epidemics.
The main question that comes as a result of this research is: Should perennial plants such
as peony be monitored for viruses prior to movement across state lines or international borders?
Viruses, particularly those that are asymptomatic in perennial, clonally propagated plants could
be readily moved inadvertently. There are no strict regulations for testing for pathogens in most
ornamentals that could be moving viruses asymptomatically. HTS allows for the rapid
identification of new pathogens, but without studying virus biology it is hard to justify the
application of restrictive regulations and testing procedures that would further limit the
movement of plant material. In the case of peony there seems to be no immediate harm to the
plant for most of the viruses found. However, as material is moved around, rapid virus evolution
could facilitate a scenario where more virulent strains emerge that could not only damage peony,
but other agricultural and horticultural crops.