the structure–activity relationship studies of binary room temperature complex electrolytes based...
TRANSCRIPT

www.elsevier.com/locate/vibspec
Vibrational Spectroscopy 44 (2007) 297–307
The structure–activity relationship studies of binary room temperature
complex electrolytes based on LiTFSI and organic compounds
with acylamino group
Renjie Chen a,c, Feng Wu b,c,*, Li Li b,c, Xinping Qiu a, Liquan Chen a, Shi Chen b,c
a Department of Chemistry, Tsinghua University, Beijing 100084, Chinab School of Chemical Engineering and the Environment, Beijing Institute of Technology, Beijing 100081, China
c National Development Center of High Technology Green Materials, Beijing 100081, China
Received 30 May 2006; received in revised form 29 December 2006; accepted 30 January 2007
Available online 9 February 2007
Abstract
Binary room temperature complex electrolytes based on lithium bis(trifluoromethane sulfone) imide (LiN(SO2CF3)2, LiTFSI) and organic
molecules with acylamino (amide) groups, such as ethyleneurea, acetamide, etc., have been synthesized and evaluated with differential scanning
calorimetry (DSC) and ac impedance spectroscopy. Most of the complex systems with proper molar ratio have excellent thermal stability and
electrochemical performance. Infrared (IR) and Raman spectroscopic studies have been carried out to understand the formation these electrolytes.
It is shown that the organic compounds with amide group can coordinate with the Li+ cation and the TFSI� anion via their polar groups (the C O
and NH groups). Such strong interactions lead to the dissociation of LiTFSI and the breaking of the hydrogen bonds among the organic molecules,
resulting in the formation of the complex systems. In order to have a comprehensive understanding of the above interactions and the structure–
activity relationship of these complex systems, the Mulliken charges on the O and N atoms, the equilibrium configuration and the bonding energy
of the systems have been determined by quantum chemistry calculations with non-local density function theory (DFT). The calculations indicate
that the structure and the substitution group of organic molecules influence the charge density and coordination strength of the carbonyl oxygen of
these molecules. In addition, the strength of hydrogen bonding between the organic molecules influences the physico-chemical properties of the
complex electrolyte.
# 2007 Elsevier B.V. All rights reserved.
Keywords: Room temperature complex electrolyte; LiTFSI; Acylamino group; Structure–activity relationship
1. Introduction
Ionic compounds are usually solid with high melting point,
boiling point and rigidity at room temperature due to their
strong electrovalent bonds. However, these properties will be
drastically changed when the big and asymmetric anions and/or
cations are introduced due to the steric hindrance of these
anions and/or cations [1,2]. Room temperature molten salts
(RTMS) have long been the subject of fundamental research in
many fields [3–14], owing to their unusual properties, such as a
* Corresponding author at: School of Chemical Engineering and the Envir-
onment, Beijing Institute of Technology, Beijing 100081, China.
Tel.: +86 10 68912657; fax: +86 10 68451429.
E-mail address: [email protected] (F. Wu).
0924-2031/$ – see front matter # 2007 Elsevier B.V. All rights reserved.
doi:10.1016/j.vibspec.2007.01.003
wide liquid-phase range, high thermal stability, low vapor
pressure, high ionic conductivity, and so forth. Previously we
reported several novel complex systems based on solid LiTFSI
and solid organic compounds with acylamino group, urea, for
example [15–19]. It is noticeable that these complex systems
possess similar characteristics as RTMS, especial low vapor
pressure and nonhygroscopic. Furthermore, these systems are
easy to prepare, and show high thermal stability and good
electrochemical properties. Electric double layer capacitors
based on the active carbon or the carbon nanotube with the
LiTFSI/acetamide complex system as electrolyte showed a
high specific capacity and rather good cycling performance,
indicating that the complex system is a promising candidate of
electrolyte for supercapacitor and other electrochemical
devices [19,20]. Preliminary analysis indicates that the
structure (chain or loop type) and the substitute in the organic

Scheme 1. Structure of organic compounds with acylamino group.
R. Chen et al. / Vibrational Spectroscopy 44 (2007) 297–307298
molecules and the activities of their hydrogen bonds strongly
influence the physico-chemical performances of these complex
electrolytes.
Clearly it is necessary to make a comprehensive study on
the interaction between lithium salt and the organic
compound, the relations between the microstructure in the
complex systems and their thermal and electrochemical
performances, especially how and why the structure of the
organic compound will influence the interactions and the
formation of the microstructures in the complex systems. It is
a pity, however, that most authors stopped at characterizing
the properties of the complex systems. Only very few of them
tried to understand the intrinsic reasons for the experimental
observations, and even fewer scientists take efforts to find out
these important questions in the scale of microstructures and
in the aspects of theoretical calculations. Maginn and co-
workers [21–23] used Monte Carlo method to study
extensively the kinetics and the related properties of
imidazolium-based ionic liquids. Liang et al. [15] thought
that the strong interactions, mainly the Li–O coordination,
between the two components, lead to the formation of the
LiTFSI/urea composite. Hu et al. [24,25] studied the
vibration spectra of the LiCF3SO3/acetamide complex and
observed obvious band shifting for the C O stretching of
acetamide.
In this paper, binary complex electrolytes based on LiTFSI
and organic compounds with amide groups are synthesized.
The thermal properties and ionic conductivities of these
complex electrolytes are characterized by DSC and ac
impedance, respectively. Most of these organic compounds,
such as acetamide and urea, are common candidates used to
lower the melting point of a composite and form a eutectic
system with different organic or inorganic compounds owing
to their ‘water-like’ physical properties (e.g., high dielectric
constant and dissociation constant) [26,27]. In order to clarify
the influence of the molecular structure on the thermal and
electrochemical properties of the binary complex electro-
lytes, ethyleneurea is chosen because it possesses a five-
membered ring. Moreover, it is interesting to find out how the
properties of the complex systems will be changed when the
NH2 groups in an organic compound are replaced with other
functional groups. Methylurea (NMU) and 1,3-dimethylurea
(DMU) are introduced to examine the effects of the
substitution of methyl for the hydrogen atom on the NH
group in urea. As the purpose of this work is to
comprehensively understand the interactions and microstruc-
tures of the complex electrolyte, vibration spectroscopy as a
powerful tool is applied. Furthermore, quantum chemistry
calculations should be a simply but appropriate method to
characterize the interactions and the structure–activity
relationship of the LiTFSI-based composites. The Cerius2
program is matured commercial software, and its DMol3
module has been popularly used for quantum chemistry
calculations. This module will be applied upon the geometry
optimization of each organic molecule before and after
coordinated with Li+ and the calculations about the total
energy, frontier energy orbital, etc.
2. Experimental
2.1. Sample preparation
LiTFSI (3 M Inc., 99%) was dried at 140 8C for 12 h in
vacuum. Acetamide (Acros Inc., AP) and ethyleneurea (Tokyo
Kasei Kogyo Co. Inc., W > 97%) were recrystallized with
chloroform and urea (Beijing Chemical Reagents Inc., 99%),
NMU (Acros Inc., 97%) and DMU (Acros Inc., 98%) were
recrystallized with anhydrous ethanol. These materials were
then dried at 55 8C for 10 h in vacuum. All the complex systems
were prepared by simply mixing LiTFSI and the organic
compound with an acylamino group at various molar ratios in
an argon-filled MBraun LabMaster 130 glovebox
(H2O < 5 ppm). Homogeneous and stable liquids for partial
composites with the proper molar ratio were obtained directly
after the mixtures were mechanically stirred at room
temperature. Moreover, the complex systems with other recipes
were prepared by slightly heating the mixture and then cooled
down to room temperature (see Scheme 1). The water content in
the complex electrolyte was determined to be less than 39 ppm
by Karl–Fischer titration method on DL37KF Coulometer,
Mettler Toledo.
2.2. Thermal and electrochemical measurement
The melting points of the complex systems were determined
on a DSC 2010 differential scanning calorimeter (TA Inc.) by

R. Chen et al. / Vibrational Spectroscopy 44 (2007) 297–307 299
sealing ca. 10 mg of the composite in an aluminum pan. The
pan and the electrolyte were first cooled to about�100 8C with
liquid nitrogen and then heated to 100 8C at a rate of 5 8C/min.
Special attention was paid to avoid exposing the hygroscopic
samples to moisture by continuous nitrogen flowing around the
sample during measurement. Ionic conductivity measurements
were carried out with an electrochemical cell with Pt electrode.
The cell constant was determined with standard KCl solution
(0.01 M) at 25 8C. The ac impedance of the samples was
measured on a CHI660a electrochemical workstation (1–
100 kHz, 0–80 8C).
2.3. IR and Raman spectra
The IR spectra of the samples were recorded on a Nicolet
Magna 750 FTIR spectrometer between 4000 and 400 cm�1
with the resolution set at 4 cm�1. The solid sample was mixed
with dry KBr and pressed into pellet while a droplet of the
liquid sample was spread on a dry KBr pellet for the IR
spectroscopic measurements. The Raman spectra of the
electrolyte sealed in a test tube were recorded on a Nicolet
950 FT-Raman spectrometer between 3700 and 100 cm�1
with the resolution set at 4 cm�1. The Raman and IR spectra
shown here were the average results of 400 and 50 scans,
respectively.
2.4. Computational methods
The configurations of the organic molecules coordinated
with the Li+ ions were optimized with the BLYP function of
non-local DFT with DNP (double numerical with polariza-
tion) basis set using the DMol3 module of the Cerius2
program. The Mulliken charges before and after the
coordination between the organic molecule and the Li+
ion, the binding energies for the interactions between the
organic molecule and the Li+ ion, and the total energy and
Frontier molecular orbital energy of each organic molecule
are calculated with this program. The sizes of the DNP basis
sets are comparable to the Gaussian 6-31G** basis sets, giving
the p polarization functions on hydrogen apart form the d
functions on the heavy atoms. In particular, the numerical
basis set is much more accurate than a Gaussian basis set of
the same size [28].
Table 1
Thermal properties and ionic conductivities of some complex systems
Composition Glass temperature at various molar ratios, Tg (8C)
1:2.0 1:3.0 1:3.3 1:4.0 1:4.5
LiTFSI/acetamide – �57.1 �60.4 �60.9 �62.2
LiTFSI/ethyleneurea �29.7 – 5.4 9.5 �35.1
LiTFSI/urea22 – �31.9 �31.0 – –
LiTFSI/NMU �34.8 �38.0 – �40.0 –
LiTFSI/DMU �28.0 – – – –
a At room temperature.b Molar ratios are the data in parenthesis.c Supercooled liquid.
3. Results and discussion
3.1. Physical chemical properties analysis
It is known that the melting points of LiTFSI and organic
compounds as studied are above 273 8C and 81 8C, respectively
(see Table 1). It is interesting that two solids can form directly a
homogeneous liquid at room temperature when they are mixed
with the proper molar ratio. For the complex systems with
LiTFSI/acetamide molar ratios between 1:2.5 and 1:7.5, the salt
and the organic molecule become wet immediately after
contact with each other and liquid drops can be observed on the
wall of the container. DSC analyses indicate the eutectic
temperatures of these complex electrolytes are as low as
�60 8C. The composites of LiTFSI and ethyleneurea with a
five-membered ring with molar ratios between 1:2.5 and 1:4.0
need heating until they become a homogeneous, and remain
liquid after cooled down to room temperature. They are
supercooled and have to be stored in a sealed container. We
previously reported that mechanically mixing LiTFSI and urea
with molar ratios between 1:3.0 and 1:3.8 led to the formation
of homogeneous liquids at room temperature [15]. In
comparison, the eutectic temperature of the LiTFSI/NMU
complex system is lower and their range of liquid phase is wider
than that of the LiTFSI/urea system. The formation of the
LiTFSI/DMU complex system is very slow at room tempera-
ture. However, slight heating leads to the quick formation of a
homogeneous liquid. The lowest eutectic temperature (�28 8C)
is reached at LiTFSI:DMU = 1:2.0.
Experimental results for the glass transition temperature,
melting point and range of liquid phase are summarized in
Table 1. In comparison, the LiTFSI/ethyleneurea composite has
higher eutectic temperature than the other complex systems.
The phenomena observed during sample preparation and the
DSC measurement results indicate that a eutectic system can be
formed by mixing different organic compounds with LiTFSI.
The acylamino group takes an important role on the interaction
between the organic compound and LiTFSI and the formation
of a complex electrolyte. In the present case, it is reasonable to
assume that acylamino groups work as a complexing agent for
both the cations and the anions due to their polarity (C O and
NH2) capable of coordinating with cations and anions,
respectively. The weakening and even breaking down the
Melting point of organic
compounds, Tm (8C)
Range of liquid
phasea
Ionic conductivity
(�10�3 S cm�1, 25 8C)
81 1:2.0–1:6.0 1.21 (1:6.0)b
132 1:2.5–1:4.0c 0.26 (1:4.0)
132.7 1:3.0–1:3.8 0.17 (1:3.3)
102 1:2.0–1:4.0 0.18 (1:4.0)
105 1:2.0–1:3.0 0.01 (1:2.0)

Fig. 2. The glass transition temperature (A) and the ionic conductivities (B)
variations as a function of salt concentration in the various complex systems.
Inset: the correlation of the glass transition temperatures Tg with the log(s)
values for the binary complex samples at a molar ratio 1:4.0.
R. Chen et al. / Vibrational Spectroscopy 44 (2007) 297–307300
bonding between the Li+ cation and TFSI� anion results in the
formation of the complex electrolyte.
In order to further find out the influence of the molecular
structure on the electrochemical performance of the molten
salts, their conductivities are examined. Of all the complex
electrolytes, the LiTFSI/acetamide system shows the highest
ionic conductivity at the same temperatures and molar ratios.
The data can be one order higher than that of other recipes as
summarized in Table 1. Fig. 1 shows the Arrhenius plots of
different binary complex systems. The temperature-dependent
conductivity plots of these complex systems are protruding
upward, indicating that their conductivity–temperature rela-
tionships do not follow the Arrhenius equation. It is well known
that the Vogel–Tammann–Fulcher (VTF) equation is valid for
polymer and glassy electrolytes and concentrated electrolyte
solutions [29–31]. The inset of Fig. 1 indicates clearly that the
conductivity–temperature relationship of the composite is
linear. As the VTF equation is closely correlated to the free-
volume model, the excellent agreement of the conductivity
versus temperature behavior with the VTF equation implies a
solvent-assisted ionic conduction mechanism for these complex
systems.
Fig. 2 clearly show that the glass transition temperature and
the ionic conductivities variations as a function of lithium salt
concentration in the various complex systems. It is noticeable to
find that there is the relativity between the thermal and the
electrochemical properties of the complex systems. The ionic
conductivities of the complex sample with the lower glass
transition temperature are higher than that of those with the
higher glass transition temperature. Accordingly, the correla-
tion of the glass transition temperatures Tg with the log(s)
values for the binary complex samples at a molar ratio 1:4.0 is
list in inset of Fig. 2. This behavior is attributed to the
differences of the structure–activity relationship of these
complex systems based on LiTFSI and the various organic
compounds as will be shown in the subsequent spectroscopic
study and quantum chemistry calculations.
Fig. 1. The Arrhenius plots of different LiTFSI-based binary complex systems
at various molar ratios. Inset: the Vogel–Tammann–Fulcher (VTF) plots of
some typical binary complex systems based on LiTFSI. T0 in the VTF equation
is ca. 50 8C lower than the glass transition temperature.
3.2. Spectroscopic characterization and quantum
chemistry calculations
In recent years, LiTFSI have been popularly used as the
source of lithium in various electrolytes for lithium ion batteries
and other electrochemical devices. The assignments of the
vibration modes of LiTFSI are very abundant but often conflict
with each other in the literature due to its complicated structure
and vibration spectrum [32–39]. It seems that the assignments
of the IR and Raman bands by Rey et al. [40] are more
reasonable than others’ attribution and will be adopted in this
paper. The recognition to the vibration spectra of several
organic compounds in this work, such as ethyleneurea with
five-membered ring, is based on the reports of molecules with
similar structures [41–43]. The vibration spectra of acetamide
[44–46] and urea [47,48] have been extensively studied in
literature. The assignments of Ganceshsrinivas et al. [49] for
acetamide and of Keuleers et al. [50] for urea will be borrowed
hereafter.
The molecules of solid ethyleneurea are all associated with
hydrogen bonding due to the coordination between the O atom
on the C O group and the H atom on the NH group (N–H� � �O).
Obvious spectral changes are also observed throughout the
spectra when solid ethyleneurea is mixed with solid LiTFSI and
a homogeneous liquid is obtained. The spectrum varies for two
reasons when the physical state of a substance changes from
solid to liquid: damage to its crystal field and the breaking of the
hydrogen bonds. The former will lead to the breakdown of the
structural symmetry of a crystal and changes the distribution of
its vibration modes. That means some new modes may appear
and some old modes may disappear. Although the hydrogen
bonding may not change the symmetry of a molecule, its
breaking will definitely result in variation of interactions
between molecules and therefore, their spectral changes. Fig. 3
shows the IR spectra of the symmetric (3282 cm�1) and
asymmetric (3406 cm�1) stretching of NH of ethyleneurea.
Both bands blue-shift gradually with increasing LiTFSI content
and reach 3423 and 3583 cm�1, respectively at LiTFSI/
ethyleneurea = 1:3.0. This demonstrates the breaking of the
hydrogen bonds among the ethyleneurea molecules and the
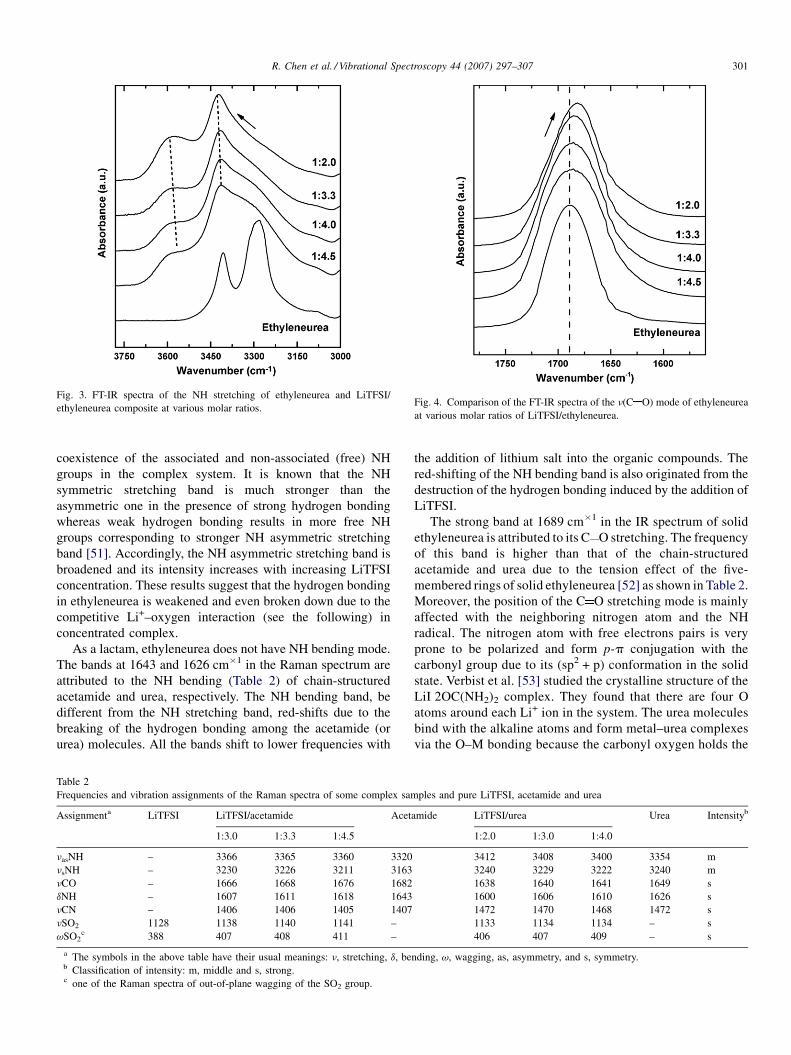
Fig. 3. FT-IR spectra of the NH stretching of ethyleneurea and LiTFSI/
ethyleneurea composite at various molar ratios.Fig. 4. Comparison of the FT-IR spectra of the n(C O) mode of ethyleneurea
at various molar ratios of LiTFSI/ethyleneurea.
R. Chen et al. / Vibrational Spectroscopy 44 (2007) 297–307 301
coexistence of the associated and non-associated (free) NH
groups in the complex system. It is known that the NH
symmetric stretching band is much stronger than the
asymmetric one in the presence of strong hydrogen bonding
whereas weak hydrogen bonding results in more free NH
groups corresponding to stronger NH asymmetric stretching
band [51]. Accordingly, the NH asymmetric stretching band is
broadened and its intensity increases with increasing LiTFSI
concentration. These results suggest that the hydrogen bonding
in ethyleneurea is weakened and even broken down due to the
competitive Li+–oxygen interaction (see the following) in
concentrated complex.
As a lactam, ethyleneurea does not have NH bending mode.
The bands at 1643 and 1626 cm�1 in the Raman spectrum are
attributed to the NH bending (Table 2) of chain-structured
acetamide and urea, respectively. The NH bending band, be
different from the NH stretching band, red-shifts due to the
breaking of the hydrogen bonding among the acetamide (or
urea) molecules. All the bands shift to lower frequencies with
Table 2
Frequencies and vibration assignments of the Raman spectra of some complex sam
Assignmenta LiTFSI LiTFSI/acetamide Acet
1:3.0 1:3.3 1:4.5
nasNH – 3366 3365 3360 3320
nsNH – 3230 3226 3211 3163
nCO – 1666 1668 1676 1682
dNH – 1607 1611 1618 1643
nCN – 1406 1406 1405 1407
nSO2 1128 1138 1140 1141 –
vSO2c 388 407 408 411 –
a The symbols in the above table have their usual meanings: n, stretching, d, beb Classification of intensity: m, middle and s, strong.c one of the Raman spectra of out-of-plane wagging of the SO2 group.
the addition of lithium salt into the organic compounds. The
red-shifting of the NH bending band is also originated from the
destruction of the hydrogen bonding induced by the addition of
LiTFSI.
The strong band at 1689 cm�1 in the IR spectrum of solid
ethyleneurea is attributed to its C O stretching. The frequency
of this band is higher than that of the chain-structured
acetamide and urea due to the tension effect of the five-
membered rings of solid ethyleneurea [52] as shown in Table 2.
Moreover, the position of the C O stretching mode is mainly
affected with the neighboring nitrogen atom and the NH
radical. The nitrogen atom with free electrons pairs is very
prone to be polarized and form p-p conjugation with the
carbonyl group due to its (sp2 + p) conformation in the solid
state. Verbist et al. [53] studied the crystalline structure of the
LiI�2OC(NH2)2 complex. They found that there are four O
atoms around each Li+ ion in the system. The urea molecules
bind with the alkaline atoms and form metal–urea complexes
via the O–M bonding because the carbonyl oxygen holds the
ples and pure LiTFSI, acetamide and urea
amide LiTFSI/urea Urea Intensityb
1:2.0 1:3.0 1:4.0
3412 3408 3400 3354 m
3240 3229 3222 3240 m
1638 1640 1641 1649 s
1600 1606 1610 1626 s
1472 1470 1468 1472 s
1133 1134 1134 – s
406 407 409 – s
nding, v, wagging, as, asymmetry, and s, symmetry.

Scheme 2. The resonance form of ethyleneurea.
Fig. 6. Raman spectra of the out-of-plane (SO2) wagging of pure LiTFSI and
LiTFSI/ethyleneurea composite at various molar ratios.
R. Chen et al. / Vibrational Spectroscopy 44 (2007) 297–307302
strongest electronegativity [54]. These studies clearly indicate
that the Li+ ions tend to coordinate with the carbonyl oxygen
rather than the nitrogen of the acylamino group of the organic
molecule. Fig. 4 compares the IR spectra of the C O stretching
mode of the LiTFSI/ethyleneurea complex with different
molar ratios. It shows that the position of this band shifts
toward low frequency with increasing LiTFSI content in the
composite.
Considering the resonance forms of ethyleneurea (Scheme
2), the position of the C–N stretching band at 1277 cm�1 in pure
ethyleneurea shifts downward slightly in frequency (Fig. 5)
owing to the influences of the Li+–oxygen coordination and the
breaking of the hydrogen bonds. With increasing LiTFSI
content in ethyleneurea, the interaction between the Li ions and
the O atom on the C O group becomes more obvious and the
C–N stretching band keeps blue-shifting. This will lead to the
increase of the amount of ethyleneurea with resonance forms
(A) or (C). As a result, the C–N bonding is more like a double
bond in the liquid composite than in solid ethyleneurea. The
spectral variation of the C–N band is in good agreement with
that of the C O band.
The Raman band at 387 cm�1 is assigned to the out-of-plane
SO2 wagging of solid LiTFSI in Fig. 6. It shifts to higher
frequencies with the decrease of LiTFSI concentration.
Similarly, the band at 1128 cm�1 for the SO2 stretching mode
Fig. 5. Evolution of the FT-IR spectra of the n(CN) mode of ethyleneurea at
various molar ratios of LiTFSI/ethyleneurea.
of solid LiTFSI red-shifts to 1134 cm�1 upon the introduction
of LiTFSI. It has been reported that these bands are sensitive to
the introduction of the salt and that the negative charges of
LiTFSI are delocalized between the N and the O atoms [40].
This means that the above spectral variations may be related to
the strong interaction between the SO2 group of TFSI� anion
and the NH group of ethyleneurea. However, the position
variation of these bands is less obvious with the salt
concentration, consistent with the previous observations [15].
This might be due to the narrower range of salt concentration
change in this work. Similar phenomena have also been
observed for the LiTFSI/acetamide and LiTFSI/urea complex
systems in Fig. 7, and relative data are listed in Table 2. It is
understandable that part of the O atoms on the SO2 group
possess negative charges when the Li ions coordinate strongly
with the C O group of ethyleneurea. Considering the
resonance forms of ethyleneurea, these O atoms tend to
interact with the partially positive-charged NH groups of
ethyleneurea. On the other hand, the Raman band of the CF3
symmetric stretching at 1245 cm�1 shows no obvious changes
with addition of ethyleneurea, consistent with the previous
report [55]. This further proves that ethyleneurea mainly
interacts with the TFSI� anion via the SO2 group in the LiTFSI/
ethyleneurea composite.
There are various ionic species in the complex system, such
as the ‘‘free’’ anions and contact ion pairs, due to the strong
interaction between LiTFSI and ethyleneurea in the complex
system and the differential concentration in the various molar
ratios. The configuration of ions (‘‘free’’ ion or ion pairs) and
the interaction between them have an important influence on
the electrochemical performance of the complex system,
especially their ionic conductivities. The possible structures of
different ionic species in the LiTFSI–ethyleneurea complex
system which were fully optimized at the BLYP/DNP level
using the Dmol3 program package are shown in Fig. 8,

Fig. 7. The observed band frequency variations of the Raman spectra as a function of lithium salt concentration for several typical binary complex systems based on
LiTFSI.
Fig. 8. Possible optimized structures of different ionic species in LiTFSI–ethyleneurea complex system calculated at the BLYP/DNP.
Fig. 9. Raman spectra of LiTFSI/ethyleneurea composite at various molar
ratios between 770 and 670 cm�1.
R. Chen et al. / Vibrational Spectroscopy 44 (2007) 297–307 303
corresponding to contact ion pairs (Fig. 8a and b) and the
‘‘free’’ anions (Fig. 8c and d). Moreover, the behavior of the
specific Raman spectra is more sensitive to the different kinds
of ionic species.
The spectral evolution of the 740 cm�1 band of LiTFSI has
been studied extensively [56–60]. Bakker et al. [61] reported
that its position is determine by the coordinate and type of
cations in the system. Edman [62] systematically studied this
band in the (PEO)n–LiTFSI system. They found that this band
blue-shifts and becomes broadened upon the increase of the
lithium salt concentration and attributed these results to the
formation of ion pairs in the system. The band at 708 cm�1 in
Fig. 9 is assigned to the out-of-plane wagging of the NH group
of ethyleneurea. It is partially superposed with the ca. 745 cm�1
band of LiTFSI when the composites are formed. OriginPro
software is used to fit the band and the results are shown in
Fig. 9. The three fitted Guassian components are located at 746,
740 and 708 cm�1, corresponding to the contact ion pairs, the
‘‘free’’ anions and the out-of-plane wagging of the NH group,
respectively.
The NH wagging band shifts to higher frequencies with the
increase of LiTFSI concentration but its relative intensity
becomes weak, clearly demonstrating that the interaction
between the NH group and TFSI- anion is enhanced in the

Table 3
Calculated Mulliken charges for various organic compounds with acylamino group and each with Li+ coordinationa
Atom Mulliken charge
O Cb Nc Li
Ethyleneurea (Li+) �0.458 (�0.573) 0.518 (0.670) �0.390 (�0.367) 0.694
CH3CO(Li+)NH2 �0.435 (�0.535) 0.357 (0.452) �0.352 (�0.276) 0.732
NH2CO(Li+)NH2 �0.479 (�0.578) 0.486 (0.595) �0.388 (�0.335) 0.713
NH2CO(Li+)NHCH3 �0.493 (�0.588) 0.535 (0.644) �0.416 (�0.380)d 0.697
CH3NHCO(Li+)NHCH3 �0.506 (�0.594) 0.588 (0.696) �0.422 (�0.389) 0.679
a Mulliken charges of each organic compounds with Li+ coordination are the data in parenthesis (excluding the H atom).b Carbon atom in acylamino group.c Nitrogen atom in acylamino group, the Mulliken charges on the two nitrogen atoms are the same for ethyleneurea, urea and 1,3-dimethylurea.d Nitrogen atom adjacent with methyl.
R. Chen et al. / Vibrational Spectroscopy 44 (2007) 297–307304
samples with high LiTFSI concentrations. The position of the
multicomponent band shifts to the high frequency side with
increasing content of LiTFSI in the system, from 741 cm�1 at
LiTFSI/ethyleneurea = 1:4.5 to 745 cm�1 at LiTFSI/ethyle-
neurea = 1:2. This slight position variation evidences that the
number of ion pairs increases with increasing salt content in the
complex system.
The above results indicate clearly that the Li+ ions
coordinate with the C O group of ethyleneurea while the
NH group of ethyleneurea interacts with the SO2 group in
TFSI� anion, consistent with the spectral variation of the
LiTFSI/acetamide and the LiTFSI/urea system (Table 2). The
breaking of the hydrogen bonding among the ethyleneurea
molecules and the dissociation of LiTFSI result in the
formation of a eutectic system. Such interactions lead to the
transition of resonance form of ethyleneurea and are reflected in
the IR and Raman spectra. In order to confirm the above
discussions, Quantum chemistry calculations are performed in
the following by optimizing the geometries of the organic
molecules and coordinating ions and by calculating the
energies.
The difference in structure and substituting group for various
organic molecules has an important influence on the
physicochemical properties of the related composite. Acet-
amide can be regarded as urea with one of its NH2 groups
substituted with a methyl group. This substitution changes the
charge density on the carbonyl oxygen and determines the
strength of coordination of Li+ ion with the carbonyl oxygen.
Quantum chemistry calculations will give the further insight
into these discussions.
Table 4
Selected bond lengths, bond angle and binding energy (BE) of various organic co
Atom Bond length (�10�1 nm)
r(C O) r(C–N)
Ethyleneurea (Li+) 1.229 (1.267) 1.405 (1.363)
CH3CO(Li+)NH2 1.234 (1.268) 1.380 (1.342)
NH2CO(Li+)NH2 1.235 (1.272) 1.387 (1.356)
NH2CO(Li+)NHCH3 1.237 (1.274) 1.385 (1.355)c
CH3NHCO(Li+)NHCH3 1.239 (1.275) 1.388 (1.360)
a The data in parenthesis represent the bond length of each organic compound cb BE = �{E(RCO(Li+)NH2) � E(RCONH2) � E(Li+)}.c Nitrogen atom adjacent with methyl.
The results of the charge distributions are shown in Table 3.
It is seen that the negative charge on the carbonyl oxygen is
larger than on the nitrogen, agreeing with the fact that the Li+
ion is prone to coordinate with the carbonyl oxygen. After
coordination with Li+ ion, the Mulliken charge of the carbonyl
oxygen becomes more negative while that of the nitrogen atom
is less negative. This explains the equilibrium of resonance
form of organic compounds, ethyleneurea, for example.
The calculated bond lengths, bond angle of various organic
compounds before and after coordination with the Li+ ion as
well as the binding energies for these coordinations are listed in
Table 4. The variation of the bond length in all the systems
exhibits the same trend, i.e. the coordination elongates the C O
bond but shortens the CN bond length. As a result, the CN bond
is more characteristic of a double bond in the complex,
consistent with the IR and Raman spectroscopic results. More
information focus on the structural parameters and geometries
optimized for ethyleneurea, acetamide and each with Li+
coordination from BLYP/DNP are shown in Fig. 10.
Based upon the molecular orbital theory, the ability to gain
and lose electrons is judged by the energy level of the highest
occupied molecular orbital (HOMO) and the lowest unoccu-
pied molecular orbital (LUMO). The total energy, the Frontier
molecular orbital energy, the energy gaps between the HOMO
and LUMO, and the dipole moment of various organic
molecules are listed in Table 5. The LUMO energy of
ethyleneurea is higher than that of other molecules. So it is
difficult for the ethyleneurea to accept electrons. On the
contrary, the LUMO energy of the acetamide is low. Therefore,
it can easily accept electrons and bears a high reaction activity.
mpounds with acylamino group and each with Li+ coordinationa
Bond angle of
Li–O–C (8)BEb (kJ/mol)
r(Li–O)
1.745 180.0 223.581
1.758 170.6 203.615
1.746 180.0 216.225
1.740 178.0 224.115
1.736 180.0 231.280
oordinated with Li+.

Fig. 10. Structural parameters and geometries optimized for ethyleneurea, acetamide and each with Li+ coordination from BLYP/DNP.
Table 5
Total energy, Frontier molecular orbital energy and dipole moment of various organic molecules
Organic molecule ET (Ha) Frontier molecular orbital energy (Ha) Dipole moment (Debye)
EHOMO ELUMO DEga
Ethyleneurea �302.7100 �0.2140 0.0086 0.2226 4.4165
CH3CONH2 �209.2479 �0.2106 �0.0171 0.1935 3.8280
NH2CONH2 �225.3175 �0.2160 �0.0085 0.2075 4.2799
NH2CONHCH3 �264.6124 �0.2115 �0.0052 0.2063 4.1850
CH3NHCONHCH3 �303.9070 �0.2007 �0.0035 0.1927 4.0804
a DEg = ELUMO � EHOMO.
R. Chen et al. / Vibrational Spectroscopy 44 (2007) 297–307 305
The HOMO energies of urea and its ramifications are in the
subsequence of 1,3-dimethylurea > methylurea > urea. The
ability of 1,3-dimethylurea to lose electrons is stronger than
other molecules due to the electron donation of the methyl. It
can be seen in Fig. 11 that the HOMO is mainly located around
the carbonyl oxygen. This indicates that the Li–O coordination
is easy to occur in the interaction between the lithium salt and
organic compounds.
Both ethyleneurea and acetamide have two N–H bonds.
However, as a cyclic organic molecule with acylamino group,
ethyleneurea has stronger intermolecular tension and larger
steric hindrance than acetamide does. As a result, the LiTFSI/
ethyleneurea complex owns the highest glass transition
temperature. Most of the complexes are in their supercooled
state partially due to the big Li–O bond length and low binding
energy between the Li ion and the ethyleneurea molecules.
Moreover, the low ionic conductivity of LiTFSI/ethyleneurea is
attributed to the high viscosity and low ionic migration rate of
this composite. Table 4 also shows that the LiTFSI/acetamide
Fig. 11. Frontier molecular orbital
system has the biggest Li–O bond length and lowest binding
energy in all the complex electrolytes. Compared with urea, the
acetamide molecule has only one NH2 radical, thus the lithium
salt is apt to be dissociate in it by weak hydrogen bonding and
forms a complex system with low melting point and plastic
viscosity. Consequently, the thermal and electrochemical
properties of the LiTFSI/acetamide complex are superior to
that of LiTFSI/urea. Comparing the molecular structures of
urea, methylurea and 1,3-dimethylurea, it seems that, as an
electron donator, the methyl group can enhance the electro-
negativity of the carbonyl oxygen. On the other hand, the
influence of the methyl on the steric hindrance of organic
molecule is negative to the coordination with the Li+ ion. The
LiTFSI/methylurea system prefers to form a liquid at room
temperature while the LiTFSI/urea system has a high ionic
conductivity due to the easy migration of the ions in them. A
balance has to be built up between the association and
dissociation of the salt in order to reach high ionic conductivity
and low eutectic temperature.
of ethyleneurea and acetamide.

R. Chen et al. / Vibrational Spectroscopy 44 (2007) 297–307306
Clearly the thermal and electrochemical properties of the
complex systems are influenced by the structure of the organic
molecules, especially their configurations and the properties of
the substituent. On one hand, forcing the coordination between
the lithium salt and organic molecules will promote the
dissociation of lithium salt and decrease the eutectic
temperature of the composite. On the other land, it is difficult
for the Li ions with very strong coordination to the carbonyl
oxygen to migrate in the complex in the viewpoint of
electrochemical applications. Consequently, the compromise
has to be made between the salt dissociation and the Li ion
migration in order to reach optimized general properties for the
complex electrolyte.
4. Conclusion
The complex systems based on the LiTFSI and the above-
mentioned organic compound with acylamino group appear as a
liquid at room temperature in the relevant range of the molar
ratios. The formation mechanism of the complex systems are due
to the strong interaction between the O atom of the C O group in
the organic compounds and the Li cations in addition to the
interaction of the TFSI� anions with the NH2 group in the organic
compounds via hydrogen bonding. Large coordinated cations are
formed and their positive charges are shielded within the organic
molecules. The charge of the TFSI� anion is partially
delocalized, leading to charge shielding in the whole molecule
due to the impact of the CF3 end-group. The coulombic forces
between the cations and anions are very weak. In this way, a
homogeneous, stable and highly ionic conductive room
temperature complex electrolyte can be obtained from two
solid components. Moreover, the calculations indicate that the
molecular structure and the substituting group affect the charge
density and coordination strength of the carbonyl oxygen in an
organic molecule and that the hydrogen bonding interaction
between the organic molecules determines the properties of the
composite, its thermal stability, for example. In addition, a
compromise has to be made between the salt dissociation and ion
migration in the complex system in order to synthesize complex
electrolytes with excellent properties.
Acknowledgements
This work was supported by the National 973 Program
(Contract No. 2002CB211800) and the National Key Program
for Basic Research of China (Contract No. 2001CCA05000).
The authors thank Prof. C.M. Hong (College of Chemistry and
Molecular Engineering, Peking University) for a critical
reading of this manuscript, and Dr. J. Weng (NeoTrident
Technology Ltd.) for quantum chemistry calculation assistance.
The authors are grateful to 3M Company for providing the
LiTFSI sample.
References
[1] J.S. Wikes, M. Zaworotko, J. Chem. Commun. 9 (1992) 965.
[2] T. Welton, Chem. Rev. 99 (1999) 2071.
[3] P. Walden, Bull. Acad. Imper. Sci. (St. Petersburg) (1914) 1800.
[4] J. Robinson, R.A. Osteryoung, J. Am. Chem. Soc. 101 (1979) 323.
[5] I. Okada, H. Horinouchi, J. Electroanal. Chem. 396 (1995) 547.
[6] K.R. Seddon, J. Chem. Biotechnol. 68 (1997) 351.
[7] A.B. McEwen, S.F. McDevitt, V.R. Koch, J. Electrochem. Soc. 144 (1997)
L84.
[8] M. Videa, C.A. Angell, J. Phys. Chem. B 103 (1999) 4185.
[9] D.R. MacFarlane, J.H. Huang, M. Forsyth, Nature 402 (1999) 792.
[10] Y. Ito, T. Nohira, Electrochim. Acta. 45 (2000) 2611.
[11] K. Xu, M.S. Ding, T.R. Jow, J. Electrochem. Soc. 148 (2001) A267.
[12] W. Lu, A.G. Fadeev, B. Qi, E. Smela, B.R. Mattes, J. Ding, G.M. Spinks, J.
Mazurkiewicz, D. Zhou, G.G. Wallace, D.R. MacFarlane, S.A. Forsyth,
M. Forsyth, Science 297 (2002) 983.
[13] R.D. Rogers, K.R. Seddon, Science 302 (2003) 792.
[14] T. Fukushima, A. Kosaka, Y. Ishimura, T. Yamamoto, T. Takigawa, N.
Ishii, T. Aida, Science 300 (2003) 2072.
[15] H.Y. Liang, H. Li, Z.X. Wang, F. Wu, L.Q. Chen, X.J. Huang, J. Phys.
Chem. B 105 (2001) 9966.
[16] Y.S. Hu, H. Li, X.J. Huang, L.Q. Chen, Electrochem. Commun. 6 (2004)
28.
[17] R.J. Chen, F. Wu, H.Y. Liang, C.Z. Zhang, Chem. J. Chinese U. 25 (2004)
2108.
[18] R.J. Chen, F. Wu, L. Li, Chem. J. Chinese U. 26 (2005) 1517.
[19] R.J. Chen, F. Wu, H.Y. Liang, L. Li, B. Xu, J. Electrochem. Soc. 152
(2005) A1979.
[20] B. Xu, F. Wu, R.J. Chen, G.P. Cao, S. Chen, G.Q. Wang, Y.S. Yang, J.
Power Sources 158 (2006) 773.
[21] J.L. Anthony, E.J. Maginn, J.F. Brennecke, J. Phys. Chem. B 105 (2001)
10942.
[22] J.L. Anthony, E.J. Maginn, J.F. Brennecke, J. Phys. Chem. B 106 (2002)
7315.
[23] T.I. Morrow, E.J. Maginn, J. Phys. Chem. B 106 (2002) 12807.
[24] Y.S. Hu, Z.X. Wang, H. Li, X.J. Huang, L.Q. Chen, Vib. Spectrosc. 37
(2005) 1.
[25] Y.S. Hu, Z.X. Wang, H. Li, X.J. Huang, L.Q. Chen, Spectochim. Acta A61
(2005) 403.
[26] M.O.S.P. Caldeira, C.A.C. Sequeira, Molten Salt Forum 1/2 (1993) 407.
[27] D.H. Kerridge, Chem. Soc. Rev. 17 (1988) 181.
[28] DMol User Guide, Biosym/MSI, San Diego, 1995, pp. E-2–E-4.
[29] J. Barthel, H.J. Gores, G. Schmeer, Ber. Bunsenges. Phys. Chem. 83
(1979) 911.
[30] H.C. Shiao, D. Chua, H.P. Lin, S. Slane, M. Salomon, J. Power Sources 87
(2000) 167.
[31] G. Adam, H. Gibbs, J. Chem. Phys. 43 (1965) 139.
[32] R. Arnaud, D. Benrabah, J.Y. Sanchez, J. Phys. Chem. 100 (1996) 10882.
[33] A. Ferry, M. Doeff, L. Jomghe, J. Electrochem. Soc. 408 (1996) 113.
[34] K. Xu, D. Day, C.A. Angell, J. Electrochem. Soc. 143 (1996) L209.
[35] P. Johansson, S.P. Gejji, J. Tegenfeldt, J. Lindgren, Electrochim. Acta 43
(1998) 1375.
[36] J.Z. Li, X.J. Huang, L.Q. Chen, J. Electrochem. Soc. 147 (2000) 2653.
[37] J.J. Golding, D.R. Macfarlane, L. Spiccia, M. Forsytn, B.W. Skelton, A.H.
White, Chem. Commun. 15 (1998) 1593.
[38] D.R. Macfarlane, P. Meakin, J. Sun, N. Amini, M. Forsyth, J. Phys. Chem.
B 103 (1999) 4164.
[39] J. Sun, M. Forsyth, D.R. Macfarlane, Ionics 3 (1997) 356.
[40] I. Rey, P. Johansson, J. Lindgren, J.C. Lassegues, J. Grondim, L. Servant, J.
Phys. Chem. A 102 (1998) 3249.
[41] R.D. Ross, D.G. Crosby, J. Agr. Food Chem. 21 (1973) 335.
[42] R.F. de Farias, Thermochim. Acta 376 (2001) 63.
[43] R.F. de Farias, L. Martınez, C. Airoldi, Thermochim. Acta 376 (2001) 91.
[44] E.A. Cutmore, H.E. Hallam, Spectrochim. Acta 25A (1969) 1767.
[45] R.A. Kydd, A.R.C. Dunham, J. Mol. Struct. 69 (1980) 79.
[46] R. Knudsen, O. Sala, Y. Hase, J. Mol. Struct. 321 (1994) 187.
[47] B.K. Banerjee, P.C. Srivastava, Fertilizer Technol. 19 (1979) 1575.
[48] R. Keuleers, G.S. Papaefstathiou, C.P. Raptopoulou, S.P. Perlepes, H.O.
Desseyn, J. Mol. Struct. 525 (2000) 173.
[49] E. Ganeshsrinivas, D.N. Sathyanarayana, K. Machida, Y.J. Miwa, J. Mol.
Struct. 361 (1996) 217.

R. Chen et al. / Vibrational Spectroscopy 44 (2007) 297–307 307
[50] R. Keuleers, H.O. Desseyn, B. Rousseau, C.V. Alesenoy, J. Phys. Chem. A
103 (1999) 4621.
[51] R.A. Nyquist, Interpreting infrared, Raman, and nuclear magnetic reso-
nance spectra, Variables in Data Interpretation of Infrared and Raman
Spectra, vol. 1, Academic Press, San Diego, 2001.
[52] R.M. Silverstein, F.X. Webster, Spectrometric Identification of Organic
Compounds, John Wiley & Sons, New York, 1998.
[53] J. Verbist, R. Meulemans, P. Piret, M.V. Meerssche, Bull. Soc. Chim.
Belges. 79 (1970) 391.
[54] T. Theophanides, P. Harvey, Coord. Chem. Rev. 76 (1987) 237.
[55] L. Edman, M.M. Doeff, A. Ferry, J. Kerr, L.C. de Jonghe, J. Phys. Chem. B
104 (2000) 3476.
[56] S. Wen, T.J. Richardson, D.J. Ghantous, K.A. Strickel, P.N. Ross, E.J.
Cairns, J. Electroanal. Chem. 408 (1996) 113.
[57] I. Rey, J.C. Lassegues, J. Grondin, L. Servant, Electrochim. Acta 43 (1998)
1505.
[58] Z.X. Wang, W.D. Gao, L.Q. Chen, Y.J. Mo, X.J. Huang, Solid State Ionics
154/155 (2002) 51.
[59] S.I. Jo, S.I. Lee, Y.B. Jeong, D.W. Kim, H.J. Sohn, Electrochim. Acta 50
(2004) 327.
[60] M. Castriota, T. Caruso, R.G. Agostino, E. Cazzanelli, W.A. Henderson, S.
Passerini, J. Phys. Chem. A 109 (2005) 92.
[61] A. Bakker, S. Gejji, J. Lindgre, K. Hermansson, Polymer 36 (1995) 4371.
[62] L. Edman, J. Phys. Chem. B 104 (2000) 7254.