the role of angiotensin-(1-7) in cardiovascular...
TRANSCRIPT

THE ROLE OF ANGIOTENSIN-(1-7) IN CARDIOVASCULAR PHYSIOLOGY
By
JUSTIN L. GROBE
A DISSERTATION PRESENTED TO THE GRADUATE SCHOOL OF THE UNIVERSITY OF FLORIDA IN PARTIAL FULFILLMENT
OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY
UNIVERSITY OF FLORIDA
2006

Copyright 2006
by
Justin L. Grobe

This document is dedicated to my wife, my family, my mentors, and my friends.

ACKNOWLEDGMENTS
I would first like to thank my graduate mentor, Michael J. Katovich, for his
guidance over the last five years. His continual support, guidance, encouragement, and
direction have been invaluable. I truly appreciate all the friendship he has shown me, and
all the attention and effort he has poured into my education.
Along with my graduate mentor, I would like to thank my undergraduate mentor,
Christopher C. Barney. His guidance and friendship are the primary reasons for my
success as an undergraduate, and my interest in animal physiology.
Next, I would like to thank my graduate committee members, Mohan K. Raizada,
Maureen Keller-Wood, William J. Millard, and Jeffrey A. Hughes, for their input into my
education and this document. Their direction over the last five years has shaped me into
a very capable modern physiologist. I would also like to thank Joanna Peris for her
continuous encouragement, and for filling in during my final defense.
I have been blessed with many other wonderful teachers and mentors at all levels of
my education, but a few deserve particular attention. I would like to thank Marjorie and
James Bullerdick, for teaching me patience and compassion. I would like to thank Trena
Thornburg for nurturing my abilities and teaching me to appreciate thinking “outside the
box.” Jeff Ehlers and David Dodendorf were wonderful coaches, and I thank them for
encouraging me and promoting my interest in the sciences. The faculty and staff of the
Hope College Biology Department welcomed me from the day I entered college, and I
am grateful for their continuing support and encouragement. Matthew J. Huentelman,
iv

both my peer and teacher, also deserves my gratitude for his excitement and
uncompromising drive.
Many people have blessed me with their friendship, and have made my years in
college and graduate school truly enjoyable. Many should be named, but Bill Murdoch
and Lane Blanchard deserve particular mention for sticking with me in both my joys and
sorrows throughout my undergraduate and graduate years.
I would like to thank my parents, Edward and Loretta Grobe, for their continual
support, energy, and encouragement. From childhood they have recognized my interests,
and provided me every conceivable opportunity to follow my dreams.
Finally, my thanks go to my wife, Connie, who continually provides me with
support, encouragement, inspiration, and direction as she also pursues her Ph.D. I look
forward to sharing my life and work with my best friend.
v

TABLE OF CONTENTS page
ACKNOWLEDGMENTS ................................................................................................. iv
LIST OF TABLES............................................................................................................. ix
LIST OF FIGURES .............................................................................................................x
ABSTRACT...................................................................................................................... xii
CHAPTER
1 GENERAL INTRODUCTION ....................................................................................1
Heart Failure .................................................................................................................1 Cardiac Remodeling .....................................................................................................2 The Renin-Angiotensin System....................................................................................4
Angiotensin Converting Enzyme 2 .....................................................................10 Angiotensin-(1-7) ................................................................................................13
Estrogenic Modulation of Heart Failure .....................................................................13 Estrogenic Modulation of the Renin-Angiotensin System .........................................14 Aims............................................................................................................................15
2 CARDIAC FIBROSIS IS PREVENTED BY CHRONIC ANGIOTENSIN-(1-7) DURING ANGIOTENSIN II INFUSION .................................................................16
Abstract.......................................................................................................................16 Introduction.................................................................................................................17 Methods ......................................................................................................................19
Animals................................................................................................................19 Indirect Blood Pressure .......................................................................................19 Cardiac Remodeling ............................................................................................20 Statistics...............................................................................................................20
Results.........................................................................................................................20 Blood Pressure.....................................................................................................20 Cardiac Hypertrophy ...........................................................................................20 Cardiac Fibrosis...................................................................................................22
Discussion...................................................................................................................22
vi

3 CHRONIC ANGIOTENSIN-(1-7) PREVENTS CARDIAC FIBROSIS IN THE DOCA-SALT MODEL OF HYPERTENSION .........................................................27
Abstract.......................................................................................................................27 Introduction.................................................................................................................28 Methods ......................................................................................................................30
Animals................................................................................................................30 Chemicals ............................................................................................................30 DOCA-salt Model ...............................................................................................30 Indirect Blood Pressure .......................................................................................31 Direct Blood Pressure/Acute Angiotensin II Responses .....................................31 Cardiac Remodeling ............................................................................................32 Statistics...............................................................................................................33
Results.........................................................................................................................33 Indirect Blood Pressure .......................................................................................33 Direct Blood Pressure/Acute Angiotensin II Responses .....................................34 Cardiac Remodeling ............................................................................................34
Discussion...................................................................................................................36
4 PROTECTIVE EFFECTS OF ANGIOTENSIN-(1-7) DURING ISOPROTERENOL-INDUCED CARDIAC REMODELING ..................................45
Abstract.......................................................................................................................45 Introduction.................................................................................................................46 Methods ......................................................................................................................47
Animals................................................................................................................47 Group A – Males with isoproterenol at 1 mg/kg..........................................47 Group B – Ovariectomized and intact females with isoproterenol at 1 and
2 mg/kg .....................................................................................................47 Group C – Males with isoproterenol at 2 mg/kg..........................................48
Indirect Blood Pressure .......................................................................................48 Cardiac Remodeling ............................................................................................49
Results.........................................................................................................................49 Cardiac Hypertrophy ...........................................................................................49 Blood Pressure.....................................................................................................52
Discussion...................................................................................................................52
5 ALTERATIONS IN AORTIC VASCULAR REACTIVITY TO ANGIOTENSIN-(1-7) IN 17-β-ESTRADIOL-TREATED FEMALE SD RATS .................................56
Abstract.......................................................................................................................56 Introduction.................................................................................................................57 Methods ......................................................................................................................59
Animal Housing and Surgery ..............................................................................59 Reagents ..............................................................................................................60 Aortic Vascular Reactivity Assay .......................................................................60 Plasma Steroid Measurement ..............................................................................61
vii

Statistics...............................................................................................................61 Results.........................................................................................................................62
Estradiol Levels and Effects on Body, Heart and Uterine Growth .....................62 Aortic Ring Properties.........................................................................................62 Aortic Relaxation to Angiotensin-(1-7)...............................................................63 Aortic Ring Integrity ...........................................................................................65
Discussion...................................................................................................................65
6 PRELIMINARY CELL CULTURE EXPERIMENTS..............................................70
Abstract.......................................................................................................................70 Introduction.................................................................................................................70 Methods ......................................................................................................................72
Isolation and Culturing of Neonatal Rat Cardiac Myocytes ...............................72 Isolation and Culturing of Neonatal Rat Cardiac Fibroblasts .............................72 Myocyte-Fibroblast Cross-Talk Paradigm ..........................................................73 Real-time RT-PCR ..............................................................................................73 Lentiviral ACE2 Gene Transfer ..........................................................................73 Hypoxia-Reperfusion Paradigm ..........................................................................74 ACE2 Activity .....................................................................................................74 Soluble Collagen .................................................................................................75 Transforming Growth Factor-β1 .........................................................................75
Results.........................................................................................................................75 Cardiac Myocyte-Fibroblast Cross-Talk is Inhibited by Ang-(1-7) at the
Myocyte ...........................................................................................................75 Lentiviral Delivery of EF1α-ACE2 Results in Increased ACE2 Activity and
Protection from Hypoxia..................................................................................77 ACE2 Gene Delivery Attenuates Hypoxia-Reperfusion Induction of
Transforming Growth Factor-β1......................................................................78 Discussion...................................................................................................................78
7 GENERAL DISCUSSION .........................................................................................82
Summary and Conclusions .........................................................................................82 Pitfalls and Shortcomings ...........................................................................................84 Future Directions ........................................................................................................91 Perspectives ................................................................................................................94
APPENDIX
A LOADING OSMOTIC MINIPUMPS........................................................................95
B MASSON’S TRICHROME STAINING AND ANALYSIS .....................................96
LIST OF REFERENCES...................................................................................................98
BIOGRAPHICAL SKETCH ...........................................................................................119
viii

LIST OF TABLES
Table page 5-1. Effects of steroid treatments on body parameters in female Sprague-Dawley rats..62
5-2. Aortic ring properties ...............................................................................................62
ix

LIST OF FIGURES
Figure page 1-1. The renin-angiotensin system.....................................................................................5
1-2. Ventricular hypertrophy with chronic Ang II is attenuated by lentiviral delivery of ACE2....................................................................................................................11
1-3. Ang II-induced cardiac interstitial fibrosis is prevented by lentiviral delivery of ACE2. .......................................................................................................................12
1-4. Lentiviral delivery of ACE2 has no effect on the blood pressure increase from chronic Ang II infusion ............................................................................................12
2-1. Chronic Ang-(1-7) has no effect on blood pressure increases with chronic Ang II infusion.....................................................................................................................21
2-2. Chronic Ang-(1-7) prevents myocyte hypertrophy due to chronic Ang II infusion 21
2-3. Chronic Ang-(1-7) prevents interstitial fibrosis due to chronic Ang II infusion .....22
3-1. Chronic Ang-(1-7) has no effect on blood pressure increases with DOCA-salt treatment...................................................................................................................33
3-2. Chronic Ang-(1-7) has no effect on acute pressor responses to Ang II in animals treated with DOCA-salt............................................................................................34
3-3. Chronic Ang-(1-7) has no effect on cardiac hypertrophy due to DOCA-salt treatement .................................................................................................................35
3-4. Chronic Ang-(1-7) prevents interstitial fibrosis with DOCA-salt treatment............36
3-5. Chronic Ang-(1-7) prevents perivascular fibrosis with DOCA-salt treatment ........37
4-1. Isoproterenol (1 mg/kg/day) induces cardiac hypertrophy in male rats...................50
4-2. Ovariectomy potentiates the isoproterenol-mediated induction of cardiac hypertrophy in female rats........................................................................................50
4-3. Chronic Ang-(1-7) prevents the hypertrophic, but not fibrotic effects of isoproterenol (2 mg/kg/day) in male rats .................................................................51
x

4-4. Isoproterenol and Ang-(1-7) had no chronic ffects on blood pressure in male rats.52
5-1. Maximal contraction to angiotensin II in aortic rings from ovariectomized rats with estrogen replacement........................................................................................63
5-2. Estradiol attenuates the aortic relaxation response to acute Ang-(1-7)....................64
6-1. Ang-(1-7) attenuates myocyte-derived pro-fibrotic signaling..................................76
6-2. Ang-(1-7) attenuates expression of TGFβ1 mRNA in cardiac myocytes. ................76
6-3. Lentiviral delivery of ACE2 attenuates the collagen production response to hypoxia in cultured cardiac fibroblasts ....................................................................77
6-4. Lentiviral delivery of ACE2 attenuates hypoxia-induced expression of transforming growth factor-β1 protein. ....................................................................78
xi

Abstract of Dissertation Presented to the Graduate School of the University of Florida in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy
THE ROLE OF ANGIOTENSIN-(1-7) IN CARDIOVASCULAR PHYSIOLOGY
By
Justin L. Grobe
May, 2006
Chair: Michael J. Katovich Major Department: Pharmacodynamics
Heart failure (HF) is a devastating condition that impacts our society emotionally,
physiologically, and fiscally, and it is a growing problem in the United States. Aberrant
activity of the renin-angiotensin hormone system (RAS) has been implicated as a
causative factor in HF. Recently, a new angiotensin converting enzyme 2 (ACE2) was
recognized, which catalyzes the degradation of the active angiotensin II (Ang II)
hormone to angiotensin-(1-7) (Ang-(1-7)). Studies into the actions of ACE2 have led to
the concept that Ang-(1-7) may act as an active, endogenous inhibitor of the RAS.
Interestingly, investigations into the mechanisms of action of ACE inhibitors and AT1R
antagonists have revealed that both of these treatments result in increased circulating
levels of Ang-(1-7). Some have therefore hypothesized that pharmacological inhibitors
of the RAS may provide cardioprotective effects through the actions of Ang-(1-7).
The present studies were designed to characterize the cardioprotective actions of
Ang-(1-7) in various models of cardiac remodeling. The protective effect of Ang-(1-7)
was evaluated in Ang II-infusion, mineralocorticoid, and β-adrenergic models of cardiac
xii

remodeling. Further, due to gender differences in the development and progression of
heart failure, the protective actions of Ang-(1-7) were evaluated in intact and
ovariectomized female rats. Cardiac remodeling was inhibited by Ang-(1-7), with
specific effects determined by the type of stimulus, and estrogens were determined to
play an important role in regulating tissue sensitivity to Ang-(1-7).
From these findings, it was concluded that Ang-(1-7) has specific actions in the
cardiovascular system, some of which oppose the actions of its precursor, Ang II. In
addition, our results lead us to hypothesize that estrogen replacement therapy during
menopause may predispose women to cardiovascular diseases through decreased tissue
sensitivity to the protective Ang-(1-7) hormone. Future studies should more directly
characterize the effects of estrogens on cardiac tissue, and further elucidate the tissue-
level and intracellular signaling mechanisms of Ang-(1-7). Our results indicate that Ang-
(1-7) may represent a new, unexploited class of targets for pharmacological intervention
before and during HF.
xiii

CHAPTER 1 GENERAL INTRODUCTION
Heart Failure
Congestive heart failure (CHF) is one of the most debilitating cardiovascular
pathologies, having both tremendous economic and emotional impacts on our society.
Approximately five million Americans, or 2.3% of the entire population of the United
States, suffers from CHF (Thom et al., 2006). The lifetime risk of developing CHF in the
fourth decade of life is 20%, and this risk doubles for individuals with blood pressure
>160/90 (Thom et al., 2006).
Congestive heart failure is a disease characterized by inadequate movement of
blood by the heart. Inadequate blood flow results in poor perfusion of critical organs,
including the brain and the heart itself, thereby leading to cellular hypoxia and death. It
was estimated that in 2005, 80% of men and 70% of women under age 65 diagnosed with
CHF will die within eight years of diagnosis. After diagnosis, survival is poorer in men
than women, but fewer than 15% of women will survive longer than eight to twelve
years. Twenty percent of people diagnosed with CHF will die within one year of
diagnosis (Thom et al., 2006).
There are two major forms of CHF, which are defined by the morphological and
functional changes that lead to inadequate blood movement. Systolic heart failure is
characterized by insufficient contraction of the ventricles (which are typically dilated),
usually resulting from idiopathic dilated or ischemic cardiomyopathies. Diastolic heart
failure is characterized by thickened, noncompliant ventricular walls and small
1

2
ventricular volumes, usually resulting from hypertension, stenotic valvular disease, or
primary hypertrophic cardiomyopathies. Clinically, patients typically present with a
combination of both systolic and diastolic heart failure, in that the heart is too weak to
maintain cardiac output (systolic dysfunction) despite thick, stiff walls (diastolic
morphology). Thus, conventional therapy is targeted at relieving symptoms of
inadequate blood flow rather than reversing or preventing the myocardial damage which
leads to CHF.
Cardiac Remodeling
Whereas systolic heart failure is typically the result of myocardial ischemia and
cellular death (which leads to a loss of muscle and force-generating ability), diastolic
heart failure is the result of inappropriate myocyte growth accompanied by a large
increase in extracellular matrix deposition. Together, myocyte hypertrophy and net
increases in extracellular matrix components are referred to as cardiac remodeling.
Cardiac remodeling can be normal and compensatory, as is observed in the
myocardium of athletes engaged in aerobic sports. In this “normal” remodeling, the
process results from increased peripheral demand for cardiac output, which provides a
mechanical stimulus for myocyte hypertrophy. Cardiac fibroblasts, which produce the
proteins of the extracellular matrix, retain relatively normal growth patterns and function.
This type of remodeling differs from pathological remodeling primarily due to the
difference in the response of the cardiac fibroblast to the stimulus (Weber, 2000; Miner
and Miller, 2006). Pathological remodeling, in contrast, can take many forms, which are
differentiated primarily by stimulus and mechanism.
Mineralocorticoids can elicit direct effects on cardiac remodeling. Aldosterone
levels are elevated during heart failure, and this elevation is related to increased mortality

3
(Swedberg et al., 1990), and inhibition of mineralocorticoid receptors by spironolactone
or eplerenone significantly reduces mortality in heart failure patients (Pitt et al., 1999,
Pitt et al., 2003). Spironolactone treatment also results in decreased extracellular matrix
remodeling (Zannad et al., 2000). In vitro, cardiac fibroblasts are stimulated by
aldosterone to increase collagen production, and to change the relative pattern of collagen
types expressed (Zhou et al., 1996; Brilla, 2000; Rombouts et al., 2001).
Angiotensin II also directly promotes cardiac remodeling. In vitro, angiotensin II
promotes proliferation of cardiac fibroblasts, collagen production, and expression of
adhesion receptors (Kim and Iwao, 2000; Schnee and Hsueh, 2000; Lijnen et al., 2001).
Inhibition of angiotensin converting enzyme improves the outcomes for heart failure
patients (Khalil et al., 2001), as does antagonism of the AT1 subtype angiotensin II
receptor (Frigerio and Roubina, 2005), thereby implicating angiotensin II in cardiac
remodeling.
Catecholamines can act upon the myocardium to cause remodeling. Epinephrine
and norepinephrine are elevated during heart failure and correlate to prognosis (Francis et
al., 1993). Inhibition of β-adrenergic receptors decreases the symptoms of heart failure,
and improves survival (Shibata et al., 2001). The actions of norepinephrine on the
extracellular matrix are not well described at the present time, though it is known that
norepinephrine induces the production of growth factors, such as transforming growth
factor β1, which are potent mediators of fibrosis (Bhambi and Eghbali, 1991; Barth et al.,
2000; Akiyama-Uchida et al., 2002; Miner and Miller, 2006).
Cytokines such as transforming growth factor-β1 are produced by cardiac cells
during heart failure, and are well-established mediators of cardiac remodeling (Sun et al.,

4
1998; Lijnen et al., 2000; Iwata et al., 2005; Tallant et al., 2005). In vitro, transforming
growth factor-β1 acts in an autocrine/paracrine fashion to modulate collagen and
fibronectin production in the cardiac fibroblast (Roberts et al., 1986). Because it is
upregulated by angiotensin II and norepinephrine, this cytokine may represent a common
mediator of the fibrotic effects of these hormones (Bhambi et al., 1991; Yoo et al., 1998).
The Renin-Angiotensin System
In 1836, Richard Bright observed a link between left ventricular hypertrophy and
renal disease, and he suggested a link among hypertrophy, increased resistance to blood
flow in small vessels, and an altered condition of the blood (Basso and Terragno, 2001).
George Johnson in 1868 and F.A. Mahomed in 1872 also reported links between renal
diseases and left ventricular hypertrophy. Finally, in 1898, Tigerstedt and Bergman
determined that extracts from rabbit renal tissue contained a pressor agent which
mediated the relationship between renal disease and cardiac hypertrophy. Tigerstedt and
Bergman named this vasoactive substance “renin.” Subsequent attempts to isolate and
fully characterize this and related blood-borne substances resulted in simultaneous
discovery of the compound “hypertensin” by Braun-Menendez et al. (1939), and
“angiotonin” by Page et al. (1939). These two would later agree to compromise on
standard nomenclature for the various components of the developing system, including
the name “angiotensin” as a combination of the two earlier names (Braun-Menendez and
Page, 1958).
Many more components of the renin-angiotensin system (RAS) have been
discovered, as have the relationships among the various components. The system is
present both systemically (in the blood plasma) and in the interstitium (as an autocrine or
paracrine system), and it is the presence or absence of the various components at the site

5
of action which determines the specific outcome of RAS activation in that location.
Figure 1-1 summarizes the presently recognized components of the RAS.
Angiotensinogen
Figure 1-1. The renin-angiotensin system. (ACE – angiotensin converting enzyme; ACE2 – angiotensin converting enzyme 2; Endopeptidases – neprilysin, prolyl endopeptidase, and thimet oligopeptidase; AP-A – aminopeptidase A; AP-B – aminopeptidase B; AT1 – angiotensin II type 1 receptor; AT2 – angiotensin II type 2 receptor; AT1-7? – possible receptor for angiotensin-(1-7), which may be the Mas receptor; AT4 – angiotensin type 4 receptor).
Release of renin from the kidney is the rate limiting step in the generation of
circulating angiotensin II (Ang II). Renin, an aspartyl protease, is synthesized and
released by juxtaglomerular cells located in the walls of the afferent arterioles of the
nephron just as the vessel enters the glomerulus. Renin is synthesized as the 406 amino
acid preprorenin, then cleaved to form prorenin which is a 373 amino acid mature, but
Angiotensin I
Angiotensin II
Angiotensin-(1-9)
Angiotensin-(1-7)
Angiotensin III
Angiotensin IV
Angiotensin-(1-5)
ACE2
Pro-Renin Renin ACE2
ACE Endo-
ACE peptidases
ACE
AT1 AT2
AT1-7?
AP-A
AP-B
AT4
Vasoconstriction Anti-AT1 effects Hypertrophy Vasodilation Vasoconstriction Anti-AT1 effects Fibrosis Differentiation Memory effects? Vasodilation

6
inactive, enzyme. Final activation of renin by an as yet uncharacterized enzyme involves
removal of the 43 amino terminal residues (Goodman and Gilman, 2001).
The only known substrate for renin is angiotensinogen. This highly abundant
globular glycoprotein is synthesized as a 452-amino acid preangiotensinogen by liver, fat,
certain nervous tissues, and the kidney. Angiotensinogen’s synthesis is stimulated by
inflammation, insulin, estrogens, glucocorticoids, thyroid hormone, and angiotensin II.
Due to the circulating levels of angiotensinogen and the Km of renin for its substrate
(approximately 1 µM), the rate of synthesis of angiotensinogen can modulate the levels of
circulating angiotensin II. Indeed, increased circulating levels of angiotensinogen are
associated with essential hypertension (Jeunemaitre et al. 1992; Goodman and Gilman,
2001). The mature peptide is only fourteen amino acids in length.
Angiotensin I represents the first ten amino acids of the amino terminus of
angiotensinogen, and is the result of renin action upon angiotensinogen. This peptide is
inactive until converted to angiotensin II by angiotensin converting enzyme.
Angiotensin converting enzyme (ACE) is also known as Kininase II and Dipeptidyl
Carboxypeptidase. This ectoenzyme is comprised of 1277 amino acids, and displays two
homologous zinc-dependent enzymatic domains. The large extracellular domain of this
enzyme can be cleaved by secretase (Beldent et al., 1995; Goodman and Gilman 2001) to
produce circulating ACE enzyme, but it has been suggested that intact, membrane-bound
ACE may act as a serine-threonine kinase extracelluar receptor (Fleming et al., 2005).
ACE is a nonspecific peptidase that removes two amino acids from substrates with
different sequences (although it prefers substrates with one free carboxyl group in the
carboxyl-terminal residue, and proline can not be the penultimate amino acid – therefore

7
it can not act on angiotensin II). Substrates for ACE include bradykinin (ACE inactivates
bradykinin and other potent vasodilators), angiotensin I, angiotensin-(1-9), and
angiotensin-(1-7).
An important genetic variation of ACE exists, in which an insertion/deletion
polymorphism in intron 16 of the gene significantly contributes to circulating ACE levels
(Rigat et al., 1990). The deletion version of the polymorphism results in higher levels of
circulating ACE and may be a risk factor for the development of heart disease (Cambien
et al., 1992; Gardeman et al., 1995; Mattu et al., 1995), vascular endothelial dysfunction
(Butler et al., 1999), left ventricular hypertrophy (Iwai et al., 1994; Schunkert et al.,
1994), and hypertension (Fornage et al., 1998; O’Donnell et al., 1998). It is interesting to
note that humans who live past 100 years of age display a high frequency of the deletion
polymorphism (Schachter et al., 1994), which may be a result of the strong association of
the deletion allele with protection from Alzheimer’s disease (Kehoe et al., 1999).
ACE-mediated cleavage of the two carboxyl amino acids of angiotensin I results in
the production of the eight-amino acid angiotensin II (Ang II). Ang II is the most active
component of the RAS, and its actions are receptor-dependent. The subtype of receptor
that is activated by Ang II determines the cellular response to this ligand.
Other enzymatic pathways exist, which can produce active angiotensin peptides
without involvement of renin and/or ACE. Nonrenin proteases can convert
angiotensinogen to angiotensin I, and cathepsin G and tonin can convert angiotensinogen
directly to angiotensin II. Angiotensin I can be converted to angiotensin II by cathepsin
G, chymostatin-selective angiotensin II-generating enzyme, and heart chymase (Dzau et
al., 1993; Goodman and Gilman, 2001). In humans, chymase is an important enzyme for

8
the conversion of angiotensin I to angiotensin II at the tissue level, especially in cardiac
(Wolny et al., 1997; Wei et al., 1999) and renal tissue (Hollenberg et al., 1998).
Regardless of its formation pathway, angiotensin II exerts its effects through two
major subtypes of G-protein coupled receptors. The first subtype of receptor, the
angiotensin II type 1 (AT1) receptor, is nearly ubiquitous, and is responsible for most of
the overall actions of angiotensin II. This 359-amino acid cell-surface receptor can
activate a large number of signal transduction pathways. These pathways can include
calcium release and influx, phospholipases, mitogen-activated protein kinase pathways,
Janus kinase pathways, serine/threonine protein kinases, nonreceptor tyrosine kinases,
small GTP-binding proteins, inducible transcription factors, factors affecting translational
efficiency, and the production of reactive oxygen species (Griendling et al., 1997; Berk,
1999; Blume et al., 1999; Inagami, 1999; Goodman and Gilman, 2001). Activation of
AT1 receptors results in endothelial dysfunction, vasoconstriction, vascular smooth and
cardiac muscle cell growth, enhanced coagulation and sympathetic nerve activity, all of
which can be detrimental to cardiovascular function and ventricular structure (Yasunari et
al., 2005). AT1 receptors on cardiac myocytes cause hypertrophy, increased protein
synthesis, overall gene reprogramming, increased production of atrial natriuretic factor,
and can induce apoptosis (Yasunari et al., 2005). These same receptors, on cardiac
fibroblasts, lead to proliferation, increased DNA and protein synthesis, and increased
gene expression including transforming growth factor β1 and fibronectin (Yasunari et al.,
2005). Pharmacological inhibition (Yasunari et al., 2005) and genetic modulation
(Raizada et al., 2000) of the AT1 receptor show great promise for inhibiting cardiac
remodeling.

9
The second subtype of angiotensin II receptor is the angiotensin II type 2 (AT2)
receptor. Much less is known about the AT2 receptor, but its actions are generally
considered to oppose those of the AT1 receptor. The AT2 receptor is highly expressed in
fetal tissues, but levels are low in adults until cardiac insult, such as myocardial
infarction, which results in increased AT2 expression (Nio et al., 1995). This G-protein
coupled receptor is 363-amino acids long, and is known to activate phosphatases,
potassium channels, and nitric oxide production, while inhibiting calcium channels
(Horiuchi et al., 1999). Most of the effects of the AT2 receptor are mediated by the Gαi
binding protein (Goodman and Gilman, 2001). AT2 receptors on cardiac myocytes
inhibit cell growth and decrease protein synthesis, while their activation on cardiac
fibroblasts inhibits AT1-mediated effects, and decreases DNA and protein synthesis
(Yasunari et al., 2005). Overexpression of this receptor subtype has been shown to
prevent cardiac remodeling (Metcalfe et al., 2004).
In addition to angiotensin II, there are multiple other angiotensin peptides that are
endogenously produced by the RAS. For example, the alanine residue at the amino
terminus of angiotensin II can be removed by aminopeptidase A, thus producing
angiotensin-(2-8), also known as angiotensin III. The role of angiotensin III is under
debate, as this peptide has been shown to have similar pharmacological actions at the AT1
and AT2 receptor as angiotensin II (Wright and Harding, 1997).
Angiotensin III is subsequently digested by aminopeptidase B to remove the
arginine residue at the amino terminus. This digestion produces angiotensin-(3-8), which
is commonly known as angiotensin IV. Angiotensin IV selectively binds the AT4
receptor, which is also known as the insulin-regulated aminopeptidase. In the heart, this

10
receptor stimulates protein synthesis in cardiac fibroblasts and inhibits mechanically-
stimulated expression of immediate early genes cfos and egr-1, although the
predominantly recognized role of the AT4 receptor at this time is in learning and memory
(Chai et al., 2004).
Finally, angiotensin IV can be cleaved by aminopeptidase N or
dipeptidylaminopeptidase to form angiotensin-(4-8), angiotensin-(5-8), or angiotensin-(6-
8), all of which are inactive fragments of the RAS (von Bohlen and Halbach, 2003).
Angiotensin Converting Enzyme 2
Recently a second angiotensin converting enzyme (ACE2) was discovered by
Tipnis (2000) and Donoghue (2000). Although this enzyme shares approximately 40%
homology to ACE within its catalytic domain, ACE2 differs from ACE in substrate
specificity and is not inhibited by classic ACE inhibitors (Tipnis et al., 2000, Guy et al.,
2003).
ACE2 has been shown to play a role in heart development and function (Boehm
and Nabel, 2002; Crackower et al., 2002; Donoghue et al., 2003; Oudit et al., 2003).
Genetic knockout of the ACE2 gene causes increased circulating angiotensin II, impaired
cardiac function, and induction of hypoxia-response genes (Crackower et al., 2002;
Donoghue et al., 2003). Overexpression of this gene in rats delays the onset of cardiac
hypertrophy (Santos et al., 2004), and transgenic overexpression in mice results in
decreased systolic blood pressure (Crackower et al., 2002). Several animal models of
hypertension exhibit depressed ACE2 levels, which suggests a relationship between
ACE2 and hypertension (Crackower et al., 2002; Danilczyk et al., 2003; Garcia et al.,
2003). Finally, the ACE2 gene maps to a defined quantitative trait locus that has been
associated with hypertension (Crackower et al., 2002). It has been suggested (Donoghue

11
et al., 2000; Tipnis et al., 2000; and Turner and Hooper, 2002) that the balance of actions
between ACE and ACE2 may determine the overall physiological / pathophysiological
(blood pressure and structural) balance within various organ systems.
To examine the hypothesis that increased activity of ACE2 would result in
protection from cardiac remodeling, we previously delivered the murine ACE2 gene to
rats by use of a lentivirus. Neonatal delivery of lenti-mACE2 to Sprague-Dawley rats
provided these animals protection from cardiac remodeling with angiotensin II infusion
during adulthood (Huentelman et al., 2005). ACE2 overexpression by these animals
attenuated cardiac hypertrophy (Figure 1-2), and completely prevented the interstitial
fibrosis (Figure 1-3) caused by angiotensin II infusion. It is interesting to note that these
effects were not mediated by changes in blood pressure (Figure 1-4), thus indicating that
ACE2 can selectively protect the myocardium from remodeling.
Figure 1-2. Ventricular hypertrophy with chronic Ang II is attenuated by lentiviral delivery of ACE2. (Reprinted with permission from Huentelman MJ, Grobe JL, Vazquez J, Stewart JM, Mecca AP, Katovich MJ, Ferrario CM, Raizada MK. Protection from angiotensin II-induced cardiac hypertrophy and fibrosis by systemic lentiviral delivery of ACE2 in rats. Experimental Physiology. 90(5):783-90. 2005.)

12
A B
Figure 1-3. Ang II-induced cardiac interstitial fibrosis is prevented by lentiviral delivery of ACE2. A) Representative sections of left ventrile wall, and B) quantified collagen density in left ventricle wall sections. (Reprinted with permission from Huentelman MJ, Grobe JL, Vazquez J, Stewart JM, Mecca AP, Katovich MJ, Ferrario CM, Raizada MK. Protection from angiotensin II-induced cardiac hypertrophy and fibrosis by systemic lentiviral delivery of ACE2 in rats. Experimental Physiology. 90(5):783-90. 2005.)
Figure 1D. Lentiviral delivery of ACE2 has no effect on the systolic blood pressure increase from chronic Ang II infusion. (Reprinted with permission from Huentelman MJ, Grobe JL, Vazquez J, Stewart JM, Mecca AP, Katovich MJ, Ferrario CM, Raizada MK. Protection from angiotensin II-induced cardiac hypertrophy and fibrosis by systemic lentiviral delivery of ACE2 in rats. Experimental Physiology. 90(5):783-90. 2005.)

13
The mechanism by which ACE2 can provide cardioprotection during hypertension
is unknown, but it is likely that increased formation of the enzyme’s product,
angiotensin-(1-7), may be important.
Angiotensin-(1-7)
ACE2 has been shown to be one of several enzymes that catalyzes the conversion
of angiotensin I and angiotensin II to the newly recognized hormone angiotensin-(1-7)
(Figure 1-1, and Ferrario and Chappell, 2004), but in the heart, ACE2 and neprilysin have
been shown to have the most significant roles in the generation of angiotensin-(1-7)
(Zisman et al., 2003). ACE, which is a key enzyme in the RAS for the conversion of
angiotensin I to angiotensin II, is the primary enzyme involved in the degradation of
angiotensin-(1-7) to angiotensin-(1-5) (Yamada et al., 1998; Ferrario and Chappell,
2004).
Estrogenic Modulation of Heart Failure
Gender differences exist in the susceptibility, onset, progression, and outcome of
heart failure. Approximately 15% of women and 20% of men have hypertension, which
is a major risk factor for the development of heart failure (Regitz-Vagrosek and
Lehmkuhl, 2005). Blood pressure tends to increase with age in both sexes, although at
menopause, the prevalence of hypertension increases greatly in women. Between the
ages of 65 and 75, approximately 45% of women and 41% of men are hypertensive
(Hayes and Taler, 1998; Gasse et al., 2001). It is important to note that systolic blood
pressure disproportionately increases in women compared to men, and the subsequent
rise in pulse pressure represents an important and independent predictor of cardiovascular
outcome (Burt et al., 1995; Regitz-Vagrosek and Lehmkuhl, 2005).

14
The effects of hypertension in women are more pronounced than in men.
Hypertension is by far the major causative factor for the development of heart failure in
women, whereas myocardial infarction is the most causative factor in men. In the
Framingham Heart Study, hypertension was the primary causative factor for the
development of heart failure in 59% of women, compared to only 39% in men (Levy et
al., 1996). Left ventricular hypertrophy (which results from chronic hypertension) also
carries a greater risk for mortality in women than in men (Liao et al., 1995).
Roles of estrogen and its receptors in the development and progression of heart
failure are fairly well established. In many animal models of cardiovascular
abnormalities, phenotypes are worse in male and ovariectomized females, and
administration of estrogens can attenuate or reverse the phenotype (Regitz-Vagrosek and
Lehmkuhl, 2005). Estrogen has been shown to reduce the size of myocardial infarcts,
and reduce apoptosis (Patten et al., 2004). Estrogen also antagonizes cardiac myocyte
hypertrophy in vitro (Van Eickels et al., 2001). Many of the cardioprotective effects of
estrogen have been attributed to the estrogen receptor (ER)-α subtype (Zhai et al., 2000),
although both α and β estrogen receptor subtypes are known to be present in cardiac
myocytes and fibroblasts from humans and rats (Grohe et al., 1997; Taylor and Al-
Azzawi, 2000). In humans, both receptor subtypes show increased expression during
cardiac hypertrophy (Nordmeyer et al., 2004).
Estrogenic Modulation of the Renin-Angiotensin System
Estrogens are known to modulate many hormonal systems involved in the
regulation of cardiac structure and function. The RAS is regulated by estrogens at
several points. Angiotensinogen and the AT2 receptor are increased with estrogen
treatment (Healy et al., 1992; Armando et al., 2002), while prorenin, ACE, neprilysin,

15
aminopeptidases, and the AT1 receptor are all decreased with estrogen treatment
(Szilagyi et al., 1995; Pinto et al., 1999; Nickenig et al., 2000; Seli et al., 2001; Turner
and Hooper, 2002). Recent studies by Brosnihan et al. (2003) have determined that
during pregnancy, renal ACE2 expression is increased, as are circulating levels of
angiotensin-(1-7). Together, these findings indicate that estrogen decreases the
production and effectiveness of angiotensin II, while it has positive effects on the
generation of angiotensin-(1-7).
Aims
It was the goal of these studies to elucidate the role, if any, of angiotensin-(1-7) in
protecting the myocardium from pathological remodeling. First, the protective effects of
angiotensin-(1-7) will be examined in both angiotensin II–dependent and –independent
forms of hypertension-induced cardiac remodeling. Second, modulation of normotensive
cardiac remodeling by angiotensin-(1-7) will be studied. Third, modulation of the effects
of angiotensin-(1-7) by estrogen will be examined. Finally, preliminary studies into the
cellular mechanism of action of angiotensin-(1-7) will be presented. Together, these
studies will demonstrate the cardioprotective actions of angiotensin-(1-7), establish the
modulatory role of estrogen on this hormone, and point the direction for future studies
into the actions of angiotensin-(1-7).

CHAPTER 2 CARDIAC FIBROSIS IS PREVENTED BY CHRONIC ANGIOTENSIN-(1-7)
DURING ANGIOTENSIN II INFUSION
Abstract
Aberrant activity of the renin-angiotensin system is well established to cause
hypertension and associated cardiac remodeling, and chronic infusion of angiotensin II
(Ang II) can be used to model these effects in animal models. Recent studies have
indicated that the angiotensin-(1-7) (Ang-(1-7)) breakdown product of angiotensin II may
act as an endogenous buffer against the actions of Ang II. Here, we examine the
hypothesis that chronic infusion of Ang-(1-7) provides cardioprotection during
hypertension due to chronic infusion of Ang II. Sprague-Dawley rats between 320-470
grams were implanted with osmotic minipumps which chronically delivered Ang II (100
ng/kg/min). A subset of these animals was also implanted with osmotic minipumps that
chronically delivered Ang-(1-7) (100 ng/kg/min). Control animals underwent sham
implantation surgery. Blood pressure was monitored by an indirect tail cuff method, and
after four weeks of treatment, hearts were harvested, sectioned and stained using
Masson’s Trichrome. Chronic infusion of Ang II resulted in significant increases in
blood pressure, ventricular and myocyte hypertrophy, and interstitial fibrosis. Chronic
infusion of Ang-(1-7) significantly decreased this myocyte hypertrophy and interstitial
fibrosis. These results lead us to conclude that Ang-(1-7) selectively attenuates cardiac
remodeling during chronic infusion of Ang II, without effect on blood pressure. These
findings complement and extend our previous findings that lentiviral delivery and
16

17
subsequent overexpression of the murine ACE2 gene prevents cardiac remodeling in Ang
II-infused rats.
Introduction
The renin-angiotensin system (RAS) is well documented as an important
endogenous system that regulates cardiac structure and function. An aberrant RAS has
been shown to be integrally involved in the development and maintenance of
hypertension and cardiac remodeling (Kim et al., 1995; Alderman et al., 1991; Dzau,
1993; Parmley, 1998; Bader et al., 2001; Ruiz-Ortega et al., 2001; Unger, 2002; Zaman
et al., 2002). Further, use of RAS inhibitors (both angiotensin converting enzyme
inhibitors and angiotensin II type 1 receptor blockers) has been shown to decrease blood
pressure, attenuate cardiac remodeling, and prevent cardiac function loss (Frigerio and
Roubina, 2005).
Chronic infusion of angiotensin II (Ang II) has long been used in animal models to
mimic the onset and progression of hypertension and hypertension-induced
pathophysiologies. Chronic infusion of Ang II induces these changes through direct
vasoconstriction of the vasculature (Rajagopalan et al., 1996), water and salt reabsorption
at the kidney (Zou et al., 2002; Lopez et al., 2003; Rugale et al., 2003), and altered
sympathetic tone, both centrally and peripherally (Zanzinger and Czachurski, 2000;
Zimmerman et al., 2002).
Within the heart, Ang II induces structural changes both directly through actions at
cardiac cells and indirectly through changes in arterial blood pressure. In culture, Ang II
stimulates growth of cardiac myocytes, fibroblasts, and smooth muscle cells (Baker et al.,
1992; Corda et al., 2000). Ang II also increases production of the extracellular matrix
proteins, including collagen and fibronectin, and inhibits the activity of collagenases

18
(Baker et al., 1992; Corda et al., 2000). These results are paralleled in vivo during Ang
II-induced hypertension and following myocardial infarction in the rat (Brilla et al., 1990;
Weber and Brilla, 1991; Lorell, 1997; Swynghedauw, 1999; Corda et al., 2000). Low
(non-pressor) doses of Ramipril, and angiotensin converting enzyme (ACE) inhibitor, can
regress cardiac fibrosis, thus highlighting the direct effects of the local RAS in regulating
cardiac structure independent of blood pressure (Nagasawa et al., 1995; Corda et al.,
2000).
The mechanism by which low-dose ACE inhibition can attenuate cardiac
remodeling is likely three-fold. First, ACE inhibition can result in decreased levels of
Ang II. Second, ACE inhibition may result in increased levels of bradykinin, as ACE is
known to degrade this protective hormone (Goodman and Gilman, 2001). Third, ACE
inhibition has been shown to result in increased levels of the newly recognized hormone
angiotensin-(1-7) (Ang-(1-7)) (Tom et al., 2003), likely due to the fact that ACE is the
principle degradation pathway for Ang-(1-7) (Ferrario and Chappell, 2004).
We have recently demonstrated that increased expression of angiotensin converting
enzyme 2 (ACE2), the primary Ang-(1-7) production enzyme in the heart (Zisman et al.,
2003; Ferrario and Chappell, 2004), prevents Ang II-induced cardiac remodeling in rats
(Huentelman et al., 2005). Specifically, lentiviral transfer of the mouse ACE2 gene to
prevented cardiac hypertrophy (Figure 1-2) and cardiac fibrosis (Figure 1-3) without
effect on blood pressure (Figure 1-4). At least two possible mechanisms involving the
renin-angiotensin system may be at work in ACE2-mediated cardioprotection during Ang
II-induced hypertension and cardiac remodeling. First, it is possible that ACE2 simply
decreases the levels of its reactant, Ang II. Second, it is possible that the increased levels

19
of Ang-(1-7) resulting from ACE2 enzymatic activity provide some active protection
from cardiac remodeling. Therefore, the following study was undertaken to examine the
hypothesis that chronic infusion of Ang-(1-7) would prevent cardiac remodeling in the
Ang II-infusion model of hypertension.
Methods
Animals
Eighteen sprague-Dawley rats, between 320-470 grams, were divided into three
groups. Animals were anesthetized by halothane inhalation before implantation of
subcutaneous osmotic minipumps (Alzet – model 2004). Eight animals received pumps
that delivered Ang II at 100 ng/kg/min. Four animals received an additional pump that
delivered Ang-(1-7) at 100 ng/kg/min. Six animals underwent sham surgery. For a
detailed method for loading osmotic minipumps, please see Appendix A.
Indirect Blood Pressure
Systolic blood pressure was determined weekly using an indirect tail cuff method
as previously described (Katovich et al., 2001). Briefly, animals were heated using a
200-watt heat lamp for 5 minutes before restraint in a heated Plexiglas restrainer to which
the animals had been previously conditioned. A pneumatic pulse sensor was then
attached to the tail distal to a pressure cuff under the control of a Programmed Electro-
Sphygmomanometer (Narco). Voltage outputs from the pressure sensor bulb and
inflation cuff were recorded and analyzed electronically using a PowerLab signal
transduction unit and associated Chart software (ADInstruments). At least three separate
indirect pressures were averaged for each animal.

20
Cardiac Remodeling
Following four weeks of chronic treatment, animals were sacrificed by overdose of
halothane inhalant, and hearts were removed by blunt dissection. Heart ventricles were
cleaned of atria and other adherent tissue before wet weight was determined. Cross-
sections of the heart ventricles were then cut at the level of the papillary muscles,
embedded in paraffin, and sectioned at 4 microns. Sections were then stained using
Masson’s Trichrome, and collagen content was determined using the ImageJ program
from NIH (Rasband, 1997), as previously described (Grobe et al., 2006 – Chapter 3).
For a detailed method, please see Appendix B.
Statistics
Indirect blood pressure data were analyzed by one-way repeated measures
ANOVA. Cardiac hypertrophy measurements were analyzed by one-way ANOVA.
Interstitial fibrosis was analyzed by nonparametric analyses, namely an overall Kruskal-
Wallis test followed by post-hoc Mann-Whitney U tests. For all analyses, P<0.05 was
considered statistically significant.
Results
Blood Pressure
Angiotensin II (100 ng/kg/min) infusion resulted in significant increases in indirect
systolic blood pressures (Figure 2-1).
Cardiac Hypertrophy
Ventricular size was significantly increased in animals infused with Ang II (Figure
2-2A). Angiotensin-(1-7) had no significant effect on this increase in ventricle size, but a
trend toward reversal was observed. Ang-(1-7) did, however, significantly prevent the
Ang II-induced increase in individual cardiac myocyte diameter (Figure 2-2B).

21
110
120
130
140
150
160
170
0 1 2 3Weeks
Syst
olic
BP
(mm
Hg)
Ang IIAng II + Ang-(1-7)Sham
Figure 2-1. Chronic Ang-(1-7) has no effect on blood pressure increases with chronic Ang II infusion. Analysis by one-way repeated measures ANOVA reveals a significant increase in BP with Ang II (P=0.0053), and Ang II + Ang-(1-7) was not different from Ang II alone (P=0.4579). Data presented as mean ± SE.
2.7
2.8
2.9
3.0
3.1
3.2
3.3
3.4
Sham Ang II Ang II +Ang-(1-7)
Hea
rt M
ass (
g/kg
)
* A
12
13
14
15
16
17
18
Sham Ang II Ang II +Ang-(1-7)
Myo
cyte
Dia
met
er (m
icro
ns) *
†
B
Figure 2-2. Chronic Ang-(1-7) prevents myocyte hypertrophy due to chronic Ang II infusion. (A) Heart weight, normalized to body weight, is significantly increased by chronic Ang II. Chronic Ang-(1-7) causes a non-significant (P=0.09) trend toward reversing this effect. (B) Chronic infusion of Ang II significantly increases myocyte cross-sectional diameter, and chronic Ang-(1-7) significantly prevents this effect. (* indicates P<0.05 vs. Sham, and † indicates P<0.05 vs. Ang II) Data presented as mean ± SE.

22
Cardiac Fibrosis
Interstitial collagen deposition was significantly increased in animals with chronic
Ang II infusion, and this increase was prevented by simultaneous Ang-(1-7) infusion
(Figure 2-3).
97
100
104
107
Sham Ang II Ang II +Ang-(1-7)
Inte
rstit
ial F
ibro
sis (
% S
ham
) *
†
A
Ang II + Ang-(1-7)Ang II B
Sham
Figure 2-3. Chronic Ang-(1-7) prevents interstitial fibrosis due to chronic Ang II infusion. (A) Infusion of Ang II significantly increased interstitial fibrosis in the left ventricle wall. Infusion of Ang-(1-7) significantly prevented this effect. (* indicates P<0.05 vs Sham, and † indicates P<0.05 vs. Ang II) Data presented as mean ± SE. (B) Representative 250x-magnification images illustrate changes in myocyte size and interstitial collagen.
Discussion
In this study, chronic Ang II infusion resulted in increased blood pressure,
hypertrophied heart ventricles and cardiac myocytes, and increased interstitial collagen

23
deposition. Simultaneous infusion of Ang-(1-7) at a nearly equal molar rate prevented
cardiac remodeling, both in terms of hypertrophy and fibrosis, without effect on blood
pressure. These findings are parallel to those observed with ACE2 overexpression during
Ang II infusion (Huentelman et al., 2005).
The finding that chronic Ang-(1-7) provides the same cardioprotection as
overexpression of ACE2 supports the concept that Ang-(1-7) mediates the cardiovascular
actions of ACE2. Further, these findings indicate that Ang-(1-7) is an active hormone
(rather than simply a degradation product of the trophic Ang II peptide).
There are several possible mechanisms by which Ang-(1-7) may inhibit
pathophysiologies induced by Ang II. First, it has been proposed that Ang-(1-7) may
antagonize the AT1 receptor (Gironacci et al. 1999; Caruso-Neves et al., 2000; Ferrario et
al., 2005; Ferrario et al., 2005; Igase et al., 2005). Second, some evidence exists that
Ang-(1-7) selectively activates the AT2 receptor (Santos et al., 2000; De Souza et al.,
2004; Gironacci et al., 2004; Roks et al., 2004), and thereby opposes the effects of AT1
receptor activation. Third, Ang-(1-7) may inhibit ACE (Donoghue et al., 2000), and
therefore inhibit endogenous production of Ang II. Fourth, Ang-(1-7) may act through its
own receptor to effect changes in cellular function directly (Santos et al., 2003; Tallant et
al., 2005).
Several groups have proposed that Ang-(1-7) may bind the AT1 receptor. For
example, Ang-(1-7) stimulates the Na+-ATPase, but not the Na+,K+-ATPase activity
present in pig kidney proximal tubules, and this stimulation is attenuated by losartan (a
selective AT1 antagonist), but not PD-123,319 (a selective AT2 antagonist) or D-Ala-
Ang-(1-7) (also known as A779, an Ang-(1-7) receptor binding antagonist) (Caruso-

24
Neves et al., 2000). Similarly, others have shown that radiolabeled Ang II binds to AT1
receptors in rat renal cortex, and that Ang-(1-7) displaces the Ang II with high affinity
(Gironacci et al., 1999). Clearly it is possible that Ang-(1-7) may bind to the AT1
receptor in target tissues. If Ang-(1-7) did interfere with Ang II binding to AT1 receptors
in the present study, though, one would hypothesize that the Ang-(1-7) would interfere
with pressor effects of chronic Ang II (which are well established to be AT1 dependent).
Indirect pressure data over time (Figure 2-1), however, fail to support this hypothesis.
Therefore, Ang-(1-7) antagonism of AT1 receptors seems unlikely as the cardioprotective
mechanism in the current study.
Activation of the AT2 receptor is another possible mechanism by which Ang-(1-7)
prevents cardiac remodeling in the present study. Ang-(1-7) inhibits the pig renal inner
cortex basolateral Na+-ATPase activity in a dose-dependent manner, and this effect is
attenuated by PD-123,319 but not losartan or A779 (De Souza et al., 2004). Ang-(1-7)-
mediated inhibition of the release of norepinephrine in the spontaneously hypertensive rat
is also AT2 dependent (Gironacci et al., 2004), as is the endothelium-dependent, non-
competitive antagonism of Ang II contraction of aortic rings (Roks et al., 2004), and the
osmotic water permeability in isolated toad skin (Santos et al., 2000). It has been shown
that AT2 receptors are cardioprotective (Metcalfe et al., 2004), and these receptors are
known to be expressed in both cardiac myocytes (Matsubara, 1998) and fibroblasts
(Lijnen and Petrov, 1999; Lijnen and Petrov, 2003), although their expression profiles
during adulthood are debated. Therefore, it is possible that AT2-mediated transduction of
Ang-(1-7) signaling could account for the present results.

25
Another possible mechanism by which Ang-(1-7) may mediate its effects is
through the inhibition of the ACE enzyme. This enzyme is responsible for most of the
endogenous production of Ang II in rats, and is also known to degrade Ang-(1-7). It has
been suggested (Tom et al., 2003) that Ang-(1-7) may work through inhibition of ACE,
and therefore decreased production of Ang II. This inhibition could represent a feedback
loop, with the degradation product of Ang II regulating the production of Ang II.
Although this mechanism seems plausible, it is unlikely that decreased production of Ang
II through ACE inhibition could account for the present results, as Ang II is chronically
infused in the current paradigm. Therefore, it is unlikely that Ang-(1-7) inhibition of Ang
II production through ACE would account for the present results. Inhibition of ACE
could also represent a mechanism of cross-talk with the bradykinin / nitric oxide system,
though, which likely could mediate the observed effects. Active (vasodilatory and
cardioprotective) bradykinin is inactivated and degraded to bradykinin-(1-7) by ACE
(Tom et al., 2003). In fact, more than 50% of the degradation of bradykinin in vivo is
mediated by ACE (Goodman and Gilman, 2001). It is plausible that Ang-(1-7) may, by
inhibiting the destruction of the cardioprotective bradykinin through inhibition of ACE,
elicit the observed cardioprotective actions. One would expect, though, that increased
survival of bradykinin through Ang-(1-7) inhibition of ACE would significantly impact
blood pressure regulation. Therefore, although it is possible that inhibition of ACE may
account for the cardioprotective actions of Ang-(1-7), the observed effects of chronic
Ang-(1-7) on blood pressure do not support this hypothesis.
A few years ago, Santos et al. (2003) established that Ang-(1-7) selectively binds
the G-protein coupled receptor Mas, and that the anti-diuretic actions of Ang-(1-7) in the

26
mouse kidney are Mas-dependent. Subsequent studies by others have established that
Ang-(1-7) mediates anti-hypertrophic (Tallant et al., 2005) and vasodilator (Lemos et al.,
2005) effects in vitro through the Mas receptor. In light of these findings, it seems
possible that chronic Ang-(1-7) effects on cardiac structure in vivo are mediated through
the Mas receptor.
While the determination that Ang-(1-7) actively opposes the trophic, fibrotic
actions of Ang II is significant, this study does not clarify the mechanism of Ang-(1-7)
action, nor does it determine if the actions of Ang-(1-7) are limited to Ang II-mediated
cardiac remodeling. Therefore, the next study was designed to determine if Ang-(1-7) is
effective in preventing cardiac remodeling in a hypertensive animal model that displays
low circulating Ang II levels.

CHAPTER 3 CHRONIC ANGIOTENSIN-(1-7) PREVENTS CARDIAC FIBROSIS IN THE DOCA-
SALT MODEL OF HYPERTENSION
Note: This chapter has already been published in the American Journal of Physiology: Heart & Circulatory Physiology. Grobe JL, Mecca AP, Mao H, and Katovich MJ. Chronic angiotensin-(1-7) prevents cardiac fibrosis in the DOCA-salt model of hypertension. Am J Physiol Heart Circ Physiol. [Epub ahead of print], 2006.
Abstract
Cardiac remodeling is a hallmark hypertension-induced pathophysiology. In the
current study, the role of the angiotensin-(1-7) fragment in modulating cardiac
remodeling was examined. Sprague-Dawley rats underwent uninephrectomy surgery and
were implanted with a deoxycorticosterone acetate pellet (DOCA). DOCA animals had
their drinking water replaced with 0.9% saline solution. A subgroup of DOCA-salt
animals was implanted with osmotic minipumps, which delivered angiotensin-(1-7)
chronically (100 ng/kg/min). Control animals underwent sham surgery and were
maintained on normal drinking water. Blood pressure (BP) was measured weekly using a
tail-cuff method, and after four weeks of treatment, BP responses to graded doses of
angiotensin II were determined by direct carotid artery cannulation. Ventricle size was
measured and cross-sections of the heart ventricles were paraffin embedded and stained
using Masson’s Trichrome to measure interstitial and perivascular collagen deposition
and myocyte diameter. DOCA-salt treatment caused significant increases in blood
pressure, cardiac hypertrophy, and myocardial and perivascular fibrosis. Angiotensin-(1-
7) infusion prevented the collagen deposition effects without any effect on blood pressure
27

28
or cardiac hypertrophy. These results indicate that angiotensin-(1-7) selectively prevents
cardiac fibrosis independent of blood pressure or cardiac hypertrophy in the DOCA-salt
model of hypertension.
Introduction
Cardiac fibrosis is a major facet of hypertensive cardiac disease, and it interferes
with the normal function and structure of the myocardium (Brilla et al. 1991; Weber and
Brilla 1991; Weber, 2000). Increased deposition of basement membrane collagen is a
hallmark of the remodeling process, and results in an increase in cardiac tissue stiffness.
This remodeling predisposes the patient to an increased risk of adverse cardiac events,
including myocardial ischemia, infarction, arrhythmias and sudden cardiac death (Weber,
2000). Thus, prevention and reversal of cardiac fibrosis are essential in the management
of hypertensive heart disease.
The renin angiotensin aldosterone system (RAAS) has been suggested to
participate in the development of end-organ damage in hypertensive patients (Brunner et
al., 1972; Alderman et al. 1991). Support for this concept comes from clinical trials that
demonstrate treatment of hypertensive patients with either ACE inhibitors (Pfeffer et al.,
1992; The SOLVD Investigators, 1992) or AT1 receptor blockers (Pitt et al., 1997;
Thurmann et al. 1998) provides significant protection from, and even reversal of, end-
organ damage. Animal studies have also demonstrated that ACE inhibitors and AT1
receptor antagonists prevent cardiovascular injury (Jalil et al., 1991; Stier et al., 1991;
Stier et al., 1993) as well as protect against renal (Anderson et al., 1986; Stier et al.,
1991; Stier et al. 1998) and cerebral (Stier et al. 1991; Stier et al., 1993; Stier et al.,
1998) injury.

29
The use of these antagonists of the RAAS not only reduce the formation and
actions of angiotensin II (Ang II), but also result in a significant elevation of another
fragment of the RAAS, angiotensin-(1-7) (Ang-(1-7)) (Ferrario et al., 2005; Keidar et al.,
2005). This peptide, which can be generated locally in the myocardium (Santos et al.,
1990), has been reported to work antagonistically to Ang II and has been implicated in
protecting against cardiac pathophysiology (Santos et al., 2004). Recently, Loot et al.
(2002) demonstrated that chronic infusion of Ang-(1-7) improved endothelial function
and coronary perfusion, and preserved cardiac function in an animal model of heart
failure. Ang-(1-7) has also been shown to significantly increase cardiac output and stroke
volume in rodents (Ferreira et al., 2002) and to improve contractile function in isolated
perfused rat hearts (Sampaio et al., 2003). Using a fusion protein to generate a transgenic
rat model that overproduces Ang-(1-7), Santos et al. (2004) showed that Ang-(1-7)
reduced the induction of cardiac hypertrophy and the duration of reperfusion arrhythmias,
and improved post-ischemic function in isolated perfused hearts. Thus, Ang-(1-7) may
be an important component of the RAAS that has opposing actions to Ang II, and it may
provide protective action(s) in the heart and possibly other end-organs. Angiotensin-(1-
7) also recently has been reported to inhibit the growth of cardiac myocytes in vitro
(Tallant et al., 2005), suggesting that elevated levels of Ang-(1-7) may also protect
against cardiac hypertrophy in hypertension. Interestingly, blockade of Ang-(1-7)
receptors has also been shown to reverse the antihypertensive effects of long term
administration of ACE inhibitors (Iyer et al., 1998). Thus, Ang-(1-7) may play a
significant role in the beneficial effects on the cardiovascular system attributed to some
antihypertensive agents.

30
Not only has Ang II been implicated in the development of cardiac fibrosis, but so
has the aldosterone component of the RAAS (Funder, 2001). It has been demonstrated in
experimental studies utilizing uninephrectomized rats, that deoxycorticosterone acetate
supplementation (DOCA-salt) treatment results in significant cardiac and renal
remodeling, including intersititial fibrosis and hypertrophy (Dobrzynski, et al, 2000;
Young et al. 1995). The purpose of this study was to determine if chronically elevated
levels of Ang-(1-7) alters the development of hypertension, cardiac hypertrophy and/or
cardiac fibrosis in the DOCA-salt model of hypertension.
Methods
Animals
Male Sprague-Dawley rats weighing between 170 and 250 grams were used for this
study. Animals were housed in a temperature and humidity controlled room in standard
shoebox cages, and maintained on a 12:12 hour light:dark schedule with free access to
food and water. All procedures were approved by the University of Florida Institutional
Animal Care and Use Committee.
Chemicals
Angiotensin-(1-7) was obtained from Bachem Bioscience, Inc.
Deoxycorticosterone acetate was purchased from Sigma Aldrich (St. Louis, MO).
DOCA-salt Model
Animals were anesthetized using an intramuscular injection of a Ketamine,
Xylazine Acepromazine, and (30, 6, and 1 mg/kg, i.m., respectively) before
uninephrectomy. During the surgery, fifteen animals were implanted with a
subcutaneous pellet of deoxycorticosterone acetate (40 mg). Seven of these animals were
additionally implanted with a subcutaneous osmotic pump that delivered Ang-(1-7) (100

31
ng/kg/min) for the duration of the experiment. After surgery (and for the remainder of
the experiment), drinking water for these animals was replaced with 0.9% saline solution.
Ten animals underwent a sham surgery, and were maintained on normal drinking water.
Four additional sham animals were implanted with only an Ang-(1-7) pump.
Indirect Blood Pressure
Systolic blood pressures were determined weekly by an indirect method utilizing a
tail-cuff and pneumatic pulse sensor as previously reported (Katovich et al., 2001).
Briefly, animals were heated using a 200-watt heat lamp for 3-5 minutes before being
restrained in a heated Plexiglas restrainer to which the animals had been conditioned
prior to the experiment. A pneumatic pulse sensor was then attached to the tail distal to a
pressure cuff under the control of a Programmed Electro-Sphygmomanometer (Narco).
Voltage outputs from the pressure sensor bulb and inflation cuff were recorded and
analyzed electronically using a PowerLab signal transduction unit and associated Chart
software (ADInstruments). At least three separate indirect pressures were averaged for
each animal.
Direct Blood Pressure/Acute Angiotensin II Responses
After four weeks of chronic treatment, animals underwent carotid artery and jugular
cannula implantation surgery as previously reported (Lu et al., 1997). Briefly, animals
were anesthetized with a mixture of Ketamine, Xylazine, and Acepromazine (30, 6, and 1
mg/kg, i.m., respectively). A polyethylene cannula (PE-50, Clay Adams) was introduced
into the carotid artery for direct blood pressure measurements, while a silicone elastomer
cannula (Helix Medical) was introduced into the descending jugular vein for acute
intravenous injections of drug. Both cannulae were filled with heparin saline (40 U/mL,
sigma), and sealed with stylets. Animals were allowed 24 hours to recover before

32
experiments were performed. Direct blood pressure responses to acute intravenous
injections of Ang II (5 to 320 ng/kg) in awake, freely moving animals were recorded by
connecting the carotid cannula to a liquid pressure transducer, which was interfaced to a
PowerLab (ADInstruments) signal transduction unit. Data were analyzed using the Chart
program that was supplied with the PowerLab system.
Cardiac Remodeling
Cardiac remodeling was determined at the end of the direct blood pressure
experiment by ventricular hypertrophy and cardiac fibrosis after four weeks of respective
treatments. Following the acute Ang II blood pressure experiment, animals were
anesthetized by inhaled halothane, then euthanized by decapitation. Ventricular
hypertrophy was determined by measuring ventricle mass normalized by body mass.
Cross-sections of the ventricles were then embedded in paraffin, sectioned at 4 microns,
and stained for collagen deposition using Masson’s Trichrome. Single sections from
each animal were then viewed and photographed with a Moticam 1000 digital camera
(Motic, Richmond, British Columbia, Canada) under 10X (whole heart) and 250X
(perivascular collagen and myocyte diameter) magnifications. The collagen content of
the left ventricle wall was determined by measuring the blue stain density of the left wall
(from the septum in the posterior to the septum in the anterior) and was normalized to the
red stain density of the same portion of the image. The area of blue-stained collagen
surrounding the left anterior descending coronary artery was measured and normalized to
the area of the lumen of the vessel. Myocyte cross-sectional diameter was also measured
to confirm ventricular hypertrophy results. Color density, perivascular collagen and
lumen areas, and myocyte diameter were determined using the ImageJ program from
NIH (Rasband, 1997).

33
Statistics
Basal direct blood pressure and ventricular hypertrophy (by mass and by myocyte
diameter) were analyzed by two-way ANOVA, while indirect blood pressures and direct
blood pressure responses to graded, acute doses of Ang II were analyzed by two-way
ANOVA with repeated measures. Cardiac fibrosis measurements were compared using
the more stringent nonparametric Kruskal-Wallis ANOVA, followed by post-hoc Mann-
Whitney U tests. Data were considered significantly different with p<0.05.
Results
Indirect Blood Pressure
Figure 3-1. Chronic Ang-(1-7) has no effect on blood pressure increases with DOCA-salt treatment. DOCA-salt treatment led to a significant increase in systolic blood pressure as measured by an indirect tail-cuff method (p=0.006). Chronic infusion of Ang-(1-7) had no effect on the systolic blood pressure increases due to DOCA-salt treatment (p=0.22). Data presented as mean +/- SE.
DOCA-salt treatment caused a significant increase (p=0.006) in systolic blood
pressure over the course of the experiment. Ang-(1-7), when infused simultaneously,
failed to prevent these increases (p=0.22) (Figure 3-1). Ang-(1-7), when infused alone,

34
did not significantly alter blood pressure when compared to sham-implanted animals
(p=0.69, data not shown).
Direct Blood Pressure/Acute Angiotensin II Responses
Direct pressure measurements confirmed that while DOCA-salt treatment
significantly increased pressure (p=0.007), Ang-(1-7) had no effect (p=0.20) on basal
systolic blood pressure (Sham, 129.2±3.7; DOCA, 160.0±5.2; DOCA + Ang-(1-7),
171.6±10.8; Ang-(1-7) alone, 143.8±21.0 mmHg). In addition, DOCA-salt treatment
significantly potentiated (p=0.02), and Ang-(1-7) infusion had no effect (p=0.41), on the
dose-response curves to acute, graded injections of Ang II (Figure 3-2).
Figure 3-2. Chronic Ang-(1-7) has no effect on acute pressor responses to Ang II in animals treated with DOCA-salt. DOCA-salt treatment significantly potentiated the dose-response curve to acute Ang II (p=0.02). Chronic Ang-(1-7) had no effect (p=0.41) on the acute responses to Ang II. Data presented as mean +/- SE.
Cardiac Remodeling
DOCA-salt treatment significantly increased ventricle mass (p=0.02), and Ang-(1-
7) failed to prevent this effect (p=0.35) (Figure 3-3A). These effects were confirmed by

35
measurement of myocyte diameter, which uncovered a significant increase in size with
DOCA-salt treatment (p<0.0001), but no effect of Ang-(1-7) (p=0.71) (Figure 3-3B).
A B
Figure 3-3. Chronic Ang-(1-7) has no effect on cardiac hypertrophy due to DOCA-salt treament. (A) DOCA-salt treatment significantly increased heart mass (p=0.02). Chronic Ang-(1-7) had no effect on the hypertrophy due to DOCA-salt treatment (p=0.35). (B) Cardiac myocyte diameter was also significantly increased by DOCA-salt treatment (p<0.0001), but chronic Ang-(1-7) had no effect (p=0.71). Data are presented as mean +/- SE. (* indicates p<0.05 vs. Sham)
A significant increase in left ventricular wall fibrosis was observed with DOCA-
salt treatment (p=0.04 vs. sham), and chronic Ang-(1-7) treatment significantly prevented
this trend (p=0.04 vs. DOCA) (Figure 3-4). While the increase in ventricular wall
fibrosis is only approximately 4%, this value is most likely diluted by our measurement
of collagen in the entire left ventricular wall at 10X magnification (as opposed to the
common method of measuring selected “representative samples” at high magnification).
Clearly, from Figure 3-4B, analysis of “representative areas” within the left wall would
lead to much larger relative changes.

36
A
Figure 3-4. Chronic Ang-(1-7) prevents interstitial fibrosis with DOCA-salt treatment. (A) DOCA-salt treatment caused a significant increase in collagen deposition in the left ventricle wall. Chronic Ang-(1-7) significantly prevented this increase. Data are presented as mean +/- SE. (* indicates p<0.05 vs. Sham, † indicates p<0.05 vs. DOCA). (B) Representative images of mid-myocardium at 250X magnification, stained with Masson’s Trichrome. Blue color indicates collagen fibers.
DOCA-salt treatment further resulted in a significant (p=0.0002 vs sham) increase
in perivascular collagen deposition around the left anterior descending coronary artery,
and Ang-(1-7) completely prevented this effect (p=0.0007 vs DOCA) (Figure 3-5).
Discussion
In this study, DOCA-salt treatment resulted in increases in blood pressure, acute
pressor responses to Ang II, cardiac hypertrophy, and mid-myocardial and perivascular
fibrosis. The Ang-(1-7) fragment had no effect on the elevated blood pressure, acute
B

37
pressor responses or cardiac hypertrophy, but significantly prevented the cardiac mid-
myocardial and perivascular fibrosis in the DOCA-salt hypertensive rat model.
Figure 3-5. Chronic Ang-(1-7) prevents perivascular fibrosis with DOCA-salt treatment. (A) DOCA-salt significantly increased perivascular collagen around the left
5 f
Deox
previo
s
A
B
anterior descending coronary artery. Chronic Ang-(1-7) significantly prevented this effect. Data are presented as mean +/- SE. (* indicates p<0.0vs. Sham, † indicates p<0.05 vs. DOCA). (B) Representative images operivascular collagen surrounding the left anterior descending coronary artery, stained with Masson’s Trichrome. Blue color indicates collagen fibers.
ycorticosterone treatment in uninephrectomized rats drinking 1.0% NaCl has
usly been shown to cause hypertension, cardiac hypertrophy and interstitial and
perivascular fibrosis (Young et al. 1995; Dobrzynski et al., 2000). These cardiac effect
are mediated via mineralocorticoid receptors (Young and Funder, 2000; Rocha and Stier,

38
2001; Weber, 2001), and are not observed in animals on a low salt diet (Brilla et al.,
1992). Others have suggested the involvement of AT1 receptors in the fibrosis associ
with DOCA-salt treatment (Kim et al. 1994; Nishikawa, 1998). Elevation of cardiac AT
ated
pertrophy response to DOCA
treatm
r than
d
ce
cts of DOCA on cardiac hypertrophy have been postulated to involve
angiotensin II or transforming growth factor β (TGFβ) (Young et al., 1994; Takeda et al.,
1
receptors associated with DOCA-salt treatment may potentiate the well described fibrotic
effect of Ang II. There is evidence that the trigger for cardiac fibrosis in this animal
model is coronary vasculitus, as the inflammatory cell infiltration occurs within a few
days of DOCA treatment (Young and Funder, 2003).
In a study by Young et al. (1995), the cardiac hy
ent was restricted to the left ventricle, whereas the collagen deposition was
indistinguishable between left and right ventricle, thus supporting a humoral, rathe
a hemodynamic, etiology. Further support for a humoral etiology for collagen deposition
comes from Weber’s (1994) laboratory, where spironolactone prevented aldosterone-
induced cardiac fibrosis without lowering blood pressure. Additionally, interstitial an
perivascular fibrosis can be produced by mineralocorticoid and salt without substantial
hypertension or cardiac hypertrophy (Young et al., 1995). Young et al. (1995) also
demonstrated that central infusion of a mineralocorticoid receptor antagonist lowered
blood pressure but not the pattern of cardiac fibrosis, further suggesting an independen
of fibrosis and blood pressure. Our results also demonstrate the fibrosis observed in this
model was found in both the right and left ventricle walls (right ventricle data not
shown), and such fibrosis appears to be independent of both blood pressure and
hypertrophy.
The effe

39
2002) ot
),
CA-
sts
ibrosis induced by coronary artery ligation in male
Wista
al
sly
)
udy,
. Whether these same mechanisms are responsible for the cardiac fibrosis are n
clear. Young and Funder (2003) demonstrated that animals treated for as few as 8 days
with DOCA/salt treatment demonstrated a significant increase in cardiac perivascular
collagen deposition, with no changes in heart weight, and the collagen formation was
inhibited by a mineralocorticiod receptor antagonist. Further evidence that the cardiac
hypertrophy and fibrosis may be independent include results from Lekgabe et al. (2005
who demonstrated that cardiac hypertrophy, and the related markers of hypertrophy that
were elevated in the SHR compared to the WKY, were not reversed with chronic relaxin
treatment, whereas the fibrosis and collagen content in the hearts of the SHR were
normalized by relaxin treatment to levels observed in the WKY. In the current study,
Ang-(1-7) also showed differential effects upon hypertrophy and fibrosis in the DO
salt model of hypertension, suggesting different mechanisms may exist for these two
endpoints of cardiac remodeling.
Nehme et al. (2005) reported that both aldosterone and Ang II receptor antagoni
significantly reduced the cardiac f
r rats. The effects of the aldosterone receptor antagonist (spironolactone) were
independent of changes in mean blood pressure. This form of experimental myocardi
infarction (MI) in rats has been shown to activate the RAAS (Pinto et al., 1993,
Schunkert et al., 1993). Spironolactone also has been shown to reduce arterial collagen
production, independent of changes in systemic blood pressure in the spontaneou
hypertensive rat (Benetos et al., 1997) and in an L-NAME (nitro-L-arginine methyl ester
plus Ang II with salt model of hypertension (Rocha et al., 2000). As in the current st
Nehme et al. (2005) also reported that these treatments did not prevent the cardiac

40
hypertrophy induced in this MI model. This is further evidence that the mechanisms for
cardiac hypertrophy and fibrosis may be independent.
Generally, the actions of Ang-(1-7) are opposite of those attributed to Ang II
(Santos et al., 2000). Ang-(1-7) has been reported to have intrinsic vasoactive effects in
some .,
the
g-
ts
no
studies (Brosnihan et al., 1996; Iyer et al., 1998; Sasaki et al., 2001; Ren et al
2002), whereas these effects are absent in other studies (Davie et al., 1999; Wilsdorf et
al., 2001; Zhu et al., 2002; Stegbauer et al., 2003). Bayorh et al. (2002) demonstrated
hypotensive effects of Ang-(1-7) were not observed in Dahl animals on a low salt diet
and Eatman et al. (2001) noted a gender difference in the vasodilatory action of Ang-(1-
7) in Dahl rats. Neves et al. (2004) recently reported that the vasodilatory effects of An
(1-7) are modulated during the estrus cycle. Thus, the contrasting findings of Ang-(1-7)
on the vasculature may be related to the differences in animal models studied, dose of
Ang-(1-7), vascular bed studied, or the experimental condition (in vitro versus in vivo) in
which the peptide is evaluated. In the current study, at the dose of Ang-(1-7) infused,
there was no effect on the basal blood pressure of normotensive SD animals or those
animals made hypertensive with DOCA-salt treatment. There are situations, such as in
models of endothelial dysfunction (Le Tran et al., 1997), where the vasodilatory effec
of Ang-(1-7) have been shown to be blunted. This may be the reason for a lack of effect
in our hypertensive model. In the current study, no reduction in the pressor response to
acute administration of Ang II was observed. Benter et al. (1995) reported that infusion
of Ang-(1-7) did not alter the pressor responses to Ang II or phenylephrine in WKY
animals; however, it did reduce the response to these agents in the SHR. A higher dose
of Ang-(1-7) was utilized in the Benter study compared to ours. Although there was

41
observable effect on blood pressure at the dose of Ang-(1-7) used in the current study,
there was a significant attenuation of cardiac fibrosis.
The mechanism of action of Ang-(1-7), an active peptide fragment of the renin
angiotensin system, is currently controversial. It has been reported to act as an
angio nd
eviewed
f hypertension. First, Ang-(1-7) could reduce blood pressure,
which ts would
ute
asts.
f various pro-
thelin-1, as
tensin receptor antagonist, an ACE inhibitor, as well as altering prostaglandins a
the bradykinin-nitric oxide system. These mechanisms have been exhaustively r
by Santos et al. (2000) and others. The mechanisms may involve the reported mas
receptor (Tallant et al., 2005) or direct interaction with other angiotensin receptors
(Santos et al., 2000).
There are several mechanisms by which Ang-(1-7) may decrease collagen
deposition in models o
would decrease hemodynamic-dependent pro-fibrotic signaling. Our resul
not support this mechanism, as changes in blood pressures and pressor responses to ac
Ang II were not observed by indirect or direct methods during chronic Ang-(1-7)
treatment (Figures 3-1 and 3-2). Second, Ang-(1-7) could directly inhibit the secretion of
collagen or the activation of the fibroblasts or their differentiation into myofibrobl
Third, Ang-(1-7) may increase collagen degradation by activating matrix
metalloproteinases (MMP’s), or by inhibiting tissue inhibitors of matrix
metalloproteinases (TIMP’s). Finally, Ang-(1-7) may inhibit the actions o
fibrotic factors such as norepinephrine, angiotensin II, TGFβ, and/or endo
Ang-(1-7) infusion has been reported to result in a reduction of cardiac Ang II levels
(Mendes et al., 2005). Future studies will be aimed at dissecting the possible

42
mechanisms by which Ang-(1-7) reduces cardiac fibrosis in hypertension and
determine if this anti-fibrotic action also occurs in non-hypertensive fibrotic cond
This study also supports the idea that angiotensin-(1-7) has direct physiological
to
itions.
action re
ptor
owth
collagen
protective
effect
s, rather than simply being the result of metabolized Ang II. Our in vivo results a
supported by a recent in vitro study (Iwata et al., 2005). In this study, the authors
examined angiotensin fragment binding in cultured adult rat cardiac fibroblasts, and
showed that Ang-(1-7) binds to two separate sites that are unaffected by the AT1 rece
antagonist valsartan and the AT2 receptor antagonist PD-123,319. Importantly, these
authors showed that within the concentration ranges studied for binding, Ang-(1-7)
inhibited collagen formation as measured by [3H]proline incorporation, and also
decreased mRNA expression of various growth factors including transforming gr
factor β1 (TGFβ1), endothelin-1 (ET-1), and leukemia inhibitory factor (LIF).
Pretreatment of the cardiac fibroblasts with Ang-(1-7) inhibited Ang II-induced
protein. Together, the current in vivo study and the in vitro study by Iwata et al. (2005)
strongy support the hypothesis that Ang-(1-7) actively regulates cardiac fibrosis. These
findings further indicate the significant role that Ang-(1-7) plays in the heart.
Our results parallel those of Loot et al. (2002), who demonstrated cardio
s of Ang-(1-7). In the current study, we utilized one fourth of the dose of Ang-(1-7)
that these authors employed. We did not determine the plasma or cardiac levels of Ang-
(1-7) in the current study. It is conceivable that utilization of higher concentrations of
Ang-(1-7) may result in greater cardioprotection, and it may also uncover effects on
hypertrophy.

43
The results of the current study fit well with the findings of our recent study, in
which systemic overexpression of ACE2 (the most important enzyme involved in
angiotensin-(1-7) production) in angiotensin-infusion hypertensive rats resulted in blood
pressure-independent decreases in cardiac fibrosis (Huentelman et al., 2005). This study
supports the concept that ACE2 mediates many (or most) of its protective effects through
the production of angiotensin-(1-7). Myocardial infarction increases ACE2 expression in
rats and humans (Burrell et al., 2005). Averill et al. (2003) also reported that Ang-(1-7)
immunoreactivity was significantly increased in the ventricular tissue surrounding an area
of myocardial infarction and absent in the remodeled area of infarction. Thus, there is
evidence suggesting that the ACE2 enzyme and the formation of Ang-(1-7) play
significant roles in modifying cardiac remodeling.
Recently, Keider et al. (2005) demonstrated that the mineralocorticoid aldosterone
significantly increased cardiac ACE and decreased ACE2 mRNA and activity levels in
both humans and mice, whereas aldosterone antagonists produced the opposite responses.
This data would suggest that ACE2 levels, and the subsequent generation of Ang-(1-7)
may be the mechanism by which mineralocorticoid receptor blockers improve cardiac
function in myocardial infarct patients (Pitt et al., 2003).
Several investigators (Abraham and Simon, 1994; Simon et al., 1995; Melargno
and Fink, 1996) have demonstrated that chronic infusion of angiotensin II results in the
development of hypertension, cardiac hypertrophy and fibrosis. Functional Ang II
receptors have been documented in cardiac fibroblasts (Crabbos et al., 1994) and Ang II-
stimulated expression and secretion of collagen is completely abolished by AT1 and not
AT2 receptor antagonism in vivo and in vitro (Lijnen and Petrov, 1999, Lijnen et al.,

44
2001). As Ang-(1-7) is significantly elevated with the use of AT1 antagonists (Ferrario et
al., 2005), it is possible that some of the anti-fibrotic effects attributed to AT1 antagonism
are mediated via actions of Ang-(1-7).
The development of cardiac fibrosis is a major complication of hypertensive
cardiac disease. The increased deposition of basement membrane collagen is a hallmark
of the remodeling process. This remodeling can predispose a patient to increased risk of
adverse cardiac events, and therefore any reduction or reversal of cardiac fibrosis would
facilitate the management of hypertensive heart disease. Our results suggest that Ang-(1-
7) may be a beneficial component of the RAAS that provides this cardioprotection during
hypertension, and any maneuver that would increase cardiac levels of ACE2 and
subsequently Ang-(1-7) may be used as an effective therapeutic intervention in
hypertensive heart disease patients.

CHAPTER 4 PROTECTIVE EFFECTS OF ANGIOTENSIN-(1-7) DURING ISOPROTERENOL-
INDUCED CARDIAC REMODELING
Abstract
β-Adrenergic stimulation of cardiac tissue leads to myocyte hypertrophy and can
result in heart failure. A recent study by Santos et al. showed that transgenic rats
overexpressing an angiotensin-(1-7) (Ang-(1-7)) fusion gene were significantly protected
from cardiac hypertrophy and arrhythmias induced by administration of the β-adrenergic
receptor agonist isoproterenol. Here, we first characterized the effects of isoproterenol in
male and female rats, then examined the hypothesis that chronic infusion of Ang-(1-7)
would prevent cardiac hypertrophy and fibrosis due to isoproterenol administration.
Intact male, intact female, and ovariectomized female Sprague-Dawley rats between 180-
280 grams were injected with isoproterenol at either 1 or 2 mg/kg daily for fourteen
consecutive days. A subset of the 2 mg/kg isoproterenol-treated male rats were
implanted with osmotic minipumps which chronically delivered Ang-(1-7) (100
ng/kg/min). Hearts were harvested and weighed, and the (2 mg/kg isoproterenol) male
rat hearts were sectioned and stained using Masson’s Trichrome. Isoproterenol injections
resulted in large increases in heart size in males at both doses, but had no effect on
cardiac fibrosis. While isoproterenol also induced large increases in heart size in intact
females, this effect was blunted in ovariectomized animals. Chronic infusion of Ang-(1-
7) in the 2 mg/kg male group resulted in significantly attenuated cardiac hypertrophy.
From these studies we conclude both that Ang-(1-7) inhibits the hypertrophic response to
45

46
isoproterenol, and that ovarian hormones are permissive to the hypertrophic effects of
isoproterenol. These findings lead us to the hypothesis that ovarian hormones may alter
cardiac tissue sensitivity to the protective Ang-(1-7).
Introduction
High blood pressure is a key risk factor for cardiac remodeling and subsequent
heart failure (Regitz-Zagrosek and Lehmkuhl, 2005; Thom et al., 2006). Others have
previously demonstrated that reductions in arterial pressure can prevent or even reverse
the cardiac remodeling associated with high blood pressure (Diez et al., 2001). To
determine whether the cardioprotective effects of Ang-(1-7) are mediated by decreased
arterial pressure (or limited to hypertensive models), the following study was carried out
using a normotensive model of cardiac remodeling.
Isoproterenol is a pharmacological catecholamine with an N-alkyl substitution,
making it almost entirely selective for β-adrenergic receptors (Jacob, 1996; Goodman and
Gilman, 2001). Administration of isoproterenol results in decreased peripheral resistance
through stimulation of β2 receptors, which leads to a decrease in diastolic pressure.
Although systolic pressure typically is normal or slightly increased, mean arterial
pressure is usually depressed. Stimulation of β1 receptors in the heart results in positive
inotropic and chronotropic effects, thus increasing cardiac output. Stimulation of cardiac
tissue by isoproterenol may lead to sinus tachycardia, palpitations, and more serious
arrhythmias, and large doses may cause myocardial necrosis (Goodman and Gilman,
2001). These properties of isoproterenol have led many investigators to use the
compound to induce cardiac remodeling without increasing blood pressure.
A recent study by Santos et al. (2004) found that increasing circulating Ang-(1-7)
levels protected rats from isoproterenol-induced hypertrophy and associated arrhythmias.

47
While the data are encouraging, the authors utilized transgenic rats that overexpressed an
Ang-(1-7) fusion protein, which confounds interpretation of the physiological data as the
role of Ang-(1-7) during development is currently unknown. Further, the authors did not
examine the effect of Ang-(1-7) on isoproterenol-induced cardiac fibrosis.
In the present study, daily injections of isoproterenol were used to induce cardiac
remodeling. Several groups of both male and female animals were tested at various
doses of isoproterenol to characterize this normotensive model of cardiac remodeling
before testing the cardioprotective effects of angiotensin-(1-7) in this model.
Methods
Animals
Group A – Males with isoproterenol at 1 mg/kg
Fourteen male Sprague-Dawley rats weighing 220-250 grams were divided into
two groups. The first group, with eight animals, was treated with daily injections of 0.9%
saline solution (1 mL/kg, s.c.). The second group, with six animals, was treated with
daily injections of isoproterenol (1 mg/kg, s.c.). Daily injections were performed
between 11:00 AM and 1:00 PM. Both groups were treated for fourteen consecutive
days, and on the fifteenth day all animals were sacrificed without a final injection.
Group B – Ovariectomized and intact females with isoproterenol at 1 and 2 mg/kg
Twenty-four female Sprage-Dawley rats weighing 180-210 grams were divided
into two groups of twelve animals each. The first group underwent ovariectomy surgery,
while the second group served as intact controls. Within each group, three subgroups
were formed, including a saline control-injection subgroup and two subgroups that
received daily isoproterenol injections at either 1 mg/kg or 2 mg/kg, s.c. Daily injections

48
were performed between 11:00 AM and 1:00 PM for fourteen days. On the fifteenth day,
animals were sacrificed without a final injection.
Group C – Males with isoproterenol at 2 mg/kg
Seven male Sprague-Dawley rats weighing 240-280 grams were divided into three
groups. Three animals served as saline-injection controls. Two animals received daily
injections of isoproterenol (2 mg/kg, s.c.). The final two animals were implanted with
subcutaneous osmotic minipumps (Alzet, model 2002), which chronically delivered
angiotensin-(1-7) at 100 ng/kg/min. This final group also received daily injections of
isoproterenol (2 mg/kg, s.c.). Again, daily injections were performed between 11:00 AM
and 1:00 PM for fourteen days. On the fifteenth day, animals were sacrificed without a
final injection.
Indirect Blood Pressure
Indirect blood pressures were recorded from animals in group C one day before
pump implantation and isoproterenol injections, and weekly during the isoproterenol
treatment using an indirect tail-cuff method as described in previous experiments.
Briefly, animals were heated using a 200-watt heat lamp for 3-5 minutes before being
restrained in a heated Plexiglas restrainer to which the animals had been conditioned
prior to the experiment. A pneumatic pulse sensor was then attached to the tail distal to a
pressure cuff under the control of a Programmed Electro-Sphygmomanometer (Narco).
Voltage outputs from the pressure sensor bulb and inflation cuff were recorded and
analyzed electronically using a PowerLab signal transduction unit and associated Chart
software (ADInstruments). At least three separate indirect pressures were averaged for
each animal.

49
Cardiac Remodeling
Following fourteen days of isoproterenol injections with or without chronic
infusion of angiotensin-(1-7), animals were sacrificed by overdose of halothane inhalant.
Hearts were removed by blunt dissection, weighed, and sectioned. Again, as in previous
experiments, cross-sections at the level of the papillary muscles were then fixed in 10%
formalin solution, embedded in paraffin, sectioned at 4 microns, and stained using
Masson’s Trichrome. Single sections from each animal were then viewed and
photographed with a Moticam 1000 digital camera (Motic, Richmond, British Columbia,
Canada) with a 10X magnification. The collagen content of the left ventricle wall was
determined by measuring the blue stain density of the left wall (from the septum in the
posterior to the septum in the anterior) and was normalized to the red stain density of the
same portion of the image. The area of blue-stained collagen surrounding the left
anterior descending coronary artery was measured and normalized to the area of the
lumen of the vessel. Color density and perivascular collagen and lumen areas were
determined using the ImageJ program from NIH (Rasband, 1997).
Results
Cardiac Hypertrophy
Male rats injected daily with 1 mg/kg isoproterenol (Group A) displayed marked
increases in cardiac hypertrophy (Figure 4-1). The increase in size was approximately
28%, which is much larger than that observed with the Ang II (5%) or DOCA-salt (14%)
models of hypertension-induced cardiac remodeling.

50
Figure 4-1. Isoproterenol (1 mg/kg/day) induces cardiac hypertrophy in male rats. Fourteen days of isoproterenol treatment resulted in a significantly increased heart size (P<0.0001). Data presented as mean ± SE.
Figure 4-2. Ovariectomy potentiates the isoproterenol-mediated induction of cardiac hypertrophy in female rats. Daily injections of isoproterenol resulted in differential effects on cardiac hypertrophy, depending on ovarian hormone status. Two-way ANOVA reveals a significant effect of isoproterenol treatment level (P<0.0001), but ovariectomy treatment only resulted in a non-signficant trend (P=0.0765). The presence of a significant interaction (P=0.0308), however, indicates some impact of ovary status. Data presented as mean ± SE.

51
Female rats injected daily with isoproterenol (Group B) showed differential
hypertrophic responses to isoproterenol injection based on ovarian hormone status
(Figure 4-2). Isoproterenol significantly increased heart size in both surgery groups
(P<0.0001). Ovariectomy appeared to attenuate the response to isoproterenol, and
although two-way analysis of variance reveals only a non-significant trend with regard to
the effect of surgery (P=0.0765), an significant interaction between surgery and
isoproterenol dose was uncovered (P=0.0308).
Figure 4-3. Chronic Ang-(1-7) prevents the hypertrophic, but not fibrotic effects of isoproterenol (2 mg/kg/day) in male rats. (A) Isoproterenol resulted in significantly increased cardiac hypertrophy (P=0.0007), and chronic infusion of angiotensin-(1-7) at 100 ng/kg/min significantly attenuated this effect (P=0.0082). (B) Isoproterenol and angiotensin-(1-7) had no effect on cardiac fibrosis. Data presented as mean ± SE.
Male rats injected daily with 2 mg/kg isoproterenol (Group C) exhibited
tremendous cardiac hypertrophy. As this combination of gender and drug dose resulted
in the greatest effect upon cardiac hypertrophy, the effect of angiotensin-(1-7) was
evaluated with this combination. Chronic infusion of angiotensin-(1-7) significantly

52
attenuated the hypertrophic response to isoproterenol (Figure 4-3A). These hearts were
then processed to determine the effect of isoproterenol and angiotensin-(1-7) on cardiac
fibrosis. Isoproterenol and angiotensin-(1-7) had no significant effects on cardiac fibrosis
(Figure 4-3B).
Blood Pressure
Figure 4-4. Isoproterenol and Ang-(1-7) had no chronic effects on blood pressure in male rats. (P=0.4207 and P=0.6615 vs. saline, respectively) Data presented as mean ± SE.
Indirect blood pressure was also determined in the male rats with 2 mg/kg
isoproterenol, as this combination of gender and dose resulted in the greatest changes in
cardiac hypertrophy. Isoproterenol had no significant effect on blood pressure, and
neither did chronic infusion of angiotensin-(1-7) (Figure 4-4).
Discussion
Daily injections of isoproterenol resulted in cardiac hypertrophy in both male and
female rats. It was determined that ovariectomy significantly attenuated the effects of

53
isoproterenol in females, and that males were slightly more sensitive to isoproterenol-
induced cardiac remodeling than intact females at the 2 mg/kg dose. Angiotensin-(1-7)
infusion significantly attenuated the hypertrophic response in these high-dose
isoproterenol male rats. Cardiac fibrosis and blood pressure were unaffected by the 2
mg/kg dose of isoproterenol in male rats. Further, angiotensin-(1-7) had no effect on
either blood pressure or cardiac fibrosis.
While the lack of effect of angiotensin-(1-7) on blood pressure is unsurprising,
given the results of both the Ang II and DOCA-salt models (Chapters 2 and 3,
respectively), the trend toward increased fibrosis in this model (Figure 4-3B) was
unanticipated. These findings appear to conflict with the results in both the Ang II and
DOCA-salt models, as Ang-(1-7) completely prevented interstitial fibrosis in both of
these models, but also give clues as to the site of Ang-(1-7) interference with pro-fibrotic
signaling. Neves et al. (2005) recently determined that mineralocorticoid receptor
activation mediates the pro-fibrotic actions of Ang II. Bos et al. (2005) found that
isoproterenol-induced fibrosis could be inhibited by antagonism of mineralocorticoid
receptors. Together, these findings suggest that the anti-fibrotic effects of Ang-(1-7) may
work though interference with the pro-fibrotic signaling pathways of Ang II and DOCA,
upstream of the convergence with isoproterenol signals.
The effect of Ang-(1-7) upon ventricular hypertrophy was substantial, which is
again in direct opposition to the effects observed in the Ang II and DOCA-salt models
(Chapters 2 and 3, respectively). Others have determined that antagonism of the
mineralocorticoid receptor (Bos et al., 2005) or AT1 receptors (Nakano et al., 1995) in
vivo can prevent isoproterenol-induced cardiac effects, thus clearly demonstrating cross-

54
talk among these signaling cascades. Ang-(1-7) likely inhibits the hypertrophy process
between the convergence of signals from Ang II or isoproterenol with signals from
mineralocorticoids.
The difference in inhibitory roles for Ang-(1-7) with regard to hypertrophy and
fibrosis in the Ang II, DOCA-salt, and isoproterenol models likely is related to the
differential effects of Ang-(1-7) in the various cardiac cell types. Tallant et al. (2005)
showed that Ang-(1-7) acts in vitro at the cardiac myocyte to modulate hypertrophy, and
Iwata et al. (2005) also showed that Ang-(1-7) acts in vitro at the cardiac fibroblast to
elicit changes in collagen turnover. Possible differences in intracellular signaling
cascades in each cell type, or differences in their in vitro models versus our in vivo
models, may account for the differential effects of Ang-(1-7) on hypertrophy and fibrosis
with mineralocorticoid or β-adrenergic stimulation. Further studies in vitro will help
elucidate the intracellular signaling cascade(s) of Ang-(1-7) in myocytes and fibrosis,
which should uncover the biochemical cause of the different outcomes.
Differences in cardiac responsiveness by gender have previously been noted, which
prompted the inclusion of intact and ovariectomized female rats in the present study.
Schwertz et al., (1999) showed that isolated female rat hearts have smaller inotropic
responses to isoproterenol administration than male hearts. In the present study, only
small differences in cardiac hypertrophy responses between males and females were
observed with 1 and 2 mg/kg isoproterenol, but ovariectomized females exhibited blunted
responses to isoproterenol. This may indicate a role for ovarian hormones in regulating
cardiac structure.

55
Previous work by Tallant et al. (2005) uncovered that the anti-hypertrophic actions
of Ang-(1-7) are mediated at the cardiac myocyte through the Mas receptor. Preliminary
in silico analysis of the Mas receptor promoter indicates that there are several possible
estrogen receptor and AP-1 binding sites within the first few kilobases 5’ of the
transcription start site, thus suggesting that estrogens may alter expression of this receptor
(data not shown). These findings, along with our present study of cardiac hypertrophy
responses in ovariectomized versus intact animals, lead us to the hypothesis that ovarian
hormones may decrease tissue sensitivity to the cardioprotective angiotensin-(1-7)
hormone.
It has been documented that the Ang-(1-7) receptor (Mas) is found in the
vasculature, and stimulation of this receptor results in a relaxation response (le Tran and
Forster, 1997). Therefore, in the subsequent study we decided to evaluate estrogenic
modulation of vascular reactivity to Ang-(1-7) in rat aorta.

CHAPTER 5 ALTERATIONS IN AORTIC VASCULAR REACTIVITY TO ANGIOTENSIN-(1-7)
IN 17-β-ESTRADIOL-TREATED FEMALE SD RATS
Note: This chapter has already been published in Regulatory Peptides. Grobe, JL and Katovich MJ. Alterations in aortic vascular reactivity to angiotensin-(1-7) in 17-β-estradiol-treated female SD rats. Reg Pept 133:62-67, 2006.
Abstract
Estrogen's suggested cardio-protective effects have come into question following
the results of recent clinical trials. Two major components of the renin–angiotensin
system (RAS) that are modulated by estrogen are angiotensin converting enzyme, and the
angiotensin II type 1 receptor. Further research has revealed several new components of
the RAS, including angiotensin converting enzyme 2, its peptide product angiotensin-(1-
7) (Ang-(1-7)), and that peptide's receptor, Mas. These components appear to oppose the
classical effects of the RAS, and may act to buffer the RAS in vivo. Recent work has
shown that during pregnancy, when estradiol levels are elevated, renal and urinary Ang-
(1-7) are greatly increased. This study examined the effects of estradiol on the efficacy of
Ang-(1-7) in the rat aorta. Female Sprague–Dawley rats were ovariectomized and a
subgroup was chronically treated with subcutaneous pellets of estradiol for 3 weeks.
Thoracic aortas were harvested for assessment of in vitro vascular reactivity to Ang-(1-
7). The results demonstrated that increased estradiol exposure attenuated the relaxation
response to Ang-(1-7) in a dose-dependent manner. These findings are in contrast to
recent work showing potentiated responses to Ang-(1-7) in mesenteric arteries from
56

57
estrogen-manipulated rats, and may suggest a regional specificity in estradiol-mediated
changes in the RAS.
Introduction
The leading cause of death in women is cardiovascular disease (CVD). Blood
pressure (BP) is higher in men than in premenopausal women at similar ages, but
following menopause, BP increases in women to levels higher than men (Tremollieres et
al., 1999; Harrison-Bernard and Raij, 2000). In the United States, nearly 38% of
postmenopausal women use some form of hormone replacement therapy (HRT) (Keating
et al., 1999). For many years, it has been thought that HRT provided women some level
of protection against CVD, specifically with regard to myocardial infarction (Grodstein
and Stampfer, 1995; Grodstein and Stampfer, 1998; Mendelsohn and Karas, 1999;
Arkhrass et al., 2003).
Recent clinical trials, including the Women's Health Initiative (WHI) and the Heart
and Estrogen–progestin Replacement Study (HERS), have cast some doubt on the
protective effects of estrogens in cardiovascular biology, as these studies failed to show a
decreased risk for stroke in postmenopausal women treated with HRT (Simon et al.,
2001). The WHI was stopped 2 years early due to an increase in risk of heart disease,
strokes, and blood clots (Hulley et al., 2002; Rossouw et al., 2002). Questions remain,
then, as to what specific harmful and/or protective effects estrogens have on the
cardiovascular system.
One endogenous hormonal system that is very important in cardiovascular
regulation and is altered by estrogen is the renin–angiotensin system. Specifically,
estrogen has been shown to stimulate the production of angiotensinogen (Healy et al.,
1992) and the angiotensin II type 2 receptor (AT2R) (Armando et al., 2002), and to

58
inhibit the production of prorenin (Szilagyi et al., 1995), angiotensin converting enzyme
(ACE) (Turner and Hooper, 2002), neprilysin (Pinto et al., 1999), aminopeptidase N (Seli
et al., 2001), and the angiotensin II type 1 receptor (AT1R) (Nickenig et al., 2000). These
changes in the RAS have the net effect of attenuating the pressor effects of the RAS, but
potentiating the depressor effects. Collectively, these effects of estrogen on components
of the renin–angiotensin system should reduce blood pressure and offer some
cardiovascular protective effects.
Angiotensin-(1-7) (Ang-(1-7)) is an endogenous product of the renin–angiotensin
system. Production of this peptide hormone is achieved by three distinct pathways,
including breakdown of angiotensin II (Ang II) by angiotensin converting enzyme 2
(ACE2) (Donoghue et al., 2000; Tipnis et al., 2000), and breakdown of angiotensin I
(Ang I) both directly by various endopeptidases and also through the intermediate
formation of angiotensin-(1-9) by ACE2 followed by digestion by ACE (Ferrario and
Chappell, 2004). Ang-(1-7) is then degraded by ACE to form an inactive product,
angiotensin-(1-5) (Ferrario and Chappell, 2004).
At least three possible physiological actions of Ang-(1-7) have been suggested.
First, it is possible that Ang-(1-7) acts as a partial agonist or antagonist of the AT1 and/or
AT2 receptors. This is supported by evidence that Ang-(1-7) can block the effects of Ang
II (Brosnihan et al., 1998; Chaves et al., 2000; Machado et al., 2001; Pinheiro et al.,
2004), and that in some tissues Ang-(1-7) can signal through the AT1R (Gironacci et al.,
1998; Caruso-Neves et al., 2000) and the AT2R (Santos et al., 2000; De Souza et al.,
2004; Gironacci et al., 2004; Roks et al., 2004). Second, it is possible that Ang-(1-7) acts
through its own receptor. The Mas receptor has been suggested as one possible Ang-(1-7)

59
receptor, and various experiments have shown changes in Ang-(1-7) signaling in Mas-
altered tissues and animals (Santos et al., 2003). Third, it has been suggested that Ang-(1-
7) may act as an endogenous ACE inhibitor. This is supported by experiments which
examined the catalytic efficiency of ACE in the presence of Ang-(1-7) (Iyer et al., 1998;
Collister and Hendel, 2003; Tom et al., 2003).
Recent work by Broshinhan and colleagues (Brosnihan et al., 2003; Neves et al.,
2003) has shown that both the production and the efficacy of Ang-(1-7) are altered in
pregnancy. Specifically, during pregnancy, plasma and urinary Ang-(1-7) increase
significantly, and mesenteric artery preparations are much more reactive to Ang-(1-7).
Because circulating estrogen increases during pregnancy, it is conceivable that estrogens
may control production and/or sensitivity to Ang-(1-7).
The current study was undertaken to further examine the effects of estrogen on
vascular reactivity to Ang-(1-7). Aortic rings were used to examine the Ang-(1-7) effects
upon the rat vasculature, as it has been shown that rat aorta relax to Ang-(1-7) in an
endothelium-dependent manner (Le Tran and Forster, 1997). As estrogen significantly
increases in pregnancy, it was hypothesized that estrogen would cause a significant
potentiation of vascular responses to Ang-(1-7).
Methods
Animal Housing and Surgery
Twelve female Sprague–Dawley rats (150–200 g) were housed doubly in shoebox
style cages with free access to standard rat chow and tap water, and were maintained on a
12 : 12 h light : dark cycle, beginning at 7:00 AM. Twenty-one days before the aortic
vascular reactivity experiment, all animals were ovariectomized while under anesthesia
by xylazine/acepromazine/ketamine injectable cocktail. Subsets of animals (four each

60
group) received estradiol replacement via subcutaneous pellet (0.05 or 25 mg). All
procedures involving animals were approved by the University of Florida Institutional
Animal Care and Use Committee.
Reagents
Subcutaneous estradiol pellets were obtained from IRA (Innovative Research of
America, Sarasota, FL). All chemicals and peptides for aortic vascular reactivity
experiments were obtained from Sigma Aldrich (St. Louis, MO). Estradiol ELISA kit
was obtained from ALPCO, Inc. (Windham, NH).
Aortic Vascular Reactivity Assay
Rats were decapitated and aortas were isolated using blunt dissection. The thoracic
aorta was cleaned of all adherent tissue and two adjacent 3.0 mm ring sections were
obtained from each animal. These rings were then placed onto pressure transducer arms
and submerged in tissue baths containing a modified Krebs solution (118 mM NaCl, 4.7
mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 25.0 mM NaHCO3, 11.1 mM Dextrose, and
2.5 mM CaCl2) with 95% O2 / 5% CO2 compressed gas mixture bubbling through.
Pressure transducers were connected to a PowerLab signal transduction unit
(ADInstruments), and data was continuously recorded on a PC computer, and further
analyzed using the Chart program (ADInstruments). Rings were pretensioned to 4 g over
1 h, as preliminary studies determined this to provide the optimal length–tension ratio
(data not shown). A single dose of potassium chloride was applied (60 mM final
concentration) to determine smooth muscle viability. After maximal contraction was
reached, the tissue baths were purged and fresh modified Krebs solution was applied.
Following relaxation back to the 4 g baseline, phenylephrine (10− 7 M final concentration)
was applied. After maximal contraction was reached, graded doses of Ang-(1-7) were

61
applied (10− 10 M through 10− 6 M final concentrations), separated by 3 min intervals.
Baths were then purged, and the contractile response to angiotensin II (10− 7 M) was
assessed. Finally, relaxation responses to acetylcholine and sodium nitroprusside were
determined in a similar manner to those of Ang-(1-7). Rings were then removed from the
tissue chambers and allowed to dry overnight in a dessicating oven. Dry ring masses were
then measured, and contractile responses to each drug were normalized to the mass of the
dried ring. Relaxation responses were determined by dividing maximal relaxation
responses (tension change from maximal contraction to phenylephrine) by total active
tension (total tension minus 4 g baseline).
Plasma Steroid Measurement
Trunk blood was collected from the animals at decapitation into EDTA-coated
collection tubes and centrifuged for 5 min at 3000 ×g in a Triac centrifuge (Becton,
Dickinson). Plasma was removed and placed into cryogenic vials for storage at −20 C
until assayed. To determine plasma estradiol levels, an ELISA kit (ALPCO, sensitivity of
4.6 pg/mL) was utilized following the manufacturer's instructions, in duplicate.
Statistics
Body, uterine and heart masses, plasma estradiol, ring masses and contractile
responses to potassium chloride and angiotensin II were analyzed using one-way
ANOVA with significance set at P<0.05. Relaxation responses to angiotensin-(1-7) were
analyzed using repeated measures ANOVA with significance set at P<0.05. All post-hoc
tests were performed using Fisher's method with significance set at P<0.05.

62
Results
Estradiol Levels and Effects on Body, Heart and Uterine Growth
Estrogen caused significant dose-related reductions in overall body mass at all
doses, but no significant effects were observed on heart size with any level of estrogen
replacement. Uterine mass was significantly increased in a dose-dependent manner with
estrogen replacement. Plasma estradiol was significantly (P < 0.05) increased over
ovariectomized levels in the 25 mg pellet group (Ovex, 8.90 ± 0.34; 0.05 mg pellet,
16.28 ± 3.99; 25 mg pellet, 95818.26 ± 3515.76 pg/mL).
Body Mass (g) Heart (g/kg) Uterus (g/kg) Ovariectomized 274.4±8.1 3.24±0.14 0.61±0.11 Estrogen, 0.05 mg 198.4±3.8 * 3.67±0.24 2.67±0.24 * Estrogen, 25 mg 171.2±5.5 * 3.25±0.15 5.36±0.80 *
Table 5-1. Effects of steroid treatments on body parameters in female Sprague-Dawley
rats. Estrogen exposure caused significant changes in body size and uterine mass at all doses. No significant changes were observed in heart size with any treatment. Data presented as means ± SE. (* indicates P<0.05 versus ovariectomized).
Aortic Ring Properties
60 mM KCl (g) Phenylephrine (g) Ring mass (g) Ovariectomized 3.31±0.09 1.96±0.50 1.35±0.02 Estrogen, 0.05 mg 3.17±0.16 1.68±0.17 1.41±0.11 Estrogen, 25 mg 2.83±0.48 3.13±0.43 1.27±0.07
Table 5-2. Aortic ring properties. Aortas were sectioned into 3 mm length rings for
vascular reactivity assay. Control contractions by potassium chloride (KCl) and phenylephrine were performed to determine treatment effects on contractile responses. Following the assay, rings were dried in a dessicator and then weighed to determine effects of treatment on protein content. Data presented as means ± SE.
Vascular smooth muscle contractions to 60 mM potassium chloride and
phenylephrine were similar across treatments, as were ring masses (Table 5-2). Estradiol
treatment, however, resulted in significant alterations of the vascular smooth muscle
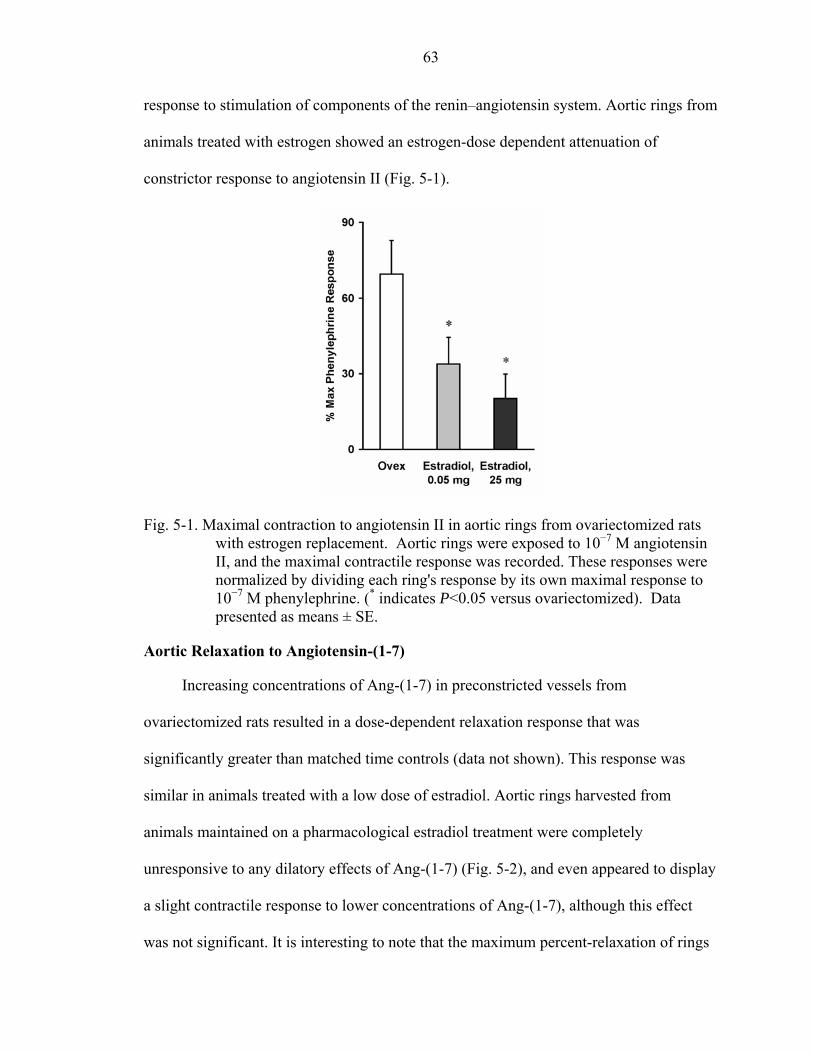
63
response to stimulation of components of the renin–angiotensin system. Aortic rings from
animals treated with estrogen showed an estrogen-dose dependent attenuation of
constrictor response to angiotensin II (Fig. 5-1).
Fig. 5-1. Maximal contraction to angiotensin II in aortic rings from ovariectomized rats with estrogen replacement. Aortic rings were exposed to 10−7 M angiotensin II, and the maximal contractile response was recorded. These responses were normalized by dividing each ring's response by its own maximal response to 10−7 M phenylephrine. (* indicates P<0.05 versus ovariectomized). Data presented as means ± SE.
Aortic Relaxation to Angiotensin-(1-7)
Increasing concentrations of Ang-(1-7) in preconstricted vessels from
ovariectomized rats resulted in a dose-dependent relaxation response that was
significantly greater than matched time controls (data not shown). This response was
similar in animals treated with a low dose of estradiol. Aortic rings harvested from
animals maintained on a pharmacological estradiol treatment were completely
unresponsive to any dilatory effects of Ang-(1-7) (Fig. 5-2), and even appeared to display
a slight contractile response to lower concentrations of Ang-(1-7), although this effect
was not significant. It is interesting to note that the maximum percent-relaxation of rings

64
from ovariectomized animals at each dose is similar in magnitude to those of the
mesenteric arteries from pregnant animals in the work from Neves et al. (2003).
Fig. 5-2. Estradiol attenuates the aortic relaxation response to acute Ang-(1-7). Increasing doses of estrogen caused dose-dependent attenuations of vascular reactivity to angiotensin-(1-7). Repeated measures ANOVA reveals a significant effect of steroid treatment. Post-hoc analyses reveal no differences between ovariectomized and the 0.05 mg estradiol treatment, but 25 mg estradiol replacement caused a significant attenuation of the relaxation response when compared to either ovariectomized or 0.05 mg estradiol treatments. Data presented as means ± SE.
In a subsequent study, the dependence of aortic relaxation responses to Ang-(1-7)
upon the Mas receptor was determined. At doses of 10−6 M and lower, aortic relaxation
responses in endothelium-intact rings were completely abolished by 10−5 M D-Ala7-
Angiotensin-(1-7) (A779), an antagonist to the Mas receptor. At 10−5 M, however, Ang-
(1-7) relaxation responses were minimally effected by this receptor antagonist (data not

65
shown). These observations have been reported previously for rat aortic rings (Le Tran
and Forster, 1997).
Aortic Ring Integrity
Endothelial integrity was examined by exposing rings pre-constricted with 10−7 M
phenylephrine to 10−4 M acetylcholine. Rings from all animals relaxed more than 60%
(with the exception of one animal, which was dropped from all analyses), and no
statistical difference exists between groups (P=0.5219). Smooth muscle integrity and
responsiveness to nitric oxide was also tested by exposing rings pre-constricted with 10−7
M phenylephrine to 10−5 M sodium nitroprusside. All rings relaxed between 80% and
100%, with no differences between groups (P=0.8397).
Discussion
This study provides some significant insight into the effects of estradiol on
cardiovascular physiology. Although it was determined that estrogen does appear to have
effects on the vasodilatory responses of the aorta to Ang-(1-7), the directions of responses
were unexpected. Estrogen was, in fact, detrimental to the vasodilatory responses to Ang-
(1-7).
Estrogen caused the expected dose-dependent attenuation of body weight, and a
potentiation of uterine mass (Table 5-1). These effects are well documented in the
literature (Harris et al., 2002), as is the dose-dependent attenuation of aortic ring
constrictor responses to Ang II (Cheng and Gruetter, 1992). These effects of estrogen on
the response to Ang II have been attributed to decreased transcription and activity of the
AT1R (Schunkert et al., 1997; Nickenig et al., 1998; Fisher et al., 2002). Other responses
to Ang II, such as the dipsogenic or in vivo pressor response are also attenuated with

66
elevated levels of estrogen (Jonklaas and Buggy, 1984; Fregly, et al, 1985). It is possible
that Ang-(1-7) works through interactions with the AT1 and/or AT2 receptors (Jaiswal et
al., 1992; Gironacci et al., 1998; Caruso-Neves et al., 2000; Santos et al., 2000; Roks et
al., 2004), and that altered levels of these receptors may account for the changes in
responsiveness to Ang-(1-7) in these aortic rings. Actions of Ang-(1-7) through the AT1
receptor seem less likely, in that the 0.05 mg estradiol pellet group displayed a
significantly decreased constrictor response to Ang II (Figure 5-1), but its relaxation
response to Ang-(1-7) was not different from ovariectomized (Figure 5-2).
Chronic estrogen treatment did not appear to alter the in vitro vascular contractile
response to a depolarizing agent (potassium chloride), or a receptor-mediated
vasoconstrictor (phenylephrine). The lack of effect on vascular ring mass also suggests
no significant effect of steroid treatment on aortic vessel musculature. Likewise, the
similar relaxation responses of the vessels to sodium nitroprusside demonstrate that the
vascular smooth muscle response to nitric oxide was not altered by estrogen treatment.
Maximal responses to acetylcholine suggest that endothelial function was not
compromised with estrogen treatment. Utilization of lower doses of acetylcholine may
have been able to distinguish if the sensitivity to acetylcholine is altered by estrogen
treatment. Other literature reports would suggest that this may be the cause, as estradiol
increases endothelial and calcium-dependent nitric oxide synthase (Saito, et al, 1999;
Morschl et al., 2000). Estradiol, however, does appear to alter the vascular response to
components of the renin–angiotensin system.
Vasodilatory responses to Ang-(1-7) were hypothesized to increase with estrogen
exposure, as the circulating level of estradiol significantly increases along with Ang-(1-7)

67
levels and sensitivity during pregnancy (Neves et al., 2003). The results of the current
study suggest that in the aorta, increased estrogen appears to have a dose-dependent
inhibitory effect on the vasodilatory response to Ang-(1-7) (Figure 5-2). These results,
obtained from aortic rings, are in contrast to the recent findings of Neves et al. (2004),
who found that estrogen mediates increased vasodilatory responses to angiotensin-(1-7)
in mesenteric artery preparations of female rats. These differential effects on the aorta
and the mesentery may suggest a regional specificity of the effects of estrogen, which has
been suggested for other estrogen-mediated physiological effects (Zubair et al., 2005).
Further, methodological differences between this study and those of Brosnihan et al. may
contribute to the conflicting results. Brosnihan's group utilizes the peptide endothelin-1 to
preconstrict their vessels before relaxation with Ang-(1-7), whereas we have used
phenylephrine. Additionally, their measurements of vascular reactivity in mesenteric
arteries are performed using a pressurized myograph system, whereas our measurements
are made using an isometric wire myograph system. Finally, our experiments utilized a
pharmacological dose of estradiol, whereas their group has used more physiological
doses.
Differential effects between vascular beds may contribute to the ongoing debate
regarding the cardio-protective effects of estrogen. Further studies in this area, examining
the different mechanisms of action of estrogen in the aorta versus the mesenteric arteries
may help to discover a mesentery-selective estrogen analog that would be able to produce
the cardio-protective effects of estrogen, but not cause the detrimental aorta-specific
effects.

68
One potential shortfall to this design was the absence of an intact control. This
particular group was not utilized as we anticipated that the normal estrous cycle could
impact the results and confound data interpretation. An intact control could be used in
future studies, provided the stage of estrous cycle was determined prior to harvesting the
vascular tissue. Another confounding effect with the estrogen treatment paradigm
incorporated was that a normally cycling estrogen was replaced with a chronic exposure.
This chronic exposure is of interest, despite its difference from normal physiology, as it
more closely approximates the estrogen dosing paradigm of women on monthly estrogen
pills. Further, this chronic exposure mimics the chronically increased levels of estradiol
observed during pregnancy. The doses of estradiol utilized were recommended by IRA to
mimic levels achieved during pregnancy. At the completion of the physiological
measurements, the results of the estradiol ELISA assay revealed much higher levels than
expected, and future studies will evaluate lower levels of estradiol that more closely
mimic those observed during normal pregnancy in the rat. The fact that such high
estrogen levels were achieved in the current study may suggest that nonspecific effects of
estrogen are mediating the observed attenuation of Ang-(1-7) responses, however the
responses observed to angiotensin II are consistent with the literature (Cheng and
Gruetter, 1992).
In a preliminary study with human coronary artery endothelial cells, we observed
that estrogen treatment significantly reduced the transcription of the Mas receptor mRNA
when examined using realtime RT-PCR. This observation would further support our
current demonstration that estrogen decreased relaxational responses to Ang-(1-7) in rat
aorta. These observations were made in cells at a relatively late passage number, and this

69
preliminary study will have to be repeated at early passages before a definitive effect of
estrogen on the Mas receptor could be validated.
In conclusion, we have found that angiotensin-(1-7) causes a dose-dependent
relaxation of the female rat aorta, and that this relaxation response is attenuated by 17-β-
estradiol. This aortic relaxation has previously been shown to be endothelium- and Mas
receptor-dependent (Le Tran and Forster, 1997). These results are in contrast to findings
from other studies on the effects of estradiol on the relaxation response to angiotensin-(1-
7), although as these other studies have been performed in mesenteric arteries, a regional
specificity of action of estradiol is suggested. Future studies will examine this regional
(vessel) specificity, the dose–response relationship of estradiol with the Mas receptor,
and the influence of 17-β-estradiol and other estrogenic compounds on Mas receptor
expression.

CHAPTER 6 PRELIMINARY CELL CULTURE EXPERIMENTS
Abstract
Angiotensin-(1-7) (Ang-(1-7)) has been previously demonstrated to provide
protection from cardiac remodeling in vivo. Here, the role of Ang-(1-7) in mediating the
production of collagen by cultured neonatal rat cardiac fibroblasts following stimulation
by both myocyte preconditioned media and by hypoxia-reperfusion injury was examined.
It was determined that Ang-(1-7) inhibits myocyte-derived pro-fibrotic stimuli, possibly
including transforming growth factor-β (TGFβ). Further, it was determined that lentiviral
delivery of the angiotensin converting enzyme 2 gene to fibroblasts prevented hypoxia-
induced collagen and TGFβ production. Together, these results indicate protective roles
for ACE2 and Ang-(1-7) in vitro, and may suggest future therapeutic avenues for
myocardial injury.
Introduction
The preceding chapters have explored the beneficial effects of angiotensin-(1-7)
(Ang-(1-7)) in vivo. While these studies provide an integrated-systems verification of the
cardioprotective role of Ang-(1-7), they fail to examine the tissue-level mechanism(s) of
these effects.
The two major components of cardiac remodeling, myocyte hypertrophy and
cardiac fibrosis, are primarily mediated by (and between) cardiac myocytes and
fibroblasts. When these cells are cultured independently, cardiac myocyte hypertrophy
can be triggered by treatment with media formerly on cardiac fibroblasts (Iwata et al.,
70

71
2005), and collagen production by cardiac fibroblasts can be potentiated by co-culture
with cardiac myocytes (Sarkar et al., 2004). Together, these findings indicate that cross-
talk between cardiac cell types is an important factor in the regulation of normal tissue-
level regulation.
Iwata et al. (2005) recently demonstrated that cultured adult rat cardiac fibroblasts
show a blunted collagen-production response to various stimuli when the culture media is
supplemented with Ang-(1-7). Tallant et al. (2005) also recently demonstrated that Ang-
(1-7) inhibits cardiac myocyte growth in vitro through the Mas receptor. These
investigators, though, did not examine the effect of Ang-(1-7) on myocyte-fibroblast
cross-talk demonstrated by Sarkar et al. (2004). Therefore, the effects of Ang-(1-7) on
myocyte-mediated stimulation of collagen production by cardiac fibroblasts were
examined.
In addition to myocyte-derived pro-fibrotic stimuli, certain pathologies can lead to
aberrant activity of cardiac fibroblasts. One pathophysiological state which leads to
excessive collagen production by cardiac fibroblasts is hypoxia, which can result from
acute or chronic myocardial ischemia (Agocha et al., 1997).
Averill et al. (2003) recently determined that Ang-(1-7) is conspicuously absent in
the infarct zone of surgically-induced myocardial ischemia in Lewis rats, although intact
myocardium surrounding the infarct zone stains with normal levels of the peptide.
Preliminary studies from our lab have also determined that cardiac myocytes have a
moderate basal activity of the angiotensin converting enzyme 2 (ACE2) enzyme, which is
the major source of Ang-(1-7) in the heart (Zisman et al., 2003). Cultured cardiac
fibroblasts, on the other hand, do not have detectable ACE2 activity. Together, these

72
findings next led us to the hypothesis that the absence of ACE2 in an infarct zone is
permissive for aberrant extracellular matrix production by fibroblasts that are able to
survive in the hypoxic local environment.
Methods
Isolation and Culturing of Neonatal Rat Cardiac Myocytes
Neonatal rat cardiac myocytes were isolated using a commercially available kit
(Worthington Biochemical Corp., Lakewood, NJ). Briefly, hearts from 5-day old rats
were removed by blunt dissection, then digested with trypsin and collagenase. Myocytes
were grown in DMEM/F-12 media supplemented with 1% penicillin/streptomycin and
10% fetal bovine serum (FBS) in a 5% CO2 / 95% air humidified incubator. Cells were
used for experiments within one week.
Isolation and Culturing of Neonatal Rat Cardiac Fibroblasts
Neonatal rat cardiac fibroblasts were isolated using a protocol adapted from Zhang
et al. (2001). Five day old Sprague-Dawley rat pups were deeply anesthetized by
inhalation of halothane. Following heart excision and mincing, heart tissue was digested
for 2 hours with 1% collagenase (Worthington Biochemical Corp, Lakewood, NJ), spun
down at 3000 rpm for 2 min, then digested for 1 hour with 1% trypsin (Worthington
Biochemical Corp, Lakewood, NJ). Cells were then spun down for 2 min at 3000 rpm,
and reconstituted in media and plated in a T-75 flask. Cells were maintained in DMEM
supplemented with 1% penicillin/streptomycin, 50 µg/mL ascorbic acid, and 10% fetal
bovine serum. Cells were split by standard trypsin procedure, and were used within the
first three passages. Cells were seeded at 3000 cells/cm2.

73
Myocyte-Fibroblast Cross-Talk Paradigm
Cultured cardiac myocytes were grown for three to five days before serum-
starvation for 24 hours. Following this starvation, cells were treated with 10% FBS-
supplemented media with or without angiotensin-(1-7) at 10-7 M for 24 hours. At the end
of this incubation, medias were transferred to confluent cultured fibroblasts, and some
(previously 10% FBS-only) media was supplemented with angiotensin-(1-7) at this time,
as indicated in Figure 6-1A. Collagen was sampled after 12 hours of incubation on the
cardiac fibroblasts.
An additional set of cardiac myocytes was treated in a parallel fashion, but
following the 24 hour incubation with or without angiotensin-(1-7), RNA from these cells
was isolated using a commercially available kit from Ambion. These RNA samples were
then used to measure transcript for transforming growth factor-β1.
Real-time RT-PCR
Real-time RT-PCR was used to determine levels of transforming growth factor-β1
mRNA. Primers and probe for the gene, along with all reagents for the reaction and
primers and probe for the loading control (the 18S gene), were obtained from Applied
Biosystems. One-step RT-PCR reactions were carried out using the manufacturer’s
instructions in a Prism 7000 thermocycler.
Lentiviral ACE2 Gene Transfer
Lentivirus harboring the gene for ACE2 under transcriptional control of the
elongation factor-1α promoter (EF1α) was obtained from the Raizada laboratory. This
batch of virus was determined to exhibit an infectious titer of 6.68x109 ifu/mL.
Cultured cardiac fibroblasts were grown to approximately 80% confluence before
infection. The virus was diluted in serum-free media for infection. Residual media was

74
removed from the fibroblasts, and the minimal volume of virus-containing media (200 µL
for a single well of a 24-well plate) was placed onto the cells. After four hours, 10% FBS
media was added to the cells to bring the media volume up to normal (500 µL total for a
single well of a 24-well plate).
Hypoxia-Reperfusion Paradigm
Upon confluence, cardiac fibroblasts were serum-starved for 48 hours. Medias
were then replaced with 10% FBS media immediately before cells were transferred to a
hypoxia chamber (or a normoxia control chamber). Cells in the hypoxia chamber were
exposed to a continual flow of 95% N2 / 5% CO2 gas for one hour. After one hour, cells
were returned to the normoxia condition for 12 or 24 hour hours before assays for ACE2
activity, collagen or transforming growth factor-β1 were performed.
ACE2 Activity
ACE2 activity was determined in cardiac fibroblasts as previously described
(Huentelman et al., 2004). Briefly, cells were scraped into a buffer consisting of 1 M
NaCl, 75 mM Tris, 0.5 mM ZnCl2, pH 7.5. Cells were then sonicated and protein content
was determined by a standard Bradford Assay. ACE2 activity was then determined by
kinetic assay using the Fluorogenic Peptide Substrate VI (FPS VI, 7Mca-Y-V-A-D-A-P-
K(Dnp)-OH, R&D Systems, Minneapolis, MN). Samples containing ACE2 enzyme (up
to 50 µl) were incubated with 100 µM FPS VI, 10 µM captopril (to inhibit ACE activity),
and reaction buffer (1 M NaCl, 75 mM Tris, 0.5 mM ZnCl2, pH 7.5) in a final reaction
volume of 100 µl at 37 °C. In the assay, ACE2 removes the C-terminal dinitrophenyl
moiety that quenches the inherent fluorescence of the 7-methoxycoumain group resulting
in an increase in fluorescence in the presence of ACE2 activity at excitation and emission
spectra of 328 and 392 nm, respectively.

75
Soluble Collagen
Soluble collagen was determined using a commercially available Sircol Soluble
Collagen kit (Accurate Chemical) according to manufacturer’s instructions.
Transforming Growth Factor-β1
Total transforming growth factor-β1 protein was determined by ELISA (R&D
Systems) according to manufacturer’s instructions.
Results
Cardiac Myocyte-Fibroblast Cross-Talk is Inhibited by Ang-(1-7) at the Myocyte
In this first series of experiments, cardiac myocytes were serum starved for twenty-
four hours before treatment with normal (10% FBS) media with or without Ang-(1-7)
supplementation. After twenty-four hours of incubation, this myocyte-preconditioned
media was transferred to cardiac fibroblasts. A preliminary study determined that
myocyte-preconditioned media caused increases in fibroblast collagen production only if
the myocytes were stimulated with FBS (data not shown). In this experiment
(summarized in Figure 6-1A), Ang-(1-7) supplementation caused a significant decrease
in collagen production by the cardiac fibroblasts if the peptide was allowed to act at the
cardiac myocyte, but had no effect if only the fibroblast was treated with Ang-(1-7)
(Figure 6-1B).
In a subsequent set of cultured cells, myocytes were treated with serum-free, 10%
FBS, or 10% FBS media supplemented with Ang-(1-7). After twenty-four hours of
incubation, cells were scraped from their growth chambers and total RNA was isolated.
One-step real-time RT-PCR measurement of mRNA for TGFβ1 demonstrated that while
10% FBS results in a significant increase in TGFβ1 mRNA, Ang-(1-7) co-incubation
significantly attenuates this effect (Figure 6-2).

76
Figure 6-1. Angiotensin-(1-7) attenuates myocyte-derived pro-fibrotic signaling. (A) Neonatal rat cardiac myocytes were incubated 24 hours with 10% fetal bovine serum-supplemented media with or without 10-7 M Ang-(1-7). Medias were then transferred to neonatal rat cardiac fibroblasts, and a subset of fibroblasts receiving Ang-(1-7)-free media were spiked with Ang-(1-7) at time of transfer. (B) Significant attenuation of collagen production (P=0.0320 vs. 10% FBS) was only achieved if Ang-(1-7) was allowed to act at the cardiac myocyte.
Figure 6-2. Angiotensin-(1-7) attenuates expression of TGFβ1 mRNA in cardiac myocytes. Neonatal rat cardiac myocytes were stimulated for 24 hours with serum-free, 10% serum, or 10% serum supplemented with 10-5 M Ang-(1-7). Real-time RT-PCR was used to measure mRNA for the TGFβ1 cytokine. Serum significantly increased transcription of TGFβ1 (P=0.0009), and Ang-(1-7) significantly attenuated this effect (P=0.0282).

77
Lentiviral Delivery of EF1α-ACE2 Results in Increased ACE2 Activity and Protection from Hypoxia
Preliminary studies determined that while cultured cardiac myocytes do exhibit a
moderate basal level of ACE2 activity, cultured cardiac fibroblasts have no detectable
basal activity (data not shown). Lentiviral delivery of the murine ACE2 gene under
transcriptional control of the elongation factor-1α (EF1α) promoter, though, results in a
viral dose-dependent increase in ACE2 activity in cultured cardiac fibroblasts (Figure 6-
3A).
Figure 6-3. Lentiviral delivery of ACE2 attenuates the collegen production response to hypoxia in cultured cardiac fibroblasts. (A) Lentiviral delivery of the EF1α-ACE2 construct results in a dose-dependent increase in ACE2 activity in cultured cardiac fibroblasts. (B) Lentiviral delivery of ACE2 attenuates the collagen production response to hypoxia in cultured cardiac fibroblasts. Fibroblasts, previously infected with various doses of lentivirus harboring the EF1α-ACE2 construct, were exposed to 1 hour of hypoxia, before being returned to normoxic conditions for 12 hours. Two-way ANOVA indicates significant effects of hypoxia (P<0.0001) and virus (P<0.0001) on collagen production, and a significant interaction between these factors (P=0.0111). The viral dose of “Low” indicates 2.36x105 ifu/cm2, “Medium” indicates 5.9x105 ifu/cm2, and “High” indicates 11.82x105 ifu/cm2.

78
Cardiac fibroblasts exhibit a strong collagen-production response to acute hypoxia
followed by normoxia. Lentiviral delivery of the murine ACE2 gene caused a significant
attenuation of this response to hypoxia (Figure 6-3B).
ACE2 Gene Delivery Attenuates Hypoxia-Reperfusion Induction of Transforming Growth Factor-β1
In addition to attenuating the collagen-production response to hypoxia, lentiviral
delivery of murine ACE2 significantly attenuated both basal and hypoxia-stimulated
media levels of TGFβ1 protein (Figure 6-4).
1000
1050
1100
1150
1200
1250
1300
1350
None Low Mid High
Viral dose
TGF-
beta
(pg/
mL)
NormoxiaHypoxia
Figure 6-4. Lentiviral delivery of ACE2 attenuates hypoxia-induced expression of transforming growth factor-β1 protein. Two-way ANOVA indicates significant effects of hypoxia (P=0.0019) and virus (P=0.0002) on TGF-β1 protein, but no interaction between these factors (P=0.5405). Viral doses are the same as above.
Discussion
In this preliminary set of experiments utilizing cultured neonatal rat cardiac
myocytes and fibroblasts, we determined that Ang-(1-7) inhibits myocyte-fibroblast
cross-talk, and that this inhibition may be due to decreased transcription of the myocyte-
derived pro-fibrotic cytokine TGFβ.

79
In addition to examining the effects of Ang-(1-7) on myocyte-fibroblast cross-talk,
the protective effects of ACE2 in fibroblasts treated with hypoxia were also examined.
Here, it was determined that while cultured neonatal rat cardiac fibroblasts show no
detectable basal ACE2 activity, lentiviral delivery of the murine ACE2 gene dose-
dependently increases ACE2 activity. This increased activity prevents hypoxia-
reperfusion induced collagen production by the fibroblasts, and this may be mediated by
decreased TGFβ protein. Finally, it was demonstrated that increased ACE2 activity, even
if temporally limited to the onset of hypoxia, is sufficient in cultured fibroblasts to
provide some anti-fibrotic protection.
The determination that ACE2 protects fibroblasts from hypoxia-induced metabolic
changes may lead to a future therapy for myocardial infarction. During the hypoxic
phase of a myocardial infarction, cardiac myocytes die and formerly quiescent fibroblasts
are stimulated by the hypoxic environment to produce large quantities of collagens I and
III, and these cells morphologically change into myofibroblasts (Stenmark et al., 2002).
The formation of myofibroblasts is vital, as this is a necessary step in wound healing.
Death of cardiac myocytes is also important, as these cells represent the endogenous
cardiac source of ACE2 and Ang-(1-7) (Averill et al., 2003; Zisman et al., 2003).
Future studies should be directed at determining the mechanism of ACE2-mediated
protection in cultured fibroblasts. Although Iwata et al. (2005) determined that Ang-(1-
7) can act upon cultured fibroblasts, our cross-talk experiment (Figure 6-1) suggests that
Ang-(1-7) did not have direct effects upon cultured fibroblasts. In light of these
conflicting findings, one can not simply assume that ACE2 mediates its effects in
cultured fibroblasts through Ang-(1-7). In addition to increasing Ang-(1-7) levels, which

80
may or may not be able to act upon fibroblasts, it is possible that ACE2 may work in this
paradigm solely through degradation of Ang II. Agocha et al. (1997) demonstrated that
during hypoxia, Ang II is a significantly more potent stimulus for cardiac fibroblasts, and
decreased Ang II levels would presumably contribute to decreased stimulation of the
fibroblasts. Lijnen et al. (2006) recently showed that Ang II mediates its actions in
cultured cardiac fibroblasts through NAD(P)H oxidase, and Byrne et al. (2003) published
a suggestive review article regarding the role of reactive oxygen species (which are
produced by NAD(P)H oxidase) in the development and progression of heart failure.
Okada et al. (2005) showed that viral delivery of a gene encoding a soluble TGFβII
receptor (thereby artificially decreasing available TGFβ cytokine in the myocardium)
resulted in significant protection from cardiac remodeling, function loss, and apoptosis of
the reparative myofibroblasts. Together with our current findings regarding the effects of
Ang-(1-7) and ACE2 on TGFβ, we conclude that Ang-(1-7) and ACE2 may mediate their
cardioprotective effects through inhibition of TGFβ, and that loss of myocytes (and
therefore the source of ACE2 and Ang-(1-7)) may permit increased TGFβ signaling.
Collectively, these preliminary studies in vitro suggest important roles for both
Ang-(1-7) and ACE2. In particular, it was determined that Ang-(1-7) likely mediates
myocyte-fibroblast cross-talk through actions on the cardiac myocyte, and that it may be
the loss of myocytes (which are the endogenous cardiac source of ACE2, and therefore
Ang-(1-7)) that permits increased TGFβ signaling and overactivity of cardiac fibroblasts
in an infarct zone. These findings lead to the conclusion that ACE2, and possibly Ang-
(1-7), may represent an as-yet unexplored therapy for myocardial infarction. Ongoing

81
studies are currently examining the utility of viral and pharmacological stimulation of
ACE2 in vivo during surgically-induced infarction.
Future studies should also repeat and expand upon the examination of Ang-(1-7)
modulation of myocyte-fibroblast cross-talk, both to confirm these results, and to
determine whether Ang-(1-7) can act directly upon cardiac fibroblasts. Interpretation of
the current findings (Figure 6-1) is hindered by the experimental design, as Ang-(1-7)
was delivered in a single bolus, and therefore the rate of degradation of the peptide in
cultures of cardiac fibroblasts may mask the effects of Ang-(1-7) upon these cells.
Ongoing studies are designed to utilize lentiviral-mediated delivery of the gene for an
Ang-(1-7) fusion protein (Santos et al., 2004) to provide a constant source of the peptide,
and thereby overcome this design limitation.

CHAPTER 7 GENERAL DISCUSSION
Summary and Conclusions
The studies presented here aimed to examine the cardioprotective actions of
angiotensin-(1-7) (Ang-(1-7)). Lentiviral-mediated increases in expression of
angiotensin converting enzyme 2 (ACE2) resulted in significant protection from cardiac
remodeling during chronic infusion of angiotensin II (Ang II) in vivo. Here, it was
demonstrated that chronic infusion of Ang-(1-7) in rats resulted in the same
cardioprotective effects during chronic Ang II infusion, thus suggesting that Ang-(1-7)
mediates the anti-trophic, anti-fibrotic actions of ACE2 on the myocardium. These
findings lead us to several important conclusions. First, this provides evidence that the
predominant mechanism of ACE2 cardioprotection during hypertension is to produce
Ang-(1-7) rather than simply degrade Ang II or metabolize ACE2’s other endogenous
ligands, including apelin-13 and -32, some of the kinin metabolites (with the exception of
bradykinin), neurotensin, and opioid peptides such as dynorphin (Vickers et al., 2002).
Second, this establishes Ang-(1-7) as an active cardioprotective hormone. The lack of
effect of Ang-(1-7) on blood pressure is also important, as this indicates a direct effect of
Ang-(1-7) on cardiac tissue, and establishes that any changes in blood pressure associated
with ACE2 overexpression may be a by-product of the degradation of Ang II.
Next, it was determined that the cardioprotective actions of Ang-(1-7) are not
limited to the Ang II-infusion model of hypertension and cardiac remodeling. In the
deoxycorticosterone acetate (DOCA)-salt hypertensive model, it was observed that
82

83
chronic infusion of Ang-(1-7) attenuated both interstitial and perivascular fibrosis,
without effect on cardiac hypertrophy. The anti-fibrotic actions of Ang-(1-7) in the
DOCA-salt (mineralocorticoid-dependent) model are potent but independent of effect
upon hypertrophy and blood pressure, which leads to two important conclusions. First,
these findings further support the notion that the signaling mechanisms of cardiac
hypertrophy and fibrosis are independent. Second, Ang-(1-7) must either only oppose the
effects of mineralocorticoids at the cardiac fibroblast, or act at the cardiac myocyte to
alter mineralocorticoid-induced paracrine pro-fibrotic signals but not intracellular (or
autocrine) hypertrophic signals.
Use of the normotensive isoproterenol-injection model of cardiac remodeling
further demonstrated that the cardioprotective effects of Ang-(1-7) are not mediated by
decreased blood pressure. In the isoproterenol model, it was observed that infusion of
Ang-(1-7) prevents cardiac hypertrophy, without effect on fibrosis. These findings, while
in contrast to those of the DOCA-salt model, establish that the effects of Ang-(1-7) are
model-dependent, and that Ang-(1-7) may have diverse effects across the cell types
within cardiac tissue. Protective effects in this model further establish that Ang-(1-7) has
an active role in the myocardium, and that this hormone is not a passive inhibitor of the
RAS. A follow-up study on the effect of ovarian hormones on isoproterenol-induced
cardiac remodeling indicated that ovarian hormones potentiate the hypertrophic response
to isoproterenol injection.
Next, it was determined that estrogen attenuates cardiovascular tissue sensitivity to
Ang-(1-7). Aortic rings from ovariectomized rats showed a dose-dependent relaxation
response to Ang-(1-7), while rings from ovariectomized rats with 17-β-estradiol

84
replacement showed an estrogen dose-dependent attenuation of this relaxation response.
Other hormones, including progesterone and testosterone were examined, but these
hormones showed little effect on vascular reactivity to Ang-(1-7) (data not shown).
Finally, the roles of ACE2 and Ang-(1-7) in cultured cardiac fibroblasts and
myocytes were examined to begin to dissect their mechanism(s) of action in these cell
types. It was determined that Ang-(1-7) inhibits myocyte-derived pro-fibrotic signaling,
which may be mediated by decreased transforming growth factor-β (TGFβ) gene
transcription. Further, it was determined that ACE2 gene delivery to cultured cardiac
fibroblasts, which have no detectable basal ACE2 activity, causes dose-dependent
increases in ACE2 activity and provides protection from hypoxia-induced collagen and
TGFβ production. Together with the literature, these findings suggest that within an
infarct zone, where myocytes (the endogenous source of ACE2 in myocardium) are
absent, lack of ACE2 (and possibly Ang-(1-7)) may be permissive for overactivity of
cardiac fibroblasts. This overactivity would result in severe cardiac remodeling, and loss
of function, eventually resulting in heart failure.
Pitfalls and Shortcomings
Few studies are ever perfect in design and execution, and the studies presented here
are no exception. There are several design flaws and shortcomings in these studies,
which should be noted so as not to repeat these errors in future studies.
In chapter 2, chronic infusion of Ang II resulted in increased blood pressure,
cardiac hypertrophy, and interstitial fibrosis. Chronic infusion of Ang-(1-7) had no effect
on the increased blood pressure, but significantly attenuated the hypertrophy and fibrosis.
In this study, a single dose of Ang II, and a single dose of Ang-(1-7) were utilized. The
dose of Ang II (100 ng/kg/min) was selected based on a long series of previous and

85
preliminary studies within the Katovich lab to provide a moderate increase in blood
pressure and moderate cardiac remodeling over four weeks, and the dose of Ang-(1-7)
was selected to represent a nearly-equimolar dose to that of the Ang II. The four-week
timeframe for the study was selected for practical limitations, namely the maximum time
afforded by the commercially available osmotic minipumps that were used to infuse both
hormones. Timeframes longer than four weeks would require removal of original pumps
and implantation of secondary pumps for each hormone after every four weeks of
treatment, and it was decided that additional anesthesia and subcutaneous “surgery” was
an unnecessary risk for the animals. Presumably, a longer treatment timecourse would
lead to overt heart failure in the Ang II-infused animals, as direct effects on cardiac
remodeling and indirect effects through increased vascular resistance and peripheral
blood pressure would result in gross morphological changes of the myocardium.
An additional problem with this first study was the use of Masson’s Trichrome to
stain the extracellular matrix of the myocardium. While this stain provides beautiful
pictures, it is not particularly well suited for the specific analysis of collagen deposition
in the matrix. This treatment results in red-stained myocytes, green-stained endothelial
cells, and blue-stained matrix. To analyze matrix deposition in the heart with two
different dominant stain colors on each slide (red and blue), pictures of the slides must be
analyzed by “splitting” the RGB data into three separate files (using the ImageJ program
from NIH – Rasband, 2005). These three individual files represent a pixel-by-pixel
color-saturation matrix for each of the primary (red, green, blue) values. Sections of the
left ventricle wall are then outlined in the red and blue files, and the average color
saturation for red and for blue are then determined for each animal’s heart-slice picture.

86
The blue saturation is then normalized to the red saturation within each animal to control
for batch-to-batch differences in the stain. Together, this process, along with the analysis
of the entire left ventricle wall (as described in chapter 2), artificially compresses the
magnitude of matrix deposition change with treatments, as is evidenced by only an
approximate 5% increase with chronic Ang II infusion (Figure 2-3). This problem is
again reflected in chapters 3 (mineralocorticoid model) and 4 (isoproterenol model). This
same extracellular matrix deposition, if analyzed using a different stain and analyzed
through a “percent-area” method, may result in more impressive numerical changes with
treatments. Regardless of the magnitude of the numerical values, though, the changes
which are visually obvious in the pictures (especially in chapter 3) are significantly
different even when analyzed by stringent non-parametric analysis when analyzed this
way. Heart slices from animals in each treatment group (from at least one future
experiment) should be stained by both Masson’s Trichrome and another more specific
stain, such as Sirius Red, to confirm the reliability of Masson’s Trichrome and to
determine the relationship between numerical results (in other words, to determine what a
5% difference when Masson’s Trichrome is used relates to if Sirius Red had been used).
In chapter 3, where deoxycorticosterone acetate (DOCA) was delivered by
subcutaneous pellet to uninephrectomized animals, similar design flaws existed. Again, a
single dose of DOCA was administered, and a single dose of Ang-(1-7) was utilized.
Once again, preliminary studies in the Katovich lab led to the use of the DOCA dose.
The Ang-(1-7) dose of 100 ng/kg/min was used because of successful results in the Ang
II study (chapter 2). Once again, four weeks was used as the timecourse, as this
represented the time limit imposed by the use of osmotic minipumps. This study was

87
also plagued by the numerically-unimpressive, yet statistically significant differences in
interstitial extracellular matrix changes with DOCA treatment. Future studies certainly
should be done with either the Ang II or DOCA models to determine the dose-response
relationship of Ang-(1-7) and its anti-remodeling effects.
Chapter 4 examined the effects of chronic Ang-(1-7) infusion on isoproterenol-
induced cardiac remodeling. Again, similar flaws in design were present as were
mentioned above. Several preliminary studies were carried out to determine the
appropriate dose of isoproterenol and timecourse, as the literature is hyper-variable with
regard to the specifics of this model. It was determined that while daily subcutaneous
injections of 1 mg/kg isoproterenol for one week resulted in moderate cardiac
hypertrophy in males, the variability between animals would require a very large sample
size to see statistical differences. Increasing the timecourse of treatment to two weeks
resulted in only slightly larger hypertrophy, and thus it was decided to increase the dose
of isoproterenol to 2 mg/kg daily. This increase in dosage resulted in a much larger
cardiac hypertrophy, but also greatly increased spontaneous deaths of animals.
Preliminary experiments led to the conclusion that in male Sprague-Dawley rats, 2 mg/kg
daily for 14 days was required to see large, reliable changes in cardiac hypertrophy.
Further, to decrease the spontaneous death rate of these animals, it was determined that
the daily injections of isoproterenol should be administered during the middle of the light
cycle of the animals. Animals should also be allowed to recover for at least one day
before the first injection of isoproterenol is administered, as the precipitous acute drop in
blood pressure is potentiated by anesthesia and minipump delivery of blood pressure
lowering compounds.

88
In chapter 5, the effect of estrogen status upon aortic vascular reactivity to Ang-(1-
7) was investigated. Several design flaws plagued this study, although their effects
probably would not change the conclusions of the study; at most these flaws likely only
limit the implications of the study. In particular, the lack of an intact (not
ovariectomized) treatment group and the use of a super-physiological dose of estradiol in
the “high estradiol” group detract from the utility of the data. An intact control group
would confirm the “normal” aortic response to graded doses of Ang-(1-7). The super-
physiological dose of estradiol in the “high estradiol” treatment group was the result of an
error by the commercial supplier of the pellets, as we requested a “low” dose which
should mimic a high-cycle level of estradiol, and a “high” dose which would mimic
circulating estradiol levels in a rat near the end of pregnancy. The estradiol ELISA at the
end of the study determined that the estradiol dose in the “high” group was approximately
1000-fold higher than desired. While this dose of estradiol was obviously too high to
mimic end-pregnancy levels, it should certainly indicate the ceiling of the hypothetical
estradiol / attenuation-of-Ang-(1-7)-effect dose-response curve.
As this study was actually carried out first in the series of experiments in this
dissertation, errors were made in the execution of the experiment despite an excellent
design. While analysis of ring responses to acetylcholine and sodium nitroprusside were
included in the design (to determine effects of estrogen modulation on endothelial and
smooth muscle integrity, respectively), these determinations should have been done at
multiple doses of each compound to more fully characterize the dose-response
relationships instead of the single (maximum) doses which were administered in the
current study. As executed, it is impossible to fully know how estradiol treatments may

89
(or may not) have affected the low-dose effects of these compounds, and thus the true
basal changes due to estradiol treatment.
Finally, this vascular reactivity experiment was carried out using aortic rings,
which limits interpretation and application of the data obtained. As was described in the
discussion section of chapter 5, other groups have examined estrogen effects upon
vascular reactivity to Ang-(1-7) in mesenteric artery preparations and have determined
the exact opposite effect of estrogens upon Ang-(1-7) vascular reactivity. Clearly, as the
goal of this work was to characterize the cardiac effects of estrogen on Ang-(1-7) actions,
future studies should be performed using cardiac tissue. This could involve the vascular
reactivity endpoint, but the vessel must be coronary arteries. This could also involve
alternate endpoints, including myocyte hypertrophy, or interstitial or perivascular
collagen deposition. Ongoing studies are currently investigating the effect of estrogen
status on myocyte-derived pro-fibrotic factors in cultured cardiac myocytes.
Chapter 6 focused on cultured cardiac cells to begin to tease apart tissue-level
effects of ACE2 and Ang-(1-7). The first and most obvious limitation of these studies
revolves around the use of a cultured tissue system, as no in vitro system can ever fully
emulate in vivo physiology. Cultures of cardiac cells are particularly plagued by a lack of
constant mechanical and electrical stimulation that is present in vivo, and is established to
affect myocyte morphology (Kira et al., 1994). Cardiac myocyte cultures are further
troublesome due to problems with cell type purity of cultures. While the isolation
procedure for cardiac fibroblasts is very selective, and other cell types die due to harsh
isolation conditions, cardiac myocytes are fragile cells which require serial selection
steps. Myocytes do not readily attach to plastic culture dishes, and they require constant

90
oxygenation. Because of these weak isolation conditions, cardiac fibroblasts, smooth
muscle, and endothelial cells typically survive the isolation procedure, and thus
contaminate myocyte cultures. Therefore, cultures of “myocytes” are typically a mixture
of many cell types. Before the preliminary experiments described in chapter 6 are
publishable in a scientific journal, a series of characterizations must be done to determine
the morphology of cell types present in the myocyte and fibroblast cultures.
The major endpoint for both the myocyte-fibroblast cross-talk and hypoxia series of
experiements was soluble collagen. In both experiments, soluble collagen in the cell
culture media was quantified using a commercially available kit that works by staining
and pelleting the free-floating collagen, then resuspending the pellet and releasing the
collagen-bound dye. The dye, Sirius Red, is thus present in the resuspended solution in a
directly proportional ratio to the amount of collagen in the original sample. This dye is
the same as used to stain heart tissue cross-sections in many in vivo experiments, and this
chemical stain is very specific for collagens. The inclusion of standardized collagen
solutions in the assay allows for the construction of a standard curve, and thus absolute
collagen values for each sample. Despite all of the advantages of this fast and simple
assay, most studies in the literature examine collagen levels through the use of a
collagenase-degradable tritiated-proline incorporation assay. This procedure, while
highly sensitive, does not allow for construction of a standard curve, and is much more
difficult and time consuming due to the use of tritiated proline. It is possible that
different results would be obtained using the proline assay versus the Sirius Red assay, as
the proline assay only determines the amount (or rate) of “new collagen” being produced
by the cells. Just as Masson’s Trichrome stains may result in different numerical values

91
than Sirius Red stains of heart sections, this assay will offer different numerical values
than the tritiated proline assay, but the interpretation of the results (that the positive
control increases collagen, and Ang-(1-7) or ACE2 reverses that effect) is unchanged.
While there are many tangential issues which could be pursued to improve the
“wow factor” of the numerical data presentation, and various tweaks to the doses and
timecourses of the experiments presented, these changes likely would have little effect on
the conclusions drawn here. The fact that several chapters have already been published
(data in chapter 1 in Experimental Physiology, chapter 3 in the American Journal of
Physiology, and chapter 5 in Regulatory Peptides) supports this view.
Future Directions
While these studies have resulted in several major advancements in our
understanding of the role of Ang-(1-7) in cardiovascular regulation, there are several
lines of investigation which have not yet been explored.
First, the mechanism of Ang-(1-7) signal transduction needs further examination.
As was described in chapter 2, there are many possible mechanisms of Ang-(1-7) action,
including antagonism of the AT1-subtype Ang II receptor, activation of the AT2-subtype
Ang II receptor, activation of the Mas receptor, and inhibition of angiotensin converting
enzyme (ACE). The current studies most strongly support actions of Ang-(1-7) at the
Mas receptor, although this support is entirely based upon indirect inference. A very
recent publication by Santos et al. (2006) describing the development and
characterization of a Mas-receptor knockout mouse represents the type of direct study
that is needed to fully characterize the mechanism of Ang-(1-7). Repeating the present
studies in a Mas-receptor knockout mouse would help elucidate which of the anti-
remodeling effects of Ang-(1-7) are mediated through this receptor.

92
The search for downstream hormone “targets” of Ang-(1-7) action presented here
was in no way exhaustive. There is a large body of literature that supports the role of
TGFβ in cardiac remodeling, and therefore the discovery that Ang-(1-7) modulates this
cytokine simultaneously solidified the importance of Ang-(1-7) in modulating cardiac
remodeling, and precluded exploration of other possible targets of Ang-(1-7). Other pro-
remodeling and anti-remodeling hormones may very well be modulated by Ang-(1-7).
Interleukins 1β and 6, and tumor necrosis factor α are all suggested to be pro-fibrotic
cytokines, while interleukin 10 may represent an anti-fibrotic hormone, and future studies
should be designed to determine if Ang-(1-7) modulates any of these hormones in the
myocardium.
While ligands, including Ang II, endothelin, catecholeamines, mineralocorticoids
and the various above-mentioned cytokines may be altered by Ang-(1-7), their receptors
may also be regulated by Ang-(1-7). Binding studies should be done to determine the
effects of Ang-(1-7) on AT1 and AT2 populations in both myocytes and fibroblasts, and
similar studies should be done on the receptors for the other pro- and anti-remodeling
hormones as well.
Two intermediaries of Ang-(1-7) action have been suggested in the literature. It
has been suggested that Ang-(1-7) may act through increasing production of local nitric
oxide, and through the production of protective prostaglandins such as prostaglandin I2
(Ferrario and Chappell, 2004). A study examining the protective actions of Ang-(1-7) in
the L-NAME model of hypertension and remodeling is currently in the planning stages,
and should be completed soon. Additionally, future studies could be directed at

93
determining the contribution of Ang-(1-7) modulation of prostaglandins on cardiac
structure and function.
It is possible that Ang-(1-7) may modulate not only the production of extracellular
matrix proteins, but also the activities of extraceullar matrix degradation enzymes. The
collagenases (including matrix metalloproteinases (MMP’s)-1, -8, and -13) and the
gelatinases (MMP-2 and -9) may be modulated by Ang-(1-7), and this modulation may
contribute to the actions of Ang-(1-7) in vivo, especially in recovery following
myocardial infarction.
Ongoing studies in the laboratory are focused on investigating the utility of ACE2
overexpression following surgically-induced myocardial infarction. These studies are
needed to verify the results obtained in cultured cardiac fibroblasts (chapter 6). Further,
these in vivo studies are needed to examine the effect of ACE2 overexpression on the
preservation of not only cardiac structure, but cardiac function. Many, or most, of the
presented studies focus on Ang-(1-7) modulation of cardiac structure, and the choice of
this endpoint was primarily based on the assumption that cardiac structure changes result
in functional changes, in addition to availability of assays for assessing changes in
cardiac function. Although no data are presented herein, a system for directly measuring
left ventricular pressure is in development, through which left ventricle end diastolic
pressure, ventricular contractility (positive dP/dt), and reflexive ventricular relaxation
(negative dP/dt) can be measured. Future experiments should examine both structural
changes and functional changes, as it will be the loss of function (which is only indirectly
related to structure) that is the clinically relevant endpoint. Timecourses of future

94
experiments will likely need to be much longer than four weeks, as a functional loss will
likely not be present until severe cardiac remodeling is present.
With the development of primary cardiac myocyte fibroblast and myocyte cultures
described here, many avenues of investigation are now possible for our laboratory. For
example, independent cultures of the various cell types represents the only direct way to
examine directional cross-talk between myocytes and fibroblasts. Further,
characterization of the effects of Ang-(1-7) in cultured cardiac fibroblasts during hypoxia
will be much faster, easier, and less expensive than if these studies were done in vivo.
Perspectives
In summary, these studies have shown that angiotensin-(1-7) is an active hormone,
which opposes cardiac fibrosis in two different models of hypertensive cardiac
remodeling, and cardiac hypertrophy in a normotensive model. It was also determined
that estrogens modulate the actions of angiotensin-(1-7). Finally, preliminary data was
shown that suggests angiotensin-(1-7) can act at the cardiac myocyte to inhibit myocyte-
fibroblast cross-talk, and that transforming growth factor-β1 may represent one target of
angiotensin-(1-7) in the cardiac myocyte.
From these findings, we conclude that angiotensin-(1-7) is a cardioprotective
hormone which warrants further study. As actions of this hormone may be mediated by a
novel receptor class, angiotensin-(1-7) may represent a new class of anti-remodeling
target that could eventually lead to an entirely new class of heart failure medication and
treatment.

APPENDIX A LOADING OSMOTIC MINIPUMPS
Alzet (Cupertino, CA) osmotic minipumps were used for all studies. Model 2004
pumps with a 0.25 µL/hr infusion rate were used for four-week studies (Ang II and
DOCA-salt), and model 2002 pumps with a 0.5 µL/hr infusion rate were used for two-
week studies (isoproterenol). The concentration of drug in the filling solution for the
pumps is determined by the following calculation:
Target Rate 1 µg Body Mass
Pumps are filled with appropriate solution, then primed for 24 hours in a 0.9%
saline solution for twenty-four hours at 37ºC before implantation. Animals are
anesthetized by inhalation of metophane, halothane, or isoflurane before shaving and
cleaning an area between the shoulder blades. A small incision is then made, and the
pump is introduced into the subcutaneous space. Wound clips are then used to close the
incision. Wound clips should be removed from the skin after 1-2 weeks.
(ng/kg/min) (kg) X 1000 ng X
= Filling Solution (µg/µL)or (mg/mL)
Pump Rate 1 hr X (µL/hr) 60 min
95

APPENDIX B MASSON’S TRICHROME STAINING AND ANALYSIS
One cross-section of the heart ventricles is cut (approximately 2 mm thick) just
above the level of the papillary muscles, and this section is immediately placed into a
10% neutral buffered formalin solution. These cross-sections (in formalin) are then
processed by the University of Florida Molecular Pathology and Immunology Core.
Briefly, the cross-section is paraffin embedded and sectioned at 5 microns. Sections are
then mounted on glass slides and stained sequentially with Weigert’s Iron Hematoxylin,
Biebrich Scarlet, and Aniline Blue stains. For the presented studies, embedding,
sectioning, and staining were performed by the University of Florida Molecular
Pathology Core.
Slides stained with Masson’s Trichrome are viewed under 10X (whole heart cross-
section), 250X (muscle bundles and interstitial collagen) and 400X (left anterior
descending coronary artery and associated perivascular collagen) magnification, and
digital photographs are taken using a Moticam 1000 digital microscope camera. Images
are stored and analyzed on a PC computer using the ImageJ program from NIH
(Rasband, 1997). It is imperative that all slides within one study are imaged with the
same lighting conditions, and therefore all images at a given magnification should be
taken in one session.
Images of slides at 250X magnification are analyzed for myocyte diameter using
the ImageJ program. Briefly, an image of a micrometer taken at the same (250X)
magnification is used to calibrate the ruler tool within the ImageJ program. At least
96

97
fifteen individual myocytes that have been cut across their short-axis in the slide are then
measured for cross-sectional diameter. Only myocytes with nearly circular shape are
used to ensure measurements are made across the short-axis of the myocyte. The fifteen
values are then averaged to give one myocyte diameter value per animal.
Images of slides viewed at 10X magnification are used to analyze left wall fibrosis.
Briefly, the digital image is split into red, blue, and green channels by the ImageJ
program. The left ventricle wall is then manually outlined and red and blue color
intensity within the outlined section is then determined by the program. These color
intensity data are then exported to a spreadsheet, and for each heart the blue color
intensity is normalized to the red color intensity. Higher ratios indicate larger blue
staining, and hence more collagen deposition. Because of differences in each batch of
stain, comparison of blue/red ratios between batches is inappropriate. Therefore, data
presentation as “% control” is necessary.
Images of slides viewed at 400X magnification of the left anterior descending
coronary artery are used to measure perivascular collagen deposition. Briefly, the
freestyle area selection tool within the ImageJ program is used to trace (A) the outline of
the outside edge of the perivascular collagen, (B) the outside edge of the vessel’s smooth
muscle layer, and (C) the lumen of the vessel. The areas enclosed within the outlines are
used to calculate the normalized area of perivascular collagen as follows; [normalized
area] = ( [A] – [B] ) / [C]. It is imperative to only use images of nearly circular (not
collapsed) blood vessels, as the lumen area is highly dependent upon the shape of the
vessel.

LIST OF REFERENCES
Abraham G, and Simon G. Autopotentiation of pressor responses by subpressor angiotensin II in rats. Am J Hypertension 7: 269-275, 1994.
Agocha A, Lee HW, Eghbali-Webb M. Hypoxia regulates basal and induced DNA synthesis and collagen type I production in human cardiac fibroblasts: effects of transforming growth factor-beta1, thyroid hormone, angiotensin II and basic fibroblast growth factor. J Mol Cell Cardiol. 29: 2233-2244, 1997.
Akhrass F, Evans AT., Wang Y, Rich S, Kannan CR, Fogelfeld L, and Mazzone T. Hormone replacement therapy is associated with less coronary atherosclerosis in postmenopausal women. J Clin Endocrinol Metab 88: 5611-5614, 2003.
Akiyama-Uchida Y, Ashizawa N, Ohtsuru A, Seto S, Tsukazaki T, Kikuchi H, Yamashita S, and Yano K. Norepinephrine enhances fibrosis mediated by TGF-beta in cardiac fibroblasts. Hypertension 40: 148-154, 2002.
Al Zubair, K, A Razak, S Bexis, and JR Docherty. Relaxations to oestrogen receptor subtype selective agonists in rat and mouse arteries. Eur J Pharmacol 513:101-108, 2005.
Alderman MH, Madhavan S, Ooi WL, Cohen H, Sealey JE, and Laragh JH. Association of the renin-sodium profile with the risk of myocardial infarction in patients with hypertension. N Eng J Med 324: 1098-1104, 1991.
Anderson S, Rennke HG, and Brenner BM. Advantage of converting enzyme inhibitors in arresting progressive renal disease associated with systemic hypertension in the rat. J Clin Invest 77: 1993-2000, 1986.
Armando I, M Jezova, AV Juorio, JA Terron, A Falcon-Neri, C Semino-Mora, H Imboden, and JM Saavedra. Estrogen upregulates renal angiotensin II receptors. Am J Physiol Renal Physiol 283: F934-F943, 2002.
Averill DB, Ishiyama Y, Chappell MC, and Ferrario CM. Cardiac angiotensin-(1-7) in ischemic cardiomyopathy. Circulation 108: 2141-2146, 2003.
Bader M, Peters J, Baltatu O, Muller DN, Luft FC, and Ganten D. Tissue renin-angiotensin systems: new insights from experimental animal models in hypertension research. J Mol Med 79: 76-102, 2001.
98

99
Baker KM, Booz GW, and Dostal DE. Cardiac actions of angiotensin: II. Role of an intracardiac renin-angiotensin system. Annu Rev Physiol 54: 227-241, 1992.
Barth W, Deten A, Bauer M, Reinohs M, Leicht M, and Zimmer HG. Differential remodeling of the left and right heart after norepinephrine treatment in rats: studies on cytokines and collagen. J Mot Cell Cardiol 32: 273-284, 2000.
Basso N, and Terragno NA. History about the discovery of the renin-angiotensin system. Hypertension 38: 1246-1249, 2001.
Bayorh MA, Eatman D, Walton M, Socci RR, Thierry-Palmer M, and Emmett N. A-779 attenuates angiotensin-(1-7) depressor response in salt-induced hypertensive rats. Peptides 23: 57-64, 2002.
Beldent V, Michaud A, Bonnefoy C, Chauvet MT, and Corvol P. Cell surface localization of proteolysis of human endothelial angiotensin I-converting enzyme. Effect of the amino-terminal domain in the solubilization process. J Biol Chem 270: 28962-28969, 1995.
Benetos A, Lacolley P, and Safar ME. Prevention of aortic fibrosis by spironolactone in spontaneously hypertensive rats. Arterioscler Thromb Vasc Biol 17: 1152-1156, 1997.
Benter IF, Diz DI, and Ferrario CM. Pressor and reflex sensitivity is altered in spontaneously hypertensive rats treated with angiotensin-(1-7). Hypertension 26: 1138-1144, 1995.
Berk BC. Angiotensin II signal transduction in vascular smooth muscle: pathways activated by specific tyrosine kinases. J Am Soc Nephrol 10: S62-S68, 1999.
Bhambi B, and Eghbali M. Effect of norepinephrine on myocardial collagen gene expression and response of cardiac fibroblasts after norepinephrine treatment. Am J Pathol 139: 1131-1142, 1991.
Blume A, Herdegen T, and Unger T. Angiotensin peptides and inducible transcription factors. J Mol Med 77: 339-357, 1999.
Boehm M, and Nabel EG. Angiotensin-converting enzyme 2-a new cardiac regulator. N Engl J Med 347: 1795-1797, 2002.
Bos R, Mougenot N, Findji L, Mediani O, Vanhoutte PM, and Lechat P. Inhibition of Catecholamine-Induced Cardiac Fibrosis by an Aldosterone Antagonist. J Cardiovasc Pharmacol 45: 8-13, 2005.
Braun-Menéndez E, Fasciolo JC, Leloir LF, and Muñoz JM. La sustancia hipertensora de la sangre del riñón isquemiado. Rev Soc Arg Biol 15: 420-425, 1939.

100
Braun-Menéndez E, and Page IH. Suggested revision of nomenclature: angiotensin. Science 127: 242, 1958.
Bright R. Tubular view of the morbid appearances in 100 cases connected with albuminous urine: With observations. Guy’s Hosp Rep 1: 380-400, 1836.
Brilla CG. Aldosterone and myocardial fibrosis in heart failure. Herz 25: 299-306, 2000.
Brilla CG, and Weber KT. Mineralocorticoid excess, dietary sodium, and myocardial fibrosis. J Lab Clin Med 120: 893-901, 1992.
Brilla CG, Janicki JS, and Weber KT. Impaired diastolic function and coronary reserve in genetic hypertension. Role of interstitial fibrosis and medial thickening of intramyocardial coronary arteries. Circ Res 69: 107-115, 1991.
Brilla CG, Pick R, Tan LB, Janicki JS, and Weber KT. Remodeling of the rat right and left ventricles in experimental hypertension. Circ Res 67: 1355-1364, 1990.
Brosnihan KB, Li P, and Ferrario CM. Angiotensin-(1-7) dilates canine coronary arteries through kinins and nitric oxide. Hypertension 27: 523-528, 1996.
Brosnihan KB, Li P, Tallant EA, and Ferrario CM. Angiotensin-(1-7): a novel vasodilator of the coronary circulation. Biol Res 31: 227-234, 1998.
Brosnihan, KB, Neves LA, Joyner J, Averill DB, Chappell MC, Sarao R, Penninger J, and Ferrario CM. Enhanced renal immunocytochemical expression of ANG-(1-7) and ACE2 during pregnancy. Hypertension 42: 749-753, 2003.
Brunner HR, Laragh JH, Baer L, Newton MA, Goodwin FT, Krakoff LR, Bard RH, and Buhler FR. Essential hypertension: renin and aldosterone, heart attack and stroke. N Eng J Med 286: 441-449, 1972.
Burrell LM, Risvanis J, Kubota E, Dean RG, MacDonald PS, Lu S, Tikellis C, Grant SL, Lew RA, Smith AI, Cooper ME, and Johnston CI. Myocardial infarction increases ACE2 expression in rat and humans. European Heart J 26: 369-375, 2005.
Burt VL, Whelton P, Roccella EJ, Brown C, Cutler JA, Higgins M, Horan MJ, and Labarthe D. Prevalence of hypertension in the US adult population. Results from the Third National Health and Nutrition Examination Survey, 1988-1991. Hypertension 25: 305-313, 1995.
Butler R, Morris AD, Burchell B, and Struthers AD. DD angiotensin-converting enzyme gene polymorphism is associated with endothelial dysfunction in normal humans. Hypertension 33: 1164-1168, 1999.

101
Byrne JA, Grieve DJ, Cave AC, Shah AM. Oxidative stress and heart failure. Arch Mal Coeur Vaiss. 96: 214-221, 2003.
Cambien F, Poirier O, Lecerf L, Evans A, Cambou JP, Arveiler D, Luc G, Bard JM, Bara L, Ricard S, Tiret L, Amouyel P, Alhenc-Gelas F, and Soubrier F. Deletion polymorphism in the gene for angiotensin-converting enzyme is a potent risk factor for myocardial infarction. Nature 359:641-644, 1992.
Caruso-Neves C, Lara LS, Rangel LB, Grossi AL, and Lopes AG. Angiotensin-(1-7) modulates the ouabain-insensitive Na+-ATPase activity from basolateral membrane of the proximal tubule. Biochim Biophys Acta 1467: 189-197, 2000.
Chai SY, Fernando R, Peck G, Ye SY, Mendelsohn FA, Jenkins TA, and Albiston AL. The angiotensin IV/AT4 receptor. Cell Mol Life Sci 61: 2728-2737, 2004.
Chaves GZ, Caligiorne SM, Santos RA, Khosla MC, and Campagnole-Santos MJ. Modulation of the baroreflex control of heart rate by angiotensin-(1-7) at the nucleus tractus solitarii of normotensive and spontaneously hypertensive rats. J Hypertens 18: 1841-1848, 2000.
Cheng, DY, and Gruetter CA. Chronic estrogen alters contractile responsiveness to angiotensin II and norepinephrine in female rat aorta. Eur J Pharmacol 215: 171-176, 1992.
Collister JP, and Hendel MD. The role of Ang (1-7) in mediating the chronic hypotensive effect of losartan in normal rats. J Renin Angiotensin Aldosterone Syst 4: 176-179, 2003.
Corda S, Samuel JL, and Rappaport L. Extracellular matrix and growth factors during heart growth. Heart Fail Rev 5: 119-130, 2000.
Crabos M, Roth M, Hahn AW, and Erne P. Characterization of angiotensin II receptors in cultured adult rat cardiac fibroblasts. Coupling to signaling systems and gene expression. J Clin Invest 93: 2372-2378, 1994.
Crackower MA, Oudit GY, Kozieradzki I, Sarao R, Sun H, Sasaki T, Hirsch E, Suzuki A, Shioi T, Irie-Sasaki J, Sah R, Cheng HY, Rybin VO, Lembo G, Fratta L, Oliveira-dos-Santos AJ, Benovic JL, Kahn CR, Izumo S, Steinberg SF, Wymann MP, Backx PH, and Penninger JM. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 417: 822-828, 2002.
Danilczyk U, Eriksson U, Crackower MA, and Penninger JM. A story of two ACEs. J Mol Med 81: 227-234, 2003.
Davie AP, and McMurray JJ. Effect of angiotensin-(1-7) and bradykinin in patients with heart failure treated with an ACE inhibitor. Hypertension 34: 457-460, 1999.

102
De Souza, AM, Lopes AG, Pizzino CP, Fossari RN, Miguel NC, Cardozo FP, Abi-Abib R, Fernandes MS, Santos DP, and Caruso-Neves C. Angiotensin II and angiotensin-(1-7) inhibit the inner cortex Na+ -ATPase activity through AT2 receptor. Regul Pept 120: 167-175, 2004.
Diez J, Gonzalez A, Lopez B, Ravassa S, and Fortuno MA. Effects of antihypertensive agents on the left ventricle: clinical implications. Am J Cardiovasc Drugs 1: 263-279, 2001.
Dobrzynski E, Wang C, Chao J, and Chao L. Adrenomedullin gene delivery attenuates hypertension, cardiac remodeling, and renal injury in deoxycorticosterone acetate-salt hypertensive rats. Hypertension 36: 995-1001, 2000.
Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, Donovan M, Woolf B, Robinson K, Jeyaseelan R, Breitbart RE, and Acton S. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angtiotensin 1-9. Circ Res 87: 1-9, 2000.
Donoghue M, Wakimoto H, Maguire CT, Acton S, Hales P, Stagliano N, Fairchild-Huntress V, Xu J, Lorenz JN, Kadambi V, Berul CI, Breitbart RE. Heart block, ventricular tachycardia, and sudden death in ACE2 transgenic mice with downregulated connexins. J Mol Cell Cardiol 35: 1043-1053, 2003.
Dzau, V.J. Tissue renin-angiotensin system in myocardial hypertrophy and failure. Arch Intern Med 153: 937-942, 1993.
Dzau VJ, Sasamura H, and Hein L. Heterogeneity of angiotensin synthetic pathways and receptor subtypes: physiological and pharmacological implications. J Hypertens 22: S1-S9, 1993.
Eatman D, Wang M, Socci RR, Thierry-Palmer M, Emmett N, and Bayorh MA. Gender differences in the attenuation of salt-induced hypertension by angiotensin (1-7). Peptides 22: 927-933, 2001.
Ferrario CM and Chappell MC. Novel angiotensin peptides. Cell Mol Life Sci 61: 2720-2727, 2004.
Ferrario CM, Jessup J, Chappell MC, Averill DB, Brosnihan KB, Tallant EA, Diz DI, and Gallagher PE. Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation 111: 2605-2610, 2005.
Ferrario CM, Jessup J, Gallagher PE, Averill DPB, Brosnihan KP, Tallant AE, Smith RD, and Chappell MC. Effects of renin-angiotensin system blockade on renal angiotensin-(1-7) forming enzymes and receptors. Kidney Int 68: 2189-2196, 2005.

103
Ferreira AJ, Santos RA, and Almeida AP. Angiotensin-(1-7) improves the post-ischemic function in isolated perfused rat hearts. Brazilian J Medical Biol Res 35: 1083-1090, 2002.
Fisher, M, Baessler A, and Schunkert H. Renin angiotensin system and gender differences in the cardiovascular system. Cardiovasc Res 53: 676-677, 2002.
Fleming I, Kohlstedt K, and Busse R. New fACEs to the renin-angiotensin system. Physiology 20: 1-95, 2005.
Fornage M, Amos CI, Kardia S, Sign CF, Turner ST, and Boerwinkle E. Variation in the region of the angiotensin-converting enzyme gene influences interindividual differences in blood pressure levels in young white males. Circulation 97: 1773-1779, 1998.
Francis GS, Cohn JN, Johnson G, Rector TS, Goldman S, Simon A, VHeFT VA Cooperative Studies Group. Plasma norepinephrine, plasma renin activity, and congestive heart failure: relations to survival and the effects of therapy in V-HeFT II. Circulation 87: VI40-V148, 1993.
Fregly MJ, Rowland NE, Sumners C, and Gordon DB. Reduced dipsogenic responsiveness to intracerebroventricularly administered angiotensin II in estrogen-treated rats. Brain Res 338: 115-121, 1985.
Frigerio M, and Roubina E. Drugs for left ventricular remodeling in heart failure. Am J Cardiol 96: 10L-18L, 2005.
Funder J. Mineralocorticoids and cardiac fibrosis: the decade in review. Clin Exp Pharmacol Physiol 28: 1002-1006, 2001.
Garcia EA, Newhouse S, Caulfield MJ, and Munroe PB. Genes and hypertension. Curr Pharm Des 9: 1679-1689, 2003.
Gardeman A, Weiss T, Schwartz O, Eberbach A, Katz N, Hehrlein FW, Tillmanns H Waas W, and Haberbosch W. Gene polymorphism but not catalytic activity of angiotensin I-converting enzyme is associated with coronary artery disease and myocardial infarction in low-risk patients. Circulation 92: 2796-2799, 1995.
Gasse C, Hense HW, Stieber J, Doring A, Liese AD, and Keil U. Assessing hypertension management in the community: trends of prevalence, detection, treatment, and control of hypertension in the MONICA Project, Augsburg 1984-1995. J Hum Hypertens 15: 27-36, 2001.
Gironacci MM, Coba MP, and Pena C. Angiotensin-(1-7) binds at the type 1 angiotensin II receptors in rat renal cortex. Regul Pept 84: 51-54, 1999.

104
Gironacci MM, Valera MS, Yujnovsky I, and Pena C. Angiotensin-(1-7) inhibitory mechanism of norepinephrine release in hypertensive rats. Hypertension 44: 783-787, 2004.
Goodman LS, and Gilman A. The pharmacological basis of therapeutics, 10th edition, edited by Hardman JG, and Limbard LE. New York, NY: McGraw-Hill, 2001.
Griendling KK, Ushio-Fukai M, Lassegue B, and Alexander RW. Angiotensin II signaling in vascular smooth muscle. New concepts. Hypertension 29: 366-373, 1997.
Grobe JL, Mecca AP, Mao H, and Katovich MJ. Chronic angiotensin-(1-7) prevents cardiac fibrosis in the DOCA-salt model of hypertension. Am J Physiol Heart Circ Physiol. [Epub ahead of print], 2006.
Grodstein F, and Stampfer M. Estrogen for women at varying risk of coronary disease. Maturitas 30: 19-26, 1998.
Grodstein F, and Stampfer M. The epidemiology of coronary heart disease and estrogen replacement in postmenopausal women. Prog Cardiovasc Dis 38: 199-210, 1995.
Grohe C, Kahlert S, Lobbert K, Stimpel M, Karas RH, Vetter H, and Neyses L. Cardiac myocytes and fibroblasts contain functional estrogen receptors. FEBS Lett 416: 107-112, 1997.
Guy JL, Jackson RM, Acharya KR, Sturrock ED, Hooper NM, and Turner AJ. Angiotensin-converting enzyme-2 (ACE2): comparative modeling of the active site, specificity requirements, and chloride dependence. Biochemistry 42: 13185-13192, 2003.
Harris HA, Katzenellenbogen JA, and Katzenellenbogen BS. Characterization of the biological roles of the estrogen receptors, ERalpha and ERbeta, in estrogen target tissues in vivo through the use of ERalpha-selective ligand. Endocrinology 143: 4172-4177, 2002.
Harrison-Bernard LM, and Raij L. Postmenopausal hypertension. Curr Hypertens Rep 2: 202-207, 2000.
Hayes SN, and Taler SJ. Hypertension in women: current understanding of gender differences. Mayo Clin Proc 73: 157-165, 1998.
Healy DP, Ye MQ, Yuan LX, and Schachter BS. Stimulation of angiotensinogen mRNA in rat pituitary by estradiol. Am J Physiol 263: E355-E361, 1992.
Hollenberg, NK, Fisher NDL, and Price DA. Pathwasy for angiotensin II generation in intact human tissue. Evidence from comparative pharmacological interruption of the renin system. Hypertension 32: 387-392, 1998.

105
Horiuchi M, Akishita M, and Dzau VJ. Recent progress in angiotensin II type 2 receptor research in the cardiovasular system. Hypertension 33: 613-621, 1999.
Huentelman MJ, Grobe JL, Vazquez J, Stewart JM, Mecca AP, Katovich MJ, Ferrario CM, and Raizada MK. Protection from angiotensin II-induced cardiac hypertrophy and fibrosis by systemic lentiviral delivery of ACE2. Exp Physiol 90: 783-790, 2005.
Huentelman MJ, Zubcevic J, Katovich MJ, and Raizada MK. Cloning and characterization of a secreted form of angiotensin-converting enzyme 2. Regul Pept. 122: 61-67, 2004.
Hulley S, Furberg C, Barrett-Connor E, Culey J, Grady D, Haskell W, Knopp R, Lowery M, Satterfield S, Schrott H, Vittinghoff E, and Hunningbake D. Noncardiovascular disease outcomes during 6.8 years of hormone therapy: Heart and Estrogen/Progestin Replacement Study Follow-up (HERS II). JAMA 288: 58-66, 2002.
Igase M, Strawn WB, Gallagher PE, Geary RL, and Ferrario CM. Angiotensin II AT1 receptors regulated ACE2 and angiotensin-(1-7) expression in the aorta of spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol 289: H1013-1019, 2005.
Inagami T. Molecular biology and signaling of angiotensin receptors: an overview. J Am Soc Nephrol 10: S2-S7, 1999.
Iwai N, Ohmichi N, Nakamura Y, and Kinoshita M. DD genotype of the angiotensin-converting enzyme gene is a risk factor for left ventricular hypertrophy. Circulation 90: 2622-2628, 1994.
Iwata M, Cowling RT, Gurantz D, Moore C, Zhang S, Yuan JX, and Greenberg BH. Angiotensin-(1-7) binds to Specific receptors on cardiac fibroblasts to initiate anti-fibrotic and anti-trophic effects. Am J Physiol Heart Circ Physiol 289: H2356-63, 2005.
Iyer SN, Chappell MC, Averill DB, Diz DI, and Ferrario CM. Vasodepressor actions of angiotensin-(1-7) unmasked during combined treatment with lisinopril and losartan. Hypertension 31: 699-705, 1998.
Iyer S, Ferrario CM, and Chappell MC. Angiotensin-(1-7) contributes to the antihypertensive effects of blockade of the renin-angiotensin system. Hypertension 31: 356-361, 1998.
Jacob LS. Pharmacology, 4th edition. Philadelphia, PA: Williams & Wilkins, 1996.
Jaiswal N, Diz DI, Chappell MC, Khosla MC, and Ferrario CM. Stimulation of endothelial cell prostaglandin production by angiotensin peptides. Characterization of receptors. Hypertension 19: 1149-1155, 1992.

106
Jalil JE, Janicki JS, Pick R, and Weber KT. Coronary vascular remodeling and myocardial fibrosis in the rat with renovascular hypertension. Response to captopril. Am J Hypertens 4: 51-55, 1991.
Jeunemaitre X, Soubrier F, Kotelevtsev YV, Lifton RP, Williams CS, Charru A, Hunt SC, Hopkins PN, Williams RR, Lalouel JM, and Corvol P. Molecular basis of human hypertension: Role of angiotensinogen. Cell 71: 169-180, 1992.
Johnson GI. On certain points in the anatomy and pathology of Bright’s diseases of the kidney, II: on the influence of the minute blood-vessels upon the circulation. Med Chir Trans 51: 57-80, 1868.
Jonklaas, J, and Buggy J. Angiotensin-estrogen interaction in female brain reduces drinking and pressor responses. Am J Physiol 247: R167-R172, 1984.
Khalil ME, Basher AW, Brown EJ Jr, and Alhaddad L. A remarkable medical story: benefits of angiotension-converting enzyme inhibitors in cardiac patients. Am Coll Cardiol 37: 1757-1764, 2001.
Katovich MJ, Reaves PY, Francis SC, Pachori AS, Wang HW, and Raizada MK. Gene therapy attenuates the elevated blood pressure and glucose intolerance in an insulin-resistant model of hypertension. J Hypertens 19: 1553-1558, 2001.
Keating NL, Cleary PD, Rossi AS, Zaslavsky AM, and Ayanian JZ. Use of hormone replacement therapy by postmenopausal women in the United States. Ann Intern Med 130: 545-553, 1999.
Kehoe PG, Russ C, McIlory S, Williams H, Holmans P, Holmes C, Liolitsa D, Vahidassr D, Powell J, McGleenon B, Liddell M, Plomin R, Dynan K, Williams N, Neal J, Cairns NJ, Wilcock G, Passmore P, Lovestone S, Williams J, and Owen MJ. Variation in DCP1, encoding ACE, is associated with susceptibility to Alzheimer disease. Nature Genetics 21: 71-72, 1999.
Keidar S, Gamliel-Lazarovich A, Kaplan M, Pavlotzky E, Hamoud S, Hayek T, Karry R, and Abassi Z. Mineralocorticoid receptor blocker increases angiotensin-converting enzyme 2 activity in congestive heart failure patients. Circ Res 97: 946-53, 2005.
Kim HS, Krege JH, Kluckman KD, Hagaman JR, Hodgin JB, Best CF, Jennette C, Coffman TM, Maeda N, and Smithies O. Genetic control of blood pressure and the angiotensinogen locus. Proc Natl Acad Sci 92: 2735-2739, 1995.
Kim S, and Iwao H. Molecular and cellular mechanisms of angiotensin II mediated cardiovascular and renal diseases. Pharmacol Rev 52: l 1-34, 2000.

107
Kim S, Ohta K, Hamaguchi A, Omura T, Yukimura T, Miura K, Inada Y, Wada T, Ishimura Y, Chatani F, and Iwao H. Role of angiotensin II in renal injury of deoxycorticosterone acetate-salt hypertensive rats. Hypertension 24: 195-204, 1994.
Kira Y, Nakaoka T, Hashimoto E, Okabe F, Asano S, Sekine I. Effect of long-term cyclic mechanical load on protein synthesis and morphological changes in cultured myocardial cells from neonatal rat. Cardiovasc Drugs Ther. 8: 251-262, 1994.
Le Tran Y, and Forster C. Angiotensin-(1-7) and the rat aorta: modulation by the endothelium. J Cardiovasc pharmacol 30: 676-682, 1997.
Lekgabe ED, Kiriazis H, Zhao C, Xu Q, Moore XL, Su Y, Bathgate RA, Du XJ, and Samuel CS. Relaxin reverses cardiac and renal fibrosis in spontaneously hypertensive rats. Hypertension 46: 412-418, 2005.
Lemos VS, Silva DM, Walther T, Alenina N, Bader M, and Santos RA. The endothelium-dependent vasodilator effect of the nonpeptide Ang(1-7) mimic AVE 0991 is abolished in the aorta of mas-knockout mice. J Cardiovasc Pharmacol 46: 274-279, 2005.
Levy D, Larson MG, Vasan RS, Kannel WB, and Ho KK. The progression from hypertension to congestive heart failure. JAMA 275: 1557-1562, 1996.
Liao Y, Cooper RS, Mensah GA, and McGee DL. Left ventricular hypertrophy has a greater impact on survival in women than in men. Circulation 92: 805-810, 1995.
Lijnen P, Papparella I, Petrov V, Semplicini A, Fagard R. Angiotensin II-stimulated collagen production in cardiac fibroblasts is mediated by reactive oxygen species. J Hypertens. 24: 757-766, 2006.
Lijnen P, and Petrov V. Antagonism of the renin-angiotensin-aldosterone system and collagen metabolism in cardiac fibroblasts. Methods Find Exp Clin Pharmacol 21: 215-27, 1999.
Lijnen P, and Petrov V. Role of intracardiac renin-angiotensin-aldosterone system in extracellular matrix remodeling. Methods Find Exp Clin Pharmacol 25: 541-564, 2003.
Lijnen PJ, Petrov VV, and Fagard RH. Angiotensin II-induced stimulation of collagen secretion and production in cardiac fibroblasts is mediated via angiotensin II subtype 1 receptors. JRAAS 2: 117-22, 2001.
Lijnen PJ, Petrov VV, and Fagard RH. Induction of cardiac fibrosis by transforming growth factor-beta(l). Mol Genet Metab 71: 418-435, 2000.

108
Loot AE, Roks AJ, Henning RH, Tio RA, Suurmeijer AJ, Boomsma F, and van Gilst WH. Angiotensin-(1-7) attenuates the development of heart failure after myocardial infarction in rats. Circulation 105: 1548-50, 2002.
Lopez B, Salom MG, Arregui B, Valero F, and Fenoy FJ. Role of superoxide in modulating the renal effects of angiotensin II. Hypertension 42: 1150-1156, 2003.
Lorell BH. Transition from hypertrophy to failure. Circulation 96: 3824-3827, 1997.
Lu D, Raizada MK, Iyer S, Reaves P, Yang H, and Katovich MJ. Losartan versus gene therapy: chronic control of high blood pressure in spontaneously hypertensive rats. Hypertension 30: 363-370, 1997.
Machado RD, Santos RA, and Andrade SP. Mechanisms of angiotensin-(1-7)-induced inhibition of angiogenesis. Am J Physiol Regul Integr Comp Physiol 280: R994-R1000, 2001.
Mahomed FA. The physiology and clinical use of the sphygmograph. Med Times Gaz. 1: 62–64, 1872.
Matsubara, H. Pathophysiological role of angiotensin II type 2 receptor in cardiovascular and renal diseases. Circ Res 83; 1182-1191, 1998.
Mattu RK, Needham EW, Galton DJ, Frangos E, Clark AJL, and Caulfield M. A DNA variant at the angiotensin-converting enzyme gene locus associates with coronary artery disease in the Caerphilly Heart Study. Circulation 91: 270-274, 1995.
Melargno M, and Fink G. Inhibition of the slow pressor effect of angiotensin II contributes to the antihypertensive effect of angiotensin converting enzyme inhibitors in renovascular hypertension. J Pharmacol Exp Ther 278: 297-303, 1996.
Mendelsohn ME, and Karas RH. The protective effects of estrogen on the cardiovascular system. N Engl J Med 340: 1801-1811, 1999.
Mendes AC, Ferreira AJ, Pinheiro SV, and Santos RA. Chronic infusion of angiotensin-(1-7) reduces heart angiotensin II levels in rats. Reg Peptides 125: 29-34, 2005.
Metcalfe BL, Huentelman MJ, Parilak LD, Taylor DG, Katovich MJ, Knot HJ, Sumners C, and Raizada MK. Prevention of cardiac hypertrophy by angiotensin II type-2 receptor gene transfer. Hypertension 43: 1233-1238, 2004.
Miner EC, and Miller WL. A look between the cardiomyocytes: the extracellular matrix in heart failure. Mayo Clin Proc 61: 71-76, 2006.

109
Morschl E, Bretus I, Nemcsik J, Laszlo F, and Pavo I. Estrogen-mediated up-regulation of the Ca-dependent constitutive nitric oxide synthase in the rat aorta and heart. Life Sci 68: 49-55, 2000.
Nagasawa K, Zimmermann R, Munkel B, Linz W, Scholkens B, and Schaper J. Extracellular matrix deposition in hypertensive hearts antifibrotic effects of ramipril. Eur Heart J 16: 33-37, 1995.
Nakano M, Kanda T, Matsuzaki S, Hasegawa S, Ma H, Imai S, Suzuki T, and Kobayashi I. Effect of losartan, an AT1 selective angiotensin II receptor antagonist, on isoproterenol-induced cardiac ornithine decarboxylase activity. Res Commun Mol Pathol Pharmacol 88: 21-30, 1995.
Nehme JA, Lacolley P, Labat C, Challande P, Robidel E, Perret C, Leenhardt A, Safar ME, Delcayre C, and Milliez P. Spironolactone improves carotid artery fibrosis and distensibility in rat post-ischaemic heart failure. J Mol Cell Cardiol 39: 511-519, 2005.
Neves LA, Averill DB, Ferrario CM, Aschner JL, and Brosnihan KB. Vascular responses to angiotensin-(1-7) during the estrous cycle. Endocrine 24: 161-165, 2004.
Neves MF, Amiri F, Virdis A, Diep QN, and Schiffrin EL. Role of aldosterone in angiotensin II-induced cardiac and aortic inflammation, fibrosis, and hypertrophy. Can J Physiol Pharmacol 83: 999-1006, 2005.
Neves, LA, Williams AF, Averill DB, Ferrario CM, Walkup MP, and Brosnihan KB. Pregnancy enhances the angiotensin (Ang)-(1-7) vasodilator response in mesenteric arteries and increases the renal concentration and urinary excretion of Ang-(1-7). Endocrinology 144: 3338-3343, 2003.
Nickenig G, Strehlow K, Wassmann S, Baumer AT, Albory K, Sauer H, and Bohm M. Differential effects of estrogen and progesterone on AT(1) receptor gene expression in vascular smooth muscle cells. Circulation 102: 1828-1833, 2000.
Nickenig, G, Baumer AT, Grohe C, Kahlert S, Strenlow K, Rosenkranz S, Stablein A, Beckers F, Smits JF, Daemon MJ, Vetter H, and Bohm M. Estrogen modulates AT1 receptor gene expression in vitro and in vivo. Circulation 97: 2197-2201, 1998.
Nio Y, Matsubara H, Murasawa S, Kanasaki M, and Inada M. Regulation of gene transcription of angiotensin II receptor subtypes in myocardial infarction. J Clin Invest. 95: 46-54, 1995.
Nishikawa K. Angiotensin AT1 receptor antagonism and protection against cardiovascular end-organ damage. J Hum Hypertens 12: 301-9, 1998.

110
Nordmeyer J, Eder S, Mahmoodzadeh S, Martus P, Fielitz J, Bass J, Bethke N, Zurbrugg HR, Pregla R, Hetzer R, and Regitz-Zagrosek V. Upregulation of myocardial estrogen receptors in human aortic stenosis. Circulation 110: 3270-3275, 2004.
O'Donnell CJ, Lindpaintner K, Larson MG, Rao VS, Ordovas JM, Schaefer EJ, Myers RH, and Levy D. Evidence for association and genetic linkage of the angiotensin-converting enzyme locus with hypertension and blood pressure in men but not women in the Framingham Heart Study. Circulation 97: 1766-1772, 1998.
Okada H, Takemura G, Kosai K, Li Y, Takahashi T, Esaki M, Yuge K, Miyata S, Maruyama R, Mikami A, Minatoguchi S, Fujiwara T, Fujiwara H. Postinfarction gene therapy against transforming growth factor-beta signal modulates infarct tissue dynamics and attenuates left ventricular remodeling and heart failure. Circulation. 111: 2430-2437, 2005.
Oudit GY, Crackower MA, Backx PH, and Penninger JM. The role of ACE2 in cardiovascular physiology. Trends Cardiovasc Med 13: 93-101, 2003.
Page IH, Helmer OM. A crystalline pressor substance: angiotonin. Proc Center Soc Clin Invest 12: 17, 1939.
Parmley WW. Evolution of angiotensin-converting enzyme inhibition in hypertension, heart failure, and vascular protection. Am J Med 105: 27s-31s, 1998.
Patten RD, Pourati I, Aronovitz MJ, Baur J, Celestin F, Chen X, Michael A, Haq S, Nuedling S, Grohe C, Force T, Mendelsohn ME, and Karas RH. 17beta-estradiol reduces cardiomyocyte apoptosis in vivo and in vitro via activation of phospho-inositide-3 kinase/Akt signaling. Circ Res 95: 692-699, 2004.
Pfeffer MA, Braunwald E, Moye LA, Basta L, Brown EJ Jr, Cuddy TE, Davis BR, Geltman EM, Goldman S, and Flaker GC. Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction. Results of the survival and ventricular enlargement trial. The SAVE Investigators. N Engl J Med 327: 669-677, 1992.
Pinheiro SV, Simoes e Silva AC, Sampaio WO, de Paula RD, Mendes EP, Bontempo ED, Pesquero JB, Walther T, Alenina N, Bader M, Bleich M, and Santos RA. Nonpeptide AVE 0991 is an angiotensin-(1-7) receptor Mas agonist in the mouse kidney. Hypertension 44: 490-496, 2004.
Pinto FM, Armesto CP, Magraner J, Trujillo M, Martin JD, and Candenas ML. Tachykinin receptor and neutral endopeptidase gene expression in the rat uterus: characterization and regulation in response to ovarian steroid treatment. Endocrinology 140: 2526-2532, 1999.

111
Pinto YM, de Smet BG, van Gilst WH, Scholtens E, Monnink S, de Graeff PA, and Wesseling H. Selective and time related activation of the cardiac renin-angiotensin system after experimental heart failure: relation to ventricular function and morphology. Cardiovasc Res 27: 1933-8, 1993.
Pitt B, Remme W, Zannad F, Neaton J, Martinez F, Roniker B, Bittman R, Hurley S, Kleiman J, and Gatlin M. Eplerenone post-acute myocardial infarction heart failure efficacy and survival study investigators. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med 348: 1309-21, 2003.
Pitt B, Segal R, Martinez FA, Meurers G, Cowley AJ, Thomas I, Deedwania PC, Ney DE, Snavely DB, and Chang PI. Randomised trial of losartan versus captopril in patients over 65 with heart failure (Evaluation of Losartan in the Elderly Study, ELITE). Lancet 349: 747-52, 1997.
Pitt B, Zannad F, Remme WJ, Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A, Palensky J, Wittes J, Randomized Aldactone Evaluation Study Investigators. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. N Engl J Med 341: 709-717, 1999.
Raizada MK, Francis SC, Wang H, Gelband CH, Reaves PY, and Katovich MJ. Targeting of the renin-angiotensin system by antisense gene therapy: a possible strategy for the long-term control of hypertension. J Hypertens 18: 353-362, 2000.
Rajagopalan S, Kurz S, Munzel T, Tarpey M, Freeman BA, Griendling KK, and Harrison DG. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J Clin Invest 97: 1916-1623, 1996.
Rasband, WS. ImageJ, U.S. National Institutes of Health, Bethesda, MD, USA, 1997-2005. [Software available at http://rsb.info.nih.gov/ij/]
Regitz-Zagrosek V, and Lehmkuhl E. Heart failure and its treatment in women. Herz 30: 356-367, 2005.
Ren Y, Garvin JL, and Carretero OA. Vasodilator action of angiotensin-(1-7) on isolated rabbit afferent arterioles. Hypertension 39: 799-802, 2002.
Rigat B, Hubert C, Alhenc-Gelas, F, Cambien F, Corvol P, and Soubrier F. An insertion/deletion polymorphism in the angiotensin I-converting enyzme gene accounting fro half the variance of serum enzyme levels. J Clin Invest 86: 1343-1346, 1990.
Roberts AB, Sporn MB, Assoian RK, Smith JM, Roche NS, Wakefield LM, Heine UI, Liotta LA, Falanga V, Kehrl JH, and Fauci AS. Transforming growth factor type beta: rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc Natl Acad Sci U S A 83: 4167-4171, 1986.

112
Rocha R, and Stier CT. Pathophysiological effects of aldosterone in cardiovascular tissues. Trends Endrocrinol Metab 12: 308-314, 2001.
Rocha R, Stier CT Jr, Kifor I, Ochoa-Maya MR, Rennke HG, Williams GH, and Adler GK. Aldosterone: a mediator of myocardial necrosis and renal arteriopathy. Endocrinology 141: 3871-8, 2000.
Roks AJ, Nijholt J, van Buiten A, van Gilst WH, de Zeeuw D, Henning RH. Low sodium diet inhibits the local counter-regulator effect of angiotensin-(1-7) on angiotensin II. J Hypertens 22: 2355-2361, 2004.
Rombouts K, Wielant A, Hellemans K, Schuppan D, and Geerts A. Influence of aldosterone on collagen synthesis and proliferation of rat cardiac fibroblasts. Br J Pharmacol 134: 224-23, 2001.
Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, Jackson RD, Beresford, SA, Howard BV, Johnson KC, Kotchen JM, and Ockene J. Risks and benefits of estrogen plus progestin in healthy menopausal women: principal results from the Women’s Health Initiative Randomized Controlled Trial. JAMA 288: 321-333, 2002.
Rugale C, Delbosc S, Cristol JP, Mimran A, and Jover B. Sodium restriction prevents cardiac hypertrophy and oxidative stress in angiotensin II hypertension. Am J Physiol Heart Circ Physiol 284: H1744-H1750, 2003.
Ruiz-Ortega M, Lorenzo O, Ruperez M, Esteban V, Suzuki Y, Mezzano S, Plaza JJ, and Egido J. Role of the renin-angiotensin system in vascular diseases: expanding the field. Hypertension 38: 1382-1387, 2001.
Saito, S, Aras RS, Lou H, Ramwell PW, and Foegh ML. Effects of estrogen on nitric oxide synthase expression in rat aorta allograft and smooth muscle cells. J Heart Lung Transplant 18: 937-945, 1999.
Sampaio WO, Nascimento AA, and Santos RA. Systemic and regional hemodynamic effects of angiotensin-(1-7) in rats. Amer J Physiol 284: H1985-H1994, 2003.
Santos JC, Jerez S, Peral de Bruno M, and Coviello A. Angiotensin-(1-7) increases osmotic water permeability in isolated toad skin. Braz J Med Biol Res 33: 1099-1104, 2000.
Santos RA, Brum JM, Brosnihan KB, and Ferrario CM. The renin-angiotensin system during acute myocardial ischemia in dogs. Hypertension 15: I121-I127, 1990.
Santos RA, Campagnole-Santos MJ, and Andrade SP. Angiotensin-(1-7): an update. Reg Peptides 91: 45-62, 2000.

113
Santos RA, Castro CH, Gava E, Pinheiro SV, Almeida AP, de Paula RD, Cruz JS, Ramos AS, Rosa KT, Irigoyen MC, Bader M, Alenina N, Kitten GT, Ferreira AJ. Impairment of in vitro and in vivo heart function in angiotensin-(1-7) receptor mas knockout mice. Hypertension. [Epub ahead of print], 2006.
Santos RA, Ferreira AJ, Nadu AP, Braga AN, de Almeida AP, Campagnole-Santos MJ, Baltatu O, Iliescu R, Reudelhuber TL, and Bader M. Expression of an angiotensin-(1-7)-producing fusion protein produces cardioprotective effects in rats. Physiol Genomics 17: 292-299, 2004.
Santos, RAS, Simoes e Silva AC, Maric C, Silva DMR, Machado RP, de Buhr I, Heringer-Walther S, Pinheiro SVB, Lopes MT, Bader M, Mendes EP, Lemos VS, Campagnole-Santos MJ, Schultheiss HP, Speth R, and Walther T. Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. PNAS 100: 8258-8263, 2003.
Sarkar S, Vellaichamy E, Young D, and Sen S. Influence of cytokines and growth factors in ANG II-mediated collagen upregulation by fibroblasts in rats: role of myocytes. Am J Physiol Heart Circ Physiol. 287: H107-H117, 2004.
Sasaki S, Higashi Y, Nakagawa K, Matsuura H, Kajiyama G, and Oshima T. Effects of angiotensin-(1-7) on forearm circulation in normotensive subjects and patients with essential hypertension. Hypertension 38: 90-94, 2001.
Schachter F, Faure-Delanef L, Guenot F, Rouger H, Froguel P, Lesueur-Ginot L, and Cohen D. Genetic associations with human longevity at the APOE and ACE loci. Nature Genetics 6: 29-32, 1994.
Schnee JM, and Hsueh WA. Angiotensin II, adhesion, and cardiac fibrosis. Cardiovasc Res 46: 264-268, 2000.
Schunkert H, Hense HW, Holmer SR, Stender M, Perz S, Keil U, Lorell BH, and Riegger GA. Association between a deletion polymorphism of the angiotensin-converting-enzyme gene and left ventricular hypertrophy. N Engl J Med 330: 1634-1638, 1994.
Schunkert H, Tang SS, Litwin SE, Diamant D, Riegger G, Dzau VJ, and Ingelfinger JR. Regulation of intrarenal and circulating renin-angiotensin systems in severe heart failure in the rat. Cardiovasc Res 27: 731-735, 1993.
Schunkert, H, Danser AH, Hense HW, Derkx FH, Kurzinger S, and Riegger GA. Effects of estrogen replacement therapy on the renin-angiotensin system in post-menopausal women. Circulation 1997;95:39-45.
Schwertz DW, Vizgirda V, Solaro RJ, Piano MR, and Ryjewski C. Sexual dimorphism in rat left atrial function and response to adrenergic stimulation. Mol Cell Biochem 200: 143-153, 1999.

114
Seli E, Senturk LM, Bahtiyar OM, Kayisli UA, and Arici A. Expression of aminopeptidase N in human endometrium and regulation of its activity by estrogen. Fertil Steril 75: 1172-1176, 2001.
Shibata MC, Flather MD, and Wang D. Systematic review of the impact of beta blockers on mortality and hospital admissions in heart failure. Eur J Heart Fail 3: 35l-351, 2001.
Simon G, Abranham G, and Cserep G. Pressor and subpressor angiotensin II administration. Two experimental models of hypertension. Am J Hypertension 8: 645-650, 1995.
Simon JA, Hsia J, Cauley JA, Richards C, Harris F, Fong J, Barrett-Connor E, and Hulley SB. Postmenopausal hormone therapy and risk of stroke: The Heart and Estrogen-progestin Replacement Study (HERS). Circulation 103: 638-642, 2001.
Stegbauer J, Vonend O, Oberhauser V, and Rump LC. Effects of angiotensin-(1-7) and other bioactive components of the renin-angiotensin system on vascular resistance and noradrenaline release in rat kidney. J Hypertens 21: 1391-1399, 2003.
Stenmark KR, Gerasimovskaya E, Nemenoff RA, Das M. Hypoxic activation of adventitial fibroblasts: role in vascular remodeling. Chest. 122: 326S-334S, 2002.
Stier CT Jr, Adler LA, Levine S, and Chander PN. Stroke prevention by losartan in stroke-prone spontaneously hypertensive rats. J Hypertens Suppl 11: S37-S42, 1993.
Stier CT Jr, Benter IF, Ahmad S, Zuo HL, Selig N, Roethel S, Levine S, and Itskovitz HD. Enalapril prevents stroke and kidney dysfunction in salt-loaded stroke-prone spontaneously hypertensive rats. Hypertension 13: 115-21, 1998.
Stier CT Jr, Chander P, Gutstein WH, Levine S, and Itskovitz HD. Therapeutic benefit of captopril in salt-loaded stroke-prone spontaneously hypertensive rats is independent of hypotensive effect. Am J Hypertens 4: 680-7, 1991.
Studies of Left Ventricular Dysfunction (SOLVD) Investigators. Effect of enalapril on mortality and the development of heart failure in asymptomatic patients with reduced left ventricular ejection fractions. N Engl J Med 327: 685-691, 1992.
Sun Y, Zhang JQ, Zhang J, and Ramires FJ. Angiotensin II, transforming growth factor-betal and repair in the infarcted heart. Mol Cell Cardiol 30: 1559-1569, 1998.
Swedberg K, Eneroth P, Kjekshus J, Wilhelmsen L, CONSENSUS Trial Study Group. Hormones regulating cardiovascular function in patients with severe congestive heart failure and their relation to mortality. Circulation 82: 1730-1736, 1990.

115
Swynghedauw B. Molecular mechanisms of myocardial remodeling. Physiol Rev 79: 215-62, 1999.
Szilagyi A, Mukhopadhyay AK, Werling J, and Szabo I. Regulation of placental prorenin secretion from the early human placenta in vitro. Early Pregnancy 1: 119-123, 1995.
Takeda Y, Yoneda T, Demura M, Usukura M, and Mabuchi H. Calcineurin inhibition attenuates mineralocorticoid-induced cardiac hypertrophy. Circulation 105: 677-679, 2002.
Tallant EA, Ferrario CM, and Gallagher PE. Angiotensin-(1-7) Inhibits growth of cardiac myocytes through activation of the mas receptor. Am J Physiol Heart Circ Physiol 289: H1560-H1566, 2005.
Taylor AH, and Al-Azzawi F. Immunolocalisation of oestrogen receptor beta in human tissues. J Mol Endocrinol 24: 145-155, 2000.
Thom T, Haase N, Rosamond W, Howard VJ, Rumsfeld J, Manolio T, Zheng ZJ, Flegal K, O'Donnell C, Kittner S, Lloyd-Jones D, Goff DC Jr, Hong Y, Adams R, Friday G, Furie K, Gorelick P, Kissela B, Marler J, Meigs J, Roger V, Sidney S, Sorlie P, Steinberger J, Wasserthiel-Smoller S, Wilson M, Wolf P; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics--2006 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 113: E85-E151, 2006.
Thurmann PA, Kenedi P, Schmidt A, Harder S, and Rietbrock N. Influence of the angiotensin II antagonist valsartan on left ventricular hypertrophy in patients with essential hypertension. Circulation 98: 2037-2042, 1998.
Tigerstedt R, and Bergman PG. Niere und Kreislauf. Skand Arch Physiol 8: 223-271, 1898.
Tipnis SR, Hooper NM, Hyde R, Karran E, and Christie, G. A human homolog of angiotensin-converting enzyme. J Biol Chem 275; 33238-33243, 2000.
Tom, B, Dendorfer A, and Danser AHJ. Bradykinin, angiotensin 1-7 and ACE inhibitors: how do they interact. Int J Biochem Cell Biol 35: 792-801, 2003.
Tremollieres FA, Pouilles JM, Cauneille C, and Ribot C. Coronary heart disease risk factors and menopause: a study in 1984 French women. Atherosclerosis 142: 415-523, 1999.
Turner AJ and Hooper NM. The angiotensin-converting enzyme gene family: genomics and pharmacology. TRENDS in Pharm Sci 23: 177-183, 2002.

116
Unger T. The role of the renin-angiotensin system in the development of cardiovascular disease. Am J Cardiol 89: 3A-9A, 2002.
van Eickels M, Grohe C, Cleutjens JP, Janssen BJ, Wellens HJ, and Doevendans PA. 17beta-estradiol attenuates the development of pressure-overload hypertrophy. Circulation 104: 1419-1423, 2001.
Vickers C, Hales P, Kaushik V, Dick L, Gavin J, Tang J, Godbout K, Parsons T, Baronas E, Hsieh F, Acton S, Patane M, Nichols A, Tummino P. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J Biol Chem. 277: 14838-14843, 2002.
von Bohlen und Halbach O. Angiotensin IV in the central nervous system. Cell Tissue Res 311: 1-9, 2003.
Weber KT. Aldosterone in congestive heart failure. N Engl J Med 345: 1689-1697, 2001.
Weber KT. Fibrosis and hypertensive heart disease. Curr Opin Cardiol 15: 264-272, 2000.
Weber KT. Hormones and fibrosis: a case for lost, reciprocal regulation. News Physiol Sci 9: 123-128, 1994.
Weber KT, and Brilla CG. Pathological hypertrophy and cardiac interstitium. Fibrosis and renin-angiotensin-aldosterone system. Circulation 83: 1849-1865, 1991.
Wei CC, Meng QC, Palmer R, Hageman GR, Durand J, Bradley WE, Farrell DM, Hankes G, Oparil S, and Dell'Italia LJ. Evidence for angiotensin-converting enzyme-and chymase-mediated angiotensin II formation in the interstitial fluid space of the dog heart in vivo. Circulation 99: 2583-2589, 1999.
Wilsdorf T, Gainer JV, Murphey LJ, Vaughan DE, and Brown NJ. Angiotensin-(1-7) does not affect vasodilator or TPA responses to bradykinin in human forearm. Hypertension 37: 1136-1140, 2001.
Wolny A, Clozel JP, Rein J, Mory P, Vogt P, Turino M, Kiowski W, and Fischli W. Functional and biochemical analysis of angiotensin II-forming pathways in the human heart. Circ Res 80: 219-227, 1997.
Wright JW, and Harding JW. Important role for angiotensin III and IV in the brain renin-angiotensin system. Brain Res Rev 25: 96-124, 1997.
Yamada K, Iyer SN, Chappell MC, Ganten D, and Ferrario CM. Converting enzyme determines plasma clearance of angiotensin-(1-7). Hypertension 32: 496-502, 1998.

117
Yasunari K, Maeda K, Nakamura M, Watanabe T, Yoshikawa J, and Hirohashi K. Left ventricular hypertrophy and angiotensin II receptor blocking agents. Curr Med Chem Cardiovasc Hematol Agents 3: 61-67, 2005.
Yoo KH, Thomhill BA, Wolstenholme JT, and Chevalier RL. Tissue specific regulation of growth factors and clusterin by angiotensin II. Am J Hypertens 11: 715-722, 1998.
Young M, and Funder J. Mineralocorticoid action and sodium-hydrogen exchange: studies in experimental cardiac fibrosis. Endocrinology 144: 3848-3851, 2003.
Young M, and Funder JW. Aldosterone and the heart. Trends Endocrinol Metab 11: 224-226, 2000.
Young M, Fullerton M, Dilley R, and Funder J. Mineralocorticoids, hypertension, and cardiac fibrosis. J Clin Invest 93: 2578-2583, 1994.
Young M, Head G, and Funder J. Determinants of cardiac fibrosis in experimental hypermineralocorticoid states. Am J Physiol 269: E657-E662, 1995.
Zaman MA, Oparil S, and Calhoun DA. Drugs targeting the renin Angiotensin-aldosterone system. Nat Rev Drug Discov 1: 621-636, 2002.
Zannad F, Alia F, Dousset B, Perez A, Pitt B, RALES Investigators. Limitation of excessive extracellular matrix turnover may contribute to survival benefit of spironolactone therapy in patients with congestive heart failure: insights from the randomized aldactone evaluation study (RALES) [published correction appears in Circulation 103: 476, 2001]. Circulation 102: 2700-2706, 2000.
Zanzinger J, and Czachurski J. Chronic oxidative stress in the RVLM modulates sympathetic control of circulation in pigs. Pflugers Arch 439: 489-494, 2000.
Zhai P, Eurell TE, Cotthaus R, Jeffery EH, Bahr JM, and Gross DR. Effect of estrogen on global myocardial ischemia-reperfusion injury in female rats. Am J Physiol Heart Circ Physiol 279: H2766-H2775, 2000.
Zhang X, Azhar G, Nagano K, and Wei JY. Differential vulnerability to oxidative stress in rat cardiac myocytes versus fibroblasts. J Am Coll Cardiol 38: 2055-2062, 2001.
Zhou G, Kandala JC, Tyagi SC, Katwa LC, and Weber KT. Effects of angiotensin II and aldosterone on collagen gene expression and protein turn., over in cardiac fibroblasts. Moll Cell Biochem 154: 171-178, 1996.
Zhu Z, Zhong J, Zhu S, Liu D, Van Der Giet M, Tepel M. Zhu Z, Zhong J, Zhu S, Liu D, Van Der Giet M, and Tepel M. Angiotensin-(1-7) inhibits angiotensin II-induced signal transduction. J Cardiov Pharmacol 40: 693-700, 2002.

118
Zimmerman MC, Lazartigues E, Lang JA, Sinnayah P, Ahmad IM, Spitz DR, and Davisson RL. Superoxide mediates the actions of angiotensin II in the central nervous system. Circ Res 91: 1038-1045, 2002.
Zisman LS, Keller RS, Weaver B, Lin Q, Speth R, Bristow MR, and Canver CC. Increased angiotensin-(1-7)-forming activity in failing human heart ventricles: evidence for upregulation of the angiotensin-converting enzyme Homologue ACE2. Circulation 108: 1707-1712, 2003.
Zou Y, Takano H, Akazawa H, Nagai T, Mizukami M, and Komuro I. Molecular and cellular mechanisms of mechanical stress-induced cardiac hypertrophy. Endocr J 49: 1-13, 2002.

BIOGRAPHICAL SKETCH
Justin L. Grobe was born November 3, 1979, to Edward E. Grobe and Loretta L.
Grobe, Ph.D. in Mason, Michigan. From a young age, he was interested in the physical,
chemical, and biological sciences. Nurtured by his parents, his caregivers Marjorie and
James Bullerdick, and the school district’s Gifted and Talented Coordinator, Trena
Thornburg, Justin had many advanced educational opportunites from a young age. His
early interests in science and mathematics were manifested in his involvement in the
Mason Middle School Science Olympiad Team, coached by teachers Jeffrey Ehlers and
David Dodendorf. During his four years on the team, Justin personally won twenty-two
regional, fifteen state, and two national awards and shared several team honors as well.
Justin’s aptitude toward science and mathematics earned him an invitation to a
newly developed mathematics program at Michigan State University. This “Cooperative
Highly Accelerated Mathematics Program (CHAMP)” allowed him the opportunity to
complete courses in algebra I and II, geometry, trigonometry, and pre-calculus by the end
of 9th grade. Early completion of these courses prepared Justin for the Honors Calculus I
and II courses at Michigan State University during his sophomore year of high school, in
addition to several courses at Lansing Community College. Dual-enrollment at these
higher education institutions and Mason High School allowed Justin to complete
requirements for graduation after only three years.
Upon graduation, Justin enrolled as an undergraduate at Hope College, in Holland,
Michigan. During his first week, Justin attended a research symposium summarizing the
119

120
previous summer’s findings by undergraduate researchers at Hope College. One of these
presenters was Bradley Andresen, a college senior working in the laboratory of
Christopher C. Barney, Ph.D. After some discussion of the project, Justin was invited to
try working in Dr. Barney’s laboratory to examine his interest in animal research.
Over the next four years, Justin continued working in Dr. Barney’s laboratory, and
their work resulted in several scientific publications, and multiple poster presentations at
both the Pew Midstates Undergraduate Research Symposium and the Experimental
Biology annual meetings. In the summer of 1998, Justin received a National Science
Foundation Research Experiences for Undergraduates (NSF-REU) Fellowship through
Hope College to work in Dr. Barney’s laboratory. In the summer of 1999, Justin received
an individual undergraduate research fellowship from the American Heart Association to
continue his work. Then, in the summer of 2000, Justin was awarded a summer NSF-
REU position at the University of Florida, Department of Psychology, and he worked in
the laboratories of Neil E. Roland, Ph.D. and Michael J. Katovich, Ph.D. Finally, in the
spring of 2001, Justin graduated from Hope College with a Bachelor of Science in
biology, and a Bachelor of Arts in chemistry. Having enjoyed working with Dr.
Katovich the previous summer, Justin applied to graduate school at the University of
Florida, College of Pharmacy’s Department of Pharmacodynamics, with the intention of
pursuing his Ph.D. under the tutelage of Dr. Katovich.