the origin of reactive oxygen species in mouse …...the origin of reactive oxygen species in mouse...
TRANSCRIPT

Development 113, 551-560 (1991)Printed in Great Britain © The Company of Biologists Limited 1991
551
The origin of reactive oxygen species in mouse embryos cultured in vitro
MOHAMMAD M. NASR-ESFAHANI and MARTIN H. JOHNSON*
Department of Anatomy, Downing Street, Cambridge CB2 3DY, UK* and Assisted Conception Research Unit, Department of Obstetrics andGynaecology, 6th Floor, Ward Block, St Thomas's Hospital, London SE1 7EH, UK
* Author and address for correspondence
Summary
The increase in production of reactive oxygen speciessuch as H2O2 at the G2/M phase of the second cell cyclemay be related to the in vitro block to development ofmouse 2-cell embryos. The occurrence of the H2O2 rise isindependent of the activation of the embryonic genomeand of passage through the S, G2 and M phases of thefirst cell cycle and Gx and M phases of the second cellcycle, but does require the activation of the unfertilizedoocyte. The H2O2 is produced via dismutation ofsuperoxlde by the enzyme superoxide dismutase. Pro-duction of superoxide via mitochondrial, NADPH-
oxidase and xanthine/xanthine oxidasc systems has beeninvestigated. The evidence suggests that superoxide, andthereby H2O2, is produced by the xanthine/xanthineoxidase system, but an involvement of the othersuperoxide generating systems has not been excluded.The relation between H2O2 and development in vitro isdiscussed.
Key words: hydrogen peroxide, superoxide, 2-cell block,embryo, oxygen radicals, reactive oxygen species.
Introduction
Embryos from most outbred and inbred mouse strainsdo not develop to blastocysts when cultured in achemically defined medium but arrest at the 2-cellstage, a phenomenon referred to as 'the two-cell block'(Whittingham, 1974). We reported recently that theperiod of in vitro development at which this blockoccurs (the mid 2-cell stage to the early 4-cell stage) isassociated with a rise in reactive oxygen species such asH2O2 or lipid peroxides (Nasr-Esfahani et al. 1990a).This rise only occurred after culture in vitro andregardless of the total time spent in vitro. It was notobserved in embryos of the same age recovered directlyfrom the female tract, nor was it observed in embryos atother preimplantation stages that had been cultured invitro. We concluded that the rise in peroxide levels wasa response to in vitro culture dependent on the stage ofdevelopment. We suggested that the presence ofelevated levels of reactive oxygen species under theseconditions might be related in some way to the 2-cellblock, a conclusion also reached recently by Noda et al.(1991).
Reactive oxygen species such as H2O2, in conjunctionwith superoxide (O2°), can cause cell damage bypromoting hydroxy radical formation via the iron-
catalysed Haber-Weiss reaction (Halliwell, 1987; Hal-liwell and Gutteridge, 1987):
(1)(2)
Net reaction: (3)
Fe3++O2°Fe2 +
2°H 2 O 2 •
O2 + Fe2+
Fe3+ + HO0 + OH"
O2° H2O2 -> O2 + HO0 + OH"
This reaction requires the presence of iron. Iron canalso act directly on lipids to magnify peroxidativedamage once this has been initiated by free hydroxyradicals (Minotti and Aust, 1989). It is thereforeimportant to note that the addition of bovine apo-transferrin and certain other iron chelators to theculture medium to sequester free traces of ironpromotes development through the two-cell block(Nasr-Esfahani et al. 1990b), further supporting theidea that the rise in reactive oxygen species may beinvolved in the block.
In this paper we examine the origin of the rise inreactive oxygen species and its dependence on the firstand second cell cycles. We use the technique describedpreviously to measure the rise (Nasr-Esfahani et al.1990a). The principle underlying this procedure may bedescribed briefly as follows: 2',7'-dichlorodihydrofluor-escein diacetate (DCHFDA), because of its non-ionized state, is membrane permeant and therefore is

552 M. Nasr-Esfahani and M. H. Johnson
able to diffuse readily into cells. Within the cell, theacetate groups are hydrolyzed by intracellular esteraseactivity forming 2',7'-dichlorodihydrofluorescein(DCHF) which is polar and thus trapped within the cell.DCHF fluoresces when it is oxidised by H2O2 or lipidperoxides to yield 2',7'-dichlorofluorescein (DCF). Thelevel of DCF produced within the cells is relatedlinearly to that of peroxides present and thus itsfluorescent emission provides a measure of the peroxidelevels.
Materials and methods
Recovery and handling of oocytes and embryosMF1 female mice (3-4 weeks; OLAC) and ¥x (3-4 weeks;C57BL/lOScSn/OLa femalesxCBA/Ca/OLa male, bred inlaboratory) were superovulated by intraperitoneal injection of5 or 10 i.u. of pregnant mare's serum gonadotrophin (PMS;Intervet) and human chorionic gonadotrophin (hCG; Inter-vet) 48 h apart. To obtain embryos, females were pairedindividually overnight with HC-CFLP males (Interfauna) andinspected for vaginal plugs the next day as an indication ofsuccessful mating.
Unfertilized oocytes were recovered from unmated femalesat about 13 h post-hCG. Fertilized eggs at the pronuclearstage were recovered from mated females at between 21 and30 h post-hCG as indicated in the Results. Oocytes andembryos were released from the oviduct into warmedH6+BSA (a Hepes buffered form of modified T6 medium;Nasr-Esfahani et al. 1990/?) and then cultured in drops ofT6+BSA or BAT6+BSA (Nasr-Esfahani et al. 19906) underparaffin oil (FSA Suppliers, Loughborough, UK) in Falcontissue culture dishes in 5 % CO2 in air. All manipulations werecarried out at 37°C on heated stages, pads or in incubators.
In some experiments, oocytes were activated by exposureto 8% ethanol in H6+BSA for 8-10min (Kaufman, 1983),rinsed, placed in culture and inspected for activation 4 h later.Non-activated and deformed oocytes were removed, and theremaining activated oocytes were divided into two groups: (i)those that had undergone immediate cleavage to two cells andin which each cell had a single pronucleus - designated'immediate cleavage', and (ii) those with a single pronucleusand a polar body plus those containing two pronuclei and nosecond polar body - together designated 'activated'.
1-cell fertilized oocytes were cultured under the conditionsspecified in the Results section and inspected at regularintervals as follows: at 47-50 h post-hCG the few 1-cell orabnormal eggs were removed and are not included in thetotals, the remaining embryos being scored as 2-, 3- or 4-cells;at 69-72 h post-hCG embryos were scored as being dead,2-cell, 3-cell, 4-cell, 5- to 8-cell precompact or compact; at96-100 h post-hCG embryos were scored as being dead,noncompact, compact, early blastocyst or expanded blasto-cyst (see Chisholm, Johnson, Warren, Fleming and Pickering,1985, for definitions of blastocyst subtypes); at 116-119 h post-hCG embryos were scored as being dead, preblastocyst, earlyblastocyst or expanded blastocyst.
Measurement of reactive oxygen speciesStock solutions of DCHFDA (Kodak, Eastman KodakCompany, Rochester, NY, USA) were prepared in acetone at1 x 10"3 M. The stock solutions were diluted in H6+PVP to therequired concentration. DCHFDA stock solutions wereprepared just before the start of each experiment and werekept in the dark and used over a maximum period of 48 h.
Oocytes, parthenogenotes and embryos were washedthoroughly in H6+PVP after their removal from H6,T6+BSA or the condition specified in the Results section, andthen loaded with the dye in cavity blocks for 15min. Theoocytes or embryos were then washed in H6+BSA to removetraces of the dye and were placed in specially designed smallchambers containing H6+BSA and covered by a coverslip(Maroetal. 1984).
The fluorescence emissions of the oocytes and embryoswere measured immediately by photocytometry, using aPerspex carrying slide for viewing with a long workingdistance x32 objective on a Leitz Ortholux II microscope withstabilized HBO100 mercury vapor lamp and filter set LI forFITC. For quantitation of fluorescence, the photomultiplierhousing of a Leitz MPV-1 was fitted to the Ortholux Uphototube (McConnell et al. 1990). The phototube containeda variable measuring diaphragm that could be adjusted tosurround the periphery of an individual oocyte or embryo,thus excluding background. A 6.25% transmission neutraldensity filter (Leitz N16) was placed in the path of the excitinglight, to minimise any potential damage to the cells.Fluorescent emission was deflected to the amplifier/discriminator (Model 1140B, SSR Instruments Co, USA) of aquantum photometer (Model 1140A, SSR Instruments) thathad been zeroed against background and set to read in actss"1 mode via a deflection meter. The individual oocyte orembryo was positioned within the adjusted diaphragm andexposed to the excitation wavelength for a period of less than10 s and the fluorescent emission recorded (ctss"1 on the 1Mscale to a maximum reading of 10). In some experiments, twoneutral density filters were used so that readings higher than10 could be measured. For each datum point in eachexperiment, the fluorescent emissions of 10 to 20 oocytes orembryos were measured and their mean values wereexpressed as the 'mean reading'.
This set-up, involving short exposure to the exciting light,was designed to avoid damage to the oocytes or embryos,which was only detected with greater than a 40 s exposure tothe exciting light or removal of the 6.25 % transmissionneutral density filter. Under these latter conditions, a rapidrise in fluorescent emission, reflecting conversion of DCHF toDCF, was detected as a secondary consequence of light-induced lipid peroxidation. This latter property was used byus as a positive control to show that the uptake anddeesterification of DCHFDA was not limiting the fluorescentsignal that we measured under any given condition. Thus,sample embryos were exposed to a brief pulse of highintensity light and the emission value recorded (see Nasr-Esfahani et al. 1990a).
Chromosomal staining1-cell fertilized zygotes, treated with nocodazole or aphidicol-in, were sampled at various times, fixed in 4% formaldhydefor 30min, rinsed in PBS, permeabilised in 0.2% TritonX-100 for lOmin, rinsed in PBS, exposed to ammoniumchloride (26 nig ml"1) for lOmin, rinsed in PBS and stainedwith Hoeschst 33342 at 0.1/jgml"1 in PBS for 30min. Thesamples were rinsed in PBS, mounted in Citiflour (CityUniversity, London, UK) and examined on the LeitzOrtholux II microscope.
One-dimensional SDS-polyacrylamide gelelectrophoresisEmbryos were cultured for lh in a dilution of 5 /A of[35S]methionine (1000-1400 CimmoP1, Amersham Inter-national Ltd) in 45 d̂ T6+BSA with or without drugs.Following the labelling period, groups of 25 embryos were

Reactive oxygen species 553
washed thoroughly in H6+PVP and stored at -70°C beforeseparation of proteins on 10% SDS-polyacrylamide gel asdescribed previously (Flach et al. 1982). Following electro-phoresis, gels were treated with Amplify (Amersham Inter-national) and exposed to preflashed Fuji X-ray film at -70°C.
ChemicalsChemicals used in these experiments were obtained andstored as follows: Q'-amanitin (Boehringer-Mannheim, FRG)made up as an 11 mg ml"1 stock in distilled water; Nocodazole(Aldrich) made up as a 10 ELM stock solution in DSMO;Hoechst dye 33342 (Sigma) stored as a stock solution at1 mg mF 1 saline; anisomycin (gift from Pfizer) was stored as astock of 10 nw in H6; aphidicolin (ICI) made up as a stocksolution of 2mgml~1 DMSO; phorbol myristate acetate(PMA) was made up as a stock of 25 fig ml"* DMSO (Sigma);hydroethidine (Polysciences) was made up at 5mM in N,N-dimethylformamide (BDH); hypoxanthine, allopurinol, oxy-purinol, 6-amino nicotinamide, antimycin-A, rotenone, dini-trophenol and azide were obtained from Sigma. Diphenyl-eneiodonium (DPI) was a gift from Dr B. Meier, ChemischesInstitut, Hannover. Cyanide and ethanol were obtained fromBDH (England).
Results
The peroxide rise does not occur in the absence ofoocyte activationThe rise in peroxides occurs between mid to late G2 andearly M phase of the 2-cell stage, which, in mostexperimental groups of embryos, covers the period 45to 53 h post-hCG, although in more slowly developing
10'
I 8S-3" I 6
II 4
E 2
0
10-1
18 22 32 34 36 39 41 44 46 49 50 51 56 60Time in hours post-hCG
Fig. 1. Unfertilized and fertilized oocytes were recoveredat 13 and 21 h post-hCG respectively and their peroxidelevels measured at various intervals as indicated.
groups it can be delayed by up to 5 h (discussed in Nasr-Esfahani et al. 1990a). To determine whether the rise atthis time depended upon activation of oocytes or simplyreflected the length of time since ovulation, two types ofcomparison were made. First, fertilised and unfertilisedoocytes were cultured in vitro and their peroxide levelscompared. Only fertilised oocytes showed the rise inH2O2, unfertilised oocytes showing declining levelsover the same period (Fig. 1). Second, sperm-activatedoocytes were compared with oocytes activated par-thenogenetically by ethanol. Two such experiments(Fig. 2) revealed a rise in H2O2 levels regardless of
22 24 27 28 33 36 42 45
Time in hours post-hCG
1 «n s 6- fti
46 48 49 50
• Fertilized
• A-IMC
H Activated
52 55
21 27 29 35 36 39 41Time in hours post-hCG
44 47 49
Fig. 2. Unfertilized and fertilized oocytes were recovered at 12 and 21 h post-hCG respectively. The unfertilized oocyteswere activated by 8 % ethanol and then sorted into those showing immediate cleavage (A-IMC) and those with 2 pronucleior 1 pronucleus and a polar body (Activated). The peroxide levels of embryos in each group were assessed at differentintervals and compared with those for fertilised oocytes in the same experiment. The results of two such experiments areshown. An asterisk indicates that some readings went off scale, so error bars are not shown.
554 M. Nasr-Esfahani and M. H. Johnson
5
45 51Time in hours post-hCG
1 • T6-Control
2 • Aphidicolin (23 h post hCG)
3 • Nocodazole (26 h post hCG)
4 0 Nocodazole (36 h post hCG)
5 • Alpha-Amanitin (42 h post hCG)
6 D Alpha-Amanitin (26 h post hCG)
7 M Anisomycin (34 h post hCG)
8 E3 Anisomycin (44 h post hCG)
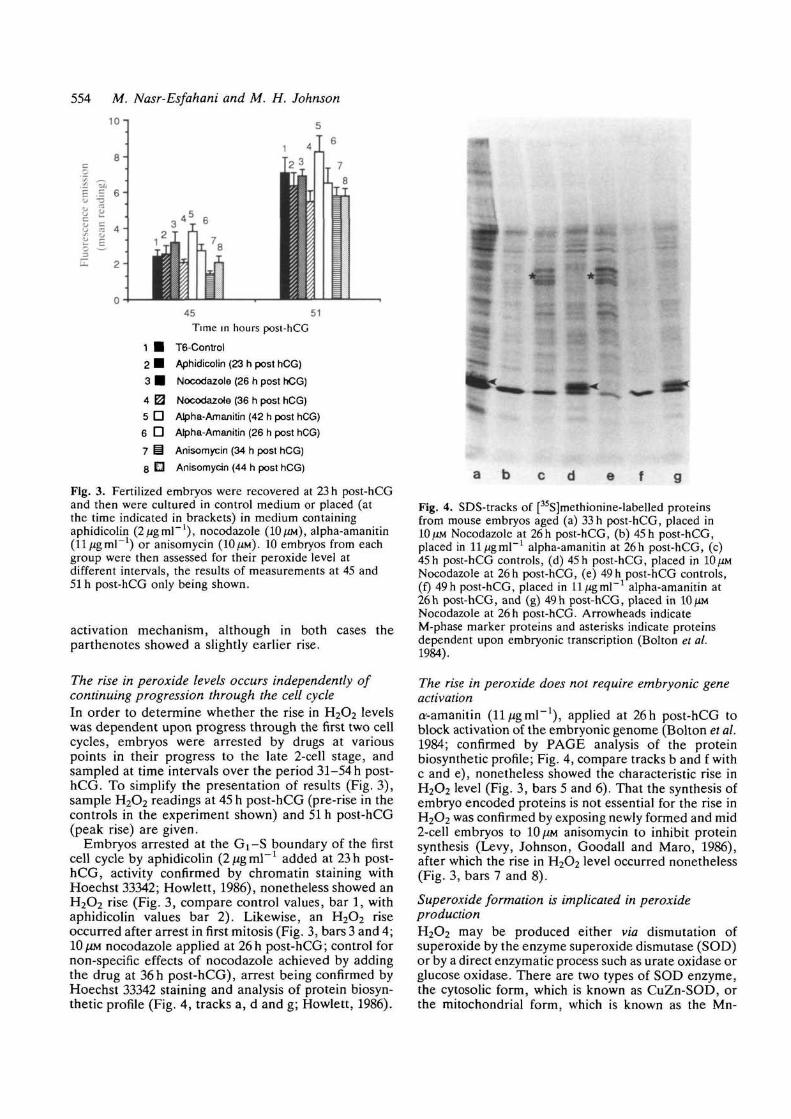
Fig. 3. Fertilized embryos were recovered at 23 h post-hCGand then were cultured in control medium or placed (atthe time indicated in brackets) in medium containingaphidicolin (2/*gml~l), nocodazole (10/^M), alpha-amanitin(ll^gml"1) or anisomycin (10/ZM). 10 embryos from eachgroup were then assessed for their peroxide level atdifferent intervals, the results of measurements at 45 and51 h post-hCG only being shown.
activation mechanism, although in both cases theparthenotes showed a slightly earlier rise.
The rise in peroxide levels occurs independently ofcontinuing progression through the cell cycleIn order to determine whether the rise in H2O2 levelswas dependent upon progress through the first two cellcycles, embryos were arrested by drugs at variouspoints in their progress to the late 2-cell stage, andsampled at time intervals over the period 31-54 h post-hCG. To simplify the presentation of results (Fig. 3),sample H2O2 readings at 45 h post-hCG (pre-rise in thecontrols in the experiment shown) and 51 h post-hCG(peak rise) are given.
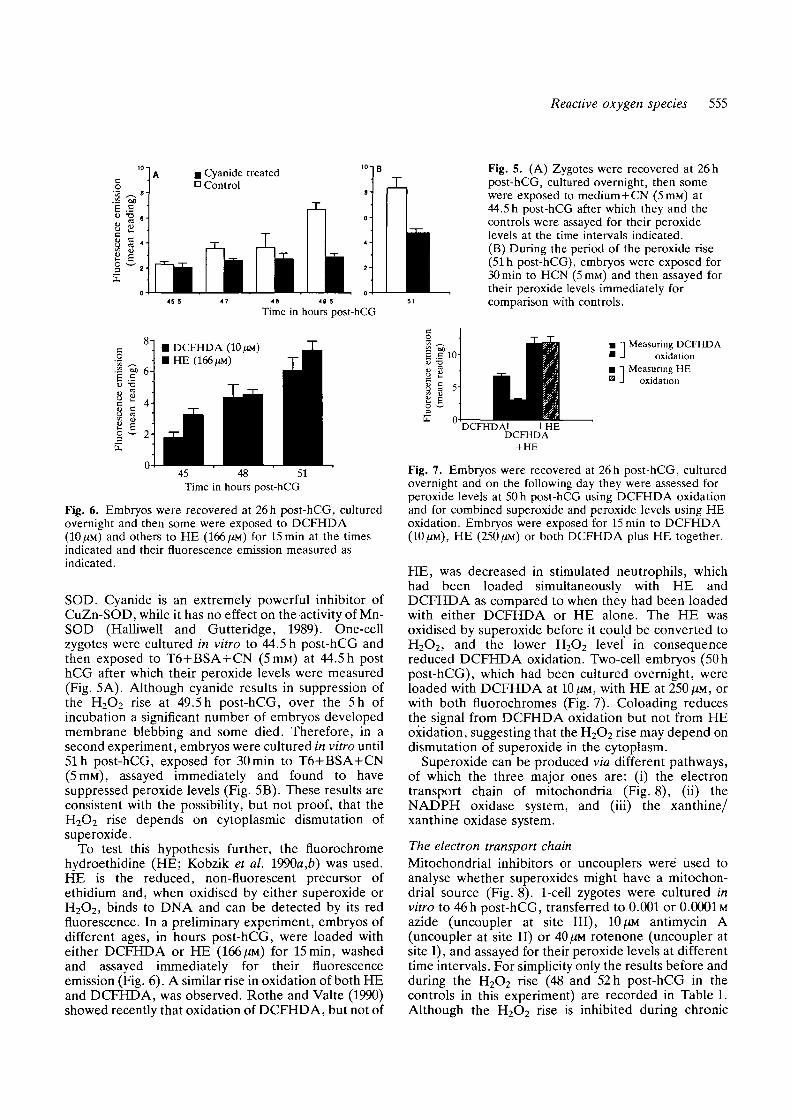
Embryos arrested at the Gi-S boundary of the firstcell cycle by aphidicolin {2ugm\~l added at 23 h post-hCG, activity confirmed by chromatin staining withHoechst 33342; Howlett, 1986), nonetheless showed anH2O2 rise (Fig. 3, compare control values, bar 1, withaphidicolin values bar 2). Likewise, an H2O2 riseoccurred after arrest in first mitosis (Fig. 3, bars 3 and 4;10/XM nocodazole applied at 26h post-hCG; control fornon-specific effects of nocodazole achieved by addingthe drug at 36h post-hCG), arrest being confirmed byHoechst 33342 staining and analysis of protein biosyn-thetic profile (Fig. 4, tracks a, d and g; Howlett, 1986).
a e f
Fig. 4. SDS-tracks of [3iS]methionine-labelled proteinsfrom mouse embryos aged (a) 33 h post-hCG, placed in10 tm Nocodazole at 26 h post-hCG, (b) 45 h post-hCG,placed in ll^gml"1 alpha-amanitin at 26 h post-hCG, (c)45 h post-hCG controls, (d) 45 h post-hCG, placed inNocodazole at 26 h post-hCG, (e) 49 h post-hCG controls,(f) 49 h post-hCG, placed in 11/igmP1 alpha-amanitin at26h post-hCG, and (g) 49 h post-hCG, placed in 10 fmNocodazole at 26 h post-hCG. Arrowheads indicateM-phase marker proteins and asterisks indicate proteinsdependent upon embryonic transcription (Bolton et al.1984).
The rise in peroxide does not require embryonic geneactivationa'-amanitin (11/igmP1), applied at 26h post-hCG toblock activation of the embryonic genome (Bolton et al.1984; confirmed by PAGE analysis of the proteinbiosynthetic profile; Fig. 4, compare tracks b and f withc and e), nonetheless showed the characteristic rise inH2O2 level (Fig. 3, bars 5 and 6). That the synthesis ofembryo encoded proteins is not essential for the rise inH2O2 was confirmed by exposing newly formed and mid2-cell embryos to 10 ^M anisomycin to inhibit proteinsynthesis (Levy, Johnson, Goodall and Maro, 1986),after which the rise in H2O2 level occurred nonetheless(Fig. 3, bars 7 and 8).
Superoxide formation is implicated in peroxideproductionH2O2 may be produced either via dismutation ofsuperoxide by the enzyme superoxide dismutase (SOD)or by a direct enzymatic process such as urate oxidase orglucose oxidase. There are two types of SOD enzyme,the cytosolic form, which is known as CuZn-SOD, orthe mitochondrial form, which is known as the Mn-
Reactive oxygen species 555
tt 48 5
Time in hours post-hCG
DCFHDA (10 /JM)HE (166 UM)
45 48 51Time in hours post-hCG
Fig. 6. Embryos were recovered at 26 h post-hCG, culturedovernight and then some were exposed to DCFHDA(10 ^M) and others to HE (166 ^M) for 15min at the timesindicated and their fluorescence emission measured asindicated.
SOD. Cyanide is an extremely powerful inhibitor ofCuZn-SOD, while it has no effect on the activity of Mn-SOD (Halliwell and Gutteridge, 1989). One-cellzygotes were cultured in vitro to 44.5 h post-hCG andthen exposed to T6+BSA+CN (5ITIM) at 44.5 h posthCG after which their peroxide levels were measured(Fig. 5A). Although cyanide results in suppression ofthe H2O2 rise at 49.5 h post-hCG, over the 5h ofincubation a significant number of embryos developedmembrane blebbing and some died. Therefore, in asecond experiment, embryos were cultured in vitro until51 h post-hCG, exposed for 30min to T6+BSA+CN(5 HIM), assayed immediately and found to havesuppressed peroxide levels (Fig. 5B). These results areconsistent with the possibility, but not proof, that theH2O2 rise depends on cytoplasmic dismutation ofsuperoxide.
To test this hypothesis further, the fluorochromehydroethidine (HE; Kobzik et al. 1990a,b) was used.HE is the reduced, non-fluorescent precursor ofethidium and, when oxidised by either superoxide orH2O2, binds to DNA and can be detected by its redfluorescence. In a preliminary experiment, embryos ofdifferent ages, in hours post-hCG, were loaded witheither DCFHDA or HE (166 (M) for 15min, washedand assayed immediately for their fluorescenceemission (Fig. 6). A similar rise in oxidation of both HEand DCFHDA, was observed. Rothe and Valte (1990)showed recently that oxidation of DCFHDA, but not of
Fig. 5. (A) Zygotes were recovered at 26 hpost-hCG, cultured overnight, then somewere exposed to medium+CN (5mM) at44.5 h post-hCG after which they and thecontrols were assayed for their peroxidelevels at the time intervals indicated.(B) During the period of the peroxide rise(51 h post-hCG), embryos were exposed for30min to HCN (5mM) and then assayed fortheir peroxide levels immediately forcomparison with controls.
Measuring DCFHDAoxidation
Measuring HEoxidation
Fig. 7. Embryos were recovered at 26h post-hCG, culturedovernight and on the following day they were assessed forperoxide levels at 50 h post-hCG using DCFHDA oxidationand for combined superoxide and peroxide levels using HEoxidation. Embryos were exposed for 15min to DCFHDA(10;UM), HE (250/iM) or both DCFHDA plus HE together.
HE, was decreased in stimulated neutrophils, whichhad been loaded simultaneously with HE andDCFHDA as compared to when they had been loadedwith either DCFHDA or HE alone. The HE wasoxidised by superoxide before it could be converted toH2O2, and the lower H2O2 level in consequencereduced DCFHDA oxidation. Two-cell embryos (50hpost-hCG), which had been cultured overnight, wereloaded with DCFHDA at 10 fm, with HE at 250 /JM, orwith both fluorochromes (Fig. 7). Coloading reducesthe signal from DCFHDA oxidation but not from HEoxidation, suggesting that the H2O2 rise may depend ondismutation of superoxide in the cytoplasm.
Superoxide can be produced via different pathways,of which the three major ones are: (i) the electrontransport chain of mitochondria (Fig. 8), (ii) theNADPH oxidase system, and (iii) the xanthine/xanthine oxidase system.
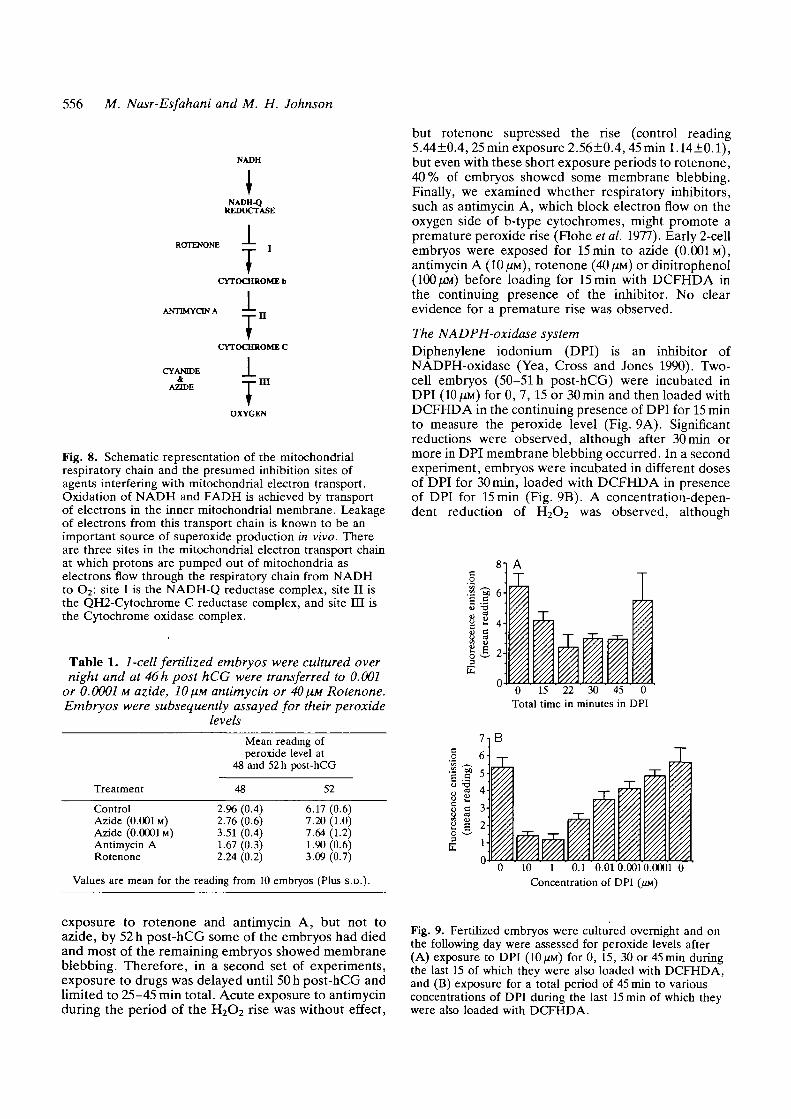
The electron transport chainMitochondrial inhibitors or uncouplers were used toanalyse whether superoxides might have a mitochon-drial source (Fig. 8). 1-cell zygotes were cultured invitro to 46 h post-hCG, transferred to 0.001 or 0.0001 Mazide (uncoupler at site III), 10^M antimycin A(uncoupler at site II) or 40 [M rotenone (uncoupler atsite I), and assayed for their peroxide levels at differenttime intervals. For simplicity only the results before andduring the H2O2 rise (48 and 52 h post-hCG in thecontrols in this experiment) are recorded in Table 1.Although the H2O2 rise is inhibited during chronic
556 M. Nasr-Esfahani and M. H. Johnson
NADH
NADH-QREDUCTASE
ROTENONE 1T
ANTIMYCINA
CYANIDE
AZIDE • in
CVTOCHROMEb
I"CYTOCHROMEC
fOXYGEN
Fig. 8. Schematic representation of the mitochondrialrespiratory chain and the presumed inhibition sites ofagents interfering with mitochondrial electron transport.Oxidation of NADH and FADH is achieved by transportof electrons in the inner mitochondrial membrane. Leakageof electrons from this transport chain is known to be animportant source of superoxide production in vivo. Thereare three sites in the mitochondrial electron transport chainat which protons are pumped out of mitochondria aselectrons flow through the respiratory chain from NADHto O2: site I is the NADH-Q reductase complex, site II isthe QH2-Cytochrome C reductase complex, and site EH isthe Cytochrome oxidase complex.
Table 1. 1-cell fertilized embryos were cultured overnight and at 46 h post hCG were transferred to 0.001
or 0.0001 M azide, 10 fiM antimycin or 40 HM Rotenone.Embryos were subsequently assayed for their peroxide
levelsMean reading ofperoxide level at
48 and 52 h post-hCG
Treatment 48 52
ControlAzide (0.001 M)Azide (0.0001 M)Antimycin ARotenone
2.96 (0.4)2.76 (0.6)3.51 (0.4)1.67 (0.3)2.24 (0.2)
6.17 (0.6)7.20 (1.0)7.64 (1.2)1.90 (0.6)3.09 (0.7)
but rotenone supressed the rise (control reading5.44±0.4, 25 min exposure 2.56±0.4, 45 mini. 14±0.1),but even with these short exposure periods to rotenone,40% of embryos showed some membrane blebbing.Finally, we examined whether respiratory inhibitors,such as antimycin A, which block electron flow on theoxygen side of b-type cytochromes, might promote apremature peroxide rise (Flohe et al. 1977). Early 2-cellembryos were exposed for 15 min to azide (0.001M),antimycin A (10 \m), rotenone (40 \m) or dinitrophenol(100 UM) before loading for 15 min with DCFHDA inthe continuing presence of the inhibitor. No clearevidence for a premature rise was observed.
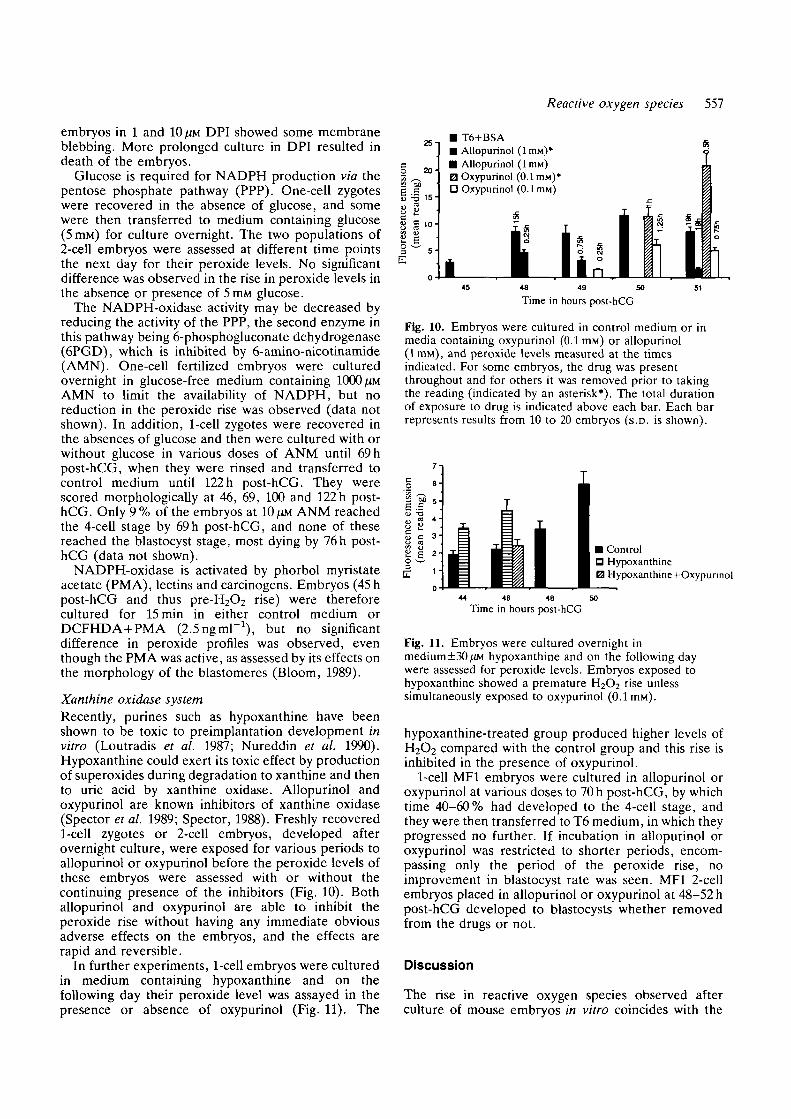
The NADPH-oxidase systemDiphenylene iodonium (DPI) is an inhibitor ofNADPH-oxidase (Yea, Cross and Jones 1990). Two-cell embryos (50-51 h post-hCG) were incubated inDPI (10 /JM) for 0, 7, 15 or 30min and then loaded withDCFHDA in the continuing presence of DPI for 15 minto measure the peroxide level (Fig. 9A). Significantreductions were observed, although after 30 min ormore in DPI membrane blebbing occurred. In a secondexperiment, embryos were incubated in different dosesof DPI for 30 min, loaded with DCFHDA in presenceof DPI for 15 min (Fig. 9B). A concentration-depen-dent reduction of H2O2 was observed, although
co
II6"1
i 1 4
ii illI0 15 22 30 45 0
Total time in minutes in DPI
T
Values are mean for the reading from 10 embryos (Plus S.D.).10 1 0.1 0.010.0010.0001 0
Concentration of DPI
exposure to rotenone and antimycin A, but not toazide, by 52 h post-hCG some of the embryos had diedand most of the remaining embryos showed membraneblebbing. Therefore, in a second set of experiments,exposure to drugs was delayed until 50 h post-hCG andlimited to 25-45 min total. Acute exposure to antimycinduring the period of the H2O2 rise was without effect,
Fig. 9. Fertilized embryos were cultured overnight and onthe following day were assessed for peroxide levels after(A) exposure to DPI (10/iM) for 0, 15, 30 or 45 min duringthe last 15 of which they were also loaded with DCFHDA,and (B) exposure for a total period of 45 min to variousconcentrations of DPI during the last 15 min of which theywere also loaded with DCFHDA.
Reactive oxygen species 557
embryos in 1 and 10/IM DPI showed some membraneblebbing. More prolonged culture in DPI resulted indeath of the embryos.
Glucose is required for NADPH production via thepentose phosphate pathway (PPP). One-cell zygoteswere recovered in the absence of glucose, and somewere then transferred to medium containing glucose(5mM) for culture overnight. The two populations of2-cell embryos were assessed at different time pointsthe next day for their peroxide levels. No significantdifference was observed in the rise in peroxide levels inthe absence or presence of 5mM glucose.
The NADPH-oxidase activity may be decreased byreducing the activity of the PPP, the second enzyme inthis pathway being 6-phosphogluconate dehydrogenase(6PGD), which is inhibited by 6-amino-nicotinamide(AMN). One-cell fertilized embryos were culturedovernight in glucose-free medium containing 1000/iMAMN to limit the availability of NADPH, but noreduction in the peroxide rise was observed (data notshown). In addition, 1-cell zygotes were recovered inthe absences of glucose and then were cultured with orwithout glucose in various doses of ANM until 69 hpost-hCG, when they were rinsed and transferred tocontrol medium until 122h post-hCG. They werescored morphologically at 46, 69, 100 and 122 h post-hCG. Only 9 % of the embryos at 10 JUM ANM reachedthe 4-cell stage by 69 h post-hCG, and none of thesereached the blastocyst stage, most dying by 76 h post-hCG (data not shown).
NADPH-oxidase is activated by phorbol myristateacetate (PMA), lectins and carcinogens. Embryos (45 hpost-hCG and thus pre-H2O2 rise) were thereforecultured for 15min in either control medium orDCFHDA+PMA (2.5ngml~1), but no significantdifference in peroxide profiles was observed, eventhough the PMA was active, as assessed by its effects onthe morphology of the blastomeres (Bloom, 1989).
Xanthine oxidase systemRecently, purines such as hypoxanthine have beenshown to be toxic to preimplantation development invitro (Loutradis et al. 1987; Nureddin et al. 1990).Hypoxanthine could exert its toxic effect by productionof superoxides during degradation to xanthine and thento uric acid by xanthine oxidase. Allopurinol andoxypurinol are known inhibitors of xanthine oxidase(Spector et al. 1989; Spector, 1988). Freshly recovered1-cell zygotes or 2-cell embryos, developed afterovernight culture, were exposed for various periods toallopurinol or oxypurinol before the peroxide levels ofthese embryos were assessed with or without thecontinuing presence of the inhibitors (Fig. 10). Bothallopurinol and oxypurinol are able to inhibit theperoxide rise without having any immediate obviousadverse effects on the embryos, and the effects arerapid and reversible.
In further experiments, 1-cell embryos were culturedin medium containing hypoxanthine and on thefollowing day their peroxide level was assayed in thepresence or absence of oxypurinol (Fig. 11). The
£ S
25"
20-
15-
• T6+BSA• Allopurinol (1 HIM)*
• Allopurinol (lmM)0 Oxypurinol (0.1 nw)1
D Oxypurinol (0.1 mM)
V. E
49 SO 51
Time in hours post-hCG
Fig. 10. Embryos were cultured in control medium or inmedia containing oxypurinol (0.1 mM) or allopurinol(lmM), and peroxide levels measured at the timesindicated. For some embryos, the drug was presentthroughout and for others it was removed prior to takingthe reading (indicated by an asterisk*). The total durationof exposure to drug is indicated above each bar. Each barrepresents results from 10 to 20 embryos (S.D. is shown).
• ControlD HypoxanthineE3 Hypoxanthine+Oxypunnol
44 46 48Time in hours post-hCG
Fig. 11. Embryos were cultured overnight inmedium±30^M hypoxanthine and on the following daywere assessed for peroxide levels. Embryos exposed tohypoxanthine showed a premature H2O2 rise unlesssimultaneously exposed to oxypurinol (0.1 mM).
hypoxanthine-treated group produced higher levels ofH2O2 compared with the control group and this rise isinhibited in the presence of oxypurinol.
1-cell MF1 embryos were cultured in allopurinol oroxypurinol at various doses to 70 h post-hCG, by whichtime 40-60% had developed to the 4-cell stage, andthey were then transferred to T6 medium, in which theyprogressed no further. If incubation in allopurinol oroxypurinol was restricted to shorter periods, encom-passing only the period of the peroxide rise, noimprovement in blastocyst rate was seen. MF1 2-cellembryos placed in allopurinol or oxypurinol at 48-52 hpost-hCG developed to blastocysts whether removedfrom the drugs or not.
Discussion
The rise in reactive oxygen species observed afterculture of mouse embryos in vitro coincides with the

558 M. Nasr-Esfahani and M. H. Johnson
period over which the developmental block occurs. Thisperiod is also marked by a number of importantdevelopmental events, notably the activation of tran-scription by the embryonic genome and the inactivationor destruction of much of the preexisting maternalmRNA, both of which are dependent on progressthrough the first two cell cycles. (Flach et al. 1982;Bolton et al. 1984; reviewed by Telford et al. 1990). Inthis paper, we demonstrate that the rise in reactiveoxygen species is dependent on the activation of theoocyte (Fig. 1 and 2). The observation that partheno-genetically activated oocytes show a slightly earlier risein peroxides may reflect their slightly acceleratedpassage through to the 4-cell stage (Kaufman, 1983).However, although the rise in reactive oxygen speciesoccurs characteristically over the end of the second cellcycle, it does not depend on passage through the firstand second DNA synthetic phases of development, thefirst mitotic division or Gi of the second cell cycle(Fig. 3). Neither is it dependent upon the transcriptionof the embryonic genes or the translation of theirmRNAs (Fig. 3). Thus, the rise in reactive oxygenspecies appears to involve the setting of a timingmechanism within the oocyte itself at or shortly afteractivation, which then can interact with in vitro cultureconditions to produce the observed and appropriatelytimed rise in peroxides independently of continuingprogress through the cell cycle. Presumably the timingmechanism involves some sort of metabolic maturationor cascade.
Although the rise in reactive oxygen species requiresthe activation of the oocyte, it is not dependent upon acontribution from the spermatozoon. Interestingly, it isknown that the genotype of the oocyte alone, regardlessof the fertilising spermatozoon, determines whetherembryos will block at the 2-cell stage when cultured invitro (Goddard and Pratt, 1983). Thus, both theperoxide rise and the in vitro block share a commonunderlying feature. Might then the two be related? Inprinciple, a rise in reactive oxygen species might be partof the mechanism by which the block is caused or mightsimply reflect underlying metabolic problems, them-selves responsible for establishing the block. Alterna-tively, the rise in reactive oxygen species and the blockmight merely be coincidental but independent events.Indeed, it is possible under certain circumstances toseparate the in vitro block from the rise in H2O2. Thus,(i) embryos from different strains of mice show a rise inH2O2 production in vitro whether or not their embryosalso block in vitro, and (ii) conditions of improved invitro culture, such as the addition of iron chelatingagents (Nasr-Esfahani et al. 1990a), may allow other-wise blocking embryos to develop to blastocysts moreeffectively, but appear nonetheless to be associatedwith a rise in reactive oxygen species at the late 2-cellstage (Nasr-Esfahani et al. 1990b). These observationsdo not prove that the rise in peroxides is unrelated tothe block, since arrest of embryonic development byperoxidative damage to cells may require furtherinteractions, for example with superoxide to producereactive oxygen species such as hydroxy radicals. It is
possible that the properties of non-blocking embryos,as well as those of culture conditions that can to someextent overcome the block in vitro, include theinhibition of superoxide production, the inhibition ofconversion of superoxide and H2O2 to hydroxy radicalsand/or the presence of scavenging activity to preventdamage by free radicals generated. Indeed, as wepointed out previously (Nasr-Esfahani etal. 1990£>), thepositive effects of iron chelators on in vitro culturecould result from the protection that they offer againstboth free radical generation and the extension of freeradical peroxidative damage to lipids by free iron.Recent evidence suggests that there are strain differ-ences in the sensitivity of cardiac lipids to peroxidation,Fi strains (which characteristically produce non-blocking embryos) being more resistant to peroxidativedamage than are those strains that generate blockingembryos (Baird and Hough, 1990). Clearly, it will beimportant to measure the dynamics of production ofsuperoxide, hydroxy radical and peroxidised lipid inblocking and non-blocking embryos under differentculture conditions.
In this context, it was important to try to identify thelikely origin of the reactive oxygen species. The findingthat the peroxide rise was sensitive to cyanide suggestedthat H2O2 might be produced via cytoplasmic dismu-tation of superoxide, a conclusion supported by use ofhydroethidine, which reacts with superoxide to depleteits availability for forming H2O2, so reducing the levelof DCF formation by embryos.
Superoxide can be produced via electron leakagefrom the electron transport chain of the mitochondriaand by various cytoplasmic systems. It is difficult toinvestigate the origin of superoxides in embryos byconventional biochemical assays, because of the quanti-tative limitations of the material, and so use of intactcells with indicator dyes is necessary. Nonetheless, wewere able to draw some tentative conclusions. First, wecould not provide clear evidence to favour a mitochon-drial origin for superoxides. Thus, although acute andchronic exposure to the mitochondrial poison rotenone,and chronic exposure to antimycin A, prevented therise in peroxide levels, acute exposure to antimycin Aand chronic exposure to azide had no effect on theH2O2 level. The depressant effect of these poisonsseemed to be related more to their toxic effects than toa specific effect on electron leakage. This conclusion issupported by the fact that respiratory inhibitorsblocking electron flow on the substrate site of theb-cytochrome (site II in Fig. 8) might be expected tosuppress H2O2 formation, while antimycin A, blockingelectron flow on the oxygen side of b-type cytochromesbetween the complex II and III, is expected to enhanceperoxide production (Flohe et al. 1977). Yet a prema-ture rise in H2O2 was not obtained after exposure toantimycin A, azide or cyanide. Thus, it seems unlikelythat the peroxide rise is due to electron leakage orsuperoxide production at sites I and II of the electrontransport chain. It is also known that oxidativephosphorylation activity via the Kreb's TCA cycle isvery low at pre-8-cell stages of mouse development.

Reactive oxygen species 559
In contrast, the pentose phosphate pathway (PPP),which produces NADPH, shows relatively high activityduring the 2-cell stage (O'Fallon and Wright, 1986).NADPH-oxidase is a membrane enzyme system, welldefined in neutrophils and known as the oxygen burstsystem (Lambeth, 1988). It consists of a flavo-haem-protein, which oxidizes NADPH in the cytosol intoNADP+. The electron produced by oxidation ofNADPH is used to reduce oxygen to the superoxideradical. NADPH is known to be involved in H2O2production via NADPH-oxidase in sea urchin oocytesat fertilization (Heinecke and Shapiro, 1989). Dipheny-lene iodonium (DPI), an analogue of NADPH, isknown to inhibit NADPH-oxidase activity in neutro-phils during the oxygen burst. Low concentrations ofDPI (0.001 [JM) inhibit the peroxide rise in mouse 2-cellembryos. However, brief exposure to DPI proved to bequite toxic to mouse embryos, which raises concernabout the specificity and mechanism of its effects.Glucose, which is required for NADPH production viathe PPP, has been reported to retard the developmentof embryos through the 2-cell stage in vitro (Chatot etal. 1989). However, omission of glucose from themedium did not prevent the H2O2 rise. Were NADPH-oxidase to be the source of peroxide production,removal of glucose on its own might not reduce theH2O2 level since the embryos are likely to obtainglucose from their glycogen stores. Therefore, in orderto reduce the intracellular formation of NADPH,embryos were cultured free of glucose in6-amino-nicotinamide, which partially inhibits theproduction of NADPH (White et al. 1988). Theseconditions neither reduced the peroxide level norimproved the embryo culture. Finally, NADPH-oxidase activity in neutrophils and other biologicalsystems is stimulated with phorbol myristate acetate(PMA). However, PMA did not trigger a prematureH2O2 rise in the early two-cell embryos, althoughevidently capable of affecting embryos at this stage(Bloom, 1989). This evidence, taken together, suggeststhat NADPH-oxidase activity is unlikely to be involvedin causing the peroxide to rise. However, the situationis complex since, NADPH production via the PPP isalso essential for the conversion of H2O2 to H2O by theglutathione anti-oxidant system, and so its neutralisa-tion might elevate peroxide levels and enhance devel-opmental damage caused by H2O2 from other sources.
Purines such as hypoxanthine have been shown toretard development through the second cell cycle ofembryos from most strains of mice and to do so by amechanism that does not involve inhibition of phospho-diesterase (Nureddin et al. 1990). Superoxide is a by-product of the degradation of purines to uric acid.Indeed, embryos cultured in medium supplementedwith hypoxanthine produced high and premature levelsof H2O2 compared with control embryos. Moreover,both the natural and hypoxanthine-induced peroxideelevations were prevented by the xanthine oxidaseinhibitors allopurinol and oxypurinol. This inhibitionwas reversible upon the removal of the inhibitor. Thefact that 1-cell embryos cultured in medium sup-
plemented with allopurinol and oxypurinol showed noimproved rates of blastocyst formation compared withcontrols may be due to other adverse effects of theseinihibitors (Miyazaki et al. 1989; Miyazaki et al. 1991).Both allopurinol and oxypurinol are also known to haveantioxidant properties (Moorhouse et al. 1987), buttheir ability to prevent the peroxide rise is more likelyto be due to the inhibition of xanthine oxidase since thehypoxanthine-induced premature rise is inhibited byoxypurinol whereas UV-induced lipid peroxidationdamage is not (data not shown). Therefore, it seemspossible that the superoxide precursor of H2O2 in earlymouse embryos may be produced via the xanthineoxidase system. A possible explanation for the rise insuperoxide output at the 2-cell stage is the accumulationof substrates for xanthine oxidase as a result of thebreakdown of maternal mRNA occurring at about thistime in activated oocytes (see Telford. et al 1990 forreview). What is not clear is why peroxides produced bythis system should accumulate after in vitro culture butnot in vivo. It also remains to be determined whetherthe accumulation is due to more active formation ofreactive oxygen species or to their less effectiveremoval.
Finally, it is important to stress that we have notproved that xanthine oxidase is the only systemgenerating peroxides in vitro, and their accumulationfrom a number of sources remains a possibility. Thus,the recovery and culture of 1-cell zygotes from theoviduct involves their exposure to light, high oxygenlevels, traces of transitional elements and disturbedconcentrations of metabolic substrates. Exposure tovisible light has been shown to be deleterious todevelopment (Daniel, 1964; Hirao and Yanagimachi,1978; Hegele-Hartung et al. 1988; Fischer et al. 1988),possibly via an effect on photosensitizer pigments, suchas the mitochondrial flavins, NADPH-oxidase or bloodpigments carried over into culture on BSA, sogenerating singlet oxygen species which then lead tosuperoxides, lipid peroxides and H2O2. The elevatedoxygen in vitro will promote these reactions as willtraces of free iron, to which embryos are unlikely to beexposed in vivo because of the presence of iron-bindingtransferrin in the genital tract secretions (Nasr-Esfahaniet al. 19906). It is possible that the accumulated insultsof mRNA turnover, light, high oxygen tension andtrace metals push endogenous peroxide production tohigher levels in vitro than in vivo and that some strainsof embryo are protected less adequately from theireffects by scavengers and endogenous reducing agentsthan are others.
We wish to thank John Aitken for valuable advice andMartin George and Brendan Doe for technical assistance.This work was supported by a grant from the MedicalResearch Council to MHJ and Dr PR Braude.
References
BAIRD, M. B. AND HOUGH, J. L. (1990). Adrinomycin inducedoxiative stress: Evidence for strain specific protection againstcardiac lipid peroxidation. In Free Radical Biology and

560 M. Nasr-Esfahani and M. H. Johnson
Medicine. 5th biennial meeting of the international society offree radical research. Abst 13.8.
BLOOM, T. (1989). The effect of phorbol esters on mouseblastomeres: role for protein kinase C in compaction?Development 106, 159-171.
BOLTON, V. N., OADES, P. J. AND JOHNSON, M. H. (1984). Therelationship between cleavage, DNA replication, and geneexpression in the mouse 2-cell embryo. /. Embryol. exp. Morph.79, 139-163.
CHATOT, C. L., ZIOMEK, C. A., BAVISTER, B. D., LEWIS, J. L. ANDTORRES, I. (1989). An improved culture medium supportsdevelopment of random-bred 1-cell mouse embryos in vitro. J.Reprod. Fert. 86, 679-688.
CHISHOLM, J. C , JOHNSON, M. H., WARREN, P. D., FLEMING, T.P. AND PICKEJUNG, S. J. (1985). Developmental variability withinand between mouse expanding blastocysts and their ICMs. /.Embryol. exp. Morph. 86, 311-336.
DANIEL, J. C. (1964). Cleavage of mammalian ova inhibited byvisible light. Nature 201, 316.
FISCHER, B., SCHUMACHER, A., HEGELE-HARTUNG, C. AND BEIER,H. M. (1988). Potential risk of light and room temperatureexposure to preimplantation embryos. Fert. Steril. 50, 938-944.
FLACH, G., JOHNSON, M. H., BRAUDE, P. R., TAYLOR, R. S. ANDBOLTON, V. N. (1982). The transition from maternal toembryonic control in the 2-cell embryo. EMBO J. 1, 681-686.
FLOHE, L., LOSCHEN, G., AZZI, A. AND RICHTER, CH. (1977).Superoxides in mitochondria. In Superoxide and SuperoxideDismutases (eds, Michelson, A. M., McCord, J. M. andFridovich, E.) Academic Press, London, pp. 323-334.
GODDAHD, M. J. AND PRATT, H. P. M. (1983). Control of eventsduring early cleavage of the mouse embryo: an analysis of the 2-cell block. J Embryol. exp. Morph. 73, 111-133.
HALUWELL, B. AND GUTTERJDGE, J. M. C. (1987). Free Radicalsin Biology and Medicine. 3rd edn. Oxford University Press.
HALLIWELL, B. (1987). Superoxide-dependent formation ofhydroxy radicals in presence of iron chelates. FEBS lett. 92,321-326.
HEGELE-HARTUNG, C , SCHUMACHER, A. AND FISCHER, B. (1988).Ultrastructure of pre-implantation rabbit embryos exposed tovisible light and room temperature. Anat. Embryol 178,229-241.
HEINECKEL, J. W. AND SHAPIRO, B. M. (1989). Respiratory burstoxidase of fertilization. Proc. natn. Acad. Sci. U.S.A. 86,1259-1263.
HIRAO, Y. AND YANAGIMACHI, R. (1987). Detrimental effect ofvisible light on meiosis of mammalian eggs in vitro. J. exp. Zool.206, 365-370.
HOWLETT, S. K. (1986a). A set of proteins showing cell cycledependent modification in the early mouse embryo. Cell 45,386-396.
HOWLETT, S. K. (19866). The effect of inhibiting DNA replicationin the one-cell embryo. Roux's. Arch. Dev. Biol. 195, 499-505.
KAUFMAN, M. H. (1983). Early Mammalian Development:Parthenogenetic Studies. Cambridge: Cambridge UniversityPress.
KOBZIL, L., GODLESH, J. J. AND BRAIN, J. D. (1990a). Oxidativemetabolism in the alveolar macrophage: Analysis by flowcytometry. J. Leuk. Biol. 47, 295-303.
KOBZIL, L., GODLESH, J. J. AND BRAJN, J. D. (19906). Selectivedown-regulation of alveolar macrophage oxidative response toopsonin-independent phagocytosis. /. Immun. 44, 4312-4319.
LAMBETH, J. D. (1988). Respiratory burst oxidase and itsregulation. J. Bioenergetics and Biomembranes 20, 633-677.
LEVY, J. B., JOHNSON, M. H., GOODALL, H. AND MARO, B. (1986).The timing of compaction: control of a major developmentaltransition in mouse early embryogenesis. J. Embryol. exp.Morph. 95, 213-237.
LOUTRADIS, D., JOHN, D. AND KIESSLING, A. A. (1987).Hypoxanthine causes a 2-cell block in random bred mouseembryos. Biol. Reprod. 37, 311-316.
MARO, B., JOHNSON, M. H., PICKERING, S. J. AND FLACH, G.
(1984). Changes in actin distribution during fertilization of themouse egg. /. Embryol. exp Morph. 81, 211-237.
MCCONNELL, J., PICKERING, S. J., JOHNSON, M. H. AND BASHFORD,J. (1990). A technique for quantifying the amount ofmacromolecule injected into cells of the early mouse embryo. /.Reprod. Fert. 88, 375-381.
MINOTTI, G. AND AUST, S. D. (1989). The role of iron m oxygenradical mediated lipid peroxidation. Chem. biol. Interactions 71,1-19.
MJYAZAKI, T . , KUO, T. C , SUEOKA, K. , DHAJtMARAJAN, M . A . ,ATLAS, S. J. AND WALLACH, E. E. (1989). In vivo administrationof allopurinol affects ovulation and early embryonicdevelopment in rabbits. Am. J. Obstet. Gynec. 161, 1709-1714.
MIYAZAKI, T., SUEOKA, K., DHARMARAJAN, M. A., ATLAS, S. J.,BULKLEY, G. B. AND WALLACH, E. E. (1991). Effect ofinhibition of oxygen radical on ovulation and progestroneproduction by the in vitro perfused rabbit ovary. /. Reprod.Fert. 91, 207-212.
MOORHOUSE, P. C , GROOTVELD, M., HALLIWELL, B., QUINLAN, J.G. AND GuTTERiDGE, J. M. C. (1987). Allopurinol andoxypurinol are hydroxy radical scavengers. FEBS Letts 213,23-28.
NASR-ESFAHANI, M. H., ATTKEN, J. R. AND JOHNSON, M. H.(1990a). Hydrogen peroxide levels in mouse oocytes and earlycleavage stage embryos developed in vitro or in vivo.Development 109, 501-507.
NASR-ESFAHANI, M. H., JOHNSON, M. H. AND AITKEN, J. R.(19906). The effect of iron and iron chelators on the in vitroblock to development of the mouse preimplantation embryo:BAT6 a new medium for improved culture of mouse embryos invitro. Hum. Reprod. 5, 997-1003.
NODA, Y., MATSUMOTO, H., UMAOKA, Y., TATSUMI, K., KISHI, J.AND MORI, T. (1991). Involvement of superoxide radicals in themouse two-cell block. Molec. Reprod. Devi. 28, 356-360.
NUREDDIN, A., EPSARO, E. AND KIESSLING, A. A. (1990). Purinesinhibit development of mouse embryos in vitro. J. Reprod. Fert.90, 455-464.
O'FALLON, J. V. AND WRIGHT, R. W. (1986). Quantitativedetermination of the pentose phosphate pathway inpreimplantation embryos. Biol. Reprod. 34, 58-64.
PRATT, H. P. M. AND MUGGLETON-HARRIS, A. L. (1988). Cyclingcytoplasmic factors that promote mitosis in the cultured 2-cellmouse embryo. Development 104, 115-120.
ROTHE, G. AND VALET, G. (1990). Flow cytometric analysis ofrespiratory burst activity in phagocytes with hydroethidine and2,7-dichlorofluorescin. /. Leuk. Biol. 47, 440-448.
SPECTOR, T. (1988). Oxypurinol as an inhibitor of xanthine oxidasecatalysed production of superoxide radical. Biochem. Pharmac.37, 349-352.
SPECTOR, T., HALL, W. W., PORTER, D. J., LAMBE, C. U., NELSON,D. J. AND KRENTTSKY, T. A. (1989). Inhibition of xanthineoxidase by 4-Hydroxy-6-Mercaptopyrazolo[3,4-D] Pyrimidine.Biochem. Pharmac. 38, 4315-4320.
TELFORD, N. A., WATSON, A. J. AND SCHULTZ, G. A. (1990).Transition from maternal to embryonic control in earlymammalian development: A comparison of several species.Molec. Reprod. Devi. 26, 90-100.
WHITE, C. W., JACKSON, J. H., MCMURTERY, I. F. AND REPINE, J.E. (1988). Hypoxia increases glutathione redox cycle andprotects rat lungs against oxidants. /. Appl. Physiol. 65,2607-2616.
WHTTTINGHAM, D. G. (1974). Fertilization, early development andstorage of mammalian ova in vitro. In The Early Developmentof Mammals (ed. M. Balls and A. E. Wild), pp. 1-24.Cambridge University Press.
YEA, C. M., CROSS, A. R. AND JONES, T. G. (1990). Purificationand some properties of the 45 kDa diphenylene iodonium-binding flavoprotein of neutrophil NADPH oxidase. Biochem. J.265, 95-100.
{Accepted 28 June 1991)