the johanson-blizzard syndrome: report of a new case with special reference to the dentition and...
TRANSCRIPT

Clinical Genetics 1986: 30: 177- I83
The Johanson-Blizzard syndrome: report of a new case with special reference to the
dentition and review of the literature
K. ZERRES' AND E. -A. HOLTGRAVE~ 'Institut fur Humangenetik der Universitat Bonn, and 2Poliklinik fur Kieferorthopadie des
Fachbereiches Zahn-, Mund- und Kierferheilkunde der Freien Universitat Berlin, FRG
We report a new case of Johanson-Blizzard syndrome. The clinical findings with special reference to the dentition are discussed, and the literature is reviewed. The reported case underlines the heterogeneity of ectodermal dysplasias mentioned by Freire-Maia (197 1).
Received 17 April 1985. revised, accepted for pubtication 3 May 1986
Key words: Autosomal recessive inheritance; ectodermal dysplasias; Johanson-Blizzard syn- drome.
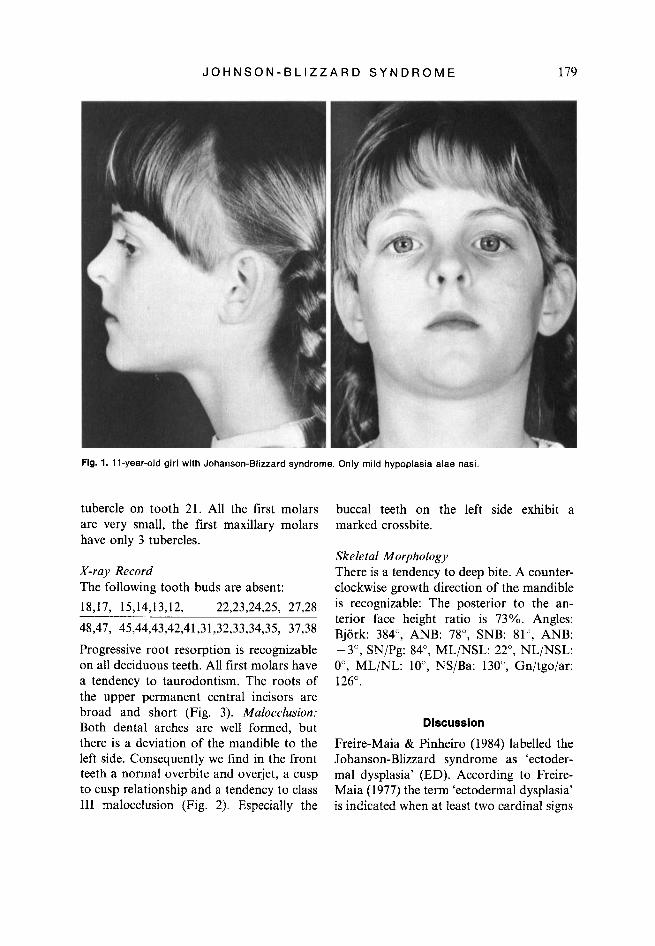
In 1971 Johanson & Blizzard described three female patients with low birth weight, dwarfism, aplastic alae nasi, midline scalp defects, microcephaly, deafness, thyroid dysfunction, rectourogenital abnormalities, motor-mental retardation, and absent per- manent tooth buds.
The patients described earlier by Mor- ris & Fisher (1967) and Townes (1969) are examples of the syndrome as well. The pa- tient of Grand et al. (1966) with an XXY karyotype, pancreatic insufficiency, hypo- thyroidism, deafness, chronic lung disease, dwarfism and microcephaly, may have the same condition. However, the authors stat- ed that the nose was slender but otherwise normal, and the pictures published do not resolve this issue.
As constant signs all reported patients with Johanson-Blizzard syndrome show hy- poplasia or aplasia alae nasi, exocrine pan- creatic insufficiency, failure to thrive and a
combination of missing and very small teeth (Table 1).
Case Report
The patient (Fig. 1) is the second daughter of healthy non-consanguineous parents, Her bone age is delayed (8.5 instead of 11 years). Her body height (138 cm) is within the normal range. Since her birth she has suffered from malabsorption as a result of an exocrine pancreatic insufficiency with failure to thrive, and fouly, fatty, bulky stools. Additional symptoms are listed in Table 1.
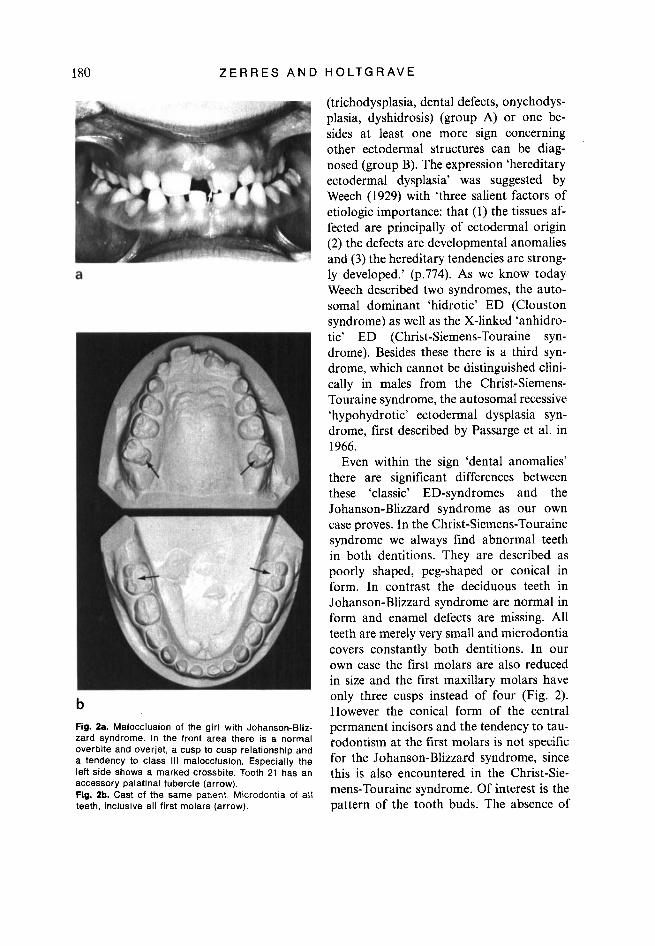
Dental Report Up to teeth 51 and 61 there is a persistent deciduous dentition with microdontia (Fig. 2). Therefore spaces between the teeth were noted. The central upper permanent inci- sors are conical in form with an accessory

178 Z E R R E S A N D H O L T G R A V E
Table 1
Clinical f ind ings in the Johanson-Bl izzard syndrome
1. 2. 3. 4. 5. 6. 7. 8. 9.
10. 11. 12. 13. 14. 15. 16. 17. 18. 19. 20. 21. 22. 23. 24.
patient sex birthweight (9) hypoplasialaplasia alae nasi pancreatic insufficiency failure to thrive aplasia of permanent teeth small teeth scalp defect abnormal hair pattern shortness of stature delayed bone age hearing loss mental retardation anemia GU anomalies imperforate anus hypothyroidism microcephaly cutaneolacrimal duct fistula congenital heart defect nipple anomalies maxillary hypoplasia age of death
frequency 10111
1 f
4099 + + +
2 3 4 5 f f f f
+ + + + + + + + + + + + + + + +
+ + + + + + + + + + + + + + + + + +
+ + + + + + + + + + + +
+ + + + + +
+ + + +
2270 2500 3000
-
- -
- -
6 m
3500 + + +
7 rn
3500 + + +
21/21 20120 1811 8 10/10 1311 4 18/20 16/17 14/17 9/12
12/17 15/19 719 7/19 8/21 5/16 7/21 416 3/10 316 415
+ + -
+ +
+ + + -
+
+ 4 m
1. 2. 3. 4. 5. 6. 7. 8. 9.
10. 11. 12. 13. 14. 15. 16. 17. 18. 19. 20. 21. 22. 23. 24.
9 m
2300 +
10 11 12 13 rn f f rn
2280 normal normal 3500 + + + + + + + + + + +
+ + + + + + + + + + + + + +
+ + + +
+ + + + + +
+
- - - - -
- - - - - - - -
- - - + + - - - -
- -
14 rn
3680 + + +
15 m
3440 + + +
16 f
3080 + + +
17 rn
2720 + + + + + + + + + + +
18 f
2810 + + +
19 20 f rn
2610 2600 + + + + + + + + + + + + +
+ + +
+ +
+ - +
+ +
+ +
+ + +
+ + +
+ 3 h 10 m 3 m 3 m
Cases: (1) Morris & Fisher (1967); (2) Townes (1969, 1972), Townes & White (1981); (3, 4, 5) Johanson & Blizzard (1971), case 3 also Park et al. (1972); (6) Schussheim et al. (1976); (7) Mardini et al. (1978); (8. 9) Day & Israel (1978) (siblings), case 8 also case CCFA 4447 of Motohashi et al. (1981); (10) Daentl et al. (1979); (11, 12) Reichart et at. (1979) (siblings): (13) Sisrnanis et al. (1979); (14) Bresson et al. (1980); (15, 16) Helin & Jodal (1981) (siblings); (17) Motohashi et al. (1981); (18) Baraitser & Hodgson (1982); (19, 20) Moeschler & Lubinsky (1985) (siblings); (21) own case.
J O H N S O N - B L I Z Z A R D S Y N D R O M E 179
Fig. 1. 1 I-year-old girl with Johanson-Blizzard syndrome. Only mild hypoplasia alae nasi.
tubercle on tooth 21. All the first molars are very small, the first maxillary molars have only 3 tubercles.
X-ray Record The following tooth buds are absent: 18,17, 15,14,13,12, 22,23,24,25, 27,28 48,47, 45,44,43,42,41,31,32,33,34,35, 37,38
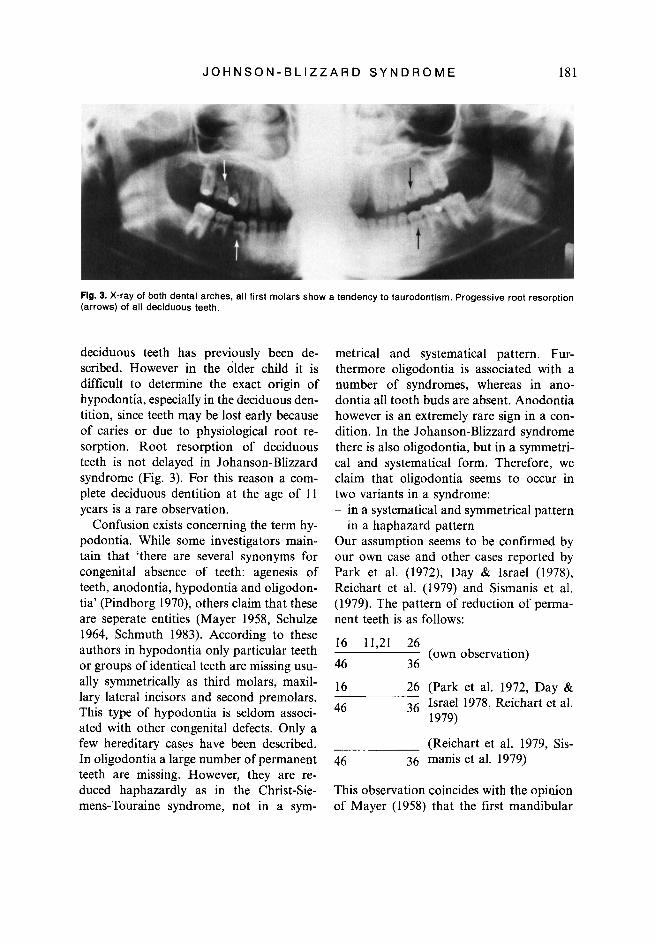
Progressive root resorption is recognizable on all deciduous teeth. All first molars have a tendency to taurodontism. The roots of the upper permanent central incisors are broad and short (Fig. 3). Malocclusion: Both dental arches are well formed, but there is a deviation of the mandible to the left side. Consequently we find in the front teeth a normal overbite and overjet, a cusp to cusp relationship and a tendency to class 111 malocclusion (Fig. 2). Especially the
buccal teeth on the left side exhibit a marked crossbite.
Skeletal Morphology There is a tendency to deep bite. A counter- clockwise growth direction of the mandible is recognizable: The posterior to the an- terior face height ratio is 73%. Angles: Bjork: 384", ANB: 78", SNB: 81", ANB:
O", ML/NL: lo", NS/Ba: 130", Gn/tgo/ar: 126".
-3", SN/Pg: 84", ML/NSL: 22", NL/NSL:
Discussion
Freire-Maia & Pinheiro (1984) labelled the Johanson-Blizzard syndrome as 'ectoder- ma1 dysplasia' (ED). According to Freire- Maia (1977) the term 'ectodermal dysplasia' is indicated when at least two cardinal signs
180 Z E R R E S A N D H O L T G R A V E
b Fig. 2a. Malocclusion of the girl with Johanson-Bliz- zard syndrome. In the front area there is a normal overbite and overjet, a cusp to cusp relationship and a tendency to class 111 malocclusion. Especially the left side shows a marked crossbite. Tooth 21 has an accessory palatinal tubercle (arrow). Fig. 2b. Cast of the same patient Microdontia of all teeth, inclusive all first molars (arrow).
(trichodysplasia, dental defects, onychodys- plasia, dyshidrosis) (group A) or one be- sides at least one more sign concerning other ectodermal structures can be diag- nosed (group B). The expression ‘hereditary ectodermal dysplasia’ was suggested by Weech (1929) with ‘three salient factors of etiologic importance: that (1) the tissues af- fected are principally of ectodermal origin (2) the defects are developmental anomalies and (3) the hereditary tendencies are strong- ly developed.’ b.774). As we know today Weech described two syndromes, the auto- soma1 dominant ‘hidrotic’ ED (Clouston syndrome) as well as the X-linked ‘anhidro- tic’ ED (Christ-Siemens-Touraine syn- drome). Besides these there is a third syn- drome, which cannot be distinguished clini- cally in males from the Christ-Siemens- Touraine syndrome, the autosomal recessive ‘hypohydrotic’ ectodermal dysplasia syn- drome, first described by Passarge et al. in 1966.
Even within the sign ‘dental anomalies’ there are significant differences between these ‘classic’ ED-syndromes and the Johanson-Blizzard syndrome as our own case proves. In the Christ-Siemens-Touraine syndrome we always find abnormal teeth in both dentitions. They are described as poorly shaped, peg-shaped or conical in form. In contrast the deciduous teeth in Johanson-Blizzard syndrome are normal in form and enamel defects are missing. All teeth are merely very small and microdontia covers constantly both dentitions. In our own case the first molars are also reduced in size and the first maxillary molars have only three cusps instead of four (Fig. 2). However the conical form of the central permanent incisors and the tendency to tau- rodontism at the first molars is not specific for the Johanson-Blizzard syndrome, since this is also encountered in the Christ-Sie- mens-Touraine syndrome. Of interest is the pattern of the tooth buds. The absence of
J O H N S O N - B L I Z Z A R D S Y N D R O M E 181
Flg. 3. X-ray of both dental arches, all first molars show a tendency to taurodontism. Progessive root resorption (arrows) of all deciduous teeth.
deciduous teeth has previously been de- scribed. However in the older child it is difficult to determine the exact origin of hypodontia, especially in the deciduous den- tition, since teeth may be lost early because of caries or due to physiological root re- sorption. Root resorption of deciduous teeth is not delayed in Johanson-Blizzard syndrome (Fig. 3). For this reason a com- plete deciduous dentition at the age of 11 years is a rare observation.
Confusion exists concerning the term hy- podontia. While some investigators main- tain that ‘there are several synonyms for congenital absence of teeth: agenesis of teeth, anodontia, hypodontia and oligodon- tia’ (Pindborg 1970), others claim that these are seperate entities (Mayer 1958, Schulze 1964, Schmuth 1983). According to these authors in hypodontia only particular teeth or groups of identical teeth are missing usu- ally symmetrically as third molars, maxil- lary lateral incisors and second premolars. This type of hypodontia is seldom associ- ated with other congenital defects. Only a few hereditary cases have been described. In oligodontia a large number of permanent teeth are missing. However, they are re- duced haphazardly as in the Christ-Sie- mens-Touraine syndrome, not in a sym-
metrical and systematical pattern. Fur- thermore oligodontia is associated with a number of syndromes, whereas in ano- dontia all tooth buds are absent. Anodontia however is an extremely rare sign in a con- dition. In the Johanson-Blizzard syndrome there is also oligodontia, but in a symmetri- cal and systematical form. Therefore, we claim that oligodontia seems to occur in two variants in a syndrome: - in a systematical and symmetrical pattern
~ in a haphazard pattern Our assumption seems to be confirmed by our own case and other cases reported by Park et al. (1972), Day & Israel (1978), Reichart et al. (1979) and Sismanis et al. (1979). The pattern of reduction of perma- nent teeth is as follows:
16 11,21 26
46 36
16 26 (Park et al. 1972, Day &
(own observation)
46 36 Israel 1978, Reichart et a]. 1979)
(Reichart et al. 1979, Sis- 46 36 manis et al. 1979)
This observation coincides with the opinion of Mayer (1958) that the first mandibular

182 Z E R R E S A N D H O L T G R A V E
molars are the most constant teeth in the dentition.
The Johanson-Blizzard syndrome has so far only been diagnosed in children with hypoplasia or aplasia alae nasi. In addition all patients, as far as mentioned in the litera- ture, had exocrine pancreatic insufficiency, failure to thrive and dental defects (small teeth, aplasia of a large number of teeth). All other symptoms as described in Table 1 are not obligatory.
Bresson et al. (1980) only mentioned an affected brother of their described patient without aplasia alae nasi. This patient, who died at the age of 13 months because of marasm, showed besides scalp defects a cu- taneolacrimal duct fistula. A second cousin of their patient, who died as a newborn, only showed an imperforate anus. Further data about the patients are not reported.
Besides the signs listed in Table 1, further abnormalities are reported only occasion- ally: cafk au lait spots (Johanson & Blizzard 1971, Sismanis et al. 1979), strabismus (Johanson & Blizzard 1971, Motohashi et al. 1981), asplenia (Day & Israel 1978), po- lysplenia (Helin & Jodal 198 l), clinodactylia (Schussheim et al. 1976), hypoplasia of the middle phalanx of the fifth fingers (Sismanis et al. 1979), situs inversus (Helin & Jodal 1981). A CNS autopsy by Daentl et al. (1 979) described focal neuronal migration disturbances that were evident in distortion of the cortical layering with a columnar pat- tern perpendicular to the surface. The au- thors suggest a true CNS developmental de- fect as a probable explanation of mental retardation. Histologic examination of the pancreas revealed absence of pancreatic pa- renchyma with replacement by fat (Daentl et al. 1979, Bresson et al. 1980).
(1978), Reichart et al. (1979), Helin & Jodal (1981), Motohashi et al. (1981) (siblings not described), Bresson et al. (1981) (affected brother and second cousin are only men- tioned) and Moeschler & Lubinsky (1985) suggest autosomal recessive inheritance as well.
Note Added in Proof
Dr. F.A. Beemer from Utrecht (Nether- lands) informed us about a further case of Johanson-Blizzard syndrome.
The male newborn showed the following signs: birthweight 2430 g, hypoplasia alae nasi, pancreatic insufficiency, failure to thri- ve, abnormal hair pattern, shortness of stat- ure, delayed bone age, microcephaly, maxil- lary hypoplasia. The patient did not show: scalp defect, anemia, GU anomalies, imper- forate anus, hypothyroidism, cutaneolacri- ma1 fistula, congenital heart defect, and nip- ple anomalies. No information can be given about the teeth, hearing, and mental devel- opment.
Acknowledgment
We thank Professor Dr. M. Becker for his clinical information about the patient.
References
Baraitser, M. & S. V. Hodgson (1982). The Johanson-Blizzard syndrome. J . Med. Genet.
Bresson, J. L., J. Schmitz, J. M. Saudubray, G. Lesec, J. A. Hummel & J. Rey (1980). Le syn-
19, 302-303.
drome de Johanson-Blizzard. Arch. Fr. Pediatr. 37. 21-24.
Daentl, D. L., J. L. Frias, E. F. Gilbert & J. M. Opitz (1979). The Johanson-Blizzard syn- drome: case report and autopsy findings. Am. J . Med. Genet. 3, 129-135.
Day, D. W. & J. N. Israel (1978). Johanson-Bliz- In the families, described by Schussheim
et al. (1976), Mardini et al. (1978), Sismanis et al. (1979) and Bresson et al. (1980) con- sanguinity is proven. The Observations Of
affected siblings described by Day & Israel
zard syndrome. Birth Defects! OAS XIV (6B), 275-287.
Freire-Maia, N. (1971). Ectodermal Dysplasias. Hum, Hered, 21, 309-312,
Freire-Maia, N. (1977). Ectodermal dysplasias re- visted. Acta Genet. Med. Gemellol. 26, 121-131.

J O H N S O N - B L I Z Z A R D S Y N D R O M E 183
Freire-Maia, N. & M. Pinheiro (1984). Ectoder- ma1 Dysplasias: A Clinical and Genetic Study. New York, Alan R. Liss, Inc.
Grand, R. J., S. W. Rosen, P. A. diSant’Agnese & W. R. Kirkham (1966). Unusual case of XXY Klinefelter’s syndrome with pancreatic insuf- ficiency, hypothyroidism, deafness, chronic lung disease, dwarfism, and microcephaly. Am. J. Med. 41, 478485.
Helin, I. & U. Jodal (1981). A syndrome of con- genital hypoplasia of the alae nasi, situs inver- sus, and severe hypoproteinemia in two sib- lings. J . Pediatr. 99, 932-934.
Johanson, A. & R. Blizzard (1971). A syndrome of congenital aplasia of the alae nasi, deafness, hypothyroidism, dwarfism, absent permanent teeth, and malabsorption. J . Pediatr. 79,
Mardini, M. K., M. Ghandour, N. A. Sakati & W. L. Nyhan (1978). Johanson-Blizzard syn- drome in a large inbred kindred with three involved members. Clin. Genet. 14, 247-250.
Mayer, W. (1958). Pathologie der Zahne und des Gebisses. In Zahn-, Mund- und Kieferheilkunde Band I. K. Haupl, W. Mayer & K. Schuchardt (eds). Miinchen/Berlin, Urban und Schwarzen- berg, p. 467.
Moeschler, J. B. & M. S. Lubinsky (1985). Brief Clinical Report: Johanson-Blizzard syndrome with normal intelligence. Am. J . Med. Genet.
Morris, M. D. & D. A. Fisher (1 967). Trypsinogen Deficiency Disease. Am. J . Dis. Child. 114,
Motohashi, N., S. Pruzansky & D. Day (1981). Roentgencephalometric analysis of craniohci- al growth in the Johanson-Blizzard syndrome. J . Craniofacial Genet. Develop. Biol. 1, 57-72.
Park, I. J., A. Johanson, H. W. Jones & R. Bliz- zard (1972). Special female hermaphroditism associated with multiple disorders. Obstet. Gy- necol. 39, 10&106.
982-987.
22, 69-13.
203-208.
Passarge, E., C. T. Nuzum & W. K. Schubert (1966). Anhidrotic ectodermal dysplasia as autosomal recessive trait in an inbred kindred. Humangenetik 3, 181-185.
Pindborg, J. J. (1970). Pathology of Dental Hard Tissues. Copenhagen, Munksgaard.
Reichart, P., S. Flatz & M. Burdelski (1979). Ek- todermale Dysplasie und exokrine Pancreasin- suffizienz - ein erblich bedingtes Syndrom. Dtsch. zahnarztl. Z . 34, 263-265.
Schmuth, G. P. F. (1983) Kieferorthopiidie - Grundzuge und Probleme. Stuttgart, Thieme.
Schulze, C. (1 964). Anomalien, MiDbildungen und Krankheiten der Ziihne, des Mundes und der Kiefer. In Humangenetik Band 11. P. E. Becker (ed). Stuttgart, Thieme.
Schussheim, A,, S. J. Choi & M. Silverberg (1976). Exocrine pancreatic insufficiency with congenital anomalies. J . Pediatr. 89, 782-784.
Sismanis, A,, I. A. Polisar, M. L. Ruffy & J. C. Lambert (1979). Rare congenital syndrome associated with profound hearing loss. Arch. Otolaryngol. 105, 222-224.
Townes, P. L. (1969). Proteolytic and lipolytic deficiency of the exocrine pancreas. J . Pediatr.
Townes, P. L. (1972). Trypsinogen deficiency and other proteolytic deficiency diseaeses. Birth De- fects: OAS, VIII (2), 95-101.
Townes, P. L. & M. R. White (1981). Identity of two syndromes. Am. J . Dis. Child. 135,
Weech, A. A. (1929). Hereditary ectodermal dy- splasia (congenital ectodermal defect). Am. J. Dis. Child. 37, 766790.
75, 221-228.
248-250.
Address: Dr. med. Klaus Zerres Institut Fur Humangenetik
Wilhelmstr. 31 0-5300 Bonn I FRG
der Universitat