the group specific component/vitamin d binding protein (gc/dbp) system in the analysis of disputed...
TRANSCRIPT

398 J. Constans and H. Cleve Electrophoresis 1988. 9, 398-403
Jacques Constans’ Hartwig Cleve
Centre National de la Recherche Scientifique, Centre de Recherches sur le Polymorphisme Genetique des Populations Humaines, Toulouse
Institut 6 r Anthropologie und Humangenetik der Universitat Munchen
The group specific component/vitamin D binding protein (GC/DBP) system in the analysis of disputed paternities
The group-specific component (GC) was discovered in 1959, and in the same year a vitamin D binding protein (DBP)in human plasma was found; however, their identity was established as late as 1975. In the GC/DBP system three common alleles, GC*lF , GC*lS, and GC*2, determine six G C phenotypes: IF, lS, 2,1F-lS, 2-1F and 2- 1 S, these common alleles having been found in all human populations studied. In addition, more than 120 G C variants have been discovered, with varying frequencies in different populations. The distribution of the common G C phenotypes and the presence of rare G C variant phenotypes render the GC/DBP system useful for the analysis of disputed paternities.
1 Introduction
In 1958 the Swedish investigator Jan Hirschfeld carried out studies to establish the reproducibility of patterns of human sera as obtained by the immunoelectrophoretic technique of Grabar and Williams [ 11. He discovered differences in the electrophoretic positions of a precipitation arc in the al-a2- globulin fraction due to a hitherto unknown antigen present in human serum 121. Since he was uncertain as to the nature of this antigen he used the term “component”. He distinguished three different phenotypes and was able to classify according- ly all human sera tested, thus differentiating three groups. He, therefore, coined the term “group specific component” ab- breviated to Gc, now written as GC [21. At the same time, Thomas et al. [31 discovered a vitamin D binding factor in the a-globulin fraction of human serum, which was called D-bin- ding protein (DBP). Although the G C system was studied in- tensively by human geneticists and by forensic scientists [4], more than 15 years passed before the identity ofthese two pro- teins was established by Daiger et al. [51. Now this system is known as the GC/DBP system.
1.1 Molecular structure of GC/DBP
GC/DBP is an a2-globulin first observed in human serum or plasma; it has been demonstrated since in several mammalian species and has also been shown in fish [61. Presumably, it is present in all vertebrates and functions as the carrier protein for vitamin D and its natural derivatives, 25-(OH)-D3, 1,25 (OH),-D3 and 24,25-(OH)2-D3. The genetics have been well established: three common alleles, GC * 1 F, GC* 1 S and GC*2, determine six common GC phenotypes, 1F- lF , 1s-lS, 2-2, IF-IS, 2-1F, and 2-1s [71. In addition, more than 120 more or less uncommon alleles have been identified [81. GC/ DBP consists of a single polypeptide chain with 458 amino acid residues. The complete amino acid sequence has been determined and structural homologies to albumin and to a- foetoprotein were revealed [9, 101. From the albumin model proposed by Brown 1 I 11 it can be deduced that GC/DBP has 14 disulfide bridges forming nine double loops that define a triple-domain structure [ 10, 12,131. The nucleotide sequence
Correspondence: Prof. Dr. Hartwig Cleve, Institut fur Anthropologie und Humangenetik der Universitat Miinchen, Richard-Wagner-Str. lO/I, D-8000 Munchen 2, FRG
Abbreviations: DBP, vitamin D binding protein; Gal, galactose; Gal Nac, N-acetylgalactosamine; GC, group specific component; IEF, isoelectric focusing; IPG, immobilized pH gradient; NANA, N-acetylneuraminic acid
0 VCH Verlagsgesellschaft mbH. D-6940 Weinheim, 1988
from cDNA has also been established [ 12,131. The structural differences between the genetic variants have not been directly determined. The sequence data available suggest that the allelic differences may involve as many as four amino acid substitutions, namely at positions 152,3 1 1,4 16 and 420 [ 10, 12, 131.
An interesting posttranslational difference between the allelic products concerns the presence or absence of a carbohydrate side chain [ 141. Only G C 1F and G C 1s molecules contain carbohydrates, GC2 is devoid of sugars. Predominant is a trisaccharide, containing galactose (Gal), N-acetylgalactos- amine (Gal Nac) and N-acetylneuramic acid (NANA), with the structure NANA a (2-3) Gal p (1-3) Gal Nac a (l+O), which is 0-glycosidically linked to the polypeptide chain. Furthermore, there is in some of the G C 1 molecules a tetrasaccharide with an additional NANA residue; this iso- protein is normally below the level of detectability, but in- creased in patients with liver diseases. The allelic products of the G C genes 1 F and 1 S and their mutants are heterogeneous when analyzed by electrophoresis or by isoelectric focusing (IEF), consisting of an anodal and a cathodal isoprotein. This heterogeneity is due to the presence or absence of the oligosaccharide side chain, attached to the anodal (GC la ) isoform but absent in the cathodal isoprotein (GC lc). G C 2 and the mutants of G C 2 do not have a carbohydrate side chain. The carbohydrate structure has been determined in a purified preparation from a G C 1 s donor [ 141. However, the effect of treatment with neuraminidase was studied in com- parison for all the common and several rare GC variants [ 151. The results indicate that there is no difference between G C 1F and G C lS, both allelic products having one NANA residue peronemoleculeoftheGC laisoformi 151.Thevastmajority of G C 1 mutants have the same characteristic. G C 2 and the G C 2 mutants, on the other hand, have no carbohydrate side chain and are devoid of sialic acid [ 151. A few exceptions to this general rule will be mentioned below.
The gene locus for GC/DBP has been mapped first by deletion mapping to the long arm of chromosome 4,4ql1-4q13 [ 161. Later, the gene family for a-foetoprotein, albumin and GC/ DBP was mapped at chromosome 4 in a narrow region within a distance of 1.55 centimorgans. The localization was con- firmed by in situ hybridization [ 17, 181.
1.2 Nomenclature of the GC/DBP system
The three phenotypes G c 1-1,2-1,2-2 were distinguishedfirst by immunoelectrophoresis [2]. Their genetic control was con-
01 73-0835/880808-0398%02.50/0

GC/DBP system 399 E/eclrvphoresis 1988, 9, 398-403
firmed through family studies which indicated the existence of two autosomal alleles, called Gc' and Gc2 [14,201. By avarie- ty of electrophoretic procedures, additional variants like Gc X, Y, Aborigine, Chippewa und Z were identified 121-231. Also, a heterogeneity of the G c l allelic product was noticed and an anodal and a cathodic isoform were distinguished 124-261, which eventually led to theclassification into double- band and single-band G C mutants [271. A major advance was made when Constans and Viau introduced IEF for the analysis of the G C system f281. They discovered G C subtypes and divided the gene GC' into suballeles called GCIF and GC", to which now the notations GC*lF and GC* 1 s are given. IEF, furthermore, allowed the recognition of numerous G C variant phenotypes. At an international workshop held in July 1978 in Paris a new nomenclature was adopted for the GC/DBP system to accomodate this increasing body ofinfor- mation [71. Double-band variants are called G C 1 and single- band variants are called G C 2 . G C 1 mutants located anodal to the position of G C IS are called G C lA, those located cathodal to G C IS are named G C 1C. Accordingly, single- band variants anodal from G C 2 are called G C 2A variants and those located cathodal to G C 2 are named G C 2C variants. Numbers (following the chronological order of their discovery) are added to the different G C mutants [71. In a series of reports from 1979,1981, and 1983 the continuously accumulating information was summarized [7, 29, 301. At present, published information is available on a total of 3 com- mon and 124 rare G C variants [81. They comprise 32 1A variants, 53 1C variants, 20 2A variants, and 19 2C variants IS]. In addition, one deficiency mutant, GCl" has been characterized [ 3 11, and four instances of a GC ''null'' allele have also been documented [32-351.
2 Methods
2.1 Immunoelectrophoresis
The genetic variations of the group specific component were originally discovered by immunoelectrophoresis [2]. The micromodification of the method of Grabar and Williams [ 11 as developed by Scheidegger [361 was employed. Hirschfeld provided a recipe for Gc classification which was used for more than 15 years in laboratories throughoutthe world [21]. In brief, sera were separated for 100 to 120 min on agar gels with a voltage gradient of 7 V/cm in a barbital buffer system, pH 8.6, containing calcium lactate [37, 21. For the immune reaction antisera were preferred that had antibodies against G C as well as against other serum proteins, so that the position of their precipitin lines could serve as reference points.
2.2 Electrophoretic procedures for classification of rare GC phenotypes
For the classification of the three common GC types known at that time, analysis by immunoelectrophoresis was sufficient as well as reliable. For the delineation of variant phenotypes, other methods were applied in addition: starch gel electro- phoresis [22], polyacrylamide gel electrophoresis [ 381, and antigen-antibody crossed gel electrophoresis [ 3 91.
2.3 Agarose gel electrophoresis and specific immunofixation
A powerful method for the separation of the common and rare genetic G C types was developed by Alper and Johnson [401. Separation was varried out on thin-layer agarose gel plates with a 0.075 M barbital buffer, pH 8.6. After electrophoresis monospecific antiserum was placed at the gel surface. Thegels were subsequently pressed, washed and finally stained. With this procedure the electrophoretic heterogeneity of the GC 1 gene product could readily be demonstrated 1271. All available rare G C variants were analyzed with this method and classified into bouble-band and single-band mutants. Double- band variants were derived through mutations from the GC* 1 gene: single-band mutants were ascribed to mutations of the GC*2 gene [271.
2.4 IEF in polyacrylarnide gels followed by immunoprinting
IEF for the analysis of the G C system was employed first in 1977 by Constans and Viau [281. With this method it is possible to recognize subtypes of G C 1 so that six common G C phenotypes can be distinguished; they are determined by three autosomal alleles. IEF has become the method of choice for G C classification. Separation is carried out in flat-bed polyacrylamide gels of 0.5 mm thickness (T = 5 %; C = 3 %) in carrier ampholyte generated pH gradients. Pharmalyte pH 4.5-5.4 (I .2 mL in a final volume of 18 mL of polymerization solution) [431. Separation time is usually4 h at electric settings of 1900 V, 20 mA and 25 W. As electrode solution 1 M phosphoric acid is used at the anode and 0.2 M NaOH at the cathode. An efficient cooling system is necessary and the cir- culating cooling fluid usually has a temperature of 10 "C. The sera to be analyzed are conventionally applied on filter paper pieces, placed on the gel surface 2 cm from the cathodic edge, and removed after 30 min. G C is visualized after IEF by an immune reaction with a monospecific G C antiserum. The procedure is known as immunoprint: cellulose acetate strips are soaked with antiserum and placed on the gel surface, care being taken not to entrap air bubbles. Depending on the an- tiserum, the cellulose acetate strip may be applied wet or air- dried and left in contact with the gel for 3-5 min. The strip is washed afterwards with running tap water for 20 min and sub- sequently stained with Coomassie Brilliant Blue R-250. Classification of GC types without visualization of the GC components by an immune reaction is, in our experience, unreliable [411.
2.5 Alternative methods for the delineation of GC phenotypes
2.5.1 IEF in the presence of 3 M urea
Polyacrylamide gels with a thickness of 1 mm are prepared with 2 % w/v carrier ampholytes, pH 4-6. Urea is added to a final concentration of 3 M. Electrode solutions are the same as for the conventional IEF (see Section 2.4). The conditions for IEF are 1200 V, 20 mA, 15 W for 2 h and 30 min at a tem- perature of 10 "C. Serum samples are applied without prior reduction and alkylation. Immunoprints are made with air- dried cellulose acetate strips, saturated with monospecific GC antiserum, with a contact time of only 1 min. Under these con- ditions urea does not interfere with the immune precipitation up to a concentration as high as 6 M.

400 J . Constans and H. Clew Ekctrophorcsis 1988. 9. 398-403
2.5.2 Two-dimensional analysis
Three different modifications have been employed for the analysis of G C variant phenotypes which have been described previously: (i) polyacrylamide gel electrophoresis and an- tigen-antibody crossed gel electrophoresis in agarose [421; (ii) IEF and antigen-antibody crossed gel electrophoresis in agarose 1311, and (iii) the combination of IEF without urea and IEF in the presence of 6 M urea [3 11. Occasionally also one-dimensional gel electrophoresis in polyacrylamide gels is used, as first described for the GC system in 1965 1381.
2.6 IEF in immobilized pH gradients (IPG)
The gels, 250 x 120 x 0.5 mm in size, are prepared, from the following two solutions, in a cassette with the aid of a micro- gradient mixer coupled to aperistaltic pump. The acid solution contains 500 pL Immobiline pK 4.6, 300 pL Immobiline pK 6.2, 1.3 mL acrylamide and N,N’-methylenebisacrylamide (28.8 % + 1.2 % w/v), 2.5 g glycerol and distilled water up to 10 mL. The basic solution has 600 pL ofeach Immobiline, pK 4.6 and pK 6.2, respectively, 1.3 mL of the acrylamide and N,N’-methylenebisacrylamide (as above) and distilled water added to 10 mL. The final pH gradient ranges from 4.78 to 5.50 [431. Polymerization is catalyzed by N,N,N’,N’-tetra- methylethylenediamine and ammonium persulfate, added to each solution. For accelerated polymerization the gels are kept at 50 “C for 2 h. Before use, they are placed in a refrigerator for 1 h. Gels are removed from the cassette and used without prewashing and without prefocusing 1441. Separation is carried out overnight for 16 h and the electrical settings are 2500 V, 25 mA and 10 W. The cooling system with circulating cooling fluid is set at 10 OC. G C is visualized on immunoprints and may also be observed on the gels after staining with Coomassie Brilliant Blue G-250. Hybrid IEF in immobilized pH gradients with added carrier ampholytes of- fers, in our experimence, no advantage for the GC/DBP system.
3 Results
3.1 Routine GC classification
G C classification in routine work is made by IEF followed by immunoprinting. In Fig. 1 the sic common G C phenotypes lF , IF-IS, IS, 2-1F, 2-1s and 2 are shown. Note the double band patterns of GC 1F and GC 1s and the single band pat- tern of GC 2 and the various combinations in the heterozy- gous phenotypes.
3.2 Less common phenotypes
A selection of less common G C 1A variants, with the common GC type 1F- 1 S as reference, is presented in Fig. 2. Six different 1A variants in the heterozygous state, together with I F or lS, are shown with their four band patterns. The variants 1A8 and 1A7 are similar under these conditions. In Fig. 3 1A variants are demonstrated with positions between the bands of I F and 1s. Shown are the variants 1Al and 1A10, which were originally named G C Aborigine and GC Chippewa, respec- tively, according to the ethnic groups in which they were
Figure 1. Six common G C phenotypes demonstrated by IEF followed by immunoprinting. Fromleft toright: GC2,2-1F, lF, IF-IS, lS,2-1S,and2. Samples applied at the cathode. In this and subsequent figures the anode is at the top.
Figure 2. GC 1A variants analyzed by IEF and subsequent immunoprint. From1efttoright:GC 1F-IS, IA9-lF, IA8-lS, 1A7-1S, 1A5-1S, 1A3-1F, 1A2-1S, and IF-1s.
Figure 3. IEF and immunoprint ofthe GC variants 1A 1 and 1A10. From left toright: GC IF-IS, IA1-2, l A l , IF-IS, IAIO-IS, and IF-IS.
originally observed, the Australian Aborigines and the Chip- pewa Indians. 1 A 1 and 1 A 10 can clearly be distinguished by polyacrylamide gel electrophoresis and IEF in the presence of 3 M urea (not shown). Two unusual 1A variants are presented in Fig. 4. They are unusual because of their anodal positions, which are related to their special form of sialilation. GC 1 F, 1 s and the vast majority of 1A and 1C variants havedouble band patterns of which the l a isoform has, per GC polypeptide chain, one single carbohydrate side chain with one terminal residue of sialic acid, whereas the l c isoform is devoid of carbohydrate [ 151. 1A 17 and 1A 1 1 of Fig. 4 apparently have several sialic acid residues and both isoforms are sialilated [45 I . GC 1 A 17 has three or four NANA residues in the 1 a isoform and three or two NANA residues in the l c isoform. G C 1A 1 1 probably has three NANA residues in the anodal and two NANA residues in the cathodic component.
In Fig. 5 a selection of less common 1C phenotypes is pre- sented. 1C3 is frequently encountered in the German popula-
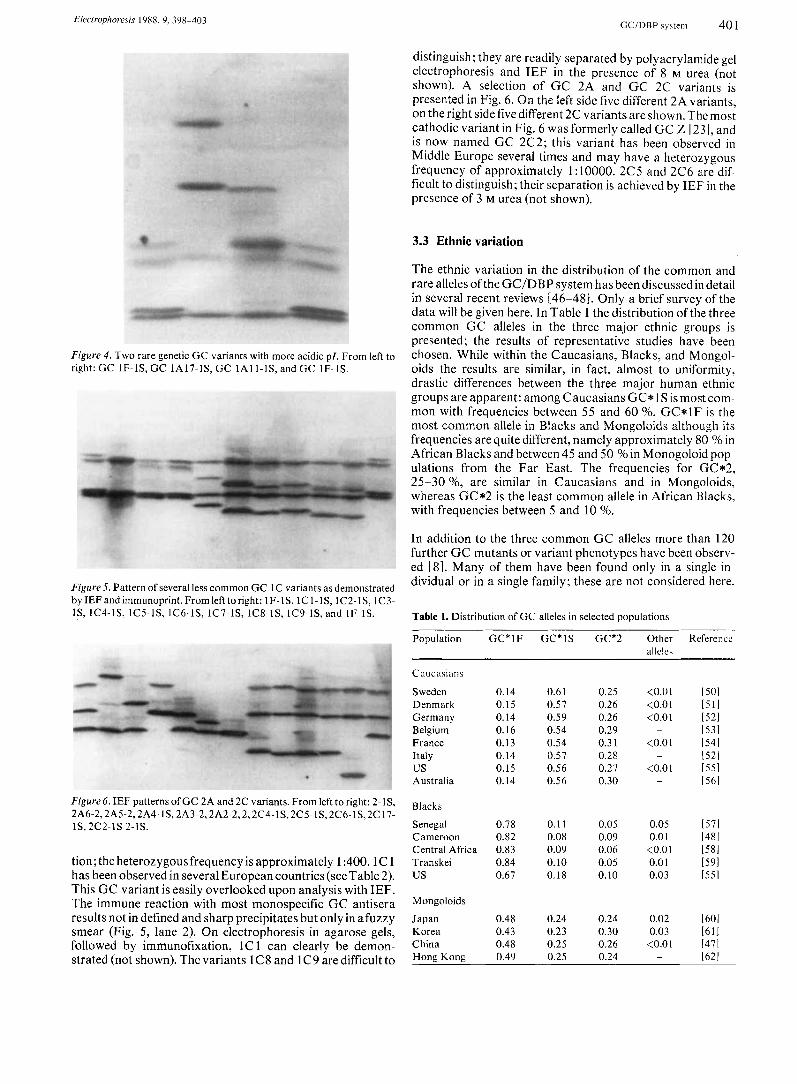
Electrophoresis 1988, 9, 398-403 GC/DBP sy\teni 401
distinguish; they are readily separated by polyacrylamide gel electrophoresis and IEF in the presence of 8 M urea (not shown). A selection of GC 2A and GC 2C variants is presented in Fig. 6. On the left side five different 2A variants, on the right side five different 2C variants are shown. The most cathodic variant in Fig. 6 was formerly called GC Z I23 I , and is now named GC 2C2; this variant has been observed in Middle Europe several times and may have a heterozygous frequency of approximately 1 : 10000. 2C5 and 2C6 are dif- ficult to distinguish; their separation is achieved by IEF in the presence of 3 M urea (not shown).
3.3 Ethnic variation
The ethnic variation in the distribution of the common and rare alleles of the GC/DBP system has been discussedin detail in several recent reviews 146-481. Only a brief survey of the data will be given here. In Table 1 the distribution of the three common GC alleles in the three major ethnic groups is presented; the results of representative studies have been chosen. While within the Caucasians, Blacks, and Mongol- oids the results are similar, in fact, almost to uniformity, drastic differences between the three major human ethnic groups are apparent: among Caucasians GC* IS is most com- mon with frequencies between 55 and 60 %. GC*lF is the most common allele in Blacks and Mongoloids although its frequencies are quite different, namely approximately 80 %in African Blacks and between 45 and 50 %in Monogoloid pop- ulations from the Far East. The frequencies for GC*2, 25-30 %, are similar in Caucasians and in Mongoloids, whereas GC*2 is the least common allele in African Blacks, with frequencies between 5 and 10 %.
Figure 4 . Two rare genetic GC variants with more acidic PI. From left to right: GC IF-IS, GC lA17-lS, GC ]All-lS, and GC IF-IS.
Figure 5 . Pattern of several less common GC 1C variants as demonstrated by IEF and immunoprint. From left to right: 1 F-l S, 1C 1- 1 S, 1 C2-lS, 1C3- lS, 1C4-1S, 1C5-lS, 1C6-1S, 1C7-1S, lCS-lS, lC9-lS, and IF-IS.
In addition to the three common G C alleles more than 120 further GC mutants or variant phenotypes have been observ- ed 181. Many of them have been found only in a single in- dividual or in a single family; these are not considered here.
Table 1. Distribution of GC alleles in selected populations
Figure 6. IEF patterns of GC 2A and 2C variants. From left to right: 2-1S, 2A6-2,2A5-2,2A4- IS, 2A3-2,2A2-2,2,2C4-lS,2CS-lS, 2C6-lS,2C 17- lS, 2c2-1s 2-1s.
tion; the heterozygous frequency is approximately 1 :400.1C 1 has been observed in several European countries (see Table 2). This GC variant is easily overlooked upon analysis with IEF. The immune reaction with most monospecific G C antisera results not in defined and sharp precipitates but onlyin afuzzy smear (Fig. 5, lane 2). On electrophoresis in agarose gels, followed by immunofixation, l C 1 can clearly be demon- strated (not shown). The variants lC8 and l C 9 aredifficult to
Population
Caucasians
Sweden Denmark Germany Belgium France Italy us Australia
GC*lF GC*lS
0.14 0.61 0.15 0.57 0.14 0.59 0.16 0.54 0.13 0.54 0.14 0.57 0.15 0.56 0.14 0.56
GC*2
0.2s 0.26 0.26 0.29 0.3 1 0.28 0.27 0.30
Other Reference allele\
(0.01 [SO1 (0.01 l 5 l l (0.01 [521
b3l (0.01 [54l
1521 (0.01 [55l
1561
-
-
-
Blacks
Senegal 0.78 0.11 0.05 0.05 [571 Cameroon 0.82 0.08 0.09 0.0 1 1481 Central Africa 0.83 0.09 0.06 (0.01 (581 Transkei 0.84 0.10 0.05 0.0 1 [591 us 0.67 0.18 0.10 0.03 [551
Mongoloids
Japan 0.48 0.24 0.24 0.02 [601 Korea 0.43 0.23 0.30 0.03 1611 China 0.48 0.25 0.26 <0.01 [471 Hone Kone 0.49 0.2s 0.24 1621 -

Elcctruphurcsis 1988. 9, 398-403 402 J . Constans and H. Clew
Others have been found repeatedly, either confined to certain populations or, also, with a more widespread geographical dis- tribution. These GC variant are presented in Table 2. The variant 1A 1 is fairly common in Australian Aborigines, Melanesians as well as in African and American Blacks. 1A2, 1A3, lA8, 1A9 and 2A4 are fairly common in Mongoloid populations from the Far East, especially among J apanese. In Europe, particularly in Central Europe, the variants 1A5, l A l l , 1C1, 1C3,1C5,1C7,1C8, 1C9,1C25,2A2 and 2C2 are observed occasionally. 2A3 and 2A5 are fairly frequent in African and American Blacks. A deficiency mutant was observed in a large kindred from Czechoslovakia 13 11. This GC variant was called GC 1”; the allele G C 1” was observed only in the heterozygous state. The G C concentrations in the sera of these persons were reduced to almost 50 % of the nor- mal concentration. A gene product of the deficiency mutant could be demonstrated only by a two-dimensional electro- phoresis technique with IEF in a conventional system in the first dimension and IEF in the presence of 8 M urea in the se- cond dimension. Instances of a so-called G C “null” allele have been reported by several authors [32-351.
4 Discussion
4.1 Paternity testing
The usefulness of the GC/DBP system for paternity testing is apparant. In Table 3 the theoretical exclusion rate for non- fathers and the probability of identity of two unrelated in- dividuals calculated for the German population are given.
Table 2. GC mutants with appreciablefrequenciesin certain populations
GC Mutantr
IA I
1A2 IA3
1A4 1A5 1A6 1A8 1A9 lAlO l A l l
1 c 1 1 c 2 1C3 1C5 1C7 1C8 1C9 l C l 0 1C24 1C25 1C27
2A2 2A3 2A4 2AS 2A8 2A9 2A10
Table 3. Discriminatory power of the GC/DBP system in cases of disputed paternitya)
Theoretical exclusion rate for non-fathers
Probability of identity of two unrelated individuals
29.4 %
35.6 %
a) Calculated from the genotype and allele frequencies of the German population
Similar figures are obtained for the other European popula- tions. In addition, the presence of a rare G C variant in a child and in an alleged father is a strong indication for the biological fatherhood of this man. In our view, it is not necessary to iden- tify precisely the uncommon G C variant. I t suffices toclassify the variant into the broad categories lA, lC , 2A and 2C, respectively, and to establish the identity of the G C variant in the child and the alleged father by repeated examinations side by side. As emphasized earlier, while IEF in polyacrylamide gels with carrier ampholytes, followed by immunoprinting with monospecific antiserum, is the method of choice for routine classification, it may be necessary -in uncertain cases - to supplement the investigation with one of the other methods outlined above. Unreliable in our experience is the reading of G C types from gels without specific immune reac- tion only after fixation of proteins with sulfosalicylic acid and methanol. With the latter procedure erroneous GC classifica- tions may occur L411. The rare GC variant most easily over- looked is G C 1C 1 since this mutant is not characterized by a sharp band but rather by a fuzzy smear. The possibility of a “null” allele in the G C system has also to be born in mind, par- ticularly if an exclusion constellation is only given in the G C system.
Fthnic o r rcnrrnpliic di\trihuticin
Australian and New Guinean Aborigines, African and American Blacks, Polynesia, Indonesia Japanese, Chinese, Koreans, Eskimos Negritos, Japanese, Chinese, Koreans, Eskimos, North American Indians Eskimos, South American Indians, Japanese Germany East Africa Asia, South American Indians, Europe Asia, South American Indians, Europe Chippewa Indians, Europe Belgium, France, Germany
France, Germany, Austria, Czechoslovakia B ali Germany, France, North and Central Africa Europe Czechoslovakia, Germany, Tibet, Brazil France, Germany Denmark, Germany North American Indians Indonesia US Caucasians, Germany Germany
Czechoslovakia, Germany, Sweden African and US Blacks Japan, China African and US Blacks Germany Scotland, Sweden, Denmark Guam
4.2 DBP-Actin complexes
Problems may also arise by the presence of DBP-actin complexes in plasma or, particularly, in blood stains, The DBP-actin complex migrates faster and has a more acidic pl. GC/DBP classification in blood stains is, therefore, routinely performed after extraction with solvents containing either 6 M urea or 4 M guanidine hydrochloride. Unusual GC patterns are also observed when the holoprotein is present in increased concentrations. Usually more than 95 % of the plasma GC/ DBP is unsaturated with vitamin D and is recognized as the apo-protein. The holoform is present in less than 5 % and escapes notice. The DBP-vitamin D complex also has a faster electrophoretic migrating rate and a more acidic p l [61. In children, following large doses of vitamin D, the holoprotein is occasionally observed on IEF as an additional band close to the anode. Furthermore, GC/DBP may be present in a dif- ferent isoform due to an unusual sialilation. This isoform ofthe G C 1 protein, either 1F or lS, has two NANA residues at- tached to the 1 a isoform istead of the ordinary 1 a isoform with one carbohydrate side chain with a single terminal NANA residue. This so-called la’isoform of DBP is recognized as an additional anodally located band, especially in patients with severe liver diseases, for instance in patients with alcoholic cir- rhosis of the liver [49].
4.3 Conclusions
2 c 2 Germany, Denmark The determination of serum protein polymorphismsh dis- 2C5 Israel, Sweden puted paternities is now routinely carried out. Various elec-

Hecrruphoresis 1988, 9. 398-403 GCIDBI' system 403
trophoretic methods are used and important new informa- tion was gained by the application of IEF. For the specific identification of genetic markers, immunologic methods are employed. GC is an abundant serum protein and its deter- mination is easy and cheap, since more than40 samples can be run on a single gel. A side-by-side analysis ofthe serais an im- portant factor for the resolution of variant phenotypes or if only very small quantities of serum are available. It should also be used when the protein is present in low concentrations in other biological fluids, such as cerebrospinal fluid, seminal li- quid and bloodstains recovered from different materials. Western blots using nitrocellulose or nylon membranes can be performed combined with sensitive immunoenzymatic stain- ing. The results appear to be reliable despite the occurrence of complexes between actin and DBP. The presence of a similar DBP protein among mammals and other vertebrates has to be kept in mind when considering the conclusions in stain analysis. Fortunately, most of the commercial GC antisera do not precipitate the GC belonging to animals other than primates.
Received June 13, 1988
5 References
Grabar, P. and Williams, C. A., Biochim. Biophvs. Acta 1953, 10,
Hirschfeld. J., Acta Path. Microbiol. Scand. 1959, 47, 160-168. Thomas, W. C., Morgan, H. G., Connor,T. B., Haddock, L., Bills, 6. E. and Hovord, J. E.,J. Clin. Invest. 1959,38, 1078-1082. Cleve, H., fsr. J. Med. Sci. 1973, 9, 1133-1 146. Daiger, S. P., Schanfield, M. S. and Cavalli-Sforza, L. L., Proc. Natl. Acad. Sci. USA 1975,72,2076-2080. Constans, J., in: Binding ProteinsofSteroidHormones, Colloque l n - serm, Vol. 149,JohnLibbey EurotextLtd., Paris 1988,pp. 357-379. Constans, J., Cleve, H., Bennet, A,, Bouillon, R., Cox, D. W., Daiger, S. P., Ehnholm, C., Fujiki, N., Johnson,A. M., Kirk, R. L., Kuhnl, P., Martin, W., Matsumoto, H., Mayr, W. R., Miyake, K., Miyazaki, T., Omoto, K., Porck, H. J.,Seger,J .,Thymann, M.,Tills, D.,Toyomasu, M., Baelen, H. van, Vavrusa, B. and Viau, M., Human Genet. 1979,
Cleve, H. and Constans, J., Vox Sang. 1988,54,2 15-225. Schoentgen, F., Metz-Boutigue, M. H., Jolles, J., Constans, J . and Jolles, P., FEBS Lett. 1985, 185,47-50. Schoentgen, F., Metz-Boutigue, M. H., Jolles, J., Constans, J. and Jolles, P., Biochim. Biophys. ACtQ 1986,871, 189-198. Brown, J. R., Fed. Proc. Fed. Am. Soc. Exp. Biol. 1976, 35,
Cooke,N. E. andDavid,E.V.,J.Clin.lnvest. 1985,76,2420-2424. Yang, F., Brune, J. L., Naylor,S. L.,Cupples,R. L.,Naberhaus,K. H. and Bowman, B. H., Proc. Natl. Acad. Sci., USA 1985, 82,
Viau, M., Constans, J., Debray, H. and Montreuil. J., Biochem. Biophys. Res. Commun. 1983, I 17, 324-33 I . Cleve. H. and Patutschnick, W., Hum. Genet. 1979, 47, 193-198. Mikkelsen, M., Jacobsen, P. and Henningsen,K., Hum. Hered. 1977, 27, 105-107. Cooke, N. E., Willard, H. F., David, E. V. andGeorge, D. L., Human Genet. 1986,73,225-229. McCombs, J.L.,Yang, F.,Bowman, B. H., McGill, J. R. and Moore, C. M.. Cytogenet. Cell Genet. 1986,42, 62-64. Hirschfeld, J., Jonsson, B. and Rasmuson, M., Nature 1960, 185, 93 1-932. Cleve,H.andBearn,A.G.,Ann.N.Y.Acad.Sci. 1961,94,218-224. Hirschfeld, J., Prog. Allergy 1962,6, 155-186. Cleve, H., Kirk, R. L., Parker, W. C., Bearn, A. G., Schacht, L. E., Kleinmann, H. and Horsfall, W. R., Am. J. Hum. Genet. 1963, 15,
193-194.
48, 143-149.
2 141-2 144.
7994-7998.
368-379.
123 I [24l 1251
Hennig, W. and Hoppe, H. H., Vox Sang. 1965,10,214-217. Reinskou, T., Acta Path. Microbiol. Scand. 1963,59, 526-528. Ackfors, K.-E. and Beckman, L., Acta Genet. Stat. Med. 1963, 13, 23 1-234.
1261 Bearn, A. G., Kitchin, F. D. and Bowman, B. H.,J. Exp. Med. 1964,
1271 Johnson,A.M.,Cleve,H.andAlper,C.,Am.J.Hum.Genet. 1975,27,
1281 Constans, J. and Viau, M., Science 1977,198, 1070-1071. [29l Cleve, H.,Constans,J., Berg, S.,Hoste,B.,Ichimoto,G.,Matsumoto,
H., Spees, E. K. and Weber, W., Hum. Genet. 1981,57,312-316. [30l Constans, J., Cleve,H.. Dykes,D.,Fischer,M., Kirk, R. L., Papiha,S.
S., Scheffrahn, W., Scherz, R., Thymann, M. and Weber, W., Hum. Genet. 1983,65, 176-180.
1311 Vavrusa, B., Cleve, H. and Constans, J., Hum. Genet. 1983, 65,
1321 Prokop, 0. and Rackwitz, A., Deutsch. Z. Gerichtl. Med. 1968,62,
1331 Patscheider,H. and Dirnhofer,R.,Z.Rechtsmed. 1979,82,243-249. [341 Brinkmann, B., Soder, R. and Janssen, W., in: Hummel, K . and Ger-
chow, J. (Eds.), Biomathematical Evidence of Paternity. Springer, Berlin 1981, pp. 127-130.
1351 Suzuki, K., Itoh, S., Kawai, N., Miyazaki, T., Matsui, K., Yanazaki, K. and Matsumoto, H., Hum. Hered. 1986,36,326-329.
1361 Scheidegger, J . J., Internat. Arch. Allergy 1955, 7, 103-1 10. 1371 Laurell,C. B.,Laurell,S. and Skoog,N., Clin. Chenz. 1956,2,99- 1 1 I . 1381 Kitchin,F.D.,Proc. Soc. Exp. Biol.Med. 1965, 119, 1153-1155. 1391 McDermid, E. M. and Cleve, H., Hum. Hered. 1972,22, 249-253. 1401 Alper, C. A. and Johnson,A. M., VoxSang. 1969, 17,445-452. [411 Weidinger, S.,Cleve,H.,Schwarzfischer,F. andpatutschnick, W.,in:
Hummel, K. and Gerchow, J. (Eds.), Biomathematicat Evidence oj Paternity, Springer, Berlin 198 1, pp. 1 13- 12 1.
1421 Constans, J . Cleve, H., Viau, M. and Gouaillard, C., Vox Sang. 1978,
1431 Constans, J. and Cleve, H., Electrophoresis 1988, 9, in press. [441 Cleve, H., Patutschnick, W., Postel, W., Weser, J. and Gorg, A.,
Eiectrophoresis, 1982, 3, 342-345. [451 Thymann, M., Hoste, B., Scheffrahn, W., Constans, J. andCleve, H.,
Hum. Genet. 1985,69, 224-227. [461 Kamboh.M.1. andFerrell,R.E., Hum. Genet. 1986,72,281-293. [471 Kaniboh, M.I., Ranford,P. R. and Kirk, R. L., Hum. Genet. 1984,67,
[481 Constans, J., Hazout, S., Garruto, R. M., Gajdusek, D. C. and Spees, E. K., Am. J . PhjJs. Anthr. 1985,68, 107-122.
1491 Constans, J., Arlet, Ph., Viau, M. and Bouissou, C., Clin. Chim. Acta, 1983,13O,219-230.
[SO] Svcnsson, M. and Hjalmarsson, K., Proceedings o f thc 9th Inter- national Congress of the Society of Forensic Hnemogenctics, Bern
120,83-9 I .
728-736.
102-107.
261-268.
34.46-50.
378-384.
1981, pp. 559-562. [5 l l Thymann, M., Hum. Hered. 1981,31,219-221. 1521 Cleve, H., Patutschnick, W., Nevo, S. and Wendt,G. G.,Hum. Genet.
1531 Hoste,B., Hum. Genet. 1979,50, 75-79. 1541 Constans, J., Viau, M., Cleve, H., Jaeger, G., Quilici, J. C. and
(551 Dykes, D. D., Crawford, M. H. and Polesky, H. F., A m . J . Phys.
1561 Nicholls, C. and Mulley, J . C., Aust. J . Exp. B i d . Med. Sci. 1982,60,
1571 Constans, J., Viau, M., Pison, G. and Langaney, A,, Jap. J . Hum.
1581 Constans, J. Lefevre-Witier, Ph., Richard, P. and Jaeger, G., Am. J .
[59] Papiha, S. S., Constans, J., White, I. and McGregor, I . A.,Ann. Hum.
1601 Ishinioto, G., Kuwata, M. and Nakajima, H., Jap. J . Hum. Genet.
[611 Matsumoto, H., Matsui, K., Ishida, H., Ohkura, K. andTeng. Y . 3 ,
1621 Kwok,K.Y.Y. andLewis,W. H.P.,Hum.Genet. 1981,59,72-74.
1978,44, 117-122.
Palisson, M. J., Hum. Genet. 1978, 41, 53-60.
Anthr. 1983,62, 137-146.
427-431.
Genet. 1978,23, 111-117.
Phys. Anthr. 1980,52,435-441.
B i d . 1985, 12, 17-26.
1979,24,75-83.
Am. J . Phys. Anthr. 1980,53, 505-508.