the coalescent and measurably evolving populations alexei drummond department of computer science...
Post on 22-Dec-2015
219 views
TRANSCRIPT

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns
The Coalescent and Measurably Evolving Populations
Alexei Drummond
Department of Computer Science
University of Auckland, NZ

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns Overview
1. Introduction to the Coalescent
2. Hepatitis C in Egypt• An example using the coalescent
3. Measurably evolving populations
4. HIV-1 evolution within and among hosts• An example using MEP concepts
5. Summary + Conclusions

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns The coalescent
• The coalescent is a model of the ancestral relationships of a small sample of individuals taken from a large background population.
• The coalescent describes a probability distribution on ancestral genealogies (trees) given a population history. – Therefore the coalescent can convert information from ancestral
genealogies into information about population history and vice versa.
• The coalescent is a model of ancestral genealogies, not sequences, and its simplest form assumes neutral evolution.

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns The history of coalescent theory
• 1930-40s: Genealogical arguments well known to Wright & Fisher
• 1964: Crow & Kimura: Infinite Allele Model
• 1966: (Hubby & Lewontin) & (Harris) make first surveys of population allele variation by protein electrophoresis
• 1968: Motoo Kimura proposes neutral explanation of molecular evolution & population variation. So do King & Jukes
• 1971: Kimura & Ohta proposes infinite sites model.
• 1975: Watterson makes explicit use of “The Coalescent”
• 1982: Kingman introduces “The Coalescent”.
• 1983: Hudson introduces “The Coalescent with Recombination”
• 1983: Kreitman publishes first major population sequences.
• 1987: Cann et al. traces human origin and migrations with mitochondrial DNA.

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns The history of coalescent theory
• 1988: Hughes & Nei: Genes with positive Darwinian Selection.
• 1989-90: Kaplan, Hudson, Takahata and others: Selection regimes with coalescent structure (MHC, Incompatibility alleles).
• 1991: MacDonald & Kreitman: Data with surplus of replacement interspecific substitutions.
• 1994-95: Griffiths-Tavaré + Kuhner-Yamoto-Felsenstein introduces sampling techniques to estimate parameters in population models.
• 1997-98: Krone-Neuhauser introduces Ancestral Selection Graph
• 1999: Wiuf & Donnelly uses coalescent theory to estimate age of disease allele
• 2000: Wiuf et al. introduces gene conversion into coalescent.
• 2000-: A flood of SNP data & haplotypes are on their way.

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns Population processes
Genealogy
COALESCENT
THEORY

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns
Randomly sample individuals from population
Obtain gene sequences from sampled individuals
Reconstruct tree / trees from sequences
Infer coalescent results from tree / trees
Infer coalescent results directly from sequences
Coalescent inference

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns Demographic History
• Change in population size through time
• Applications include– Estimating history of human populations– Conservation biology– Reconstructing infectious disease epidemics– Investigating viral dynamics within hosts

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns Idealized Wright-Fisher populations
Now
Parents
Grand parents
Haploid Diploid

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns Random mating in an ideal population
•A constant population size of N individuals•Each individual in the new generation “chooses” its parent from the previous generation at random

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns Genetic drift: extinction and ancestry
If you trace the ancestry of a sample of individuals back in time you inevitably reach a single most recent common ancestor.If you pick a random individual and trace their descendents forward in time, all the descendents of that individual will with high probability eventually die out.

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns
Dis
cret
e G
ener
atio
ns
Present
PastA sample genealogy of 3 sequences from a population (N =10).
Present
Past
A sample genealogy from an idealized Wright-Fisher population

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns
The coalescent: distributions and expectations on a sample genealogy
Present
Past
t2 ~ Exp(N)
t3 ~ Exp N /3
E[t3]N /3
E[t2]N
E[tk ]2N
k(k 1)
E[troot ]2N 11
n

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns
The coalescent: probability density distribution
Present
Past
P(t2 | N) 1
Nexp
t2
N
P(t3 | N) 3
Nexp
3t3
N
fG (g | N) k(k 1)
Nexp
k(k 1)tk
N
k2
n
dg
t2 ~ Exp(N)
t3 ~ Exp N /3
Kingman (1982a,b)
g Eg ,t The genealogy is an edge graph Eg and a vector of times t.

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns
The coalescent: estimating population size from a sample genealogy
Present
Past
ˆ N k k(k 1)
2tk
ˆ N 3 24
ˆ N 2 7
ˆ N 1
n 1
k(k 1)
2tk
k2
n
ˆ N 15.5
t2 7
t3 8
Felsenstein (1992)

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns
The coalescent: estimating population size confidence limits via ML
Maximum likelihood can be used to estimate population size by choosing a population size that maximizes the probability of the observed coalescent waiting times.
The confidence intervals are calculated from the curvature of the likelihood. For a single parameter model the 95% confidence limits are defined by the points where the log-likelihood drops 1.92 log-units below the maximum log-likelihood.
-20
-18
-16
-14
-12
-10
-8
-6
1 10 100 1000
Population size (N)
rela
tive log lik
elihood
ˆ N 15.5 (5.1, 93.1)

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns
Exponential growth Constant size
The coalescent: shapes of gene genealogies
The coalescent can be used to convert coalescent times into knowledge about population size and its change though time.
Th
e C
oal
esce
nt

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns
smallsmall NN00 large large NN00
TIMETIME
Constant population size: N(t)=N0
Th
e C
oal
esce
nt

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns Coalescent and serial samples
Constant population Exponential growth
Th
e C
oal
esce
nt

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns Uncertainty in Genealogies
Th
e C
oal
esce
nt
How similar are these two trees? Both of them are plausible given the data.
We can use MCMC to get the average result over all plausible trees,

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns Coalescent Summary
• The coalescent provides a theory of how population size is related to the distribution of coalescent events in a tree.
• Big populations have old trees• Exponentially growing populations have star-like trees• Given a genealogy the most likely population size can be
estimated.• MCMC can be used to get a distribution of trees from
which a distribution of population sizes can be estimated.
Th
e C
oal
esce
nt

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns Markov chain Monte Carlo (MCMC)
MC
MC
• Imagine you would like to estimate two parameters (,) from some data (D).
• You want to find values of and that have high probability given the data: p(,|D)
• Say you have a likelihood function of the form: Pr{D| ,}
• Bayes rule tells us that:– p(,|D) = Pr{D| ,}p(,) / Pr{D}– So that p(,|D) Pr{D| ,}p(,)

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns Markov chain Monte Carlo (MCMC)
MC
MC
• p(,|D) is called the posterior probability (density) of , given D• In an ideal world we want to know the posterior density for all
possible values of ,.• Then we could pick a “credible region” in two dimensions that
contained values of , that account for the majority of the posterior probability mass.
• This credible region would serve as an estimate that includes incorporates our uncertainty and this credible set could be used to address hypotheses like: is greater than x.
• In reality we have to make due with a “sample” of the posterior - so that we evaluate p(,|D) for a finite number (say 10,000,000) pairs of ,.
• So which pairs should we choose?

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns Markov chain Monte Carlo (MCMC)
MC
MC
• Lets construct a random walk in 2-dimensional space• In each step of the random walk we propose to make an (unbiased)
small jump from our current position (,) to a new position (’,’) • If p(’,’|D) > p(,|D) then we make the proposed jump• However, if p(’,’|D) < p(,|D), then we make the proposed jump
with probability = p(’,’|D) / p(,|D), otherwise we stay where we are.
• It can be shown (trust me!) that if you proceed in this fashion for an infinite time then the equilibrium distribution of this random walk will be p(,|D)!
• That is, the random walk will visit a particular region [0, 1] x [0, 1] of the state space this often:
p(, | D)dd 0
1
0
1

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns Markov chain Monte Carlo (MCMC)
MC
MC
p(, | D) Z Pr{D |,g} f ( | g) f (,)g

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns Hepatitis C Virus (HCV)
Po
pu
lati
on
gen
etic
s o
f H
epat
itis
C in
Eg
ypt
• Identified in 1989• 9.6kb single-stranded RNA genome • Polyprotein cleaved by proteases• No efficient tissue culture system

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns How important is HCV?
• 170m+ infected• ~80% infections are chronic• Liver cirrhosis & cancer risk• 10,000 deaths per year in
USA• No protective immunity?
Po
pu
lati
on
gen
etic
s o
f H
epat
itis
C in
Eg
ypt

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns
Percutaneous exposure to infected bloodPercutaneous exposure to infected blood
• Blood transfusion / blood products
• Injecting & nasal drug use
• Sexual & vertical transmission
• Unsafe injections
• Unidentified routes
HCV Transmission
Po
pu
lati
on
gen
etic
s o
f H
epat
itis
C in
Eg
ypt

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns
Estimating demographic history of HCV using the coalescent
• Egyptian HCV gene sequences
• n=61
• E1 gene, 411bp
• All sequence contemporaneous
• Egypt has highest prevalence of HCV worldwide (10-20%)
• But low prevalence in neighbouring states
• Why is Egypt so seriously affected?
• Parenteral antischistosomal therapy (PAT)
Po
pu
lati
on
gen
etic
s o
f H
epat
itis
C in
Eg
ypt

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns Demographic model
• The coalescent can be extended to model deterministically varying populations.
• The model we used was a const-exp-const model.
• A Bayesian MCMC method was developed to sample the gene genealogy, the substitution model and demographic function simultaneously.
N(t)NC if t x
NC exp[ r(t x)] if x t y
NA if t y
Po
pu
lati
on
gen
etic
s o
f H
epat
itis
C in
Eg
ypt

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns Estimated demographic history
Po
pu
lati
on
gen
etic
s o
f H
epat
itis
C in
Eg
ypt
Averaged over all trees
Based on a single tree

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns Parameter estimates
Po
pu
lati
on
gen
etic
s o
f H
epat
itis
C in
Eg
ypt

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns Uncertainty in parameter estimates
Growth rate of the growth phase
Grey box is the prior
Rates at different codon positions,
All significantly different
Demographic parameters Mutational parameters
Po
pu
lati
on
gen
etic
s o
f H
epat
itis
C in
Eg
ypt

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns Full Bayesian Estimation
• Marginalized over uncertainty in genealogy and mutational processes
• Yellow band represents the region over which PAT was employed in Egypt
Po
pu
lati
on
gen
etic
s o
f H
epat
itis
C in
Eg
ypt

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns
Measurably evolving populations (MEPs)
Present time point (n = 5)
Earlier time point (n = 5)
• MEP pathogens:– HIV – Hepatitis C– Influenza A
• MEPs from ancient DNA– Bison– Brown Bears– Adelie penguins– Anything cold and numerous
• Even over short periods (less than a year) HIV sequences can exhibit measurable evolutionary change
• Time-structure can not be ignored in our models
Mea
sura
bly
evo
lvin
g p
op
ula
tio
ns

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns Time structure in samples
timeContemporary sampleno time structure
Serial samplewith time structure
2000
1980
1990
Mea
sura
bly
evo
lvin
g p
op
ula
tio
ns

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns
Ne
time
AB
CD
E
Molecular evolution and population genetics of MEPs
• Given sequence data that is time-structured estimate true values of:– substitution parameters
• Overall substitution rate and relative rates of different substitutions
– population history: N(t)– Ancestral genealogy
• Topology• Coalescent times
Mea
sura
bly
evo
lvin
g p
op
ula
tio
ns

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns
Molecular evolutionary model: Felsenstein’s likelihood (1981)
b1
b2 b5
b4
b3
AAAC
GA
GCThe probability of the sequence alignment,
can be efficiently calculated given a tree and branch lengths (T), and a probabilistic model of mutation represented by an instantaneous rate matrix (Q). In phylogenetics, branch lengths are usually unconstrained.
Pr{D | T,Q}

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns
Combining the coalescent with Felsenstein’s likelihood
b1
b2 b5
b4
b3
AA
AC
GA
GC
t2
t4
t3
AA
GA AC GC
The “molecular clock” constraint
2n–3 branch lengths n–1 waiting times
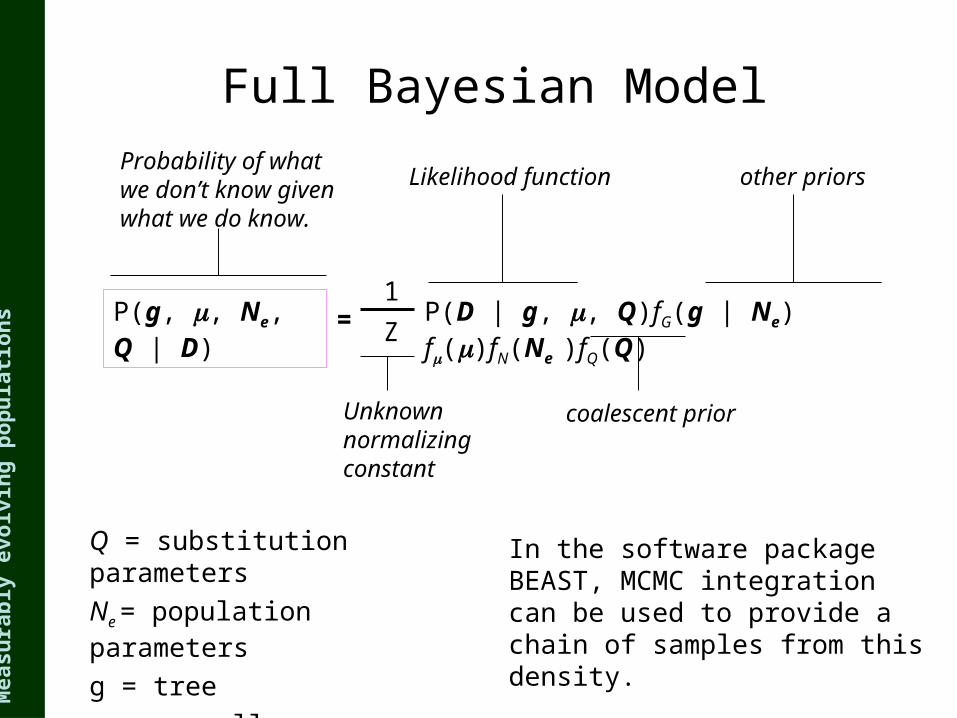
p(N,g,Q | D)Pr{D |g,Q} fG (g | N) fN (N) fQ (Q)
The joint posterior probability of the population history (N), the genealogy (g) and the mutation matrix (Q) are estimated using Markov chain Monte Carlo (Drummond et al, Genetics, 2002)
Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns Full Bayesian Model
P(g, , Ne, Q | D) Z
1P(D | g, , Q)fG(g | Ne) f()fN(Ne )fQ(Q)
Probability of what we don’t know given what we do know.
Unknown normalizing constant
Likelihood function
coalescent prior
other priors
=
In the software package BEAST, MCMC integration can be used to provide a chain of samples from this density.
Mea
sura
bly
evo
lvin
g p
op
ula
tio
ns
Q = substitution parameters
Ne = population parameters
g = tree
= overall substitution rate

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns
Pt.7
Pt.9
HIV1U36148
HIV1U36015HIV1U35980
HIV1U36073
HIV1U35926
HIVU95460
Pt.2
Patient #6 fromWolinsky et al.
Pt.5
Pt.3Pt.1Pt.8
Pt.6
10%
Shankarappa et al (1999)
HIV-1 (env) evolution in nine infected individuals
Mea
sura
bly
evo
lvin
g p
op
ula
tio
ns

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns
Molecular clock: HIV-1 (env) evolution in 9 individuals
0 2 4 6 8 10
Years Post Seroconversion
Viral Divergence
2%
4%
6%
8%
10%
Shankarappa et al (1999)
Mea
sura
bly
evo
lvin
g p
op
ula
tio
ns

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns MEP Summary
• Most RNA viruses, including HCV and HIV are measurably evolving
• Most vertebrate populations that have well-preserved recent fossil records are MEPs.
• If sequence data comes from different times the time-structure can’t be ignored
• Time structure permits the direct estimation of:– substitution rate– Concerted changes in substitution rate– coalescent times in calendar units– Demographic function N(t) in calendar units
Mea
sura
bly
evo
lvin
g p
op
ula
tio
ns

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns
Intermission
My brain is fried!

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns
• HIV is a retrovirus.
• Within infected individuals HIV exhibits extremely high genetic variability due to:
– Error-prone reverse transcriptase (RT) that converts RNA to DNA (error rate is about one mutation per genome per replication cycle).
– DNA-dependent polymerase also error-prone
– High turnover of virus within infected individual throughout infection.
HIV virion
HIV gp120 binds to CD4 T cell surface receptors
viral core inserted into cell
replication of virus genome by reverse transcription (ssRNA to dsDNA)
integration of proviral DNA into DNA of infected cell
viral RNA transcription
regulatory genes
migration of dsDNA to nucleus
viral genomic RNA
RNA packaging and virion assembly
structural proteins and viral enzymes
translation
LTRLTR
budding of virus from cell and maturation
nucleus
host cell
What is HIV?
Po
pu
lati
on
gen
etic
s o
f H
IV
Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
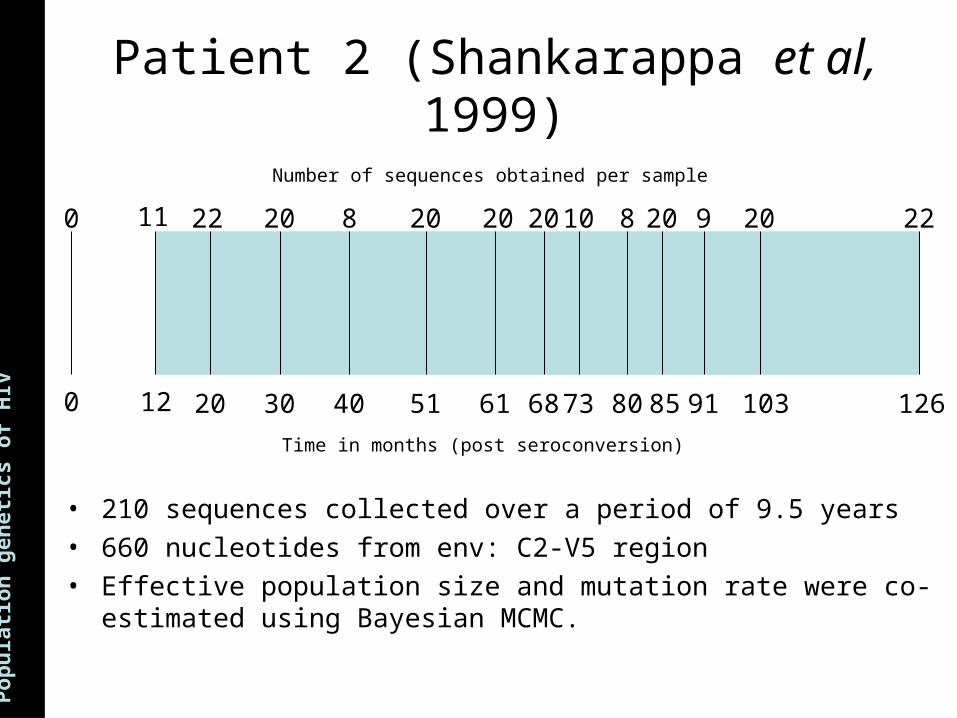
ns Patient 2 (Shankarappa et al, 1999)
0
0
12 20 30 40 51 61 68 73 80 85 91 103 126
11 22 20 8 20 20 20 10 8 20 9 20 22
Time in months (post seroconversion)
Number of sequences obtained per sample
• 210 sequences collected over a period of 9.5 years• 660 nucleotides from env: C2-V5 region• Effective population size and mutation rate were co-estimated using
Bayesian MCMC.Po
pu
lati
on
gen
etic
s o
f H
IV

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns
A tree sampled from the posterior distribution
Lineage A
Lineage B
‘Ladder-like’ appearance
Po
pu
lati
on
gen
etic
s o
f H
IV
Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
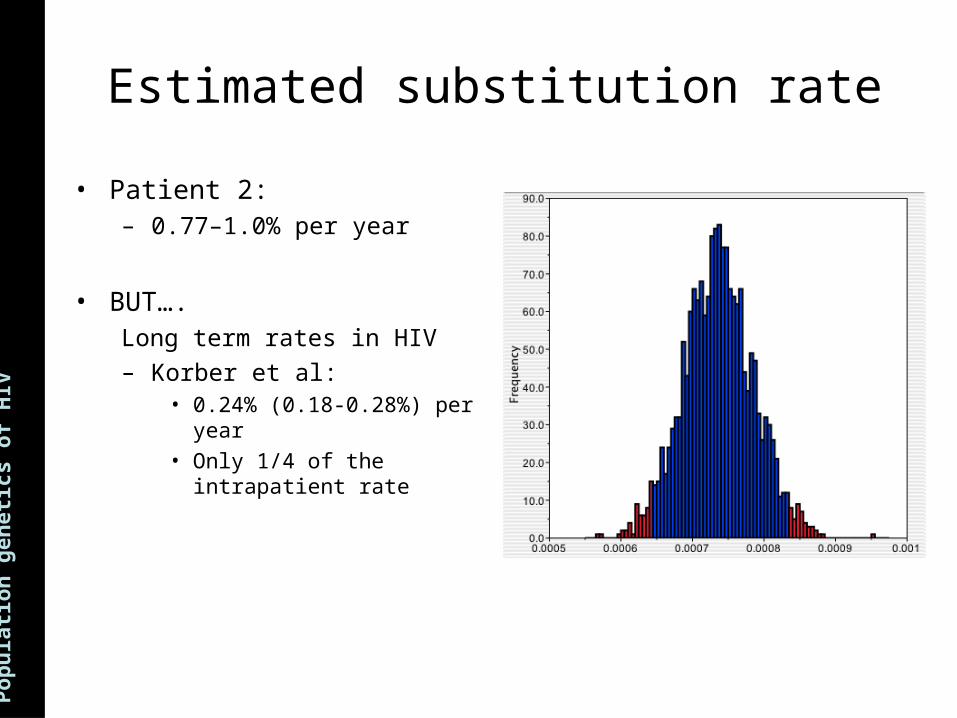
ns Estimated substitution rate
• Patient 2:– 0.77–1.0% per year
• BUT….Long term rates in HIV
– Korber et al:• 0.24% (0.18-0.28%) per year• Only 1/4 of the intrapatient
rate
Po
pu
lati
on
gen
etic
s o
f H
IV

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns Bayesian MCMC of Shankarappa data
estimate upper limitp1 Logistic 0.0123 882 0.278 1.57% 6.68%p2 plasma Logistic 0.0166 1708 0.242 0.63% 3.34%p2 provirus Logistic 0.0090 2798 0.278 0.04% 0.18%p3 Logistic 0.0175 620 0.237 1.80% 4.97%p5 plasma Exponential 0.0223 938 0.192 11.80% 27.50%p5 provirus Logistic 0.0215 1345 0.293 8.19% 15.20%p6 Logistic 0.0195 581 0.221 0.52% 1.35%p7 Logistic 0.0085 3320 0.322 2.94% 13.60%p8 Exponential 0.0162 2309 0.455 28.70% 48.50%p9 Logistic 0.0071 2757 0.346 17.90% 44.80%p11 Logistic 0.0128 2502 0.239 0.15% 0.53%
Overall Logistic 0.0148 1796 0.282 6.75% 15.15%* At the time of last sample assuming a generation length of 2.6 days
Bottleneck (at seroconversion)
Patient
Best-fitting demographic
modelEstimated rate
(per site per year)
Effective population
size*
Rate heterogeneity
(alpha parameter)
Mea
sura
bly
evo
lvin
g p
op
ula
tio
ns

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns
Intra- and inter- patient rate estimates (C2V3 envelope)
0.00E+00
5.00E-03
1.00E-02
1.50E-02
2.00E-02
2.50E-02
3.00E-02
0 50 100 150 200 250
Sampling interval (months)
rate
(p
er
sit
e p
er
year)
Intrapatient estimates
Interpatient estimates
BAC
p1 - p11
Po
pu
lati
on
gen
etic
s o
f H
IV

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns Summary: HIV intra-patient evolution
• HIV evolutionary rates appear to be faster intra-patient then across pandemic– Different selection pressure at transmission?– Transmitted viruses undergoing less rounds of
replication?– Latent viruses?– Reversion of escape mutants?
• Effective population size is changing over time (bottleneck in envelope at least)
Po
pu
lati
on
gen
etic
s o
f H
IV

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns But how good is our best model?
• We can use standard statistical model-choice criteria to choose between different models of substitution and demography, but are any of the models we consider any good at all?
• One way to look at this is ask the following question:– Does our real data look anything like what we would
expect data from our model to look like?
• So what aspect of the data should we look at?
• And what should we expect?
Go
od
nes
s-o
f-fi
t te
sts

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns
We could look at branch length distributions…
E[Ln ]2Ne
E[Jn ]2Ne
1
kk1
n 1
E[troot ]2Ne 11
n
troot
Ln
Jn Ln
Go
od
nes
s-o
f-fi
t te
sts

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns
Tree imbalance measures might also be interesting…
4 cherries 3 cherries 2 cherries
Ic 0
Ic 0.24
Ic 0.81
N 3
N 3.125
N 4.125
Go
od
nes
s-o
f-fi
t te
sts

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns Posterior predictive simulation
• A method of testing the goodness-of-fit of a Bayesian model.
1. Run a Bayesian MCMC analysis on the data2. Calculate the value of your favourite summary statistic, T(.)
from the data, D3. For each state in the chain
1. Simulate a synthetic dataset, Di, using the parameter values of state i.
2. Calculate T(Di) from the simulated data set.
4. Compare the T(D) value with predictive distribution of T(Di)
Po
ster
ior
pre
dic
tive
sim
ula
tio
n

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns So we need some summary statistics
• Summary statistics that can be measured directly from sequence alignment:– Mean pairwise distance
()– Tajima’s D– Fu & Li’s D– Number of segregating
sites (S)– …
• Summary statistics that can be measured directly from an genealogy:– Genealogical mean
pairwise distance ()– Genealogical Tajima’s D– Genealogical Fu & Li’s D– Tree-imbalance statistics– Age of the root– Length of the treeP
ost
erio
r p
red
icti
ve s
imu
lati
on

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns Posterior predictive simulation (2)
• Testing the goodness-of-fit of the neutral coalescent model under variable demographic functions.
1. Run a Bayesian MCMC analysis on the data2. For each state in the chain
1. Simulate a coalescent genealogy (GiS) using the population parameter
values of state i.
2. Calculate T(GiS) from the ith simulated genealogy
3. Calculate T(GiP) from the ith posterior genealogy
3. Calculate the predictive probability by comparing the posterior distribution of T(.) with predictive distribution of T(.):
Po
ster
ior
pre
dic
tive
sim
ula
tio
n

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns Human influenza A (HA gene) trees
Posterior genealogy Predictive simulations
State 5m
State 10m
Ne 9.12
t2 11.03 years
Ne 5.00
t2 15.29 years
Go
od
nes
s-o
f-fi
t te
sts

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns
Human influenza A trees: Genealogical Fu & Li’s D statistic
Go
od
nes
s-o
f-fi
t te
sts
Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns
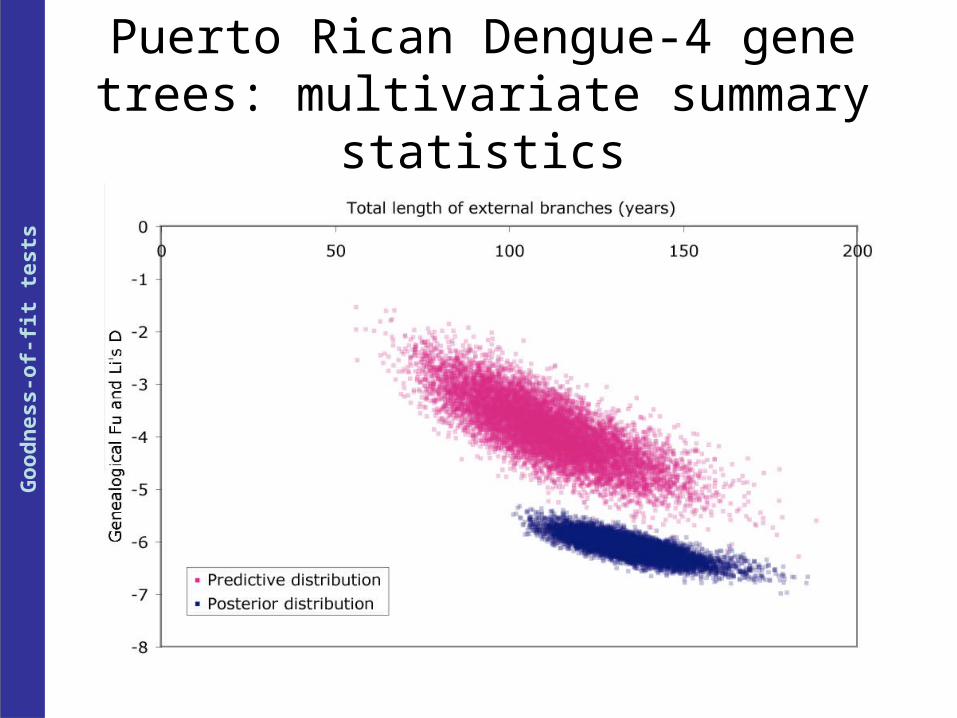
Puerto Rican Dengue-4 gene trees: multivariate summary statistics
Go
od
nes
s-o
f-fi
t te
sts

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns Results of test of neutrality
Table 2. The predictive probabilities (
PT) for summary statistics on each of the example
data sets are shown. Significant departures from neutrality are marked (*) and marginallysignificant departures (x < 0.05 or x > 0.95) are marked with (†). Significant departureson the best fitting model for each data set are in bold.
Predictive probabilitiesDataset Demographic
modelT troot DFL IC Cn B1
Brown bear Constant 0.739 0.815 0.863 0.693 0.163 0.103
(d-loop) Exponential growth 0.615 0.623 0.800 0.679 0.163 0.111
RSVA Constant 0.956† 0.964† 0.946 0.163 0.152 0.134
(g gene) Exponential growth 0.693 0.656 0.884 0.206 0.149 0.134
Dengue-4 Constant 0.9574† 0.9958* 0.9997* 0.562 0.608 0.427
(E gene) Exponential growth 0.745 0.809 0.9792* 0.559 0.653 0.505
Human influenza A Constant 0.9510† 0.900 0.9999* 0.0462† 0.605 0.610
(HA) Exponential growth 0.910 0.620 0.9995* 0.0866 0.575 0.677
Go
od
nes
s-o
f-fi
t te
sts

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns
Results for 28 HIV-1 infected individuals
Go
od
nes
s-o
f-fi
t te
sts
Fu & Li's D
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
0.0 0.2 0.4 0.6 0.8 1.0
proportion of data sets
P*
Sim Constant sizeSim Exponential GrowthTargetConstant sizeExponential Growth

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns Is the population size constant?
Patient 2
Po
pu
lati
on
gen
etic
s o
f H
IV
Pop size
10
100
1000
0 20 40 60 80 100 120
months (post seroconversion)
Ne /
30
mean
lower
upper

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns
Virus population dynamics
Measles virus
Human influenza virus
Ph
ylo
dyn
amic
s

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns
Dengue-4: Modeling complex demography
Hospital case data courtesy of Shannon Bennett
N(t) = N0exp(-rt): -10566.421N(t) = scaled translated case data: -10478.572
Ph
ylo
dyn
amic
s

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns Population size changes
Po
pu
lati
on
siz
e ch
ang
es

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns The generalized skyline plot
• Visual framework for exploring the demographic history of sampled DNA sequences
• Input: a single estimated ancestral genealogy (a tree)• Output: nonparametric plot of the population size through time
– Groups adjacent coalescent intervals
– Converts information within these intervals to estimates of population size
Po
pu
lati
on
siz
e ch
ang
es
ˆ N k k(k 1)
2tk
ˆ N k,l k(k l)
2lti
ik l1
k
Estimate of population size from single coalescent interval
Estimate of population size from l adjacent coalescent intervals.

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns Examples
I: Constant population size
N(t)=N(0)
Gen
eral
ized
Sky
line
Plo
t

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns Skyline Plot
I: Constant population size
N(t)=N(0)
II: Exponential growth
N(t)=N(0)e-rt
Gen
eral
ized
Sky
line
Plo
t

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns Skyline Plot
III: HIV-1 group M(tree estimated in Yusim et al (2001) Phil. Trans. Roy. Soc. Lond. B 356: 855-866)
– Black curve is a parametric estimate obtained from the same data under the “expansion model”
– Results follow accepted demographic pattern for the HIV pandemic
Gen
eral
ized
Sky
line
Plo
t

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns The Bayesian skyline plot
Estimate a demographic function that has a certain fixed number of steps (in this example 15) and then integrate over all possible positions of the break points.
Explains the Dengue data quite well (test of neutrality do not reject the data if we use the Bayesian skyline plot to describe the demographic history.
Po
pu
lati
on
siz
e ch
ang
es

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns
Prior/Model: population is auto-correlated through time
In addition to this model we also introduce a simple smoothing on
which representsour belief that effective population size is auto-correlated through time. The priordistribution we assume in all subsequent simulations and analyses is that, going back intime, each new population size is drawn from an exponential distribution with a meanequal to the previous population size:
j ~ Exp( j 1), 2 j m . (5)
In addition we introduce a scale-invariant prior (Jeffreys 1946) on the first element
f1(1)
1
1
to signify that our prior belief is invariant to changes in time scale. This
results in a following simple prior distribution on
:
f ()1
1
j 1 exp j / j 1 j2
m
. (6)
Po
pu
lati
on
siz
e ch
ang
es

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns Validating the Bayesian skyline plot (1)
Simulated data: Constant population Simulated data: Exponential growth
Po
pu
lati
on
siz
e ch
ang
es

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns Validating the Bayesian skyline plot (2)
Bayesian skyline (49 or 12 epochs)
0.00001
0.0001
0.001
0.01
0.1
1
10
100
0 0.002 0.004 0.006 0.008
Time (mutations)
Th
eta
Median (49)lower (49)upper (49)truthMedian (12)lower (12)upper (12)P
op
ula
tio
n s
ize
chan
ges

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns
Comparing Bayesian skyline plot of Dengue-4 with incidence data
1
10
100
1000
10000
100000
0 50 100 150 200Months (before November 1998)
Eff
ect
ive n
um
ber
of
infe
ctio
ns
Median
Upper
Lower
Smoothed translated incidence
Po
pu
lati
on
siz
e ch
ang
es

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns Example of Bayesian skyline plot
(1920-1980) Anti-schistosomal needle-based treatment
Effective population size jumped from 300 to 100 to 10,000
Po
pu
lati
on
siz
e ch
ang
es

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns Comparison to parametric model
Po
pu
lati
on
siz
e ch
ang
es

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns
http://evolve.zoo.ox.ac.uk/BEAST

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns Coalescent with population structure
Str
uct
ure
d p
op
ula
tio
ns

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns
Str
uct
ure
d p
op
ula
tio
ns
Population subdivision - two demes

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns
Str
uct
ure
d p
op
ula
tio
ns
Population subdivision - two demes

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns Stepping stone model of subdivision
Str
uct
ure
d p
op
ula
tio
ns
Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns
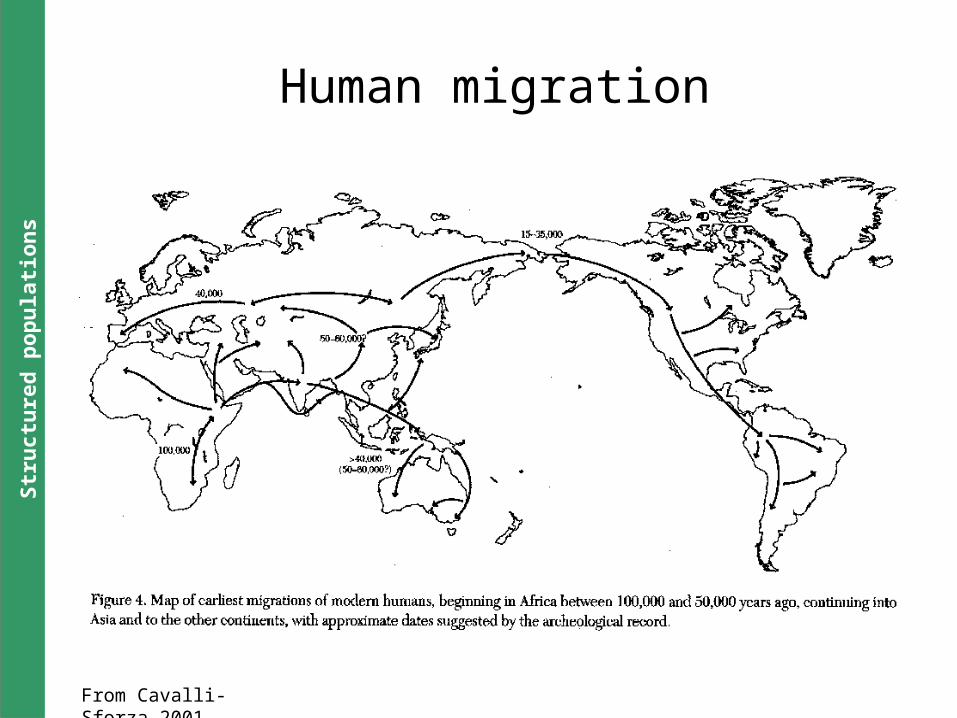
From Cavalli-Sforza,2001
Human migration
Str
uct
ure
d p
op
ula
tio
ns
Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns
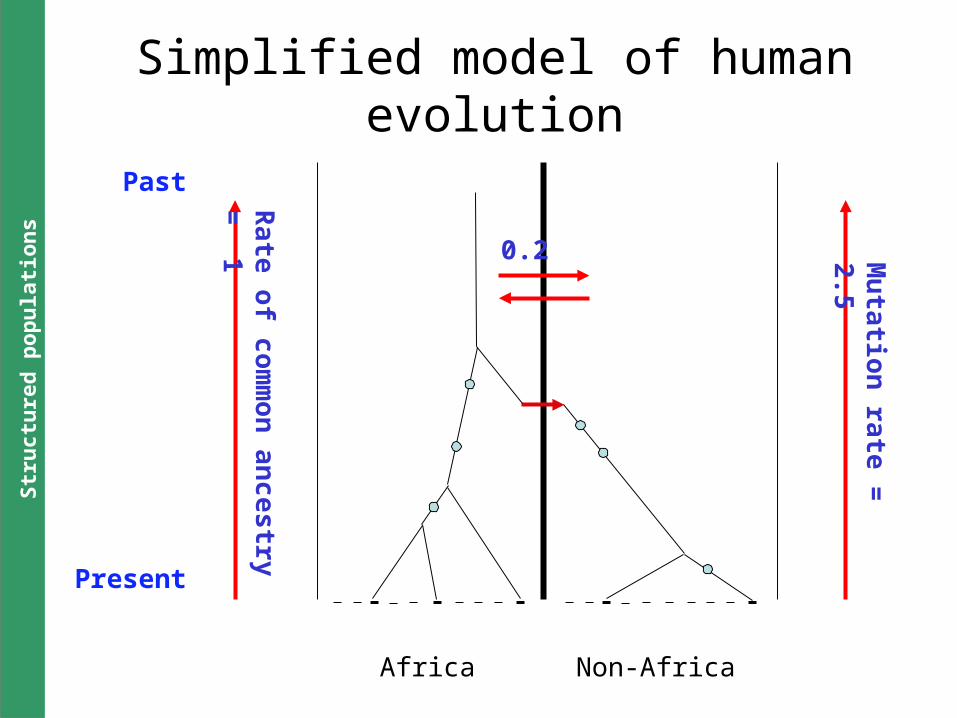
Africa Non-Africa
0.2 Mu
tatio
n ra
te =
2.5
Ra
te o
f co
mm
on
an
ce
stry =
1
Past
Present
Simplified model of human evolution
Str
uct
ure
d p
op
ula
tio
ns

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns Why Bayesian?
• Probabilistic model-based inference– Can make simple statements about the probability of alternative
hypotheses given the data
• Markov chain Monte Carlo– Convenient computational technique– Allows for complex models: “if you can simulate you can sample”
• Incorporates prior probabilities – P(|D) P(D| )P()– Convenient means of assessing alternative sets of assumptions– Allows incorporation of independent sources of information
• Easy to include sources of uncertainty– Don’t need to assume perfect knowledge of tree (for example)– Can treat the tree and a nuisance parameter and focus on parameters
of interest (strength of selection, mutation rate, growth rate, etc)

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns Conclusions & cautionary remarks
• Bayesian MCMC has advantages– a useful tool for exploring prior hypotheses– Good for assessing levels of uncertainty– Complex models can be investigated on practical datasets
• Bayesian MCMC has disadvantages– Diagnostics are difficult, and it is essentially impossible to
guarantee correctness– Model comparison can be difficult– Requires large programs that are difficult to optimize and debug.

Th
e C
oal
esce
nt
and
Mea
sura
bly
Evo
lvin
g P
op
ula
tio
ns Conclusions & cautionary remarks (2)
• Population genetics has advantages– provides a framework for objective analysis of genetic data– Allows interpretation of genetic data in terms of biological
properties of virus– Can be extended to include selection, recombination et cetera
• Population genetics has disadvantages– Models are still too simple– Assumptions are too strong– Extending to complex models that include changing selection
pressures and recombination are possible in MCMC but still very difficult!