the analytical possibilities of vapour hydride generation ... · nr 5/2010 • tom 64 • 1 the...
TRANSCRIPT

nr 5/2010 • tom 64 • 1
scie
nce
• te
chni
queThe analytical possibilities of vapour hydride
generation coupled with optical emission spectrometry (HG-ICP-OES) Part.2. Arsenic determination in aluminosilicate materials
Magdalena PIĄTKOWSKA - Analytical Laboratory at the Institute for Inorganic Chemistry, Gliwice
Please cited as: CHEMIK 2010, 64, 5, xx-xx
IntroductionIn modern world many important opinions and decisions depend
on the results of chemical analysis. This applies to determination of both macro components, as well as micro components present in trace amounts. These concentrations must remain under strict control. However, the problem of reliability of the results of inorganic trace analysis, despite the use of sophisticated instruments and application of well-defined procedures, still exists [1, 2].
Modern analytical methods enable direct multi element analysis of samples, when concentration of the determined component is above the quantitation limit of the method. This condition is not met in many analytical tasks. In such cases the sample is pre-treated using various methods of concentration or separation of the determined component from the principal constituents of the matrix. In the end the concentration of the analytes in the sample matrix is increased to above the quantitation limit of the analytical instrument, the matrix composition is made less complex and interfering components are removed from the sample, which allows the sensitivity of the method to be increased.
For these reasons the CVG (Chemical Vapour Generation) tech-nique in combination with two variations of atomic absorption spec-trometry: cold vapour generation (CV-AAS) for Hg determination, and volatile hydrides generation (HG-AAS), as well as in combination with optical emission spectrometry (ICP-OES), atomic fluorescence spec-trometry (AFS) and mass spectrometry (MS) for elements that form volatile hydrides (As, Bi, Ge, In, Pb, Sb, Sn, Se, Te and Tl) has become the basic analytical technique for determining the elements mentioned [3÷5]. By enabling the isolation of analytes from the environmental or organic matrix, which often strongly interferes or distorts analytical results, it provides low detectability limits and allows for direct and selective determination of analytes in the samples tested [6]. Hydride generation is a technique of introducing analytes in gaseous form into an excitation source, applied in spectrometric methods to determine chemical elements, the ions of which react with a reducing agent in an acidic medium to form volatile gaseous hydrides stable at ambient temperature [18]. In 1978 the volatile hydride generation technique was combined with inductively coupled plasma (HG-ICP-OES). Since then a rapid development of this technique applied in trace analysis is observed, and the great interest taken in it is reflected in the large number of review articles [6÷17].
Chemical analysis of materials with an aluminosilicate matrix, in-cluding various complex minerals, is still not a simple analytical proce-dure. The main component, silica, constitutes 50 to 70% of the com-position, whereas the content of aluminium oxide is 20 to 40%. Oxides of other elements, such as: calcium, magnesium, titanium, iron, potas-sium, sodium, manganese, carbon and phosphorus are present in small amounts, while still other occur in trace amounts (less than 0.01%): oxides of zirconium, vanadium, chromium, barium, strontium, sulphur, nickel, copper, etc. The standards in force impose limits on the content of main components at 10-2–10-1%. Determination of trace amounts
of selected elements by means of conventional methods (colorimetry, flame photometry) is laborious and not very precise.
Arsenic is one of the elements the significance of which for the human organism is rising. Its compounds constitute trace chemical components of ecosystems, the concentrations of which rarely attain toxic levels. However, the closeness of the range of dozes absorbed by organisms and of the toxic dose requires, when one considers the widespread occurrence thereof, strict control [18, 19].
Direct spectrometric determination of trace amounts of arsenic using optical emission spectrometry in solutions of inorganic ma-trixes studied, is characterised by relatively low sensitivity due to low efficiency of ionisation in plasma. The work presented included at-tempts to develop and optimise analytical procedures of determining trace amounts ofAs in selected aluminosilicate materials using optical emission spectrometry coupled with hydride generation (HG-ICP-OES). The quality of the method developed was assessed through validation by analysing the features of the method, such as: accu-racy, trueness, precision, repeatability, reproducibility, determinable concentration range and linearity, detection and quantitation limit, selectivity, specificity, sensitivity, ruggedness and measurement un-certainty.
Research methodologyCharacteristics of the substances investigated
The materials on which experiments were conducted comprised samples of layered aluminosilicates: − kaolin (Egypt)- china clay, containing mainly kaolinite (basic alu-
minium silicate with chemical formula of Al4[Si4O10](OH)8, and also quartz and mica
− halloysite - hydrated aluminium silicate with general chemical for-mula of Al4(OH) 8/Si4O10·10 H2O
− bentonite - (Jelsowy Potok) - mineral of the smectite group com-posed of one or mostly one component: montmorillonite - hy-drated aluminium, magnesium and sodium silicate.
Additionally, in order to validate the analytical procedure, two refer-ence materials were used, both with known and certified content of the analytes determined in aluminosilicate matrix (Tab. 1):
− loessial soil RTH 912, (LGC Promochem - Warsaw)− kaolin KK 194, (MBH Analytical Limited - Kraków).
Measuring instruments and auxiliary materials used in the study
The equipment used for determinations and validation of the analytical procedure comprised an inductively coupled plasma optical emission spectrometer (ICP-OES) iCAP 6300 MFC Duo (Thermo Scien-tific), with Echelle type optical system, CID solid-state matrix detector, fitted with a dedicated hydride generator - Enhanced Vapour System (with separate separator and regulator) (Fig. 1). The main component of the generator was a compact acrylic chamber, of low dead volume, with a reaction vessel arranged inside.
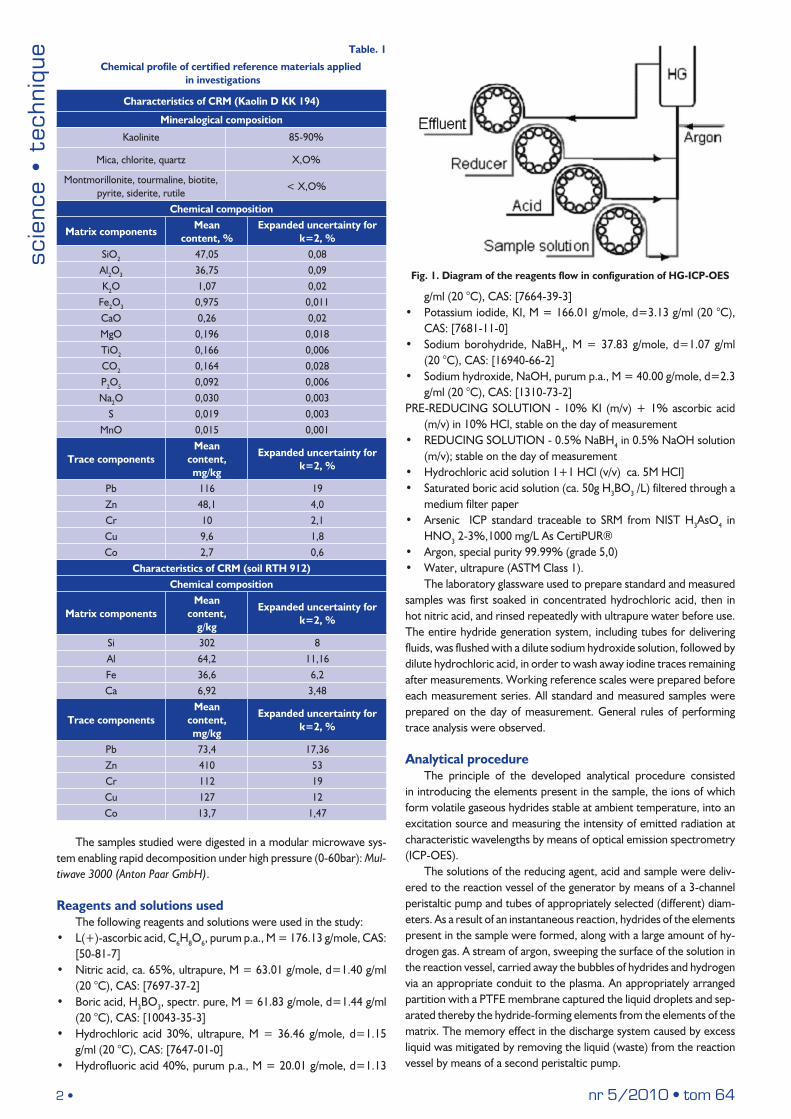
2 • nr 5/2010 • tom 64
scie
nce
• te
chni
que
The samples studied were digested in a modular microwave sys-tem enabling rapid decomposition under high pressure (0-60bar): Mul-tiwave 3000 (Anton Paar GmbH).
Reagents and solutions usedThe following reagents and solutions were used in the study:
• L(+)-ascorbic acid, C6H8O6, purum p.a., M = 176.13 g/mole, CAS: [50-81-7]
• Nitric acid, ca. 65%, ultrapure, M = 63.01 g/mole, d=1.40 g/ml (20 °C), CAS: [7697-37-2]
• Boric acid, H3BO3, spectr. pure, M = 61.83 g/mole, d=1.44 g/ml (20 °C), CAS: [10043-35-3]
• Hydrochloric acid 30%, ultrapure, M = 36.46 g/mole, d=1.15 g/ml (20 °C), CAS: [7647-01-0]
• Hydrofluoric acid 40%, purum p.a., M = 20.01 g/mole, d=1.13
g/ml (20 °C), CAS: [7664-39-3]• Potassium iodide, KI, M = 166.01 g/mole, d=3.13 g/ml (20 °C),
CAS: [7681-11-0] • Sodium borohydride, NaBH4, M = 37.83 g/mole, d=1.07 g/ml
(20 °C), CAS: [16940-66-2]• Sodium hydroxide, NaOH, purum p.a., M = 40.00 g/mole, d=2.3
g/ml (20 °C), CAS: [1310-73-2]PRE-REDUCING SOLUTION - 10% KI (m/v) + 1% ascorbic acid
(m/v) in 10% HCl, stable on the day of measurement • REDUCING SOLUTION - 0.5% NaBH4 in 0.5% NaOH solution
(m/v); stable on the day of measurement• Hydrochloric acid solution 1+1 HCl (v/v) ca. 5M HCl]• Saturated boric acid solution (ca. 50g H3BO3 /L) filtered through a
medium filter paper • Arsenic ICP standard traceable to SRM from NIST H3AsO4 in
HNO3 2-3%,1000 mg/L As CertiPUR®• Argon, special purity 99.99% (grade 5,0)• Water, ultrapure (ASTM Class 1).
The laboratory glassware used to prepare standard and measured samples was first soaked in concentrated hydrochloric acid, then in hot nitric acid, and rinsed repeatedly with ultrapure water before use. The entire hydride generation system, including tubes for delivering fluids, was flushed with a dilute sodium hydroxide solution, followed by dilute hydrochloric acid, in order to wash away iodine traces remaining after measurements. Working reference scales were prepared before each measurement series. All standard and measured samples were prepared on the day of measurement. General rules of performing trace analysis were observed.
Analytical procedureThe principle of the developed analytical procedure consisted
in introducing the elements present in the sample, the ions of which form volatile gaseous hydrides stable at ambient temperature, into an excitation source and measuring the intensity of emitted radiation at characteristic wavelengths by means of optical emission spectrometry (ICP-OES).
The solutions of the reducing agent, acid and sample were deliv-ered to the reaction vessel of the generator by means of a 3-channel peristaltic pump and tubes of appropriately selected (different) diam-eters. As a result of an instantaneous reaction, hydrides of the elements present in the sample were formed, along with a large amount of hy-drogen gas. A stream of argon, sweeping the surface of the solution in the reaction vessel, carried away the bubbles of hydrides and hydrogen via an appropriate conduit to the plasma. An appropriately arranged partition with a PTFE membrane captured the liquid droplets and sep-arated thereby the hydride-forming elements from the elements of the matrix. The memory effect in the discharge system caused by excess liquid was mitigated by removing the liquid (waste) from the reaction vessel by means of a second peristaltic pump.
Table. 1
Chemical profile of certified reference materials applied in investigations
Characteristics of CRM (Kaolin D KK 194)
Mineralogical composition
Kaolinite 85-90%
Mica, chlorite, quartz X,O%
Montmorillonite, tourmaline, biotite, pyrite, siderite, rutile
< X,O%
Chemical composition
Matrix componentsMean
content, %Expanded uncertainty for
k=2, %SiO2 47,05 0,08Al2O3 36,75 0,09K2O 1,07 0,02
Fe2O3 0,975 0,011CaO 0,26 0,02MgO 0,196 0,018TiO2 0,166 0,006CO2 0,164 0,028P2O5 0,092 0,006Na2O 0,030 0,003
S 0,019 0,003MnO 0,015 0,001
Trace componentsMean
content, mg/kg
Expanded uncertainty for k=2, %
Pb 116 19Zn 48,1 4,0Cr 10 2,1Cu 9,6 1,8Co 2,7 0,6
Characteristics of CRM (soil RTH 912)Chemical composition
Matrix componentsMean
content, g/kg
Expanded uncertainty for k=2, %
Si 302 8Al 64,2 11,16Fe 36,6 6,2Ca 6,92 3,48
Trace componentsMean
content, mg/kg
Expanded uncertainty for k=2, %
Pb 73,4 17,36Zn 410 53Cr 112 19Cu 127 12Co 13,7 1,47
Fig. 1. Diagram of the reagents flow in configuration of HG-ICP-OES

nr 5/2010 • tom 64 • �
scie
nce
• te
chni
queAluminosilicate material samples were solubilised using microwave
energy. To this end, 0.25-0.3 g samples were weighed on an analytical balance to the nearest 0.0001 g and digested by microwave irradiation in a closed pressurised system, in PTFE vessels, containing concen-trated mineral acids ( HNO3+HCl+HF 2:5:2 v/v; P1=1400W). The F– fluoride ions present in the solution were complexed in a sepa-rate step of digesting with 12 ml of saturated boric acid H3BO3 (6mL H3BO3/1mL HF; P2=1000W) in the following reaction:
H3BO3 + 3HF = BF3 + 3H2OBF3 + HF = HBF4
Upon completion of the program, the vessels were cooled and the products were transferred quantitatively to 100 ml measuring flasks, without diluting to the mark. In addition to measured sample solu-tions, a blind sample was prepared in such manner, as to undergo the same operations and to contain the same reagents in concentrations comparable to those in the measured samples.
Conditions of arsenic pre-reductionAs arsenic hydrides generation depends on the acidity of the reac-
tion medium and on the valency of the element, tests were carried out in the first stage of the study in order to establish optimum con-ditions for the pre-reduction reaction to attain the highest possible hydride yield. A procedure was proposed, where arsenic in the sample was reduced outside of the reaction system. To this end, the digested product in the 100mL measuring samples was diluted using 50 ml of solution of 10% KI (m/v) + 1% ascorbic acid (m/v) in 10% HCl (v/v) and made up to the mark with water. Sample solutions were left for ca. 3 h. at ambient temperature, in a dark place, for arsenic to be com-pletely reduced to lower oxidation states, and afterwards they were transferred to the generator system together with the reducing agent (0.5% NaBH4 stabilised in 0.5% NaOH solution, m/v) and 5M HCl solution. The solutions for the arsenic reference scale were prepared in similar manner.
Optimisation of ICP measurement conditionsDuring the optimisation tests the effect of the power applied on
the arsenic detectability and measurement sensitivity in the HG-ICP-OES technique was established. Model standard solutions were used in the tests: 0.00, 0.01, 0.05, 0.10 and 0.20 µ g/ml As. It was found that increasing the power applied (in the 1000-1300 W range) resulted in increased sensitivity (expressed as the slope b of the standardisation curves) and in markedly improved detectability (increased limit of de-tection of arsenic, LOD=3s) (Fig. 2,3).
Optimisation of reagent concentrations in the HG-ICP-OES system
In the next stage of optimisation, the effect of sodium borohydride (the reducing agent) concentration within NaBH4 content range of 0.1 to 1.0%, on the As measurement sensitivity and detectability was stud-ied using model standard solutions containing 0.0, 0.01, 0.05, 0.10 and 0.20 µg /ml As, respectively. It was found that increasing the concentra-tion of the reducing agent (within the adopted range of concentrations) had a beneficial effect on As measurement sensitivity and detectability (LOD=3s) (Fig. 4,5).
Optimisation of reagent flow rate in the HG-ICP-OES system
The next step included the determination of the effect of the flow rate of the individual reagents (solution of sample, reducing agent and acid) in the hydride generator system HG, within the range of 25 to 45 rpm, on the measurement sensitivity and analyte detectability. Model standard solutions were used in the tests: 0.00, 0.01, 0.05, 0.10 and 0.20 µ g/ml As. It was found that the increase in the flow rate of all three reagents, within the defined pump speed range, resulted in a marked increase in arsenic determination sensitivity (in the slopes b of
Fig. 2. The effect of applied power on the sensitivity of arsenic measurement by HG- ICP-OES
Fig. 3. The effect of applied power on arsenic detectability by HG- ICP-OES
Fig. 4. The effect of reducing agent concentration on the sensitivity of arsenic measurement by HG-ICP-OES
Fig. 5. The effect of reducing agent concentration on arsenic detectability by HG- ICP-OES

4 • nr 5/2010 • tom 64
scie
nce
• te
chni
que
the regression lines); however, when reagent feed rate increased, de-tectability (LOD=3s) improved to a certain fixed value only (35 rpm), after which it continually deteriorated (Fig. 6,7).
The adopted optimum flow rate of solutions (of the reducing agent, acid and sample) fed to the generator system HG by pump no. 1, under plasma generation conditions set beforehand (P=1200 W) and sodium borohydride concentration of 0.5%, was 35 rpm. Having set the speed of pump no. 1, the rate of withdrawing unreacted liquid (waste) from the reaction system by means of peristaltic pump no. 2 was also optimised: a double discharge system operating at a rate of 50 rpm was found to be sufficient to prevent the excess unreacted liquid and by-products from being transferred together with gaseous hydrides to the excitation source and from interfering with the spec-trometric measurement. Optimum operating parameters of HG-ICP-OES spectrometer are given in Table 2.
Interferent effectsThe next stage of the study included assessment of the resistance
of the developed analytical procedure to potential interacting interfer-ents. To this end, five fortified model mixtures were prepared, these mixtures having increasing content of interferents (Fe, Mn, Cr, Zn, Co, Cd, Cu, Ni, Pb) which, according to the literature, had the strongest impact on spectrometric measurements. These model mixtures had fixed and known content of arsenic (0.01 mg/L). Thereafter As recov-ery in these mixtures was measured by HG-ICP-OES using a defined procedure. It was found, that within the adopted range of interferent concentrations, arsenic recovery was within the permissible range of 95 to 105%, which indicated the absence of any potential spectral coincidences and interference from matrix components (Tab. 3.).
Results and discussion The tests and studies carried out have confirmed the feasibility of
spectrometric determination of arsenic (As) in selected aluminosili-cate matrixes (soil, bentonite, kaolin) with the use of volatile hydride
generation technique (HG-ICP-OES) under relatively simple chemical conditions and by employing relatively simple equipment (Tab. 4, 5, 6). As the kinetics of hydride generation are strongly dependent on the form of element occurrence in the sample and as hydride gen-eration proceeds in a different way for the different oxidation states of the analytes, determination of total arsenic content required the pre-treatment of the analytical sample (pre-reduction to As(III)) and
Fig. 6. The effect of flow rate of the reagents on the sensitivity of arse-nic measurement by HG- ICP-OES
Fig. 7. The effect of flow rate of the reagents on arsenic detectability by HG- ICP-OES
Table 2
Optimum operating parameters of ICP-OES spectrometer (iCAP 6300 Duo, Thermo) coupled with vapour hydride generation
Operating parameter Parameter value
Excitation frequency 27.12 MHz
Excitation power 1200 W
Observation Axial
Solid state detector CID (Charge Injection Device)
Burner type Burner HF Duo, Thermo
Central tube Ceramic 2.0 mm
Spray chamber type none
Nebuliser type none
Hydride generator yes
-nebuliser argon pressure
-auxiliary gas flow
-plasma gas flow
-coolant gas flow
-
0.5 L/min
12.0 L/min
14.0 L/minPumping rate (rpm):
-sample (pump no. 1)
-acid (pump no. 1)
-reducer (pump no. 1)
- effluent (pump no. 2)
1x35 rpm
1x35 rpm
1x35 rpm
2x50 rpmSample washing time 60 s
Stabilisation time 5 s
Analysis rate 40 rpm
Emission line integration time
-Low (166-230 nm)
-High (230-847 nm)
15 s
5 sNo. of repetitions/measurement 3 - 6
Background correction yes
Table 3
The effect of matrix components on As recovery in fortified model mixtures
Analyte: interferent ratio
(A/I)
Arsenic recovery, %As
189.0 nm
As
193.7 nm
As
197.2 nm
As
228.8 nm1:0 103 102 103 1031:1 104 104 104 103
1:10 98 98 99 981:100 103 103 104 103
1:1000 105 105 105 105
Table 4
Investigation of CRM LGC Promochem Swiss Loess Soil RTH 912
As_189.0 nm As_193.7 nm As_197.2 nm As_228.8 nm
Nominal concentration, mg/kg
21.6 ± 3.5
Measured mean concentration, mg/kg (n=9)
22.56 22.30 22.42 22.43
Mean recovery, %
104.4 103.3 103.8 103.9

nr 5/2010 • tom 64 • 5
scie
nce
• te
chni
que
optimisation of a number of parameters of the hydride generation process and of measurement conditions to ensure maximum yield of the hydrides.
It was found that the pre-reduction of As(V) to As(III), carried out on-line with the use of potassium iodide added to the solution of the reducing agent (sodium borohydride), does not ensure satisfactory yield of hydrides. This process, however, can be successfully accom-plished by using a reducing mixture (10% KI (m/v)+1% C6H8O6 (m/v) in 10% HCl) added to the measured sample solution in a proportion of 1:1, ca. 3 hours before taking the measurement. It was observed that transforming elements to their lower oxidation states requires time, and that potassium iodide shows reducing abilities only in a solution of strong mineral acid (ca. 5M HCl), whereas ascorbic acid protects iodine against oxidation by oxygen in air.
The experimentally determined reagent concentrations and flow rates that ensure appropriate arsenic measurement sensitivity and de-tectability in plasma were as follows: flow rate of 35 rpm for the solu-tions of sample, acid (5M HCl) and reducing agent (0.5% NaBH4) and 100 rpm for waste liquid, at plasma power of 1200 W.
The proposed procedure of decomposing the matrix of an alumi-nosilicate sample was simple and comprised a relatively short micro-wave digestion program in a closed pressurised system, with the use of concentrated mineral acids and complexing of F– fluoride ions in a separate digestion step involving the use of saturated boric acid H3BO3. It was found that the addition of large amounts of boric acid (in the case of high SiO2 content) caused deleterious precipitation of: potassium fluoroborate (solubility: 0.44g/100g H2O at 20°C) and hydroiodic acid during pre-reduction of digested sample with potassium iodide:
KI + HBF4 = KBF4 + HIThis effect was particularly disadvantageous in the case of kaolin, in
which the silica content is relatively high, resulting in higher uncertainty of arsenic determination.
In a series of experiments with enriched model solutions and during analysis of certified reference materials (CRM) it was confirmed that interference caused by chemical elements that form volatile species
under the conditions of hydride generation (transition group metals: Fe, Mn, Cr, Zn, Co, Cd, Cu, Ni, Pb) do not pose a significant analytical problem during As determination, even if present in concentrations up to 1000 times higher than that of arsenic. Moreover, determinations can be carried out using standardisation curves obtained by standard series method of calibration, the standards being prepared in the same manner as solutions of the measured samples (including the pre-reduc-tion step).
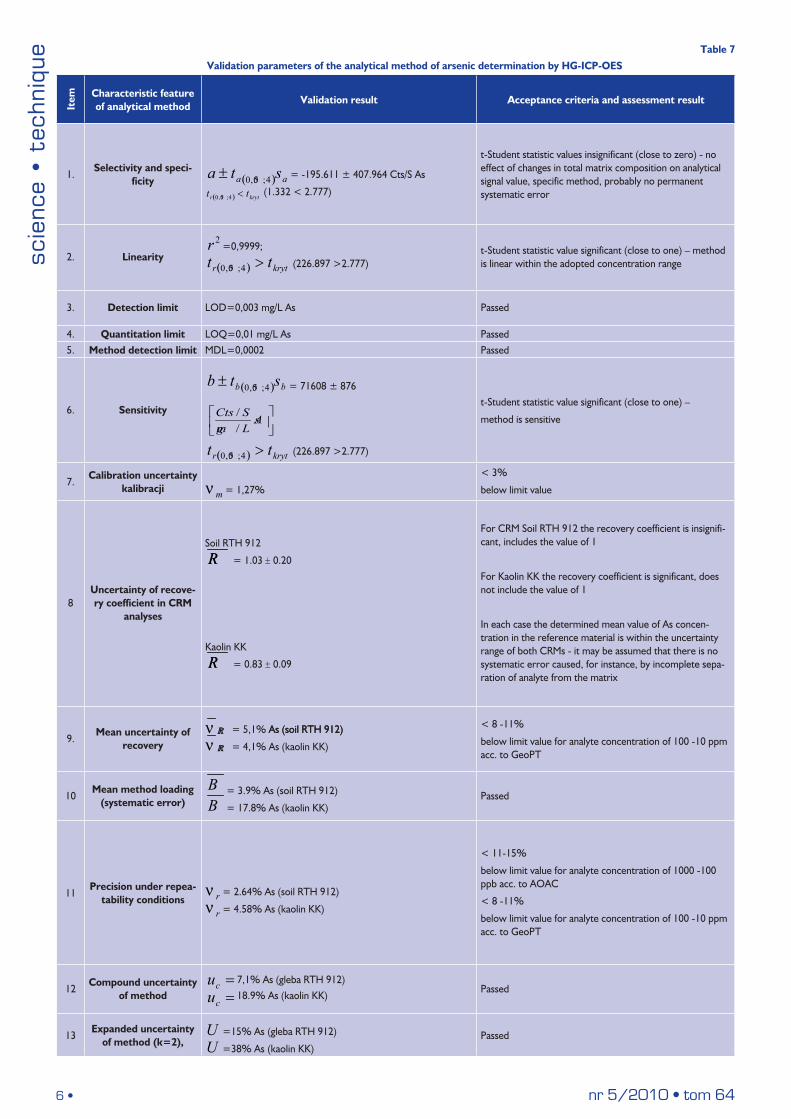
The feasibility of the method of determining trace amounts of As by HG-ICP-OES was finally verified and assessed through validation by determining the features of the method, such as: detection and quantitation limit, determinable concentration range and linearity, precision, accuracy and uncertainty of measurement using certified reference materials, enriched samples and retention real samples of appropriate matrix (Tab. 7). Confidence level of P=0.95 was adopted as the criterion for the verification of statistical evaluation, whereas for the verification of statistical hypotheses the results with gross error were discarded (by checking raw measurement data and applying the Dixon’s Q test for rejecting outliers).
It was found that within the adopted working concentration ranges (0.01–1.00 mg/l) the calibration curves for the various arsenic emission lines showed very good linearity: the linear correlation coefficients r were in every case larger than 0.9995, and the values of their t statistics were significantly different from zero. The calculated values of statisti-cal parameters of the regression lines, that is: the intercepts of the line
aa ∆± and the slopes of the line bb ∆± , have confirmed that the method developed is sensitive and specific (absence of a systematic er-ror independent of concentration and absence of a proportional error indicated a high probability of the lack of effect of changes in the overall matrix composition on analysis results). The calibration uncertainty, expressed by the variation coefficient, was in every case lower than the limit value of 3%. The analysis of residuals (differences between the measured value of intensity of the standard solution and the value calculated from the calibration curve for this solution) indicated the uniform distribution thereof around the zero value and the absence of any trend related to concentration, which supported the linearity of the curves determined. The obtained values of standard solution con-centrations were within the permissible limits of error, which indicated the reliability thereof (Fig. 8).
Four emission lines (189.0 nm, 193.7 nm, 197.2 nm and 228.8 nm) were used for spectrometric determination of arsenic. These lines are relatively free from spectral and matrix interference (Fig.9). Un-fortunately, the variances and variation coefficients of the limits of the working range (of concentrations) differed in a statistically significant manner for all of these emission lines. This indicated that the precision of measurements in the adopted range of concentrations was variable (the correlation between the standard deviation and the concentration was significant). In view of measurements made in the vicinity of quan-titation limits of the method, the working concentration ranges were accepted. However, in some cases (e.g. for very sensitive emission lines of arsenic: 189.0 and 193.7 nm) a further lowering of the limit of the proposed range of concentrations might be considered.
The method developed also featured relatively low limits of de-tection and quantitation, which were determined on the basis of the so-called blind digestion samples subjected to analytical procedure in parallel to the measured samples (LOD = 0.01 mg/L As). The accuracy and trueness of the analytical method, verified by determining recov-ery, on one (average) analyte concentration level using two certified reference materials (soil, treated as aluminosilicate material containing organic substances, and kaolin) was correct and within limits (0,8–1,1) according to criteria for geochemical research proficiency defined by GeoPT (Geostandards and Geoanalytical Research). Apart from that, the average recovery value determined on the basis of repeated analysis of reference material, was within the interval of uncertainty
Table 5
Investigation of CRM MBH Analytical Ltd. Kaolin KK
As_189.0 nm As_193.7 nm As_197.2 nm As_228.8 nmNominal concentration, mg/kg
15.2 ± 3.4
Measured mean concentration, mg/kg (n=9)
12.62 12.50 12.70 12.64
Mean recovery, %
83.00 82.27 83.52 83.12
Table 6
Results of arsenic determinations in selected layered aluminosilicates by HG-ICP-OES (x ± ts, v %)
As_189.0 nm As_193.7 nm As_197.2 nm As_228.8 nm
Measured mean concentration, mg/kg (n=9)
Bentonite (Jelsowy Potok)
48.45 ±1.432
5.34
49.19±1.664
6.16
49.11±1.277
4.67
49.23±1.548
5.71
Kaolin (Egypt)
<MDL <MDL <MDL <MDL
Halloysite
<MDL <MDL <MDL <MDL
6 • nr 5/2010 • tom 64
scie
nce
• te
chni
que Table 7
Validation parameters of the analytical method of arsenic determination by HG-ICP-OESIt
em Characteristic feature of analytical method
Validation result Acceptance criteria and assessment result
1.Selectivity and speci-
ficity ( ) aa sta 4;05,0± = -195.611 ± 407.964 Cts/S As
( ) krytr tt <4;05,0 (1.332 < 2.777)
t-Student statistic values insignificant (close to zero) - no effect of changes in total matrix composition on analytical signal value, specific method, probably no permanent systematic error
2. Linearity
2r =0,9999;
( ) krytr tt >4;05,0 (226.897 >2.777)t-Student statistic value significant (close to one) – method is linear within the adopted concentration range
3. Detection limit LOD=0,003 mg/L As Passed
4. Quantitation limit LOQ=0,01 mg/L As Passed 5. Method detection limit MDL=0,0002 Passed
6. Sensitivity
( ) bb stb 4;05,0± = 71608 ± 876
AsLmgSCts
//
( ) krytr tt >4;05,0 (226.897 >2.777)
t-Student statistic value significant (close to one) –
method is sensitive
7.Calibration uncertainty
kalibracji mν = 1,27%
< 3%
below limit value
8Uncertainty of recove-ry coefficient in CRM
analyses
Soil RTH 912
RR = 1.03 ± 0.20
Kaolin KK
RR = 0.83 ± 0.09
For CRM Soil RTH 912 the recovery coefficient is insignifi-cant, includes the value of 1
For Kaolin KK the recovery coefficient is significant, does not include the value of 1
In each case the determined mean value of As concen-tration in the reference material is within the uncertainty range of both CRMs - it may be assumed that there is no systematic error caused, for instance, by incomplete sepa-ration of analyte from the matrix
9.Mean uncertainty of
recoveryRRν = 5,1% As (soil RTH 912)As (soil RTH 912)
RRν = 4,1% As (kaolin KK)
< 8 -11%
below limit value for analyte concentration of 100 -10 ppm acc. to GeoPT
10Mean method loading
(systematic error)
B = 3.9% As (soil RTH 912)
B = 17.8% As (kaolin KK)Passed
11Precision under repea-
tability conditions rν = 2.64% As (soil RTH 912)
rν = 4.58% As (kaolin KK)
< 11-15%
below limit value for analyte concentration of 1000 -100 ppb acc. to AOAC
< 8 -11%
below limit value for analyte concentration of 100 -10 ppm acc. to GeoPT
12Compound uncertainty
of method=cu 7,1% As (gleba RTH 912)
=cu 18.9% As (kaolin KK)Passed
13Expanded uncertainty
of method (k=2),U =15% As (gleba RTH 912)
U =38% As (kaolin KK)Passed

nr 5/2010 • tom 64 • �
scie
nce
• te
chni
que
specified by the CRM manufacturer and indicated that there was no constant systematic error in the method.
The repeatability of the method determined on the basis of the scatter of measurement results obtained by one operator on one real sample within a short period of time, confirmed that the precision was acceptable under repeatability conditions (11%, GeoPT criteria). However, repeatability deteriorated when the measured analyte con-centration values approached the quantitation limit of the method. Series of analyte determinations conducted in parallel did not differ, in terms of accuracy, in a statistically significant manner.
The expanded uncertainty of the method, expressed in ac-cordance with the law of propagation of uncertainty as the sum of standard uncertainties of: calibration, analyte recovery from sample matrix, method loading and precision under repeatability conditions, determined on the basis of CRM analyses, was equal to: 15% As (soil RTH 912) and 38% As (kaolin KK), respectively.
In conclusion, it should be stated that the application of ICP emis-sion spectrometer with horizontal arrangement of the plasma burner and with hydride generation had substantially improved arsenic deter-minability as compared to conventional methods used so far, and that the method developed has a number of advantages, most important being: good repeatability of analytical results, high sensitivity and se-lectivity, and low quantitation limit. It should, however, be borne in mind that irrespective of all of the advantages of the hydride genera-tion method, its application will always require optimising a number of parameters in order to attain maximum hydride generation efficiency. Such optimisation may include: finding optimum conditions associated
with the concentration of the acid and with the quantity and concen-tration of the reducing agent (valency of the elements determined has a significant effect on measurement sensitivity); providing efficient means of hydride transfer to the excitation source, with consideration given to the stability of the hydride; and finding measurement conditions that will ensure appropriate sensitivity and precision of determination (plasma discharge power, plasma gas flow rate, plasma burner position, selection of analytical lines, quantitation limit).
Bibliography1. Jakubowska M.: Metody Sprawdzania Wiarygodności Wyników Analizy Che-
micznej. AGH Kraków 2001.
2. Sargent, MacKay G.: Guidelines for Achieving Quality in Trace Analysis. ISSN:ISSN:
0 85404 402 7, RSC 1995.
3. PN-EN 14546:2005: Artykuły żywnościowe – Oznaczanie pierwiastków
śladowych – Oznaczanie całkowitej zawartości arsenu metodą atomowej
spektrometrii absorpcyjnej z generacją wodorków HGAAS) po mineralizacji
suchej.
4. PN-EN 14627:2005: Artykuły żywnościowe – Oznaczanie pierwiastków
śladowych – Oznaczanie całkowitej zawartości arsenu i selenu metodą
atomowej spektrometrii absorpcyjnej z generacją wodorków (HGAAS)
po mineralizacji ciśnieniowej.
5. Garboś S., Wojtczak M.: Zastosowanie atomowej spektrometrii i technik łą-
czonych w analizie śladowej i specjacyjnej z elementami walidacji. Aktualne
zagadnienia związane z kontrolą jakości wody, PZH Warszawa 2003.
6. Niedzielski P.: Źródła błędów w technice generowania wodorków. GBC Ślesin
2002.
7. Thompson M., Pahlavanpour B., Walton S.J., Krikbright G.F.: Analyst 1978,
102, 568.
8. Nakahara T.: Hydride Generation Techniques in Atomic Spectroscopy [w]: Ad-
vances in Atomic Spectroscopy 2, Greenwich 1995, 139-153.
9. Narasaki H.: Hydride Generation Sample Introduction for Spectroscopic Ana-
lysis in Environmental Samples. [w]: Encyclopedia of Analytical Chemistry,
Wiley 2000, 2643-266.
10. Sturgeon R.E., Mester Z.: Anal. Chem. 2002, 56, 202A.
11. Takase I., Pereira H.B., Luna A.S., Grinbergi P., Calixto de Campos R.: Quim.
Nova 2002, 25, 1132.
12. Bolea E., Laborda F., Castillo J.R., Sturgeon R.E.: Spectrochimica Acta 2004,
B 59, 505-513.
13. Pohl P.: Hydride generation – recent advances in atomic emission spectrometry.
Trends in Analytical Chemistry 2004, 23, 87.
14. Ribeiro A.S., Vieira M.A., Curtius A.J.: Spectrochimica Acta 2004, B 59,
243.
15. Zoltan T., Benzo Z., Murillo M., Marcano E., Gómez C., Salas J. and Quintal
M.: Analytical and Bioanalytical Chemistry 2005, 382, 1419.
16. Narasaki H., Cao Jun-Yan.: Atomic Spectroscopy 1996, 17(2), 77.
17. Hong-Bing H., Hisatake N.: Atomic Spectroscopy 1998, 19(1), 23.
18. Kabata-Pendias A., Szteke B.: Problemy jakości analizy śladowej w badaniach
środowiska przyrodniczego. Wyd. Eduk. Zofii Dobkowski, Warszawa, 1998.
19. Niedzielski P., Siepak M., Siepak J.: Rocznik Ochrony Środowiska 2000, 2,
317-341.
Fig. 8. As 189.042 nm calibration curve
Fig. 9. As 189.042 nm sub array plots for blank, sample and standard
Magdalena PIĄTKOWSKA, M.Sc. - graduated from the Silesian Technical University in Gliwice, Faculty of Chemistry (1999). She is a research and technical specialist and manager of an Analytical Laboratory at the Institute of Inorganic Chemistry in Gliwice.
Specialization – analytical chemistry, instrumental analysis.