the adaptor protein p66shc: roles in cell signaling ... · roles in cell signaling, metabolism and...
TRANSCRIPT

The Adaptor Protein p66SHC:
Roles in Cell Signaling, Metabolism and Growth
by
Mohamed Ahmed Mohamed El-Badry Soliman
A thesis submitted in conformity with the requirements
for the Degree of Philosophy
Graduate Department of Molecular Genetics
University of Toronto
© Copyright by Mohamed Soliman 2014

ii
The Adaptor Protein p66SHC: Roles in Cell Signaling,
Metabolism and Growth
Mohamed Soliman
Doctor of Philosophy
Department of Molecular Genetics
University of Toronto
2014
Abstract
Adaptor proteins link surface receptors to intracellular signaling pathways and control the way
cells respond to nutrient availability. Mice deficient in p66Shc, the most recently evolved
isoform of the Shc1 adaptor proteins and a mediator of receptor tyrosine kinase signaling,
display resistance to diabetes and obesity. Using quantitative mass spectrometry, I found that
p66Shc inhibited glucose metabolism. Depletion of p66Shc enhanced glycolysis and increased
the allocation of glucose-derived carbon into anabolic metabolism, characteristics of a metabolic
shift called the Warburg effect. This change in metabolism was mediated by the mammalian
target of rapamycin (mTOR) as inhibition of mTOR partly reversed the glycolytic phenotype
caused by p66Shc deficiency. Thus, unlike the other isoforms of Shc1, p66Shc appears to
antagonize insulin and mTOR signaling, which limits glucose uptake and metabolism. This study
identifies a critical inhibitory role for p66Shc in anabolic metabolism and insulin-mTOR
signaling.

iii
Happy is the one who has been able to learn the causes of things.
- Virgil, Georgics (II, 490)

iv
To the memory of my PhD advisor, Dr. Tony Pawson (1952-2013)
No longer mourn for me when I am dead
Then you shall hear the sullen surly bell
Give warning to the world that I am fled
- Shakespeare, Sonnet 71

v
Acknowledgments
Foremost is my gratitude to God for the shower of blessings throughout my PhD to complete my
research successfully. Especially, the blessings of health and determination were vital in
providing the physical and mental abilities to see this project through.
This work is dedicated to the memory of my PhD advisor, the late Dr. Tony Pawson, who passed
away months before the completion of this effort. I am grateful for the opportunity to train in one
of the best labs in the world in the field of cell signaling. I was inspired by his humbleness and
collegiality, and by his contagious enthusiasm about science. I am thankful for the exceptional
freedom and independence he gave me as a graduate student. I learned patience, perseverance,
and dedication; it was indeed a maturing journey.
I would like to express my deepest gratitude to my current PhD advisor, Dr. Jim Dennis, whose
expertise, understanding and knowledge added considerably to my graduate experience; to my
supervisory committee members, Drs. Charlie Boone, Anne-Claude Gingras, and Jane McGlade,
for the continuous advice they provided over the years at all levels of my research project; Drs.
Brendan Manning (Harvard University), Fritz Roth (University of Toronto) and Linda Penn
(University of Toronto) for taking the time out to serve as my examiners. My gratitude goes to
Dr. David Sabatini (MIT) for accommodating me in his lab as a visiting scientist to do the
mTOR experiments of my project.

vi
I have had the good fortune of keeping company with the members of the Pawson, Dennis and
Sabatini labs; they have provided me with novel perspectives on my project. Thank you all for
your advice, ideas, friendship and support during the certain and uncertain times in the lab. In
particular, I would like to thank Dr. Jerry Gish who has been an essential part of my education
and entertainment, Dr. Anas Abdel Rahman and Ms. Judy Pawling for all their effort with my
project, and Drs. Karen Colwill and Melissa Stacey for their insight and help over the years.
My gratitude goes to my research funding resouces: the University of Toronto, the Canadian
Institute of Heath Research, the Government of Ontario and the Vanier Canada Graduate
Scholarship program. Without their support, it would not have been possible to solely focus on
my research during my PhD tenure.
I am deeply indebted to my parents for always being supportive of my education. Although they
are so far away, they always provided the absolute encouragement that inspires me to follow my
career. Special thanks to my friends Drs. Hamza Jalal and Omer Yilmaz; your support and
understanding make you formidable friends.
Finally, my thanks go to each of those – unmentioned – who have made it possible for me to
reach this stage of my career, who shared my triumphs and frustration, who shaped my days and
continue to do so: I am grateful for having you in my life.

vii
List of Abbreviations ............................................................................................... x
List of Figures and Tables ................................................................................... xiii
Chapter 1. Introduction ........................................................................................... 1
1.1. Adaptor proteins in signal transduction ........................................................................... 2
1.1.1. Shc1: a prototype of adaptor proteins ....................................................................................4
1.1.2. p66Shc: an integrator of mitogenic and metabolic signaling ................................................9
1.1.3. Role of p66Shc in regulating oxidative stress .....................................................................10
1.1.4. p66Shc and energy metabolism ...........................................................................................12
1.2. Cell metabolism in normal and cancer cells................................................................... 13
1.2.1. The Warburg effect: glucose metabolism and anabolic demands of cell growth ................14
1.2.2. Glutamine: a metabolic fuel for proliferating cells ..............................................................17
1.3. Signaling Pathways and Regulation of Cellular Metabolism......................................... 18
1.3.1. Tyrosine kinase signaling and selective metabolic regulation in dividing cells ..................18
1.3.2. The PI3K-Akt-mTOR pathway ...........................................................................................20
1.3.3. Transcriptional regulators of anabolic metabolism .............................................................22
1.3.3.1. HIF .......................................................................................................................................22
1.3.3.2. Myc ......................................................................................................................................24
1.3.3.3. p53 .......................................................................................................................................24
1.3.4. Metabolic enzymes as oncogenes ........................................................................................25
1.4. mTOR: from signaling to metabolism ........................................................................... 28
1.4.1. Molecular components of mTOR ........................................................................................28
1.4.2. mTORC1 .............................................................................................................................31
1.4.2.1. Upstream regulators of mTORC1 ........................................................................................31
1.4.2.2. Downstream effectors of mTORC1 .....................................................................................35
1.4.3. mTORC2 .............................................................................................................................36
1.4.3.1. Upstream regulators of mTORC2 ........................................................................................37
1.4.3.2. Downstream effectors of mTORC2 .....................................................................................37
1.4.4. mTOR and metabolism ........................................................................................................38
1.4.4.1. Glucose metabolism ............................................................................................................38
1.4.4.2. Lipid synthesis .....................................................................................................................39
1.4.4.3. Protein synthesis ..................................................................................................................39
1.4.4.4. Nucleotide metabolism ........................................................................................................40

viii
1.5. Rationale and objectives of the study ............................................................................. 41
Chapter 2. Materials and Methods ....................................................................... 43
2.1. Cell culture and treatments ............................................................................................. 44
2.2. Freezing and thawing of cells......................................................................................... 44
2.3. Cell culture ..................................................................................................................... 45
2.4. Plasmid preparation and DNA constructs ...................................................................... 45
2.5. Cell transfection ............................................................................................................. 46
2.6. Retroviral production and cell infection......................................................................... 46
2.7. Metabolite extraction...................................................................................................... 47
2.8. Isotope labeling and kinetic profiling............................................................................. 48
2.9. [3H]-2-deoxy-D-glucose uptake assay ........................................................................... 48
2.10. Oxygen consumption rate measurement ........................................................................ 49
2.11. Cell size determination ................................................................................................... 49
2.12. Cell lysis and immunoblotting ....................................................................................... 49
2.13. Western blotting ............................................................................................................. 50
2.14. Mass spectrometry analysis of the p66Shc protein-interactions .................................... 51
2.15. RNA-seq ......................................................................................................................... 52
Chapter 3. p66Shc Inhibits Anabolic Metabolism .............................................. 53
3.1. Background .................................................................................................................... 54
3.2. Loss of p66Shc enhances glycolytic metabolism ........................................................... 55
3.3. Loss of p66Shc promotes glucose metabolism through the pentose phosphate and
hexosamine biosynthesis pathways ........................................................................................... 55
3.4. Restoring p66Shc expression inhibits glycolytic metabolism ....................................... 56
3.5. p66Shc expression inhibits amino acid biosynthesis and pyrimidine metabolism ........ 57
3.6. p66Shc regulates redox homeostasis .............................................................................. 58
3.7. p66Shc is necessary and sufficient to alter glucose uptake and metabolism ................. 59
3.8. Lack of p66Shc enhances glycolytic flux and anabolic metabolism ............................. 60
Chapter 4. p66Shc Inhibits Signaling to The Metabolic Sensor mTOR ........... 83
4.1. Background .................................................................................................................... 84
4.2. p66Shc inhibits growth factor signaling to the metabolic sensor mTOR ...................... 85
4.3. p66Shc expression causes a decrease in cell size ........................................................... 86

ix
4.4. Effects of p66Shc on glycolytic metabolism are mediated through mTOR .................. 86
Chapter 5. Discussion and Future Directions .................................................... 101
5.1. p66Shc signaling to mTOR: an open question ............................................................. 104
5.2. Competition between Shc1 isoforms in regulating PI3K-mTOR signaling ................. 106
5.3. Regulation of receptor and glucose transporter glycosylation by p66Shc ................... 107
5.4. p66Shc and fatty acid signaling to mTOR ................................................................... 108
5.5. Genes regulated through p66Shc expression ............................................................... 109
5.6. Negative regulation of insulin signaling by adaptor proteins ...................................... 110
5.7. Summary ...................................................................................................................... 111
Chapter 6. Appendix ............................................................................................ 113
6.1. LC-MS/MS transitions for the metabolites measured in this study ............................. 114
6.2. LC-MS/MS transitions for 1,2-13
C2 Glucose intermediates. ........................................ 121
6.3. List of gene differentially regulated by p66Shc expression ......................................... 122
6.4. List of identified p66Shc-interacting proteins .............................................................. 138

x
List of Abbreviations
1,3BPG 1,3-bisphosphoglycerate
2-DG 2-deoxy-D-glucose
2-HG 2-hydroxyglutarate
3PG 3-phosphoglycerate
4E-BP Eukaryotic translation initiation factor 4E-binding protein
ACoA Acetyl-CoA
AMPK AMP-activated protein kinase
Ang II Angiotensin II
CAD Carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and
dihydroorotase
CH1 Collagen homology 1
CH2 Collagen homology 2
CTP Cytidine triphosphate
Deptor DEP domain-containing mTOR-interacting protein
DHAP Dihydroxyacetone phosphate
E4P Erythrose-4-phosphate
EGFR Epidermal growth factor receptor
ErbB2 V-erb-b2 avian erythroblastic leukemia viral oncogene homolog 2
Erk Extracellular signal-regulated kinase
F1,6BP Fructose-1,6-bisphosphate
F6P Fructose-6-phosphate
Fes Feline sarcoma oncogene
FoxO Forkhead box O
G3P Glycerol-3-phosphate
G6P Glucose-6-phosphate
Gab Grb2-associated binding protein
GADP Glyceraldehyde-3-phosphate
GAP GTPase-activating protein
GAPDH Glyceraldehyde-3-phosphate dehydrogenase

xi
GlcNAcP N-acetylglucosamine-6-phosphate
GN6P Glucosamine-6-phosphate
Glut Glucose transporter
Grb2 Growth factor receptor-bound protein 2
GSH Glutathione (reduced)
GSK3β Glycogen synthase kinase 3β
GSSG Glutathione (oxidized)
HIF Hypoxia-inducible factor
IDH Isocitrate dehydrogenase
IGF-1 Insulin-like growth factor-1
IMP Inositol polyphosphate multikinase
IRS Insulin receptor substrate
KO Knockout
LC-MS/MS Liquid chromatography–tandem mass spectrometry
MAPK Mitogen-activated protein kinases
MCoA Malonyl-CoA
MEFs Murine embryonic fibroblasts
Mgat5 Mannosyl (α-1,6-)-Glycoprotein
β-1,6-N-Acetyl-Glucosaminyltransferase
mTORC1 Mammalian target of rapamycin complex 1
mTORC2 Mammalian target of rapamycin complex 2
NDRG N-myc downstream regulated gene
OAA Oxaloacetate
PEP Phosphoenolpyruvate
PI3K Phosphoinositide-3-kinase
PIP3 Phosphatidylinositol (3,4,5)-triphosphate
PK Pyruvate kinase
PKC Protein kinase C
PPP Pentose phosphate pathway
PRAS40 40 kDa pro-rich akt substrate
Protor Protein observed with rictor

xii
PTB Phospho-tyrosine binding
PTEN Phosphatase and tensin homolog
PTPN12 Protein tyrosine phosphatase, non-receptor type 12
R5P Ribose-5-phosphate
Rac1 Ras-related C3 botulinum toxin substrate 1
Raptor Regulatory-associated protein of mTOR
Ras Rat sarcoma oncogene
Rheb Ras homolog enriched in brain
Rictor Rapamycin-insensitive companion of mTOR
ROS Reactive oxygen species
RTK Receptor tyrosine kinase
SGK Serum- and glucocorticoid-induced protein kinase
SH2 Src homology 2
SH3 Src homology 3
SHC Src-homology collagen-containing protein
shRNA short hairpin RNA
SIN1 Stress-activated map-kinase-interacting protein 1
SOD Superoxide dismutase
Sos Son of sevenless homolog
SREBP Sterol regulatory element-binding protein
TCA cycle Tricarboxylic acid cycle
TSC1/2 Tuberous sclerosis 1 and 2
UDP-GlcNAc Uridine-diphosphate N-acetylglucosamine
UTP Uridine triphosphate
VHL Von Hippel–Lindau
X5P Xylulose-5-phosphate

xiii
List of Figures and Tables
Fig. 1.1 Schematic diagram of the Shc1 proteins. 6
Fig. 1.2 Role of Shc1 in signaling downstream of RTK. 7
Fig. 1.3 Cancer metabolism: the Warburg effect. 16
Fig. 1.4 mTOR signaling pathway. 30
Fig. 3.1 Effect of p66Shc on glycolytic metabolism. 61
Fig. 3.2 Lack of p66Shc enhances glycolytic metabolism. 62
Fig. 3.3 p66Shc deficiency increases the levels of the pentose phosphate
and the hexosamine pathway intermediates. 63
Fig. 3.4 Levels of Shc1 isoforms in p66Shc KO and p66+ MEFs. 64
Fig. 3.5 Unsupervised principal component analysis for targeted metabolomics
screen in p66Shc KO and p66+ MEFs. 65
Fig. 3.6 p66Shc expression decreases the levels of glycolytic intermediates. 66
Fig. 3.7 p66Shc inhibits fatty acid biosynthesis. 67
Fig. 3.8 p66Shc expression decreases the levels of the pentose phosphate
and the hexosamine pathway intermediates. 68
Fig. 3.9 Deficiency of p66Shc inhibits oxygen consumption rate and lowers
AMP/ATP ratio. 69
Fig. 3.10 p66Shc expression inhibits the synthesis of nonessential amino acids. 70
Fig. 3.11 Tracing of 15
N-labeled-amino acids in p66Shc KO and p66+ cells. 71
Fig. 3.12 p66Shc inhibits de novo pyrimidine synthesis intermediates. 72
Fig. 3.13 p66Shc regulates redox homeostasis. 73

xiv
Fig. 3.14 Lack of p66Shc enhances 2-DG uptake. 74
Fig. 3.15 p66Shc inhibits cellular lactate secretion. 75
Fig. 3.16 Abundance of Glut1 in p66Shc-deficient and p66Shc-competent cells. 76
Fig. 3.17 Isotope-tracing of 13
C-labeled glucose in p66Shc-deficient and
p66Shc-competent HeLa cells. 77
Fig. 3.18 Isotope-tracing of 13
C-labeled hexosamine pathway intermediates in
p66Shc-deficient and p66Shc-competent HeLa cells. 78
Fig. 3.19 Isotope-tracing of 13
C-labeled glycolytic intermediates in p66Shc KO
and p66+ MEFs. 79
Fig. 3.20 Isotope-tracing of 13C-labeled nonessential amino acids in p66Shc KO
and p66+ MEFs 80
Fig. 3.21 Flux analysis of 13
C-labeled glucose in p66Shc KO and p66+ MEFs. 81
Table 3.1 Fold change of the most significantly p66Shc-inhibited metabolites 82
Fig. 4.1 p66Shc inhibits mTORC1 and mTORC2 activation following serum
stimulation. 88
Fig. 4.2 p66Shc inhibits insulin signaling to mTOR. 89
Fig. 4.3 p66Shc inhibits mTORC1 activation in response to 90
amino acid stimulation.
Fig. 4.4 p66Shc expression inhibits mTOR activation in response to insulin 91
and IGF1, but not to EGF, stimulation.
Fig. 4.5 p66Shc expression inhibits mTOR activation in 92
response to serum stimulation.
Fig. 4.6 p66Shc expression inhibits mTOR activation in 93
response to amino acid stimulation.
Fig. 4.7 Stable expression of p66Shc, but not p52Shc, in p66Shc KO cells
inhibits the mTOR pathway. 94
Fig. 4.8 Stable expression of p66Shc, but not p52Shc, in p66Shc KO 95
cells inhibits glycolytic metabolism.

xv
Fig. 4.9 p66Shc expression decreases cell size. 96
Fig. 4.10 p66Shc mediates cell growth. 97
Fig. 4.11 Effect of rapamycin on mTOR signaling in p66Shc KO and
p66+ MEFs. 98
Fig. 4.12 mTOR mediates the effects of p66Shc on glycolytic
and pyrimidine metabolism. 99
Fig. 4.13 Effect of Akt inhibition on the abundance of glycolytic
metabolites in p66Shc-competent and p66Shc-deficient MEFs. 100

1
Chapter 1. Introduction

2
1.1. Adaptor proteins in signal transduction
Membrane receptors sample the extracellular environment. When activated by threshold levels of
cognate ligands, receptors stimulate a signaling cascade that leads to precise biological
responses. Signaling proteins contain catalytic and adaptor functions that typically reside in
discrete, independently folded domains. Most adaptor domains display binding specificity for
peptide motifs in other signaling proteins that transmit and codify the signals (Pawson, 2007).
For example, Src kinase has a canonical kinase fold as well as a Src homology 2 (SH2) domain
that bind to specific phosphotyrosine residues on activated receptors, and a Src homology 3
(SH3) domain that binds to polyproline (Pro-X-X-Pro) motifs. SH2 and SH3 domains are found
in many other proteins and function to recruit signaling proteins into complexes where catalytic
efficiency is greatly enhanced.
Analysis of the molecular evolution of genomes suggests that phenotypic diversity can
frequently be attributed to new combinations of existing protein domains, rather than from the
creation of completely new proteins. Gene duplication and shuffling of modular domains, also
results in the emergence of novel connectivity between existing proteins, expanding the
information flow through regulatory pathways and the complexity of responses. This could
explain the less-than-expected number of protein-coding genes and protein domains upon
sequencing of animal genomes more than a decade ago (Bhattacharyya et al., 2006). Thus
evolutionary innovation is achieved by domain duplication, recombination and adaptation of
domain affinities for novel binding partners (Jin and Pawson, 2012). As independent folds within
the same protein sequence, the catalytic and recruitment domains can evolve at different rates.
Disconnecting catalytic and recruitment domains into separate genes allows independent

3
regulation by transcription, splicing and translation, and thereby more diversity and evolutionary
possibilities for increased complexity in signaling networks (Bhattacharyya et al., 2006).
The terms adaptors and scaffolds are used interchangeably in the literature for non-catalytic
proteins that cross-link and promote the assembly of specific signaling complexes (Pawson and
Scott, 1997). Herein, we refer to these proteins as “adaptor proteins.” In mammals they include
the growth factor receptor-bound protein 2 (Grb2)/ Grb2-related adapter protein (Grap)/ Grb2-
related adaptor downstream of Shc (Gads), Grb7/10/14, SH2B/ adapter protein with pleckstrin
homology (APS)/ lymphocyte adaptor protein (Lnk), SH2D1-4, Shc1-4, SHB/SHD/SHE/SHF,
and the non catalytic region of tyrosine kinase (Nck)1/2 gene families, among others.
Differential expression or posttranslational modifications of an adaptor can determine whether a
pathway will function in a particular cell type. For example, the SH2-containing collagen-related
(Shc), alternative splice variants can have different interaction and pathway output, depending on
their levels of expression in various tissues. In addition, relocalization of adaptors to a specific
cellular compartment in a timely fashion is often a requirement of signal transduction fidelity
(Scott and Pawson, 2009). For example, Shc1 very rapidly recruits proteins associated with acute
stimulation of epidermal growth factor receptor (EGFR), such as Grb2-associated binding
protein 1/2 (Gab1/2) and Grb2-son of sevenless homolog 1 (Sos1) complex which promotes
exchange of Rat sarcoma oncogene (Ras)-bound GDP by GTP, while slowly recruits signaling
proteins that mediate negative regulation of EGFR signaling, such as protein tyrosine
phosphatase, non-receptor type 12 (PTPN12) (Zheng et al., 2013).

4
1.1.1. Shc1: a prototype of adaptor proteins
The Shc adaptor proteins were first identified by screening a human cDNA library for sequences
complementary to the SH2 domain of the feline sarcoma oncogene (c-fes) tyrosine kinase
(Pelicci et al., 1992). Following this initial screen, three sequence-related Shc-like transcripts and
proteins were identified (Luzi et al., 2000). The mammalian Shc gene family comprises four
members: ShcA, B, C and D. In addition, alternative splicing of ShcA and ShcC transcripts
results in multiple protein isoforms. While ShcA is expressed in almost all tissues, ShcB
(Sck/Sli/Shc2) and ShcC (Rai/N-Shc/Shc3) are found predominantly in the brain and ShcD is
expressed mainly in brain and muscle tissues (Wills and Jones, 2012). All of the Shc members
are structurally characterized by the unique modular arrangement of a phospho-tyrosine binding
(PTB) domain, a collagen homology 1 (CH1) region followed by a SH2 domain (Luzi et al.,
2000). The PTB and SH2 domains independently bind motifs containing phosphorylated tyrosine
residues. The domains are separated by the CH1 region which contains three consensus tyrosine
residues that are phosphorylated by tyrosine kinases (Luzi et al., 2000). The phosphotyrosine
residues subsequently serve as recognition motifs for the SH2 domain of proximal signaling
molecules including Grb2. The C. elegans Shc homologue lacks the phosphotyrosine sites in the
CH1 region, whereas the D. melanogaster Shc homologue contains two of the three consensus
sites, consistent with an expanded role for phosphotyrosine signaling with metazoan evolution
(Lim and Pawson, 2010).
The ShcA gene (hereafter named Shc1) locus in mammals encodes three isoforms; p46Shc,
p52Shc and p66Shc (Fig. 1.1) (Ravichandran, 2001). p52Shc is the most extensively studied
isoform and the one traditionally referred to as Shc1 in the literature. p46Shc and p52Shc

5
originate from different translation initiation sites within the same mRNA (Pelicci et al., 1992).
p52/p46Shc bind to autophosphorylated tyrosine residues in activated receptor and cytoplasmic
tyrosine kinases, which in turn phosphorylate p52/p46Shc on three tyrosine residues (239, 240,
and 317) located in the CH1 region, enabling Shc proteins to recruit the Grb2-SOS complex that
activates the GTPase Ras and the Grb2-Gab2-phosphoinositide-3-kinase (PI3K) complex (Fig.
1.2) (Ravichandran, 2001; Wills and Jones, 2012). Rapid tyrosine phosphorylation of these three
residues is necessary for transmitting the RTK-mediated mitogenic and cell survival signals to
downstream targets. For example, recent quantitative proteomic analysis of Shc1 signaling
showed that phosphorylation of EGFR Tyr1148 and Tyr1173, the main binding sites of Shc1,
occurs within seconds of EGF ligand binding, allowing rapid recruitment and tyrosine
phosphorylation of Shc1 (Dengjel et al., 2007). The SH2 domain of Grb2 binds to the
phosphorylated tyrosine residues in the CH1 region of Shc1, promoting signaling to the Ras-
mitogen-activated protein kinases (MAPK) and the PI3K-protein kinase B (PKB/Akt) cascades.
Shc1, however, is not necessary to activate the Ras-MAPK pathway as Grb2 can bind directly to
EGFR (Batzer et al., 1994). Shc1 mainly functions to sensitize cells to EGF stimulation and
MAPK activation as deletion of all Shc1 isoforms diminishes, but does not abolish, MAPK
phorphorylation (Lai and Pawson, 2000). Since EGFR has been extensively studied as a model
for RTK-Shc1 signaling to Ras-MAPK activation, it will be discussed in further detail.

6
Fig. 1.1. Schematic diagram of the Shc1 proteins.
All three Shc1 isoforms share the same domain architecture. The PTB and SH2 domains of Shc1
bind to phosphotyrosine-containing sequences. The PTB preferentially binds to NPXpY motifs
and specificity is determined by residues N-terminal to the pY. The SH2 domain binds to pY-
hydrophobic-X-hydrophobic motif and the specificity is determined by residues C-terminal to the
pY. The CH1 region contains three phospho-tyrosine sites: The Y239/Y240 twin tyrosines and
Y317. The SH2 domain of Grb2 binds to the CH1 pY residues, coupling Shc1 to MAPK
activation.
p52Shc is the isoform most extensively studied in the context of growth factor signaling. p46Shc
lacks the first 46 amino acids within the PTB domain and its function is not clearly understood.
The p66Shc isoform possesses an additional N-terminus CH2 region of 110 amino acids
containing a S36 phosphorylation site that has been implicated in mediating oxidative stress
signaling.

7
Fig. 1.2: Role of Shc1 in signaling downstream of RTK.
Ligand binding causes RTKs to dimerize and autophosphorylate specific tyrosine residues in their
cytoplasmic tail, which serve as docking sites for the Shc1 PTB domain. The EGFR kinase
phosphorylates tyrosine in the CH1 region, to which the Grb2 adaptor bind and initiate the Ras/MAPK
cascade. Shc1 can also be phosphoryalted by cytoplamic tyrosine kinases such as Src. Grb2 and its
constitutive binding partner, the guanine nucleotide exchange factor (GEF) SOS, activates the
membrane-associated Ras GTPase, which in turn activates the Raf Ser/Thr kinase which activates the
Erk MAPK. The Ras-MAPK pathway promotes proliferation, differentiation and survival. Like
p52Shc, p66Shc also interacts with Grb2, however it exerts an inhibitory or no effect on the MAPK
pathway. Shc1 can also activate the PI3k/Akt pathway through binding to the adaptor Gab2. Following
RTK activation, Gab2 interacts with the p85 regulatory subunit of PI3K. The catalytic p110 subunit of
PI3K can then phosphorylate membrane phospholipids, generating PIP3, which recruits the Ser/Thr
kinase Akt. Akt activates several downstream targets to enhance cell survival and growth.

8
The Shc1 proteins mediate signal transduction to downstream effectors through complex
phosphorylation/dephosphorylation dynamics. For example, following EGF stimulation, p52Shc
is phosphorylated on both Tyr and Ser/Thr residues but at different time points. Initially, as
mentioned above, p52Shc is phosphorylated on Tyr239, Tyr240 and Tyr317 (1–2 minutes after
EGF stimulation). Afterwards, Ser29 (3 minutes), Thr214 (5 minutes) and Ser335 (20 minutes)
undergo phosphorylation (Zheng et al., 2013). Each phosphorylation event leads to the
association of p52Shc with specific proteins (Zheng et al., 2013). The first phosphorylation event
leads to the recruitment of the Grb2 adaptor to p52Shc1 pTyr sites, activating pro-mitogenic and
survival pathways. Ser29/Thr214 phosphorylation recruits the tyrosine phosphatase PTPN12
which dephosphorylates p52Shc1 leading to loss of Grb2 binding. Ser335 phosphorylation of
p52Shc is required for cytoskeletal reorganization through recruiting phosphatases and GTPase-
activating proteins which downregulate Ras-MAPK signaling (Zheng et al., 2013).
Deletion of all Shc1 isoforms (p66Shc, p52Shc and p46Shc) is embryonic lethal by E11.5 due to
cardiovascular defects (Lai and Pawson, 2000). Knock-in embryos expressing Shc1 with a non-
functional PTB domain (R175Q substitution) die at approximately E11.5 and exhibit cardiac
defects reminiscent of full Shc1 KO, indicating that Shc1 regulates heart development by a PTB-
dependent mechanism (Hardy et al., 2007). However, Knock-in embryos with a non-
phosphotyrosine binding SH2 domain mutation (R397K) or all three phosphotyrosine residues in
the CH1 region substituted by phenylalanine (3Y to 3F) can survive to birth, but demonstrate
impaired motor coordination due to abnormal development of muscle spindles (Hardy et al.,
2007). Thus it appears that heart morphogenesis requires Shc1 with a functional PTB domain,
but occurs independently of phosphorylation of the CH1 tyrosine residues, the primary Grb2

9
binding sites. Biochemical data from experiments in D. melanogaster suggest that this process is
conserved as the Drosophila Shc1 ortholog, dShc, requires the PTB domain for signaling
downstream of the RTKs DER and Torso (Lai et al., 1995; Li et al., 1996).
1.1.2. p66Shc: an integrator of mitogenic and metabolic signaling
The p66Shc isoform has emerged with vertebrates (Luzi et al., 2000; Migliaccio et al., 1997). It
has an additional unique N-terminal 110-amino-acid collagen-homology region 2 (CH2). While
p46Shc and p52Shc are ubiquitously expressed in various tissues and developmental stages,
p66Shc is expressed in a tissue-specific fashion, mainly in liver, lungs, skin and heart. It is
notably absent from some tissues, including haematopoietic cells and brain (Lebiedzinska et al.,
2009). p66Shc fails to enhance MAPK phosphorylation upon EGF stimulation, despite being
phosphoryaled by EGFR and binding Grb2 (Migliaccio et al., 1997). Unlike p52Shc,
overexpressing p66Shc does not transform NIH3T3 mouse fibroblasts (Migliaccio et al., 1997).
In contrast, stable knockdown of p66Shc in L6 skeletal muscle myoblasts caused increased basal
activation of the Erk MAPK (Natalicchio et al., 2004). This observation is intriguing given that
p66Shc shares identical PTB sequences, phosphotyrosine sites on the CH1 region and SH2
sequence with the shorter isoforms. Negative regulation of EGFR signaling by p66Shc has been
attributed to MAPK-mediated phosphorylation of Ser36 in the CH2 region unique to p66Shc
(Okada et al., 1997). This serine phosphorylation event is thought to destabilize EGFR-p66Shc
interaction, thereby decoupling p66Shc from Ras-MAPK activation (Okada et al., 1997). Since
p66Shc and p52Shc compete for a limited pool of Grb2, p66Shc may sequester the Grb2-SOS
complex away from RTKs at the membrane, inhibiting Ras-MAPK activation.

10
Unlike p52Shc, the abundance of p66Shc is substantially decreased in breast cancer cell lines
expressing the oncogene v-erb-b2 avian erythroblastic leukemia viral oncogene homolog 2
(ErbB2), suggesting that p66Shc may function as a tumor suppressor (Stevenson and Frackelton,
1998). p66Shc expression correlates with a favorable outcome for breast cancer, particularly in
the context of the ratio of total phospho-Tyr317 Shc1 to the levels of p66Shc isoform (pTyr
Shc/p66Shc); higher levels of phosphorylated Shc1 relative to the abundance of the p66Shc
isoform are associated with aggressive neoplasms and increased risk of relapse (Davol et al.,
2003). This antagonistic effect of p66Shc on RTK signaling occurs, most likely, due to the
competition between p66Shc and p52Shc for common effectors of RTK signaling (Okada et al.,
1997).
1.1.3. Role of p66Shc in regulating oxidative stress
p66Shc has been portrayed as both a sensor and a proponent of reactive oxygen species (ROS)
production, promoting oxidative stress and pro-apoptotic signaling (Pinton and Rizzuto, 2008).
The role of p66Shc in the generation of ROS involves at least three mechanisms: a) p66Shc
activation leads to inhibition of the Forkhead box O (FoxO) transcription factors via Akt-
mediated phosphorylation. This results in a decrease in FoxO-dependent expression of ROS
scavenging enzymes, such as superoxide dismutase (SOD) and catalase (Nemoto and Finkel,
2002), b) At the plasma membrane, p66Shc promotes the activation of the Ras-related C3
botulinum toxin substrate 1 (Rac1) GTPase, triggering ROS production by the membrane-bound
NADPH oxidase (Khanday et al., 2006), and c) p66Shc acts in the mitochondrial intermembrane
space (IMS) where it interacts with cytochrome c (Giorgio et al., 2005). While all three isoforms
of Shc1 are predominantly cytoplasmic and a fraction translocate to the plasma membrane
following growth factor stimulation, approximately 10-20% of p66Shc also relocates to the

11
mitochondrial intermembrane space in response to oxidative stress (Orsini et al., 2004). Under
stress conditions (e.g. hydrogen peroxide treatment), Ser36 in the CH2 region is phosphorylated
by protein kinase C β (PKCβ) resulting in a phospho-Ser36-Pro37 motif recognized by the prolyl
isomerase Pin1 which induces isomerization around the Ser-Pro bond, targeting p66Shc to the
mitochondria (Pinton et al., 2007). Inside the IMS, p66Shc interacts with cytochrome c,
promoting transfer of electrons to oxygen and generating hydrogen peroxide (Giorgio et al.,
2005). The increase in ROS production leads to swelling and rupture of mitochondria and release
of pro-apoptotic factors into the cytoplasm. In this context, p66Shc uses the reducing equivalents
of the mitochondrial electron transfer chain through the oxidation of cytochrome c, leading to the
activation of programmed cell death (Giorgio et al., 2005). Factors regulating p66Shc expression
under stress conditions are still unknown. One potential mechanism involves the tumor
suppressor protein p53, where activated p53 upregulates p66Shc expression to induce apoptosis
in murine embryonic fibroblasts (MEFs) and endothelial cells under oxidative stress conditions
(Kim et al., 2008a; Trinei et al., 2002).
Consistent with its role in mediating oxidative stress and programmed cell death, p66Shc
knockout MEFs show increased resistance to apoptosis in response to various conditions,
including treatment with UV, taxol or amyloid β-peptide (Migliaccio et al., 1999; Smith et al.,
2005b; Yang and Horwitz, 2000). Additionally, MEFs lacking p66Shc show lower levels of
systemic and tissue oxidative stress markers, such as 8-oxoguanine, compared to p66Shc-
competent cells (Napoli et al., 2003; Nemoto and Finkel, 2002; Trinei et al., 2002). p66Shc
mediates cell death in cardiac cells following chronic exposure to angiotensin II (Ang II), a
secreted factor that shows high abundance in patients with hypertension, atherosclerosis, and

12
diabetes. Selective disruption of the p66Shc isoform in mice caused resistance to Ang II-
mediated hypertrophy and apoptosis in cardiomyocytes and endothelial cells (Graiani et al.,
2005). Deletion of p66Shc also reduces vascular cell apoptosis and early atherogenesis in mice
fed a high-fat diet (Napoli et al., 2003). These phenotypes have been partly attributed to
increased resistance of the p66Shc KO cells to oxidative stress-mediated cell damage (Migliaccio
et al., 1999).
1.1.4. p66Shc and energy metabolism
p66Shc fulfills a function in energy metabolism, as indicated by its role in oxidative stress and
insulin signaling. p66Shc has been shown to inhibit insulin-like growth factor-1 (IGF-1)-Akt
signaling in vascular smooth muscle cells (Xi et al., 2010a; Xi et al., 2010b). In addition, L6
myoblasts overexpressing p66Shc displayed reduced rates of basal glucose uptake and a
reduction in the protein abundance of glucose transporters (Natalicchio et al., 2009). In animal
models, inactivation of p66Shc in mice improved glucose tolerance and insulin sensitivity
(Ranieri et al., 2010; Tomilov et al., 2011). In obese mice lacking the hormone leptin (ob/ob
mice), fasting glycemia values were significantly lower in p66Shc KO than in p66Shc WT mice,
despite no difference in plasma insulin levels (Ranieri et al., 2010). These studies suggest that
p66Shc may suppress glucose metabolism by dampening insulin signaling (Giorgio et al., 2012)

13
1.2. Cell metabolism in normal and cancer cells
The metabolic program of resting non-dividing cells provides the energy required for
maintaining ATP production. On the contrary, proliferating cells must generate sufficient energy
to support cell division, cellular biosynthesis and maintain cellular redox homeostasis (Cairns et
al., 2011). Reprogramming of cellular metabolism towards anabolism, in response to growth
factor signaling, is crucial for supplying nucleotides, proteins and lipids needed for a cell to
double its size before dividing into two daughter cells (Ward and Thompson, 2012).
The first observation regarding the atypical metabolic demands of rapidly dividing cells can be
traced back to the pioneering work of Otto Warburg (Warburg, 1956). In the presence of oxygen,
most normal tissues break down glucose to generate pyruvate, which is largely oxidized to
carbon dioxide in the mitochondria through the tricarboxylic acid (TCA) cycle followed by
oxidative phosphorylation. This process produces a net of 36 ATPs. However, under anaerobic
conditions, pyruvate is shunted away from mitochondria and gets reduced to lactate, generating
only 2 ATPs. Warburg’s studies on rapidly dividing ascites cancer cells showed that tumors
displayed unusually high rates of glycolysis despite the low ATP yield of converting glucose to
lactate. He proposed that tumor cells have impaired mitochondria and, hence, depend on
fermenting glucose to meet their energy demand. This phenomenon, named “the Warburg
effect,” has been confirmed in other cancer types and has been extended beyond glycolysis to
include increased deployment of carbon into anabolic metabolism that supports cell growth
(Vander Heiden et al., 2009). The reliance of cancer on higher glucose uptake has been clinically
used for tumor detection and monitoring through the implementation of 18
F-deoxyglucose-
positron emission tomography (PDG-PET) imaging.

14
1.2.1. The Warburg effect: glucose metabolism and anabolic demands of cell
growth
The selective advantage that glycolytic metabolism provides for proliferating tumor cells has
been debatable for years (Ward and Thompson, 2012). The initial proposal by Warburg that
mitochondrial dysfunction in tumor cells required aerobic glycolysis to cope with low
competence of mitochondrial ATP generation was not fully correct. Mitochondrial respiration
occurs in cancer cells and remains the primary source of ATP production in most tumors (Ward
and Thompson, 2012). For example, an oncogenic mutant of KRas induced aerobic glycolysis as
evidenced by ~ 2-fold increase in glucose uptake and lactate secretion. Yet, most cellular ATP (~
60%) is still produced by mitochondrial oxidation in these cells (Fan et al., 2013). In addition,
mitochondrial function in KRas-mediated tumorigenesis may be crucial for transformation and
tumor progression (Weinberg et al., 2010). Another possible explanation for the Warburg effect
is that heightened glycolytic metabolism takes place as an adaptive response to hypoxic
conditions in the early phases of tumor development prior to vascularization, driven in part by
acidic microenvironment resulting from excess lactate production (Gatenby and Gillies, 2004).
An alternative and more plausible explanation is that the preferential metabolism of glucose
through glycolysis serves to provide precursor molecules, such as amino acids, for biomass
synthesis and to maintain redox balance in proliferating cells (Fig. 1.3) (Cairns et al., 2011;
Vander Heiden et al., 2009). There are several examples supporting this concept. The glycolytic
intermediates fructose-6-phosphate (F6P) and glyceraldehyde-3-phosphate (GADP) can be
shunted into the non-oxidative branch of the pentose phosphate pathway, generating ribose-5-
phosphate (R5P), a critical component of nucleotide biosynthesis. Similarly, glucose-6-

15
phosphate (G6P), the first intermediate in the glycolytic pathway, could feed the oxidative arm of
the pentose phosphate pathway to generate NADPH, a coenzyme used in anabolic reactions,
such as lipid and nucleic acid synthesis, and in maintaining the reductive environment of the cell.
In addition to their role in glycolysis, G6P and F6P are precursors of the hexosamine
biosynthesis pathway which provides uridine-diphosphate N-acetylglucosamine (UDP-GlcNAc)
as a substrate for O-GlcNAcylation of cytosolic proteins and N-linked and O-linked
glycosylation of proteins produced in the secretory pathway. N-glycans branching provides
positive feedback to enhance nutrient transport and receptors residency at the cell surface (Lau et
al., 2007; Ohtsubo et al., 2005; Partridge et al., 2004). The glycolytic intermediates also serve as
precursors for protein synthesis: 3-phosphoglycerate (3PG) can be converted to serine and
pyruvate can be transaminated to alanine. In addition, glycolytic metabolism provides the
building blocks for lipid synthesis. Reduction of the glucose-derived dihydroxyacetone (DHAP)
to glycerol-3-phosphate (G3P) endows proliferating cells with an essential precursor for
phospholipids and triacylglycerol biosynthesis. In addition, pyruvate can contribute to the
production of mitochondrial citrate, which can then be exported to the cytoplasm to be utilized in
de novo synthesis of fatty acids (Hatzivassiliou et al., 2005).

16
Fig. 1.3. Cancer metabolism: the Warburg effect.
Rapidly dividing cells needs sufficient energy to support cell division, increase cell biomass and maintain
redox homeostasis. Reprogramming of cellular metabolism towards glycolysis provides the building
blocks required for the synthesis of nucleotides, proteins and lipids. Preferential diversion of pyruvate into
lactate production allows proliferating cells to shunt glycolytic intermediates into branching anabolic
pathways, including the pentose phosphate and hexosamine pathways, and lipid and protein biosynthesis.

17
1.2.2. Glutamine: a metabolic fuel for proliferating cells
Glucose metabolism supplies cancer cells with essential anabolic building blocks (Hanahan and
Weinberg, 2011), yet cannot explain all the metabolic changes needed to support cell growth.
The significance of other nutrients, such as the amino acid glutamine, as essential fuel for cancer
growth and survival has become better understod in recent years (DeBerardinis and Cheng,
2010). Cancer cells are in high demand of nitrogenous compounds, including nucleotides,
nonessential amino acids, and hexosamines. Glutamine, the most abundant free amino acid in
human blood, acts as an obligate nitrogen donor in purine and pyrimidine synthesis, and as the
primary nitrogen source in anabolism of nonessential amino acids from α-keto acids
(DeBerardinis and Cheng, 2010). Glutamine is not an essential amino acid, but high growth rates
in embryonic and cancer cells depend on glutamine as a nitrogen source and also in conversion
to α-ketoglutarate which supports the TCA cycle and ATP production. This allows more of the
glycolytic intermediates to be utilized in anabolic pathways. Real-time 13
C NMR demonstrates
that glutamine carbon can be converted into lactic acid and secreted from cancer cells in a
process termed “glutaminolysis” (DeBerardinis et al., 2007).
Glutamine also functions as an essential carbon source for replenishing depleted TCA cycle
intermediates, a process known as “anaplerosis.” It gets deaminated to the amino acid glutamate
which is converted to the mitochondrial intermediate α-ketoglutarate and then to citrate.
Glutamine contributes to the carbons of mitochondrial citrate, which can be exported to the
cytoplasm and converted to acetyl-CoA, a precursor for fatty acid synthesis, and oxaloacetate
(OAA). OAA can be shuttled back to the mitochondria and metabolized through multiple steps

18
to yield NADPH, providing the reducing substrates required for lipid synthesis and regeneration
of reduced glutathione needed in rapidly dividing cells (DeBerardinis et al., 2007). In addition,
OAA can be transaminated to aspartate, which can be utilized as a carbon source in nucleotide
biosynthesis. Further studies are needed to dissect the exact mechanisms regulating glutamine
metabolism and its impact on proliferating cells.
1.3. Signaling pathways and regulation of cellular metabolism
Signaling downstream of growth factor receptors not only mediates cell proliferation, but also
alters cellular metabolism. As stated earlier, this metabolic rewiring is critical for the cell to meet
the energy and anabolic requirements associated with cell growth and division. For example,
receptor tyrosine kinase signaling mediated by the PI3K/Akt pathway plays a central role in
regulating glucose uptake, expression and activation of glycolytic enzymes, as well as regulating
glucose carbon flux into lipid synthesis (Elstrom et al., 2004). In addition, tyrosine kinase-
dependent regulation of glycolytic enzymes, such as pyruvate kinase, reroutes glucose
metabolism into anabolic pathways (Christofk et al., 2008a; Christofk et al., 2008b). These
signaling pathways, among others, allow cells to coordinate growth and division with their
metabolic activity.
1.3.1. Tyrosine kinase signaling and selective metabolic regulation in
dividing cells
Several metabolic enzymes become tyrosine phosphorylated following growth factor stimulation,
where signaling is initiated by phosphorylation of activated receptors and downstream effectors

19
on specific tyrosine residues. An example is pyruvate kinase (PK), the enzyme that catalyzes the
final irreversible step of glycolysis, converting phosphoenolpyruvate (PEP) to pyruvate with the
concomitant generation of ATP. There are four members of the PK family of proteins: PKL,
PKR, PKM1 and PKM2. PKL and PKR expression is restricted to liver and red blood cells,
respectively. PKM1 and PKM2 are splice variants encoded by the PKM gene. These two
isoforms differ only by one exon: inclusion of exon 9 and exclusion of exon 10 for PKM1 and
vice versa for PKM2, rsulting in a difference of 23 amino acids at their carboxy terminal. Most
cells in adult tissues predominantly express PKM1. PKM2, on the other hand, is found in cells
with self-renewal capacity, including stem cells and embryonic cells. In addition, this isoform is
highly expressed in many tumors (Mazurek et al., 2005).
Despite the seemingly paradoxical observation that PKM2 possesses lower specific activity than
PKM1, cells expressing PKM2 have a selective growth advantage over cells expressing PKM1
(Christofk et al., 2008a; Christofk et al., 2008b). Unlike the constitutively active PKM1 and
other PK variants, PKM2 is sensitive to growth factor signaling. This isoform binds to tyrosine
phosphorylated proteins and is tyrosine phosphorylated, leading to inhibition of PKM2 activity
(Hitosugi et al., 2009), allowing PKM2 to act as a gatekeeper for the glucose carbon metabolic
fate. By slowing glycolysis, PKM2 enables cells to shunt glycolytic intermediates into branching
anabolic pathways, such as serine and glycine synthesis, pentose phosphate and hexosamine
pathways, rather than sending pyruvate to the TCA cycle.
The oncoprotein Myc promotes the expression of PKM2 at the expense of PKM1 by altering
exon splicing through upregulating the expression of heterogeneous nuclear ribonucleoproteins

20
(hnRNPs) (David et al., 2010). This observation is well in line with the role of Myc in
stimulating glycolysis and inducing the expression of hypoxia-inducible factor 1 (HIF-1) to
support cell growth of rapidly dividing cells (Gordan et al., 2007). PKM2 may also have roles
beyond regulating PK activity. For example, PKM2 interacts with HIF-1α, stimulating HIF-1-
mediated transactivation of glycolytic genes (Luo et al., 2011). In addition, PKM2, upon
translocation to the nucleus, activates β-catenin (Yang et al., 2011) where PKM2 binds to
tyrosine-phosphorylated β-catenin in the nucleus and contributes to β-catenin-mediated
transactivation of cyclin D and Myc, promoting tumor progression (Yang et al., 2011). Further
investigation is needed to determine how the glycolytic and non-glycolytic functions of PKM2
are regulated in both resting and proliferating cells.
1.3.2. The PI3K-Akt-mTOR pathway
One of the most highly conserved signal transduction pathways downstream of growth factor
receptors is PI3K-Akt-mTOR (Hemmings and Restuccia, 2012). PI3K is activated when growth
factors bind to receptor tyrosine kinases, G-protein coupled receptors and cytokine receptors
(Vanhaesebroeck et al., 2012). PI3K not only provides growth and survival signals, but redirects
cellular metabolism to meet the cellular demands for growth. Not surprisingly, the PI3k-Akt
pathway is one of the most commonly mutated signaling nodes in human cancers (Rodon et al.,
2013). Activation can occur due to mutations in PI3K, its negative regulator phosphatase and
tensin homolog (PTEN), or abnormal signaling from upstream receptors (Engelman et al., 2006).

21
When activated, PI3K phosphorylates membrane lipids to generate phosphatidylinositol (3,4,5)-
triphosphate (PIP3) which subsequently leads to recruitment and activation of Ser/Thr kinases
with PH domains that bind PIP3 (Engelman et al., 2006). The best characterized effector
downstream of PI3K is Akt (also known as PKB). Akt drives glycolytic metabolism by
increasing the expression of the glucose transporter 1 (Glut1) and membrane translocation of
adipose- and striated muscle-specific glucose transporter 4 (Glut4) (Robey and Hay, 2009). In
addition, Akt phosphorylates and activates key glycolytic enzymes, such as hexokinase (Gottlob
et al., 2001). Akt also directly phosphorylates the enzyme phosphofructokinase 2, increasing the
levels of fructose 2,6-bisphosphate which enhances the activity of the rate-limiting glycolytic
enzyme phosphofructokinase 1 (Deprez et al., 1997).
The prolonged Akt signaling associated with transformation inhibits the FoxO transcription
factors, resulting in transcriptional changes that lead to enhanced glycolytic metabolism (Khatri
et al., 2010). When FoxO1 is phosphorylated by Akt on Thr24, Ser256, and Ser319, it is
excluded from the nucleus, ubiquitilated and degraded. Phosphorylation of FoxO1 by Akt
decreases the hepatic glucose production through decreasing the transcription of glucose 6-
phosphatase (Nakae et al., 2001). Akt also stimulates anabolic pathways downstream of
glycolysis. For example, Akt phosphorylates and activates ATP citrate lyase, an enzyme that
catalyzes the conversion of citrate to acetyl-CoA required for fatty acid synthesis (Berwick et al.,
2002). Furthermore, Akt activates the mammalian target of rapamycin complex 1 (mTORC1) by
phosphorylating and inhibiting its negative regulator tuberous sclerosis 2 (TSC2) (Manning et
al., 2002; Tee et al., 2002). mTOR functions as a key metabolic regulator, stimulating glycolytic
metabolism, ribogenesis, protein and lipid synthesis, and cell growth in response to growth

22
signals and nutrient availability (Laplante and Sabatini, 2012). In addition, mTOR activates
transcription factors, such as HIF1, which increases the capacity of cells to carry out glycolysis
(Hudson et al., 2002). Upstream and downstream modulators of the mTOR signaling pathway
will be discussed in a separate section.
1.3.3. Transcriptional regulators of anabolic metabolism
Several transcriptional regulators have been implicated in regulating cell metabolism. Due to
their fundamental roles in controlling cell proliferation, the metabolic functions of HIF, Myc and
p53 will be covered in this section.
1.3.3.1. HIF
Under hypoxic conditions, mammalian cells undergo a metabolic shift towards increasing
glucose consumption and redirecting glycolytic pyruvate to lactate (Greer et al., 2012). This
adjustment is mediated by the HIF1 and HIF2 complexes which are the major transcription
factors expressed in response to low oxygen conditions. The two exist as heterodimers of the
constitutively expressed HIF1β subunit and either HIF1α or HIF2α subunits that are stabilized
under hypoxic conditions (Bertout et al., 2008). Under normoxic conditions, the alpha subunits
of HIF are hydroxylated at conserved proline residues by HIF prolyl hydroxylases, resulting in
their recognition and subsequent ubiquitilation by the Von Hippel–Lindau (VHL) tumor
suppressor E3 ubiquitin ligase which tags them for rapid degradation by the proteasome
(Maxwell et al., 1999). HIF prolyl hydroxylases are inhibited in hypoxic environments, since
they utilize oxygen as a cosubstrate (Semenza, 2004). While HIF1α is ubiquitously expressed,
HIF2α is limited to specific tissues (Bertout et al., 2008). Although both transcription factors

23
activate overlapping sets of genes, most studies have focused on the role of HIF1 in regulating
metabolism.
When oxygen is limited, continuous mitochondrial oxidative phosphorylation might cause
mitochondrial redox stress. Under such conditions, HIF1 induces expression of genes supporting
anaerobic glucose metabolism, including glucose transporters and glycolytic enzymes to increase
the capacity of cells to carry out glycolysis at the expense of the TCA cycle. The HIF1-mediated
increased expression of lactate dehydrogenase and pyruvate dehydrogenase kinase 1 divert
pyruvate from entering the mitochondria to lactate production. This reduction in pyruvate flux
into the TCA cycle decreases oxidative phosphorylation and oxygen consumption, enabling ATP
production in rapidly dividing cells in an oxygen-independent mechanism (Greer et al., 2012).
In addition, HIF1 can be stabilized in proliferating cells under normoxic conditions. For
example, activated Akt, through mTOR signaling, increases HIF1α mRNA translation (Hudson
et al., 2002). Furthermore, in the presence of normal oxygen levels, the increased glucose uptake
in proliferating cells can also inhibit the hydroxylation and subsequent degradation of HIF1α.
This occurs through enhanced glucose-mediated production of ROS which act as strong
inhibitors of the HIF-targeting prolyl hydroxylases (Chandel et al., 2000). Mutations in the TCA
cycle enzymes succinate dehydrogenase and fumarate hydratase lead to the accumulation of their
substrates, succinate and fumarate, respectively, which can also inhibit prolyl hydroxylases
(Isaacs et al., 2005; Selak et al., 2005). Inhibition of prolyl hydroxylases by ROS, succinate or
fumarate represents a feedback mechanism decreasing the flow of glucose carbon into
mitochondria and reinforcing a glycolytic phenotype in tumors.

24
1.3.3.2. Myc
Like HIF1, Myc increases the expression of genes involved in glucose uptake and glycolytic
metabolism (Osthus et al., 2000). For example, Myc induction of lactate dehydrogenase diverts
pyruvate away from mitochondria, enhancing the glycolytic pathway (Osthus et al., 2000). It also
stimulates the expression of genes supporting anabolic utilization of glutamine, where Myc
directly induces the expression of glutamine transporters SLC5A1 and SLC7A1 (Gao et al.,
2009). In addition, Myc increases the levels of glutaminase 1, the first enzyme of glutaminolysis,
by inhibiting the expression of its negative regulators mir-23A and mir-23B (Gao et al., 2009).
Myc-transformed cells cannot survive in the absence of exogenous glutamine which functions as
a critical carbon source for anapleuretic reactions in these cells (Wise et al., 2008; Yuneva et al.,
2007). Myc also induces the expression of amino acid (serine hydroxymethyltransferase) and
fatty acid (fatty acid synthase) biosynthesis genes (O'Connell et al., 2003).
1.3.3.3. p53
The transcriptional factor and tumor suppressor p53 is a vital gatekeeper against cellular stresses.
It activates a myriad of cell defense pathways, including cell cycle arrest, DNA damage repair
and apoptosis (Bieging and Attardi, 2012). More recently, several lines of evidence suggest a
critical role for p53 in regulating metabolism (Vousden and Ryan, 2009). p53 inhibits glycolytic
metabolism by repressing the expression of the glucose transporters Glut1 and Glut4
(Schwartzenberg-Bar-Yoseph et al., 2004) and the glycolytic enzyme phosphoglycerate mutase
(Kondoh et al., 2005). In addition, p53 increases the levels of the TP-53-induced glycolysis and
apoptosis regulator gene (TIGER), a negative regulator of glycolytic metabolism (Bensaad et al.,
2006). Importantly, p53 mutated at three critical Lys sites (p533KR/3KR
) is deficient in p53-

25
dependent regulation of cell-cycle arrest, apoptosis and senescence, yet retains its tumor
suppressor activity. The p533KR/3KR
mutation continues to regulate a subset of metabolic genes,
notably suppressing the expression of Glut3 (Li et al., 2012).
p53 promotes oxidative phosphorylation by activating the expression of cytochrome oxidase c 2
(SCO2) which is required for the assembly of the cytochrome c complex in the electron transport
chain (Matoba et al., 2006). p53 can also alter metabolism in a transcription-independent
manner. It has been reported that p53 directly binds to and inactivates the enzyme glucose-6-
phosphate dehydrogenase, inhibiting glucose metabolism through the pentose phosphate
pathway. Indeed, p53-deficient cells have higher levels of R5P and NADPH compared to p53-
competent cells (Jiang et al., 2011). These observations suggest that p53 mutation or deletion
could act as a driving force supporting a glycolytic phenotype in cancer.
1.3.4. Metabolic enzymes as oncogenes
Not all cancer-associated mutations influencing cellular metabolism are connected to augmenting
anabolic demands of dividing cells (Gottlieb and Tomlinson, 2005). Oncogenic mutations could
provide a metabolic enzyme with a neomorphic activity to drive tumorigenesis.
An example is the driving mutation discovered in the cytosolic isocitrate dehydrogenase 1
(IDH1). Whole genome sequencing identified somatic mutations in IDH1 in a subset of gliomas
and acute myeloid leukemia (AML). These mutations are remarkably restricted to specific
arginine residues required for IDH binding to its substrate isocitrate (Mardis et al., 2009; Parsons

26
et al., 2008). All affected patients were heterozygous for the mutations, retaining a wild-type
IDH1 allele. The heterozygous nature of the mutation has been shown to dominantly inhibit
wild-type IDH1 in cells (Zhao et al., 2009). The IDH1 R132 mutation endows IDH1 with a new
reductive activity to convert α-ketoglutarate to 2-hydroxyglutarate (2-HG), a rare metabolite
found in trace amounts in cells under normal conditions (Dang et al., 2009; Ward et al., 2010).
Studies of mutations in IDH2, the mitochondrial homologue of IDH1, demonstrated that 2-HG is
a pathogenic “oncometabolite” rather than a byproduct of a loss-of-function mutation. Mutations
in R172 of IDH2, the analogous residue of R132 in IDH1, also resulted in elevated levels of 2-
HG in AML and glioma patients (Yan et al., 2009). However, not all AML samples with
elevated 2-HG have neomorphic mutations in either IDH1 R132 or IDH2 R172 (Ward et al.,
2010). A more comprehensive sequencing of these samples revealed an additional critical
mutation of IDH2, R140 (Ward et al., 2010), emphasizing that the main feature selected for by
this mutation is 2-HG production. While much work is needed to utilize 2-HG measurement in
diagnostic tests, recent data showed that 1H NMR can be applied for 2-HG detection of glioma in
vivo (Andronesi et al., 2012; Choi et al., 2012).
The observations that IDH mutations are selected for early during tumor progression (Watanabe
et al., 2009), and that 2-HG is not a mutagen (Mardis et al., 2009) suggested a specific role of 2-
HG in altering cancer metabolism. Several reports showed that 2-HG can inhibit the ten-eleven
translocation (TET) family of enzymes which oxidize 5-methylcytosine to 5-
hydroxymethylcytosine, a key intermediate in DNA demethylation (Figueroa et al., 2010; Turcan
et al., 2012; Xu et al., 2011). TET2 loss-of-function mutations and IDH1 or IDH2 neomorphic

27
mutations are mutually exclusive in AML (Figueroa et al., 2010). Furthermore, knocking down
TET2 recapitulated the effect of overexpressing IDH mutants in preventing hematopoietic cell
differentiation (Figueroa et al., 2010).
Inhibition of histone lysine demethylases is a potential mechanism by which 2-HG mediates its
oncogenic effect (Chowdhury et al., 2011). Expression of mutant IDH or treatment with cell-
permeable 2-HG repressed the expression of lineage-specific differentiation genes in 3T3-L cells
and blocked cellular differentiation. This correlated with a significant increase in repressive
histone methylation marks (Lu et al., 2012). The 2-HG-mediated alterations in histone and DNA
methylation are likely synergistic. The precise mechanisms connecting the effect of 2-HG on
DNA and histone methylation are still unknown. Further investigation is needed to clarify
whether epigenetic regulation is the only mechanism mediating the oncogenic effects of 2-HG.

28
1.4. mTOR: from signaling to metabolism
The mTOR pathway integrates signals for cell growth and energy metabolism. It enables
unicellular organisms such as yeast to sense nutrient availability and to support cell growth under
favorable environmental conditions. With emerging complexity in multicellullar organisms,
mTOR acquired additional roles such as regulation of immunity and neurogenesis. Hence,
dysregulation of mTOR signaling has been implicated in health conditions, including cancer,
Alzheimer’s and metabolic syndrome (Zoncu et al., 2011).
1.4.1. Molecular components of mTOR
mTOR was discovered in a yeast genetic screen for molecular targets of the immunosuppressant
rapamycin, yielding two genes, TOR1 and TOR2 that mediated the toxic effects of the drug
(Cafferkey et al., 1993; Kunz et al., 1993). Shortly afterwards, the mammalian homolog of TOR
(mTOR) was isolated as the direct target of rapamycin (Brown et al., 1994; Sabatini et al., 1994;
Sabers et al., 1995). mTOR is a Ser/Thr protein kinase that belongs to the PI3K-related kinases
(PIKK) family. It interacts with several distinct proteins to form complexes named mTOR
complex 1 (mTORC1) and 2 (mTORC2). The two complexes respond differently to rapamycin
and have different upstream regulators and downstream effectors (Zoncu et al., 2011). The
regulatory-associated protein of mTOR (Raptor) and the rapamycin-insensitive companion of
mTOR (Rictor) characterize mTORC1 and mTORC2, respectively. These two proteins function
as scaffolds for substrate binding and complex assembly (Zoncu et al., 2011).

29
Both mTOR complexes share 2 proteins: GβL (mLST8) (Jacinto et al., 2004; Kim et al., 2003)
and DEP domain-containing mTOR-interacting protein (Deptor) (Peterson et al., 2009) (Fig.
1.4). Unique components of mTORC1 include a negative regulator, 40 kDa proline-rich Akt
substrate (PRAS40) (Sancak et al., 2007), whereas mTORC2 contains stress-activated map-
kinase-interacting protein 1 (mSin1) (Frias et al., 2006; Jacinto et al., 2006; Yang et al., 2006)
and protein observed with rictor 1 and 2 (Protor 1 and 2) (Pearce et al., 2007). Structural and
biochemical studies suggest that mTORC1 functions as an obligate dimer where the dimeric
interfaces are formed by interactions between the mTOR and raptor subunits. Biochemical
analysis suggest that mTORC2 functions as oligomer, most likely as a TORC2-TORC2 dimer
(Wullschleger et al., 2005; Yip et al., 2010).
Rapamycin binds the prolyl isomerase FK506-binding protein (FKBP12) forming the rapamycin
-FKBP12 complex which binds to and inhibits mTOR (Brown et al., 1994; Chen et al., 1995;
Sabatini et al., 1994). It has been suggested that rapamycin might disrupt mTOR-Raptor
association, hindering mTOR from binding to its substrates (Kim et al., 2002; Yip et al., 2010).
However, the exact mechanism of mTORC1 inhibition by the FKBP12-rapamycin complex is
still unclear. Prolonged treatment with rapamycin can lead to a complete loss of intact mTORC2
in a subset of cell lines (Sarbassov et al., 2006). The effect of rapamycin on mTORC2 could be
attributed to rapamycin-FKBP12 mediated sequestration of the mTOR pool in the cell, thus
decreasing the availability of free mTOR for assembly into mTORC2.

30
Fig 1.4. mTOR signaling pathway.
Growth factors, such as insulin, stimulate PI3K to generate PIP3 to which PH domain-containing
Ser/Thr kinases, such as Akt and PDK1 bind. Activated by mTORC2 and PDK1, Akt
phosphorylates TSC1/2 on multiple sites and inhibits its GAP activity towards the GTPase Rheb.
GTP-loaded Rheb then activates mTORC1 which in turn phosphorylates several downstream targets
including 4E-BP1 to enhance protein synthesis, ULK1 to inhibit autophagy and S6K1 to enhance
ribogenesis and cell growth. Kinases other than Akt, such as Erk1/2 and RSK1, can also
phosphorylate TSC1/2 and inhibit its activity. Low energy (high AMP/ATP ratio) activates AMPK
which phosphorylates Raptor and TSC1/2, inhibiting mTORC1 activation. In addition to
phosphorylating Akt, mTORC2 also phosphorylates PKCα and SGK1. The number of phosphate
groups in the figure does not represent the actual number of phosphorylation sites on the proteins
indicated.

31
1.4.2. mTORC1
mTORC1 is the more extensively characterized of the two complexes. It can sense inputs mainly
from growth factors, amino acids, stress and energy status of the cell (Dibble and Manning,
2013). This allows mTORC1 to control several cellular functions including autophagy, protein
synthesis and cell survival (Dibble and Manning, 2013).
1.4.2.1. Upstream regulators of mTORC1
1) Growth factor signaling to mTORC1: One key component of growth factor signaling to
mTORC1 is the heterodimer of TSC1 and TSC2. The TSC1/2 dimer stably associates with TBC1
domain family member 7 (TBC1D7), the third core subunit of the complex, which is needed for
stabilization and full GTPase-activating protein (GAP) activity of TSC1/2 (Dibble et al., 2012).
The complex functions as a GAP for the Ras homolog enriched in brain (Rheb) GTPase (Garami
et al., 2003; Inoki et al., 2003; Tee et al., 2003; Zhang et al., 2003). The active GTP-loaded form
of Rheb directly interacts with and activates mTORC1 (Long et al., 2005). Indeed, Rheb is
essential for mTORC1 activation as its loss prevents mTOR activation by growth factors and
nutrients. Conversely, overexpression of Rheb constitutively activates mTORC1 even in the
absence of growth factors and nutrients (Saucedo et al., 2003; Stocker et al., 2003). By
converting Rheb into an inactive GDP-bound state, the TSC1/2 complex inhibits mTORC1
activation.

32
The TSC1/2 complex integrates signals from several mTORC1 upstream, including those arising
from growth factors, such as insulin. Akt, activated downstream of the insulin receptor, directly
phosphorylates and inactivates TSC1/2 leading to mTORC1 activation (Fig. 1.4) (Inoki et al.,
2002; Manning et al., 2002; Potter et al., 2002). Akt can also activate mTORC1 in a TSC1/2-
independent manner by phosphorylating the mTOR negative regulator PRAS40, causing its
dissociation from mTORC1 (Kovacina et al., 2003). Kinases other than Akt, such as Erk1/2 (Ma
et al., 2005) and ribosomal S6 kinase (RSK1) (Roux et al., 2004), can also phosphorylate TSC1/2
to inhibit it. Moreover, TSC1/2 can be phosphorylated and inactivated by glycogen synthase
kinase 3β (GSK3β) downstream of Wnt signaling causing mTORC1 activation (Castilho et al.,
2009; Inoki et al., 2006; Yang et al., 2006). The convergence of multiple growth signaling inputs
on mTORC1 allows it to act as a central signaling hub in several developmental stages. This
might explain the absolute requirement of mTORC1 signaling in early embryonic development
(Guertin et al., 2006).
2) Stress signaling to mTORC1: In addition to receiving inputs from growth factors, TSC1/2
also integrates signals from cell energy status associated with nutrient deprivation, low oxygen
and DNA damage (Dibble and Manning, 2013). mTORC1 indirectly senses low ATP levels
through the AMP-activated protein kinase (AMPK). When AMP/ATP ratio is high, under
conditions of nutrient deprivation, AMPK phosphorylates TSC2 (Corradetti et al., 2004; Dibble
and Manning, 2013; Inoki et al., 2006; Inoki et al., 2003). AMPK-mediated phosphorylation
activates TSC2 towards Rheb, inhibiting mTORC1. Under low energy stress conditions, AMPK
also phosphorylates the mTORC1 scaffold protein Raptor, inducing its binding to the regulatory
14-3-3 proteins which leads to inhibition of mTORC1 signaling (Gwinn et al., 2008).

33
In contrast to energy depletion, hypoxia can also promote TSC1/2 activation and, hence,
mTORC1 suppression in an AMPK-independent manner. Hypoxia-induced HIF1 stabilization
increases the expression of the gene regulated in development and DNA damage response 1
(REDD1), which promotes TSC1/2 activation through a mechanism that is yet to be elucidated
(Brugarolas et al., 2004; DeYoung et al., 2008; Reiling and Hafen, 2004). The HIF1-mediated
induction of REDD1 is controlled by signaling from the DNA damage repair kinase ataxia
telangiectasia mutated (ATM) (Cam et al., 2010). Like hypoxia, DNA damage inhibits mTORC1
activation. This occurs through p53-dependent induction of sestrin 1 and sestrin 2 which activate
AMPK (Budanov and Karin, 2008; Jones et al., 2005). DNA damage also induces the expression
of TSC2, PTEN and AMPK, suppressing the PI3K-mTORC1 pathway (Feng et al., 2007).
3) Nutrients signaling to mTORC1: Amino acids, particularly leucine and arginine, are
indispensable for mTORC1 signaling regardless of the upstream stimulating signal (Hara et al.,
1998; Wang et al., 1998). The most studied connection between amino acid stimulation and
mTORC1 is the Rag family of GTPases (Kim et al., 2008b; Sancak et al., 2008). Rags form
obligate heterodimers of either RagA/RagB with either RagC/D. The two units of the
heterodimer have opposite nucleotide loading states: when RagA/B is GDP-bound, RagC/D is
bound to GTP and vice versa. The yeast Gtr1 (RagA/B homolog) and Gtr2 (RagC/D homolog)
have been shown to functionally and genetically interact with mTORC1 (Urano et al., 2000).
Through currently uncharacterized mechanisms, amino acids cause the Rag heterodimer to
switch to the active form, where RagA/B becomes loaded with GTP and the RagC/D is bound to
GDP. This allows the Rags to interact with Raptor, promoting mTORC1 clustering at the surface
of lysosomes and late endosomes where the mTOR activators, Rag and Rheb GTPases, reside

34
(Sancak et al., 2008). Expression of constitutively GTP-bound RagA/B mutants renders
mTORC1 resistant to amino acid starvation (Kim et al., 2008b; Sancak et al., 2010). Inhibition of
glutaminolysis prevented GTP loading of RagB and subsequent lysosomal translocation and
mTORC1 activation (Duran et al., 2012). The mTORC1-Rag complex then anchors to a protein
complex called Ragulator, essential for Rag tethering to the lysosomal surface and amino acids-
mediated activation of mTORC1 (Sancak et al., 2010). In addition to its role as a scaffold
protein, Ragulator serves as a GEF, activating RagA/B following amino acid stimulation (Bar-
Peled et al., 2012).
After the Rag-Ragulator complex brings mTORC1 to the lysosomal surface, Rheb can bind to
and activate mTORC1 (Kim et al., 2008b; Sancak et al., 2008). RagA/B gets inactivated by a
tumor suppressor protein complex named GATOR that has a GAP activity (Bar-Peled et al.,
2013; Panchaud et al., 2013) while RagC/D are deactivated by the tumor suppressor folliculin1/2
which possesses GAP activity (Tsun et al., 2013). While amino acids can activate mTORC1
independently of TSC1/2 complex (Smith et al., 2005a), new reports show that TSC2 plays an
essential role in deactivation of mTORC1 following amino acid withdrawal (Demetriades et al.,
2014; Menon et al., 2014).
In addition to the Rag GTPases, several pathways have been linked to amino acid signaling
upstream of mTORC1, including mitogen-activated protein kinase kinase kinase kinase 3
(MAP4K3) (Findlay et al., 2007; Yan et al., 2010), the PI3K catalytic subunit type 3 (VPS34)
(Gulati et al., 2008; Nobukuni et al., 2005), the autophagy protein p62 (Duran et al., 2011),
leucyl-tRNA synthetase (Han et al., 2012) and inositol polyphosphate multikinase (IMP) (Kim et

35
al., 2011). The mechanisms by which these proteins signal to and interact with the Rag-
Ragulator complex are not fully characterized.
Amino acids are not the only nutrients that can activate mTORC1. Genetic studies suggest a role
for Rags in mTORC1 sensing of glucose. Knock-in embryos expressing constitutively GTP-
bound RagA (RagAGTP
) are resistant to amino acid or glucose withdrawal effects on mTORC1
signaling (Efeyan et al., 2013), and glucose, like amino acids, controls mTORC1 recruitment to
the lysosomal surface (Efeyan et al., 2013). In addition, phosphatidic acid has been reported to
activate mTORC1 (Fang et al., 2001). Indeed, over expressing the phosphatidic acid-producing
enzymes phospholipase D1 and D2 activates mTORC1 (Fang et al., 2003).
1.4.2.2. Downstream effectors of mTORC1
mTORC1 phosphorylates a myriad of downstream targets (Hsu et al., 2011; Yu et al., 2011). For
the sake of conciseness, the focus will be on the most characterized effectors of mTORC1 and
how they mediate their cellular functions.
mTORC1 enhances protein synthesis through phosphorylating S6K1 and the eukaryotic
translation initiation factor (eIF4E)-binding protein (4E-BP1). Both proteins associate with the
control of mRNA translation initiation and regulation of the protein synthesis rate (Ma and
Blenis, 2009). Following its phosphorylation by mTORC1, 4E-BP1 dissociates from eIF4E,
enabling it to form the eIF4E initiation complex necessary for initiation of cap-dependent
translation (Gingras et al., 1999; Haghighat et al., 1995; Hara et al., 1997). On the other hand,

36
S6K phosphorylates ribosomal protein S6 (component of the 40S ribosomal subunit) at five
residues (Ferrari et al., 1991). Knock-in mice with the five Ser targets in S6 mutated to Ala are
defective for ribosomal biogenesis and mRNA expression (Ruvinsky et al., 2005). In addition,
phosphorylated S6K1 enhances mRNA translation initiation and progression by associating with
several downstream targets including eukaryotic elongation factor 2 kinase (eEF2K) (Wang et
al., 2001), S6K1 Aly/REF-like target (SKAR) (Ma et al., 2008), eIF4B (Wilson et al., 2000),
among others. In addition, S6K1 upregulates the transcriptional activity of RNA polymerase I
and, hence, increases the expression of ribosomal RNA (rRNA) (Mayer et al., 2004).
In addition to protein synthesis, mTORC1 regulates autophagy, a catabolic cellular recycling
process that involves cell degradation of dysfunctional cellular components through the actions
of lysosomes (Rabinowitz and White, 2010). In mammalian cells, mTORC1 phosphorylates and
inhibits ULK1/Atg13/FIP200 kinase complex required for autophagy initiation (Ganley et al.,
2009; Hosokawa et al., 2009; Jung et al., 2009). In addition, mTORC1 directly phosphorylates
and inhibits the transcription factor EB (TFEB), a key regulator of lysosomal biogenesis (Pena-
Llopis et al., 2011; Settembre et al., 2012). By inhibiting autophagy, mTORC1 ensures the
utilization of available cellular energy resources towards cell growth.
1.4.3. mTORC2
mTORC2 functions as an important regulator of the actin cytoskeleton through activating the
Rac1 and Rho GTPases, and PKCα (Jacinto et al., 2004; Laplante and Sabatini, 2012; Sarbassov

37
et al., 2004). In addition, mTORC2 phosphorylates Akt at Ser473 (Sarbassov et al., 2005) as well
as Thr450 (Oh et al., 2010), leading to full activation of Akt.
1.4.3.1. Upstream regulators of mTORC2
Compared to mTORC1, little is known about upstream activators of mTORC2. The complex
responds to stimulation by growth factors such as insulin, but not to amino acid treatment
(Nicklin et al., 2009; Nobukuni et al., 2005). Insulin-dependent activation involves a physical
interaction of mTORC2 with ribosomes in a PI3K-dependent fashion (Zinzalla et al., 2011).
TSC1/2 might also be involved in proper activation of mTORC2 and its downstream target Akt
(Huang et al., 2008).
1.4.3.2. Downstream effectors of mTORC2
The mTORC2 complex phosphorylates and activates several members of the AGC family
kinases including Akt (Sarbassov et al., 2005), PKC (Ikenoue et al., 2008), and serum- and
glucocorticoid-induced protein kinase 1 (SGK1) (Garcia-Martinez and Alessi, 2008) in a motif
required for maximum kinase activation. Knocking down mTORC2 inhibits the phosphorylation
of some Akt targets, such as FoxO1/3a, but not other targets including TSC1/2 (Guertin et al.,
2006; Jacinto et al., 2006). Mechanisms regulating mTORC2-Akt substrate specificity are
currently unknown. Unlike Akt, SGK1 activity is completely abolished by mTORC2 loss, and
phosphorylation of NDRG1/2 (N-myc downstream regulated gene 1 and 2), physiological
substrates of SGK1, is dimished in Rictor- or Sin1-deficient fibroblasts (Garcia-Martinez and

38
Alessi, 2008). Finally, mTORC2-mediated phosphorylation of PKC plays a role in regulating
actin cytoskeleton and cell morphology (Jacinto et al., 2004; Sarbassov et al., 2004).
1.4.4. mTOR and metabolism
As mentioned earlier, mTOR integrates nutrient availability and signals from growth factors,
playing a central role in regulating metabolism in cells (Shimobayashi and Hall, 2014). In
animals, the transition between fasting and fed states changes the circulating amounts of
nutrients and growth factors. In turn, these changes affect how tissues balance catabolic and
anabolic processes. In fed states, high levels of nutrients and growth factors increase lipogenesis
in adipose tissues, glycogenesis in liver and muscle, and inhibit gluconeogenesis in liver.
1.4.4.1. Glucose metabolism
Transcriptomic analysis of Tsc1 and Tsc2 KO MEFs revealed that mTORC1 increases
transcription of genes involved in glycolysis, the pentose phosphate pathway and lipogenesis
(Duvel et al., 2010). Metabolomic analysis of the same cells mirrored changes observed at the
transcriptional levels. Chronic activation of mTORC1 in Tsc2 KO MEFs enhanced glucose
uptake, lactate secretion, glycolytic metabolism, and biosynthesis of the pentose phosphate
pathway and lipid intermediates. Rapamycin treatment of Tsc2 KO MEFs identified nine
metabolites positively regulated by mTORC1 (Ben-Sahra et al., 2013). Consistent with previous
observations (Duvel et al., 2010), most of these metabolites were part of the pentose phosphate
pathway. mTORC2 has also been shown to regulate glucose metabolism. Liver-specific Rictor
knockout mice displayed reduced glucokinase activity in the liver, leading to constitutive

39
gluconeogenesis and impaired glycolysis, and expression of constitutively active Akt2 in
mTORC2-deficient hepatocytes restored both glucose flux and lipogenesis (Hagiwara et al.,
2012). This suggests that mTORC2 regulates hepatic glucose via insulin-induced Akt signaling.
In addition, mTORC2-Akt signaling is required for Wnt3A-mediated reprogramming of glucose
metabolism towards glycolysis (Esen et al., 2013). In summary, these observations confirm that
both mTORC1 and mTORC2 are involved in regulating glucose metabolism.
1.4.4.2. Lipid synthesis
mTORC1 promotes lipogenesis through activating the sterol regulatory element-binding protein
1/2 (SREBP1/2) transcription factors which control expression of several fatty acid and
cholesterol synthesis genes. In response to insulin or sterol deficiency, SREBPs undergo
proteolytic cleavage and the active form translocates to the nucleus to activate transcription.
mTORC1 induces SREBP1/2, activating the expression of lipogenesis genes in both S6K-
dependent and independent mechanisms (Duvel et al., 2010; Porstmann et al., 2008). mTORC2
also plays a role in lipogenesis. SREBP1c activity is reduced in the liver-specific Rictor
knockout mice and is restored following the expression of constitutively active Akt2 (Hagiwara
et al., 2012). Therefore, mTORC1, mTORC2 and Akt are required for activation of SREBP and
lipogenesis.
1.4.4.3. Protein synthesis
As discussed in a previous section, mTORC1 plays an integral role in ribosomal biogenesis and
mRNA translation (Ma and Blenis, 2009). In contract to mTORC1, the role of mTORC2 in

40
protein synthesis is less-defined. Recently, it has been shown that mTORC2 associates with
actively translating ribosomes to phosphorylate its substrates. For example, mTORC2 co-
translationally phosphorylates Akt at Thr450 which prevents ubiquitilation and increases the
stability of Akt (Oh et al., 2010). mTORC2 also co-translationaly phosphorylates IGF2 mRNA-
binding protein 1 (IMP1), strongly enhancing IMP1 binding to the IGF2 mRNA translational
initiation region. This increases IGF2 translation, stimulating cell growth (Dai et al., 2013). The
molecular mechanism of mTORC2-ribosome association is still unclear.
1.4.4.4. Nucleotide metabolism
mTORC1 stimulates the expression of the pentose phosphate pathway which provides the ribose
moiety for nucleotide synthesis (Duvel et al., 2010). Metabolomic and phosphoproteomic
analyses revealed that mTORC1 stimulates de novo pyrimidine biosynthesis in S6K1-dependent
mechanism (Ben-Sahra et al., 2013; Robitaille et al., 2013). This occurs through mTORC1-
mediated phosphorylation and activation of the carbamoyl-phosphate synthetase 2, aspartate
transcarbamylase, and dihydroorotas (CAD) protein, which catalyzes the first steps in the
synthesis of the pyrimidine ring. The phosphorylation of CAD at Ser1859 by mTOR stimulates
its oligomerization and activation (Robitaille et al., 2013). Whether mTORC1 also controls
purine synthesis and whether mTORC2 plays a role in purine and pyrimidine metabolism need
further investigation.

41
1.5. Rationale and objectives of the study
A common mechanism through which activated growth factor receptors control signaling
specificity is by recruiting adaptor proteins. In this thesis, I address the biological role of the
p66Shc adaptor isoform in connecting growth factor signaling to metabolim.
p52Shc is a well-characterized adaptor protein that acts downstream of receptor tyrosine kinases
to amplify signaling to the Ras-Erk and PI3K-Akt pathways. However, the Shc1 locus also
encodes a p66Shc isoform, identical to p52Shc with the exception of an N-terminal extension.
This isoform is only found in vertebrates and it regulates insulin sensitivity. In mammals, lack of
p66Shc confers resistance to oxidative stress-induced vascular damage, hyperglycemia-induced
endothelial dysfunction, diabetes and obesity. These observations raise the possibility that
p66Shc provides negative feedback regulation to insulin signaling, and here I test the hypothesis
that, in this capacity, p66Shc also suppresses glucose metabolism.
The objectives of this thesis are to:
1) Analyze the effect of p66Shc on cellular metabolism using a targeted mass spectrometry-
based metabolomics approach.
2) Determine metabolic pathways affected by p66Shc expression.
3) Explore potential molecular mechanisms by which p66Shc affects energy metabolism.

42
My data suggest that loss of p66Shc redirects carbon metabolism in favor of glycolysis in a
manner reminiscent of the Warburg effect. Specifically, silencing of p66Shc improves glucose
uptake, increases carbon flux towards lactate production, and redirects glucose carbon towards
anabolic pathways, including fatty acid biosynthesis, the pentose phosphate and hexosamine
pathways, resulting in increased cell size. Mechanistically, the heightened glycolytic metabolism
in p66Shc-deficient cells is partly rescued upon mTOR inhibition by rapamycin. These results
indicate that the Shc1 locus not only regulates mitogenic signaling, but also modulates anabolic
metabolism in an mTOR-dependent manner through the p66Shc isoform. The approach used in
this thesis could be of general utility in dissecting the roles of adaptor proteins regulating
metabolism in normal and disease states.

43
Chapter 2. Materials and Methods

44
2.1. Cell culture and treatments
HeLa cells and p66Shc knockout MEFs were cultured in Dulbecco’s Modified Eagle’s Medium
(Gibco) supplemented with 10% fetal bovine serum (HyClone), 4mM L-glutamine and 1mM
sodium pyruvate. Cells were maintained in a humidified atmosphere of 5% CO2 and 95% air at
37°C and media were changed every 2 days. Plastic tissue culture plates were supplied by Fisher.
For growth factor treatment, cells were serum-starved for 4 h and treated with a final
concentration of 100 ng/ml of EGF, IGF (Peprotech) or insulin (Novo Nordisk) or 10% serum
for 10 min. For amino acid stimulation, almost confluent cell cultures in 6-well plates were
rinsed once with amino acid-free RPMI (US Biological), incubated in amino acid-free RPMI for
50 min and stimulated with 10X amino acid mixture for 10 min before lysis. For rapamycin
treatment, rapamycin (Calbiochem) was dissolved in ethanol and cell were treated with a final
concentration of 20 nM for the times indicated in the figures. For Akt inhibition, cells were
treated with Akt Inhibitor IV (EMD Millipore) at a final concentration of 10 nM for the times
indicated in the experiments.
2.2. Freezing and thawing of cells
Cryopreservation was done by harvesting cells using trypsin-EDTA (Gibco-BRL) treatment
followed by centrifugation at 1200 rpm for 5 min. Cells were then resuspended in a medium
containing 10% FBS and 5-10% sterile dimethylsulfoxide (Sigma) to yield approximately l-2 X
l06 cells/ml. 1 ml aliquots of cell suspension were transferred to cryovials (Corning) and vials
were placed at -80°C overnight. Frozen cells were then placed in liquid nitrogen for long-term
storage. To thaw cells, vials of frozen cells were removed from liquid nitrogen and placed in a
37°C water bath for 2 min. The thawed cell suspension was quickly transferred to 15 ml tubes
(BD Falcon) containing fresh culture media supplemented with 10% FBS, centrifuged at 1200

45
rpm for 5 min, resuspended in 10 ml of culture media/10% FBS, transferred to 10 cm plates
(Corning) then incubated at 37°C in an atmosphere of 5% CO2.
2.3. Cell culture
Cells reaching confluence were incubated in a trypsin-EDTA solution at 37°C in a 5% CO2
incubator for 2-5 min until they lose anchorage from the tissue culture plates. Cells were
harvested by triturating with an appropriate volume of DMEM containing 10% FBS. Cells were
seeded onto fresh tissue culture dishes. Uniform spreading of cells was achieved by gently
shaking plates containing medium and cells in cross-shape direction.
2.4. Plasmid preparation and DNA constructs
Murine p66Shc cDNA (NCBI refseq number NP_001106802) was cloned in a gateway-
compatible pMX retroviral puromycin-resistant destination vector according to the
manufacture’s recommendation (Invitrogen). For p66Shc knockdown in HeLa cells, the
following hairpin sequence was cloned into a pMSCV retroviral puromycin-resistant vector
(with 3' LTR removed): sense 5'-cgg aat gag tct ctg tca tcg ct (tt)-3' and anti-sense 5'-ag cga tga
cag aga ctc att ccg (tt)-3'. Small scale and large scale plasmid preparations were done using the
Invitrogen plasmid preparation kits. These plasmid purification protocols are based on a
modified alkaline lysis procedure, followed by binding of plasmid DNA to an anion-exchange
resin under appropriate low-salt and pH conditions. RNA, proteins, dyes and low molecular
weight impurities were removed by a medium salt wash. Plasmid DNA was then eluted in a
high-salt buffer and then concentrated and desalted by isopropanol precipitation.

46
2.5. Cell transfection
Cells were seeded onto tissue culture plates at 40-50% confluence. 18-24 h later, when cells
reached 80-90% confluence, cells were transiently transfected using Lipofectamine 2000
transfection reagent (Invitrogen) or Fugene 6 (Roche). For Lipofectamine transfections, cells
were washed with PBS and media was replaced with serum-free medium (Opti-MEM media).
Plasmid DNA and Lipofectamine reagent were then mixed separately in antibiotic- and serum-
free medium for 5 min at room temperature then mixed together for 20 min with agitation at
room temperature. The ratio of µg of DNA to µL of Lipofectamine was 1:2. After 20 min of
incubation, the DNA: Lipofectamine complexes were added drop-wise to cells. Growth medium
was then replaced after incubation for 4-6 h at 37°C with fresh media containing 10% FBS. For
Fugene 6 transfections, media were replaced with fresh antibiotic free FBS-containing media.
Fugene 6 was added to antibiotic and serum-free media and incubated for 5 min. DNA was
added to the Fugene 6-media mixture and incubated for 20 min at room temperature. The ratio of
DNA: Fugene 6 used was 1:2. The mixture was added drop-wise to the plate of cells containing
media without antibiotics and distributed evenly by gently shaking the plate forth and back
several times. The cells were incubated at 37°C in a 5% CO2 incubator and harvested at the time
points indicated in each experiment. Amounts of DNA and Lipofectamine used were according
to the manufacture’s recommendation.
2.6. Retroviral production and cell infection
Vectors were packaged as recombinant retrovirus and pseudotyped – where the enveloped virus
particle gets assembled with a foreign viral glycoprotein - with VSV-G protein to enhance
infectivity. Either control shGFP or shp66Shc was co-transfected with pCMV-VSV-G into the
retroviral packaging cell line Platinum E (modified HEK293 cells) (Morita et al., 2000). Viral

47
supernatants from 10 cm plates were collected at 48 h posttransfection and filtered using 0.45
µm filters. Cells were infected with viral supernatant where media were supplemented with 10
μg/ml of hexadimethrine bromide (Sigma-Aldrich). 48 h post-infection, infected cells were
treated with puromycin (1 μg/ml) to generate cell lines stably expressing the constructs of
interest.
2.7. Metabolite extraction
The relative levels of metabolites were determined in HeLa cells and MEF using the protocol
indicated in (Abdel Rahman et al., 2013). Cells were seeded in 6-well plate in 6 replicates. After
24 h, the media were removed and the cells were washed 2X with warm PBS, then were placed
on dry ice. The metabolites were immediately extracted by adding 1 ml of dry ice-cold extraction
solution (40% acetonitrile, 40% methanol, and 20% water) containing internal standards
(500µg/ml and 300µg/ml of D7-Glucose and 13C915N-Tyrosine, respectively). Cells were
scraped and collected in 1.5 ml eppendorff tubes, and shaken for 1 h at 4°C/1400 rpm in a
Thermomixer (Eppendorf). Samples were centrifuged at 14000 rpm, for 10 min at 4°C, and
supernatants were transferred into fresh tubes to be evaporated to dryness in a CentreVap
concentrator at 40°C (Labconco). The dry extract samples were stored at -80°C until LC-MS
analysis. The dry metabolite extracts were reconstituted in 100 µL of water. Samples were
injected twice through the HPLC (Dionex Corporation) in gradient reversed phase column
Inertsil ODS-3, 4.6 mm internal diameter, 150 mm length, and 3 µM particle size for positive
and negative mode analysis. Automated washing procedure was used between samples. Eluted
metabolites were analyzed in MRM mode on electrospray ionization (ESI) triple-quadrupole
mass spectrometer (ABSciex4000Qtrap). Signals were normalized to both internal standard and
cell number. Metaboanalyst was used to analyze the data (Xia and Wishart, 2011). The LC-

48
MS/MS system does not resolve hexose and hexosamine isomers including glucose/galactose
and GlcNAc/GalNAc. To monitor trends in metabolic pathways, we referred to these isomers in
their glucose (Glc) forms.
2.8. Isotope labeling and kinetic profiling
Cells were seeded a day before the experiment in 6-well plates. Media of cell cultures in 6-well
dishes were replaced with unlabeled fresh DMEM containing 5% dialyzed FBS 2 h before
exposure to labeled glucose to allow equilibration to the new conditions. The media was then
replaced with DMEM/5% dialyzed FBS containing 25 mM [1,2-13
C2]-labeled glucose or 15
N2-
labeled glutamine (Cambridge Isotope Laboratories)for the times indicated in the figures
legends. Metabolites were immediately extracted with dry ice-cold extraction solution indicated
previously and analyzed using different MRM transitions that were developed for 1,2-13
C2-
glucose as detailed in Appendix 6.2.
2.9. [3H]-2-deoxy-D-glucose uptake assay
HeLa cells and MEF grown in 6-well plates were rinsed with HEPES-buffered saline [140 mM
NaCl, 20 mM HEPES, 5 mM KCl, 2.5 mM MgSO4, 1 mM CaCl2 (pH 7.4)]. [3H]-2-deoxy-D-
glucose (2-DG) uptake was performed for 3, 5, and 10 min in HEPES-buffered saline containing
2 mM unlabeled 2-DG and 0.2 μCi/ml [3H] 2-DG at room temperature. The reaction was
terminated by washing three times in ice-cold 0.9% NaCl (w/v). To quantify the radioactivity
incorporated, cells were lysed with 0.05 N NaOH and lysates were counted with scintillation
fluid in a β-counter. Nonspecific uptake was determined in the presence of cytochalasin B (10
μM) during the assay. The results are expressed as pmole 2-DG transported per min/mg of

49
protein. This experiment was done by Huogen Lu in Dr. George Fantus’ lab (Lunenfeld Research
Institute, University of Toronto).
2.10. Oxygen consumption rate measurement
Cells were seeded at 50,000 cells/well in XF-24 cell culture plates. The day after cell seeding,
cell culture media was replaced with RPMI and oxygen consumption was measured using an XF-
24 Flux Analyzer as described in (Birsoy et al., 2013). This experiment was done by Kıvanç
Birsoy in Dr. David Sabatini’s lab (Whitehead Institute, MIT).
2.11. Cell size determination
Cells were grown to 100% confluence in 10 cm plates, washed with 1X PBS, treated with 1 ml
of trypsin, and diluted 1:20 with counting solution (Isoton II Diluent, Beckman Coulter). Cell
diameters and volumes were determined using a particle size counter (Coulter Z2, Beckman
Coulter) with Coulter Z2 AccuComp software. For cell size determination using flow cytometry,
cells were washed with 1X PBS, treated with 1 ml of trypsin, suspended in 9 ml of DMEM/10%
FBS media, and centrifuged at 1200 rpm for 5 min. Media was discarded and cells were
resuspended in a mixture of 1 ml of 2% FBS/ 5 mM EDTA in PBS, filtered through the mesh cap
of BD Falcon polypropylene tubes. Cells were analyzed by flow cytometry for differences in
forward scatter (FSC correlates with cell size).
2.12. Cell lysis and immunoblotting
Cells growing in 6-well plates were rinsed once with 1X cold PBS and lysed on ice with 300 μl
of ice-cold Lysis Buffer (40 mM HEPES pH 7.5, 120 mM NaCl, 1 mM EDTA, 10 mM
pyrophosphate, 10 mM glycerophosphate, 10 mM NaF, 1 mM orthovanadate, and EDTA-free
protease inhibitors (Roche)) containing 0.5% NP-40. Cell suspensions were then transferred into

50
1.5 ml eppendorf tubes, centrifuged at 13,000 rpm for 10. 250 μl of cell supernatent was
transferred to a new tube and protein content was measured used Bradford reagent according to
the manufacturer’s instructions (Pierce). After normalization of protein concentration, 250 µL of
2X SDS sample buffer (2% SDS, 20% glycerol, 20 mM TrisCl pH 8.0, 4% β-mercaptoethanol,
2mM EDTA, and 0.02% bromophenol blue) was added and cell lysate was resolved by 8%-16%
SDS-PAGE gels.
2.13. Western blotting
For western blots, denatured whole cell lysates in sample buffer were electrophoresed at 100 V
for 2 h (8%-16% SDS-PAGE, Bio-Rad apparatus) and transferred to polyvinylidene difluoride
(PVDF) membranes for 2 h at 50 mAmp/gel using semi-dry transfer apparatus (Bio-Rad).
Membranes were blocked in 5% w/v milk in TBS buffer/0.1% tween (TBST) or 5% w/v BSA in
TBST for 1 h followed by overnight incubation with primary antibody in the blocking buffer.
Antibodies used for western blots in the course of this study include phospho-T389 S6K1 (CST,
#9234), phosho-T308 Akt (CST, #2965), phospho-S473 Akt (CST, #4058), phospho-T346
NDRG1 (CST, #3217), Akt (CST, #9272), S6K1 (CST, #2708) from Cell Signaling Technology
(1:1000 dilution); mouse (BD, #610878) and rabbit (BD, # 610082) Shc1 antibodies from BD
Biosciences (1:1000 dilution); FLAG M2 (#F3165, 1:1000 dilution) and tubulin (#T6793,
1:10,000 dilution) antibodies from Sigma-Aldrich. Horseradish peroxidase (HRP)-conjugated
secondary antibodies were used for western blotting (Santa Cruz Biotechnology, 1:2000 for goat
α-rabbit and 1:5000 for goat α-mouse) and blots were developed using chemiluminescent
substrate (Thermo Scientific).

51
2.14. Mass spectrometry analysis of the p66Shc protein-interactions
The p66Shc protein interaction network was determined following a protocol described in
(Zheng et al., 2013). Briefly, p66+ MEFs (expressing 3XFLAG-p66Shc) were washed with ice-
cold PBS and lysed in NP40 lysis buffer (50 mM HEPES pH 8, 150 mM NaCl, 1 mM EGTA,
0.5% NP40, 100 mM NaF, 2.5 mM MgCl2, 10 mM Na4P2O7, 1 mM DTT, 10% glycerol)
supplemented with protease and phosphatase inhibitors (50 mM β-glycerolphosphate, 10 µg/ml
aprotinin, 10 µg/ml leupeptin, 1 mM Na3VO4, 100 nM calyculin A, 1 mM PMSF
(phenylmethylsulphonyl fluoride)). The total cell lysates were centrifuged at 10,000 rpm for 15
min to pellet the nuclei and insoluble material. Nuclear-free lysates were pre-cleared by 1 h
incubation with protein A sepharose and normalized for total protein concentration using the
Bio-Rad protein assay. 3xFLAG-p66Shc was immunoprecipitated by incubating lysates with 5 µl
(bed volume) anti-Flag M2 antibody-conjugated agarose for 4 h at 4 °C. Beads were washed 3X
with lysis buffer, then 2X with 50 mM ammonium bicarbonate. After aspiration of washing
buffers, proteins on beads were treated with 0.5 µg trypsin (Promega) overnight at 37ºC. Beads
were centrifuged at 10,000 rpm for 2 min and trypsinized peptides were transferred to new 0.5
ml Eppendorf tubes and dried in a CentriVap at 40°C for 3 h (Labconco). Dried peptides were
kept at -80 ºC until LC-MS analysis. Dried tryptic samples were reconstituted with 3% formic
acid. Samples were loaded onto a 75 mm inner diameter (ID)/360 mm outer diameter (OD)
pulled tip packed with 3 mm ReproSil C18 and analysed on an TripleTOF 5600 mass
spectrometer (AB SCIEX) coupled to an Eksigent nano LC Ultra 1D plus pump with a flow rate
of 200 nl/min and a gradient of 2% to 35% acetonitrile over 90 min. A cycle time of 1.3 sec was
employed using a survey TOF scan of 250 msec at 30,000 resolution followed by selection of the
top 20 most intense peptides for MS/MS for 50 msec each with high sensitivity (at 18,000

52
resolution). Only peptides with a charge state above 11 were selected for MS/MS and dynamic
exclusion was set to 15 sec for all ions within 20 ppm.
2.15. RNA-seq
cDNA library was prepared using Illumina TruSeq RNA Sample Prep Kit v2 (Cat#RS-122-
2001). Briefly, 1 µg of high quality total RNA was isolated from p66Shc KO and p66+ MEFs to
generate the cDNA library according to the Illunima kit protocol. The generated barcoded cDNA
library has an average fragment size of 350-400 bp. This bar-coded library is quality checked
with Agilent Bioanalyzer and quantified with qPCR using KAPA SYBR FAST Universal 2x
qPCR Master Mix (Kapa Biosystem, Cat#KK4602). The quality checked libraries are then
loaded on a flowcell for cluster generation using Illumina c-Bot and TruSeq PE Cluster Kit v3
(Cat#: PE-401-3001). Sequencing was done on HiSeq2000 with TruSeq SBS Kit v3 (pair-ended
200 cycles, Cat#: FC401-3001). The real-time base call (.bcl) files are converted to fastq files
using CASAVA 1.8.2 (on CentOS 6.0 data storage and computation linux servers).

53
Chapter 3. p66Shc Inhibits Anabolic Metabolism
A version of this chapter appeared in the following article:
The Adaptor Protein p66Shc Inhibits mTOR-Dependent Anabolic Metabolism.
Science Signaling, 7, ra17 (2014).
Mohamed A. Soliman, Anas M. Abdel Rahman, Dudley W. Lamming, Kivanç Birsoy, Judy
Pawling, Maria E. Frigolet, Huogen Lu, I. George Fantus, Adrian Pasculescu, Yong Zheng,
David M. Sabatini, James W. Dennis, and Tony Pawson.
Experiments in Fig. 3.9 were done by Kivanç Birsoy
Experiments in Fig. 3.14 and 3.16 were done by Huogen Lu

54
3.1. Background
There are three isoforms of Shc1 with molecular masses of 46, 52, and 66 kDa. Shc1 adaptor
proteins mediate signal transduction, linking multiple tyrosine kinase growth factor receptors to
activation of the Ras-MAPK and PI3K-Akt pathway (Ravichandran, 2001). Like p52/p46shc,
p66Shc is tyrosine-phosphorylated following EGF stimulation, binds to activated EGFRs and
forms stable complexes with the adaptor protein Grb2 (Migliaccio et al., 1997). However, unlike
p52/46Shc, p66Shc is unable to transform mouse fibroblasts in vitro, and does not increase
MAPK activation following EGF stimulation (Migliaccio et al., 1997). In addition, it competes
with p52Shc for Grb2 binding, inhibiting signaling downstream of IGF-1 receptor in vascular
smooth muscle cells (Xi et al., 2008). p66Shc has a unique role amongst the other Shc1 isoforms
in promoting oxidative stress and pro-apoptotic signaling. p66Shc-deficient MEFs shows
enhanced resistance to apoptosis in response to hydrogen peroxide treatment (Migliaccio et al.,
1999). Knockout studies suggest a pivotal role for p66Shc in regulating insulin signaling and
glucose metabolism. Deletion of p66Shc in mice improves glucose tolerance and insulin
sensitivity at organism and tissue levels (Tomilov et al., 2011). Suggestive of an inhibitory role
in insulin signaling and glucose metabolism, deletion of p66Shc caused a marked increase in
basal glucose transport in skeletal muscle cell lines (Natalicchio et al., 2009). These observations
led me to investigate the role of p66Shc as a repressor of glucose metabolism in particular and
anabolic metabolism in general. To test this possibility, I generated HeLa cell lines stably
expressing shRNA against control or p66Shc and examined the change in glycolytic metabolism
in cells that are competent or deficient in p66Shc expression. To test whether p66Shc is
sufficient to alter glycolytic metabolism, I did “add-back” experiments where I performed
metabolomic analysis of p66Shc KO MEFs stably expressing GFP or 3xFLAG-p66Shc.

55
3.2. Loss of p66Shc enhances glycolytic metabolism
To elucidate the function of p66Shc in cellular metabolism, I performed a targeted metabolomic
analysis using liquid chromatography-tandem mass spectrometry (LC-MS/MS) in multiple
reaction monitoring (MRM) mode. Approximately 250 metabolites were measured in positive or
negative mode runs (Appendix 6.1). Metabolite spectral patterns were validated using standard
mixture of metabolites. To assess the role of p66Shc in cancer cell metabolism, HeLa cells were
stably transfected with either a short hairpin shRNA that specifically targets the isoform p66Shc
or a control hairpin (Fig. 3.1) (Kisielow et al., 2002). The profiles of metabolites extracted from
the two cell types were analyzed. Loss of p66Shc resulted in increased abundance of
intermediates of glucose metabolism (Fig. 3.1). Specifically, p66Shc deficiency was
accompanied by significant increases in glucose-6-phosphate (G6P), and downstream glycolysis
intermediates including fructose-1,6-bisphosphate (F1,6BP), phosphoenolpyruvate (PEP), and
pyruvate (Fig. 3.2). p66Shc-deficient HeLa cells produced more lactate than control cells,
consistent with the Warburg effect. The metabolic shift also redirects glucose-derived citrate
from the TCA cycle into lipid synthesis to generate biomass (Hatzivassiliou et al., 2005).
Consistent with this effect, p66Shc-deficient cells had higher citrate concentrations (Fig. 3.2).
These data suggest that depletion of p66Shc is sufficient to enhance anabolic metabolism in
HeLa cells.
3.3. Loss of p66Shc promotes glucose metabolism through the pentose phosphate and
hexosamine biosynthesis pathways
In addition to the role of these molecules in glycolysis, G6P and F6P are precursors of the
hexosamine biosynthesis and the pentose phosphate pathways, which are essential anabolic

56
pathways in proliferating cells. The hexosamine biosynthesis pathway provides UDP-GlcNAc as
a substrate for O-GlcNAcylation of cytosolic proteins, and N-linked and O-linked glycosylation
of proteins produced in the secretory pathway (Dennis et al., 2009). The pentose phosphate
pathway provides ribose for nucleic acid synthesis and NADPH to maintain the reductive
environment of the cell. In HeLa cells lacking p66Shc, a ~ 4-fold increase in N-
acetylglucosamine-6-phosphate (GlcNAcP), and a ~ 2-fold increase in UDP-GlcNAc abundance,
the major products of the hexosamine biosynthesis pathway, was observed (Fig. 3.3A). A 2-3
fold increase in UDP-GlcNAc enhances N-glycosylation of growth factor receptors, thereby
promoting positive feedback to signaling (Lau et al., 2007; Wellen et al., 2010). Concentrations
of ribose-5-phosphate (R5P) and xylulose-5-phosphate (X5P) were also increased, which may
involve the oxidative pentose phosphate pathway that contributes to redox balance (Fig. 3.3B).
We could not determine whether p66Shc loss enhances either or both of the oxidative and non-
oxidative branches of the pentose phosphate pathway as the mass spectrometry signals for 6-
phosphogluconolactone or 6-phosphogluconate, the main metabolites of the oxidative branch,
were too weak to be accurately measured.
3.4. Restoring p66Shc expression inhibits glycolytic metabolism
To test whether p66Shc was sufficient for enhanced glycolytic metabolism, immortalized p66Shc
KO MEFs were stably infected with either GFP or 3xFLAG-p66Shc retroviral vectors (hereafter
referred to as KO and p66+ MEFs, respectively) (Fig. 3.4). Principal component analysis (PCA)
was used to evaluate replicate consistency, and revealed a notable difference in the overall
metabolic profiles of the p66Shc KO and p66+ cells (Fig. 3.5). Consistent with the p66Shc-
mediated effects in HeLa cells, F6P and G6P were among the metabolites with the most

57
significant changes, and the abundance of glycolytic intermediates was generally lower in p66+
MEFs compared to the KO cells (Table 1). The abundance of glycolysis intermediates changed
to a similar extent in MEFs as in HeLa cells; we observed ~ 3 fold decrease in G6P
concentrations, and a concomitant decrease in downstream three-carbon glycolytic metabolites
including PEP and lactate in p66+ cells (Fig. 3.6).
The lower concentration of citrate in p66+ cells was accompanied by a concomitant decrease in
the amounts of the fatty acid synthesis precursors acetyl-CoA and malonyl-CoA (Fig. 3.7).
Malonyl-CoA is a high energy and committed intermediate in the fatty acid biosynthesis
pathway, thus exclusively an anabolic metabolite. These results suggest that p66Shc inhibits de
novo lipid biosynthesis. In addition, the abundance of intermediates in the hexosamine
biosynthesis and pentose phosphate pathways was decreased in p66+ MEFs (Fig. 3.8). This
profile confirms that p66Shc deficiency in non-transformed cells enhanced glycolysis at the
expense of decreasing oxidative mitochondrial metabolism. Indeed, p66Shc deficient MEFs
displayed lower oxygen consumption (Fig. 3.9A) but improved energy utilization (AMP/ATP)
(Fig. 3.9B).
3.5. p66Shc expression inhibits amino acid biosynthesis and pyrimidine metabolism
The levels of amino acids were measured in p66Shc-competent and p66Shc-deficient cells.
Higher amounts of non-essential amino acids, including alanine, serine and aspartate, were
detected in p66Shc-deficient MEFs (Fig. 3.10). To determine whether the effect of p66Shc
signaling on the steady state abundance of nonessential amino acids reflects regulation of
metabolic flux through de novo synthesis, we measured the relative flux with a pulse of stable-
isotope labeled 15
N2-glutamine. Increased incorporation of labeled nitrogen into nonessential

58
amino acids was detected in p66Shc KO MEFs (Fig. 3.11). Increased flux of nitrogen from
extracellular glutamine into nonessential amino acids in p66Shc KO MEFs is consistent with
utilization catabolism of glutamine and reprogramming that favors anabolism. Pyrimidine
derivatives (dCTP and UTP) were among the top 10 metabolites whose abundance was
significantly decreased in p66+ MEFs (Table 3.1). Pyrimidine nucleotides are required as high
energy donors for phospholipid and glycoconjugate biosynthesis. p66+ MEFs showed lower
amounts of the pyrimidine synthesis intermediates orotate and dihydroorotate as compared to
knockout MEFs (Fig. 3.12). These results confirm that p66Shc inhibits anabolic metabolic
pathways, and demonstrate a prominent role for p66Shc in reprogramming cell metabolism
towards glucose catabolism and oxidative respiration.
3.6. p66Shc regulates redox homeostasis
By associating with cytochrome c, p66Shc promotes the generation of ROS (Giorgio et al., 2005;
Pinton et al., 2007). Consistent with these findings, a higher ratio of reduced to oxidized
glutathione (GSH/GSSG) was observed in p66Shc KO MEFs compared to p66+ MEFs (Fig.
3.13A). The ratio of NADH/NAD+ also reflects the redox state of the cell (Fisher-Wellman and
Neufer, 2012), and a ~ 3-fold increase in NADH/NAD+ indicates a more reducing environment
in p66Shc-deficient cells (Fig. 3.13B). NADH is produced in the second half of glycolysis
during the glyceraldehyde-3-phosphate dehydrogenase (GAPDH)-catalyzed step, and this may
contribute to the altered NADH/NAD+ ratio. The higher GSH/GSSG ratio in KO MEFs is
consistent with the reported lower amounts of ROS in p66Shc KO MEFs (Giorgio et al., 2005).
A previous report has shown that ROS contribute to insulin resistance and decrease glucose
metabolism (Houstis et al., 2006).

59
3.7. p66Shc is necessary and sufficient to alter glucose uptake and metabolism
To directly test whether p66Shc alters glucose uptake, we measured 3H-labeled 2-deoxy-D-
glucose (2-DG) uptake at different time points. The rate of 2-DG uptake in p66Shc-deficient
HeLa cells and in p66Shc KO MEFs was greater than in p66Shc-expressing cells (Fig. 3.14).
Consistent with the enhanced glucose consumption and higher glycolytic metabolism in p66Shc-
deficient cells, increased concentrations of extracellular lactate were observed in the media of
HeLa cells and MEFs lacking p66Shc (Fig. 3.15). However, western blots showed no apparent
difference in the abundance of glucose transporter 1 (Glut1) between p66Shc-competent and
p66Shc-deficient cells (Fig. 3.16).
To monitor the effect of p66Shc on glucose catabolism, the metabolites of 13
C-labeled glucose in
control and p66Shc-deficient HeLa cells were examined. The metabolism of [1,2-13
C2] glucose
generates M0, and M2 and M4 mass isotopomers corresponding to ion fragments that contain
zero, two or four labeled carbons, respectively (Fig. 3.17A and Appendix 6.2). Rapid transfer of
cells from unlabeled medium to identical medium containing [1,2-13
C2]-labeled glucose ensures
that metabolism is minimally disturbed (Munger et al., 2008). Consistent with enhanced
glycolytic metabolism in p66Shc-depleted cells, the amounts of the M2-labeled form of G6P and
downstream intermediates including M2 pyruvate were increased in p66Shc-depleted HeLa cells
(Fig. 3.17B). p66Shc silencing enhanced the amount of labeled glucose-derived citrate nearly 2
fold, consistent with the redirection of glucose-derived carbons towards lipid biosynthesis (Fig.
3.17B). Furthermore, the levels of M2-labeled form of hexosamine pathway intermediates were
also higher in p66Shc-deficient HeLa cells (Fig. 3.18). Conversely, re-expressing p66Shc in
p66Shc KO MEFs inhibited the heightened abundance of [1,2-13
C2] labeled glycolytic

60
metabolites and downstream anabolic intermediates, including pyruvate and citrate, observed in
p66Shc-deficient cells (Fig. 3.19). Additionally, p66Shc inhibited the de novo synthesis of amino
acids (Fig. 3.20). Collectively, our data suggest that p66Shc is necessary and sufficient to
reprogram glucose utilization into anabolic pathways.
3.8. Lack of p66Shc enhances glycolytic flux and anabolic metabolism
To study the dynamics of incorporation of isotope-labeled glucose into downstream anabolic
metabolites, kinetics flux profiling in p66Shc KO and p66+ MEFs using [1,2-
13C2] labeled
glucose was performed by measuring the relative amounts of labeled glycolytic intermediates
over 6 time points (0, 1, 2.5, 5, 10, 15 min). Consistent with the steady state data, p66Shc
inhibited labeled-glucose flux into glycolytic intermediates such as pyruvate in p66+ MEFs (Fig.
3.21A). In addition, the amounts of labeled citrate, UDP-GlcNAc and R5P were lower in cells
expressing p66Shc (Fig. 3.21, B-D, Appendix 6.2). UDP-GlcNAc is a high-energy donor
required for protein glycosylation and an example of an anabolic metabolite where the GlcNAc
portion is not catabolized back to glucose (Wellen et al., 2010). Overall, these results suggest
that increased glycolytic flux in p66Shc-deficient cells accounts for the observed increase in
metabolites at steady state, consistent with a role for p66Shc as an inhibitor of glucose
catabolism.

61
A B
Fig. 3.1. Effect of p66Shc on glycolytic metabolism.
(A) Abundance of the three Shc1 isoforms in HeLa cells stably expressing shRNAs targeting
GFP or p66Shc. (B) Summary of the changes in intracellular metabolite amounts associated with
p66Shc knockdown in HeLa cells. Glucose-6-phosphate (G6P); fructose-6-phosphate (F6P);
fructose-1,6-phosphate (F1,6BP); dihydroxyacetone phosphate (DHAP); glyceraldehyde 3-
phosphate (GADP); 1,3-bisphosphoglycerate (1,3BPG); 3-phosphoglycerate (3PG);
phosphoenolpyruvate (PEP); acetyl-CoA(ACoA); ribose-5-phosphate (R5P); xylulose 5-
phosphate (X5P); glucosamine-6-phosphate (GlcN6P); N-acetylglucosamine phosphate
(GlcNAcP); uridine diphosphate N-acetylglucosamine (UDP-GlcNAc).
Glucose transporter

62
Fig. 3.2. Lack of p66Shc enhances glycolytic metabolism.
Fold change of glycolytic intermediates in p66Shc-competent and p66Shc-deficient HeLa cells
as measured by LC-MS/MS. Error bars represent SD of at least three biological replicates (* p <
0.05, ** p < 0.01, *** p < 0.001). Glucose-6-phosphate (G6P); fructose-1,6-phosphate (F1,6BP);
phosphoenolpyruvate (PEP).

63
A
B
Fig. 3.3. p66Shc deficiency increases the levels of the pentose phosphate and the
hexosamine pathway intermediates.
Fold change of the hexosamine biosynthesis (A) and the pentose phosphate (B) pathway
intermediates in p66Shc-competent and p66Shc-deficient HeLa cells as measured by LC-
MS/MS. Error bars represent SD of at least three biological replicates (* p < 0.05, ** p < 0.01,
*** p < 0.001). Fructose-6-phosphate (F6P); glucosamine-6-phosphate (GlcN6P); uridine
diphosphate N-acetylglucosamine (UDP-GlcNAc); ribose-5-phosphate (R5P); xylulose 5-
phosphate (X5P).

64
Fig. 3.4. Levels of Shc1 isoforms in p66Shc KO and p66+ MEFs.
Abundance of the three Shc1 isoforms in p66Shc KO MEFs stably infected with GFP (KO) or
3xFLAG-p66Shc (p66+).

65
Fig. 3.5. Unsupervised principal component analysis for targeted metabolomics screen in
p66Shc KO and p66+ MEFs.
Each dot represents a biological replicate. Red dots indicate replicates for p66Shc KO MEFs, and
green dots indicate replicates for p66+ MEFs. The analysis demonstrates clear separation of
overall metabolomic profiles of the two genotypes with statistical significance 99.7% (PC1) and
0.2% (PC2) (p < 0.05). The metabolites with the most significant changes in p66+ MEFs
compared to KO MEFs are summarized in Table 3.1.

66
Fig. 3.6. p66Shc expression decreases the levels of glycolytic intermediates.
Fold change of glycolytic metabolites in p66Shc-deficient (white) and p66Shc-competent (black)
MEFs as measured by LC-MS/MS. Error bars represent SD of at least three biological replicates
(* p < 0.05, ** p < 0.01, *** p < 0.001). Glucose-6-phosphate (G6P); fructose-1,6-phosphate
(F1,6BP); phosphoenolpyruvate (PEP).

67
Fig. 3.7. p66Shc inhibits fatty acid biosynthesis
Fold change of acetyl-CoA (ACoA) and malonyl-CoA (MCoA) in p66Shc-deficient (white) and
p66Shc-competent (black) MEFs as measured by LC-MS/MS. Error bars represent SD of at least
three biological replicates (* p < 0.05, ** p < 0.01, *** p < 0.001).

68
A
B
Fig. 3.8. p66Shc expression decreases the levels of the pentose phosphate and the
hexosamine pathway intermediates.
Fold change of the hexosamine biosynthesis (A) and the pentose phosphate (B) pathways
metabolic intermediates in p66Shc-deficient (white) and p66Shc-competent (black) MEFs as
measured by LC-MS/MS. Error bars represent SD of at least three biological replicates (* p <
0.05, ** p < 0.01, *** p < 0.001). Fructose-6-phosphate (F6P); glucosamine-6-phosphate
(GlcN6P); uridine diphosphate N-acetylglucosamine (UDP-GlcNAc); ribose-5-phosphate (R5P);
xylulose 5-phosphate (X5P).

69
A B
Fig. 3.9. Deficiency of p66Shc inhibits oxygen consumption rate and lowers AMP/ATP
ratio.
(A) Oxygen consumption rate in p66Shc-deficient (white) and p66Shc-competent (black) MEFs
were measured by XF-24 Flux Analyzer. Error bars represent SD of three biological replicates (*
p < 0.05). (B) Relative amounts of AMP/ATP ratio in p66Shc KO and p66+ MEFs by LC-
MS/MS. Error bars represent SD of three biological replicates (* p < 0.05).
Oxygen consumption rate
KO p66+ KO p66+

70
Fig. 3.10. p66Shc expression inhibits the synthesis of nonessential amino acids
Fold change of the amino acids alanine, serine and aspartate in p66Shc-deficient (white) and
p66Shc-competent (black) MEFs as measured by LC-MS/MS. Error bars represent SD of at least
three biological replicates (*** p < 0.001).

71
Fig. 3.11. Tracing of 15
N-labeled-amino acids in p66Shc KO and p66+ cells.
Fold change of intracellular amounts of 15
N-labeled alanine, serine and aspartate in p66Shc KO
(white) and p66+ (black) MEFs grown in media containing
15N-glutamine for 2h. Error bars
represent SD of at least three biological replicates (*** p < 0.001)

72
Fig. 3.12. p66Shc inhibits de novo pyrimidine synthesis intermediates.
Fold change of de novo pyrimidine synthesis intermediates, N-carbamoyl aspartate, orotate and
uridine monophosphate (UMP) in p66Shc KO (white) and p66+ (black) MEFs. Error bars
represent SD of at least three biological replicates (* p < 0.05).

73
A B
Fig. 3.13. p66Shc regulates redox homeostasis.
Relative amounts of the ratio of reduced/oxidized glutathione (GSH/GSSG) (A) and
reduced/oxidized NAD+ (NADH/NAD
+) (B) in p66Shc KO (white) and p66
+ (black) MEFs. Error
bars represent SD of at least three biological replicates (*** p < 0.001).

74
A
B
Fig. 3.14. Lack of p66Shc enhances 2-DG uptake.
Tracing of 3H-labeled-2-deoxy glucose (2-DG) uptake over time in p66Shc-competent and
p66Shc-deficient HeLa cells stably expressing shRNAs targeting GFP and p66Shc (A) and
p66Shc KO and p66+
MEFs (B). Error bars represent SD of at least three biological (p < 0.05).

75
A
B
Fig. 3.15. p66Shc inhibits cellular lactate secretion.
Fold change of extracellular lactate in media from p66Shc-competent and p66Shc-deficient
HeLa (A) and MEFs (B). Error bars represent SD of at least three independently prepared
samples (*** p < 0.001).

76
A
B
Fig. 3.16. Abundance of Glut1 in p66Shc-deficient and p66Shc-competent cells.
p66Shc-competent and p66Shc-deficient HeLa and MEFs express similar levels of Glut1. (A)
HeLa cells stably transfected with indicated shRNAs were lysed and cell lysates were analyzed
for the indicated proteins by immunoblotting. (B) Lysates of p66Shc KO and p66+ MEFs were
examined as in (A).
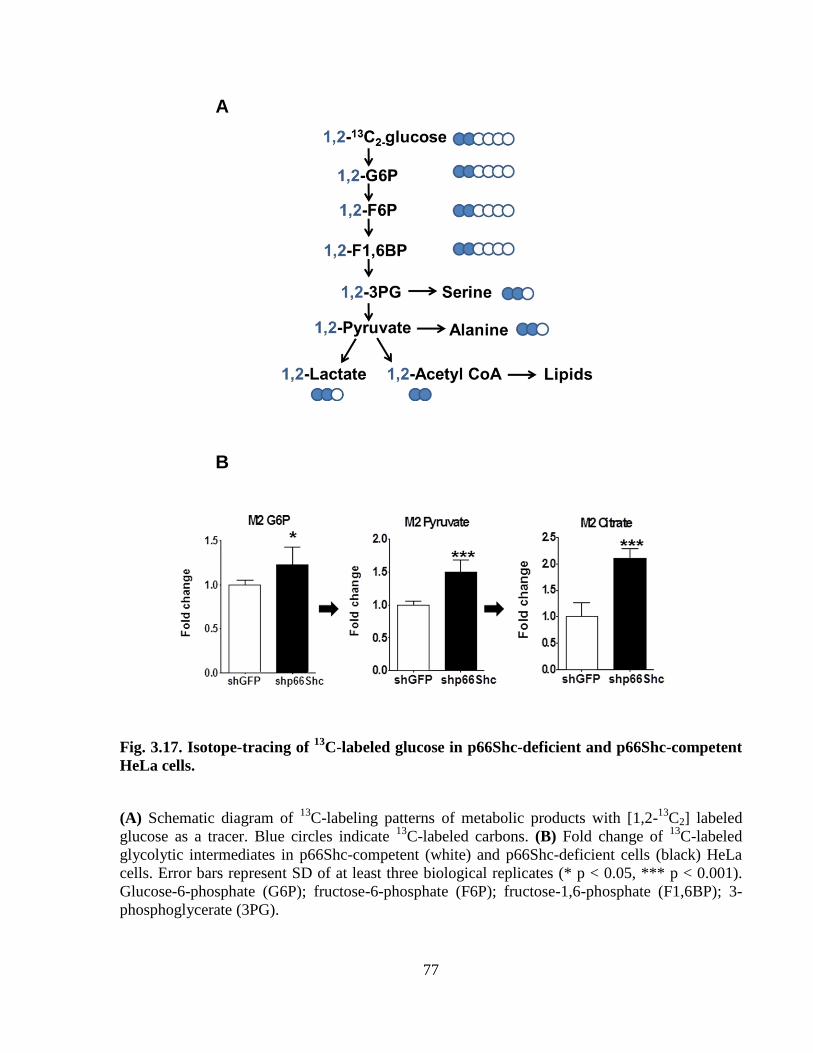
77
A
B
Fig. 3.17. Isotope-tracing of 13
C-labeled glucose in p66Shc-deficient and p66Shc-competent
HeLa cells.
(A) Schematic diagram of 13
C-labeling patterns of metabolic products with [1,2-13
C2] labeled
glucose as a tracer. Blue circles indicate 13
C-labeled carbons. (B) Fold change of 13
C-labeled
glycolytic intermediates in p66Shc-competent (white) and p66Shc-deficient cells (black) HeLa
cells. Error bars represent SD of at least three biological replicates (* p < 0.05, *** p < 0.001).
Glucose-6-phosphate (G6P); fructose-6-phosphate (F6P); fructose-1,6-phosphate (F1,6BP); 3-
phosphoglycerate (3PG).

78
Fig. 3.18. Isotope-tracing of 13
C-labeled hexosamine pathway intermediates in p66Shc-
deficient and p66Shc-competent HeLa cells.
Fold change of 13
C-labeled hexosamine biosynthesis pathway intermediates in p66Shc-
competent (white) and p66Shc-deficient cells (black) HeLa cells. Error bars represent SD of at
least three biological replicates (** p < 0.01, *** p < 0.001). Fructose-6-phosphate (F6P);
glucosamine-6-phosphate (GlcN6P); uridine diphosphate N-acetylglucosamine (UDP-GlcNAc).

79
Fig. 3.19. Isotope-tracing of 13
C-labeled glycolytic intermediates in p66Shc KO and p66+
MEFs.
Fold change of 13
C-labeled glycolytic glycolytic intermediates in p66Shc KO (white) and p66+
(black) MEFs. Error bars represent SD of at least three biological replicates (** p < 0.01, *** p <
0.001). Glucose-6-phosphate (G6P); fructose-6-phosphate (F6P).

80
Fig. 3.20. Isotope-tracing of 13
C-labeled nonessential amino acids in p66Shc KO and p66+
MEFs.
Fold change of 13
C-labeled serine (A) and alanine (B) in p66Shc KO (white) and p66+ (black)
MEFs. Error bars represent SD of at least three biological replicates (*** p < 0.001).
M2 Alanine
KO p66+
0.0
0.5
1.0
1.5
Fo
ld c
han
ge
M2 Serine
KO p66+
0.0
0.5
1.0
1.5
Fo
ld c
han
ge
A B
*** ***

81
A B
C D
Fig. 3.21. Flux analysis of 13
C-labeled glucose in p66Shc KO and p66+ MEFs.
Relative amounts of 13
C-labeled pyruvate (A), citrate (B), and the hexosamine pathway
intermediates UDP-GlcNAc (C) and R5P (D) after incubating cells with [1,2-13
C2] labeled
glucose for the indicated time points. Data are represented as the ratio of 13
C-labeled metabolites
to an internal standard (D7-glucose). Error bars represent SD of at least three biological
replicates (p < 0.05). Uridine diphosphate N-acetylglucosamine (UDP-GlcNAc); ribose-5-
phosphate (R5P).
M4 UDP-GlcNAc
0 5 10 150.000
0.001
0.002
0.003
0.004
0.005KO
p66+
Time (min)
Are
a R
ati
o
KO
p66+
Time (min)
M4 UDP-GlcNAC M2 R5P
M2 Pyruvate M2 Citrate

82
Table 3.1: Fold change of the most significantly p66Shc-inhibited metabolites.
The 10 metabolites showing the most statistically significant changes in p66+
over
p66Shc KO MEFs. FDR: False discovery rate.
Metabolite Fold change p-value FDR
Dihydroxyacetone phosphate (DHAP) 0.23 2.44E-05 0.00024
Fructose 6-phosphate (F6P) 0.29 7.64E-05 0.00027
Glucose 6-phosphate (G6P) 0.36 8.11E-05 0.00027
Fructose 1,6-bisphosphate (F1,6BP) 0.29 0.000207 0.00045
Malonyl Co-enzyme A (MCoA) 0.30 0.000226 0.00045
Erythrose-4-phosphate (E4P) 0.26 0.000598 0.00091
NADH/NAD+ 0.34 0.000641 0.00091
Acetyl Co-enzyme A (ACoA) 0.29 0.001544 0.00193
Deoxycytidine triphosphate (dCTP) 0.41 0.00265 0.00294
Uridine 5-triphosphate (UTP) 0.45 0.003853 0.003853

83
Chapter 4. p66Shc Inhibits Signaling to The Metabolic Sensor
mTOR
A version of this chapter appeared in the following article:
The Adaptor Protein p66Shc Inhibits mTOR-Dependent Anabolic Metabolism.
Science Signaling, 7, ra17 (2014).
Mohamed A. Soliman, Anas M. Abdel Rahman, Dudley W. Lamming, Kivanç Birsoy, Judy
Pawling, Maria E. Frigolet, Huogen Lu, I. George Fantus, Adrian Pasculescu, Yong Zheng,
David M. Sabatini, James W. Dennis, and Tony Pawson.

84
4.1. Background
The evolutionarily conserved Ser/Thr kinase TOR integrates signals from growth factors and
nutrients to activate anabolic pathways driving cell growth. Chronic activation of mTORC1 in
Tsc2 KO MEFs enhances glucose uptake, lactate production and the biosynthesis of lipid
metabolites, and rapamycin treatment inhibits the heightened glycolytic phenotype in Tsc2 KO
MEFs (Duvel et al., 2010). The same study showed that mTORC1 increases the levels of the
pentose phosphate pathway intermediates, which provide the ribose sugar component for the
pyrimidine nucleotides (Duvel et al., 2010). In line with this study, recent metabolomic and
phosphoproteomic analyses described an essential role for mTORC1-S6K1 in de novo
pyrimidine biosynthesis (Ben-Sahra et al., 2013; Robitaille et al., 2013). mTORC2 also plays a
role in cell metabolism. In gliobastoma tumors, mTORC2 controls glycolytic metabolism by
regulating the cellular level of Myc. The enhancement of glucose metabolism correlated with
shorter survival of glioblastoma patients (Masui et al., 2013).
My data suggest that silencing of p66Shc stimulated glycolytic and lipid metabolism (Chapter
3). Given the central role of mTOR in coordinating growth signaling and cellular metabolism
(Shimobayashi and Hall, 2014), I hypothesized that deficiency of p66Shc enhances insulin
signaling to mTOR, hence increasing the utilization of glucose into branching anabolic pathways
to meet the growth demands of proliferating cells. This chapter addresses the effect of p66Shc
expression on mTORC1 and mTORC2 activation to provide insight into the molecular
mechanism by which p66Shc regulates cellular metabolism.

85
4.2. p66Shc inhibits growth factor signaling to the metabolic sensor mTOR
Several aspects of the metabolic profile of p66Shc KO MEFs resemble that of cells with chronic
mTOR activation; these include enhanced amounts of glycolytic and pentose phosphate pathway
intermediates (G6P, F6P, and R5P) (Duvel et al., 2010), increased amounts of de novo
pyrimidine metabolites (orotate) (Ben-Sahra et al., 2013; Robitaille et al., 2013) and increased
lipid biosynthesis precursors (acetyl-CoA and malonyl-CoA) (Duvel et al., 2010). To monitor
mTOR signaling, phosphorylation of Thr389 in the ribosomal protein S6 kinase (S6K1), a
substrate of mTORC1, Ser473 in Akt which is a direct target of mTORC2 (Sarbassov et al.,
2005) and Thr346 NDRG1, an indirect target of mTORC2 (Garcia-Martinez and Alessi, 2008),
were examined (Zoncu et al., 2011). Following serum stimulation, p66Shc-deficient HeLa cells
displayed markedly increased phosphorylation of both mTORC1 and mTORC2 targets compared
to control cells, despite having equal abundance of the p52Shc and p46Shc isoforms (Fig. 4.1).
Similar results were obtained with insulin stimulation (Fig. 4.2). We also found that amino acid
activation of the mTORC1 pathway was enhanced in p66Shc-deficient HeLa cells (Fig. 4.3). In
p66+ MEFs, p66Shc inhibited the activation of mTOR targets following stimulation with IGF-1
and insulin, but not with EGF (Fig. 4.4). This observation is consistent with previous reports
showing that p66Shc has little effect on EGF signaling (Migliaccio et al., 1997). The
phosphorylation of mTORC1 and mTORC2 targets was decreased in p66+ MEFs following
serum (Fig. 4.5) and amino acid stimulation (Fig. 4.6). In contrast to the inhibitory effects of
p66Shc expression on mTOR signaling in KO MEFs, mTOR activation was sustained in KO
MEFs stably overexpressing the p52Shc isoform (p52+ cells) (Fig. 4.7). In line with this,
elevated glycolytic metabolism was maintained in p52+ cells (Fig. 4.8). This confirms earlier

86
reports that the p66Shc and p52Shc isoforms have opposing effects attributable to the unique N-
terminal CH2 region of p66Shc (Migliaccio et al., 1997; Xi et al., 2008).
4.3. p66Shc expression causes a decrease in cell size
mTOR functions as a central regulator of cell growth. The inhibitory effect of p66Shc on mTOR
activation therefore predicted that p66Shc deficiency would lead to a larger cell size. Indeed,
stable re-expression of p66Shc in p66Shc KO MEFs caused a decrease in cell size as measured
by FACS forward scatter analysis (Fig. 4.9). These findings were confirmed using Coulter
Counter measurement (Fig. 4.10A). Conversely, HeLa cells depleted of p66Shc displayed an
increase in median cell size, consistent with redirection of glucose-derived carbon towards
biomass synthesis (Fig. 4.10B). Together, these results suggest that p66Shc acts as a negative
regulator of the nutrient-sensing mTOR signaling pathway leading to inhibition of cell growth
and anabolic metabolism.
4.4. Effects of p66Shc on glycolytic metabolism are mediated through mTOR
To determine which mTOR complex contributes to the metabolic phenotype observed in p66Shc
KO cells, we treated p66Shc KO and p66+ MEFs with rapamycin for 16 h, a time frame that
inhibits both mTORC1 and mTORC2 enzymes (Fig. 4.11). In line with reports linking mTORC1
and mTORC2 to glycolytic metabolism (Duvel et al., 2010; Hagiwara et al., 2012; Lamming et
al., 2014), we found that inhibition of both mTOR complexes with rapamycin reversed the
metabolic phenotype of p66Shc KO MEFs, notably diminishing the increase in the pentose
phosphate pathway (R5P) and the hexosamine biosynthesis pathway (UDP-GlcNAc) (Fig. 4.12).

87
Furthermore, inhibition of Akt, a kinase that is upstream of mTORC1 and downstream of
mTORC2 (Zoncu et al., 2011), significantly decreased the amounts of glycolytic metabolites in
p66Shc-deficient MEFs (Fig. 4.13).

88
Fig. 4.1. p66Shc inhibits mTORC1 and mTORC2 activation following serum stimulation.
HeLa cells stably transfected with indicated shRNAs were serum starved, treated with either
DMSO or Torin for 1 h, and then stimulated with 10% serum (10 min). Cell lysates were
analyzed for the indicated proteins and phosphorylation states by immunoblotting. Pre-treatment
of cells with the mTOR inhibitor Torin1 (Thoreen et al., 2009) abolished the phosphorylation of
mTOR targets in both cell types confirming the specificity of the signal.

89
Fig. 4.2. p66Shc inhibits insulin signaling to mTOR.
HeLa cells stably transfected with indicated shRNAs were serum starved, treated with either
DMSO or Torin for 1 h, and then stimulated with 100 nM Insulin (10 min). Cell lysates were
analyzed for the indicated proteins and phosphorylation states by immunoblotting. Pre-treatment
of cells with the mTOR inhibitor Torin1 (Thoreen et al., 2009) abolished the phosphorylation of
mTOR targets in both cell types confirming the specificity of the signal.

90
Fig. 4.3. p66Shc inhibits mTORC1 activation in response to amino acid stimulation.
HeLa cells stably transfected with shRNAs targeting the indicated genes were starved of amino
acids for 50 min and stimulated either with dialyzed serum or normal serum for 10 min.
Phosphorylation of S6K1 was analyzed by Western blotting.

91
Fig. 4.4. p66Shc expression inhibits mTOR activation in response to insulin and IGF1, but
not to EGF, stimulation.
p66Shc KO and p66+ MEFs were serum starved, then stimulated with 100 nM EGF, IGF, or
insulin (10 min). Cell lysates were analyzed for the indicated proteins and phosphorylation states
by immunoblotting.

92
Fig. 4.5. p66Shc expression inhibits mTOR activation in response to serum stimulation.
p66Shc KO and p66+ MEFs were serum starved for 4 h, then stimulated with 10% serum for 10
min. Cell lysates were analyzed for the indicated proteins and phosphorylation states by
immunoblotting. Pre-treatment of cells with the mTOR inhibitor Torin1 (Thoreen et al., 2009)
abolished the phosphorylation of mTOR targets in both cell types confirming the specificity of
the signal.

93
Fig. 4.6. p66Shc expression inhibits mTOR activation in response to amino acid
stimulation.
p66Shc KO and p66+ MEFs were starved of amino acids for 50 min and stimulated either with
dialyzed serum or normal serum for 10 min. Cell lysates were analyzed for the indicated proteins
and phosphorylation states by immunoblotting.

94
Fig. 4.7. Stable expression of p66Shc, but not p52Shc, in p66Shc KO cells inhibits the
mTOR pathway.
p66Shc KO, p66+ and p52
+ MEFs were serum starved, then stimulated with 10% serum for 10
min. Cell lysates were analyzed for the indicated proteins and phosphorylation states by
immunoblotting.

95
A B
Fig. 4.8. Stable expression of p66Shc, but not p52Shc, in p66Shc KO cells inhibits glycolytic
metabolism.
Fold change of G6P (A) and F6P (B) in p66Shc KO (white), p66+ (black) and p52
+ MEFs
(srtiped). Error bars represent SD of at least three independently prepared samples (** p < 0.01).
** **
G6P F6P

96
A B
Fig. 4.9. p66Shc expression decreases cell size.
Cell size distribution of p66Shc KO and p66+ MEFs using flow cytometry forward scattering
(A). Average cell size of KO and p66+ from three independent experiments is represented (B),
*** p < 0.001.

97
A
B
Fig. 4.10. p66Shc mediates cell growth.
(A) Cell size measurement of p66Shc KO and p66+
cells. Cell size was measured using coulter
counter. (B) Cell size measurement of p66Shc–deficient and p66Shc-competent HeLa cells.
Experiment was done as in (A).

98
Fig. 4.11. Effect of rapamycin on mTOR signaling in p66Shc KO and p66+ MEFs.
Rapamycin treatment for 16 h is sufficient to inhibit both mTORC1 and mTORC2 signaling in
p66Shc KO and p66+ MEFs. Cell lysates were analyzed for the indicated proteins and
phosphorylation states by immunoblotting.

99
A
Fig. 4.12. mTOR mediates the effects of p66Shc on glycolytic and pyrimidine metabolism.
p66Shc KO and p66+ MEFs were treated with vehicle or rapamycin for 16 h, and amounts of
glycolysis (A), pentose phosphate (B), hexosamine biosynthesis (C), and pyrimidine
biosynthesis (D) pathways metabolic intermediates were quantified by LC-MS/MS. Error bars
represent SD of three independently prepared samples (ANOVA analysis, * p < 0.05, ** p <
0.01, *** p < 0.001). Glucose-6-phosphate (G6P); fructose-6-phosphate (F6P); ribose-5-
phosphate (R5P); uridine diphosphate N-acetylglucosamine (UDP-GlcNAc); uridine
monophosphate (UMP); cytidine monophosphate (CMP).
B
C
D

100
Fig. 4.13. Effect of Akt inhibition on the abundance of glycolytic metabolites in p66Shc-
deficient and p66Shc-competent MEFs.
Fold change of 3PG (A), PEP (B), R5P (C), and citrate (D) in p66Shc KO MEFs (white), p66Shc
KO MEFs treated with Akt inhibitor (black), p66+ (grey) and p66
+ treated with Akt inhibitor
(stripped) MEFs. Error bars represent SD of at least three independently prepared samples (* p <
0.05, *** p < 0.001). 3-phosphoglycerate (3PG); phosphoenolpyruvate (PEP); ribose-5-
phosphate (R5P).
A B
C D
***
***
***
*
**
* *
***
***

101
Chapter 5. Discussion and Future Directions

102
Understanding the biological role of the adaptor protein p66Shc in regulating metabolic
homeostasis was the primary motivation in undertaking this study. Since the discovery of the
adaptor protein Shc1 in 1992 (Pelicci et al., 1992), extensive research has confirmed an essential
role for Shc1 in development, growth and diseases such as cancer (Ravichandran, 2001). The
most abundantly expressed Shc1 isoform, p52Shc, acts principally downstream of receptor
tyrosine kinases to amplify the Ras-MAPK (Rozakis-Adcock et al., 1992) and the PI3K-Akt
pathways signaling (Gu et al., 2000). However, my curiosity was piqued by the p66Shc isoform
(Migliaccio et al., 1997), identical to p52Shc1 but containing an N-terminal extension that has
been reported to posess unique properties affecting metabolism and oxidative stress (Luzi et al.,
2000). For example, in mammals, lack of p66Shc confers resistance to hyperglycemia-induced
endothelial dysfunction (Camici et al., 2007) and early atherogenesis in mice fed a high-fat diet
(Napoli et al., 2003). Indeed, deletion of p66Shc in mice improves glucose tolerance and insulin
sensitivity (Ranieri et al., 2010; Tomilov et al., 2011). These observations raised the possibility
that p66Shc evolved in mammals in response to variations in nutrient availability as a suppressor
for insulin signaling and metabolism, although a thorough study of the role of p66Shc in
metabolism using modern mass spectrometry-based metabolomics has not been done.
My results confirmed that p66Shc expression is associated with a dampening in the cellular
signaling response to insulin stimulation and a reduction in glycolytic metabolism. This
suggested that p66Shc mediates feedback inhibition of both growth factor signaling and, in turn,
glucose metabolism. The metabolomic analysis of p66Shc-competent and p66Shc-deficient cells
shows an increase in glucose-6-phosphate and downstream intermediates, including fructose-6-
phosphate and pyruvate, in cells lacking p66Shc. The kinetic flux analysis using 13
C-labeled

103
glucose shows faster carbon incorporation and higher steady state levels in p66Shc-deficient
cells. No statistically significant differences are found between p66Shc-deficient and wild-type
cells for other metabolites, including GTP, ADP, ATP, adenine, flavin adenine dinucleotide
(FAD), CoA, and glyoxalic acid. It should be noted that our analytical method targeted a specific
set of metabolites, including intermediates of glycolysis, glucosamine, pentose phosphate and
hexosamine pathways, amino acids, nucleic acids, fatty acids synthesis and oxidation, TCA and
urea cycles, and bile acid biosynthesis. Hence, I cannot exclude the possibility that p66Shc
affects other metabolic pathways that were not included in our targeted approach.
p66Shc silencing in both transformed and non-transformed immortalized cells improves glucose
uptake and enhances lactate production. Furthermore, p66Shc deficiency increases metabolite
abundance for fatty acid biosynthesis, the hexosamine pathway, pentose phosphate pathway and
increases cell size. This metabolic shift depends in part on the mTOR pathway, because
rapamycin treatment partly reversed the glycolytic shift caused by p66Shc loss. However, I could
not exclude the possibility that other signaling pathways might mediate p66Shc metabolic
effects, especially the Wnt signaling pathway. Wnt signaling can modulate glucose homeostasis
and insulin sensitivity (Yoon et al., 2010) and Wnt3A can induce aerobic glycolysis by
increasing the level of key glycolytic enzymes (Esen et al., 2013). This metabolic regulation is
mediated through mTORC2-Akt signaling (Esen et al., 2013). Given that p66Shc also signals to
mTORC2, it will be of interest to see if knocking down Wnt3a will inhibit the heightened
glycolytic metabolism observed in p66Shc-deficient cells.

104
My data suggest that depletion of p66Shc is sufficient to enhance the Warburg effect in cancer
cell lines, demonstrating that p66Shc may function as a potential tumor suppressor. Previous
reports showed that Shc1 signaling is essential for breast tumour progression, where tyrosine
phosphorylation sites Tyr239/240/317 on p52Shc are essential for ErbB2‐induced mammary
tumour outgrowth (Ahn et al., 2013; Ursini-Siegel et al., 2008; Ursini-Siegel and Muller, 2008).
However, p66Shc expression has been shown to inversely correlate to tumorigenesis (Stevenson
and Frackelton, 1998). This is consistent with my results in which p66Shc loss enhances mTOR
signaling and anabolic metabolism to support cell growth. Clinical studies report that both an
increase in tyrosine phosphorylated Shc and a decrease in the expression of p66Shc correlated
with breast tumor recurrence (Davol et al., 2003). Unlike p52Shc, the abundance of p66Shc is
substantially decreased in ErbB2-overexpressing breast cancer cell lines (Stevenson and
Frackelton, 1998). This antagonistic effect of p66Shc on RTK signaling might arise from a
competition between p66Shc and p52Shc for common targets downstream of activated receptors
(Migliaccio et al., 1997; Okada et al., 1997).
5.1. p66Shc signaling to mTOR: an open question
My results show that p66Shc inhibits insulin signaling to the mTOR pathway. Identifying
upstream and downstream regulators of p66Shc could provide valuable insight into how p66Shc
regulates mTOR activation and the associated changes in glycolytic metabolism. A limitation of
studying the functional interaction between p66Shc and mTOR is the difficulty of preserving the
p66Shc protein complexes. Using Co-IP and MS analysis, interactions between p66Shc and
mTOR, Raptor and Rictor were undetectable. The Scansite Motif bioinformatics tool analysis of
the unique CH2 region of the p66Shc isoform suggests putative binding of GSK3 to the SPSASS

105
(SxxxS) motif. This is reasonable as GSK3 inhibits mTOR signaling via phosphorylation of the
TSC2 tumor suppressor (Inoki et al., 2006). In addition, GSK3 regulates glucose transport in
several cell types, reducing glucose uptake by almost two-fold in an mTOR-mediated manner
(Buller et al., 2008). GSK3-mediated inhibition of glucose uptake is restored in TSC2 null cells
expressing wild-type TSC2, but not by a TSC2 construct with mutated GSK3 phosphorylation
sites (Buller et al., 2008). However, I was unable to detect an interaction between GSK3 and
p66Shc using co-immunoprecipitation, LC-MS or BioID (proximity-dependent biotin
identification). As well, attempts using milder cell lysis conditions failed. In future work,
chemical cross-linking might capture this putative transient interaction. If an interaction can be
confirmed, then understanding the functional role of p66Shc-GSK3 interaction in regulating
mTOR signaling and glycolytic metabolism will be essential.
Importantly, the contribution of the p66Shc PTB and SH2 domains and the three conserved Tyr
residues in the CH1 region to the observed glycolytic phenotype warrants further investigation.
Despite the fact that all Shc1 isoforms share the same PTB and SH2 domains, it is possible that
these domains and conserved Tyr residues may not bind the same proteins at the same time for
the three isoforms. Domain binding to specific partners will also depend on the local molecular
environment surrounding each isoform. Due to the high percentage of Pro and Gly, it is predicted
that the 110 amino acid peptide at the N-terminus of p66Shc is highly unstructured. It is possible
that this region can form a weak intramolecular interaction with the PTB domain, the SH2
domain or the CH1 region, blocking access of substrates to p66Shc binding surfaces. It will be
interesting to test whether expressing a p66Shc mutant with either a non-functional PTB
(R285Q), a non-phosphotyrosine binding SH2 domain (R507K) or with all three CH1 region

106
phosphotyrosine residues substituted by phenylalanine (3Y to 3F) would be able to rescue the
glycolytic phenotype in p66Shc KO MEFs.
Inhibition of PI3K is a potential mechanism by which p66Shc might suppress mTOR activation.
As PI3K acts upstream of mTORC1 and mTORC2, inhibition of PI3K would affect both mTOR
complexes. It has been reported that p66Shc-deficient vascular smooth muscle cells display
enhanced IGF-I-stimulated PI3K activity, as measured by an increase in the levels of PIP3 (Xi et
al., 2010b). Mechanistically, silencing of p66Shc enhanced IGF-I-stimulated association between
Grb2 and p85, the regulatory unit of PI3K, which led to PI3K-Akt activation (Xi et al., 2010b).
Membrane fractionation studies showed that IGF-I-stimulated membrane recruitment of Akt was
inhibited by overexpression of p66Shc and enhanced by knockdown of p66Shc (Xi et al.,
2010b).
5.2. Competition between Shc1 isoforms in regulating PI3K-mTOR signaling
In contrast to the inhibitory effects of p66Shc, mTOR pathway activation was sustained in
p66Shc KO MEFs stably overexpressing the p52Shc isoform. These effects are particularly
striking since the p52Shc isoform is identical to p66Shc in all but the N-terminal CH2 region.
Signaling downstream of p66Shc and p52Shc to the mTOR pathway might vary depending on
the cell type and the relative abundance of each protein. My proteomic analysis shows that
p66Shc has common binding proteins to those known for p52Shc (Appendix 6.4) (Zheng et al.,
2013). Thus, it is probable that p66Shc competes with p52Shc for binding partners in a dynamic
manner that disrupts downstream signaling to mTOR. Future studies could include the mass-

107
spectrometry based analysis of the time-dependent recruitment of p52Shc versus p66Shc
signaling complexes by receptors, which could reveal the molecular basis for p66Shc action on
signaling and anabolic metabolism. I cannot exclude the possibility that additional independent
pathways could be signaling to p66Shc and p52Shc independently, allowing cells to fine tune
mTOR activity in response to nutrient and environmental cues. Given the complexity of insulin-
Shc-mTOR signaling, and the feedback loops among the components of mTOR pathway, a better
understanding of the physiological consequences of altering the p66Shc-p52Shc signaling
balance will require generating mouse models with inducible overexpression or deletion of
p66Shc.
5.3. Regulation of receptor and glucose transporter glycosylation by p66Shc
UDP-GlcNAc and hexosamine pathway intermediates were among the metabolites showing the
largest increases in p66Shc knockout cells, and may contribute to the Warburg-like phenotype in
cells lacking p66Shc. UDP-GlcNAc is an essential substrate for the Golgi N-glycan branching
pathway and consequently for galectin binding, which enhances the residency of cytokine
receptors at the cell surface (Lau et al., 2007; Partridge et al., 2004). More recently, regulation by
N-glycan branching has been extended to surface residency of glucose transporters, as reported
for Glut2 in β-cells (Ohtsubo et al., 2005), Glut1 in tumor cells (Kitagawa et al., 1995), and
Glut4 in cells simulated by insulin or UDP-GlcNAc (Haga et al., 2011).
The N-glycans are remodeled in the Golgi by the branching N-acetylglucosaminyltransferases
enzymes encoded by the genes Mgat1-5. Previous work showed that Mgat5-deficient mice

108
display hypoglycemia, lower body weight and fail to gain weight on a high fat diet (Cheung et
al., 2007). Since p66Shc knockout mice are also insulin sensitive (Tomilov et al., 2011) and
show resistance to high fat diet (Napoli et al., 2003), it is possible that p66Shc functionally
interacts with the HBP and N-glycan branching to affect the level of glycosylation of nutrient
transporters. We expect that silencing HBP and N-glycan branching enzymes, such as Mgat5,
would suppress the p66Shc-mediated metabolic phenotype. Conversely, forced expression of N-
glycan branching enzymes would be expected to oppose p66Shc-mediated suppression of
glycolytic metabolism.
5.4. p66Shc and fatty acid signaling to mTOR
Higher levels of malonyl-CoA were observed in p66Shc-deficient MEFs. Malonyl Co-A is a
strictly anabolic metabolite that supplies and commits the 2-carbon building blocks to fatty acid
chain biosynthesis. Although the scope of the project did not allow for investigating lipid
biosynthesis, it would be important to examine the effect of p66Shc expression on the abundance
of various lipids using a non-targeted metabolomics approach. Earlier reports have suggested
that mTOR binds lipids, in particular phosphatidic acid, and that this binding increases mTOR
kinase activity (Fang et al., 2001). In addition, one of the enzymes responsible for the production
of phosphatidic acid, phospholipase D, acts as an upstream regulator in the mTOR pathway
(Fang et al., 2003). Hence, high intracellular levels of particular lipid species might activate
mTOR directly, bypassing the normal requirement of growth factor and amino acid signaling.
This raises the possibility that p66Shc could be playing an indirect role in regulating mTOR
activity through its effect on modulating lipid metabolism.

109
5.5. Genes regulated through p66Shc expression
RNA-seq analysis showed that p66Shc-competent and p66Shc-deficient cells have comparable
expression levels of genes encoding metabolic enzymes and glucose transporters (Appendix
6.3). These results are surprising given that mTOR has been shown to regulate the transcription
of metabolic genes in a HIF1- and SREBP-dependent manner (Duvel et al., 2010). The
transcriptome of Tsc1 and Tsc2 KO MEFs revealed that mTORC1 promotes the transcription of
genes involved in glycolysis, the pentose phosphate pathway and de novo lipogenesis (Duvel et
al., 2010).
Regulation by allostery and posttranslational modifications to metabolic enzymes and
transporters, rather than transcriptional regulation, may account for much of the observed
p66Shc-depdendent metabolic reprogramming. However, the stable cell lines used in our
experiments have adapted to p66Shc expression or loss, and the associated changes in signaling
and metabolic flux may be reinforced by other changes in gene expression and posttranslational
modifications. Importantly, transcripts showing a change in abundance were mainly clustered in
the category of secretory proteins. The list of genes showing differential expression in p66Shc-
competent cells included transcripts that have been reported to be involved in regulating insulin
signaling, such as Wnt4 (Heller et al., 2011). Additional studies are required to assess their
contribution to the p66Shc metabolic phenotype.

110
5.6. Negative regulation of insulin signaling by adaptor proteins
In addition to p66Shc, other adaptor proteins, including IRS1, Grb10 and Grb14, facilitate
negative feedback inhibition of the insulin/IGF-1 signaling pathway. Major regulatory
mechanisms include phosphorylation and O-GlcNAcylation of Ser/Thr residues of the insulin
receptor substrate 1 (IRS1) (D'Alessandris et al., 2004; Harrington et al., 2004; Shah et al.,
2004). Repression of IRS1 gene expression and direct phosphorylation of IRS1 by S6K1, where
IRS1 becomes limiting for signal transmission from the insulin receptor to the PI3K-mTOR
pathway, drives constitutive activation of the mTORC1-S6K1 axis (Harrington et al., 2004; Shah
et al., 2004). In addition to p66Shc and IRS1, other adaptor proteins including the family of
growth factor receptor-bound proteins Grb10 and Grb14, and SH2B2 have been strongly linked
to the inhibition of insulin signaling.
Grb10 and Grb14 have emerged as major negative regulators of insulin/IGF-1 action (Holt and
Siddle, 2005). Various studies have provided links between these adaptors, the pathways they are
involved in and metabolic diseases such as diabetes. For example, phosphoproteomic analysis of
mTOR targets has revealed that Grb10 suppresses insulin sensitivity through feedback inhibition
of insulin-PI3K-mTOR signaling (Hsu et al., 2011; Yu et al., 2011). These reports showed that
overexpression of Grb10 and Grb14 inhibited insulin-stimulated tyrosine phosphorylation of
substrates, including IRS1, and downstream PI3K-Akt-mTORC1, while deficiency of Grb10
through gene silencing potentiated insulin-induced substrate phosphorylation (Hsu et al., 2011;
Kasus-Jacobi et al., 1998; Yu et al., 2011). Consistent with the cell culture data, disruption of the
maternal allele of Grb10 in mice improved insulin sensitivity and glucose tolerance, and
enhanced insulin-induced Akt activation (Smith et al., 2007; Wang et al., 2007). Similarly,

111
Grb14 knockout mice showed increased insulin sensitivity, glucose tolerance and activation of
PI3K-Akt in skeletal muscles and liver (Cooney et al., 2004). Deletion of the Sh2b2 gene in mice
led to an increase in insulin sensitivity and insulin-stimulated glucose transport in adipocytes
(Minami et al., 2003).
Further evidence of the link between adaptor proteins and metabolic diseases was provided from
human genome-wide association studies. Genome sequencing revealed an association between
single nucleotide polymorphisms (SNPs) at the GRB10 (Di Paola et al., 2006; Rampersaud et al.,
2007) and the GRB14 (Kooner et al., 2011; Morris et al., 2012) gene loci and incidence of type 2
diabetes, and with an increase in fasting blood glucose and insulin levels (Manning et al., 2012;
Scott et al., 2012). Overall, the role of these adaptor proteins as negative regulators of
physiological insulin/IGF1 signaling represents an exciting avenue for further investigation.
Much remains to be done to identify new binding partners and functions for p66Shc, Grb10 and
Grb14, to identify transcriptional factors that regulate their expression, and to assess the
implications of their altered signaling in human metabolic diseases.
5.7. Summary
Deficiency of the adaptor protein p66Shc improves glucose tolerance of mice. My results
indicate that the Shc1 proteins not only regulate mitogenic signaling, but also modify anabolic
metabolism in an mTOR-dependent manner. Using high-throughput metabolomic approaches, I
show that p66Shc silencing enhanced glucose metabolism, increased the abundance of
intermediates in various biosynthetic pathways and increased cell size. These changes in

112
metabolism required the mTOR pathway, which couples energy, nutrient, and growth factor
(including insulin) signals to processes that mediate cellular growth and metabolism. To my
knowledge, this is the first study that directly assesses the role of an adaptor-type protein in
regulating energy metabolism using a metabolomic approach. My data points to the prospect that
monitoring or modulating p66Shc abundance could be important in the management of
pathological conditions in which metabolic signaling is dysregulated, such as cancer and
diabetes.

113
Chapter 6. Appendix

114
6.1. LC-MS/MS transitions for the metabolites measured in this study
Name RT Q1 Q3 DP CE HMDB
(-)-Riboflavin 2.2 377 63 31 75 HMDB00244
1,4-diaminobutane 2.4 87 45 41 21 HMDB01414
1-Methylhistamine 3.3 126 109 30 20 HMDB00898
2`-Deoxyadenosine 6.2 252 136 40 20 HMDB00101
2`-Deoxycytidine 2.8 228 112 111 53 HMDB00014
2`-Deoxyuridine 2.8 229 113 50 20 HMDB00012
2'-Deoxy-D-ribose 3.9 134 117 31 15 HMDB03224
Tryptophanol 4.5 162 144 61 19 HMDB03447
3-OH-anthranilate 6.3 154 136 30 18 HMDB01476
4-guanidinobutyrate 2.5 146 87 76 25 HMDB03464
Acetoacetate 1.9 85 70 40 20 HMDB00060
Acetylcholine 2.5 147 87 25 21 HMDB00895
Adenosine 6.1 268 136 40 30 HMDB00050
(A)symetric dimethyl arginine 1.9 203 70 50 40 HMDB01539
Allantoin 6.5 159 116 50 11 HMDB00462
Aminoisobutyrate 2.4 104 86 40 16 HMDB01906
Anthranilate 9.4 138 120 25 18 HMDB01123
Argininosuccinate 3.2 291 70 50 54 HMDB00052
Betaine 10.3 118 58 40 41 HMDB00043
Carnitine 2.5 163 85 25 29 METPA0048
Carnosine 2.1 227 110 40 33 HMDB00033
Choline 2.8 105 60 50 27 HMDB00097
Cobalamin 18.1 678 147 60 52 HMDB02174
Creatine 3.2 132 90 50 17 HMDB00064
Creatinine 2.3 114 44 25 28 HMDB00562
Cysteamine 2.2 78 61 40 16 HMDB02991
Cysteine 3.2 122 76 25 20 METPA0075
Cytidine 2.8 244 112 25 17 HMDB00089
Cytosine 2.3 112 95 40 26 HMDB00630
D-alpha-Aminobutyrate 3.3 104 58 30 17 HMDB00452
Dimethylglycine 3.3 104 58 30 20 HMDB00092
DL-2-Aminoadipate 3.8 162 98 46 25 HMDB00510
DL-Homocystine 3.4 136 90 50 20 HMDB00575
Dopamine 3.9 154 137 50 16 HMDB00073
Epinephrine 11.5 184 166 27 13 HMDB00068
gamma-Aminobutyrate 2.4 104 87 41 15 HMDB00112
gamma-Aminoisobutyrate 2.7 104 86 40 16 HMDB00112
Glucosamine 2.2 180 162 50 10 HMDB01514
Gluconolactone 5.8 179 133 46 15 HMDB00150

115
Glycerol 3.6 93 57 30 10 HMDB00131
Glycine 3.1 76 30 20 21 HMDB00123
Guanidinoacetate 3.7 118 72 25 18 HMDB00128
Guanine 4.5 152 135 40 30 HMDB00132
Guanosine 6.2 284 152 40 25 HMDB00133
Histamine 2.3 112 95 40 26 HMDB00870
HO-Tryptophan 7.5 221 204 40 18 HMDB03447
Hydroxybutyrate 4.6 105 87 70 10 HMDB00008
Inosine 6 269 137 1 15 HMDB00195
Isobutyrylglycine 1.9 146 72 50 22 HMDB00730
Kynurenine 3.1 209 146 115 45 HMDB00684
L-Alanine 3.2 90 44 25 17 HMDB00161
L-alpha-Aminoadipate 3.9 163 73 41 37 HMDB00510
L-alpha-Aminobutyrate 3.3 104 58 30 17 HMDB00452
L-Arabinose 10.1 151 107 51 35 HMDB00646
L-Arginine 2.8 175 70 25 32 HMDB00517
L-Asparagine 3.2 133 74 30 23 HMDB00168
L-Aspartate 6.3 134 74 25 21 HMDB00191
L-Canavanine 2.1 177 76 56 35 HMDB02706
L-Carnitine 2.5 162 103 86 25 HMDB00062
L-Citrulline 3.2 176 70 41 27 HMDB00904
L-Cystathyonine 17.3 223 149 31 29 HMDB00099
L-Cystine 2.1 241 109 46 37 HMDB00192
L-Glutamate 3.8 148 84 25 23 HMDB00148
L-Glutamine 3.2 147 84 25 25 HMDB00641
L-Histidine 2.1 156 110 25 21 HMDB00177
L-Homoserine 3.2 120 74 50 40 HMDB00719
Lipoamide 7.5 206 189 41 15 HMDB00962
L-Isoleucine 4.8 132 86 50 18 HMDB00172
L-Leucine 5.6 132 86 50 18 HMDB00687
L-Lysine 2.1 147 84 25 25 HMDB00182
L-Methionine 4.6 150 61 40 31 HMDB00696
N-Monomethyl-L-arginine 3.2 189 70 60 40 NA
L-Phenylalanine 6.7 166 120 50 19 HMDB00159
L-Proline 3.5 116 70 50 20 HMDB00162
L-Sarcosine 2.9 91 73 46 17 HMDB00271
L-Serine 3.2 106 60 25 18 HMDB00187
L-Threonine 3.2 120 74 50 20 HMDB00167
L-Tryptophan 7.5 205 188 25 16 HMDB00929
L-Tyrosine 5.6 182 136 25 19 HMDB00158
L-Valine 3.7 118 72 25 18 HMDB00883
Melatonin 5.4 233 174 35 18 HMDB01389
Metanephrine 3.4 198 181 40 13 HMDB04063

116
N-Carbamoyl-Beta-Alanine 3.4 133 115 40 12 HMDB00026
Niacinamide 6 123 80 30 30 HMDB01406
Normetanephrine 11.5 184 166 27 13 HMDB00819
Hydroxy proline 3.3 132 86 50 18 HMDB00725
Ornithine 2 133 70 16 31 HMDB00214
Phosphocholine 4.2 184 125 66 20 HMDB01565
phosphoethanoloamine 3.2 142 44 50 20 HMDB00224
Phosphoserine 7.8 186 130 56 22 HMDB00272
Protoporphyrin IX 0.2 563 129 251 27 HMDB00241
Pyridoxal 3 168 150 51 79 HMDB01545
Pyridoxamine 11.7 169 123 21 23 HMDB01431
Pyridoxine 3 170 152 40 17 HMDB00239
S-(5`-Adenosyl)-L-homocysteine 5.4 385 136 60 25 HMDB00939
Serotonin 3.2 177 160 50 32 HMDB00259
Spermidine 1.9 146 72 46 18 HMDB01257
Spermine 1.9 203 129 40 25 HMDB01256
Taurine 3.3 126 108 50 20 HMDB00251
Thiamine 2.8 265 122 46 18 HMDB00235
Thiamine monophosphate 2.3 345 122 66 29 HMDB02666
Thymidine 6.8 243 127 30 35 HMDB00273
Thymine 6.5 127 110 40 16 HMDB00262
Trans-4-hydroxy-L-Proline 3.3 132 68 66 23 HMDB00725
Trimethylamine-N-oxide 2.9 76 58 25 35 HMDB00925
Uracil 5.3 113 70 71 29 HMDB00300
Uridine 5.9 245 113 96 25 HMDB00296
Xanthine 5.9 153 110 40 21 HMDB00292
Xanthosine 14.6 285 63 41 27 HMDB00299
2,3-Dihydroxybenzoate 8.8 153 109 -45 -30 HMDB00397
2,3-Pyridinedicarboxylate 5 166 122 -110 -54 HMDB00232
2-Aminoadipate 6.4 160 116 -50 -18 HMDB00510
2'-Deoxy cytidine 5'-
monophosphate (dCMP)
7.6 306 79 -60 -54 HMDB01202
2'-Deoxy cytidine diphosphate
(dCDP)
8.2 386 79 -30 -60 HMDB01245
2'-Deoxy thymidine
monophosphate (dTMP)
7 321 195 -60 -18 HMDB01227
2'-Deoxy uridine monophosphate
(dUMP)
7.5 307 79 -60 -50 HMDB01409
2'-Deoxyadenosine 8.6 250 134 -60 -24 HMDB00101
2'-Deoxycytidine 5-triphosphate
(dCTP)
8.5 466 159 -60 -30 HMDB00998
2'-dexyuridine 5-triphosphate
(dUTP)
11 467 159 -40 -40 HMDB01191
2-Oxobutyrate 8.1 101 57 -30 -10 HMDB00005
3-Indoleacetate 8.1 174 130 -25 -14 HMDB00197

117
3-Phosphoglycerate 8.3 185 97 -40 -22 HMDB00807
4-Aminobenzoate 7.8 136 92 -40 -14 HMDB01392
4-Hydroxy-3-
methoxyphenylglycolaldehyde
9.5 183 150 -40 -30 HMDB04061
4-Hydroxybenzoate 7.9 137 93 -20 -20 HMDB00500
4-Hydroxyphenylpyruvate 3.3 179 107 -110 -54 HMDB00707
4-Pyridoxate 8.8 182 138 -40 -30 HMDB00017
5-hydroxyindole-3-acetate 7.2 190 146 -50 -16 HMDB00763
5-Methyl THF 9.5 485 329 -70 -30 HMDB01396
Acetoacetate 8.1 101 57 -40 -15 HMDB00060
Acetyl Co-enzyme A (ACoA) 8.7 808 159 -150 -88 HMDB01206
Adenine 5.5 134 107 -65 -24 HMDB00034
Adenosine 5'-monophosphate
(AMP)
8.3 346 79 -30 -66 HMDB00045
Adenosine diphosphate (ADP) 8.6 426 159 -75 -36 HMDB01341
Adenosine triphosphate (ATP) 8.6 506 159 -100 -44 HMDB00538
Adenylosuccinate 8.3 462 79 -60 -48 HMDB00536
Adipate 9.1 145 101 -110 -54 HMDB00448
α-ketoglutarate 8.2 145 101 -20 -12 HMDB00208
Ascorbate 8.1 175 115 -40 -17 HMDB00044
Bilirubin 11.1 583 285 -30 -36 HMDB00054
Biotin 4.8 243 200 -60 -18 HMDB00030
Butyrate 7.9 87 45 -30 -10 HMDB00039
Chenodeoxycholate 13.4 391 374 -80 -50 HMDB00518
Cholate 4.3 407 343 -100 -20 HMDB00619
Aconitate 8.3 173 85 -25 -16 HMDB00072
Citrate 8.7 191 111 -25 -18 HMDB00094
Co-enzyme A (CoA) 8.7 766 159 -135 -86 HMDB01423
Creatine phosphate 8 210 79 -35 -24 HMDB01511
Cyclic adenosine monophosphate
(cAMP)
8.6 328 134 -80 -42 HMDB11616
Cyclic guanosine monophosphate
(cGMP)
8.8 344 150 -40 -34 HMDB11616
Cytidine 5-diphosphate (CDP) 8.5 402 158 -60 -30 HMDB01546
Cytidine 5-triphosphate (CTP) 8.5 482 159 -60 -40 HMDB00082
Cytidine monophosphate (CMP) 7.5 322 139 -70 -32 HMDB00095
D7-Glucose (Internal Standard) 3.4 186 124 -55 -12 NA
D-Arabino-1,4-lactone 7.7 147 59 -55 -18 METPA0132
Dihydrofolate 9.5 442 176 -50 -40 HMDB01056
Dihydroxyacetone phosphate
(DHAP)
7.4 169 97 -40 -15 HMDB01473
Isocitrate 8.7 191 111 -35 -20 HMDB00193
D-Maltose 3.2 341 161 -40 -12 HMDB00163
D-Pantothenate 7.5 218 88 -30 -16 HMDB00210

118
D-Rib(ul)ose-5-Phosphate (R5P) 7.5 229 79 -15 -58 HMDB01548
D-Xylose 3.4 149 89 -50 -8 HMDB00098
Erythrose-4-Phosphate (E4P) 7.3 199 97 -40 -16 HMDB01321
Folate 8 440 311 -80 -30 HMDB00121
Folinate 13.4 472 315 -40 -40 HMDB01562
Fructose 1,6-bisphosphate (Fru 1,6
DP)
8.3 339 241 -30 -22 HMDB01058
Fructose 6-phosphate (Fru-6P) 6.9 259 169 -50 -18 HMDB00124
Fumarate 8.1 115 71 -30 -10 HMDB00134
Geranyl pyrophosphate 7.3 313 79 -35 -37 METPA0034
Glucoronate 8.6 193 113 -60 -22 HMDB00127
Glucosamine (GlcN) 3.5 178 145 -30 -12 HMDB01514
Glucosamine 6-Phosphate (GlcNP) 4.1 258 97 -75 -18 HMDB01254
Glucose (Glc) 3.3 179 119 -60 -12 HMDB00122
Glucose 6-Phosphate 6.9 259 199 -55 -16 HMDB01401
Glyceraldehyde (GA) 3.4 89 59 -35 -10 HMDB01051
Glycerate 2-P 8.3 185 79 -40 -30 HMDB00362
Glycerol-3-Phosphate (G3P) 7.2 171 79 -50 -22 HMDB00126
Glycochenodeoxycholate 10 448 74 -80 -58 HMDB00637
Glycocholate 13.9 464 74 -30 -60 HMDB00138
Glyoxylate 7.4 73 45 -45 -10 HMDB00119
Guanosine diphosphate (GDP) 8.7 442 159 -75 -38 HMDB01201
Guanosine diphosphate- Fucose
(GDP-Fuc)
8 588 159 -100 -60 HMDB01095
Guanosine diphosphate-Mannose
(GDP-Man)
8.7 604 159 -120 -70 HMDB01163
Guanosine monophosphate (GMP) 7.5 362 79 -70 -60 HMDB01397
Guanosine triphosphate (GTP) 8.8 522 159 -95 -48 HMDB01273
Hippurate 3.5 178 134 -50 -16 HMDB00714
Homocystine 8.9 267 132 -30 -22 HMDB00575
Homogentisate 6.2 167 123 -50 -23 HMDB00130
Homovanillate 5.7 181 137 -50 -12 HMDB00118
Hypoxanthine 4.8 135 92 -30 -22 HMDB00157
Inosine 5'-monophosphate (IMP) 7.7 347 79 -65 -100 HMDB00175
Inositol 7.2 179 81 -50 -15 HMDB00211
Itaconate 8.2 129 85 -40 -12 HMDB02092
Kynurenate 8.1 188 144 -30 -20 HMDB00715
L-(-)-Sorbose 3.3 179 89 -40 -12 HMDB01266
Lactose (L) 3.3 341 161 -50 -11 HMDB00186
L-Dihydroorotate 5.7 157 113 -35 -12 HMDB03349
L-Fucose 3.7 163 103 -55 -10 HMDB00174
L-Lactate 7.41 89 71 -55 -16 HMDB00190
L-Malate 9 133 115 -40 -14 HMDB00156
Maleate 7.4 115 71 -110 -54 HMDB00176

119
Malonate 7.3 103 59 -40 -15 HMDB00691
Malonyl Co-enzyme A (MCoA) 8.7 852 159 -130 -96 HMDB01175
Melibiose 3.2 341 89 -50 -28 HMDB00048
Methylmalonate 2.7 117 73 -110 -54 HMDB00202
Mevalonate 7.4 147 59 -40 -19 HMDB00227
myo-inositol 3.2 179 87 -65 -26 HMDB00211
N-acetylglucosamine (GlcNAc) 3.5 220 119 -60 -10 HMDB00215
N-acetylglucosamine phosphate
(GlcNAcP)
7 300 199 -75 -20 HMDB02817
N-Acetylglutamate 8.1 188 143 -35 -18 HMDB01138
NADH 8.1 664 408 -130 -46 HMDB01487
NADPH 11.6 372 134 -60 -30 HMDB00221
Neopteron 5.8 252 192 -50 -22 HMDB00845
Nicotinamide adenine dinucleotide
(NAD+)
6.9 662 540 -90 -28 HMDB00902
Nicotinamide adenine dinucleotide
phosphate (NADP)
9 371 309 -40 -10 HMDB00217
Nicotinate ribonucleotide 9.9 334 290 -50 -13 HMDB01132
Nicotinate 7.8 122 78 -35 -18 HMDB01488
hydroxyphenylpyruvate 3.3 179 107 -40 -12 HMDB00205
o-Nitrophenol 4.9 138 108 -50 -22 HMDB01232
o-Phosphoryl-ethanol-amine 4.7 140 79 -110 -54 HMDB00224
Orotate 8.8 155 111 -50 -22 HMDB00226
Orotidine 5'-monophosphate
(OMP)
3.6 367 323 -50 -17 HMDB00218
Oxaloacetate 8.7 133 87 -30 -12 HMDB00223
Oxidized glutathione (GSSG) 7.8 611 306 -60 -60 HMDB03337
Palmitate 6.5 255 69 -75 -80 HMDB00220
Pantothenate 7.5 218 88 -55 -22 HMDB00210
Phenylpyruvate 9.5 163 91 -25 -14 HMDB00205
Phosphoenolpyruvate 8.5 167 79 -40 -31 HMDB00263
Phosphotyrosine 11.1 260 79 -50 -30 HMDB06049
Phytate 11.6 329 79 -40 -98 HMDB03502
PPA 9.5 163 91 -40 -15 HMDB04110
Propionate 4 73 55 -30 -20 HMDB00237
Prostaglandin E2 14.3 351 315 -40 -20 HMDB01220
Pyridoxal-5-Phosphate 7.4 246 97 -60 -20 HMDB01491
Pyruvate 7.7 87 43 -35 -10 HMDB00243
Quinolinate 9.1 166 122 -30 -13 HMDB00232
Reduced glutathione (GSH) 7 306 143 -40 -32 HMDB00125
Salicylurate 12.3 194 150 -60 -20 HMDB00840
Shikimate 8.9 173 93 -25 -20 HMDB03070
Sialate 7 308 170 -50 -22 HMDB00230
Sorbitol 3.4 181 89 -60 -20 HMDB00247

120
Succinate 8 117 73 -30 -16 HMDB00254
Succinyl Co-enzyme A (SCoA) 8.8 866 159 -135 -90 HMDB01022
Taurochenodeoxycholate 14.3 498 80 -90 -88 HMDB00951
Taurocholate 10.5 514 80 -50 -90 HMDB00036
Thiamine pyrophosphate 3.6 424 382 -10 -24 HMDB01372
Trehalose 3.2 341 59 -80 -52 HMDB00975
UDP-GlcNAc 8 606 159 -110 -66 HMDB00290
UDP-glucuronate (UDP-G) 8.5 579 403 -50 -28 HMDB00935
Urate 6.2 167 124 -60 -22 HMDB00289
Uridine 5'-monophosphate (UMP) 7.9 323 79 -65 -66 HMDB00288
Uridine 5-triphosphate (UTP) 8.6 483 159 -75 -45 HMDB00285
Uridine diphosphate (UDP) 8.6 403 159 -90 -36 HMDB00295
Uridine diphosphate-glucose 8.1 565 323 -85 -30 HMDB00286
Xanthosine 5-monophosphate
(XMP)
7.4 363 779 -40 -60 METPA1379
Xanthurenate 8.3 204 160 -30 -33 HMDB00881
Xyl(ul)ose-5P 7.5 229 97 -30 -10 HMDB00868

121
6.2. LC-MS/MS transitions for 1,2-13
C2 Glucose intermediates.
DP: de-clustering potential, EP: entrance potential, CE: Collision energy, CXP: collision exit
potential
Q1 Q3 RT ID DP CE CXP
87 43 7.41
Pyruvate -35 -14 -3
89 45 [1,2]13
C-Pyruvate [M2] -35 -14 -3
89 43
7.41
Lactate -55 -16 -1
90 44 [1]13
C-lactate [M1] -55 -16 -1
91 45 [1,2]13
C-lactate [M2] -55 -16 -1
191 111 8.6
Citrate -30 -18 -7
193 112 [1,2]13
C-Citrate [M2] -30 -18 -7
259 97 7.33 G6P -40 -28 -5
261 79 7.33
[1,2]13
C-G6P [M2] -40 -28 -5
261 97 [1,2]13
C-G6P [M2] -40 -28 -5
259 79 8.26 D-Fructose-6P -55 -72 -11
261 79 8.26 [1,2]13
C-Fructose-6P [M2] -55 -72 -11
300 79 7.7
GlcNAcP -45 -76 -15
302 79 [1,2]13
C-GlcNAcP [M2] -60 -76 -15
606 159
8.5
UDP-GlcNAc -110 -66 -1
608 159 UDP-[1,2]13
C-GlcNAc [M2] -110 -66 -1
610 159 UDP-[1,2]13
C-GlcNAc [M4] -110 -66 -1
808 159
8.5
Acetyl Co-enzyme A (ACoA) -150 -88 -9
809 159 [2]13
C-Acetyl Co-enzyme A (ACoA) [M1] -150 -88 -9
810 159 [1,2]13
C-Acetyl Co-enzyme A (ACoA) [M2] -150 -88 -9

122
6.3. List of gene differentially regulated by p66Shc expression
Gene Gene full Name p66Shc KO -
p66+(average log 10)
p-value SD
Lrp2 low density lipoprotein receptor-
related protein 2
-1.915792811 0.00053 0.265341548
Lingo4 leucine rich repeat and Ig domain
containing 4
-1.811543741 0.00053 0.08564836
Fam19a2 family with sequence similarity 19,
member A2
-1.635935145 0.00053 0.094915819
Fras1 Fraser syndrome 1 homolog (human) -1.632833069 0.00053 0.088349326
Epha7 Eph receptor A7 -1.617034208 0.00053 0.265811188
2810405K02Rik RIKEN cDNA 2810405K02 gene -1.545497146 0.00053 0.104072241
Pcdhgb8 protocadherin gamma subfamily B, 8 -1.505347374 0.00053 0.134934987
Nol4 nucleolar protein 4 -1.491293872 0.00053 0.147634015
Elavl2 ELAV (embryonic lethal, abnormal
vision, Drosophila)-like 2 (Hu antigen
B)
-1.475961437 0.00053 0.172756757
Slc38a4 solute carrier family 38, member 4 -1.450549921 0.00053 0.082508643
Slc16a7 solute carrier family 16
(monocarboxylic acid transporters),
member 7
-1.422033949 0.00053 0.180917562
Fam132b family with sequence similarity 132,
member B
-1.408791619 0.00053 0.067851145
Fndc3c1 fibronectin type III domain containing
3C1
-1.317312614 0.00053 0.082532254
Cpa6 carboxypeptidase A6 -1.311506175 0.00053 0.39594187
Lrrtm1 leucine rich repeat transmembrane
neuronal 1
-1.30232534 0.00053 0.261829654
Tmem178 transmembrane protein 178 -1.229814213 0.00053 0.252731011
Pcdh9 protocadherin 9 -1.209521591 0.00053 0.06012591
Rnf128 ring finger protein 128 -1.200148584 0.00053 0.295306529
Mfap3l microfibrillar-associated protein 3-like -1.184275636 0.00053 0.471271669

123
Nr3c2 nuclear receptor subfamily 3, group C,
member 2
-1.179205418 0.00053 0.245215328
Epha3 Eph receptor A3 -1.141945788 0.00053 0.18094104
Pde3b phosphodiesterase 3B, cGMP-
inhibited
-1.126724114 0.00053 0.058435767
Armcx4 armadillo repeat containing, X-linked
4
-1.118182406 0.00053 0.009426341
Pcsk5 proprotein convertase subtilisin/kexin
type 5
-1.110683672 0.00053 0.122133709
Mycn v-myc myelocytomatosis viral related
oncogene, neuroblastoma derived
(avian)
-1.109619722 0.00053 0.085221891
Fibin fin bud initiation factor homolog
(zebrafish)
-1.075052764 0.00053 0.053954081
Mei4 meiosis-specific, MEI4 homolog (S.
cerevisiae)
-1.027144733 0.00054 0.404083607
Slc26a7 solute carrier family 26, member 7 -1.018616084 0.00053 0.265390359
Pmaip1 phorbol-12-myristate-13-acetate-
induced protein 1
-0.991734978 0.00053 0.180923943
Sfrp2 secreted frizzled-related protein 2 -0.974846673 0.00053 0.172783959
Tll1 tolloid-like -0.957443259 0.00053 0.175013394
Tmem151b transmembrane protein 151B -0.952357544 0.00053 0.148567314
Tnfrsf21 tumor necrosis factor receptor
superfamily, member 21
-0.92950432 0.00053 0.067073319
Lef1 lymphoid enhancer binding factor 1 -0.929191993 0.00054 0.311455724
Hunk hormonally upregulated Neu-
associated kinase
-0.925378588 0.00054 0.488512276
Tdrkh tudor and KH domain containing
protein
-0.924298141 0.00053 0.016466993
Plcl1 phospholipase C-like 1 -0.922570232 0.00053 0.050416995
Pkia protein kinase inhibitor, alpha -0.917178399 0.00053 0.145255127
Tm6sf2 transmembrane 6 superfamily member
2
-0.909526811 0.00054 0.265433652
Mtap7d3 MAP7 domain containing 3 -0.905721395 0.00054 0.265381819

124
Zfp583 zinc finger protein 583 -0.89964319 0.00054 0.231461609
Fam38b family with sequence similarity 38,
member B
-0.886354776 0.00054 0.393893458
Sort1 sortilin 1 -0.883128964 0.00053 0.036959691
Hist1h2bg histone cluster 1, H2bg -0.870775698 0.00054 0.08807413
Snca synuclein, alpha -0.850845519 0.00054 0.101000827
Dok5 docking protein 5 -0.829479412 0.00054 0.098944847
Rell2 RELT-like 2 -0.824736054 0.00054 0.261803243
Gabre gamma-aminobutyric acid (GABA) A
receptor, subunit epsilon
-0.824104256 0.00054 0.134844879
Hist1h4k histone cluster 1, H4k -0.821945916 0.00054 0.296458294
Gm10406 predicted gene 10406 -0.808209064 0.00054 0.180904117
Cacna1b calcium channel, voltage-dependent, N
type, alpha 1B subunit
-0.805835164 0.00054 0.143616773
Hoxb9 homeobox B9 -0.797828438 0.00054 0.198343907
Asxl3 additional sex combs like 3
(Drosophila)
-0.791955705 0.00054 0.054836257
Pcdhb2 protocadherin beta 2 -0.787980234 0.00054 0.160613469
Snx10 sorting nexin 10 -0.784827431 0.00054 0.100231686
Pcdhb3 protocadherin beta 3 -0.779873929 0.00054 0.114090528
4921528I01Rik RIKEN cDNA 4921528I01 gene -0.777509771 0.00054 0.192939047
Wnt2b wingless related MMTV integration
site 2b
-0.755864987 0.00054 0.0903634
Peg10 paternally expressed 10 -0.744421658 0.00054 0.10331376
Fndc5 fibronectin type III domain containing
5
-0.744227916 0.00054 0.074817703
Ntng2 netrin G2 -0.741595235 0.00054 0.121176342
Klf12 Kruppel-like factor 12 -0.738830144 0.00054 0.041258524
Npy1r neuropeptide Y receptor Y1 -0.736048621 0.00054 0.125603467
Gpr137c G protein-coupled receptor 137C -0.73215916 0.00059 0.534608914
8430408G22Rik RIKEN cDNA 8430408G22 gene -0.724087278 0.00055 0.222465861

125
Bhlhe22 basic helix-loop-helix family, member
e22
-0.718249387 0.00054 0.150545533
Nlrx1 NLR family member X1 -0.713254896 0.00054 0.030703712
Elovl4 elongation of very long chain fatty
acids (FEN1/Elo2, SUR4/Elo3, yeast)-
like 4
-0.711290071 0.00055 0.223211263
Chdh choline dehydrogenase -0.705962742 0.00056 0.361115226
Sema6a sema domain, transmembrane domain
(TM), and cytoplasmic domain,
(semaphorin) 6A
-0.703651997 0.00054 0.059363453
Cthrc1 collagen triple helix repeat containing
1
-0.701838551 0.00055 0.173849535
Enox1 ecto-NOX disulfide-thiol exchanger 1 -0.699117306 0.00054 0.114088181
Fbxl7 F-box and leucine-rich repeat protein 7 -0.698448854 0.00054 0.044721972
Snrpn small nuclear ribonucleoprotein N -0.696287165 0.00055 0.123683742
1700010I14Rik RIKEN cDNA 1700010I14 gene -0.687747012 0.00056 0.2527405
Tmem169 transmembrane protein 169 -0.686456758 0.00057 0.395917183
Palm3 paralemmin 3 -0.684145211 0.00059 0.50659648
Tnfaip8l1 tumor necrosis factor, alpha-induced
protein 8-like 1
-0.683349349 0.00055 0.029609797
Cd200 CD200 antigen -0.681445884 0.00055 0.046093045
Pcdh8 protocadherin 8 -0.680995051 0.00056 0.27531672
Dclk2 doublecortin-like kinase 2 -0.676081914 0.00055 0.134788259
Igf2 insulin-like growth factor 2 -0.674359709 0.00055 0.036664029
Fut4 fucosyltransferase 4 -0.672636529 0.00057 0.404091035
F2rl1 coagulation factor II (thrombin)
receptor-like 1
-0.664956186 0.00055 0.016152716
Tmem200a transmembrane protein 200A -0.663179322 0.00055 0.036468114
Tbx2 T-box 2 0.663640332 0.00055 0.021998753
Gm1661 predicted gene 1661 0.665248537 0.00055 0.108950542
Diras2 DIRAS family, GTP-binding RAS-
like 2
0.667441057 0.00056 0.246125103

126
Rab3b RAB3B, member RAS oncogene
family
0.667673045 0.00055 0.087214478
Col28a1 collagen, type XXVIII, alpha 1 0.668464877 0.00055 0.103303578
Agt angiotensinogen (serpin peptidase
inhibitor, clade A, member 8)
0.668787912 0.00055 0.059258563
Slc22a23 solute carrier family 22, member 23 0.670209459 0.00055 0.036892269
Il13ra2 interleukin 13 receptor, alpha 2 0.670898039 0.00055 0.077902592
Syt5 synaptotagmin V 0.673105204 0.00055 0.134876587
Cfh complement component factor h 0.673403902 0.00055 0.014587192
Cdh26 cadherin-like 26 0.674748099 0.00055 0.171407963
1700034H15Rik RIKEN cDNA 1700034H15 gene 0.676046005 0.00056 0.339792831
Lgals9 lectin, galactose binding, soluble 9 0.678298366 0.00055 0.051080452
Gm14393 predicted gene 14393 0.680908099 0.00058 0.403850786
Adh7 alcohol dehydrogenase 7 (class IV),
mu or sigma polypeptide
0.68327306 0.00055 0.152894719
Cygb cytoglobin 0.683903613 0.00055 0.157736829
4-Sep septin 4 0.684369152 0.00061 0.376028174
Nsg1 neuron specific gene family member 1 0.68443605 0.00055 0.027139644
Tmem117 transmembrane protein 117 0.6868859 0.00055 0.123397066
Siglecg sialic acid binding Ig-like lectin G 0.687365882 0.00055 0.072380418
Cbln3 cerebellin 3 precursor protein 0.688270313 0.00055 0.066008871
Islr immunoglobulin superfamily
containing leucine-rich repeat
0.692047166 0.00054 0.019871733
Adamtsl1 ADAMTS-like 1 0.695033204 0.00054 0.104145348
Lama3 laminin, alpha 3 0.69513384 0.00054 0.108472544
Acy3 aspartoacylase (aminoacylase) 3 0.696801941 0.00054 0.086061403
Atf3 activating transcription factor 3 0.69773903 0.00054 0.024900306
Rsph1 radial spoke head 1 homolog
(Chlamydomonas)
0.701830671 0.00055 0.207766755
Phactr1 phosphatase and actin regulator 1 0.703918435 0.00054 0.055588964

127
9930023K05Rik RIKEN cDNA 9930023K05 gene 0.704413698 0.00058 0.462960081
Atp8b1 ATPase, class I, type 8B, member 1 0.704855288 0.00054 0.066155661
Timp3 tissue inhibitor of metalloproteinase 3 0.705980282 0.00054 0.009519119
Ticam2 toll-like receptor adaptor molecule 2 0.706980197 0.00055 0.216094637
Nhsl2 NHS-like 2 0.708152301 0.00055 0.187820581
Tcf7 transcription factor 7, T cell specific 0.710676527 0.00054 0.079743011
Asgr1 asialoglycoprotein receptor 1 0.710908571 0.00054 0.102195858
Gem GTP binding protein (gene
overexpressed in skeletal muscle)
0.711129665 0.00054 0.074775012
Acss1 acyl-CoA synthetase short-chain
family member 1
0.713136814 0.00056 0.332812454
Adrbk2 adrenergic receptor kinase, beta 2 0.71389249 0.00054 0.017370486
Mapkapk3 mitogen-activated protein kinase-
activated protein kinase 3
0.717198672 0.00054 0.11169346
Map3k5 mitogen-activated protein kinase
kinase kinase 5
0.722403796 0.00054 0.060276991
Islr2 immunoglobulin superfamily
containing leucine-rich repeat 2
0.723526789 0.00054 0.137727559
Ggt7 gamma-glutamyltransferase 7 0.724810409 0.00054 0.121828837
Zfp296 zinc finger protein 296 0.726254956 0.00059 0.440252288
Sh2d1b1 SH2 domain protein 1B1 0.726373223 0.00054 0.086700286
B3gnt8 UDP-GlcNAc:betaGal beta-1,3-N-
acetylglucosaminyltransferase 8
0.729573708 0.00057 0.303349205
Tnni3 troponin I, cardiac 3 0.731254977 0.00058 0.514112037
9130019O22Rik RIKEN cDNA 9130019O22 gene 0.732389495 0.00054 0.098879186
Ccbe1 collagen and calcium binding EGF
domains 1
0.733942913 0.00054 0.137946031
Gstt1 glutathione S-transferase, theta 1 0.734574564 0.00054 0.095911655
Rtp4 receptor transporter protein 4 0.737249757 0.00054 0.050153752
Abi3 ABI gene family, member 3 0.739334553 0.00054 0.101934592
Aldh1a3 aldehyde dehydrogenase family 1,
subfamily A3
0.742734742 0.00054 0.085524474

128
Rasl11a RAS-like, family 11, member A 0.743417171 0.00054 0.069964518
Epb4.1l4a erythrocyte protein band 4.1-like 4a 0.743720766 0.00054 0.067778459
Ica1 islet cell autoantigen 1 0.746449159 0.00058 0.532783978
Hyal3 hyaluronoglucosaminidase 3 0.746670131 0.00058 0.532544673
Ifi27l2a interferon, alpha-inducible protein 27
like 2A
0.748805838 0.00054 0.128921448
Matn4 matrilin 4 0.751296432 0.00055 0.309099514
Afap1l2 actin filament associated protein 1-like
2
0.754643517 0.00054 0.040200803
Abcc3 ATP-binding cassette, sub-family C
(CFTR/MRP), member 3
0.755485412 0.00055 0.246203752
Deptor DEP domain containing MTOR-
interacting protein
0.758137134 0.00054 0.116223933
Smoc1 SPARC related modular calcium
binding 1
0.759456057 0.00056 0.432661364
Dcn decorin 0.759916709 0.00054 0.063105273
Cldn1 claudin 1 0.761833728 0.00054 0.04408684
Traf1 TNF receptor-associated factor 1 0.764048952 0.00054 0.065079109
Cfb complement factor B 0.76455062 0.00054 0.088308557
Vtcn1 V-set domain containing T cell
activation inhibitor 1
0.765821423 0.00055 0.267925708
Acta1 actin, alpha 1, skeletal muscle 0.765964795 0.00054 0.043835532
Gm12216 predicted gene 12216 0.76644448 0.00054 0.135900469
1700003F12Rik RIKEN cDNA 1700003F12 gene 0.768055495 0.00054 0.089105998
Saa3 serum amyloid A 3 0.773580491 0.00054 0.173053452
Cdhr1 cadherin-related family member 1 0.774388432 0.00054 0.045177575
Ptgfr prostaglandin F receptor 0.775513945 0.00054 0.090611927
Susd2 sushi domain containing 2 0.775594375 0.00054 0.097255044
Cdkn1c cyclin-dependent kinase inhibitor 1C
(P57)
0.776172936 0.00054 0.06379327
Mndal myeloid nuclear differentiation antigen
like
0.779296909 0.00055 0.379651909

129
Clec11a C-type lectin domain family 11,
member a
0.780058561 0.00054 0.055581965
Apol10b apolipoprotein L 10B 0.782834626 0.00054 0.137041661
Cyp26b1 cytochrome P450, family 26,
subfamily b, polypeptide 1
0.782836703 0.00054 0.117528419
Kctd14 potassium channel tetramerisation
domain containing 14
0.784766049 0.00056 0.360430947
AI428936 expressed sequence AI428936 0.787294696 0.00054 0.099279262
Adam33 a disintegrin and metallopeptidase
domain 33
0.788839092 0.00054 0.085605242
Aox1 aldehyde oxidase 1 0.790952518 0.00054 0.028503273
Blnk B cell linker 0.790963356 0.00054 0.301575058
Ecscr endothelial cell surface expressed
chemotaxis and apoptosis regulator
0.793488682 0.00054 0.035874271
Rarres2 retinoic acid receptor responder
(tazarotene induced) 2
0.793997302 0.00054 0.137749917
Epas1 endothelial PAS domain protein 1 0.794441813 0.00054 0.093825829
Fam179a family with sequence similarity 179,
member A
0.797152443 0.00054 0.085500996
Sncg synuclein, gamma 0.797621076 0.00054 0.089113939
Angptl7 angiopoietin-like 7 0.79815192 0.00054 0.044478057
Eln elastin 0.798869667 0.00054 0.018790951
Gprin3 GPRIN family member 3 0.800688631 0.00054 0.128589472
Rhox5 reproductive homeobox 5 0.801127842 0.00055 0.265045427
Pcdhga2 protocadherin gamma subfamily A, 2 0.802342483 0.00055 0.4339809
Ppl periplakin 0.806596571 0.00054 0.017650785
Pdzrn4 PDZ domain containing RING finger
4
0.816216272 0.00054 0.03802449
Bdh1 3-hydroxybutyrate dehydrogenase,
type 1
0.818013017 0.00054 0.13315328
Gbp6 guanylate binding protein 6 0.820104623 0.00054 0.059587685
Auts2 autism susceptibility candidate 2 0.823419711 0.00054 0.017035484

130
Peg3 paternally expressed 3 0.824601124 0.00054 0.095374777
Slfn2 schlafen 2 0.828174735 0.00054 0.180890243
Espnl espin-like 0.829729488 0.00054 0.187549219
Raet1d retinoic acid early transcript delta 0.831300419 0.00054 0.084477615
Col10a1 collagen, type X, alpha 1 0.834622555 0.00054 0.043812899
Usp18 ubiquitin specific peptidase 18 0.841243699 0.00054 0.33200192
Rab40b Rab40b, member RAS oncogene
family
0.845386805 0.00054 0.216744132
Ddit4l DNA-damage-inducible transcript 4-
like
0.846972376 0.00054 0.038527775
Panx1 pannexin 1 0.847590486 0.00054 0.159315477
Isg15 ISG15 ubiquitin-like modifier 0.856119102 0.00054 0.114701267
Lxn latexin 0.85697897 0.00054 0.065921894
Cercam cerebral endothelial cell adhesion
molecule
0.860725077 0.00054 0.094089887
Ankrd6 ankyrin repeat domain 6 0.861965839 0.00054 0.148669382
Naalad2 N-acetylated alpha-linked acidic
dipeptidase 2
0.863873157 0.00054 0.043166595
Ceacam1 carcinoembryonic antigen-related cell
adhesion molecule 1
0.868657678 0.00054 0.09876767
Nptx1 neuronal pentraxin 1 0.868733414 0.00055 0.432899662
Tnfrsf9 tumor necrosis factor receptor
superfamily, member 9
0.869749964 0.00054 0.0822082
Apol6 apolipoprotein L 6 0.870822486 0.00054 0.096801623
Thsd4 thrombospondin, type I, domain
containing 4
0.874631254 0.00054 0.375134475
Pcdhb13 protocadherin beta 13 0.877924773 0.00054 0.085463636
Acsbg1 acyl-CoA synthetase bubblegum
family member 1
0.878723314 0.00054 0.30885885
Serpina3n serine (or cysteine) peptidase inhibitor,
clade A, member 3N
0.879110484 0.00053 0.041690783
Aoc3 amine oxidase, copper containing 3 0.881136113 0.00053 0.018828905

131
5430407P10Rik RIKEN cDNA 5430407P10 gene 0.881624059 0.00053 0.072724759
Serpina3m serine (or cysteine) peptidase inhibitor,
clade A, member 3M
0.883754442 0.00053 0.060574541
Bst1 bone marrow stromal cell antigen 1 0.886785141 0.00056 0.540574601
Stac2 SH3 and cysteine rich domain 2 0.892804709 0.00054 0.147549381
Gjb3 gap junction protein, beta 3 0.893450437 0.00055 0.431657445
Nap1l3 nucleosome assembly protein 1-like 3 0.893947626 0.00054 0.139544026
Kng2 kininogen 2 0.895607628 0.00053 0.074813929
Il33 interleukin 33 0.896838083 0.00054 0.144757383
Ngfr nerve growth factor receptor (TNFR
superfamily, member 16)
0.898674402 0.00053 0.085524089
Sorl1 sortilin-related receptor, LDLR class
A repeats-containing
0.899089977 0.00054 0.152335302
H2-T10 histocompatibility 2, T region locus 10 0.902292843 0.00054 0.380676837
Insc inscuteable homolog (Drosophila) 0.90417236 0.00053 0.112156662
Spink2 serine peptidase inhibitor, Kazal type
2
0.905591442 0.00054 0.14676946
Clstn3 calsyntenin 3 0.908201748 0.00053 0.089411025
Gpr4 G protein-coupled receptor 4 0.909466117 0.00054 0.216203667
Cxcl17 chemokine (C-X-C motif) ligand 17 0.909828619 0.00055 0.537563025
Abcg1 ATP-binding cassette, sub-family G
(WHITE), member 1
0.91071001 0.00054 0.274435175
Plcd4 phospholipase C, delta 4 0.913182402 0.00054 0.171352782
Ifit1 interferon-induced protein with
tetratricopeptide repeats 1
0.918782586 0.00053 0.175317001
Cd74 CD74 antigen (invariant polypeptide
of major histocompatibility complex,
class II antigen-associated)
0.926534365 0.00053 0.095906798
Rhpn2 rhophilin, Rho GTPase binding
protein 2
0.927138782 0.00053 0.08551548
Dgkg diacylglycerol kinase, gamma 0.927520067 0.00053 0.100662246
Adh1 alcohol dehydrogenase 1 (class I) 0.928183835 0.00054 0.359441834

132
Edar ectodysplasin-A receptor 0.928254958 0.00053 0.072900312
Il2rb interleukin 2 receptor, beta chain 0.928805896 0.00054 0.267954993
Aim1 absent in melanoma 1 0.929351782 0.00053 0.057430612
Prex2 phosphatidylinositol-3,4,5-
trisphosphate-dependent Rac exchange
factor 2
0.93719502 0.00053 0.08997535
Ptk2b PTK2 protein tyrosine kinase 2 beta 0.938135831 0.00053 0.027228824
Ldhd lactate dehydrogenase D 0.93871423 0.00054 0.439647844
1700024P16Rik RIKEN cDNA 1700024P16 gene 0.938819604 0.00054 0.360716271
Gstt3 glutathione S-transferase, theta 3 0.948569948 0.00053 0.024634112
Sim2 single-minded homolog 2
(Drosophila)
0.953991468 0.00053 0.143385491
Efhd1 EF hand domain containing 1 0.956252223 0.00053 0.103256292
Oasl2 2'-5' oligoadenylate synthetase-like 2 0.959954864 0.00053 0.023324485
Neurl3 neuralized homolog 3 homolog
(Drosophila)
0.964136118 0.00053 0.134849684
Kng1 kininogen 1 0.977131516 0.00053 0.049991385
Sp7 Sp7 transcription factor 7 0.978273933 0.00054 0.309076084
Parm1 prostate androgen-regulated mucin-
like protein 1
0.980508708 0.00053 0.058662293
Foxf2 forkhead box F2 0.981038713 0.00053 0.059632285
C1qtnf5 C1q and tumor necrosis factor related
protein 5
0.982903387 0.00054 0.290011907
Spib Spi-B transcription factor (Spi-1/PU.1
related)
0.983279881 0.00053 0.157542051
Fgd4 FYVE, RhoGEF and PH domain
containing 4
0.984260836 0.00053 0.139318882
D14Ertd668e DNA segment, Chr 14, ERATO Doi
668, expressed
0.988864035 0.00053 0.127634024
Dll1 delta-like 1 (Drosophila) 0.992361465 0.00054 0.432629062
Rnase1 ribonuclease, RNase A family, 1
(pancreatic)
0.993085648 0.00054 0.285009166
Acox2 acyl-Coenzyme A oxidase 2, branched 0.993184757 0.00053 0.094325878

133
chain
Cybrd1 cytochrome b reductase 1 0.994028526 0.00053 0.225943878
Sult1a1 sulfotransferase family 1A, phenol-
preferring, member 1
0.995373222 0.00053 0.12120022
Tfap2a transcription factor AP-2, alpha 0.997539572 0.00053 0.135302675
Pdgfb platelet derived growth factor, B
polypeptide
0.998420792 0.00053 0.163272181
Hck hemopoietic cell kinase 0.999070587 0.00053 0.250495191
C2cd4a C2 calcium-dependent domain
containing 4A
0.999639773 0.00053 0.208161313
Pcdhgb1 protocadherin gamma subfamily B, 1 1.005347499 0.00053 0.080214417
9030224M15Rik RIKEN cDNA 9030224M15 gene 1.013271323 0.00054 0.359742465
Mustn1 musculoskeletal, embryonic nuclear
protein 1
1.015777183 0.00053 0.127381615
Tbxa2r thromboxane A2 receptor 1.015799552 0.00053 0.044372957
Rapsn receptor-associated protein of the
synapse
1.017472248 0.00053 0.106670772
Mgst1 microsomal glutathione S-transferase
1
1.018475319 0.00053 0.0203985
Gas6 growth arrest specific 6 1.018597236 0.00053 0.012972127
Ptprq protein tyrosine phosphatase, receptor
type, Q
1.023762028 0.00053 0.09260246
Mrvi1 MRV integration site 1 1.024551664 0.00053 0.047280673
Selenbp1 selenium binding protein 1 1.027124543 0.00053 0.136774659
H28 histocompatibility 28 1.027624929 0.00053 0.114131969
Pcdhga6 protocadherin gamma subfamily A, 6 1.027721531 0.00053 0.273393221
Serpina3f serine (or cysteine) peptidase inhibitor,
clade A, member 3F
1.031536232 0.00053 0.178581046
Ccl27a chemokine (C-C motif) ligand 27A 1.03329066 0.00054 0.484304318
Iigp1 interferon inducible GTPase 1 1.036102758 0.00053 0.065017448
Apol9b apolipoprotein L 9b 1.039598534 0.00053 0.153952589
Mmp13 matrix metallopeptidase 13 1.040888988 0.00053 0.083124011

134
Fmo4 flavin containing monooxygenase 4 1.046495119 0.00053 0.216815067
Steap4 STEAP family member 4 1.052428065 0.00053 0.002584317
Cln3 ceroid lipofuscinosis, neuronal 3,
juvenile (Batten, Spielmeyer-Vogt
disease)
1.058161466 0.00053 0.309026893
C1qtnf3 C1q and tumor necrosis factor related
protein 3
1.062870795 0.00053 0.085885934
Dio2 deiodinase, iodothyronine, type II 1.07779171 0.00053 0.209791746
Tmprss6 transmembrane serine protease 6 1.079375769 0.00053 0.033600766
Xdh xanthine dehydrogenase 1.085912708 0.00053 0.079133761
Pcdhgb6 protocadherin gamma subfamily B, 6 1.088637837 0.00053 0.059579985
Alox5ap arachidonate 5-lipoxygenase
activating protein
1.09044588 0.00053 0.166524406
Notum notum pectinacetylesterase homolog
(Drosophila)
1.10405039 0.00053 0.094076015
Lbp lipopolysaccharide binding protein 1.113722934 0.00053 0.03739157
Clec3b C-type lectin domain family 3,
member b
1.116350535 0.00053 0.076252528
Enpp2 ectonucleotide
pyrophosphatase/phosphodiesterase 2
1.117753237 0.00053 0.347134136
Acpp acid phosphatase, prostate 1.120588722 0.00053 0.137820192
Rragb Ras-related GTP binding B 1.125262389 0.00053 0.309078515
Gdnf glial cell line derived neurotrophic
factor
1.132545993 0.00053 0.109641032
Hp haptoglobin 1.13791102 0.00053 0.43130165
Lcn2 lipocalin 2 1.139064261 0.00053 0.08005039
Hspa12b heat shock protein 12B 1.14116058 0.00053 0.085648791
Syt13 synaptotagmin XIII 1.143486055 0.00053 0.027679077
Slc16a2 solute carrier family 16
(monocarboxylic acid transporters),
member 2
1.159110478 0.00053 0.398843774
Sod3 superoxide dismutase 3, extracellular 1.167418756 0.00053 0.056023357
Ugt1a6b UDP glucuronosyltransferase 1 1.170555987 0.00053 0.128208789

135
family, polypeptide A6B
Slc22a18 solute carrier family 22 (organic cation
transporter), member 18
1.174411334 0.00053 0.145295089
Entpd3 ectonucleoside triphosphate
diphosphohydrolase 3
1.180120689 0.00053 0.1037217
Cxcl10 chemokine (C-X-C motif) ligand 10 1.193671012 0.00053 0.140252908
Zfp941 zinc finger protein 941 1.194086034 0.00053 0.085421735
Elfn1 leucine rich repeat and fibronectin
type III, extracellular 1
1.194848088 0.00053 0.020379081
Apol9a apolipoprotein L 9a 1.197174371 0.00053 0.246164347
Gpbar1 G protein-coupled bile acid receptor 1 1.198285099 0.00053 0.135408239
Cda cytidine deaminase 1.205054123 0.00053 0.237088505
Fam107a family with sequence similarity 107,
member A
1.205425335 0.00053 0.039519945
Nr1h3 nuclear receptor subfamily 1, group H,
member 3
1.206406134 0.00053 0.091558614
Plcg2 phospholipase C, gamma 2 1.206711745 0.00053 0.398864245
Trim30a tripartite motif-containing 30A 1.208785913 0.00053 0.144143522
Csmd1 CUB and Sushi multiple domains 1 1.210273167 0.00053 0.057302114
Psca prostate stem cell antigen 1.212806007 0.00053 0.091731027
Psmb9 proteasome (prosome, macropain)
subunit, beta type 9 (large
multifunctional peptidase 2)
1.213436685 0.00053 0.1345473
Pcsk6 proprotein convertase subtilisin/kexin
type 6
1.216391075 0.00053 0.032624856
Prelp proline arginine-rich end leucine-rich
repeat
1.216786271 0.00053 0.051151785
A4galt alpha 1,4-galactosyltransferase 1.22310685 0.00053 0.061975306
Vnn1 vanin 1 1.22704685 0.00053 0.016872923
Fxyd1 FXYD domain-containing ion
transport regulator 1
1.231112772 0.00053 0.523075014
Trpv2 transient receptor potential cation
channel, subfamily V, member 2
1.248179355 0.00053 0.216250134

136
Renbp renin binding protein 1.248781807 0.00053 0.017383204
Grhl1 grainyhead-like 1 (Drosophila) 1.253278296 0.00053 0.270603819
Msl3l2 male-specific lethal 3-like 2
(Drosophila)
1.25986456 0.00053 0.137146309
Arhgap6 Rho GTPase activating protein 6 1.266639625 0.00053 0.180941556
Ccl5 chemokine (C-C motif) ligand 5 1.300214635 0.00053 0.131616389
H60b histocompatibility 60b 1.301586423 0.00053 0.311046603
H2-Q4 histocompatibility 2, Q region locus 4 1.303535724 0.00053 0.084559364
C3 complement component 3 1.322856021 0.00053 0.063090714
AI607873 expressed sequence AI607873 1.346856052 0.00053 0.137647255
Cyp2j9 cytochrome P450, family 2, subfamily
j, polypeptide 9
1.359477206 0.00053 0.086144262
Tnfsf10 tumor necrosis factor (ligand)
superfamily, member 10
1.375656394 0.00053 0.185884401
C130074G19Rik RIKEN cDNA C130074G19 gene 1.377624185 0.00053 0.13747556
Serping1 serine (or cysteine) peptidase inhibitor,
clade G, member 1
1.386331531 0.00053 0.095017679
Nfe2l3 nuclear factor, erythroid derived 2,
like 3
1.389071601 0.00053 0.032589724
Gda guanine deaminase 1.400864825 0.00053 0.026537201
Crabp1 cellular retinoic acid binding protein I 1.404406104 0.00053 0.242432756
Htatip2 HIV-1 tat interactive protein 2,
homolog (human)
1.407270603 0.00053 0.057078139
Plb1 phospholipase B1 1.44324694 0.00053 0.269786455
Oas2 2'-5' oligoadenylate synthetase 2 1.443352045 0.00053 0.137723246
Inmt indolethylamine N-methyltransferase 1.460243408 0.00053 0.310783802
Pik3r5 phosphoinositide-3-kinase, regulatory
subunit 5, p101
1.467004844 0.00053 0.08535067
Pdzk1ip1 PDZK1 interacting protein 1 1.474467624 0.00053 0.042997429
Mmp3 matrix metallopeptidase 3 1.479116108 0.00053 0.058432453
Tril TLR4 interactor with leucine-rich
repeats
1.50993966 0.00053 0.137914316

137
Tubg2 tubulin, gamma 2 1.527010062 0.00053 0.309441049
Pdpn podoplanin 1.555027038 0.00053 0.4399842
Sp140 Sp140 nuclear body protein 1.570560246 0.00053 0.250312212
Apod apolipoprotein D 1.61198472 0.00053 0.221408291
Chi3l1 chitinase 3-like 1 1.636684153 0.00053 0.583086161
Unc93b1 unc-93 homolog B1 (C. elegans) 1.66968256 0.00053 0.360200982
H2-DMb1 histocompatibility 2, class II, locus
Mb1
1.67565171 0.00053 0.359347716
Slc15a2 solute carrier family 15 (H+/peptide
transporter), member 2
1.700204294 0.00053 0.385075496
Wnt4 wingless-related MMTV integration
site 4
1.713137287 0.00053 0.360869216
Fmo2 flavin containing monooxygenase 2 1.815536475 0.00053 0.308692687
Sparcl1 SPARC-like 1 1.816996676 0.00053 0.063385442
Cyp2f2 cytochrome P450, family 2, subfamily
f, polypeptide 2
1.83370284 0.00053 0.078208358
Klhl29 kelch-like 29 (Drosophila) 1.88351542 0.00053 0.138028824
Eml2 echinoderm microtubule associated
protein like 2
1.970482521 0.00053 0.027241317
Slpi secretory leukocyte peptidase inhibitor 2.08642332 0.00053 0.441915633
Lgals6 lectin, galactose binding, soluble 6 5.671892314 0.00053 0.198517766

138
6.4. List of identified p66Shc-interacting proteins
Identified protein No. of peptides p66+
MEF –
serum starvation
No. of peptides
p66+
MEF – serum
stimulation / 10 min
Shc1 900 920
Sgk269 22 23
FAM59A 4 27
Beta2-adaptin 5 23
Alpha1-adaptin 4 22
Grb2 0 12
Lrrk1 0 9
Anks1 1 7
PTPN12 1 5
Gab1 0 2
PP2C-beta 8 14
PP1A 2 2

139
References
Abdel Rahman, A.M., Ryczko, M., Pawling, J., and Dennis, J.W. (2013). Probing the
hexosamine biosynthetic pathway in human tumor cells by multitargeted tandem mass
spectrometry. ACS chemical biology 8, 2053-2062.
Ahn, R., Sabourin, V., Ha, J.R., Cory, S., Maric, G., Im, Y.K., Hardy, W.R., Zhao, H., Park, M.,
Hallett, M., et al. (2013). The ShcA PTB domain functions as a biological sensor of
phosphotyrosine signaling during breast cancer progression. Cancer research 73, 4521-4532.
Andronesi, O.C., Kim, G.S., Gerstner, E., Batchelor, T., Tzika, A.A., Fantin, V.R., Vander
Heiden, M.G., and Sorensen, A.G. (2012). Detection of 2-hydroxyglutarate in IDH-mutated
glioma patients by in vivo spectral-editing and 2D correlation magnetic resonance spectroscopy.
Sci Transl Med 4, 116ra114.
Bar-Peled, L., Chantranupong, L., Cherniack, A.D., Chen, W.W., Ottina, K.A., Grabiner, B.C.,
Spear, E.D., Carter, S.L., Meyerson, M., and Sabatini, D.M. (2013). A Tumor suppressor
complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1.
Science 340, 1100-1106.
Bar-Peled, L., Schweitzer, L.D., Zoncu, R., and Sabatini, D.M. (2012). Ragulator is a GEF for
the rag GTPases that signal amino acid levels to mTORC1. Cell 150, 1196-1208.
Batzer, A.G., Rotin, D., Urena, J.M., Skolnik, E.Y., and Schlessinger, J. (1994). Hierarchy of
binding sites for Grb2 and Shc on the epidermal growth factor receptor. Molecular and cellular
biology 14, 5192-5201.
Ben-Sahra, I., Howell, J.J., Asara, J.M., and Manning, B.D. (2013). Stimulation of de Novo
Pyrimidine Synthesis by Growth Signaling Through mTOR and S6K1. Science 339, 1323-1328.
Bensaad, K., Tsuruta, A., Selak, M.A., Vidal, M.N., Nakano, K., Bartrons, R., Gottlieb, E., and
Vousden, K.H. (2006). TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 126,
107-120.
Bertout, J.A., Patel, S.A., and Simon, M.C. (2008). The impact of O2 availability on human
cancer. Nature reviews Cancer 8, 967-975.
Berwick, D.C., Hers, I., Heesom, K.J., Moule, S.K., and Tavare, J.M. (2002). The identification
of ATP-citrate lyase as a protein kinase B (Akt) substrate in primary adipocytes. The Journal of
biological chemistry 277, 33895-33900.

140
Bhattacharyya, R.P., Remenyi, A., Yeh, B.J., and Lim, W.A. (2006). Domains, motifs, and
scaffolds: the role of modular interactions in the evolution and wiring of cell signaling circuits.
Annual review of biochemistry 75, 655-680.
Bieging, K.T., and Attardi, L.D. (2012). Deconstructing p53 transcriptional networks in tumor
suppression. Trends in cell biology 22, 97-106.
Birsoy, K., Wang, T., Possemato, R., Yilmaz, O.H., Koch, C.E., Chen, W.W., Hutchins, A.W.,
Gultekin, Y., Peterson, T.R., Carette, J.E., et al. (2013). MCT1-mediated transport of a toxic
molecule is an effective strategy for targeting glycolytic tumors. Nature genetics 45, 104-108.
Brown, E.J., Albers, M.W., Shin, T.B., Ichikawa, K., Keith, C.T., Lane, W.S., and Schreiber,
S.L. (1994). A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature
369, 756-758.
Brugarolas, J., Lei, K., Hurley, R.L., Manning, B.D., Reiling, J.H., Hafen, E., Witters, L.A.,
Ellisen, L.W., and Kaelin, W.G., Jr. (2004). Regulation of mTOR function in response to
hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes & development 18,
2893-2904.
Budanov, A.V., and Karin, M. (2008). p53 target genes sestrin1 and sestrin2 connect genotoxic
stress and mTOR signaling. Cell 134, 451-460.
Buller, C.L., Loberg, R.D., Fan, M.H., Zhu, Q., Park, J.L., Vesely, E., Inoki, K., Guan, K.L., and
Brosius, F.C., 3rd (2008). A GSK-3/TSC2/mTOR pathway regulates glucose uptake and GLUT1
glucose transporter expression. American journal of physiology Cell physiology 295, C836-843.
Cafferkey, R., Young, P.R., McLaughlin, M.M., Bergsma, D.J., Koltin, Y., Sathe, G.M.,
Faucette, L., Eng, W.K., Johnson, R.K., and Livi, G.P. (1993). Dominant missense mutations in
a novel yeast protein related to mammalian phosphatidylinositol 3-kinase and VPS34 abrogate
rapamycin cytotoxicity. Molecular and cellular biology 13, 6012-6023.
Cairns, R.A., Harris, I.S., and Mak, T.W. (2011). Regulation of cancer cell metabolism. Nature
reviews Cancer 11, 85-95.
Cam, H., Easton, J.B., High, A., and Houghton, P.J. (2010). mTORC1 signaling under hypoxic
conditions is controlled by ATM-dependent phosphorylation of HIF-1alpha. Molecular cell 40,
509-520.
Camici, G.G., Schiavoni, M., Francia, P., Bachschmid, M., Martin-Padura, I., Hersberger, M.,
Tanner, F.C., Pelicci, P., Volpe, M., Anversa, P., et al. (2007). Genetic deletion of p66(Shc)
adaptor protein prevents hyperglycemia-induced endothelial dysfunction and oxidative stress.
ProcNatlAcadSciUSA 104, 5217-5222.

141
Castilho, R.M., Squarize, C.H., Chodosh, L.A., Williams, B.O., and Gutkind, J.S. (2009). mTOR
mediates Wnt-induced epidermal stem cell exhaustion and aging. Cell stem cell 5, 279-289.
Chandel, N.S., McClintock, D.S., Feliciano, C.E., Wood, T.M., Melendez, J.A., Rodriguez,
A.M., and Schumacker, P.T. (2000). Reactive oxygen species generated at mitochondrial
complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2
sensing. The Journal of biological chemistry 275, 25130-25138.
Chen, J., Zheng, X.F., Brown, E.J., and Schreiber, S.L. (1995). Identification of an 11-kDa
FKBP12-rapamycin-binding domain within the 289-kDa FKBP12-rapamycin-associated protein
and characterization of a critical serine residue. Proceedings of the National Academy of
Sciences of the United States of America 92, 4947-4951.
Cheung, P., Pawling, J., Partridge, E.A., Sukhu, B., Grynpas, M., and Dennis, J.W. (2007).
Metabolic homeostasis and tissue renewal are dependent on beta1,6GlcNAc-branched N-
glycans. Glycobiology 17, 828-837.
Choi, C., Ganji, S.K., DeBerardinis, R.J., Hatanpaa, K.J., Rakheja, D., Kovacs, Z., Yang, X.L.,
Mashimo, T., Raisanen, J.M., Marin-Valencia, I., et al. (2012). 2-hydroxyglutarate detection by
magnetic resonance spectroscopy in IDH-mutated patients with gliomas. Nat Med 18, 624-629.
Chowdhury, R., Yeoh, K.K., Tian, Y.M., Hillringhaus, L., Bagg, E.A., Rose, N.R., Leung, I.K.,
Li, X.S., Woon, E.C., Yang, M., et al. (2011). The oncometabolite 2-hydroxyglutarate inhibits
histone lysine demethylases. EMBO Rep 12, 463-469.
Christofk, H.R., Vander Heiden, M.G., Harris, M.H., Ramanathan, A., Gerszten, R.E., Wei, R.,
Fleming, M.D., Schreiber, S.L., and Cantley, L.C. (2008a). The M2 splice isoform of pyruvate
kinase is important for cancer metabolism and tumour growth. Nature 452, 230-233.
Christofk, H.R., Vander Heiden, M.G., Wu, N., Asara, J.M., and Cantley, L.C. (2008b). Pyruvate
kinase M2 is a phosphotyrosine-binding protein. Nature 452, 181-186.
Cooney, G.J., Lyons, R.J., Crew, A.J., Jensen, T.E., Molero, J.C., Mitchell, C.J., Biden, T.J.,
Ormandy, C.J., James, D.E., and Daly, R.J. (2004). Improved glucose homeostasis and enhanced
insulin signalling in Grb14-deficient mice. The EMBO journal 23, 582-593.
Corradetti, M.N., Inoki, K., Bardeesy, N., DePinho, R.A., and Guan, K.L. (2004). Regulation of
the TSC pathway by LKB1: evidence of a molecular link between tuberous sclerosis complex
and Peutz-Jeghers syndrome. Genes & development 18, 1533-1538.
D'Alessandris, C., Andreozzi, F., Federici, M., Cardellini, M., Brunetti, A., Ranalli, M., Del
Guerra, S., Lauro, D., Del Prato, S., Marchetti, P., et al. (2004). Increased O-glycosylation of
insulin signaling proteins results in their impaired activation and enhanced susceptibility to

142
apoptosis in pancreatic beta-cells. FASEB journal : official publication of the Federation of
American Societies for Experimental Biology 18, 959-961.
Dai, N., Christiansen, J., Nielsen, F.C., and Avruch, J. (2013). mTOR complex 2 phosphorylates
IMP1 cotranslationally to promote IGF2 production and the proliferation of mouse embryonic
fibroblasts. Genes & development 27, 301-312.
Dang, L., White, D.W., Gross, S., Bennett, B.D., Bittinger, M.A., Driggers, E.M., Fantin, V.R.,
Jang, H.G., Jin, S., Keenan, M.C., et al. (2009). Cancer-associated IDH1 mutations produce 2-
hydroxyglutarate. Nature 462, 739-744.
David, C.J., Chen, M., Assanah, M., Canoll, P., and Manley, J.L. (2010). HnRNP proteins
controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature 463, 364-368.
Davol, P.A., Bagdasaryan, R., Elfenbein, G.J., Maizel, A.L., and Frackelton, A.R., Jr. (2003).
Shc proteins are strong, independent prognostic markers for both node-negative and node-
positive primary breast cancer. Cancer research 63, 6772-6783.
DeBerardinis, R.J., and Cheng, T. (2010). Q's next: the diverse functions of glutamine in
metabolism, cell biology and cancer. Oncogene 29, 313-324.
DeBerardinis, R.J., Mancuso, A., Daikhin, E., Nissim, I., Yudkoff, M., Wehrli, S., and
Thompson, C.B. (2007). Beyond aerobic glycolysis: transformed cells can engage in glutamine
metabolism that exceeds the requirement for protein and nucleotide synthesis. Proceedings of the
National Academy of Sciences of the United States of America 104, 19345-19350.
Demetriades, C., Doumpas, N., and Teleman, A.A. (2014). Regulation of TORC1 in Response to
Amino Acid Starvation via Lysosomal Recruitment of TSC2. Cell 156, 786-799.
Dengjel, J., Akimov, V., Olsen, J.V., Bunkenborg, J., Mann, M., Blagoev, B., and Andersen, J.S.
(2007). Quantitative proteomic assessment of very early cellular signaling events. Nature
biotechnology 25, 566-568.
Dennis, J.W., Nabi, I.R., and Demetriou, M. (2009). Metabolism, cell surface organization, and
disease. Cell 139, 1229-1241.
Deprez, J., Vertommen, D., Alessi, D.R., Hue, L., and Rider, M.H. (1997). Phosphorylation and
activation of heart 6-phosphofructo-2-kinase by protein kinase B and other protein kinases of the
insulin signaling cascades. The Journal of biological chemistry 272, 17269-17275.
DeYoung, M.P., Horak, P., Sofer, A., Sgroi, D., and Ellisen, L.W. (2008). Hypoxia regulates
TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling.
Genes & development 22, 239-251.

143
Di Paola, R., Ciociola, E., Boonyasrisawat, W., Nolan, D., Duffy, J., Miscio, G., Cisternino, C.,
Fini, G., Tassi, V., Doria, A., et al. (2006). Association of hGrb10 genetic variations with type 2
diabetes in Caucasian subjects. Diabetes care 29, 1181-1183.
Dibble, C.C., Elis, W., Menon, S., Qin, W., Klekota, J., Asara, J.M., Finan, P.M., Kwiatkowski,
D.J., Murphy, L.O., and Manning, B.D. (2012). TBC1D7 is a third subunit of the TSC1-TSC2
complex upstream of mTORC1. Molecular cell 47, 535-546.
Dibble, C.C., and Manning, B.D. (2013). Signal integration by mTORC1 coordinates nutrient
input with biosynthetic output. Nature cell biology 15, 555-564.
Duran, A., Amanchy, R., Linares, J.F., Joshi, J., Abu-Baker, S., Porollo, A., Hansen, M., Moscat,
J., and Diaz-Meco, M.T. (2011). p62 is a key regulator of nutrient sensing in the mTORC1
pathway. Molecular cell 44, 134-146.
Duran, R.V., Oppliger, W., Robitaille, A.M., Heiserich, L., Skendaj, R., Gottlieb, E., and Hall,
M.N. (2012). Glutaminolysis activates Rag-mTORC1 signaling. Molecular cell 47, 349-358.
Duvel, K., Yecies, J.L., Menon, S., Raman, P., Lipovsky, A.I., Souza, A.L., Triantafellow, E.,
Ma, Q., Gorski, R., Cleaver, S., et al. (2010). Activation of a metabolic gene regulatory network
downstream of mTOR complex 1. MolCell 39, 171-183.
Efeyan, A., Zoncu, R., Chang, S., Gumper, I., Snitkin, H., Wolfson, R.L., Kirak, O., Sabatini,
D.D., and Sabatini, D.M. (2013). Regulation of mTORC1 by the Rag GTPases is necessary for
neonatal autophagy and survival. Nature 493, 679-683.
Elstrom, R.L., Bauer, D.E., Buzzai, M., Karnauskas, R., Harris, M.H., Plas, D.R., Zhuang, H.,
Cinalli, R.M., Alavi, A., Rudin, C.M., et al. (2004). Akt stimulates aerobic glycolysis in cancer
cells. Cancer research 64, 3892-3899.
Engelman, J.A., Luo, J., and Cantley, L.C. (2006). The evolution of phosphatidylinositol 3-
kinases as regulators of growth and metabolism. Nature reviews Genetics 7, 606-619.
Esen, E., Chen, J., Karner, C.M., Okunade, A.L., Patterson, B.W., and Long, F. (2013). WNT-
LRP5 signaling induces Warburg effect through mTORC2 activation during osteoblast
differentiation. Cell Metab 17, 745-755.
Fan, J., Kamphorst, J.J., Mathew, R., Chung, M.K., White, E., Shlomi, T., and Rabinowitz, J.D.
(2013). Glutamine-driven oxidative phosphorylation is a major ATP source in transformed
mammalian cells in both normoxia and hypoxia. Molecular systems biology 9, 712.

144
Fang, Y., Park, I.H., Wu, A.L., Du, G., Huang, P., Frohman, M.A., Walker, S.J., Brown, H.A.,
and Chen, J. (2003). PLD1 regulates mTOR signaling and mediates Cdc42 activation of S6K1.
Current biology : CB 13, 2037-2044.
Fang, Y., Vilella-Bach, M., Bachmann, R., Flanigan, A., and Chen, J. (2001). Phosphatidic acid-
mediated mitogenic activation of mTOR signaling. Science 294, 1942-1945.
Feng, Z., Hu, W., de Stanchina, E., Teresky, A.K., Jin, S., Lowe, S., and Levine, A.J. (2007).
The regulation of AMPK beta1, TSC2, and PTEN expression by p53: stress, cell and tissue
specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways.
Cancer research 67, 3043-3053.
Ferrari, S., Bandi, H.R., Hofsteenge, J., Bussian, B.M., and Thomas, G. (1991). Mitogen-
activated 70K S6 kinase. Identification of in vitro 40 S ribosomal S6 phosphorylation sites. The
Journal of biological chemistry 266, 22770-22775.
Figueroa, M.E., Abdel-Wahab, O., Lu, C., Ward, P.S., Patel, J., Shih, A., Li, Y., Bhagwat, N.,
Vasanthakumar, A., Fernandez, H.F., et al. (2010). Leukemic IDH1 and IDH2 mutations result
in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic
differentiation. Cancer Cell 18, 553-567.
Findlay, G.M., Yan, L., Procter, J., Mieulet, V., and Lamb, R.F. (2007). A MAP4 kinase related
to Ste20 is a nutrient-sensitive regulator of mTOR signalling. The Biochemical journal 403, 13-
20.
Fisher-Wellman, K.H., and Neufer, P.D. (2012). Linking mitochondrial bioenergetics to insulin
resistance via redox biology. Trends EndocrinolMetab 23, 142-153.
Frias, M.A., Thoreen, C.C., Jaffe, J.D., Schroder, W., Sculley, T., Carr, S.A., and Sabatini, D.M.
(2006). mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct
mTORC2s. Current biology : CB 16, 1865-1870.
Ganley, I.G., Lam du, H., Wang, J., Ding, X., Chen, S., and Jiang, X. (2009).
ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. The
Journal of biological chemistry 284, 12297-12305.
Gao, P., Tchernyshyov, I., Chang, T.C., Lee, Y.S., Kita, K., Ochi, T., Zeller, K.I., De Marzo,
A.M., Van Eyk, J.E., Mendell, J.T., et al. (2009). c-Myc suppression of miR-23a/b enhances
mitochondrial glutaminase expression and glutamine metabolism. Nature 458, 762-765.
Garami, A., Zwartkruis, F.J., Nobukuni, T., Joaquin, M., Roccio, M., Stocker, H., Kozma, S.C.,
Hafen, E., Bos, J.L., and Thomas, G. (2003). Insulin activation of Rheb, a mediator of
mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Molecular cell 11, 1457-1466.

145
Garcia-Martinez, J.M., and Alessi, D.R. (2008). mTOR complex 2 (mTORC2) controls
hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein
kinase 1 (SGK1). The Biochemical journal 416, 375-385.
Gatenby, R.A., and Gillies, R.J. (2004). Why do cancers have high aerobic glycolysis? Nature
reviews Cancer 4, 891-899.
Gingras, A.C., Gygi, S.P., Raught, B., Polakiewicz, R.D., Abraham, R.T., Hoekstra, M.F.,
Aebersold, R., and Sonenberg, N. (1999). Regulation of 4E-BP1 phosphorylation: a novel two-
step mechanism. Genes Dev 13, 1422-1437.
Giorgio, M., Berry, A., Berniakovich, I., Poletaeva, I., Trinei, M., Stendardo, M., Hagopian, K.,
Ramsey, J.J., Cortopassi, G., Migliaccio, E., et al. (2012). The p66Shc knocked out mice are
short lived under natural condition. Aging Cell 11, 162-168.
Giorgio, M., Migliaccio, E., Orsini, F., Paolucci, D., Moroni, M., Contursi, C., Pelliccia, G.,
Luzi, L., Minucci, S., Marcaccio, M., et al. (2005). Electron transfer between cytochrome c and
p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell 122, 221-
233.
Gordan, J.D., Thompson, C.B., and Simon, M.C. (2007). HIF and c-Myc: sibling rivals for
control of cancer cell metabolism and proliferation. Cancer Cell 12, 108-113.
Gottlieb, E., and Tomlinson, I.P. (2005). Mitochondrial tumour suppressors: a genetic and
biochemical update. Nature reviews Cancer 5, 857-866.
Gottlob, K., Majewski, N., Kennedy, S., Kandel, E., Robey, R.B., and Hay, N. (2001). Inhibition
of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and
mitochondrial hexokinase. Genes & development 15, 1406-1418.
Graiani, G., Lagrasta, C., Migliaccio, E., Spillmann, F., Meloni, M., Madeddu, P., Quaini, F.,
Padura, I.M., Lanfrancone, L., Pelicci, P., et al. (2005). Genetic deletion of the p66Shc adaptor
protein protects from angiotensin II-induced myocardial damage. Hypertension 46, 433-440.
Greer, S.N., Metcalf, J.L., Wang, Y., and Ohh, M. (2012). The updated biology of hypoxia-
inducible factor. The EMBO journal 31, 2448-2460.
Gu, H., Maeda, H., Moon, J.J., Lord, J.D., Yoakim, M., Nelson, B.H., and Neel, B.G. (2000).
New role for Shc in activation of the phosphatidylinositol 3-kinase/Akt pathway. MolCell Biol
20, 7109-7120.
Guertin, D.A., Stevens, D.M., Thoreen, C.C., Burds, A.A., Kalaany, N.Y., Moffat, J., Brown,
M., Fitzgerald, K.J., and Sabatini, D.M. (2006). Ablation in mice of the mTORC components

146
raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and
PKCalpha, but not S6K1. Developmental cell 11, 859-871.
Gulati, P., Gaspers, L.D., Dann, S.G., Joaquin, M., Nobukuni, T., Natt, F., Kozma, S.C.,
Thomas, A.P., and Thomas, G. (2008). Amino acids activate mTOR complex 1 via Ca2+/CaM
signaling to hVps34. Cell Metab 7, 456-465.
Gwinn, D.M., Shackelford, D.B., Egan, D.F., Mihaylova, M.M., Mery, A., Vasquez, D.S., Turk,
B.E., and Shaw, R.J. (2008). AMPK phosphorylation of raptor mediates a metabolic checkpoint.
Molecular cell 30, 214-226.
Haga, Y., Ishii, K., and Suzuki, T. (2011). N-glycosylation is critical for the stability and
intracellular trafficking of glucose transporter GLUT4. The Journal of biological chemistry 286,
31320-31327.
Haghighat, A., Mader, S., Pause, A., and Sonenberg, N. (1995). Repression of cap-dependent
translation by 4E-binding protein 1: competition with p220 for binding to eukaryotic initiation
factor-4E. The EMBO journal 14, 5701-5709.
Hagiwara, A., Cornu, M., Cybulski, N., Polak, P., Betz, C., Trapani, F., Terracciano, L., Heim,
M.H., Ruegg, M.A., and Hall, M.N. (2012). Hepatic mTORC2 activates glycolysis and
lipogenesis through Akt, glucokinase, and SREBP1c. Cell Metab 15, 725-738.
Han, J.M., Jeong, S.J., Park, M.C., Kim, G., Kwon, N.H., Kim, H.K., Ha, S.H., Ryu, S.H., and
Kim, S. (2012). Leucyl-tRNA synthetase is an intracellular leucine sensor for the mTORC1-
signaling pathway. Cell 149, 410-424.
Hanahan, D., and Weinberg, R.A. (2011). Hallmarks of cancer: the next generation. Cell 144,
646-674.
Hara, K., Yonezawa, K., Kozlowski, M.T., Sugimoto, T., Andrabi, K., Weng, Q.P., Kasuga, M.,
Nishimoto, I., and Avruch, J. (1997). Regulation of eIF-4E BP1 phosphorylation by mTOR. The
Journal of biological chemistry 272, 26457-26463.
Hara, K., Yonezawa, K., Weng, Q.P., Kozlowski, M.T., Belham, C., and Avruch, J. (1998).
Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a common
effector mechanism. The Journal of biological chemistry 273, 14484-14494.
Hardy, W.R., Li, L., Wang, Z., Sedy, J., Fawcett, J., Frank, E., Kucera, J., and Pawson, T.
(2007). Combinatorial ShcA docking interactions support diversity in tissue morphogenesis.
Science 317, 251-256.

147
Harrington, L.S., Findlay, G.M., Gray, A., Tolkacheva, T., Wigfield, S., Rebholz, H., Barnett, J.,
Leslie, N.R., Cheng, S., Shepherd, P.R., et al. (2004). The TSC1-2 tumor suppressor controls
insulin-PI3K signaling via regulation of IRS proteins. The Journal of cell biology 166, 213-223.
Hatzivassiliou, G., Zhao, F., Bauer, D.E., Andreadis, C., Shaw, A.N., Dhanak, D., Hingorani,
S.R., Tuveson, D.A., and Thompson, C.B. (2005). ATP citrate lyase inhibition can suppress
tumor cell growth. Cancer Cell 8, 311-321.
Heller, C., Kuhn, M.C., Mulders-Opgenoorth, B., Schott, M., Willenberg, H.S., Scherbaum,
W.A., and Schinner, S. (2011). Exendin-4 upregulates the expression of Wnt-4, a novel regulator
of pancreatic beta-cell proliferation. American journal of physiology Endocrinology and
metabolism 301, E864-872.
Hemmings, B.A., and Restuccia, D.F. (2012). PI3K-PKB/Akt pathway. Cold Spring Harbor
perspectives in biology 4, a011189.
Hitosugi, T., Kang, S., Vander Heiden, M.G., Chung, T.W., Elf, S., Lythgoe, K., Dong, S.,
Lonial, S., Wang, X., Chen, G.Z., et al. (2009). Tyrosine phosphorylation inhibits PKM2 to
promote the Warburg effect and tumor growth. Science signaling 2, ra73.
Holt, L.J., and Siddle, K. (2005). Grb10 and Grb14: enigmatic regulators of insulin action--and
more? The Biochemical journal 388, 393-406.
Hosokawa, N., Hara, T., Kaizuka, T., Kishi, C., Takamura, A., Miura, Y., Iemura, S., Natsume,
T., Takehana, K., Yamada, N., et al. (2009). Nutrient-dependent mTORC1 association with the
ULK1-Atg13-FIP200 complex required for autophagy. Molecular biology of the cell 20, 1981-
1991.
Houstis, N., Rosen, E.D., and Lander, E.S. (2006). Reactive oxygen species have a causal role in
multiple forms of insulin resistance. Nature 440, 944-948.
Hsu, P.P., Kang, S.A., Rameseder, J., Zhang, Y., Ottina, K.A., Lim, D., Peterson, T.R., Choi, Y.,
Gray, N.S., Yaffe, M.B., et al. (2011). The mTOR-regulated phosphoproteome reveals a
mechanism of mTORC1-mediated inhibition of growth factor signaling. Science 332, 1317-
1322.
Huang, J., Dibble, C.C., Matsuzaki, M., and Manning, B.D. (2008). The TSC1-TSC2 complex is
required for proper activation of mTOR complex 2. Molecular and cellular biology 28, 4104-
4115.
Hudson, C.C., Liu, M., Chiang, G.G., Otterness, D.M., Loomis, D.C., Kaper, F., Giaccia, A.J.,
and Abraham, R.T. (2002). Regulation of hypoxia-inducible factor 1alpha expression and
function by the mammalian target of rapamycin. Molecular and cellular biology 22, 7004-7014.

148
Ikenoue, T., Inoki, K., Yang, Q., Zhou, X., and Guan, K.L. (2008). Essential function of TORC2
in PKC and Akt turn motif phosphorylation, maturation and signalling. The EMBO journal 27,
1919-1931.
Inoki, K., Li, Y., Zhu, T., Wu, J., and Guan, K.L. (2002). TSC2 is phosphorylated and inhibited
by Akt and suppresses mTOR signalling. Nature cell biology 4, 648-657.
Inoki, K., Ouyang, H., Zhu, T., Lindvall, C., Wang, Y., Zhang, X., Yang, Q., Bennett, C.,
Harada, Y., Stankunas, K., et al. (2006). TSC2 integrates Wnt and energy signals via a
coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell 126, 955-968.
Inoki, K., Zhu, T., and Guan, K.L. (2003). TSC2 mediates cellular energy response to control
cell growth and survival. Cell 115, 577-590.
Isaacs, J.S., Jung, Y.J., Mole, D.R., Lee, S., Torres-Cabala, C., Chung, Y.L., Merino, M., Trepel,
J., Zbar, B., Toro, J., et al. (2005). HIF overexpression correlates with biallelic loss of fumarate
hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell 8,
143-153.
Jacinto, E., Facchinetti, V., Liu, D., Soto, N., Wei, S., Jung, S.Y., Huang, Q., Qin, J., and Su, B.
(2006). SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation
and substrate specificity. Cell 127, 125-137.
Jacinto, E., Loewith, R., Schmidt, A., Lin, S., Ruegg, M.A., Hall, A., and Hall, M.N. (2004).
Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nature
cell biology 6, 1122-1128.
Jiang, P., Du, W., Wang, X., Mancuso, A., Gao, X., Wu, M., and Yang, X. (2011). p53 regulates
biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nature cell
biology 13, 310-316.
Jin, J., and Pawson, T. (2012). Modular evolution of phosphorylation-based signalling systems.
Philosophical transactions of the Royal Society of London Series B, Biological sciences 367,
2540-2555.
Jones, R.G., Plas, D.R., Kubek, S., Buzzai, M., Mu, J., Xu, Y., Birnbaum, M.J., and Thompson,
C.B. (2005). AMP-activated protein kinase induces a p53-dependent metabolic checkpoint.
Molecular cell 18, 283-293.
Jung, C.H., Jun, C.B., Ro, S.H., Kim, Y.M., Otto, N.M., Cao, J., Kundu, M., and Kim, D.H.
(2009). ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery.
Molecular biology of the cell 20, 1992-2003.

149
Kasus-Jacobi, A., Perdereau, D., Auzan, C., Clauser, E., Van Obberghen, E., Mauvais-Jarvis, F.,
Girard, J., and Burnol, A.F. (1998). Identification of the rat adapter Grb14 as an inhibitor of
insulin actions. The Journal of biological chemistry 273, 26026-26035.
Khanday, F.A., Santhanam, L., Kasuno, K., Yamamori, T., Naqvi, A., Dericco, J., Bugayenko,
A., Mattagajasingh, I., Disanza, A., Scita, G., et al. (2006). Sos-mediated activation of rac1 by
p66shc. The Journal of cell biology 172, 817-822.
Khatri, S., Yepiskoposyan, H., Gallo, C.A., Tandon, P., and Plas, D.R. (2010). FOXO3a
regulates glycolysis via transcriptional control of tumor suppressor TSC1. The Journal of
biological chemistry 285, 15960-15965.
Kim, C.S., Jung, S.B., Naqvi, A., Hoffman, T.A., DeRicco, J., Yamamori, T., Cole, M.P., Jeon,
B.H., and Irani, K. (2008a). p53 impairs endothelium-dependent vasomotor function through
transcriptional upregulation of p66shc. Circulation research 103, 1441-1450.
Kim, D.H., Sarbassov, D.D., Ali, S.M., King, J.E., Latek, R.R., Erdjument-Bromage, H., Tempst,
P., and Sabatini, D.M. (2002). mTOR interacts with raptor to form a nutrient-sensitive complex
that signals to the cell growth machinery. Cell 110, 163-175.
Kim, D.H., Sarbassov, D.D., Ali, S.M., Latek, R.R., Guntur, K.V., Erdjument-Bromage, H.,
Tempst, P., and Sabatini, D.M. (2003). GbetaL, a positive regulator of the rapamycin-sensitive
pathway required for the nutrient-sensitive interaction between raptor and mTOR. Molecular cell
11, 895-904.
Kim, E., Goraksha-Hicks, P., Li, L., Neufeld, T.P., and Guan, K.L. (2008b). Regulation of
TORC1 by Rag GTPases in nutrient response. Nature cell biology 10, 935-945.
Kim, S., Kim, S.F., Maag, D., Maxwell, M.J., Resnick, A.C., Juluri, K.R., Chakraborty, A.,
Koldobskiy, M.A., Cha, S.H., Barrow, R., et al. (2011). Amino acid signaling to mTOR
mediated by inositol polyphosphate multikinase. Cell Metab 13, 215-221.
Kisielow, M., Kleiner, S., Nagasawa, M., Faisal, A., and Nagamine, Y. (2002). Isoform-specific
knockdown and expression of adaptor protein ShcA using small interfering RNA. BiochemJ 363,
1-5.
Kitagawa, T., Tsuruhara, Y., Hayashi, M., Endo, T., and Stanbridge, E.J. (1995). A tumor-
associated glycosylation change in the glucose transporter GLUT1 controlled by tumor
suppressor function in human cell hybrids. Journal of cell science 108 ( Pt 12), 3735-3743.
Kondoh, H., Lleonart, M.E., Gil, J., Wang, J., Degan, P., Peters, G., Martinez, D., Carnero, A.,
and Beach, D. (2005). Glycolytic enzymes can modulate cellular life span. Cancer research 65,
177-185.

150
Kooner, J.S., Saleheen, D., Sim, X., Sehmi, J., Zhang, W., Frossard, P., Been, L.F., Chia, K.S.,
Dimas, A.S., Hassanali, N., et al. (2011). Genome-wide association study in individuals of South
Asian ancestry identifies six new type 2 diabetes susceptibility loci. Nature genetics 43, 984-989.
Kovacina, K.S., Park, G.Y., Bae, S.S., Guzzetta, A.W., Schaefer, E., Birnbaum, M.J., and Roth,
R.A. (2003). Identification of a proline-rich Akt substrate as a 14-3-3 binding partner. The
Journal of biological chemistry 278, 10189-10194.
Kunz, J., Henriquez, R., Schneider, U., Deuter-Reinhard, M., Movva, N.R., and Hall, M.N.
(1993). Target of rapamycin in yeast, TOR2, is an essential phosphatidylinositol kinase homolog
required for G1 progression. Cell 73, 585-596.
Lai, K.M., Olivier, J.P., Gish, G.D., Henkemeyer, M., McGlade, J., and Pawson, T. (1995). A
Drosophila shc gene product is implicated in signaling by the DER receptor tyrosine kinase.
Molecular and cellular biology 15, 4810-4818.
Lai, K.M., and Pawson, T. (2000). The ShcA phosphotyrosine docking protein sensitizes
cardiovascular signaling in the mouse embryo. Genes & development 14, 1132-1145.
Lamming, D.W., Demirkan, G., Boylan, J.M., Mihaylova, M.M., Peng, T., Ferreira, J., Neretti,
N., Salomon, A., Sabatini, D.M., and Gruppuso, P.A. (2014). Hepatic signaling by the
mechanistic target of rapamycin complex 2 (mTORC2). FASEB journal : official publication of
the Federation of American Societies for Experimental Biology 28, 300-315.
Laplante, M., and Sabatini, D.M. (2012). mTOR signaling in growth control and disease. Cell
149, 274-293.
Lau, K.S., Partridge, E.A., Grigorian, A., Silvescu, C.I., Reinhold, V.N., Demetriou, M., and
Dennis, J.W. (2007). Complex N-glycan number and degree of branching cooperate to regulate
cell proliferation and differentiation. Cell 129, 123-134.
Lebiedzinska, M., Duszynski, J., Rizzuto, R., Pinton, P., and Wieckowski, M.R. (2009). Age-
related changes in levels of p66Shc and serine 36-phosphorylated p66Shc in organs and mouse
tissues. ArchBiochemBiophys.
Li, S.C., Lai, K.M., Gish, G.D., Parris, W.E., van der Geer, P., Forman-Kay, J., and Pawson, T.
(1996). Characterization of the phosphotyrosine-binding domain of the Drosophila Shc protein.
The Journal of biological chemistry 271, 31855-31862.
Li, T., Kon, N., Jiang, L., Tan, M., Ludwig, T., Zhao, Y., Baer, R., and Gu, W. (2012). Tumor
suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell
149, 1269-1283.

151
Lim, W.A., and Pawson, T. (2010). Phosphotyrosine signaling: evolving a new cellular
communication system. Cell 142, 661-667.
Long, X., Lin, Y., Ortiz-Vega, S., Yonezawa, K., and Avruch, J. (2005). Rheb binds and
regulates the mTOR kinase. Current biology : CB 15, 702-713.
Lu, C., Ward, P.S., Kapoor, G.S., Rohle, D., Turcan, S., Abdel-Wahab, O., Edwards, C.R.,
Khanin, R., Figueroa, M.E., Melnick, A., et al. (2012). IDH mutation impairs histone
demethylation and results in a block to cell differentiation. Nature 483, 474-478.
Luo, W., Hu, H., Chang, R., Zhong, J., Knabel, M., O'Meally, R., Cole, R.N., Pandey, A., and
Semenza, G.L. (2011). Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-
inducible factor 1. Cell 145, 732-744.
Luzi, L., Confalonieri, S., Di Fiore, P.P., and Pelicci, P.G. (2000). Evolution of Shc functions
from nematode to human. CurrOpinGenetDev 10, 668-674.
Ma, L., Chen, Z., Erdjument-Bromage, H., Tempst, P., and Pandolfi, P.P. (2005).
Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis
and cancer pathogenesis. Cell 121, 179-193.
Ma, X.M., and Blenis, J. (2009). Molecular mechanisms of mTOR-mediated translational
control. Nature reviews Molecular cell biology 10, 307-318.
Ma, X.M., Yoon, S.O., Richardson, C.J., Julich, K., and Blenis, J. (2008). SKAR links pre-
mRNA splicing to mTOR/S6K1-mediated enhanced translation efficiency of spliced mRNAs.
Cell 133, 303-313.
Manning, A.K., Hivert, M.F., Scott, R.A., Grimsby, J.L., Bouatia-Naji, N., Chen, H., Rybin, D.,
Liu, C.T., Bielak, L.F., Prokopenko, I., et al. (2012). A genome-wide approach accounting for
body mass index identifies genetic variants influencing fasting glycemic traits and insulin
resistance. Nature genetics 44, 659-669.
Manning, B.D., Tee, A.R., Logsdon, M.N., Blenis, J., and Cantley, L.C. (2002). Identification of
the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the
phosphoinositide 3-kinase/akt pathway. Molecular cell 10, 151-162.
Mardis, E.R., Ding, L., Dooling, D.J., Larson, D.E., McLellan, M.D., Chen, K., Koboldt, D.C.,
Fulton, R.S., Delehaunty, K.D., McGrath, S.D., et al. (2009). Recurring mutations found by
sequencing an acute myeloid leukemia genome. N Engl J Med 361, 1058-1066.

152
Masui, K., Tanaka, K., Akhavan, D., Babic, I., Gini, B., Matsutani, T., Iwanami, A., Liu, F.,
Villa, G.R., Gu, Y., et al. (2013). mTOR complex 2 controls glycolytic metabolism in
glioblastoma through FoxO acetylation and upregulation of c-Myc. Cell Metab 18, 726-739.
Matoba, S., Kang, J.G., Patino, W.D., Wragg, A., Boehm, M., Gavrilova, O., Hurley, P.J., Bunz,
F., and Hwang, P.M. (2006). p53 regulates mitochondrial respiration. Science 312, 1650-1653.
Maxwell, P.H., Wiesener, M.S., Chang, G.W., Clifford, S.C., Vaux, E.C., Cockman, M.E.,
Wykoff, C.C., Pugh, C.W., Maher, E.R., and Ratcliffe, P.J. (1999). The tumour suppressor
protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399,
271-275.
Mayer, C., Zhao, J., Yuan, X., and Grummt, I. (2004). mTOR-dependent activation of the
transcription factor TIF-IA links rRNA synthesis to nutrient availability. Genes & development
18, 423-434.
Mazurek, S., Boschek, C.B., Hugo, F., and Eigenbrodt, E. (2005). Pyruvate kinase type M2 and
its role in tumor growth and spreading. Seminars in cancer biology 15, 300-308.
Menon, S., Dibble, C.C., Talbott, G., Hoxhaj, G., Valvezan, A.J., Takahashi, H., Cantley, L.C.,
and Manning, B.D. (2014). Spatial Control of the TSC Complex Integrates Insulin and Nutrient
Regulation of mTORC1 at the Lysosome. Cell 156, 771-785.
Migliaccio, E., Giorgio, M., Mele, S., Pelicci, G., Reboldi, P., Pandolfi, P.P., Lanfrancone, L.,
and Pelicci, P.G. (1999). The p66shc adaptor protein controls oxidative stress response and life
span in mammals. Nature 402, 309-313.
Migliaccio, E., Mele, S., Salcini, A.E., Pelicci, G., Lai, K.M., Superti-Furga, G., Pawson, T., Di
Fiore, P.P., Lanfrancone, L., and Pelicci, P.G. (1997). Opposite effects of the p52shc/p46shc and
p66shc splicing isoforms on the EGF receptor-MAP kinase-fos signalling pathway. EMBO J 16,
706-716.
Minami, A., Iseki, M., Kishi, K., Wang, M., Ogura, M., Furukawa, N., Hayashi, S., Yamada, M.,
Obata, T., Takeshita, Y., et al. (2003). Increased insulin sensitivity and hypoinsulinemia in APS
knockout mice. Diabetes 52, 2657-2665.
Morita, S., Kojima, T., and Kitamura, T. (2000). Plat-E: an efficient and stable system for
transient packaging of retroviruses. Gene therapy 7, 1063-1066.
Morris, A.P., Voight, B.F., Teslovich, T.M., Ferreira, T., Segre, A.V., Steinthorsdottir, V.,
Strawbridge, R.J., Khan, H., Grallert, H., Mahajan, A., et al. (2012). Large-scale association
analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes.
Nature genetics 44, 981-990.

153
Munger, J., Bennett, B.D., Parikh, A., Feng, X.J., McArdle, J., Rabitz, H.A., Shenk, T., and
Rabinowitz, J.D. (2008). Systems-level metabolic flux profiling identifies fatty acid synthesis as
a target for antiviral therapy. NatBiotechnol 26, 1179-1186.
Nakae, J., Kitamura, T., Silver, D.L., and Accili, D. (2001). The forkhead transcription factor
Foxo1 (Fkhr) confers insulin sensitivity onto glucose-6-phosphatase expression. The Journal of
clinical investigation 108, 1359-1367.
Napoli, C., Martin-Padura, I., de Nigris, F., Giorgio, M., Mansueto, G., Somma, P., Condorelli,
M., Sica, G., De Rosa, G., and Pelicci, P. (2003). Deletion of the p66Shc longevity gene reduces
systemic and tissue oxidative stress, vascular cell apoptosis, and early atherogenesis in mice fed
a high-fat diet. Proceedings of the National Academy of Sciences of the United States of
America 100, 2112-2116.
Natalicchio, A., De Stefano, F., Perrini, S., Laviola, L., Cignarelli, A., Caccioppoli, C.,
Quagliara, A., Melchiorre, M., Leonardini, A., Conserva, A., et al. (2009). Involvement of the
p66Shc protein in glucose transport regulation in skeletal muscle myoblasts. American journal of
physiology Endocrinology and metabolism 296, E228-237.
Natalicchio, A., Laviola, L., De Tullio, C., Renna, L.A., Montrone, C., Perrini, S., Valenti, G.,
Procino, G., Svelto, M., and Giorgino, F. (2004). Role of the p66Shc isoform in insulin-like
growth factor I receptor signaling through MEK/Erk and regulation of actin cytoskeleton in rat
myoblasts. The Journal of biological chemistry 279, 43900-43909.
Nemoto, S., and Finkel, T. (2002). Redox regulation of forkhead proteins through a p66shc-
dependent signaling pathway. Science 295, 2450-2452.
Nicklin, P., Bergman, P., Zhang, B., Triantafellow, E., Wang, H., Nyfeler, B., Yang, H., Hild,
M., Kung, C., Wilson, C., et al. (2009). Bidirectional transport of amino acids regulates mTOR
and autophagy. Cell 136, 521-534.
Nobukuni, T., Joaquin, M., Roccio, M., Dann, S.G., Kim, S.Y., Gulati, P., Byfield, M.P., Backer,
J.M., Natt, F., Bos, J.L., et al. (2005). Amino acids mediate mTOR/raptor signaling through
activation of class 3 phosphatidylinositol 3OH-kinase. Proceedings of the National Academy of
Sciences of the United States of America 102, 14238-14243.
O'Connell, B.C., Cheung, A.F., Simkevich, C.P., Tam, W., Ren, X., Mateyak, M.K., and Sedivy,
J.M. (2003). A large scale genetic analysis of c-Myc-regulated gene expression patterns. The
Journal of biological chemistry 278, 12563-12573.
Oh, W.J., Wu, C.C., Kim, S.J., Facchinetti, V., Julien, L.A., Finlan, M., Roux, P.P., Su, B., and
Jacinto, E. (2010). mTORC2 can associate with ribosomes to promote cotranslational
phosphorylation and stability of nascent Akt polypeptide. The EMBO journal 29, 3939-3951.

154
Ohtsubo, K., Takamatsu, S., Minowa, M.T., Yoshida, A., Takeuchi, M., and Marth, J.D. (2005).
Dietary and genetic control of glucose transporter 2 glycosylation promotes insulin secretion in
suppressing diabetes. Cell 123, 1307-1321.
Okada, S., Kao, A.W., Ceresa, B.P., Blaikie, P., Margolis, B., and Pessin, J.E. (1997). The 66-
kDa Shc isoform is a negative regulator of the epidermal growth factor-stimulated mitogen-
activated protein kinase pathway. The Journal of biological chemistry 272, 28042-28049.
Orsini, F., Migliaccio, E., Moroni, M., Contursi, C., Raker, V.A., Piccini, D., Martin-Padura, I.,
Pelliccia, G., Trinei, M., Bono, M., et al. (2004). The life span determinant p66Shc localizes to
mitochondria where it associates with mitochondrial heat shock protein 70 and regulates trans-
membrane potential. The Journal of biological chemistry 279, 25689-25695.
Osthus, R.C., Shim, H., Kim, S., Li, Q., Reddy, R., Mukherjee, M., Xu, Y., Wonsey, D., Lee,
L.A., and Dang, C.V. (2000). Deregulation of glucose transporter 1 and glycolytic gene
expression by c-Myc. The Journal of biological chemistry 275, 21797-21800.
Panchaud, N., Peli-Gulli, M.P., and De Virgilio, C. (2013). Amino acid deprivation inhibits
TORC1 through a GTPase-activating protein complex for the Rag family GTPase Gtr1. Science
signaling 6, ra42.
Parsons, D.W., Jones, S., Zhang, X., Lin, J.C., Leary, R.J., Angenendt, P., Mankoo, P., Carter,
H., Siu, I.M., Gallia, G.L., et al. (2008). An integrated genomic analysis of human glioblastoma
multiforme. Science 321, 1807-1812.
Partridge, E.A., Le Roy, C., Di Guglielmo, G.M., Pawling, J., Cheung, P., Granovsky, M., Nabi,
I.R., Wrana, J.L., and Dennis, J.W. (2004). Regulation of cytokine receptors by Golgi N-glycan
processing and endocytosis. Science 306, 120-124.
Pawson, T. (2007). Dynamic control of signaling by modular adaptor proteins. Current opinion
in cell biology 19, 112-116.
Pawson, T., and Scott, J.D. (1997). Signaling through scaffold, anchoring, and adaptor proteins.
Science 278, 2075-2080.
Pearce, L.R., Huang, X., Boudeau, J., Pawlowski, R., Wullschleger, S., Deak, M., Ibrahim, A.F.,
Gourlay, R., Magnuson, M.A., and Alessi, D.R. (2007). Identification of Protor as a novel
Rictor-binding component of mTOR complex-2. The Biochemical journal 405, 513-522.
Pelicci, G., Lanfrancone, L., Grignani, F., McGlade, J., Cavallo, F., Forni, G., Nicoletti, I.,
Pawson, T., and Pelicci, P.G. (1992). A novel transforming protein (SHC) with an SH2 domain
is implicated in mitogenic signal transduction. Cell 70, 93-104.

155
Pena-Llopis, S., Vega-Rubin-de-Celis, S., Schwartz, J.C., Wolff, N.C., Tran, T.A., Zou, L., Xie,
X.J., Corey, D.R., and Brugarolas, J. (2011). Regulation of TFEB and V-ATPases by mTORC1.
The EMBO journal 30, 3242-3258.
Peterson, T.R., Laplante, M., Thoreen, C.C., Sancak, Y., Kang, S.A., Kuehl, W.M., Gray, N.S.,
and Sabatini, D.M. (2009). DEPTOR is an mTOR inhibitor frequently overexpressed in multiple
myeloma cells and required for their survival. Cell 137, 873-886.
Pinton, P., Rimessi, A., Marchi, S., Orsini, F., Migliaccio, E., Giorgio, M., Contursi, C., Minucci,
S., Mantovani, F., Wieckowski, M.R., et al. (2007). Protein kinase C beta and prolyl isomerase 1
regulate mitochondrial effects of the life-span determinant p66Shc. Science 315, 659-663.
Pinton, P., and Rizzuto, R. (2008). p66Shc, oxidative stress and aging: importing a lifespan
determinant into mitochondria. Cell Cycle 7, 304-308.
Porstmann, T., Santos, C.R., Griffiths, B., Cully, M., Wu, M., Leevers, S., Griffiths, J.R., Chung,
Y.L., and Schulze, A. (2008). SREBP activity is regulated by mTORC1 and contributes to Akt-
dependent cell growth. Cell Metab 8, 224-236.
Potter, C.J., Pedraza, L.G., and Xu, T. (2002). Akt regulates growth by directly phosphorylating
Tsc2. Nature cell biology 4, 658-665.
Rabinowitz, J.D., and White, E. (2010). Autophagy and metabolism. Science 330, 1344-1348.
Rampersaud, E., Damcott, C.M., Fu, M., Shen, H., McArdle, P., Shi, X., Shelton, J., Yin, J.,
Chang, Y.P., Ott, S.H., et al. (2007). Identification of novel candidate genes for type 2 diabetes
from a genome-wide association scan in the Old Order Amish: evidence for replication from
diabetes-related quantitative traits and from independent populations. Diabetes 56, 3053-3062.
Ranieri, S.C., Fusco, S., Panieri, E., Labate, V., Mele, M., Tesori, V., Ferrara, A.M., Maulucci,
G., De, S.M., Martorana, G.E., et al. (2010). Mammalian life-span determinant p66shcA
mediates obesity-induced insulin resistance. ProcNatlAcadSciUSA 107, 13420-13425.
Ravichandran, K.S. (2001). Signaling via Shc family adapter proteins. Oncogene 20, 6322-6330.
Reiling, J.H., and Hafen, E. (2004). The hypoxia-induced paralogs Scylla and Charybdis inhibit
growth by down-regulating S6K activity upstream of TSC in Drosophila. Genes & development
18, 2879-2892.
Robey, R.B., and Hay, N. (2009). Is Akt the "Warburg kinase"?-Akt-energy metabolism
interactions and oncogenesis. Seminars in cancer biology 19, 25-31.

156
Robitaille, A.M., Christen, S., Shimobayashi, M., Cornu, M., Fava, L.L., Moes, S., Prescianotto-
Baschong, C., Sauer, U., Jenoe, P., and Hall, M.N. (2013). Quantitative phosphoproteomics
reveal mTORC1 activates de novo pyrimidine synthesis. Science 339, 1320-1323.
Rodon, J., Dienstmann, R., Serra, V., and Tabernero, J. (2013). Development of PI3K inhibitors:
lessons learned from early clinical trials. Nature reviews Clinical oncology 10, 143-153.
Roux, P.P., Ballif, B.A., Anjum, R., Gygi, S.P., and Blenis, J. (2004). Tumor-promoting phorbol
esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90
ribosomal S6 kinase. Proceedings of the National Academy of Sciences of the United States of
America 101, 13489-13494.
Rozakis-Adcock, M., McGlade, J., Mbamalu, G., Pelicci, G., Daly, R., Li, W., Batzer, A.,
Thomas, S., Brugge, J., Pelicci, P.G., et al. (1992). Association of the Shc and Grb2/Sem5 SH2-
containing proteins is implicated in activation of the Ras pathway by tyrosine kinases. Nature
360, 689-692.
Ruvinsky, I., Sharon, N., Lerer, T., Cohen, H., Stolovich-Rain, M., Nir, T., Dor, Y., Zisman, P.,
and Meyuhas, O. (2005). Ribosomal protein S6 phosphorylation is a determinant of cell size and
glucose homeostasis. Genes & development 19, 2199-2211.
Sabatini, D.M., Erdjument-Bromage, H., Lui, M., Tempst, P., and Snyder, S.H. (1994). RAFT1:
a mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is
homologous to yeast TORs. Cell 78, 35-43.
Sabers, C.J., Martin, M.M., Brunn, G.J., Williams, J.M., Dumont, F.J., Wiederrecht, G., and
Abraham, R.T. (1995). Isolation of a protein target of the FKBP12-rapamycin complex in
mammalian cells. The Journal of biological chemistry 270, 815-822.
Sancak, Y., Bar-Peled, L., Zoncu, R., Markhard, A.L., Nada, S., and Sabatini, D.M. (2010).
Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its
activation by amino acids. Cell 141, 290-303.
Sancak, Y., Peterson, T.R., Shaul, Y.D., Lindquist, R.A., Thoreen, C.C., Bar-Peled, L., and
Sabatini, D.M. (2008). The Rag GTPases bind raptor and mediate amino acid signaling to
mTORC1. Science 320, 1496-1501.
Sancak, Y., Thoreen, C.C., Peterson, T.R., Lindquist, R.A., Kang, S.A., Spooner, E., Carr, S.A.,
and Sabatini, D.M. (2007). PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein
kinase. Molecular cell 25, 903-915.
Sarbassov, D.D., Ali, S.M., Kim, D.H., Guertin, D.A., Latek, R.R., Erdjument-Bromage, H.,
Tempst, P., and Sabatini, D.M. (2004). Rictor, a novel binding partner of mTOR, defines a

157
rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Current
biology : CB 14, 1296-1302.
Sarbassov, D.D., Ali, S.M., Sengupta, S., Sheen, J.H., Hsu, P.P., Bagley, A.F., Markhard, A.L.,
and Sabatini, D.M. (2006). Prolonged rapamycin treatment inhibits mTORC2 assembly and
Akt/PKB. MolCell 22, 159-168.
Sarbassov, D.D., Guertin, D.A., Ali, S.M., and Sabatini, D.M. (2005). Phosphorylation and
regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098-1101.
Saucedo, L.J., Gao, X., Chiarelli, D.A., Li, L., Pan, D., and Edgar, B.A. (2003). Rheb promotes
cell growth as a component of the insulin/TOR signalling network. Nature cell biology 5, 566-
571.
Schwartzenberg-Bar-Yoseph, F., Armoni, M., and Karnieli, E. (2004). The tumor suppressor p53
down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer research 64,
2627-2633.
Scott, J.D., and Pawson, T. (2009). Cell signaling in space and time: where proteins come
together and when they're apart. Science 326, 1220-1224.
Scott, R.A., Lagou, V., Welch, R.P., Wheeler, E., Montasser, M.E., Luan, J., Magi, R.,
Strawbridge, R.J., Rehnberg, E., Gustafsson, S., et al. (2012). Large-scale association analyses
identify new loci influencing glycemic traits and provide insight into the underlying biological
pathways. Nature genetics 44, 991-1005.
Selak, M.A., Armour, S.M., MacKenzie, E.D., Boulahbel, H., Watson, D.G., Mansfield, K.D.,
Pan, Y., Simon, M.C., Thompson, C.B., and Gottlieb, E. (2005). Succinate links TCA cycle
dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 7, 77-85.
Semenza, G.L. (2004). Hydroxylation of HIF-1: oxygen sensing at the molecular level.
Physiology (Bethesda) 19, 176-182.
Settembre, C., Zoncu, R., Medina, D.L., Vetrini, F., Erdin, S., Huynh, T., Ferron, M., Karsenty,
G., Vellard, M.C., Facchinetti, V., et al. (2012). A lysosome-to-nucleus signalling mechanism
senses and regulates the lysosome via mTOR and TFEB. The EMBO journal 31, 1095-1108.
Shah, O.J., Wang, Z., and Hunter, T. (2004). Inappropriate activation of the
TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival
deficiencies. Current biology : CB 14, 1650-1656.
Shimobayashi, M., and Hall, M.N. (2014). Making new contacts: the mTOR network in
metabolism and signalling crosstalk. Nature reviews Molecular cell biology 15, 155-162.

158
Smith, E.M., Finn, S.G., Tee, A.R., Browne, G.J., and Proud, C.G. (2005a). The tuberous
sclerosis protein TSC2 is not required for the regulation of the mammalian target of rapamycin
by amino acids and certain cellular stresses. The Journal of biological chemistry 280, 18717-
18727.
Smith, F.M., Holt, L.J., Garfield, A.S., Charalambous, M., Koumanov, F., Perry, M., Bazzani,
R., Sheardown, S.A., Hegarty, B.D., Lyons, R.J., et al. (2007). Mice with a disruption of the
imprinted Grb10 gene exhibit altered body composition, glucose homeostasis, and insulin
signaling during postnatal life. MolCell Biol 27, 5871-5886.
Smith, W.W., Norton, D.D., Gorospe, M., Jiang, H., Nemoto, S., Holbrook, N.J., Finkel, T., and
Kusiak, J.W. (2005b). Phosphorylation of p66Shc and forkhead proteins mediates Abeta toxicity.
The Journal of cell biology 169, 331-339.
Stevenson, L.E., and Frackelton, A.R., Jr. (1998). Constitutively tyrosine phosphorylated p52
Shc in breast cancer cells: correlation with ErbB2 and p66 Shc expression. Breast Cancer
ResTreat 49, 119-128.
Stocker, H., Radimerski, T., Schindelholz, B., Wittwer, F., Belawat, P., Daram, P., Breuer, S.,
Thomas, G., and Hafen, E. (2003). Rheb is an essential regulator of S6K in controlling cell
growth in Drosophila. Nature cell biology 5, 559-565.
Tee, A.R., Fingar, D.C., Manning, B.D., Kwiatkowski, D.J., Cantley, L.C., and Blenis, J. (2002).
Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian
target of rapamycin (mTOR)-mediated downstream signaling. Proceedings of the National
Academy of Sciences of the United States of America 99, 13571-13576.
Tee, A.R., Manning, B.D., Roux, P.P., Cantley, L.C., and Blenis, J. (2003). Tuberous sclerosis
complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-
activating protein complex toward Rheb. Current biology : CB 13, 1259-1268.
Thoreen, C.C., Kang, S.A., Chang, J.W., Liu, Q., Zhang, J., Gao, Y., Reichling, L.J., Sim, T.,
Sabatini, D.M., and Gray, N.S. (2009). An ATP-competitive mammalian target of rapamycin
inhibitor reveals rapamycin-resistant functions of mTORC1. JBiolChem 284, 8023-8032.
Tomilov, A.A., Ramsey, J.J., Hagopian, K., Giorgio, M., Kim, K.M., Lam, A., Migliaccio, E.,
Lloyd, K.C., Berniakovich, I., Prolla, T.A., et al. (2011). The Shc locus regulates insulin
signaling and adiposity in mammals. Aging Cell 10, 55-65.
Trinei, M., Giorgio, M., Cicalese, A., Barozzi, S., Ventura, A., Migliaccio, E., Milia, E., Padura,
I.M., Raker, V.A., Maccarana, M., et al. (2002). A p53-p66Shc signalling pathway controls
intracellular redox status, levels of oxidation-damaged DNA and oxidative stress-induced
apoptosis. Oncogene 21, 3872-3878.

159
Tsun, Z.Y., Bar-Peled, L., Chantranupong, L., Zoncu, R., Wang, T., Kim, C., Spooner, E., and
Sabatini, D.M. (2013). The folliculin tumor suppressor is a GAP for the RagC/D GTPases that
signal amino acid levels to mTORC1. Molecular cell 52, 495-505.
Turcan, S., Rohle, D., Goenka, A., Walsh, L.A., Fang, F., Yilmaz, E., Campos, C., Fabius, A.W.,
Lu, C., Ward, P.S., et al. (2012). IDH1 mutation is sufficient to establish the glioma
hypermethylator phenotype. Nature 483, 479-483.
Urano, J., Tabancay, A.P., Yang, W., and Tamanoi, F. (2000). The Saccharomyces cerevisiae
Rheb G-protein is involved in regulating canavanine resistance and arginine uptake. The Journal
of biological chemistry 275, 11198-11206.
Ursini-Siegel, J., Hardy, W.R., Zuo, D., Lam, S.H., Sanguin-Gendreau, V., Cardiff, R.D.,
Pawson, T., and Muller, W.J. (2008). ShcA signalling is essential for tumour progression in
mouse models of human breast cancer. The EMBO journal 27, 910-920.
Ursini-Siegel, J., and Muller, W.J. (2008). The ShcA adaptor protein is a critical regulator of
breast cancer progression. Cell Cycle 7, 1936-1943.
Vander Heiden, M.G., Cantley, L.C., and Thompson, C.B. (2009). Understanding the Warburg
effect: the metabolic requirements of cell proliferation. Science 324, 1029-1033.
Vanhaesebroeck, B., Stephens, L., and Hawkins, P. (2012). PI3K signalling: the path to
discovery and understanding. Nature reviews Molecular cell biology 13, 195-203.
Vousden, K.H., and Ryan, K.M. (2009). p53 and metabolism. Nature reviews Cancer 9, 691-700.
Wang, L., Balas, B., Christ-Roberts, C.Y., Kim, R.Y., Ramos, F.J., Kikani, C.K., Li, C., Deng,
C., Reyna, S., Musi, N., et al. (2007). Peripheral disruption of the Grb10 gene enhances insulin
signaling and sensitivity in vivo. MolCell Biol 27, 6497-6505.
Wang, X., Campbell, L.E., Miller, C.M., and Proud, C.G. (1998). Amino acid availability
regulates p70 S6 kinase and multiple translation factors. The Biochemical journal 334 ( Pt 1),
261-267.
Wang, X., Li, W., Williams, M., Terada, N., Alessi, D.R., and Proud, C.G. (2001). Regulation of
elongation factor 2 kinase by p90(RSK1) and p70 S6 kinase. The EMBO journal 20, 4370-4379.
Warburg, O. (1956). On the origin of cancer cells. Science 123, 309-314.
Ward, P.S., Patel, J., Wise, D.R., Abdel-Wahab, O., Bennett, B.D., Coller, H.A., Cross, J.R.,
Fantin, V.R., Hedvat, C.V., Perl, A.E., et al. (2010). The common feature of leukemia-associated

160
IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-
hydroxyglutarate. Cancer Cell 17, 225-234.
Ward, P.S., and Thompson, C.B. (2012). Metabolic reprogramming: a cancer hallmark even
warburg did not anticipate. Cancer Cell 21, 297-308.
Watanabe, T., Nobusawa, S., Kleihues, P., and Ohgaki, H. (2009). IDH1 mutations are early
events in the development of astrocytomas and oligodendrogliomas. Am J Pathol 174, 1149-
1153.
Weinberg, F., Hamanaka, R., Wheaton, W.W., Weinberg, S., Joseph, J., Lopez, M.,
Kalyanaraman, B., Mutlu, G.M., Budinger, G.R., and Chandel, N.S. (2010). Mitochondrial
metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proceedings of
the National Academy of Sciences of the United States of America 107, 8788-8793.
Wellen, K.E., Lu, C., Mancuso, A., Lemons, J.M., Ryczko, M., Dennis, J.W., Rabinowitz, J.D.,
Coller, H.A., and Thompson, C.B. (2010). The hexosamine biosynthetic pathway couples growth
factor-induced glutamine uptake to glucose metabolism. Genes Dev 24, 2784-2799.
Wills, M.K., and Jones, N. (2012). Teaching an old dogma new tricks: twenty years of Shc
adaptor signalling. The Biochemical journal 447, 1-16.
Wilson, K.F., Wu, W.J., and Cerione, R.A. (2000). Cdc42 stimulates RNA splicing via the S6
kinase and a novel S6 kinase target, the nuclear cap-binding complex. The Journal of biological
chemistry 275, 37307-37310.
Wise, D.R., DeBerardinis, R.J., Mancuso, A., Sayed, N., Zhang, X.Y., Pfeiffer, H.K., Nissim, I.,
Daikhin, E., Yudkoff, M., McMahon, S.B., et al. (2008). Myc regulates a transcriptional program
that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proceedings of the
National Academy of Sciences of the United States of America 105, 18782-18787.
Wullschleger, S., Loewith, R., Oppliger, W., and Hall, M.N. (2005). Molecular organization of
target of rapamycin complex 2. The Journal of biological chemistry 280, 30697-30704.
Xi, G., Shen, X., and Clemmons, D.R. (2008). p66shc negatively regulates insulin-like growth
factor I signal transduction via inhibition of p52shc binding to Src homology 2 domain-
containing protein tyrosine phosphatase substrate-1 leading to impaired growth factor receptor-
bound protein-2 membrane recruitment. MolEndocrinol 22, 2162-2175.
Xi, G., Shen, X., and Clemmons, D.R. (2010a). p66shc inhibits insulin-like growth factor-I
signaling via direct binding to Src through its polyproline and Src homology 2 domains, resulting
in impairment of Src kinase activation. JBiolChem 285, 6937-6951.

161
Xi, G., Shen, X., Radhakrishnan, Y., Maile, L., and Clemmons, D. (2010b). Hyperglycemia-
Induced p66shc Inhibits Insulin-Like Growth Factor I-Dependent Cell Survival via Impairment
of Src Kinase-Mediated Phosphoinositide-3 Kinase/AKT Activation in Vascular Smooth Muscle
Cells. Endocrinology.
Xia, J., and Wishart, D.S. (2011). Web-based inference of biological patterns, functions and
pathways from metabolomic data using MetaboAnalyst. NatProtoc 6, 743-760.
Xu, W., Yang, H., Liu, Y., Yang, Y., Wang, P., Kim, S.H., Ito, S., Yang, C., Xiao, M.T., Liu,
L.X., et al. (2011). Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-
ketoglutarate-dependent dioxygenases. Cancer Cell 19, 17-30.
Yan, H., Parsons, D.W., Jin, G., McLendon, R., Rasheed, B.A., Yuan, W., Kos, I., Batinic-
Haberle, I., Jones, S., Riggins, G.J., et al. (2009). IDH1 and IDH2 mutations in gliomas. N Engl
J Med 360, 765-773.
Yan, L., Mieulet, V., Burgess, D., Findlay, G.M., Sully, K., Procter, J., Goris, J., Janssens, V.,
Morrice, N.A., and Lamb, R.F. (2010). PP2A T61 epsilon is an inhibitor of MAP4K3 in nutrient
signaling to mTOR. Molecular cell 37, 633-642.
Yang, C.P., and Horwitz, S.B. (2000). Taxol mediates serine phosphorylation of the 66-kDa Shc
isoform. Cancer research 60, 5171-5178.
Yang, Q., Inoki, K., Ikenoue, T., and Guan, K.L. (2006). Identification of Sin1 as an essential
TORC2 component required for complex formation and kinase activity. Genes & development
20, 2820-2832.
Yang, W., Xia, Y., Ji, H., Zheng, Y., Liang, J., Huang, W., Gao, X., Aldape, K., and Lu, Z.
(2011). Nuclear PKM2 regulates beta-catenin transactivation upon EGFR activation. Nature 480,
118-122.
Yip, C.K., Murata, K., Walz, T., Sabatini, D.M., and Kang, S.A. (2010). Structure of the human
mTOR complex I and its implications for rapamycin inhibition. Molecular cell 38, 768-774.
Yoon, J.C., Ng, A., Kim, B.H., Bianco, A., Xavier, R.J., and Elledge, S.J. (2010). Wnt signaling
regulates mitochondrial physiology and insulin sensitivity. Genes & development 24, 1507-1518.
Yu, Y., Yoon, S.O., Poulogiannis, G., Yang, Q., Ma, X.M., Villen, J., Kubica, N., Hoffman,
G.R., Cantley, L.C., Gygi, S.P., et al. (2011). Phosphoproteomic analysis identifies Grb10 as an
mTORC1 substrate that negatively regulates insulin signaling. Science 332, 1322-1326.

162
Yuneva, M., Zamboni, N., Oefner, P., Sachidanandam, R., and Lazebnik, Y. (2007). Deficiency
in glutamine but not glucose induces MYC-dependent apoptosis in human cells. The Journal of
cell biology 178, 93-105.
Zhang, Y., Gao, X., Saucedo, L.J., Ru, B., Edgar, B.A., and Pan, D. (2003). Rheb is a direct
target of the tuberous sclerosis tumour suppressor proteins. Nature cell biology 5, 578-581.
Zhao, S., Lin, Y., Xu, W., Jiang, W., Zha, Z., Wang, P., Yu, W., Li, Z., Gong, L., Peng, Y., et al.
(2009). Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce
HIF-1alpha. Science 324, 261-265.
Zheng, Y., Zhang, C., Croucher, D.R., Soliman, M.A., St-Denis, N., Pasculescu, A., Taylor, L.,
Tate, S.A., Hardy, W.R., Colwill, K., et al. (2013). Temporal regulation of EGF signalling
networks by the scaffold protein Shc1. Nature 499, 166-171.
Zinzalla, V., Stracka, D., Oppliger, W., and Hall, M.N. (2011). Activation of mTORC2 by
association with the ribosome. Cell 144, 757-768.
Zoncu, R., Efeyan, A., and Sabatini, D.M. (2011). mTOR: from growth signal integration to
cancer, diabetes and ageing. NatRevMolCell Biol 12, 21-35.