tga presentation: medical devices - manufacturer evidence and applications for inclusion in the artg
TRANSCRIPT

Manufacturer Evidence and applications for
inclusion in the ARTG
Medical Devices
Susan Barker
Devices Application and Verification Section
Medical Devices Branch
Devices Sponsor Information Day
11 October 2017

Purpose
Facilitate better understanding of the regulatory requirements for medical devices
ARTG inclusion – explain the process and give some examples
2

Overview
• Key definitions and regulatory framework
• Manufacturer’s Evidence
• Information in an application
• Avoiding common problems in applications
3

What is a medical device?
• Used for human beings
• Intended purpose
– Diagnosis, prevention, monitoring, treatment or alleviation of disease or compensation for injury
or disability
– Investigation, replacement or modification of anatomy or physiological processes
– Control of conception
• Principal intended action
– Not by pharmacological, immunological or metabolic means
• Definition of IVD in the MD Regulations
• Products declared to be or not to be a medical device
– Therapeutic Goods (Articles that are Medical Devices) Specification and/or
Therapeutic Goods (Articles that are not Medical Devices) Orders
• Accessory to a medical device as described aboveTherapeutic Goods Act 1989, section 41BD 4

What is an IVD?
• A reagent, calibrator, control material, kit, specimen receptacle, instrument,
software, equipment or system
• Intended for the in vitro examination of human specimens for
– giving information about a physiological or pathological state
– giving information about a congenital abnormality
– determining safety and compatibility with a potential recipient
– monitoring therapeutic measures
Therapeutic Goods (Medical Devices) Regulations 2002, Dictionary5

ARTG inclusion
• Any medical device must be included in ARTG
• Except:
- Exempt devices (e.g. custom-made medical devices)
• Sponsor is responsible for ARTG inclusion
• Before you start
• TGA Business Services (TBS)
– Register and get your Client ID
6

Process for ARTG inclusion
Basics - process through TGA
7

Manufacturer evidence• Manufacturer must apply appropriate conformity assessment procedure to the device
(quality management system and control over the design of the device)
• Sponsor must lodge the manufacturer’s certification of the conformity assessment with the
TGA
- Except for Class I medical devices (no measuring function and/or not supplied sterile),
Class 1 IVD, and/or Export Only
• TGA conformity assessment certificate
• EC Certificate issued under MDD 93/42/EEC, AIMDD 90/385/EEC, IVD 98/79/EC
• MRA certificates issued by EU Notified Body (with certain exceptions)
• Declarations of conformity made under Clause 7.5 of Schedule 3 (systems or procedure
packs)
• ISO 13485 for IVDs only 8

Manufacturer Evidence
Submitted
Accepted
−What does it mean?
Go to the next step - lodging application
for ARTG inclusion
9

Application for ARTG inclusion
• Must be made for a kind of device and
• Must be made in accordance with a form and manner approved (via TBS) and
• Application fee must be paid and
• For the devices that must have TGA conformity assessment certificate – such certificate is in force and
• Must not contain information that is false or misleading in a material particular
Therapeutic Goods Act 1989, sections 41FC and 41EA, and Therapeutic Goods (Medical Devices) Regulations 2002, regulation 4.1
10

What we look at
Kind of device
Intended Purpose
GMDN Code
Device Product Characteristics
Classification
Conformity Assessment Procedures
11

Kind of device
• Sponsor
• Manufacturer
• Device nomenclature system code (GMDN)
• Classification
• Unique product identifier (UPI)(for Class III and active implantable medical devices (AIMD), and Class 4IVD, except immunohaematology reagent)
a medical device is taken to be of the same kind as
another medical device if they have the same:
Therapeutic Goods Act 1989, section 41BE and Therapeutic Goods (Medical
Devices) Regulations 2002, regulations 1.6 and 1.7 12

Intended purpose of a kind of device• Means the purpose for which the manufacturer of the device intends
it to be used, as stated in the information provided with the device
(labelling, instructions for use, advertising material and technical
documentation)
• Intended purpose stated in the application must be consistent with
the purpose for which the manufacturer intends the devices of the kind
to be used
Note: The manufacturer must have evidence that the device
performs as intended (refer essential principles)
13

Global Medical Device Nomenclature (GMDN)
- One of the characteristics that defines the kind of device
- Is to be consistent with the intended purpose of the device
- Must be the code that best describes the kind of device
- For medical devices (not-IVD) – relevant preferred term (except Class
I – relevant template term)
- For IVDs - collective terms (Level 1, 2 or 3) (except
Class 4IVD that is not immunohaematology reagent - relevant
preferred term)
Manufacturer’s responsibility14

Device Product Characteristics• Ensure ‘Device Product Characteristics’ section in the application is
correctly filled in
15

Medical devices are classified having regard to the intended purpose of the device
Class I Class Is and
Class Im
Class IIa Class IIb Class III and
AIMD
The lowest level The highest level
Therapeutic Goods (Medical Devices) Regulations 2002, Part 3
Division 3.1 and Schedules 216

IVD medical devices are classified having regard to the
intended purpose of the device and its risk to public health and/or personal risk
• Class 1 IVD – no public health risk or low personal risk
• Class 2 IVD – low public health risk or moderate personal risk
• Class 3 IVD – moderate public health risk or high personal risk
• Class 4 IVD – high public health riskRIS
K
Therapeutic Goods (Medical Devices) Regulations 2002, Schedules 2A17

Conformity assessment procedure
• Minimum conformity assessment procedures for different Classes of devices
• Sufficient information to demonstrate application of the appropriate conformity assessment procedures to the kind of device
Part 3 Division 3.2 and Schedule 3, Therapeutic Goods (Medical Devices) Regulations 2002
18

Conformity assessment procedures• Minimum conformity assessment procedures for different Classes of
devices
Class Is device
(e.g. Sterile surgical gown)
Class III device
supplied sterile
(e.g. Knee Femur)
Annex II.3 - Full Quality Assurance
Annex V – Production Quality
Plus
Declaration of conformity
(Part 6, Schedule 3)
Annex III - Type Examination
Plus:
Annex V - Production Quality Assurance
Annex II.3 - Full Quality Assurance
Plus
Annex II.4 - Examination of Design
OR
OR
19

One page document• Describe the device and intended purpose if needed in more detail
• Cite the classification rules in accordance with
Schedule 2 or 2A of the Regulations and provide justification where
required
• Explain how the kind of device is covered under the scope of certificate
included in Manufacturer’s Evidence
• Make sure all the information is complete and correct
• Do not attach more than one page
20

Information provided in the application• Do not attach information that is not relevant with the
application, for example:
– Declaration of conformity made under EU Medical Device Directive
– Audit and/or technical data reports
• Ensure intended purpose is clear and correct
• Ensure the device is classified correctly
• Ensure the Manufacturer’s Evidence stated in the application contains correct certificate
21

Examples of common mistakes
Incorrect Device
Product Characteristics
GMDN code not most
relevant for the kind of
device
Incorrect Classification
Scope of EC certificate
does not correctly cover
the kind of device
Attaching documents not
relevant, which do not
meet requirements
22

Where do we go from here?
Matters certified under s.41FD Ensure all information provided is correct
Any application may be selected for audit
Some applications must be selected for audit
23

SME Assist
• Guidance on TGA website
• Interactive online tool
24

Further information
• Medical devices (http://www.tga.gov.au/medical-devices-ivds)
• SME Assist (https://www.tga.gov.au/sme-assist)
TGA website
• Therapeutic Goods Act 1989 Chapter 3, Part 4-5
• Therapeutic Goods (Medical Devices) Regulations 2002
Federal Register of Legislation
• [email protected] ph.: 1800 141 144
Contact the TGA Medical Devices Branch
25

Thank you!
26


Juliana WilliamSr RA Associate
Baxter Healthcare
11/10/2017 Devices Sponsor Information Day 28

AgendaSuccessful and Efficient Submission
Preparation
Checks to do
Additional information
Non-mandatory audit submissions
This presentation will not cover details of:
• Systems and Procedure Packs
• Conformity Assessment
• Audit Assessment
• Class III/AIMD, UPI and Variants
• Clinical Evidence & Risk Assessment compliance
11/10/2017 Devices Sponsor Information Day 29

Common DeficienciesManufacturer Evidence
(ME)
• Two or more
certificates are
provided
• EU declaration of
Conformity
• ISO 13485 certificate
• Incomplete certificate
(missing attachments)
• Incorrect annex route
• Incorrect directive
• Not in English!
Device Application (DA)
• GMDN code does no
match the ‘Intended
purpose’
• Wrong classification
• Device is not covered in
the EC Certificate scope
• Route of conformity (EC
Certificate Annex) is not
appropriate for the
proposed Class
• Wrong manufacturer
evidence!
Administrative
• Missing documents or
pages!
• TGA evaluator unable to
find information easily
• Poor hyperlinking and
bookmarking
• Spelling errors
• Poor English Translation
• Missing signatures!
• Application fees not
processed
11/10/2017 Devices Sponsor Information Day 30

Preparation Ensure a legal/quality/distribution agreement is/are in place which outline
clearly roles and responsibilities for post-market monitoring, vigilance and reporting obligations
Obtain Australian Declaration of Conformity from manufacturer Make sure you have the right template for class and conformity route. See
http://www.tga.gov.au/industry/devices-forms-declaration-conformity.htm
Ensure the codes listed are the code to be registered and marketed
Obtain Australian Essential Principles Checklist from manufacturer● Ensure compliance with all standards applied and compliance with the Australian EP. Section 8 of the ARGMD
explain the differences between EU ER and Australia EP.
Product Labelling and Instruction for Use
Risk Management Report and Clinical Evaluation Report
11/10/2017 Devices Sponsor Information Day 31

Checks to Do
Check whether the device: Sterile? What is the sterilisation method
Measuring function?
Invasive?
Reusable or Single Use?
Active?
Single product or System or Procedure pack?
Contains a medicine?
Contains material or ingredients of microbial/ recombinant / GMO/ animal/ human origin? Country of origin?
Is the device classified correctly according to the TGA Classification Rules? (Schedule 2 of The Regulations)
11/10/2017 Devices Sponsor Information Day 32
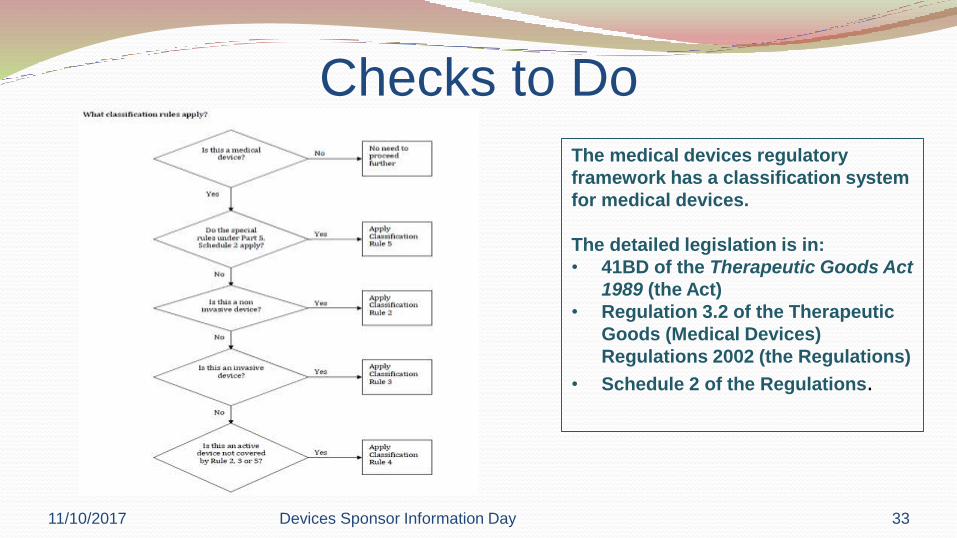
Checks to Do
The medical devices regulatory
framework has a classification system
for medical devices.
The detailed legislation is in:
• 41BD of the Therapeutic Goods Act
1989 (the Act)
• Regulation 3.2 of the Therapeutic
Goods (Medical Devices)
Regulations 2002 (the Regulations)
• Schedule 2 of the Regulations.
11/10/2017 Devices Sponsor Information Day 33

Checks to Do• Is the assigned GMDN code appropriate? (Section 10 of the Regulation)
Remember that manufacturer assign the GMDN code.
Check if the GMDN is on the TGA database. If the GMDN code is not in the TGA database, ask for it to be added in advance by email to [email protected]
There may be differences between the GMDN Agency code table database and the TGA code table database.
Based on the Device information, is the GMDN appropriate and reflects the intended purpose?
Must be a preferred term
Does the device perform according to its intended purpose? This should be a detailed description of the manufacturers intended purpose and should closely
align with the relevant GMDN description Remember: Intended purpose is different from Functional description (describes the
operation of the medical device, not its composition)
11/10/2017 Devices Sponsor Information Day 34

Checks to Do
● Is the device same kind of medical device (Section 41BE of The Act)?
A medical device is of the same ‘kind’ if it has the same: Intended purpose
Classification
GMDN code
Legal manufacturer
Sponsor
Does it have appropriate evidence of Conformity Assessment? ARGMD Section 5 Conformity Assessment Overview Need to check the appropriate level and route is held by the manufacturer
according to the classification
11/10/2017 Devices Sponsor Information Day 35

Checks to Do
11/10/2017 Devices Sponsor Information Day 36

Checks to Do
There are a number of
important details that a
sponsor should check to
ensure that the certificate
is valid for particular
devices.
Refer to Section 7 of the
ARGMD
11/10/2017 Devices Sponsor Information Day 37

Additional InformationIf there is additional information that could help the assessor, then it would be beneficial to attach a ONE PAGE ONLY document with the application illustrating:
A Picture/diagram of the device
A description of the device (not the intended purpose)
A description of the mechanism of action (how it works)
How the device is included in the scope of the CE Certificate
Evidence of the same kind of medical device (for multiple devices)
11/10/2017 Devices Sponsor Information Day 38

Non-mandatory audit Any Device Application is subject to non-mandatory audit.
Be prepared to submit any documents from Level 1 or 2 audit list, as well as anything else you may
need to show compliance with any of the Essential Principles or Conformity Assessment
Procedures.
Examples: Compliance with Essential Principles
Notified Body audit reports to verify the validity of the EC Certificate
Product labelling and IFU
Explanation and justification of Classification
Recent Clinical Evaluation Report
Recent Risk Management File
Compliance with particular standards (or justification why it is not complied with)
Remember you declare you can submit in 20 days!
11/10/2017 Devices Sponsor Information Day 39

Non-mandatory audit Provides all items requested by the TGA
Electronic and easy to navigate (entire document):
Clear table of content (hyperlinking and bookmarking work!)
Text readable
Page numbers throughout
Up-to-date Clinical Evidence Report (signed by a clinical expert!)
Up-to-date Risk Management File
Well prepared submission will:
Allow the delegate to easily navigate and find information quickly.
Reduce the number of questions raised by the delegate
Speed approval
The better quality the submission, the easier it is to read and evaluate
11/10/2017 Devices Sponsor Information Day 40

Wrap Up Make sure you understand your product.
Do your preparation and verify information before submission! Time spent before submitting is well worth it and will increase your success rate
Engage your cross function team (R&D/Quality/Medical/Clinical..etc) well in advance of your submission plan. Discuss application requirements and TGA’s expectation.
Submit meaningful, helpful and well-set out information. Perform second check before sending application and electronic files Clearly explain the scope of your application in the cover letter
Provide comprehensive Table of Content
Ensure all hyperlinks and bookmarks are actually working
Ensure all documents and attachments are provided with the application!
Don’t submit extra documents that aren’t useful or haven’t been requested
11/10/2017 Devices Sponsor Information Day 41

References
The Act
Therapeutic Goods Act 1989
The Regulations
Therapeutic Goods (Medical Device) Regulations 2002
ARGMD
Australian Regulatory Guidance for Medical Devices
11/10/2017 Devices Sponsor Information Day 42

Questions?
11/10/2017 Devices Sponsor Information Day 43