tesis doctoral · albert selva o'callaghan. doctor en medicina departamento de medicina...
TRANSCRIPT

NUEVAS TERAPIAS EN CçNCER COLORRECTAL METASTçSICO
REFRACTARIO: PAPEL DE LOS FçRMACOS
QUE INTERFIEREN EN LA VêA DE MET E INTEGRINAS
M» Elena �lez Fern�ndez
TESIS DOCTORAL
Facultat de Medicina - Departament de Medicina l 2015

Facultad de MedicinaDepartamento de Medicina
2015
NUEVAS TERAPIAS DIRIGIDAS EN CçNCER COLORRECTAL METASTçSICO:
PAPEL DE LOS FçRMACOS QUE INTERFIEREN EN LA VêA DE MET E INTEGRINAS
Tesis Doctoral presentada para optar al grado de Doctor en Medicina:Mar�a Elena �lez Fern�ndez
Programa de Doctorado en Medicina
Director de la TesisJosep Mar�a Tabernero Caturla. Doctor en Medicina
Departamento de MedicinaUniversitat Aut�noma de Barcelona
Tutor de la TesisAlbert Selva O'Callaghan. Doctor en Medicina
Departamento de MedicinaUniversitat Aut�noma de Barcelona

A mis padres F�lix y Teo por tantos a�os de duro trabajo. Por ellos he llegado hasta aqu�.
A mi marido Sebasti�n por su apoyo y respeto constantes desde el primer d�a.
A mi hermano F�lix y a Carmen por sus eternos buenos consejos.A mis sobrinas Adriana y Laura por su imaginaci�n
y gran capacidad para sorprenderme.A Leo por su incondicionalidad.
A todos los pacientes que de forma desinteresada han participado en estos estudios y que son la raz�n principal
por la que seguimos investigando.

4
NUEVAS TERAPIAS EN CçNCER COLORRECTAL METASTçSICO REFRACTARIO: PAPEL DE LOS FçRMACOS QUE INTERFIEREN EN LA VêA DE MET E INTEGRINAS
on muchas las personas que han marcado la trayectoria que he seguido para llegarhasta aqu�. Familia, amigos, compa�eros, docentes y profesionales que de un modo uotro han influido en cada una de las decisiones que han acabado condicionando mifuturo profesional y cient�fico. Sin embargo, no veo mejor forma para encabezar esta
tesis doctoral que dedic�ndosela al mayor ejemplo de nobleza, tenacidad y perseveranciaque he tenido durante todos estos a�os; mis padres. Sumo adem�s la brillante inteligencia ygenialidad de mi hermano F�lix que seguro �l mismo no sabe hasta qu� punto me ha ayuda-do y que comparten Mamen, Adriana y Laura como no pod�a ser de otra manera. Le agradez-co todo a Sebasti�n; el d�a a d�a, su apoyo constante, que entienda y respete mi trabajo y queme entienda a mi.
De mis primeros a�os de formaci�n, me gustar�a mencionar al Dr Francisco Franch que desper-t� en mi la curiosidad por la medicina, a Teresa Prat que me ayud� a tomar una decisi�n quehoy creo fue definitiva, a Mabeli y Maite que sin duda me ayudaron en mis primeros a�os aca-d�micos, a Julia por estar siempre ah� y a Marta porque no se me ocurre a nadie mejor paracompartir 6 a�os de carrera.
Le quiero dar las gracias a Antonieta Salud porque adora esta especialidad y as� lo ha transmiti-do a sus alumnos a lo largo de los a�os. Es un ejemplo de vocaci�n y humanidad.
A todos los m�dicos adjuntos y residentes que han contribuido de alg�n modo en mis primerosa�os de formaci�n profesional. Gracias a Toni Juan, Juan Ramon Perez Mas, Merc� Biosca eIsabela D�az de Corcuera. Gracias a Berta Bernad Jordi Rodon que siempre ser�n mis Rs mayo-res. A mis Co-Rs Pili y Laura porque nos hemos re�do mucho juntas y me han ayudado a ser elm�dico que soy hoy. A Ivan Castellv� porque ha sido compa�ero y amigo desde el primer d�a.
A todos los adjuntos de l'Institut Catal� d'Oncologia Duran i Reynals que me formaron comoresidente de Oncolog�a M�dica, gracias en particular a Xavier Garcia del Muro, Ricard Mesia yRam�n Salazar por despertar en mi la curiosidad que creo tiene que ser la base que motiva lainvestigaci�n.
S
AGRADECIMIENTOS

AGRADECIMIENTOS
5
A Ana Oaknin por conocerme, creer en mi y aconsejarme.
A Teresa Macarulla, Javier Ramos y Jaume Capdevila por ser compa�eros y profesores. A Mar�ay Guillem por su motivaci�n, entusiasmo, ayuda y amistad.
A Manolo, Cristina, V�ctor, Tamara y Julieta por su ayuda en el d�a a d�a y porque todav�a tene-mos ganas de re�rnos.
A Mar�a Herranz por la ayuda inestimable en el desarrollo de este trabajo y porque me encan-ta trabajar con ella.
A Albert Selva, porque sin �l esta tesis no hubiera sido posible. Porque por m�dicos y profeso-res como �l nos sigue gustando el arte de la medicina.
Quisiera agradecer por igual a los doctores Josep Baselga y Josep Tabernero su contribuci�n alcampo de la oncolog�a no s�lo desde un punto de vista cient�fico e investigacional sino tambi�nhumano ya que su capacidad de lucha y objetivo de cura en cada uno de nuestros pacientes esexcepcional y nos han llevado hasta aqu�.
A Josep Tabernero le quiero dar las gracias particularmente por darme esta oportunidad y porla confianza depositada en mi. Por haberme ense�ado que se puede ser cient�fico sin dejar deser m�dico. Por su capacidad docente y haberme inculcado su tenacidad y rigurosidad sin ago-tar su paciencia. Per respondre sempre, gr�cies.

6
NUEVAS TERAPIAS EN CçNCER COLORRECTAL METASTçSICO REFRACTARIO: PAPEL DE LOS FçRMACOS QUE INTERFIEREN EN LA VêA DE MET E INTEGRINAS
LISTA DE ABREVIACIONES . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 8
êNDICE DE FIGURAS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9
1. INTRODUCCIîN . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10
1.1. PREçMBULO . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10
1.2. ANTECEDENTES . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11
1.2.1. Base molecular del c�ncer colorrectal . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 111.2.1.1. Generalidades . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 111.2.1.2. V�a de se�alizaci�n de WNT/APC/§-catenina . . . . . . . . . . . . . . . . . . . . . 121.2.1.3. V�a de se�alizaci�n del factor de crecimiento epid�rmico . . . . . . . . . . 14
1.2.1.3.1. Se�alizaci�n mediada por RAS/RAF/MEK/MAPK . . . . . . . . . . . . . . .151.2.1.3.2. Se�alizaci�n mediada por PI3K/AKT/MTOR . . . . . . . . . . . . . . . . . . . .16
1.2.1.4. V�a de se�alizaci�n de MET . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 171.2.1.5. V�a de se�alizaci�n del factor de crecimiento transformante § . . . . . 181.2.1.6. Angiog�nesis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 19
1.2.1.6.1. Generalidades . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 191.2.1.6.2. Papel de las Integrinas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 20
1.2.2.Tratamiento convencional del c�ncer colorrectal . . . . . . . . . . . . . . . . . . . . . 211.2.2.1. Papel de los quimioter�picos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 211.2.2.2. F�rmacos antiangiog�nicos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 211.2.2.3. Anticuerpos dirigidos contra el receptor del factor
de crecimiento epid�rmico . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 221.2.2.3.1. Papel de los anticuerpos dirigidos contra el recetor del factor
de crecimiento epid�rmico en el tratamiento del c�ncer colorrectal metast�sico . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22
1.2.2.3.2. Factores de resistencia a terapias frente el factor de crecimiento epid�rmico . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23
1.2.3. Nuevas v�as de investigaci�n en c�ncer colorrectal metast�sico . . . . . . . . 27
êNDICE

êNDICE
7
2. ARTêCULOS DE TESIS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29
2.1. ARTêCULO DE TESIS I . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29
2.1.1. A pharmacodynamic/pharmacokinetic study of ficlatuzumab in patients with advanced solid tumors and liver metastases . . . . . . . . . . . . 29
2.1.2. Conclusiones art�culo del art�culo de tesis I . . . . . . . . . . . . . . . . . . . . . . . . . 43
2.2. ARTêCULO DE TESIS II . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 45
2.2.1. Abituzumab combined with cetuximab plus irinotecan versus cetuximab plus irinotecan alone for patients with KRAS wild-type metastatic colorectal cancer: the randomised phase I/II POSEIDON trial . . . . . . . . . . . . . . . . . . . 45
2.2.2. Conclusiones art�culo de tesis II . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 55
3. DISCUSIîN . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 57
4. CONCLUSIONES . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 60
5. REFERENCIAS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 62
6. AP�NDICE: OTRAS PUBLICACIONES . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 67
6.1. Publicaciones relacionadas con nuevas terapias dirigidas en c�ncer colorrectal metast�sico y biomarcadores pron�sticos y de resistencia a tratamiento sist�mico. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 67
6.2. Publicaciones relacionadas con estudios fase I para tumores s�lidos avanzados con f�rmacos que interfieren en el metabolismo epitelio-mes�nquima. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 136

8
NUEVAS TERAPIAS EN CçNCER COLORRECTAL METASTçSICO REFRACTARIO: PAPEL DE LOS FçRMACOS QUE INTERFIEREN EN LA VêA DE MET E INTEGRINAS
LISTA DE ABREVIACIONES
5-FU: 5-FluorouraciloADN: çcido desoxirribonucleico
ARNm: çcido ribonucleico mensajeroAKT: Prote�na quinasa B
ALFA-V: αυAPC: Adenomatous polyposis coliATP: Adenosin trifosfatoCCR: C�ncer colorrectal
CCRm: C�ncer colorrectal metast�sicoCMS: Clinical molecular subtypeDSH: DishevelledEGF: Epidermal growth factor
EGFR: Epidermal growth factor receptorEMT: Epithelial mesenchymal transitionERK: Quinasas reguladas por se�alizaci�n extracelular
Ev: EndovenosoFISH: Fluorescence in situ hybridation
FOXO3: Forkhead box O3FP: Fluoropirimidina
GEPs: Guanine Exchanging FactorsGTP: Guanina trifosforiladasHGF: Factor de crecimiento de los hepatocitosIMS: Inestabilidad de microsat�lites
IgG1: Inmunoglobulina 1Kg: Kilogramos
LEF: Potenciador del factor linfoideoLV: Leucovorin
MAPK: Prote�na quinasa activadora de mit�genosMg: Miligramos
mTOR: Mammalian target of rapamicyn NGS: Next generation sequencingPDK1 Prote�na quinasa 1 dependiente de fosfoin�sitoPI3K: Fosfo-Inositol 3 quinasaPIP3: Fosfo-inositol trifosfato
PLGF: Factor de crecimiento placentarioPTEN: Fosfatasa hom�loga del tensin�geno
RAS: Rat SarcomaSG: Supervivencia global
SLP: Supervivencia libre de progresi�nTCF: Factor de c�lulas TTGF: Factor de crecimiento transformante
TR: Tasa de respuestaVEGF: Vascular endotelial growth factor
VEGFR: Vascular endotelial growth factor receptorWIF: Factor inhibidor de WNTWT: Wild type

9
LISTA DE FIGURAS
Figura 1Alteraciones gen�ticas en la progresi�n
del c�ncer colorrectal
Figura 2V�a de se�alizaci�n de Wnt/§-catenina
Figura 3Receptores EGFR
Figura 4Principales v�as de se�alizaci�n celulares implicadas en la proliferaci�n
y progresi�n tumoral y su interacci�n
Figura 5V�a de se�alizaci�n de Met
Figura 6V�a de se�alizaci�n de VEGF
Figura 7Mecanismos moleculares de resistencia primaria
y secundaria a terapias anti-EGFR

10
NUEVAS TERAPIAS EN CçNCER COLORRECTAL METASTçSICO REFRACTARIO: PAPEL DE LOS FçRMACOS QUE INTERFIEREN EN LA VêA DE MET E INTEGRINAS
1. INTRODUCCIîN
1.1. PREçMBULO
l c�ncer colorrectal (CCR) sigue constituyendo una de las primeras causas de muerte anivel mundial(1). El mejor conocimiento de la biolog�a tumoral de cada tipo de tumorespec�fico ha sido crucial para el desarrollo e incorporaci�n de nuevos f�rmacos biol�gi-cos en el tratamiento de esta entidad llegando a alcanzar una supervivencia global supe-
rior a 30 meses en enfermedad diseminada seg�n datos presentados recientemente (2). Sinembargo, pese a todos estos progresos; la quimioterapia sigue siendo la base del tratamientode esta enfermedad puesto que es la herramienta terap�utica que ha demostrado mayor efica-cia. Sabemos que estos tumores presentan una alta tasa de respuesta a dicho tratamiento y tie-nen impacto en t�rminos de supervivencia global (SG) y supervivencia libre de progresi�n (SLP);no obstante, esta eficacia se acaba perdiendo y la enfermedad adquiere resistencia al mismo.De este modo, el CCR ha sido uno de los primeros tumores s�lidos en someterse a una clasifi-caci�n desde un punto de vista molecular, dando lugar a extensas investigaciones que han pues-to de manifiesto distintos genes y v�as de se�alizaci�n cruciales para el desarrollo y progresi�nde estos tumores as� como la identificaci�n de nuevas potenciales dianas terap�uticas y estra-tegias para revertir resistencia a tratamientos.
As�, los ensayos cl�nicos m�s recientes y los nuevos que se encuentran en v�as de desarrollo,est�n incorporando los �ltimos avances cient�ficos con el fin de pasar de una oncolog�a basadaexclusivamente en el tipo histol�gico para cada subtipo tumoral a una nueva pr�ctica cl�nicafundamentada en la clasificaci�n molecular.
Las terapias dirigidas deber�an ser �ptimas cuando se indiquen en un contexto molecular apro-piado. Esto supondr�a un tipo de f�rmaco que interferir�a en la funci�n de una mol�cula previa-mente identificada en la c�lula tumoral pero no en las c�lulas integrantes del tejido sano. Estadiana, desempe�ar�a un papel fundamental en el crecimiento anormal y/o la capacidad de inva-si�n y met�stasis del tumor. La potencial especificidad que aportar�an este tipo de f�rmacosconllevar�a una mayor eficiencia terap�utica respecto la quimioterapia est�ndar y adem�s evi-tar�a una toxicidad innecesaria a aquellos pacientes que presumiblemente, no tuvieran poten-cial beneficio del tratamiento.
Por tanto, �ste ser� un objetivo a perseguir en el desarrollo de nuevas terapias en el caso con-creto del c�ncer colorrectal metast�sico (CRCm). En definitiva, posibilitar� contribuir a validarposibles biomarcadores predictivos para terapias moleculares dirigidas, generar informaci�nvaliosa en lo que se refiere a la biolog�a tumoral y ganar eficacia en el potencial beneficio deltratamiento para el propio paciente.
E

INTRODUCCIîN
11
Este proceso ha incrementado la complejidad de la investigaci�n cl�nica en el sentido que, lasdosis de administraci�n y esquemas terap�uticos de cada uno de estos tratamientos; vendr�supeditada no s�lo a su perfil de toxicidad y seguridad sino tambi�n a ciertos requisitos biol�-gicos. Esto significa que es preciso alcanzar un equilibrio que nos permita administrar los trata-mientos a una dosis y pauta que sea segura para el paciente pero que a su vez, logre nuestraintenci�n terap�utica bloqueando la v�a de se�alizaci�n concreta que persigamos. Los estudioscl�nicos fase I son el marco id�neo para este objetivo ya que, paralelamente a los procedimien-tos realizados para testar distintos esquemas de tratamiento y dosis administradas de la tera-pia a estudio; se pueden asociar estudios de farmacodin�mica en que se testa la diana biol�gi-ca y respuesta en las c�lulas tumorales y/o sanas. La correlaci�n de estos hallazgos con par�me-tros de farmacocin�tica, nos conducen a la interpretaci�n de la dosis �ptima de tratamiento.
A lo largo de esta memoria se tratar� la situaci�n actual del c�ncer CCRm as� como la justifica-ci�n de la incorporaci�n de nuevas drogas en esta enfermedad. Se tratar�n las principales v�asde se�alizaci�n molecular implicadas en el desarrollo del CCR profundizando en aquellas impli-cadas en la proliferaci�n celular, particularmente en la v�a de se�alizaci�n del factor de creci-miento epid�rmico (EGFR). Partiendo de esta base, se trataran de manera concreta el papel quedesempe�a MET y las integrinas en este campo. A continuaci�n, se presentan los dos art�culosde tesis que siguen estas l�neas de investigaci�n en lo que se refiere a interferir en dichas v�asde se�alizaci�n para el tratamiento del CCRm.
En la primera publicaci�n, se caracteriza el desarrollo de Flicatuzumab o SCH900105, un anti-cuerpo monoclonal humanizado de tipo inmunoglobulina 1 (IgG1) dirigido contra el factor decrecimiento de los hepatocitos (HGF) con efectos antitumorales debido a su capacidad de uni�ny neutralizaci�n de HGF libre, inhibici�n de la fosforilaci�n de c-MET, inhibici�n de la prolifera-ci�n, inducci�n de apoptosis, angiog�nesis e inhibici�n de la capacidad de invasi�n y motilidadcelulares (3). En el segundo trabajo se describe la caracterizaci�n del f�rmaco Abituzumab oEMD525797 en combinaci�n con quimioterapia est�ndar en segunda l�nea de tratamiento paraCCRm. Abituzumab es anticuerpo monoclonal humanizado de tipo IgG2 que act�a contra lasubunidad alfa-v (αυ) de los receptores de la integrina humana. De este modo, el f�rmaco tieneel potencial de inhibir la progresi�n tumoral mediante la inhibici�n de la angiog�nesis inducidapor el tumor y la inhibici�n de la proliferaci�n tumoral actuando directamente sobre las c�lulastumorales y afectando a la migraci�n y la extravasaci�n de las c�lulas tumorales (4). Cada unade las dos publicaciones, se acompa�a de una discusi�n de los resultados presentados y poten-ciales l�neas de investigaci�n asociadas.
1.2. ANTECEDENTES
1.2.1. BASE MOLECULAR DEL CçNCER COLORRECTAL
1.2.1.1. Generalidades
A finales de la d�cada de los 80 y principios de los 90 se sentaron las bases gen�ticas del CCRmediante la descripci�n de dos modelos tumorig�nicos complementarios que conjuntamenteargumentaban la g�nesis de la mayor�a de los tumores de este origen: la secuencia p�lipo-ade-noma-adenocarcinoma de Vogelstein (5, 6), responsable de la g�nesis de aproximadamente el90% de los tumores de colon y la inestabilidad de microsat�lites (MSI) responsable del 10% res-tante descrita por Perucho (7) . Estos modelos posteriormente, se han ido completandomediante la sedimentaci�n progresiva de nuevo conocimiento hasta llegar al actual panoramamolecular de la enfermedad al inicio de la era de la ultrasecuenciaci�n tumoral.
As� pues, la mayor parte de tumores colorrectales son fruto de un proceso progresivo de even-tos que dan lugar a la ganancia de mutaciones acumulativas en ciertos oncogenes y la p�rdidade funci�n de genes supresores de tumores que llevan a la transici�n de lesiones benignas pre-

12
NUEVAS TERAPIAS EN CçNCER COLORRECTAL METASTçSICO REFRACTARIO: PAPEL DE LOS FçRMACOS QUE INTERFIEREN EN LA VêA DE MET E INTEGRINAS
existentes a la formaci�n de un carcinoma. El inicio de esta cascada de carcinog�nesis parte dealteraciones del gen que codifica para una prote�na llamada APC (del ingl�s; Adenomatous poly-posis coli) cuya p�rdida de funci�n conlleva la concatenaci�n de dicha sucesi�n de eventosmoleculares. Uno de los puntos fundamentales en la adquisici�n de un fenotipo agresivo vienedado por la incorporaci�n de activaciones de las v�as de transducci�n que se inician en losreceptores de membrana a trav�s de prote�nas intermediarias con actividad GTPasa que provo-can una se�al de inducci�n de proliferaci�n mediada por MAPK/ERK. Histol�gicamente, estosupone un evoluci�n del adenoma desde sus fases m�s incipientes hasta las m�s tard�as con unadisplasia que va alcanza progresivamente etapas de mayor severidad (5) (figura 1, adaptado deVogeltestein et cols (5)).
Estas v�as de se�alizaci�n han sido, por tanto; objeto de estudio no s�lo para el mejor conoci-miento de la biolog�a molecular del CCR sino tambi�n por su potencial aplicabilidad desde unpunto de vista terap�utico. A continuaci�n se detallar� los procesos moleculares concretos quetiene mayor impacto en la g�nesis y supervivencia tumoral en el caso concreto del CCR viendoadem�s, como estos han llegado y pueden llegar a constituir potenciales dianas farmacol�gicas.
1.2.1.2. V�a de se�alizaci�n de WNT/APC/§-CATENINA
Hasta en un 80% de los adenomas y tumores CCR, el gen supresor de tumoral APC se encuen-tra defectuoso siendo esta una de las alteraciones moleculares m�s frecuentes y precoces en lasecuencia de alteraciones gen�ticas acumuladas que llevan al CCR. En este tipo de tumores, lamayor parte de mutaciones a nivel de APC suponen un truncamiento de la prote�na conllevan-do su p�rdida de funci�n (8).
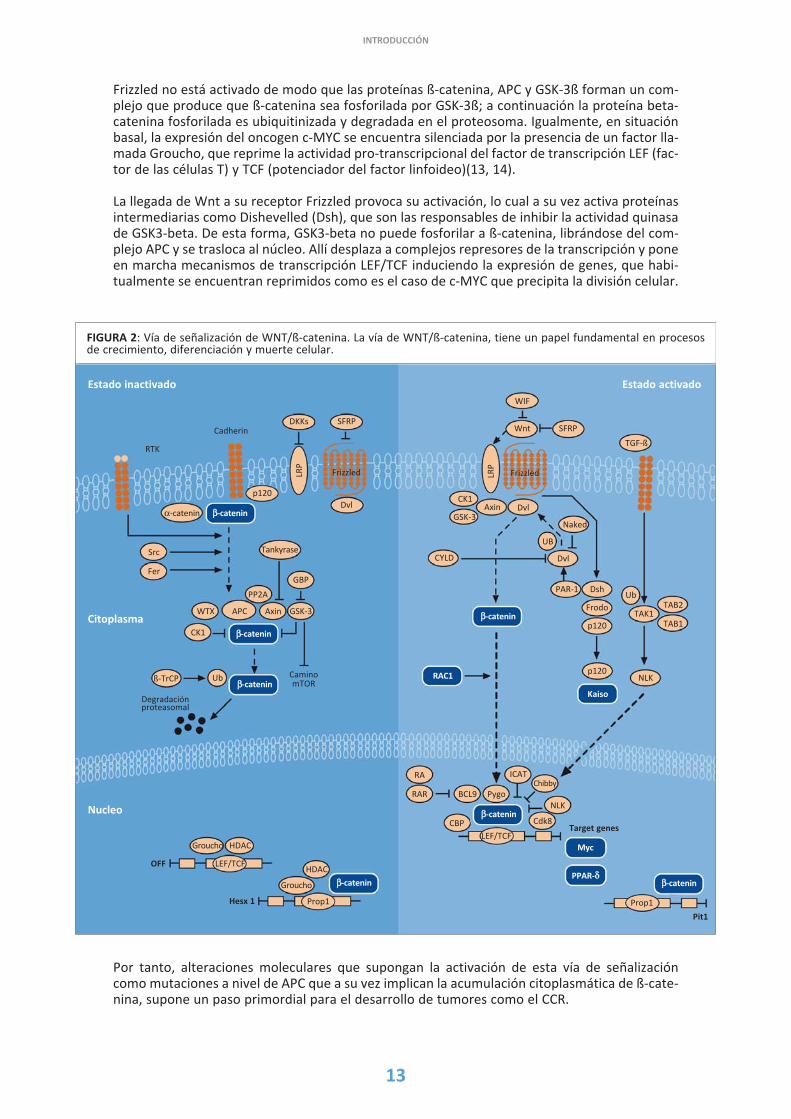
En condiciones normales, la prote�na APC degrada la §-catenina citoplasm�tica una vez �sta seune al complejo APC/axina/GSK-3§. GSK-3§ act�a fosforilando a §-catenina haciendo que seconjugue con ubiquitina y sea finalmente degradada (9, 10). Por tanto, la presencia de una pro-te�na APC no funcionante supone la acumulaci�n de §-catenina en el citoplasma que a su vez seacaba traslocando al n�cleo. Esta mol�cula no solo tiene un papel fundamental en la adhesi�ncelular sino que tiene una importante interrelaci�n con la v�a de se�alizaci�n de WNT pudien-do llegar a activarla tal y como se describe a continuaci�n (11, 12) (Figura 2).
El gen WNT1 codifica para una glicoprote�na que desempe�a un papel fundamental en la se�a-lizaci�n extracelular y, a su vez, forma parte de una familia de factores de crecimiento (13). Nos�lo WNT1, sino tambi�n otros miembros de la familia de WNT, intervienen en procesos de cre-cimiento, diferenciaci�n y muerte celular. Esta v�a de se�alizaci�n se inicia con la uni�n de WNTa un receptor espec�fico denominado Frizzled. Para que esto no ocurra, se encuentra tambi�nun factor extracelular denominado WIF (factor inhibidor de WNT). En esta situaci�n fisiol�gica,
FIGURA 1: Alteraciones gen�ticas en la progresi�n del CRC. Las v�as de se�alizaci�n m�s relevantes que llevan a latumorig�nesis se muestran en la transici�n de cada estadio evolutivo. Uno o diversos genes que codifican componentesde cada una de estas v�as pueden verse alterados en cada tumor en particular. La edad indica los intervalos de tiempodurante los cuales dichos genes suelen verse mutados.
V�as de se�alizaci�n APC RASPI3K
Ciclo celularTGF§
Edad cronol�gica (a�os) 30-50 40-60 50-70
Epitelio col�nico normal Adenoma incipiente Adenoma avanzado Carcinoma
INTRODUCCIîN
13
Frizzled no est� activado de modo que las prote�nas §-catenina, APC y GSK-3§ forman un com-plejo que produce que §-catenina sea fosforilada por GSK-3§; a continuaci�n la prote�na beta-catenina fosforilada es ubiquitinizada y degradada en el proteosoma. Igualmente, en situaci�nbasal, la expresi�n del oncogen c-MYC se encuentra silenciada por la presencia de un factor lla-mada Groucho, que reprime la actividad pro-transcripcional del factor de transcripci�n LEF (fac-tor de las c�lulas T) y TCF (potenciador del factor linfoideo)(13, 14).
La llegada de Wnt a su receptor Frizzled provoca su activaci�n, lo cual a su vez activa prote�nasintermediarias como Dishevelled (Dsh), que son las responsables de inhibir la actividad quinasade GSK3-beta. De esta forma, GSK3-beta no puede fosforilar a §-catenina, libr�ndose del com-plejo APC y se trasloca al n�cleo. All� desplaza a complejos represores de la transcripci�n y poneen marcha mecanismos de transcripci�n LEF/TCF induciendo la expresi�n de genes, que habi-tualmente se encuentran reprimidos como es el caso de c-MYC que precipita la divisi�n celular.
Por tanto, alteraciones moleculares que supongan la activaci�n de esta v�a de se�alizaci�ncomo mutaciones a nivel de APC que a su vez implican la acumulaci�n citoplasm�tica de §-cate-nina, supone un paso primordial para el desarrollo de tumores como el CCR.
FIGURA 2: V�a de se�alizaci�n de WNT/§-catenina. La v�a de WNT/§-catenina, tiene un papel fundamental en procesosde crecimiento, diferenciaci�n y muerte celular.
Estado inactivado
Citoplasma
Nucleo
Estado activado
DKKs
p120
α-catenin
Tankyrase
ββ-catenin
ββ-catenin
ββ-catenin
ββ-catenin
ββ-catenin
ββ-catenin
ββ-catenin
Myc
RAC1
Kaiso
PPAR-δδ
SFRP
WIF
Wnt SFRP
TGF-§
Dvl
GBP
PP2A
GSK-3Axin
mTOR
RTK
Cadherin
Camino
Degradaci�nproteasomal
APCWTX
Ub
LEF/TCFOFF
Hesx 1
HDAC
HDAC
RA
RAR BCL9 Pygo
ICAT
NLKp120
p120
Frodo
PAR-1 Dsh
Naked
Dvl
DvlAxin
LRP
CK1
GSK-3
FrizzledFrizzled
UB
CYLD
Ub
TAK1TAB1
TAB2
NLK
Cdk8
LEF/TCF
Prop1
Pit1
Target genesCBP
Chibby
Prop1
Groucho
Groucho
§-TrCP
CK1
Src
Fer
LRP

14
NUEVAS TERAPIAS EN CçNCER COLORRECTAL METASTçSICO REFRACTARIO: PAPEL DE LOS FçRMACOS QUE INTERFIEREN EN LA VêA DE MET E INTEGRINAS
1.2.1.3. V�a de se�alizaci�n del factor de crecimiento epid�rmico
El c�ncer de colon, como neoplasia epitelial, presenta una expresi�n muy significativa de EGF(del ingl�s; epidermal growth factor-EGF o factor de crecimiento epid�rmico) y/o su receptor;EGFR. EGFR se encuentra sobreexpresado en un 75-90% de los tumores y se ha postulado quedicho aumento en el n�mero de receptores extracelulares podr�a conferir peor pron�stico (15).Se trata de una prote�na de membrana con funci�n tirosin-quinasa que representa una impor-tante diana para el tratamiento contra el c�ncer dado que su activaci�n estimula una cascadade procesos implicados en el crecimiento y progresi�n tumoral incluyendo mecanismos de pro-liferaci�n, angiog�nesis, invasi�n y met�stasis (16). La familia del receptor EGFR comprende 4mol�culas: EGFR (HER1), HER2, HER3 y HER4. Estos receptores, tienen la habilidad de poderunirse a distintos ligandos que inician la activaci�n de la v�a una vez se da la uni�n al receptor(17). Se pueden definir sistem�ticamente tres pasos en la activaci�n de la se�alizaci�n intrace-lular dependiente de EGFR (Figura 3, adaptado de Ciardiello et cols (18)). En primer lugar, tienelugar la uni�n espec�fica de un ligando al receptor en la porci�n extracelular del receptor EGFRo en uno de los receptores relacionados con el mismo. A continuaci�n, se da la formaci�n deuna homodimerizaci�n o heterodimerizaci�n entre los mencionados receptores que supone lafosforilaci�n dependiente de adenosin-trifosfato (ATP) de residuos tirosina espec�ficos en el
FIGURA 3: Receptores EGFR. La activaci�n del receptor EGFR consiste en tres pasos fundamentales en que se da la uni�nespec�fica de un ligando a la porci�n extracelular del receptor, ocurre una homodimerizaci�n o heterodimerizaci�n entrelos receptores y se fosforilan los residuos tirosina espec�ficos en el dominio intracelular activando un programa de se�al-izaci�n intracelular desde el citoplasma hasta el n�cleo.
Ligandos receptor-espec�ficos
EGFTGFα
AnfiregulinaHB-EGF
Epiregulina
NRGsHB-EGF
NRGs
EGFR
Membrana plasm�tica
HER2
Dominios tirosin-quinasa
HER4
HER3
P
PP
PAkt
MAPK MEK
N�cleo
Transcripci�n
Proliferaci�n celularSupervivencia celularInvasi�n y met�stasis
Angiog�nesis inducida por tumor
Citoplasma
RAF
RAS
SOS
VEGFEGFR
EGFR, HER2,HER3 or HER4
TGFαInterleuquin-8
bFGF
Pl3K

INTRODUCCIîN
15
dominio intracelular del receptor EGFR (19). Finalmente, esta fosforilaci�n de los residuos espe-c�ficos tirosina activa un complejo programa de se�alizaci�n intracelular desde el citoplasmahasta el n�cleo. Una de las v�as de se�alizaci�n primordial mediada por la activaci�n del recep-tor EGFR es RAS-RAF-MEK-MAPK (20). Esta v�a, media la transcripci�n gen�mica, progresi�n delciclo celular desde G1 a S y proliferaci�n celular.
1.2.1.3.1. V�a de se�alizaci�n de RAS/RAF/MEK/MAPK
La sobre-activaci�n de la v�a de las MAPK es uno de los principales fen�menos en la g�nesis delCCR. Como ya se ha mencionado, la activaci�n de la cascada de se�alizaci�n comienza cuandoel dominio tirosina-quinasa de alguno de los receptores efectores de membrana es fosforilado.Tras este proceso, los grupos fosfato incorporados son capaces de atraer los dominios SH2 dealgunas de las prote�nas activadoras de KRAS que reciben el nombre en ingl�s de GuanineExchanging Factors (GEPs). Cuando esto sucede, los GEPs son a su vez fosforilados produci�n-dose un cambio en su conformaci�n que les permite atraer y unirse a las prote�nas de la fami-lia RAS. Las prote�nas RAS (KRAS, HRAS y NRAS) son GTPasas, cuya funci�n consiste en unirmol�culas de guanina tri-fosforiladas (GTP) y defosforilarlas, proceso mediante el cual sufren uncambio en su conformaci�n que les permite interactuar con otras prote�nas. Una vez esta inte-racci�n se ha realizado, la prote�na RAS recupera su conformaci�n inicial mediante la defosfori-laci�n de la mol�cula de GTP.
RAS es una de las prote�nas que podemos encontrar m�s frecuentemente mutada en tumoreshumanos (21). La mayor parte de mutaciones oncog�nicas de RAS se encuentran se dan en elcod�n 12 mediante la sustituci�n de una glicina por cualquier otro amino�cido en esta situa-ci�n. Esto implica un bloqueo de la uni�n GAP al complejo RAS-GTP haciendo que RAS seencuentre constitutivamente activado en la situaci�n RAS-GTP (22). La activaci�n continua deRAS genera una estimulaci�n celular constante de diversas funciones oncog�nicas tales como elcrecimiento, progresi�n del ciclo celular a trav�s de la fase G1-S y estimulaci�n de la angiog�-nesis. Adem�s, este fen�meno favorece la transformaci�n del metabolismo celular hacia pro-gramas predominantemente anab�licos que sustentan los nuevos y elevados requerimientosderivados del comportamiento tumoral, inmortalizaci�n celular o el incremento de la motilidady potencial metast�sico (23).
A continuaci�n, el complejo activo RAS-GTP se une a una prote�na serina-treonina quinasa lla-mada RAF mediante su fragmento N-terminal. La hidr�lisis de RAS-GTP a RAS-GDP hace que RAFsea activado mediante fosforilaci�n y posteriormente liberado (24). BRAF, miembro de la fami-lia de prote�nas RAF (A,B,C-RAF) es la principal efectora de RAS en la v�a de las MAPK. CuandoBRAF es activada por RAS, �sta interact�a con CRAF para activar a MEK, que a su vez extiendela se�al mitog�nica a la activaci�n de ERK que finalmente se trasloca al n�cleo celular dondeactiva los programas gen�ticos que resultan en los efectos previamente descritos (25). Hasta lafecha, se han identificado mutaciones en tres prote�nas RAF diferentes (ARAF, BRAF y CRAF) queest�n implicadas en tumores humanos. De �stas, las mutaciones en BRAF son las m�s frecuen-tes. Se han llegado a describir mutaciones de BRAF en el 13% de los tumores originados en colony recto de las cuales, el 99% presentaban transici�n a V600E. Este fen�meno es mutuamenteexcluyente con la presencia de mutaciones en RAS en una misma c�lula. La alteraci�n activado-ra m�s frecuente descrita es la V600E y consiste en una sustituci�n de la valina en su posici�n600 por un �cido glut�mico (26). Ya existe evidencia que indica que las mutaciones BRAF podr�-an actuar como un factor de resistencia a terapia anti-EGFR (27). Esto implicar�a que las muta-ciones BRAF activadas puedan suprimir la inhibici�n del crecimiento inducida normalmente porel agente terap�utico. De igual modo, tambi�n constituir�a un marcador pron�stico negativo enpacientes con CCRm y su efecto, al contrario de las mutaciones RAS, parece no estar limitado alos resultados del tratamiento con anti-EGFR (28). Por tanto, los f�rmacos inhibidores de BRAFpodr�an suponer una interesante alternativa terap�utica para los pacientes cuyos tumores pre-senten este tipo de mutaciones.

16
NUEVAS TERAPIAS EN CçNCER COLORRECTAL METASTçSICO REFRACTARIO: PAPEL DE LOS FçRMACOS QUE INTERFIEREN EN LA VêA DE MET E INTEGRINAS
1.2.1.3.2. V�a de se�alizaci�n de PI3K/AKT/mTOR
La v�a de PI3K-AKT-mTOR constituye un eje central en la se�alizaci�n celular tanto en tejidossanos como en la mayor�a de tipos tumorales. La desregulaci�n de esta v�a de se�alizaci�n esuna de las alteraciones m�s frecuentes en neoplasias humanas y se considera crucial para eldesarrollo tumoral (29). La se�al se desencadena con la estimulaci�n del receptor de membra-na, el dominio tirosina-quinasa del cual; una vez fosforilado atrae hacia s� el dominio SH2 de laparte p85 de PI3K (siglas en ingl�s de Fosfo-Inositol quinasa). PI3K es una quinasa lip�dica for-mada por dos subunidades, p85 parte reguladora y p110 subunidad catal�tica. Cuando la subu-nidad p85 est� unida al grupo fosfato del receptor transmembrana, se produce un cambio con-formacional sobre p110 que le posibilita interactuar con las mol�culas de fosfo-inositol bifosfa-to (PIP2) fosforil�ndolas para dar fosfo-inositol trifosfato (PIP3), mecanismo que tambi�n puedeocurrir por la uni�n de KRAS directamente a la subunidad p110. El PIP3 puede ser reciclado aPIP2 por acci�n de la fosfatasa hom�loga del tensin�geno (PTEN) que antagoniza la funci�n dePI3K actuando como represor de la v�a. PIP3 es capaz de fosforilar AKT (prote�na quinasa B) yPDK1 (prote�na quinasa 1 dependiente de fosfoinositol), dos prote�nas situadas por debajo enla v�a, que a su vez tienen como funciones m�s importantes la activaci�n de los complejos mTOR1 y 2 (del ingl�s; mammalian target of rapamycin), P70S6 quinasa y tambi�n la supresi�n deFOXO3a (del ingl�s; Forkhead box O3), provocando, entre otras cosas, la activaci�n del metabo-lismo, adquisici�n de resistencia a la apoptosis, un incremento de la motilidad celular, unaumento de la angiog�nesis y la progresi�n del ciclo celular.
La prote�na PI3K se encuentra alterada por mutaci�n en un 10-20% de los CCR siendo menos fre-cuente que se encuentre amplificada (30). Por otro lado, PTEN, principal represor de la v�a, puedeverse mermado por mutaciones inactivadoras o por disminuci�n de su expresi�n ya sea por p�r-didas al�licas o metilaci�n de su promotor (31). Entorno al 19-36% de los CCR presentan una dis-minuci�n en la expresi�n de PTEN, alteraci�n que por la relaci�n con la v�a de EGFR podr�a tenerun impacto en lo que se refiere al tratamiento con terapia especificas dirigidas (26).
Cabe destacar adem�s, que existen numerosas interacciones entre diferentes v�as de se�aliza-ci�n involucradas en la proliferaci�n y la progresi�n tumoral (Figura 4). Estas interacciones esta-blecer�an mecanismos de escape que permitir�an al tumor escaparse del bloqueo farmacol�gi-co. Como ya se ha mencionado, las v�as de RAS-RAF-MEK-MAPK y la de PI3K-Akt-mTOR jueganun papel importante en la tumorig�nesis a trav�s de la fosforilizaci�n de distintas prote�nas yfactores de transcripci�n que directamente controlan el crecimiento , la diferenciaci�n y laapoptosis (32). Mutaciones en los genes KRAS, NRAS, BRAF o PI3K resulta en una activaci�ncontinua de sus prote�nas de cascada RAS cascada RAS-MAPK o PI3K, de forma independientea la inhibici�n producida por f�rmacos que bloquean el receptor del EGFR.
FIGURA 4: Principales v�as de se�alizaci�n celulares involucradas en la proliferaci�n y laprogresi�n tumoral y su interacci�n.
Factores de crecimiento
C�ncer colorrectal

INTRODUCCIîN
17
1.2.1.4. V�a de se�alizaci�n de MET
El proto-oncogen MET se identific� por primera vez en l�neas celulares de osteosarcoma en1984 y su prote�na resultante fue posteriormente descrita como un receptor de tipo tirosin-qui-nasa cuyo ligando es el factor de crecimiento de los hepatocitos conocido en ingl�s como HGF(33, 34). La v�a de MET/HGF juega un papel fundamental en la formaci�n de los hepatocitos y laplacenta durante la embriog�nesis y sus efectos son cr�ticos para la regeneraci�n de tejidos (35,36). As� mismo, favorece la proliferaci�n de los queratinocitos y su migraci�n hacia zonas no epi-telizadas (37). Por tanto, la desregulaci�n en esta v�a de se�alizaci�n favorecer�a procesos deproliferaci�n, apoptosis y migraci�n celular que facilitar�an el proceso de crecimiento tumoral ymetastatizaci�n generando neoplasias dependientes de esta v�a.
La activaci�n dependiente de la uni�n de MET a HGF da lugar a la dimerizaci�n del receptor y lafosforilaci�n de tres residuos de dominio tirosin-quinasa dando lugar a un proceso de autofos-forilaci�n que favorece el reclutamiento de prote�nas efectoras citoplasm�ticas y activando lase�alizaci�n transmembrana. Los efectos de esta cascada de se�alizaci�n se transmitenmediante la v�a de las MAP-quinasas, PI3K/AKT, STAT y el factor nuclear k_ (Figura 5). Como seha mencionado previamente, la consecuencia final de la activaci�n de estas v�as de se�alizaci�n,es la activaci�n de procesos citoplasm�ticos y nucleares que incrementan la proliferaci�n celu-lar, supervivencia, movilizaci�n y capacidad de invasi�n (38, 39). Se ha observado que el desa-rrollo de modelos murinos con una mutaci�n activadora a nivel de MET, da lugar a la aparici�nde distintos tipos de neoplasias siendo esto una evidencia mas de la actividad oncog�nica quetendr�a este tipo de alteraci�n molecular (40). Aunque la amplificaci�n de MET (cromosoma7q31) se ha descrito en ciertos tipos de tumores como es el caso de CCR (41); la sobreexpresi�ndel receptor es m�s frecuentemente observada (34).
FIGURA 5: V�a de se�alizaci�n de MET. La uni�n de MET a HGF da lugar a la dimerizaci�n del receptor y fosforilaci�n deldominio tirosin-quinasa dando lugar a un proceso de autofosforilaci�n que favorece el reclutamiento de prote�nas efec-toras citoplasm�ticas entre las que se encuentran y activando la se�alizaci�n transmembrana.
APC RASPI3K
Ciclo celularTGF§
Citoplasma
E-cadherinaIntegrina
N�cleo

18
NUEVAS TERAPIAS EN CçNCER COLORRECTAL METASTçSICO REFRACTARIO: PAPEL DE LOS FçRMACOS QUE INTERFIEREN EN LA VêA DE MET E INTEGRINAS
Tambi�n cabe destacar la interacci�n de la v�a de MET con otras mecanismos celulares pro-oncog�nicos como HER2 y HER3. Por ejemplo, las c�lulas que expresan EGFR y MET han demos-trado ser independientes de la activaci�n de MET mediante su uni�n propia a ligando siendoposible este fen�meno gracias a la activaci�n a trav�s de EGFR (42, 43). Por otro lado, en algu-nos tipos de tumores en que la v�a de EGFR es particularmente relevante; se han hecho obser-vaciones particulares al respecto. Se ha observado que en el c�ncer de pulm�n de c�lula nopeque�a cuyas c�lulas presentan mutaci�n a nivel de EGFR presumiblemente sensibles a inhi-bidores tirosin-quinasa EGFR, la amplificaci�n de MET seria un mecanismo de resistencia a estetipo de terapias mediante la activaci�n de la v�a de PI3K a trav�s de HER3 (43, 44).
En el caso concreto de CCR, se ha observado en l�neas celulares que en ausencia de mutaci�n anivel de RAS y cuando existe sobreexpresi�n del factor de crecimiento tumoral alfa (TGF-§)como ligando de EGFR, da lugar a la activaci�n de la v�a de MET de modo que esto conferir�aresistencia a las terapias anti-EGFR como cetuximab o panitumumab (45, 46).
La se�alizaci�n mediada por MET tambi�n favorece la progresi�n tumoral mediante la angiog�-nesis. MET/HGF es un inductor potente de la producci�n del factor de crecimiento del endote-lio vascular (del ingl�s; vascular endothelial growth factor, VEGF) tipo A y un inhibidor de latrombospondina-1 a trav�s de mol�culas de se�alizaci�n comunes dando lugar al incrementode la vascularizaci�n (47, 48).
Finalmente, cabe destacar que tambi�n hay estudios que apuntan a un posible papel de METfavoreciendo el mantenimiento del nicho de las c�lulas madre o pluripotentes. HGF ser�a secre-tado por miofibroblastos del nicho favoreciendo su estabilizaci�n (49-52). Esto tiene particularrelevancia si tenemos en consideraci�n que la v�a de WNT tiene un papel fundamental en CCR.
1.2.1.5. V�a de se�alizaci�n del factor de crecimiento transformante §
El factor de crecimiento transformante § (TGF-§) forma parte de una amplia familia de p�ptidoscon funci�n de factores de crecimiento que incluyen 5 genes altamente conservados (TGF-§1 aTGF-§5) los productos de los cuales forman d�meros de 25 kDa. Se han identificado tres prote�-nas de la superficie celular capaces de unirse a TGF§. Estas ser�an los receptores de tipo 1, tipo2 y tipo 3 de TGF§ (TGF§1, TGF§2, TGF§3) que se unen a TGF§1 y TGF§2 con una alta afinidad.El dominio extracelular rico en ciste�nas de estas mol�culas presenta una regi�n con alta iden-tidad con la mol�cula DAF-1 y con el receptor de activina pero no tiene homolog�a con EGFR oel factor de crecimiento derivado de las plaquetas. Los receptores TGF§1 y TGF§2 se localizanen la membrana citoplasm�tica y se distribuyen de forma ubicua a nivel tisular. Ambos recep-tores median la acci�n de los miembros de la familia y son serina-treonina quinasas transmem-brana. TGF§ se une directamente al receptor 2 de TGF§, el cual es directamente fosforilado.
Las distintas isoformas de TGF§ son potentes inhibidores de la proliferaci�n en muchos tiposcelulares . Sin embargo, TGF§ tambi�n es secretado por una amplia variedad de c�lulas tumo-rales y su el incremento en su producci�n frecuentemente se correlaciona con propiedadesan�malas de adhesi�n celular pudiendo incrementar la capacidad metast�sica. TGF§ es capazde estimular al mismo tiempo la proliferaci�n de una l�nea celular de adenocarcinoma y promo-cionar la invasividad tumoral como ocurre tambi�n en ciertos tipos de tumores como el c�ncerde pulm�n. Sin embargo, en otro tipo de entidades como el c�ncer de mama, TGF§ inhibe laproliferaci�n posiblemente a trav�s de la estimulaci�n de la prote�na de uni�n al factor de cre-cimiento similar a la insulina (53, 54).
Cabe destacar que la sobreactivaci�n del eje de TGF§ es de particular relevancia en la enferme-dad avanzada puesto que contribuyen al mantenimiento y supervivencia de las c�lulas madrepluripotenciales a la vez que contribuye a la transici�n epitelio-mes�nquima (del ingl�s, epite-lial mesenchymal transition, EMT) favoreciendo as� el potencial de metastatizaci�n (55).

INTRODUCCIîN
19
1.2.1.6. Angiog�nesis
1.2.1.6.1. Generalidades
Es bien sabido que la angiog�nesis juega un papel crucial en el desarrollo del CCR. Se trata deun proceso mediante el cual se da lugar a la formaci�n de nuevos capilares en un lecho vascu-lar previo favoreciendo as� la supervivencia y crecimiento tumoral. Cuando el tumor adquiereun tama�o cr�tico que imposibilita la llegada de nutrientes y ox�geno, se da una situaci�n dehipoxia que es el desencadenante fundamental que da lugar a la liberaci�n de factores angio-g�nicos. VEGF es uno de los principales activadores angiog�nicos. Las c�lulas endotelialesexpresan en su membrana receptores con actividad tirosin-quinasa espec�ficos para VEGF (delingl�s; vascular endotelial growth factor receptor, VEGFR). Se conocen tres tipos de receptoresVEGF: VEGFR1, VEGFR2 y VEGFR3 (Figura 6, adaptado de Kerbel et cols (56)). El receptor de tipo1 es necesario para mediar la migraci�n de monocitos y macr�fagos y el reclutamiento de pre-cursores hematopoy�ticos, mientras que los receptores de tipo 2 y 3 est�n m�s implicados enlos procesos de linfangio y vasculog�nesis. VEGF se expresa en la mayor parte de neoplasiashumanas asoci�ndose generalmente a un peor pron�stico. Existen distintos factores que pue-den incrementar la expresi�n de VEGF como factores microambientales tipo hipoxia, pH bajo,
FIGURA 6: V�a de se�alizaci�n de VEGF: La activaci�n del receptor da lugar al inicio de una cascada de se�alizaci�n en laque se est�n implicadas otras v�as intracelulares de vital importancia para la proliferaci�n y supervivencia tumoral comoes el caso de la v�a de las MAP-quinasa o la v�a de PI3K.
VEGF-B
VEGF-A121 VEGF-A165
VEGF-CVEGF-D
PIGF
C�lula endotelial
Membrana plasm�tica
VEGFR-2(flk-1/KDR)NRP-2NRP-1
VEGFR-1(flt-1)
VEGFR-3(flt-4)
SÑS SÑS
MEK
Movilizaci�n
Migraci�nSupervivenciaProliferaci�n
MAPK
Akt
Pl3K
HostVEGF
MEK
Raf
PKC
PLCγ
Permeaci�nvascular

20
NUEVAS TERAPIAS EN CçNCER COLORRECTAL METASTçSICO REFRACTARIO: PAPEL DE LOS FçRMACOS QUE INTERFIEREN EN LA VêA DE MET E INTEGRINAS
citoquinas inflamatorias o factores de crecimiento. Otras causas, incluyen cambios gen�ticosque suponen la activaci�n de distintos oncogenes o p�rdida de funci�n de genes supresorestumorales. La uni�n de VEGF al receptor desencadena una cascada de se�alizaci�n que pone enmarcha mecanismos que dan lugar a un incremento de la permeabilidad celular que posibilitala entrada de otros factores de crecimiento, inducen la proliferaci�n celular, aumentan la capa-cidad de migraci�n de la c�lula, inhiben la apoptosis y finalmente aumentan la supervivencia(56). De este modo, bloquear los procesos que la angiog�nesis constituye una interesante v�apara tratar el CCR siendo bevacizumab, anticuerpo monoclonal dirigido a VEGF-A; el primer f�r-maco biol�gico incorporado al tratamiento est�ndar de la enfermedad avanzada (57, 58).
1.2.1.6.2. Papel de las integrinas
Las integrinas son receptores transmembrana de tipo glicoprote�na formadas por una subuni-dad α y β que median la interacci�n intercelular y con la matriz extracelular. El entorno extra-celular juega un papel fundamental en el comportamiento celular. De este modo, las integrinasactuar�an como receptores que median la uni�n y migraci�n celular mediante el reconocimien-to de distintas mol�culas de la matriz extracelular de modo que esto tiene particular relevanciaen la angiog�nesis tumoral mediante la interacci�n de las c�lulas tumorales con las c�lulasendoteliales. Las integrinas tambi�n tiene un efecto directo sobre las c�lulas tumorales estimu-lando la se�alizaci�n intercelular que puede afectar la expresi�n g�nica y alterar la regulaci�nde la supervivencia celular, diferenciaci�n y proliferaci�n (59-61).
Tal y como se ha descrito previamente, el rol de factores de crecimiento como VEGF y sus recep-tores como componentes fundamentales en el desarrollo de la angiog�nesis tumoral est�n bienestablecidos. Sin embargo, tambi�n sabemos que los factores de crecimiento secretados en ellecho tumoral tambi�n son de gran importancia la activaci�n de mol�culas de implicadas eninvasi�n y adhesi�n relevantes para la angiog�nesis. La supervivencia de las c�lulas tumorales,depende en gran parte de la uni�n c�lula-c�lula y de su contacto con la matriz extracelular demodo que las integrinas actuar�an permitiendo estas dos funciones. Existen varios mecanismosmediante los cuales la interacci�n de la matriz extracelular con las c�lulas tumorales puedenpromover la proliferaci�n celular, supervivencia e invasi�n incluyendo aquellos mediados pormetalopoteinas-3 (MMP-3) y las integrinas (62). La interacci�n que median las integrinas con lamatriz r�gida extracelular y la c�lula tumoral resultar�a en la activaci�n de la v�a de se�alizaci�nde RHO de modo que esto estimular�a la contractilidad y la EMT, la v�a de ERK y; por tanto, seinducir�a la proliferaci�n celular. De igual modo, las integrinas alfa§3/alfa§5 podr�an ejercerefectos antiangiog�nicos interaccionando con vibrinectina, fibrin�geno y otras mol�culas en lasc�lulas endoteliales tras la activaci�n de MMP-2 o trombospondina-1. Por otro lado, tambi�nexisten trabajos en que se ha observado que la sobreexpresi�n de integrinas en ciertos tipostumorales puede conferir un pron�stico desfavorable (63). As� pues, estas mol�culas podr�anconsiderarse tambi�n potenciales dianas terap�uticas en el CCR.
Base Molecular del C�ncer Colorrectal: Puntos Clave
¥ El modelo tradicional de desarrollo de CCR se basa en la acumulaci�n progresiva de alteracio-nes gen�ticas y epigen�ticas que llevan al desarrollo de una secuencia de progresi�n adeno-ma-carcinoma.
¥ Este modelo ha llevado a identificar los principales oncogenes y genes supresores de tumo-res implicados en este proceso como KRAS, NRAS, BRAF, PI3K y APC, PTEN respectivamente.
¥ Las alteraciones moleculares descritas conllevan una desregulaci�n en v�as de se�alizaci�ncruciales para la proliferaci�n y supervivencia tumorales siendo las m�s relevantes: WNT/§-catenina, TGF§, EGFR, v�a de las MAPK y PI3K.
¥ La angiog�nesis y los mecanismos de adhesi�n intercelular y con la matriz extracelular tienenun rol fundamental en la progresi�n tumoral y potencial de metastatizacion.
¥ Dichas alteraciones moleculares no s�lo han sido base para la incorporaci�n de nuevos agen-tes terap�uticos en el tratamiento del CCRm como anticuerpos anti-EGFR y antiangiog�nicos,sino que siguen una v�a de investigaci�n fundamental para la identificaci�n de mecanismosde resistencia a los mismos y desarrollo de nuevas terapias dirigidas.

INTRODUCCIîN
21
1.2.2. TRATAMIENTO CONVENCIONAL DEL CçNCER COLORRECTAL METASTçSICO
1.2.2.1. Papel de los quimioter�picos
Si bien al principio de la d�cada de los 80 la supervivencia media de los pacientes con CCRmera de 6 meses, �sta se prolong� con la incorporaci�n de quimioter�picos como el 5-Fluorouracilo (5-FU) que, en combinaci�n con leucovor�n (LV) ofreci� datos de SG de 12meses (64). El desarrollo de reg�menes infusionales de 5-FU en combinaci�n con LV supusouna mejor�a en el perfil de toxicidad del tratamiento as� como en t�rminos de eficacia. Porotro lado, la incorporaci�n de las fluoropirimidinas (FP) orales como capecitabina, tambi�n hapermitido ofrecer a estos pacientes otras alternativas terap�uticas con un buen perfil de toxi-cidad y eficacia (65, 66). La asociaci�n de 5-FU infusional a LV e irinotecan (FOLFIRI) u oxali-platino (FOLFOX) supuso una mejor�a en el pron�stico de estos enfermos alcanzando mayorTR, SLP y SG que 5-FU/LV (65, 67-69).
FOLFOX y FOLFIRI ofrecen una actividad similar aunque tienen distinto perfil de toxicidad fun-damentalmente neuropat�a sensorial atribuible a oxaliplatino y alopecia y mayor incidencia dediarrea en el caso de irinotecan (68). Cabe destacar que, cuatro estudios aleatorizados handemostrado que no hay mayores diferencias en t�rminos de SG de un tratamiento de combina-ci�n de inicio respecto una opci�n secuencial. As�, una pauta secuencial inici�ndose con FP seriauna opci�n v�lida en determinados pacientes considerados fr�giles o de riesgo para toxicidad(70-72). De hecho, ciertos estudios retrospectivos han demostrado que la exposici�n a los trescitost�ticos en distintas secuencias supone mayor SG. Tambi�n existe evidencia acerca de lastriples combinaciones de 5-FU, oxaliplatino e irinotecan en pauta FOLFOXIRI mostrando benefi-cio en todos los t�rminos de eficacia comparado con un doblete de FOLFIRI aunque a expensasde mayor toxicidad. �ste es un r�gimen de tratamiento, por tanto; que se reserva a pacientesseleccionados (73). Las segundas l�neas de tratamiento se basaran en la opci�n tomada en laprimera l�nea y no hay en la actualidad evidencia de que una opci�n preferente (68).
Sin embargo, lo que ha supuesto el mayor avance en el tratamiento de esta entidad ha sido laincorporaci�n de f�rmacos biol�gicos como es el caso de los f�rmacos antiangiog�nicos comobevacizumab y aflibercept y los anticuerpos anti-EGFR como cetuximab y panitumumab (28, 58,74). La exposici�n a todos estos agentes a lo largo de la historia oncol�gica del paciente es loque parece haber ofrecido mejores resultados de supervivencia. Recientemente se han presen-tado estudios fase III que enfrentan combinaciones que incluyen anti-EGFR o antiangiog�nicosy que ofrecen SGs entorno a los 30 meses siendo este beneficio mayor en poblaciones molecu-lares seleccionadas (75, 76).
1.2.2.2. F�rmacos antiangiog�nicos
Bevacizumab es un anticuerpo monoclonal que se une a VEGF evitando as� su uni�n a VEGFR1y VEGFR2 que se encuentra en las c�lulas endoteliales. Esto implica una reducci�n de la vascu-larizaci�n tumoral y por tanto inhibe el crecimiento tumoral.
Bevacizumab ha demostrado un incremento en t�rminos de SG, SLP y TR en primera l�nea detratamiento en combinaci�n con 5FU/LV e irinotecan (57). En asociaci�n con oxaliplatino y FPha evidenciado mejor�a en SLP tambi�n en primera l�nea de tratamiento (58). La combinaci�nde FOLFOXIRI con bevacizumab ha demostrado tambi�n tener impacto en SLP y TR en compa-raci�n con FOLFIRI y bevacizumab llegando a alcanzar unos de los mejores resultados de super-vivencia hasta el momento (77). La mol�cula tambi�n ofrece beneficios significativos en SG ySLP en asociaci�n con FOLFOX en segunda l�nea de tratamiento (78). Sin embargo, y de formam�s interesante; tambi�n se ha demostrado que mantener bevacizumab una vez se cambia elesquema de quimioterapia tras progresi�n a una primera l�nea que incluya el anticuerpo;impacta en SG y SLP (79). La toxicidad caracter�stica de bevacizumab es la derivada de su meca-nismo de su acci�n como antiangiog�nico, de modo que puede ocasionar hipertensi�n, protei-nuria y mayor incidencia de trombosis y sangrado entre otros. Cabe decir que a d�a de hoy toda-v�a no existen factores moleculares predictivos de respuesta para este tipo de agentes.

22
NUEVAS TERAPIAS EN CçNCER COLORRECTAL METASTçSICO REFRACTARIO: PAPEL DE LOS FçRMACOS QUE INTERFIEREN EN LA VêA DE MET E INTEGRINAS
Aflibercept es una mol�cula de fusi�n recombinante dirigida a bloquear VEGF-A, VEGF-B y elfactor de crecimiento placentario (PLGF). Esta mol�cula ha demostrado mejorar la SG, SLP y TRen combinaci�n con FOLFIRI en segunda l�nea de tratamiento para CCRm independientementedel tratamiento previo con bevacizumab (80, 81). Aflibercept presenta un perfil de toxicidadmuy similar a bevacizumab puesto que tambi�n se trata de un f�rmaco antiangiog�nico aunquese ha observado que se asocia a cierto incremento de los efectos secundarios de la quimiotera-pia presentando mayor incidencia de diarrea, neutropenia o mucositis.
Regorafenib es un f�rmaco oral inhibidor multiquinasa que ha demostrado incrementar SG ySLP respecto a tratamiento exclusivo de soporte en aquellos enfermos en situaci�n de refracta-riedad a tratamiento est�ndar (81). Los efectos adversos m�s frecuentemente observados en elestudio regulatorio s�ndrome mano-pie, astenia, diarrea, hipertensi�n y eritema o descama-ci�n.
1.2.2.3. F�rmacos dirigidos frente al receptor del factor de crecimiento epid�rmico
1.2.2.3.1. Papel de los anticuerpos dirigidos frente al factor de crecimiento epid�rmico en el tratamiento del c�ncer colorrectal metast�sico
La indicaci�n de tratamiento con anti-EGFR en CCRm supone el paradigma de la terapia perso-nalizada en esta neoplasia. Cetuximab y panitumumab son los dos f�rmacos anti-EGFR aproba-dos para el tratamiento de CCRm en distintas l�neas de tratamiento y bajo distintas combinacio-nes. Ambos f�rmacos son anticuerpos monoclonales de tipo murino IgG1 en el caso de cetuxi-mab y, humanizado e IgG2 en el caso de panitumumab. La potencial eficacia de los mismos, sinembargo; viene supeditada a aquellos pacientes cuyos tumores no presentan mutaci�n a nivelde los exones 2, 3 y 4 de KRAS y NRAS. Se ha demostrado que la presencia de mutaci�n a unode estos niveles no solo constituye un factor predictor de no respuesta a tratamiento con aestas drogas sino que, el empleo de las mismas en estas poblaciones; puede implicar incluso unefecto detrimental (74, 82). De este modo, la ausencia de mutaci�n a nivel de RAS es requisitopara la prescripci�n de ambos anticuerpos de acuerdo con la Agencia Europea delMedicamento.
En lo que se refiere a datos concretos de eficacia, ambos anticuerpos han demostrado actividaddesde su administraci�n en monoterapia hasta distintas combinaciones con esquemas citost�-ticos. Cetuximab ha demostrado beneficio en t�rminos de SG en pacientes refractarios a trata-miento quimioter�pico est�ndar comparado con tratamiento exclusivo de soporte (83, 84).Panitumumab impacta en SLP cuando se administra en monoterapia tambi�n en este subgrupode pacientes aunque probablemente no se lleg� a demostrar beneficio en SG debido a la posi-bilidad de administraci�n del f�rmaco una vez se constataba progresi�n en la poblaci�n quehab�a sido asignada a tratamiento exclusivamente de soporte (85, 86). Cabe decir, que ambosanticuerpos han demostrado eficacia similar cuando se han comparado directamente (87). Encombinaci�n con irinotecan, existen datos favorables acerca de la asociaci�n a cetuximab enque los resultados son superiores a la administraci�n del anticuerpo en monoterapia siendoesta una opci�n est�ndar en pacientes con tumores RAS wild type (del ingl�s; de tipo salvaje onativo, WT) y quimiorrefractarios (83-86, 88).
Los estudios de segunda l�nea han demostrado tambi�n beneficio en SLP y TR de combinacio-nes basadas en irinotec�n y anticuerpos anti-EGFR aunque sin llegar a alcanzar los mismosresultados para SG probablemente por la administraci�n de anti-EGFRs en l�neas posteriores detratamiento (89-91).
En lo que se refiere a la primera l�nea de tratamiento, ambos anticuerpos han demostradobeneficio en todos los t�rminos de eficacia. En el caso de cetuximab, la combinaci�n con FOLFI-RI supera a la quimioterapia en cuanto a SG, SLP y TR en la poblaci�n RAS WT (28, 92). En com-binaci�n con FOLFOX ha demostrado beneficio en t�rminos de TR y SLP (93) aunque esto no seha reproducido en estudios que exploraban otras combinaciones basadas en oxaliplatino ya seacon la FP oral capecitabina (CAPOX) o con bolus de 5-FU (FLOX) (93, 94). Panitumumab en com-binaci�n con FOLFOX ha demostrado impacto en SG, SLP y TR en la poblaci�n RAS WT (24, 25).

INTRODUCCIîN
23
De este modo, la combinaci�n de f�rmacos anti-EGFR no se recomienda en reg�menes que aso-cien capecitabina o bolus de 5-FU.
La toxicidad caracter�stica de estos medicamentos es una erupci�n cut�nea de tipo acneiformeas� como la hipomagnesemia. En el caso de cetuximab, al tratarse de un anticuerpo murino, laincidencia de reacciones infusionales es mayor que con panitumumab que se trata de un anti-cuerpo monoclonal humanizado.
A d�a de hoy todav�a no se ha establecido si hay una secuencia establecida para optar por untipo de biol�gico u otro en primera l�nea de tratamiento en la poblaci�n RAS no mutada. Existentres estudios cuyos resultados han sido presentados recientemente que comparan ambas estra-tegias. El estudio CALGB80405 exploraba la combinaci�n de cetuximab o bevacizumab con qui-mioterapia en primera l�nea de tratamiento en poblaci�n KRAS ex�n 2 no mutado (76). Losenfermos se aleatorizaban a recibir cetuximab o bevacizumab asociado al esquema de quimio-terapia escogido por el investigador siendo el objetivo principal del estudio SG. Los resultadosde este estudio se han presentado recientemente sin encontrar diferencias significativas entrelas combinaciones que inclu�an anti-EGFR o antiangiog�nico obteniendo una SG de 29'9 mesespara la poblaci�n tratada con bevacizumab y 29'0 meses para los tratados con el anticuerpoanti-EGFR. Cuando nos centramos en el an�lisis en base al esquema de quimioterapia emplea-do, los pacientes tratados con FOLFOX o FOLFIRI en combinaci�n con cetuximab o bevacizumabtampoco observamos diferencias relevantes entre la asociaci�n de uno u otro anticuerpo.Queda pendiente el an�lisis de dichos resultados en base al estudio completo del estado muta-cional de RAS.
El estudio FIRE-3 evaluaba la combinaci�n de FOLFIRI con cetuximab o bevacizumab en pobla-ci�n WT para ex�n 2 de KRAS (95). Este estudio fase III incluy� 592 pacientes sin encontrar dife-rencias significativas en TR, objetivo principal del estudio, aunque se observ� beneficio signifi-cativo en t�rminos de SG para los pacientes tratados con cetuximab siendo particularmenterelevante al analizar dichos resultados en base al resultados de RAS. Se ha postulado que estasdiferencias se puedan deber la profundidad y precocidad de respuesta al tratamiento que esmayor para la poblaci�n tratada con el anti-EGFR implicando esto, una traducci�n en t�rminosde SG tal y como ha mostrado el an�lisis radiol�gico centralizado del estudio.
Finalmente, el estudio PEAK se trata de un ensayo fase II que explora la combinaci�n de FOL-FOX con panitumumab o bevacizumab para poblaci�n KRAS WT ex�n 2 (96). Con el an�lisis com-pleto de RAS se objetiv� beneficio en t�rminos de SLP para los enfermos que se trataron con lacombinaci�n que inclu�a panitumumab .
En definitiva, todav�a queda por establecer la secuencia �ptima de tratamiento en la poblaci�nRAS WT y, lo que pueda llegar a ser m�s relevante; cuales son los mecanismos por los cualesuna poblaci�n ultraseleccionada para respuesta a tratamiento basado en anti-EGFRs como sonestos pacientes, pueden acabar desarrollando resistencia a dichos tratamientos.
1.2.2.3.2. Factores de resistencia a terapia dirigida frente al factor de crecimiento epid�rmico
Tal y como se ha descrito anteriormente, el bloqueo de EGFR mediante anticuerpos monoclo-nales como cetuximab y panitumumab, constituye una estrategia eficaz particularmente cuan-do se indica basada desde un punto de vista molecular en aquellos pacientes cuyos tumores notienen mutaci�n a nivel de RAS. Sin embargo, cabe destacar que en algunos casos, aun trat�n-dose de tumores RAS nativos, existen pacientes que no responden a dicho tratamiento.
Se han descrito distintos mecanismos potenciales de resistencia como la posibilidad de muta-ciones a nivel de BRAF o PI3K (30), amplificaci�n de HER2, MET o p�rdida de expresi�n de PTEN(45, 97, 98). Todos estos factores tienen como caracter�stica com�n su interacci�n en la v�a deEGFR en mayor o menor medida. Adem�s de estas alteraciones moleculares como posibles fac-tores predicciones negativos a terapia anti-EGFR, tambi�n se ha apuntado al n�mero de copiasdel gen que codifica para EGFR como un posible biomarcador candidato para predecir respues-ta a estos tratamientos (99). Sin embargo, la evaluaci�n del n�mero de copias del gen median-

24
NUEVAS TERAPIAS EN CçNCER COLORRECTAL METASTçSICO REFRACTARIO: PAPEL DE LOS FçRMACOS QUE INTERFIEREN EN LA VêA DE MET E INTEGRINAS
te FISH (del ingl�s; in situ fluorescence hybridation) es una t�cnica que difiere entre los distin-tos laboratorios de patolog�a molecular de modo que no hay un consenso acerca del punto decorte que debe considerarse para definirlo como un factor de predictor de resistencia a trata-miento (100). La sobreexpresi�n de HER3 y el factor de crecimiento de insulina tipo 1 tambi�nse ha postulado que pueden condicionar resistencia a la terapia anti-EGFR (101, 102). Por otrolado, incluso aquellos enfermos que responden a estas terapias, alcanzan refractariedad a lasmismas en alg�n momento de su historia oncol�gica. Esto supondr�a lo que podr�amos conside-rar resistencias adquiridas.
La resistencia a los anticuerpos monoclonales anti-EGFR suele aparecer despu�s de variosmeses de tratamiento. Se desconoce el mecanismo subyacente, pero datos recientes han iden-tificado las mutaciones de KRAS como impulsores potenciales y frecuentes de la resistenciaadquirida a ambos anticuerpos monoclonales anti-EGFR, panitumumab y cetuximab. La expan-si�n de los subclones con KRAS mutado as� como las mutaciones de KRAS de novo en clones conKRAS inicialmente no mutado parecen contribuir a la aparici�n de resistencia mediada por KRASdespu�s de iniciado el tratamiento con anticuerpos monoclonales anti-EGFR. Y lo que es m�simportante, los trabajo publicados por Misale et al y Diaz et al, indicaron que la incorporaci�nde inhibidores MEK a los anticuerpos monoclonales anti-EGFR pueden representar una estrate-gia racional para revertir la resistencia a los anticuerpos monoclonales anti-EGFR (103, 104).Este potencial para el tratamiento combinado con inhibidores MEK y anticuerpos monoclona-les anti-EGFR para revertir la resistencia adquirida est� siendo investigado para revertir la resis-tencia primaria a los anticuerpos monoclonales anti-EGFR en tumores con KRAS mutado.
Otra aproximaci�n que nos podr�amos plantear para aumentar la efectividad en el bloqueo dela v�a de se�alizaci�n mediada por EGFR seria el bloquear otros receptores distintos de la mismafamilia. Los receptores EGFR 2 a 4 juegan un papel fundamental mediando interacciones entrereceptores que procuran distintos mecanismos funcionales incluyendo co-activaci�n y/o activa-ci�n de otras v�as alternativas de se�alizaci�n. HER3 se encuentra mutado s�lo ocasionalmenteen CRCm, sin embargo de pueden detectar niveles elevados de mRNA y prote�na (105-107). Seha observado una alta expresi�n de ERBB3 en ciertos tumores como en el caso del CCR motivopor el que se ha pensado en el mismo como una potencial diana terap�utica fundamentalmen-te por la interacci�n que podr�a tener este receptor con otros de la misma familia. Adem�s, dis-tintos estudios han demostrado que ERBB3 podr�a tener un papel en la resistencia tumoral aterapias que pretenden bloquear EGFR y HER2.
En la actualidad existen numerosos compuestos en desarrollo frente a ERBB3 como ciertos anti-cuerpos monoclonales con gran afinidad espec�fica para unirse a EGFR y ERBB3. Una vez estetipo de mol�culas se unen a los mencionados receptores, se consigue evitar la uni�n de suscorrespondientes ligandos dando ligar a la inhibici�n de la se�alizaci�n mediada por la dimeri-zaci�n de EGFR/EGFR, EGFR/HER2, EGFR/HER3 y HER2/HER3. Distintos estudios precl�nicos hanpuesto de manifiesto la potencial eficacia aportada por estos medicamentos en tumores decolon, pulm�n, p�ncreas, mama u ovario (108, 109).
En relaci�n a HER2 como diana en CRCm, tambi�n existe evidencia precl�nica del potencial rolque podr�a jugar esta diana (97, 110). En el estudio publicado por Yonesaka et cols no s�lo seevidencia la amplificaci�n de HER2 como factor de resistencia intr�nseca y adquirida a anti-EGFRs en l�neas celulares sino que tambi�n se testa en pacientes (110). Se observ� que la super-vivencia era mayor para aquellos pacientes que no presentaban amplificaci�n de HER2 y recib�-an cetuximab mostrando una SG de 515vs 307 d�as, y SLP de 149 vs 89 d�as. Es m�s, la amplifi-caci�n de HER2 se observ� en una muestra tumoral obtenida posterior al tratamiento con cetu-ximab y no en la basal en dos de los pacientes que desarrollaron resistencia al mismo. El an�li-sis de una cohorte adicional de 70 enfermos puso de manifiesto que aquellos pacientes que pre-sentaban niveles pl�sticos menores de heregulina (ligando HER3/4) ten�an mayor probabilidadde respuesta a cetuximab. Estos resultados fueron consistentes con la determinaci�n de laexpresi�n de �cido ribonucleico mensajero (ARNm) en la muestra tumoral de 44 pacientes. En7 casos en los que se hab�a alcanzado resistencia a cetuximab, los niveles de heregulina eranmayores tras el tratamiento que antes del mismo. Por tanto, estos resultados nos indican quelos mecanismo de resistencia intr�nseca tambi�n pueden serlo de resistencia adquirida. Dehecho, la amplificaci�n de HER2 como un mecanismo de resistencia intr�nseca tambi�n ha sido

INTRODUCCIîN
25
descrita en un estudio basado en modelos marinos de CCR. Bertotti et al pusieron de manifies-to que aquellos espec�menes que no presentaban respuesta al tratamiento aun teniendo unestado nativo de KRAS, NRAS, BRAF y PIK3CA, presentaban amplificaci�n de HER2 (45). En untrabajo posterior llevado a cabo, se confirmo este hallazgo en una cohorte de pacientes condiagn�stico de CRCm, encontr�ndose la amplificaci�n en un 5'1% de casos. Estos hallazgos hansido la base para el desarrollo de estudios cl�nicos dirigidos a esta poblaci�n tras progresi�n atratamiento con cetuximab o panitumumab.
En otra l�nea, las alteraciones a nivel del propio receptor tirosin-quinasa tambi�n pueden con-tribuir a la resistencia adquirida a terapias anti-EGFR. En el trabajo publicado por Montagut etal, se evidencia la presencia de la mutaci�n a nivel del cod�n 494 (S492R) del gen de EGFR enl�neas celulares que alcanzan resistencia a cetuximab. De forma interesante, estos clones s� pre-sentaron respuesta a la introducci�n de gefitinib y panitumumab. De este modo, la mutaci�ndel receptor impedir�a la uni�n de cetuximab al receptor (111). La determinaci�n de esta alte-raci�n molecular se llev� a cabo en 10 pacientes diagnosticados de CRCm que presentaron pro-gresi�n de la enfermedad tras respuesta inicial a tratamiento con cetuximab. En 2 de 10 enfer-mos, se detect� la mutaci�n S492R en las biopsias tumorales obtenidas tras el tratamiento y laprogresi�n de la enfermedad. Uno de estos dos enfermos presentaba tambi�n mutaci�n a nivelde BRAF (V600E) en las muestras basales y post-tratamiento pese a que se documento respues-ta al anti-EGFR. Cabe destacar que en ambos casos se detecto amplificaci�n de EGFR en las dosmuestras obtenidas. Uno de estos dos enfermos recibi� tratamiento con panitumumab poste-riormente llegando a alcanzar respuesta al mismo. Sin embargo, no se pudieron llevar a cabonuevos an�lisis de potencial resistencia a la progresi�n de panitumumab tras 5 meses de trata-miento.
Este mismo grupo de trabajo, curs� la determinaci�n de esta mutaci�n de EGFR en 156 enfer-mos que no hab�an recibido tratamiento previo con cetuximab o panitumumab sin encontrar laalteraci�n en ning�n caso. Estos hallazgos fueron confirmados posteriormente en una cohortede enfermos de iguales caracter�sticas por un grupo de trabajo distinto lo cual parece indicarque dicho evento se tratara de un factor de resistencia adquirida (112).
Como se ha comentado previamente, HGF tiene la capacidad de unirse al receptor de membra-na de tipo tirosin-quinasa conocido como c-Met. Esta uni�n, da lugar a la estimulaci�n de deter-minadas funciones celulares relacionadas con su desarrollo, regeneraci�n tisular y homeostasiscomo puedan ser la mitog�nesis o la morfog�nesis. Esto sucede en una amplia variedad de estir-pes celulares incluyendo c�lulas epiteliales, endoteliales, hematopoy�ticas, neuronas, melano-citos y hepatocitos. Asimismo, la se�alizaci�n mediada por HGF tambi�n contribuye a la onco-g�nesis y progresi�n tumoral en distintos tipos de tumores favoreciendo la capacidad de inva-si�n celular asociada al potencial de metastatizaci�n. La v�a de c-Met se encuentra desreguladaen varios tipos de c�ncer ya sea por sobreexpresi�n, amplificaci�n o mutaci�n. La alteraci�nm�s frecuentemente observada en CRCm es la sobreexpresi�n y/o amplificaci�n. De este modo,la activaci�n de c-Met por HGF jugar�a un papel fundamental en la capacidad de metastatiza-ci�n de las c�lulas de CRC a nivel hep�tico cooperando con la mutaci�n de KRAS e incrementan-do el potencial oncol�gico de las c�lulas de CRCm (113). Tambi�n cabe destacar que la sobre-expresi�n del receptor c-Met y su ligando HGF se ha correlacionado con estadios avanzados deCRC y pron�stico desfavorable (114). Existen, adem�s, trabajos en l�neas celulares de CRC quepostulan la sobreexpresi�n de c-Met constituir�a un factor de resistencia a terapias anti-EGFR(45, 115, 116). La activaci�n del receptor tirosin-quinasa c-MET resultar�a en una autofosforila-ci�n y uni�n del mismo a prote�nas adaptadoras que llevar�a a reclutar a otras mol�culas aso-ciadas a la cascada de se�alizaci�n y activaci�n de la v�a que dar�a lugar a un incremento de laproliferaci�n, supervivencia, potencial de migraci�n y metastacizaci�n.
Convergencia de las v�as de resistencia
Como se ha descrito, la evoluci�n de la resistencia adquirida a terapias anti-EGFR se puede defi-nir como la consecuencia de una perturbaci�n en un sistema en el que el equilibrio esta basa-do en c�lulas altamente dependientes de EGFR. El hallazgo de que la mayor parte de mutacio-nes que emergen tras el tratamiento, involucran genes que son miembros directos de la v�a dese�alizaci�n de EGFR (EGFR, KRAS; NRAS o BRAF) indica que para escapar de esta perturbaci�n

26
NUEVAS TERAPIAS EN CçNCER COLORRECTAL METASTçSICO REFRACTARIO: PAPEL DE LOS FçRMACOS QUE INTERFIEREN EN LA VêA DE MET E INTEGRINAS
las c�lulas deben establecer un nuevo equilibrio que deber�a ser de nuevo basado en salir encierto modo de la v�a de se�alizaci�n de EGFR. Esta hip�tesis viene soportada por el an�lisis bio-qu�mico de modelos de l�neas celulares de CCR que desarrollan resistencia a terapia anti-EGFRindependientemente de genes/mutaciones que confieren resistencia, puesto que el desarrollodel mismo depende de una activaci�n basada en la activaci�n de MEK y ERK definiendo portanto un ejemplo de una evoluci�n convergente. De este modo, se podr�a postular que cuandose bloquea EGFR en un paciente con CCR con m�ltiples lesiones metast�sicas, el efecto que ejer-ce la droga, desencadena la evoluci�n bioqu�mica convergente de distintos clones cada uno delos cuales reactivar�a la v�a de EGFR. As�, pese a que cada met�stasis desarrollar�a lo que podr�-an considerarse factores de resistencia heterog�neos en realidad estar�an altamente relaciona-dos puesto que el objetivo final ser� reactivar la v�a de EGFR.
Esto tendr�a cierto impacto en el desarrollo de drogas para este tipo de tumores. En primerlugar, este escenario homogeneizar�a el perfil de resistencia �nter e intrapaciente. Por ejemplo,en pacientes con CCR que recibe terapias anti-EGFR, el abanico de alteraciones que pueden sur-gir tras la reca�da converger�an a la activaci�n del eje EGFR-RAS-MAPK. As�, en el momento dela progresi�n los distintos mecanismos de resistencia podr�an ser interceptados interfiriendo enla cascada de se�alizaci�n que es precisamente donde converger�an los distintos eventos gen�-ticos, en este caso a nivel de MAP-ERK. En segundo lugar, se podr�a postular que se podr�aretrasar la aparici�n de resistencias desde el inicio se bloquearan aquellas mol�culas o genesdiana que sabemos est�n implicados en dichos mecanismos de resistencia como pudiera ser laasociaci�n de un inhibidor de MEK a la terapia anti-EGFR. �sta seria pues, la base para el desa-rrollo de nuevas drogas y combinaciones designadas no s�lo a optimizar la respuesta a estostratamientos sino tambi�n a prolongar su eficacia y retrasar la situaci�n de progresi�n.
FIGURA 7: Mecanismos moleculares de resistencia primaria y secundaria a terapias anti-EGFR. Los mecanismos gen�ti-cos responsables de la resistencias de novo y adquiridas se solapan ampliamente a excepci�n de las mutaciones a niveldel receptor EGFR. Como punto com�n, convergen en la activaci�n del eje de las MAP-quinasas, MEK y ERK.
EGFR
MET, HER2,o KRASamplificaci�n
MET, HER2,o KRASamplificaci�n
Resistencias adquiridasResistencias de novo
Exon 2 Exon 2 Exon 2 Exon 2
Exon 3 Exon 3 Exon 3 Exon 3
Exon 4 Exon 4 Exon 4 Exon 4
Exon 15 Exon 15
MEK MEK
ERK ERK
Mutaciones BRAF Mutaciones BRAF
Mutaciones NRAS Mutaciones NRASMutaciones KRAS Mutaciones KRAS
EGFR mutaciones
Dominio extracelular
Dominio Tyrosina kinase

INTRODUCCIîN
27
Tratamiento convencional del c�ncer colorrectal metast�sico: Puntos Clave
¥ La quimioterapia sigue siendo base del tratamiento del CCRm. Las opciones de tratamientopara esta poblaci�n han evolucionado desde el empleo del 5-FU en monoterapia hasta lacombinaci�n de reg�menes con oxaliplatino e irinotec�n.
¥ El empleo de f�rmacos biol�gicos dirigidos frente a dianas moleculares como los anticuerposantiangiog�nicos: bevacizumab y aflibercept; anticuerpos anti-EGFR: cetuximab y panitumu-mab y el inhibidor multiquinasa regorafenib, es la maniobra que ha ofrecido mayor impactoen la supervivencia de estos pacientes.
¥ La evidencia derivada de estudios cl�nicos fase III prospectivos y aleatorizados todav�a no handemostrado una secuencia preferente en lo que se refiere al empleo de anticuerpos antian-giog�nicos o anti-EGFR y viceversa. La elecci�n del tratamiento debe basarse en el objetivodel mismo y perfil de toxicidad del esquema seleccionado.
¥ El beneficio de los anticuerpos anti-EGFR viene supeditado a una subpoblaci�n molecularconcreta basada en la ausencia de mutaci�n a nivel de los exones 2, 3 y 4 de KRAS y NRAS yposiblemente a la ausencia de mutaci�n a nivel de BRAF.
¥ La eficacia de los f�rmacos anti-EGFR est� condicionada a determinados factores de resisten-cia entre los que destacan, alteraciones gen�ticas a nivel de la v�a de se�alizaci�n de RAS-RAF-MEK, mutaciones del receptor EGFR, se�alizaci�n mediada por otros receptores EGFR oalteraciones que conllevan una desregulaci�n en la v�a de MET. Estos factores de resistenciaconstituyen, a su vez, base para el desarrollo de nuevos agentes dirigidos o nuevas estrate-gias terap�uticas investigacionales.
1.2.3. NUEVAS VêAS DE INVESTIGACIîN EN CçNCER COLORRECTAL METASTçSICO
En la introducci�n de esta memoria se ha descrito la base molecular del CCR, las principales v�asde se�alizaci�n implicadas en la misma y alteraciones que llevan al desarrollo de la entidad, as�como la identificaci�n de dianas moleculares base para el desarrollo de nuevos agentes tera-p�uticos. Sin embargo, el empleo de nuevas t�cnicas de ultrasecuenciaci�n han permitido unan�lisis molecular m�s exhaustivo del CCR tal y como se llev� a cabo por The Cancer GenomeAtlas Network (117). As�, teniendo en cuenta la tasa de mutaciones, el CCR se podr�a clasificaren dos grupos: tumor hipermutados o no hipermutados. Los tumores hipermutados presenta-r�an eventos som�ticos ya sea en los genes reparadores o genes relacionados con la reparaci�ndel ADN lo que caracteriza a los tumores con IMS. Por contra, los tumores no hipermutados ten-dr�an m�s alteraciones en el n�mero de copias de genes y m�s incidencia de mutaciones a nivelde TP53. Cabe destacar que hasta en un 50% de los tumores hipermutados se encontraronmutaciones a nivel de BRAF o TGF§R2 mientras que s�lo se observ� en un 5% de las muestrasque hab�an sido clasificadas como no tumores no hipermutados. No s�lo esto, sino que adem�s,las mutaciones a nivel de KRAS y PI3K condicionan la activaci�n de la v�a las MAPK y PI3K en lostumores no hipermutados. Sin embargo, pese a estas diferencias la inactivaci�n de la v�a dese�alizaci�n de WNT y la inactivaci�n de la v�a de TGF§ que resultan en un incremento de latranscripci�n de MYC son elementos pr�cticamente ubicuos en CCR. Otro tipo de alteracionescomo a nivel de EGFR incluyendo la amplificaci�n de EGFR2 tambi�n se describen en un gruporeducido de muestras de ambos subtipos. Por tanto, la complejidad del CRC va m�s all� de losbiomarcadores hasta ahora descritos y sobretodo aquellos validados para decisi�n terap�uticaque, a d�a de hoy; ser�an exclusivamente KRAS y NRAS.

28
NUEVAS TERAPIAS EN CçNCER COLORRECTAL METASTçSICO REFRACTARIO: PAPEL DE LOS FçRMACOS QUE INTERFIEREN EN LA VêA DE MET E INTEGRINAS
Recientemente, varios grupos de investigaci�n han aplicado m�todos de agrupaci�n no super-visada de datos gen�micos para intentar definir los subtipos intr�nsecos de CCR llegando a iden-tificarse hasta 6 potenciales subgrupos. Sin embargo, existen tres subgrupos que se constatanindependientemente del grupo de trabajo. De este modo, existir�a un subtipo de tumores queser�an hipermutados, con IMS, con mayor �ndice de mutaciones de BRAF que ser�an aquellostumores que podr�amos considerar de perfil inflamatorio. Un segundo subtipo ser�an tumoressin IMS de tipo epitelial proliferativo con una sobreactivaci�n de la v�a de WNT. Finalmente,existir�a un tercer subtipo com�n identificado que considerar�amos de tipo mesenquimal carac-terizado por ausencia de IMS y enriquecido por c�lulas madre pluripotenciales. Cabe decir, quepese a que existen ciertos subtipos bien identificados y caracterizados como los previamentedescritos, las caracter�sticas cl�nicas y moleculares se solapan en mayor o menor medida depen-diendo del trabajo publicado. Por este motivo, el Colorectal Cancer Subtyping Consortium, surgecon objetivo de consensuar una clasificaci�n molecular definitiva del CCR que sirva adem�scomo base para el desarrollo de nuevas v�as de investigaci�n y terapias dirigidas para cada sub-tipo molecular concreto. As�, podr�amos hipotetizar que las alteraciones que hasta el momentohemos venido testando en la pr�ctica cl�nica rutinaria es un modo muy grosero de clasificar estetipo de tumores. La clasificaci�n molecular puede dar explicaci�n al perfil pron�stico de cadauno de ellos as� como de la respuesta o resistencia al tratamiento instaurado.
En cualquier caso, si bien esta es la direcci�n en la que est� evolucionando la investigaci�n enCCR; el modelo actual se desarrollo farmacol�gico se basa en la identificaci�n de alteracionesmoleculares concretas que vehiculicen una se�alizaci�n aberrante subsidiaria de bloqueo far-macol�gico espec�fico.
En el momento del desarrollo de los estudios cl�nicos objetivo de esta memoria, esta �ltima fuela base para su dise�o, hip�tesis y objetivos tal y como se describe a continuaci�n.

ARTICULOS DE TESIS
29
2. ARTICULOS DE TESIS

30
NUEVAS TERAPIAS EN CçNCER COLORRECTAL METASTçSICO REFRACTARIO: PAPEL DE LOS FçRMACOS QUE INTERFIEREN EN LA VêA DE MET E INTEGRINAS
2.1. Art�culo de tesis (I)
2.1.1. A PHARMACODYNAMIC/PHARMACOKINETIC STUDY OF FICLATUZUMAB IN PATIENTS WITH ADVANCED SOLID
TUMORS AND LIVER METASTASES

ARTICULOS DE TESIS
45
2.2. Art�culo de tesis (2)
2.2.1. ABITUZUMAB COMBINED WITH CETUXIMAB PLUS IRINOTECAN VERSUS CETUXIMABPLUS IRINOTECAN ALONE FOR PATIENTS WITH KRAS WILD-TYPE METASTATIC COLORECTALCANCER: THE RANDOMISED PHASE I/II POSEIDON TRIAL

ARTICULOS DE TESIS
55
2.2.2. CONCLUSIONES ARTêCULO DE TESIS (II)
Abituzumab o EMD 525797 es un antagonista del anticuerpo monoclonal IgG2 que act�a contra lasubunidad αυ de los receptores de la integrina humana y que se ha desarrollado para el tratamien-to del c�ncer. Las integrinas son una familia de mol�culas de adherencia celular que intervienen enuna amplia variedad de interacciones entre las c�lulas y la matriz extracelular (MEC) y entre lasc�lulas entre s� (118). Se trata de receptores transmembranarios heterodim�ricos para las prote�-nas de la MEC, formados por una subunidad α y otra β y se activan uni�ndose a sus respectivosligandos de la MEC. Despu�s de esta interacci�n, las integrinas pueden poner en marcha cascadasde quinasas intracelulares para modular la proliferaci�n y la supervivencia celulares, y asociarse alcitoesqueleto de actina para dirigir la adhesi�n y la locomoci�n celulares (119).
EMD525797 se une espec�ficamente a la cadena αυ, de forma que inhibe la uni�n de ligandosa todos los heterod�meros de dicha cadena (aυ1β, aυβ3, aυβ5, aυβ6, aυβ8). Las integrinas αυse expresan en los vasos sangu�neos de los tumores angiog�nicos proliferantes y en determina-dos tipos de c�lulas tumorales. As� mismo, en ciertos tumores; el aumento de la expresi�n deαυβ3 se asocia a un incremento de la invasi�n y la met�stasis celulares (120). Se ha demostra-do que los miembros de la familia de integrinas aυ desempe�an una funci�n directa en la pro-gresi�n tumoral, la angiog�nesis tumoral y la met�stasis (121). Los datos de inmunohistoqu�mi-ca y los ensayos in vitro realizados con l�neas celulares de CCR y con c�lulas endoteliales handemostrado que la integrina αυ se expresa en la vasculatura tumoral y en c�lulas de CCR (122).Por consiguiente, abituzumab tiene el potencial de inhibir la progresi�n tumoral mediante lainhibici�n de la angiog�nesis inducida por el propio tumor, la inhibici�n del crecimiento tumo-ral actuando directamente sobre las c�lulas neopl�sicas y afectando a la migraci�n y la extra-vasaci�n de las c�lulas tumorales metast�sicas.
Se ha demostrado que las integrinas diana αυ de EMD525797 se expresan en la vasculatura yc�lulas tumorales CCR. En muestras congeladas de CRC se ha detectado expresi�n de αυβ3 mode-rada a intensa en las c�lulas estromales y m�s pronunciada a nivel de los vasos sangu�neos, y unaexpresi�n moderada a intensa de αυβ5 en las c�lulas tumorales (122). Como se ha descrito pre-viamente, tambi�n se ha propuesto que la expresi�n de las integrinas podr�a tener implicacionespron�sticas para el CCR. El an�lisis de la expresi�n de αυβ6 en pacientes con CCR puso ha pues-to de manifiesto que los pacientes con expresi�n elevada de αυβ6 presentan menor SG (63). Porotro lado, abituzumab ofrece tambi�n potencial antiangiog�nico. De este modo, la actividadantiangiog�nica del f�rmaco aumentar�a su actividad en el tratamiento del CCRm.
Tras considerar los datos obtenidos de los modelos precl�nicos mencionados y la eficacia de lacombinaci�n de irinotec�n y cetuximab en pacientes con diagn�stico de CCRm KRAS ex�n 2 nomutado en el momento del desarrollo de dicho estudio, este protocolo se dise�� para evaluarla eficacia de la combinaci�n de abituzumab, cetuximab e irinotec�n en sujetos con CCRm KRASex�n 2 no mutado.
Los objetivos principales en la parte de seguridad del estudio fueron caracterizar el perfil deseguridad y tolerabilidad de la administraci�n de diferentes niveles de dosis de EMD525797 encombinaci�n con cetuximab e irinotec�n en pacientes con CCRm de tipo KRAS wt a la progre-si�n del tratamiento de primera l�nea basado en oxaliplatino. En la parte aleatorizada, se eva-lu� la actividad antineopl�sica de dos dosis de abituzumab en lo que se refiere al tiempo desupervivencia sin progresi�n en pacientes con CCRm de tipo KRAS ex�n 2 no mutado despu�sdel fracaso del tratamiento de primera l�nea con una pauta con oxaliplatino. Como objetivossecundarios se estudi� la eficacia de dos dosis de EMD525797 con respecto a la duraci�n de laSG, el SLP, TR y el tiempo hasta el fracaso del tratamiento (TFT); el perfil farmacocin�tico deEMD525797 y sus efectos sobre cetuximab, y el perfil farmacocin�tica de cetuximab y su efec-to sobre EMD525797. Cabe destacar que el estudio contemplaba como objetivos exploratoriosel identificar posibles marcadores de predicci�n de la respuesta al tratamiento en investigaci�n,estudiar la relaci�n existente entre las prote�nas candidatas circulantes en la sangre y/o expre-sadas por el tumor y los criterios de valoraci�n de la eficacia y la seguridad, explorar el efectode las variaciones gen�ticas en el genoma del anfitri�n y/o en el genoma tumoral sobre los cri-terios de valoraci�n de la eficacia y la seguridad y finalmente establecer una correlaci�n entremodelos de expresi�n g�nica en muestras de sangre y/o en muestras tumorales y los criteriosde valoraci�n de la eficacia y la seguridad

56
NUEVAS TERAPIAS EN CçNCER COLORRECTAL METASTçSICO REFRACTARIO: PAPEL DE LOS FçRMACOS QUE INTERFIEREN EN LA VêA DE MET E INTEGRINAS
Los resultados del estudio POSEIDON demuestran que la mol�cula a estudio, abituzumab; tieneun adecuado perfil de seguridad en combinaci�n con irinotec�n y cetuximab en la poblaci�n aestudio llegando a alcanzar dosis de 1000 mg. En la parte aleatorizada del estudio fase II delestudio en que se incluyeron entorno a 70 pacientes con caracter�sticas cl�nicas bien balancea-das en cada una de las ramas de tratamiento, no se encontraron diferencias estad�sticamentesignificativas en lo que se refiere a SLP, objetivo principal del estudio. Tampoco se encontrarondiferencias en t�rminos de TR aunque s� se evidenci� una tendencia a la mejor�a en lo que a SGse refiere para los pacientes tratados con la combinaci�n investigacional respecto a los que reci-bieron s�lo irinotec�n y cetuximab. De forma interesante, aquellos pacientes cuyos tumorespresentaban una alta expresi�n de integrina αυβ6 y que recibieron tratamiento dentro de algu-nas de las ramas que contenida abituzumab, presentaron mejor SG (rama A; HR 0.55 [0.30-1.00], rama B; HR 0.41 [0.21-0.81]).
Este es el primer estudio cl�nico publicado dirigido a pacientes con diagn�stico de CCRm queexplora el papel de un anticuerpo anti-integrina. Como se ha descrito previamente, irinotecanen combinaci�n con cetuximab a la progresi�n a terapia previa con oxaliplatino, tiene una acti-vidad limitada en CCRm. Sin embargo, el an�lisis exploratorio pre-especificado en el estudiosugiere que abituzumab puede impactar en t�rminos de SG en aquellos pacientes con tumoresque expresen integrina αυβ6 constituyendo esta, una l�nea de investigaci�n futura. Adem�s, laexpresi�n de αυβ6 constituir�a un factor pron�stico negativo a tenor de la media de SG en lospacientes con expresi�n elevada de integrina αυβ6 que resulta ser entorno a 4 meses inferiorrespecto a los enfermos que con niveles bajos de la misma tal y como se hab�a observado enestudios previos para enfermedad limitada. Sin embargo, los resultados obtenidos del estudio,sugieren que a�adir abituzumab al tratamiento est�ndar en los pacientes con αυβ6 elevadareducir�a el riesgo de muerte entorno a un 56% comparado con la rama de tratamiento est�n-dar. La SLP y TR tambi�n tienden a mejorar cuando se selecciona la poblaci�n en base a dichobiomarcador.
Cabe destacar que existen trabajos que postulan que la integrina αυβ6 est� implicada en tran-sici�n epitelio-mes�nquima (TEM) que, a su vez, intensificar�a el fenotipo agresivo de tumoresepiteliales como el CCRm. El proceso de TEM tambi�n puede contribuir a la enfermedad metas-t�sica, y los altos niveles de αυβ6 a nivel de las met�stasis hep�ticas y del sistema linf�ticosugieren que esta integrina conferir�a mayor agresividad al CCRm. Por otro lado, esta integrinatambi�n es una factor activador de TGF-β que, como se ha comentado previamente, tambi�nestimula el crecimiento e invasi�n tumorales durante la progresi�n del CCR. Por tanto, las tera-pias cuya diana sea αυβ6, tendr�an el potencial de interferir en la capacidad de invasi�n ymetastatizaci�n de las c�lulas tumorales justificando as� la mejor�a obtenida en OS y PFS paralos pacientes tratados con abituzumab y niveles altos de αυβ6. Adem�s, TGF-β tambi�n tieneun papel clave en la regulaci�n de la inmunidad y sabemos que las integrinas son elementos cr�-ticos para este proceso. Adem�s, la capacidad de abituzumab para bloquear αυβ6 disminuir�ala actividad inhibitoria de TGF-β sobre la respuesta antitumoral siendo este otro potencialmecanismo de acci�n adicional para la mol�cula.
Cabe decir, que si bien los resultados particularmente obtenidos en esta poblaci�n molecularseleccionada son muy alentadores, los datos deben interpretarse con cautela. Una de las limi-taciones del estudio es que se trata de un estudio fase III y que los datos referentes a los bio-marcadores explorados se basan en un an�lisis pre-especificado aunque puramente explorato-rio. Se necesitan estudios adicionales para conformar el potencial valor pron�stico y predictivode las integrinas αυ en pacientes con CRCm tratados con abituzumab.
En definitiva, nuestros datos indican que las terapias dirigidas a integrinas pueden tener unpapel en el tratamiento del CRCm en poblaciones moleculares seleccionadas. Los an�lisis explo-ratorios desarrollados en el marco del estudio POSEIDON indican que el tratamiento basado enabituzumab puede ser de particular inter�s en aquellos pacientes con CCRm con niveles eleva-dos de expresi�n de αυβ6 que, a su vez, se tratar�a de pacientes con particular mal pron�sti-co. Filiar el impacto real de esta potencial estrategia terap�utica, implica, sin embargo, el desa-rrollo y estandarizaci�n de una t�cnica diagnostica adecuada y una poblaci�n particularmenteseleccionada.

DISCUSION
57
La investigaci�n dirigida a profundizar los mecanismos moleculares de desarrollo y progresi�ndel CCR ha permitido conocer eventos gen�ticos y epigen�ticos fundamentales para su desarro-llo as� como v�as de se�alizaci�n celular cruciales para su progresi�n, supervivencia y potencialde metastacizaci�n. La investigaci�n traslacional ha logrado, por tanto, acelerar la integraci�nde la investigaci�n b�sica y cl�nica permitiendo poner a disposici�n del paciente los �ltimosavances en investigaci�n molecular, abordando la enfermedad desde todos los �ngulos posiblesy creando sinergias entre la investigaci�n molecular y cl�nica a la vez que se emplean estosmedios como plataforma clave en el proceso de desarrollo de f�rmacos. En el caso concreto delCCR, el empleo de biomarcadores se inici� mediante la determinaci�n del estado mutacionaldel ex�n 2 de KRAS como factor predictor de respuesta negativo para anticuerpos anti-EGFR.Hoy sabemos, que aquellos pacientes cuyos tumores presentan una mutaci�n no s�lo a niveldel ex�n 2 de KRAS sino tambi�n en los exones 2, 3 y 4 de KRAS y NRAS no obtienen beneficiodel tratamiento con estos f�rmacos siendo incluso detrimental su empleo. En el desarrollo deesta memoria se han tratado temas de particular relevancia en el marco de la investigaci�n delCCRm en lo que se refiere a biolog�a molecular, principales mecanismos de desarrollo y v�as dese�alizaci�n para supervivencia. Se han descrito las drogas empleadas en el tratamiento deesta entidad enfatizando aquellos dirigidos a dianas moleculares concretas como son los f�rma-cos antiangiog�nicos, inhibidores multi-quinasa y anticuerpos anti-EGFR incluyendo potencialesmecanismos moleculares de resistencia a los mismos. La evidencia presentada constata quecuando la indicaci�n de las terapias diana se basa en la selecci�n de pacientes con caracter�sti-cas moleculares concretas, el impacto en t�rminos de TR, SLP y SG es mayor.
Las dos publicaciones aportadas exploran el papel de exploran el papel de dos anticuerposmonoclonales dirigidos a potenciales dianas relevantes en CCRm como es el caso de flicatuzu-mab como anticuerpo anti-HGF capaz de interferir en la v�a de se�alizaci�n de MET, y abituzu-mab como anticuerpo anti-integrina αυ con capacidad antiproliferativa, antiangiog�nica ymoduladora de la v�a de TGFβ. Ambos estudios podr�an considerarse paradigma del desarrollofarmacol�gico de las terapias diana. Empezando por el dise�o del estudio, es preciso mencio-nar que los estudios fase I permiten no s�lo testar distintos esquemas y dosis de tratamiento,sino que paralelamente a los procedimientos realizados con este objetivo, se pueden asociarestudios de farmacodin�mica en que se testa la diana biol�gica y respuesta en las c�lulas tumo-rales. La correlaci�n de estos hallazgos con par�metros de farmacocin�tica, nos conducen a lainterpretaci�n de la dosis �ptima de tratamiento tal y como se ha descrito para ambas drogasy en ambos casos con un perfil similar al tratarse de anticuerpos monoclonales. Y esto nos llevaa una segunda caracter�stica com�n para ambos estudios: la selecci�n poblacional. En ambosestudios la selecci�n poblacional se basa en caracter�sticas moleculares concretas siendo en elestudio POSEIDON la ausencia de mutaci�n a nivel del ex�n 2 de KRAS, y la sobreexpresi�n dep-MET determinada por inmunohistoqu�mica en el estudio de ficlatuzumab. Una de las cr�ticasque podemos apuntar acerca del estudio POSEIDON es que, en el momento de su dise�o, laindicaci�n de los anticuerpos anti-EGFR ven�a supeditada a la ausencia de mutaci�n en el ex�n2 de KRAS. Como se ha mencionado previamente, se precisa el estado salvaje de los exones 2,3 y 4 de KRAS y NRAS para obtener beneficio de estos f�rmacos. As�, los resultados obtenidospodr�an verse condicionados por entorno a un 17% adicional de mutaciones que pudieran inter-ferir en la eficacia del anticuerpo anti-EGFR tal y como se ha descrito previamente. El estudio
3. DISCUSION

58
NUEVAS TERAPIAS EN CçNCER COLORRECTAL METASTçSICO REFRACTARIO: PAPEL DE LOS FçRMACOS QUE INTERFIEREN EN LA VêA DE MET E INTEGRINAS
de ficlatuzumab basaba la inclusi�n de pacientes en histolog�as en que la v�a de se�alizaci�n deHGF/c-Met juega un papel relevante para su desarrollo, entre ellas CCR. De nuevo, las publica-ciones m�s relevantes focalizadas en CCR que describen la amplificaci�n o sobreexpresi�n deMET como una factor de resistencia a terapias anti-EGFR son posteriores al desarrollo de dichoestudio en que idealmente se deber�a haber restringido la poblaci�n CCR a enfermos sin muta-ci�n a nivel de RAS, tratados previamente con f�rmacos anti-EGFR y con posterior constataci�nde alteraci�n a nivel de Met en biopsia tumoral obtenida a la progresi�n de cetuximab o pani-tumumab. Alternativamente se podr�a haber seleccionado en base a presencia de mutaci�nnivel de KRAS. Precisamente siguiendo esta l�nea de investigaci�n se ha desarrollado el estudioMercuric del que el Vall d'Hebron Institut d'Oncologia es uno del los centros investigadoresprincipales (123). Mercuric se trata de una colaboraci�n paneuropea, financiada por el progra-ma marco EU FP7, que se basa en usar una estrategia de ensayos cl�nicos basada en la medici-na personalizada particularmente focalizada en dos prote�nas clave para el CCR como son MEKy MET, que como se ha comentado est�n desregulados tanto en CCR RAS no mutado como RASmutado. El estudio postula que una doble inhibici�n de MEK y MET en combinaci�n puedeimpactar en supervivencia en pacientes con CCRm refractarios a tratamiento est�ndar. El estu-dio incluir� dos cohortes de pacientes. Por un lado, se reclutar�n pacientes afectos de CCRmRAS no mutado en progresi�n a terapia anti-EGFR y con sobreexpresi�n o amplificaci�n de METen una biopsia tumoral obtenida previamente a la inclusi�n en el estudio. De este modo, lacombinaci�n investigacional trata uno de los potenciales mecanismos de resistencia a terapiaanti-EGFR tal y como se ha constatado mediante biopsia. Por otro lado, existir� una cohorte depacientes con CCRm RAS mutado. En este segundo grupo, sabemos que la inhibici�n de MEK1/2como efector conocido en la cascada de se�alizaci�n mediada por EGFR y RAS, es una potencialestrategia terap�utica de particular inter�s de inter�s. Existen distintos modelos precl�nicos invivo que demuestran que los inhibidores de MEK pueden frenar el crecimiento de tumores CRCRAS mutado (124, 125). Sin embargo, el empleo del f�rmaco en monoterapia tiene una eficacialimitada debido a la activaci�n de otras v�as alternativas de resistencia o supervivencia, de ah�el racional de combinarlo con un inhibidor de MET (124). El ensayo cl�nico consta de un progra-ma de investigaci�n traslacional asociado que incluir� el empleo de tecinas de secuenciaci�n denueva generaci�n (del ingles; next generation sequencing, NGS), asociadas a estrategias diag-n�sticas de micro-RNA con el objetivo de identificar subgrupos de pacientes que mas puedanbeneficiarse de estrategia terap�utica.
Si bien efectivamente los estudios presentan las mencionadas limitaciones, cabe decir que enambos casos hemos constatado de una forma directa o indirecta el efecto que ejerce el f�rma-co dirigido frente a su diana. Esto lleva a preguntarnos el motivo por el que no hay una directaen t�rminos de eficacia. A modo de ejemplo, en el caso de los pacientes con CCRm sin eviden-cia de mutaci�n a nivel de RAS, BRAF o incluso otros biomarcadores no tan concluyentes comoes el caso de PIK3CA o PTEN, seguimos sin identificar la causa por la que estos enfermos no lle-gan incluso ni a responder al tratamiento con anti-EGFRs. O por el contrario, en el caso de tumo-res con mutaci�n a nivel de BRAF, podemos llegar a observas respuestas. Un fen�meno similarlo observamos en el caso de los estadios m�s precoces de CCR. Existen CCR en estadio II quepresentan recurrencias precoces y estadios m�s avanzados como un CCR en estadio III, conafectaci�n ganglionar N2 que no llegan a recaer nunca aun habiendo aplicado en ambos casosun tratamiento �ptimo quir�rgico y sist�mico con el mismo esquema de quimioterapia (126).De este modo, cuando clasificamos los tumores desde un punto de vista histopatol�gico, segui-mos sin poder ciertos tumores de alto riesgo o bien, podr�amos incluso llegar a sobretratar CCRsin apenas riesgo de recurrencia. Precisamente en el campo de la adyuvancia se han llevado acabo estudios de firmas de expresi�n g�nica que predecir�an el riesgo de recurrencia de esta-dios precoces de CCR. Oncotype DX¨ se basa en la expresi�n de 12 genes determinados enmuestras de tumor parafinado que clasifica los tumores en alto, intermedio o bajo riesgo derecurrencia. ColoPrint¨ determina 18 genes en tejido congelado estableciendo alto o bajo ries-go de reca�da. Ambos han sido validados en grandes series de pacientes con CCR. Cabe desta-car que un hallazgo com�n en estos estudios es el alto grado de discordancia entre el riesgo deestratificaci�n entre el perfil de expresi�n f�nica y los factores pron�sticos anatomo-patol�gi-cos convencionales (127, 128). Por tanto, en la enfermedad mestast�sica podr�a darse un fen�-meno similar en que la determinaci�n y resultado determinados biomarcadores puede no sersuficiente para etiquetar el comportamiento de cada tipo de tumor concreto. Como se ha des-crito previamente, varios grupos de investigaci�n han aplicado m�todos de agrupaci�n no

DISCUSIîN
59
supervisada de datos gen�micos para intentar definir los subtipos intr�nsecos de CCR. ElColorectal Cancer Subtyping Consortium, surge con objetivo de consensuar una clasificaci�nmolecular definitiva del CCR mediante el intercambio de datos a gran escala y un meta-an�lisis.As�, el CRCSC consiste en 6 grupos de investigaci�n formados por m�s de 15 instituciones refe-rentes en CCR que analizaron m�s de 30 cohorte de pacientes con datos de expresi�n abarcan-do m�ltiples plataformas y m�todos de preparaci�n de muestras. Cada uno de los 6 clasificado-res que establec�an de 3 a 6 subtipos moleculares distintos de CCR, se aplic� a una colecci�n de4000 muestras de CCR principalmente estadios II y III. La concordancia entre cada subtipo mole-cular y las caracter�sticas cl�nicas, moleculares y vinculadas a v�as de se�alizaci�n concretas seanalizaron centralmente por un equipo independiente. El an�lisis de dichos datos mostr� que,pese a la heterogeneidad en las cohortes y los m�todos, el an�lisis de concordancia por subti-pos estableci� cuatro subtipos moleculares claros de CCR (del ingl�s; clinical molecular subty-pes: CMS). De este modo, CMS1 o inflamatorio se tratar�an de tumores hipermutados, con IMSy activaci�n potente del sistema inmune. CMS2 o can�nico, se tratar�a de un subtipo de tumo-res de tipo epitelial, sin IMS y con una activaci�n pronunciada de la v�a de WNT y MYC. El grupoCMS3 o metab�lico estar�a constituido por neoplasias epiteliales con una evidente desregula-ci�n metab�lica. Finalmente, el subtipo CMS4 o mesenquimal en que tendr�a un papel funda-mental la activaci�n de la v�a de TGFβ, presentar�a invasi�n estromal y la angiog�nesis tendr�aun papel relevante. En este �ltimo subtipo, el an�lisis prote�mico objetiva la representaci�nque tendr�an mol�culas como NOTCH VEGFR o las integrinas. Por tanto, este trabajo pondr�ade manifiesto que, efectivamente; la clasificaci�n del CCR ir�a m�s all� de los biomarcadoresactualmente validados. Los resultados de este trabajo han sido base para dise�ar el proyectoMoTriColor (del ingles; Molecularly guided trials with specific treatment strategies in patientswith advanced newly molecular defined subtypes of colorectal cancer) (129). Se trata de un pro-yecto de investigaci�n que pretende aplicar la clasificaci�n molecular propuesta etiquetando alos pacientes en CSM1-4 de modo que en funci�n del perfil diagnosticado el paciente tendr�aacceso a estudios cl�nicos dise�ados espec�ficamente para el subtipo molecular concreto al quepertenece. En el estudio POSEIDON, se consigue identificar una subpoblaci�n de pacientes que,a la vez que parece presentar peor pron�stico, es la subgrupo de enfermos que m�s beneficioobtiene del tratamiento. Estos enfermos serian aquellos cuyos tumores exhiben una expresi�nalta de integrina αυβ6. Como se ha mencionado, el subtipo molecular CMS4 o mesenquimal, secaracteriza por la activaci�n de la v�a de TGF_, con importante componente de invasi�n estro-mal y procesos angiog�nicos de los que las integrinas forman parte. Como centro coordinadordel proyecto MoTriColor, se ha iniciado un proyecto paralelo que va a tener como objetivo tipi-ficar por CMS los pacientes del estudio POSEIDON con alta expresi�n de αυβ6. En caso de cons-tatar que los pacientes con expresi�n elevada de dicha integrina presentan un perfil mesenqui-mal, el tratamiento con abituzumab ser�a una opci�n de tratamiento dentro de ensayo cl�nicopara el subtipo CMS4.
En definitiva, ambas mol�culas ofrecen un atractivo mecanismo de acci�n y base molecularpara su empleo en el tratamiento del CCRm. Sin embargo, la indicaci�n de las mismas puedeoptimizarse mediante una mejor selecci�n poblacional como en pacientes que presenten unperfil molecular de tipo mesenquimal en que ambas v�as de se�alizaci�n (HGF/MET, Integrinas)tienen un papel fundamental por su interrelaci�n con TGFβ.

60
NUEVAS TERAPIAS EN CçNCER COLORRECTAL METASTçSICO REFRACTARIO: PAPEL DE LOS FçRMACOS QUE INTERFIEREN EN LA VêA DE MET E INTEGRINAS
4. CONCLUSIONES
1. El conocimiento de la biolog�a molecular del CCR y en concreto las v�as metab�licas y dese�alizaci�n involucradas en la diferenciaci�n, crecimiento y supervivencia celular han per-mitido el dise�o de f�rmacos dirigidos a dianas clave en este tipo de tumor e identificarmecanismos de resistencia a los mismos.
2. La v�a de se�alizaci�n HGF/c-Met contribuye a la oncog�nesis y progresi�n tumoral en dis-tintos tipos de neoplasias favoreciendo la capacidad de invasi�n y met�stasis. Esta v�a, seencuentra desregulada en varios tipos tumorales por sobreexpresi�n, amplificaci�n omutaci�n. La alteraci�n m�s frecuentemente observada en CRCm es la sobreexpresi�n y/oamplificaci�n constituyendo una potencial diana terap�utica para el tratamiento de estaentidad.
3. Ficlatuzumab (SCH900105) es un anticuerpo humanizado que neutraliza la uni�n de HGF ac-Met inhibiendo as� la fosforilaci�n de c-Met. Ficlatuzumab explorado a dosis de 2 mg (6pacientes), 10 mg (7 pacientes) y 20 mg (6 pacientes) ev en ciclos de 14 d�as ha demostra-do tener un perfil de seguridad aceptable presentando como toxicidad m�s habitual aste-nia, edema perif�rico, tos e hipoalbuminemia. Ninguno de estos eventos adversos lleg� acumplir criterios de toxicidad limitante de dosis reproduciendo los resultados previamentereportados para la mol�cula. La dosis recomendada de 20 mg/kg ev cada dos semanas sebasa en criterios de tolerancia y, fundamentalmente; par�metros de farmacodin�mica.
4. El mecanismo de acci�n de ficlatuzumab ha sido corroborado en el estudio mediante laobtenci�n de biopsias tumorales hep�ticas antes y despu�s de del tratamiento con el f�r-maco mediante la modulaci�n de los biomarcadores p-Met, p-ERK, p-AKT y HGF. La admi-nistraci�n de ficlatuzumab a 20 mg/kg ev cada dos semanas disminuye los niveles tumora-les de p-Met en un 53%, p-ERK en un 43% y p-AKT en un 2% e incrementa los niveles deHGF en un 33%. Los niveles s�ricos de HGF se correlacionan con la dosis y numero de ciclosde tratamiento administrados. Por todos estos motivos, la dosis biol�gica recomendada deficlatuzumab es 20 mg/kg ev cada dos semanas.
5. En el estudio P05670 no se llegaron a evidenciar respuestas parciales con ficlatuzumab enCCR. Esto puede ser bien por no haber seleccionado correctamente la poblaci�n a estudioo por requerir de la combinaci�n con un agente dirigido que procure vencer mecanismosde supervivencia celular o resistencia asociados. Por tanto, el desarrollo de f�rmacos diri-gidos a la v�a de se�alizaci�n de Met constituye una l�nea de investigaci�n garantizada enel campo del CCR particularmente en aquellos pacientes que desarrollan resistencia a tera-pias anti-EGFR o con resistencias primarias a modo de mutaci�n a nivel de RAS en cuyo casola asociaci�n a inhibidores de MEK tendr�a particular inter�s.

CONCLUSIONES
61
6. Las integrinas se encuentran sobreexpresadas de forma variable en CCR y est�n implicadasen fen�menos de progresi�n tumoral y met�stasis confiriendo mal pron�stico a estas enti-dad.
7. Abituzumab (EMD525797) es un anticuerpo monoclonal dirigido a las integrinas αυ que hademostrado presentar acci�n sin�rgica con irinotec�n y cetuximab en modelos precl�nicosde CCR. La combinaci�n de abituzumab con irinotec�n y cetuximab explorado a dosis fijasde 250 mg, 500 mg, 750 mg o 1000 mg ha demostrado tener un adecuado perfil de seguri-dad siendo los efectos secundarios m�s frecuentemente observados; diarrea, mucositis yastenia sin llegar a cumplir criterios de toxicidad limitante de dosis. Las dosis recomenda-das de 500 mg y 1000 mg para la parte aleatorizada del estudio POSEIDON se basaron endatos de farmacocin�tica y eficacia.
8. La triple combinaci�n de cetuximab, irinotecan y abituzumab en segunda l�nea de trata-miento para pacientes con CCRm KRAS ex�n 2 no mutado a dosis de 500 mg o 1000 mg noha demostrado un beneficio significativo en t�rminos de SLP, objetivo principal del estudio; oSG. Sin embargo, se observa una tendencia a la mejor�a en SG para aquellos pacientes trata-dos con abituzumab y con tumores con expresi�n alta de integrina αυβ6 indicando por unlado signos indirectos de la actividad del f�rmaco en cuanto a mecanismo de acci�n e indi-cando una posible poblaci�n de pacientes que podr�an beneficiarse de dicho tratamiento.
9. Incorporar pruebas de determinaci�n de biomarcadores en los estudios cl�nicos permite,no s�lo corroborar el mecanismo de acci�n del f�rmaco; sino tambi�n identificar posiblessubpoblaciones de pacientes que m�s puedan beneficiarse del mismo. Sin embargo, losbiomarcadores empleados de rutina como es el caso de RAS/BRAF en la pr�ctica cl�nica ruti-naria, o la determinaci�n aislada de otros candidatos en el �rea investigacional como se hadescrito en esta memoria para p-Met o la integrina αυβ6; siguen presentando limitacionespuesto que aun constatando la acci�n del f�rmaco sobre la diana a estudio, no se obtienetraducci�n en t�rminos de eficacia.
10. La selecci�n poblacional basada en la caracterizaci�n molecular es fundamental paraimpactar en supervivencia cuando escogemos una terapia dirigida. Las caracter�sticas cl�ni-cas, histol�gicas y moleculares explican s�lo parcialmente el comportamiento de estostumores ya sea en cuanto a pron�stico o sensibilidad/resistencia a tratamiento. Por tanto,la clasificaci�n molecular del CRC debe ser un objetivo primordial en la investigaci�n deesta entidad puesto que las aplicaciones terap�uticas podr�an suponer un nuevo paradig-ma en el tratamiento de la misma.

62
NUEVAS TERAPIAS EN CçNCER COLORRECTAL METASTçSICO REFRACTARIO: PAPEL DE LOS FçRMACOS QUE INTERFIEREN EN LA VêA DE MET E INTEGRINAS
5. REFERENCIAS
1. Siegel R, DeSantis C, Virgo K, Stein K, Mariotto A, Smith T, etal. Cancer treatment and survivorship statistics, 2012. CA: a can-cer journal for clinicians. 2012;62(4):220-41.
2. Lenz H ND, Innocenti F et al. CALGB/SWOG 80405: Phase IIItrial of irinotecan/5-FU/leucovorin (FOLFIRI) or oxaliplatin/5-FU/leucovorin (mFOLFOX6) with bevacizumab (BV) or cetuximab(CET) for patients (pts) with untreated metastatic adenocarcino-ma of the colon or rectum (MCRC): Expanded RAS analyses. .European Society of Medical Oncology Congress 2014;madrid2014.
3. Tabernero J, Elez ME, Herranz M, Rico I, Prudkin L, Andreu J,et al. A pharmacodynamic/pharmacokinetic study of ficlatuzu-mab in patients with advanced solid tumors and liver metasta-ses. Clinical cancer research : an official journal of the AmericanAssociation for Cancer Research. 2014;20(10):2793-804.
4. Elez E, Kocakova I, Hohler T, Martens UM, Bokemeyer C, VanCutsem E, et al. Abituzumab combined with cetuximab plus iri-notecan versus cetuximab plus irinotecan alone for patients withKRAS wild-type metastatic colorectal cancer: the randomisedphase I/II POSEIDON trial. Annals of oncology : official journal ofthe European Society for Medical Oncology / ESMO.2015;26(1):132-40.
5. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA,Jr., Kinzler KW. Cancer genome landscapes. Science.2013;339(6127):1546-58.
6. Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC,Leppert M, et al. Genetic alterations during colorectal-tumordevelopment. The New England journal of medicine.1988;319(9):525-32.
7. Shibata D, Peinado MA, Ionov Y, Malkhosyan S, Perucho M.Genomic instability in repeated sequences is an early somaticevent in colorectal tumorigenesis that persists after transforma-tion. Nature genetics. 1994;6(3):273-81.
8. Metzger U. Recent developments in molecular biology, scree-ning, and treatment of colorectal cancer. Current opinion inoncology. 1990;2(4):738-46.
9. Worthley DL, Whitehall VL, Spring KJ, Leggett BA. Colorectalcarcinogenesis: road maps to cancer. World journal of gastroen-terology : WJG. 2007;13(28):3784-91.
10. Ponten J. Tumor biology--colorectal cancer as an illuminatingexample. The European journal of surgery Supplement : = Actachirurgica Supplement. 1991(561):9-13.
11. Kitisin K, Mishra L. Molecular biology of colorectal cancer:new targets. Seminars in oncology. 2006;33(6 Suppl 11):S14-23.
12. Mader RM. Links between biology, prognosis and predictionof response to chemotherapy in colorectal cancer. Onkologie.2006;29(7):334-41.
13. Walker J, Quirke P. Biology and genetics of colorectal cancer.European journal of cancer. 2001;37 Suppl 7:S163-72.
14. Barbier M, Attoub S, Calvez R, Laffargue M, Jarry A, MareelM, et al. Tumour biology. Weakening link to colorectal cancer?Nature. 2001;413(6858):796.
15. Hemming AW, Davis NL, Kluftinger A, Robinson B, QuenvilleNF, Liseman B, et al. Prognostic markers of colorectal cancer: anevaluation of DNA content, epidermal growth factor receptor,and Ki-67. Journal of surgical oncology. 1992;51(3):147-52.
16. Mendelsohn J. Targeting the epidermal growth factor recep-tor for cancer therapy. Journal of clinical oncology : official jour-nal of the American Society of Clinical Oncology. 2002;20(18Suppl):1S-13S.
17. Hynes NE, Lane HA. ERBB receptors and cancer: the comple-xity of targeted inhibitors. Nat Rev Cancer. 2005;5(5):341-54.
18. Ciardiello F, Tortora G. EGFR antagonists in cancer treat-ment. The New England journal of medicine. 2008;358(11):1160-74.
19. Burgess AW, Cho HS, Eigenbrot C, Ferguson KM, Garrett TP,Leahy DJ, et al. An open-and-shut case? Recent insights into theactivation of EGF/ErbB receptors. Mol Cell. 2003;12(3):541-52.
20. Weinberg RA. The biology of cancer. Second edition. ed. NewYork, NY, US: Garland Science; 2014. xx, 875 pages p.
21. Malumbres M, Barbacid M. RAS oncogenes: the first 30years. Nature reviews Cancer. 2003;3(6):459-65.
22. Schlessinger J. Cell signaling by receptor tyrosine kinases.Cell. 2000;103(2):211-25.
23. Prior IA, Lewis PD, Mattos C. A comprehensive survey of Rasmutations in cancer. Cancer research. 2012;72(10):2457-67.
24. Avruch J, Khokhlatchev A, Kyriakis JM, Luo Z, Tzivion G,Vavvas D, et al. Ras activation of the Raf kinase: tyrosine kinaserecruitment of the MAP kinase cascade. Recent Prog Horm Res.2001;56:127-55.
25. Marais R, Marshall CJ. Control of the ERK MAP kinase casca-de by Ras and Raf. Cancer surveys. 1996;27:101-25.
26. Forbes SA, Bhamra G, Bamford S, Dawson E, Kok C, ClementsJ, et al. The Catalogue of Somatic Mutations in Cancer (COSMIC).Curr Protoc Hum Genet. 2008;Chapter 10:Unit 10 1.

REFERENCIAS
63
27. Di Nicolantonio F, Martini M, Molinari F, Sartore-Bianchi A,Arena S, Saletti P, et al. Wild-type BRAF is required for responseto panitumumab or cetuximab in metastatic colorectal cancer.Journal of clinical oncology : official journal of the AmericanSociety of Clinical Oncology. 2008;26(35):5705-12.
28. Van Cutsem E, Kohne CH, Lang I, Folprecht G, Nowacki MP,Cascinu S, et al. Cetuximab plus irinotecan, fluorouracil, and leu-covorin as first-line treatment for metastatic colorectal cancer:updated analysis of overall survival according to tumor KRAS andBRAF mutation status. Journal of clinical oncology : official jour-nal of the American Society of Clinical Oncology.2011;29(15):2011-9.
29. Cantley LC. The phosphoinositide 3-kinase pathway. Science.2002;296(5573):1655-7.
30. De Roock W, Claes B, Bernasconi D, De Schutter J, BiesmansB, Fountzilas G, et al. Effects of KRAS, BRAF, NRAS, and PIK3CAmutations on the efficacy of cetuximab plus chemotherapy inchemotherapy-refractory metastatic colorectal cancer: a retros-pective consortium analysis. The Lancet Oncology.2010;11(8):753-62.
31. Alberts B. Molecular biology of the cell. 5th ed. New York:Garland Science; 2008.
32. Yarden Y, Sliwkowski MX. Untangling the ErbB signalling net-work. Nature reviews Molecular cell biology. 2001;2(2):127-37.
33. Cooper CS, Park M, Blair DG, Tainsky MA, Huebner K, CroceCM, et al. Molecular cloning of a new transforming gene from achemically transformed human cell line. Nature.1984;311(5981):29-33.
34. Park M, Dean M, Kaul K, Braun MJ, Gonda MA, Vande WoudeG. Sequence of MET protooncogene cDNA has features charac-teristic of the tyrosine kinase family of growth-factor receptors.Proceedings of the National Academy of Sciences of the UnitedStates of America. 1987;84(18):6379-83.
35. Bladt F, Riethmacher D, Isenmann S, Aguzzi A, Birchmeier C.Essential role for the c-met receptor in the migration of myoge-nic precursor cells into the limb bud. Nature.1995;376(6543):768-71.
36. Dietrich S, Abou-Rebyeh F, Brohmann H, Bladt F,Sonnenberg-Riethmacher E, Yamaai T, et al. The role of SF/HGFand c-Met in the development of skeletal muscle. Development.1999;126(8):1621-9.
37. Huelsken J, Vogel R, Erdmann B, Cotsarelis G, Birchmeier W.beta-Catenin controls hair follicle morphogenesis and stem celldifferentiation in the skin. Cell. 2001;105(4):533-45.
38. Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF.Met, metastasis, motility and more. Nature reviews Molecularcell biology. 2003;4(12):915-25.
39. Lai AZ, Abella JV, Park M. Crosstalk in Met receptor oncoge-nesis. Trends in cell biology. 2009;19(10):542-51.
40. Ma PC, Tretiakova MS, MacKinnon AC, Ramnath N, JohnsonC, Dietrich S, et al. Expression and mutational analysis of MET inhuman solid cancers. Genes, chromosomes & cancer.2008;47(12):1025-37.
41. Knudsen BS, Vande Woude G. Showering c-MET-dependentcancers with drugs. Current opinion in genetics & development.2008;18(1):87-96.
42. Khoury H, Naujokas MA, Zuo D, Sangwan V, Frigault MM,Petkiewicz S, et al. HGF converts ErbB2/Neu epithelial morpho-genesis to cell invasion. Molecular biology of the cell.2005;16(2):550-61.
43. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C,Park JO, et al. MET amplification leads to gefitinib resistance inlung cancer by activating ERBB3 signaling. Science.2007;316(5827):1039-43.
44. Turke AB, Zejnullahu K, Wu YL, Song Y, Dias-Santagata D,Lifshits E, et al. Preexistence and clonal selection of MET ampli-fication in EGFR mutant NSCLC. Cancer cell. 2010;17(1):77-88.
45. Bardelli A, Corso S, Bertotti A, Hobor S, Valtorta E, SiravegnaG, et al. Amplification of the MET receptor drives resistance toanti-EGFR therapies in colorectal cancer. Cancer discovery.2013;3(6):658-73.
46. Troiani T, Martinelli E, Napolitano S, Vitagliano D, CiuffredaLP, Costantino S, et al. Increased TGF-alpha as a mechanism ofacquired resistance to the anti-EGFR inhibitor cetuximabthrough EGFR-MET interaction and activation of MET signaling incolon cancer cells. Clinical cancer research : an official journal ofthe American Association for Cancer Research.2013;19(24):6751-65.
47. Zhang YW, Wang LM, Jove R, Vande Woude GF. Requirementof Stat3 signaling for HGF/SF-Met mediated tumorigenesis.Oncogene. 2002;21(2):217-26.
48. Sulpice E, Ding S, Muscatelli-Groux B, Berge M, Han ZC,Plouet J, et al. Cross-talk between the VEGF-A and HGF signallingpathways in endothelial cells. Biology of the cell / under the aus-pices of the European Cell Biology Organization.2009;101(9):525-39.
49. Klaus A, Birchmeier W. Wnt signalling and its impact on deve-lopment and cancer. Nature reviews Cancer. 2008;8(5):387-98.
50. Boon EM, van der Neut R, van de Wetering M, Clevers H, PalsST. Wnt signaling regulates expression of the receptor tyrosinekinase met in colorectal cancer. Cancer research.2002;62(18):5126-8.
51. Liu Y, Chattopadhyay N, Qin S, Szekeres C, Vasylyeva T,Mahoney ZX, et al. Coordinate integrin and c-Met signaling regu-late Wnt gene expression during epithelial morphogenesis.Development. 2009;136(5):843-53.
52. Brembeck FH, Schwarz-Romond T, Bakkers J, Wilhelm S,Hammerschmidt M, Birchmeier W. Essential role of BCL9-2 inthe switch between beta-catenin's adhesive and transcriptionalfunctions. Genes & development. 2004;18(18):2225-30.
53. O'Brien BL, DiPalma JA. Molecular biology and colorectal can-cer: genetic alterations, inherited syndromes, and applicationsto colon cancer screening. Digestive diseases. 1995;13(3):182-9.
54. Allen JI. Molecular biology of colon polyps and colon cancer.Seminars in surgical oncology. 1995;11(6):399-405.
55. Bellam N, Pasche B. Tgf-beta signaling alterations and coloncancer. Cancer treatment and research. 2010;155:85-103.
56. Kerbel RS. Tumor angiogenesis. N Engl J Med.2008;358(19):2039-49.
57. Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T,Hainsworth J, Heim W, et al. Bevacizumab plus irinotecan, fluo-rouracil, and leucovorin for metastatic colorectal cancer. N EnglJ Med. 2004;350(23):2335-42.
58. Saltz LB, Clarke S, Diaz-Rubio E, Scheithauer W, Figer A, WongR, et al. Bevacizumab in combination with oxaliplatin-based che-motherapy as first-line therapy in metastatic colorectal cancer: arandomized phase III study. Journal of clinical oncology : officialjournal of the American Society of Clinical Oncology.2008;26(12):2013-9.

64
NUEVAS TERAPIAS EN CçNCER COLORRECTAL METASTçSICO REFRACTARIO: PAPEL DE LOS FçRMACOS QUE INTERFIEREN EN LA VêA DE MET E INTEGRINAS
59. Mitra SK, Schlaepfer DD. Integrin-regulated FAK-Src signalingin normal and cancer cells. Current opinion in cell biology.2006;18(5):516-23.
60. Guo W, Giancotti FG. Integrin signalling during tumour pro-gression. Nature reviews Molecular cell biology. 2004;5(10):816-26.
61. Assoian RK, Klein EA. Growth control by intracellular tensionand extracellular stiffness. Trends in cell biology.2008;18(7):347-52.
62. Hynes RO. A reevaluation of integrins as regulators of angio-genesis. Nature medicine. 2002;8(9):918-21.
63. Bates RC, Bellovin DI, Brown C, Maynard E, Wu B, KawakatsuH, et al. Transcriptional activation of integrin beta6 during theepithelial-mesenchymal transition defines a novel prognosticindicator of aggressive colon carcinoma. The Journal of clinicalinvestigation. 2005;115(2):339-47.
64. Petrelli N, Douglass HO, Jr., Herrera L, Russell D, Stablein DM,Bruckner HW, et al. The modulation of fluorouracil with leucovo-rin in metastatic colorectal carcinoma: a prospective randomi-zed phase III trial. Gastrointestinal Tumor Study Group. Journalof clinical oncology : official journal of the American Society ofClinical Oncology. 1989;7(10):1419-26.
65. de Gramont A, Figer A, Seymour M, Homerin M, Hmissi A,Cassidy J, et al. Leucovorin and fluorouracil with or without oxa-liplatin as first-line treatment in advanced colorectal cancer.Journal of clinical oncology : official journal of the AmericanSociety of Clinical Oncology. 2000;18(16):2938-47.
66. Cassidy J, Clarke S, Diaz-Rubio E, Scheithauer W, Figer A,Wong R, et al. Randomized phase III study of capecitabine plusoxaliplatin compared with fluorouracil/folinic acid plus oxalipla-tin as first-line therapy for metastatic colorectal cancer. Journalof clinical oncology : official journal of the American Society ofClinical Oncology. 2008;26(12):2006-12.
67. Douillard JY, Cunningham D, Roth AD, Navarro M, James RD,Karasek P, et al. Irinotecan combined with fluorouracil comparedwith fluorouracil alone as first-line treatment for metastaticcolorectal cancer: a multicentre randomised trial. Lancet.2000;355(9209):1041-7.
68. Tournigand C, Andre T, Achille E, Lledo G, Flesh M, Mery-Mignard D, et al. FOLFIRI followed by FOLFOX6 or the reversesequence in advanced colorectal cancer: a randomized GERCORstudy. Journal of clinical oncology : official journal of theAmerican Society of Clinical Oncology. 2004;22(2):229-37.
69. Grothey A, Sargent D, Goldberg RM, Schmoll HJ. Survival ofpatients with advanced colorectal cancer improves with theavailability of fluorouracil-leucovorin, irinotecan, and oxaliplatinin the course of treatment. Journal of clinical oncology : officialjournal of the American Society of Clinical Oncology.2004;22(7):1209-14.
70. Seymour MT, Maughan TS, Ledermann JA, Topham C, JamesR, Gwyther SJ, et al. Different strategies of sequential and com-bination chemotherapy for patients with poor prognosis advan-ced colorectal cancer (MRC FOCUS): a randomised controlledtrial. Lancet. 2007;370(9582):143-52.
71. Koopman M, Antonini NF, Douma J, Wals J, Honkoop AH,Erdkamp FL, et al. Sequential versus combination chemotherapywith capecitabine, irinotecan, and oxaliplatin in advanced colo-rectal cancer (CAIRO): a phase III randomised controlled trial.Lancet. 2007;370(9582):135-42.
72. Ducreux M, Malka D, Mendiboure J, Etienne PL, Texereau P,Auby D, et al. Sequential versus combination chemotherapy forthe treatment of advanced colorectal cancer (FFCD 2000-05): anopen-label, randomised, phase 3 trial. The Lancet Oncology.2011;12(11):1032-44.
73. Falcone A, Ricci S, Brunetti I, Pfanner E, Allegrini G, BarbaraC, et al. Phase III trial of infusional fluorouracil, leucovorin, oxa-liplatin, and irinotecan (FOLFOXIRI) compared with infusionalfluorouracil, leucovorin, and irinotecan (FOLFIRI) as first-line tre-atment for metastatic colorectal cancer: the Gruppo OncologicoNord Ovest. Journal of clinical oncology : official journal of theAmerican Society of Clinical Oncology. 2007;25(13):1670-6.
74. Douillard JY, Oliner KS, Siena S, Tabernero J, Burkes R,Barugel M, et al. Panitumumab-FOLFOX4 treatment and RASmutations in colorectal cancer. The New England journal ofmedicine.369(11):1023-34.
75. Heinemann V, Stintzing S. FOLFIRI with cetuximab or bevaci-zumab: FIRE-3-authors' reply. The Lancet Oncology.2014;15(13):e583-4.
76. Venook A ND, Lenz H et al. CALGB/SWOG 80405: Phase IIItrial od irinotecan/5-FU/leucovorin (FOLFIRI) or oxaliplatin/5-FU/LV (mFOLFOX6) with bevacizumab or cetuximab for patientswith KRAS wild type untreaed metastatic adenocarcinoma of thecolon or rectum. . Journal of Clinical Oncology2014. p. 32:5s,2014 (suppl; abstr LBA3).
77. Loupakis F, Cremolini C, Masi G, Lonardi S, Zagonel V,Salvatore L, et al. Initial therapy with FOLFOXIRI and bevacizu-mab for metastatic colorectal cancer. The New England journalof medicine. 2014;371(17):1609-18.
78. Giantonio BJ, Catalano PJ, Meropol NJ, O'Dwyer PJ, MitchellEP, Alberts SR, et al. Bevacizumab in combination with oxalipla-tin, fluorouracil, and leucovorin (FOLFOX4) for previously trea-ted metastatic colorectal cancer: results from the EasternCooperative Oncology Group Study E3200. Journal of clinicaloncology : official journal of the American Society of ClinicalOncology. 2007;25(12):1539-44.
79. Bennouna J, Sastre J, Arnold D, Osterlund P, Greil R, VanCutsem E, et al. Continuation of bevacizumab after first progres-sion in metastatic colorectal cancer (ML18147): a randomisedphase 3 trial. The Lancet Oncology. 2013;14(1):29-37.
80. Van Cutsem E, Tabernero J, Lakomy R, Prenen H, Prausova J,Macarulla T, et al. Addition of aflibercept to fluorouracil, leuco-vorin, and irinotecan improves survival in a phase III randomizedtrial in patients with metastatic colorectal cancer previously tre-ated with an oxaliplatin-based regimen. Journal of clinical onco-logy : official journal of the American Society of ClinicalOncology.30(28):3499-506.
81. Grothey A, Van Cutsem E, Sobrero A, Siena S, Falcone A,Ychou M, et al. Regorafenib monotherapy for previously treatedmetastatic colorectal cancer (CORRECT): an international, multi-centre, randomised, placebo-controlled, phase 3 trial.Lancet.381(9863):303-12.
82. Douillard JY, Siena S, Cassidy J, Tabernero J, Burkes R, BarugelM, et al. Randomized, phase III trial of panitumumab with infu-sional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versusFOLFOX4 alone as first-line treatment in patients with previouslyuntreated metastatic colorectal cancer: the PRIME study.Journal of clinical oncology : official journal of the AmericanSociety of Clinical Oncology. 2010;28(31):4697-705.
83. Jonker DJ, O'Callaghan CJ, Karapetis CS, Zalcberg JR, Tu D, AuHJ, et al. Cetuximab for the treatment of colorectal cancer. TheNew England journal of medicine. 2007;357(20):2040-8.

REFERENCIAS
65
84. Karapetis CS, Khambata-Ford S, Jonker DJ, O'Callaghan CJ, TuD, Tebbutt NC, et al. K-ras mutations and benefit from cetuximabin advanced colorectal cancer. The New England journal of medi-cine. 2008;359(17):1757-65.
85. Van Cutsem E, Peeters M, Siena S, Humblet Y, Hendlisz A,Neyns B, et al. Open-label phase III trial of panitumumab plusbest supportive care compared with best supportive care alonein patients with chemotherapy-refractory metastatic colorectalcancer. Journal of clinical oncology : official journal of theAmerican Society of Clinical Oncology. 2007;25(13):1658-64.
86. Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S,Freeman DJ, et al. Wild-type KRAS is required for panitumumabefficacy in patients with metastatic colorectal cancer. Journal ofclinical oncology : official journal of the American Society ofClinical Oncology. 2008;26(10):1626-34.
87. Price TJ, Peeters M, Kim TW, Li J, Cascinu S, Ruff P, et al.Panitumumab versus cetuximab in patients with chemotherapy-refractory wild-type KRAS exon 2 metastatic colorectal cancer(ASPECCT): a randomised, multicentre, open-label, non-inferio-rity phase 3 study. The Lancet Oncology. 2014;15(6):569-79.
88. Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H,Santoro A, et al. Cetuximab monotherapy and cetuximab plus iri-notecan in irinotecan-refractory metastatic colorectal cancer.The New England journal of medicine. 2004;351(4):337-45.
89. Sobrero AF, Maurel J, Fehrenbacher L, Scheithauer W,Abubakr YA, Lutz MP, et al. EPIC: phase III trial of cetuximab plusirinotecan after fluoropyrimidine and oxaliplatin failure inpatients with metastatic colorectal cancer. Journal of clinicaloncology : official journal of the American Society of ClinicalOncology. 2008;26(14):2311-9.
90. Peeters M, Price TJ, Cervantes A, Sobrero AF, Ducreux M,Hotko Y, et al. Randomized phase III study of panitumumab withfluorouracil, leucovorin, and irinotecan (FOLFIRI) compared withFOLFIRI alone as second-line treatment in patients with metasta-tic colorectal cancer. Journal of clinical oncology : official journalof the American Society of Clinical Oncology. 2010;28(31):4706-13.
91. Seymour MT, Brown SR, Middleton G, Maughan T, RichmanS, Gwyther S, et al. Panitumumab and irinotecan versus irinote-can alone for patients with KRAS wild-type, fluorouracil-resistantadvanced colorectal cancer (PICCOLO): a prospectively stratifiedrandomised trial. The Lancet Oncology. 2013;14(8):749-59.
92. Van Cutsem E, Kohne CH, Hitre E, Zaluski J, Chang Chien CR,Makhson A, et al. Cetuximab and chemotherapy as initial treat-ment for metastatic colorectal cancer. The New England journalof medicine. 2009;360(14):1408-17.
93. Bokemeyer C, Bondarenko I, Makhson A, Hartmann JT,Aparicio J, de Braud F, et al. Fluorouracil, leucovorin, and oxali-platin with and without cetuximab in the first-line treatment ofmetastatic colorectal cancer. Journal of clinical oncology : officialjournal of the American Society of Clinical Oncology.2009;27(5):663-71.
94. Tveit KM, Guren T, Glimelius B, Pfeiffer P, Sorbye H,Pyrhonen S, et al. Phase III trial of cetuximab with continuous orintermittent fluorouracil, leucovorin, and oxaliplatin (NordicFLOX) versus FLOX alone in first-line treatment of metastaticcolorectal cancer: the NORDIC-VII study. Journal of clinical onco-logy : official journal of the American Society of ClinicalOncology. 2012;30(15):1755-62.
95. Heinemann V, von Weikersthal LF, Decker T, Kiani A, Vehling-Kaiser U, Al-Batran SE, et al. FOLFIRI plus cetuximab versus FOL-FIRI plus bevacizumab as first-line treatment for patients withmetastatic colorectal cancer (FIRE-3): a randomised, open-label,phase 3 trial. The Lancet Oncology. 2014;15(10):1065-75.
96. Schwartzberg LS, Rivera F, Karthaus M, Fasola G, Canon JL,Hecht JR, et al. PEAK: a randomized, multicenter phase II studyof panitumumab plus modified fluorouracil, leucovorin, and oxa-liplatin (mFOLFOX6) or bevacizumab plus mFOLFOX6 in patientswith previously untreated, unresectable, wild-type KRAS exon 2metastatic colorectal cancer. Journal of clinical oncology : officialjournal of the American Society of Clinical Oncology.2014;32(21):2240-7.
97. Bertotti A, Migliardi G, Galimi F, Sassi F, Torti D, Isella C, et al.A molecularly annotated platform of patient-derived xenografts("xenopatients") identifies HER2 as an effective therapeutic tar-get in cetuximab-resistant colorectal cancer. Cancer discovery.2011;1(6):508-23.
98. Frattini M, Saletti P, Romagnani E, Martin V, Molinari F,Ghisletta M, et al. PTEN loss of expression predicts cetuximabefficacy in metastatic colorectal cancer patients. British journalof cancer. 2007;97(8):1139-45.
99. Moroni M, Veronese S, Benvenuti S, Marrapese G, Sartore-Bianchi A, Di Nicolantonio F, et al. Gene copy number for epider-mal growth factor receptor (EGFR) and clinical response toantiEGFR treatment in colorectal cancer: a cohort study. TheLancet Oncology. 2005;6(5):279-86.
100. Sartore-Bianchi A, Fieuws S, Veronese S, Moroni M,Personeni N, Frattini M, et al. Standardisation of EGFR FISH incolorectal cancer: results of an international interlaboratoryreproducibility ring study. Journal of clinical pathology.2012;65(3):218-23.
101. Scartozzi M, Mandolesi A, Giampieri R, Bittoni A, PierantoniC, Zaniboni A, et al. The role of HER-3 expression in the predic-tion of clinical outcome for advanced colorectal cancer patientsreceiving irinotecan and cetuximab. The oncologist.2011;16(1):53-60.
102. Scartozzi M, Mandolesi A, Giampieri R, Pierantoni C,Loupakis F, Zaniboni A, et al. Insulin-like growth factor 1 expres-sion correlates with clinical outcome in K-RAS wild type colorec-tal cancer patients treated with cetuximab and irinotecan.International journal of cancer Journal international du cancer.2010;127(8):1941-7.
103. Diaz LA, Jr., Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, etal. The molecular evolution of acquired resistance to targetedEGFR blockade in colorectal cancers. Nature.2012;486(7404):537-40.
104. Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, LiskaD, et al. Emergence of KRAS mutations and acquired resistanceto anti-EGFR therapy in colorectal cancer. Nature.2012;486(7404):532-6.
105. Hu YP, Venkateswarlu S, Sergina N, Howell G, St Clair P,Humphrey LE, et al. Reorganization of ErbB family and cell survi-val signaling after Knock-down of ErbB2 in colon cancer cells.The Journal of biological chemistry. 2005;280(29):27383-92.
106. Kountourakis P, Pavlakis K, Psyrri A, Rontogianni D, Xiros N,Patsouris E, et al. Prognostic significance of HER3 and HER4 pro-tein expression in colorectal adenocarcinomas. BMC cancer.2006;6:46.
107. Baselga J, Swain SM. Novel anticancer targets: revisitingERBB2 and discovering ERBB3. Nature reviews Cancer.2009;9(7):463-75.
108. Schaefer G, Haber L, Crocker LM, Shia S, Shao L, DowbenkoD, et al. A two-in-one antibody against HER3 and EGFR has supe-rior inhibitory activity compared with monospecific antibodies.Cancer cell. 2011;20(4):472-86.

66
NUEVAS TERAPIAS EN CçNCER COLORRECTAL METASTçSICO REFRACTARIO: PAPEL DE LOS FçRMACOS QUE INTERFIEREN EN LA VêA DE MET E INTEGRINAS
109. Huang S, Li C, Armstrong EA, Peet CR, Saker J, Amler LC, etal. Dual targeting of EGFR and HER3 with MEHD7945A overco-mes acquired resistance to EGFR inhibitors and radiation. Cancerresearch. 2013;73(2):824-33.
110. Yonesaka K, Zejnullahu K, Okamoto I, Satoh T, Cappuzzo F,Souglakos J, et al. Activation of ERBB2 signaling causes resistan-ce to the EGFR-directed therapeutic antibody cetuximab.Science translational medicine. 2011;3(99):99ra86.
111. Montagut C, Dalmases A, Bellosillo B, Crespo M, Pairet S,Iglesias M, et al. Identification of a mutation in the extracellulardomain of the Epidermal Growth Factor Receptor conferringcetuximab resistance in colorectal cancer. Nature medicine.2012;18(2):221-3.
112. Esposito C, Rachiglio AM, La Porta ML, Sacco A, Roma C,Iannaccone A, et al. The S492R EGFR ectodomain mutation isnever detected in KRAS wild-type colorectal carcinoma beforeexposure to EGFR monoclonal antibodies. Cancer biology & the-rapy. 2013;14(12):1143-6.
113. Seiden-Long IM, Brown KR, Shih W, Wigle DA, Radulovich N,Jurisica I, et al. Transcriptional targets of hepatocyte growth fac-tor signaling and Ki-ras oncogene activation in colorectal cancer.Oncogene. 2006;25(1):91-102.
114. Van Cutsem E, Eng C, Nowara E, Swieboda-Sadlej A, TebbuttNC, Mitchell E, et al. Randomized phase Ib/II trial of rilotumu-mab or ganitumab with panitumumab versus panitumumabalone in patients with wild-type KRAS metastatic colorectal can-cer. Clinical cancer research : an official journal of the AmericanAssociation for Cancer Research.20(16):4240-50.
115. Diaz LA, Jr., Sausen M, Fisher GA, Velculescu VE. Insightsinto therapeutic resistance from whole-genome analyses of cir-culating tumor DNA. Oncotarget. 2013;4(10):1856-7.
116. Mohan S, Heitzer E, Ulz P, Lafer I, Lax S, Auer M, et al.Changes in colorectal carcinoma genomes under anti-EGFR the-rapy identified by whole-genome plasma DNA sequencing. PLoSgenetics. 2014;10(3):e1004271.
117. Cancer Genome Atlas N. Comprehensive molecular charac-terization of human colon and rectal cancer. Nature.2012;487(7407):330-7.
118. Arnaout MA, Goodman SL, Xiong JP. Structure and mecha-nics of integrin-based cell adhesion. Current opinion in cell bio-logy. 2007;19(5):495-507.
119. Goel HL, Languino LR. Integrin signaling in cancer. Cancertreatment and research. 2004;119:15-31.
120. Felding-Habermann B, Fransvea E, O'Toole TE, Manzuk L,Faha B, Hensler M. Involvement of tumor cell integrin alpha vbeta 3 in hematogenous metastasis of human melanoma cells.Clinical & experimental metastasis. 2002;19(5):427-36.
121. Nemeth JA, Nakada MT, Trikha M, Lang Z, Gordon MS,Jayson GC, et al. Alpha-v integrins as therapeutic targets in onco-logy. Cancer investigation. 2007;25(7):632-46.
122. Max R, Gerritsen RR, Nooijen PT, Goodman SL, Sutter A,Keilholz U, et al. Immunohistochemical analysis of integrin alphavbeta3 expression on tumor-associated vessels of human carci-nomas. International journal of cancer Journal international ducancer. 1997;71(3):320-4.
123. SCHAEYBROECK SV. European Comission. 2014.
124. Little AS, Balmanno K, Sale MJ, Newman S, Dry JR, HampsonM, et al. Amplification of the driving oncogene, KRAS or BRAF,underpins acquired resistance to MEK1/2 inhibitors in colorectalcancer cells. Science signaling. 2011;4(166):ra17.
125. Yoon J, Koo KH, Choi KY. MEK1/2 inhibitors AS703026 andAZD6244 may be potential therapies for KRAS mutated colorec-tal cancer that is resistant to EGFR monoclonal antibody therapy.Cancer research. 2011;71(2):445-53.
126. Andre T, Boni C, Mounedji-Boudiaf L, Navarro M, TaberneroJ, Hickish T, et al. Oxaliplatin, fluorouracil, and leucovorin asadjuvant treatment for colon cancer. The New England journalof medicine. 2004;350(23):2343-51.
127. Gray RG, Quirke P, Handley K, Lopatin M, Magill L, BaehnerFL, et al. Validation study of a quantitative multigene reversetranscriptase-polymerase chain reaction assay for assessment ofrecurrence risk in patients with stage II colon cancer. Journal ofclinical oncology : official journal of the American Society ofClinical Oncology. 2011;29(35):4611-9.
128. Salazar R, Roepman P, Capella G, Moreno V, Simon I,Dreezen C, et al. Gene expression signature to improve progno-sis prediction of stage II and III colorectal cancer. Journal of clini-cal oncology : official journal of the American Society of ClinicalOncology. 2011;29(1):17-24.
129. Consortium CCS. European Comission Horizon 2020. 2015.

APENDICE: OTRAS PUBLICACIONES
67
6. APENDICE: OTRAS PUBLICACIONES
6.1. Publicaciones relacionadas con nuevas terapias
dirigidas en c�ncer colorrectal metast�sico y biomarcadores pron�sticos y de resistencia
a tratamiento sist�mico


NUEVAS TERAPIAS EN CçNCER COLORRECTAL METASTçSICO REFRACTARIO: PAPEL DE LOS FçRMACOS QUE INTERFIEREN EN LA VêA DE MET E INTEGRINAS
M» Elena �lez Fern�ndez