teaching module luini - embl european molecular … introduction this module describes a newly...
TRANSCRIPT

Teaching module for: CORRELATIVE VIDEO-LIGHT ELECTRON MICROSCOPY
SUMMARY
In studies of dynamic cellular processes, it would be ideal to be able to combine the capability of in
vivo fluorescence video microscopy with the resolution power of electron microscopy (EM). This
module describes an approach based on the combination of these two techniques, by which an
individual intracellular structure can be monitored in vivo, typically through the use of markers fused
with green fluorescent protein (GFP), and then analyzed by EM and three-dimensional (3D)
reconstruction methods. This results in a "snapshot" of its fine structure at any chosen time during its
life cycle. The potential of this approach is discussed in relation to various aspects of cell biology.

2
INTRODUCTION
This module describes a newly developed method that allows the examination of living cells by
time-lapse analysis, with the subsequent EM identification and examination of the particular
organelle under observation at a specific moment in its life-cycle.
Many cellular functions, such as intracellular traffic, cytokinesis and cell migration, crucially
depend on rapid translocations and/or shape changes of specific intracellular organelles. To
understand how such functions are organized and executed in vivo, it is important to be able to
observe in real time in living cells such dynamic structures as a budding transport carrier, an
elongating microtubule or a developing mitotic spindle, with the degree of spatial resolution
afforded by EM.
We describe here the most suitable method for this, which is conceptually simple, yet
powerful, and which we have called correlative video-light EM (CVLEM). This indeed allows
observations of the in vivo dynamics and ultrastructure of intracellular objects to be combined to
achieve this result. We also illustrate the type of questions that the CVLEM approach was designed
to address, as well as the particular know-how that is important for successful application of this
technique.
BASIC PROTOCOL
Strategy
The CVLEM procedure includes several stages (see Figure 1): A) observation of structures labelled
with GFP in living cells; B) immunolabelling for EM, embedding, and identification of the cell on
the resin block; C) cutting; D) EM analysis; and E) 3D reconstruction. During the first step, the
cells are transfected with cDNA encoding the GFP fusion protein of choice, and the structure of
interest is observed in the living cells. In this way, it is possible to gain information about its
dynamic properties (e.g. motility, speed and direction, changes in size and shape). At the end of this
stage, it is necessary to kill the cells by the addition of fixative, and in this way to capture the

3
fluorescent object at the moment of interest. As GFP is not
visible under EM, immunostaining provides the possibility
of identifying the GFP-labelled structure at the EM level.
Here, we describe the immunogold and
immunoperoxidase protocols used for staining for EM.
The immunogold protocol (Burry et al., 1992) is suitable
for the labelling of the large majority of antigens, while
the immunoperoxidase protocol allows only the labelling
of antigens residing within small membrane-enclosed
compartments because the electron dense product of the
peroxidase reaction tends to diffuse from the place of
antibody binding (Brown and Farquhar, 1989; Deerinck et
al., 1994). Once stained, the cells must be prepared for EM
by traditional epoxy embedding, and the cell and structure
of interest must be identified in sections under the EM.
Finding an individual subcellular structure in single thin
sections can be complex, or even impossible, simply
because most of the cellular organelles are bigger than the
thickness of the section and lie in a plane that is different
from that of the section. So an analysis of serial sections of
the whole cell is required to identify the structure observed
previously in vivo. An example of this identification
procedure is given in Figure 3. Finally, the EM analysis of
serial sections can be supported by high voltage EM
tomography and/or digital 3D reconstruction.
E. Digital 3D reconstruction.
D. Identification of the structure in sections for EM.
A. GFP-based time-lapse confocal microscopy
B. Immunoperoxidase labelling and embedding in resin.
C. Cutting of serial sections.
resin block
knife
Figure 1. The CVLEM stages

4
Materials
Cells of interest
MatTek petri dishes with CELLocate coverslip (MatTek Corporation, Ashland, MA, USA)
0.2 M HEPES buffer:
Dissolve 4.77 g of HEPES in 100 ml distilled water; add 1 N HCl to a pH of ~7.2-7.4.
Fixative (0.1% glutaraldehyde - 8% paraformaldehyde):
Dissolve 4 g of paraformaldehyde powder in 50 ml HEPES buffer, stirring while heating the
solution to 60 °C. Add drops of 1 N NaOH until the solution clears. Add 1.25 ml 8%
glutaraldehyde and 50 ml of HEPES buffer.
4% paraformaldehyde:
Dissolve 4 g of paraformaldehyde powder in 100 ml HEPES buffer, stirring while heating the
solution to 60°C. Add drops of 1 N NaOH until the solution clears.
Blocking solution:
0.5 g BSA
0.1 g saponin
0.27 g NH4Cl
100 ml phosphate-buffered saline (PBS)
The Fab fragments of secondary antibodies conjugated with horse radish peroxidase (HRP)
(Rockland, Gilbertsville, PA, USA)
NANOGOLD conjugated Fab fragments of secondary antibodies (Nanoprobes Inc., Yaphank,
NY, USA)
0.1 M TRIS-HCl buffer:
Dissolve 1.21 g TRIZMA base in 100 ml distilled water; add 1 N HCl to a pH of ~7.2-7.4.
Diaminobenzidine (DAB) solution:
Dissolve 0.01 g DAB in 20 ml TRIS-HCl. Add 13.3 µl 30% H2O2 solution just before use.
Gold-enhance mixture:

5
Use the Gold-enhance kit from Nanoprobes with equal amounts of each of the four
components (Solutions A, B, C and D); prepare about 40 µl of reagent per grid. (a convenient
method is to use an equal number of drops from each bottle).
a. First, mix Solution A (enhancer: green cap) and Solution B (activator: yellow cap)
b. Wait 5 min.
c. Add Solution C (initiator: purple cap) and then Solution D (buffer: white cap), and mix.
0.2 M Cacodylate buffer:
Dissolve 2.12 g sodium cacodylate in 100 ml distilled water; add 1N HCl to a pH of ~7.2-7.4.
OsO4 (Electron Microscopy Sciences, Fort Washington, PA, USA)
Potassium ferrocyanide
EPON
To the same test tube, add 20 g EPON, 13.0 g DDSA and 11.5 g MNA. Heat the tube in an
oven for 2-3 min at 60 °C and then vortex it well. Add 0.9 g of DMP-30 and immediately
vortex the tube again.
It is possible to freeze the EPON in aliquots at –20 °C and store it for a long time before use.
Hydrofluoric acid (HF)
Pick-up loop (Agar, Cambridge, England)
Slot grids covered with carbon-formvar supporting film (Electron Microscopy Sciences, Fort
Washington, PA, USA)
Basic protocol.
1. Observation and fixation of the living cells.
a) Plate the cells for CVLEM in a MatTek petri dish with a CELLocate coverslip attached to
the bottom of it. The CELLocate coverslips contain an etched grid with coordinates,
allowing the cell of interest to be found easily during all steps of the procedure.
b) Transfect the cells with the cDNA of the GFP fusion protein using your method of choice.

6
c) Find a transfected cell of interest, and determine its position on the CELLocate grid, as
shown in Figure 2.
Fig. 2. GFP-transfected cells on a CELLocate grid
d) Draw the position of the cell on the map of the CELLocate grid (available from MatTek).
e) Observe the GFP-labelled structures in this living cell on a confocal or light-microscope
stage that allows the grabbing of a time-lapse series of images by the computer.
f) At the moment of interest, add the fixative to the cell culture medium while continuing to
grab images (the volume ratio of fixative:medium is 1:1). Fixation usually induces the
rapid fading of GFP fluorescence, and blocks the motion of labelled structures in the cell.
g) Stop grabbing the time-lapse images and leave the cells in the fixative for 10 min.
h) Wash the cells with 4% paraformaldehyde once, and then leave them in 4%
paraformaldehyde for 30 min.
2. Immunolabelling for electron miscroscopy.
Immunolabelling for EM with NANOGOLD:
a) Wash the cells for 3x 5 min with PBS.

7
b) Incubate the cells with blocking solution for 30 min.
c) Incubate the cells overnight with the primary antibodies diluted in blocking solution.
d) Wash the cells for 6x 2 min with PBS.
e) Dilute the NANOGOLD conjugated Fab fragments of the secondary antibodies ~50 times
in blocking solution and add it to the cells; incubate for 2 h.
f) Wash the cells for 6x 2 min with PBS.
g) Fix the cells for 5 min with 1% glutaraldehyde in 0.2 M HEPES buffer.
h) Wash the cells for 3x 5 min with PBS.
i) Wash the cells for 3x 5 min in distilled water.
j) Incubate the cells with freshly prepared gold-enhancement mixture for 6-10 min.
k) Wash the cells for 3x 5 min with distilled water.
Immunolabelling for EM with HRP:
a) Wash the cells for 3x 2 min with PBS.
b) Incubate the cells with blocking solution for 30 min.
c) Incubate the cells overnight with the primary antibodies diluted in blocking solution.
d) Wash the cells for 6x 2 min with PBS.
e) Incubate the cells with HRP-conjugated Fab fragments of the secondary antibody for 2 h.
f) Wash the cells for 6x 2 min with PBS.
g) Fix the cells for 5 min with 1% glutaraldehyde in 0.2 M HEPES buffer.
h) Wash the cells for 3x 5 min with PBS.
i) Incubate the cells with the DAB-solution.
j) Wash the cells for 3x 2 min with PBS.
Find the cell of interest again on the CELLocate grid and compare its pattern of GFP and EM
labelling, which should be identical (Figure 3A-C).

8
3. Embedding.
a) Incubate the cells in the 1:1 mixture of 2% OsO4 and 3% potassium ferrocyanide in 0.2 M
cacodylate buffer for 1 h on ice.
b) Wash the cells once with distilled water.
c) To dehydratation the cells, incubate them (in order) with the following ethanol solutions:
50% (once), 70% (once), 90% (once), 100% (3x); each for 10 min.
d) Keep the cells in the mixture of EPON and 100% ethanol (1:1) for 1-2 h.
e) Keep the cells in EPON at the room temperature for 1-2 h, and then leave the specimens
overnight in an oven at 60 °C.
4. Location of the cell in the resin block.
a) After 12 h of polymerization of EPON, place a small droplet of fresh resin on the site
where the examined cell is located (Figure 3C) and insert a resin cylinder (prepared before
by polymerization of resin in a cylindrical mold) with a flat lower surface; leave the
sample for an additional 18 h in an oven at 60 °C.
b) Carefully pick up the resin block from the Petri dish and glass. This is easy to do by the
gentle to and fro bending of the cylinder. If the glass with a coordinated grid cannot be
detached from the cells in the resin, the resin should be placed in commercially available
HF (do not use a glass container) for 30 – 60 min to remove glass remnants.
c) Control the completeness of the glass dissolution under a stereomicroscope.
d) After the complete removal of the glass, wash the sample in water.
e) Leave the sample for 1 h in 0.1 M PBS or HEPES buffer (pH 7.3).
f) Wash the sample in water and leave it to dry.
5. Cutting.

9
a) Trim the resin block into a pyramid of ~2x 2 mm in size with the cell of interest at its
centre (Figure 3C).
b) Put the holder into the ultratome such that the segment arc is in a vertical position.
c) Bring the sample as close as possible to the glass knife.
d) Align the bottom edge of the pyramid parallel to the knife edge.
e) Tilting the segment arc and the knife, adjust the bright gap between the knife edge and the
surface of the sample. It has to be identical in width during the full up and down movement
of the resin block. This will ensure that every point of the surface containing the cell of
interest is the same distance from the knife edge.
f) Turn the specimen holder 90° to the left or the right, and trim the edges of the resin block
using a glass knife to form a narrow pyramid with its long axis parallel to the knife edge.
The pyramid should be as narrow as possible (practically, no more than 100 µm).
g) Turn the specimen holder 90° back and lock it in exactly the same position as before (this
is very important).
h) Bring the specimen towards the knife again.
i) When it very close, replace the glass knife with a diamond one.
j) Make serial sections according to the instructions with the ultratome (e.g. Leica).
k) For the picking up of the sections, stop the motor and using two eyelashes divide the
section ribbon into pieces suitable in size for their collecting with a perfect loop.
l) Touch the surface of the water with the band of sections with the perfect loop in such a
way that the band is completely inside the inner circle of the loop; do not touch the loop.
m) Raise the loop with the droplet of water with the sections on it and place it inside the tripod
under the microscope.
n) Take the slot grid coated with formvar (or preferaby butvar)/carbon supporting the film
and gently touch the sections on the water (do not touch the loop) with the carbon coated
surface of the grid.

10
o) Move the grid very slowly laterally, until as much water as possible is eliminated from the
surface of the supporting film. If the movement is slow enough, then only a very small
droplet of water remains on the grid and this does not present an obstacle for the placement
of the grid directly into the grid container.
6. EM analysis.
a) Place the slot grid under the electron microscope and find the cell of interest using the
traces of the coordinates on the first few sections.
b) Take consecutive photographs (or grab the images with the computer) from the serial
sections until the organelle of interest (observed under the LSCM) is no longer seen (see
example in Figure 3D-R).
c) Using software for 3D reconstruction, align the images and then construct a 3D model
according to the software instructions (see example in Figure 3D-R).
11
s
li j
A B
C
D E F G
H I J K Lli j
M N O P R
S
T
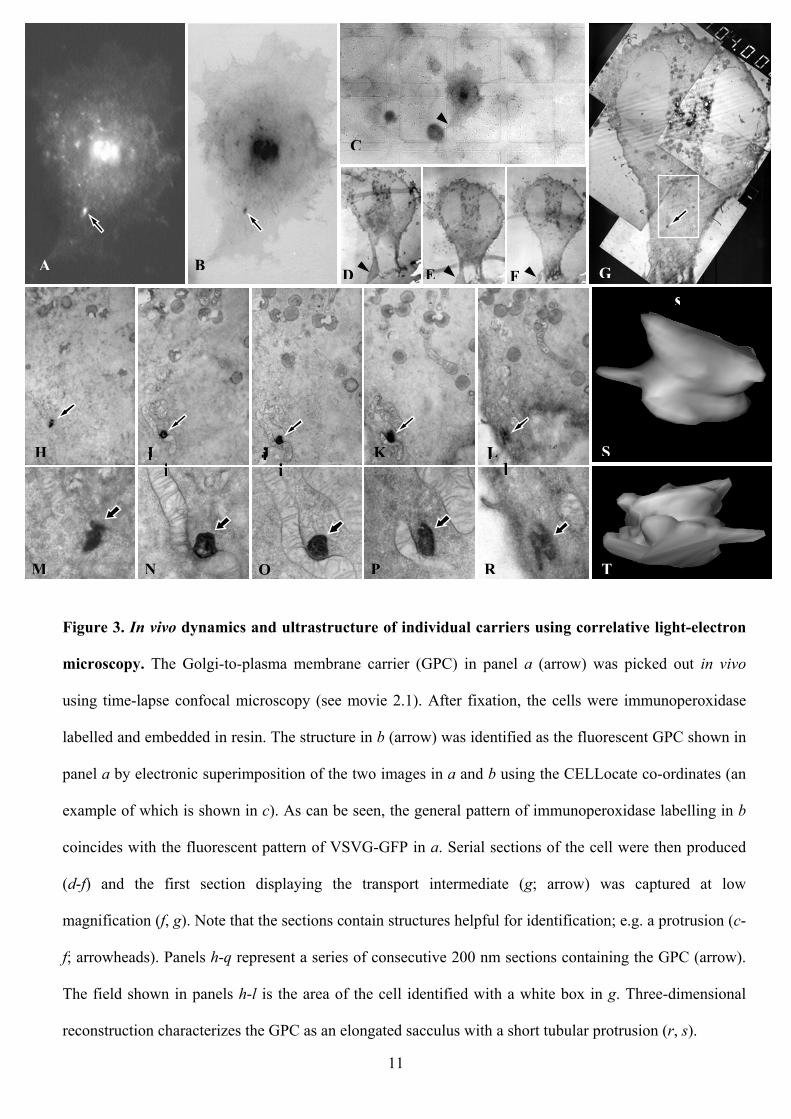
Figure 3. In vivo dynamics and ultrastructure of individual carriers using correlative light-electron
microscopy. The Golgi-to-plasma membrane carrier (GPC) in panel a (arrow) was picked out in vivo
using time-lapse confocal microscopy (see movie 2.1). After fixation, the cells were immunoperoxidase
labelled and embedded in resin. The structure in b (arrow) was identified as the fluorescent GPC shown in
panel a by electronic superimposition of the two images in a and b using the CELLocate co-ordinates (an
example of which is shown in c). As can be seen, the general pattern of immunoperoxidase labelling in b
coincides with the fluorescent pattern of VSVG-GFP in a. Serial sections of the cell were then produced
(d-f) and the first section displaying the transport intermediate (g; arrow) was captured at low
magnification (f, g). Note that the sections contain structures helpful for identification; e.g. a protrusion (c-
f; arrowheads). Panels h-q represent a series of consecutive 200 nm sections containing the GPC (arrow).
The field shown in panels h-l is the area of the cell identified with a white box in g. Three-dimensional
reconstruction characterizes the GPC as an elongated sacculus with a short tubular protrusion (r, s).

12
Critical parameters and troubleshooting.
Taken all together, the stages of CVLEM represent quite a long procedure (see below) and require
significant effort of a researcher or a technician. It would thus be particularly disappointing to lose
such tour de force experiments because of small problems in specimen handling. To apply CVLEM
successfully, several important parameters should always be taken into account by the experimenter.
First, preliminary trials should be carried out to determine if the antibodies selected for the
labelling of a GFP-fusion protein work with the immuno-EM protocol. Many antibodies that give
perfect results with immunofluorescence do not work with immuno-EM staining. This is because the
glutaraldehyde used in most EM fixatives tends to cross-link amino groups of antigen epitopes, and
therefore to decrease the antigenicity of the target protein. However, a decrease in, or absence of,
glutaraldehyde in the fixative can result in the poor preservation of ultrastructure of intracellular
organelles. So, if this produces problems with immuno-EM labelling, it is possible to optimize the
concentration of glutaraldehyde in the fixative, or to use a periodate-lysine-paraformaldehyde
fixative (Brown and Farquhar, 1989). It is also important to use the immunoperoxidase or
immunogold protocols for the labelling of a structure of interest. We would advise the immunogold
protocol to be used only for the labelling of epitopes of a GFP-fusion protein located in the cytosol
(see Strategy section), while for other epitopes HRP labelling is also suitable.
Secondly, it is extremely important to be able to find the cell of interest at all the stages of the
CVLEM procedure. Thus, only cells located on the grid in the MatTek petri dish can be selected for
time-lapse observations. The position of the cell of interest on the grid should be noted, otherwise it
will be difficult to find it again. The low magnification images showing the field surrounding the
cell of interest can help greatly both in the trimming of the resin block around the correct cells, and
in the finding of the cell of interest under EM. In this case, neighbouring cells can be used as
landmarks for the identification of the cell of interest. For this reason, the cells for these experiments
should be plated at a low confluence (50-60%). During the analysis of the serial sections under EM,

13
it is also useful to have the fluorescence and phase-contrast images of the target cell, as particular
structures (e.g. microvilli, pseudopodia, inclusions) can help greatly in the finding of both the cell
and the structure of interest.
Thirdly, during the cutting of the specimen, the thickness of the serial sections has to be
selected. This should be about 80 nm for routine work, 50 nm (or less) for very precise 3D
reconstruction, and 250 nm for EM tomography.
Time considerations
The entire CVLEM procedure requires a reasonable time for its completion (4-5 days). During the
first day, the cells need to be transfected with the cDNA. The next day, the observations can be
made on the living cells, they can be fixed, and the immunolabelling can be started. The third day is
required to complete the immunolabelling and resin embedding of the cells. During the fourth day,
the cell of interest can be identified in the resin block and cut in serial sections. However, it is also
possible to store already embedded speciments for a long time, and hence to cut them later.
RELEVANT LITERATURE
Brown, W.J. and Farquhar, M.G. 1989. Immunoperoxidase methods for the localization of antigens
in cultured cells and tissue sections by electron microscopy. Methods Cell Biol. 31: 553-569.
Deerinck, T.J. et al. 1994. Fluorescence photooxidation with eosin: a method for high resolution
immunolocalization and in situ hybridization detection for light and electron microscopy. J.
Cell Biol. 126: 901-910.
Burry, R.W., Vandre, D.D. and Hayes, D.M. 1992. Silver enhancement of gold antibody probes in
pre-embedding electron microscopic immunocytochemistry. J. Histochem. Cytochem. 40:
1849-1856.
Mironov, A.A., Polishchuk, R.S. and Luini, A. 2000. Visualising membrane traffic in vivo by
combined video fluorescence and 3-D-electron microscopy. Trends Cell Biol. 10: 349-353.

14
Polishchuk, R.S. and Mironov, A.A. (2001). Correlative video/light electron microscopy. Current
Protocols in Cell Biology. Bonifacino, J.S. , Dasso, M., Harford, J.B., Lippincott-Schwartz,
J. and Yamada, K.M. (eds). John Wiley & Sons, New York. 4.8.1–4.8.9.
Polishchuk, R.S., Polishchuk, E.V., Marra, P., Buccione, R., Alberti, S., Luini, A. and Mironov, A.A.
2000. GFP-based correlative light-electron microscopy reveals the saccular-tubular ultrastructure of
carriers in transit from the Golgi apparatus to the plasma membrane. J. Cell Biol. 148: 45-58.
Polishchuk, R.S., Polishchuk, E.V., Trucco, A. and Mironov A.A. (2003) Pushing forward the
resolution of time-lapse imaging. Imag. Microsc. 5: 38-40.
Powell, R.D. et al. 1998. Combined fluorescent and gold immunoprobes: reagents and methods for
correlative light and electron microscopy. Microsc. Res. Tech. 42: 2-12.