systematic identification of signaling pathways with
TRANSCRIPT

R E S E A R C H A R T I C L E
C A N C E R
Systematic identification of signaling pathwayswith potential to confer anticancer drug resistanceColin A. Martz,1* Kathleen A. Ottina,2,3* Katherine R. Singleton,1* Jeff S. Jasper,1
Suzanne E. Wardell,1 Ashley Peraza-Penton,1 Grace R. Anderson,1 Peter S. Winter,1
Tim Wang,2,3,4 Holly M. Alley,1 Lawrence N. Kwong,5 Zachary A. Cooper,5 Michael Tetzlaff,5
Pei-Ling Chen,5 Jeffrey C. Rathmell,1 Keith T. Flaherty,6 Jennifer A. Wargo,5
Donald P. McDonnell,1 David M. Sabatini,2,3,4† Kris C. Wood1†
http://stke.sciencD
ownloaded from
Cancer cells can activate diverse signaling pathways to evade the cytotoxic action of drugs.We created andscreened a library of barcoded pathway-activating mutant complementary DNAs to identify those thatenhanced the survival of cancer cells in the presence of 13 clinically relevant, targeted therapies. We foundthat activation of the RAS-MAPK (mitogen-activated protein kinase), Notch1, PI3K (phosphoinositide3-kinase)–mTOR (mechanistic target of rapamycin), and ER (estrogen receptor) signaling pathways oftenconferred resistance to this selection of drugs. Activation of the Notch1 pathway promoted acquiredresistance to tamoxifen (an ER-targeted therapy) in serially passaged breast cancer xenografts in mice,and treating mice with a g-secretase inhibitor to inhibit Notch signaling restored tamoxifen sensitivity.Markers of Notch1 activity in tumor tissue correlated with resistance to tamoxifen in breast cancer patients.Similarly, activationofNotch1signalingpromotedacquired resistance toMAPK inhibitors inBRAFV600Emel-anoma cells in culture, and the abundance of Notch1 pathwaymarkerswas increased in tumors froma sub-set ofmelanoma patients. Thus, Notch1 signalingmay be a therapeutic target in somedrug-resistant breastcancers and melanomas. Additionally, multiple resistance pathways were activated in melanoma cell lineswith intrinsic resistance to MAPK inhibitors, and simultaneous inhibition of these pathways synergisticallyinduceddrugsensitivity. Thesedata illustrate thepotential for systematic identificationof thesignalingpath-ways controlling drug resistance that could inform clinical strategies and drug development for multipletypes of cancer. This approach may also be used to advance clinical options in other disease contexts.
ema
on December 23, 2014
g.org/
INTRODUCTION
Clinical resistance to anticancer therapies is a persistent problem that can becaused by genetic or epigenetic events occurring within cancer cells or byextracellular cues such as soluble factors or cell-cell contacts (1–6). Ulti-mately, these diverse events lead to the activation of growth and survivalsignaling pathwayswithin cancer cells that enable them to survive otherwiselethal pharmacological insults (1–6). By blocking these drug resistancepathways, it may be possible to improve the efficacy and durability of anti-cancer drugs. However, for most drugs, the identities of potential resistancepathways are unknown (1).
We sought to develop a strategy to systematically identify the signalingpathways that, when activated, have the potential to confer resistance to ther-apeutic agents. If successful, such an effort could lead to a more completeunderstanding of the repertoire of signaling events that can render a cancercell drug-resistant, potentially resulting in improvements in our ability to(i) stratify patients intogroupsmore and less likely to respond to therapy and
1Department of Pharmacology and Cancer Biology, Duke University, 450 Re-search Drive, Durham, NC 27710, USA. 2Whitehead Institute for Biomedical Re-search, 9 Cambridge Center, Cambridge, MA 02142, USA. 3Howard HughesMedical Institute, Department of Biology, Massachusetts Institute of Technology,77 Massachusetts Avenue, Cambridge, MA 02139, USA. 4Broad Institute, 7Cambridge Center, Cambridge, MA 02142, USA. 5University of Texas M. D.Anderson Cancer Center, 1515 Holcombe Boulevard, Houston, TX 77030,USA. 6Massachusetts General Hospital Cancer Center, 55 Fruit Street, Boston,MA 02114, USA.*These authors contributed equally to this work and are listed alphabetically.†Corresponding author. E-mail: [email protected] (D.M.S.); [email protected] (K.C.W.)
www.SC
(ii) design multicomponent combination therapies that simultaneously acton cancer cell dependencies and resistance pathways.
RESULTS
Screening to identify potential drug resistance pathwaysWith the objective of identifying key drug resistance pathways, we con-structed a list of 17 signaling pathways that are frequently implicated incancer cell proliferation, survival, differentiation, and apoptosis (7). Foreach pathway, a set of one to three mutant complementary DNAs (cDNAs)were identified representing core nodes in each pathway that, when over-expressed, constitutively activated or inactivated the pathway (Fig. 1 and ta-ble S1). Pathway-activating mutants were used for those pathways thattypically have tumor-promoting roles, whereas pathway-inhibiting mutantswere used for those that have tumor-suppressive roles. All cDNAs in thecollection were obtained, barcoded, and cloned into a PGK (phospho-glycerate kinase 1) promoter–driven lentiviral expression vector. Constructswere then fully sequenced (data file S1) and produced as vesicular stomatitisvirus G (VSV-G) pseudotyped lentiviruses (8), 86% of which (31 of 36)were functionally validated in cells by Western blotting, reporter gene as-says, or immunofluorescence to ensure proper engagement of targeted path-ways (table S1). Finally, to screen library constructs for pathways withpotential to confer resistance to anticancer drugs, we developed a modified,positive selection, pooled screening protocol with sequencing-based de-convolution that is analogous to those previously described (fig. S1) (9).The abundance of each cDNA in cells infected with the pooled librarywas assessed immediately after infection and again after 4 weeks in culture.In all cases, cDNA abundance was relatively stable (fig. S2).
IENCESIGNALING.org 23 December 2014 Vol 7 Issue 357 ra121 1

R E S E A R C H A R T I C L E
on Decem
ber 23, 2014http://stke.sciencem
ag.org/D
ownloaded from
To validate this screening approach, we first screened a BRAF-mutantmelanoma cell line (UACC-62) to identify pathways of resistance to aMEK1/2 (mitogen-activated or extracellular signal–regulated protein kinasekinase 1/2) inhibitor (AZD6244). Resistancemechanisms toMAPK (mitogen-activated protein kinase) pathway inhibitors have been studied intensively inthis setting. Our results corroborated the findings of these studies, showingthat five major pathways are capable of conferring resistance to MEK1/2inhibition in cultured cells. Three of these pathways—RAS-MAPK, phos-phoinositide 3-kinase (PI3K)–mechanistic target of rapamycin (mTOR),and nuclear factor kB (NF-kB)—have been previously identified and are,in fact, frequently exploited by resistant tumors and cell lines thus far re-ported (10–13). Additionally, we found that two other previously un-identified pathways, those mediated by Notch1 and estrogen receptor(ER), were also capable of conferring resistance to MEK1/2 inhibition inthis primary screen (fig. S3).
On the strength of this technological validation, we then performedscreens covering a total of 13 targeted therapies, most of which are eitherclinically approved, in clinical trials, or in late-stage preclinical develop-ment. Each drug was screened at two or three concentrations in one to threecell lines that had the appropriate drug-sensitizing genetic mutations andnanomolar drug sensitivity (Fig. 2A and tables S2 and S3). Individualconstructs whose expression conferred resistance to a given drugwere iden-tified as those yielding an enrichment score (the relative abundance of eachconstruct in the presence of drug normalized to the same value in the ab-sence of drug) of ≥1.5 and scoring in at least two of the three drug concen-trations screened. This value was set because more than 80% of constructsthat scored at or above this level in pilot screens were successfully validatedin subsequent eight-point GI50 (half-maximal growth inhibition) assays.Twelve of 17 pathways scored as providing resistance to at least one drug,
www.SC
with the RAS-MAPK, Notch1, PI3K-mTOR, and ER pathways eachscoring in more than 30% of all screens (Fig. 2A and fig. S4A). Further,we also found that the cellular sensitivity to 11 of 13 drugs screened couldbe partially decreased by the activation of five or fewer pathways (fig. S4B).Finally, we noted that the manipulation of some pathways, such as the inhi-bition of apoptosis through the expression of dominant-negative caspases,rarely conferred resistance to targeted therapies in these assays, despite theirdemonstrable roles in modulating sensitivity to cytotoxic chemotherapeutics(fig. S5) (14).
A common theme in the targeted therapy of oncogene-driven cancers isthe emergence of acquired resistance mediated by pathway reactivation,which can occur through copy number changes, alternative splicing events,mutations in members within the pathway, or second-site mutations in thedrug target itself (1–4, 15). Thus, current efforts aim to inhibit pathwaynodes downstream of the driving oncogene, on the assumption that pathwayreactivation ismore difficult to achieve after downstreampathway inhibitioncompared with upstream inhibition. This notion led to the use of MEK andextracellular signal–regulated kinase (ERK) inhibitors to complement RAFinhibitors in the treatment of BRAF-mutant melanomas (10, 11, 16). Bycombining resistance screening results with immunoblotting in melano-ma cells that were treated with drugs targeting multiple nodes in theRAF-MEK-ERK pathway, we found that whereas cDNAs that reactivatethe MAPK pathway at the level of ERK phosphorylation can drive potentresistance when the pathway is inhibited upstream of ERK, alternativeresistance pathways that do not reactivate ERK phosphorylation dominatethe resistance landscapewhen the pathway is inhibited “downstream” at thelevel of ERK (Fig. 2B and fig. S6). To our knowledge, these are the firstevolutionary experiments demonstrating a shift in the resistance landscapeon the basis of the location of a pathway node targeted by a drug. These
KRASG12V HRASG12V
MEK1S218D/S222D
myr-PIK3CA
myr-AKT1
RHEBQ64L IKK S176E/S180E
IKK S177E/S181E
GSK3 K85A
-Catenin4A/S33Y
JNK2WT
Mkk7-JNK2
MEK5S311D/T315D
myr-MEK5
NOTCH1ICD NOTCH3ICD
p38WT
MKK6S207E/T211E
TGF RIT204D/GSD
BCL-2WT BCL-XLWT
CASP3C360A CASP8C163A
ER Y537S AR-V7
YAP25SA
LATS2K697R
p53R175H
HRASG12V/E37G
Rgl2-CAAX
RALAG23V
Ras-MAPK
PI3K-mTOR
Cytoplasm
Ral
NF- B
ERK5
JNK
p38 Hippo
TGF-
ER AR Notch
Wnt
Jak-Stat
Hedgehog
p53
Mitochondria
Death receptors
Apoptosis
mTOR
SMADs
Nucleus
JAK2V617F
STAT3A662C/N664C/V667L
GLI2trunc
SMO-M2W535L
Fig. 1. Strategy for manipulating oncogenic signaling pathways. Pathway names are in bold and situated in the cellular context in which they function.The engineered cDNA constructs in each pathway are denoted as either wild type (WT; black), constitutively active mutants (red), or dominant-negative mutants (blue).
IENCESIGNALING.org 23 December 2014 Vol 7 Issue 357 ra121 2

R E S E A R C H A R T I C L E
www.SCIENCESIGNALING.org 23 Decemb
on Decem
ber 23, 2014http://stke.sciencem
ag.org/D
ownloaded from
findings demonstrate that althoughdownstream pathway inhibition maybe an effective tactic for suppressingresistance that is driven by pathway re-activation, it is also likely to encour-age the evolution of resistance drivenby alternative pathways, a phenomenonthat may have undesired consequences.
Notch1-driven resistance totargeted therapies in breastcancer cellsTo identify findings from our screenswith potential translational relevance,we first examined our data in breastcancer cells. We screened tamoxifenand fulvestrant (ER inhibitors) inMCF-7 estrogen-responsive (ER+)cells; lapatinib (HER2 inhibitor) inHER2-amplified SkBr3 cells; andthe PI3K-mTOR pathway inhibitorsBKM-120 (PI3K inhibitor), MK-2206(AKT inhibitor), Torin1 (mTORC1/2inhibitor), and rapamycin (mTORC1inhibitor) in breast cancer cell lineswith activating mutations in PIK3CA(BT20, MDA-MB-453, and T47D)(Fig. 2A). The RAS-MAPK and PI3K-mTOR pathways frequently scored ashits against these drugs, as expected(6, 17, 18). Additionally, the Notch1pathway scored against these drugs, anobservation that we validated throughGI50 assays in cells that either expressedthe Notch1 intracellular domain (ICD;Fig. 3A) or were treated with the DSLpeptide (residues 188 to 204 of theNotch ligandJagged-1), a solubleNotchagonist (fig. S7) (19). Notch1-mediatedresistance did not involve proximalreactivation of targeted signaling path-ways, but was associated with the in-duction of canonical Hes and Heyfamily Notch1 target genes; the induc-tion of a dedifferentiation process thatresembled the epithelial-to-mesenchymaltransition (EMT); and the acquisi-tion of enhanced migration, sphereformation, and apoptosis resistanceproperties as well as cross-resistanceto cytotoxic agents, like doxorubicin(Fig. 3, A and B, and figs. S8 and S9)(7, 20). These findings are consistentwith the established role of the Notch1pathway in stimulating EMT in epithe-lial cells and its prior association withresistance to cytotoxic chemotherapiesand targeted inhibitors of EGFR (epi-dermalgrowth factor receptor) andPI3Kin epithelial cancer cells (21–24).
A
B
17 pathways 13
targ
eted
ther
apie
s
Percentage of screens in which construct scores
Number of pathways scoring
Enrichment score = 1.5 - 3.0 Enrichment score greater than 3.0
Genotype (pathway
dependency) Drug
(target) Cell line KR
AS
(G
12V
) H
RA
S (
G12
V)
ME
K1
(DD
)
HR
AS
(G
12V
/E37
G)
RG
L2-C
AA
X
Ral
A (
G23
V)
myr
-PIK
3CA
myr
-AK
T1
RH
EB
(Q
64L)
Sm
oM2
(W53
5L)
GLI
2-tr
unc
JAK
2 (V
617F
)
STA
T3
(CC
)
IKK
(E
E)
IKK
(E
E)
GS
K3
(K
85A
)
-Cat
enin
(4A
)
-Cat
enin
(S
33Y
)
myr
-ME
K5
ME
K5
(DD
)
MK
K7-
JNK
2
JNK
2 (W
T)
MK
K6
(EE
)
P38
(W
T)
LAT
S2
(KD
)
YA
P2
(5S
A)
TG
FR
I (G
SD
)
CA
SP
3 (C
361A
)
CA
SP
8 (C
360A
)
BC
L-2
(WT
)
BC
L-X
L (W
T)
P53
(R
175H
)
NO
TC
H1
(IC
D)
NO
TC
H3
(IC
D)
ER
(Y
537S
)
AR
-V7
BRAF (V600) melanomas (Ras-MAPK
pathway)
PLX4720 (RAF)
A375
Colo679
UACC62
AZD6244 (MEK)
A375
Colo679
UACC62
VX-11E (ERK)
A375
Colo679
UACC62
PIK3CA (H1047R) breast cancers (PI3K-mTOR
pathway)
BKM-120 (PI3K)
T47D
MD-453
BT-20
MK-2206 (AKT)
T47D
MD-453
BT-20
Torin1 (mTOR)
T47D
MD-453
BT-20
Rapamycin (mTORC1)
T47D
MD-453
BT-20
ER+ breast cancer (ER pathway)
Tamoxifen (ER)
MCF-7
Fulvestrant (ER)
MCF-7
HER2-amplified breast cancer
(HER2)
Lapatinib (HER2)
SkBr3
FGFR-amplified gastric carcinoma
(FGFR)
PD173074 (FGFR)
Kato III
PDGFR-amplified NSCLC
(PDGFR)
Sunitinib (PDGFR)
NCI-H1703
BCL-dependent lymphoma
(BCL-2, BCL-XL)
ABT-737 (BCL-2, BCL-XL)
DHL4
Ly.1
Pathway: Ras-MAPK
RAL
PI3K-mTOR
Hedgehog
JAK-STAT
NF- B
WNT
ERK5
JNK
p38
Hippo
TGF
Apoptosis
p53
Notch
ER
AR
3
1
3
1
2
5
2
5
5
2
4
2
5
3
3
4
4
3
4
3
4
4
6
5
4
4
7
3
0
36
0
64
0
0
4
4
4
7
4
0
0
14
0
0
7
4
4
0
7
7
7
14
21
11
14
11
50
25
0
0
0
21
86
25
µ
µ
MAPK PI3K NF B Notch ER Controls Pathway:
KRAS HRAS
MEK
ERK
BRAFV600E
Growth survival
Pathway nodes Pathway inhibitors Resistance pathways
Pathway reactivation
Pathway reactivation +
alternative pathways
Alternative pathways only
AZD6244
VX-11E
PLX4720
Fig. 2. Global analysis of screening results. (A) Results of each screen color-coded according to enrichmentscore (the relative abundance of each construct in the presence of drug normalized to the same value in theabsence of drug). (B) Scoring of pathway-activating constructs in screens involving UACC-62 BRAFV600E
melanoma cells treated with PLX4720 [RAF inhibitor, red (top)], AZD6244 [MEK inhibitor, green (middle)],or VX-11E [ERK inhibitor, blue (bottom)].
er 2014 Vol 7 Issue 357 ra121 3

R E S E A R C H A R T I C L E
on Decem
ber 23, 2014http://stke.sciencem
ag.org/D
ownloaded from
0.0001
0.001
0.01
0.1
1
10
100
Luciferase
Notch1 ICD
Ful Tam Lap Rapamycin MK-2206 BEZ-235 Doxorubicin
MCF7 MCF7 MCF7 MCF7 MCF7 MCF7 SkBr3 T47D T47D T47D T47D
GI 5
0 (
M)
Cell line:
Drug:
**
**
**
*** *** *** **
***
** **
***
A B
C
0 50 100 150
Months
Dis
tant
rec
urre
nce–
fr
ee s
urvi
val
0
0.2
0.4
0.6
0.8
1.0 P = 9.4e-06, N = 458
200 250 300
0.0
2.0
4.0
MCF7 T47D
N-Cadherin *
*
0.0
2.0
4.0
MCF7 T47D
Vimentin
***
***
0
1000
2000
3000
MCF7 T47D
Slug
**
**
0.0
2.0
4.0
MCF7 T47D
CD44
*** *
D
E F
tum
or
vo
lum
e (
cm
3)
0 1 0 2 0 3 0
0 .0
0 .2
0 .4
0 .6
T a m c o n tro l
T a m + R O 4 9 2 9 0 9 7
* p < 0 .0 0 0 1
R O a lo ne
n o tre a tm e n t*
Tum
or v
olum
e (c
m3 )
Time (days on treatment)
0 10 20 30
* P < 0.0001
*
0.0
0.2
0.4
0.6
0.8 Tamoxifen only
Tamoxifen + RO4929097
RO4929097 only
Vehicle
0.0
2.0
4.0
MCF7 T47D
Fo
ld c
han
ge
(Tra
nsc
rip
t) Hey1
*
**
0.0
2.0
4.0
MCF7 T47D
Fo
ld c
han
ge
(Tra
nsc
rip
t) Hes4
*
**
ESR1
HES1
MYC
HEY1
HES2
HEYL
HEY2
CDH1
VIM
SNAI2
SNAI1
CDH2
TWIST1
NOTC
H1
JAG1
PSEN1
APH1A
PSEN2
NCSTN
ADAM10
J AG2
ADAM17
PSENEN
DLL
3
DLK
2
DLK
1
Activators Targets Dedifferentiation
Low
High
0 .5 1–1 –.5
*** *** *** ***
Med
ian-
cent
ered
exp
ress
ion
Fol
d ch
ange
(tr
ansc
ript)
Hey1 N-cadherin Vimentin
CD44 Slug Hes4
Median-centered log2 expression Notch1 activity (treatment)
Low (no tamoxifen) Low (tamoxifen) High (no tamoxifen) High (tamoxifen)
Notch1 signature
Fig. 3. Notch1 pathway activation confers resistance to targeted therapies inbreast cancer, including tamoxifen. (A) GI50 values for the indicated breastcancer cell lines expressing Notch1 ICD or luciferase and treated with theindicated drugs. Data are means ± SD from three experiments. (B) Quanti-tative real-time PCR (qRT-PCR) analysis of Notch1 target genes andmesen-chymal markers in breast cancer cell lines expressing Notch1 ICD (red) orluciferase (gray). Data are means ± SD from three experiments. (C) Expres-sion of the indicated genes in parental MCF-7 and TamR cells. Data aremeans ± SD from three experiments. (D) Growth of TamR xenografts treated
www.SC
with tamoxifen, RO4929097 (g-secretase inhibitor), or the combination. Dataaremeans±SEM from9 to13miceper group. Exponential growth curveandtwo-way analysis of variance analyses were performed using GraphPadPrism 6. (E) Kaplan-Meier plot depicting survival in breast cancer patientstreatedwith tamoxifen as a function ofNotch1 pathway gene expression sig-nature level (first and third tertiles). P value denotes significance betweendrug-treated patients in high and low Notch1 groups. (F) Expression heat-map of the indicated genes in Notch1 low and high groups from patients inpart (E). *P < 0.1; **P < 0.05; ***P < 0.01.
IENCESIGNALING.org 23 December 2014 Vol 7 Issue 357 ra121 4

R E S E A R C H A R T I C L E
on Decem
ber 23, 2014http://stke.sciencem
ag.org/D
ownloaded from
Resistance to endocrine therapy (such as tamoxifen) is a major clinicalproblem that eventually occurs in most patients with ER+ metastatic breastcancer (17). Given our finding thatNotch1 can drive resistance to tamoxifenin cultured cells, and the fact that Notch1 and ER reciprocally regulate eachother in ER+ cells (25), we reasoned that this pathway may play a role inacquired resistance. To test this hypothesis, we used a mouse model of ta-moxifen resistance in whichMCF-7 xenografts acquired resistance throughselective pressure induced by serial passaging in the presence of tamoxifenin vivo (26). In this model, as in patients with tamoxifen-resistant (TamR)disease, tamoxifen exerts pronounced growth agonist activity to the tumorsin mice. This model also retains sensitivity to fulvestrant. The expression ofNOTCH1, Notch1 target genes, and genes encoding mesenchymal markerswere significantly increased in TamR tumors relative to tamoxifen-sensitiveparental tumors (Fig. 3C and fig. S10), consistent with potential Notch1pathway activation. Further, cotreatment of TamR tumors with tamoxifenand RO4929097, a g-secretase inhibitor that blocks Notch1 signaling(27), blocked tamoxifen-stimulated growth in these tumors (Fig. 3D). Cor-roborating these data, we also found that in a meta-analysis of 458 womenwith ER+ breast cancer enrolled acrossmultiple studies, Notch1 activation(evident as induction of a Notch1-associated gene expression signature)correlated with the acquisition of a dedifferentiated phenotype and lossof the beneficial effects of tamoxifen on distant recurrence–free survival.This effect was specific to tamoxifen because outcomes in women nottreated with tamoxifen were not correlated with Notch1 activation (Fig. 3,E and F, and fig. S11) (28–30). Together, these data suggest that Notch1activationmay drive resistance to tamoxifen in vivo and that patientswithTamR disease may benefit from combination therapy with g-secretaseinhibitors.
Notch1-driven resistance to targeted therapies inmelanoma cellsIn melanoma, we screened three inhibitors of the RAS-MAPK pathway:PLX4720 (a RAF inhibitor), AZD6244 (a MEK1/2 inhibitor), and VX-11E(anERK2 inhibitor), each inMAPK inhibitor–sensitivemelanoma cell linesharboring activating mutations in BRAF (A375, Colo679, and UACC-62)(Fig. 2A). Consistent with our pilot screen involving AZD6244, thesescreens revealed that resistance can be driven by MAPK reactivation orcompensatory activation of the PI3K-mTOR,NF-kB,Notch1, andERpath-ways. Notch1 and ER signaling have not been previously implicated inMAPK inhibitor resistance in melanoma. GI50 assays revealed that Notch1activation conferred robust GI50 shifts of 0.5 to 2 orders of magnitude inmultiple cell lines treated with MAPK inhibitors (Fig. 4A), whereas ER ac-tivation conferred more modest GI50 shifts of 2.5- to 3-fold for the samedrugs (fig. S12). Because of the strength of Notch1-mediated resistance,we chose to focus our follow-up efforts in melanoma primarily on thispathway. Analogous to our findings in breast cancer cells, expression ofthe Notch1 ICD increased the expression of Notch1 target genes and stimu-lated the dedifferentiation ofmelanoma cells, resulting in the loss ofmelanoma-specific antigens tyrosinase (TYR) and DCT (TRP-2) (31) and increasedexpression of markers associated with melanocyte dedifferentiation, suchas NGFR and N-cadherin (32) (Fig. 4B and fig. S13). Furthermore,Notch1-activated cells exhibited increased migration, melanosphere forma-tion, and viability and did not reactivate ERK activation (phosphorylation)in the presence of drug treatment, suggesting that the Notch1 pathway op-erates independently, or possibly downstream, of ERK and supports themaintenance of a dedifferentiated state (figs. S14 and S15).
To explore the potential role of Notch1 in the setting of acquiredresistance, which occurs in most patients who initially respond to RAFand/or MEK inhibitor therapy, we first induced resistance to RAF, MEK,and ERK inhibitors in a panel of six cell lines by stepwise selection (33).
www.SC
In 7 of 18 (39%) of these derivatives, Notch1 knockdown by two inde-pendent short hairpinRNAs (shRNAs) fully resensitized drug-resistant cellsto MAPK inhibitors (Fig. 4C, fig. S16, and table S4). Eighty-four clonalderivatives of these lines were also established, 24 of which were resensi-tized to MAPK inhibitors by Notch1 knockdown (29%) (fig. S17). In celllines whose resistance appeared to be Notch1-dependent, resistance toMAPK inhibitors was associated with the increased expression of Notch1,Notch1 target genes, andmarkers of dedifferentiation, aswell as the absenceof ERKpathway reactivation (evident in the loss of ERKphosphorylation inthe presence ofMAPK pathway inhibitors) (Fig. 4D and fig. S17). To iden-tify potential clinical correlates of these findings, we examined twoindependent cohorts of matched pretreatment/post-relapse tumor samplesfrom BRAF-mutant melanoma patients. In each cohort, we searched for ev-idence of Notch1 activation in relapsed tumors that is consistent with theactivation observed in engineered and evolved Notch1-activated cells:increased Notch1 and Notch1 target gene expression (more than twofold)with concomitant decreased expression of differentiation markers orincreased expression of dedifferentiation markers. In cohort 1, we queriedgene expression and targeted sequencing data from a published set of 29matched pretreatment/post-relapse tumors from patients treated initiallywith the RAF inhibitors vemurafenib or dabrafenib (34). Fifteen of 29(52%) relapsed tumors showed evidence of canonical acquired mutationsin MAPK pathway genes (NRAS, BRAF, MEK1, and MEK2), and 1 of 29had an activating mutation in AKT1 (34). Twelve of 29 (41%) relapsed tu-mors lacked evidence of canonical MAPK or PI3K pathway alterations.Among this latter group, four (14% of total) exhibited expression patternsconsistent with Notch1 activation (Fig. 4E and fig. S18). Additionally, onerelapsed tumor exhibited evidence of coincident Notch1 activation andinsulin-like growth factor (IGF-1) receptor overexpression (fig. S18). To ver-ify these findings, we obtained a second, independent cohort of matchedpretreatment/post-relapse samples from seven patients with acquiredresistance to RAF or RAF +MEK inhibitor combinations. Consistent withcohort 1, 3 of 7 (43%) relapsed tumors harbored acquired MAPK pathwayalterations (BRAF,MEK2), 1 of 7 (14%) harbored a PTENmutation, and 3of 7 (43%) lacked canonical resistance alterations. Two of the tumors in thelatter group (29% of total) exhibited Notch1 activation on relapse as judgedby the criteria above, whereas none of the four tumors with MAPK/PI3Kpathway alterations showed evidence of Notch1 activation (Fig. 4, F and G,fig. S19, and table S5). Together, these data demonstrate that Notch1 acti-vation can drive acquired resistance to MAPK inhibitors in engineered andevolved cellular models and suggest that Notch1 signaling is likely to play arole in the minority of patients whose relapsed tumors lack canonicalMAPKor PI3K pathway–mediated resistance mechanisms. These findingsare also consistent with emerging data suggesting that the Notch1 pathwaypromotes melanoma tumor growth and may act as an essential mediator oftumorigenesis downstream of BRAF (35, 36).
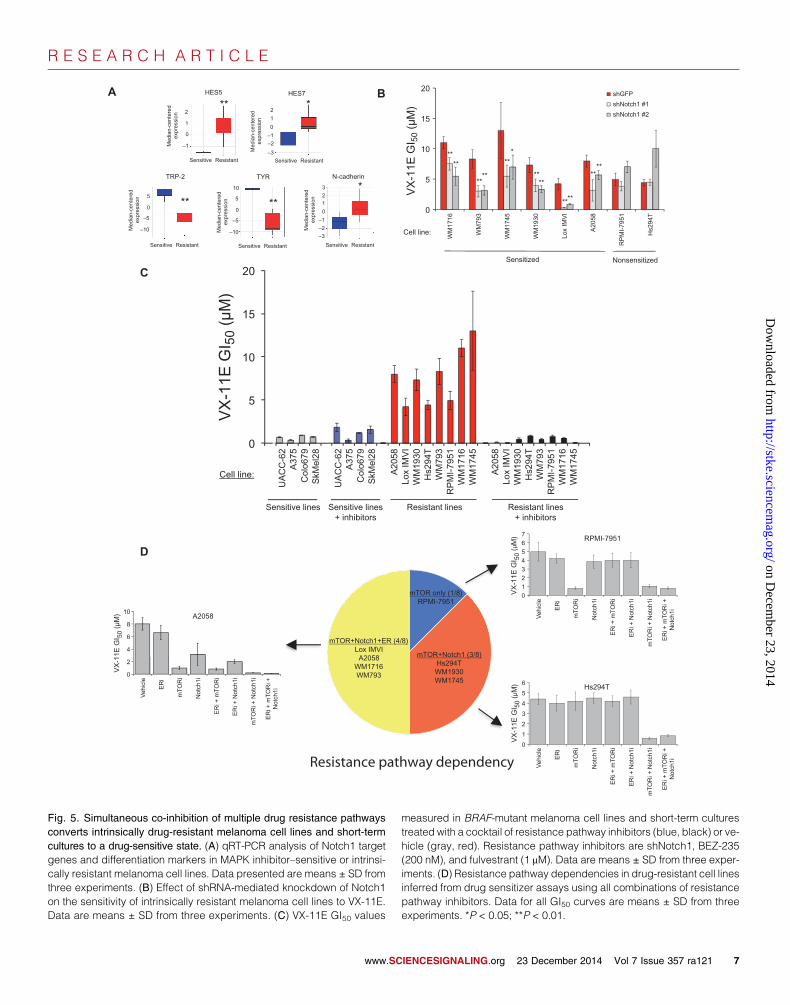
Finally, we sought to assess the potential relevance of our findings in thesetting of intrinsic resistance to MAPK inhibitors, which occurs in 10 to20% of BRAF-mutant melanoma patients (37). To assess whether Notch1activation may be responsible for this phenomenon, we obtained eightBRAF-mutantmelanoma cell lines and short-term cultures exhibiting intrin-sic resistance to MAPK inhibitors (GI50 > 4 mM) through uncharacterizedmechanisms (37, 38). Although these cultures exhibited higher expressionof certainNotch1 target genes, lower expression ofmelanocyte differentiationmarkers, and higher expression of dedifferentiation markers than a referencecohort of MAPK inhibitor–sensitive cell lines (Colo679, UACC-62, andMalme3M) (Fig. 5A), knocking downNotch1 by two independent shRNAsonly modestly sensitized six of eight of these cell lines to MAPK inhibitors(Fig. 5B). Thus, we concluded that although Notch1 appears to play a func-tional role in the maintenance of intrinsic resistance in these cells, other
IENCESIGNALING.org 23 December 2014 Vol 7 Issue 357 ra121 5

R E S E A R C H A R T I C L E
on Decem
ber 23, 2014http://stke.sciencem
ag.org/D
ownloaded from
A B
C D
E F
Other (2)
0.0
1.0
2.0
3.0
4.0
Luciferase N1 ICD
N-Cad
**
0.0
2.0
4.0
6.0
Luciferase N1 ICD
NGFR **
0.0
0.5
1.0
1.5
Luciferase N1 ICD
TYR
**
0.0
0.5
1.0
1.5
Luciferase N1 ICD
TRP-2
**
0.0
1.0
2.0
Luciferase N1 ICD
Hes1 **
0.0
2.0
4.0
6.0
Luciferase N1 ICD
Hey2 *
Putative resistance alterations in 29 relapsed BRAFV600 melanomas (cohort 1)
Putative resistance alterations in 7 relapsed BRAFV600 melanomas (cohort 2)
0.0
2.0
3a 3b 8a 12
Fo
ld c
han
ge
(Rel
apse
/Pre
-Rx)
Notch1
0.0
2.0
4.0
6.0
8.0
3a 3b 8a 12
Fo
ld c
han
ge
(Rel
apse
/Pre
-Rx)
NGFR
0.0
0.5
1.0
1.5
3a 3b 8a 12
Fo
ld c
han
ge
(Rel
apse
/Pre
-Rx)
TRP-2
MAPK pathway (3)
(BRAF, MEK2)
PTEN mutation
(1)
0.0
2.0
4.0
6.0
8.0
3 24
Notch1 **
**
0.0
0.5
1.0
1.5
3 24
TYR
**
**
0.0
2.0
4.0
6.0
8.0
3 24
NGFR ** **
Pretreatment
Relapse Pretreatment
Relapse
shGFP shNotch1 #1 shNotch1 #2
0.0
2.0
4.0
Parental Resistant
Notch1 **
0 500
1000 1500 2000 2500
Parental Resistant
NGFR **
0.0
0.5
1.0
1.5
2.0
Parental Resistant
HES1 *
0
5
10
15
20
Parental Resistant
HES2 *
Unknown mechanisms (12)
MAPK pathway (15) (NRAS, BRAF, MEK1/2)
Notch1 activation (4)
Unknown mechanisms (3)
Notch1 activation (2)
G Pretreatment Relapse
Total = 18
Notch1-dependent resistanceNotch1-independent resistance
MAPKi-resistant cell lines
11
7
5
0.0
0.5
1.0
1.5
2.0
GI7
5 (µ
M)
* **** **** **
0
2
4
6
8
10
GI7
5 (µ
M)
** ***
**
*****
**
Fol
d ch
ange
(tr
ansc
ript)
Hes1
Hes2 Hes1
Notch1 NGFR
Fol
d ch
ange
(tr
ansc
ript)
Hey2 NGFR
TYR TRP-2 N-cadherin
MAPKi-resistant cell lines
Fol
d ch
ange
(re
laps
e/pr
etre
atm
ent)
Fol
d ch
ange
(re
laps
e/pr
etre
atm
ent)
Notch1
TRP-2
NGFR
Notch1
TYR
NGFR
Pretreatment Relapse
Patient 24
GI 7
5 (µ
M)
A37
5
(par
enta
l)
A37
5
PLX
-R (
F)
A37
5
AZ
D-R
(S
)
A37
5
(par
enta
l)
A37
5
PLX
-R (
F)
A37
5
AZ
D-R
(S
)
Cell line:
Drug: PLX4720 AZD6244
GI 7
5 (µ
M)
0.1
1
10
Luciferase
Notch1 ICD
VX-11E
A375 Colo679 UACC62
GI 5
0 (µ
M)
Cell line:
Drug:
A375 A375 Colo679 Colo679 UACC62 UACC62
AZD6244 PLX4720
*** ***
*** ***
**
** **
** **
Fig. 4. Notch1 pathway activation in BRAF-mutant, MAPK pathway inhibitor–resistantmelanoma. (A) GI50 values for the indicatedmelanoma cell lines expressing Notch1 ICD(N1 ICD) or luciferase and treated with theindicated drugs. Data are means ± SD fromthree experiments. (B) qRT-PCR analysis ofNotch1 target genes and differentiation markersin UACC-62 cells expressing Notch1 ICD (red)or luciferase (gray). Data are means ± SD fromthree experiments. (C) Notch1-dependent re-
www.SCIENCESIGNALING.org 23
sistance in cell lines with evolved resistance to MAPK inhibitors (left) as identified by resensitization in the presence of Notch1 knockdown(examples, right). Data are means ± SD from three experiments. (D) qRT-PCR analysis of Notch1, Notch1 target genes, and NGFR in Colo679 cellswith evolved Notch1-dependent resistance to PLX4720. Data are means ± SD from three experiments. (E) Putative mechanisms of resistance in acohort of 29 relapsed tumors from BRAFV600 melanoma patients. “Other” indicates AKT1 mutation (one patient) or IGF-1 overexpression (one pa-tient). Inset shows fold changes in expression levels of the indicated genes on relapse in the Notch1 activation group. (F) Putative mechanisms ofresistance in a second cohort of seven relapsed tumors from BRAFV600 melanoma patients. Inset shows fold changes in expression levels of theindicated genes on relapse in the Notch1 activation group. Data are means ± SD from three experiments. (G) Analysis of Notch1 nuclear localizationby immunohistochemistry in matched pretreatment and relapse samples from patient 24. Examples of nuclear Notch1 are indicated by arrowheads.*P < 0.1; **P < 0.05; ***P < 0.01.
December 2014 Vol 7 Issue 357 ra121 6
R E S E A R C H A R T I C L E
on Decem
ber 23, 2014http://stke.sciencem
ag.org/D
ownloaded from
Resistance pathway dependency
mTOR+Notch1+ER (4/8) Lox IMVI A2058
WM1716 WM793
mTOR+Notch1 (3/8) Hs294T WM1930 WM1745
mTOR only (1/8) RPMI-7951
0
2
4
6
8
10
Veh
icle
ER
i
mT
OR
i
Not
ch1i
ER
i + m
TO
Ri
ER
i + N
otch
1i
mT
OR
i + N
otch
1i
ER
i + m
TO
Ri +
N
otch
1i
VX
-11E
GI 5
0 (µ
M) A2058
C
D
0
5
10
15
20 U
AC
C-6
2 A
375
Col
o679
S
kMel
28
UA
CC
-62
A37
5 C
olo6
79
SkM
el28
A20
58
Lox
IMV
I W
M19
30
Hs2
94T
W
M79
3 R
PM
I-79
51
WM
1716
W
M17
45
A20
58
Lox
IMV
I W
M19
30
Hs2
94T
W
M79
3 R
PM
I-79
51
WM
1716
W
M17
45
Resistant lines + inhibitors
VX
-11E
GI 5
0 (µ
M)
Cell line:
Sensitive lines + inhibitors
Resistant lines
Sensitive lines
A B
Med
ian-
cent
ered
ex
pres
sion
N-cadherin TYR
HES7
3
21
0
–1
–2
–3
Sensitive Resistant
Med
ian-
cent
ered
ex
pres
sion
10
5
0
–5
–10
Sensitive Resistant
Med
ian-
cent
ered
ex
pres
sion
TRP-2
Sensitive Resistant
5
0
–5
–10
Med
ian-
cent
ered
ex
pres
sion
HES5
Sensitive Resistant
2
1
0
–1
Sensitive Resistant
Med
ian-
cent
ered
ex
pres
sion
2
1
0
–3
–2
–1
** *
** **
*
0
5
10
15
20
WM
1716
WM
793
WM
1745
WM
1930
Lox
IMV
I
A20
58
RP
MI-
7951
Hs2
94T
shGFP
shNotch1 #1
shNotch1 #2
Nonsensitized
VX
-11E
GI 5
0 (µ
M)
Cell line:
Sensitized
**
**
**
**
**
** **
**
*
**
**
**
RPMI-7951
0 1 2 3 4 5 6 7
Veh
icle
ER
i
mT
OR
i
Not
ch1i
ER
i + m
TO
Ri
ER
i + N
otch
1i
mT
OR
i + N
otch
1i
ER
i + m
TO
Ri +
N
otch
1i
VX
-11E
GI 5
0 (µ
M)
VX
-11E
GI 5
0 (µ
M)
RPMI-7951
0
1
2
3
4
5
6
Veh
icle
ER
i
mT
OR
i
Not
ch1i
ER
i + m
TO
Ri
ER
i + N
otch
1i
mT
OR
i + N
otch
1i
ER
i + m
TO
Ri +
N
otch
1i
Hs294T
Fig. 5. Simultaneous co-inhibition of multiple drug resistance pathwaysconverts intrinsically drug-resistant melanoma cell lines and short-termcultures to a drug-sensitive state. (A) qRT-PCR analysis of Notch1 targetgenes and differentiation markers in MAPK inhibitor–sensitive or intrinsi-cally resistant melanoma cell lines. Data presented are means ± SD fromthree experiments. (B) Effect of shRNA-mediated knockdown of Notch1on the sensitivity of intrinsically resistant melanoma cell lines to VX-11E.Data are means ± SD from three experiments. (C) VX-11E GI50 values
www.SC
measured in BRAF-mutant melanoma cell lines and short-term culturestreated with a cocktail of resistance pathway inhibitors (blue, black) or ve-hicle (gray, red). Resistance pathway inhibitors are shNotch1, BEZ-235(200 nM), and fulvestrant (1 mM). Data are means ± SD from three exper-iments. (D) Resistance pathway dependencies in drug-resistant cell linesinferred from drug sensitizer assays using all combinations of resistancepathway inhibitors. Data for all GI50 curves are means ± SD from threeexperiments. *P < 0.05; **P < 0.01.
IENCESIGNALING.org 23 December 2014 Vol 7 Issue 357 ra121 7

R E S E A R C H A R T I C L E
on Decem
ber 23, 2014http://stke.sciencem
ag.org/D
ownloaded from
factors are likely to contribute to this state as well. Given that our screeningresults suggested that the PI3K-mTOR, NF-kB, and ER pathways are alsocapable of driving resistance toMAPKpathway inhibitors,we hypothesizedthat full sensitization of intrinsically resistant cellsmay require simultaneousinhibition of more than one of these pathways. To test this hypothesis, wecotreated these cultures with the combination of an ER inhibitor (fulves-trant), a dual PI3K-mTOR inhibitor (BEZ-235), and shRNAagainst Notch1(IkB kinase a/b inhibitors for the NF-kB pathway had no effect on MAPKinhibitor sensitivity and were thus excluded). This combination convertedall eight drug-resistant cultures to a drug-sensitive state,with submicromolarVX-11E GI50 values comparable to those typically found in drug-sensitivecell lines (Fig. 5C). Although this drug cocktail exerted backgroundeffects on cell growth rate (~50%), its effects onVX-11E sensitivity werenevertheless specific and not due to a general “sick + sick” phenomenonbecause (i) background toxicity was removed before GI50 analysis (referto Materials and Methods), (ii) numerous control drugs and drug com-binations with similar overall toxicity had no effect on VX-11E sensitiv-ity (fig. S20), and (iii) the cocktail had only modest effects on VX-11Esensitivity in MAPK inhibitor–sensitive cell lines (Fig. 5C). Finally, byapplying all possible combinations of resistance pathway inhibitors to eachresistant culture, we were able to deconvolute the particular set of pathwaysmaintaining the resistant state in that culture. Four of eight cultures showedevidence of resistance mediated by the combination of all three resistancepathways (ER,Notch1, andPI3K-mTOR); three of eight cultures relied onlyon the Notch1 and PI3K-mTOR pathways; and one culture (RPMI-7951)relied exclusively on the PI3K-mTOR pathway for the maintenance of itsresistant state (Fig. 5D).
DISCUSSION
By enabling the direct screening of signaling pathways instead of individualgenes (8, 39), the methods described here may significantly accelerate thesystematic mapping of pathways to the oncogenic phenotypes, such as drugresistance, which they control. In the future, these methods may be furtherimproved by using expression vectors that normalize transgene expressionto physiological levels and mutants that reflect naturally occurring mecha-nisms of pathway activation in human cancers, steps that may minimize thepotential for nonphysiological signaling effects. The lack of these features inthe current screening library may explain, for example, why HRASG12V
scored more frequently in screens thanKRASG12Vor whymyr-AKT1 scoredmore frequently than myr-PIK3CA. Further, although the pathway-activatinglibrary described here encompasses many of the pathways most frequentlyimplicated in cancer physiology, it will be improved by the addition of con-structs representing additional signaling nodes and biological pathways, aprocess that is currently under way.
In the area of drug resistance, our findings begin to reconcile two dis-parate notions of how this phenomenonmay emerge: one based on reactiva-tion of drug-inhibited signaling pathways and the other based on thereversion of cancer cells to a dedifferentiated state (1, 40). Our findings sug-gest that both may occur simultaneously, and that the latter, mediated by theNotch1 pathway, may be a particularly effective strategy for generatingbroad-spectrum resistance to drugs with varying mechanisms of action.Whereas the induction of an EMT-like process has been described exten-sively as a mode of therapeutic resistance in epithelial cancers, these resultsprovide evidence of an analogous, Notch1-driven process in melanoma thatmay also contribute to therapeutic resistance. The precise molecular mech-anisms by which Notch1 is activated in tumors resistant to tamoxifen andMAPK inhibitors, the identification of biomarkers of Notch1-driven re-sistance, and clarification of the cellular mechanisms bywhich this pathwaydrives therapeutic resistance will each be important areas for future study.
www.SC
Finally, this work describes a straightforward approach for designing com-binations of drugs that block or reverse resistance by simultaneously in-hibiting multiple resistance pathways. Although such combinations mayfrequently require the use of three or more drugs in parallel, an abundanceof clinical evidence suggests that multidrug therapies can sometimes besafely tolerated by patients (41–44).
MATERIALS AND METHODS
Cell lines and drugsA375, Colo679, UACC-62, Malme3M, WM793, WM1716, WM1745,WM1930, and SkBr3 cells were grown in RPMIwith 10% fetal bovine ser-um (FBS) and 1% penicillin/streptomycin. BT20, T47D, SkMel28, LoxIMVI, Hs294T, A2058, MCF-7, MDA-MB-453, Kato III, NCI-H1703,293T, and RPMI-7951 cells were grown in Dulbecco’s modified Eagle’smedium (DMEM) with 10% FBS and 1% penicillin/streptomycin. Ly.1and DHL4 cells were grown in Iscove’s modified DMEM supplementedwith 10% FBS, Hepes, glutamine, b-mercaptoethanol, and 1% penicillin/streptomycin. Malme3M, WM793, WM1716, WM1745, WM1930,SkMel28, Lox IMVI, Hs294T, A2058, and RPMI-7951 cells were obtainedfromL.Garraway (HarvardUniversity,Dana-FarberCancer Institute). All oth-er cell lineswerepurchased from theAmericanTypeCultureCollection.Drugswere purchased from Selleck Chemicals, ChemieTek, and Sigma-Aldrich.Torin1 was obtained from N. S. Gray (Harvard University, Dana-FarberCancer Institute). DSL peptide was obtained from Anaspec.
Library construction and validationcDNA templates obtained from the sources listed in table S1were barcodedand transferred into the Gateway system via polymerase chain reaction (PCR)and recombinational cloning (8).Donor vector pDONR223 anddestination/expressionvector pcw107were donated by J. Doench (Broad Institute) (fig.S21). A C-terminal V5 epitope tag was added to pcw107 to tag constructslacking functionally validated N-terminal tags. Barcoded attB1 primersweregenerated containing a 4-nucleotide (nt) barcode assigned to individualconstructs followed by a 14-nt common linker sequence containing aKozaksequence and ~21 nt of the open reading frame (ORF) of interest. AttB2primers were generated to contain the final 21 or 24 nt of the ORF depend-ing onwhether a C-terminalV5 tagwas desired. In general, constructs alreadycontaining an N-terminal epitope tag were cloned without a C-terminal V5tag. Constructs not containing an N-terminal epitope tag were cloned bothwith and without the C-terminal V5 tag, and both constructs were function-ally validated. For details, refer to tables S1, S6, and S7. The PCR fragmentswere gel-purified and used in the BP recombination reaction using BP clonase(Invitrogen) with pDONR223 to generate entry clones. Individual clones weresequence-verified using primers M13-F and M13-R to ensure proper inte-gration of the N-terminal barcode linker sequence and the presence or ab-sence of the C-terminal stop codon (table S7). Two of the constructs, SMOand LATS2, were further manipulated using Agilent’s QuikChange II XLSite-Directed Mutagenesis Kit and appropriate primers to generate thedesired activatingmutations (tables S1 and S7). Entry cloneswere then usedin the LR recombination reaction using LR clonase (Invitrogen) withpcw107-V5 to obtain lentiviral expression clones. All expression clones werefully sequenced using primers PGK-FandWPRE-R and gene-specific internalsequencing primers as needed to verify the presence of the N-terminal bar-code, the linker sequence, the desired activatingmutations, and the presenceor absence of the V5 tag (table S7).
Functional validation of library clones was performed as follows. First,expression clones were used to produce lentivirus particles by transfecting293T cells, using a three-plasmid system (expression clone +VSV-G+∂VPR)
IENCESIGNALING.org 23 December 2014 Vol 7 Issue 357 ra121 8

R E S E A R C H A R T I C L E
on Decem
ber 23, 2014http://stke.sciencem
ag.org/D
ownloaded from
as previously described (8). Each virus was titered via infection withlimiting dilution in UACC-62 cells (8). ORF transgene expression wasmeasured by Western blot by probing for the epitope tag present in eachconstruct in 293T cell lysates stably infected with each lentivirus. Pathwayactivation was assessed using the assays summarized in table S1 in 293Tcells after infection and puromycin selection. All infections were performedby adding a 1:10 to 1:20 dilution of lentivirus to 293T cells in six-well platesin the presence of polybrene (7.5 mg/ml). After virus addition, plates werecentrifuged at 1200g for 1 hour at 37°C. Twenty-four hours after infection,puromycin (2 mg/ml) was added for selection and cells were incubated for48 hours.
Primary screensAll pathway-activating constructs and controls (table S1) were produced inlentiviral form, individually titered, and pooled together in approximatelyequal representation. The mixed pool was aliquoted and stored at −80°C.Screens were performed by seeding each cell line at 500,000 cells per wellin a single well of a six-well plate. Twenty-four hours later, the cells wereinfected with the mixed virus pool at a multiplicity of infection of 0.3 toensure that most infected cells contained only a single viral integration.The lentiviral pool was added directly to cells in the presence of poly-brene (7.5 mg/ml), and then plates were centrifuged at 1200g for 1 hour at37°C. Twenty-four hours after infection, medium was replaced with puro-mycin (2 mg/ml) for selection. After 48 hours of selection, cells were splitand divided into (typically) seven equal portions, one ofwhichwas frozen at−80°C (t = 0 infected cell pool), and the other six were added to thewells ofa six-well plate. Twenty-four hours later, two to three of the wells receiveddrugs at the indicated doses (typically in the range of the drug’sGI20 toGI80)and the remaining three wells received vehicle [dimethyl sulfoxide (DMSO)]only. Drugs and vehicle were refreshed every 3 days for 2 to 3 weeks, splitting(1:10) as necessary. After this culture period, all drug- and vehicle-treatedsampleswere trypsinized andwashed, and genomicDNAwas isolated usingthe Qiagen DNeasy Blood and Tissue Kit.
GenomicDNA sampleswere prepared for Illumina Sequencing by PCRamplification of individual construct barcodes, using a common P5 Illumi-na adapter primer (PGK-Illumina-F) and a unique P7 Illumina barcodedadapter primer (P7-Illumina–RIP-Index-X where X is a unique numericalidentifier for each indexed primer) (table S7). Each drug- or vehicle-treatedscreen sample was associated with a unique P7-Illumina–RIP-Index-Xprimer to facilitate pooling of samples and sequencing in a single Illuminalane. The P5 primer targets the PGK promoter upstream of the constructbarcode, and the P7-reverse index primers target the ATG of the ORFdownstream of the construct barcode. P7-reverse index primers contain a6-nt index barcode and the reverse complement of theATGplus 14-nt linkersequence just downstream of, but not including, the 4-nt construct barcode.When paired, these Illumina adapter primers generate a ~250-nt DNA frag-ment, a fraction of which was visualized on a 2% agarose gel. Band inten-sities,which are proportional to the amount of barcodeDNAamplified fromeach sample, were quantified, and a normalized sample pool containing allsamples to be sequencedwas submitted for Illumina sequencing. A custom-designed Illumina Sequencing Primer (ISP) that targets the region of ge-nomic DNA just upstream of the 4-nt barcode was used, yielding 27-ntIllumina HiSeq reads with the following structure: 4-nt construct barcode +17-nt linker sequence/ATG + 6-nt index barcode (table S7). Each samplewas prepared with two different P7-Illumina–RIP-Index barcodes, generat-ing technical replicates. Both technical replicates (that is, a single samplewhose barcodes were PCR-amplified using two unique sets of primers)and biological replicates (that is, two replicate vehicle-treated screen sam-ples) yielded comparable data (R2 > 0.9), and replicateswere averaged in thedata sets reported here.
www.SC
Analysis of Illumina sequencing data proceeded as follows. First, thenumber reads associated with each unique sequence (4-nt construct barcode+ 6-nt index barcode) were counted, and the fractional representation of eachconstruct barcode in each screen was determined by normalizing the numberof reads associated with that construct barcode to the total number of reads inthat sample (that is, all samples containing the same index barcode). Next,fractional representation of each construct in technical replicates was aver-aged, and the fractional representation of each construct in three biologicalreplicate, vehicle-treated samples was averaged. Finally, the fractional repre-sentation of each construct in drug-treated samples was normalized to thesame quantity in vehicle-treated samples of the same cell line. Hitswere iden-tified as constructs selectively enriched in drug-treated samples relative tovehicle-treated samples. For a signaling pathway to be considered a resistancepathway for a given drug, we required that at least one activating construct forthat pathway must confer >50% enrichment above controls and that it mustscore in two or more drug concentrations in primary screens.
Secondary GI50 assaysORF- or shRNA-expressing lentiviruses were produced as previously de-scribed (13) and used to infect cells at a 1:10 to 1:20 dilution in six-wellplates in the presence of polybrene (7.5 mg/ml). After virus addition, plateswere centrifuged at 1200g for 1 hour at 37°C. Twenty-four hours after in-fection, puromycin (2 mg/ml) was added for selection, and cells were incu-bated for 48 hours. Cells were then trypsinized, counted, and seeded in96-well plates at 5000 cells per well. Twenty-four hours later, DMSO orconcentrated serial dilutions of the indicated drugs (in DMSO) were addedto cells (1:1000) to yield final drug concentrations of 100, 10, 1, 0.1, 0.01,0.001, 0.0001, and 0.00001 mM. The CellTiter-Glo luminescent viabilityassay (Promega) was used to measure cell viability 4 days after drug addi-tion. Viability was calculated as the percentage of control (untreated cells)after background subtraction with a minimum of three replicates for eachcell line/ORF (or shRNA)/drug/concentration. GI50 valueswere determinedas the drug dose corresponding to half-maximal growth inhibition (13).GI50 values for unmodified parental cells were determined using the pro-tocol above by seeding cells directly into 96-well plates without the initialinfection step. Similarly, GI50 values for parental cells treated with DSLpeptide were determined using the protocol above by seeding cells di-rectly into 96-well plates in medium containing DSL peptide at the indi-cated concentrations.
Quantitative real-time PCRRNAwas extracted using QIAshredder Homogenizers and RNEasy MiniKits according to the manufacturer’s specifications (Qiagen). RNA purityas measured by absorbance at 260 nm (A260)/A280 was confirmed uponquantification. cDNAswere synthesized using iScript cDNASynthesis Kitswith at least 1 mg of RNA template as directed by the protocol provided bythe manufacturer (Bio-Rad).
qRT-PCRs were carried out using iQ SYBR Green Supermix and aCFX384 Touch Real-Time PCR Detection System according to the manu-facturer’s specifications (Bio-Rad). PCR primers were purchased fromIntegrated DNATechnologies and processed to 10 mM working stocks foruse in quantitative PCR reactions (table S7). Average cycle threshold (Ct)values were calculated for each gene, and the maximum Ct value was set at40 cycles. Ct values were normalized to the reference genes GAPDH andb-actin, and relative gene expression was determined using the DDCt
method.
Xenograft tumor analysesAll procedures were approved by the Duke University Institute forAnimal Care and Use Committee. Tamoxifen-stimulated TamR tumors
IENCESIGNALING.org 23 December 2014 Vol 7 Issue 357 ra121 9

R E S E A R C H A R T I C L E
on Decem
ber 23, 2014http://stke.sciencem
ag.org/D
ownloaded from
were initiated orthotopically by serial transfer into female NU/NU mice(~6 weeks old, Duke breeding colony). Briefly, ovariectomized recipientmice received no treatment or tamoxifen treatment via a timed releasepellet (5 mg of tamoxifen/60 days, Innovative Research of America) im-planted subcutaneously. Two days later, TamR tumors (~0.8-cm3 volume)were sterilely excised from euthanized tamoxifen-treated donor mice, dicedinto ~2-mm3 sections, and implanted into the axial mammary gland of re-cipient mice under anesthesia (10-gauge trocar). Tumor growth wasmeasured three times weekly by caliper [tumor volume = (A2 × B)/2, whereA is the longer axis]. When tumor volume reached ~0.15 cm3 (~20 days),mice (n = 9 to 13) were randomized to vehicle or RO4929097 treatment(10 mg/kg oral gavage in 0.1 ml of 1% hydroxypropyl cellulose/0.2%Tween 80/2.5% DMSO). Tumor growth was monitored over 4 weeksof daily treatment.
Gene expression analysisSurvival analysis of Notch1 and Notch1 gene sets in breast cancer ispresented in Fig. 3, E and F. A breast cancer metaset was compiled from26 publicly available expression data sets comprising 4886 patients.Breast cancer data from GPL570 and GPL96 platforms were down-loaded from GEO (Gene Expression Omnibus), normalized with fRMA(frozen robust multiarray), and batch-corrected using the COMBATalgorithm within R. Associated clinical features and patient responsedata were combined, and tumor subtypes were called using the PAM50algorithm. Gene expression was split into tertiles, and only the low (firsttertile) and high (third tertile) categories were plotted. Survival curveswere generated using the survival package in R, and all P values werecalculated using the log-rank method. Gene sets representing the Notch1pathway and Notch1 target genes were downloaded from the GSEA(Gene Set Enrichment Analysis) Web site, and the R package genefuwas used to assign signature scores of each gene set to patients by map-ping Affymetrix probes to Entrez gene IDs. Signature scores were thencategorized into low and high groups for each treatment condition andplotted similarly as above. For GSEA, a ranked list of pairwise gene cor-relation of Notch1 and all other probe sets on the HGU133A platformfrom luminal B tumor samples were submitted to the GSEAPrerankedtool for pathway enrichment. Metaplot (fig. S11A) was generated usingluminal B patients from the assembled breast cancer metaset in R withthe rmeta and survcomp packages. Correlation matrix (fig. S11B) wasclustered using Ward’s method and visualized using the corrplot pack-age in R.
Patient samplesDetails on patient sample acquisition, quality control, total RNA extraction,cDNA preparation, and mRNA sequencing are described elsewhere (31).Immunohistochemistry on patient tumor sampleswas performed as follows.Sections (5 mm) were deparaffinized in xylene, followed by serial hydrationin 100, 95, 70, and 50% ethanol, then distilled water. Sections were thenplaced in sodium citrate buffer (10 mM, pH 6.0) and boiled in a pressurechamber at 95°C for 30 min, 90°C for 10 s, and then cooled to room tem-perature. Immunohistochemistry was performed using an antibody to acti-vated Notch1 (Abcam, ab8925; 1:100) at 4°C overnight, and the tissuesections were incubated with secondary antibody (Abcam, ab64256) atroom temperature for 30 min, then washed with TBST (tris-buffered salinewith Tween 20) for 5min three times. Immunoreactivity was detected usingthe Dako liquid DAB+ substrate chromogen system.
Small-molecule sensitization assaysSensitization assays using small-molecule signaling pathway inhibitorswere performed to assess the effect of resistance pathway inhibition on
www.SCI
the sensitivities of drug-resistant melanoma cell lines to VX-11E. VX-11EGI50 values for parental cell lines were determined as above with DMSOadded to medium at a 1:1000 dilution as a vehicle-only control. GI50 valuesfor parental cell lines in the presence of inhibitorswere obtained using amod-ified version of theGI50 protocol above,where pathway inhibitorswere addedto allwells containingVX-11E aswell as one set of triplicatewells containingmedium only (pathway inhibitors were in DMSO at a 1:1000 dilution in me-dium). As a reference, a second set of medium-only wells contained nopathway inhibitors (medium and DMSO only). By comparing the viabilityof cells in medium-only wells to the viability of cells in wells containingmedium plus pathway inhibitors (no VX-11E), the toxicity of the inhibitorsalone was ascertained. GI50 values were then determined as the VX-11Edose corresponding to half-maximal growth inhibition relative to the viabil-ity in inhibitor-only wells.
ImmunoblottingImmunoblottingwas performed as previously described (13), and blotswereprobed with primary antibodies (1:1000 dilution) recognizing N-cadherin(BD Transduction), vimentin (Cell Signaling), Slug (Cell Signaling), phos-phorylated AKT (Thr308, Cell Signaling), AKT (Cell Signaling), tyrosinase(Santa Cruz Biotechnology), TRP-2 (Santa Cruz Biotechnology), b-actin(Cell Signaling), Notch1 (Cell Signaling), phosphorylated ERK1/2 (Thr202/Tyr204, Cell Signaling), phosphorylated MEK1/2 (Ser217/Ser221, CellSignaling), ERK1/2 (Cell Signaling), and Na/K adenosine triphosphatase(Cell Signaling).
Notch shRNAsTRC shRNA clones for Notch1were obtained from Sigma-Aldrich as glyc-erol stocks. They were prepared in lentiviral form and used to infect targetcells via the procedures described above for ORF clones. shNotch1 #1 isTRCN0000350330 with target sequence CCGGGACATCACGGATCA-TAT, and shNotch1 #2 is TRCN0000003360 with target sequenceCGCTGCCTGGACAAGATCAAT.
In vitro adaptation of MAPK inhibitor–resistant cellsCell lines resistant to PLX4720, AZD6244, or VX-11E were generated bytwomethods as previously described (45). Briefly, either cells were culturedin gradually increasing concentrations of the inhibitor starting with a con-centration of 1 nM [slow method, denoted with (S)] or 3 × 106 cells weretreated with 3 mM inhibitor and resistant clones were cultured [fast method,denoted with (F)]. Cell lines were deemed resistant when they could beconsistently cultured in medium containing 3 mM inhibitor. Parental controlcell lines were cultured concurrently with the resistant cell lines in growthmedium containing DMSO.
Clonogenic growth assayTo measure the effect of inhibitors and shRNA-mediated knockdown oncell growth, cells were seeded at 10,000 cells per well in six-well tissueculture plates in complete medium. After 24 hours, cells were infected with1 × 105 infectious units (IFUs) per well of viral medium containing shRNAvectors targeted to either GFP or Notch1 and polybrene (8 mg/ml). Twelvehours after infection, cells were treated with growth medium containing in-hibitors as indicated and puromycin (2 mg/ml). Freshmedium and inhibitorswere added every 7 days, and the assays were cultured for 10 to 20 days.Plates were then rinsed with phosphate-buffered saline (PBS) and fixed andstained with 0.5% (w/v) crystal violet in 6.0% (v/v) glutaraldehyde solution(Thermo Fisher Scientific) for 30 min at room temperature. Plates wererinsed in distilled water and scanned. The ImageJ software program and theColonyArea plugin (46) were used to quantify colony area as a percentageof the well covered.
ENCESIGNALING.org 23 December 2014 Vol 7 Issue 357 ra121 10

R E S E A R C H A R T I C L E
on Decem
ber 23, 2014http://stke.sciencem
ag.org/D
ownloaded from
Sphere formation assayThe ability of pathway-activated cells to form spheres on low-attachmentplates was analyzed using previously described assays (47, 48). A single-cell suspension was created by passing cell suspensions through a 70-mmstrainer (GreinerBioOne). Two thousand singlemelanoma cellswere platedin triplicate in 2ml ofRPMI supplementedwithN2 (Invitrogen) in ultralow-attachment six-well plates (Corning). Breast cancer cell lines were platedsimilarly in DMEM supplemented with B27 (Invitrogen), hydrocortisone,insulin, recombinant EGF, and gentamicin (Lonza). After 5 days, sphereswere counted. The number of spheres was divided by the number of cellsplated to derive the sphere formation efficiency percentage.
Wound healing assayTo quantify the ability of pathway activators to affect the ability of cellsto migrate, 5 × 105 cells were seeded in six-well plates in triplicate. After24 hours, the medium was changed to growth medium containing 1%FBS. The cells were cultured for an additional 24 hours and then evenlywounded. Thewoundwas rinsed twicewith PBS, and then freshmediumcontaining 1% FBS was added. The wound was photographed at 0 hourand every 12 hours subsequently.
Annexin V apoptosis assayTo quantify the induction of apoptosis in inhibitor-treated cells, cells wereplated in triplicate at 300,000 cells per well in six-well plates. Twenty-fourhours later, cells were treated with the indicated dose of drug or diluent(DMSO). After a 72-hour incubation, cells were washed twice with PBSand resuspended in Annexin V binding buffer composed of 10 mMHepes,140 mMNaCl, and 2.5 mM CaCl2 (BD Biosciences). Surface exposure ofphosphatidylserine was measured using allophycocyanin-conjugated An-nexin V (BD Biosciences). 7-Amino-actinomycin D (BD Biosciences)was used to quantify viability. All treatments were evaluated at 20,000counts per sample using BD FACSVantage SE. Gatings were defined usingappropriate untreated/unstained cells.
StatisticsResults are expressed asmeans ± SD. For comparisons between twogroups,P values were calculated with unpaired, two-tailed Student’s t tests.
SUPPLEMENTARY MATERIALSwww.sciencesignaling.org/cgi/content/full/7/357/ra121/DC1Fig. S1. Schematic of the screen.Fig. S2. Stable transfection of constructs.Fig. S3. Results of a screen in UACC-62 melanoma cells (BRAFV600E) for pathwaysconferring resistance to MEK1/2 inhibitor AZD6244.Fig. S4. Meta-analysis of screening results across 13 targeted therapies.Fig. S5. Resistance to etoposide mediated by stable expression of non-cleavable cas-pases in MCF-7 breast cancer cells.Fig. S6. Immunoblotting of BRAFV600 melanoma cells expressing pathway-activatingconstructs and treated with MAPK pathway inhibitors.Fig. S7. Resistance to targeted and cytotoxic drugs in breast cancer cells treated with asoluble Notch agonist.Fig. S8. Differentiation markers and signaling in MCF-7 breast cancer cells expressingNotch1.Fig. S9. Characterization of dedifferentiation-associated phenotypes in breast cancer cellswith activated Notch1.Fig. S10. Analysis of Notch1 pathway members and EMT markers in a mouse model oftamoxifen-resistant breast cancer (TamR).Fig. S11. Analysis of Notch1 pathway members and EMT markers in human breast cancerpatients.Fig. S12. Estrogen receptor–driven resistance to MAPK inhibitors.Fig. S13. Notch1 target gene expression in melanoma cells with activated Notch1.Fig. S14. Western blot analysis of differentiation markers and signaling in BRAF-mutantmelanoma cells expressing Notch1.Fig. S15. Characterization of dedifferentiation-associated phenotypes in melanoma cellswith activated Notch1.
www.SCI
Fig. S16. Notch1 hairpin validation.Fig. S17. Characterization of evolved MAPK inhibitor–resistant, BRAF-mutant melanomacell lines.Fig. S18. Analysis of patient tumors in cohort 1.Fig. S19. Analysis of patient tumors in cohort 2.Fig. S20. Resistance pathway inhibitors sensitize intrinsically resistant melanoma cells toVX-11E independently of inhibitor effects on cell viability.Fig. S21. Vector maps for vectors used in this study.Table S1. List of pathway-activating constructs and controls used in this study.Table S2. List of all drugs, drug concentrations, and cell lines screened.Table S3. Results of primary screens.Table S4. Characterization of cell lines and clonal derivates with evolved resistance toMAPK inhibitors.Table S5. Metastatic melanoma patient characteristics (cohorts 1 and 2).Table S6. List of attB1/B2 primers used to barcode and amplify constructs by PCR.Table S7. Sequences of additional primers used in this study.Data file S1. Nucleotide sequences.
REFERENCES AND NOTES1. M. S. Glickman, C. L. Sawyers, Converting cancer therapies into cures: Lessons from
infectious diseases. Cell 148, 1089–1098 (2012).2. M. Azam, R. R. Latek, G. Q. Daley, Mechanisms of autoinhibition and STI-571/
imatinib resistance revealed by mutagenesis of BCR-ABL. Cell 112, 831–843(2003).
3. C. M. Johannessen, J. S. Boehm, S. Y. Kim, S. R. Thomas, L. Wardwell, L. A. Johnson,C. M. Emery, N. Stransky, A. P. Cogdill, J. Barretina, G. Caponigro, H. Hieronymus,R. R. Murray, K. Salehi-Ashtiani, D. E. Hill, M. Vidal, J. J. Zhao, X. Yang, O. Alkan, S. Kim,J. L. Harris, C. J. Wilson, V. E. Myer, P. M. Finan, D. E. Root, T. M. Roberts, T. Golub,K. T. Flaherty, R. Dummer, B. L. Weber, W. R. Sellers, R. Schlegel, J. A. Wargo,W. C. Hahn, L. A. Garraway, COT drives resistance to RAF inhibition through MAP ki-nase pathway reactivation. Nature 468, 968–972 (2010).
4. J. A. Engelman, K. Zejnullahu, T. Mitsudomi, Y. Song, C. Hyland, J. O. Park, N. Lindeman,C. M. Gale, X. Zhao, J. Christensen, T. Kosaka, A. J. Holmes, A. M. Rogers, F. Cappuzzo,T. Mok, C. Lee, B. E. Johnson, L. C. Cantley, P. A. Jänne, MET amplification leadsto gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 316,1039–1043 (2007).
5. R. Straussman, T. Morikawa, K. Shee, M. Barzily-Rokni, Z. R. Qian, J. Du, A. Davis,M. M. Mongare, J. Gould, D. T. Frederick, Z. A. Cooper, P. B. Chapman, D. B. Solit,A. Ribas, R. S. Lo, K. T. Flaherty, S. Ogino, J. A. Wargo, T. R. Golub, Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature487, 500–504 (2012).
6. T. R. Wilson, J. Fridlyand, Y. Yan, E. Penuel, L. Burton, E. Chan, J. Peng, E. Lin,Y. Wang, J. Sosman, A. Ribas, J. Li, J. Moffat, D. P. Sutherlin, H. Koeppen, M. Merchant,R. Neve, J. Settleman, Widespread potential for growth-factor-driven resistance to anti-cancer kinase inhibitors. Nature 487, 505–509 (2012).
7. R. A. Weinberg, The Biology of Cancer (Garland Science, New York, 2007).8. X. Yang, J. S. Boehm, X. Yang, K. Salehi-Ashtiani, T. Hao, Y. Shen, R. Lubonja,
S. R. Thomas, O. Alkan, T. Bhimdi, T. M. Green, C. M. Johannessen, S. J. Silver,C. Nguyen, R. R. Murray, H. Hieronymus, D. Balcha, C. Fan, C. Lin, L. Ghamsari,M. Vidal, W. C. Hahn, D. E. Hill, D. E. Root, A public genome-scale lentiviral expressionlibrary of human ORFs. Nat. Methods 8, 659–661 (2011).
9. R. Possemato, K. M. Marks, Y. D. Shaul, M. E. Pacold, D. Kim, K. Birsoy, S. Sethumadhavan,H. K. Woo, H. G. Jang, A. K. Jha, W. W. Chen, F. G. Barrett, N. Stransky, Z. Y. Tsun,G. S. Cowley, J. Barretina, N. Y. Kalaany, P. P. Hsu, K. Ottina, A. M. Chan, B. Yuan,L. A. Garraway, D. E. Root, M. Mino-Kenudson, E. F. Brachtel, E. M. Driggers,D. M. Sabatini, Functional genomics reveal that the serine synthesis pathway is essentialin breast cancer. Nature 476, 346–350 (2011).
10. A. M. Alcalá, K. T. Flaherty, BRAF inhibitors for the treatment of metastatic mela-noma: Clinical trials and mechanisms of resistance. Clin. Cancer Res. 18, 33–39(2012).
11. P. I. Poulikakos, N. Rosen, Mutant BRAF melanomas—Dependence and resistance.Cancer Cell 19, 11–15 (2011).
12. V. C. Gray-Schopfer, M. Karasarides, R. Hayward, R. Marais, Tumor necrosis factor-ablocks apoptosis in melanoma cells when BRAF signaling is inhibited. Cancer Res.67, 122–129 (2007).
13. K. C. Wood, D. J. Konieczkowski, C. M. Johannessen, J. S. Boehm, P. Tamayo,O. B. Botvinnik, J. P. Mesirov, W. C. Hahn, D. E. Root, L. A. Garraway, D. M. Sabatini,MicroSCALE screening reveals genetic modifiers of therapeutic response in melanoma.Sci. Signal. 5, rs4 (2012).
14. C. W. Benjamin, R. R. Hiebsch, D. A. Jones, Caspase activation in MCF7 cells re-sponding to etoposide treatment. Mol. Pharmacol. 53, 446–450 (1998).
15. C. M. Emery, K. G. Vijayendran, M. C. Zipser, A. M. Sawyer, L. Niu, J. J. Kim,C. Hatton, R. Chopra, P. A. Oberholzer, M. B. Karpova, L. E. MacConaill, J. Zhang,N. S. Gray, W. R. Sellers, R. Dummer, L. A. Garraway, MEK1 mutations confer
ENCESIGNALING.org 23 December 2014 Vol 7 Issue 357 ra121 11

R E S E A R C H A R T I C L E
on Decem
ber 23, 2014http://stke.sciencem
ag.org/D
ownloaded from
resistance to MEK and B-RAF inhibition. Proc. Natl. Acad. Sci. U.S.A. 106, 20411–20416(2009).
16. K. T. Flaherty, J. R. Infante, A. Daud, R. Gonzalez, R. F. Kefford, J. Sosman, O. Hamid,L. Schuchter, J. Cebon, N. Ibrahim, R. Kudchadkar, H. A. Burris III, G. Falchook,A. Algazi, K. Lewis, G. V. Long, I. Puzanov, P. Lebowitz, A. Singh, S. Little, P. Sun,A. Allred, D. Ouellet, K. B. Kim, K. Patel, J. Weber, Combined BRAF and MEK inhibitionin melanoma with BRAF V600 mutations. N. Engl. J. Med. 367, 1694–1703 (2012).
17. A. Ring, M. Dowsett, Mechanisms of tamoxifen resistance. Endocr. Relat. Cancer 11,643–658 (2004).
18. J. T. Garrett, C. L. Arteaga, Resistance to HER2-directed antibodies and tyrosine ki-nase inhibitors: Mechanisms and clinical implications. Cancer Biol. Ther. 11, 793–800(2011).
19. B. J. Nickoloff, J. Z. Qin, V. Chaturvedi, M. F. Denning, B. Bonish, L. Miele, Jagged-1mediated activation of notch signaling induces complete maturation of human keratino-cytes through NF-kB and PPARg. Cell Death Differ. 9, 842–855 (2002).
20. R. Kalluri, R. A. Weinberg, The basics of epithelial-mesenchymal transition. J. Clin.Invest. 119, 1420–1428 (2009).
21. Z. Wang, Y. Li, A. Ahmad, A. S. Azmi, S. Banerjee, D. Kong, F. H. Sarkar, TargetingNotch signaling pathway to overcome drug resistance for cancer therapy. Biochim.Biophys. Acta 1806, 258–267 (2010).
22. M. K. Muellner, I. Z. Uras, B. V. Gapp, C. Kerzendorfer, M. Smida, H. Lechtermann,N. Craig-Mueller, J. Colinge, G. Duernberger, S. M. Nijman, A chemical-geneticscreen reveals a mechanism of resistance to PI3K inhibitors in cancer. Nat. Chem.Biol. 7, 787–793 (2011).
23. S. Cho, M. Lu, X. He, P. L. Ee, U. Bhat, E. Schneider, L. Miele, W. T. Beck, Notch1regulates the expression of the multidrug resistance gene ABCC1/MRP1 in culturedcancer cells. Proc. Natl. Acad. Sci. U.S.A. 108, 20778–20783 (2011).
24. M. Xie, L. Zhang, C. S. He, F. Xu, J. L. Liu, Z. H. Hu, L. P. Zhao, Y. Tian, Activation ofNotch-1 enhances epithelial-mesenchymal transition in gefitinib-acquired resistantlung cancer cells. J. Cell. Biochem. 113, 1501–1513 (2012).
25. P. Rizzo, H. Miao, G. D’Souza, C. Osipo, L. L. Song, J. Yun, H. Zhao, J. Mascarenhas,D. Wyatt, G. Antico, L. Hao, K. Yao, P. Rajan, C. Hicks, K. Siziopikou, S. Selvaggi,A. Bashir, D. Bhandari, A. Marchese, U. Lendahl, J. Z. Qin, D. A. Tonetti, K. Albain,B. J. Nickoloff, L. Miele, Cross-talk between Notch and the estrogen receptor in breastcancer suggests novel therapeutic approaches. Cancer Res. 68, 5226–5235 (2008).
26. C. E. Connor, J. D. Norris, G. Broadwater, T. M. Willson, M. M. Gottardis, M. W. Dewhirst,D. P. McDonnell, Circumventing tamoxifen resistance in breast cancers using antiestro-gens that induce unique conformational changes in the estrogen receptor. Cancer Res.61, 2917–2922 (2001).
27. L. Luistro, W. He, M. Smith, K. Packman, M. Vilenchik, D. Carvajal, J. Roberts, J. Cai,W. Berkofsky-Fessler, H. Hilton, M. Linn, A. Flohr, R. Jakob-Røtne, H. Jacobsen,K. Glenn, D. Heimbrook, J. F. Boylan, Preclinical profile of a potent g-secretaseinhibitor targeting notch signaling with in vivo efficacy and pharmacodynamic prop-erties. Cancer Res. 69, 7672–7680 (2009).
28. S. Loi, B. Haibe-Kains, C. Desmedt, F. Lallemand, A. M. Tutt, C. Gillet, P. Ellis, A. Harris,J. Bergh, J. A. Foekens, J. G. Klijn, D. Larsimont, M. Buyse, G. Bontempi, M. Delorenzi,M. J. Piccart, C. Sotiriou, Definition of clinically distinct molecular subtypes in estrogenreceptor–positive breast carcinomas through genomic grade. J. Clin. Oncol. 25, 1239–1246(2007).
29. S. Loi, B. Haibe-Kains, C. Desmedt, P. Wirapati, F. Lallemand, A. M. Tutt, C. Gillet,P. Ellis, K. Ryder, J. F. Reid, M. G. Daidone, M. A. Pierotti, E. M. Berns, M. P. Jansen,J. A. Foekens, M. Delorenzi, G. Bontempi, M. J. Piccart, C. Sotiriou, Predicting prog-nosis using molecular profiling in estrogen receptor-positive breast cancer treatedwith tamoxifen. BMC Genomics 9, 239 (2008).
30. S. Loi, B. Haibe-Kains, S. Majjaj, F. Lallemand, V. Durbecq, D. Larsimont,A. M. Gonzalez-Angulo, L. Pusztai, W. F. Symmans, A. Bardelli, P. Ellis, A. N. Tutt,C. E. Gillett, B. T. Hennessy, G. B. Mills, W. A. Phillips, M. J. Piccart, T. P. Speed,G. A. McArthur, C. Sotiriou, PIK3CA mutations associated with gene signature of lowmTORC1 signaling and better outcomes in estrogen receptor-positive breast cancer.Proc. Natl. Acad. Sci. U.S.A. 107, 10208–10213 (2010).
31. D. T. Frederick, A. Piris, A. P. Cogdill, Z. A. Cooper, C. Lezcano, C. R. Ferrone, D. Mitra,A. Boni, L. P. Newton, C. Liu, W. Peng, R. J. Sullivan, D. P. Lawrence, F. S. Hodi,W. W. Overwijk, G. Lizée, G. F. Murphy, P. Hwu, K. T. Flaherty, D. E. Fisher, J. A. Wargo,BRAF inhibition is associated with enhanced melanoma antigen expression and a morefavorable tumor microenvironment in patients with metastatic melanoma. Clin. CancerRes. 19, 1225–1231 (2013).
32. J. Landsberg, J. Kohlmeyer, M. Renn, T. Bald, M. Rogava, M. Cron, M. Fatho, V. Lennerz,T. Wölfel, M. Hölzel, T. Tüting, Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature 490, 412–416 (2012).
33. J. A. Engelman, T. Mukohara, K. Zejnullahu, E. Lifshits, A. M. Borrás, C. M. Gale,G. N. Naumov, B. Y. Yeap, E. Jarrell, J. Sun, S. Tracy, X. Zhao, J. V. Heymach,B. E. Johnson, L. C. Cantley, P. A. Jänne, Allelic dilution obscures detection of a biolog-ically significant resistance mutation in EGFR-amplified lung cancer. J. Clin. Invest. 116,2695–2706 (2006).
www.SCI
34. H. Rizos, A. M. Menzies, G. M. Pupo, M. S. Carlino, C. Fung, J. Hyman, L. E. Haydu,B. Mijatov, T. M. Becker, S. C. Boyd, J. Howle, R. Saw, J. F. Thompson, R. F. Kefford,R. A. Scolyer, G. V. Long, BRAF inhibitor resistance mechanisms in metastatic melanoma:Spectrum and clinical impact. Clin. Cancer Res. 20, 1965–1977 (2014).
35. L. A. Garraway, A Notch for noncoding RNA in melanoma. N. Engl. J. Med. 370,1950–1951 (2014).
36. M. Forloni, S. K. Dogra, Y. Dong, D. Conte Jr., J. Ou, L. J. Zhu, A. Deng, M. Mahalingam,M. R. Green, N. Wajapeyee, miR-146a promotes the initiation and progression of mela-noma by activating Notch signaling. Elife 3, e01460 (2014).
37. D. J. Konieczkowski, C. M. Johannessen, O. Abudayyeh, J. W. Kim, Z. A. Cooper, A. Piris,D. T. Frederick, M. Barzily-Rokni, R. Straussman, R. Haq, D. E. Fisher, J. P. Mesirov,W. C. Hahn, K. T. Flaherty, J. A. Wargo, P. Tamayo, L. A. Garraway, A melanoma cellstate distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov. 4,816–827 (2014).
38. W. M. Lin, A. C. Baker, R. Beroukhim, W. Winckler, W. Feng, J. M. Marmion, E. Laine,H. Greulich, H. Tseng, C. Gates, F. S. Hodi, G. Dranoff, W. R. Sellers, R. K. Thomas,M. Meyerson, T. R. Golub, R. Dummer, M. Herlyn, G. Getz, L. A. Garraway, Modelinggenomic diversity and tumor dependency in malignant melanoma. Cancer Res. 68,664–673 (2008).
39. D. E. Root, N. Hacohen, W. C. Hahn, E. S. Lander, D. M. Sabatini, Genome-scale loss-of-function screening with a lentiviral RNAi library. Nat. Methods 3, 715–719 (2006).
40. P. B. Gupta, T. T. Onder, G. Jiang, K. Tao, C. Kuperwasser, R. A. Weinberg, E. S. Lander,Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell138, 645–659 (2009).
41. K. Dunleavy, S. Pittaluga, L. S. Maeda, R. Advani, C. C. Chen, J. Hessler, S. M. Steinberg,C. Grant, G. Wright, G. Varma, L. M. Staudt, E. S. Jaffe, W. H. Wilson, Dose-adjustedEPOCH-rituximab therapy in primary mediastinal B-cell lymphoma. N. Engl. J. Med. 368,1408–1416 (2013).
42. E. H. Romond, E. A. Perez, J. Bryant, V. J. Suman, C. E. Geyer Jr., N. E. Davidson,E. Tan-Chiu, S. Martino, S. Paik, P. A. Kaufman, S. M. Swain, T. M. Pisansky,L. Fehrenbacher, L. A. Kutteh, V. G. Vogel, D. W. Visscher, G. Yothers, R. B. Jenkins,A. M. Brown, S. R. Dakhil, E. P. Mamounas, W. L. Lingle, P. M. Klein, J. N. Ingle,N. Wolmark, Trastuzumab plus adjuvant chemotherapy for operable HER2-positivebreast cancer. N. Engl. J. Med. 353, 1673–1684 (2005).
43. E. Van Cutsem, C. H. Köhne, I. Láng, G. Folprecht, M. P. Nowacki, S. Cascinu, I. Shchepotin,J. Maurel, D. Cunningham, S. Tejpar, M. Schlichting, A. Zubel, I. Celik, P. Rougier,F. Ciardiello, Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatmentfor metastatic colorectal cancer: Updated analysis of overall survival according to tumorKRAS and BRAF mutation status. J. Clin. Oncol. 29, 2011–2019 (2011).
44. M. H. Cohen, J. Gootenberg, P. Keegan, R. Pazdur, FDA drug approval summary:Bevacizumab plus FOLFOX4 as second-line treatment of colorectal cancer. Oncologist12, 356–361 (2007).
45. K. E. Ware, T. K. Hinz, E. Kleczko, K. R. Singleton, L. A. Marek, B. A. Helfrich,C. T. Cummings, D. K. Graham, D. Astling, A. C. Tan, L. E. Heasley, A mechanism ofresistance to gefitinib mediated by cellular reprogramming and the acquisition of anFGF2-FGFR1 autocrine growth loop. Oncogenesis 2, e39 (2013).
46. C. Guzmán, M. Bagga, A. Kaur, J. Westermarck, D. Abankwa, ColonyArea: An ImageJplugin to automatically quantify colony formation in clonogenic assays. PLOS One 9,e92444 (2014).
47. M. Perego,M. Tortoreto, G. Tragni, L. Mariani, P. Deho, A. Carbone,M. Santinami, R. Patuzzo,P. D. Mina, A. Villa, G. Pratesi, G. Cossa, P. Perego, M. G. Daidone, M. R. Alison,G. Parmiani, L. Rivoltini, C. Castelli, Heterogeneous phenotype of human melanomacells with in vitro and in vivo features of tumor-initiating cells. J. Invest. Dermatol. 130,1877–1886 (2010).
48. F. L. Shaw, H. Harrison, K. Spence, M. P. Ablett, B. M. Simões, G. Farnie, R. B. Clarke,A detailed mammosphere assay protocol for the quantification of breast stem cell ac-tivity. J. Mammary Gland Biol. Neoplasia 17, 111–117 (2012).
Acknowledgments: We thank C. Johannessen, R. Gunaratne, D. Frederick, A. Nichols,E. Hodis, andE.VanAllenand themembersof theMcDonnell, Sabatini, andWood laboratoriesfor helpful discussions and technical assistance. Funding: This work was supported by DukeUniversity School ofMedicine start-up funds andby theDukeCancer Institute (K.C.W.), theNIHBuilding InterdisciplinaryResearchCareers inWomen’sHealth Program (K.C.W.), aHarry J. LloydTrustTranslationalResearchAward (K.C.W.), aGolfersAgainstCancerResearchAward (K.C.W.),a Stewart Trust Fellowship (K.C.W.), a V Scholar Award from the V Foundation for CancerResearch (K.C.W.), and NIH grants CA103866 (D.M.S.), AI07389 (D.M.S.), and R37DK048807(D.P.M.). D.M.S. is an investigator of the Howard Hughes Medical Institute. All cDNA constructsdescribed here have been deposited in Addgene. Author contributions: K.C.W., K.A.O.,T.W., and D.M.S. designed and created the barcoded screening library. K.C.W., K.A.O., J.C.R.,and C.A.M. performed drug resistance screens. K.C.W. and T.W. analyzed screening data.K.R.S., K.C.W., C.A.M.,G.R.A., P.S.W., andA.P.-P. performed follow-up experiments onNotch1in breast cancer and melanoma as well as pathway co-inhibition studies. J.S.J. and D.P.M. per-formed computational analyses of Notch1 pathway activation in breast cancer patients. S.E.W.planned, directed, and conducted animal experiments with assistance from H.M.A. J.A.W.
ENCESIGNALING.org 23 December 2014 Vol 7 Issue 357 ra121 12

R E S E A R C H A R T I C L E
and K.T.F. provided matched pretreatment and progression-stage melanoma patientsamples, and L.N.K., Z.A.C., M.T., and P.-L.C. analyzed these tumors for mutations in can-didate resistance genes and expression of markers of interest by immunohistochemistry. Allauthors contributed to the design of experiments and analysis of results. K.C.W.wrote theman-uscript, and all authors provided editorial input.Competing interests:K.A.O., T.W., D.M.S., andK.C.W. are inventors on a U.S.patent application describing the barcoded pathway-centricscreening technology. The other authors declare no competing financial interests. Data andmaterials availability: The screen data can be accessed from table S3 and theWood labora-tory Web site (http://wood.duhs.duke.edu/publications/index.html).
www.SCI
Submitted 29 October 2014Accepted 4 December 2014Final Publication 23 December 201410.1126/scisignal.aaa1877Citation: C. A. Martz, K. A. Ottina, K. R. Singleton, J. S. Jasper, S. E. Wardell, A. Peraza-Penton,G. R. Anderson, P. S. Winter, T. Wang, H. M. Alley, L. N. Kwong, Z. A. Cooper, M. Tetzlaff,P.-L. Chen, J. C. Rathmell, K. T. Flaherty, J. A. Wargo, D. P. McDonnell, D. M. Sabatini,K. C. Wood, Systematic identification of signaling pathways with potential to conferanticancer drug resistance. Sci. Signal. 7, ra121 (2014).
ENCESIGNALING.org 23 December 2014 Vol 7 Issue 357 ra121 13
on Decem
ber 23, 2014http://stke.sciencem
ag.org/D
ownloaded from

(357), ra121. [doi: 10.1126/scisignal.aaa1877]7Science Signaling 2014) P. McDonnell, David M. Sabatini and Kris C. Wood (December 23, Jeffrey C. Rathmell, Keith T. Flaherty, Jennifer A. Wargo, DonaldN. Kwong, Zachary A. Cooper, Michael Tetzlaff, Pei-Ling Chen, Anderson, Peter S. Winter, Tim Wang, Holly M. Alley, LawrenceJasper, Suzanne E. Wardell, Ashley Peraza-Penton, Grace R. Colin A. Martz, Kathleen A. Ottina, Katherine R. Singleton, Jeff S.confer anticancer drug resistanceSystematic identification of signaling pathways with potential to
This information is current as of December 23, 2014. The following resources related to this article are available online at http://stke.sciencemag.org.
Article Tools
http://stke.sciencemag.org/content/7/357/ra121article tools: Visit the online version of this article to access the personalization and
MaterialsSupplemental
http://stke.sciencemag.org/content/suppl/2014/12/19/7.357.ra121.DC1.html"Supplementary Materials"
Related Content
http://stke.sciencemag.org/content/sigtrans/7/357/pc33.full.htmlhttp://stke.sciencemag.org/content/sigtrans/4/166/pe16.full.htmlhttp://stke.sciencemag.org/content/sigtrans/5/224/rs4.full.htmlhttp://stke.sciencemag.org/content/sigtrans/4/166/ra18.full.htmlhttp://stke.sciencemag.org/content/sigtrans/4/166/pc6.full.htmlhttp://stke.sciencemag.org/content/sigtrans/6/294/eg5.full.htmlhttp://stke.sciencemag.org/content/sigtrans/7/357/ra122.full.html
's sites:ScienceThe editors suggest related resources on
Referenceshttp://stke.sciencemag.org/content/7/357/ra121#BIBLThis article cites 47 articles, 20 of which you can access for free at:
Glossaryhttp://stke.sciencemag.org/cgi/glossarylookupLook up definitions for abbreviations and terms found in this article:
Permissionshttp://www.sciencemag.org/about/permissions.dtlObtain information about reproducing this article:
reserved. DC 20005. Copyright 2014 by the American Association for the Advancement of Science; all rightsAmerican Association for the Advancement of Science, 1200 New York Avenue, NW, Washington,
(ISSN 1937-9145) is published weekly, except the last December, by theScience Signaling
on Decem
ber 23, 2014http://stke.sciencem
ag.org/D
ownloaded from