synthesis of all-silica and high-silica molecular sieves in
TRANSCRIPT

1
The original publication is available at www.springerlink.com http://springerlink.com/content/rr22r7220l3178t2/ D.O.I.: 10.1023/A:1019154304344 ISSN: 1022-5528 (Print) 1572-9028 (Online) Topics in Catalysis, 1999, vol. 9, pp. 59-76
Synthesis of all-silica and high-silica molecular sieves in fluoride media
M.A. Camblor,* L.A. Villaescusa, M.J. Díaz-Cabañas
Corresponding Autor: Miguel A. Camblor, current address: Instituto de Ciencia de
Materiales de Madrid (CSIC), c/Sor Juana Inés de la Cruz, 3, 28049 Madrid, Spain
Current email: [email protected]

2
Synthesis of all-silica and high-silica molecular sieves in fluoride media
M.A. Camblor,* L.A. Villaescusa, M.J. Díaz-Cabañas
Instituto de Tecnología Química, Avda. Los Naranjos s/n, 46022 Valencia, Spain
Corresponding Author: M.A. Camblor. [email protected], Phone: 34-96-3877811;
Fax: 34-96-3877809.
Short title: Synthesis of silica molecular sieves
Keywords: synthesis; silica zeolites; framework density; phase selectivity; connectivity
defects.
Abstract
Recent advances in the synthesis of all-silica and high-silica crystalline
molecular sieves in fluoride media, with special regard to low framework density
phases, are presented. The fundamental differences between the synthesis in hydroxide
and fluoride media with respect to the properties of the materials obtained and the phase
selectivity of the crystallization are discussed.
Introduction
Zeolites exist in nature as high-alumina materials (Si/Al < 5), while natural pure
silica crystalline phases are dense non-porous solids (Table 1), with the only exception
of the rare mineral melanophlogite (MEP). However, crystalline microporous high-
silica and all-silica phases consisting of 3D, 4-connected frameworks can be
synthesized by the use of suitable organic (or metallorganic) additives. The role of such
additives (called structure-directing agents, SDA) in the synthesis of high-silica zeolites

3
in hydroxide medium has been recently reviewed and discussed1 and in the most simple
formulation relies on the ability of the additive to select between different possible
phases of similar thermodynamic stability.
The aim of this paper is to discuss on several phase selectivity issues in the
crystallization of all-silica molecular sieves in fluoride media in the presence of organic
SDA’s. Some fundamental differences between the synthesis in alkaline media and
fluoride media with regard to defect concentration and in relation to the synthesis of low
framework density phases are discussed. We also try to rationalize an apparent
relationship between the framework density of the silica phase obtained in fluoride
media and the degree of dilution of the synthesis mixture.2 Upon incorporation of active
centers (Al, Ti or funtionalized organic moieties) these phases may be active catalysts,
and their activity and selectivity may be determined in some extent by their
hydrophobic nature.
Experimental
The pure silica materials were synthesized hydrothermally in teflon lined
stainless steel autoclaves under slow rotation (60 rpm). Typically, tetraethylorthosilicate
(TEOS) was hydrolyzed in an aqueous solution of the appropriate structure-directing
agent in its hydroxide form and the resulting mixture was left under stirring until
complete evaporation of the ethanol produced (scheme 1). Within the detection limit of
1H MAS NMR no ethanol remained in the synthesis mixture after evaporation (Fig. 1).
This suggests that these water/alcohol/silica/SDA mixtures do not form azeotropic
compositions, as opposed to the known behavior of water/ethanol mixtures. On the
other hand the consumption of two water molecules per TEOS has to be considered

4
when defining the important H2O/SiO2 molar ratio of the final synthesis mixture
(scheme 1).
After ethanol evaporation HF (48% aqueous solution) was added and the
mixture was homogenized by hand stirring. The resulting mixture is always a slurry
with a viscosity that depends on the specific SDA used and the final water content. We
never obtained a homogeneous clear solution and this is an important difference with
the OH method, for which in some cases it is possible to prepare such solutions. Unless
otherwise stated, the final reaction mixtures had a composition
SiO2 : 0.5 HF : 0.5 SDAOH : w H2O
In some case, HF was substituted by NH4F and/or the SDAOH was increased to rise the
pH. Throughout this paper the H2O/SiO2 ratio will be w in the above formula, although
the actual ratio will generally be 0.5 mole larger because of the neutralization reaction
between HF and SDAOH.
When commercially unavailable, the SDA’s were prepared by quaternization of
the parent amine using the proper halide and the product was subsequently anion
exchanged to produce the hydroxide form. We have used a large number of SDA’s for
the synthesis of pure silica phases in fluoride media and those relevant to this report are
shown in scheme 2. These SDA’s were chosen primarily according to the availability of
the cation or of the parent amine and to the criteria recently outlined as important in
determining a high structure-directing ability (rigidity, size, shape, C/N+ ratio).3 Thus,
SDA’s with polycyclic moieties (giving rise to SDA’s with relatively large and rigid
portions) predominate. The C/N+ ratios have been varied in some extent to investigate
the influence of this parameter in fluoride media in comparison with the observations in
hydroxide media.

5
The samples were characterized by conventional techniques, including powder
XRD, chemical analysis (C, H, N, F) and 29Si MAS NMR. The 29Si MAS NMR was
recorded on a Varian VXR 400SWB spectrometer with a spinning rate of 5.5 kHz at a
29Si frequency of 79.459 MHz using a 55.4º pulse length of 4.0 µs. The recycle delay
was typically 60 s for the calcined samples and 240 s for the as-made ones. Through
experiments at different recycle delays with as-made ITQ-4 we have confirmed that as-
made samples typically require such long recycle delays in order to get reliable spectra.
This is attributed to the lack of O2 in the pores of as-made zeolites, which causes an
increase in the 29Si spin-lattice relaxation time4 in these samples compared to the
calcined ones. For the same reason the spectrum of octadecasil (a clathrasil with
openings too small to allow diffusion of O2) was recorded also with 240 s recycle
delays.
Results and discussion
Synthetic efforts have produced a number of microporous pure silica phases in
the last two decades (Fig. 2). These phases may be denoted by the three-letter code
assigned by the International Zeolite Association to every accepted zeolite topology.5
Throughout this paper the framework density (i.e., the number of tetrahedra per nm3,
FD) will be used to characterize the silica phases. It is important to note that the FD
reported here are experimental framework densities calculated from the actual structural
data of materials with composition SiO2 and that they may differ largely from both the
FD reported for the “type materials” (which are frequently low silica or even
aluminophosphate materials)5 and the FDSi derived from the DLS refined topologies.6
As shown in Fig. 2, the ability to produce low density silica phases by direct
hydrothermal synthesis appears to be increasing. Until 1996 and irrespective of the

6
synthesis route (OH- or F-) the framework density of known silica phases was relatively
high (FD≥17.3). However, we have been able to prepare new pure silica phases with
lower framework densities (FD≤17) during the past 3 years. Only one of them (ITQ-1,
structure code MWW, FD=16.5)7 was prepared in hydroxide medium.8 The synthesis of
ITQ-1 will not be discussed here because of the peculiarities of the mechanism of
formation of its structure: the as-made phase is probably a layered material containing
“structural”, not randomly distributed defects which are annealed upon calcination with
formation of the three-dimensional 4-connected MWW SiO2 framework.7,9,10 All the
other low framework density phases were prepared in fluoride medium and are the
result of a systematic investigation in which a large number of organic cations and
synthesis conditions were screened for the synthesis of pure silica phases. The aim of
this paper is to show that the fluoride route is specially well suited for the synthesis of
low density silica phases, and to discuss on the reasons for this and on some factors that
determine the nature of the phase obtained.
Framework density and the H2O/SiO2 ratio of the synthesis mixture
Pure silica phases can be prepared hydrothermally using either hydroxide or
fluoride as mineralizers and both methods (and the materials obtained) will be denoted
here according to the mineralizer as the OH and F routes (and OH- and F-materials).
The hydrothermal synthesis of microporous silica phases using fluoride anions instead
of hydroxide anions as mineralizers has been known since the pioneering work by
Flanigen and Patton11 and extensively used by others during the last two decades.12,13
Interestingly, the F route yielded only relatively high density phases (FD>17.3) until
very recently. The successful synthesis in our laboratory of pure silica low density
phases such as chabazite (CHA, FD=15.4),14 Beta (15.6),15 ITQ-3 (ITE, 16.3)16 and

7
ITQ-4 (IFR, 17.0)17 and several other unreported phases heavily relied on a
modification of the fluoride route which will be described in this section. Table 2 lists
some interesting silica phases that we have prepared in fluoride aqueous media,
including materials with very different channel systems (1D to 3D, 8MR to 14MR).
It can be inferred from Table 2 that frequently different phases can be prepared
using the same SDA by just varying the H2O/SiO2 ratio. Thus, the effect of the water
content of the synthesis mixture can modify in some extent the structure-directing
ability of the SDA and very different pore architectures can be made by using the same
SDA. We have very recently reported on an apparent empirical relation between the
H2O/SiO2 ratio of the synthesis mixture and the FD of the phase obtained in fluoride
aqueous media: most frequently, the lower the H2O/SiO2 ratio the lower the framework
density of the silica phase obtained.2 The successful synthesis of pure silica CHA
(FD=15.4),14 the less dense SiO2 polymorph ever reported (possessing a void fraction of
nearly 50%: 0.46 cm3/cm3) illustrates well this observation (Fig. 3). The synthesis of
SiO2 CHA using TMAda+ at 150ºC heavily depends on a very low H2O/SiO2 ratio in the
reaction mixture (3.0), as higher degrees of dilution favor the more dense SSZ-23 (FD=
17.0). At a ratio of 3 CHA is fully crystallized before two days and SSZ-23 starts to
compete only after one week of heating at the crystallization temperature. By contrast,
when the H2O/SiO2 ratio is increased to 5.8 a mixture of phases, containing SSZ-23 and
Chabazite, is formed. The amount of SSZ-23 in this mixture increases with the heating
time. When the H2O/SiO2 ratio is further increased to 7.5 or 10 SSZ-23 is the only
phase that crystallizes and no CHA is observed. At still higher water contents
(H2O/SiO2= 15 and 20) some SSZ-31 (FD= 18.7) is also observed at the initial stages of
the crystallization, although SSZ-23 is finally obtained as a pure phase. At a
temperature of 175ºC the same effects are still observed, but the increase in temperature

8
favors SSZ-23 with respect to CHA (Fig. 3). We note that the H2O/SiO2 ratios we used
to produce low density phases are normally lower than those typically used in the
hydrothermal synthesis of zeolites in F- or OH- medium, and sometimes approach the
reagent rather than solvent level. This is the key modification of the known fluoride
route12 to the synthesis of zeolites and silica phases that allowed us to prepare the low
density silica phases shown in the first entries of Table 2. In occasions, the phase
selectivity change appears for very close H2O/SiO2 ratio values. This is the case
encountered for benzylquinuclidinium (BQ+) at 175ºC where ITQ-4 is formed for
H2O/SiO2 ratios equal or above 4.5, while at a ratio of 3.6 pure silica Beta crystallizes
instead (Table 2). By contrast, there are also instances in which there are not phase
selectivity changes even when the water content is reduced to zero, as exemplified by
the “dry synthesis” of silicalite using TPA+ and F-.19
Some further examples of the influence of the H2O/SiO2 ratio on the density of
the phase obtained are illustrated in the next Figures and in references 2 and 20. For
example, elongated bulky diquats with an approximately linear shape (as M8BQ2+,
M6BQ2+, p-BBQ2+, M10BTM2+) may yield both pure silica Beta or MTW (Fig. 4). The
phase selectivity of these diquats depends on the H2O/SiO2 ratio, the less dense Beta
material being obtained at the lower H2O contents. By contrast, when the
quinuclidinium moieties in benzylbisquinuclidinium diquats are not in para- position to
each other only Beta was obtained and the phase selectivity change caused by dilution
of the reaction mixture was not observed. In the case of o-BBQ2+ this is most likely due
to the bent shape of the SDA, which probably precludes accommodation of this cation
inside the straight 12MR channels of MTW. In the case of m-BBQ2+, three
conformations (one linear, two bent) appear feasible but only one could fit into the
MTW straight channels. Thus, in the case of o- and m-BBQ2+ the shape of the SDA

9
apparently determines the phase selectivity and the water content is a less important
parameter.
Figure 5 provides another example of the effect of the degree of dilution of the
synthesis mixture on the phase selectivity of the crystallization. Using N
methylsparteinium (MSPT+) as SDA the one-dimensional materials SSZ-24 (AFI,
12MR)21,22 and CIT-5 (CFI, 14MR)23 may be crystallized. For pure silica compositions
in fluoride media, the phase of lower framework density (AFI) crystallizes at the lower
water content.20 We note that this effect vanishes if CFI crystals are added as seeds,20
suggesting that the effect of the water content on the phase selectivity may be related to
the nucleation step and hence is, probably, a kinetic effect.
Actually, we have rationalized this apparent rule as a kinetic control of the
crystallization: more metastable phases occur at the most concentrated conditions (low
H2O/SiO2 ratios, for which higher supersaturation rates are speculated).2 In our opinion,
the observed trend appears to relate density and synthesis conditions as a mere
consequence of the generally more metastable character of the low framework density
phases (see below). There is some resemblance between the phase sequences obtained
when increasing the H2O/SiO2 ratio and the phase sequences obtained with increasing
time (Ostwald ripening) or temperature which suggests the kinetic control outlined
above. We warn, however, that Ostwald ripening is normally very slow in these
conditions, probably due to the low solubility of the crystalline phases. Thus, examples
of full transformations are not frequent. However, the case of CHA and STT (for which
full transformations have been found) can be used again to illustrate this effect:
increasing water content, temperature or time favors the denser STT material (Fig. 3).
Similarly, in the synthesis with M8BQ2+ Beta and MTW may crystallize, and the denser
MTW is favored when increasing water content, temperature or time (Fig. 6). Phase

10
sequences obtained by Ostwald ripening give an order of increasing thermodynamic
stability. On the other hand, the phase sequences obtained with increasing temperature
may reflect both an order of increasing stability and an order of increasing activation
energy of crystallization. The resemblance alluded to above suggests that the most
stable phases appear at high H2O/SiO2 ratios and that those appearing at low ratios are
metastable and appear through a kinetic control.
The observed trend suggests that, generally, low density phases are metastable
with respect to denser phases. It has been shown very recently that the differences in the
enthalpy of formation of silica phases are small24 and that they may show some
tendency to increase with the decrease in FD.25 This would make low density phases
(slightly) metastable towards higher density phases, although it has to be noted that this
refers to the pure silica frameworks rather than to the SDA/F/SiO2 composite actually
synthesized (see below). As suggested by Lobo et al., the role of the SDA is to select
amongst different silica phases with rather similar energies.1 This may occur through a
kinetic or through a thermodynamic effect, the last one occurring if the additional
stabilization provided by the interaction of the SDA (and/or F-, see below) and the
framework is large enough to surpass the small difference in energy between the phases.
As the observation we are trying to rationalize refers to syntheses in which a given SDA
produces different phases, a relatively low specificity in the interaction between the
SDA and the silica host may be anticipated. This low specificity suggests in the cases
discussed here the interaction between the SDA and different silica phases may
generally be not different enough to afford a thermodynamic control.
However, we would like to make another point for discussion: it could be
possible that even for relatively unspecific SDA’s there could be differences in the
stabilization due to the interaction between silica hosts of varying density and the guest

11
species (SDA+ or F-). This is not claimed to be due to an increased energy in the
interaction between the SDA and the silica host but to the increased concentration of
guest species inside the zeolitic cavities. As the framework density decreases the
concentration of occluded guests (defined as SDA/SiO2 and F/SiO2 in the final material)
generally increases, so the energy of the interaction (per mole of SiO2) could in some
cases increase without an increase in the specificity of the interaction. This appears to
be the case in the synthesis of pure silica Beta and MTW zeolites using p-
benzylbisquinuclidinium (pBBQ2+). As shown in Table 3 (last entry, last column), the
most dense phase appears to be metastable towards the low density phase, as suggested
by the Ostwald ripening sequence of phases: ZSM-12 → Beta. This example is puzzling
since zeolite Beta (the less dense but, apparently, more stable phase in the presence of
p-BBQ2+) is still favored at low H2O/SiO2 ratios (Fig. 4).
We note that the phase sequence found with time with p-BBQ2+ (MTW→Beta,
Table 3, last column) is the opposite to that found with M8BQ2+ (Beta→MTW, Fig. 5).
This suggests that the relative stabilities of Beta and MTW are different in the presence
of each SDA. To further confirm this, we designed experiments aimed to discriminate
the relative stabilities by Ostwald ripening transformations. As-made Beta and MTW
zeolites prepared with p-BBQ2+ and M8BQ2+ were used as only silica sources in
conditions in which (using hydrolyzed TEOS) the opposite phase crystallizes. As shown
in Table 3, only ZSM-12 transformed partially to Beta, while Beta never transformed to
MTW (only a decrease in crystallinity or transformation to tridymite was observed,
possibly due to decomposition of the SDA after the long crystallization times used).
While this experiments suggest that Beta is more stable than MTW (despite this phase
being denser) with both SDA’s, the Ostwald ripening sequence found with M8BQ2+ at
135ºC and H2O/SiO2=15 (Fig. 5) suggests the opposite. Apparently, there is not a single

12
answer about the relative stabilities (in the synthesis conditions) of Beta and MTW, as
this seems to depend not only on the SiO2 framework but also on the SDA, the
concentration of the solution and the temperature. In our opinion, the relative stabilities
of zeolite frameworks alone may not allow to extract directly meaningful conclusions
on their synthesis.
As we already suggested,2 if the apparent relation between density and synthesis
conditions is just a consequence of the generally lower stability of low density phases
we may expect exceptions to the observed trend. The very recent synthesis of ITQ-9 (a
phase isomorphous to SSZ-35,26 whose structure was first reported by Wagner et al.27
and independently solved by us)18 appears to provide such an exception. This phase
(FD= 17.3) has been obtained by drastically reducing the water content in a reaction
mixture otherwise yielding ITQ-3 (FD= 16.3) or SSZ-31 (FD= 18.7) at intermediate and
high water contents, respectively (Fig. 7). We speculate that the order of increasing
stability (in the synthesis conditions) of these phases may be ITQ-9<ITQ-3<SSZ-31,
and thus does not correlate with the density of the phases (ITQ-3<ITQ-9<SSZ-31). This
example further illustrates the impressive influence of the water content on the phase
selectivity of the crystallization.
Finally, we would like to argue on still another conceivable effect contributing
to the observed trend between the water content and the FD of the phase obtained. In
our system, soluble silicate species will most probably be oxofluorosilicates. As the
H2O/SiO2 ratio decreases (and correspondingly the F- concentration increases) the
fluoride to oxygen ratio of silicate species in solution could likely increase. If we
assume that the crystallization takes place by condensation of silicate species in
solution, as the water content decreases and F- concentration increases a higher
concentration of occluded F- may be expected, requiring a higher concentration of SDA

13
cations in the material and, hence, a lower density phase able to accommodate a higher
concentration of cations. However, this unique effect can not explain the observed
formation of ITQ-9 instead of ITQ-3 in the experiments commented in the previous
paragraph, because both materials occlude the same concentration of SDA cations and
F- anions. Clearly, attempts to rationalize the phenomena described in this section are
much limited by the comparatively small amount of experimental data and the very
complex nature of the systems under investigation. Thus, the main conclusions that can
be extracted from the results presented here is that the water/silica ratio may have a
strong influence on the phase selectivity of the crystallization (beyond other structure-
directing effects that are also operative) and that this can be used as an useful strategy to
discover new silica zeolites, specially low density ones.
It would be interesting to check the effect of the H2O/SiO2 ratio on the phase
selectivity of crystallizations using the OH- method. We have found very few reports of
systematic investigations of this parameter. In the case of silica-based materials, the
investigation of this open issue is much limited by the stability of quaternary
ammonium cations in alkaline medium, which is dramatically reduced as the
concentration of the reacting mixture, and hence the pH, increases. Thus, although there
are some few reports on the effect of this ratio on the crystallization of zeolites,
apparently they covered a range of H2O/SiO2 ratios at relatively large dilutions
(H2O/SiO2 =15-30 for zeolite Beta28 and 37 to 100 for ZSM-12)29 and only changes in
the crystallization kinetics but not on the phase selectivity were observed.
By contrast, the synthesis of phosphate-based materials can be an interesting
field to explore the influence of the concentration of the reacting mixture on the phase
selectivity in the absence of fluoride anions, since in this case the stability of the
organic additives in a very concentrated hydrothermal system is not a problem of much

14
concern because the typical pH during the crystallization is close to neutral. Such
syntheses are usually carried out in relatively concentrated systems, with H2O/T (T=P,
Al, ....) typically around 10. Actually, there exist at least one report in which the H2O/T
ratio has a clear effect on the phase selectivity of the crystallization: in the synthesis of
Mg-substituted AlPO4 phases using triethylamine at 150ºC a decrease on the water
content results in an increase in the amount of the CHA product relative to the AFI
product after 16 hours of crystallization. Since CHA has a lower FD than AFI (both for
SiO2 and for AlPO4 phases) this result resembles those most frequently found by us in
the synthesis of SiO2 phases.30 By contrast, VPI.-5 is typically synthesized at H2O/T =
1031 but it can be prepared also in highly concentrated systems (H2O/(Al+P) = 3.26).32
Incorporation of aluminum
Aluminum may be incorporated into the framework of high-silica materials in
fluoride medium, giving rise to active acid catalysts (see below). However, and
similarly to previously reported observations in alkaline media,1,33 sometimes
introduction of Al changes the phase selectivity of the crystallization. We will use Fig. 8
to illustrate this effect. As seen in this Figure, introduction of Al in a reaction mixture
containing benzylquinuclidinium (BQ+) may change the phase selectivity from a one-
dimensional large pore material (ITQ-4) to a three-dimensional one (zeolite Beta). For a
relatively high water/silica ratio (15) the effect could be solely attributed to a charge
balance effect: when the [AlO4]- content per unit cell surpasses the SDA content of the
zeolite (2 tightly packed cations per unit cell in ITQ-4),34 charge balance in ITQ-4 is no
longer possible because the SDA is the only positively charged species inside the

15
channels in these synthesis conditions (absence of alkali cations). Thus, forcing the
limits of Al concentration induces a change to a less dense, multidimensional large pore
zeolite (Beta). The results obtained at lower water/silica ratios are perhaps more
interesting because the phase selectivity change occurs for Al contents well below the
limit imposed by charge balance in ITQ-4 (2Al per 32 T atoms or Al/(Al+Si)=0.0625).
We have not a clear explanation for this effect but, as seen in Fig. 8, the phase
selectivity change induced by increasing the Al content is the same as that induced by
decreasing the water content. The main process involved in this phase selectivity
change may be nucleation, because when a reaction mixture leading to zeolite Beta
(H2O/SiO2 = 7.5, Al/(Al+Si)= 0.026) is seeded with ITQ-4 crystals the phase obtained
is almost pure ITQ-4 (with a small zeolite Beta impurity). Fig. 8 also suggests that the
effect of the water content on the phase selectivity of the crystallization discussed in the
previous section applies to the crystallization not only of pure silica phases but also of
aluminosilicate zeolites.
The effect shown in Fig. 8 is similar to some observations reported in alkaline
medium, where introduction of B or Al frequently induces a change in phase selectivity
from 1D to 3D pore materials.1,33 A clear rationalization of this trend is lacking but
structural relationships have been noted:1 phase selectivity changes upon isomorphous
substitution of Si by B or Al frequently involve topologies with a common projection
(like MTW and BEA or SSZ-33 and AFI), or similar structural subunits (like SSZ-33
and BEA) and with no change in the number of tetrahedra in the channel window. By
contrast, we cannot see any structural relationship between BEA and IFR (Fig. 8),
except that they both have 12MR channels. Moreover, Fig. 9 shows that, in some
conditions, Al can induce phase selectivity changes between structures with no common
projection, no similar structural subunits and different size of the channel window (like

16
the 1D/12MR SSZ-31 and the 2D/8MR ITQ-3). Here again introduction of Al leads to a
less dense phase and, very interestingly, the effect is similar to that observed when
reducing the water content (SSZ-31 to ITQ-3, Fig. 9). The same is also true in the
synthesis with TMAda+, where introduction of Al clearly favors CHA instead of SSZ-
23.
It has been noted that phase selectivity changes upon substitution of Si by B or
Al in alkaline medium usually consists of changes not only to more porous
multidimensional materials but also to structures containing a larger population of four-
rings.33 By contrast, our observations in fluoride medium do not show any clear
tendency towards 4MR as the Al content increases: the concentration of 4MR is larger
in IFR (0.44 4MR per Si) than in BEA (0.31) while is much smaller in SSZ-23 (0.37)
than in CHA (0.75). The difference with the observations in hydroxide media could
possibly be due to 4MR being favored by both Al (as implied in ref. 33) and F- (see
below) and to the fact that, generally, increasing the Al content in fluoride media causes
a decrease in the fluoride content of the zeolite.
The C/N+ ratio in fluoride media
According to Zones and coworkers,1,3,33,35 the C/N+ ratio of the SDA is a critical
parameter in determining the ability of the organic additive in producing high silica
phases in hydroxide medium: SDA cations with C/N+ ratios between 11 and 15 “work
well” in the synthesis of very high silica phases. These authors also showed that the
partition of quaternary ammonium compounds between water and chloroform is highly
dependent on the C/N+ ratio, the percentage of transfer to the organic phase being very
low for C/N+<11 and very large for C/N+>15. An optimum structure-direction
performance of cations with intermediate C/N+ ratios (11-15) was found and considered

17
a consequence of their moderate hydrofobicity. Smaller cations show a large charge-to-
volume ratio and, consequently, their interaction with water is mainly hydrophilic. By
contrast, too large cations interact too weakly with water and may segregate into a
separate phase. This rationalization may explain why small cations like TMA+ and
TEA+ have a much limited ability to produce pure silica phases in hydroxide medium.
However, when SDA cations with C/N+ ratios below 11 are used in the synthesis
of pure silica phases in fluoride aqueous media a completely different scenario appears.
For example, TMA (C/N+=4) and TEA+ (8) which in hydroxide medium are not
adequate SDA cations for pure silica phases work well in fluoride medium. Here we
will consider that an SDA “works well” if the synthesis conditions necessary for it to
direct the crystallization to a microporous phase are not very restricted. We think that,
in addition to the hydrophobic properties of the SDA, charge balance and the
concentration of defects have to be considered in order to understand the importance of
the C/N+ ratio.
In the synthesis of pure silica phases in OH- medium the charge of the SDA (and
alkali cations, if present) need to be counterbalanced by Si-O- connectivity defects and,
consistently, the obtained materials are usually highly defective (see below). Typically,
the amount of Si-O- and Si-OH species outnumbers the amount of SDA charges by a
factor of about 4 or above.36 By contrast, in fluoride medium charge balance of the SDA
is generally achieved by occluded fluoride and the concentration of Si-O- and Si-OH
groups is typically very low (see next section for a discussion on defects in silica
zeolites). Thus, cations with a small C/N+ ratio, i.e. cations with a large charge to
volume ratio, may require in hydroxide medium a very large concentration of defects of
connectivity. For example, and we will assume in the following discussion that Q3 =
4×SDA+, using TMA+ the synthesis of pure silica sodalite (2TMA+ per unit cell of 12

18
Si) and octadecasil (2 TMA+ per unit cell of 20 Si) would require 50% and 40%,
respectively, of the Si species to be partly unconnected. Similarly, the synthesis of
zeolite Beta using TEA+ would require around 37-50% defects (6-8 TEA+ per unit cell
of 64 Si). Actually, we have observed that in as-made high silica Beta synthesized in
hydroxide medium in the absence of alkali cations the concentration of defects increases
as the Al content decreases and that the excess concentration of defects (defined as
Q3/(TEA-Al)) increases from 3.6 for Si/Al = 14 to 5.0 for Si/Al = 76.37
We speculate that silica phases containing such a large concentration of
connectivity defects may be generally unstable under hydrothermal synthesis conditions
at high pH, making difficult the formation of stable nuclei. It is generally accepted that
the formation of nuclei goes through a thermodynamically hindered stage until reaching
the so called “critical radius”, for which the rate of dissolution of nuclei equals the rate
of growing.38 Nuclei larger than the critical radius are stable and their growth is
thermodynamically favored, while smaller nuclei are unstable and may dissolve before
reaching the critical size. We think nuclei presenting a large concentration of defects
may be more prone to dissolution, since a highly defective silica framework may be
regarded as partly depolimerized silica. This does not mean that the synthesis of pure
silica phases in hydroxide medium using SDA cations with small C/N+ ratios are not
possible. What it means is that the required conditions for the crystallization (specially
for the nucleation) will be narrowly restricted.
As an example, the synthesis of pure silica Beta using TEA+ is very difficult in
hydroxide medium (37-50% defects required) while occurs spontaneously (without
recourse to seeding techniques) in fluoride medium.15 Thus, for over 30 years and up to
very recently pure silica Beta was never produced in hydroxide medium. Interestingly,
recent procedures affording this synthesis made use of either SDA cations with larger

19
C/N+ ratios (15.539 and 16,40 requiring probably less than 25% defects, sometimes with
seeding required)27 or a different synthetic method under very specific conditions (the
so called “dry gel conversion technique”,41 which possibly yields very high
supersaturation rates). By contrast, pure silica zeolite Beta may be readily synthesized
without seeding by using TEA+ in fluoride aqueous medium under a very wide range of
conditions: static or under rotation, in the 100 to 175ºC temperature range and with a
rather large tolerance with respect to pH and H2O/SiO2 and F-/SiO2 ratios (7-11.8, 2.5-
14 and 0.4-2, respectively, for T=140ºC).53 Thus, we must consider that TEA+ “works
very well” in the synthesis of pure silica zeolites in fluoride medium while “works bad”
in hydroxide medium.
Another example of the different behavior of small SDA cations in fluoride and
hydroxide media is provided by t-butyltrimethylammonium (t-BTMA+, C/N+ = 7). This
SDA affords the synthesis of octadecasil in fluoride medium under a very wide range of
conditions (T=135-175ºC, H2O/SiO2= 2.5-15, synthesis times: 20 h to 12 days,
depending on conditions). By contrast, we have not been able to prepare this material in
hydroxide medium (expected concentration of defects: 40%) and only upon introduction
of aluminum (Si/Al = 30, expected concentration of defects: 27%) the synthesis yielded
octadecasil with low crystallinity (ca. 15%) after a very long crystallization time (54
days).
With regard to this issue, it is interesting to consider here the synthesis of IFR
phases (ITQ-4, SSZ-4242 and MCM-58)43 in hydroxide and fluoride media: while this
structure may be readily prepared as a SiO2 polymorph in fluoride medium, it has never
been prepared by the hydroxide route in the absence of F-. We have recently shown that
the important parameter here is the presence of F- rather than the synthesis pH, as we
prepared the phase at the rather basic pH of 11.2.34 The final material contained one F-

20
per unit cell (and a slightly increased concentration of defects, see below) and we
speculate that, even for the relatively large BQ+ cation (C/N+ = 14), the synthesis of the
pure silica form may be hindered by the relatively large concentration of defects (25%
expected) because of the high packing of the SDA inside the material.34 Additionally, it
has to be considered that in IFR there are relatively dense columns of cages which are
bound to each other by single Si-O-Si bridges (Fig. 10) and thus the stability of this
phase may be more critically affected by the presence of a high concentration of
connectivity defects than other phases. As a matter of fact, the single Si-O-Si bridges
referred to above represent as much as 12.5 % of the Si-O-Si bridges. The stabilization
of this structure by the presence of fluoride (see below) may be another parameter to be
considered, which is difficult to evaluate at present.
Thus, the synthesis of pure silica phases of very low framework density in
hydroxide medium may be hindered in some extent by the necessity of a relatively large
concentrations of defect groups to counterbalance the organic cations used as SDA.
This could make difficult the formation of stable nuclei or their growth under the
hydrothermal conditions of the synthesis. To diminish this effect one may consider the
possibility of using larger cations, although this will be limited by the decreased
solubility in water of too large organic cations. On the other hand, the fluoride route
could be a very feasible alternative, since here the concentration of connectivity defects
is drastically reduced and, possibly, the interaction of F- with framework Si (see below)
may provide additional stabilization of silica frameworks.
Connectivity defects in pure silica frameworks
We have argued above on the importance of connectivity defects in the synthesis
of high silica materials and commented on the different concentration of defects when

21
the synthesis is carried out in hydroxide or fluoride aqueous media. We will focus now
on the reasons for this difference, which is an empirical, well contrasted observation.
Previous reasoning on such a difference argued that in a fluoride medium lower
supersaturations are achieved, hence providing a slower crystal growth rate which
affords more perfect crystals to be formed.44,45 However, we have observed very fast
crystallizations in fluoride media (time scale of hours) yielding materials with no
defects and also very slow crystallizations in hydroxide media (time scale of weeks)
producing very defective materials. We think that supersaturation and crystal growth
rate are not the key factors determining the concentration of defects and that this issue
may be rationalized on the basis of more chemically sound arguments.
Pure silica materials synthesized in hydroxide medium in the presence of
organic cations typically present a large concentration of Q3 species, i.e. Si(OSi)3OH
groups, which can be detected by MAS NMR and IR spectroscopies. We call these
moieties “connectivity defects” as they arise from a lack of connection between
adjacent [SiO4] tetrahedra which, ideally, should have condensed with elimination of
H2O. Typically, the concentration of Q3 species is ca. four times larger than the
concentration of positive charges in the channels (see above and ref. 36). By contrast,
pure silica materials prepared in fluoride medium at near to neutral pH typically present
a very low concentration of such species and in this sense are essentially defect-free.
This is demonstrated by the lack of significant resonances assignable to Q3 species (-90
to -104 ppm chemical shift range) in the 29Si MAS NMR spectra ( see ref. 46 and Fig.
11). We think there are several reasons contributing to these differences between OH-
and F-materials:
(1).- in the absence of other negatively charged species charge balance of the occluded
SDA cations shall be balanced by defects in OH-materials, while are generally balanced

22
by occluded F- in F-materials. However, this could only account, in the most favorable
case (i.e., the case in which a positive charge is balanced by a couple Si-O- HO-Si), for
half the typical total concentration of defects in OH-materials.
(2).- the synthesis pH in hydroxide media is generally high (typically above 11) while it
is generally low (7-9) in fluoride media. This would certainly affect the protonation
state of the condensing species and, thus, the likeliness of a complete condensation: for
a pH above the pKa of the condensing species Si-O- groups will predominate and
condensation will require a prior protonation of at least one of the condensing groups.
By contrast, at pH < pKa Si-OH groups will predominate, readily yielding full
condensation with formation of water (see scheme 1 in ref. 34). The true condensing
species and their pKa values are not known (and may be expected to vary with the pH
and other conditions). However, we may expect a pKa in a wide range by comparison
with the known values of monosilicic acid (first pKa= 9.8-9.9, second to fourth pKa
=11.7), and of the surface of amorphous silica (pKa= 6.8-9.5, depending on the extent to
which the surface is ionized).47
We recently showed that for calcined pure silica ITQ-4 the concentration of
defects depends on the synthesis pH and that the dependence is not linear: we found no
defects for pH≤10 while for higher pH the concentration of defects was essentially
constant (≅6%) and the F- content of the as-made materials decreased from 2F/uc to
around 1F/uc.34 This gave the basis for our argument on the dependence between
defects and synthesis pH in relation to the pKa of the condensing species. However, for
full consistency of the argument it was necessary to determine the defect concentration
on the as-made rather than calcined materials through very time-consuming 29Si BD
MAS NMR experiments (see experimental section). We can now present these results
(Fig. 12) which are fully consistent with the results on the calcined materials: there is a

23
low concentration of residual defects in the as-made material prepared at low pH (<
3%), while those prepared at pH ≥10.4 present a constant defect concentration of
around 9%.
(3).-Defect Si(OSi)3OH sites may be stabilized by strong hydrogen bonding to nearby
Si-O- species. The presence of such very strong bonds in high-silica materials prepared
in OH- media was recently demonstrated in a very pertinent paper by Koller et al.36 1H
MAS NMR of the as-made samples showed very low field resonances of 10.2 ppm
which correspond to very short O···O distances of 2.7 Å, hence revealing the presence
of very strong hydrogen bonds. This may yield an stabilization energy permitting the
presence of a high concentration of Q3 species in materials prepared in OH- medium. By
contrast, for F-materials this effect will be really minor because of the lack or very low
concentration of Si-O- groups in this case (see point 1 above and Fig. 2 in ref. 48).
(4).- Finally, in materials prepared in F- medium calcination to remove organic cations
also removes F-: contrary to recent predictions based on theoretical calculations,49 but
much in line with expectations, F- does leave the materials even when occluded in the
smallest cage in which it has bee found in zeolites.48 It may be speculated that at least
some of the fluoride could leave the material as HF, the proton being provided by the
thermal decomposition of the SDA cation.48 If this is the case, HF may help the healing
of residual connectivity defects in F-materials. Actually, we sometimes found a small
concentration of Q3 sites (typically below 3%) in the 29Si MAS NMR spectra of the as-
made materials but they almost completely disappear upon calcination. This is the case
of ITQ-4 synthesized at pH =8, which presents <3% defects in the as-made material,
Fig. 12, and no defects detectable by 29Si MAS NMR in the calcined one.34 A similar
effect is observed in SSZ-23.50 We have tried to detect HF and NH4F during the thermal
treatment of F-zeolites by mass spectrometry coupled with thermal analysis and had no

24
success, possibly due to the reaction of these species with the gas transfer line, which is
made of silica. However, we have been able to detect acidic species containing F- by
using a “chemical trap” during the calcination: a mixture of octadecasil and Ca(OH)2
heated at 550ºC was then washed with diluted HCl; CaF2 was then detected by XRD in
the final solid (around 5% CaF2, in rough agreement with a 2.7% F- content in as-made
octadecasil).48
Thus, silica materials prepared in OH- medium using SDA cations usually have a
large concentration of Q3 defect sites due to charge balance of the SDA and to the high
pH of the synthesis, while the formation of very strong Si-O-···HO-Si hydrogen bonds
may reduce the instability of the defective silica network. By contrast, silica materials
prepared in fluoride media have a small concentration of Q3 sites because charge
balance is generally achieved with occluded F- and the low synthesis pH favors a more
complete condensation of the silica. Additionally, in this case the stabilization of Si-OH
groups by hydrogen bonding to Si-O- must be minor (because the concentration of SiO-
is low) and fluoride-containing species developed upon calcination may help the
annealing of residual defects.
The 29Si MAS NMR spectra of a number of calcined new pure silica phases
(Fig. 11) demonstrate that the F route generally yields after calcination SiO2 phases
essentially free of connectivity defects. The spectra show no Q3 species and a very high
resolution of Si(OSi)4 species in different crystallographic sites. Only for CHA (see
below), STT and SSZ-31 a small concentration of defects (<8%) could be detected by
29Si MAS NMR. The resolution of Q4 resonances in the spectra of Fig. 11 allows to
extract a wealth of structural information, complementary to that obtained by diffraction
methods. The agreement between these spectra and the reported structures (number and
relative intensity of Si sites and average Si-O-Si angles calculated from the spectra

25
using the equation of Thomas et al.)51 is outstanding, as we recently showed for most of
these phases.14,16,18,20,48,50,52,53 In the case of MTW, the spectrum shows seven Si sites
with about equal intensities in the -108 to -112.8 ppm chemical shift range, also in good
agreement with the reported refined structure (seven sites with equal multiplicities and
average Si-O-Si angles for each site in the 145-153º range compared to 142.5-150.8º
range deduced51 from the chemical shift range). In the case of SSZ-31, a lower
resolution of Si(OSi)4 sites may be due both to the presence of defects and to the very
complex intergrown structure of this material.54
Finally, there are some few points that are interesting to be addressed with
respect to the above arguments. First, point 2 above and scheme 1 in ref. 34 suggest
silica condensation at pH > pKa may be hindered by the necessity of protonation of at
least one of the condensing Si-O- groups. If this is so, increasing the pH should slow
down the crystallization. We have checked this by following the crystallization kinetics
of pure silica ITQ-4 in the presence of benzylquinuclidinium (BQ+) as a function of the
synthesis pH (Fig. 13). The pH was increased by substituting HF by NH4F in the
reaction mixture and further increased by increasing also the (BQ+OH-)/NH4F ratio. As
shown in Fig. 13 as the alkalinity increases the crystallization slows down, as expected
from the above arguments.
Second, we have found some instances in which the concentration of defects in
F-materials synthesized at different pH is higher or lower than expected. Thus, defects
are not detected in calcined zeolite Beta materials even when synthesized at a relatively
high pH of 11.4.53 This may reflect a very high pKa of the condensing species in this
system. On the contrary, we have been unable to obtain a completely defect-free SiO2
chabazite sample. Typically, around 6-8 % Q3 sites are detected in the 29Si MAS NMR
of calcined SiO2 CHA.14 When trying to decrease the pH by addition of HCl (HCl/SiO2

26
= 0.25) SSZ-23 instead of CHA was formed. Interestingly, there is relatively little F-
occluded in as-made SiO2 Chabazite (ca. 1 weight %, affording charge balance of about
half the organic cations). The reasons for these observations are not clear at present.
F- occluded in SiO2 materials
Most frequently, pure and high-silica phases synthesized in fluoride medium
contain occluded F-. Previously, the location of fluoride was unambiguously known
only in two silica phases: octadecasil (AST)55 and nonasil (NON) synthesized with an
organometallic cation.56 In both cases F- was located in small cages within the SiO2
framework: [46] in AST and [415462] in NON. Here, cages are denoted according to the
number m of windows of n tetrahedra limiting the cage as [nmn’m’...]. Very recently, the
location of F- in small cages in two new silica phases has also been reported: [435261] in
ITQ-434 and [4354] in SSZ-23.50 In the case of MFI, contradictory reports proposed F-
was located in the main channel57 or in a small “interstice” within the framework
(actually a [415262] cage).58 However, very recent multinuclear MAS NMR experiments
demonstrated F- is very close to Si in MFI,59 supporting its location in a small cage
rather than in the channel. Thus, in five silica phases F- appears to be occluded in rather
small cages within the zeolite framework (Fig. 14). Each of these cages contains at least
one 4MR window and, very interestingly, the location of F- is always closer to this than
to any other window. Although no definitive conclusions can be drawn from these few
F- locations it may be argued that F- may exert some kind of structure-direction in the
synthesis of high and pure silica phases.60 This directing effect could be towards
structures containing small cages or a high density of 4MR windows.
On the other hand, very recent investigations using multinuclear MAS and
CPMAS NMR have shown that F- strongly interacts with framework Si, giving rise to

27
pentacoordinated [SiO4/2F]- in a large number of pure silica phases.59,61 In some cases F-
bonds to a single Si site at room temperature, while in some others there is a dynamic
situation with F- alternatively bonding to different Si atoms. On the other hand, recent
calculations suggest that the inclusion of F- in the [46] cage of octadecasil is
energetically favorable and that there is significant electron transfer from F- to the Si
atoms of the framework.62 The interaction between F- and the framework may thus
provide an additional stabilization energy that, together with the coulombic and van der
Waals interactions between the charged framework and the SDA cation, must be
considered when discussing on the relative stabilities of as-made silica phases. Thus, as
discussed above low framework density phases, usually presenting larger F-/SiO2 and
SDA+/SiO2 ratios, could in some cases be more stable than denser phases, even in the
absence of a very specific (geometrically and energetically) interaction between the
SDA and the framework. Clearly, a quantitation of the magnitude of these additional
interactions is necessary to understand many of the issues discussed in this paper.
Catalytic Applications
The fluoride route may afford the incorporation of elements other than silicon
into the framework, and this may give rise to active catalysts. Using the F method we
have introduced Al in relatively large amounts into the framework of, for instance,
zeolite Beta (Si/Al=6 to ∞),63 ITQ-4 (15 to ∞), ITQ-3 (20 to ∞),64 SSZ-23 (50 to ∞),
CHA (15 to ∞), ITQ-9 (40 to ∞) and CIT-5 (30 to ∞). We have also incorporated Ti in
several SiO2 phases by direct synthesis in fluoride medium and obtained active catalysts
for oxidation reactions using H2O2.65
Pure silica phases prepared in fluoride media are strictly hydrophobic due to its
SiO2 composition and the very low concentration of Si-OH groups. This has been

28
demonstrated in the case of SiO2 Beta by water adsorption measurements.66 The
superior hydrophobicity of this material compared to Beta materials containing Si-OH
groups was also shown by competitive adsorption of water/toluene and
water/methylcyclohexane.67 Introduction of Al or Ti in the framework increases the
hydrophilicity but the samples still show a far superior hydrophobicity than their
analogues prepared in hydroxide medium.66,68 The polarity of the catalyst may be of
great importance since a strong adsorption of a reactive or product may determine the
activity and/or the selectivity of the reaction.
For example, aluminosilicate zeolite Beta is an active and selective catalyst in
the acetalization of glucose to form alkylglucoside nonionic surfactants.68 In this
reaction two reactants with rather different polar character (glucose and the alcohol)
must diffuse inside the zeolite and, thus, the activity depends not only on the strength
and concentration of the acid sites but also on the adsorption properties of the catalyst.
The activity of the catalysts shows a maximum for intermediate Si/Al ratios and the
position of the maximum depends on the method of preparation of the zeolite. When
materials prepared in F- medium were compared with zeolites prepared in hydroxide
medium and dealuminated with HNO3 acid the maximum occurred at lower Si/Al ratios
and hence gave much higher activity for the former (Fig. 15). This was attributed to the
different polar character of the materials prepared by the different methods since those
prepared in hydroxide medium present a much higher concentration of Si-OH defect
sites (as demonstrated by 29 Si MAS NMR) and a much higher hydrophilicity (as shown
by the weight loss up to 523 K of the calcined hydrated materials) than those prepared
in fluoride medium.68
In the case of titanosilicate zeolite Beta, Si-OH sites are abundant in the OH-
materials and scarce in the F-materials as shown by 29Si MAS and CPMAS NMR

29
experiments. The OH-materials are correspondingly much more hydrophilic as
demonstrated by water adsorption measurements.66 This is believed to be the cause of
the enhanced activity and H2O2 selectivity of the F-zeolite in the epoxidation of
unsaturated fatty acids, since the performance of the hydrophilic OH-zeolite is limited
by the strong adsorption of the reactant through its polar end.66
Finally, silica materials may yield new catalyts by using a completely different
approach than introducing active heteroatoms into the structure. The versatile and
highly innovative approach was envisaged and demonstrated by Jones et al.69 who,
taking our method to synthesize SiO2 Beta with TEA+ as a basis, substituted a small
fraction of this SDA by a suitable organosilicon compound. This was incorporated into
the final product with its silicon atoms as a part of the tetrahedral framework. Due to
this, the organic moiety remained anchored to the structure within the hydrophobic
pores after elimination of TEA+ by chemical methods, and was subsequently
sulphonated to yield shape selective catalysts. The concept is important since appears as
a highly versatile route to prepare new catalysts using different organic moieties and
funtionalities and silica materials with different pore architectures and, hence, could
possible yield new shape-selective catalysis. Although the concept can be in principle
extended to materials containing Al or materials synthesized through the OH- route, the
use of synthetic methods as those discussed in previous sections would permit the
absence of other active sites ([AlO4]-H+, [AlO4]-Na+, Si-OH,...) which could show an
undesired selectivity. In essence, these materials may be regarded as organic catalysts
inside a molecular-sized SiO2 reactor providing a hydrophobic and spatially constrained
environment.
Which zeolite topologies will form SiO2 analogs?

30
We want to make a final point on the currently apparent incapacity to predict
which zeolites can possibly crystallize as SiO2 analogs. There seems to be a tendency to
look for topological factors that may hinder or allow the synthesis of SiO2 analogs. A
common belief just few years ago was that high silica materials need to have a low
density of 4MR and a high density of 5MR within each structures (like is the case in the
so-called “pentasils”). This belief is now discredited as several pure silica materials
completely lacking 5MR and with a high concentration of 4MR have been synthesized:
pure silica sodalite (SOD, first synthesized in ethylenglycol70 and soon after in water71),
SSZ-24 (AFI),21 octadecasil (AST)55 and now chabazite (CHA).14
Another attempt to derive a likelihood for SiO2 analogs from topological factors
was based on the loop configuration of tetrahedral atoms (that is, the way in which rings
are connected to a tetrahedral atom).72 It was proposed that some loop configurations
(as the ones depicted in Fig. 16, top left) were more favorable to be present in SiO2
analogs than others. Apparently, it was considered that loop configurations with a high
density of fused four rings would be less favorable for a SiO2 structure. This translated
into the prediction of a high improbability of several SiO2 analogs, including the
technologically important FAU and the CHA phase recently synthesized by us, both
presenting the same “unfavorable” loop configuration (Fig. 16). The synthesis of pure
silica CHA may be regarded then as good news not only because of its very low
framework density but also because the direct synthesis of pure and high-silica FAU
should not longer be considered as improbable on the basis of those topological
considerations. Furthermore, such syntheses may not require very specific SDA cations,
since CHA has been prepared by using an SDA which can also produce several other
phases, like STT, MWW,7 AFI21 and VET.74

31
Conclusions
We have observed that, frequently, if a structure-directing agent in F- medium
permits the crystallization of several phases a decrease in the H2O/SiO2 ratio favors the
crystallization of the least dense phase. We think that the observed trend is probably a
kinetic effect favoring metastable phases (which are, generally, the less dense phases) at
high supersaturations (presumably achieved at low water/silica ratios). However, we
have shown examples (ITQ-9/ITE and Beta/MTW) suggesting that this is not always
the case and that the relative stabilities which are of importance for the synthesis are
those of the SiO2/F/SDA composites embedded in a solution containing F and SDA at a
given temperature, rather than the stability of the isolated pure silica frameworks.
While much has been written concerning the structure-directing ability of
organic and inorganic cations,1 fluoride anions60 and heteroatoms (B, Al, Zn),1 to our
knowledge the degree of dilution of the starting mixture has never been considered as a
fundamental parameter in determining the phase selectivity in zeolite synthesis. We
proved that the systematic variation of the H2O/SiO2 ratio for given SDA cations is a
promising strategy for the discovery of new SiO2 phases. The synthesis of porous silica
phases reported here show that new opportunities have been opened up by our fluoride
method in concentrated reaction mixtures, which in turn may yield new low-density
phases in the near future.
Materials prepared in F medium show a low density of Si-OH groups
(connectivity defects). This is likely due to a combination of factors, like charge balance
between F- and SDA+, low synthesis pH, absence of significant Si-O-···HO-Si hydrogen
bonds and, possibly, HF produced on calcination. The low density of connectivity
defects in F-materials may have important implications on both the synthesis and
applications of pure and high silica porous materials. From the point of view of

32
synthesis, SDA with low C/N+ ratios may show a low ability to produce high silica
phases in OH medium because of the unstability in hydrothermal conditions of the
highly defective crystals produced. By contrast, the F route appears as a very feasible
route to prepare SiO2 phases even when using SDA with small C/N+ ratios.
The defect-free pure SiO2 materials are strictly hydrophobic. Introduction of Al
or Ti in the framework yield active catalysts that may show distinct activity and/or
selectivity in reactions involving moieties with different polarities. Active and shape
selective catalysts may also be produced by a very recent approach based on the
tethering of organic moieties to the silica framework during the synthesis.
Acknowledgments.- The authors greatly acknowledge financial support by the Spanish
CICYT (project MAT97-0723).

33
[1] R.F. Lobo, S.I. Zones and M.E. Davis, J. Inclusion Phenom. Mol. Recogn. Chem. 21 (1995) 47. [2] P.A. Barrett, E.T. Boix, M.A. Camblor, A. Corma, M.J. Díaz-Cabañas, S. Valencia and L.A. Villaescusa, in: Proceedings of the 12th International Zeolite Conference, eds. M.M.J. Treacy, B. Marcus, J.B. Higgins and M.E. Bisher (Materials Research Society, 1998) pp. 1495–1502. [3] Y. Kubota, M.M. Helmkamp, S.I. Zones and M.E. Davis, Microporous Mater. 6 (1996) 213. [4] J. Klinowski, T. Adrian Carpenter and J.M. Thomas, J. Chem. Soc. Chem. Commun. (1986) 956. [5] W.M. Meier, D.H. Olson and Ch. Baerlocher, Atlas of Zeolite StructureTypes (Elsevier, London, 1996). [6] IZA web site, http://www.iza-sc.ethz.ch/IZA-SC/. [7] M.A. Camblor, A. Corma, M.J. Díaz-Cabañas and Ch. Baerlocher, J. Phys. Chem. B 102 (1998) 44. [8] M.A. Camblor, C. Corell, A. Corma, M.J. Díaz-Cabañas, S. Nicolopoulos, J.M. González-Calbet and M. Vallet-Regí, Chem. Mater. 8 (1996) 2415. [9] W.J. Roth, C.T. Kresge, J.C. Vartuli, M.E. Leonowicz, A.S. Fung and S.B. McCullen, Stud. Surf. Sci. Catal. 94 (1995) 301. [10] R. Millini, G. Perego, W.O. Parker, G. Bellussi and L. Carluccio, Microporous Mater. 4 (1995) 221. [11] E.M. Flanigen and R.L. Patton, US Pat. 4,073,865 (1978). [12] H. Kessler, J. Patarin and C. Schott-Darie, Stud. Surf. Sci. Catal. 85 (1994) 75. [13] J.L. Guth, H. Kessler and R. Wey, in: New Developments in Zeolite Science and Technology, eds. Y. Murakami, A. Iijima and J.W. Ward (Elsevier, Amsterdam, 1986) p. 121. [14] M.J. Díaz-Cabañas, P.A. Barrett and M.A. Camblor, Chem. Commun. (1998) 1881. [15] M.A. Camblor, A. Corma and S. Valencia, Chem. Commun. (1996) 2365. [16] M.A. Camblor, A. Corma, P. Lightfoot, L.A. Villaescusa and P.A. Wright, Angew. Chem. Int. Ed. Engl. 36 (1997) 2659. [17] M.A. Camblor, A. Corma and L.A. Villaescusa, Chem. Commun. (1997) 749. [18] L.A. Villaescusa, P.A. Barrett and M.A. Camblor, Chem. Commun. (1998) 2329. [19] R. Althoff, K. Unger and F. Schüth, Microporous Mater. 2 (1994) 557. [20] P.A. Barrett, M.J. Díaz-Cabañas, M.A. Camblor and R.H. Jones, J. Chem. Soc. Faraday Trans. 94 (1998) 2475. [21] R.A. Van Nordstrand, D.S. Santilli and S.I. Zones, in: Perspectives in Molecular Sieve Science, ACS Symp. Series 368, eds. W.H. Flank and T.E. Whyte Jr., 1988, p. 236. [22] R.F. Lobo and M.E. Davis, Microporous Mater. 4 (1994) 61. [23] P. Wagner, M. Yoshikawa, M. Lovallo, K. Tsuji, M. Tsapatsis and M.E. Davis, Chem. Commun. (1997) 2179. [24] I. Petrovic, A. Navrotsky, M.E. Davis and S.I. Zones, Chem. Mater. 5 (1993) 1805. [25] N.J. Henson, A.K Cheetham and J.D. Gale, Chem. Mater. 6 (1994) 1647. [26] Y. Nakagawa, US Pat. 5,268,161 (1993). [27] P. Wagner, S.I. Zones, R.C. Medrud and M.E. Davis, 12th International Zeolite Conference, Baltimore, 1998. [28] M.A. Camblor, A. Mifsud and J. Pérez-Pariente, Zeolites 11 (1991) 792. [29] S. Ernst, P.A. Jacobs, J.A. Martens and J. Weitkamp, Zeolites 7 (1987) 458. [30] P. Concepción, J.M. López Nieto, A. Mifsud and J. Pérez-Pariente, Zeolites 16 (1996) 56.

34
[31] H.-X. Li and M.E. Davis, Catal. Today 19 (1994) 61. [32] M.E. Davis, C. Montes, P.E. Hathaway and J.M. Garces, Stud. Surf. Sci. Catal. 49 A (1989) 199. [33] S.I. Zones, Y. Nakagawa, G.S. Lee, C.Y. Chen and L.T. Yuen, Microporous and Mesoporous Mater. 21 (1998) 199. [34] P.A. Barrett, M.A. Camblor, A. Corma, R.H. Jones and L.A. Villaescusa, J. Phys. Chem. B 102 (1998) 4147. [35] Y. Nakagawa, G.S. Lee, T.V. Harris, L.T. Yuen and S.I. Zones, Microporous and Mesoporous Mater. 22 (1998) 69. [36] H. Koller, R.F. Lobo, S.L. Burkett and M.E. Davis, J. Phys. Chem. 99 (1995) 12588. [37] M.A. Camblor, A. Corma and S. Valencia, Microporous and Mesoporous Mater. 25 (1998) 59. [38] R.M. Barrer, Hydrothermal Chemistry of Zeolites (Academic Press, London, 1982) ch. 4. [39] R.J. Saxton, US Pat. 5,453,511 (1995). [40] J.C. van der Waal, M.S. Rigutto and H. van Bekkum, J. Chem. Soc. Chem. Commun. (1994) 1241. [41] P.R. Hari Prasad Rao, K. Ueyama, E. Kikuchi and M. Matsukata, Proceedings of the 12th International Zeolite Conference, eds. M.M.J. Treacy, B. Marcus, J.B. Higgins and M.E. Bisher (Materials Research Society, 1998) pp. 1515–1522. [42] S.I. Zones and A. Rainis, WOP 95/908793 (1995). [43] E.W. Valyocsik, US Patent 5,441,721 (1995). [44] J.L. Guth, H. Kessler, J.M. Higel, J.M. Lamblin, J. Patarin, A. Seive, J.M. Chezeau and R. Wey, in: Zeolite Synthesis, ACS Symp. Ser. 398, eds. M.L. Occelli and H. Robson, 1989, p. 176. [45] S.A. Axon and J. Klinowski, App. Catal. 81 (1992) 27. [46] J.M. Chézeau, L. Delmotte, J.L. Guth and M. Soulard, Zeolites 9 (1989) 78. [47] R.K. Iler, The Chemistry of Silica (Wiley, New York, 1979) ch. 3. [48] L.A. Villaescusa, P.A. Barrett and M.A. Camblor, Chem. Mater. 10 (1998) 3966. [49] A.R. George and C.R.A. Catlow, Zeolites 18 (1997) 67. [50] M.A. Camblor, M.J. Díaz-Cabañas, J. Pérez-Pariente, S.J. Teat, W. Clegg, I.J. Shannon, P. Lightfoot, P.A. Wright and R.E. Morris, Angew. Chem. Int. Ed. 37 (1998) 2122. [51] J.M. Thomas, J. Klinowski, S. Ramdas, B.K. Hunter and D.T.B. Tennakoon, Chem. Phys. Lett. 102 (1983) 158. [52] P.A. Barrett, M.A. Camblor, A. Corma, R.H. Jones and L.A. Villaescusa, Chem. Mater. 9 (1997) 1713. [53] S. Valencia, Ph.D. thesis, Universidad Politécnica de Valencia (1997). [54] R.F. Lobo, M. Tsapatsis, C.C. Freyhardt, I. Chan, C.Y. Chen, S.I. Zones and M.E. Davis, J. Am. Chem. Soc. 119 (1997) 3732. [55] P. Caullet, J.L. Guth, J. Hazm, J.M. Lamblin and H. Gies, Eur. J. Solid State Inorg. Chem. 28 (1991) 345. [56] G. van de Goor, C.C. Freyhardt and P. Behrens, Z. Anorg. Allg. Chem. 621 (1995) 311. [57] G.D. Price, J.J. Pluth, J.V. Smith, J.M. Bennett and R.L. Patton, J. Am. Chem. Soc. 104 (1982) 5971. [58] B.F. Mentzen, M. Sacerdote-Peronnet, J.L. Guth and H. Kessler, C. R. Acad. Sci. Paris 313 (1991) 177.

35
[59] H. Koller, A. W¨olker, H. Eckert, C. Panz and P. Behrens, Angew. Chem. Int. Ed. Engl. 36 (1997) 2823. [60] J.L. Guth, H. Kessler, P. Caullet, J. Hazm, A. Merrouche and J. Patarin, in: Proceedings 9th International Zeolite Conference, eds. R. von Ballmoos, J.B. Higgins and M.M.J. Treacy (Butterworth-Heinemann, London, 1993) pp. 215–222. [61] H. Koller, A. Wölker, L.A. Villaescusa, M.J. Díaz-Cabañas, S. Valencia and M.A. Camblor, J. Am. Chem. Soc. 121 (1999) 3368. [62] A.R. George and C.R.A. Catlow, Chem. Phys. Lett. 247 (1995) 408. [63] M.A. Camblor, A. Corma and S. Valencia, J. Mater. Chem. 8 (1998) 2137. [64] L.A. Villaescusa and M.A. Camblor, to be published. [65] M.J. Díaz-Cabañas, L.A. Villaescusa and M.A. Camblor, to be published. [66] T. Blasco, M.A. Camblor, A. Corma, P. Esteve, J.M. Guil, A. Martínez, J.A. Perdigón-Melón and S. Valencia, J. Phys. Chem. B 102 (1998) 75. [67] J. Stelzer, M. Paulus, M. Hunger and J. Weitkamp, Microporous and Mesoporous Mater. 22 (1998) 1. [68] M.A. Camblor, A. Corma, S. Iborra, S. Miquel, J. Primo and S. Valencia, J. Catal. 172 (1997) 76. [69] C.W. Jones, K. Tsuji and M.E. Davis, Nature 393 (1998) 52. [70] D.M. Bibby and M.P. Dale, Nature 317 (1985) 157. [71] J. Keijsper, C.J.J. Den Ouden and M.F.M. Post, Stud. Surf. Sci. Catal. 49 (1989) 237. [72] G.O. Brunner, Zeolites 13 (1993) 592. [73] J.A. Hriljac, M.M. Eddy, A.K. Cheetham, J.A. Donohue and G.J. Ray, J. Solid State Chem. 106 (1993) 66. [74] M.A. Camblor, M. Yoshikawa, S.I. Zones and M.E. Davis, in: Synthesis of Microporous Materials, eds. M.L. Occelli and H. Kessler (Marcel Dekker, New York, 1996) pp. 243–261.

36
Table 1. Crystalline SiO2 polymorphs found in nature.
Name (Code) FD (Si/1000Å3) Comments
Quartz 26.52a low pressure phase, most stable phase at T<870ºC
Tridymite 22.61a low pressure, most stable phase at T = 870-1470ºC
Cristobalite 23.33a low pressure, most stable phase at T> 1710ºC
Moganite 26.23a low pressure phase
Keatite 25.04a high pressure phase
Coesite 29.16b high pressure phase
Stishovite 42.95b high pressure, rutile-like structure (6-coordinated Si)
Melanophlogite (MEP) 18.9a SiO2/hydrocarbon host/guest compound
a from crystallographic data; b from the experimental density.

37
Table 2.- Some pure silica phases synthesized by our group through the fluoride route
Material Code FD (Si/nm3) Channel system Vμ (cm3/g)a SDA and representative synthesis conditions for the pure
SiO2 analog (T, H2O/SiO2)b
Chabazite CHA 15.4 3D, 8MR 0.30 TMAda+ (150ºC, 3.0)
Beta -c 15.6 3D, 12MR 0.20 TEA+, (140ºC, 2.5-14), M8BQ2+, M6BQ2+, M10BTM2+, p-
BBQ2+ (175-150ºC, 7.5), o-BBQ2+ (175ºC, 6.8), m-BBQ2+
(175ºC,7.5), BQ+ (175ºC, 3.6)
ITQ-3 ITE 16.3 3D, 8MR 0.23 DMABO+ (150ºC, 7.5), DEcDMP+ (150ºC, 14)
ITQ-4 IFR 17.0 1D, 12MR 0.22 BQ+ (150ºC, 15; 175ºC, 4.5), BQol+ (150ºC, 2.1), BDABCO+
(175ºC, 7.1)
SSZ-23 STT 17.0 2D, 7 + 9 MR 0.20 TMADa+ (150ºC, 7.5-10)
ITQ-9 -c 17.3 1D, 10MR 0.21 DMABO+ (150ºC, 3.75), DMTMP+ (135ºC, 3.5)
Octadecasil AST 17.3 only cages - t-BTMA+,b (175ºC, 2.4-15)
SSZ-24 AFI 17.8 1D, 12MR 0.12 MSPT+ (175ºC, 5-7.5)
CIT-5 CFI 18.2 1D, 14MR 0.13 MSPT+ (175ºC, 10-15)
SSZ-31 -c 18.7 1D, 12MR 0.08 DMABO+ (135-150ºC, 15), DEABO+ (150ºC, 15)
ZSM-12 MTW 19.4 1D, 12MR 0.09 M8BQ2+, M6BQ2+, p-BBQ2+ (175ºC, 15), M10BTM2+ (175-
150ºC, 14) aMicropore volume determined from the N2 adsorption isotherms by the t-plot method;b The reported conditions are not exhaustive but particular
examples in which the phase was obtained. c Beta and SSZ-31 are disordered intergrown structures and, as such, do not have a zeolite structure
code (the code *BEA only refers to one of the polymorphs present in zeolite Beta and, thus, shall not be used to name this material). No code has
been assigned yet to ITQ-9.

38
Table 3.- Synthesis using as-made zeolites as only silica sourcea
SDA Silica sourceb H2O/SiO2 Results Comparisonc
M8BQ2+ MTW (M8BQ2+) 7.5 MTW (+Beta), 48 days Beta, 7 days
M8BQ2+ Beta (M8BQ2+) 15 Beta + amorphous, 48 days MTW, 7 days
p-BBQ2+ MTW (p-BBQ2+) 7.5 MTW + Beta + amorphous, 30 days Beta, 4 days
p-BBQ2+ Beta (p-BBQ2+) 15 Tridymite, 30 days MTW, 5 days → Beta 9 days
a 175ºC, composition as shown in the experimental section. b The SDA used to produce the as-made silica phase used as silica source is given in
parentheses. c.- Results obtained using the same synthesis conditions but with hydrolyzed TEOS as silica source.

39
H2ORT
H2OHF
H2OΔ [SDAF]y[SiO2]SiO(2-x)(OH,F)x (gel) + F- + SDA+
OSi O
OO
+ 2 H2O + SDA++ OH- + SDA++ OH-SiO(2-x)(OH)x (gel) + 4 EtOH
Scheme 1.- Steps in the synthesis of pure silica microporous materials.

40
N+
N+
p-BBTM2+
N+
t-BTMA+
TEA+
N+
M8BQ2+
N+
N+
(CH2 )8
M6BQ2+
N+
N+
(CH2 )6
DMABO+
N+
DEABO+
N+
TMAda+
N+
M10BTM2+
N+
N+
(CH2 )10
BQ+
N+
o-BBQ+
N+
N+
p-BBQ+
N+
N+
MSPT+
N+
N
N+
DMTMP+BQol+
N+
HO
BDABCO+
N+
N
m-BBQ+
N+
N+
N+
DEcDMP+ Scheme 2.- Some structure-directing agents (SDA) used to produce new open SiO2 structures in fluoride media.

41
Captions to the figures.
Fig. 1.- 1H MAS NMR spectra in D2O of synthesis mixtures prepared by hydrolysis of
TEOS in aqueous solutions of four different structure-directing agents in hydroxide
form, after ethanol evaporation at room temperature. No resonances assignable to
protons in the methylene and methyl groups of ethanol (3.59ppm (c), 1.18 ppm (t)
respectively) are detected.
Fig. 2.- The framework density (FD, determined from experimental data) of synthetic
silica phases plotted against the year of the synthesis report. The phases are denoted by
the structural code assigned by the International Zeolite Association to accepted zeolite
topologies (except for ITQ-9, Beta and SSZ-31, see footnote c in Table 2).
Fig. 3.- Selectivity to silica CHA and STT as a function of water content, crystallization
temperature and time (horizontal arrows representing time). SDA = TMAda+.
Fig. 4.- Selectivity to pure silica phases using several diquats as SDA at 175ºC. The
more elongated diquats (top) may yield both Beta and MTW, depending on the water
content of the synthesis mixture. The SDA diquats at the bottom only give Beta,
presumably due to the bent shape of the diquats.
Fig. 5.- Selectivity to pure silica phases using N methylsparteinium as SDA at 175ºC.
Apparently, the observed effect is mainly related to the nucleation step, as seeding with
CFI produces CFI for any water/silica ratio. Actually, in order to observe the effect of
the water to silica ratio it is necessary to use “virgin” teflon liners, i.e. liners which have
never been used before to produce the CFI phase.20
Fig. 6.- The effect of the water content of the synthesis gel and the crystallization
temperature on the phase selectivity for pure silica structures using M8BQ2+ as SDA in
F- medium. Note that at 135ºC and H2O/SiO2 = 15 a mixture of Beta and MTW is

42
obtained but completely transforms to MTW by increasing the crystallization time
(Ostwald ripening). Also note, that this is not observed at higher temperatures.
Fig. 7.- Changes in the selectivity of DMABO+ to pure silica phases at 150ºC as a
function of the degree of dilution of the crystallizing mixture.
Fig. 8.- Crystallization fields at 175ºC for zeolites ITQ-4 and Beta as a function of the
Al and water content of the synthesis gel. SDA= BQ+.
Fig. 9.- An example of a phase selectivity change induced by Al in fluoride medium at
175ºC. The change occurs for Al contents well below the limit for Al incorporation in
SSZ-31 according to charge balance of this SDA (around Si/Al=20). Note that the
phases involved have no common structure projection, no common structural subunits
and a different pore window size.
Fig. 10.- The IFR topology formally regarded as made by relatively dense columns (two
of them depicted at the left) which are joined to each other by single Si-O-Si bridges,
with formation of single 4MR and 6MR. This yields the 3D, 4-connected framework
represented at the right side of the picture, where the channels run along the sequence of
single 4MR + 6MR formed. Topologies like IFR, where large portions of the structure
are joined together by single Si-O-Si bridges, may be unstable in the presence of a
comparatively small concentration of defects. This could make difficult its synthesis by
the OH- route (see text). The figure is not intended to represent the actual mechanism of
formation of IFR phases.
Fig. 11.- 29Si MAS NMR spectra of several calcined pure silica phases prepared by the
F- route. Note the low intensity or absence or Q3 resonances (-90 to -104 ppm region)
and the generally high resolution of Q4 resonances corresponding to Si[OSi]4 species in
different crystallographic sites.

43
Fig. 12.- 29Si MAS NMR of as-made ITQ-4 synthesized at 150ºC using different
compositions of the synthesis mixtures to obtained different final pH of the mother
liquors. The compositions (per mol of SiO2) were: 0.5 BQ+OH- : 0.5 HF : 15 H2O (final
pH = 8.5), 0.5BQ+OH- : 0.5 NH4F : 15H2O (final pH = 10.5) and 0.7 BQ+OH- : 0.5
NH4F : 15 H2O (final pH = 11.2).
Fig. 13.- The influence of the synthesis pH of the synthesis mixture on the
crystallization kinetics of pure silica ITQ-4 at 150ºC. The composition (per mol of
SiO2) and pH of the mother liquors at the end of the crystallization were: 0.5 BQ+OH- :
0.5 HF : 15 H2O, final pH = 8.5 ( ), 0.5BQ+OH- : 0.5 NH4F : 15H2O, final pH = 10.5
( ) and 0.7 BQ+OH- : 0.5 NH4F : 15 H2O, final pH = 11.2 ( ).
Fig. 14.- The known locations of fluoride in porous SiO2 phases, denoted by their
structural codes. In all the five cases F- is located inside a small cage within the SiO2
framework. The cage has been denoted by the number m of windows of n Si-Si edges as
[nmn’m’...]. F- is always nearer to a 4MR window (drawn with thicker lines) than to any
other window in the cage. In the case of NON56 at room temperature and STT50 at 160
K, direct F-Si bonds (as depicted) have been determined by diffraction techniques. For
STT only one of three very close locations of F- at 160 K have been drawn. See text for
references and for discussion on the location of F in MFI.
Fig. 15.- Initial rate in the acetalization of D-glucose with n-butanol of two zeolite Beta
series as a function of the Al content, according to reference 68, Table 7. The
hydrophilic Beta series ( , prepared by synthesis in OH- medium and dealumination
with HNO3), shows a maximum at lower Al contents and hence with lower activity than
the more hydrophobic series ( , prepared by the F- route).
Fig. 16.- Loop configurations (left) in relation to the likelihood of forming SiO2
analogs, the likelihood decreasing from top to bottom, according to reference 72. The

44
presence of bonds belonging to two 4MR (“fused rings”, common bonds depicted as
thicker lines) were considered to be unfavorable for SiO2 analogs and the direct
synthesis of some SiO2 phases such as FAU and CHA (right) were deemed improbable.
The recent synthesis of SiO2 CHA,14 containing C32 as the only one loop configuration,
demonstrates the inadequacy of such analysis. Thus, the predicted improbability of SiO2
FAU (containing the same loop configuration) may be questioned. The framework
density of SiO2 FAU has been calculated using the structural data of the SiO2 material
produced by dealumination procedures.73

45
N+
N+
N+
N
N+
MSPT
TMADa
BQ
DMABO
Fig.1 .- 1H MAS NMR spectra in D2O of mixtures prepared by hydrolysis of TEOS in aqueous solutions of four different structure-directing agents in hydroxide form, after ethanol evaporation at room temperature. No resonances assignable to protons in the methylene and methyl groups of ethanol (3.59ppm (c), 1.18 ppm (t) respectively) are detected.

46
1981 1986 1991 1996 2001
16
18
20
MFIMEL
MTW
MTN
MTTTON
DOH
SOD
NON
DDRSGT AFI
AST
FER
CFI
STT
SSZ-31
MWW
Beta
IFR
ITE
CHA
ITQ-9
FD
(S
i/nm
3 )
Year of first report
Fig. 2. The framework density (FD, determined from experimental data) of synthetic silica phases plotted against the year of the synthesis report. The phases are denoted by the structural code assigned by the International Zeolite Association to accepted zeolite topologies (except for ITQ-9, Beta and SSZ-31, see footnote c in Table 2).

47
Fig. 3.- Selectivity to silica CHA and STT as a function of water content, crystallization temperature and time (horizontal arrows representing time). SDA = TMAda+.

48
Fig. 4.- Selectivity to pure silica phases using diquats as SDA at 175ºC. The more ellongated diquats (top) may yield both Beta and MTW, depending on the water content of the synthesis mixture. The SDA diquats at the bottom only give Beta, presumably due to the bent shape of the diquats.

49
N+
N
AFI, 1D 12MR CFI, 1D 14MR
5-7.5 10-15H2O/SiO2=
Fig. 5.- Selectivity to pure silica phases using N methyl sparteinium as SDA at 175ºC. Apparently, the observed effect is mainly related to the nucleation step, as seeding with CFI produces CFI for any water/silica ratio. Actually, in order to observe the effect of the water to silica ratio it is necessary to use “virgin” teflon liners, i.e. liners which have never been used before to produce the CFI phase.20

50
Beta
Beta
Beta
MTW
MTW
Beta+MTW-->MTW
130
135
140
145
150
155
160
165
170
175
180
4 6 8 10 12 14 16 18 20
Tem
pera
ture
(ºC
)
H2O/SiO
2 ratio
Fig. 6.- The effect of the water content of the synthesis gel and the crystallization temperature on the phase selectivity for pure silica structures using M8BQ2+ as SDA in F medium. Note that at 135ºC and H2O/SiO2 = 15 a mixture of Beta and MTW is obtained but completely transforms to MTW by increasing the crystallization time (Ostwald ripening). Also note, that this is not observed at higher temperatures.

51
ITQ-3, 2D 8MRITQ-9, 1D 10MR
DMABO+
N+
H2O/SiO2 = 3.75 7.5 15
SSZ-31, 1D 12MR
Fig. 7.- Changes in the selectivity of DMABO+ to pure silica phases at 150ºC as a function of the degree of dilution of the crystallizing mixture.

52
2 4 6 8 10 12 14 16
0.00
0.02
0.04
0.06
0.08
0.10
0.12
ITQ-4 (IFR)
1D, 12MR
Beta
3D 12MR
Al/(
Al+
Si)
H2O/SiO
2 Fig. 8.- Crystallization fields at 175ºC for zeolites ITQ-4 and Beta as a function of the Al and water content of the synthesis gel. SDA= BQ+.

53
N+
DMABO
H2O/SiO2 = 15
Si/Al=∞
Si/Al=38
SSZ-31
ITQ-3
1D,12MR
2D, 8MR
Fig. 9.- An example of a phase selectivity change induced by Al in fluoride medium at 175ºC. The change occurs for Al contents well below the limit for Al incorporation in SSZ-31 according to charge balance of this SDA (around Si/Al=20). Note that the phases involved have no common structure projection, no common structural subunits and a different pore window size.

54
Si-OH + Si-OH Si-O-Si + H2O
Fig. 10.- The IFR topology may be formally regarded as made by relatively dense columns (two of them depicted at the left) which are joined to each other by single Si-O-Si bridges, with formation of single 4MR and 6MR. This yields the 3D, 4-connected framework represented at the right side of the picture, where the channels run along the sequence of single 4MR + 6MR formed. Topologies like IFR, where large portions of the structure are joined together by single Si-O-Si bridges, may be unstable in the presence of a relatively small concentration of defects. This could make difficult its synthesis by the OH- route (see text). The figure is not intended to represent the actual mechanism of formation of IFR phases.

55
-100 -110 -120
STT
SSZ-31
*MTW
CFI
AST
ITQ-9
IFR
ITE
Beta
CHA
Fig. 11.- 29Si MAS NMR spectra of several calcined pure silica phases prepared by the F-route. Note the low intensity or absence or Q3 resonances (-90 to -104 ppm region) and the generally high resolution of Q4 resonances corresponding to Si[OSi]4 species in different crystallographic sites.

56
-80 -90 -100 -110 -120 -130
8.5
10.5
11.2
pH
29Si δ /ppm from TMS
Fig. 12.- 29Si MAS NMR of as-made ITQ-4 synthesized at 150ºC using different compositions of the synthesis mixtures to obtained different final pH of the mother liquors. The compositions (per mol of SiO2) were: 0.5 BQ+OH- : 0.5 HF : 15 H2O (final pH = 8.5), 0.5BQ+OH- : 0.5 NH4F : 15H2O (final pH = 10.5) and 0.7 BQ+OH- : 0.5 NH4F : 15 H2O (final pH = 11.2).
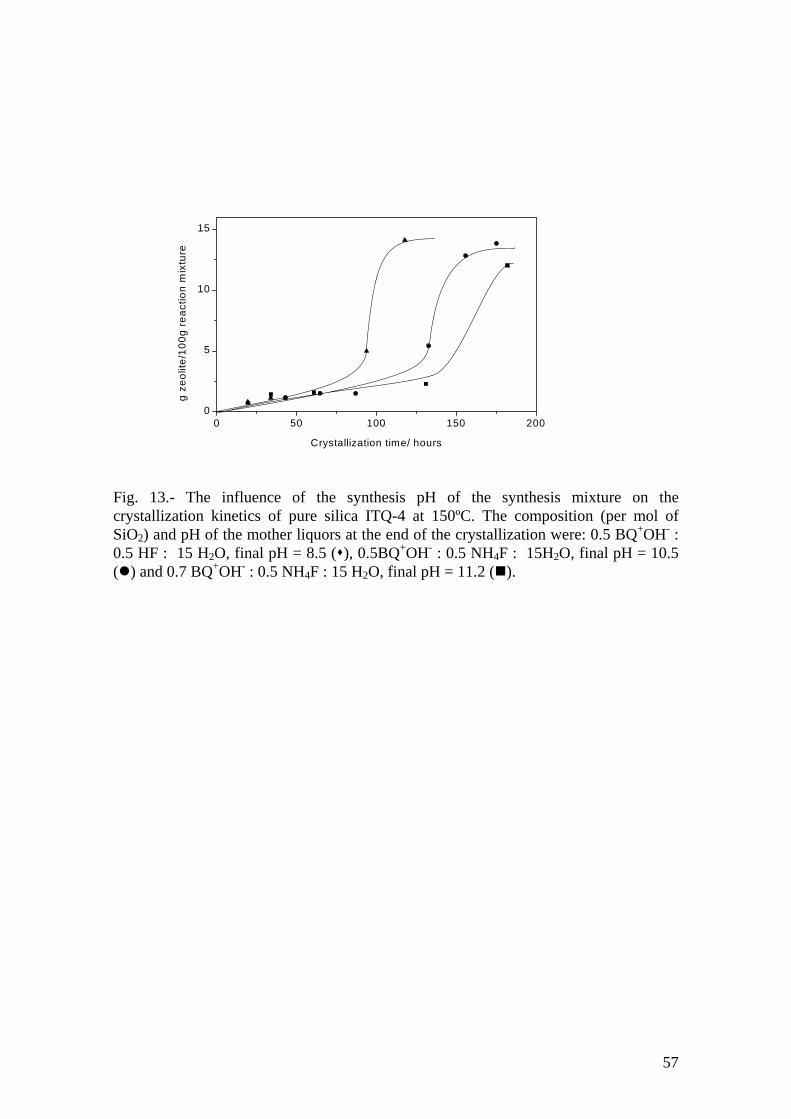
57
0 50 100 150 2000
5
10
15
g ze
olite
/100
g re
actio
n m
ixtu
re
Crystallization time/ hours
Fig. 13.- The influence of the synthesis pH of the synthesis mixture on the crystallization kinetics of pure silica ITQ-4 at 150ºC. The composition (per mol of SiO2) and pH of the mother liquors at the end of the crystallization were: 0.5 BQ+OH- : 0.5 HF : 15 H2O, final pH = 8.5 ( ), 0.5BQ+OH- : 0.5 NH4F : 15H2O, final pH = 10.5 ( ) and 0.7 BQ+OH- : 0.5 NH4F : 15 H2O, final pH = 11.2 ( ).

58
F
IFR [435261]
F
STT [4354]
F
F
F
NON [415462] MFI [415262]AST [46]
Fig. 14.- The known locations of fluoride in porous SiO2 phases, denoted by their structural codes. In all the five cases F- is located inside a small cage within the SiO2 framework. The cage has been denoted by the number m of windows of n Si-Si edges as [nmn’m’...]. F- is always nearer to a 4MR window (drawn with thicker lines) than to any other window in the cage. In the case of NON at room temperature and STT at 160 K, direct F-Si bonds (as depicted) have been determined by diffraction techniques. For STT only one of three very close locations of F- at 160 K have been drawn. See text for references and for discussion on the location of F in MFI.

59
0 1 2 3 4 5
2
3
4
5
6
7
8
r 0 x 1
04 (m
ol/m
in g
)
Al/uc
Fig. 15.- Initial rate in the acetalization of D-glucose with n-butanol of two zeolite Beta series as a function of the Al content, according to reference XX, Table 7. The hydrophilic Beta series ( , prepared by synthesis in OH- medium and dealumination with HNO3), shows a maximum at lower Al contents and hence with lower activity than the more hydrophobic series ( , prepared by the F- route).

60
B2 A Z
B21
C33 C32
D43 C34 D24
FAU 3D, 12MRFD=13.5 Si nm-3
CHA 3D, 8MRFD=15.4 Si nm-3
Fig. 16.- Loop configurations (left) in relation to the likelihood of forming SiO2 analogs, the likelihood decreasing from top to bottom, according to reference 72. The presence of bonds belonging to two 4MR (“fused rings”, commom bonds depicted as thicker lines) were considered to be unfavorable for SiO2 analogs and the direct synthesis of some SiO2 phases such as FAU and CHA (right) were deemed improbable. The recent synthesis of SiO2 CHA (14), containing C32 as the only one loop configuration, demonstrates the inadequacy of such analysis. Thus, the predicted improbability of SiO2 FAU (containing the same loop configuration) may be questioned. The framework density of SiO2 FAU has been calculated using the structural data of the SiO2 material produced by dealumination procedures.73