synthese und strukturoptimierung konformativ fixierter ... · 3 assignment of tasks 19 4...
TRANSCRIPT

Synthese und Strukturoptimierung konformativ fixierter Dopamin- und
Serotoninmimetika
Den Naturwissenschaftlichen Fakultäten
der Friedrich-Alexander-Universität Erlangen-Nürnberg
zur
Erlangung des Doktorgrades
vorgelegt von
Miriam Dörfler aus Bayreuth

Als Dissertation genehmigt von den Naturwissen-
schaftlichen Fakultäten der Universität Erlangen-Nürnberg
Tag der mündlichen Prüfung: 19.05.2008
Vorsitzender der
Promotionskommission: Prof. Dr. Eberhard Bänsch
Erstberichterstatter: Prof. Dr. Peter Gmeiner
Zweitberichterstatter: Prof. Dr. Reinhard Troschütz

Die vorliegende Arbeit entstand im Department Chemie und Pharmazie der Friedrich-
Alexander-Universität Erlangen-Nürnberg unter der Leitung von
Herrn Professor Dr. Peter Gmeiner. Ihm danke ich für das mir entgegengebrachte Vertrauen und die hervorragende Be-
treuung. Besonders dankbar bin ich ihm für die Motivation, die mir vor allem in den
ersten Monaten meiner Promotion geholfen hat, an meine Fähigkeiten zu glauben
und mit der Promotion fortzufahren. Seine Unterstützung, sowie zahlreiche Anre-
gungen und fachliche Diskussionen haben das Entstehen dieser Arbeit erst möglich
gemacht.
Herrn Prof. Dr. R. Troschütz danke ich für die Übernahme des Zweitgutachtens und
Herrn Prof. Dr. A. Ludwig für die Übernahme der Nebenfachprüfung in Pharmako-
logie.
Dr. Stefan Löber und Dr. Jürgen Einsiedel danke herzlich ich ihre Ratschläge und
Bereitschaft zu Diskussionen bei synthetischen Fragestellungen sowie ihre
Hilfsbereitschaft bei Problemen mit den HPLC-Anlagen. Sie nahmen sich immer Zeit
für mich, auch wenn sie selbst viel Arbeit hatten.
Bei Dr. Harald Hübner bedanke ich mich für die hilfreichen Diskussionen sowie die
Durchführung und Auswertung der Rezeptorbindungsstudien. Dr. Reiner Waibel
danke ich für seine Hilfsbereitschaft bei der Lösung spektroskopischer und chromato-
graphischer Probleme. Dr. Wolfgang Utz und Steffen Härterich möchte ich für die
schnelle und kompetente Hilfe bei Rechner-Problemen aller Art danken.
Meinen Laborkollegen Dr. Stefan Löber, Dr. Christian Kormann, Dr. Marika Skultety,
Dani Huber, Julia Kühhorn und Dr. Ismail Salama sowie Roswitha Höfner-Stich,
Melanie Hertel, Anja Eckert und Franzi Fischer gebührt besonderer Dank. Ich habe
unheimlich gerne mit ihnen zusammengearbeitet und die freundliche Atmosphäre in
unserem Labor genossen. Für ihre Hilfsbereitschaft, ihre Freundschaft und ihre auf-
munternden Worte (und Glückskekse) danke ich Dr. Marika Skultety ganz herzlich.
Meinen Kollegen bei der Betreuung des 3. Semesters, Dr. Stefan Löber, Dr. Holger
Bittermann, Dr. Marika Skultety, Stefan Bollinger und Dani Huber danke ich für die
gute Zusammenarbeit.

Herzlicher Dank gebührt auch meinen Kondoktoranden und Kollegen für die
freundschaftliche und harmonische Atmosphäre sowie die schöne Zeit, die wir
miteinander verbringen durften. Ich werde mich immer gerne an unsere Kaffee- und
Tee-Pausen im Kaffeelabor erinnern. Namentlich soll hier die ehemalige Teerunde
mit Dr. Holger Bittermann, Dr. Jürgen Einsiedel, Steffen Härterich, Dr. Susanne
Lochner, Dr. Karin Schlotter und Dr. Marika Skultety sowie die Senseo-Connection
mit Stefan „Bärchen“ Bollinger, Angela Götz, Matthias Horner, Daniela Huber, Laurin
Leeb, Dr. Stefan Löber, Susanne Koschatzky, Julia Kühhorn, Andrea Pinsker und
Igor Usai sowie Nicole „Mausi“ Wenzel genannt werden.
Herzlicher Dank gilt auch Dr. Susanne Lochner und Dr. Karin Schlotter, bei denen ich
schon als Studentin ins Labor hineinschnuppern durfte, die mich motiviert haben zu
promovieren und die ich auch während meiner Arbeit gerne um Rat fragen durfte.
Für das geduldige Korrekturlesen danke ich besonders Dr. Stefan Löber, Dani Huber,
meinem Freund Flo und meiner Mama.
Für die Aufnahme von Massen- und NMR-Spektren bedanke ich mich bei Sonja
Burkhardt, Roswitha Höfner-Stich und Anke Seitz. Außerdem danke ich den
Mitarbeiterinnen der Bio-Abteilung, Petra Hübner, Beatrix Jung, Jenny Schenker,
Petra Schmitt, Heike Szczepanek und Sabine Würdemann, für die Durchführung der
pharmakologischen Tests. Großer Dank geht auch an Claus Fischer und Alfred
Zillich-Balthasar für ihre schnelle Hilfe bei technischen Problemen sowie an Rainer
Köppl und Karlheinz Thomas für die Bestellung und Beschaffung von Materialien.
Monika Bögelein und Martina Schaper danke ich für ihre Hilfsbereitschaft und die
netten Unterhaltungen.
Meinem lieben Flo gilt besonders großer Dank für die uneingeschränkte Unter-
stützung, den Trost bei Misserfolgen, das Mitfreuen bei Durchbrüchen, die Motivation
durchzuhalten und das Nähen meines Daumens.
Der größte Dank gilt meiner Familie, deren bedingungsloser und uneingeschränkter
Unterstützung ich mir schon mein ganzes Leben lang sicher sein konnte. Ohne Euch
wäre das hier nicht möglich gewesen. Es gibt nicht viele Worte, die ausdrücken
können, was ich Euch verdanke. Ich liebe Euch von ganzem Herzen!

Teile dieser Arbeit wurden bereits veröffentlicht:
Dörfler, M., Hübner, H., Gmeiner, P.
Dopamine Receptor Ligands Featuring Enynes As Nonaromatic Benzene Bioiso-
steres
DPhG/GDCh Annual Meeting. Erlangen, October 10-13, 2007
Dörfler, M., Hübner, H., Gmeiner, P.
Enynes And Triazoles As Bioisostere In Dopamine Receptor Ligands
DPhG/GDCh Annual Meeting: Frontiers in Medicinal Chemistry. Regensburg, March
2-5, 2008


Meinen Eltern in Liebe und Dankbarkeit


Inhaltsverzeichnis
Inhaltsverzeichnis
1 Einleitung 1 2 Entwicklung D3-selektiver Liganden 5 1. Aminotetraline und Analoge 5
2. Aminoindane 10
3. Arylcarboxamid-substituierte Aminotetraline und deren Analoge 11
4. Benzamide und Analoge 13
5. Phenylpiperazine 14
6. Hybride 18
3 Aufgabenstellung 19 4 Arylcarboxamidoalkyl-substituierte Aminoindane und –tetraline 22 1. Strukturvariationen der Arylcarboxamid-Einheit 22
2. Strukturvariationen der Aminoindan-Einheit 34
3. Strukturvariationen durch bioisosteren Ersatz der Carboxamid-
Funktionalität 38
5 Alkinylcyclohexen als Tetralin-Bioisoster 41 1. Alkinylcyclohexene mit Amid-Partialstruktur 41
2. Alkinylcyclohexene mit Triazol-Partialstruktur 46
6 Austausch der Indan- bzw. Tetralin-Einheit durch „Dummy- Substituenten“ 50 7 2-Arylethyl-substituierte Aminoindane 54 8 Zusammenfassung 57 9 Experimenteller Teil 81 10 Literaturverzeichnis 176 11 Abkürzungsverzeichnis 185 12 Lebenslauf 188

Index of contents
Index of contents
1 Introduction 1 2 Development of D3-selektive ligands 5 1. Aminotetralines and analogues 5
2. Aminoindans 10
3. Arylcarboxamido-substituted aminotetralines and analogues 11
4. Benzamides and analogues 13
5. Phenylpiperazines 14
6. Hybrids 18
3 Assignment of tasks 19 4 Arylcarboxamidoalkyl-substituted aminoindans and –tetralines 22 1. Structural variations of the arylcarboxamide moiety 22
2. Structural variations of the aminoindane moiety 34
3. Structural variations by bioisosteric replacement of the carboxamide
moiety 38
5 Alkynylcyclohexene as a bioisostere for tetraline 41 1. Alkynylcyclohexenes with amide substructure 41
2. Alkynylcyclohexenes with Triazol substructure 46
6 Replacement of the indan or tetraline moiety by “dummy substituents” 50 7 2-Arylethyl-substituted aminoindans 54 8 Summary 57 9 Experimental part 81 10 Bibliography 176 11 Table of abbreviations 185 12 Curriculum vitae 188

Einleitung
1
1. Einleitung Wirkstoffe, die in das dopaminerge Neurotransmittersystem eingreifen, werden, je
nachdem, ob sie Dopamin-Rezeptoren aktivieren (Agonisten) oder blockieren (Ant-
agonisten), zur Behandlung verschiedener Krankheiten eingesetzt.
Mit den Agonisten (Abb. 1) Apomorphin 1, den Ergolin-Derivaten Lisurid 2, Bromo-
criptin 3 und Pergolid 5 sowie Rotigotin 6, Pramipexol 7 und Roponirol 8 behandelt
man die Parkinson’sche Krankheit und Dyskinesien.1 Bromocriptin 3 wird außerdem
als Prolaktin-Inhibitor zum Abstillen eingesetzt1, während Apomorphin 1 als
Emetikum1 bzw. in den letzten Jahren auch bei erektiler Dysfunktion2 angewandt
wird. Zur Behandlung des Restless-Legs-Syndroms wird Pramipexol 7 eingesetzt.3
Auch Rotigotin 6 scheint für diese Indikation geeignet zu sein.4 Quineloran 9,
Quinpirol 10 und PD128907 4 werden als pharmakologische Tools genutzt, wobei
der Vollagonist Quinpirol 10 als Referenzsubstanz in Funktionstests eingesetzt wird.
N
OHOH
H N
NHNH
N
OH N
NH
Br
NH
O
H
ON
N
O
OH
OH
N
NHS
H
H
N
SNH2
NH
NH
ON
N
N
N
NH2
H
H
N
NNH
H
H
N
OH
S
O
NO
OHH
HPr
10 QuinpirolD1: >1000(f)
D2: 3,3(g) / 39 + 4000(c)
D3: 5,0(h) / 25 + 600(c)
D4: 18(i) / 1,8 + 53(c)
5-HT1A: >1000(j)
α1: >1000(k)
2 LisuridD1: 62(f)
D2: 0,3(g) / 1,9(c)
D3: 1,7(h) / 0,14(c)
D4: 3,2(i)
5-HT1A: 0,5(j)
α1: 55(k)
1 ApomorphinD1: 101(a)
D2: 32(b) / 590(c)
D3: 26(b) / 59(c)
D4: 2,6(b)
5-HT1A: 121(d)
α1: 3000(e)
4 (R)-(+)-PD128907D2: 18 + 540(c)
D3: 1,4 + 160(c)
5 PergolidD1: 1200(f)
D2: 4,0(g) / 1,2 + 28(c)
D3: 4,0(h) / 0,85 + 7,8(c)
D4: 6,2(i)
5-HT1A: 5,7(j)
α1: 890(k)
6 RotigotinD1: 360(l)
D2: 11(l) / 20(m)
D3: 0,94(l) / 4,0(n)
D4: 55(o)
5-HT1A: 100(p)
7 Pramipexol D1: >1000(f)
D2: 6,9(g) / 28 + 2500(c)
D3: 0,9(h) / 1,1 + 41(c)
D4: 15(i) / 6,9 + 110(c)
5-HT1A: >1000(j)
α1: >1000(k)
8 RopinirolD1: >1000(f)
D2: 7,2(g)
D3: 19(h)
D4: >1000(i)
5-HT1A: >1000(j)
α1: >1000(k)
9 QuineloranD2: 11 + 550(c)
D3: 0,93 + 15(c)
3 Bromocriptin D1: 3400(f)
D2: 10(g)
D3: 87(h)
D4: 370(i)
5-HT1A: 24(j)
α1: 18(k)
Abb. 1: Dopamin-Agonisten, Ki-Werte in nM (a) gemessen gegen [3H]SCH23390, D1-Rezeptoren5 (b) gemessen gegen [3H]Spiperon, D2-, D3 und D4-Rezeptoren5 (c) gemessen gegen [3H]Spiperon, humane D2short-, D3 und D4.4-Rezeptoren (eigene Daten)

Einleitung
2
(d) gemessen gegen [3H]8-OH-DPAT, 5-HT1A-Rezeptoren5 (e) gemessen gegen [3H]Prazosin, α1-Rezeptoren5 (f) gemessen gegen [3H]SCH23390, Ratten D1-Rezeptoren in CHO-Zellen6 (g) gemessen gegen [3H]U-86170, Ratten D2l-Rezeptoren in CHO-Zellen6 (h) gemessen gegen [3H](R)-(+)-7-OH-DPAT, Ratten D3-Rezeptoren in CHO-Zellen6 (i) gemessen gegen [3H]Spiperon, humane D4.2-Rezeptoren in HEK 293 Zellen6 (j) gemessen gegen [3H]8-OH-DPAT, humane 5-HT1A-Rezeptoren in CHO-Zellen6 (k) gemessen gegen [3H]Prazosin, α1-Rezeptoren in homogenisiertem Ratten-Cortex6 (l) gemessen am Ratten-Rezeptor in CHO Zellen7 (m) gemessen gegen [3H]N-0437 am humanen D2L-Rezeptor in CHO K-1 Zellen8 (n) gemessen gegen [3H]Spiperon am humanen Rezeptor in CHO K-1 Zellen8 (o) gemessen gegen [3H]Spiperon am humanen D4.2-Rezeptor in CHO K-1 Zellen8 (p) gemessen an Rinder-Caudatum-Membranen7
Antagonisten (Abb. 2) sind als Neuroleptika bei Schizophrenien und Angstzuständen
wirksam. Dabei unterscheidet man, je nachdem welche Symptome der
Schizophrenie von den Arzneistoffen gelindert werden können, zwischen „klassi-
schen“ und atypischen Neuroleptika.1 Zu den Ersteren zählen die Substanzklassen
der Phenothiazine (z. B. Chlorpromazin 11), Thioxanthene (z. B. Chlorprothixen 12),
Butyrophenone (z. B. Haloperidol 13) und die Diphenylbutylpiperidine (z. B. Pimozid
15). Diese Stoffe vermögen vor allem die Positiv-Symptome der Psychose, wie
Halluzinationen, Wahn, motorische Unruhe, Bewegungsstereotypien, Bewegungs-
und Sprachlosigkeit sowie Antriebshemmung zu lindern.1 Als Nebenwirkungen treten
oft extrapyramidalmotorische Symptome auf.1 Atypische Neuroleptika rufen diese
Nebenwirkungen kaum oder in deutlich geringerem Umfang hervor.1 Außerdem
greifen sie nicht nur in die Positiv- sondern auch in die Negativsymptomatik, mit
Störungen des Denkens und der Sprache sowie Affektivitäts- und
Persönlichkeitsstörungen, ein.1 Zu den atypischen Neuroleptika gehören die
tricyclischen Verbindungen Clozapin 17 und Olanzapin 18, die neben D4- und D2-
Rezeptoren auch serotoninerge 5-HT2-, Muscarin-, adrenerge α1- und Histamin H1-
Rezeptoren blockieren. Risperidon 19, zeigt ebenfalls an mehreren
Rezeptorsystemen antagonistische Eigenschaften.1 Aripiprazol 16 werden stabilisie-
rende Eigenschaften für dopaminerge und serotoninerge Systeme zugeschrieben.9
Dabei ist es als Agonist für präsynaptische und als Antagonist für postsynaptische
D2-Rezeptoren beschrieben.10, 11 Ebenfalls als atypische Neuroleptika im Handel
befindliche Arzneistoffe sind die Benzamide Sulpirid 20 und Amisulprid 21.1 Weitere
Benzamide wie Racloprid 22, Nemonaprid 23 und Nafadotrid 24 sowie das

Einleitung
3
Butyrophenon Spiperon 14 sind nicht als Medikamente im Handel, sondern werden
als pharmakologische Tools eingesetzt. Racloprid 22 und Spiperon 14 werden
beispielsweise als Radioliganden in Bindungstests eingesetzt.
N
NH
ClN
N
N
NH
ClN
N
S
N
N
ON
N
ON
F
O
SNH
O
NH2
O ON
O
SNH
OO ON
NH2
O
ClNH
O
N
Cl
OH
O
ClNH
O
NH
NNH
OO
N
CN
F
ON
OH
ClN
S
Cl
N
S
Cl
N
F
ON
N
NHO
F
N
F
N
NHO
NH
ON
O
N
ClCl
13 HaloperidolD1: 78(c)
D2: 0,87(d)
D3: 8,4(d)
D4: 5,9(d)
5-HT1A: 1202(a)
α1: 16(b)
12 ChlorprothixenD2: 3,3(a)
D4: 0,64(a)14 SpiperonD2: 0,16(e)
α1: 1,2(f)
17 Clozapin D1: 420(c)
D2: 28(d)
D3: 960(d)
D4: 16(d)
5-HT1A: 460(g)
α1: 10(h)
18 Olanzapin D2: 58(a)
D3: 72(a)
D4: 63(a)
5-HT1A: 2063(a)
α1: 25(b)
19 RisperidonD1: 60,6(a)
D2: 4,9(a)
D3: 12,2(a)
D4: 7,12(a)
5-HT1A: 427(a)
α1: 1,6(b)
20 SulpiridD1: 50000(c)
D2: 51(d)
D3: 120(d)
D4: 2100(d)
21 Amisulprid D2: 1,3(a)
D3: 2,4(a)
D4:>1000(a)
5-HT1A: 5,6(a)
α1: inaktiv(b)
22 RaclopridD2: 2,6(d)
D3: 14(d)
D4: 7000(d)
23 NemonapridD2: 0,16(i)
D3: 0,16(i)
D4: 0,21(i)
24 NafadotridD2: 3,0(j)
D3: 0,31(j)
15 PimozidD1: >10000(a)
D2: 2,51(a)
D3: 2,84(a)
D4: 1,8(a)
5-HT1A: 650(a)
α1: 76(f)
11 ChlorpromazinD1: 112(a)
D2: 5,4(a)
D3: 5,0(a)
D4: 15,9(a)
5-HT1A: 3115(a)
α1: 1,6(b)
16 AripirazolD1: 1100(c)
D2: 2,7(d)
D3: 4,5(d)
D4: 57(d)
5-HT1A: 66(g)
α1: 17(h)
Abb. 2: Dopamin-Antagonisten, Ki in nM (a) aus Review12 (b) gemessen gegen [3H]Prazosin, α1-Rezeptoren in Ratten-Cortex13 (c) gemessen gegen [3H]SCH23390, bovine D1-Rezeptoren in CHO-Zellen (eigene Daten) (d) gemessen gegen [3H]Spiperon, jeweils humaner D2s-, D3- und D4.4-Rezeptor in CHO-Zellen (eigene
Daten) (e) gemessen gegen [3H]Spiperon, humaner Caudate nucleus14, 15 (f) gemessen gegen [3H]Prazosin, humanes Gehirn14, 15 (g) gemessen gegen [3H]8-OH-DPAT, porcine 5-HT1A-Rezeptoren (eigene Daten)

Einleitung
4
(h) gemessen gegen [3H]Prazosin, porcine α1-Rezeptoren (eigene Daten) (i) gemessen gegen [125I]Iodsulpirid, Ratten D2- und D3-Rezeptoren in CHO Zellen16 (j) gemessen gegen [125I]Iodsulprid, D2long- und D3-Rezeptoren in CHO Zellen17
Die sich heute auf dem Markt befindlichen Dopamin-Agonisten und –Antagonisten
verhalten sich relativ unselektiv. Sie sprechen sowohl mehrere Dopamin-Rezeptor-
Subtypen als auch andere G-Protein gekoppelte Rezeptoren (GPCRs), wie beispiels-
weise Serotonin- (5-HT-) oder adrenerge α1-Rezeptoren, an.2
Um Nebenwirkungen, die durch die unerwünschte Bindung an anderen Zielstrukturen
bedingt sind, zu vermeiden, versucht man heute möglichst selektive Liganden zu
entwickeln. Diese Bemühungen beziehen sich auch auf den D3-Rezeptor.
Man nimmt an, dass die extrapyramidalmotorischen Nebenwirkungen von Neuro-
leptika vor allem auf die Blockade von D2-Rezeptoren zurückzuführen sind, ihre
antipsychotische Wirkung aber auf ihrem Antagonismus an D3-Rezeptoren beruht.18
Daher möchte man besonders D3-selektive Liganden herstellen, was aufgrund der
großen strukturellen Ähnlichkeit der Bindungstaschen der Dopamin D2- und D3-
Rezeptoren19, 20 jedoch sehr schwierig ist. Trotzdem ist in den letzten Jahren eine
Entwicklung hin zu selektiven D3-Liganden gelungen, die im folgenden Kapitel
beschrieben werden soll.

Entwicklung D3-selektiver Liganden
5
2. Entwicklung D3-selektiver Liganden 2.1. Aminotetraline und Analoge 2.1.1. Aminotetraline Der natürliche Neurotransmitter Dopamin kann laut Cannon21 als α- oder als β-
Konformer vorliegen (Abb. 3), wobei nur die meta-Hydroxy-Funktion für die dopami-
nerge Aktivität essentiell ist und unter den N-Alkyl-substituierten Verbindungen
diejenigen mit einer n-Propyl-Gruppe am aktivsten sind.
NH2
OH
OH NH2
OHOH
NR2OH NR2
OH
α-Konformer 25a β-Konformer 25b
26a 26b Abb. 3: Dopamin-Konformere
Die formale konformative Fixierung der Ethylen-Kette des Dopamins in einem
annelierten Sechsring führt zur Substanzklasse der Aminotetraline 27 (Abb. 4).
NRR'
X
NRR'
X
27 Abb. 4: formale Entstehung der Aminotetraline
Dabei entspricht das α-Konformer dem 6,7-Dihydroxy- und das β-Konformer dem
5,6-Dihydroxy-Derivat des 2-Aminotetralins. Letzteres zeigt im Tierversuch eine deut-
lich höhere dopaminerge Aktivität.22, 23
O-Methylierte Aminotetraline zeigen im Vergleich zu den Hydroxy-Analogen eine
verminderte Aktivität.8, 22, 24 Da Letzere aber bei peripherer Applikation wirksam sind,
wird eine Bioaktivierung hin zum aktiven Hydroxylderivat vermutet.22, 24

Entwicklung D3-selektiver Liganden
6
Unter den mono-hydroxylierten Dipropylaminotetralinen (DPATs) ist das 7-substi-
tuierte Derivat (7-OH-DPAT; 27b) am D3-selektivsten25, wobei das (R)-(+)-Enan-
tiomer deutlich aktiver und somit das Eutomer ist.8 Im Gegensatz dazu ist bei 5-OH-
DPAT 27a das (S)-(-)-Isomer die aktivere Form mit gemischtem D2/D3-Affinitätsprofil8
(Tab. 1). Karlsson et al.26 ordnen dem Eutomer starke D2-agonistische und dem (R)-
Enantiomer schwache D2-antagonistische Eigenschaften zu. Das unsubstituierte
DPAT hat je nach absoluter Konformation gemischte agonistische bzw. partial-
agonistische Aktivität an Rezeptoren aus der D2-Familie sowie an 5-HT1A-Rezep-
toren.27
NPr2
X
X D2L D3 D2/D3
(S)-27a (S)-(-)-5-OH 6 0,54 11
(R)-27b (R)-(+)-7-OH 56 0,57 98
Tab. 1: Rezeptoraffinitäten von 5-OH- und 7-OH-DPAT; gemessen gegen [3H] Spiperon; jeweils humaner
Rezeptor in CHO K-1 Zellen, Ki in nM8
Auch die Substitution der 2-Aminofunktion spielt für die dopaminerge Aktivität eine
entscheidende Rolle: Die Di-n-propyl-Substitution ist am effektivsten, während
kleinere Reste wie Methyl und Ethyl schwächer aktiv22, 24, 25 und Butyl, Pentyl,
i-Propyl sowie cyclische Amine sogar inaktiv sind.22, 24 Primäre und sekundäre Amine
sind schwächer wirksam8 oder inaktiv22. Durch Ersetzen der primären Aminfunktion
durch ein N,N-di-n-propyl-substituiertes Amin kommt es beim 5-Hydroxy-
Aminotetralin zu einem deutlichen Anstieg der D2- und D3-Aktivität, während beim
7-Hydroxyderivat keine entsprechend deutliche Veränderung zu sehen ist.8 Zusam-
menfassend kann festgestellt werden, dass sich ein tertiäres Amin mit mindestens
einer Propylseitenkette positiv auf die dopaminerge Aktivität auswirkt. Diese
Propylseitenkette füllt eine bestimmte Struktur innerhalb der Bindungstasche optimal
aus, die auch als „Propyl-Cleft“ bezeichnet wird.25
Weitere Variationen einer der n-Propyl-Seitenketten führen zu Substituenten mit
terminalen π-Systemen wie in den Verbindungen 2828, 29 und 2930 (Abb. 5).

Entwicklung D3-selektiver Liganden
7
NR
X
I
S
28
296: X = 5-OH: Rotigotin
Abb. 5: Seitenkettenvariationen
Unter den Liganden der Struktur 28 ist das (R)-(+)-trans-7-Hydroxy-2-[N-propyl-N-(3’-
iodpropen-2’-yl)amino]tetralin (7-OH-PIPAT) D3-selektiv, während 5-OH-PIPAT eine
höhere D3-Affinität, aber keine Selektivität gegenüber D2 aufweist.28, 29
Rotigotin 6 ähnelt als D3/D2/D1-Agonist in seinem Bindungsprofil dem natürlichen
Neurotransmitter Dopamin. Die höchste Affinität besitzt es an D3- und D2-Rezep-
toren. Es interagiert aber auch mit D1-, D4-, 5-HT1A- und α2-Rezeptoren (Abb. 1).7, 30
2.1.2. Bioisostere DPATs Da die phenolischen Hydroxyl-Gruppen der OH-DPATs in vivo sehr schnell durch
Konjugation mit Glucuronsäure metabolisiert und anschließend über die Nieren
ausgeschieden werden, besitzen die OH-DPATs nur eine sehr geringe orale
Bioverfügbarkeit und eine begrenzte Wirkdauer.31 Um die schnelle Metabolisierung in
vivo zu vermeiden, versucht man, diese Strukturen durch bioisosteren Austausch zu
ersetzen.31 Dies sollte möglichst ohne Verlust der biologischen Aktivität aber mit
verbesserten pharmakokinetischen Eigenschaften erfolgen.
2.1.2.1. Heterocyclische Bioisostere Der bioisostere Austausch der Hydroxyphenyl-Struktur kann durch substituierte und
unsubstituierte, annelierte Sechs- und Fünfring Heteroaromaten erfolgen und resul-
tiert in Strukturen mit unterschiedlicher Affinität und Selektivität (Abb. 1; Abb. 6)
sowie intrinsischer Aktivität.
Quineloran 9 und Quinpirol 10 (beide Abb. 1) haben eine ausgeprägte D3-Selektivität
gegenüber D2, die je nach Testsystem unterschiedlich groß ausfällt.18, 32-36 Eine
Übersicht zeigt Levant37 in ihrem Review.
Pramipexol 7 (Abb. 1) verhält sich an D2L, D3 und D4 als Agonist mit einer Präferenz
für D338, wobei auch hier verschiedene Werte aus unterschiedlichen Testsystemen
beschrieben sind.34, 37-39 Außerdem werden Pramipexol 7 agonistische Effekte an

Entwicklung D3-selektiver Liganden
8
Autorezeptoren zugeschrieben.40 Ersetzt man die sekundäre Aminstruktur durch ein
primäres Amin, kommt es zum völligen Aktivitätsverlust.41
NPr2
N
NPr2
NNH2
NPr2
N
OH
NPr2
NH
NPr2
N
NPr2
NHNPr2
NN
NPr2
NN
NPr2
NN
Cl
NPr2
S
NPr2S
(S) (S) (S)
(S) (S) (S)
30a(a)
D2: 272 nMD3: 4,97 nM
30b(a)
D2: 17,1 nMD3: 0,87 nM
31 (FAUC 54)(b)
D2: 90 nMD3: 6,0 nM
32a(c)
D2: 9 700 nMD3: 38 nM
32b(c)
D2: 62 nMD3: 34 nM
33(c)
D2: 92 nMD3: 33 nM
34 (FAUC 725)(c)
D2: 35 nMD3: 0,54 nM
35a(c)
D2: 250 nMD3: 4,0 nM
35b(c)
D2: 210 nMD3: 6,1 nM
36(d)
D2: 20 nMD3: 40 nM
37(d)
D2: 27 nMD3: 28 nM
Abb. 6: Rezeptoraffinitäten verschiedener bioisosterer DPATs, Ki in nM (a) gemessen gegen [3H]Spiperon, jeweils humaner Rezeptor in CHO K-1 Zellen8, 31 (b) gemessen gegen [3H]Spiperon, boviner D2-Rezeptor in Striatum-Membran bzw. humaner D3-
Rezeptor in CHO Zellen, Wert für hochaffine Bindungsstelle42 (c) gemessen gegen [3H]Spiperon, jeweils humaner Rezeptor (D2short und D3) in CHO Zellen, jeweils
Wert für hochaffine Bindungsstelle (Ausnahme: D2 bei 32a, K0,5 angegeben)43-45 (d) gemessen gegen [3H]N-0437 für D2L bzw. [³H]Spiperon für D3, jeweils humaner Rezeptor (D2L und
D3) in CHO K-1 Zellen46
Beide Pyridin-Analogen 30a und 30b besitzen eine deutliche D3-Selektivität sowie
agonistische Eigenschaften an Autorezeptoren.31
Die Verbindungen FAUC 54 3142, FAUC 725 3444 und 35a45, die einstellig-nano-
molare bzw. subnanomolare D3-Affintiäten aufweisen, zeigen im Mitogenese-Experi-
ment mit 86 %45, 82 %44 und 82 %45 hohe intrinsische Aktivitäten, während die
Liganden 32a43 und 35b45 mit 55 %43 und 54 %45 intrinsischer Aktivität Partialago-
nisten sind (bezogen auf Quinpirol als vollen Agonisten).
Entwicklung D3-selektiver Liganden
9
Die Tetrahydro-Benzothiophen-Derivate 36 und 37 sind Agonisten mit einer im Ver-
gleich zu 5-OH-DPAT 27a deutlich gesteigerten oralen Bioverfügbarkeit von 10 %
versus 1 %.46, 47
2.1.2.2. Nicht-aromatische Bioisostere Neben heteroaromatischen Bioisosteren sind auch nicht-aromatische bioisostere
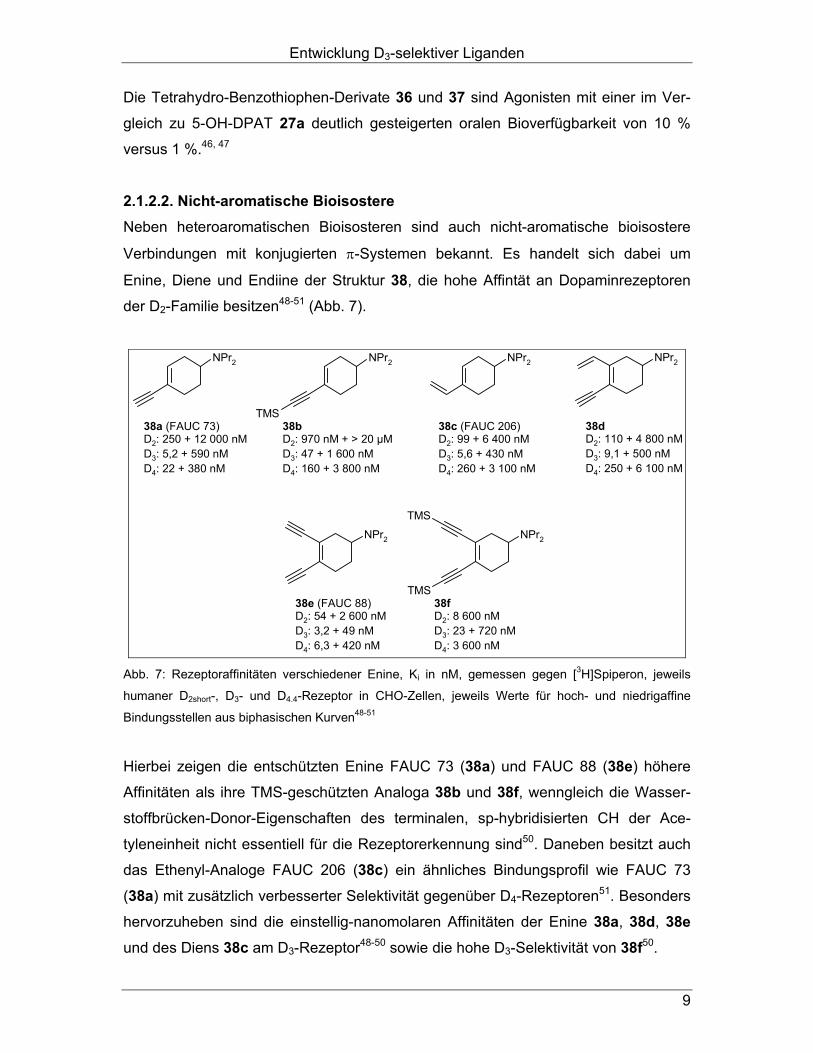
Verbindungen mit konjugierten π-Systemen bekannt. Es handelt sich dabei um
Enine, Diene und Endiine der Struktur 38, die hohe Affintät an Dopaminrezeptoren
der D2-Familie besitzen48-51 (Abb. 7).
NPr2 NPr2
TMS
NPr2
NPr2 NPr2
TMS
TMS
NPr2
38a (FAUC 73)D2: 250 + 12 000 nMD3: 5,2 + 590 nMD4: 22 + 380 nM
38bD2: 970 nM + > 20 µMD3: 47 + 1 600 nMD4: 160 + 3 800 nM
38c (FAUC 206)D2: 99 + 6 400 nMD3: 5,6 + 430 nMD4: 260 + 3 100 nM
38e (FAUC 88)D2: 54 + 2 600 nMD3: 3,2 + 49 nMD4: 6,3 + 420 nM
38fD2: 8 600 nMD3: 23 + 720 nMD4: 3 600 nM
38dD2: 110 + 4 800 nMD3: 9,1 + 500 nMD4: 250 + 6 100 nM
Abb. 7: Rezeptoraffinitäten verschiedener Enine, Ki in nM, gemessen gegen [3H]Spiperon, jeweils
humaner D2short-, D3- und D4.4-Rezeptor in CHO-Zellen, jeweils Werte für hoch- und niedrigaffine
Bindungsstellen aus biphasischen Kurven48-51
Hierbei zeigen die entschützten Enine FAUC 73 (38a) und FAUC 88 (38e) höhere
Affinitäten als ihre TMS-geschützten Analoga 38b und 38f, wenngleich die Wasser-
stoffbrücken-Donor-Eigenschaften des terminalen, sp-hybridisierten CH der Ace-
tyleneinheit nicht essentiell für die Rezeptorerkennung sind50. Daneben besitzt auch
das Ethenyl-Analoge FAUC 206 (38c) ein ähnliches Bindungsprofil wie FAUC 73
(38a) mit zusätzlich verbesserter Selektivität gegenüber D4-Rezeptoren51. Besonders
hervorzuheben sind die einstellig-nanomolaren Affinitäten der Enine 38a, 38d, 38e
und des Diens 38c am D3-Rezeptor48-50 sowie die hohe D3-Selektivität von 38f50.

Entwicklung D3-selektiver Liganden
10
Die terminalen Enine FAUC 73 (38a) und FAUC 88 (38e) zeigen im Mitogeneseex-
periment hohe intrinsische Aktivitäten für alle Rezeptoren der D2-Familie50.
2.2. Aminoindane Durch die formale Verkleinerung des Aminotetralin-Sechsrings zu einem Fünfring
entsteht die Substanzklasse der Aminoindane 39 (Abb. 8). Wie bei den Aminotetra-
linen spielt auch bei den Aminoindanen das Substitutionsmuster des Aromaten sowie
das der Aminofunktion eine entscheidende Rolle für die dopaminerge Aktivität.
NRR'
X NRR'X
27 39 Abb. 8: formale Entstehung der Aminoindane
Dabei zeigt das 4-hydroxysubstituierte Dipropylaminoindan 39b gemischte
D2/5-HT1A-Affinität (Tab. 2; 5-HT1A: Ki=19,5 nM52), was sowohl die Analogie zum
dopaminerg aktiven 5-OH-DPAT als auch zum 5-HT1A-Liganden 8-OH-DPAT
wiederspiegelt. Dagegen bindet das 5-OH-Isomer 39a mit einer etwa dreifachen
Selektivität an D3-Rezeptoren.52 Eine höhere D3-Selektivität wird durch 5,6-
Dimethoxy-Substitution erreicht (U 99194, 39c, 30- bzw-20-fache Selektivität).52, 53
Dabei kommt es zum Verlust der intrinsischen Aktivität, was zu antagonistischen
Eigenschaften führt.52, 53
NPr2X
X D2 D3 D2/D3
39a 5-OH 53 14 3,7
39b 4-OH 5,2 42 0,12
39c 5,6-di-OMe 992 31(a) 32
Tab. 2: Rezeptoraffinitäten verschiedener DPAIs, gemessen gegen [3H]-PNU-86170 (außer (a): [3H]-7-OH-
DPAT), Ratten D2-Rezeptoren in CHO-Zellen bzw. gemessen gegen [3H]Spiperon, Ratten D3-Rezeptoren in
CHO-Zellen 52

Entwicklung D3-selektiver Liganden
11
2.3. Arylcarboxamidoalkyl-substituierte Aminotetraline und deren Analoge 2.3.1. Arylcarboxamide mit Butyl-Linker Durch den Austausch einer der n-Propyl-Seitenketten der DPATs durch eine N-Butyl-
4-biphenylamido-Struktur kommt es zu einer höheren D3-Affinität und –Selektivität.54
Je nach Rest X in Struktur 40 (Abb. 9) verhalten sich die Verbindungen als Agonisten
oder Antagonisten.54
Wie bei den DPATs gibt es auch bei den Amid-Analogen heterocyclische Bioisostere
der allgemeinen Struktur 42, die sich vom Quineloran-, Quinpirol- und Pramipexol-
Grundgerüst ableiten.55 Diese Verbindungen sind Agonisten und zeigen hohe D3-
Affinitäten und Selektivitäten (Abb. 9).55
NNH
O
NNH
O
X
NNH
Ar
O
NNH
Ar
O
N
OH
O
N
NC
NZ
NHN
N
NNH2
S
NNH2
42Het
40
43
Y
41
a: X = H D3-Ki = 0,65 nM; D2/D3 = 60b: X = OH D3-Ki = 0,20 nM; D2/D3 = 125c: X = OTf D3-Ki = 1,6 nM; D2/D3 = 250
a: Ar = Naphthyl D3-Ki = 5,7 nM; D2/D3 = 14b: Ar = Cinnamyl D3-Ki = 8,5 nM; D2/D3 = 38
Z = H, OTf
Y:
Ar: Biphenyl, Cinnamyl
a: Ar =
D3-Ki = 0,79 nM; D2/D3 = 200
b: Ar =
D3-Ki = 3,2 nM; D2/D3 = 150
c: Ar =
D3-Ki = 0,50 nM; D2/D3 = 340
Abb. 9: Rezeptoraffinitäten verschiedener Amide mit Butylen-Linker, Ki in nM
gemessen gegen [125I]Iodsulprid, humane D2- und D3-Rezeptoren in CHO Zellen54-59
außer 40a: gemessen gegen [3H]Spiperon, jeweils humaner D2long- und D3-Rezeptor in CHO-Zellen
(eigene Daten)

Entwicklung D3-selektiver Liganden
12
Auch die Aminoindane 41 sind affine und selektive D3-Rezeptor-Liganden (Abb. 9).59
Da die Verbindungen des Typs 40 in vivo über Depropylierung metabolisiert
werden58, versucht man, über Einbindung der Aminofunktion in das Ringsystem,
ähnlich wie im D3-selektiven Agonisten PD 128907 (4; Abb. 1)60, die metabolische
Stabilität zu erhöhen, was zu Verbindungen der allgemeinen Struktur 43 führt (Abb.
9). Diese Liganden weisen ebenfalls eine mittlere bis hohe D3-Selektivität bei einer
D3-Affinität im niedrigen nanomolaren Bereich auf.56-59
2.3.2. Amide mit Ethyl-trans-cyclohexyl-Linker Neben Dopamin-Liganden mit flexiblem Butyl-Linker gibt es auch solche mit einem
konformativ stärker fixierten Ethyl-trans-cyclohexyl-Linker (allgemeine Struktur 44,
Abb. 10). Diese Substanzen zeichnen sich, vergleicht man sie mit den entspre-
chenden Derivaten mit flexiblem Butyl-Linker, durch eine höhere Selektivität gegen-
über den 5-HT1B/D-Rezeptoren61 sowie verbesserten pharmakokinetischen Eigen-
schaften aus.61-64
N
NH
Ar
ON
X NX NX
44
YY:
Abb. 10: Amide mit Ethylcyclohexyl-Linker
2.3.3. Weitere Linkertypen
N
SN
NN
NN N
NNH
Ar
O
OMe
N
SN
NN
Ar
Het N
SN
NN
Ar
Het
46
47 48
45
Abb. 11: Derivate mit weiteren Linkertypen

Entwicklung D3-selektiver Liganden
13
Weitere Linkertypen sind der verkürzte Ethyl-Linker, der zu Verbindungen der
allgemeinen Struktur 45 mit gemischter D2-, D3-, 5-HT1A-Aktivität führt (BPATs =
2-[N-(2-Benzamidoethyl)-N-n-propylamino]tetraline),65-67 sowie der relativ neue
1,2,4-Triazol-3-ylthiopropyl-Linker 46-48 (alle Abb. 11). Die beschriebenen Liganden
mit diesem Strukturelement sind verschiedene Benzazepine, die hohe Affinitäten und
Selektivitäten für den D3-Rezeptor besitzen.68-70 Neben heterocyclisch substituierten
Benzazepinen 4668, toleriert der D3-Rezeptor auch die heterocyclisch [h]- und [g]-
anellierten Derivate 4769 und 4870.
2.4. Benzamide und Analoge Neben Arylamid-verlinkten Tetralinen hat sich eine zweite Substanzfamilie mit Amid-
Funktion unter den dopaminerg aktiven Liganden herausgebildet, die als Benzamide
(allgemeine Formel 49; Abb. 12) bezeichnet werden.
O
X
Y
NH
OZ N
H
R
O
X
Y
X NH
NR
YOH
49 50 51 Abb. 12: Benzamide und Bioisostere
Die prominentesten Vertreter dieser Gruppe sind die Dopamin-Antagonisten Sulpirid
20, Amisulprid 21 und Racloprid 22, mit leichter D2-Präferenz32, 37, 71, sowie Nemo-
naprid 23 mit etwa gleicher Affinität an D2 und D316 (alle Abb. 2, Kapitel 1.). Vor allem
Variationen an den Molekülteilen Y und Z (Abb. 12) führen zu verbesserten D3-
Selektivitäten. Daneben gibt es Derivate, bei denen der Phenylring durch Naphthyl
ersetzt ist, was zu einer gesteigerten D3-Selektivität gegenüber D2- und den beiden
σ-Rezeptoren führt17, 72 (vgl. auch Nafadotrid 24, Abb. 2, Kapitel 1.).
Durch bioisosteren Austausch der Amidstruktur mit bestimmten fünfgliedrigen
Heteroaromaten kann die D3-Affinität und –Selektivität zusätzlich gesteigert werden.
Verglichen mit den entsprechenden Amiden weisen Pyrrole 50 (Abb. 12) eine
geringere D2-Affinität bei etwa gleich bleibender D3-Affinität auf, was zur Erhöhung
der Selektivität führt.73

Entwicklung D3-selektiver Liganden
14
Neben den Pyrrolen 50 sind auch Imidazole 51 als Amid-Isostere beschrieben (Abb.
12), die allerdings an D2- und D3–Rezeptoren weniger affin und weniger selektiv
binden als die entsprechenden Pyrrole 50.74, 75 Daher ist das Pyrrol als besseres
Bioisoster zu werten.75
Weitere fünfgliedrige Heterocyclen, die als Amid-Bioisostere getestet wurden, sind
Oxazole mit gemischten D3/D4-Affinitäten und D4-Präferenz76 sowie Oxadiazole, bei
denen allerdings keine D2/D3-Aktivität messbar war.74
2.5. Phenylpiperazine Dopaminerg aktive Phenylpiperazine bestehen aus einem Arylamid, das über einen
Linker mit einem 4-arylsubstituierten Piperazin verbunden ist (52, Abb. 13).
NH
O
N N
52π 1
π 2Linker
Abb. 13: allgemeine Formel Phenylpiperazine
2.5.1. Variationen an π2
O Cl Cl O Cl
NN
CF3
π 2
π 2a π 2b π 2c π 2d π 2e π2f Abb. 14: aromatische Reste π2
Der aromatische Rest π2 in der Struktur 52 stellt in den meisten Phenylpiperazinen
einen substituierten Phenylring dar (Abb. 14), wobei die D3-Affinität in der Reihen-
folge ortho>meta>para abnimmt.77 Am häufigsten kommen 2-Methoxy- (π2b) und
2,3-Dichlor-Phenylpiperazine (π2c) vor. Letztere haben oft eine höhere D3-Affinität
verglichen mit den unsubstituierten (π2a)78 oder o-methoxysubstituierten Derivaten
(π2b).79, 80 Hybride mit 2-Methoxy-3-chlor-Substitution (π2d) liegen mit ihren Bin-
dungseigenschaften in der Regel zwischen denen von π2b und π2c.81, 82 Neben den
schon genannten Substituenten zeigen auch 2,3-dimethyl-substituierte Verbindungen

Entwicklung D3-selektiver Liganden
15
(π2e) hohe D3-Selektivitäten gegenüber D2-, D4-, 5-HT1A-, und α1-Rezeptoren.83
Geneste et al. beschreiben auch heterocyclische Derivate (π2f).84, 85
2.5.2. Variationen am Linker Der in 52 enthaltene Linker ist in den meisten Fällen ein gesättigter oder ungesättig-
ter aliphatischer oder cyclischer Alkyl-Linker (Abb. 15). Für die D3-Affinität hat unter
den gesättigten aliphatischen Spacern der Butyl-Linker L1 mit 5 Einfachbindungen
die optimale Länge.78, 79, 83, 85, 86
Der Austausch des gesättigen Linkers durch trans-But-2-enyl L2 führt zu Antago-
nisten.87 Ähnliche Bindungseigenschaften besitzen auch Phenylpiperazine mit trans-
Cyclopropyl-Spacer L4.88 Im Gegensatz dazu nimmt die D2- und D3-Affinität bei But-
2-inyl L3 stark ab.87
Wird die Butyl-Kette durch einen Cyclohexyl-Ring L5 ersetzt, sinkt die D3-Affinität im
Vergleich zur offenkettigen Form ab.89 Verlängert man den Cyclohexyl-Spacer aber
um 2 Methylen-Gruppen, setzt man also den aus Struktur 44 bekannten Ethyl-trans-
cyclohexyl-Linker L6 ein, nehmen sowohl D3-Affinität als auch –Selektivität zu.90
Hackling et al.91 untersuchten verschiedene disubstituierte Phenyle als Linker für
Phenylpiperazine und kamen zu dem Schluss, dass lineare Linker D3- und
abgewinkelte Spacer D2-Rezeptoren bevorzugen, was sich in der D3-Affinität von L7
und L8 wiederspiegelt.
Linker
L1 L2 L3 L4
L5 L6 L7 L8 Abb. 15: Linker
2.5.3. Variationen an π1
Als Molekülteil π1 wurden eine Vielzahl verschiedener carbo- und heterocylischer,
annelierter und nicht-annelierter, substituierter und unsubstituierter Aromaten einge-
setzt (Abb.16).

Entwicklung D3-selektiver Liganden
16
Ar NH
ON
NX
N
O
N N
Y
NN NN
Fe
52a(a)
X = 2,3-diClD2: > 1 000 nMD3: 1,2 nMD4: 280 nM
52b(b)
X = 2,3-diClD2: 25 nMD3: 0,5 nM
52c (GR 103691)(c)
X = 2-OMeD2: 40 nMD3: 0,3 nM
52d (BP-897)(d)
X = 2-OMeD2: 220 nMD3: 1,3 nMD4: 44 nM
52e(a)
X = 2,3-diClD2: 5 200 nMD3: 0,58 nMD4: 370 nM
52f (NGB 2849)(e)
X = 2,3-diClD2: 260 nMD3: 0,9 nMD4: > 5 000 nM
52g (NGB 2904)(e)
X = 2,3-diClD2: 220 nMD3: 1,4 nMD4: > 5 000 nM
52h (FAUC 329)(d)
X = 2-OMeD2: 310 nMD3: 4,3 nMD4: 130 nM
52i(d)
X = 2-OMeD2: 190 nMD3: 2,8 nMD4: 67 nM
52j(d)
X = 2-OMeD2: 140 nMD3: 4,3 nMD4: 40 nM
52k(d)
Y = OX = 2-OMeD2: 110 nMD3: 1,1 nMD4: 30 nM
52l(d)
Y = OX = 2,3-diClD2: 320 nMD3: 1,5 nMD4: 93 nM
52m (FAUC346)(d)
Y = SX = 2-OMeD2: 87 nMD3: 0,23 nMD4: 15 nM
52n (FAUC 365)(d)
Y = SX = 2,3-diClD2: 3 600 nMD3: 0,50 nMD4: 340 nM
52o(f)
Y = NHX = 2-OMeD2: 37 nMD3: 0,3 nMD4: 480 nM
52p (FAUC 382)(d)
X = 2,3-diClD2: 31 nMD3: 0,64 nMD4: 0,63 nM
(R)-52q (FAUC 418)(d)
X = 2- OMeD2: 11 nMD3: 0,19 nMD4: 5,4 nM
(S)-52q(d)
X = 2-OMeD2: 15 nMD3: 3,0 nMD4: 9,5 nM
Abb. 16: Rezeptoraffinitäten verschiedener Phenylpiperazine mit Butyl-Linker, Ki in nM (a) gemessen gegen [3H]Spiperidol, humane D2L-Rezeptoren in Sf9-Zellen, Ratten-D3-Rezeptoren in
Sf9-Zellen und humane D4.4-Rezeptoren in CHO Zellen83 (b) gemessen gegen [125I]IABN, humane D2L-und D3-Rezeptoren in HEK 293 Zellen92 (c) gemessen gegen [3H]Spiperon, humane D2short-Rezeptoren in Ltk- Zellen, humane D3-Rezeptoren in
CHO Zellen86 (d) gemessen gegen [3H]Spiperon, humane D2long-, D3- und D4.4-Rezeptoren in CHO Zellen79, 81, 82 (e) primaten D2- und humane D3- Rezeptoren in CHO Zellen93 (f) gemessen gegen [125I]IABN, humane D2long-, D3- und D4-Rezeptoren in CHO Zellen94

Entwicklung D3-selektiver Liganden
17
Neben den substituierten Phenylen und Biphenylen (52a-c)83, 86, 87, 92 finden auch
annelierte carbo- und heterocyclische Systeme als Molekülteil π1 Verwendung.
Die bekanntesten Vertreter dieser Klasse sind der D3-selektive Partialagonist BP-897
(52d), der bei der Behandlung der Cocain-Sucht Erfolge zeigt95 und die beiden
tricyclischen D3-Antagonisten NGB 2849 (52f) und NGB 2904 (52g).93
Beim 2,3-Dichlor-Analogen von BP-897 52e83 ist die D3-Selektivität gegenüber D2
und D4 weiter erhöht.
Untersuchte Heterocyclen sind die an verschieden Positionen substituierten
Pyrazolopyridine, wobei gute D3-Affinitäten und -Selektivitäten bei Substitution in
Position 2 (FAUC 329, 52h), 5 (52i) und 6 (52j) erreicht wurden79 (Affinität nimmt in
der Reihenfolge 5>2=6>3>4>7 ab).96 Ebenfalls gute Bindungsprofile zeigen 2-substi-
tuierte Indole94 (52o), Benzofurane (52k, 52l) und Benzothiophene (52m, 52n).79 Bei
den letzteren besitzt das 2,3-Dichlor-Phenylpiperazin (52n, FAUC 365) verglichen mit
seinem 2-Methoxy-Analogen (52m, FAUC 346) eine höhere D3-Selektivität.79
Auch doppellagige Systeme wie Metallocene und Paracyclophane zeigen als Rest π1
in Phenylpiperazinen Affinität an Dopamin-Rezeptoren (Abb. 16). Je nach komple-
xiertem Metall und Subsitutionsmuster im Rest π2 werden neben D4- auch D3-
Rezeptoren angesprochen.81 Das Ferrocen-Derivat mit Dichlorsubstitution am
Phenylpiperazin 52p ist ein gemischter D3/D4-Ligand mit partialagonistischen
Eigenschaften.81
Unter den antagonistischen Paracyclophanen zeigt das (R)-Enatiomer (R)-52q die
größte Affinität an D3 (0,19 nM versus 3,0 nM), während das (S)-Enantiomer (S)-52q
die besseren Bindungseigenschaften an 5-HT1A besitzt (25 nM versus 58 nM).82
2.5.4. Konformativ fixierte Phenylpiperazine Ähnlich wie bei den Aminotetralinen, versuchte man auch bei den Phenylpiperazinen
Affinitäten und Selektivitäten durch konformative Fixierung zu modulieren (Abb. 17).
Verbindung 53 weist eine ähnliche D3-Selektivität über D2 auf wie BP-897 52d, ist
aber weniger D3-affin und weniger selektiv gegenüber D1.97 Sehr hohe Selektivitäten
konnten, bei ein- bis zweistellig nanomolaren Affinitäten, bei den Verbindungen des
Typs 54 erzielt werden.90, 98

Entwicklung D3-selektiver Liganden
18
NNH
ON
N
X
NH
O
53
(R)
Linker
54
Linker: Butyl, Cyclohexylethyl Abb. 17: konformativ fixierte Phenylpiperazine
2.5.5. Phenylpiperazine mit bioisoster ausgetauschtem Amid Analog zu den Benzamiden gibt es bei den Phenylpiperazinen ebenfalls Derivate, bei
denen die Amidfunktion gegen einen fünfgliedrigen Heterocyclus ausgetauscht ist 55
(Abb. 18). Auch bei diesen Substanzen besitzt der Butyl-Linker die optimale Länge
für eine hohe D3-Aktivität.99
NN
OON
π 1 55
Abb. 18: Phenylpiperazine mit Amid-Bioisoster
2.6. Hybride Durch Vereinigung des Aminotetralin- bzw. Pramipexol-Grundgerüsts und der
Phenylpiperazin-Einheit in einem Molekül, entsteht eine Art Hybrid der Struktur 56
bzw. 57 (Abb. 19). Beide Verbindungen sind Partialagonisten mit einer hohen intrin-
sischen Aktivität (73 % bzw. 86 % an D3 und 86 % bzw 90 % and D2), einstellig-
nanomolaren Affinitäten an D3 (1,75 nM bzw. 4,1nM) und hohen Selektivitäten über
D2.100, 101
NN
N
OH NN
NN
SNH2
56 57 Abb. 19: Hybride
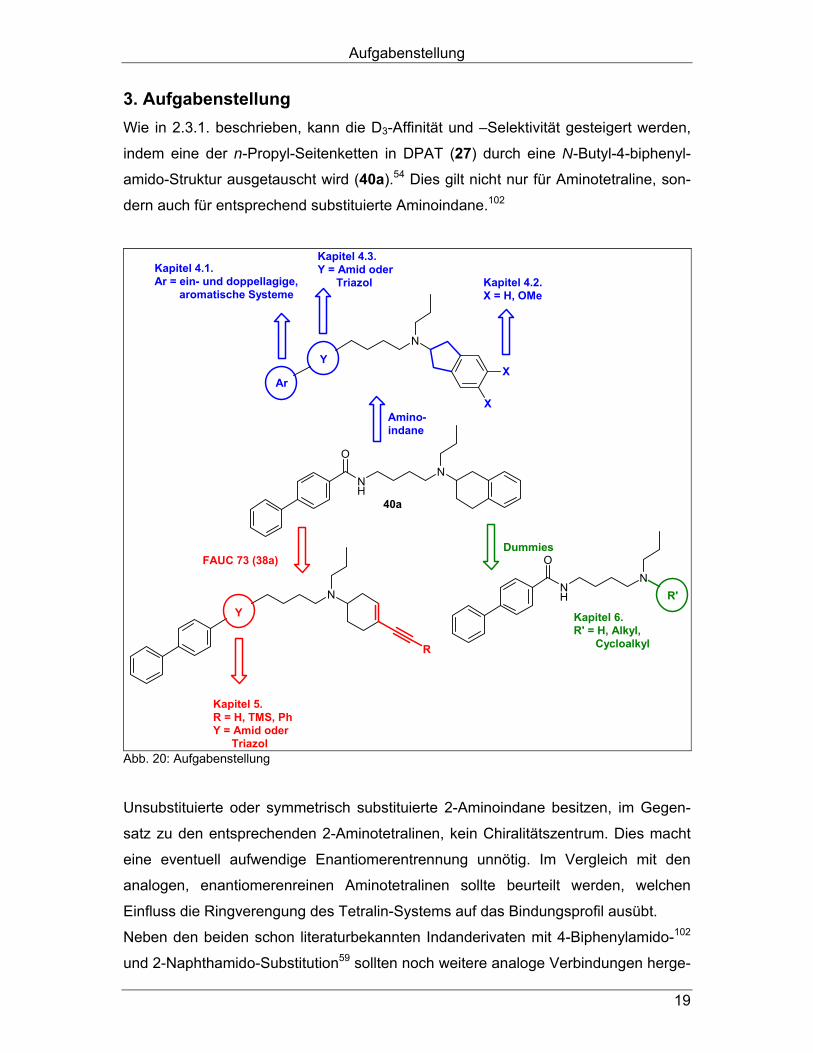
Aufgabenstellung
19
3. Aufgabenstellung Wie in 2.3.1. beschrieben, kann die D3-Affinität und –Selektivität gesteigert werden,
indem eine der n-Propyl-Seitenketten in DPAT (27) durch eine N-Butyl-4-biphenyl-
amido-Struktur ausgetauscht wird (40a).54 Dies gilt nicht nur für Aminotetraline, son-
dern auch für entsprechend substituierte Aminoindane.102
NH
ON
N
X
X
N
R
NH
ON
40a
Kapitel 4.1.Ar = ein- und doppellagige, aromatische Systeme
Kapitel 4.3.Y = Amid oder Triazol Kapitel 4.2.
X = H, OMe
Ar
Y
Y
Kapitel 5.R = H, TMS, PhY = Amid oder Triazol
R'
Kapitel 6.R' = H, Alkyl, Cycloalkyl
Amino-indane
FAUC 73 (38a)Dummies
Abb. 20: Aufgabenstellung
Unsubstituierte oder symmetrisch substituierte 2-Aminoindane besitzen, im Gegen-
satz zu den entsprechenden 2-Aminotetralinen, kein Chiralitätszentrum. Dies macht
eine eventuell aufwendige Enantiomerentrennung unnötig. Im Vergleich mit den
analogen, enantiomerenreinen Aminotetralinen sollte beurteilt werden, welchen
Einfluss die Ringverengung des Tetralin-Systems auf das Bindungsprofil ausübt.
Neben den beiden schon literaturbekannten Indanderivaten mit 4-Biphenylamido-102
und 2-Naphthamido-Substitution59 sollten noch weitere analoge Verbindungen herge-

Aufgabenstellung
20
stellt werden, deren Arylcarboxamid-Partialstrukturen verschieden großen Rauman-
spruch besitzen (Abb. 20; Kapitel 4.1.). Da 2-Dipropylamino-5,6-dimethoxyindan 39c als sehr D3-selektiv gilt52, sollte neben
dem unsubstituierten Indan auch das 5,6-Dimethoxyindangrundgerüst (X = OMe)
verwendet werden. Dadurch sollte überprüft werden, ob das Substitutionsmuster
auch bei den N-Butyl-4-biphenylamiden zu erhöhter D3-Selektivität führt (Kapitel 4.2.). Außerdem erschien ein bioisosterer Austausch der Amidstruktur durch einen Triazol-
Ring als sinnvoll, um, in Anlehnung an die Benzamide und deren Amidbioisostere
(vgl. 2.4.), die D3-Selektivität zu steigern (Kapitel 4.3.). Durch den analogen Austausch einer der Propyl-Seitenketten des D3-selektiven
Liganden FAUC 73 (38a) und seiner Derivate48 (vgl. 2.1.2.2.) mit der oben genannten
N-Butyl-4-biphenylamido-Struktur gelangt man zu nicht-aromatischen Bioisosteren
(Abb. 20). Diese Enine müssten, wären unsere Überlegungen richtig, eine höhere D3-
Affinität besitzen als ihre dipropyl-substituierten Ausgangsverbindungen. In dieser
Substanzklasse sollten neben den Amiden (Kapitel 5.1.) ebenfalls die entsprechen-
den Triazole (Kapitel 5.2.) synthetisiert werden.
Um herauszufinden, welchen Einfluss das π-System, das sich in Nachbarschaft zum
basischen Stickstoff befindet, auf die Rezeptorerkennung hat, oder ob die N-Butyl-4-
biphenylamido-Struktur auch alleine für eine hohe D3-Affinität sorgen kann, sollten
Liganden hergestellt werden, die kein solches π-System besitzen. Dafür sollten die
ehemaligen Aminoindan- bzw –tetralingerüste schrittweise weiter abgebaut werden.
Da man annimmt, dass dieses nun fehlende π-System für die Rezeptorerkennung
essentiell ist, sollten die hergestellten, „Dummy-Liganden“ (Kapitel 6) keine
dopaminerge Aktivität besitzen. In der in Abb. 20 dargestellten, allgemeinen Struktur
bilden aliphatische oder cyclische Alkylgruppen den Rest R’.
Schließlich sollten Indan-Analoge des Antiparkinson-Wirkstoffs Rotigotin 6 hergestellt
werden, um zu analysieren, in wieweit die Bindungseigenschaften des achiralen
Hybrids denen des Rotigotin 6 ähneln (Abb. 21; Kapitel 7.). Alle synthetisierten Verbindungen sollten auf ihre Affinitäten an Dopamin D1-, D2long-,
D2short-, D3- und D4.4-Rezeptoren, an 5-HT1A- und 5-HT2- sowie an α1- und α2-Re-
zeptoren untersucht werden. Besonders interessante Derivate sollten außerdem in
Funktionsexperimenten auf ihre intrinsische Aktivität getestet werden.

Aufgabenstellung
21
N
S
OH
NN
Ar Rotigotin 6Kapitel 7.Typ 39
Abb. 21: Aufgabenstellung Rotigotin-Analoge
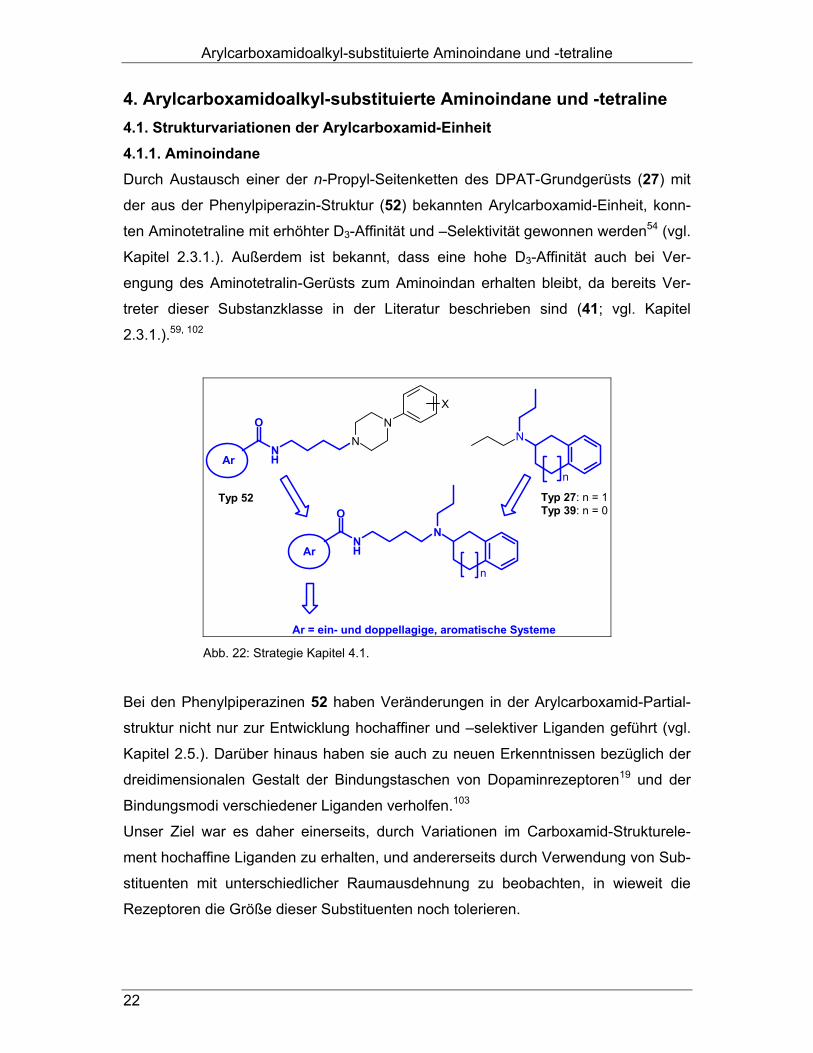
Arylcarboxamidoalkyl-substituierte Aminoindane und -tetraline
22
4. Arylcarboxamidoalkyl-substituierte Aminoindane und -tetraline 4.1. Strukturvariationen der Arylcarboxamid-Einheit 4.1.1. Aminoindane Durch Austausch einer der n-Propyl-Seitenketten des DPAT-Grundgerüsts (27) mit
der aus der Phenylpiperazin-Struktur (52) bekannten Arylcarboxamid-Einheit, konn-
ten Aminotetraline mit erhöhter D3-Affinität und –Selektivität gewonnen werden54 (vgl.
Kapitel 2.3.1.). Außerdem ist bekannt, dass eine hohe D3-Affinität auch bei Ver-
engung des Aminotetralin-Gerüsts zum Aminoindan erhalten bleibt, da bereits Ver-
treter dieser Substanzklasse in der Literatur beschrieben sind (41; vgl. Kapitel
2.3.1.).59, 102
N
n
NH
ON
N
X
NH
ON
n
Ar
Ar
Typ 52 Typ 27: n = 1Typ 39: n = 0
Ar = ein- und doppellagige, aromatische Systeme Abb. 22: Strategie Kapitel 4.1.
Bei den Phenylpiperazinen 52 haben Veränderungen in der Arylcarboxamid-Partial-
struktur nicht nur zur Entwicklung hochaffiner und –selektiver Liganden geführt (vgl.
Kapitel 2.5.). Darüber hinaus haben sie auch zu neuen Erkenntnissen bezüglich der
dreidimensionalen Gestalt der Bindungstaschen von Dopaminrezeptoren19 und der
Bindungsmodi verschiedener Liganden verholfen.103
Unser Ziel war es daher einerseits, durch Variationen im Carboxamid-Strukturele-
ment hochaffine Liganden zu erhalten, und andererseits durch Verwendung von Sub-
stituenten mit unterschiedlicher Raumausdehnung zu beobachten, in wieweit die
Rezeptoren die Größe dieser Substituenten noch tolerieren.

Arylcarboxamidoalkyl-substituierte Aminoindane und -tetraline
23
Synthese: Anders als in der Patentliteratur beschrieben, in der Biphenyl-4-carbonsäure-(4-oxo-
butyl)amid in einer reduktiven Aminierungsreaktion mit 2-Propylaminoindan umge-
setzt wird102, sollte in dieser Verbindungsserie die Diversität erst im letzten Schritt in
die Zielstruktur eingebracht werden. Dies gelang über den folgenden Reaktionsweg
(Abb. 23):
Zunächst wurde Indan-2-on 58 reduktiv mit Propylamin aminiert104 und das sekun-
däre Amin 59 als Hydrochlorid gefällt, welches im Rahmen einer nukleophilen
Substitution mit 4-Brombutyronitril umgesetzt wurde. Es folgte die Reduktion der
Nitrilfunktion des Zwischenprodukts 60 mit LiAlH4 zum primären Amin 61 mit nahezu
quantitativer Ausbeute.
O NH
N
N
N
NH2
58
a b
c
x HCl
59 x HCl 60
61Abb. 23: Reaktionsweg Aminoindane Teil 1
a (1) PrNH2, NaBH(OAc)3, HOAc, THF, RT, 1 h / Reflux, 17,5 h ; (2) HCl in Et2O (32,2 %)
b 4-Brombutyronitril, KI, K2CO3, CH3CN, Reflux, 24 h (77,8%)
c LiAlH4 in Et2O, Et2O, RT, 1 h (93,1 %)
Aus dem primären Amin 61 konnten anschließend die Amide 62a-g dargestellt
werden (Abb. 24). Dabei wurden das 4-Biphenylderivat 62a durch Aminolyse des
entsprechenden Säurechlorids und die Analogen 62b-g unter Zuhilfenahme von
Kupplungsreagenzien wie TBTU oder HATU hergestellt. Dabei konnten Ausbeuten
zwischen 56 und 100 % erzielt werden.

Arylcarboxamidoalkyl-substituierte Aminoindane und -tetraline
24
BiPh Cl
OBiPh N
H
ON
NH2
N
Ar OH
OAr N
H
ON
NH2
N
+
62a
62b-g
a
b-d+
61
61
Abb. 24: Reaktionsweg Aminoindane Teil 2
a Et3N, CH2Cl2, RT, 21 h (56 %)
b Ar = einlagige Carbocyclen: TBTU, DIPEA, DMF, CH2Cl2, RT (quant.)
c Ar = Paracyclophan : HATU, DIPEA, NMP, CH2Cl2, 0 °C (65,6 – 88,9 %)
d Ar = Ferrocen: HATU, DIPEA, DMF, RT (quant.)
Als aromatische Carbonsäuren wurden Carbocyclen mit unterschiedlichem Rauman-
spruch, wie die linear angeordnete 4- und die gewinkelte 2-Biphenylcarbonsäure,
1- und 2-Naphthoesäure und 3-Iodbenzoesäure, sowie als doppellagige Systeme
Ferrocencarbonsäure und Paracyclophancarbonsäure (65) eingesetzt.
Die Herstellung der Letzteren erfolgte nach dem in Abb. 25 beschriebenen Synthese-
schema:105, 106 Zunächst wurde Paracyclophan (63) zu Bromparacyclophan 64
bromiert, wobei neben dem monosubstituierten Produkt vermutlich auch ein
Dibromparacyclohan entstand, was die Reinigung erschwerte und die Ausbeute
verringerte. Nach einem Lithium-Halogen-Austausch mit n-BuLi und Zugabe von
frisch hergestelltem Trockeneis wurde die Paracyclophancarbonsäure 65 erhalten.
Abschließend erfolgte die Enantiomerentrennung durch Bildung diastereomerer
α-(p-Nitrophenylethyl)ammonium-Salze und Kristallisation.105, 106

Arylcarboxamidoalkyl-substituierte Aminoindane und -tetraline
25
BrOH
O
NH2
NO2
NH2
NO2
OH
O
OH
O
(R)
(S)
a b c
63 64 65
(S)-65
(R)-65
Abb. 25: Enantiomerentrennung von Paracyclophancarbonsäure
a Br2, Fe-Pulver, CCl4, CH2Cl2, Reflux, o.n. (53,2 %)
b (1) n-BuLi in Hexan, Et2O, RT, 1 h, (2) frisches Trockeneis (76,5 %)
c CHCl3, RT, 1 h / 50 °C, 2 h / - 5 °C (50,8 % bzw. 21,7 %)
Um die hergestellten Arylcarboxamide mit dem dipropylamino-substituierten Amino-
indan 66 vergleichen zu können, sollte auch Letzteres hergestellt werden. Dazu er-
folgte die reduktive Aminierung des sekundären Amins 59 (Herstellung vgl. Abb. 23)
mit Propionaldehyd (Abb. 26).
NH
Na
x HCl
59 x HCl 66 Abb. 26: Synthese des 2-Di-n-propylaminoindans 66 a Propionaldehyd, NaBH(OAc)3, HOAc, THF, Reflux, 20,5 h (37,3 %)
Bindungsdaten: Alle synthetisierten Endverbindungen wurden in Radioligand-Bindungsstudien auf
ihre Affinitäten an Dopamin D1-, D2long-, D2short-, D3- und D4.4-Rezeptoren, sowie an
Serotonin 5-HT1A- und 5-HT2- und adrenergen α1-Rezeptoren getestet.
Wie erhofft, ist es gelungen, bei allen Arylcarboxamiden des Typs 62, verglichen mit
dem Di-n-propyl-Analogen 66 eine deutliche Affinitätssteigerung an den getesteten
Rezeptoren zu erzielen (Tab. 3): Alle Arylcarboxamide 62 besitzen hohe Affinität an
den Rezeptoren der D2-Rezeptor-Familie sowie am 5-HT1A-Rezeptor und weisen
eine hohe Selektivität gegenüber D1 (1 800- bis 23 000-fach) und 5-HT2 (690- bis
9 700-fach) auf.
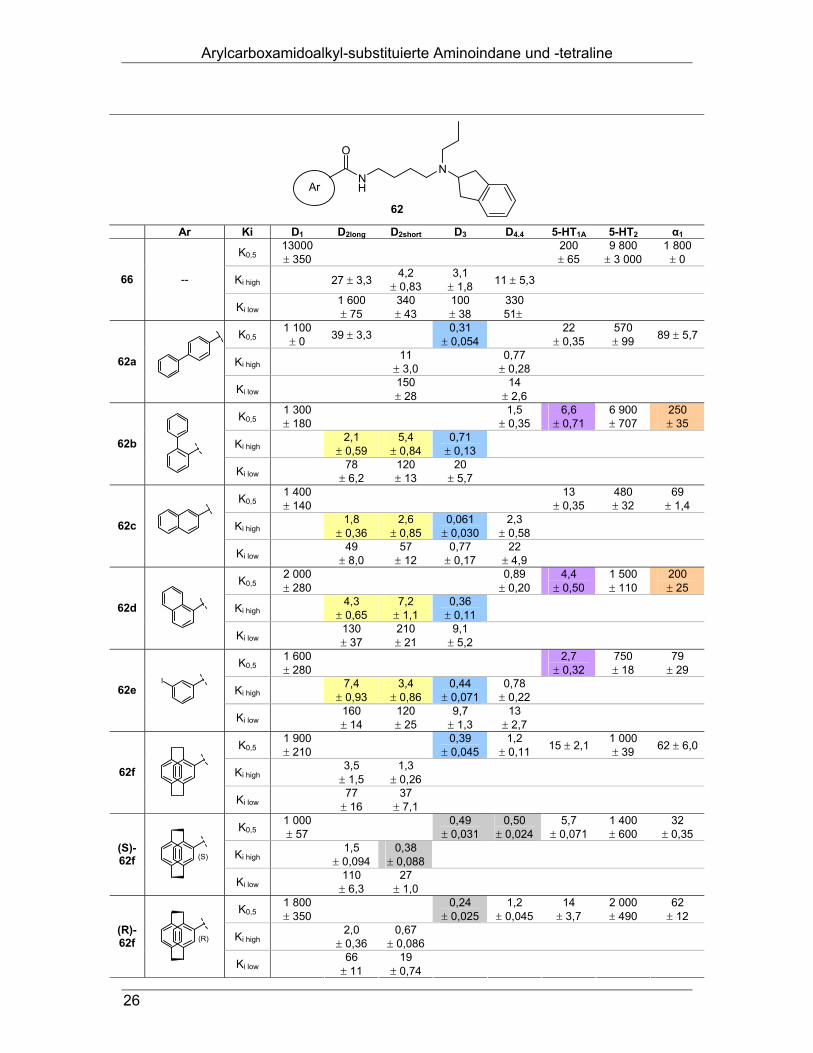
Arylcarboxamidoalkyl-substituierte Aminoindane und -tetraline
26
NH
ON
Ar
62
Ar Ki D1 D2long D2short D3 D4.4 5-HT1A 5-HT2 α1
K0,5 13000 ± 350 200
± 65 9 800
± 3 000 1 800
± 0
Ki high 27 ± 3,3 4,2 ± 0,83
3,1 ± 1,8 11 ± 5,3 66 --
Ki low 1 600 ± 75
340 ± 43
100 ± 38
330 51±
K0,5 1 100
± 0 39 ± 3,3 0,31 ± 0,054 22
± 0,35 570 ± 99 89 ± 5,7
Ki high 11 ± 3,0 0,77
± 0,28 62a
Ki low 150 ± 28 14
± 2,6
K0,5 1 300 ± 180 1,5
± 0,35 6,6
± 0,71 6 900 ± 707
250 ± 35
Ki high 2,1 ± 0,59
5,4 ± 0,84
0,71 ± 0,13 62b
Ki low 78 ± 6,2
120 ± 13
20 ± 5,7
K0,5 1 400 ± 140 13
± 0,35 480 ± 32
69 ± 1,4
Ki high 1,8 ± 0,36
2,6 ± 0,85
0,061 ± 0,030
2,3 ± 0,58 62c
Ki low 49 ± 8,0
57 ± 12
0,77 ± 0,17
22 ± 4,9
K0,5 2 000 ± 280 0,89
± 0,20 4,4
± 0,50 1 500 ± 110
200 ± 25
Ki high 4,3 ± 0,65
7,2 ± 1,1
0,36 ± 0,11 62d
Ki low 130 ± 37
210 ± 21
9,1 ± 5,2
K0,5 1 600 ± 280 2,7
± 0,32 750 ± 18
79 ± 29
Ki high 7,4 ± 0,93
3,4 ± 0,86
0,44 ± 0,071
0,78 ± 0,22 62e
I
Ki low 160
± 14 120 ± 25
9,7 ± 1,3
13 ± 2,7
K0,5 1 900 ± 210 0,39
± 0,045 1,2
± 0,11 15 ± 2,1 1 000 ± 39 62 ± 6,0
Ki high 3,5 ± 1,5
1,3 ± 0,26 62f
Ki low 77 ± 16
37 ± 7,1
K0,5 1 000 ± 57 0,49
± 0,031 0,50
± 0,024 5,7
± 0,071 1 400 ± 600
32 ± 0,35
Ki high 1,5 ± 0,094
0,38 ± 0,088 (S)-
62f (S)
Ki low 110 ± 6,3
27 ± 1,0
K0,5 1 800 ± 350 0,24
± 0,025 1,2
± 0,045 14
± 3,7 2 000 ± 490
62 ± 12
Ki high 2,0 ± 0,36
0,67 ± 0,086 (R)-
62f (R)
Ki low 66 ± 11
19 ± 0,74

Arylcarboxamidoalkyl-substituierte Aminoindane und -tetraline
27
Ar Ki D1 D2long D2short D3 D4.4 5-HT1A 5-HT2 α1
K0,5 4 300 ± 71 0,52
± 0,044 0,51
± 0,039 0,70
± 0,10 360
± 7,1 38
± 1,1
Ki high 2,0 ± 0,76
1,5 ± 0,49 62g Fe
Ki low 100 ± 18
55 ± 11
Tab. 3: Bindungsdaten der Aminoindane, Ki in nM ± SEM
Innerhalb der D2-Familie verhalten sie sich nicht sehr selektiv, sind aber super-
potente Liganden des D3-Rezeptors mit subnanomolaren Affinitäten (Tab. 3, blau).
Gegenüber D4 besteht für 62a, b, d und e eine etwa zweifache Präferenz. Die D2-
Rezeptorerkennung rangiert für 62b-e im einstellig nanomolaren Bereich mit ver-
schiedenen Selektivitäten gegenüber den anderen Rezeptoren der Familie (Tab. 3,
gelb). Von den einlagigen Systemen zeigen 62b, d und e einstellig nanomolare
Affinität an 5-HT1A (Tab. 3, violett). Die größte D3-Selektivität besitzt der literaturbe-
kannte Ligand 62c59 (30-fach über D2long; 38-fach über D4).
Bemerkenswert ist die erhöhte Selektivität gegenüber α1 bei den Derivaten 62b und
62d (Tab. 3, orange), die möglicherweise durch die gewinkelte Struktur dieser beiden
Verbindungen bedingt ist.
Bei den doppellagigen Paracyclophanen 62f zeigt das (S)-Enantiomer etwa gleiche
Affinität an D2short, D3 und D4, während das (R)-Enantiomer den D3-Rezeptor leicht
bevorzugt (Tab. 3, grau). Für Letzteres ist an D2, D4 und 5-HT1A ein Affinitätsrück-
gang zu beobachten. Erwartungsgemäß besitzt das Racemat am D3-Rezeptor eine
Affinität, die zwischen den Werten der Enantiomere liegt. Die Bindungsdaten an den
anderen Rezeptoren ähneln denen des (R)-Enantiomers oder liegen knapp darüber.
Das Ferrocenderivat 62g weist an D3-, D4- und 5-HT1A-Rezeptoren etwa gleiche
Affinität und eine geringe Selektivität gegenüber D2 (3-fach) auf (Tab. 3, grün).
Funktionstests: Für die Verbindungen 62a, c und d wurden zusätzliche Funktionstests durchgeführt
(Tab. 4).
Im Mitogenese-Experiment wurde ausgenutzt, dass die Aktivierung von Rezeptoren
zu einer Proliferationssteigerung führt. Bei der Replikation der DNA wird radioaktiv
markiertes [3H]Thymidin eingebaut und dessen Einbaurate gemessen. Als Ver-
gleichssubstanz diente der Vollagonist Quinpirol. Beim 5-HT1A-Rezeptor wurde die
Bindung von radioaktiv markiertem [35S]GTPγS an das G-Protein gemessen, die bei

Arylcarboxamidoalkyl-substituierte Aminoindane und -tetraline
28
Aktivierung des Rezeptors erhöht ist. Als Vergleichssubstanz wurde der Vollagonist
und natürliche Ligand Serotonin verwendet.
Die Arylcarboxamide 62a, c und d besitzen mit 73 – 83 % intrinsischer Aktitivät hohe
D3-partialagonistische Eigenschaften. Das Ferrocen-Analoge 62g weist an D3- und
D4-Rezeptoren geringere partialgonistische Aktivität auf, ist aber ein voller 5-HT1A-
Agonist.
Auffällig ist, dass die EC50-Werte der Aminoindane 62a, c und d über den jeweiligen
Ki-Werten liegen (1,5 nM versus 0,31 nM, 0,51 nM versus 0,061 nM und 2,1 nM
versus 0,36 nM), während der EC50-Wert des Ferrocen-Derivats 62g am D3-Rezeptor
sowohl niedriger ist als sein entsprechender Ki-Wert (0,32 nM versus 0,52 nM) als
auch niedriger ist als die EC50-Werte der analogen Verbindungen.
NH
ON
Ar
62
D2long(a) D3
(a) D4.4(a) 5-HT1A
(b)
EC50 Aktivität EC50 Aktivität EC50 Aktivität EC50 Aktivität
62a
1,5 80 %
62c
0,51 83 %
62d
2,1 73 %
62g Fe
5,2 72 % 0,32 54 % 2,0 40 % 2,3 101 %
Tab. 4: Aktivität der Aminoindane, EC50 in nM, intrinsische Aktivität bezogen auf die Vollagonisten Quinpirol und Serotonin (a) Mitogenese-Experiment (b) GTPγS-Bindung
Zusammenfassend lässt sich feststellen, dass es durch den Austausch einer der
n-Propyl-Seitenketten von DPAT mit der N-Butyl-4-arylamido-Struktur gelungen ist,

Arylcarboxamidoalkyl-substituierte Aminoindane und -tetraline
29
superaffine und hochpotente Liganden zu synthetisieren, die hinsichtlich ihrer
Bindungseigenschaften der di-n-propyl-substituierten Leitstruktur überlegen sind.
4.1.2. Aminotetraline Aufgrund der interessanten Bindungsprofile der Paracyclophanamide 62f, erschien
es lohnenswert, auch die vier entsprechenden stereoisomeren Aminotetraline herzu-
stellen.
Synthese: Bei der Synthese wurde analog zu der für die Aminoindane beschriebenen Reak-
tionssequenz (Abb. 23 und 24, Kapitel 4.1.1.) vorgegangen mit dem Unterschied,
dass als Ausgangsverbindungen die enantiomerenreinen 2-Propylaminotetraline ein-
gesetzt wurden.107 Die primären Amine 67 wurden mit HATU als Kupplungsreagenz
zu den Amiden 68 umgesetzt (Abb. 27). Da die Reinigung der entstandenen Para-
cyclophancarboxamide nicht mittels Flashchromatographie möglich war, mussten die
Substanzen mit Hilfe präparativer HPLC gereinigt und die freie Base ausgeschüttelt
werden. Durch die mehrfache und aufwendige Reinigung sind die Ausbeuten ver-
glichen mit den Aminoindanen reduziert.
NNH2
NNH
O
NNH2
NNH
O
(R)-67
(S)-67 (R,S)-68(S,S)-68
(R,R)-68(S,R)-68
a
a
Abb. 27: Reaktionsweg Aminotetraline
a: Parcyclophancarbonsäure, HATU, DIPEA, NMP, CH2Cl2, RT, 21 h (28,5-35,5 %)

Arylcarboxamidoalkyl-substituierte Aminoindane und -tetraline
30
Bindungsdaten: Radioligand-Bindungsstudien (Tab. 5) ergaben, dass die vier Diastereomere mit etwa
1 nM alle ähnliche D4-Affintät besitzen. Die Verbindungen mit (R)-Konfiguration an
Position 2 des Aminotetralins zeigen einstellig-nanomolare Bindung an 5-HT1A-
Rezeptoren (Tab. 5, orange), während die Werte ihrer jeweiligen Enantiomeren circa
um das 10-fache erhöht sind. Bei den Isomeren mit (R)-Paracyclophancarboxamid-
Struktur ist der D3-Rezeptor bevorzugt (Tab. 5, blau). Dies bestätigen die p-Werte
aus einem einseitigen, heteroskedatischen Student’schen T-Test mit 0,048 für (R,R)
vesus (S,S), 0,00086 für (R,R) vesus (S,R), 0,049 für (R,S) vesus (S,S) und 0,00088
für (S,R) vesus (R,S). In Gegensatz dazu ist beim (S,S)-Derivat die größte Affinität
am D3- und D2long- bzw. beim (S,R)-Analogen am D4-Rezeptor zu beobachten. Wie
auch bei den Aminoindanen besteht eine hohe Selektivität gegenüber D1 und 5-HT2.
NNH
O
68
Ki D1 D2long D2short D3 D4.4 5-HT1A 5-HT2 α1
K0,5 1 200 ±°250 0,22
±°0,025 0,95
±°0,057 4,5
±°0,53 7 800
±°1 200 77 ±°2,1
Ki high 1,1 ±°0,31
1,9 ±°0,44 (R,R)68
Ki low 32 ±°6,7
53 ±°6,0
K0,5 1 500 ±°240 0,41
±°0,067 0,94
±°0,083 44 ±°2,8 5 600 ±°35 19 ±°1,5
Ki high 0,57 ±°0,10
1,2 ±°0,16 (S,S)68
Ki low 26 ±°2,0
47 ±°3,5
K0,5 2 700 ±°180 0,22
±°0,014 1,0
±°0,11 62
±°8,8 2 300 ±°740
20 ±°0,71
Ki high 0,73±° 0,12
1,3 ±°0,28 (R,S)68
Ki low 32 ±°1,8
40 ±°29
K0,5 1 100 ±°270 2,4
±°0,26 1,1
±°0,12 5,8
±°0,91 5 300 ±°660 67 ±°1,2
Ki high 11 ±°5,5
6,1 ±°1,5 (S,R)68
Ki low 130 ±°24
140 ±°6,5
Tab. 5: Bindungsdaten der Aminotetraline, Ki in nM ± SEM

Arylcarboxamidoalkyl-substituierte Aminoindane und -tetraline
31
4.1.3. Vergleich verschiedener Arylcarboxamide mit doppellagigen Systemen Im Folgenden sollen die gerade beschriebenen doppellagigen Systeme mit den
bereits literaturbekannten analogen Phenylpiperazinen 52p81 und 52q82 (Abb. 16 und
28) sowie dem Ferrocen-gekuppelten Aminotetralinderivat 69107 verglichen werden
(Abb. 28).
NH
ON
N
X
NH
ON
N
X
Fe
NH
ON
Fe
52r: X = 2-OMe52p: X = 2,3-di-Cl52s: X = 2-OMe-3-Cl
52q: X = 2-OMe52t: X = 2,3-di-Cl52u: X = 2-OMe-3-Cl
69
Abb. 28: weitere doppellagige Systeme mit dopaminerger Aktivität
Bindungsdaten: Unter den ferrocen-substituierten Phenylpiperazinen besitzt das 2-Methoxy-Derivat
52r subnanomolare Bindungseigenschaften am D4- und am 5-HT1A-Rezeptor,
während das 2,3-Dichlor-Analoge 52p subnanomolare Affinitäten an D3- und D4-
Rezeptoren zeigt. Bei den Aminotetralinen ist eine Bevorzugung des D3-Rezeptors
gegenüber den anderen Rezeptoren der D2-Familie zu erkennen, wenngleich das
(S)-Enantiomer (S)-69 auch subnanomolare Affinität an D4-Rezeptoren besitzt. Das
Bindungsprofil des Indanderivates 62g kann man daher, aufgrund der subnano-
molaren Affinitäten an D3, D4 und 5-HT1A, der Selektivität gegenüber D1 und 5-HT2
sowie der zweistellig nanomolaren Affinität an α1, als Mischung der Bindungs-
eigenschaften der beiden Phenylpiperazine 52r und 52p betrachten, obwohl die
Selektivität gegenüber den beiden D2–Rezeptoren deutlich geringer ist (Tab. 6; blau).
Bei den Paracyclophanen ist in der Klasse der Phenylpiperazine eine deutliche D3-
Präferenz zu beobachten, die weder bei den Aminoindanen noch bei den Aminotetra-
linen zu sehen ist. Lediglich (R,S)-68 zeigt eine geringe D3-Selektivität. In den
Bindungsprofilen der Aminoindane und der Aminotetraline, mit hohen Affinitäten an

Arylcarboxamidoalkyl-substituierte Aminoindane und -tetraline
32
allen Rezeptoren der D2-Familie und zum Teil auch an 5-HT1A, sind Ähnlichkeiten zu
erkennen (Tab. 6; orange).
Ar NH
OR
N N
NN
X
PhPip AT AI
R:
Ar R D1 D2long D2short D3 D4.4 5-HT1A 5-HT2 α1
52r PhPip 2-OMe 1500 110 78 6,5 0,52 0,50 310 9,7
52p PhPip 2,3-diCl 630 31 19 0,64 0,63 27 250 73
52s PhPip
2-OMe-3-Cl
2200 99 72 3,5 0,91 14 110 35
(S)-69 AT 1,1 + 65
1,8 + 53
0,088+ 0,72 0,67
(R)-69 AT 4,1 + 79
7,4 + 98
0,21 + 2,5 1,2
Fe
62g AI 4300 2,0 + 100
1,5 + 55 0,52 0,51 0,70 360 38
(S)-52q PhPip 2-OMe 660 15 13 3,0 9,5 25 2 300 3,9
(R)-52q PhPip 2-OMe 490 11 5,6 0,19 5,4 58 1 600 2,5
52t PhPip 2,3-diCl 2800 220 140 3,6 120 660 3 500 480
52u PhPip
2-OMe-3-Cl
970 42 28 0,46 33 22 470 80
(R,R)68 AT 1200 1,1 + 32
1,9 + 53 0,22 0,95 4,5 7800 77
(S,S)68 AT 1500 0,57 + 26
1,2 + 47 0,41 0,94 44 5600 19
(R,S)68 AT 2700 3,4 9,2 0,25 1,4 62 2300 20
(S,R)68 AT 1100 11 + 130
6,1 + 140 2,4 1,1 5,8 5300 67
(S)- 62f AI 1000 1,5 + 110
0,38 + 27 0,49 0,50 5,7 1400 32
(R)- 62f AI 1800 2,0 + 66
0,67 + 19 0,24 1,2 14 2000 62
Tab. 6: Vergleich der Bindungsdaten verschiedener doppellagiger Systeme, Ki in nM, bei biphasischer Kurve (Ki high + Ki low)
Funktionstests: Funktionstests ergaben, dass die intrinsische Aktivität des Ferrocen-substituierten
Aminoindans 62g für den D4-Rezeptor mit 40 % etwa im gleichen Bereich wie die der
Phenylpiperazine 52p und 52s liegt (Tab. 7, grün), wenngleich der EC50-Wert mit
2,0 nM mehr denen der Aminotetraline 69 ähnelt (Tab. 7, gelb). Am D3-Rezeptor
besitzt das Aminoindan 62g mit 54 % höhere Aktivität verglichen mit den Phenyl-
piperazinen 52r, p und s und rangiert auf einer Stufe mit dem (R)-konfigurierten
Arylcarboxamidoalkyl-substituierte Aminoindane und -tetraline
33
Aminotetralin (R)-69 (Tab. 7, blau). Der EC50-Wert des hochpotenten Ferrocen-Deri-
vats 62g ist denen aller verglichenen doppellagigen Verbindungen überlegen (Tab. 7,
orange).
Für die Paracyclophan-Derivate der Aminoindane 62f und Aminotetraline 68 liegen
keine Daten aus Funktionstests vor. Der biphasische Kurvenverlauf lässt aber auf
partialagonistische Eigenschaften schließen, womit sie sich von den antagonis-
tischen Phenylpiperazinen 52q, t und u abgrenzen.
Ar NH
OR
N N
NN
X
PhPip AT AI
R:
D3
(a) D4.4(a)
Ar R EC50 Aktivität EC50 Aktivität
52r PhPip 2-OMe
2,0 38 % 0,55 67 %
52p PhPip 2,3-diCl
3,5 28 % 7,6 37 %
52s PhPip 2-OMe-3-Cl
9,8 38 % 22 45 %
(S)-69 AT 1,6 74 % 0,4 78 %
(R)-69 AT 3,7 53 % 1,9 69 %
Fe
62g AI 0,32 54 % 2,0 40 %
(S)-52q PhPip 2-OMe
0 %
(R)-52q PhPip 2-OMe
0 %
52t PhPip 2,3-diCl
0 %
52u PhPip 2-OMe-3-Cl
0 %
Tab. 7: Aktivität verschiedener doppellagiger Systeme, bestimmt im Mitogenese-Experiment, EC50 in nM,
intrinsische Aktivität bezogen auf den Vollagonisten Quinpirol

Arylcarboxamidoalkyl-substituierte Aminoindane und -tetraline
34
4.2. Strukturvariationen der Aminoindan-Einheit Der D3-Antagonist 2-Dipropylamino-5,6-dimethoxyindan 39c weist, wie in Kapitel 2.2.
beschrieben, eine große D3-Selektivität auf.52 Um herauszufinden, ob diese
Selektivität auch bei dem entsprechenden N-Butyl-4-biphenylamid erhalten bleibt,
bzw. ob die intrinsische Aktivität des neuen Liganden von den antagonistischen
Eigenschaften von 39c oder der hohen partialagonistischen Aktivtät von 62a
dominiert wird, sollte das 5,6-Dimethoxyanaloge von 62a hergestellt werden
(Abb. 29).
N
OMe
OMe
NH
ON
NH
ON
OMe
OMe
62aU 99194, 39c
Abb. 29: Strategie Kapitel 4.2.
Synthese: Die Synthese sollte analog zu der der unsubstituierten Aminoindane verlaufen, wofür
das 5,6-Dimethoxyindanon 72 als Ausgangsverbindung benötigt wurde.
In der Literatur ist im Zusammenhang mit der Synthese von Alkaloiden die
Herstellung von 72 beschrieben,108, 109 die, wie im Folgenden dargestellt, leicht
abgewandelt wurde, um die Verwendung des hochexplosiven Diazomethans zu
umgehen.
Zunächst wurde die 3,4-Dimethoxyphenylessigsäure 70 mit Oxalylchlorid zum Säure-
chlorid aktiviert und mit TMS-geschütztem Diazomethan110, 111 zum (3,4-Dimethoxy-
phenylmethyl)diazomethylketon 71 umgesetzt (Abb. 30). Dann folgte der Ringschluss
durch Rühren in einer Lösung von Trifluoressigsäure in Dichlormethan zum Indanon
72. Leider gelang es nicht, die reduktive Aminierung des Ketons mit Propylamin
durchzuführen: Es wurden vergeblich verschiedene Natriumborhydride in verschie-
denen Lösungsmitteln mit und ohne Säurezusatz verwendet. Außerdem wurde die

Arylcarboxamidoalkyl-substituierte Aminoindane und -tetraline
35
Strategie verfolgt, zunächst das Imin zu bilden, welches dann reduziert werden sollte.
Diese Bemühungen führten allerdings nicht zum Erfolg.
O
OH
MeO
MeO
O
N2
MeO
MeO
OMeO
MeO
NH
MeO
MeO
a b
70 71 72
Abb. 30: 1. Reaktionsweg Dimethoxyaminoindane
a (1) (COCl)2, Toluol, RT, 3 h, (2) TMS-Diazomethan-Lösung in Et2O, Toluol, 0 °C, 2,5 h (58,1 %)
b TFA, CH2Cl2, - 20 °C, 30 min (43,7 %)
Daher war es notwendig, auf eine alternative, längere Syntheseroute, ausgehend
von 5,6-Dimethoxyindan-1-on 73, auszuweichen.
Die ersten beiden Schritte, nämlich die Herstellung des Oxims 74 und die
nachfolgende katalytische Hydrierung zum primären Amin 75 wurden in Anlehnung
an eine Vorschrift von Haadsma-Svensson et al.52 durchgeführt (Abb. 31). Die
anschließende Acylierung zum Amid 76 erfolgte nicht wie in der Literatur mit
Propionsäure und DEPC als Kupplungsreagenz52, sondern unter Verwendung von
Propionylchlorid. Nach Reduktion mit LiAlH4 in THF52 zum sekundären Amin 77
konnte die für die unsubstituierten Aminoindane bekannte Reaktionsfolge mit
nukleophiler Substitution, Reduktion des entstandenen Nitrils 78 zum primären Amin
79 und Amidkupplung hin zur Zielverbindung 80 fortgeführt werden. Hierbei konnte
eine im Vergleich zur Reaktionssequenz der unsubstituierten Aminoindane (Abb. 23)
deutlich erhöhte Ausbeute bei der nukleophilen Substitution erzielt werden (77,8 %
versus 96,1%).

Arylcarboxamidoalkyl-substituierte Aminoindane und -tetraline
36
OMeO
MeO
O
NOH
MeO
MeO
NH2
MeO
MeO
NH
OMeO
MeO
NH
MeO
MeO
N
N
MeO
MeO
N
NH2
MeO
MeO
ClBiPh
O
NH
ON
OMe
OMe
x HCla b
c
d
73 74 x HCl 75
7677e
g
7978
f
80
Abb. 31: 2. Reaktionsweg Dimethoxyaminoindane
a n-Butylnitrit, H2SO4konz., MeOH, RT, 1,5 h + 1,75 h (79,3 %)
b HOAc, H2SO4konz., Pd/C, H2, 25 °C, 10-15 bar, 4,75 h (67,8 %)
c Propionylchlorid, Et3N, CHCl3, RT, 16 h (53,1 %)
d LiAlH4, THF, RT, 3,5 h (roh)
e 4-Brombutyronitril, NaI, K2CO3, CH3CN, Reflux, 25 h (96,1%)
f LiAlH4 in Et2O, Et2O, RT, 4,5 h (91,4 %)
g Et3N, CHCl3, RT, 5,5 h (19,5 %)
Bindungsdaten : Die Werte in Tab. 8 zeigen, dass die Affinität der Dimethoxyaminoindan-Verbindung
80 im Vergleich zum unsubstituierten Derivat 62a an allen untersuchten Rezeptoren
abnimmt. Dieser Affinitätsverlust ist an den verschiedenen Rezeptoren in unter-
Arylcarboxamidoalkyl-substituierte Aminoindane und -tetraline
37
schiedlichem Ausmaß zu beobachten, wobei die D2-Affinität in den mikromolaren
Bereich absinkt, während die D3-Rezeptorerkennung immer noch im niedrig
nanomolaren Bereich rangiert (Tab. 8, blau). Dadurch kommt es zu einer sehr hohen,
250-, 120- bzw. 190-fachen Selektivität gegenüber den D1-, D2long- und D2short-Rezep-
toren, sowie einer 11- und 21-fachen D3-Präferenz gegenüber D4- und α1-Rezep-
toren.
NH
ON
X
X X Ki/Sel D1 D2long D2short D3 D4.4 α1
K0,5 1 100
± 0 39
± 3,3 0,31 ± 0,054 89
± 5,7
Ki high 11 ± 3,0 0,77 ± 0,28
Ki low 150 ± 28 14
± 2,6
62a H
x/D3 3 500 130 35 2,5 290
K0,5 2 800 ± 350
1 300 ± 450
2 100 ± 0
11 ± 1,4
120 ± 36
230 ± 21
Ki high Ki low
80 OMe
x/D3 250 120 190 11 21 Tab. 8: Bindungsdaten von unsubstituierten und dimethoxy-substituierten Aminoindanen, Ki in nM ± SEM und D3-Selektivität als Verhältnis x/D3
Unser Vorhaben, die D3-Selektivität deutlich zu steigern, ist durch 5,6-Dimethoxy-
substitution des Aminoindan-Grundgerüsts und die Synthese des D3-affinen und
selektiven Liganden 80 am D2short- und am D4-Rezeptor gelungen. Die D3-Selektivität
gegenüber dem D2long-Rezeptor ist etwa gleich geblieben, während die sie gegen-
über D1 und α1 um etwa das 10-Fache reduziert ist.
Arylcarboxamidoalkyl-substituierte Aminoindane und -tetraline
38
4.3. Strukturvariationen durch bioisosteren Ersatz der Carboxamid- Funktionalität Wie in Kapitel 2. beschrieben, konnten durch bioisosteren Austausch der Amidfunk-
tion mit verschiedenen fünfgliedrigen Heteroaromaten Liganden mit hoher D3-Affinität
und –Selektivität hergestellt werden. Dabei wurden neben Pyrrolen und Pyrazolen
bei den Benzamiden (Abb. 12, Kapitel 2.4.) auch Isoxazole (Abb. 18, Kapitel 2.5.5.)
und 1,2,4-Triazole (Abb. 11, Kapitel 2.3.3.) bei den Phenylpiperazinen bzw.
arylcarboxamid-substituierten Aminotetralinen eingesetzt.
Um diesen selektivitätssteigernden Effekt auszunutzen, sollte auch die Amidstruktur
in Verbindung 62a durch einen Fünfring-Heteroaromaten ersetzt werden (Abb. 32).
Dazu sollte die 1H-1,2,3-Triazol-Partialstruktur genutzt werden, da sie einerseits
bisher nicht als Amid-Bioisoster in Dopamin-Liganden genutzt wurde und anderer-
seits als synthetisch besonders gut zugänglich erschien.
NH
ON
NNN
N
NH
O
N
O
EtSO2
O
EtSO2 NH
N
62a
(S)-(-)-SultopridD2/D3 = 1,0
DU 122290 (56a)D2/D3 = 5,0
81
Abb. 32: Strategie Kapitel 4.3.

Arylcarboxamidoalkyl-substituierte Aminoindane und -tetraline
39
Synthese: In der bekannten Synthesesequenz für die Aminoindane mit Carboxamid-Partial-
struktur wurde ein Teil der späteren Amid-Funktion, nämlich das primäre Amin in
geschützter Form, an Position 4 einer Butyl-Kette in das Molekülgerüst einführt.
Analog dazu sollte auch ein Teil des späteren Triazols, in diesem Fall die Azid-
Funktion, in die Struktur eingebaut werden. Anschließend sollte das Azid dann mit
einem Acetylen zur erwünschten Zielstruktur cycloaddiert werden.
Daher wurde das sekundäre Amin 59 (vgl. Abb. 23, Kapitel 4.1.1.) zunächst mit
4-Azidobutanol-mesylat 85 (Herstellung siehe Abb. 35, Kapitel 5.1.) nukleophil zum
Azid 82 substituiert (Abb. 33). Darauf folgte die Cu(I)-katalysierte 1,3-dipolare Cyclo-
addition mit Biphenylacetylen zum Triazol 81, die mit 58,4-prozentiger Ausbeute
verlief.
NH
N
N3
NNN
N
a
b
82
81
59
Abb. 33: Aminoindane mit bioisoster ersetzter Amidfunktion
a 4-Azidobutanol-mesylat 85 (vgl. Abb. 36, Kapitel 5.1.), NaI, K2CO3, CH3CN, Reflux, 22 h (11,2%)
b Biphenylacetylen, CuSO4 x 5 H2O, Na-Ascorbat, CH2Cl2, H2O, tert-BuOH, 40 °C, on (58,4 %)
Bindungsdaten: Durch Einführung der Triazol-Struktur ist es gelungen, verglichen mit dem
Carboxamid-Derivat 62a, unter geringfügiger Abnahme der D3-Affinität (Tab. 9, blau),
den super-affinen und hochselektiven Liganden 81 herzustellen. Mit Ausnahme der
Selektivität gegenüber D2-Rezeptoren, die im Fall des D2long-Rezeptors abgenommen

Arylcarboxamidoalkyl-substituierte Aminoindane und -tetraline
40
hat und bei D2short etwa gleichgeblieben ist, konnte bei allen getesteten Rezeptoren
eine Selektivitätserhöhung beobachtet werden (Tab. 9, orange). Dabei konnte die D3-
Selektivität gegenüber D1- (5700-fach versus 3500-fach) und D4- (107-fach versus
21-fach; zur Berechnung wurde der K0,5-Wert des Carboxamids 62a verwendet)
sowie den getesteten Serotonin- (5-HT1A: 3300-fach versus 71-fach; 5-HT2: 7200-
fach versus 1800-fach) und α-Rezeptoren (1100-fach versus 290-fach) deutlich
gesteigert werden.
NX
X Ki/Sel D1 D2long D2short D3 D4.4 5-HT1A 5-HT2 α1
K0,5 1 100
± 0 39
± 3,3 0,31 ± 0,054 22
± 0,35 570 ± 99
89 ± 5,7
Ki high 11 ± 3,0 0,77
± 0,28
Ki low 150 ± 28 14
± 2,6
62a Amid
x/D3 3 500 130 35 2,5 71 1 800 290
K0,5 4 700 ± 530 0,83
± 0,094 89
± 7,8 2 700 ± 140
6 000 ± 390
920 ± 130
Ki high 37 ± 1,6
30 ± 6,2
Ki low 1 200 ± 260
2 600 ± 1200
81 Triazol
x/D3 5 700 45 36 110 3 300 7 200 1 100 Tab. 9: Bindungsdaten von Aminoindanen mit Amid- oder Triazol-Partialstruktur, Ki in nM ± SEM und D3-Selektivität als Verhältnis x/D3
Die 1,4-substituierte 1H-1,2,3-Triazol-Partialstruktur kann daher als Bioisoster für
eine Amidfunktion angesehen werden.

Alkinylcyclohexen als Tetralin-Bioisoster
41
5. Alkinylcyclohexen als Tetralin-Bioisoster 5.1. Alkinylcyclohexene mit Amid-Partialstruktur In den letzten Jahren konnte von unserer Arbeitsgruppe gezeigt werden, dass der
aromatische Anteil des Aminotetralins bioisoster durch ein konjugiertes π-System
ersetzt werden kann (Abb. 7, Kapitel 2.1.2.2.).48-51 Das terminale Enin FAUC 73 (38a)
wies hohe D3-Selektivität bei einstellig-nanomolarer D3-Affinität auf.48
Um die Affinität und Selektivität weiter zu steigern, sollte, wie schon bei den Amino-
indanen, eine der n-Propyl-Gruppen durch das N-Butyl-4-biphenylamid-Motiv (vgl.
40a) ersetzt werden (Abb. 34).
NH
ON N
NH
ON
R
40a FAUC 73 (38a)
Abb. 34: Strategie Kapitel 5.1.
Synthese: Zunächst war es wichtig, eine geeignete Schutzgruppen-Strategie zu finden, um
einen orthogonalen Syntheseverlauf zu gewährleisten.
Es war nötig, die Amid-Kupplung am Ende der Synthese durchzuführen, da die
Amid-Funktion sowohl während der anstehenden Ketalspaltung als auch bei der
Reaktion mit Triflat-Anhydrid (Abb. 36) angegriffen werden könnte. Das für die
Amidsynthese notwendige primäre Amin der Seitenkette musste somit bis zum
letzten Reaktionsschritt geschützt werden. Der Einsatz von Schutzgruppen, die
hydrogenolytisch abgespalten werden wie z. B. Benzyl, war aufgrund einer mölichen
Hydrierung des konjugierten π-Systems bei der Entschützung nicht möglich. Auch
Gruppen, die im stark Sauren oder Basischen entfernt werden, waren nicht geeignet,

Alkinylcyclohexen als Tetralin-Bioisoster
42
da während der Synthese in extremen pH-Bereichen gearbeitet werden sollte. Daher
erschien die Azid-Funktion, die später in einer Staudinger-Reaktion selektiv zum
primären Amin reduziert werden konnte, als Struktur der Wahl.
Um die spätere Butyl-Seitenkette herzustellen (Abb. 35), wurde 4-Chlorbutanol 83 in
einer nukleophilen Substitution zum Azid 84 umgesetzt und anschließend die Alko-
holfunktion zum Mesylat 85 aktiviert (entsprechend einer Vorschrift für 6-Azido-
hexanol112).
OHCl
OHN3 MsO
N3a b
84 8583 Abb. 35: Synthese des Mesylats 85
a NaN3, NaI, CH3CN/H2O 9:1, Reflux, 40 h (74,7 %, roh)
b MsCl, Et3N, THF, RT, 5,25 h (81,0 %, roh)
Das Monoethylenketal des 1,4-Cyclohexandions 86 wurde mit Propylamin reduktiv
aminiert (Abb. 36)113 und das entstehende sekundäre Amin 87 mit dem Mesylat 85
nukleophil substituiert. Direkt im Anschluss wurde das Ketals durch Rühren in 6N-
HCl/Aceton gespalten.114 Substitution und Ketalspaltung ergaben das tertiäre Amin
88 mit einer Ausbeute von 21,7 %. Die Enolform der Carbonylgruppe in Verbindung
88 wurde zum Triflat 89 aktiviert (20 %) und in einer Cacchi-Kupplung mit TMS- bzw.
Phenylacetylen zu den Eninen 90 und 91 umgesetzt. Dabei verlief die Bildung des
TMS-substituierten Enins 90 mit einer hohen Ausbeute von 72,2 %, während die
Reinigung des Phenyl-substituierten Produktes 91 Schwierigkeiten bereitete. Des-
halb wurde das Enin 91 nach einer groben Reinigung für den nächsten Reaktions-
schritt eingesetzt. Abschließend folgte die Reduktion der Azid-Funktion nach Stau-
dinger zu den jeweiligen primären Aminen und deren Acylierung mit 4-Biphenyl-
carbonsäurechlorid zu den Zielverbindungen 92 und 93. Die Abspaltung der TMS-
Gruppe des Enins 92 erfolgte durch Rühren mit Tetrabutylammoniumfluorid-Lösung
und führte zum terminalen Alkin 94.

Alkinylcyclohexen als Tetralin-Bioisoster
43
O O
O
O O
NH NN3
O
NN3
OTf
NN3
R
NNH
R
O
a b c
d
e
90 R = TMS91 R = Ph
92 R = TMS 94 R = H93 R = Ph
f
87 88 89
+ 85
86
Abb. 36: Synthese der Enine mit Amidstruktur
a PrNH2, NaBH(OAc)3, HOAc, THF, RT, 15 h (92,9 %, roh)113
b (1) K2CO3, KI, CH3CN, Reflux, 3 d, (2) 6N-HCl, Aceton, RT, 19,5 h (21,7 %)
c Tf2O, 2,6-Di-tert-butyl-4-methylpyridin, 1,2-Dichlorethan, Reflux, 4,5 h (20,0 %)
d R-Acetylen, Cu(I)I, Pd(PPh3)2Cl2, Me2EtN, THF, RT, 40 min (R = TMS) / 35 min (R = Ph) (R = TMS:
72,2 %/R = Ph: roh)
e (1) PPh3, THF, H2O, RT, 2 d, (2) Biphenylcarbonsäurechlorid, Et3N, CHCl3, RT, 3 h (R = TMS) / 24 h
(R = Ph) (R = TMS: 33,2 %, R = Ph: 3,9 % über 2 Stufen)
f Bu4NF, THF, - 15 °C, 2,75 h (64,8 %)
Um das Propin-Analoge Enin zu erhalten, sollte, analog zu den di-n-propyl-
substituierten Derivaten51, die Cacchi-Kupplung unter Einleiten von Propin-Gas
durchgeführt werden (Abb. 37). Leider reagierte unter diesen Bedingungen nicht nur,
wie gewünscht, das Enoltriflat, sondern es kam auch zu einer Cu-(I)-katalysierten
1,3-dipolaren Cycloaddition zwischen dem Alkin und der Seitenketten-Azid-Funktion.
Hierbei findet die C-C-Kupplung wahrscheinlich zuerst statt, da im HPLC-MS-
Chromatogramm ein kleiner Anteil des erwünschten Produkts 95 zu sehen war. Der
große Überschuss an Alkin, der beim gasförmigen Propin schwer zu dosieren war,
Alkinylcyclohexen als Tetralin-Bioisoster
44
führte dann zur Nebenreaktion in der Seitenkette unter Bildung des Triazols 96. Die
Erfahrung mit den flüssigen Acetylen-Derivaten, bei denen selektiv lediglich die
Cacchi-Kupplung ablief, zeigt, dass es wichtig ist, nur einen geringen Überschuss (2-
bis 3-fach) der Acetylen-Komponente einzusetzen.
NN3
OTf
NN3 N
NN
N
8995 96
a a
Abb. 37: Nebenreaktion bei der Enin-Synthese
a Propin (bei – 45 °C eingeleitet), Cu(I)I, Pd(PPh3)2Cl2, Piperidin, THF, RT, 2 h (35,0 %)
Bindungsdaten: Vergleicht man die Bindungsdaten der Enine 94 und 92 mit denen ihrer di-n-propyl-
substituierten Leitstrukturen FAUC 73 (38a) und 38b, ist eine Affinitätssteigerung an
allen Rezeptoren der D2-Familie zu beobachten (Tab. 10). Am besten wird weiterhin
der D3-Rezeptor mit Affinitätswerten im subnanomolaren bzw. einstellig nanomolaren
Bereich angesprochen (Tab. 10, orange), wobei das super-affine terminale Alkin 94
mit einem Ki-Wert von 0,21 nM sowohl seinem TMS-geschützten Analogen 92 (Ki =
1,9 nM) als auch seinen Leitstrukturen FAUC 73 (38a; Ki high = 5,2 nM) und dem
Aminotetralin 40a (Ki = 0,65 nM) überlegen ist. Bezüglich der D3-Selektivität kehrt
sich dieses Verhältnis um, da sich die TMS-geschützte Form 92 (mit D2short/D3 = 210
bzw. D4/D3 = 23) sowohl selektiver als das entschützte Enin 94 (mit D2short/D3 = 23
bzw. D4/D3 = 2,8) als auch als sein di-n-propyl-substituieres Analoges 38b (mit
D2short/D3 = 21 bzw. D4/D3 = 3,4) verhält. Das terminale Enin 94 besitzt im Vergleich
zu FAUC 73 (38a; mit D2short/D3 = 48 bzw. D4/D3 = 4,2) eine geringere D3-Selektivität.
Die Bindungsdaten des phenyl-substituierten Enins 93, zeigen, analog zum Di-n-
propylderivat48, einen fast vollständigen Verlust der Rezeptorerkennung.
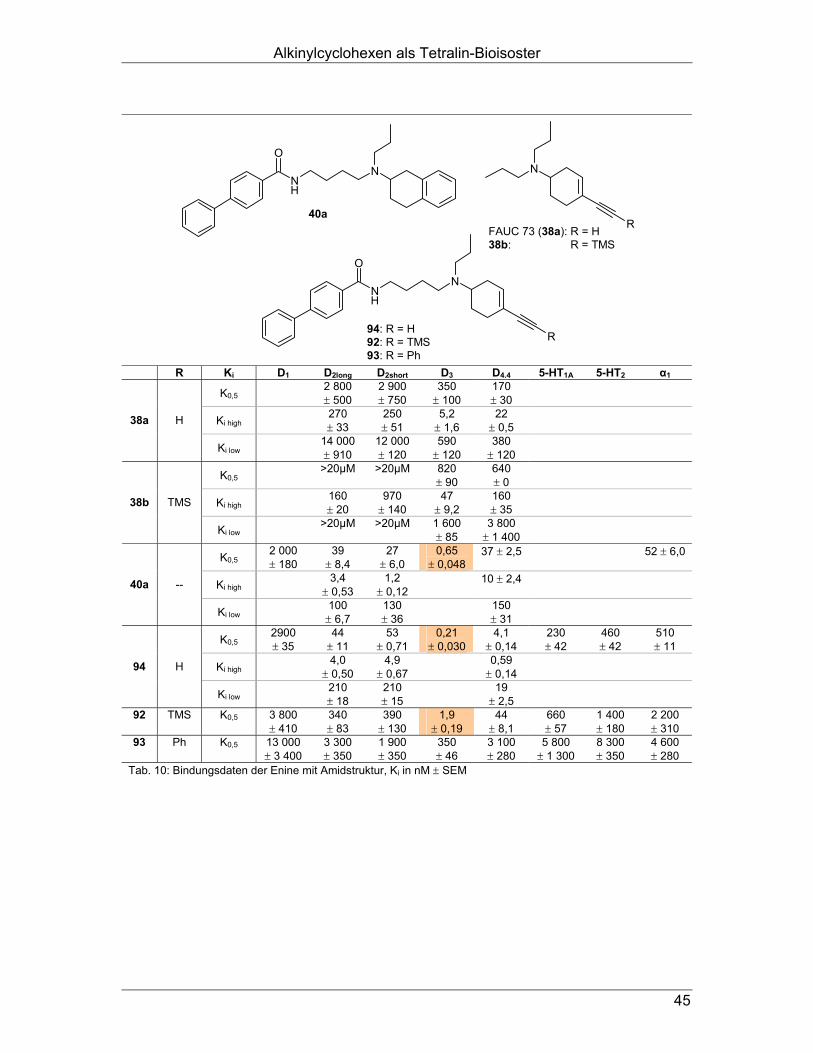
Alkinylcyclohexen als Tetralin-Bioisoster
45
NH
ON N
R
NH
ON
R
40aFAUC 73 (38a): R = H38b: R = TMS
94: R = H92: R = TMS93: R = Ph
R Ki D1 D2long D2short D3 D4.4 5-HT1A 5-HT2 α1
K0,5 2 800
± 500 2 900 ± 750
350 ± 100
170 ± 30
Ki high 270
± 33 250 ± 51
5,2 ± 1,6
22 ± 0,5
38a H
Ki low 14 000 ± 910
12 000 ± 120
590 ± 120
380 ± 120
K0,5 >20µM >20µM 820
± 90 640 ± 0
Ki high 160
± 20 970
± 140 47
± 9,2 160 ± 35
38b TMS
Ki low >20µM >20µM 1 600 ± 85
3 800 ± 1 400
K0,5 2 000 ± 180
39 ± 8,4
27 ± 6,0
0,65 ± 0,048
37 ± 2,5 52 ± 6,0
Ki high 3,4
± 0,53 1,2
± 0,12 10 ± 2,4 40a --
Ki low 100 ± 6,7
130 ± 36
150 ± 31
K0,5 2900 ± 35
44 ± 11
53 ± 0,71
0,21 ± 0,030
4,1 ± 0,14
230 ± 42
460 ± 42
510 ± 11
Ki high 4,0
± 0,50 4,9
± 0,67 0,59
± 0,14 94 H
Ki low 210 ± 18
210 ± 15
19 ± 2,5
92 TMS K0,5 3 800 ± 410
340 ± 83
390 ± 130
1,9 ± 0,19
44 ± 8,1
660 ± 57
1 400 ± 180
2 200 ± 310
93 Ph K0,5 13 000 ± 3 400
3 300 ± 350
1 900 ± 350
350 ± 46
3 100 ± 280
5 800 ± 1 300
8 300 ± 350
4 600 ± 280
Tab. 10: Bindungsdaten der Enine mit Amidstruktur, Ki in nM ± SEM
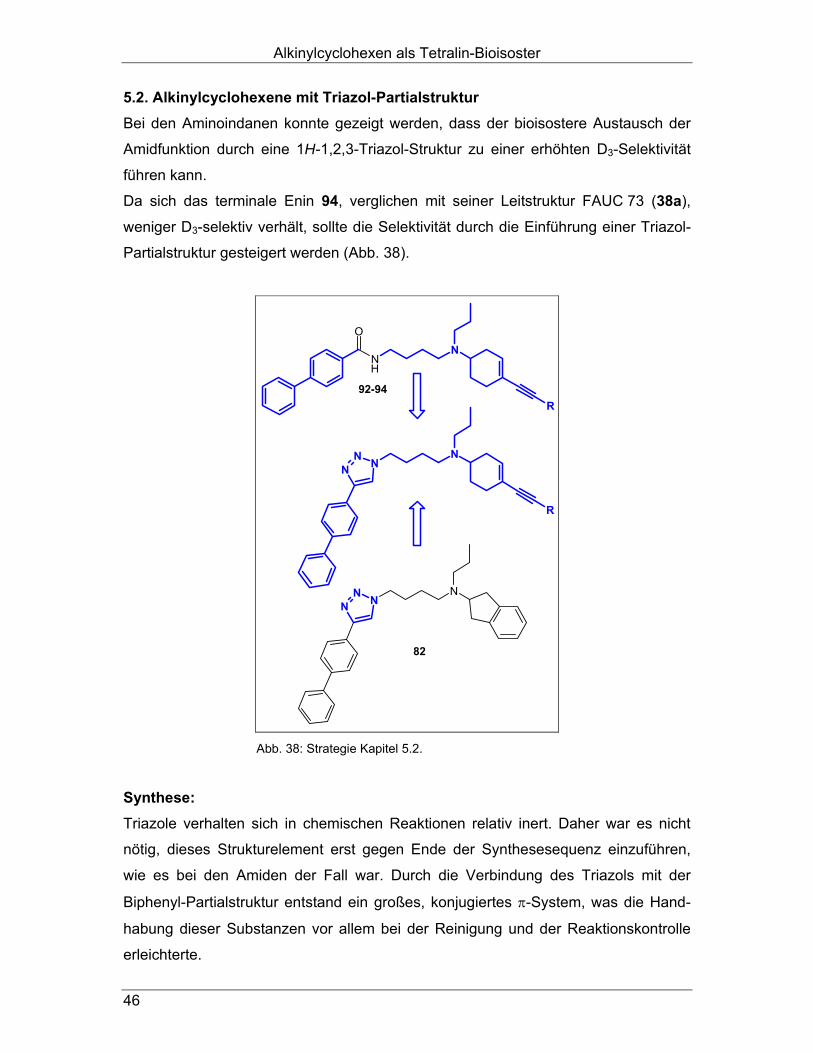
Alkinylcyclohexen als Tetralin-Bioisoster
46
5.2. Alkinylcyclohexene mit Triazol-Partialstruktur Bei den Aminoindanen konnte gezeigt werden, dass der bioisostere Austausch der
Amidfunktion durch eine 1H-1,2,3-Triazol-Struktur zu einer erhöhten D3-Selektivität
führen kann.
Da sich das terminale Enin 94, verglichen mit seiner Leitstruktur FAUC 73 (38a),
weniger D3-selektiv verhält, sollte die Selektivität durch die Einführung einer Triazol-
Partialstruktur gesteigert werden (Abb. 38).
NH
ON
R
NNN
N
R
NNN
N
92-94
82
Abb. 38: Strategie Kapitel 5.2.
Synthese: Triazole verhalten sich in chemischen Reaktionen relativ inert. Daher war es nicht
nötig, dieses Strukturelement erst gegen Ende der Synthesesequenz einzuführen,
wie es bei den Amiden der Fall war. Durch die Verbindung des Triazols mit der
Biphenyl-Partialstruktur entstand ein großes, konjugiertes π-System, was die Hand-
habung dieser Substanzen vor allem bei der Reinigung und der Reaktionskontrolle
erleichterte.

Alkinylcyclohexen als Tetralin-Bioisoster
47
Die Zwischenstufe 88 wurde zunächst mit einer Ausbeute von 76,1 % Cu(I)-kataly-
siert mit Biphenylacetylen zum 1,4-substituierten Triazol 97 cycloaddiert (Abb. 39).
Dabei wurde als Cu(I)-Quelle ein Kupfer-Komplex der Form [Cu(CH3CN)4]PF6
gewählt, da dieser in den gleichen Lösungsmitteln gut löslich war wie die beiden
Edukte. In Lösungsmittelsystemen, wie sie beim CuSO4/Na-Ascorbat-System nötig
wären, hatte Biphenylacetylen eine geringe Löslichkeit und flotierte an der Flüssig-
keitsoberfläche.
NN
OTf
NN
BiPh
NN
R
NN
BiPh
NN
O
NN
BiPh
NN3
O
99 R = TMS 101 R = H100 R = Ph
d
88 98
ba
97
c
Abb. 39: Synthese der Enine mit Triazolstruktur
a Biphenylacetylen, [Cu(CH3CN)4]PF6, CH2Cl2/MeOH 3:1, 40 °C, 25,5 h (76,1 %)
b Tf2O, 2,6-Di-tert-butyl-4-methylpyridin, 1,2-Dichlorethan, Reflux, on (roh)
c R-Acetylen, Cu(I)I, Pd(PPh3)2Cl2, Me2EtN, THF, RT, 1,5 h (R = TMS: 41,3 %/R = Ph: 14,1 %)
d Bu4NF, THF, - 15 °C, 2,45 h (42,8 %)
Die Enolform des Click-Produkts 97 wurde dann, analog zur vorher beschriebenen
Reaktionssequenz, mit Trifalt-Anhydrid zum Enoltriflat 98 aktiviert und nach grober
Reinigung in Cacchi-Kupplungen mit TMS- und Phenylacetylen zu den Eninen 99
und 100 umgesetzt. Dabei verlief die C-C-Kupplung zum TMS-geschützten Enin mit

Alkinylcyclohexen als Tetralin-Bioisoster
48
geringerer Ausbeute als in der Reaktionsfolge der Carboxamid-substituierten Enine
(41,3 % versus 72,2 %). Das TMS-substituierte Enin 99 wurde anschließend mit Hilfe
von Tetrabutylammoniumfluorid-Lösung zum terminalen Alkin 101 entschützt.
Bindungsdaten: Der bioisostere Austausch der Amidfunktion führte, wie in Tab. 11 zu sehen ist, zur
ausgesprochen D3-selektiven Verbindung 101 mit 100- und 630-facher Selektivität
über D2short und D2long, und 260-facher Selektivität über D4-Rezeptoren (Tab. 11,
grün). Die Affinität ist im Vergleich zum carboxamid-analogen Enin 94 etwa 9-mal
geringer, liegt aber immer noch bei dem sehr guten Ki-Wert von 1,9 nM. Auch beim
TMS-substituierten Enin 99 ist eine ausgesprochene D3-Selektivität bei niedrig-
nanomolarer Affinität zu beobachten (Tab. 11, grün). Verglichen mit dem Analogen
Carboxamid 92 verzehnfacht sich die Selektivität gegenüber D4 (200-fach versus 23-
fach), während die Selektivität gegenüber den D2-Rezeptoren geringer ist (D2short: 23-
fach versus 210-fach; vgl. Tab. 10 und 11).
Das Phenyl-Analoge 100 besitzt, verglichen mit dem entsprechenden Amid-Derivat,
höhere Affinität an allen getesteten Rezeptoren.
Die aus der Propin-Kupplung hervorgegangene Verbindung 96 (Abb. 37, Kapitel 5.1.)
ist kein affiner Dopamin-Ligand, wobei wahrscheinlich zwei Faktoren eine Rolle
spielen. Zum einen kann es daran liegen, dass das Triazol keinen weiteren aroma-
tischen Substituenten trägt, wie er in den Verbindungen 99-101 enthalten ist.
Andererseits kann die Methylsubstitution des Alkins ein entscheidender Grund sein,
da beim analogen di-n-propyl-substituierten Derivat51 ebenfalls, verglichen mit dem
terminalen Alkin FAUC 73 (38a), eine Abnahme in der D3-Affinität (Ki = 53 + 2 600
nM) zu beobachten ist. Dieses Di-n-propyl-Analoge zeigt allerdings kaum D4-
Rezeptorerkennung (Ki = 4100 nM), während sie bei 96 deutlich erkennbar ist und
sogar stärker ausfällt als für den D3-Rezeptor.
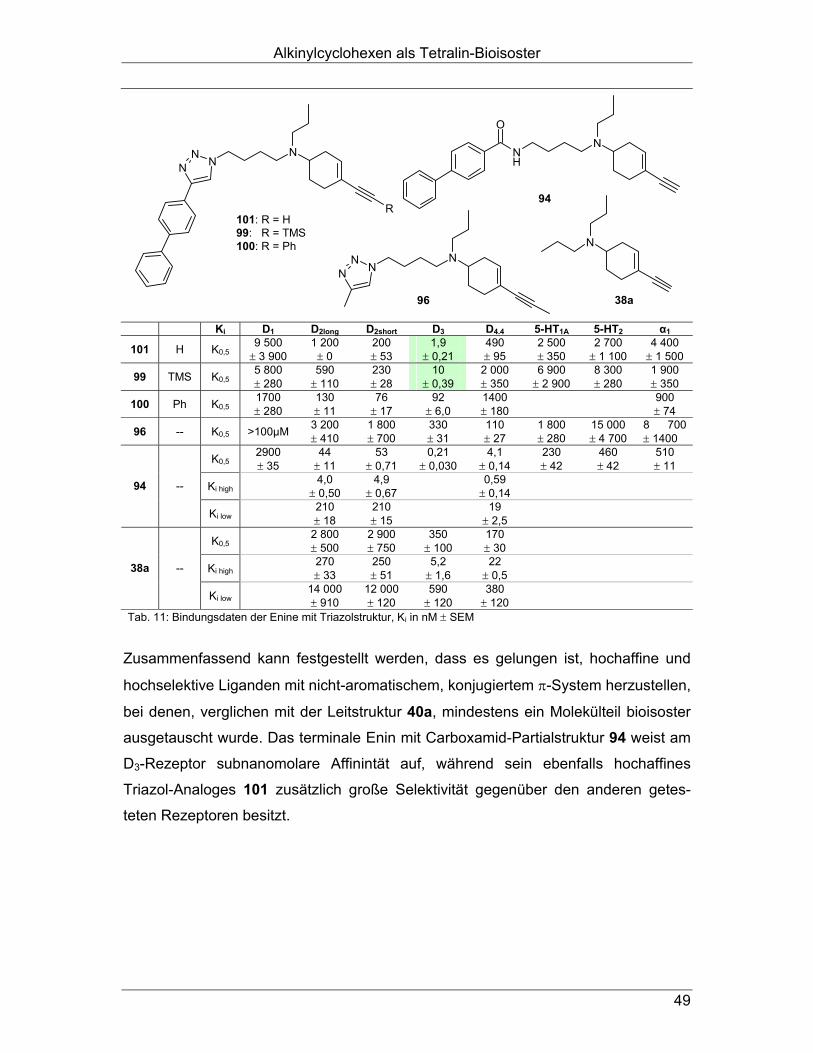
Alkinylcyclohexen als Tetralin-Bioisoster
49
NH
ON
NNN
N
R
N
NNN
N
94
38a
101: R = H99: R = TMS100: R = Ph
96
Ki D1 D2long D2short D3 D4.4 5-HT1A 5-HT2 α1
101 H K0,5 9 500
± 3 900 1 200
± 0 200 ± 53
1,9 ± 0,21
490 ± 95
2 500 ± 350
2 700 ± 1 100
4 400 ± 1 500
99 TMS K0,5 5 800 ± 280
590 ± 110
230 ± 28
10 ± 0,39
2 000 ± 350
6 900 ± 2 900
8 300 ± 280
1 900 ± 350
100 Ph K0,5 1700 ± 280
130 ± 11
76 ± 17
92 ± 6,0
1400 ± 180 900
± 74
96 -- K0,5 >100µM 3 200 ± 410
1 800 ± 700
330 ± 31
110 ± 27
1 800 ± 280
15 000 ± 4 700
8 700 ± 1400
K0,5 2900 ± 35
44 ± 11
53 ± 0,71
0,21 ± 0,030
4,1 ± 0,14
230 ± 42
460 ± 42
510 ± 11
Ki high 4,0
± 0,50 4,9
± 0,67 0,59
± 0,14 94 --
Ki low 210 ± 18
210 ± 15
19 ± 2,5
K0,5 2 800
± 500 2 900 ± 750
350 ± 100
170 ± 30
Ki high 270
± 33 250 ± 51
5,2 ± 1,6
22 ± 0,5
38a --
Ki low 14 000 ± 910
12 000 ± 120
590 ± 120
380 ± 120
Tab. 11: Bindungsdaten der Enine mit Triazolstruktur, Ki in nM ± SEM
Zusammenfassend kann festgestellt werden, dass es gelungen ist, hochaffine und
hochselektive Liganden mit nicht-aromatischem, konjugiertem π-System herzustellen,
bei denen, verglichen mit der Leitstruktur 40a, mindestens ein Molekülteil bioisoster
ausgetauscht wurde. Das terminale Enin mit Carboxamid-Partialstruktur 94 weist am
D3-Rezeptor subnanomolare Affinintät auf, während sein ebenfalls hochaffines
Triazol-Analoges 101 zusätzlich große Selektivität gegenüber den anderen getes-
teten Rezeptoren besitzt.

Austausch der Indan- bzw. Tetralin-Einheit durch „Dummy-Substituenten“
50
6. Austausch der Indan- bzw. Tetralin-Einheit durch „Dummy- Substituenten“
NH
ON
n
NH
ON
NH
ON
n
NH
ONH
NH
ON
NH
ONH
Aminoindane (n = 0)Aminotetraline (n = 1)
Enine
Liganden mit nicht-aromatischem, konjugiertem π-System
Liganden mit aromatischem π-System
Liganden ohne π-System ("Dummy-Liganden")
Abb. 40: Abbau der Aminotetralin- bzw. indanstruktur hin zu den „Dummy-Liganden“
Für alle Liganden, in denen eine frühere n-Propyl-Seitenkette durch die N-Butyl-4-
biphenylamido-Struktur ersetzt wurde, war eine erhöhte D3-Affinität charakteristisch.
Deshalb stellte sich die Frage, ob dieses Biphenylamid in Kombination mit einem

Austausch der Indan- bzw. Tetralin-Einheit durch „Dummy-Substituenten“
51
basischen Stickstoff in der Seitenkette auch ohne weiteres π-System (Indan, Tetralin
oder Enin) für eine effiziente Rezeptorerkennung ausreicht.
Um dies herauszufinden, sollte das Tetralin- bzw. Indangrundgerüst in 40a bzw. 62a
schrittweise abgebaut werden (Abb. 40), so dass weder aromatische Partial-
strukturen, noch solche mit konjugiertem π-System erhalten bleiben. Nach dem
heutigen Verständnis von Dopamin-Liganden sollte die dopaminerge Aktivität durch
diese Veränderungen völlig verloren gehen, da man ein aromatisches oder
konjugiertes π-System in einem gewissen Abstand zu einem basischen Stickstoff im
Liganden als essentiell für die Rezeptorerkennung erachtet.
Synthese:
NH
NH
BiPh
O
R
OHNH2
OHNH
BiPh
O
O
Cl
MsONH
BiPh
O
Pr2NNH
BiPh
ON
NH
BiPh
OR'
a
b
c
d
102
103104: R = n-Pr105: R = cy-Hex
RNH2
Pr2NH
106107: R = cy-Pent108: R = cy-Hex
e
Abb. 41: Synthese der „Dummy-Liganden“
a NaOH, H2O, CH2Cl2, RT, 1,75 h (92,0 %)
b MsCl, Et3N, THF, RT, on (roh weiter eingesetzt)
c RNH2, NaI, K2CO3, CH3CN, Reflux, 6 h (R = n-Pr) / on (R = cy-Hex) (R = n-Pr: 55,1 % ; Pr =
cy-Hex : 49,4 %)
d Pr2NH, NaI, K2CO3, CH3CN, Reflux, on (53,4 %)
e R’ = cy-Pent: Cyclopentanon, NaBH(OAc)3, CH2Cl2, RT, 3 d (29,1 %); R’ = cy-Hex:
Propionaldehyd, NaBH(OAc)3, CH2Cl2, RT, 7 h (55,1 %)

Austausch der Indan- bzw. Tetralin-Einheit durch „Dummy-Substituenten“
52
Auch bei dieser Synthesefolge sollte die Diversität möglichst spät in das Molekül-
gerüst eingeführt werden. Daher sollte zunächst das Biphenylcarboxamid aufgebaut
werden. Daraus ergab sich der Vorteil, dass vom ersten Syntheseschritt an ein π-
System im Molekül vorhanden war, das die Detektion der Verbindungen bei
Reaktionskontrollen und Reinigung erleichterte.
4-Aminobutanol und Biphenylcarbonsäurechlorid wurden als erstes nach einer
Literaturvorschrift102, 115 zum Amid 102 umgesetzt (Abb. 41), was in sehr hohen
Ausbeuten gelang. Dann wurde die primäre Alkoholfunktion von 102 zum Mesylat
103 aktiviert, welches roh weiter eingesetzt wurde. Darauf folgten nukleophile
Substitutionen zu den sekundären und tertiären Aminen 104-106, bei denen
Ausbeuten von 49,4 – 55,1 % erzielt werden konnten. Die sekundären Amine wurden
anschließend reduktiv zu den Zielsubstanzen 107 und 108 alkyliert.
Bindungsdaten : Radioligand-Bindungsstudien ergaben, dass die die D3-Affinität der „Dummy-Ligan-
den“ umso geringer ist, je weiter das ursprüngliche Tetralin- und Indan-Gerüst
abgebaut wird:
Während die cyclisch subsituierten tertiären Amine 107 und 108 noch eine geringe
Affinität am D3-Rezeptor zeigen (Tab. 12, blau), geht die Rezeptorerkennung beim
Di-n-Propyl-Derivat 106 und den beiden sekundären Aminen 104 und 105 nahezu
völlig verloren.
NH
ON
R'
R
R R’ D1 D2long D2short D3 D4.4 α1
104 n-Pr H 7 300 ± 1 500
21 000 ± 7 800
> 100 000 11 000 ± 1 600
6 100 ± 210
2 500 ± 180
105 H cy-Hex 6 200 ± 390
1 500 ± 280
820 ± 200
440 ± 64
1 100 ± 190
1 400 ± 110
106 n-Pr n-Pr 6 800 ± 490
9 700 ± 2 300
8 800 ± 850
950 ± 460
1 400 ± 410
2 400 ± 460
107 n-Pr cy-Pent 5 500 ± 490
> 100 000 > 100 000 90 ± 14
1 500 ± 210
1 800 ± 35
108 n-Pr cy-Hex 4 600 ± 210
4 700 ± 570
6 300 ± 3 400
140 ± 3,5
670 ± 140
1 100 ± 35
Tab. 12: Bindungsdaten der „Dummy-Liganden“, Ki in nM ± SEM

Austausch der Indan- bzw. Tetralin-Einheit durch „Dummy-Substituenten“
53
Durch den starken Affinitätsverlust der „Dummy-Liganden“ wurde die Vermutung be-
stätigt, dass ein konjugiertes π-System in Nachbarschaft zu einem basischen Stick-
stoff für die Rezeptorerkennung notwendig ist.
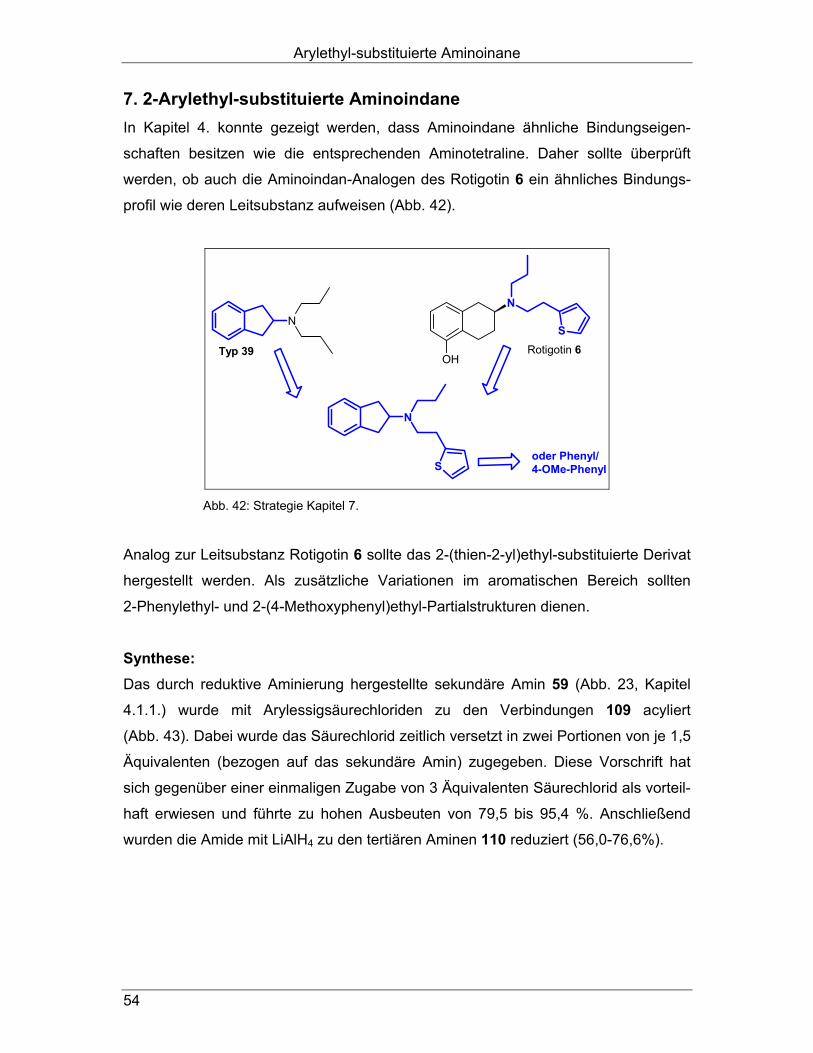
Arylethyl-substituierte Aminoinane
54
7. 2-Arylethyl-substituierte Aminoindane In Kapitel 4. konnte gezeigt werden, dass Aminoindane ähnliche Bindungseigen-
schaften besitzen wie die entsprechenden Aminotetraline. Daher sollte überprüft
werden, ob auch die Aminoindan-Analogen des Rotigotin 6 ein ähnliches Bindungs-
profil wie deren Leitsubstanz aufweisen (Abb. 42).
N
S
OH
N
S
N
Rotigotin 6Typ 39
oder Phenyl/4-OMe-Phenyl
Abb. 42: Strategie Kapitel 7.
Analog zur Leitsubstanz Rotigotin 6 sollte das 2-(thien-2-yl)ethyl-substituierte Derivat
hergestellt werden. Als zusätzliche Variationen im aromatischen Bereich sollten
2-Phenylethyl- und 2-(4-Methoxyphenyl)ethyl-Partialstrukturen dienen.
Synthese: Das durch reduktive Aminierung hergestellte sekundäre Amin 59 (Abb. 23, Kapitel
4.1.1.) wurde mit Arylessigsäurechloriden zu den Verbindungen 109 acyliert
(Abb. 43). Dabei wurde das Säurechlorid zeitlich versetzt in zwei Portionen von je 1,5
Äquivalenten (bezogen auf das sekundäre Amin) zugegeben. Diese Vorschrift hat
sich gegenüber einer einmaligen Zugabe von 3 Äquivalenten Säurechlorid als vorteil-
haft erwiesen und führte zu hohen Ausbeuten von 79,5 bis 95,4 %. Anschließend
wurden die Amide mit LiAlH4 zu den tertiären Aminen 110 reduziert (56,0-76,6%).

Arylethyl-substituierte Aminoinane
55
NH
N
ArO
N
Ar
a b
59 109 110 Abb. 43: Synthese der Rotigotin-Analogen
a Ar-Essigsäurechlorid, Et3N, CHCl3, RT, etwa 4 h (79,5-95,4 %)
b LiAlH4, Et2O, RT, 4,5 h (56,0-76,6 %)
Bindungsdaten :
N
S
OH
N
Rotigotin 6110Ar
Cy Ki D1 D2long D2short D3 D4.4 5-HT1A 5-HT2 α1
K0,5 4 400 ± 210 310
± 14 27
± 4,6 4 400 ± 810
920 ± 57
Ki high 41 ± 13
15 ± 2,5
10 ± 5,6 110a Thien-2-
yl
Ki low 1 500 ± 210
950 ± 110
320 ± 100
K0,5 4 600 ± 420 220
± 14 20
± 1,4 2 500 ± 71
480 ± 88
Ki high 41 ± 4,2
19 ± 2,1
3,4 ± 0,035 110b Phenyl
Ki low 920 ± 32
540 ± 21
76 ± 8,1
K0,5 4 400
± 0 77 ± 13
6 200 ± 350
490 ± 39
Ki high 13 ± 2,7
5,1 ± 0,96
4,7 ± 1,6
18 ± 5,2 110c
4-Methoxyphenyl
Ki low 470 ± 59
280 ± 17
62 ± 8,3
180 ± 46
6 360 20 4 55 100 Tab. 13: Bindungsdaten der Aminoindane, Ki in nM ± SEM (Messdaten von 6 siehe Abb. 1)
Die 2-arylethyl-substituierten Aminoindane 110 weisen an den hochaffinen Bindungs-
stellen von D2- und D3-Rezeptoren Affinitäten im niedigen nanomolaren Bereich auf
(Tab. 13, blau). Dies ist auch bei der Leitstruktur Rotigotin 6 zu beobachten. Ver-
gleicht man die Bindungseigenschaften am 5-HT1A-Rezeptor, so besitzen die
Analogen 110a und b eine höhere Affinität als das Methoxyphenyl-Derivat 110c und
Rotigotin 6. Gegenüber D1-, 5-HT2 und α1-Rezptoren besteht für alle Substanzen des
Typs 110 eine hohe Selektivität. Das Thien-2-yl- (110a) und das Phenyl-Derivat 110b

Arylethyl-substituierte Aminoinane
56
zeigen gegenüber D4 eine höhere Selektivität als das 4-methoxysubstituierte Analoge
110c.
Zusammenfassend kann festgestellt werden, dass die Bindungsprofile der
2-arylethyl-substituierten Aminoindane 110 zwar gewisse Parallelen mit dem Bin-
dungsprofil der Leitstruktur Rotigotin 6 aufweisen, an manchen Rezeptoren aber
stark abweichen. Die Affinitäten konnten bei den Aminoindanen nicht gesteigert
werden.

Zusammenfassung
57
8. Zusammenfassung Ausgehend von der Leitstruktur 40a (Abb. 44), einem hochaffinen Dopamin-Rezep-
tor-Liganden, sollten durch strukturelle Veränderungen Liganden mit erhöhter
dopaminerger und serotoninerger Affinität bzw. verbesserter D3-Selektivität herge-
stellt werden.
NH
ON
N
X
X
N
R
NH
ON
40a
Kapitel 4.1.Ar = ein- und doppellagige, aromatische Systeme
Kapitel 4.3.Y = Amid oder Triazol Kapitel 4.2.
X = H, OMe
Ar
Y
Y
Kapitel 5.R = H, TMS, PhY = Amid oder Triazol
R'
Kapitel 6.R' = H, Alkyl, Cycloalkyl
Amino-indane
FAUC 73 (38a)Dummies
Abb. 44: Aufgabenstellung
Dabei wurde die Struktur 40a wie folgt variiert (Abb. 44):
• Austausch des Aminotetralins gegen Aminoindan und
o Variation der Arylcarboxamid-Einheit
o Variation der Aminoindan-Einheit (Dimethoxy-Substitution)
o Variation durch bioisosteren Ersatz der Carboxamid-Funktionalität mit
einer 1H-1,2,3-Triazol-Einheit

Zusammenfassung
58
• Bioisosterer Austausch des aromatischen Aminotetralin-Anteils mit einem
konjugiertem π-System (Alkinylcyclohexene)
o Alkinylcyclohexene mit Amid-Partialstruktur
o Alkinylcyclohexene mit bioisoster ersetzter Carboxamid-Funktionalität
(1H-1,2,3-Triazol-Einheit)
• Austausch des aromatischen Aminoindan- bzw. -tetralin-Anteils mit Substi-
tuenten ohne π-System, um den Einfluss der ursprünglichen pharmakophoren
Einheiten auf die Rezeptorerkennung zu untersuchen
Schließlich sollten Aminoindan-Analoge des Antiparkinson-Wirkstoffes Rotigotin (6)
hergestellt werden (Abb. 45), um zu beobachten, inwieweit die Bindungseigenschaf-
ten des achiralen Hybrids denen der Leitstruktur 6 ähneln.
N
S
OH
NN
Ar Rotigotin 6Kapitel 7.Typ 39
Abb. 45: Aufgabenstellung Rotigotin-Analoge
8.1. Arylcarboxamidoalkyl-substituierte Aminoindane und -tetraline
Durch Austausch einer der n–Propyl-Seitenketten des Dipropylaminoindans sowie
des Dipropylaminotetralins mit einer N-Butyl-4-arylamido-Partialstruktur und anschlie-
ßender Variation des aromatischen Anteils sollten hochaffine Dopamin- und Sero-
tonin-Liganden hergestellt werden. Außerdem sollte im Aminoindan-Grundgerüst
eine 5,6-Dimethoxy-Substitution erfolgen, um analog zum bekannten, selektiven
D3-Antagonisten U99194 (39c) eine verbesserte D3-Selektivität zu erzielen. Die
Steigerung der D3-Selektivität war auch das Ziel des bioisosteren Austauschs der
Amid-Funktion durch den fünfgliedrigen Heterocyclus Triazol.
Synthese: Die Synthese der Aminoindane und –tetraline mit Carboxamid-Partialstruktur erfolgte
nach der im Folgenden beschriebenen Synthesesequenz (Abb. 46):

Zusammenfassung
59
Das unsubstituierte Indan-2-on 58 wurde zunächst mit Propylamin reduktiv zu einem
sekundären Amin 59 aminiert, das anschließend im Rahmen einer nukleophilen
Substitution mit Brombutyronitril umgesetzt wurde. Nach der Reduktion der
Nitrilfunktion des Zwischenprodukts 60 und Bildung des primären Amins 61, wurde
die Amidkupplung mit dem entsprechenden Carbonsäurechlorid oder unter
Zuhilfenahme von Kupplungsreagenzien wie TBTU und HATU durchgeführt, was zu
den Carboxamiden 62a-g führte.
NH
n
N
Nn
N
NH2
n
Ar NH
ON
n
X X
X
X
O
n
X
n
OX
(x HCl)
58: n = 1 X = H
73: n = 1 X = 5,6-di-OMe
red.Aminierung
4 Schritte
59 x HCl: n = 1; X = H77: n = 1; X = 5,6-di-OMegekauft: n = 2; X = H
SN
Reduktionmit LiAlH4
60: n = 1; X = H78: n = 1; X = 5,6-di-OMe n = 2; X = H
61: n = 1; X = H79: n = 1; X = 5,6-di-OMe(R)-/(S)-67: n = 2; X = H
Acylierung
62: n = 1; X = Ha: Ar = 4-Biphenylb: Ar = 2-Biphenylc: Ar = 2-Naphthyld: Ar = 1-Naphthyle: Ar = 3-Iod-Phenylf : Ar = (R)-/(S)-Paracyclophang: Ar = Ferrocen
80: n = 1; X = 5,6-di-OMe Ar = 4-Biphenyl
(R)-/(S)-68: n = 2; X = H Ar = (R)-/(S)-Paracyclophan
Abb. 46: Synthese der Aminoindane und –tetraline mit Amidfunktion
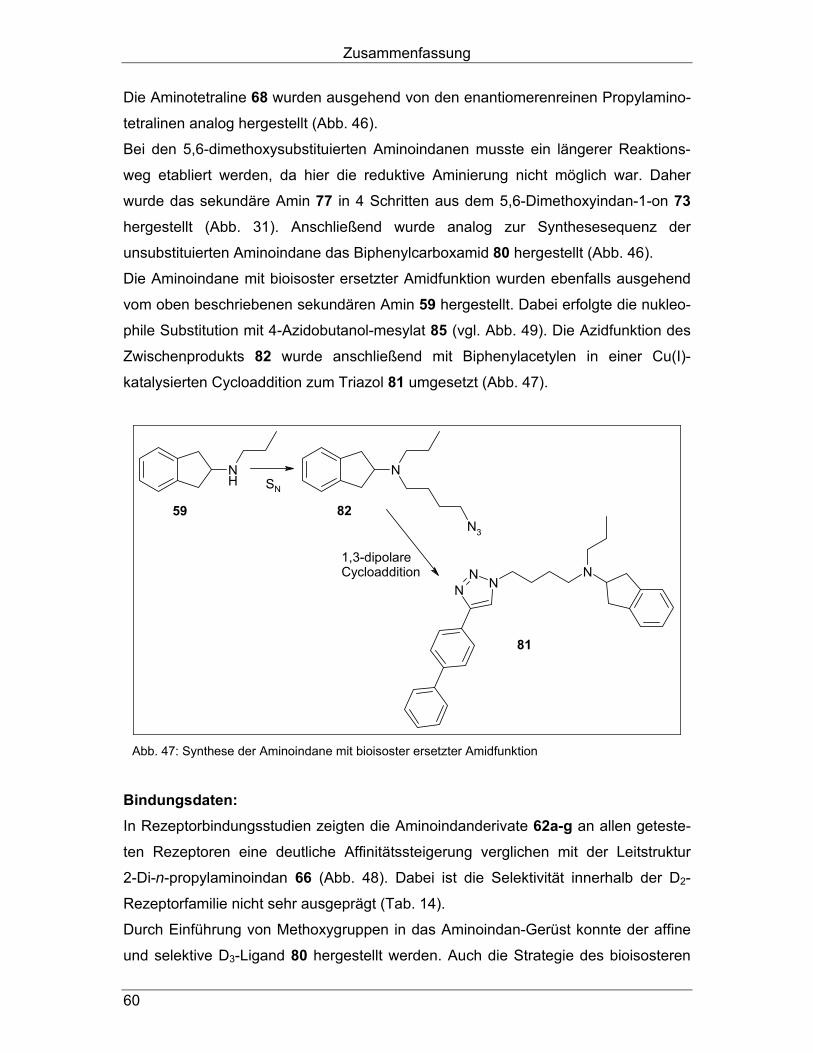
Zusammenfassung
60
Die Aminotetraline 68 wurden ausgehend von den enantiomerenreinen Propylamino-
tetralinen analog hergestellt (Abb. 46).
Bei den 5,6-dimethoxysubstituierten Aminoindanen musste ein längerer Reaktions-
weg etabliert werden, da hier die reduktive Aminierung nicht möglich war. Daher
wurde das sekundäre Amin 77 in 4 Schritten aus dem 5,6-Dimethoxyindan-1-on 73
hergestellt (Abb. 31). Anschließend wurde analog zur Synthesesequenz der
unsubstituierten Aminoindane das Biphenylcarboxamid 80 hergestellt (Abb. 46).
Die Aminoindane mit bioisoster ersetzter Amidfunktion wurden ebenfalls ausgehend
vom oben beschriebenen sekundären Amin 59 hergestellt. Dabei erfolgte die nukleo-
phile Substitution mit 4-Azidobutanol-mesylat 85 (vgl. Abb. 49). Die Azidfunktion des
Zwischenprodukts 82 wurde anschließend mit Biphenylacetylen in einer Cu(I)-
katalysierten Cycloaddition zum Triazol 81 umgesetzt (Abb. 47).
NH
N
N3
NNN
N
59 82
81
SN
1,3-dipolareCycloaddition
Abb. 47: Synthese der Aminoindane mit bioisoster ersetzter Amidfunktion
Bindungsdaten: In Rezeptorbindungsstudien zeigten die Aminoindanderivate 62a-g an allen geteste-
ten Rezeptoren eine deutliche Affinitätssteigerung verglichen mit der Leitstruktur
2-Di-n-propylaminoindan 66 (Abb. 48). Dabei ist die Selektivität innerhalb der D2-
Rezeptorfamilie nicht sehr ausgeprägt (Tab. 14).
Durch Einführung von Methoxygruppen in das Aminoindan-Gerüst konnte der affine
und selektive D3-Ligand 80 hergestellt werden. Auch die Strategie des bioisosteren

Zusammenfassung
61
Austauschs der Amid-Funktionalität durch eine 1H-1,2,3-Triazol-Struktur war von
Erfolg gekrönt und resultierte im Aminoindan 81 mit nochmals gesteigerter D3-
Selektivität (Tab. 14, Abb. 48).
Affinität Selektivität Intrinsische Aktivität
D2short
0,38-7,2
(Ki high; außer
62a)
D1/D3 1 800- 23 000einlagige Systeme
(62a, 62c, 62d) D3: 73-83 %
D3 0,061-0,71 5-HT2/D3 690-9 700 doppellagiges
Ferrocen (62g)
D2-4: 40-72 %
5-HT1A: 101 %
D4 0,50-2,3 innerhalb der
D2-Familie
keine
(außer 62a)
62a-g
5-HT1A 0,70-22
D3 11
80 andere
Rezeptoren 120-2 800
für D3 11- bis 250-
fach
D3 0,83
D2long/short 37/30 (Ki high) 81
andere
Rezeptoren 89-6 000
für D3 36- bis 7 200-
fach
Tab. 14: Übersicht über die Bindungseigenschaften der Aminoindane, Ki in nM
NH
ON
OMe
OMe
NNH
ON
NH
ON
Fe
NNN
N
66: D2s Ki = 4,2 + 340 nM D3 Ki = 3,1 + 100 nM D4 Ki = 11 + 330 nM
(S)- 62f: D2s Ki = 0,38 + 27 nM D3 Ki = 0,49 nM D4 Ki = 0,50 nM(R)-62f: D2s Ki = 0,67 + 19 nM D3 Ki = 0,24 nM D4 Ki = 1,2 nM
62g: D2s Ki = 1,5 + 55 nM D3 Ki = 0,52 nM D4 Ki = 0,51 nM 5-HT1A Ki = 0,70 nM
80: D2s Ki = 2 100 nM D3 Ki = 11 nM D4 Ki = 120 nM
81: D2s Ki = 30 + 2 600 nM D3 Ki = 0,83 nM D4 Ki = 89 nM 5-HT1A Ki = 2 700 nM
Abb. 48: Bindungsdaten einiger Aminoindane, Ki in nM, bei biphasischer Kurve (Ki high + Ki low)

Zusammenfassung
62
8.2. Alkinylcyclohexen als Tetralin-Bioisoster In den letzten Jahren konnte von unserer Arbeitsgruppe gezeigt werden, dass der
aromatische Anteil des Aminotetralins als dopaminerges Pharmakophor bioisoster
durch ein nicht-aromatisches, konjugiertes π-System ersetzt werden kann (Abb. 7,
Kapitel 2.1.2.2.).48-51 Das terminale Enin FAUC 73 (38a) wies hohe D3-Selektivität bei
einstellig-nanomolarer D3-Affinität auf.48
Um die Affinität und Selektivität weiter zu steigern, sollte, wie schon bei den Amino-
indanen, eine der n-Propyl-Gruppen durch das N-Butyl-4-biphenylamid-Motiv ersetzt
werden.
Synthese: Zunächst war es wichtig, eine geeignete Schutzgruppen-Strategie zu finden, um
einen orthogonalen Syntheseverlauf zu gewährleisten. Als Struktur der Wahl
erschien als Amin-Schutzgruppe die Azid-Funktion, da sie gegen Ende der Synthese
in einer Staudinger-Reaktion reduziert werden konnte, ohne dass die Enin-Partial-
struktur Schaden nehmen würde.
Um die spätere Butylen-Brücke aufzubauen, wurde 4-Chlorbutanol 83 in einer nuk-
leophilen Substitution zum Azid umgesetzt und anschließend die Alkoholfunktion
zum Mesylat 85 aktiviert (Abb. 49).
Das Monoethylenketal des 1,4-Cyclohexandion 86 wurde mit Propylamin reduktiv
aminiert113 und das entstehende sekundäre Amin 87 mit dem Mesylat 85 nukleophil
substituiert (Abb. 49). Direkt im Anschluss wurde das Ketals durch Rühren in 6N-
HCl/Aceton gespalten114. Das entstandene Keton 88 wurde zum Enoltriflat 89
aktiviert und in einer Cacchi-Kupplung mit TMS- bzw. Phenylacetylen zu den Eninen
90 und 91 umgesetzt. Um während der Cacchi-Kupplung eine 1,3-dipolare
Cycloaddition des Seitenketten-Azids mit den eingestzten Acetylenen zu vermeiden,
war es besonders wichtig, die Stöchiometrie der Reaktionspartner zu optimieren.
Abschließend folgte die Reduktion der Azid-Funktion nach Staudinger zu den
jeweiligen primären Aminen und deren Acylierung mit 4-Biphenylcarbonsäurechlorid
zu den Zielverbindungen 92 und 93. Die Abspaltung der TMS-Schutzgruppe des
geschützten Enins 92 führte zu einem terminalen Alkin 94.

Zusammenfassung
63
O O
O
O O
NH N
O
N3 N
OTf
N3 N
R
N3
NNH
R
O
OHCl
MsON3
NN
O
NN
BiPh
NN
OTf
NN
BiPh
NN
R'
NN
BiPh
90: R = TMS91: R = Ph
92: R = TMS 94: R = H93: R = Ph
99: R' = TMS 101: R' = H100: R' = Ph
83 85
86 87 88 89
9798
SN Tf2O Cacchi-Kupplung
red.Amin.
1) Staudinger- Reduktion2) Acylierung
1,3-dipolareCycloaddition
Cacchi-Kupplung
Tf2O
Abb. 49: Synthese der Enine
Um die entsprechenden Triazole herzustellen, wurde die Keton-Zwischenstufe 88
zunächst mit Biphenylacetylen in einer Cu(I)-katalysierten 1,3-dipolaren Cycloaddi-
tion zum 1,4-substituierten Triazol 97 umgesetzt (Abb. 49). Das Keton 97 wurde
dann, analog zur vorher beschriebenen Reaktionssequenz, zum Enoltriflat 98 akti-
viert und in Cacchi-Kupplungen mit TMS- und Phenylacetylen zu den Eninen 99 und
100 umgesetzt. Das TMS-geschützte Enin 99 wurde anschließend zum terminalen
Enin 101 entschützt.
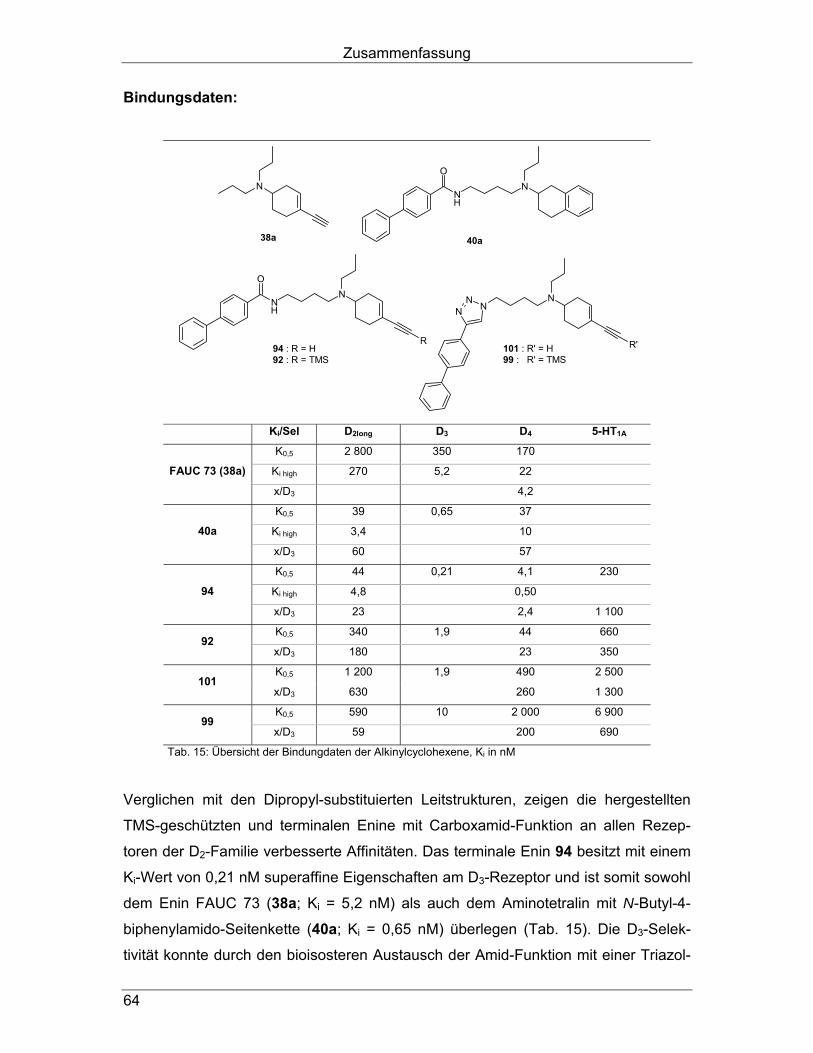
Zusammenfassung
64
Bindungsdaten:
NH
ON
R
N
NNN
N
R'
NH
ON
38a
94 : R = H92 : R = TMS
40a
101 : R' = H99 : R' = TMS
Ki/Sel D2long D3 D4 5-HT1A
K0,5 2 800 350 170
Ki high 270 5,2 22 FAUC 73 (38a)
x/D3 4,2
K0,5 39 0,65 37
Ki high 3,4 10 40a
x/D3 60 57
K0,5 44 0,21 4,1 230
Ki high 4,8 0,50 94
x/D3 23 2,4 1 100
K0,5 340 1,9 44 660 92
x/D3 180 23 350
K0,5 1 200 1,9 490 2 500 101
x/D3 630 260 1 300
K0,5 590 10 2 000 6 900 99
x/D3 59 200 690
Tab. 15: Übersicht der Bindungdaten der Alkinylcyclohexene, Ki in nM
Verglichen mit den Dipropyl-substituierten Leitstrukturen, zeigen die hergestellten
TMS-geschützten und terminalen Enine mit Carboxamid-Funktion an allen Rezep-
toren der D2-Familie verbesserte Affinitäten. Das terminale Enin 94 besitzt mit einem
Ki-Wert von 0,21 nM superaffine Eigenschaften am D3-Rezeptor und ist somit sowohl
dem Enin FAUC 73 (38a; Ki = 5,2 nM) als auch dem Aminotetralin mit N-Butyl-4-
biphenylamido-Seitenkette (40a; Ki = 0,65 nM) überlegen (Tab. 15). Die D3-Selek-
tivität konnte durch den bioisosteren Austausch der Amid-Funktion mit einer Triazol-
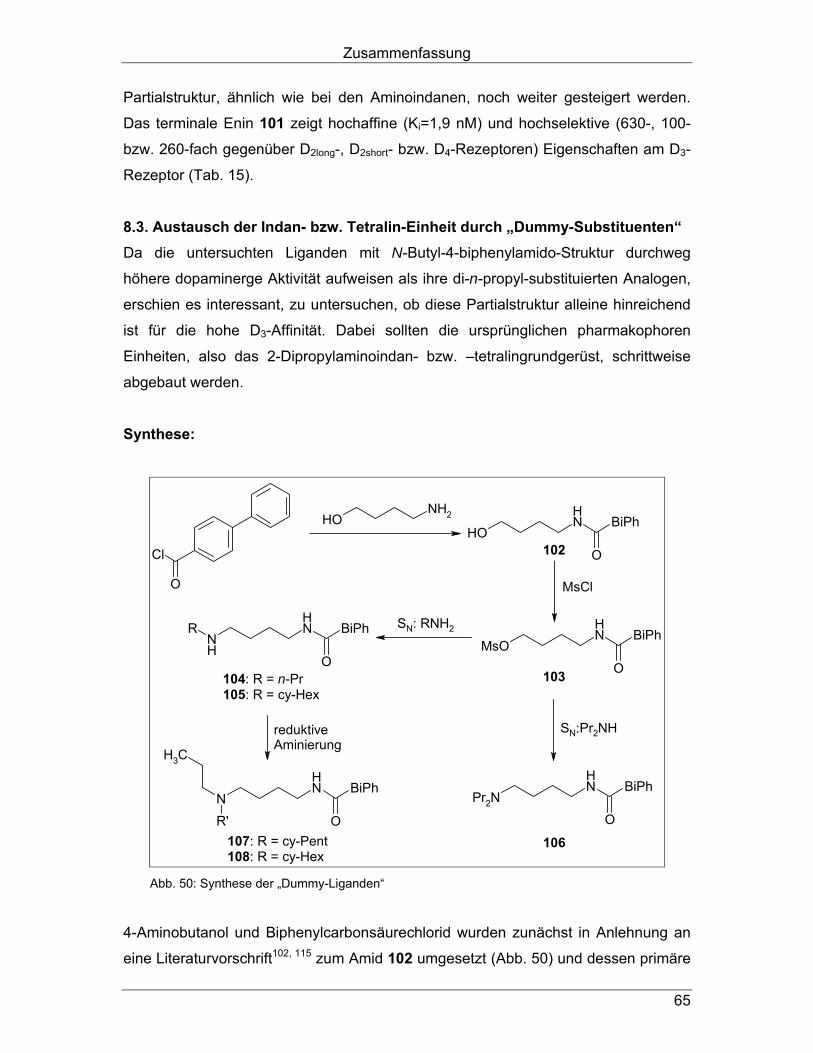
Zusammenfassung
65
Partialstruktur, ähnlich wie bei den Aminoindanen, noch weiter gesteigert werden.
Das terminale Enin 101 zeigt hochaffine (Ki=1,9 nM) und hochselektive (630-, 100-
bzw. 260-fach gegenüber D2long-, D2short- bzw. D4-Rezeptoren) Eigenschaften am D3-
Rezeptor (Tab. 15).
8.3. Austausch der Indan- bzw. Tetralin-Einheit durch „Dummy-Substituenten“
Da die untersuchten Liganden mit N-Butyl-4-biphenylamido-Struktur durchweg
höhere dopaminerge Aktivität aufweisen als ihre di-n-propyl-substituierten Analogen,
erschien es interessant, zu untersuchen, ob diese Partialstruktur alleine hinreichend
ist für die hohe D3-Affinität. Dabei sollten die ursprünglichen pharmakophoren
Einheiten, also das 2-Dipropylaminoindan- bzw. –tetralingrundgerüst, schrittweise
abgebaut werden.
Synthese:
NH
NH
BiPh
O
R
OHNH2
OHNH
BiPh
O
O
Cl
MsONH
BiPh
O
Pr2NNH
BiPh
ON
NH
BiPh
OR'
CH3
104: R = n-Pr105: R = cy-Hex
SN: RNH2
SN:Pr2NH
107: R = cy-Pent108: R = cy-Hex
MsCl
reduktiveAminierung
106
103
102
Abb. 50: Synthese der „Dummy-Liganden“
4-Aminobutanol und Biphenylcarbonsäurechlorid wurden zunächst in Anlehnung an
eine Literaturvorschrift102, 115 zum Amid 102 umgesetzt (Abb. 50) und dessen primäre

Zusammenfassung
66
Alkoholfunktion dann zum Mesylat 103 aktiviert. Darauf folgten nukleophile Substitu-
tionen zu den sekundären und tertiären Aminen 104-106. Die sekundären Amine 104
und 105 wurden anschließend reduktiv zu den cycloalkyl-propyl-substituierten
Zielsubstanzen 107 und 108 alkyliert.
Bindungsdaten : Die Bindungsdaten zeigen, dass die D3-Affinität der „Dummy-Liganden“ umso stärker
reduziert ist, je weiter das ursprüngliche Tetralin- und Indan-Gerüst abgebaut wurde
(Tab. 16). Durch diesen Affinitätsverlust konnte gezeigt werden, dass ein konjugier-
tes π-System in Nachbarschaft zu einem basischen Stickstoff für die Rezeptorerken-
nung nötig zu sein scheint.
NH
ON
R
R D2short D3 D4
106 n-Pr 8 800 950 1 400
107 cy-Pent 87 000 90 1 500
108 cy-Hex 6 300 140 670
Tab. 16: Bindungsdaten der „Dummy-Liganden“, Ki in nM
8.4. 2-Arylethyl-substituierte Aminoindane Für Aminoindane mit einer N-Butyl-4-biphenylamido-Seitenkette konnte gezeigt
werden, dass sie ähnliche Bindungseigenschaften besitzen wie die entsprechenden
Aminotetraline. Daher sollte überprüft werden, ob auch die Aminoindan-Analogen
des Antiparkinson Wirkstoffes Rotigotin 6 ein ähnliches Bindungsprofil wie deren
Leitsubstanz aufweisen.
Analog zur Leitsubstanz Rotigotin 6 sollte das 2-(thien-2-yl)ethyl-substituierte Derivat
110a hergestellt werden. Als zusätzliche Variationen im aromatischen Bereich sollten
2-Phenylethyl- und 2-(4-Methoxyphenyl)ethyl-Partialstrukturen dienen.

Zusammenfassung
67
Synthese: Das Propylaminoindan 59 wurde mit Arylessigsäurechloriden zu den Amiden 109
acyliert, die anschließend mit LiAlH4 zu den tertiären Aminen 110a-c reduziert
wurden (Abb. 51).
NH
CH3
N
CH3
ArO
N
CH3
Ar
Ar = Thienyl Phenyl 4-Methoxyphenyl
59 109 110
Acylierung Red.
Abb. 51: Synthese der Rotigotin-Analogen
Bindungsdaten:
N
Ar
N
S 110a-cRotigotin 6
Ar Ki/Sel D2long D3 D4
6 20 4 55
K0,5 310
Ki high 41 10 110a Thien-2-yl
Ki low 1 500 320
K0,5 220
Ki high 41 3,4 110b Phenyl
Ki low 920 76
Ki high 13 4,7 18 110c
4-Methoxy-
phenyl Ki low 470 62 180
Tab. 17: Bindungsdaten der 2-arylethyl-substituierten Aminoindane, Ki
in nM
Die 2-arylethyl-substituierten Aminoindane 110a-c weisen an den hochaffinen Bin-
dungsstellen von D2- und D3-Rezeptoren Affinitäten im niedrigen nanomolaren
Bereich auf (Tab. 17). Dies ist auch bei der Leitstruktur Rotigotin 6 zu beobachten.
Vergleicht man die Bindungseigenschaften am 5-HT1A-Rezeptor, so besitzen die

Zusammenfassung
68
thien-2-yl- und phenyl-substituierten Derivate eine höhere Affinität als das
p-Methoxyphenyl-Analoge und Rotigotin 6.
Die Bindungsprofile der 2-arylethyl-substituierten Aminoindane 110a-c weisen an
den Dopamin D1-3-Rezeptoren zwar gewisse Parallelen mit dem Bindungsprofil der
Leitstruktur Rotigotin 6 auf, an anderen Rezeptoren, wie den Dopamin D4- und Sero-
tonin 5-HT1A-Rezeptoren weichen sie aber stark ab. Eine grundsätzliche Affinitäts-
steigerung konnte bei den Aminoindanen nicht beobachtet werden.

Summary
69
8. Summary Starting from dopamine receptor ligand 40a (scheme 44), structural variations should
be carried out to get ligands displaying increased dopaminergic and serotoninergic
affinity and improved D3 selectivity.
NH
ON
N
X
X
N
R
NH
ON
40a
chapter 4.1.Ar = single- and doubble- layered aromatic Systems
chapter 4.3.Y = amide or triazole chapter 4.2.
X = H, OMe
Ar
Y
Y
chapter 5.R = H, TMS, PhY = amide or triazole
R'
chapter 6.R' = H, alkyl, cycloalkyl
amino-indans
FAUC 73 (38a)dummies
Scheme 44: Assignment of tasks
To achieve this, structure 40a was diversified as follows (scheme 44):
• Exchange of the aminotetraline scaffold by an aminoindan motif and
o Variation of the arylcarboxamide moiety
o Variation of the aminoindan moiety (dimethoxy substitution)
o Variation by bioisosteric exchange of the carboxamide functionality by
an 1H-1,2,3-triazole moiety
• Bioisosteric exchange of the aromatic part of aminotetraline by a conjugated
π-system (alkynylcyclohexenes)

Summary
70
o Alkynylcyclohexenes with amide structure
o Alkynylcyclohexenes with bioisosteric exchanged carboxamide function
(1H-1,2,3-triazol moiety)
• Exchange of the aromatic part of aminoindan and -tetraline by substituents
without any π-system to investigate the impact of the originally pharmaco-
phoric elements on receptor recognition
Finally, aminoindan analogues of the anti-Parkinson drug rotigotine 6 should be
synthesized (scheme 45) to observe if the binding properties of the achiral hybrid
resemble those of lead structure 6.
N
S
OH
NN
Ar rotigotine 6chapter 7.Type 39
Scheme 45: Assignment of tasks for rotigotine analogues
8.1. Arylcarboxamidoalkyl substituted Aminoindans and -tetralines
High affinity dopaminergic and serotininergic ligands should be created by exchange
of one n-propyl sidechain of dipropylaminoindan and -tetraline by a N-butyl-4-
arylamido motif and subsequent variation of the aromatic residue. Besides, modelled
on the D3 selective antagonist U99194 (39c), a 5,6-dimethoxy substitution should be
introduced into the aminoindan scaffold to improve D3-selectivity. Increase in D3
selectivity was also the objective of the bioisosteric exchange of the amide function
by the five-membered, heterocyclic triazole.
Synthesis: Synthesis of the aminoindans and –tetralines bearing a carboxamide partial structure
was performed according to the following sequence (scheme 46):
First of all, the unsubstituted indan-2-one 58 was reductively aminated with
propylamine to obtain the secondary amine 59 which was then substituted in a
nucleophilic manner with bromobutyronitrile. After reduction of the product’s (60)
nitrile functionality and formation of the primary amine 61, amide coupling was

Summary
71
conducted employing the appropriate acid chloride or by using coupling reagents like
TBTU and HATU to get the carboxamides 62a-g.
NH
n
N
Nn
N
NH2
n
Ar NH
ON
n
X X
X
X
O
n
X
n
OX
(x HCl)
58: n = 1 X = H
73: n = 1 X = 5,6-di-OMe
reductiveamination
4 steps
59 x HCl: n = 1; X = H77: n = 1; X = 5,6-di-OMecommercially available:n = 2; X = H
SN
reductionwith LiAlH4
60: n = 1; X = H78: n = 1; X = 5,6-di-OMe n = 2; X = H
61: n = 1; X = H79: n = 1; X = 5,6-di-OMe(R)-/(S)-67: n = 2; X = H
acylation
62: n = 1; X = Ha: Ar = 4-biphenylb: Ar = 2-biphenylc: Ar = 2-naphthyld: Ar = 1-naphthyle: Ar = 3-iodo-phenylf : Ar = (R)-/(S)-paracyclophaneg: Ar = ferrocene
80: n = 1; X = 5,6-di-OMe Ar = 4-biphenyl
(R)-/(S)-68: n = 2; X = H Ar = (R)-/(S)-paracyclophane
Scheme 46: Synthesis of the aminoindans and –tetralines with an amide functionality
The aminotetralines 68 were synthesized in an analogous manner starting from the
enantiomeric pure propylaminotetralines (scheme 46).
In case of the 5,6-dimethoxy substituted aminoindans, a longer reaction sequence
had to be established as the reductive amination wasn’t possible. For this reason, the
secondary amine 77 was synthesized within 4 steps starting from 5,6-dimethoxy-

Summary
72
indan-1-one 73 (scheme 31). Afterwards the synthesis could be proceeded in the
same way as described for the unsubstituted aminoindans which resulted in the
biphenylcarboxamide 80 (scheme 46).
The secondary amine 59 described above was also employed as educt for the
synthesis of the aminoindans with bioisosteric exchanged amide function. Therefore,
the nucleophilic substitution reaction was performed with 4-azidobutanol-mesylate 85
(compare scheme 49). Afterwards, the azide function of the ternary amine 82 was
reacted with biphenylacetylene in a Cu(I)-catalyzed cycloaddition to get the triazole
81 (scheme 47).
NH
CH3
N
CH3
N3
NN
CH3
NN59 82
81
SN
clickreaction
Scheme 47: Synthesis of aminoindans with bioisosteric exchanged amide function
Binding data: In receptor binding studies the aminoindan derivatives 62a-g showed a clearly
enhanced affinity towards all the receptors tested when compared to the lead
structure 2-di-n-propylaminoindan 66 (scheme 48). Within the D2 receptor family the
ligands do not bind very selectively (table 14).
By implementation of methoxy groups into the aminoindan scaffold, the affin and
selective D3 ligand 80 could be created.
The strategy of bioisosteric replacement of the amide functionality by a
1H-1,2,3-triazole moiety was fruitful as well and resulted in aminoindan 81
possessing additionally increased D3 selectivity (table 14, scheme 48).

Summary
73
affinity selectivity intrinsic activity
D2short
0,38-7,2
(Ki high; except
62a)
D1/D3 1 800- 23 000
single-layered
systems
(62a, 62c, 62d)
D3: 73-83 %
D3 0,061-0,71 5-HT2/D3 690-9 700 double-layered
Ferrocene (62g)
D2-4: 40-72 %
5-HT1A: 101 %
D4 0,50-2,3 within the D2
family
none
(except 62a)
62a-g
5-HT1A 0,70-22
D3 11
80 other
receptors 120-2 800
for D3 11- bis 250-
fach
D3 0,83
D2long/short 37/30 (Ki high) 81
other
receptors 89-6 000
for D3 36- bis 7 200-
fach
Table 14: Overview over the binding properties of the aminoindans (affinity: Ki in nM)
NH
ON
OMe
OMe
NNH
ON
NH
ON
Fe
NNN
N
66: D2s Ki = 4,2 + 340 nM D3 Ki = 3,1 + 100 nM D4 Ki = 11 + 330 nM
(S)- 62f: D2s Ki = 0,38 + 27 nM D3 Ki = 0,49 nM D4 Ki = 0,50 nM(R)-62f: D2s Ki = 0,67 + 19 nM D3 Ki = 0,24 nM D4 Ki = 1,2 nM
62g: D2s Ki = 1,5 + 55 nM D3 Ki = 0,52 nM D4 Ki = 0,51 nM 5-HT1A Ki = 0,70 nM
80: D2s Ki = 2 100 nM D3 Ki = 11 nM D4 Ki = 120 nM
81: D2s Ki = 30 + 2 600 nM D3 Ki = 0,83 nM D4 Ki = 89 nM 5-HT1A Ki = 2 700 nM
Scheme 48: Binding data of some aminoindans, Ki in nM, for biphasic curves (Ki high + Ki low)

Summary
74
8.2. Alkinylcyclohexene as tetraline bioisostere
During the last years our work group could show that the aromatic part of amino-
tetraline as dopaminergic pharmacophore can be replaced bioisosterically by a non-
aromatic, conjugated π-system (scheme. 7, chapter 2.1.2.2.).48-51 The terminal enyne
FAUC 73 (38a) exhibited high D3 selectivity and single-digit nanomolar D3 affinity.48
To further increase affinity and selectivity, one of it’s n-propyl groups should be
replaced by the N-butyl-4-biphenylamido motif, as seen with the aminoindans.
Synthesis: First of all, it was important to find an appropriate strategy concerning protecting
groups to ensure an orthogonal synthesis. As protecting group for amines, the azide
function seemed to be the structure of choice as it could be removed in the synthesis’
final stages via Staudinger reduction without damaging the enyne partial structure.
To built the later butylene bridge, 4-chlorobutanol 83 was substituted in a nucleophilic
manner to obtain an azide. Subsequently, the alcohol function was activated to get
the mesylate 85 (scheme 49).
1,4-Cyclohexandion-monoethyleneketale 86 was reductively aminated with propyl-
amine113 (scheme 49). The formed secondary amine 87 was then substituted with the
mesylate 85 followed by the decomposition of the ketal by stirring with 6N-
HCl/acetone114. The ketone 88 formed was activated to get the enoletriflate 89 and
subsequently reacted via a Cacchi’s coupling with TMS- and phenylacetylene to
obtain the enynes 90 and 91, respectively. To avoid a 1,3-dipolar cycloaddition of the
sidechain’s azide with the acetylene employed during Cacchi’s coupling, it was
particularly important to optimize the reaction partner’s stoichiometry. Afterwards the
reduction of the azide according to Staudinger’s procedure was carried out to get the
corresponding primary amines which were subsequently acylated with 4-biphenyl-
carboxylic acid chloride to the enynes 92 and 93. Removement of the TMS protecting
group of the enyne 92 resulted in the terminal alkyne 93.

Summary
75
O O
O
O O
NH N
O
N3 N
OTf
N3 N
R
N3
NNH
R
O
OHCl
MsON3
NN
O
NN
BiPh
NN
OTf
NN
BiPh
N
CH3
N
R'
NN
BiPh
90: R = TMS91: R = Ph
92: R = TMS 94: R = H93: R = Ph
99: R' = TMS 101: R' = H100: R' = Ph
83 85
86 87 88 89
9798
SN Tf2O Cacchi'scoupling
red.amin.
1) Staudinger's reduction2) acylation
1,3-dipolarcycloaddition
Tf2O
Cacchi'scoupling
Scheme 49: Synthesis of the enynes
To synthesize the corresponding triazoles, the ketone intermediate 88 was reacted
with biphenylacetylene to the 1,4-substituted triazole 97 using a Cu(I)-catalyzed
1,3-dipolar cycloaddition (scheme 49). Ketone 97 was then, according to the reaction
sequence described above, activated to the enoletriflate 98 and subsequently
coupled according to Cacchi’s conditions with TMS- and phenylacetylene to obtain
the enynes 99 and 100. Finally, the TMS group of the enyne 99 was removed getting
the terminal enyne 101.

Summary
76
Binding data:
NH
ON
R
N
NNN
N
R'
NH
ON
38a
94 : R = H92 : R = TMS
40a
101 : R' = H99 : R' = TMS
Ki/sel D2long D3 D4 5-HT1A
K0,5 2 800 350 170
Ki high 270 5,2 22 FAUC 73 (38a)
x/D3 4,2
K0,5 39 0,65 37
Ki high 3,4 10 40a
x/D3 60 57
K0,5 44 0,21 4,1 230
Ki high 4,8 0,50 94
x/D3 23 2,4 1 100
K0,5 340 1,9 44 660 92
x/D3 180 23 350
K0,5 1 200 1,9 490 2 500 101
x/D3 630 260 1 300
K0,5 590 10 2 000 6 900 99
x/D3 59 200 690
Table 15: Binding data of the alkynylcyclohexenes, Ki in nM
Compared to the dipropyl substituted lead structures, the synthesized TMS protected
und terminal enynes with carboxamide function show improved affinities to all
receptors of the D2 familiy. The terminal enyne 94 possesses super-affin properties at
the D3 receptor (Ki = 0,21 nM) and thus is superior both to the enyne FAUC 73 (38a,
Ki = 5,2 nM) and to the aminotetraline with N-butyl-4-biphenylamido sidechain (40a;
Ki = 0,65 nM, table 15). D3 selectivity could be further increased by bioisosteric
exchange of the amide function by a triazole motif, which is in accordance to the
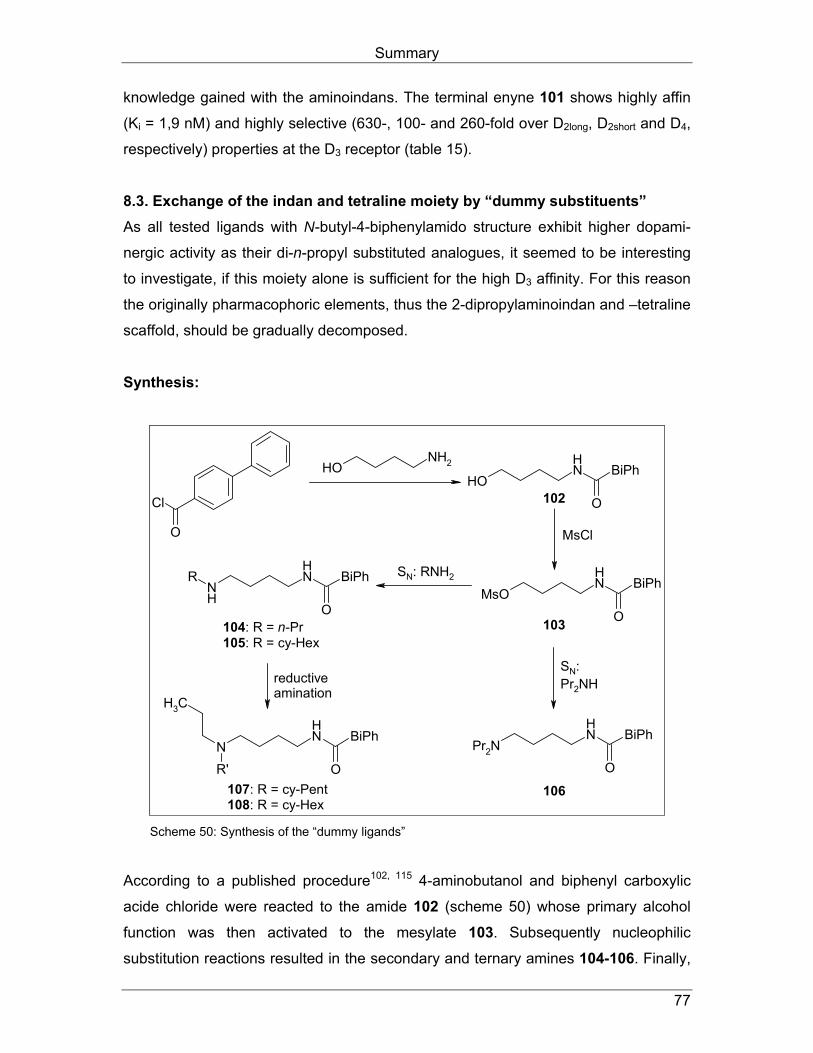
Summary
77
knowledge gained with the aminoindans. The terminal enyne 101 shows highly affin
(Ki = 1,9 nM) and highly selective (630-, 100- and 260-fold over D2long, D2short and D4,
respectively) properties at the D3 receptor (table 15).
8.3. Exchange of the indan and tetraline moiety by “dummy substituents”
As all tested ligands with N-butyl-4-biphenylamido structure exhibit higher dopami-
nergic activity as their di-n-propyl substituted analogues, it seemed to be interesting
to investigate, if this moiety alone is sufficient for the high D3 affinity. For this reason
the originally pharmacophoric elements, thus the 2-dipropylaminoindan and –tetraline
scaffold, should be gradually decomposed.
Synthesis:
NH
NH
BiPh
O
R
OHNH2
OHNH
BiPh
O
O
Cl
MsONH
BiPh
O
Pr2NNH
BiPh
ON
NH
BiPh
OR'
CH3
104: R = n-Pr105: R = cy-Hex
SN: RNH2
SN:Pr2NH
107: R = cy-Pent108: R = cy-Hex
MsCl
reductiveamination
106
103
102
Scheme 50: Synthesis of the “dummy ligands”
According to a published procedure102, 115 4-aminobutanol and biphenyl carboxylic
acide chloride were reacted to the amide 102 (scheme 50) whose primary alcohol
function was then activated to the mesylate 103. Subsequently nucleophilic
substitution reactions resulted in the secondary and ternary amines 104-106. Finally,

Summary
78
the secondary amines 104 and 105 were reductively alkylated to obtain the
cycloalkyl-propyl substituted test compounds 107 and 108.
Binding data : Binding data show that D3 affinity of the “dummy ligands” is diminished all the more
the more the original tetraline and indan scaffold is decomposed (table 16). By this
loss of affinity it could be demonstrated that a conjugated π-system adjacent to a
basic nitrogen seems to be crucial for receptor recognition.
NH
ON
R
R D2short D3 D4
106 n-Pr 8 800 950 1 400
107 cy-Pent 87 000 90 1 500
108 cy-Hex 6 300 140 670
Table 16: Binding data of the „dummy ligands”, Ki in nM
8.4. 2-Arylethyl substituted aminoindans It could be demonstrated for the aminoindans with N-butyl-4-biphenylamido sidechain
that they display binding properties similar to those of the corresponding aminotetra-
lines. Hence, it should be investigated if the aminoindan analogues of the anti-
Parkinson drug rotigotine 6 also exhibit binding profiles that resemble that of the lead
structure.
Analogous to the lead compound rotigotine 6 the 2-(thien-2-yl)ethyl substituted
derivative 110a should be synthesized. 2-Phenylethyl and 2-(4-methoxyphenyl)ethyl
moieties should serve as additional variations of the aromatic part.
Synthesis: Propylaminoindan 59 was acylated with arylacetic acid chlorides to get the amides
109 (scheme 51) which were then reduced with LiAlH4 resulting in the ternary amines
110 a-c.

Summary
79
NH
CH3
N
CH3
ArO
N
CH3
Ar
Ar = thien-2-yl phenyl 4-methoxyphenyl
59 109a-c 110a-c
acylation reduction.
Scheme 51: Synthesis of the aminoindan analogues of rotigotine
Binding data:
N
Ar
N
S 110a-crotigotine 6
Ar Ki/sel D2long D3 D4
6 20 4 55
K0,5 310
Ki high 41 10 110a Thien-2-yl
Ki low 1 500 320
K0,5 220
Ki high 41 3,4 110b Phenyl
Ki low 920 76
Ki high 13 4,7 18 110c
4-Methoxy-
phenyl Ki low 470 62 180
Table 17: Binding data of the 2-arylethyl-substituted aminoindans, Ki in
nM
The 2-arylethyl substituted aminoindans 110a-c display affinities in the low
nanomolar range for the high affinity binding sites of D2 and D3 receptors (table 17).
This holds also true for the lead structure rotigotine 6. Comparing the binding
properties at the 5-HT1A receptor, the thien-2-yl and phenyl substituted derivatives
possess higher affinity than the p-methoxyphenyl analogue and rotigotine 6.
To conclude, it could be shown that the binding profiles of the 2-arylethyl substituted
aminoindans 110a-c exhibit some similarities to that one of the lead compound
rotigotine 6 concerning the dopamine D1-3 receptors. On the other hand, there are
targets like the dopamine D4 and serotonine 5-HT1A receptors at which the binding

Summary
80
profiles differ clearly. A general increase of affinity could not be found for the
aminoindans.

Experimenteller Teil
81
9. Experimenteller Teil Allgemeine Angaben zu den chemischen Untersuchungen Lösungsmittel wurden bei den Firmen FLUKA, ACROS, ALDRICH und VWR in verschiedenen
Qualitäten erworben. Zu den Synthesen wurden meist trockene und über Molsieb
gelagerte Lösungsmittel verwendet (jeweils angegeben).
Chemikalien wurden bei den Firmen FLUKA, ACROS, ALDRICH, ALFA AESAR; CAMBREX;
VWR und MERCK in der höchsten verfügbaren Reinheit erworben und in dieser
Qualität eingesetzt.
Schmelzpunkte wurden nach der Kapillarmethode mit dem Gerät BÜCHI 510 bestimmt und wurden
nicht korrigiert.
EI-Massen wurden mit einem FINNIGAN MAT TSQ 700 Spektrometer gemessen.
Hochaufgelöste EI-Massen
wurden mit dem Modell JOEL GCmatell bei einer Auflösung von M/ΔM > 5000 ge-
messen.
Elementaranalysen wurden von dem Institut für organische Chemie bzw. von der Firma BEETZ,
Mirkroanalytisches Laboratorium Kronach (nur Substanzen 99, 101, 106) angefertigt
IR-Spektren wurden mit einem FT/IR-Spektrometer der Firma JASCO (FT/IR 410) aufgenommen,
wobei die Proben entweder auf einen NaCl-Kristall aufgetragen oder mit KBr zu
einem Pressling verarbeitet wurden.
NMR-Spektren wurden entweder mit einem BRUKER AVANCE 360- oder einem BRUKER AVANCE
600-Spektrometer in Lösung mit Tetramethylsilan als internem Standard aufge-
nommen.
Dünnschichtchromatographie wurde auf DC-Fertigplatten Kieselgel 60 F254 der Firma MERCK durchgeführt.

Experimenteller Teil
82
Flashchromatographie wurde, wenn nicht anders angegeben, mit Kieselgel 60, Korngröße 40-63 µm der
Firma MERCK durchgeführt. Außerdem wurden neutrales Aluminiumoxid Type 507C
neutral der Firma FLUKA oder Kieselgel-Fertigsäulen für die BIOTAGE SP-1-Anlage
verwendet.
Falls nichts anders beschrieben, wurde die Flash-Chromatographie in Glassäulen
durchgeführt, wobei der Druck durch N2-Gas erzeugt wurde. Ein anderes
verwendetes Verfahren war die Reinigung an der BIOTAGE SP-1-Anlage.
Präparative HPLC wurde an Anlagen der Firma AGILENT (1100 Series) mit ZORBAX ECLIPSE XDB-
C8 (21.2 mm x 250 mm, 5 µm) bei verschiedenen H2O/CH3CN-Gradienten unter
Zusatz von 0,1 % TFA durchgeführt. Die Flussrate betrug 20 ml/min und die Wellen-
länge des Detektors 210 und 254 nm.
HPLC-Analysen wurden an Anlagen der Firma AGILENT (1100 Series) mit VWL-Detektor und
ZORBAX ECLIPSE XDB-C8 (4,6 mm x 150 mm, 5 µm) durchgeführt. Die Flussrate
betrug jeweils 0,5 ml/min.
Analysesysteme zur Reinheitsbestimmung: M1: MeOH/H2O mit Zusatz von 0,1 % TFA
Gradient: 10 → 100 % in 21 min, 100 % 3 min
M2: MeOH/H2O mit Zusatz von 0,1 % TFA
Gradient: 10 → 100 % in 15 min, 100 % 3 min
A1: CH3CN/H2O mit Zusatz von 0,1 % TFA
Gradient: 10 % 2 min, 10 → 50 % in 13 min, 50 % 10 min
A2: CH3CN/H2O mit Zusatz von 0,1 % TFA
Gradient: 10 % 2 min, 10 → 80 % in 15 min, 80 % 7 min
A3: CH3CN/H2O mit Zusatz von 0,1 % TFA
Gradient: 25 % 2 min, 25 → 90 % in 21 min, 90 % 3 min
A4: CH3CN/H2O mit Zusatz von 0,1 % TFA
Gradient: 25 % 2 min, 25 → 90 % in 16 min, 90 % 5 min
A5: CH3CN/H2O mit Zusatz von 0,1 % TFA
Gradient: 10 % 2 min, 10 → 50 % in 16 min, 50 % 6 min

Experimenteller Teil
83
HPLC-MS-Analysen wurden an Anlagen der Firma AGILENT (1100 Series) mit VWL-Detektor und
ZORBAX ECLIPSE XDB-C8 (4,6 mm x 150 mm, 5 µm), gekoppelt an ein BRUKER
ECQUIRE 2000-Massenspektrometer mit APC- und ES-Ionisation durchgeführt. Als
Fließmittelsystem dienten Wasser-Methanol-Gradienten unter Zusatz von 0,1 %
HCOOH (in H2O). Die Wellenlänge des Detektors betrug 210 bzw. 254 nm.
Analysesystem zur Reinheitsbestimmung: M3: MeOH/H2O mit Zusatz von 0,1 % HCOOH
Gradient: 10 % 3 min, 10 → 100 % in 15 min, 100 % 6 min
Flussrate 0,1 ml/min
Allgemeine Angaben zu den biologischen Untersuchungen
Rezeptorbindungsstudien D1-Rezeptor:
Die IC50-Werte wurden durch Verdrängungsexperimente an Membranpräparationen
aus Schweinehirnen ermittelt. Als Radioligand wurde [3H]SCH23390 verwendet. Die
unspezifische Bindung wurde mit (+)-Butaclamol bestimmt.
D2- , D3- und D4-Rezeptoren:
Die IC50-Werte wurden durch Verdrängungsexperimente an geklonten, in CHO-
Zelllinien stabil exprimierten, humanen D2long-, D2short-, D3- und D4.4-Rezeptoren
ermittelt. Als Radioligand wurde [3H]Spiperon verwendet. Die unspezifische Bindung
wurde mit Haloperidol bestimmt.
5-HT1A-Rezeptor:
Die IC50-Werte wurden durch Verdrängungsexperimente an Membranpräparationen
aus Schweinehirnen ermittelt. Als Radioligand wurde [3H]WAY100635 verwendet.
5-HT2-Rezeptor:
Die IC50-Werte wurden durch Verdrängungsexperimente an Membranpräparationen
aus Schweinehirnen ermittelt. Als Radioligand wurde [3H]Ketanserin verwendet.
α1-Rezeptor:
Die IC50-Werte wurden durch Verdrängungsexperimente an Membranpräparationen
aus Schweinehirnen ermittelt. Als Radioligand wurde [3H]Prazosin verwendet.

Experimenteller Teil
84
Funktionstests Es wurde ein Mitogenese-Assay mit CHO-Zellen, die die entsprechenden
dopaminergen Rezeptoren stabil exprimierten, verwendet. Dabei wurde die
Einbaurate von [3H]Thymidin gemessen. Als Vergleichssubstanz diente der Agonist
Quinpirol.
Zur Bestimmung der intrinsischen Aktivität am 5-HT1A-Rezeptor wurde die Bindung
von [35S]GTPγS an das G-Protein gemessen, die durch Agonisten gesteigert wird.
Auch hier wurden CHO-Zellen verwendet. Als Vergleichssubstanz diente 5-HT.
Datenanalyse wurde mittels nichtlinearer Regression und dem Programm PRISM durchgeführt. Die
ermittelten IC50-Werte wurden mit Hilfe der Cheng-Prussof-Gleichung in Ki-Werte um-
gerechnet. Die in den Tabellen der Kapitel 4-7 angegebenen Daten sind die
Mittelwerte aus mindestens zwei Einzelexperimenten (± SEM). Dabei stellen Anga-
ben der Form x + y die jeweiligen Ki-Werte der hoch- bzw. niedrigaffinen Bindungs-
stelle dar.

Experimenteller Teil
85
59: N-(Indan-2-yl)-N-propylamin Hydrochlorid
NH x HCl
10,39 g 2-Indanon (58; 78,61 mmol) und 23,57 g NaBH(OAc)3 (111,21 mmol) werden
in 80 ml trockenem THF gelöst bzw. suspendiert, bevor 10,5 ml n-Propylamin
(127,72 mmol) und 4,5 ml Eisessig (78,53 mmol) zugetropft werden. Dann wird 1 h
bei RT und anschließend 17,5 h unter Reflux gerührt.
Das Lösungsmittel wird am Rotationsverdampfer entfernt, der Rückstand in
konzentrierter HCl aufgenommen und mehrmals mit Et2O gewaschen. Die orga-
nischen Phasen werden verworfen. Die wässrige Phase wird mit 5N-NaOH basifiziert
und anschließend mehrmals mit Et2O ausgeschüttelt. Die organischen Phasen
werden über MgSO4 getrocknet und am Rotationsverdampfer reduziert. Dann
werden 58 ml einer 2-molaren Lösung von HCl in Et2O (116,0 mmol) zugegeben und
die Lösung kühl gestellt.
Der ausgefallene, dunkel gefärbte Niederschlag wird abgefrittet und in einer
Mischung aus EtOAc und MeOH umkristallisiert.
Ausbeute: 5,37 g (32,2 %), weißer Feststoff
Analytische Daten:
Schmp.: 189-191 °C (Lit.: 196-199 °C116)
EI-MS: m/z 175
IR:
(KBr-Pressling) ν in cm-1: 3273 s, 2966 s, 2792 s, 2736 s, 2432 s, 1633 s, 1608 s,
1460 s, 1047 s, 741 s

Experimenteller Teil
86
1H-NMR: 1H (360 MHz, CD3OD): 1,05 (t, J = 7,4 Hz, 3 H, Pr-3), 1,75 (m, 2 H, Pr-2), 3,05 (m,
2 H, Pr-1), 3,13 (dd, J = 16,3 Hz, 6,4 Hz, 2 H, Indan H-1/H-3), 3,42 (dd, J = 16,3 Hz,
7,9 Hz, 2 H, Indan H-1/H-3), 4,07 (m, 1 H, Indan H-2), 7,18-7,30 (m, 4 H, Indan H-4
bis H-7)
13C-NMR: 13C (150 MHz, CD3OD): 11,30 (Pr-3), 20,96 (Pr-2), 37,19 (Indan C-1/C-3), 49,25 (Pr-
1), 59,61 (Indan C-2), 125,73 (Indan C-4/C-7), 128,56 (Indan C-5/C-6), 140,09 (Indan
C-3a/C-7a)
60: (N-Indan-2-yl-N-propyl)-4-aminobutyronitril
N
N
2,89 g des sekundären Amins 59 (13,65 mmol), 10,62 g K2CO3 (76,83 mmol) und
2,06 g KI (12,36 mmol) werden in 70 ml trockenem CH3CN gelöst bzw. suspendiert.
Dann werden 3,27 ml 4-Brombutyronitril (32,7 mmol) zugetropft und für 24 h unter
Rückfluss gerührt.
Nach dem Abkühlen wird das Lösungsmittel am Rotationsverdampfer entfernt, der
Rückstand in Wasser aufgenommen und mit 2N-NaOH basifiziert, bevor mehrmals
mit CH2Cl2 ausgeschüttelt wird. Die organischen Phasen werden über Na2SO4
getrocknet und am Rotationsverdampfer eingeengt.
Der Rückstand wird mittels Flash-Chromatographie gereinigt (Hexan/EtOAc 9/1 +
0,5 % NMe2Et).
Ausbeute: 2,57 g (77,8 %), gelb-braune Flüssigkeit

Experimenteller Teil
87
Analytische Daten:
EI-MS: m/z 242
IR:
(NaCl) ν in cm-1: 2958 s, 2245 m (Nitril), 1462 m, 1084 m, 746 s
1H-NMR: 1H (360 MHz, CDCl3): 0,88 (t, J = 7,4 Hz, 3 H, Pr-3), 1,48 (m, 2 H, Pr-2), 1,79 (m,
2 H, But-2), 2,39-2,49 (m, 4 H, Pr-1 & But-3), 2,61 (m, 2 H, But-4), 2,85 (dd, J = 15,4
Hz, 8,4 Hz, 2 H, Indan H1/H-3), 3,01 (dd, J = 15,4 Hz, 7,8 Hz, 2 H; Indan H-1/H-3),
3,67 (m, 1H, Indan H-2), 7,09-7,19 (m, 4H, Indan H-4 bis H-7),
13C-NMR: 13C (150 MHz, CDCl3): 11,84 (Pr-3), 14,69 (But-3), 20,40 (Pr-2), 23,84 (But-2), 36,11
(Indan C-1/C-3), 49,27 (But-4), 53,21 (Pr-1), 62,50 (Indan C-2), 119,94 (But-1),
124,42 (Indan C-4/C-7), 126,31 (Indan C-5/C-6), 141,68 (Indan C-3a/C-7a)
CHN (%): C16H22N2 x 0,4 H2O
Ber: C 77,00 H 9,21 N 11,22
Gef: C 77,27 H 9,30 N 11,25

Experimenteller Teil
88
61: (N-Indan-2-yl-N-propyl)butan-1,4-diamin
NH2
N
Zu einer gekühlten Lösung von 1,21 g des Nitrils 60 (4,96 mmol) in 35 ml trockenem
Et2O werden 12,4 ml einer 1-molaren Lösung von LiAlH4 in Et2O (12,4 mmol) zuge-
tropft. Anschließend wird das Eisbad entfernt und das Reaktionsgemisch 1 h bei RT
gerührt.
Es wird unter Kühlung mit gesättigter NaHCO3-Lösung gequencht, die Lösung durch
eine Fritte mit Celite-MgSO4-Celite gegeben und mit CH2Cl2 und EtOAc nachgespült.
Die Lösung wird am Rotationsverdampfer eingeengt und getrocknet.
Rohausbeute: 1,14 g (93,1 %), rote Flüssigkeit
Analytische Daten:
EI-MS: m/z 246
IR:
(NaCl) ν in cm-1: 3363 m, 2935 s, 2864 m, 1577 m, 1464 m, 1309 m, 1076 m, 742 s
1H-NMR: 1H (360 MHz, CDCl3): 0,88 (t, J = 7,4 Hz, 3 H, Pr-3), 1,37-1,57 (m, 6 H, Pr-2 & But-2/-
3), 2,49, 2,54 (m, je 2 H, Pr-1, But-1), 2,71 (m, 2 H, But-4), 2,87 (dd, J = 15,3 Hz,
8,9 Hz, 2 H, Indan H1/H-3), 3,01 (dd, J = 15,3 Hz, 7,7 Hz, 2 H; Indan H-1/H-3), 3,65
(m, 1H, Indan H-2), 7,08-7,20 (m, 4H, Indan H-4 bis H-7),

Experimenteller Teil
89
13C-NMR: 13C (150 MHz, CDCl3): 11,97 (Pr-3), 20,22 (Pr-2), 24,58 , 31,79 (But-2/3), 36,58
(Indan C-1/C-3), 42,17 (But-4), 51,24 (But-1), 53,35 (Pr-1), 63,11 (Indan C-2), 124,39
(Indan C-4/C-7), 126,30 (Indan C-5/C-6), 141,90 (Indan C-3a/C-7a)
CHN (%): C16H26N2 x 0,5 H2O
Ber: C 75,24 H 10,66 N 10,97
Gef: C 75,09 H 10,50 N 10,55
62a: N-[(N’-Indan-2-yl-N’-propyl)-4-aminobutyl]-4-biphenylcarboxamid
NH
O
N
104,6 mg des primären Amins 61 (0,42 mmol) werden in 5 ml trockenem CH2Cl2
gelöst und mit 0,18 ml Et3N (1,30 mmol) versetzt. Die Lösung wird im Eisbad
abgekühlt. Dann wird eine Lösung von 92,0 mg 4-Biphenylcarbonsäurechlorid (0,42
mmol) in 5 ml trockenem CH2Cl2 zugetropft. Das Eisbad wird entfernt und das
Reaktionsgemisch für 21 h bei RT gerührt.
Anschließend wird mit gesättigter NaHCO3-Lösung gewaschen und die wässrige
Phase mit CH2Cl2 ausgeschüttelt. Die vereinigten organischen Phasen werden über
MgSO4 getrocknet, bevor sie am Rotationsverdampfer eingeengt werden.
Der Rückstand wird mittels Flashchromatographie gereinigt (CH2Cl2/MeOH 95/5).
Ausbeute: 101,4 mg (56 %), grau-weißer Feststoff

Experimenteller Teil
90
Analytische Daten: Vgl. Literatur102
Schmp.: 78 °C
EI-MS: m/z 426
IR:
(NaCl) ν in cm-1: 3321 s, 2933 s, 2866 m, 1637 s, 1545 s, 1070 s, 1043 s, 746s
1H-NMR: 1H (360 MHz, CDCl3): 0,87 (t, J = 7,3 Hz, 3 H, Pr-3), 1,54 (m, 2 H, Pr-2), 1,67 (m,
4 H, But-2/3), 2,58 (m, 2 H, Pr-1), 2,66 (m, 2 H, But-4), 2,92-3,09 (m, 4 H, Indan
H1/H-3), 3,48 (m, 2 H, But-1), 3,71 (m, 1 H, Indan H-2), 7,05-7,19 (m, 4 H, Indan H-4
bis H-7), 7,35 (m, 1 H, Biphenyl H-4’), 7,42 (m, 2 H, Biphenyl H-3’, H-5’), 7,56 (m,
4 H, Biphenyl H-3, H-5, H-2’, H-6’), 7,88 (m, 2 H, Biphenyl H-2, H-6)
13C-NMR: 13C (150 MHz, CDCl3): 11,73 (Pr-3), 19,10 (Pr-2), 24,14, 27,39 (But-2/-3), 36,02
(Indan C-1/C-3), 39,65 (But-1), 50,81 (But-4), 52,92 (Pr-1), 63,01 (Indan C-2), 124,31
(Indan C-4/C-7), 126,45 (Indan C-5/C-6), 126,93, 127,02, 127,48, 127,78 (Biphenyl
C-2/C-6, C-3/C-5, C-2’/C-6’, C-4’), 128,74 (Biphenyl C-3’/C-5’), 133,34 (Biphenyl C-
1), 139,90 (Biphenyl C-4 oder 1’), 140,97 (Indan C-3a/C-7a), 143,85 (Biphenyl C-4
oder 1’), 167,29 (Carbonyl)

Experimenteller Teil
91
62b: N-[(N’-Indan-2-yl-N’-propyl)-4-aminobutyl]-2-biphenylcarboxamid
NH
O
N
40,0 mg 2-Biphenylcarbonsäure (0,20 mmol) werden in 5 ml trockenem CH2Cl2
gelöst und mit 0,13 ml DIPEA (0,79 mmol) versetzt. Die Lösung wird im Eisbad
abgekühlt und anschließend die in 1 ml trockenem DMF gelösten 92,2 mg TBTU
(0,29 mmol) zugegeben. Dann wird eine Lösung von 110,5 mg des primären Amins
61 (0,45 mmol) in 4 ml trockenem CH2Cl2 zugetropft. Das Eisbad wird entfernt und
das Reaktionsgemisch für 3 h bei RT gerührt.
Anschließend wird mit gesättigter NaHCO3-Lösung gewaschen und die wässrige
Phase mehrmals mit CH2Cl2 ausgeschüttelt. Die vereinigten organischen Phasen
werden mit gesättigter NaCl-Lösung gewaschen und dann über MgSO4 getrocknet,
bevor sie am Rotationsverdampfer eingeengt werden.
Der Rückstand wird mittels Flashchromatographie gereinigt (CH2Cl2/MeOH 9/1).
Ausbeute: 97,9 mg (quant.), farbloses Öl
Analytische Daten:
EI-MS: m/z 426
IR:
(NaCl) ν in cm-1: 3286 m, 2937 s, 2870 m, 1643 s, 1531 s, 1473 m, 1308 m, 746 s,
700 m

Experimenteller Teil
92
1H-NMR: 1H (360 MHz, CDCl3): 0,87 (t, J = 7,3 Hz, 3 H, Pr-3), 1,19-1,38 (m, 4 H, But-2/3), 1,48
(m, 2 H, Pr-2), 2,46-2,53 (m, 4 H, Pr-1, But-4), 2,88 (dd, J = 15,6 Hz, 8,3 Hz, 2 H,
Indan H-1/H-3), 2,99 (dd, J = 15,4 Hz, 7,8 Hz, 2 H, Indan H-1/H-3), 3,16 (m, 2 H, But-
1), 3,63 (m, 1H, Indan H-2), 5,70 (m, 1 H, NH), 7,10-7,18 (m, 4H, Indan H-4 bis H-7),
7,30-7,47 (m, 8 H, Biphenyl H-3, H-4, H-5; H-2’ bis H-6’), 7,65 (m; 1H, Biphenyl H-6)
13C-NMR: 13C (90 MHz, CDCl3): 11,73 (Pr-3), 19,33 (Pr-2), 23,93, 26,96 (But-2/-3), 36,09 (Indan
C-1/C-3), 39,50 (But-1), 50,71 (But-4), 52,91 (Pr-1), 62,93 (Indan C-2), 124,33 (Indan
C-4/C-7), 126,42 (Indan C-5/C-6), 127,45, 127,60, 128,46, 128,56, 128,64, 129,85,
130,02 (Biphenyl C-3, C-4, C-5, C-6, C-2’/C-6’, C-3’/C-5’, C-4’), , 135,95 (Biphenyl C-
1), 139,25 (Biphenyl C-2), 140,23 (Indan C-3a/C-7a) , 141,19 (Biphenyl C-1’), 169,51
(Carbonyl)
CHN (%): C29H34N2O x 0,4 H2O
Ber: C 80,29 H 8,09 N 6,46
Gef: C 80,37 H 8,40 N 6,55
62c: N-[(N’-Indan-2-yl-N’-propyl)-4-aminobutyl]-2-naphthoesäureamid
NH
O
N
20,6 mg 2-Naphthoesäure (0,12 mmol) werden in 5 ml trockenem CH2Cl2 gelöst und
mit 79 µl DIPEA (0,48 mmol) versetzt. Die Lösung wird im Eisbad abgekühlt und

Experimenteller Teil
93
anschließend die in 1 ml trockenem DMF gelösten 47,5 mg TBTU (0,15 mmol)
zugegeben. Dann wird eine Lösung von 58,9 mg des primären Amins 61 (0,24 mmol)
in 4 ml trockenem CH2Cl2 zugetropft. Das Eisbad wird entfernt und das Reaktions-
gemisch für 2 h bei RT gerührt.
Anschließend wird mit gesättigter NaHCO3-Lösung gewaschen und die wässrige
Phase mehrmals mit CH2Cl2 ausgeschüttelt. Die vereinigten organischen Phasen
werden mit gesättigter NaCl-Lösung gewaschen und dann über MgSO4 getrocknet,
bevor sie am Rotationsverdampfer eingeengt werden.
Der Rückstand wird mittels Flashchromatographie gereinigt (CH2Cl2/MeOH 9/1).
Ausbeute: 50,2 mg (quant.), gelbes Öl
Literatur: Vgl. Literatur59
Analytische Daten:
EI-MS: m/z 400
IR:
(NaCl) ν in cm-1: 3303 m, 2937 s, 2868 m, 1641 s, 1541 s, 1306 m, 744 m 1H-NMR: 1H (360 MHz, CDCl3): 0,88 (t, J = 7,4 Hz, 3 H, Pr-3), 1,57 (m, 2 H, Pr-2), 1,66-1,80
(m, 4 H, But-2/3), 2,62 (m, 2 H, Pr-1) 2,70 (m, 2 H, But-4), 2,95-3,11 (m, 4 H, Indan
H1/H-3), 3,55 (m, 2 H, But-1), 3,75 (m, 1H, Indan H-2), 7,03 (m, 1 H, NH), 7,06-7,18
(m, 4 H, Indan H-4 bis H-7), 7,52 (m, 2 H, Naphthyl H-6/H-7), 7,82-7,91 (m, 4 H,
Naphthyl H-3/H-4/H-5/-8), 8,34 (m, 1 H, Naphthyl H-1)
13C-NMR: 13C (90 MHz, CDCl3): 11,67 (Pr-3), 18,81 (Pr-2), 23,78, 27,27 (But-2/-3), 35,81 (Indan
C-1/C-3), 39,60 (But-1), 50,84 (But-4), 52,86 (Pr-1), 63,09 (Indan C-2), 123,77
(Naphthyl C-3), 124,33 (Indan C-4/C-7), 126,52 (Indan C-5/C-6), 126,61 (Naphthyl C-
7), 127,39, 127,42, 127,59, 128,19 (Naphthyl C-2, C-4, C-5, C-6), 128,86 (Naphthyl

Experimenteller Teil
94
C-8), 131,87 (Naphthyl C-1), 132,57 (Naphthyl C-8a), 134,58 (Naphthyl C-4a),
140,62 (Indan C-3a/C-7a), 167,70 (Carbonyl)
62d: N-[(N’-Indan-2-yl-N’-propyl)-4-aminobutyl]-1-naphthoesäureamid
NH
O
N
31,5 mg 1-Naphthoesäure (0,18 mmol) werden in 5 ml trockenem CH2Cl2 gelöst und
mit 0,12 ml DIPEA (0,73 mmol) versetzt. Die Lösung wird im Eisbad abgekühlt und
anschließend die in 1 ml trockenem DMF gelösten 89,9 mg TBTU (0,28 mmol) zuge-
geben. Dann wird eine Lösung von 104,0 mg des primären Amins 61 (0,42 mmol) in
4 ml trockenem CH2Cl2 zugetropft. Das Eisbad wird entfernt und das Reaktionsge-
misch für 3 h bei RT gerührt.
Anschließend wird mit gesättigter NaHCO3-Lösung gewaschen und die wässrige
Phase mehrmals mit CH2Cl2 ausgeschüttelt. Die vereinigten organischen Phasen
werden mit gesättigter NaCl-Lösung gewaschen und dann über MgSO4 getrocknet,
bevor sie am Rotationsverdampfer eingeengt werden.
Der Rückstand wird mittels Flashchromatographie gereinigt (CH2Cl2/MeOH 9/1).
Ausbeute: 73,7 mg (quant.), gelbes Öl
Analytische Daten:
EI-MS: m/z 400

Experimenteller Teil
95
IR:
(NaCl) ν in cm-1: 3282 m, 2935 s, 2868 m, 1641 s, 1537 s, 1304 m, 781 s, 744 s
1H-NMR: 1H (360 MHz, CDCl3): 0,85 (t, J = 7,4 Hz, 3 H, Pr-3), 1,50 (m, 2H, Pr-2), 1,67-1,76 (m,
4H, But-2/3), 2,53 (m, 2H, Pr-1), 2,67 (m, 2H, But-4), 2,81-2,99 (m, 4 H, Indan H1/H-
3), 3,55 (m, 2 H, But-1), 3,64 (m, 1 H, Indan H-2), 6,94 (m, 1 H, NH), 7,06-7,14 (m,
4H, Indan H-4 bis H-7), 7,39 (dd, J = 8,2 Hz, 7,0 Hz, 1H, Naphthyl H-3), 7,49 (m; 2H,
Naphthyl H-6, H-7), 7,57 (dd, J = 7,0 Hz, 1,2 Hz, 1H, Naphthyl H-4), 7,80-7,87 (m,
2H, Naphthyl H-2, H-5), 8,25-8,29 (m, 1H, Naphthyl H-8)
13C-NMR: 13C (150 MHz, CDCl3): 11,79 (Pr-3), 18,93 (Pr-2), 24,41, 27,74 (But-2/-3), 36,01
(Indan C-1/C-3), 39,79 (But-1), 50,98 (But-4), 52,91 (Pr-1), 63,01 (Indan C-2), 124,36
(Indan C-4/C-7), 124,68, 124,78 (Naphthyl C-3, C-8), 125,35 (Naphthyl C-6), 126,29,
126,48, 126,93 (Indan C-5/C-6,Naphthyl C-1, C-7), 128,23 (Naphthyl C-5), 130,12
(Naphthyl C-8a), 130,26 (Naphthyl C-2), 133,62 (Naphthyl C-4), 134,93 (Naphthyl C-
4a), 141,02 (Indan C-3a/C-7a), 169,72 (Carbonyl)
CHN (%): C27H32N2O x ¼ H2O
Ber: C 80,06 H 8,09 N 6,92
Gef: C 80,32 H 8,40 N 6,91

Experimenteller Teil
96
62e: N-[(N’-Indan-2-yl-N’-propyl)-4-aminobutyl]-3-iodbenzoesäureamid
NH
O
N
I
46,9 mg 3-Iodbenzoesäure (0,19 mmol) werden in 5 ml trockenem CH2Cl2 gelöst und
mit 125 µl DIPEA (0,76 mmol) versetzt. Die Lösung wird im Eisbad abgekühlt und
anschließend die in 1 ml trockenem DMF gelösten 77,3 mg TBTU (0,24 mmol)
zugegeben. Dann wird eine Lösung von 88,6 mg des primären Amins 61 (0,36 mmol)
in 4 ml trockenem CH2Cl2 zugetropft. Das Eisbad wird entfernt und das Reaktionsge-
misch für 3 h bei RT gerührt.
Anschließend wird mit gesättigter NaHCO3-Lösung gewaschen und die wässrige
Phase mehrmals mit CH2Cl2 ausgeschüttelt. Die vereinigten organischen Phasen
werden mit gesättigter NaCl-Lösung gewaschen und dann über MgSO4 getrocknet,
bevor sie am Rotationsverdampfer eingeengt werden.
Der Rückstand wird mittels Flashchromatographie gereinigt (CH2Cl2/MeOH 9/1).
Ausbeute: 95,1 mg (quant.), gelbes Öl
Analytische Daten:
EI-MS: m/z 476
IR:
(NaCl) ν in cm-1: 3303 m, 2935 s, 2866 m, 1639 s, 1541 s, 1464 s, 1311 m, 1068 m,
744 s

Experimenteller Teil
97
1H-NMR: 1H (360 MHz, CDCl3): 0,89 (t, J = 7,4 Hz, 3 H, Pr-3), 1,58 (m, 2H, Pr-2), 1,65-1,73 (m,
4H, But-2/3), 2,65 (m, 2H, Pr-1), 2,72 (m, 2H, But-4), 2,99-3,14 (m, 4H; Indan H-1/H-
3), 3,45 (m, 2 H, But-1), 3,77 (m, 1H, Indan H-2), 7,10 (t, J = 7,8 Hz, 1 H, Phenyl H-
5), 7,11-7,18 (m, 4H, Indan H-4 bis H-7), 7,42 (m, 1H, NH), 7,77-7,81 (m, 2H, Phenyl
H-4/H-6), 8,15-8,18 (m, 1H, Phenyl H-2)
13C-NMR: 13C (90 MHz, CDCl3): 11,66 (Pr-3), 18,81 (Pr-2), 23,72, 27,11 (But-2/-3), 35,74 (Indan
C-1/C-3), 39,55 (But-1), 50,73 (But-4), 52,89 (Pr-1), 63,00 (Indan C-2), 93,97 (Phenyl
C-3), 124,31 (Indan C-4/C-7), 126,25 (Indan C-5/C-6), 126,58 (Phenyl C-6), 129,97
(Phenyl C-5),136,04, 136,69 (Phenyl C-1/C-2), 139,89 (Phenyl C-4), 140,58 (Indan
C-3a/C-7a), 166,04 (Carbonyl)
CHN (%): C23H29IN2O x ¾ H2O
Ber: C 56,39 H 6,28 N 5,72
Gef: C 56,49 H 6,19 N 5,77
62f: N-[(N’-Indan-2-yl-N’-propyl)-4-aminobutyl]-4-paracyclophancarboxamid
NH
O
N
61,2 mg Paracyclophancarbonsäure (0,24 mmol) werden in 5 ml trockenem CH2Cl2
gelöst, im Eisbad abgekühlt und mit 0,1 ml DIPEA (0,61 mmol) versetzt.
Anschließend werden die in 1 ml trockenem NMP gelösten 97,2 mg HATU

Experimenteller Teil
98
(0,26 mmol) zugegeben, bevor eine Lösung von 80,5 mg des primären Amins 61
(0,33 mmol) in 5 ml trockenem CH2Cl2 zugetropft wird. Das Reaktionsgemisch wird
für 2,5 h bei 0 °C gerührt.
Anschließend wird die Lösung einrotiert und der Rückstand mit gesättigter NaHCO3-
Lösung und Et2O ausgeschüttelt. Die vereinigten organischen Phasen werden mit
gesättigter NaCl-Lösung gewaschen und dann über MgSO4 getrocknet, bevor sie am
Rotationsverdampfer eingeengt werden.
Der Rückstand wird mittels Flashchromatographie gereinigt (CH2Cl2/MeOH 98/2).
Ausbeute: 94,9 mg (81,4 %), weißer Feststoff
Analytische Daten:
Schmp: 65-68 °C
EI-MS: m/z 480
IR:
(NaCl) ν in cm-1: 3363 s, 2927 s, 2850 m, 1527 m, 1635 s, 1435 m, 1041s
1H-NMR: 1H (360 MHz, CDCl3): 0,87 (t, J = 7,4 Hz, 3 H, Pr-3), 1,49 (m, 2 H, Pr-2), 1,62 (m,
4 H, But-2/3), 2,50 (m, 2 H, Pr-1), 2,59 (m, 2 H, But-4), 2,79-3,70 (m, 15 H; 4 x
Paracyclophan -CH2-, Indan H-1/H-2/H-3, But-1), 5,95 (m, 1 H, N-H), 6,40 (m, 1 H,
Aromat Paracyclophan), 6,44 (m, 1 H, Aromat Paracyclophan), 6,50-6,65 (m, 4 H,
Aromat Paracyclophan), 6,81 (m, 1 H, Aromat Paracyclophan), 7,09-7,18 (m, 4 H,
Indan H-4 bis H-7)
13C-NMR: 13C (150 MHz, CDCl3): 11,94 (Pr-3), 19,68 (Pr-2), 24,82, 27,85 (But-2/-3), 34,74,
35,05, 35,24, 35,42 (Paracyclophan -CH2-), 36,37 (Indan C-1/C-3), 39,72 (But-1),
50,97 (But-4), 53,19 (Pr-1), 63,05 (Indan C-2), 124,39 (Indan C-4/C-7), 126,31 (Indan
C-5/C-6), 131,54 (Aromat Paracyclophan C-5), 131,97, 132,38, 132,51, 132,53

Experimenteller Teil
99
(Aromat Paracyclophan C-12/-13/-15/-16), 134,80 (Aromat Paracyclophan C-7),
135,14 (Aromat Paracyclophan C-4), 135,80 (Aromat Paracyclophan C-8), 138,93
(Aromat Paracyclophan C-3), 139,10 (Aromat Paracyclophan C-11), 139,80 (Aromat
Paracyclophan C-14), 140,08 (Aromat Paracyclophan C-6), 141,62 (Indan C-3a/C-
7a), 169,33 (Carbonyl)
CHN (%): C33H40N2O x 0,2 H2O
Ber: C 81,69 H 8,41 N 5,77
Gef: C 81,76 H 8,52 N 5,62
(R)-62f: N-[(N’-Indan-2-yl-N’-propyl)-4-aminobutyl]-4-(S)-paracyclophancarbox-amid
NH
O
N
38,9 mg (R)-Paracyclophancarbonsäure (0,15 mmol) werden in 5 ml trockenem
CH2Cl2 gelöst, im Eisbad abgekühlt und mit 0,05 ml DIPEA (0,30 mmol) versetzt.
Anschließend werden die in 1 ml trockenem NMP gelösten 63,2 mg HATU
(0,17 mmol) zugegeben, bevor eine Lösung von 49,9 mg des primären Amins 61
(0,20 mmol) in 5 ml trockenem CH2Cl2 zugetropft wird. Das Reaktionsgemisch wird
für 2,75 h bei 0 °C gerührt.
Anschließend wird die Lösung einrotiert und der Rückstand mit gesättigter NaHCO3-
Lösung und Et2O ausgeschüttelt. Die vereinigten organischen Phasen werden mit
gesättigter NaCl-Lösung gewaschen und dann über MgSO4 getrocknet, bevor sie am
Rotationsverdampfer eingeengt werden.

Experimenteller Teil
100
Der Rückstand wird mittels Flashchromatographie gereinigt (CH2Cl2/MeOH 98/2).
Ausbeute: 65,9 mg (88,9 %), weißer Feststoff
Analytische Daten:
[α]D25,5
+ 68,2 °
EI-MS: m/z 480
IR:
(NaCl) ν in cm-1: 3348 s, 2927 s, 2854 m, 1639 s, 1523 m, 1435 m, 1072 s, 1045 s
1H-NMR: 1H (360 MHz, CDCl3): 0,87 (t, J = 7,4 Hz, 3 H, Pr-3), 1,49 (m, 2 H, Pr-2), 1,62 (m,
4 H, But-2/3), 2,50 (m, 2 H, Pr-1), 2,60 (m, 2 H, But-4), 2,80-3,70 (m, 15 H; 4 x
Paracyclophan -CH2-, Indan H-1/H-2/H-3, But-1), 5,95 (m, 1 H, N-H), 6,40 (m, 1 H,
Aromat Paracyclophan), 6,44 (m, 1 H, Aromat Paracyclophan), 6,52-6,65 (m, 4 H,
Aromat Paracyclophan), 6,81 (m, 1 H, Aromat Paracyclophan), 7,09-7,17 (m, 4 H,
Indan H-4 bis H-7)
13C-NMR: 13C (90 MHz, CDCl3): 11,95 (Pr-3), 19,75 (Pr-2), 24,89, 27,89 (But-2/-3), 34,77,
35,11, 35,28, 35,46 (Paracyclophan -CH2-), 36,38 (Indan C-1/C-3), 39,75 (But-1),
51,03 (But-4), 53,25 (Pr-1), 63,10 (Indan C-2), 124,43 (Indan C-4/C-7), 126,35 (Indan
C-5/C-6), 131,57 (Aromat Paracyclophan C-5), 132,01, 132,41, 132,55, 132,58
(Aromat Paracyclophan C-12/-13/-15/-16), 134,84 (Aromat Paracyclophan C-7),
135,20 (Aromat Paracyclophan C-4), 135,84 (Aromat Paracyclophan C-8), 138,97
(Aromat Paracyclophan C-3), 139,13 (Aromat Paracyclophan C-11), 139,84 (Aromat
Paracyclophan C-14), 140,13 (Aromat Paracyclophan C-6), 141,63 (Indan C-3a/C-
7a), 169,36 (Carbonyl)

Experimenteller Teil
101
(S)-62f: N-[(N’-Indan-2-yl-N’-propyl)-4-aminobutyl]-4-(R)-paracyclophancarbox-amid
NH
O
N
30,3 mg (S)-Paracyclophancarbonsäure (0,12 mmol) werden in 5 ml trockenem
CH2Cl2 gelöst, im Eisbad abgekühlt und mit 0,04 ml DIPEA (0,24 mmol) versetzt.
Anschließend werden die in 1 ml trockenem NMP gelösten 48,0 mg HATU
(0,13 mmol) zugegeben, bevor eine Lösung von 34,9 mg des primären Amins 61
(0,14 mmol) in 5 ml trockenem CH2Cl2 zugetropft wird. Das Reaktionsgemisch wird
für 3 h bei 0 °C gerührt.
Anschließend wird die Lösung einrotiert und der Rückstand mit gesättigter NaHCO3-
Lösung und Et2O ausgeschüttelt. Die vereinigten organischen Phasen werden mit
gesättigter NaCl-Lösung gewaschen und dann über MgSO4 getrocknet, bevor sie am
Rotationsverdampfer eingeengt werden.
Der Rückstand wird mittels Flashchromatographie gereinigt (CH2Cl2/MeOH 98/2).
Ausbeute: 37,9 mg (65,6 %), weicher, weißer Feststoff
Analytische Daten:
[α]D27
- 67,9 °
EI-MS: m/z 480

Experimenteller Teil
102
IR:
(NaCl) ν in cm-1: 3317 m, 2929 s, 2854 m, 1643 s, 1518 s, 1298 m, 908 m, 737 s
1H-NMR: 1H (360 MHz, CDCl3): 0,87 (t, J = 7,4 Hz, 3 H, Pr-3), 1,49 (m, 2 H, Pr-2), 1,61 (m,
4 H, But-2/3), 2,50 (m, 2 H, Pr-1), 2,59 (m, 2 H, But-4), 2,80-3,70 (m, 15 H; 4 x
Paracyclophan -CH2-, Indan H-1/H-2/H-3, But-1), 5,97 (m, 1 H, NH), 6,40 (m, 1 H,
Aromat Paracyclophan), 6,44 (m, 1 H, Aromat Paracyclophan), 6,50-6,65 (m, 4 H,
Aromat Paracyclophan), 6,80 (m, 1 H, Aromat Paracyclophan), 7,09-7,17 (m, 4 H,
Indan H-4 bis H-7)
13C-NMR: 13C (150 MHz, CDCl3): 11,91 (Pr-3), 19,58 (Pr-2), 24,71, 27,81 (But-2/-3), 34,71,
35,03, 35,22, 35,40 (Paracyclophan -CH2-), 36,32 (Indan C-1/C-3), 39,67 (But-1),
50,93 (But-4), 53,13 (Pr-1), 63,02 (Indan C-2), 124,37 (Indan C-4/C-7), 126,31 (Indan
C-5/C-6), 131,52 (Aromat Paracyclophan C-5), 131,94, 132,36, 132,48, 132,51
(Aromat Paracyclophan C-12/-13/-15/-16), 134,78 (Aromat Paracyclophan C-7),
135,10 (Aromat Paracyclophan C-4), 135,78 (Aromat Paracyclophan C-8), 138,90
(Aromat Paracyclophan C-3), 139,07 (Aromat Paracyclophan C-11), 139,77 (Aromat
Paracyclophan C-14), 140,05 (Aromat Paracyclophan C-6), 141,54 (Indan C-3a/C-
7a), 169,31 (Carbonyl)

Experimenteller Teil
103
62g: N-[(N’-Indan-2-yl-N’-propyl)-4-aminobutyl]ferrocencarboxamid
NH
O
N
Fe
76,2 mg Ferrocencarbonsäure (0,33 mmol) werden in 8 ml trockenem DMF gelöst
und mit 109 µl DIPEA (0,66 mmol) versetzt. Die Lösung wird im Eisbad abgekühlt
und anschließend die in 2 ml trockenem DMF gelösten 160,8 mg HATU (0,42 mmol)
zugegeben. Dann wird eine Lösung von 122,2 mg des primären Amins 61
(0,50 mmol) in 5 ml trockenem DMF zugetropft. Das Eisbad wird entfernt und das
Reaktionsgemisch für 3 h bei RT gerührt.
Anschließend wird mit gesättigter NaHCO3-Lösung gewaschen und die wässrige
Phase mehrmals mit CH2Cl2 ausgeschüttelt. Die vereinigten organischen Phasen
werden über MgSO4 getrocknet, bevor sie am Rotationsverdampfer eingeengt
werden.
Der Rückstand wird mittels Flashchromatographie gereinigt (CH2Cl2/MeOH 98/2).
Ausbeute: 162,1 mg (quant.), orangefarbener Feststoff
Analytische Daten:
Schmp.: 96 °C Zers.
EI-MS: m/z 460

Experimenteller Teil
104
IR:
(NaCl) ν in cm-1: 3643 w, 3431 w, 2974 w, 1618 m, 1541 m, 1460 m, 1298 m, 1026
m, 841 s, 752 m
1H-NMR: 1H (600 MHz, CDCl3): 0,99 (t, J = 7,3 Hz, 3 H, Pr-3), 1,72 (m, 2 H, Pr-2), 1,75-1,86
(m, 4 H, But-2/3), 3,03 (m, 2 H, Pr-1), 3,22 (m, 2 H, But-4), 3,27 (dd, J = 16,1 Hz,
7,5 Hz, 2 H, Indan H-1/H-3), 3,35-3,42 (m, 4 H, Indan H-1/H-3 & But-1), 4,13-4,19
(m, 6 H, Indan H-2 & Ferrocen), 4,33 (t, J = 1,89 Hz, 2 H, Ferrocen), 4,70 (t, J =
1,89 Hz, 2 H, Ferrocen), 6,61 (m, 1 H, NH), 7,20-7,24 (m, 4 H, Indan H-4 bis H-7)
13C-NMR: 13C (90 MHz, CDCl3): 11,05 (Pr-3), 17,69 (Pr-2), 21,35, 26,81 (But-2/-3), 34,89 (Indan
C-1/C-3), 38,25 (But-1), 52,02 (But-4), 53,27 (Pr-1), 63,98 (Indan C-2), 68,22, 69,83,
70,89, 74,96 (Ferrocen), 124,58 (Indan C-4/C-7), 127,71 (Indan C-5/C-6), 138,42
(Indan C-3a/7a), 172,34 (Carbonyl)
HPLC: (254 nm)
System A1: tR = 13,8 min, Reinheit: 99 %
System M3: tR = 18.5 min, Reinheit: 100 %
64: Brom[2.2]paracyclophan
Br
94,2 mg Eisenpulver (1,69 mmol) werden in 20 ml CH2Cl2 suspendiert. Dann werden
etwa 3 ml einer Lösung von 0,56 ml Brom (10,90 mmol) in 40 ml CCl4 zugetropft und
2 h bei RT gerührt. Anschließend werden 100 ml CH2Cl2 zugegeben und zum
Rückfluss erhitzt, bevor 2,01 g [2.2]Paracyclophan (63, 9,66 mmol) zugesetzt

Experimenteller Teil
105
werden. Die Brom/CCl4-Lösung wird nun über 6 h langsam zugetropft. Nach been-
deter Zugabe wird über Nacht weiter unter Reflux gerührt.
Nach dem Abkühlen wird mehrmals mit Na2SO3-Lösung, Wasser und gesättigter
NaCl-Lösung gewaschen. Die organische Phase wird dann über Na2SO4 getrocknet
und am Rotationsverdampfer eingeengt.
Der Rückstand wird mittels Flash-Chromatographie gereinigt (Hexan).
Ausbeute: 1,48 g (53,2 %), weißer Feststoff
Analytische Daten: Vgl. Literatur105, 106
Schmp: 137 °C
EI-MS: m/z 287
IR:
(NaCl) ν in cm-1: 3394 m, 2924 s, 2858 m, 1631 m, 1435 m, 1068 s, 1038 s
1H-NMR: 1H (600 MHz, CDCl3): 2,79-2,85 (m, 1 H, CH2-Cyclophan), 2,87-2,94 (m, 1 H, CH2-
Cyclophan), 3,01-3,14 (m, 4 H, CH2-Cyclophan), 3,17-3,23 (m, 1 H, CH2-Cyclophan),
3,43-3,49 (m, 1 H, CH2-Cyclophan), 6,43-6,58 (m, 6 H, Aromat-Cyclophan), 7,16 (d,
J = 7,84 Hz, 1,89 Hz, 1 H, Aromat Cyclophan)

Experimenteller Teil
106
65: [2.2]Paracyclophan-4-carbonsäure
OH
O
Zu einer Lösung von 1,43 g Bromparacyclophan 64 (4,97 mmol) in trockenem Et2O
werden unter Rühren 3 ml einer 2,5-molaren Lösung von n-Butyllithium in Hexan
zugegeben und das Reaktionsgemisch 2,5 h bei RT gerührt. Dann wird frisch
hergestelltes Trockeneis unter einem N2-Gegenstrom zum Ansatz gegeben.
Nachdem das Reaktionsgemisch wieder RT erreicht hat, wird es am Rotations-
verdampfer eingeengt, der Rückstand in gesättigter NaHCO3-Lösung aufgenommen
und mehrmals mit CH2Cl2 gewaschen. Anschließend wird die wässrige Phase mit
2N-HCl angesäuert und mehrmals mit CHCl3 ausgeschüttelt. Die organischen Pha-
sen werden dann mit gesättigter NaCl-Lösung gewaschen, über MgSO4 getrocknet
und am Rotationsverdampfer eingeengt.
Der Rückstand wird mittels Flash-Chromatographie gereinigt (Hexan/EtOAc 9/1 +
1 % HCOOH).
Ausbeute: 959,6 mg (76,5 %), weißer Feststoff
Analytische Daten: Vgl. Literatur105, 106
Schmp.: 223 °C (Lit.: 223-224 °C106)
EI-MS: m/z 252
IR:
(NaCl) ν in cm-1: 3361 m, 2924 s, 2852 m, 1687 s, 1417 m, 1304 s, 1275 s, 1074 m

Experimenteller Teil
107
1H-NMR: 1H (600 MHz, CDCl3/CD3OD): 2,83-2,90 (m, 1 H; CH2-Cyclophan), 3,00-3,20 (m, 6 H,
CH2-Cyclophan), 4,11-4,18 (m, 1 H, CH2-Cyclophan), 6,45-6,48 (m, 1 H, Aromat
Cyclophan), 6,50-6,53 (m, 1H, Aromat Cyclophan), 6,54-6,58 (m, 3 H, Aromat
Cyclophan), 6,66-6,99 (m, 1 H, Aromat Cyclophan), 7,18 (d, J = 1,89 Hz, 1 H, Aromat
Cyclophan)
(S)-65: (S)-[2.2]Paracyclophan-4-carbonsäure
OH
O
884,3 mg Paracyclophancarbonsäure 65 (3,50 mmol) werden zusammen mit
605,4 mg der frisch mit Ag2O freigesetzten Base (S)-α-(p-Nitrophenyl)ethylamin
(3,60 mmol) in 35 ml CHCl3 gelöst, für 1,5 h bei RT und für 2 h bei 50 °C gerührt,
bevor auf – 5 °C abgekühlt wird und über Nacht bei dieser Temperatur gerührt wird.
Der ausgefallene Niederschlag wird abgefrittet, in 10 ml 2N-HCl suspendiert und
mehrmals mit Et2O ausgeschüttelt. Die organische Phase wird über MgSO4 getrock-
net und am Rotationsverdampfer eingeengt.
Ausbeute: 224,6 mg (50,8 %), weißer Feststoff
Analytische Daten: Vgl. Literatur106
[α]D19
+ 187,7 °

Experimenteller Teil
108
(R)-65: (R)-[2.2]Paracyclophan-4-carbonsäure
OH
O
989,7 mg Paracyclophancarbonsäure 65 (3,92 mmol) werden zusammen mit
683,9 mg der frisch mit Ag2O freigesetzten Base (R)-α-(p-Nitrophenyl)ethylamin
(4,07 mmol) in 35 ml CHCl3 gelöst, für etwa 1 h bei RT und für 2 h bei 50 °C gerührt,
bevor auf – 5 °C abgekühlt wird und über Nacht bei dieser Temperatur gerührt wird.
Da zunächst nichts ausgefallen ist, wird die Lösung mehrere Tage kühl gestellt.
Der ausgefallene Niederschlag wird abgefrittet, in 10 ml 2N-HCl suspendiert und
mehrmals mit Et2O ausgeschüttelt. Die organische Phase wird über MgSO4 getrock-
net und am Rotationsverdampfer eingeengt.
Diese Kristallisation wird nochmals mit 679,1 mg (R)-α-(p-Nitrophenyl)ethylamin
(4,04 mmol) wiederholt.
Ausbeute: 214,6 mg (21,7 %), weißer Feststoff
Analytische Daten: Vgl. Literatur106
[α]D25,7
- 172,4 °

Experimenteller Teil
109
66: N-(Indan-2-yl)-N,N-dipropylamin
N
158,3 mg des sekundären Amins 59 (0,75 mmol) und 417,9 mg NaBH(OAc)3
(1,97 mmol) werden in 10 ml trockenem THF gelöst bzw. suspendiert, bevor 0,1 ml
Propionaldehyd (1,37 mmol) und 0,04 ml HOAc (0,70 mmol) zugespritzt werden. Das
Reaktionsgemisch wird erwärmt und 20,5 h unter Reflux gerührt.
Nach dem Abkühlen wird das Lösungsmittel am Rotationsverdampfer entfernt, der
Rückstand in konzentrierter Salzsäure aufgenommen und mehrmals mit Et2O gewa-
schen. Anschließend wird die wässrige Phase mit 5N-NaOH basifiziert und mehrmals
mit Et2O ausgeschüttelt. Die organischen Phasen werden über Na2SO4 getrocknet
und am Rotationsverdampfer eingeengt.
Der Rückstand wird mittels Flash-Chromatographie gereinigt (Hexan/EtOAc 99/1 +
1 % NMe2Et).
Ausbeute: 60,6 mg (37,3 %), gelbe Flüssigkeit
Analytische Daten: Vgl. Literatur116
EI-MS: m/z 217
IR:
(NaCl) ν in cm-1: 2958 s, 2871 m, 2806 m, 1462 m, 1377 m, 1076 m, 742 s
1H-NMR: 1H (360 MHz, CDCl3): 0,88 (t, J = 7,35 Hz, 6 H, Pr-3), 1,44-1,56 (m, 4 H, Pr-2), 2,46-
2,53 (m, 4 H, Pr-1), 2,83-3,06 (m, 4 H; Indan H-1/H-3), 3,59-3,70 (m, 1 H, Indan H-2),
7,08-7,19 (m, 4 H, Indan H-4 bis H-7)

Experimenteller Teil
110
13C-NMR: 13C (150 MHz, CDCl3): 12,00 (Pr-3), 20,28 (Pr-2), 36,71 (Indan C-1/C-3), 53,52 (Pr-
1), 63,32 (Indan C-2), 124,42 (Indan C-4/C-7), 126,21 (Indan C-5/C-6), 142,03 (Indan
C-3a/C-7a)
(R)-67: (N-Tetralin-2-yl-N-propyl)butan-1,4-diamin
NH2
N
194,7 mg (R)-2-Aminotetralin x HCl (1,06 mmol) und 110,7 mg NaCNBH3
(1,76 mmol) werden in 20 ml trockenem Methanol gelöst, bevor 1,85 ml einer
Propionaldehyd-Stammlösung in Methanol (1,27 mmol) zugetropft werden. Dann
wird 24 h bei RT gerührt, bevor weitere 0,46 ml der Propionaldehyd-Stammlösung
(0,32 mmol) zugetropft werden. Nachdem weitere 24 h gerührt wurde, wird das
Lösungsmittel am Rotationsverdampfer entfernt.
Der Rückstand wird in gesättigter NaHCO3-Lösung aufgenommen und mehrmals mit
CH2Cl2 ausgeschüttelt. Die organischen Phasen werden über Na2SO4 getrocknet und
das Lösungsmittel am Rotationsverdampfer entfernt. Nach dem der Rückstand in
Et2O aufgenommen wurde, werden 5,0 ml einer 2-molaren Lösung von HCl in Et2O
(10,0 mmol) zugegeben und die Lösung kühl gestellt.
Der ausgefallene Niederschlag wird abgefrittet und getrocknet.
146,1 mg dieses Niederschlags (0,65 mmol), 272,5 mg K2CO3 (1,97 mmol) und
23,4 mg KI (0,14 mmol) werden in 10 ml trockenem CH3CN gelöst bzw. suspendiert.
Dann werden 1,04 ml 4-Brombutyronitril (0,52 mmol) zugetropft und für 24 h unter
Rückfluss gerührt.
Nach dem Abkühlen wird das Lösungsmittel am Rotationsverdampfer entfernt, der
Rückstand in Wasser aufgenommen und mit 2N-NaOH basifiziert, bevor mehrmals
mit CH2Cl2 ausgeschüttelt wird. Die organischen Phasen werden über MgSO4
getrocknet und am Rotationsverdampfer eingeengt.

Experimenteller Teil
111
Der Rückstand wird mittels Flash-Chromatographie gereinigt (CHCl3/EtOAc 4/1).
Zu einer gekühlten Lösung von 43,0 mg dieses Rückstandes (0,17 mmol) in 4 ml
trockenem Et2O werden 0,2 ml einer 1-molaren Lösung von LiAlH4 in Et2O
(0,2 mmol) zugetropft. Anschließend wird das Eisbad entfernt und das Reaktionsge-
misch 3,5 h bei RT gerührt.
Es wird unter Kühlung mit gesättigter NaHCO3-Lösung gequencht, die Lösung durch
eine Fritte mit Celite-MgSO4-Celite gegeben und mit CH2Cl2 und EtOAc nachgespült.
Die Lösung wird am Rotationsverdampfer eingeengt und getrocknet.
Ausbeute: 20,9 %
Analytische Daten: Vgl. Literatur107
(S)-67: (N-Tetralin-2-yl-N-propyl)butan-1,4-diamin
NH2
N
Vorgehensweise analog zum (R)-Enantiomer
Analytische Daten: Vgl. Literatur107

Experimenteller Teil
112
(R,R)-68: N-[(N’-(R)-Tetralin-2-yl-N’-propyl)-4-aminobutyl]-(R)-paracyclophan-4-carboxamid
NH
ON
31,5 mg (R)-Paracyclophancarbonsäure ((R)-65; 0,12 mmol) werden in 5 ml trocken-
em CH2Cl2 gelöst und mit 0,05 ml DIPEA (0,30 mmol) versetzt. Die Lösung wird im
Eisbad abgekühlt und anschließend die in 1 ml trockenem NMP gelösten 53,6 mg
HATU (0,14 mmol) zugegeben. Dann wird eine Lösung von 40,7 mg des primären
Amins (R)-67 (0,16 mmol) in 5 ml trockenem CH2Cl2 zugetropft. Das Eisbad wird
entfernt und das Reaktionsgemisch für 21 h bei RT gerührt.
Anschließend wird das Reaktionsgemisch einrotiert, der Rückstand in gesättigter
NaHCO3-Lösung aufgenommen und mehrmals mit Et2O ausgeschüttelt. Die vereinig-
ten organischen Phasen werden mit gesättigter NaCl-Lösung gewaschen und dann
über MgSO4 getrocknet, bevor sie am Rotationsverdampfer eingeengt werden.
Der Rückstand wird mittels präparativer HPLC gereinigt (H2O/CH3CN + 0,1 % TFA).
Ausbeute: 17,6 mg (28,5 %), weißlich-gelber Feststoff
Analytische Daten:
[α]D19
- 32,8 °
EI-MS: m/z 494
IR:
(NaCl) ν in cm-1: 3321 br. m, 2926 s, 2853 s, 2811 w, 1636 s, 1519 m, 1452 m, 1289
m, 1261 m, 1089 m, 1024 m, 801 m, 743 m, 636 m

Experimenteller Teil
113
1H-NMR: 1H (360 MHz, CDCl3): 0,88 (t, J = 7,3 Hz, 3 H, Pr-3), 1,46 (m, 2 H, Pr-2), 1,51-1,70
(m, 5 H, Aminotetralin H-3, But-2/3), 1,99 (m, 1 H, Aminotetralin H-3), 2,47 (m, 2 H,
Pr-1), 2,58 (m, 2 H, But-4), 2,68-3,71 (m, 15 H; Aminotetralin H-1/H-2/H-4, But-1,
Paracyclophan –CH2-), 5,83 (m, 1 H, N-H), 6,41 (d, J = 7,9 Hz, 1 H, Paracyclophan),
6,46 (d, J = 7,8 Hz, 1 H, Paracyclophan), 6,53 (t, J = 1,1 Hz, 2 H, Paracyclophan),
6,56 (dd, J = 7,9 Hz, 1,7 Hz, 1 H, Paracyclophan), 6,64 (d, J = 1,7 Hz, 1 H, Para-
cyclophan), 6,81 (d, J = 7,8 Hz, 1 H, Paracyclophan), 7,07 (m, 4 H, Aminotetralin H-5
bis H-8)
13C-NMR: 13C (90 MHz, CDCl3): 11,91 (Pr-3), 21,95 (Pr-2), 25,84, 26,57 (But-2/-3), 27,73,
29,86, 31,89 (Aminotetralin C-1/C-3/C-4), 34,77, 35,11, 35,28, 35,44 (Paracyclophan
-CH2-), 39,78 (But-1), 50,16 (But-4), 52,58 (Pr-1), 56,77 (Aminotetralin C-2), 125,56,
125,64, 128,54, 129,44 (Aminotetralin C-5/C-6/C-7/C-8), 131,56 (Aromat
Paracyclophan C-5), 132,00, 132,40, 132,54, 132,56 (Aromat Paracyclophan C-12/-
13/-15/-16), 134,84 (Aromat Paracyclophan C-7), 135,25 (Aromat Paracyclophan C-
4), 135,84 (Aromat Paracyclophan C-8), 136,42 (Aminotetralin C-4a/C-8a), 138,96
(Aromat Paracyclophan C-3), 139,11 (Aromat Paracyclophan C-11), 139,83 (Aromat
Paracyclophan C-14), 140,12 (Aromat Paracyclophan C-6), 169,29 (Carbonyl)

Experimenteller Teil
114
(S,S)-68: N-[(N’-(S)-Tetralin-2-yl-N’-propyl)-4-aminobutyl]-(S)-paracyclophan-4-carboxamid
NH
ON
38,1 mg (S)-Paracyclophancarbonsäure ((S)-65; 0,15 mmol) werden in 5 ml tro-
ckenem CH2Cl2 gelöst und mit 0,05 ml DIPEA (0,30 mmol) versetzt. Die Lösung wird
im Eisbad abgekühlt und anschließend die in 1 ml trockenem NMP gelösten 64,5 mg
HATU (0,17 mmol) zugegeben. Dann wird eine Lösung von 47,2 mg des primären
Amins (S)-67 (0,18 mmol) in 5 ml trockenem CH2Cl2 zugetropft. Das Eisbad wird
entfernt und das Reaktionsgemisch für 21 h bei RT gerührt.
Anschließend wird das Reaktionsgemisch einrotiert, der Rückstand in gesättigter
NaHCO3-Lösung aufgenommen und mehrmals mit Et2O ausgeschüttelt. Die vereinig-
ten organischen Phasen werden mit gesättigter NaCl-Lösung gewaschen und dann
über MgSO4 getrocknet, bevor sie am Rotationsverdampfer eingeengt werden.
Der Rückstand wird mittels präparativer HPLC gereinigt (H2O/CH3CN + 0,1 % TFA).
Ausbeute: 26,5 mg (35,5 %), weißlich-gelber Feststoff
Analytische Daten:
Schmp.: 89 °C
[α]D27
+ 41,1 °
EI-MS: m/z 494

Experimenteller Teil
115
IR:
(NaCl) ν in cm-1: 3413 s, 2927 m, 2854 w, 1643 m, 1516 w, 1454 w, 1292 w, 1068 w
1H-NMR: 1H (600 MHz, CDCl3): 0,87 (t, J = 7,3 Hz, 3 H, Pr-3), 1,45 (m, 2 H, Pr-2), 1,52-1,67
(m, 5 H, Aminotetralin H-3, But-2/3), 1,98 (m, 1 H, Aminotetralin H-3), 2,46 (m, 2 H,
Pr-1), 2,57 (m, 2 H, But-4), 2,69-3,70 (m, 15 H; Aminotetralin H-1/H-2/H-4, But-1,
Paracyclophan –CH2-), 5,84 (m, 1 H, N-H), 6,41 (d, J = 7,8 Hz, 1 H, Paracyclophan),
6,46 (d, J = 7,8 Hz, 1 H, Paracyclophan), 6,53 (m, 2 H, Paracyclophan), 6,56 (dd, J =
7,8 Hz, 1,7 Hz, 1 H, Paracyclophan), 6,64 (m, 1 H, Paracyclophan), 6,81 (d, J =
7,8 Hz, 1 H, Paracyclophan), 7,07 (m, 4 H, Aminotetralin H-5 bis H-8)
13C-NMR: 13C (90 MHz, CDCl3): 11,90 (Pr-3), 21,97 (Pr-2), 25,84, 26,60 (But-2/-3), 27,73,
29,85, 31,89 (Aminotetralin C-1/C-3/C-4), 34,77, 35,12, 35,28, 35,45 (Paracyclophan
-CH2-), 39,78 (But-1), 50,16 (But-4), 52,58 (Pr-1), 56,78 (Aminotetralin C-2), 125,56,
125,64, 128,54, 129,44 (Aminotetralin C-5/C-6/C-7/C-8), 131,56 (Aromat
Paracyclophan C-5), 132,00, 132,40, 132,54, 132,56 (Aromat Paracyclophan C-12/-
13/-15/-16), 134,84 (Aromat Paracyclophan C-7), 135,25 (Aromat Paracyclophan C-
4), 135,84 (Aromat Paracyclophan C-8), 136,42 (Aminotetralin C-4a/C-8a), 138,96
(Aromat Paracyclophan C-3), 139,11 (Aromat Paracyclophan C-11), 139,83 (Aromat
Paracyclophan C-14), 140,11 (Aromat Paracyclophan C-6), 169,29 (Carbonyl)
CHN (%): C34H42N2O x ¼ H2O
Ber: C 81,80 H 8,58 N 5,61
Gef: C 81,76 H 8,44 N 5,45

Experimenteller Teil
116
(S,R)-68: N-[(N’-(S)-Tetralin-2-yl-N’-propyl)-4-aminobutyl]-(R)-paracyclophan-4-carboxamid
NH
ON
32,0 mg (R)-Paracyclophancarbonsäure ((R)-65; 0,13 mmol) werden in 5 ml tro-
ckenem CH2Cl2 gelöst und mit 0,05 ml DIPEA (0,30 mmol) versetzt. Die Lösung wird
im Eisbad abgekühlt und anschließend die in 1 ml trockenem NMP gelösten 55,5 mg
HATU (0,15 mmol) zugegeben. Dann wird eine Lösung von 45,9 mg des primären
Amins (S)-67 (0,18 mmol) in 5 ml trockenem CH2Cl2 zugetropft. Das Eisbad wird
entfernt und das Reaktionsgemisch für 21 h bei RT gerührt.
Anschließend wird das Reaktionsgemisch einrotiert, der Rückstand in gesättigter
NaHCO3-Lösung aufgenommen und mehrmals mit Et2O ausgeschüttelt. Die vereinig-
ten organischen Phasen werden mit gesättigter NaCl-Lösung gewaschen und dann
über MgSO4 getrocknet, bevor sie am Rotationsverdampfer eingeengt werden.
Der Rückstand wird mittels präparativer HPLC gereinigt (H2O/CH3CN + 0,1 % TFA).
Ausbeute: 20,9 mg (32,5 %), weißlich-gelber Feststoff
Analytische Daten:
[α]D19
- 96,8 °
EI-MS: m/z 494
IR:
(NaCl) ν in cm-1: 3437 s, 2927 m, 2854 w, 1643 m, 1516 w, 1454 w, 1296 w, 1068 w

Experimenteller Teil
117
1H-NMR: 1H (360 MHz, CDCl3): 0,87 (t, J = 7,4 Hz, 3 H, Pr-3), 1,43 (m, 2 H, Pr-2), 1,50-1,69
(m, 5 H, Aminotetralin H-3, But-2/3), 1,98 (m, 1 H, Aminotetralin H-3), 2,46 (m, 2 H,
Pr-1), 2,56 (m, 2 H, But-4), 2,67-3,71 (m, 15 H; Aminotetralin H-1/H-2/H-4, But-1,
Paracyclophan –CH2-), 5,81 (m, 1 H, N-H), 6,40 (m, 1 H, Paracyclophan), 6,46 (m,
1 H, Paracyclophan), 6,51-6,58 (m, 3 H, Paracyclophan), 6,64 (m, 1 H, Paracyclo-
phan), 6,81 (m, 1 H, Paracyclophan), 7,03-7,11 (m, 4 H, Aminotetralin H-5 bis H-8)
(S,R)-68: N-[(N’-(R)-Tetralin-2-yl-N’-propyl)-4-aminobutyl]-(S)-paracyclophan-4-carboxamid
NH
ON
34,8 mg (S)-Paracyclophancarbonsäure ((S)-65; 0,14 mmol) werden in 5 ml trocke-
nem CH2Cl2 gelöst und mit 0,05 ml DIPEA (0,30 mmol) versetzt. Die Lösung wird im
Eisbad abgekühlt und anschließend die in 1 ml trockenem NMP gelösten 58,6 mg
HATU (0,15 mmol) zugegeben. Dann wird eine Lösung von 51,4 mg des primären
Amins (R)-67 (0,20 mmol) in 5 ml trockenem CH2Cl2 zugetropft. Das Eisbad wird
entfernt und das Reaktionsgemisch für 21 h bei RT gerührt.
Anschließend wird das Reaktionsgemisch einrotiert, der Rückstand in gesättigter
NaHCO3-Lösung aufgenommen und mehrmals mit Et2O ausgeschüttelt. Die vereinig-
ten organischen Phasen werden mit gesättigter NaCl-Lösung gewaschen und dann
über MgSO4 getrocknet, bevor sie am Rotationsverdampfer eingeengt werden.
Der Rückstand wird mittels präparativer HPLC gereinigt (H2O/CH3CN + 0,1 % TFA).
Ausbeute: 22,4 mg (32,8 %), weißlich-gelber Feststoff

Experimenteller Teil
118
Analytische Daten:
[α]D27
+ 102,3 °
EI-MS: m/z 494
IR:
(NaCl) ν in cm-1: 3394 m, 2927 s, 2854 m, 1639 s, 1516 m, 1450 m, 1296 w, 1065 m
1H-NMR: 1H (600 MHz, CDCl3): 0,87 (t, J = 7,3 Hz, 3 H, Pr-3), 1,44 (m, 2 H, Pr-2), 1,52-1,69
(m, 5 H, Aminotetralin H-3, But-2/3), 2,00 (m, 1 H, Aminotetralin H-3), 2,47 (m, 2 H,
Pr-1), 2,58 (m, 2 H, But-4), 2,70-3,70 (m, 15 H; Aminotetralin H-1/H-2/H-4, But-1,
Paracyclophan –CH2-), 5,84 (m, 1 H, N-H), 6,41 (d, J = 7,8 Hz, 1 H, Paracyclophan),
6,46 (d, J = 7,8 Hz, 1 H, Paracyclophan), 6,53 (m, 2 H, Paracyclophan), 6,56 (dd, J =
7,8 Hz, 1,7 Hz, 1 H, Paracyclophan), 6,65 (s, 1 H, Paracyclophan), 6,81 (d, J =
7,8 Hz, 1 H, Paracyclophan), 7,08 (m, 4 H, Aminotetralin H-5 bis H-8)
13C-NMR: 13C (90 MHz, CDCl3): 11,90 (Pr-3), 21,90 (Pr-2), 25,84, 26,49 (But-2/-3), 27,71,
29,85, 31,87 (Aminotetralin C-1/C-3/C-4), 34,77, 35,11, 35,27, 35,44 (Paracyclophan
-CH2-), 39,77 (But-1), 50,15 (But-4), 52,58 (Pr-1), 56,76 (Aminotetralin C-2), 125,56,
125,64, 128,54, 129,44 (Aminotetralin C-5/C-6/C-7/C-8), 131,55 (Aromat
Paracyclophan C-5), 131,99, 132,40, 132,53, 132,56 (Aromat Paracyclophan C-12/-
13/-15/-16), 134,83 (Aromat Paracyclophan C-7), 135,22 (Aromat Paracyclophan C-
4), 135,83 (Aromat Paracyclophan C-8), 136,40 (Aminotetralin C-4a/C-8a), 138,97
(Aromat Paracyclophan C-3), 139,10 (Aromat Paracyclophan C-11), 139,83 (Aromat
Paracyclophan C-14), 140,11 (Aromat Paracyclophan C-6), 169,30 (Carbonyl)

Experimenteller Teil
119
71: 1-Diazo-3-(3,4-dimethoxyphenyl)propan-3-on
O
N2
MeO
MeO
Eine Lösung von 9 ml Oxalylchlorid (104,8 mmol) in 20 ml trockenem Toluol wird zu
einer Lösung von 5,06 g (3,4-Dimethoxyphenyl)essigsäure (70; 25,8 mmol) in 60 ml
des gleichen Lösungsmittels getropft. Das Reaktionsgemisch wird für 3 h bei RT
gerührt, bevor es am Rotationsverdampfer eingeengt wird.
Der Rückstand wird in 20 ml trockenem Toluol aufgenommen. Die Lösung wird im
Eisbad abgekühlt, bevor 100 ml einer 6,0-molaren Lösung von Trimethylsilyldiazo-
methan in Et2O zugetropft werden. Es wird 2,5 h bei 0 °C gerührt und anschließend
am Rotationsverdampfer eingeengt.
Der Rückstand wird mittels Flash-Chromatographie über ALOX Type 507C neutral
gereinigt (Hexan/Et2O 6/4).
Ausbeute: 3,33 g (58,1 %), gelbes Öl
Analytische Daten: Vgl. Literatur108, 109
IR:
(NaCl) ν in cm-1: 2835 w (OCH3), 2104 s (Diazo), 1635 s (Carbonyl), 1514 s
(Aromat), 1358 s, 1261 s, 1142 m, 1026 m
1H-NMR: 1H (600 MHz, CDCl3): 3,56 (s, 2 H; -CH2-), 3,87 (s, 3 H, Methoxy), 3,88 (s, 3 H,
Methoxy), 5,14 (s, 1 H, -CH=), 6,74-6,78 (m, 2 H, Phenyl H), 6,82-6,84 (m, 1 H,
Phenyl H)

Experimenteller Teil
120
72: 5,6-Dimethoxyindan-2-on
OMeO
MeO
Eine Lösung von 59,5 mg des Diazoketons 71 (0,27 mmol) in 3 ml CH2Cl2 wird über
70 min zu einer -20 °C kalten Mischung von 3 ml trockenem CH2Cl2 und 4 ml Trifluor-
essigsäure (TFA) getropft. Anschließend rührt man das Reaktionsgemisch weitere
30 min bei -20 °C, bevor man CH2Cl2 und TFA am Rotationsverdampfer evaporiert.
Die Reinigung erfolgt mittels Flash-Chromatographie (Toluol/EtOAc 20/1)
Ausbeute: 22,5 mg (43,7 %), gelber Feststoff
Analytische Daten: Vgl. Literatur108, 109
Schmp.: 122 °C (Lit.: 120 °C Zers.)
IR:
(NaCl) ν in cm-1: 2924 m (OCH3), 1747s (Carbonyl), 1508 s (Aromat), 1230 s, 1086 s
1H-NMR: 1H (360 MHz, CDCl3): 3,50 (s, 4 H; Indan H-1/H-3), 3,88 (s, 6 H, Methoxy), 6,82 (s,
2 H, Indan H-4/H-7)

Experimenteller Teil
121
74: 5,6-Dimethoxy-2-hydroximinoindan-1-on
O
NOH
MeO
MeO
x HCl
Zu einer Lösung von 973,4 mg 5,6-Dimethoxyindan-1-on (73; 5,06 mmol) in 20 ml
MeOH werden 0,86 ml n-Butylnitrit (7,59 mmol) und 0,6 ml konzentrierte HCl
zugegeben. Anschließend rührt man das Reaktionsgemisch 1,5 h bei RT, bevor man
erneut 0,29 ml n-Butylnitrit (2,56 mmol) und 0,2 ml konzentrierte HCl zugibt. Nach
weiteren 1,75 h Rühren bei RT wird der ausgefallene Feststoff abgefrittet und
getrocknet.
Ausbeute: 1034,4 mg (79,3 %), gelblicher Feststoff
Analytische Daten: Vgl. Literatur52
75: 5,6-Dimethoxyindan-2-amin
NH2
MeO
MeO
105,7 mg des Oxims 74 (0,48 mmol) werden in einer Mischung von 4,8 ml HOAc und
0,4 ml konzentrierter H2SO4 gelöst. Dann wird Paladium auf Kohle zugegeben und
bei 25 °C und etwa 10-15 bar unter Rühren für 4,75 h hydriert. Anschließend wird
zweimal über Celite abgefrittet und mit MeOH nachgespült, das Filtrat mit 5N-NaOH
basisch gemacht und mehrmals mit CH2Cl2 ausgeschüttelt. Die organischen Phasen
werden vereinigt, über MgSO4 getrocknet und am Rotationsverdampfer eingeengt.
Der Rückstand wird mittels Flash-Chromatographie gereinigt (CH2Cl2/MeOH 98/2 +
1 % NMe2Et).
Ausbeute: 62,6 mg (67,8 %), gelbliches Öl

Experimenteller Teil
122
Analytische Daten: Vgl. Literatur52
76: N-(5,6-Dimethoxyindan-2-yl)propionsäureamid
NH
O
MeO
MeO
432,9 mg des primären Amins 75 (2,24 mmol) werden in einer Mischung von 40 ml
trockenem CHCl3 und 0,93 ml NEt3 (6,69 mmol) gelöst. Die Lösung wird im Eisbad
abgekühlt und 0,9 ml Propionylchlorid (10,31 mmol) zugetropft. Nach dem Entfernen
des Eisbades wird etwa 16 h bei RT gerührt.
Anschließend wird mehrmals mit gesättigter NaHCO3-Lösung und CH2Cl2 ausge-
schüttelt. Die vereinigten organischen Phasen werden über MgSO4 getrocknet, bevor
sie am Rotationsverdampfer eingeengt werden.
Der Rückstand wird mittels Flash-Chromatographie gereinigt (Hexan/EtOAc 1/2 +
0,5 % NMe2Et).
Ausbeute: 296,6 mg (53,1 %), weißer Feststoff
Analytische Daten: Vgl. Literatur52
EI-MS: m/z 249
Schmp: 127 °C
IR:
(NaCl) ν in cm-1: 3313 m, 2927 m, 2850 m, 1639 s, 1543 m, 1504 m, 1223 m, 1099 m

Experimenteller Teil
123
1H-NMR: 1H (360 MHz, CDCl3): 1,14 (t, J = 7,6 Hz, 3 H, Pr-3), 2,16 (q, J = 7,6 Hz, 2 H, Pr-2),
2,71 (dd, J = 15,8 Hz, 4,3 Hz, 2 H, Indan H-1/H-3), 3,27 (dd, J = 15,8 Hz, 7,1 Hz, 2 H,
Indan H-1/H-3), 3,85 (s, 6 H, 2 x OCH3), 4,75 (m, 1 H, Indan H-2), 5,68 (m, 1 H, NH),
6,76 (s, 2 H, Indan H-4/H-7)
13C-NMR: 13C (150 MHz, CDCl3): 9,79 (Pr-3), 29,73 (Pr-2), 40,27 (Indan C-1/C-3), 50,79 (Indan
C-2), 56,04 (Methoxy), 108,03 (Indan C-4/C-7), 132,42 (Indan C-3a/C-7a), 148,38
(Indan C-5/C-6), 173,38 (Carbonyl)
77: N-(5,6-Dimethoxyindan-2-yl)-N-propylamin
NH
MeO
MeO
Zu einer Lösung von 269,4 mg des Amids 76 (1,08 mmol) in trockenem THF werden
2,6 ml einer 2,4-molaren Lösung von LiAlH4 in THF (6,24 mmol) zugetropft. Das
Reaktionsgemisch wird 3,5 h bei RT gerührt.
Anschließend wird mit 2N-NaOH und Wasser gequencht, EtOAc zugegeben und
gerührt. Die organische Phase wird abgetrennt und die wässrige Phase mit EtOAc
ausgeschüttelt. Dann werden die organischen Phasen vereinigt, mit gesättigter NaCl-
Lösung gewaschen, über MgSO4 getrocknet und am Rotationsverdampfer eingeengt.
Das Rohprodukt wird weiter umgesetzt.
Analytische Daten: Vgl. Literatur52
EI-MS: m/z 235

Experimenteller Teil
124
IR:
(NaCl) ν in cm-1: 3379 s, 2924 m, 1647 m, 1450 s, 1045 s, 636 s
1H-NMR: 1H (360 MHz, CDCl3): 0,94 (t, J = 7,4 Hz, 3 H, Pr-3), 1,54 (m, 2 H, Pr-2), 2,64 (m,
2 H, Pr-1), 2,71 (dd, J = 15,3 Hz, 6,3 Hz, 2 H, Indan H-1/H-3), 3,12 (dd, J = 15,3 Hz,
7,2 Hz, 2 H, Indan H-1/H-3), 3,65 (m, 1 H, Indan H-2), 3,84 (s, 6 H, 2 x Methoxy),
6,74 (s, 2 H, Indan H-4/H-7)
13C-NMR: 13C (90 MHz, CDCl3): 11,87 (Pr-3), 23,37 (Pr-2), 39,99 (Indan C-1/C-3), 50,16 (Pr-1),
56,09 (Methoxy), 60,03 (Indan C-2), 108,21 (Indan C-4/C-7), 133,28 (Indan C-3a/C-
7a), 148,15 (Indan C-5/C-6)
78: [N-(5,6-Dimethoxyindan-2-yl)-N-propyl]-4-aminobutyronitril
N
N
OMeOMe
Eine Lösung von 191,1 mg des sekundären Amins 77 (0,81 mmol) in 60 ml CH3CN
wird zu 570,0 mg K2CO3 (4,12 mmol) und 292,8 mg NaI (1,95 mmol) gegeben. Dann
werden 0,41 ml 4-Brombutyronitril (4,10 mmol) zugetropft und für 25 h unter
Rückfluss gerührt.
Nach dem Abkühlen wird das Lösungsmittel am Rotationsverdampfer entfernt, der
Rückstand in Wasser aufgenommen und mit 5N-NaOH basifiziert, bevor mehrmals
mit CH2Cl2 ausgeschüttelt wird. Die organischen Phasen werden über MgSO4 ge-
trocknet und am Rotationsverdampfer eingeengt.

Experimenteller Teil
125
Der Rückstand wird mittels Flash-Chromatographie gereinigt (Hexan/EtOAc 2/1 bzw.
1/1).
Ausbeute: 236,1 mg (96,1 %), rot-braunes Öl
Analytische Daten:
EI-MS: m/z 302
IR:
(NaCl) ν in cm-1: 3425 s, 2931 m, 2854 w, 1643 m, 1504 m, 1462 m, 1099 m
1H-NMR: 1H (360 MHz, CDCl3): 0,89 (t, J = 7,3 Hz, 3 H, Pr-3), 1,48 (m, 2 H, Pr-2), 1,80 (m,
2 H, But-2), 2,40-2,47 (m, 4 H, Pr-1, But-3), 2,60 (m, 2 H, But-4), 2,81 (dd, J =
15,3 Hz, 7,9 Hz, 2 H, Indan H1/H-3), 2,96 (dd, J = 15,0 Hz, 7,9 Hz, 2 H; Indan H-1/H-
3), 3,71 (m, 1H, Indan H-2), 3,84 (s, 6 H, 2 x Methoxy), 6,72 (s, 4H, Indan H-4/H-7)
13C-NMR: 13C (150 MHz, CDCl3): 11,86 (Pr-3), 14,71 (But-3), 20,51 (Pr-2), 23,84 (But-2), 35,95
(Indan C-1/C-3), 49,18 (But-4), 53,17 (Pr-1), 56,01 (Methoxy), 62,69 (Indan C-2),
107,83 (Indan C-4/C-7), 119,96 (But-1), 133,20 (Indan C-3a/C-7a), 148,05 (Indan C-
5/C-6)
CHN (%): C18H26N2O2 x 0,2 H2O
Ber: C 70,65 H 8,70 N 9,15
Gef: C 70,66 H 8,57 N 9,06

Experimenteller Teil
126
79: [N-(5,6-Dimethoxyindan-2-yl)-N-propyl]-butan-1,4-diamin
N
NH2
OMeOMe
Zu einer gekühlten Lösung von 146,5 mg des Nitrils 78 (0,48 mmol) in 10 ml trocke-
nem Et2O werden 1,5 ml einer 1-molaren Lösung von LiAlH4 in Et2O (1,5 mmol) zu-
getropft. Anschließend wird das Eisbad entfernt und das Reaktionsgemisch 4,5 h bei
RT gerührt.
Es wird mit gesättigter NaHCO3-Lösung gequencht, die Lösung durch eine Fritte mit
Celite-MgSO4-Celite gegeben und mit CH2Cl2 und EtOAc nachgespült. Die Lösung
wird am Rotationsverdampfer eingeengt und getrocknet.
Rohausbeute: 134,4 g (91,4 %), rote Flüssigkeit
Analytische Daten:
EI-MS: m/z 306
IR:
(NaCl) ν in cm-1: 3371 m, 2931 s, 2858 m, 1662 m, 1608 m, 1504 s, 1308 m, 1223 m,
1099 s
1H-NMR: 1H (360 MHz, CDCl3): 0,88 (t, J = 7,4 Hz, 3 H, Pr-3), 1,39-1,56 (m, 6 H, Pr-2, But-2/-
3), 2,48 (m, 2 H, Pr-1), 2,53 (m, 2 H, But-1), 2,71 (m, 2 H, But-4), 2,83 (dd, J =
15,2 Hz, 8,6 Hz, 2 H, Indan H1/H-3), 2,95 (dd, J = 14,8 Hz, 7,9 Hz, 2 H; Indan H-1/H-
3), 3,69 (m, 1H, Indan H-2), 3,84 (s, 6 H, 2 x Methoxy), 6,72 (s, 4H, Indan H-4/H-7),

Experimenteller Teil
127
13C-NMR: 13C (90 MHz, CDCl3): 12,01 (Pr-3), 20,42 (Pr-2), 24,83 , 31,77 (But-2/3), 36,37 (Indan
C-1/C-3), 42,17 (But-4), 51,27 (But-1), 53,36 (Pr-1), 56,08 (Methoxy), 63,34 (Indan
C-2), 107,96 (Indan C-4/C-7), 133,47 (Indan C-3a/C-7a), 148,05 (Indan C-5/C-6)
HPLC: (220 nm)
System A5: tR = 7.9 min, Reinheit: 98 %
System M3: tR = 6.5 min, Reinheit: 100 %
80: N-{[N’-(5,6-Dimethoxyindan-2-yl)-N’-propyl]-4-aminobutyl}-4-biphenylcar-boxamid
NH
O
N
OMeOMe
Zu einer Lösung von 33,9 mg des primären Amins 79 (0,11 mmol) in 5 ml trockenem
CHCl3 werden 0,05 ml Et3N (0,36 mmol) gegeben. Dann wird eine Lösung von
86,1 mg Biphenylcarbonsäurechlorid (0,40 mmol) in 5 ml trockenem CHCl3 zugetropft
und für 5,5 h bei RT gerührt.
Anschließend wird gesättigte NaHCO3-Lösung zugegeben, die Phasen getrennt und
die wässrige Phase mehrmals mit CH2Cl2 ausgeschüttelt. Die vereinigten organi-
schen Phasen werden über MgSO4 getrocknet und am Rotationsverdampfer einge-
engt.
Der Rückstand wird mittels präparativer HPLC gereinigt (CH3CN/H2O + 0,1 % TFA,
Gradient)
Ausbeute: 10,5 mg (19,5 %), gelbliches Öl

Experimenteller Teil
128
Analytische Daten:
HR-EI-MS: Ber.: m/z 486,2883
Gef.: m/z 486,2882
IR:
(NaCl) ν in cm-1: 3305 br. m, 3059 w, 2962 m, 2936 m, 1639 s, 1545 m, 1507 s, 1448
m, 1309 s, 1220 m, 1098 m, 749 s 1H-NMR: 1H (360 MHz, CDCl3): 0,88 (t, J = 7,4 Hz, 3 H, Pr-3), 1,50 (m, 2 H, Pr-2), 1,58-1,74
(m, 4 H, But-2/-3), 2,50 (m, 2 H, Pr-1), 2,58 (m, 2 H, But-4), 2,83 (dd, J = 15,1 Hz,
8,3 Hz, 2 H, Indan H-1/H-3), 2,95 (dd, J = 15,1 Hz, 7,9 Hz, 2 H, Indan H-1/H-3), 3,51
(m, 2 H, But-1), 3,70 (m, 1 H, Indan H-2), 3,82 (s, 6 H, 2 x Methoxy), 6,69 (s, 2 H,
Indan H-4/H-7), 6,77 (m, 1 H, NH), 7,38 (m, 1 H, Biphenyl H-4’), 7,46 (m, 2 H,
Biphenyl H-3’/H-5’), 7,56-7,64 (m, 4 H, Biphenyl H-3/H-5/H-2’/H-6’), 7,83 (m, 2 H,
Biphenyl H2/H-6)
13C-NMR: 13C (150 MHz, CDCl3): 11,98 (Pr-3), 19,76 (Pr-2), 25,10, 27,77 (But-2/-3), 36,26
(Indan C-1/C-3), 40,06 (But-1), 50,91 (But-4), 53,18 (Pr-1), 56,00 (Methoxy), 63,25
(Indan C-2), 107,81 (Indan C-4/C-7), 127,11, 127,14, 127,42, 127,92 (Biphenyl C-
2/C-6, C-3/C-5, C-2’/C-6’, C-4’), 128,88 (Biphenyl C-3’/C-5’), 133,20, 133,62 (Indan
C-3a/C-7a, Biphenyl C-1), 140,02, 144,02 (Biphenyl C-1’, C-4), 148,07 (Indan C-5/C-
6), 167,37 (Carbonyl)
HPLC: (254 nm)
System A1: tR = 14.2 min, Reinheit: 98 %
System M3: tR = 16.9 min, Reinheit: 99 %

Experimenteller Teil
129
81: N-{4-[4-(4-Biphenyl)triazol-1-yl]butyl}-N-propyl-N-(indan-2-yl)amin
N NN
N
54,0 mg des Azids 81 (0,20 mmol), 113,0 mg Biphenylacetylen (0,63 mmol), 3,5 mg
CuSO4 x 5 H2O (0,01 mmol) und 14,6 mg Na-ascorbat (0,07 mmol) werden in 2 ml
CH2Cl2, 2 ml H2O und 4 ml tert-BuOH gelöst und über Nacht bei 40 °C gerührt.
Nach dem Abkühlen wird mit gesättigter NaHCO3-Lösung und CH2Cl2 ausge-
schüttelt. Die wässrige Phase wird mehrmals mit CH2Cl2 extrahiert. Die vereinigten
organischen Phasen werden über MgSO4 getrocknet und am Rotationsverdampfer
eingeengt.
Der Rückstand wird mittels präparativer HPLC gereinigt (CH3CN/H2O + 0,1 % TFA,
Gradient)
Ausbeute: 52,2 mg (58,4 %), weißer Feststoff
Analytische Daten:
Schmp.: 112 °C
EI-MS: m/z 450
IR:
(NaCl) ν in cm-1: 3425 br. m, 3101 w, 2954 s, 2802 w, 1639 m, 1485 s, 1460 m, 1355
m, 1221 m, 1076 m, 838 m, 764 s, 742 s, 728 s, 689 s, 644 s
1H-NMR: 1H (360 MHz, CDCl3): 0,87 (t, J = 7,4 Hz, 3 H, Pr-3), 1,41-1,60 (m, 4 H, Pr-2, But-2),
2,00 (m, 2 H, But-3), 2,47 (m, 2 H, Pr-1), 2,56 (m, 2 H, But-1), 2,86 (dd, J = 15,5 Hz,

Experimenteller Teil
130
8,5 Hz, 2 H, Indan H-1/H-3), 3,00 (dd, J = 15,5 Hz, 7,7 Hz, 2 H, Indan H-1/H-3), 3,66
(m, 1 H, Indan H-2), 4,44 (m, 2 H, But-4), 7,09-7,19 (m, 4 H, Indan H-4 - H-7), 7,35
(m, 1 H, Biphenyl H-4’), 7,45 (m, 2 H, Biphenyl H-3’/H-5’), 7,61-7,69 (m, 4 H,
Biphenyl H-3/H-5, H-2’/H-6’), 7,78 (s, 1 H, Triazol H-5), 7,90 (m, 2 H, Biphenyl H-2/H-
6)
13C-NMR: 13C (150 MHz, CDCl3): 11,94 (Pr-3), 20,29 (Pr-2), 24,55, 28,40 (But-2/-3), 36,38
(Indan C-1/C-3), 50,38, 50,46 (But-1/-4), 53,31 (Pr-1), 63,81 (Indan C-2), 119,38
(Triazol C-5), 124,43 (Indan C-4/C-7), 126,05 (Biphenyl C-2/C-6), 126,30 (Indan C-
5/C-6), 126,96, 127,40, 127,49 (Biphenyl C-3/C-5, C-2’/C-6’, C-4’), 128,80 (Biphenyl
C-3’/C-5’), 129,67 (Biphenyl C-1), 140,59, 140,82 (Biphenyl C-1’, C-4), 141,79 (Indan
C-3a/C-7a), 147,45 (Triazol C-4)
HPLC: (254 nm)
System A1: tR = 15.2 min, Reinheit: 100 %
System M3: tR = 17.9 min, Reinheit: 100 %
82: N-(4-Azidobutyl-N-indan-2-yl)-N-propylamin
NN3
523,2 mg des sekundären Amins 59 (freie Base; 2,98 mmol), 2,08 g K2CO3
(15,08 mmol) und 486,7 mg NaI (3,25 mmol) werden in 8 ml trockenem CH3CN
gelöst bzw. suspendiert. Dann wird eine Lösung von 887,2 mg des Azidobutanol-
mesylats 85 (4,54 mmol) in 12 ml trockenem CH3CN zugegeben und für 22 h unter
Rückfluss erhitzt.
Nach dem Abkühlen wird die Reaktionslösung am Rotationsverdampfer eingeengt,
der Rückstand in etwas H2O aufgenommen, mit 2N-NaOH basifiziert und mehrmals

Experimenteller Teil
131
mit CH2Cl2 ausgeschüttelt. Die organischen Phasen werden über MgSO4 getrocknet
und am Rotationsverdampfer eingeengt.
Der Rückstand wird mittels präparativer HPLC gereinigt (CH3CN/H2O + 0,1 % TFA,
Gradient)
Ausbeute: 90,6 mg (11,2 %), gelbliche Flüssigkeit
Analytische Daten:
HR-EI-MS: Ber.: m/z 272,2001
Gef.: m/z 272,2001
IR:
(NaCl) ν in cm-1: 3425 br. m, 2955 m, 2873 w, 2802 w, 2094 s, 1659 w, 1460 m, 1256
m, 1072 m, 1020 w, 772 m, 743 m, 657 m
1H-NMR: 1H (360 MHz, CDCl3): 0,89 (t, J = 7,4 Hz, 3 H, Pr-3), 1,44-1,67 (m, 6 H, Pr-2, But-2/-
3), 2,49 (m, 2 H, Pr-1), 2,55 (m, 2 H, But-1), 2,88 (dd, J = 15,4 Hz, 8,7 Hz, 2 H, Indan
H-1/H-3), 3,02 (dd, J = 15,4 Hz, 7,7 Hz, 2 H, Indan H-1/H-3), 3,30 (m, 2 H, But-4),
3,66 (m, 1 H, Indan H-2), 7,11-7,20 (m, 4 H, Indan H-4 - H-7)
13C-NMR: 13C (90 MHz, CDCl3): 11,96 (Pr-3), 20,29 (Pr-2), 24,46, 26,95 (But-2/-3), 36,49 (Indan
C-1/C-3), 50,72, 51,46 (But-1/-4), 53,31 (Pr-1), 62,98 (Indan C-2), 124,44 (Indan C-
4/C-7), 126,28 (Indan C-5/C-6), 141,86 (Indan C-3a/C-7a)
HPLC: (254 nm)
System A1: tR = 13.0 min, Reinheit: 100 %
System M3: tR = 14.1 min, Reinheit: 100 %

Experimenteller Teil
132
84: 4-Azidobutanol
OHN3
14,71 g NaN3 (226,35 mmol) und 11,83 g NaI (78,93 mmol) werden in 225 ml CH3CN
und 25 ml H2O gelöst bzw. suspendiert. Dann werden 7,5 ml 4-Chlorbutanol (83;
75,16 mmol) zugegeben und für etwa 40 h unter Rückfluss gerührt.
Nach dem Abkühlen wird mit Et2O verdünnt, über MgSO4 getrocknet und am Rota-
tionsverdampfer eingeengt.
Der erhaltene Rückstand wird für den nächsten Reaktionsschritt eingesetzt.
Rohausbeute: 6,58 g (74,7 %), orangefarbene Flüssigkeit
Analytische Daten: Vgl. Literatur117
IR:
(NaCl) ν in cm-1: 3376 br. s, 2943 m, 2872 m, 2098 s, 1453 w, 1352 w, 1273 m, 1253
m, 1059 m, 1037m
1H-NMR: 1H (600 MHz, CDCl3): 1,56-1,69 (m, 4 H, H-2/H-3), 3,32, 3,57 (2 x m, je 2 H, H-1,
H-4)
85: 4-Azidobutanolmesylat
MsON3
2,88 g des Azidobutanols 84 (24,56 mmol) werden in 40 ml trockenem THF gelöst.
Zur Lösung werden 4,8 ml Et3N (34,53 mmol) gegeben. Dann wird die Lösung im
Eisbad abgekühlt und es werden 2,8 ml Mesylchlorid (36,18 mmol) zugetropft. Das
Reaktionsgemisch wird für 5,25 h bei RT gerührt.
Anschließend werden 100 ml einer H2O/Eis-Mischung zugegeben und so lange
weitergerührt, bis das Eis geschmolzen ist. Man schüttelt aus und extrahiert dann die

Experimenteller Teil
133
wässrige Phase mehrmals mit Et2O. Die vereinigten organischen Phasen werden
einmal mit H2O, einmal mit 2N-HCl, einmal mit H2O, zweimal mit gesättigter
NaHCO3-Lösung und abschließend noch einmal mit H2O gewaschen. Dann wird über
MgSO4 getrocknet und am Rotationsverdampfer eingeengt.
Der erhaltene Rückstand wird für den nächsten Reaktionsschritt eingesetzt.
Rohausbeute: 3,88 g (81,0 %), rotbraune Flüssigkeit
Analytische Daten:
IR:
(NaCl) ν in cm-1: 3389 br. w, 3033 w, 2942 w, 2878 w, 2100 s, 1354 s, 1255 w, 1173
s, 976 m, 933 m, 810 w
1H-NMR: 1H (360 MHz, CDCl3): 1,64-1,90 (m, 4 H, H-2/H-3), 3,02 (s, 3 H, Methyl), 3,36 (t, J =
6,5 Hz, 2 H, H-4), 4,27 (t, J = 6,2 Hz, 2 H, H-1)
87: N-Propyl-4-aminocyclohexan-1-on-ethylenketal
NH
O O
2,13 g NaBH(OAc)3 (10,06 mmol) werden in 25 ml trockenem THF suspendiert und
die Suspension auf – 10 °C abgekühlt. Dazu wird langsam eine Lösung von
564,6 mg 1,4-Diazaspiro[4.5]decan-8-on (86; 3,62 mmol), 0,33 ml Propylamin
(4,01 mmol) und 0,23 ml HOAc (4,01 mmol) in 15 ml trockenem THF gegeben. Der
Kolben dieser Lösung wird mit 5 ml trockenem THF nachgespült. Dann wird der
Kryomat entfernt und 15 h bei RT gerührt.
Anschließend wird mit 20 ml einer 10%igen NaOH gerührt und ausgeschüttelt. Die
wässrige Phase wird mehrmals mit CH2Cl2 extrahiert. Dann werden die vereinigten

Experimenteller Teil
134
organischen Phasen mit gesättigter NaCl-Lösung gewaschen und über MgSO4 ge-
trocknet.
Die entstandene Lösung wird im Eisbad abgekühlt, mit einer Lösung von 686,8 mg
Oxalsäure x 2 H2O in 50 ml MeOH versetzt und für 25 min bei 0 °C gerührt.
Dann wird mit 150 ml gesättigter NaHCO3-Lösung versetzt und gerührt, bis die
Gasbildung aufgehört hat, bevor man ausschüttelt. Die wässrige Phase wird mehr-
mals mit CH2Cl2 extrahiert. Die vereinigten organischen Phasen werden dann über
MgSO4 getrocknet und am Rotationsverdampfer eingeengt.
Der erhaltene Rückstand wird für den nächsten Reaktionsschritt eingesetzt.
Rohausbeute: 669,0 mg (92,9 %), gelbliche Flüssigkeit
Analytische Daten: Vgl. Literatur113
EI-MS: m/z 199
IR:
(NaCl) ν in cm-1: 3359 m, 2939 s, 2873 s, 2812 w, 1446 m, 1373 m, 1130 s, 1103 s,
1034 m, 945 m, 926 m
1H-NMR: 1H (600 MHz, CDCl3): 0,91 (t, J = 7,4 Hz, 3 H, Pr-3), 1,38-1,46 (m, 2 H, Sp-7a/Sp-
9a), 1,49 (m, 2 H, Pr-2), 1,56 (ddd, J = 12,9 Hz, 12,9 Hz, 4,1 Hz, 2 H, Sp-6a/Sp-10a),
1,77 (m, 2 H, Sp-6b/Sp-10b), 1,87 (m, 2 H, Sp-7b/Sp-9b), 2,52 (m, 1 H, Sp-8), 2,58
(m, 2 H, Pr-1), 3,93 (s, 4 H, Sp-2/Sp-3)
13C-NMR: 13C (150 MHz, CDCl3): 11,80 (Pr-3), 23,53 (Pr-2), 30,29 (Sp-7/Sp-9), 33,03 (Sp-6/Sp-
10), 49,15 (Pr-1), 55,14 (Sp-8), 64,19, 64,22 (Sp-2/Sp-3), 108,69 (Sp-5)

Experimenteller Teil
135
88: N-(4-Azidobutyl)-N-propyl-4-aminocyclohexan-1-on
NN3
O
2,51 g des sekundären Amins 87 (12,60 mmol), 8,89 g K2CO3 (64,29 mmol) und
4,07 g KI (27,13 mmol) werden in 140 ml trockenem CH3CN gelöst bzw. suspendiert.
Dann werden 6,41 g des Azidobutanolmesylats 85 (32,85 mmol) in 60 ml trockenem
CH3CN gelöst und zugegeben. Es wird übers Wochenende unter Rückfluss gerührt.
Nach dem Abkühlen wird das Lösungsmittel am Rotationsverdampfer entfernt, der
Rückstand in Wasser aufgenommen und mit 6N-NaOH basifiziert, bevor mehrmals
mit CH2Cl2 ausgeschüttelt wird. Die organischen Phasen werden über MgSO4
getrocknet und am Rotationsverdampfer eingeengt.
Der Rückstand wird in 80 ml Aceton gelöst und mit 150 ml 6N-HCl für 19,5 h bei RT
gerührt.
Anschließend wird mit 6N-NaOH basifiziert und mehrmals mit CH2Cl2 ausgeschüttelt.
Die vereinigten organischen Phasen werden über MgSO4 getrocknet und am
Rotationsverdampfer eingeengt.
Der Rückstand wird mittels Flashchromatographie gereinigt (Hexan/EtOAc 20/1 +
0,5 % NMe2Et)
Ausbeute: 689,9 mg (21,7 %), farblose bis gelbliche Flüssigkeit
Analytische Daten:
EI-MS: m/z 252
IR:
(NaCl) ν in cm-1: 3363 m, 2954 s, 2870 s, 2811 m, 2094 s, 1716 s, 1462 m, 1254 m,
1072 m, 949 m

Experimenteller Teil
136
1H-NMR: 1H (600 MHz, CDCl3): 0,88 (t, J = 7,4 Hz, 3 H, Pr-3), 1,44 (m, 2 H, Pr-2), 1,51 (m,
2 H, But-2), 1,59-1,66 (m, 2 H, But-3), 1,67-1,75 (m, 2 H, Cy-3a/Cy-5a), 2,02 (m, 2 H,
Cy-3b/Cy-5b), 2,31-2,38 (m, 2 H, Cy-2a/Cy-6a), 2,39-2,46 (m, 4 H, Pr-1, Cy-2b/Cy-
6b), 2,48 (m, 2 H, But-1), 2,95 (m, 1 H, Cy-4), 3,29 (m, 2 H, But-4)
13C-NMR: 13C (150 MHz, CDCl3): 11,75 (Pr-3), 21,98 (Pr-2), 25,99 (But-2), 26,69 (But-3), 27,93
(Cy-3/Cy-5), 39,97 (Cy-2/Cy-6), 50,01 (But-1), 51,45 (But-4), 52,51 (Pr-1), 57,46 (Cy-
4), 211,29 (Cy-1)
CHN (%): C13H24N4O
Ber: C 61,87 H 9,59 N 22,20
Gef: C 61,99 H 9,58 N 22,16
89: O-[N-(4-Azidobutyl)-N-propyl-(4-aminocyclohex-1-enyl)]-trifluormethylsulfo-nat
NN3
OTf
Eine Lösung von 614,9 mg des Ketons 88 (2,44 mmol) in 40 ml 1,2-Dichlorethan
werden zu 1032,8 mg 2,6-Di-tert-butyl-4-methylpyridin (5,03 mmol) gegeben. Dann
werden langsam 0,85 ml Trifluorsulfonsäureanhydrid (5,12 mmol) zugespritzt und die
entstandene Lösung für 4,5 h unter Reflux gerührt.
Nachdem der Ansatz abgekühlt ist, wird gesättigte NaHCO3-Lösung zugegeben, die
Phasen getrennt und die wässrige Phase mehrmals mit CH2Cl2 ausgeschüttelt. Die
vereinigten organischen Phasen werden über MgSO4 getrocknet und am Rotations-
verdampfer eingeengt.

Experimenteller Teil
137
Der Rückstand wird mittels Flashchromatographie gereinigt (Hexan/EtOAc 20/1 +
0,5 % NMe2Et)
Ausbeute: 186,9 mg (20,0 %), gelbliches Öl
Analytische Daten:
HR-EI-MS: Ber.: m/z 384,1443
Gef.: m/z 384,1444
IR:
(NaCl) ν in cm-1: 3377 br. m, 2932 m, 2873 w, 2096 s, 1417 s, 1247 m, 1209 s, 1143
m, 1054 m, 1024 m, 873 m
1H-NMR: 1H (600 MHz, CDCl3): 0,87 (t, J = 7,4 Hz, 3 H, Pr-3), 1,38-1,52 (m, 4 H, Pr-2, But-2),
1,56-1,68 (m, 3 H, But-3, Cy-5a), 1,82 (m, 1 H, Cy-5b), 2,10-2,26 (m, 2 H, Cy-3),
2,34-2,51 (m, 6 H, Cy-6, Pr-1, But-1), 2,81 (m, 1 H, Cy-4), 3,28 (m, 2 H, But-4), 5,72
(m, 1 H, Cy-2)
13C-NMR: 13C (150 MHz, CDCl3): 11,75 (Pr-3), 22,22 (Pr-2), 24,83 (Cy-5), 25,83 (Cy-3), 26,22
(But-2), 26,67 (But-3), 28,01 (Cy-6), 49,95 (But-1), 51,46 (But-4), 52,43 (Pr-1), 57,73
(Cy-4), 117,76 (Cy-2), 118,51 (quart, J = 320,3 Hz, CF3), 148,41 (Cy-1)
HPLC: (220 nm)
System A1: tR = 13.9 min, Reinheit: 89 %
System M3: tR = 14.2 min, Reinheit: 89 %

Experimenteller Teil
138
90: N-(4-Azidobutyl)-N-propyl-N-(4-(2-Trimethylsilyl)-ethinylcyclohex-3-enyl)-amin
NN3
TMS
Eine Lösung von 158,5 mg des Triflats 89 (0,41 mmol) in 20 ml trockenem THF
werden zu 14,5 mg Cu(I)I (0,08 mmol) und 15,0 mg Pd(PPh3)2Cl2 (0,02 mmol)
gegeben. Dann werden 0,45 ml NMe2Et (4,15 mmol) und 0,15 ml TMS-Acetylen
(1,05 mmol) zugespritzt und für 40 min bei RT gerührt.
Anschließend wird gesättigte NaHCO3-Lösung zugegeben, die Phasen getrennt und
die wässrige Phase mehrmals mit Et2O ausgeschüttelt. Die vereinigten organischen
Phasen werden über MgSO4 getrocknet und am Rotationsverdampfer eingeengt.
Der Rückstand wird mittels Flashchromatographie gereinigt (Hexan/EtOAc 40/1 +
0,5 % NMe2Et)
Ausbeute: 99,0 mg (72,2 %), gelbliches Öl
Analytische Daten:
HR-EI-MS: Ber.: m/z 332,2396
Gef.: m/z 332,2398
IR:
(NaCl) ν in cm-1: 3388 br. m, 2953 m, 2928 m, 2870 w, 2146 m, 2095 s, 1438 m,
1249 m, 1222 w, 1075 m, 1044 m, 860 m, 842 m, 772 s

Experimenteller Teil
139
1H-NMR: 1H (600 MHz, CDCl3): 0,17 (m, 9 H, CH3-TMS), 0,86 (t, J = 7,4 Hz, 3 H, Pr-3), 1,37-
1,50 (m, 4 H, Pr-2, But-2), 1,57-1,64 (m, 3 H, But-3, Cy-6a), 1,82 (m, 1 H, Cy-6b),
2,00-2,08 (m, 1 H, Cy-2a), 2,14-2,31 (m, 3 H, Cy-2b, Cy-5), 2,37 (m, 2 H, Pr-1), 2,44
(m, 2 H, But-1), 2,74 (m, 1 H, Cy-1), 3,27 (m, 2 H, But-4), 6,13 (m, 1 H, Cy-3)
13C-NMR: 13C (150 MHz, CDCl3): 0,05 (CH3-TMS), 11,83 (Pr-3), 22,16 (Pr-2), 25,06, 26,15,
26,75, 28,43, 30,29 (But-2/-3, Cy-2/-5/-6), 49,80 (But-1), 51,50 (But-4), 52,37 (Pr-1),
55,23 (Cy-1), 91,43 (Ethinyl C-2), 106,57 (Ethinyl C-1), 120,572 (Cy-4), 135,44 (Cy-
3)
HPLC: (220 nm)
System A1: tR = 15.5 min, Reinheit: 96 %
System M3: tR = 15.8 min, Reinheit: 98 %
92: N-{[N’-(4-(2-Trimethylsilyl)-ethinylcyclohex-3-enyl)-N’-propyl]-4-aminobutyl}-4-biphenylcarboxamid
NNH
TMS
O
Eine Lösung von 32,5 mg des Enins 90 (0,10 mmol) in 4 ml THF werden zu
130,7 mg PPh3 (0,50 mmol) gegeben. Dann wird 1 ml H2O zugespritzt und für 2 Tage
bei RT gerührt.

Experimenteller Teil
140
Anschließend wird mit Et2O auf etwa 100 ml verdünnt, die Lösung über Na2SO4
getrocknet und am Rotationsverdampfer eingeengt.
Der Rückstand wird in 5 ml trockenem CHCl3 gelöst, 0,05 ml Et3N (0,36 mmol) zuge-
geben und im Eisbad abgekühlt. Dann wird eine Lösung von 92,1 mg Biphenyl-
carbonsäurechlorid (0,43 mmol) in 5 ml trockenem CHCl3 zugetropft, das Eisbad
entfernt und für etwa 3 h bei RT gerührt.
Anschließend wird gesättigte NaHCO3-Lösung zugegeben, die Phasen getrennt und
die wässrige Phase mehrmals mit CH2Cl2 ausgeschüttelt. Die vereinigten organi-
schen Phasen werden über Na2SO4 getrocknet und am Rotationsverdampfer ein-
geengt.
Der Rückstand wird mittels Flashchromatographie gereinigt (CH2Cl2/MeOH 98/2 bzw.
Hexan/EtOAc 4/1 + 0,5 % NMe2Et)
Ausbeute: 15,8 mg (33,2 %), farbloses Öl
Analytische Daten:
EI-MS: m/z 487
IR:
(NaCl) ν in cm-1: 3352 s, 2954 m, 2631 m, 2866 w, 2141 m, 1635 s, 1547 m, 1435 m,
1250 m, 1130 m, 1003 s, 841 s
1H-NMR: 1H (600 MHz, CDCl3): 0,17 (s, 9 H, TMS), 0,85 (t, J = 7,3 Hz, 3 H, Pr-3), 1,38-1,49
(m, 3 H, Pr-2, Cy-6a), 1,53 (m, 2 H, But-2), 1,66 (m, 2 H, But-3), 1,83 (m, 1 H, Cy-
6b), 2,02-2,10 (m, 1 H, Cy-2a), 2,15-2,30 (m, 3 H, Cy-2b, Cy-5), 2,39 (m, 2 H, Pr-1),
2,49 (m, 2 H, But-4), 2,77 (m, 1 H, Cy-1), 3,48 (m, 2 H, But-1), 6,12 (m, 1 H, Cy-3),
6,43 (m, 1 H, NH), 7,39 (m, 1 H, Biphenyl H-4’), 7,46 (m, 2 H, Biphenyl H-3’, H-5’),
7,60-7,67 (m, 4 H, Biphenyl H-3, H-5, H-2’, H-6’), 7,83 (m, 2 H, Biphenyl H-2, H-6)
13C-NMR: 13C (150 MHz, CDCl3): 0,03 (TMS), 11,83 (Pr-3), 21,61 (Pr-2), 24,87, 26,08, 27,44,
28,02, 30,13 (But-2/-3, Cy-2/-5/-6), 39,86 (But-1), 50,00 (But-4), 52,46 (Pr-1), 55,68

Experimenteller Teil
141
(Cy-1), 91,80 (Ethinyl C-2), 106,27 (Ethinyl C-1), 120,67 (Cy-4), 127,18, 127,19,
127,43, 127,91 (Biphenyl C-2/C-6, C-3/C-5, C-2’/C-6’, C-4’), 128,88 (Biphenyl C-3’/C-
5’), 133,49 (Biphenyl C-1), 135,30 (Cy-3), 140,10, 144,12 (Biphenyl C-4, C-1’),
167,29 (Carbonyl)
CHN (%): C31H42N2OSi
Ber: C 76,49 H 8,70 N 5,75
Gef: C 76,54 H 8,76 N 5,77
93: N-{[N’-(4-(2-Phenyl)-ethinylcyclohex-3-enyl)-N’-propyl]-4-aminobutyl}-4-bi-phenylcarboxamid
NNH
O
Eine Lösung von 194,3 mg des Triflats 89 (0,51 mmol) in 10 ml trockenem THF
werden zu 11,6 mg Cu(I)I (0,06 mmol) und 20,6 mg Pd(PPh3)2Cl2 (0,03 mmol)
gegeben. Dann werden 0,55 ml NMe2Et (5,08 mmol) und 0,11 ml Phenylacetylen
(1,0 mmol) zugespritzt und für 35 min bei RT gerührt.
Anschließend wird gesättigte NaHCO3-Lösung zugegeben, die Phasen getrennt und
die wässrige Phase mehrmals mit Et2O ausgeschüttelt. Die vereinigten organischen
Phasen werden über MgSO4 getrocknet und am Rotationsverdampfer eingeengt.

Experimenteller Teil
142
Der Rückstand wird mittels Flashchromatographie grob gereinigt (CH2Cl2/MeOH
100/0 bis 98/2).
Eine Lösung von 37,0 mg des erhaltenen Enins (91; etwa 0,11 mmol) in 4 ml THF
werden zu 154,8 mg PPh3 (0,59 mmol) gegeben. Dann wird 1 ml H2O zugespritzt
und für 2 Tage bei RT gerührt.
Anschließend wird mit Et2O auf etwa 75 ml verdünnt, die Lösung über Na2SO4
getrocknet und am Rotationsverdampfer eingeengt.
Der Rückstand wird in 5 ml trockenem CHCl3 gelöst, 0,06 ml Et3N (0,43 mmol) zuge-
geben und im Eisbad abgekühlt. Dann wird eine Lösung von 125,0 mg Biphenyl-
carbonsäurechlorid (0,58 mmol) in 5 ml trockenem CHCl3 zugetropft, das Eisbad
entfernt und für etwa 24 h bei RT gerührt.
Anschließend wird gesättigte NaHCO3-Lösung zugegeben, die Phasen getrennt und
die wässrige Phase mehrmals mit CH2Cl2 ausgeschüttelt. Die vereinigten organi-
schen Phasen werden über MgSO4 getrocknet und am Rotationsverdampfer einge-
engt.
Der Rückstand wird mittels präparativer HPLC gereinigt (CH3CN/H2O + 0,1 % TFA,
Gradient)
Ausbeute: 2,6 mg (3,9 %), weißer Feststoff (TFA-Salz)
Analytische Daten:
HR-EI-MS: Ber.: m/z 490,2984
Gef.: m/z 490,2984
IR:
(NaCl) ν in cm-1: 3745 m, 3645 m, 3531 m, 3126 w, 2779 w, 2738 w, 2434 m, 2306 s,
2214 s, 1647 s, 1612 s, 1574 s, 1558 s, 1390 m, 1138 s, 1078 s
1H-NMR: 1H (360 MHz, CD3OD, als TFA-Salz): 1,04 (t, J = 7,4 Hz, 3 H, Pr-3), 1,71-1,96 (m,
7 H, Pr-2, But-2, But-3, Cy-6a), 2,19 (m, 1 H, Cy-6b), 2,44-2,66 (m, 4 H, Cy-2/Cy-5),
3,04-3,28 (m, 4 H, Pr-1, But-4), 3,51 (m, 2 H, But-1), 3,63 (m, 1 H, Cy-1), 6,09 (m,
1 H, Cy-3), 7,29-7,41 (m, 6 H, Biphenyl H-4’, Phenyl), 7,45 (m, 2 H, Biphenyl H-3’, H-

Experimenteller Teil
143
5’), 7,62-7,45 (m, 4 H, Biphenyl H-3, H-5, H-2’, H-6’), 7,92 (m, 2 H, Biphenyl H-2, H-
6)
13C-NMR: 13C (90 MHz, CD3OD, als TFA-Salz): 11,24 (Pr-3), 19,49, 19,69 (Pr-2), 23,08, 23,26
(But-2), 24,15, 24,24, 27,04, 27,15, 27,75, 29,91 (But-3, Cy-2/-5/-6), 39,46 (But-1),
51,77, 52,09 (But-4), 53,61, 53,94 (Pr-1), 60,42 (Cy-1), 89,54, 89,64 (Ethinyl C-1/-2),
122,28, 124,27 (Cy-4, Phenyl C-1), 128,02, 128,05, 128,75, 129,05, 129,31, 129,39,
129,93 (Biphenyl C-2/C-6, C-3/C-5, C-2’/C-6’, C-3’/C-5’, C-4’, Phenyl C-3/C-5, C-4),
130,32 (Biphenyl C-1), 132,31 (Phenyl C-2/C-6), 133,92 (Cy-3), 141,05, 145,79
(Biphenyl C-4, 1’), 170,04 (Carbonyl)
HPLC: (254 nm)
System A1: tR = 22.7 min, Reinheit: 99 %
System M1: tR = 19.2 min, Reinheit: 99 %
94: N-{[N’-(4-Ethinylcyclohex-3-enyl)-N’-propyl]-4-aminobutyl}-4-biphenylcar-boxamid
NNH
O
23,9 mg des TMS-geschützten Enins 92 (0,05 mmol) werden in 5 ml trockenem THF
gelöst und die Lösung anschließend auf – 15 °C abgekühlt. Dann werden 0,1 ml
einer 1,0-molaren Lösung von Tetrabutylammoniumfluorid (0,1 mmol) zugegeben
und für 2,75 h bei – 15 °C gerührt.

Experimenteller Teil
144
Es wird gesättigte NaHCO3-Lösung zugegeben. Nachdem sich der Ansatz auf RT
erwärmt hat, werden die Phasen getrennt und die wässrige Phase mehrmals mit
CH2Cl2 ausgeschüttelt. Die vereinigten organischen Phasen werden über MgSO4
getrocknet und am Rotationsverdampfer eingeengt.
Der Rückstand wird mittels Flash-Chromatographie gereinigt (CH2Cl2/MeOH 98/2).
Ausbeute: 13,2 mg (64,8 %), farbloses Öl
Analytische Daten:
EI-MS: m/z 414
IR:
(NaCl) ν in cm-1: 3491 m, 3352 s, 1655 w, 1427 s, 1346 s, 1257 m, 1130 s, 999 s,
945 s, 825 s
1H-NMR: 1H (360 MHz, CDCl3): 0,87 (t, J = 7,3 Hz, 3 H, Pr-3), 1,41-1,73 (m, 7 H, Pr-2, But-2,
But-3, Cy-6a), 1,90 (m, 1 H, Cy-6b), 2,04-2,34 (m, 4 H, Cy-2/Cy-5), 2,46 (m, 2 H, Pr-
1), 2,56 (m, 2 H, But-4), 2,79-2,91 (m, 2 H, Cy-1, Ethin CH), 3,49 (m, 2 H, But-1),
6,11-6,16 (m, 1 H, Cy-3), 6,56 (m, 1 H, NH), 7,35-7,50 (m, 3 H, Biphenyl H-3’, H-4’,
H-5’), 7,58-7,68 (m, 4 H, Biphenyl H-3, H-5, H-2’, H-6’), 7,85 (m, 2 H, Biphenyl H-2,
H-6)
13C-NMR: 13C (90 MHz, CDCl3): 11,82 (Pr-3), 21,56 (Pr-2), 24,79, 25,96, 27,44, 27,99, 30,03
(But-2/-3, Cy-2/-5/-6), 39,84 (But-1), 50,03 (But-4), 52,46 (Pr-1),55,67 (Cy-1), 75,06
(Ethinyl C-2), 84,78 (Ethinyl C-1), 119,72 (Cy-4), 127,17, 127,19, 127,43, 127,92
(Biphenyl C-2/C-6, C-3/C-5, C-2’/C-6’, C-4’), 128,88 (Biphenyl C-3’/C-5’), 133,49
(Biphenyl C-1), 135,05 (Cy-3), 140,08, 144,13 (Biphenyl C-4, C-1’), 167,29
(Carbonyl)

Experimenteller Teil
145
CHN (%): C28H34N2O
Ber: C 81,12 H 8,27 N 6,76 O 3,86
Gef: C 80,73 H 8,36 N 6,83 O 3,94
96: N-[4-(4-Methyltriazol-1-yl)butyl]-N-propyl-N-(4-propin-1-ylcyclohex-3-enyl)-amin
NN N
N
Eine Lösung von 333,7 mg des Triflats 89 (0,87 mmol) in 3 ml trockenem THF wird
zu 28,6 mg Cu(I)I (0,15 mmol) und 63,0 mg Pd(PPh3)2Cl2 (0,09 mmol) in ein
Druckrohr gegeben. Dann werden 0,45 ml Piperidin (4,56 mmol) zugespritzt und auf
– 45 °C abgekühlt, bevor für 20 min Propingas eingeleitet und das Druckrohr
verschlossen wird. Anschließend lässt man den Ansatz auf RT erwärmen und 2 h bei
RT rühren.
Nachdem das Druckrohr vorsichtig aufgeschraubt wurde (Gas entweicht), wird
gesättigte NaHCO3-Lösung zugegeben, gerührt, die Phasen getrennt und die
wässrige Phase mehrmals mit EtOAc ausgeschüttelt. Die vereinigten organischen
Phasen werden über MgSO4 getrocknet und am Rotationsverdampfer eingeengt.
Der Rückstand wird mittels präparativer HPLC gereinigt (CH3CN/H2O + 0,1 % TFA,
Gradient)
Ausbeute: 95,6 mg (35,0 %), gelblicher, weicher Feststoff

Experimenteller Teil
146
Analytische Daten:
Schmp.: Nicht möglich wegen weicher Konsistenz
EI-MS: m/z 314
IR:
(NaCl) ν in cm-1: 2927 s, 2866 m, 1554 m, 1454 m, 1433 m, 1373 w, 1336 w, 1213
m, 1049 m, 640 m
1H-NMR: 1H (360 MHz, CDCl3): 0,84 (t, J = 7,4 Hz, 3 H, Pr-3), 1,33-1,49 (m, 5 H, Pr-2, But-2,
Cy-6a), 1,78 (m, 1 H, Cy-6b), 1,85-1,95 (m, 5 H, Propin-CH3, But-3), 1,95-2,25 (m, 4
H, Cy-2, Cy-5), 2,31-2,39 (m, 5 H, Pr-1, Triazol-CH3), 2,44 (m, 2 H, But-1), 2,72 (m,
1 H, Cy-1), 4,31 (m, 2 H, But-4), 5,94 (m, 1 H, Cy-3), 7,24 (Triazol H-5)
13C-NMR: 13C (150 MHz, CDCl3): 4,12 (Propin C-3), 10,82 (Triazol-CH3), 11,81 (Pr-3), 22,09
(Pr-2), 25,10, 25,87, 28,21, 29,69, 30,64 (But-2/-3, Cy-2/-5/-6), 49,53, 50,08 (But-
1/But-4), 52,36 (Pr-1), 55,32 (Cy-1), 80,79 (Propin C-2), 83,28 (Propin C-1), 120,84,
120,88 (Triazol C-5, Cy-4), 132,18 (Cy-3), 143,27 (Triazol C-4)
CHN (%): C19H30N4
Ber: C 72,57 H 9,62 N 17,82
Gef: C 72,12 H 9,41 N 17,48

Experimenteller Teil
147
97: N-{4-[4-(4-Biphenyl)triazol-1-yl]butyl}-N-propyl-4-aminocyclohexan-1-on
NN
O
NN
766,5 mg des Ketons 88 (3,04 mmol), 1085,0 mg Biphenylacetylen (6,09 mmol) und
115,4 mg [Cu(CH3CN)4]PF6 (0,31 mmol) werden in einer Mischung von 60 ml CH2Cl2
und 20 ml MeOH gelöst bzw. suspendiert und für 17 h bei 40 °C gerührt. Dann
werden noch einmal 1106,6 mg Biphenylacetylen (6,21 mmol) und 140,8 mg
[Cu(CH3CN)4]PF6 (0,38 mmol) zugegeben und weitere 8,5 h bei 40 °C gerührt.
Nach dem Abkühlen wird das Lösungsmittel am Rotationsverdampfer entfernt und
der Rückstand mittels Flashchromatographie gereinigt (EtOAc + 0,5 % NMe2Et bzw.
CH2Cl2/MeOH 98/2 bis 95/5)
Ausbeute: 995,7 mg (76,1 %), weißlich-gelblicher Feststoff
Analytische Daten:
Schmp.: 73 °C
HR-EI-MS: Ber.: m/z 430,2733
Gef.: m/z 430,2733

Experimenteller Teil
148
IR:
(NaCl) ν in cm-1: 2804 s, 2744 s, 2318 m, 2285 m, 2256 s, 1641 s, 1390 s, 1022 m,
808 m, 733 s
1H-NMR: 1H (360 MHz, CDCl3): 0,86 (t, J = 7,4 Hz, 3 H, Pr-3), 1,36-1,55 (m, 4 H, Pr-2, But-2
oder But-3), 1,69 (m, 2 H, Cy-3a, Cy-5a), 1,95-2,05 (m, 4 H, But-2 oder But-3, Cy-3b,
Cy-5b), 2,26-2,46 (m, 6 H, Pr-1, Cy-2, Cy-6), 2,50 (m, 2 H, But-1), 2,92 (m, 1 H, Cy-
4), 4,44 (m, 2 H, But-4), 7,35 (m, 1 H, Biphenyl H-4’), 7,45 (m, 2 H, Biphenyl H-3’, H-
5’), 7,60-7,69 (m, 4 H, Biphenyl H-3, H-5, H-2’, H-6’), 7,78 (s, 1 H, Triazol H-5), 7,88-
7,93 (m, 2 H, Biphenyl H-2, H-6)
13C-NMR: 13C (150 MHz, CDCl3): 11,76 (Pr-3), 21,99 (Pr-2), 25,69 (But-2 oder But-3), 27,85,
28,16 (But-2 oder But-3, Cy-3/Cy-5), 39,97 (Cy-2/Cy-6), 49,75 (But-1), 50,35 (But-4),
52,45 (Pr-1), 57,42 (Cy-4), 119,32 (Triazol C-5), 126,02 (Biphenyl C-2/C-6), 126,94,
127,42, 127,49 (Biphenyl C-3/C-5, C-2’/C-6’, C-4’), 128,79 (Biphenyl C-3’/C-5’),
129,62 (Biphenyl C-1), 140,54, 140,85 (Biphenyl C-4, C-1’), 147,483 (Triazol C-4),
211,16 (Cy-1)
HPLC: (254 nm)
System A3: tR = 10.3 min, Reinheit: 99 %
System M1: tR = 16.1 min, Reinheit: 95 %

Experimenteller Teil
149
98: O-[N-{4-[4-(4-Biphenyl)triazol-1-yl]butyl}-N-propyl-(4-aminocyclohex-1-en-yl)]-trifluormethylsulfonat
NN
OTf
NN
Eine Lösung von 556,0 mg des Triazols 97 (1,29 mmol) in 20 ml trockenem
1,2-Dichlorethan werden zu 820,5 mg 2,6-Di-tert-butyl-4-methylpyridin (4,00 mmol)
gegeben. Nachdem sich die Base gelöst hat, werden 0,65 ml Triflatanhydrid
(3,92 mmol) zugespritzt und über Nacht unter Rückfluss erhitzt.
Nach dem Abkühlen wird gesättigte NaHCO3-Lösung zugegeben, die Phasen ge-
trennt und die wässrige Phase mehrmals mit CH2Cl2 ausgeschüttelt. Die vereinigten
organischen Phasen werden über MgSO4 getrocknet und am Rotationsverdampfer
eingeengt.
Der Rückstand wird grob mittels Flashchromatographie gereinigt (Hexan/EtOAc 1/1 +
0,5 % NMe2Et) und anschließend weiter umgesetzt.

Experimenteller Teil
150
99: N-{4-[4-(4-Biphenyl)triazol-1-yl]butyl}-N-propyl-N-(4-(2-trimethylsilyl)-ethin-ylcyclohex-3-enyl)amin
NN N
N
TMS
Eine Lösung von 17,6 mg des Triflats 98 (0,03 mmol) in 5 ml trockenem THF werden
zu 10,4 mg Cu(I)I (0,05 mmol) und 17,1 mg Pd(PPh3)2Cl2 (0,02 mmol) gegeben.
Dann werden 0,05 ml NMe2Et (0,46 mmol) und 15 µl TMS-Acetylen (0,11 mmol)
zugespritzt und 1,5 h bei RT gerührt.
Anschließend wird gesättigte NaHCO3-Lösung zugegeben, die Phasen getrennt und
die wässrige Phase mehrmals mit Et2O ausgeschüttelt. Die vereinigten organischen
Phasen werden über MgSO4 getrocknet und am Rotationsverdampfer eingeengt.
Der Rückstand wird mittels Flashchromatographie gereinigt (CH2Cl2/MeOH 98/2 und
Hexan/EtOAc 4/1 + 0,5 % NMe2Et).
Ausbeute: 6,6 mg (41,3 %), gelber Feststoff
Analytische Daten:
EI-MS: m/z 510

Experimenteller Teil
151
IR:
(NaCl) ν in cm-1: 3766 m, 2841 s, 2646 w, 1676 s, 1583 m, 1502 s, 1412 m, 1030 m,
818 s
1H-NMR: 1H (360 MHz, CDCl3): 0,17 (s, 9 H, TMS), 0,85 (t, J = 7,3 Hz, 3 H, Pr-3), 1,33-1,53
(m, 5 H, Pr-2, But-2, Cy-6a), , 1,81 (m, 1 H, Cy-6b), 1,91-2,10 (m, 3 H, Cy-2a, But-3),
2,12-2,28 (m, 3 H, Cy-2b, Cy-5), 2,36 (m, 2 H, Pr-1), 2,47 (m, 2 H, But-1), 2,73 (m,
1 H, Cy-1), 4,43 (m, 2 H, But-4), 6,12 (m, 1 H, Cy-3), 7,35 (m, 1 H, Biphenyl H-4’),
7,45 (m, 2 H, Biphenyl H-3’, H-5’), 7,61-7,69 (m, 4 H, Biphenyl H-3, H-5, H-2’, H-6’),
7,77 (Triazol H-5), 7,91 (m, 2 H, Biphenyl H-2, H-6)
13C-NMR: 13C (90 MHz, CDCl3): 11,82 (Pr-3), 21,15 (Pr-2), 25,03, 25,95, 28,24, 28,40, 30,29
(But-2/-3, Cy-2/-5/-6), 49,53 (But-1), 50,43 (But-4), 52,39 (Pr-1), 55,23 (Cy-1), 94,50
(Et-2), 106,53 (Et-1), 119,33 (Triazol C-5), 120,64 (Cy-4), 126,08 (Biphenyl C-2/C-6),
126,98, 127,41, 127,51 (Biphenyl C-3/C-5, C-2’/C-6’, C-4’), 128,81 (Biphenyl C-3’/C-
5’), 129,72 (Biphenyl C-1), 135,28 (Cy-3), 140,64, 140,85 (Biphenyl C-4, C-1’),
147,47 (Triazol C-4)
CHN (%): C32H42N4Si
Ber: C 75,25 H 8,29 N 10,97
Gef: C 75,21 H 8,29 N 10,93

Experimenteller Teil
152
100: N-{4-[4-(4-Biphenyl)triazol-1-yl]butyl}-N-propyl-N-(4-(2-phenyl)-ethinyl-cyclohex-3-enyl)amin
NN N
N
Eine Lösung von 11,4 mg des Triflats 98 (0,02 mmol) in 5 ml trockenem THF werden
zu 7,8 mg Cu(I)I (0,04 mmol) und 12,2 mg Pd(PPh3)2Cl2 (0,02 mmol) gegeben. Dann
werden 0,05 ml NMe2Et (0,46 mmol) und 10 µl Phenylacetylen (0,09 mmol)
zugespritzt und 1,5 h bei RT gerührt.
Anschließend wird gesättigte NaHCO3-Lösung zugegeben, die Phasen getrennt und
die wässrige Phase mehrmals mit Et2O ausgeschüttelt. Die vereinigten organischen
Phasen werden über MgSO4 getrocknet und am Rotationsverdampfer eingeengt.
Der Rückstand wird mittels präparativer HPLC gereinigt (CH3CN/H2O + 01 % TFA).
Ausbeute: 5,8 mg (14,1 %), weißer Feststoff (TFA-Salz)
Analytische Daten:
HR-EI-MS: Ber.: m/z 514,3097
Gef.: m/z 514,3096

Experimenteller Teil
153
IR:
(NaCl) ν in cm-1: 3409 br. w, 2924 m, 2852 m, 1683 s, 1483 m, 1441 m, 1200 s, 1136
s, 1034 m, 845 m, 802 m, 765 m, 726 m, 692 m
1H-NMR: 1H (360 MHz, CD3OD/CHCl3 1:1, als TFA-Salz): 1,04 (t, J = 7,3 Hz, 3 H, Pr-3), 1,69-
1,87 (m, 5 H, Pr-2, But-2, Cy-6a), 2,06-2,20 (m, 3 H, But-3, Cy-6b), 2,38-2,62 (m,
4 H, Cy-2, Cy-5), 3,01-3,21 (m, 4 H, Pr-1, But-1), 3,52 (m, 1 H, Cy-1), 4,59 (m, 2 H,
But-4), 6,08 (m, 1 H, Cy-3), 7,29-7,41 (m, 6 H, Phenyl, Biphenyl H-4’), 7,45 (m, 2 H,
Biphenyl H-3’, H-5’), 7,61-7,73 (m, 4 H, Biphenyl H-3, H-5, H-2’, H-6’), 7,90 (m, 2 H,
Biphenyl H-2, H-6), 8,25 (Triazol H-5),
13C-NMR: 13C (90 MHz, CD3OD/CHCl3 1:1, als TFA-Salz): 11,25 (Pr-3), 19,30 (Pr-2), 22,73
(But-2), 23,99 (Cy-6), 26,83 (Cy-5), 27,74 (But-3), 29,66 (Cy-2), 50,00 (But-4), 50,97
(But-1), 53,46 (Pr-1), 59,92 (Cy-1), 89,23 (Et-1/-2), 121,80, 122,00, 123,76 (Triazol
C-5, Cy-4, Ph-1),126,75 (Biphenyl C-2/C-6), 127,53, 128,25 (Biphenyl C-3/C-5, C-
2’/C-6’, Ph-3/5), 129,03 (Biphenyl C-4’, Ph-4), 129,53 (Biphenyl C-3’/C-5’), 129,85
(Cy-3), 132,08 (Ph-2/-6), 141,18, 142,16 (Biphenyl C-4, C-1’), 148,61 (Triazol C-4)
HPLC: (254 nm)
System A4: tR = 14.5 min, Reinheit: 100 %
System M1: tR = 19.4 min, Reinheit: 100 %

Experimenteller Teil
154
101: N-{4-[4-(4-Biphenyl)triazol-1-yl]butyl}-N-propyl-N-(4-ethinylcyclohex-3-en-1-yl)amin
NN N
N
13,4 mg des TMS-geschützten Enins 99 (0,03 mmol) werden in 5 ml trockenem THF
gelöst und die Lösung anschließend auf – 15 °C abgekühlt. Dann werden 0,05 ml
einer 1,0-molaren Lösung von Tetrabutylammoniumfluorid (0,05 mmol) zugegeben
und für 2,45 h bei – 15 °C gerührt.
Es wird gesättigte NaHCO3-Lösung zugegeben. Nachdem sich der Ansatz auf RT er-
wärmt hat, werden die Phasen getrennt und die wässrige Phase mehrmals mit
CH2Cl2 ausgeschüttelt. Die vereinigten organischen Phasen werden über MgSO4
getrocknet und am Rotationsverdampfer eingeengt.
Der Rückstand wird mittels Flash-Chromatographie (CH2Cl2/MeOH 98/2) bzw.
präparativer HPLC (H20/CH3CN, Gradient) gereinigt.
Ausbeute: 6,2 mg (42,8 %), weißer Feststoff (TFA-Salz)
Analytische Daten:
HR-EI-MS: Ber.: m/z 438,2783
Gef.: m/z 438,2783

Experimenteller Teil
155
IR:
(NaCl) ν in cm-1: 3433 w, 3290 w, 2966 w, 2941 w, 2652 w, 2513 w, 1682 s, 1481 m,
1462 m, 1437 m, 1412 w, 1200 s, 1130 s, 831 w, 768 m
1H-NMR: 1H (360 MHz, CD3OD, als TFA-Salz): 1,01 (t, J = 7,4 Hz, 3 H, Pr-3), 1,67-1,88 (m,
5 H, Pr-2, But-2, Cy-6a), 2,03-2,15 (m, 3 H, But-3, Cy-6b), 2,34-2,58 (m, 4 H, Cy-2,
Cy-5), 3,00-3,24 (m, 4 H, Pr-1, But-1), 3,26 (s, 1 H, Ethin CH), 3,56 (m, 1 H, Cy-1),
4,57 (m, 2 H, But-4), 6,06 (m, 1 H, Cy-3), 7,35 (m, 1 H, Biphenyl H-4’), 7,45 (m, 2 H,
Biphenyl H-3’, H-5’), 7,63-7,74 (m, 4 H, Biphenyl H-3, H-5, H-2’, H-6’), 7,90 (m, 2 H,
Biphenyl H-2, H-6), 8,39 (Triazol H-5),
13C-NMR: 13C (150 MHz, CD3OD): 11,18 (Pr-3), 19,52, 19,73 (Pr-2), 23,06, 23,26 (But-2),
24,09, 26,96, 28,14, 29,82 (But-3, Cy-2/-5/-6), 50,43 (But-4), 51,34, 51,63 (But-1),
53,72, 54,05 (Pr-1), 60,47 (Cy-1), 78,07 (Et-2), 84,19 (Et-1), 121,74, 122,43 (Triazol
C-5, Cy-4), 127,11, 127,86, 128,55, 128,65 (Biphenyl C-2/C-6, C-3/C-5, C-2’/C-6’, C-
4’), 129,97 (Biphenyl C-3’/C-5’), 130,56 (Biphenyl C-1), 131,61 (Cy-3), 141,70,
142,55 (Biphenyl C-4, C-1’), 148,88 (Triazol C-4)
HPLC: (254 nm)
System A1: tR = 19.0 min, Reinheit: 100 %
System M1: tR = 17.3 min, Reinheit: 100 %

Experimenteller Teil
156
102: N-(4-Hydroxybutyl)-4-biphenylcarboxamid
OHNH
O
Zu einer Lösung von 0,5 ml 4-Aminobutanol (5,42 mmol) in 6 ml CH2Cl2 werden 2 ml
einer 10 %igen NaOH-Lösung in H2O gegeben und der Reaktionsansatz im Eisbad
abgekühlt. Dann wird langsam eine Lösung von 1122,0 mg 4-Biphenylcarbonsäure-
chlorid (5,18 mmol) in 8 ml CH2Cl2 zugetropft, das Eisbad entfernt und 5,5 h bei RT
gerührt.
Anschließend wird mit H2O gequencht und mit H2O ausgeschüttelt. Die wässrige
Phase wird dann mehrmals mit CH2Cl2 extrahiert, die organischen Phasen vereinigt,
über MgSO4 getrocknet und am Rotationsverdampfer eingeengt.
Ausbeute: 1342,5 mg (92,0 %), weißer Feststoff
Analytische Daten: Vgl. Literatur102, 115
1H-NMR: 1H (600 MHz, CD3OD): 1,60-1,66 1,68-1,74 (2 x m, 2 x 2 H, But-2/-3), 3,43 (t, J =
7,0 Hz, 2 H, But-1), 3,61 (t, J = 6,4 Hz, 2 H, But-4), 7,37 (m, 1 H, Biphenyl H-4’), 7,46
(m, 2 H, Biphenyl H-3’, H-5’), 7,64-7,73 (m, 4 H, Biphenyl H-3, H-5, H-2’, H-6’), 7,89
(m, 2 H, Biphenyl H-2, H-6)

Experimenteller Teil
157
103: N-(4-Hydroxybutyl)-4-biphenylcarboxamid-mesylat
MsONH
O
1,34 g des Amids 102 (4,98 mmol) werden in 40 ml trockenem THF suspendiert.
Zum Reaktionsansatz werden 1,0 ml Et3N (7,19 mmol) gegeben. Dann wird die
Lösung im Eisbad abgekühlt und es werden 0,6 ml Mesylchlorid (7,75 mmol)
zugetropft. Das Reaktionsgemisch wird für 2,5 h bei RT gerührt, bevor erneut 0,6 ml
Mesylchlorid (7,75 mmol) zugetropft werden. Dann wird über Nacht weiter bei RT
gerührt.
Anschließend werden 50 ml einer H2O/Eis-Mischung zugegeben und so lange weiter-
gerührt, bis das Eis geschmolzen ist. Man schüttelt aus und extrahiert dann die
wässrige Phase mehrmals mit CH2Cl2. Die vereinigten organischen Phasen werden
über MgSO4 getrocknet und am Rotationsverdampfer eingeengt.
Der erhaltene Rückstand wird für den nächsten Reaktionsschritt eingesetzt.
Ein kleiner Teil wird zu Analytik-Zwecken mittels Flash-Chromatographie gereinigt
(Hexan/EtOAc-Gradient an SP1-Anlage)
Analytische Daten:
Schmp.: 133 °C
HR-EI-MS: Ber.: m/z 347,1191
Gef.: m/z 347,1191

Experimenteller Teil
158
IR:
(NaCl) ν in cm-1: 3314 m, 2941 w, 2861 w, 1632 s, 1539 m, 1449 w, 1432 w, 1347 m,
1168 m, 978 m, 938 m, 852 w, 831 w, 749 m
1H-NMR: 1H (360 MHz, CDCl3): 1,74-1,93 (m, 4 H, But-2/-3), 3,03 (s, 3 H, Methyl), 3,54 (m,
2 H, But-4), 4,31 (m, 2 H, But-1), 6,33 (m, 1 H, NH), 7,39 (m, 1 H, Biphenyl H-4’),
7,47 (m, 2 H, Biphenyl H-3’, H-5’), 7,59-7,69 (m, 4 H, Biphenyl H-3, H-5, H-2’, H-6’),
7,85 (m, 2 H, Biphenyl H-2, H-6)
13C-NMR: 13C (90 MHz, CDCl3): 25,95, 26,59 (But-2/-3), 37,43, 39,18 (But-1/-4), 69,60 (Mesyl-
CH3), 127,17, 127,24, 127,36, 128,00 (Biphenyl C-2/C-6, C-3/C-5, C-2’/C-6’, C-4’),
128,91 (Biphenyl C-3’/C-5’), 133,04 (Biphenyl C-1), 139,94, 144,33 (Biphenyl C-4, C-
1’), 167,34 (Carbonyl)
HPLC: (254 nm)
System A1: tR = 14.7 min, Reinheit: 99 %
System M3: tR = 19.2 min, Reinheit: 100 %
104: N-[(N’-Propyl)-4-aminobutyl]-4-biphenylcarboxamid
NH
NH
O
227,8 mg des Mesylats 103 (0,66 mmol), 110,2 mg NaI (0,74 mmol) und 252,4 mg
K2CO3 (1,83 mmol) werden in 20 ml trockenem CH3CN gelöst bzw. suspendiert.
Dann werden 0,15 ml Propylamin (1,82 mmol) zugespritzt, der Reaktionsansatz
erwärmt und für 6 h unter Rückfluss gerührt.

Experimenteller Teil
159
Nach dem Abkühlen wird das Reaktionsgemisch am Rotationsverdampfer eingeengt,
der Rückstand in etwas H2O aufgenommen, mit 2N-NaOH basifiziert und mehrmals
mit CH2Cl2 ausgeschüttelt. Die organischen Phasen werden über MgSO4 getrocknet
und am Rotationsverdampfer eingeengt.
Der Rückstand wird mittels präparativer HPLC gereinigt (CH3CN/H2O + 0,1 % TFA,
Gradient).
Ausbeute: 112,9 mg (55,1 %), weißer Feststoff
Analytische Daten:
Schmp.: 124 °C
HR-EI-MS: Ber.: m/z 310,2045
Gef.: m/z 310,2046
IR:
(NaCl) ν in cm-1: 3332 s, 2958 m, 2929 m, 2871 w, 2810 w, 1633 s, 1543 s, 1485 m,
1448 m, 1307 w, 1124 w, 851 m, 744 s, 689 m
1H-NMR: 1H (360 MHz, CDCl3): 0,91 (t, J = 7,4 Hz, 3 H, Pr-3), 1,45-1,78 (m, 6 H, Pr-2, But-2/-
3), 2,58 (m, 2 H, Pr-1), 2,69 (m, 2 H, But-4), 3,45 (m, 2 H, But-1), 7,18 (m, 1 H, NH),
7,38 (m, 1 H, Biphenyl H-4’), 7,46 (m, 2 H, Biphenyl H-3’/H-5’), 7,58-7,66 (m, 4 H,
Biphenyl H-3/H-5, H-2’/H-6’), 7,84 (m, 2 H, Biphenyl H-2/H-6)
13C-NMR: 13C (90 MHz, CDCl3): 11,41 (Pr-3), 23,04 (Pr-2), 27,46, 27,77 (But-2/-3), 40,00 (But-
1), 49,32 (But-4), 51,89 (Pr-1), 127,11, 127,19, 127,45, 127,89 (Biphenyl C-2/C-6, C-
3/C-5, C-2’/C-6’, C-4’), 128,88 (Biphenyl C-3’/C-5’), 133,69 (Biphenyl C-1), 140,15,
144,01 (Biphenyl C-4, C-1’), 167,25 (Carbonyl)

Experimenteller Teil
160
HPLC: (254 nm)
System A1: tR = 12.2 min, Reinheit: 100 %
System M3: tR = 16.2 min, Reinheit: 100 %
105: N-[(N’-Cyclohexyl)-4-aminobutyl]-4-biphenylcarboxamid
NHNH
O
492,5 mg des Mesylats 103 (1,42 mmol), 228,2 mg NaI (1,52 mmol) und 410,1 mg
K2CO3 (2,97 mmol) werden in 20 ml trockenem CH3CN gelöst bzw. suspendiert.
Dann werden 0,50 ml Cyclohexylamin (4,37 mmol) zugespritzt, der Reaktionsansatz
erwärmt und über Nacht unter Rückfluss gerührt.
Nach dem Abkühlen wird das Reaktionsgemisch am Rotationsverdampfer eingeengt,
der Rückstand in etwas H2O aufgenommen, mit 2N-NaOH basifiziert und mehrmals
mit CH2Cl2 ausgeschüttelt. Die organischen Phasen werden über MgSO4 getrocknet
und am Rotationsverdampfer eingeengt.
Der Rückstand wird mittels Flashchromatographie gereinigt (CH2Cl2/MeOH, Gra-
dient, SP1-Anlage).
Ausbeute: 245,8 mg (49,4 %), weiß-gelblicher Feststoff
Analytische Daten:
Schmp.: 103 °C
EI-MS: m/z 350

Experimenteller Teil
161
IR:
(NaCl) ν in cm-1: 3316 s, 2925 s, 2852 m, 2807 w, 1633 s, 1582 m, 1447 m, 1309 w,
1126 w, 853 w, 746 m
1H-NMR: 1H (600 MHz, CDCl3/CD3OD): 1,05 (m, 2 H, Cy-3ax/Cy-5ax), 1,14 (m, 1 H, Cy-4 ax),
1,25 (m, 2 H, Cy-2ax/Cy-6ax), 1,53-1,70 (m, 5 H, But-2/-3, Cy-4eq), 1,74 (m, 2 H, Cy-
3eq/Cy-5eq), 1,89 (m, 2 H, Cy-2eq/Cy-6eq), 2,42 (m, 1 H, Cy-1), 2,65 (m, 2 H, But-
4), 3,45 (m, 2 H, But-1), 7,38 (m, 1 H, Biphenyl H-4’), 7,46 (m, 2 H, Biphenyl H-3’, H-
5’), 7,59-7,68 (m, 4 H, Biphenyl H-3, H-5, H-2’, H-6’), 7,88 (m, 2 H, Biphenyl H-2, H-
6)
13C-NMR: 13C (150 MHz, CDCl3/CD3OD): 24,85, 25,78, 26,85, 27,09 (But-2/-3, Cy-3-/-5, Cy-4),
32,86 (Cy-2/-6), 39,55 (But-1), 45,86 (But-4), 56,70 (Cy-1), 126,89, 126,97, 127,42,
127,76 (Biphenyl C-2/C-6, C-3/C-5, C-2’/C-6’, C-4’), 128,71 (Biphenyl C-3’/C-5’),
133,05 (Biphenyl C-1), 139,89, 143,98 (Biphenyl C-4, C-1’), 167,96 (Carbonyl)
HPLC: (254 nm)
System A1: tR = 13.5 min, Reinheit: 98 %
System M3: tR = 16.7 min, Reinheit: 99 %

Experimenteller Teil
162
106: N-[(N’,N’-Dipropyl)-4-aminobutyl]-4-biphenylcarboxamid
NNH
O
265,9 mg des Mesylats 103 (0,77 mmol), 186,6 mg NaI (1,24 mmol) und 258,9 mg
K2CO3 (1,87 mmol) werden in 20 ml trockenem CH3CN gelöst bzw. suspendiert.
Dann werden 0,30 ml Dipropylamin (2,19 mmol) zugespritzt, der Reaktionsansatz
erwärmt und über Nacht unter Rückfluss gerührt.
Nach dem Abkühlen wird das Reaktionsgemisch am Rotationsverdampfer eingeengt,
der Rückstand in etwas H2O aufgenommen, mit 2N-NaOH basifiziert und mehrmals
mit CH2Cl2 ausgeschüttelt. Die organischen Phasen werden über MgSO4 getrocknet
und am Rotationsverdampfer eingeengt.
Der Rückstand wird mittels Flashchromatographie gereinigt (CH2Cl2/MeOH, Gra-
dient, SP1-Anlage).
Ausbeute: 145,0 mg (53,4 %), weiße, weiche Masse
Analytische Daten:
HR-EI-MS: Ber.: m/z 352,2515
Gef.: m/z 352,2515
IR:
(NaCl) ν in cm-1: 3416 br. s, 3320 s, 3057 w, 3033 w, 2959 m, 2937 m, 2874 w, 2795
w, 1633 s, 1547 s, 1486 m, 1308 m, 1160 w, 856 m, 748 s, 697 m
1H-NMR: 1H (360 MHz, CDCl3): 0,94 (t, J = 7,4 Hz, 6 H, 2 x Pr-3), 1,63-1,90 (m, 8 H, 2 x Pr-2,
But-2/-3), 2,77 (m, 4 H, 2 x Pr-1), 2,86 (m, 2 H, But-4), 3,54 (m, 2 H, But-1), 7,37 (m,

Experimenteller Teil
163
1 H, Biphenyl H-4’), 7,45 (m, 2 H, Biphenyl H-3’, H-5’), 7,59-7,68 (m, 4 H, Biphenyl H-
3, H-5, H-2’, H-6’), 8,00 (m, 2 H, Biphenyl H-2, H-6)
13C-NMR: 13C (90 MHz, CDCl3): 11,41 (Pr-3), 17,67 (Pr-2), 22,31, 26,59 (But-2/-3), 38,62 (But-
1), 52,78 (But-4), 54,58 (Pr-1), 127,07, 127,15, 127,74, 127,83 (Biphenyl C-2/C-6, C-
3/C-5, C-2’/C-6’, C-4’), 128,84 (Biphenyl C-3’/C-5’), 133,04 (Biphenyl C-1), 140,13,
144,01 (Biphenyl C-4, C-1’), 167,36 (Carbonyl)
HPLC: (254 nm)
System A1: tR = 13.6 min, Reinheit: 99 %
System M3: tR = 16.3 min, Reinheit: 100 %
107: N-[(N’-Cyclopentyl-N’-propyl)-4-aminobutyl]-4-biphenylcarboxamid
NNH
O
50,1 mg des sekundären Amins 104 (0,16 mmol) und 78,1 mg NaBH(OAc)3
(0,37 mmol) werden in 10 ml trockenem CH2Cl2 gelöst bzw. suspendiert. Dann
werden 0,03 ml Cyclopentanon (0,34 mmol) zugegeben und über Nacht bei RT
gerührt. Dann werden weitere 0,1 ml Cyclopentanon (1,13 mmol) zugegeben, 24 h
bei RT gerührt und anschließend wieder 0,1 ml Cyclopentanon (1,13 mmol)
zugespritzt und über Nacht weiter gerührt.
Anschließend wird das Reaktionsgemisch am Rotationsverdampfer eingeengt, der
Rückstand in konzentrierter HCl aufgenommen und mehrmals mit Et2O gewaschen.
Die wässrige Phase wird dann mit 2N-NaOH basifiziert und mehrmals mit Et2O
ausgeschüttelt. Die organischen Phasen werden über MgSO4 getrocknet und am
Rotationsverdampfer reduziert.

Experimenteller Teil
164
Der Rückstand wird mittels präparativer HPLC gereinigt (CH3CN/H2O + 0,1 % TFA,
Gradient).
Ausbeute: 17,6 mg (29,1 %), weiß-gelblicher Feststoff
Analytische Daten:
Schmp.: 93 °C
HR-EI-MS: Ber.: m/z 378,2671
Gef.: m/z 378,2671
IR:
(NaCl) ν in cm-1: 3318 m, 2955 m, 2868 m, 2807 w, 1633 s, 1539 m, 1448 w, 1308 w,
1074 w, 1041 w, 853 w, 746 m
1H-NMR: 1H (360 MHz, CDCl3): 0,85 (t, J = 7,4 Hz, 3 H, Pr-3), 1,34-1,83 (m, 14 H, Pr-2, But-2/-
3, Cy-2/-3/-4/-5), 2,48 (m, 2 H, Pr-1), 2,56 (m, 2 H, But-4), 3,01 (m, 1 H, Cy-1), 3,49
(m, 2 H, But-1), 6,67 (m, 1 H, NH), 7,38 (m, 1 H, Biphenyl H-4’), 7,46 (m, 2 H,
Biphenyl H-3’/H-5’), 7,59-7,67 (m, 4 H, Biphenyl H-3/H-5, H-2’/H-6’), 7,84 (m, 2 H,
Biphenyl H-2/H-6)
13C-NMR: 13C (90 MHz, CDCl3): 11,97 (Pr-3), 19,55 (Pr-2), 24,03, 24,89, 27,73, 29,63 (But-2,
But-3, Cy-2/-5, Cy-3/-4), 40,05 (But-1), 51,40 (But-4), 53,74 (Pr-1), 63,89 (Cy-1),
127,15, 127,19, 127,45, 127,91 (Biphenyl C-2/C-6, C-3/C-5, C-2’/C-6’, C-4’), 128,89
(Biphenyl C-3’/C-5’), 133,69 (Biphenyl C-1), 140,13, 144,07 (Biphenyl C-4, C-1’),
167,35 (Carbonyl)

Experimenteller Teil
165
HPLC: (254 nm)
System A1: tR = 14.0 min, Reinheit: 100 %
System M3: tR = 16.7 min, Reinheit: 100 %
108: N-[(N’-Cyclohexyl-N’-propyl)-4-aminobutyl]-4-biphenylcarboxamid
NNH
O
117,8 mg des sekundären Amins 105 (0,34 mmol) und 160,5 mg NaBH(OAc)3
(0,76 mmol) werden in 20 ml trockenem CH2Cl2 gelöst bzw. suspendiert. Dann
werden 0,05 ml Propionaldehyd (0,67 mmol) zugegeben und für 7 h bei RT gerührt.
Anschließend wird das Reaktionsgemisch am Rotationsverdampfer eingeengt, der
Rückstand in konzentrierter HCl aufgenommen und mehrmals mit Et2O gewaschen.
Die wässrige Phase wird dann mit 2N-NaOH basifiziert und mehrmals mit Et2O
ausgeschüttelt. Die organischen Phasen werden über MgSO4 getrocknet und am
Rotationsverdampfer reduziert. In die verbleibende Lösung werden dann 2 ml einer
2-molaren Lösung von HCl in Et2O getropft und kühl gestellt. Da sich statt einem
Niederschlag nur ein Öl gebildet hat, wird das Lösungsmittel am
Rotationsverdampfer eingeengt, der Rückstand in etwas H2O aufgenommen, mit 2N-
NaOH basifiziert und mehrmals mit Et2O ausgeschüttelt. Die organischen Phasen
werden über MgSO4 getrocknet und am Rotationsverdampfer reduziert.
Der Rückstand wird mittels präparativer HPLC gereinigt (CH3CN/H2O + 0,1 % TFA,
Gradient).
Ausbeute: 112,9 mg (55,1 %), weißer Feststoff

Experimenteller Teil
166
Analytische Daten:
Schmp.: 87 °C
HR-EI-MS: Ber.: m/z 392,2828
Gef.: m/z 392,2832
IR:
(NaCl) ν in cm-1: 3323 s, 2928 s, 2854 m, 2802 w, 1634 s, 1542 m, 1448 m, 1307 w,
1074 m, 852 m, 745 m
1H-NMR: 1H (360 MHz, CDCl3): 0,85 (t, J = 7,3 Hz, 3 H, Pr-3), 1,14-1,28 (m, 5 H, Cy-2ax/Cy-
6ax, Cy-3ax/Cy-5ax, Cy-4 ax), 1,40-1,85 (m, 11 H, But-2/-3, Pr-2, Cy-2eq/Cy-6eq,
Cy-3eq/Cy-5eq Cy-4eq), 2,46 (m, 2 H, Pr-1), 2,50-2,61 (m, 3 H, But-4, Cy-1), 3,50
(m, 2 H, But-1), 6,78 (m, 1 H, NH), 7,38 (m, 1 H, Biphenyl H-4’), 7,46 (m, 2 H,
Biphenyl H-3’/H-5’), 7,59-7,67 (m, 4 H, Biphenyl H-3/ H-5, H-2’/H-6’), 7,86 (m, 2 H,
Biphenyl H-2/H-6)
13C-NMR: 13C (90 MHz, CDCl3): 11,89 (Pr-3), 21,57 (Pr-2), 26,12, 26,27, 27,37, 28,64 (But-2/-3,
Cy-2/-6, Cy-3-/-5, Cy-4), 39,86 (But-1), 50,11, 52,69 (But-4, Pr-1), 60,34 (Cy-1),
127,14, 127,19, 127,47, 127,89 (Biphenyl C-2/C-6, C-3/C-5, C-2’/C-6’, C-4’), 128,88
(Biphenyl C-3’/C-5’), 133,59 (Biphenyl C-1), 140,14, 144,03 (Biphenyl C-4, C-1’),
167,32 (Carbonyl)
HPLC: (254 nm)
System A1: tR = 14.4 min, Reinheit: 100 %
System M3: tR = 17.3 min, Reinheit: 100 %

Experimenteller Teil
167
109a: N-Propyl-N-(indan-2-yl)-2(2-thienyl)acetamid
N
SO
Zu einer Lösung von 48,5 mg des sekundären Amins 59 (0,28 mmol) in 5 ml
trockenem CHCl3, werden 0,12 ml Et3N (0,86 mmol) zugegeben. Die Lösung wird im
Eisbad abgekühlt und 0,05 ml 2-Thienylessigsäurechlorid (0,41 mmol) zugetropft.
Nach dem Entfernen des Eisbades wird für 2,5 h bei RT gerührt, bevor noch einmal
0,05 ml 2-Thienylessigsäurechlorid (0,41 mmol) zugetropft werden. Das Reaktions-
gemisch wird weitere 1,5 h bei RT gerührt.
Dann wird mehrmals mit gesättigter NaHCO3-Lösung und CH2Cl2 ausgeschüttelt, die
organischen Phasen über MgSO4 getrocknet und am Rotationsverdampfer einge-
engt.
Der Rückstand wird mittels Flash-Chromatographie gereinigt (Hexan/EtOAc 20/1).
Ausbeute: 73,7 mg (93,3 %), gelbes Öl
Analytische Daten:
EI-MS: m/z 299
IR:
(NaCl) ν in cm-1: 3390 s, 2924 m, 2854 m, 1639 s, 1435 s, 1045 s
1H-NMR: 1H (360 MHz, CDCl3): 0,84 (m, 3 H, Pr-3), 1,61 (m, 2 H, Pr-2), 2,94-3,24 (m, 6 H, Pr-
1 & Indan H-1/H-3), 3,90/3,99 (2 x s, 2 H, HOAc-2), 4,80/5,12 (2 x m, 1 H, Indan H-
2), 6,87-6,97 (m, 2 H, Thienyl H-3/H-4), 7,11-7,22 (m, 5 H, Indan H 4 – H 7 & Thienyl
H-5)

Experimenteller Teil
168
13C-NMR: 13C (90 MHz, CDCl3): 11,28, 11,54 (Pr-3), 21,98 (Pr-2), 35,62, 36,06, 36,55, 37,03
(Indan C-1/C-3, HOAc C-2), 44,77, 47,87 (Pr-1), 56,17, 58,61 (Indan C-2), 124,42,
124,51 (Indan C-4/C-7), 124,70, 124,75 (Thienyl C-5), 125,85, 126,57, 126,65,
126,74, 126,97 (Indan C-5/C-6, Thienyl C-3/C-4), 137,00 (Thienyl C-2), 140,26,
141,17 (Indan C-3a/C-7a), 169,64, 170,07 (Carbonyl)
CHN (%): C18H21NOS x 1/8 H2O
Ber: C 71,66 H 7,10 N 4,64 S 10,63
Gef: C 71,98 H 7,56 N 4,17 S 10,15
109b: N-Propyl-N-(indan-2-yl)-2-phenalacetamid
N
O
Zu einer Lösung von 37,5 mg des sekundären Amins 59 (0,21 mmol) in 5 ml trocke-
nem CHCl3, werden 0,09 ml Et3N (0,65 mmol) zugegeben. Die Lösung wird im Eis-
bad abgekühlt und 0,04 ml Phenylessigsäurechlorid (0,30 mmol) zugetropft. Nach
dem Entfernen des Eisbades wird für 2,5 h bei RT gerührt, bevor noch einmal
0,04 ml Phenylessigsäurechlorid (0,30 mmol) zugetropft werden. Das Reaktions-
gemisch wird weitere 1,5 h bei RT gerührt.
Dann wird mehrmals mit gesättigter NaHCO3-Lösung und CH2Cl2 ausgeschüttelt, die
organischen Phasen über MgSO4 getrocknet und am Rotationsverdampfer einge-
engt.
Der Rückstand wird mittels Flash-Chromatographie gereinigt (Hexan/EtOAc 10/1).
Ausbeute: 49,9 mg (79,5 %), farbloses Öl

Experimenteller Teil
169
Analytische Daten:
EI-MS: m/z 293
IR:
(NaCl) ν in cm-1: 3394 m, 2927 m, 2850 w, 1639 s, 1458 m, 1419 m, 1030 m
1H-NMR: 1H (360 MHz, CDCl3): 0,78-0,88 (m, 3 H, Pr-3), 1,51-1,72 (m, 2 H, Pr-2), 2,77-2,97
(m, 2 H, Indan H-1/H-3) 3,03-3,21 (m, 4 H, Pr-1 & Indan H-1/H-3), 3,73/3,82 (2 x s,
2 H, HOAc-2), 4,75/5,09 (2 x m, 1 H, Indan H-2), 7,10-7,36 (m, 9 H, Indan H 4 – H 7,
Phenyl)
13C-NMR: 13C (90 MHz, CDCl3): 11,28, 11,57 (Pr-3), 22,01 (Pr-2), 36,58, 36,85 (Indan C-1/C-3),
41,48, 42,03 (HOAc C-2), 44,57, 47,87 (Pr-1), 56,13, 58,52 (Indan C-2), 124,41,
124,48 (Indan C-4/C-7), 126,53, 126,72, 126,89 (Indan C-5/C-6, Phenyl C-4), 128,51,
128,64, 128,72, 128,78 (Phenyl C-2/C-3/C-5/C-6), 135,48 (Phenyl C-1), 140,34,
141,25 (Indan C-3a/C-7a), 170,75, 171,14 (Carbonyl)
CHN (%): C20H23NO x 0,1 H2O
Ber: C 81,37 H 7,92 N 4,74
Gef: C 81,42 H 8,11 N 4,64

Experimenteller Teil
170
109c: N-Propyl-N-(indan-2-yl)-2(4-methoxyphenyl)acetamid
N
OCH3
O
Zu einer Lösung von 46,0 mg des sekundären Amins 59 (0,26 mmol) in 5 ml trocke-
nem CHCl3, werden 0,11 ml Et3N (0,79 mmol) zugegeben. Die Lösung wird im Eis-
bad abgekühlt und 0,06 ml (4-Methoxyphenyl)essigsäurechlorid (0,39 mmol)
zugetropft. Nach dem Entfernen des Eisbades wird für 2 h bei RT gerührt, bevor
noch einmal 0,06 ml (4-Methoxyphenyl)essigsäurechlorid (0,39 mmol) zugetropft
werden. Das Reaktionsgemisch wird weitere 2,5 h bei RT gerührt.
Dann wird mehrmals mit gesättigter NaHCO3-Lösung und CH2Cl2 ausgeschüttelt, die
organischen Phasen über MgSO4 getrocknet und am Rotationsverdampfer einge-
engt.
Der Rückstand wird mittels Flash-Chromatographie gereinigt (Hexan/EtOAc 10/1).
Ausbeute: 81,0 mg (95,4 %), gelbes Öl
Analytische Daten:
EI-MS: m/z 323
IR:
(NaCl) ν in cm-1: 3359 m, 2962 m, 2931 m, 1639 s, 1581 m, 1512 s, 1423 s, 1246 m,
1030 m
1H-NMR: 1H (360 MHz, CDCl3): 0,83 (t, J = 7,4 Hz, 3 H, Pr-3), 1,49-1,67 (m, 2 H, Pr-2), 2,80-
2,98 (m, 2 H, Indan H-1/H-3), 3,02-3,21 (m, 4 H, Pr-1 & Indan H-1/H-3), 3,66/3,75
(2 x s, 2 H, HOAc-2), 3,80 (s, 3 H, Methoxy), 4,69-5,16 (m, 1 H, Indan H-2), 6,82-

Experimenteller Teil
171
6,89 (m, 2 H, Phenyl H-3, H-5), 7,11-7,22 (m, 6 H, Indan H 4 – H 7 & Phenyl H-2, H-
6)
13C-NMR: 13C (150 MHz, CDCl3):11,29, 11,56 (Pr-3), 22,00 (Pr-2), 36,55, 36,85 (Indan C-1/C-
3), 40,51, 41,07 (HOAc C-2), 44,53, 47,76 (Pr-1), 55,27 (Methoxy), 56,05, 58,45
(Indan C-2), 114,07, 114,13 (Phenyl C-3/C-5), 124,40, 124,49 (Indan C-4/C-7),
126,51, 126,87 (Indan C-5/C-6), 127,46, 129,50, 129,77 (Phenyl C-1/C-2/C-6),
140,36, 141,25 (Indan C-3a/7a), 158,38, 158,43 (Phenyl C-4), 171,07, 141,46
(Carbonyl)
CHN (%): C21H25NO2 x 0,2 H2O
Ber: C 77,13 H 7,83 N 4,28
Gef: C 77,23 H 7,73 N 4,36
110a: N-(Indan-2-yl)-N-propyl-N-[2-(2-thienyl)ethyl]amin
N
S
Zu einer Lösung von 65,2 mg des Amids 109a (0,22 mmol) in 5 ml trockenem Et2O
werden 0,57 ml einer 1-molaren Lösung von LiAlH4 in Et2O (0,57 mmol) zugetropft.
Anschließend wird das Reaktionsgemisch für 4,5 h bei RT gerührt.
Es wird mit gesättigter NaHCO3-Lösung gequencht, die Lösung durch eine Fritte mit
Celite-MgSO4-Celite gegeben und mit CH2Cl2 und EtOAc nachgespült. Die Lösung
wird am Rotationsverdampfer eingeengt.
Der Rückstand wird mittels Flash-Chromatographie gereinigt (Hexan/EtOAc 20/1 +
0,5 % NMe2Et)
Ausbeute: 34,8 mg (56,0 %), gelbes Öl

Experimenteller Teil
172
Analytische Daten:
EI-MS: m/z 285
IR:
(NaCl) ν in cm-1: 3384 m br, 2956 s, 1460 m, 1437 m, 1076 s, 744 s, 692 s
1H-NMR: 1H (360 MHz, CDCl3): 0,91 (t, J = 7,4 Hz, 3 H, Pr-3), 1,54 (m, 2 H, Pr-2), 2,57 (m,
2 H, Pr-1), 2,83-3,08 (m, 8 H, Ethyl H-1/H-2 & Indan H-1/H-3), 3,71 (m, 1 H, Indan H-
2), 6,80 (m, 1 H, Thienyl H-3), 6,91 (dd, J = 5,2 Hz, 3,4 Hz, 1 H, Thienyl H-4), 7,09-
7,20 (m, 5 H, Indan H 4 – H 7 & Thienyl H-5)
13C-NMR: 13C (90 MHz, CDCl3): 11,98 (Pr-3), 20,65 (Pr-2), 28,00 (Et-2), 36,73 (Indan C-1/C-3),
53,29, 53,55 (Et-1, Pr-1), 63,29 (Indan C-2), 123,22 (Thienyl C-5), 124,43, 124,48
(Indan C-4/C-7, Thienyl C-3), 126,28, 126,61 (Indan C-5/C-6, Thienyl C-4), 141,84
(Indan C-3a/C-7a), 143,10 (Thienyl C-2)
CHN (%): C18H23NS x 0,1 H2O
Ber: C 75,27 H 8,14 N 4,88 S 11,16
Gef: C 75,61 H 8,13 N 4,91 S 10,80

Experimenteller Teil
173
110b: N-(Indan-2-yl)-N-propyl-N-(2-phenylethyl)amin
N
Zu einer Lösung von 44,7 mg des Amids 109b (0,15 mmol) in 5 ml trockenem Et2O
werden 0,38 ml einer 1-molaren Lösung von LiAlH4 in Et2O (0,38 mmol) zugetropft.
Anschließend wird das Reaktionsgemisch für 4,5 h bei RT gerührt.
Es wird mit gesättigter NaHCO3-Lösung gequencht, die Lösung durch eine Fritte mit
Celite-MgSO4-Celite gegeben und mit CH2Cl2 und EtOAc nachgespült. Die Lösung
wird am Rotationsverdampfer eingeengt.
Der Rückstand wird mittels Flash-Chromatographie gereinigt (Hexan/EtOAc 20/1 +
0,5 % NMe2Et)
Ausbeute: 32,6 mg (76,6 %), gelbliches Öl
Analytische Daten:
EI-MS: m/z 279
IR:
(NaCl) ν in cm-1: 3388 m br, 2954 m, 1458 m, 1074 m, 744 s
1H-NMR: 1H (360 MHz, CDCl3): 0,91 (t, J = 7,4 Hz, 3 H, Pr-3), 1,56 (m, 2 H, Pr-2), 2,59 (m,
2 H, Pr-1), 2,73-2,94 (m, 6 H, Ethyl H-1/H-2 & Indan H-1/H-3), 3,05 (dd, J = 15,1 Hz,
7,7 Hz, 2 H, Indan H-1/H-3), 3,71 (m, 1 H, Indan H-2), 7,09-7,21 (m, 7 H, Indan H 4 –
H 7, Phenyl H-2, H-4, H-6), 7,24-7,31 (m, 2 H, Phenyl H-3, H-5)

Experimenteller Teil
174
13C-NMR: 13C (150 MHz, CDCl3): 12,02 (Pr-3), 20,50 (Pr-2), 33,51 (Et-2), 36,91 (Indan C-1/C-
3), 53,45, 53,51 (Et-1, Pr-1), 63,34 (Indan C-2), 124,43 (Indan C-4/C-7), 125,91
(Phenyl C-4), 126,28 (Indan C-5/C-6), 128,34 (Phenyl C-3/C-5), 128,70 (Phenyl C-
2/C-6), 140,74 (Phenyl C-1) 141,85 (Indan C-3a/C-7a)
CHN (%): C20H25N x 0,1 H2O
Ber: C 85,42 H 9,03 N 4,98
Gef: C 85,37 H 9,18 N 4,92
110c: N-(Indan-2-yl)-N-propyl-N-[2-(4-methoxyphenyl)ethyl]amin
N
OCH3
Zu einer Lösung von 49,9 mg des Amids 109c (0,15 mmol) in 5 ml trockenem Et2O
werden 0,40 ml einer 1-molaren Lösung von LiAlH4 in Et2O (0,40 mmol) zugetropft.
Anschließend wird das Reaktionsgemisch für 4,5 h bei RT gerührt.
Es wird mit gesättigter NaHCO3-Lösung gequencht, die Lösung durch eine Fritte mit
Celite-MgSO4-Celite gegeben und mit CH2Cl2 und EtOAc nachgespült. Die Lösung
wird am Rotationsverdampfer eingeengt.
Der Rückstand wird mittels Flash-Chromatographie gereinigt (Hexan/EtOAc 20/1 +
0,5 % NMe2Et)
Ausbeute: 29,8 mg (62,4 %), gelbes Öl

Experimenteller Teil
175
Analytische Daten:
EI-MS: m/z 309
IR:
(NaCl) ν in cm-1: 3394 m br, 2954 m, 2937 m, 1512 s, 1246 s, 744 m
1H-NMR: 1H (360 MHz, CDCl3): 0,91 (t, J = 7,4 Hz, 3 H, Pr-3), 1,55 (m, 2 H, Pr-2), 2,58 (m,
2 H, Pr-1), 2,75 (m, 4 H, Ethyl H-1/H-2), 2,88 (dd, J = 15,3 Hz, 8,9 Hz, 2 H, Indan H-
1/H-3), 3,04 (dd, J = 15,3 Hz, 7,7 Hz, 2 H, Indan H-1/H-3), 3,70 (m, 1 H, Indan H-2),
3,78 (s, 3 H, Methoxy), 6,83 (m, 2 H, Phenyl H-3, H-5) 7,07-7,20 (m, 6 H, Indan H 4 –
H 7, Phenyl H-2, H-6)
13C-NMR: 13C (90 MHz, CDCl3): 12,01 (Pr-3), 20,52 (Pr-2), 32,70 (Et-2), 36,88 (Indan C-1/C-3),
53,50, 53,74 (Et-1, Pr-1), 55,24 (Methoxy), 63,35 (Indan C-2), 113,80 (Phenyl C-3/C-
5), 124,43 (Indan C-4/C-7), 126,27 (Indan C-5/C-6), 129,57, 132,80 (Phenyl C-1/C-
2), 141,88 (Indan C-3a/C-7a), 157,89 (Phenyl C-4)
CHN (%): C21H27NO
Ber: C 81,51 H 8,79 N 4,53
Gef: C 81,31 H 9,17 N 4,53

Literaturverzeichnis
176
10. Literaturverzeichnis
1. Mutschler, E.; Geisslinger, G.; Kroemer, H. K.; Schäfer-Korting, M. Arzneimittelwirkungen; Lehrbuch der Pharmakologie und Toxikologie. Stuttgart, 2001; Vol. 8. 2. Boeckler, F. Konkurrenz für den "Goldstandard" L-DOPA? - Rezeptor-vermittelte Parkinson-Therapeutika. Pharmazie in unserer Zeit 2006, 35, 204-216. 3. Merlino, G.; 'Serafini, A.; Robiony, F.; Valente, M.; Gigli, G. L. Clinical experience with pramipexole in the treatment of restless legs syndrome. Expert Opinion on Drug Metabolism & Toxicology 2008, 4, 225-235. 4. Kenney, C.; Jankovic, J. Rotigotine transdermal patch in the treatment of Parkinson's disease and restless legs syndrome. Expert Opinion on Drug Metabolism & Toxicology 2007, 8, 1329-1335. 5. Hsieh, G. C.; Hollingsworth, P. R.; Martino, B.; Chang, R.; Terranova, M. A.; O'Neill, A. B.; Lynch, J. J.; Moreland, R. B.; Donnelly-Roberts, D. L.; Kolasa, T.; Mikusa, J. P.; McVey, J. M.; Marsh, K. C.; Sullivan, J. P.; Brioni, J. D. Central Mechanisms Regulating Penile Erection in Conscious Rats: The Dopaminergic Systems Related to the Proerectile Effect of Apomorphine. The Journal of Pharmacology and Experimental Therapeutics 2004, 308, 330-338. 6. Piercey, M. F.; Hoffmann, W. E.; Smith, M. W.; Hyslop, D. K. Inhibition of dopamine neuron firing by pramipexole, a dopamine D3 receptor-preferring agonist: comparison to other dopamine receptor agonists. European Journal of Pharmacology 1996, 312, 35-44. 7. Reynolds, N. A.; Wellington, K.; Easthope, S. E. Rotigotine In Parkinson's Disease. CNS Drugs 2005, 19, 973-981. 8. van Vliet, L. A.; Tepper, P. G.; Dijkstra, D.; Damsma, G.; Wikström, H.; Pugsley, T. A.; Akunne, H. C.; Heffner, T. G.; Glase, S. A.; Wise, L. D. Affinity for Dopamine D2, D3, and D4 Receptors of 2-Aminotetralins. Relevance of D2 Agonist Binding for Determination of Receptor Subtype Selectivity. Journal of Medicinal Chemistry 1996, 39, 4233-4237. 9. Grady, M. A.; Gasperoni, T. L.; Kirkpatrick, P. Aripiprazole. Nature Reviews - Drug Discovery 2003, 2, 427-428. 10. Burris, K. D.; Molski, T. F.; Xu, C.; Ryan, E.; Tottori, K.; Kikuchi, T.; Yocca, F. D.; Molinoff, P. B. Aripiprazole, a Novel Anitpsychotic, Is a High-Affinity Partial Agonist at Human Dopamine D2 Receptors. The Journal of Pharmacology and Experimental Therapeutics 2002, 302, 381-389. 11. Lawler, C. P.; Piroleau, C.; Lewis, M. M.; Mak, C.; Jiang, D.; Schetz, J. A.; Gonzales, A. M.; Sibley, D. R.; Mailman, R. B. Interactions of the Novel Anitpsychotic Aripiprazole (OPC-14597) with Dopamine and Serotonin Receptor Subtypes. Neuropsychopharmacology 1999, 20, 612-627. 12. Richtand, N. M.; Welge, J. A.; Logue, A. D.; Keck, P. E. J.; Strakowski, S. M.; McNamara, R. K. Dopamine and Serotonin Receptor Binding and Antipsychotic Efficacy. Neuropsychopharmacology 2007, 32, 1715-1726. 13. Lange, J. H. M.; Reinders, J.-H.; Tolboom, J. T. B. M.; Glennon, J. C.; Coolen, H. K. A. C.; Kruse, C. G. Pricipal Component Analysis Differentiates the Receptor Binding Profiles of Three Antipsychotic Drug Candidates from Current Antipsychotic Drugs. Journal of Medicinal Chemistry 2007, 50, 5103-5108. 14. Richelson, E.; Nelson, A. Antagonism by Neuroleptics of Neurotransmitter Receptors of Normal Human Brain in Vitro. European Journal of Pharmacology 1984, 103, 197-204.

Literaturverzeichnis
177
15. Richelson, E.; Souder, T. Binding of antipsychotic drugs to human brain receptors. Focus on newer generation compounds. Life Sciences 2000, 68, 29-39. 16. Ohmori, J.; Maeno, f.; Hidaka, K.; Nakato, K.; Matsumoto, M.; Tada, S.; Hattori, f.; Sakamoto, S.; Tsukamoto, S.-i.; Usuda, S.; Mase, T. Dopamine D3 and D4 Receptor Antagonists: Synthesis and Structure-Activity Relationships of (S)-(+)-N-(1-Benzyl-3-pyrrolidinyl)-5-chloro-4-[(cyclopropylcarbonyl)amino]-2-methoxybanzamide (YM-43611) and Related Compounds. Journal of Medicinal Chemistry 1996, 39, 2764-2772. 17. Sautel, F.; Griffon, N.; Sokoloff, P.; Schwartz, J.-C.; Launay, C.; Simon, P.; Costentin, J.; Schoenfelder, A.; Garrido, F.; Mann, A.; Wermuth, C. G. Nafadotride, a Potent Preferential Dopamine D3 Receptor Antagonist, Activates Locomotion in Rodents. The Journal of Pharmacology and Experimental Therapeutics 1995, 275, 1239-1246. 18. Sokoloff, P.; Andrieux, M.; Besancon, R.; Pilon, C.; Martres, M.-P.; Giros, B.; Schwartz, J.-C. Pharmacology of human dopamine D3 receptor expressed in a mammalian cell line: comparison with D2 receptor. European Journal of Pharmacology; Molecular Pharmacology Section 1992, 225, 331-337. 19. Boeckler, F.; Lanig, H.; Gmeiner, P. Modeling the Similarity and Divergence of Dopamine D2-like Receptors and Identification of Validated Ligand-Receptor Complexes. Journal of Medicinal Chemistry 2005, 48, 694-709. 20. Boeckler, F.; Gmeiner, P. The structural evolution of dopamine D3 receptor ligands: Structure-activity relationships and selected neuropharmacological aspects. Pharmacology & Therapeutics 2006, 112, 281-333. 21. Cannon, J. G. Structure-Activity Relationships of Dopamine Agonists. Annual Reviews of Pharmacology and Toxicology 1983, 23, 103-130. 22. McDermed, J. D.; McKenzie, G. M.; Phillips, A. P. Synthesis and Pharmacology of Some 2-Aminotetralins. Dopamine Receptor Agonists. Journal of Medicinal Chemistry 1975, 18, 362-367. 23. Cannon, J. G.; Lee, T.; Goldman, H. D.; Costall, B.; Naylor, R. J. Cerebral Dopamine Agonist Properties of Some 2-Aminotetralin Derivatives after Peripheral and Intracerebral Administration. Journal of Medicinal Chemistry 1977, 20, 1111-1116. 24. Hacksell, U.; Svensson, U.; Nilsson, J. L. G. N-Alkylated 2-Aminotetralins: Central Dopamine-Receptor Stimulating Activity. Journal of Medicinal Chemistry 1979, 22, 1469-1475. 25. Malmberg, A.; Nordvall, G.; Johansson, A. M.; Mohell, N.; Hacksell, U. Molecular Basis for the Binding of 2-Aminotetralins to Human Dopamine D2A and D3 Receptors. Molecular Pharmacology 1994, 46, 299-312. 26. Karlsson, A.; Björk, L.; Pettersson, C.; Andén, N.-E.; Hacksell, U. (R)- and (S)-5-Hydroxy-2-(dipropylamino)tetralin (5-OH DPAT): Assessment of Optical Purities and Dopaminergic Activities. Chirality 1990, 2, 90-95. 27. Yu, H.; Liu, Y.; Malmberg, A.; Mohell, N.; Hacksell, U.; Lewander, T. Differential serotoninergic and dopaminergic activities of the (R)- and the (S)-enantiomeres of 2-(di-n-propylamino)tetralin. European Journal of Pharmacology 1996, 303, 151-162. 28. Chumpradit, S.; Kung, M.-P.; Kung, H. F. Synthesis and Optical Resolution of (R)- and (S)-trans-7-Hydroxy-2-[N-propyl-N-(3'-iodo-2'-propenyl)amino]tetralin: A New D3 Dopamine Receptor Ligand. Journal of Medicinal Chemistry 1993, 36, 4308-4312. 29. Chumpradit, S.; Kung, M.-P.; Vessotskie, J.; Foulon, C.; Mu, M.; Kung, H. F. Iodinated 2-Aminotetralins and 3-Amino-1-benzopyrans: Ligands for Dopamine D2 and D3 Receptors. Journal of Medicinal Chemistry 1994, 37, 4245-4250.

Literaturverzeichnis
178
30. Cusack, N. J.; Peck, J. V. N-0923 Dopamine D2 Agonist. Drugs of the Future 1993, 18, 1005-1008. 31. Glase, S. A.; Corbin, A. E.; Pugsley, T. A.; Heffner, T. G.; Wise, L. D. Synthesis and Dopaminergic Activity of Pyridine Analogs of 5-Hydroxy-2-(di-n-propylamino)tetralin. Journal of Medicinal Chemistry 1995, 38, 3132-3137. 32. Sokoloff, P.; Giros, B.; M.-P., M.; Bouthenet, M.-L.; Schwartz, J.-C. Molecular cloning and characterization of a novel dopamine receptor (D3) as a target for neuroleptics. Nature 1990, 347, 146-151. 33. Tang, L.; Todd, R. D.; Heller, A.; O'Malley, K. L. Pharmacological and Functional Characterization of D2, D3 and D4 Dopamine Receptors in Fibroblast an dDopaminergic Cell Lines. The Journal of Pharmacology and Experimental Therapeutics 1994, 268, 495-502. 34. Kreiss, D. S.; Bergstrom, D. A.; Gonzales, A. M.; Huang, K.-X.; Sibley, D. R.; Walters, J. R. Dopaminergic receptor agonist potencies for inhibition of cell firing correlate with dopmaine D3 receptor binding afinities. European Journal of Pharmacology 1995, 277, 209-214. 35. Burris, K. D.; Pacheco, M. A.; Filtz, T. M.; Kung, M.-P.; Kung, H. F.; Molinoff, P. B. Lack of Discrimination by Agents for D2 and D3 Dopamine Receptors. Neuropsychopharmacology 1995, 12, 335-345. 36. Millan, M. J.; Peglion, J.-L.; Vian, J.; Rivet, J.-M.; Brocco, M.; Gobert, A.; Newman-Tancredi, A.; Dacquet, C.; Bervoets, K.; Girardon, S.; Jaques, V.; Chaput, C.; Audinot, V. Functional Correlates of Dopamine D3 Receptor Activation in the Rat In Vivo and Their Modulation by Selective Antagonist, (+)-S 14297: I. Activation of Postsynaptic D3 Receptors Mediates Hypothermia, Whereas Blockade of D2 Receptors Elicits Prolactin Secretion and Catalepsy. The Journal of Pharmacology and Experimental Therapeutics 1995, 275, 885-898. 37. Levant, B. The D3 Dopamine Receptor: Neurobiology and Potential Clinical Relevance. Pharmacological Reviews 1997, 49, 231-252. 38. Mierau, J.; Schneider, F. J.; Ensinger, H. A.; Chio, C. L.; Lajiness, M. E.; Huff, R. M. Pramipexole binding and activation of cloned and expressed dopamine D2, D3 and D4 receptors. European Journal of Pharmacology; Molecular Pharmacology Section 1995, 290, 29-36. 39. Perachon, S.; Schwartz, J.-C.; Sokoloff, P. Functional potencies of new antiparkinsonian drugs at recombinant human dopamine D1, D2 and D3 receptors. European Journal of Pharmacology 1999, 366, 293-300. 40. Mierau, J.; Schingnitz, G. Biochemical and pharmacological studies on pramipexole, a potent and selective dopamine D2 receptor agonist. European Journal of Pharmacology 1992, 215, 161-170. 41. Schneider, C. S.; Mierau, J. Dopamine Autoreceptor Agonists: Resolution and Pharmacological Activity of 2,6-Diaminotetrahydrobenzothiazole and an Aminothiazole Analogue of Apomorphine. Journal of Medicinal Chemistry 1987, 30, 494-498. 42. Lehmann, T.; Hübner, H.; Gmeiner, P. Dopaminergic 7-Aminotetrahydroindolizines: Ex-Chiral Pool Synthesis and Preferential D3 Receptor Binding. Bioorgganic & Medicinal Chemistry Letters 2001, 11, 2863-2866. 43. Bergauer, M.; Hübner, H.; Gmeiner, P. Practical ex-chiral-pool methodology for the synthesis of dopaminergic tetrahydroindoles. Tetrahedron 2004, 60, 1197-1204. 44. Löber, S.; Hübner, H.; Gmeiner, P. Fused Azaindole Derivatives: Molecular Design, Synthesis and In Vitro Pharmacology Leading to the Preferential Dopamine

Literaturverzeichnis
179
D3 Receptor Agonist FAUC 725. Bioorgganic & Medicinal Chemistry Letters 2002, 12, 2377-2380. 45. Elsner, J.; Boeckler, F.; Heinemann, F. W.; Hübner, H.; Gmeiner, P. Pharmacophore-Guided Drug Discovery Investigations Leading to Bioactive 5-Aminotetrahydropyrazolopyridines. Implications for the Binding Mode of Heterocyclic Dopmaine D3 Receptor Agonists. Journal of Medicinal Chemistry 2005, 48, 5771-5779. 46. Dijkstra, D.; Rodenhuis, N.; Vermeulen, E. S.; Pugsley, T. A.; Wise, L. D.; Wikström, H. V. Further Characterization of Structural Requirements for Ligands at the Dopamine D2 and D3 Receptor: Exploring the Thiophene Moiety. Journal of Medicinal Chemistry 2002, 45, 3022-3031. 47. Rodenhuis, N.; Timmerman, W.; Wikström, H. V.; Dijkstra, D. Thiophene analogs of naphthoxazines and 2-aminotetralins: bioisosteres with improved relative oral bioavailability, as compared to 5-OH-DPAT. European Journal of Pharmacology 2000, 394, 255-263. 48. Hübner, H.; Haubmann, C.; Utz, W.; Gmeiner, P. Conjugated Enynes as Nonaromatic Catechol Bioisosteres: Synthesis, Binding Experiments, and Computational Studies of Novel Dopamine Receptor Agonists Recognizing Preferentially the D3 Subtype. Journal of Medicinal Chemistry 2000, 43, 756-762. 49. Lenz, C.; Boeckler, F.; Hübner, H.; Gmeiner, P. Fancy bioisosteres: Synthesis, SAR, and pharmacological investigations of novel nonaromatic dopamine D3 receptor ligands. Bioorgganic & Medicinal Chemistry 2005, 13, 4434-4442. 50. Lenz, C.; Haubmann, C.; Hübner, H.; Boeckler, F.; Gmeiner, P. Fancy bioisosteres: synthesis and dopaminergic properties of the endiyne FAUC 88 as a novel non-aromatic D3 agonist. Bioorgganic & Medicinal Chemistry 2005, 13, 185-191. 51. Lenz, C.; Boeckler, F.; Hübner, H.; Gmeiner, P. Analogues of FAUC 73 revealing new insights into the structural requirements of nonaromatic dopamine D3 receptor agonists. Bioorgganic & Medicinal Chemistry 2004, 12, 113-117. 52. Haadsma-Svensson, S. R.; Cleek, K. A.; Dinh, D. M.; Duncan, J. N.; Haber, C. L.; Huff, R. M.; Laijness, M. E.; Nichols, N. F.; Smith, M. W.; Svensson, K. A.; Zaya, M. J.; Carlsson, A.; Lin, C.-H. Dopamine D3 Receptor Antagonists. 1. Synthesis and Sturcture-Activity Relationships of 5,6-Dimethoxy-N-Alkylaryl-Substituted 2-Aminoindanes. Journal of Medicinal Chemistry 2001, 44, 4716-4732. 53. Waters, N.; Svensson, K.; Haadsma-Svensson, S. R.; Smith, M. W.; Carlsson, A. The dopamine D3-receptor: a postsynaptic receptor inhibitory on rat locomotor activity. Journal of Neural Transmission 1993, 94, 11-19. 54. Boyfield, I.; Coldwell, M. C.; Hadley, M. S.; Johnson, C. N.; Riley, G. J.; Scott, E. E.; Stacey, R.; Stemp, G.; Thewlis, K. M. A novel series of 2-aminoteralins with high affinity and selectivity for the dopamine D3 receptor. Bioorgganic & Medicinal Chemistry Letters 1997, 7, 1995-1998. 55. Avenell, K. Y.; Boyfield, I.; Hadley, M. S.; Johnson, C. N.; Nash, D. J.; Riley, G. J.; Stemp, G. Heterocyclic analogues of 2-aminotetralins with high affinity and selectivity for the dopamine D3 receptor. Bioorgganic & Medicinal Chemistry Letters 1999, 9, 2715-2720. 56. Dubuffet, T.; Newman-Tancredi, A.; Cussac, D.; Audinot, V.; Loutz, A.; Millan, M. J.; Lavielle, G. Novel Benzopyrano[3,4-c]pyrrole Derivatives as Potent and Selective Dopamine D3 Receptor Antagonists. Bioorgganic & Medicinal Chemistry Letters 1999, 9, 2059-2064. 57. Austin, N. E.; Avenell, K. Y.; Boyfield, I.; Branch, C. L.; Coldwell, M. C.; Hadley, M. S.; Jeffrey, P.; A., J.; Johnson, C. N.; Nash, D. J.; Riley, G. J.; Smith, S.

Literaturverzeichnis
180
A.; Stacey, R. C.; Stemp, G.; Thewlis, K. M.; Vong, A. K. K. Novel 1,2,3,4-Tetrahydroisoquinolines with High Affinity and Selectivity for the Dopamine D3 Receptor. Bioorgganic & Medicinal Chemistry Letters 1999, 9, 179-184. 58. Avenell, K. Y.; Boyfield, I.; Coldwell, M. C.; Hadley, M. S.; Healy, M. A. M.; Jeffrey, P. M.; Johnson, C. N.; Nash, D. J.; Riley, G. J.; Scott, E. E.; Smith, S. A.; Stacey, R.; Stemp, G.; Thewlis, K. M. Fused aminotetralins: Novel antagonists with high selectivity for the dopamine D3 receptor. Bioorgganic & Medicinal Chemistry Letters 1998, 8, 2859-2864. 59. Mach, U. R.; Hackling, A. E.; Perachon, S.; Ferry, S.; Wermuth, C. G.; Schwartz, J.-C.; Sokoloff, P.; Stark, H. Development of Novel 1,2,3,4-Tetrahydroisoquinoline Derivatives and Closely Related Compounds as Potent and Selective Dopamine D3 Receptor Ligands. ChemBioChem 2004, 5, 508-518. 60. Jones, J. H.; Anderson, P. S.; Baldwin, J. J.; Clineschmidt, B. V.; McClure, D. E.; Lundell, G. F.; Randall, W. C.; Martin, G. E.; Williams, M.; Hirshfield, J. M.; Smith, G.; Lumma, P. K. Synthesis of 4-Substituted 2H-Naphth[1,2-b]-1,4-oxazines, a New Class of Dopamine Agonists. Journal of Medicinal Chemistry 1984, 27, 1607-1613. 61. Stemp, G.; Ashmeade, T.; Branch, C. L.; Hadley, M. S.; Hunter, A. J.; Johnson, C. N.; Nash, D. J.; Thewlis, K. M.; Vong, A. K. K.; Austin, N. E.; Jeffrey, P.; Avenell, K. Y.; Boyfield, I.; Hagan, J. J.; Middlemiss, D. N.; Reavill, C.; Riley, G. J.; Routledge, C.; Wood, M. Design and Synthesis of trans-N-[4-[2-(6-Cyano-1,2,3,4-tetrahydroisoquinolin-2-yl)ethyl]cyclohexyl]-4-quinolinecarboxamide (SB-277011): A Potent and Selective Dopamine D3 Receptor Antagonist with High Oral Bioavailability and CNS Penetration in the Rat. Journal of Medicinal Chemistry 2000, 43, 1878-1885. 62. Austin, N. E.; Avenell, K. Y.; Boyfield, I.; Branch, C. L.; Hadley, M. S.; Jeffrey, P.; Johnson, C. N.; Macdonald, G. J.; Nash, D. J.; Riley, G. J.; Smith, A. B.; Stemp, G.; Thewlis, K. M.; Vong, A. K. K.; Wood, M. Novel 2,3,4,5-Tetrahydro-1H-3-benzazepines with High Affinity and Selectivity for the Dopamine D3 Receptor. Bioorgganic & Medicinal Chemistry Letters 2000, 10, 2553-2555. 63. Austin, N. E.; Avenell, K. Y.; Boyfield, I.; Branch, C. L.; Hadley, M. S.; Jeffrey, P.; Johnson, C. N.; Macdonald, G. J.; Nash, D. J.; Riley, G. J.; Smith, A. B.; Stemp, G.; Thewlis, K. M.; Vong, A. K. K.; Wood, M. Design and Synthesis of Novel 2,3-Dihydro-1H-isoindoles with High Affinity and Selectivity for the Dopamine D3 Receptor. Bioorgganic & Medicinal Chemistry Letters 2001, 11, 685-688. 64. Macdonald, G. J.; Branch, C. L.; Hadley, M. S.; Johnson, C. N.; Nash, D. J.; Smith, A. B.; Stemp, G.; Thewlis, K. M.; Vong, A. K. K.; Austin, N. E.; Jeffrey, P.; Winborn, K. Y.; Boyfield, I.; Hagan, J. J.; Middlemiss, D. N.; Reavill, C.; Riley, G. J.; Watson, J. M.; Wood, M.; Parker, S. G.; Ashby, J., C. R. Design and Synthesis of trans-3-(2-(4-((3-(3-(5-Methyl-1,2,4-oxadiazolyl))-phenyl)carboxamido)cyclohexyl)ethyl)-7-methylsulfonyl-2,3,4,5-tetrahydro-1H-3-benzazepine (SB-414796): A Potent and Selective Dopamine D3 Receptor Antagonist. Journal of Medicinal Chemistry 2003, 46, 4952-4964. 65. Homan, E. J.; Copinga, S.; Elfström, L.; van der Veen, T.; Hallema, J.-P.; Mohell, N.; Unelius, L.; Johansson, R.; Wikström, H. V.; Grol, C. J. 2-Aminotetralin-derived Substitued Benzamides with Mixed Dopamine D2, D3, and Serotonin 5-HT1A Receptor Binding Properties: A Novel Class of Potential Atypical Antipsychotic Agents. Bioorgganic & Medicinal Chemistry 1998, 6, 2111-2126. 66. Homan, E. J.; Kroodsma, E.; Copinga, S.; Unelius, L.; Mohell, N.; Wikström, H. V.; Grol, C. J. Structural Analogues of 5-OMe-BPAT: Synthesis and Interactions with Dopamine D2, D3, and Serotonin 5-HT1A Receptors. Bioorgganic & Medicinal Chemistry 1999, 7, 1111-1121.

Literaturverzeichnis
181
67. Homan, E. J.; Copinga, S.; Unelius, L.; Jackson, D. M.; Wikström, H. V.; Grol, C. J. Synthesis and Pharmacology of the Enantiomers of the Potential Atypical Antipsychotic Agents 5-OMe-BPAT and 5-OMe-(2,9-di-OMe)-BPAT. Bioorgganic & Medicinal Chemistry 1999, 7, 1263-1271. 68. Micheli, F.; Bonanomi, G.; Blaney, F. E.; Braggio, S.; Capelli, A. M.; Checchia, A.; Curcuruto, O.; Damiani, F.; Di Fabio, R.; Donati, D.; Gentile, G.; Gribble, A.; Hamprecht, D.; Tedesco, G.; Terreni, S.; Tarsi, L.; Lightfoot, A.; Stemp, G.; MacDonald, G.; Smith, A.; Pecoraro, M.; Petrone, M.; Perini, O.; Piner, J.; Rossi, T.; Worby, A.; Pilla, M.; Valerio, E.; Griffante, C.; Mugnaini, M.; Wood, M.; Scott, C.; Andreoli, M.; Lacroix, L.; Schwarz, A.; Gozzi, A.; Bifone, A.; Ashby, J., C. R.; Hagan, J. J.; Heidbreder, C. 1,2,4-Triazol-3-yl-thiopropyl-tetrahydrobenzazepines: A Series of Potent and Selective Dopamine D3 Receptor Antagonists. Journal of Medicinal Chemistry 2007, 50, 5076-5089. 69. Micheli, F.; Bonanomi, G.; Braggio, S.; Capelli, A. M.; Celestini, P.; Damiani, F.; Di Fabio, R.; Donati, D.; Gagliardi, S.; Gentile, G.; Hamprecht, D.; Petrone, M.; Radaelli, S.; Tedesco, G.; Terreni, S.; Worby, A.; Heidbreder, C. New fused benzazepine as selective D3 receptor antagonists. Synthesis and biological evaluation. Part one: [h]-fused tricyclic systems. Bioorgganic & Medicinal Chemistry Letters 2008, 18, 901-907. 70. Micheli, F.; Bonanomi, G.; Braggio, S.; Capelli, A. M.; Damiani, F.; Di Fabio, R.; Donati, D.; Gentile, G.; Hamprecht, D.; Perini, O.; Petrone, M.; Tedesco, G.; Terreni, S.; Worby, A.; Heidbreder, C. New fused benzazepine as selective D3 receptor antagonists. Synthesis and biological evaluation. Part 2: [g]-Fused and hetero-fused systems. Bioorgganic & Medicinal Chemistry Letters 2008, 18, 908-912. 71. Thomas, C.; Hübner, H.; Gmeiner, P. Enantio- and Diastereocontrolled Dopamine D1, D2, D3 and D4 Receptor Binding of N-(3-Pyrrolidinylmethyl)benzamides Synthesized from Aspartic Acid. Bioorgganic & Medicinal Chemistry Letters 1999, 9, 841-846. 72. Huang, Y.; Luedtke, R. R.; Freeman, R. A.; Wu, L.; Mach, R. H. Synthesis and Structure-Activity Relationships of Naphthamides as Dopamine D3 Receptor Ligands. Journal of Medicinal Chemistry 2001, 44, 1815-1826. 73. Bolton, D.; Boyfield, I.; Coldwell, M. C.; Hadley, M. S.; Healy, M. A. M.; Johnson, C. N.; Markwell, R. E.; Nash, D. J.; Riley, G. J.; Stemp, G.; Wadsworth, H. J. Novel 2,5-Disubstituted-1H-Pyrroles with high affinity fpr the Dopamine D3 Receptor. Bioorgganic & Medicinal Chemistry Letters 1996, 6, 1233-1236. 74. Huang, Y.; Luedtke, R. R.; Freeman, R. A.; Wu, L.; Mach, R. H. Synthesis of 2-(2,3-dimethoxyphenyl)-4-(aminomethyl)imidazole Analogues and their Binding Affinities for Dopamine D2 and D3 Receptors. Bioorgganic & Medicinal Chemistry 2001, 9, 3113-3122. 75. Mach, R. H.; Huang, Y.; Freeman, R. A.; Wu, L.; Blair, S.; Luedtke, R. R. Synthesis of 2-(5-Bromo-2,3-dimethoxyphenal)-5-(aminomehtyl)-1H-pyrrole Analogues and Their Binding Affinities for Dopamine D2, D3, and D4 Receptors. Bioorgganic & Medicinal Chemistry 2003, 11, 225-233. 76. Einsiedel, J.; Thomas, C.; Hübner, H.; Gmeiner, P. Phenyloxazoles and Phenylthiazoles as Benzamide Bioisosteres: Synthesis and Dopamine Receptor Binding Profiles. Bioorgganic & Medicinal Chemistry Letters 2000, 10, 2041-2044. 77. Glase, S. A.; Akunne, H. C.; Heffner, T. G.; Johnson, S. J.; Kesten, S. R.; MacKenzie, R. G.; Manley, P. J.; Pugsley, T. A.; Wright, J. L.; Wise, L. D. 4-Bromo-1-Methoxy-N-[2-(4-Aryl-1-Piperazinyl)ethyl]-2-Naphthalencarboxamides: Selective Dopamine D3 Receptor Partial Agonists. Bioorgganic & Medicinal Chemistry Letters 1996, 6, 1361-1366.

Literaturverzeichnis
182
78. Robarge, M. J.; Husbands, S. M.; Kieltyka, A.; Brodbeck, R.; Thurkauf, A.; Newman, A. H. Design and Synthesis of [(2,3-Dichlorophenyl)piperazin-1-yl]alkylfluorenylcarboxamides as Novel Ligands Selective for the Dopamine D3 Receptor Subtype. Journal of Medicinal Chemistry 2001, 44, 3175-3186. 79. Bettinetti, L.; Schlotter, K.; Hübner, H.; Gmeiner, P. Interactive SAR Studies: Rational Discovery of Super-Potent and Highly Selective Dopamine D3 Receptor Antagonists and Partial Agonists. Journal of Medicinal Chemistry 2002, 45, 4594-4597. 80. Hocke, C.; Prante, O.; Löber, S.; Hübner, H.; Gmeiner, P.; Kuwert, T. Synthesis and evaluation of 18F-labeled dopamine D3 receptor ligands as potantial PET imaging agents. Bioorgganic & Medicinal Chemistry Letters 2005, 15, 4819-4823. 81. Schlotter, K.; Boeckler, F.; Hübner, H.; Gmeiner, P. Fancy Bioisosteres: Metallocene-Derived G-Protein-Coupled Receptor Ligands with Subnanomolar Binding Affinity and Novel Selectivity Profiles. Journal of Medicinal Chemistry 2005, 48, 3696-3699. 82. Schlotter, K.; Boeckler, F.; Hübner, H.; Gmeiner, P. Fancy Bioisosteres: Novel Paracyclophane Derivatives As Super-Affinity Dopamine D3 Receptor Antagonists. Journal of Medicinal Chemistry 2006, 49, 3628-3635. 83. Leopoldo, M.; Berardi, F.; Colabufo, N. A.; De Giorgio, P.; Lacivita, E.; Perrone, R.; Tortorella, V. Structure-Affinity Relationship Study on N-[4-(4-Arylpiperazin-1-yl)butyl]arylcarboxamides as Potent and Selective Dopamine D3 Receptor Ligands. Journal of Medicinal Chemistry 2002, 45, 5727-5735. 84. Geneste, H.; Backfisch, G.; Braje, W.; Delzer, J.; Haupt, A.; Hutchins, C. W.; King, L. J.; Kling, A.; Teschendorf, H.-J.; Unger, L.; Wernet, W. Synthesis and SAR of highly potent and selective dopamine D3-receptor antagonists: 1H-Pyrimidin-2-one derivatives. Bioorgganic & Medicinal Chemistry Letters 2006, 16, 490-494. 85. Geneste, H.; Backfisch, G.; Braje, W.; Delzer, J.; Haupt, A.; Hutchins, C. W.; King, L. L.; Lubisch, W.; Steiner, G.; Teschendorf, H.-J.; Unger, L.; Wernet, W. Synthesis and SAR of highly potent and selective dopamine D3-receptor antagonists: Quinolin(di)one and benzazepin(di)one derivatives. Bioorgganic & Medicinal Chemistry Letters 2006, 16, 658-662. 86. Murray, P. J.; Harrison, L. A.; Johnson, M. R.; Robertson, G. M.; Scopes, D. I. C.; Bull, D. R.; Graham, E. A.; Hayes, A. G.; Kilpatrick, G. J.; Den Daas, I.; Large, C.; Sheehan, M. J.; Stubbs, C. M.; Turpin, M. P. A Novel Series of Arylpiperazines with High Affinity and Selectivity for the Dopamine D3 Receptor. Bioorgganic & Medicinal Chemistry Letters 1995, 5, 219-222. 87. Newman, A. H.; Cao, J.; Bennett, C. J.; Robarge, M. J.; Freeman, R. A.; Luedtke, R. R. N-{4-[4-(2,3-Dichlorophenyl)piperazin-1-yl]butyl, Butenyl and Butynyl}arylcarboxamides as Novel Dopamine D3 Receptor Antagonists. Bioorgganic & Medicinal Chemistry Letters 2003, 13, 2179-2183. 88. Grundt, P.; Prevatt, K. M.; Cao, J.; Taylor, M.; Floresca, C. Z.; Choi, J.-K.; Jenkins, B. G.; Luedtke, R. R.; Newman, A. H. Heterocyclic Analogues of N-(4-(4-(2,3-Dichlorophenyl)piperazin-1-yl)butyl)arylcarboxamides with Functionalized Linking Chains as Novel Dopamine D3 Receptor Ligands: Potential Substance Abuse Therapeutic Agents. Journal of Medicinal Chemistry 2007, 50, 4135-4146. 89. Leopoldo, M.; Enza, L.; Colabufo, N. A.; Berardi, F.; Perrone, R. Synthesis and binding profile of constrained analogues of N-[4-(4-arylpiperazin-1-yl)butyl]-3-methoxybenzamides, a class of potent dopamine D3 receptor ligands. Journal of Pharmacy and Pharmacology 2006, 58, 209-218.

Literaturverzeichnis
183
90. Ding, K.; Chen, J.; Ji, M.; Wu, X.; Varady, J.; Yang, C.-Y.; Lu, Y.; Deschamps, J. R.; Levant, B.; Wang, S. Enantiomerically Pure Hexahydropyrazinoquinolines as Potent and Selective Dopamine 3 Subtype Receptor Ligands. Journal of Medicinal Chemistry 2005, 48, 3171-3181. 91. Hackling, A.; Ghosh, R.; Perachon, S.; Mann, A.; Höltje, H.-D.; Wermuth, C. G.; Schwartz, J.-C.; Sippl, W.; Sokoloff, P.; Stark, H. N-(ω-(4-(Methoxyphenyl)piperazin-1-yl)alkyl)carboxamides as Dopamine D2 and D3 Receptor Ligands. Journal of Medicinal Chemistry 2003, 46, 3883-3899. 92. Grundt, P.; Carlson, E. E.; Cao, J.; Bennett, C. J.; McElveen, E.; Taylor, M.; Luedtke, R. R.; Newman, A. H. Novel Heterocyclic Trans Olefin Analogues of N-{4-[4-(2,3-Dichlorophenyl)piperazin-1-yl]butyl}arylcarboxamides as Selective Probes with High Affinity for the Dopamine D3 Receptor. Journal of Medicinal Chemistry 2005, 48, 839-848. 93. Yuan, J.; Chen, X.; Brodbeck, R.; Primus, R.; Braun, J.; Wasley, J. W. F.; Thurkauf, A. NGB 2904 and NGB 2849: Two Highly Selective Sopamine D3 Receptor Antagonists. Bioorgganic & Medicinal Chemistry Letters 1998, 8, 2715-2718. 94. Chu, W.; Tu, Z.; McElveen, E.; Xu, J.; Taylor, M.; Luedtke, R. R.; Mach, R. H. Synthesis and in vitro binding of N-phenyl piperazine analogs as potential dopamine D3 receptor ligands. Bioorgganic & Medicinal Chemistry 2005, 13, 77-87. 95. Pilla, M.; Perachon, S.; Sautel, F.; Garrido, F.; Mann, A.; Wermuth, C. G.; Schwartz, J.-C.; Everitt, B. J.; Sokoloff, P. Selective inhibiton of cocaine-seeking behavior by a partial dopamine D3 receptor agonist. Nature 1999, 400, 371-375. 96. Boeckler, F.; Gmeiner, P. Dopamine D3 receptor ligands - Recent advances in the control of subtype selectivity and intrinsic activity. Biochimica et Biophysica Acta - Biomembranes 2007, 1768, 871-887. 97. Wu, X.; Chen, J.; Ji, M.; Varady, J.; Levant, B.; Wang, S. Design, synthesis, and evaluation of hexahydrobenz[f]isoquinolines as a novel class of dopamine 3 receptor ligands. Bioorgganic & Medicinal Chemistry Letters 2004, 14, 5813-5816. 98. Ji, M.; Chen, J.; Ding, K.; Wu, X.; Varady, J.; Levant, B.; Wang, S. Design, synthesis and structure-activity relationship studies of hexahydropyrazinoquinolines as a novel class of potent and selective dopmaine receptor 3 (D3) ligands. Bioorgganic & Medicinal Chemistry Letters 2005, 15, 1701-1705. 99. Cha, M. Y.; Choi, B. C.; Kang, K. H.; Pae, A. N.; Choi, K. I.; Cho, Y. S.; Koh, H. Y.; Lee, H.-Y.; Jung, D.; Kong, J. Y. Design and Synthesis of a Piperazinylalkylisoxazole Library for Subtype Selective Dopamine Receptor Ligands. Bioorgganic & Medicinal Chemistry Letters 2002, 12, 1327-1330. 100. Dutta, A. K.; X.-S., F.; Reith, M. E. A. A Novel Series of Hybrid Compounds Derived by Combining 2-Aminotetralin and Piperazine Fragments: Binding Activity at D2 and D3 Receptors. Bioorgganic & Medicinal Chemistry Letters 2002, 12, 619-622. 101. Dutta, A. K.; Venkataraman, S. K.; Fei, X.-S.; Kolhatkar, R.; Zhang, S.; Reith, M. E. A. Synthesis and biological characterization of novel hybrid 7-{[2-(4-phenyl-piperazin-1-yl)-ethyl]-propyl-amino}-5,6,7,8-tetrahydronaphthalen-2-ol and their heterocyclic bioisosteric analogues for dopamine D2 and D3 receptors. Bioorgganic & Medicinal Chemistry 2004, 12, 4361-4373. 102. Stemp, G.; Johnson, C. N. Bicyclic amine derivatives and their use as anti-psychotic agents. 1996. 103. Boeckler, F.; Ohnmacht, U.; Lehmann, T.; Utz, W.; Hübner, H.; Gmeiner, P. CoMFA and CoMSIA Investigations Revealing Novel Insights into the Binding Modes of Dopamine D3 Receptor Agonists. Journal of Medicinal Chemistry 2005, 48, 2493-2508.

Literaturverzeichnis
184
104. Andersson, B. R.; Carlsson, P. A. E.; Hansson, L. O.; Sonesson, C. A.; Stjernlof, N. P.; Svensson, K. A. I.; Waters, R. N.; Haadsma-Svensson, S. R. Indoletetralins having dopaminergic activity. 1997. 105. Cram, D. J.; Day, A. C. Macro Rings. XXXI. Quinone Derived from [2.2]Paracyclophane, an Intramolecular-Molecular Complex. Journal of Organic Chemistry 1966, 31, 1227-1232. 106. Rozenberg, V.; Dubrovina, N.; Sergeeva, E.; Antonov, D.; Belokon, Y. An improved synthesis of (S)-(+)- and (R)-(-)-[2.2]paracyclophane-4-carboxylic acid. Tetrahedron: Asymmetry 1998, 9, 653-656. 107. Wenzel, N. Synthese und SAR-Studien selektiver, dopaminerger D3-Agonisten mit Arencarboxamidstruktur. Friedrich-Alexander-Universität Erlangen-Nürnberg, Erlangen, 2005. 108. Pandit, U. K.; Das, B.; Chatterjee, A. Synthetic Entry into Yohimbinoid Alkaloids and Novel Synthesis of (+-)-17-Methoxy-Hexadehydroyohimabne. Tetrahedron 1987, 43, 4235-4239. 109. Chatterjee, A.; Ghosh, S. Syntheses of (+-)-2,3-Dimethoxyberbine, (+-)-Norcoralyine, and (+-)-Demethoxycarbonyldihydrogambirtannine. Synthesis 1981, 818-820. 110. Aoyama, T.; Shioiri, T. New Methods and Reagents in Organic Synthesis. 17. Trimethylsilyldiazomethane (TMSCHN2) as a Stable and Safe Substitute for Hazardous Diazomethane. Its Application to the Arndt-Eistert Synthesis. Chem. Pharm. Bull. 1981, 29, 3249-3255. 111. Cesar, J.; Sollner Dolenc, M. Trimethylsilyldiazomethane in the preparation of diazoketones via mixed anhydride and coupling reagent methods: a new approach to the Arndt-Eistert synthesis. Tetrahedron Letters 2001, 42, 7099-7102. 112. Shon, Y.-S.; Kelly, K. F.; Halas, N. J.; Lee, T. R. Fullerene-Terminated Alkanethiolate SAMs on Gold Generated from Unsymmetrical Disulfides. Langmuir 1999, 15, 5329-5332. 113. Pascual Coca, G.; Martín Juárez, J. Method of obtaining 2-amino-6-alkyl-amino-4,5,6,7-tetrahydrobenzothiazoles. 2006. 114. Lenz, C. Dopaminerge Enine als nichtaromatische Katecholbioisostere. Friedrich-Alexander-Universität Erlangen-Nürnberg, Erlangen, 2003. 115. Elsner, J. 7a-Azaindole aus Pyridinderivaten: Synthese, Bioisosterie und Struktur-Wirkungs-Untersuchungen. Friedrich-Alexander-Universität Erlangen-Nürnberg, Erlangen, 2005. 116. Cannon, J. G.; Perez, J. A.; Pease, J. P.; Long, J. P.; Flynn, J. R.; Rusterholz, D. B.; Dryer, S. E. Comparison of Biological Effects of N-Alkylated Congeners of β-Phenethylamine Derived from 2-Aminotetralin, 2-Aminoindan, and 6-Aminobenzocycloheptene. Journal of Medicinal Chemistry 1980, 23, 745-749. 117. Gomes Faraco, A. A.; Prado, M. A. F.; Alves, R. J.; Souza Filho, J. D.; Brondi Alves, R.; Prado Faraco, R. F. Synthesis of Benzomacrolactam by 12-endo Selective Aryl Radical Cyclization of N-(4-Allyloxybutyl)-2-iodobenzamide. Synthetic Communications 2003, 33, 463-474.

Abkürzungsverzeichnis
185
11. Abkürzungsverzeichnis
5-HT 5-Hydroxytryptamin, Serotonin
[α]Dx Drehwert gemessen bei der Temperatur x
µ mikro
Abb. Abbildung
APCI atmospheric pressure chemical ionization
Ber berechnet
BPAT 2-[N-(2-Benzamidoethyl)-N-n-propylamino]tetraline
BP-897 N-[4-[4-(2-methoxyphenyl)-1-piperazinyl]butyl]-2-
Naphthylcarboxamide
br. breit
n-BuLi n-Butyllithium
tert-BuOH tert-Butanol
bzw. Beziehungsweise
CH2Cl2 Dichlormethan
CH3CN Acetonitril
CHN Elementaranalyse
CHO-Zellen chinese hamster ovary cells
d Duplett
D Dopamin
dd doppeltes Duplett
DEPC Diethylcyanophosphonat
DC Dünnschicht-Chromatographie
DIPEA Diisopropylethylamin
DPAT 2-Di-n-propylaminotetralin
DTG 1,3-Di-(2-tolyl)-guanidin
DMF Dimethylformamid
EC50 mittlere effektive Konzentration
EI Elektronenstoßinoisation
EtOAc Ethylacetat, Essigsäureethylester
Et2O Diethylether
Gef gefunden
FT/IR Fourier-Transformation-Infrarotspektroskopie

Abkürzungsverzeichnis
186
GPCR G-Protein gekoppelter Rezeptor
h Stunde
HATU O-(7-Azabenzotriazol-1-yl)-N,N,N',N'-tetramethyl-
uronium hexafluorophosphate
HCOOH Ameisensäure
HEK humane Embryo-Nieren(kidney)-Zellen
HOAc Essigsäure
HPLC High-Performance-Liquid-Chromatography
HR high resolution, Hochauflösung
IABN Iod-2,3-dimethoxy-N-[9-(4-fluorobenzyl)-9-azabicyclo
[3.3.1]-nonan-3�-yl]benzamide
IR Infrarotspektroskopie
IC50 mittlere inhibitorische Konzentration
Lit. Literatur
Ltk- Thymidin-Kinase-frei Maus-L-Zellen
m Multiplett (NMR), medium, mittel-stark (IR)
MeOH Methanol
MHZ Megahertz
min Minute
Ms Mesyl-, Methansulfonyl-
MS Massenspektrometrie
n nano
nM nanomolar
N-0437 Rotigotin (6)
Na Natrium
nM nanomolar
NMP N-Methylpyrrolidon
NMR Nuclear-Magnetic-Resonance Spektroskopie
NPA N-Propylapomorphin
OMe Methoxy-Gruppe
PD 128907 3,4,4a,10b-tetrahydro-4-propyl-2H,5H-(1)benzopyrano-
(4,3-b)-1,4-oxazin-9-ol
Ph Phenyl
PIPAT (R)-(+)-trans-2-[N-propyl-N-(3’-iodpropen-2’-yl)amino]-

Abkürzungsverzeichnis
187
Tetralin
quant. quantitativ
Reflux Rückflussbedingungen
RT Raumtemperatur
s strong, stark
SCH23390 R-(+)-8-chloro-2,3,4,5-tetrahydro-3-methyl-5-phenyl-1H-
3-benzazepine-7-ol
Sf9-Zellen Zellen aus dem Ovargewebe von Spodoptera frugi
perda (Nachtfalter)
Schmp. Schmelzpunkt
t Triplett
tR Retentionszeit
Tab. Tabelle
TBTU O-(Benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium
tetrafluoroborate
Tf Triflyl-, Trifluormethansulfonyl-
TFA Trifluoressigsäure
THF Tetrahydrofuran
TM transmembranäre Domäne/Helix
TMS Trimethylsilyl-
U-86170 / PNU 86170 5-(Dipropylamino)-5,6-dihydro-4H-imidazo-(5,1ij)quino
lin-2(1H)-on
U 99194 5,6-Dimethoxy-2-(dipropylamino)-indan
vgl. vergleiche
vs. versus
w weak, schwach

Lebenslauf
188
12. Lebenslauf Persönliche Daten Vornamen Miriam Silke
Nachname Dörfler
Geburtstag 01. August 1979
Geburtsort Bayreuth
Familienstand ledig
Schulischer Werdegang 09/1986 – 08/1990 Graser-Grundschule, Bayreuth
09/1990 – 06/1999 Städtisches Wirtschaftswissenschaftliches und Mathema-
tisch-naturwissenschaftliches Gymnasium Bayreuth (mathe-
matisch-naturwissenschaftlicher Zweig)
06/1999 Abitur
Hochschulstudium
10/1999 – 09/2003 Studium der Pharmazie an der Friedrich-Alexander-Univer-
sität Erlangen-Nürnberg
08/2001 Erster Abschnitt der Pharmazeutischen Prüfung
10/2003 Zweiter Abschnitt der Pharmazeutischen Prüfung
Praktisches Jahr
11/2003 – 04/2004 1. PJ-Hälfte in der Markt-Apotheke, Bayreuth
05/2004 – 10/2004 2. PJ-Hälfte in der medizinisch-wissenschaftlichen Abteilung
der HEXAL Pharmaforschung GmbH, Holzkirchen
12/2004 Dritter Abschnitt der Pharmazeutischen Prüfung
und Approbation als Apothekerin
Promotion Seit 01/2005 Wissenschaftliche Mitarbeiterin am Lehrstuhl für Pharma-
zeutische Chemie der Friedrich-Alexander-Universität
Erlangen-Nürnberg unter der Leitung von Herrn Prof. Dr. P.
Gmeiner