symposium-in-print : bioluminescence: similar chemistries but many different evolutionary origins
TRANSCRIPT

Photochemistry and Photobiology, Vol. 62, No. 4, pp. 599600, 1995 Printed in the United States. All rights reserved 003 1-8655/95 $05.00+0.0C
0 1995 American Society for Photobiologj
SYMPOSIUM-IN-PRINT
BIOLUMINESCENCE: SIMILAR CHEMISTRIES BUT MANY DIFFERENT EVOLUTIONARY ORIGINS
J. WOODLAND HASTINGS Department of Molecular and Cellular Biology, Harvard University,
16 Divinity Avenue, Cambridge, MA 02138-2020, USA
Light emission occurs in many different species in phylo- genetically diverse groups.’,2 Among the different groups, the type and method of display of the light, its color3 and its function may be very different. These facts, along with biochemical studies, led workers already 50 years ago to conclude what is now firmly established: bioluminescent systems in different major groups are not evolutionarily con- served: genes coding for the proteins (e.g. luciferases) are not homol~gous .~ Luciferase (the enzyme) and luciferin (the substrate) are thus generic terms, such that the organism from which they are obtained must be specified.
However, at the chemical level all bioluminescent systems have a great deal in common. All known luciferases are oxy- genases that utilize molecular oxygen to oxidize a luciferin with formation of an intermediate enzyme-bound peroxide, whose breakdown then results in the production of an inter- mediate or product directly in its excited singlet state. A general mechanism for this chemiexcitation step remains elusive. In the first paper, Wilson confronts some of the dif- ficulties encountered in the interpretation of model chemical systems on the basis of electron transfer processes, as pro- posed in the popular chemically initiated electron exchange (CIEEL) hypothe~is .~ ,~
Mager and Tu consider CIEEL and the mechanisms of chemiexcitation in bioluminescence in some detail, based mostly on an extensive consideration of the flavin-aldehyde bacterial luciferase system. In many respects this system is certainly the most extensively studied one. Luciferase sub- units7 and the flavin-peroxide intermediateX were isolated some years ago, and the emitter has been identified9; a crys- tal structure has been recently reported.IO In the third paper T u and Mager provide an extensive review and critical eval- uation of the biochemistry and enzymology of this system.
The observation that bioluminescence and luciferase are expressed only in denser cultures led to the discovery of the phenomenon of autoinduction,” now known to occur as a more general mechanism for the regulation of specific genes in several different species, both luminous and nonlumi- nous.I2 With the cloning of the lux operon, insight into the genetic and molecular regulation of bacterial biolumines- cence has emerged, as summarized by Ulitzur and Dunlap.
Most species of luminous bacteria have been found to occur as symbionts, providing a light system for higher or- ganisms, so whether or not they have a “life of their own” can be debated.I3 Most such species are also marine. By contrast, no luminous bacterium has ever been isolated from
a soil sample. But a terrestrial (low salt-requiring) species (Photorhabdus luminescens) does occur, and with the ex- ception of a few isolates obtained from human wounds,I4 all of the known P. luminescens strains have been obtained as symbionts of soil nematodes. These in turn parasitize insects, using the luminous bacterium as the infective agent. Hos- seini and Nealson briefly review this fascinating system and report an intriguing new finding: the inhibition of mFWA synthesis by rifampicin leads to an increase in luminescence in growing cells.
Several different luminescence systems have been devel- oped and exploited for analytical purposes. The earliest such applications involved using a luciferase system for the sim- ple (albeit very sensitive and very specific) detection of sub- stance required in the reaction or linked chemically to some- thing required in the rea~t i0n . l~ The firefly ATP assay gained wide acceptance, and aequorin from jellyfish became widely used for the intracellular measurement of calcium and its changes in real time (e.g. during muscle contraction). The use of luciferase systems for reporting gene expression rep- resents a very different approach, and several luminescence genes have been engineered for such purposes. Chatterjee and Meighen review the current status of work in this area with bacterial lux genes.
The green fluorescent protein (GFP), recently developed as a reporter for gene expression,16 is not a luciferase but is an accessory fluorescent protein that serves as the emitter for an associated luminescence system in coelenterates.”,’” Its unique and powerful feature is that it requires no substrate and can be visualized by its fluorescence in both live and fixed material. The chromophore is a part of the protein it- self, being derived from the primary amino acid sequence i t ~ e l f ~ ~ . ~ ~ and is formed even when the protein is expressed in a heterologous system.I6 Chalfie provides an overview and an update on this now widely used reporter gene.
Although luciferases from different groups possess no se- quence similarities, two (coelenterate and beetle) have been found to have relationships to other known proteins. In the coelenterate system, as reviewed by Tsuji et al., the Ca2+ binding sites of several hydrozoan luciferases are highly conserved and similar in sequence to other Ca2+ binding pro- teins such as calmodulin. Aequorin and other similar pro- teins, and also several proteins isolated from other unrelated organisms, were named ‘‘photoproteins.”2/ Aequorin and re- lated proteins can be isolated from the organism in an active form in the presence of a calcium chelator and will give

J. WOODLAND HASTINGS 600
bioluminescence simply upon the addition of calcium; in- deed oxygen is not required. This unusual situation and other considerations led to the incorrect view that aequorin is not an enzyme.zz In later experiments it was shown that if fresh substrate is mixed with spent aequorin (apoaequorin) in the absence of calcium, aequorin is regenerated in a reaction requiring oxygen.23 Aequorin may thus be viewed as a stable enzyme-bound peroxide intermediate whose further reaction is triggered by calcium. The quasistable bacterial luciferase peroxide intermediate is analogous and has all the properties of a “photoprotein”24; instead of Ca2+ it requires aldehyde for its further reaction. The view expressed in the paper by Tsuji e t al. that in the course of the reaction ‘ I . . . aequorin undergoes a conformational change, converting itself into an oxygenase (luciferase) . . .” should thus be viewed with cau- tion. Enzymes can have different conformational states pos- sessing different properties.
Wood concludes the symposium with a review of the beetle system: the luciferase reaction mechanism, its evolu- tionary origin from nonluminescent enzymes, and the bio- chemical basis for differences in the color of light emitted. A key discovery in this system is that natural polymorphism in the luminescent color is caused by amino acid substitu- tions within the structures of the luciferases. Wood et u Z . ~ ~ isolated cDNA clones from a luminous elaterid beetle en- coding four luciferase isozymes; expressed in Escherichia cnli and presented with substrate, the four emitted lumines- cence with spectral maxima at 545, 560, 578 and 592 nm. With regard to evolutionary origins, amino acid sequences are known for luciferases of the three major families of lu- minous beetles. These sequences have significant similarity with six groups of nonluminescent enzymes, all of which catalyze thiol ester formation with coenzyme A (CoA). Al- though beetle luciferase does not require CoA, the unex- plained effect of CoA on the luminescent reaction is prob- ably due to the evolution of this system from ancestral CoA synthetases.
I hope that these several contributions in this exciting field will be useful and stimulating both to workers directly in the field and to those in related and other areas.
REFERENCES
1. Hastings, J . W. and J. G. Morin (1991) Bioluminescence. In Neural and Integrative Animal Physiology (Edited by C. L. Prosser), pp. 131-170. Wiley Interscience, New York.
2. Hastings. J. W. (1995) Bioluminescence. In CelZ Physiology (Edited by N. Sperelakis), pp. 665481 . Academic Press, New York.
3. Hastings, J. W. (1995) Chemistries and colors of bioluminescent reactions. Gene. (In press)
4. Hastings, J. W. (1983) Biological diversity, chemical mecha- nisms and evolutionary origins of bioluminescent systems. J . Mol. Evol. 19, 309-32 1.
5. Schuster, G. B. (1979) Chemiluminescence of organic perox- ides. Conversion of ground-state reactants to excited-state prod- ucts by the chemically initiated electron-exchange luminescence mechanism. Acct. Chem. Res. 12, 366373.
6. Catalini, L. H. and T. Wilson (1989) Electron transfer and che-
7 .
8.
9.
10.
11.
12
13
14
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
miluminescence. Two inefficient systems: 1,4-dimethoxy-9, 10- diphenylanthracene peroxide and diphenoyl peroxide. J . Am. Chem. Soc. 111, 2633-2639. Friedland, J. M. and J. W. Hastings (1967) The non-identical subunits of bacterial luciferase: their isolation and recombina- tion to form active enzyme. Proc. Natl. Acad. Sci. USA 58, 2336-2342. Hastings, J. W., C. Balny, C. Le Peuch and P. Douzou (1973) Spectral properties of an oxygenated luciferase-flavin interme- diate isolated by low-temperature chromatography. Proc. Natl. Acad. Sci. USA 70, 3468-3472. Kurfurst, M., S. Ghisla and J. W. Hastings (1984) Characteriza- tion and postulated structure of the primary emitter in the bac- terial luciferase reaction. Proc. Natl. Acad. Sci. USA 81, 2990- 2994. Fisher, A. J. , F. M. Raushel, T. 0. Baldwin and I . Rayment (1995) Three dimensional structure of bacterial luciferase from Vibrio harveyi at 2.4 A resolution. Biochemistry 34, 6581-6586. Nealson, K., T. Platt and J. W. Hastings (1970) The cellular control of the synthesis and activity of the bacterial luminescent system. J . Bacteriol. 104, 313-322. Fuqua, W. C., S. C. Winans and E. P. Greenberg (1994) Quo- rum sensing in bacteria: the IuxR-lux1 family of cell density- responsive transcriptional regulators. J. Bacreriol. 176, 269- 275. Nealson, K. and J. W. Hastings (1991) The luminous bacteria. In The Prokaq>ores, 2nd ed., Vol. 1, part 2, chapter 25 (Edited by A. Balows, H. G. Triiper, M. Dworkin, W. Harder and K. H. Schleifer), pp. 625-639. Springer-Verlag, New York. Farmer, J. J . 111, J. H. Jorgensen, P. A. D. Grimont, R. J. Ak- hurst, G. 0. Poinar, Jr., G . V. Pierce, I. A. Smith, G. P. Carter, K. Wilson and F. W. Hickman-Brenner (1989) Xenorhabdus luminescence (DNA) hybridization group 5 from human clinical specimens. J. Clin. Microbiol. 27, 1594-1600. Kricka, L. J. and T. J. N. Carter (eds.) (1982) Clinical and Biochemical Luminescence. Marcel Dekker, New York. Chalfie, M., Y. Tu, G. G. Euskirchen, W. W. Ward and D. C. Prasher (1994) Green fluorescent protein as a marker for gene expression. Science 263, 802-805. Morin, J. G. and J. W. Hastings (1971) Energy transfer in a bioluminescent system. J . Cell. Physiol. 77, 3 13-3 18. Wampler, J. E., K. Hori, J. W. Lee and M. J. Cormier (1971) Structured bioluminescence. Two emitters during both the in uitro and the in viuo bioluminescence of the sea pansy, Renilla. Biochemistry 10, 2903-2909. Prasher, D. C., V. K. Eckenrode, W. W. Ward, F. G. Prendergast and M. J. Cormier (1992) Primary structure of the Aequorea victoria green-fluorescent protein. Gene 111, 229-233. Cody, C. W., D. C. Prasher, W. M. Westler, F. G . Prendergast and W. W. Ward (1993) Chemical structure of the hexapeptide chromophore of the Aequorea green-fluorescent protein. Bio- chemistry 32, 1212-1218. Shimomura, 0. and F. H. Johnson (1966) Partial purification and properties of the Chaetopterus luminescence system. In Bio- luminescence in Progress (Edited by F. H. Johnson and Y. Ha- neda), p p . 495-52 l . Princeton University Press, Princeton, NJ. Johnson, F. H. and 0. Shimomura (1972) Enzymatic and nonen- zymatic bioluminescence. In Photophysiology, Vol. VII (Edited by A. C. Giese), pp. 275-334. Academic Press, New York. Shimomura, 0. and F. H. Johnson (1975) Regeneration of the photoprotein aequorin. Nature 256, 236-238. Hastings, J. W. and Q. H. Gibson (1963) Intermediates in the bioluminescent oxidation of reduced flavin mononucleotide. J. Biol. Chem. 238, 2537-2554. Wood, K. V., Y. A. Lam, H. H. Seliger and W. D. McElroy (1989) Complementary DNA coding click beetle luciferases can elicit bioluminescence of different colors. Science 244, 700- 702.

Photochemistry and Photobiology, Vol. 62, No. 4, pp. 601606, 1995 Printed in the United States. All rights reserved 0031-8655195 $05.00+0.00
0 1995 American Society for Photobiology
COMMENTS ON THE MECHANISMS OF CHEMI- AND BIOLUMINESCENCE
THERESE WILSON Department of Cellular and Molecular Biology, Harvard University, Cambridge, MA 02138, USA
(Received 5 May 1995; accepted 2 June 1995)
INTRODUCTION
Although there is an astonishing diversity of bioluminescent organisms, from bacteria and fungi to mollusks, crustaceans, insects and fishes, there is a degree of unity at the chemical level: bioluminescences are enzyme-catalyzed reactions of molecular oxygen with appropriate substrates, and in all probability all involve the formation and breakdown of a peroxide or hydroperoxide. Going one step further, one might classify the bioluminescence systems into two groups, those that are considered to proceed via the intermediacy of a four-member ring peroxide and those that involve linear peroxides. I will mainly review here what has been learned from chemiluminescence regarding the first group, focusing on still unanswered questions that bear also on the mecha- nism of bacterial bioluminescence, the foremost example of a system involving a linear peroxide. The discussion will attract attention to a mechanistic dilemma posed by the widely quoted CIEEL* (chemically initiated electron ex- change luminescence) proposal.
INTERMEDIACY OF CYCLIC PEROXIDES
Chemists have lavished attention on four membered cyclic peroxides, the so-called dioxetanes. Hundreds of such com- pounds have been synthesized over the past 25 years and studied in organic solvents in the hope of understanding why one of the two carbonyl fragments is generated in an excited state (in yields from 6 to 40%).1-3 Equation 1 shows the decomposition of 3,3,4,4-tetramethyldioxetane (TMD) into two molecules of acetone.
The first answer to that question is immediately obvious. The required excitation energy (on the order of 4G70 kcal for visible emission) is indeed available, for the reason basic to the ubiquitous role of peroxides in bioluminescence: littlee- nergy is needed to break the peroxide bond, one of the weak- est of chemical bonds, while a great deal of energy is gained by the formation of two strong carbonyl bonds, so the bal- ance is sufficient for the excitation of one of the carbonyl fragments. In other words, it takes a small activation energy
*Abbreviations: CIEEL, chemically initiated electron exchange lu- minescence; DBA, 9,lO-dibromoanthracene; DMD, dimethyldiox- etanone; DPA, 9,lO-diphenylanthracene; FMNH,, reduced flavin mononucleotide; TMD, 3,3,4,4-tetramethyldioxetane.
to release the large exothermicity of the reaction, including the ring strain. The cleavage of the dioxetane ring, which involves the rearrangement of only four bonding electrons, should favor chemiexcitation, if the reaction energy can be chanelled into an electronically excited state of the carbonyls instead of being degraded ~ibrationally.~ This argument is supported by semiempirical and ab initio calculations,5 which suggest that the ground-state dioxetane surface and a triplet surface are close in energy in the potential energy region corresponding to a diradical-like transition state, where the 0-0 bond is significantly elongated. When heat- ed, a dioxetane moves in energy up to that point, crosses over with a good probability to the triplet surface and ends up generating one carbonyl in the triplet state (Fig. 1). A few singlet excited products are also formed, but their very low yield may simply be due to energetic reasons, if the excited singlet surface is too high near the transition state to be accessible.
Dioxetanes illustrate the distinction between chemiexci- tation and chemiluminescence. They generate lots of mole- cules in the excited triplet state, which have long lifetimes and are therefore usually quenched in solution before they emit. In that sense, dioxetanes are poor models for intense bioluminescences. They are, however, excellent sources of excited states. It should be emphasized that in the dioxetane case the creation of an excited state from a ground-state reactant proceeds without the development of charge.
Dioxetane solutions also generate a weak luminescence, due to the fluorescence of the few singlet excited carbonyls generated in Eq. 1 and to the phosphorescence of the triplet carbonyls. This emission decays exponentially as a function of time. The addition of a suitable fluorescent molecule can amplify it via energy transfer (Eq. 2): this requires that the singlet excited S, state of such a “sensitizer” (DPA, 9 , lO- diphenylanthracene, for example) be lower than that of the singlet excited carbonyl, here acetone, and its fluorescence quantum yield be higher. Moreover, some sensitizers are able to capture the energy of the more abundant triplet car- bonyls and use it to populate quite efficiently their own sin- glet excited state; as a rule, such sensitizers are molecules with heavy atom substituents (such as 9, lo-dibromoanthra- cene, DBA), which facilitate the mixing of triplet and singlet statest (Eq. 3 )
‘A* + DPA -+ A + ’DPA* (-+hYDpA)
3A* + DBA -+ A + ‘DBA* (+huDBA).
(2)
( 3 )
+For the same reason the fluorescence quantum yield of DBA is low (0.14.2, depending on solvent).
60 1
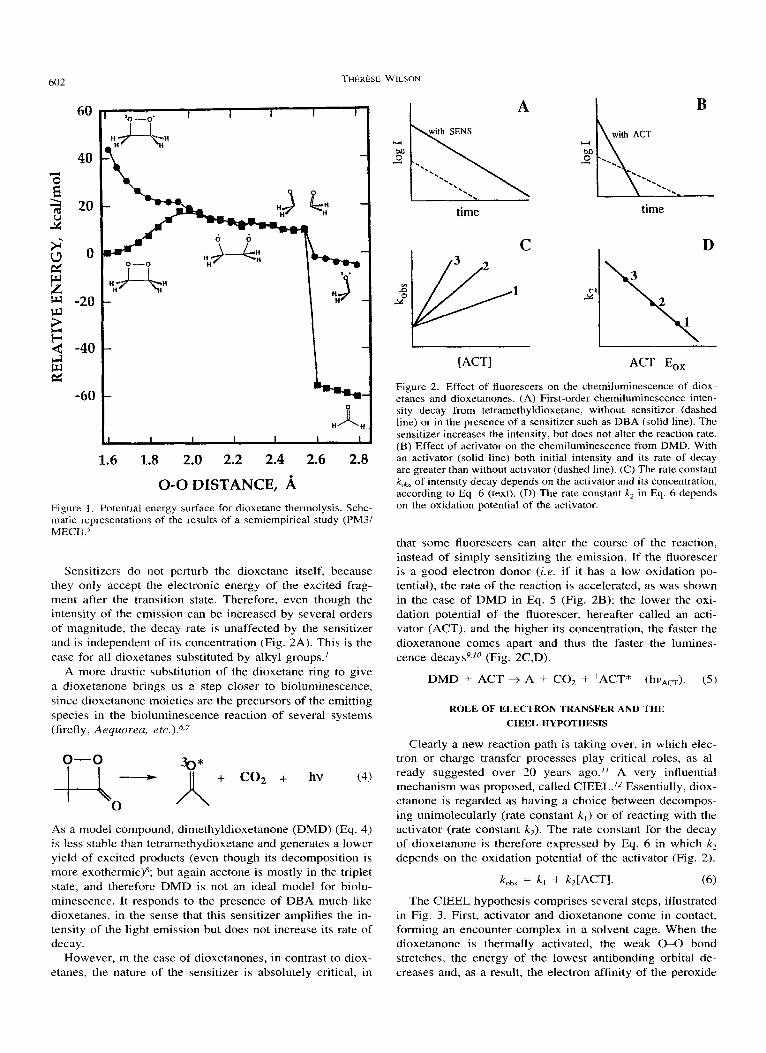
602
60
40 &
0 ti 3 20 Y $ 0
5
E
&
w -20 w
4 -40 4
2 -60
THERESE WILSON
1.6 1.8 2.0 2.2 2.4 2.6 2.8
0-0 DISTANCE, A Figure 1 . Potential energy surface for dioxetane thermolysis. Sche- matic representations of the results of a semiempirical study (PM3/ MECI).s
Sensitizers do not perturb the dioxetane itself, because they only accept the electronic energy of the excited frag- ment after the transition state. Therefore, even though the intensity of the emission can be increased by several orders of magnitude, the decay rate is unaffected by the sensitizer and is independent of its concentration (Fig. 2A). This is the case for all dioxetanes substituted by alkyl groups.'
A more drastic substitution of the dioxetane ring to give a dioxetanone brings us a step closer to bioluminescence, since dioxetanone moieties are the precursors of the emitting species in the bioluminescence reaction of several systems (firefly, Aequorea, e t ~ . ) . " ~
As a model compound, dimethyldioxetanone (DMD) (Eq. 4) is less stable than tetramethydioxetane and generates a lower yield of excited products (even though its decomposition is more exothermic)8; but again acetone is mostly in the triplet state, and therefore DMD is not an ideal model for biolu- minescence. It responds to the presence of DBA much like dioxetanes, in the sense that this sensitizer amplifies the in- tensity of the light emission but does not increase its rate of decay.
However, in the case of dioxetanones, in contrast to diox- etanes. the nature of the sensitizer is absolutelv critical. in
Do 0
'. -. '. time
C
B
time
D
\ ACT E,,
Figure 2. Effect of fluorescers on the chemiluminescence of diox- etanes and dioxetanones. (A) First-order chemiluminescence inten- sity decay from tetramethyldioxetane, without sensitizer (dashed line) or in the presence of a sensitizer such as DBA (solid line). The sensitizer increases the intensity, but does not alter the reaction rate. (B) Effect of activator on the chemiluminescence from DMD. With an activator (solid line) both initial intensity and its rate of decay are greater than without activator (dashed line). ( C ) The rate constant koba of intensity decay depends on the activator and its concentration, according to Eq. 6 (text). (D) The rate constant k, in Eq. 6 depends on the oxidation potential of the activator.
that some fluorescers can alter the course of the reaction, instead of simply sensitizing the emission. If the fluorescer is a good electron donor (i .e. if it has a low oxidation po- tential), the rate of the reaction is accelerated, as was shown in the case of DMD in Eq. 5 (Fig. 2B): the lower the oxi- dation potential of the fluorescer, hereafter called an acti- vator (ACT), and the higher its concentration, the faster the dioxetanone comes apart and thus the faster the lumines- cence decay^^,'^ (Fig. 2C,D).
DMD + ACT + A + CO, + IACT* (hvAcT). ( 5 )
ROLE OF ELECTRON TRANSFER AND THE CIEEL HYPOTHESIS
Clearly a new reaction path is taking over, in which elec- tron or charge transfer processes play critical roles, as al- ready suggested over 20 years ago." A very influential mechanism was proposed, called CIEEL.'2 Essentially, diox- etanone is regarded as having a choice between decompos- ing unimolecularly (rate constant k , ) or of reacting with the activator (rate constant k2) . The rate constant for the decay of dioxetanone is therefore expressed by Eq. 6 in which k, depends on the oxidation potential of the activator (Fig. 2).
The CIEEL hypothesis comprises several steps, illustrated in Fig. 3. First, activator and dioxetanone come in contact, forming an encounter complex in a solvent cage. When the dioxetanone is thermally activated, the weak 0-0 bond stretches, the energy of the lowest antibonding orbital de- creases and. as a result. the electron affinitv of the Deroxide

S ymposium-in-Print
ACT
1 ET-2
ACT -t hvA,
Figure 3. The CIEEL mechanism proposed for the activated che- miluminescence of DMD. Note two successive electron transfers, ET-1 and ET-2 (see text).
therefore increases to the point where the jump of an elec- tron from the activator to the peroxide becomes energetically feasible. The energy needed to reach this point is lower than that required to break the 0-0 bond or to form the radical cation of the isolated activator.
Electron transfer (ET-1, Fig. 3) from activator to peroxide then occurs, probably simultaneously with 0-0 bond cleav- age. This triggers a fast cascade of events, the rupture of the C-C bond and the formation of CO,? and acetone, leaving a pair of radical ions still in contact in the solvent cage. One is the radical cation of the fluorescer, the other the radical anion of either acetone or CO,, both good reductants (better reductants than dioxetanone itself, a critical point in the mechanism). Finally, the charges on these two radical ions annihilate (ET-2), a process that liberates enough energy to promote the activator to its singlet excited state. This is the excitation step, a well-understood chemiexcitation process taken from the field of electrochemiluminescence.13
Because all these events are considered to happen ex- tremely fast, with the final electron transfer returning the electron to the activator that had first donated it, the idea is that there will be no chance for spin inversion because the electrons remain correlated. Therefore, singlet excited states should be generated, contrary to the electrochemiluminesc- ence situation where the partition between singlet and triplet states is statistical.
The CIEEL hypothesis was first proposed in the case of diphenoyl peroxide14 (1 in Fig. 4). This peroxide has the advantage of being more stable than DMD and therefore easier to study. However, the reaction follows the same gen- eral uattern: both luminescence intensity and rate of Deroxide
B0 \ 0
/ y
benzocoumarin t co2 t ACT * 1
Ph i)Me 2
Ph OMe
Figure 4. Reactions of two peroxides discussed as possible examples of CIEEL chemiluminescence.
decomposition (or intensity decay) depend on the oxidation potential of the activator and on its concentration. The quan- tum yield was initially claimed to be remarkably high: 10% of the activators that had initially reduced and broken the peroxide bond were reported to come out of the process in their singlet excited state.I4 Unfortunately, this high efficien- cy was not ~0nfirmed.I~ In fact, the reaction quantum yield is disappointingly low, only on the order of quanta per peroxide decomposed, and the same is true for the chemiluminescence of an anthracene peroxide (1,4-di- methoxy-9,10-diphenylanthracene, 2 in Fig. 4),‘5 which also reacts according to the pattern described by Eq. 6.
or
A QUESTION OF EFFICIENCY
This is, to my knowledge, where the situation presently rests, with CIEEL continuing to enjoy its status as the par- adigm for efficient chemi- or bioluminescence. The question then is why are the two model systems (diphenoyl peroxide and the anthracene peroxide) so inefficient? There are es- sentially two possible reasons. The first is that the initial electron transfer (ET-1, Fig. 3), if it takes place, is imme- diately followed by back transfer and therefore not produc- tive. The second is that a pair of activator radical ions is indeed produced, but that they diffuse apart and out of the solvent cage before the back electron transfer (ET-2) can take place to create the excited state. Neither of these ex- planations is satisfactory, in my opinion, but both deserve a closer look.
The transfer of a single electron from the activator to the peroxide bond is an endergonic process. Because an electron jump is a hundred times faster than nuclear motions (molec- ular vibrations), it will happen only when the 0-0 bond is already appropriately elongated.I6 It should be followed im- mediately by back electron transfer, from the 0 atom that received it to the activator molecule that had donated it, un-

THERESE WILSON 604
b
cn
5 X = Sl(CH3)t-Bu
Figure 5 . Three chemiluminescences of high quantum yields, where electron transfer is considered to play a critical role. N-methylacri- dan 3, stabilized by an adamantyl substituent, has a half-life of a year in xylene at 25°C and the quantum yield is ca 10%. But on alumina, which is assumed to polarize the 0-0 bond and cause a shift to an electron transfer mechanism, the lifetime is seconds and most of the catalysis leads to excited states, since the yield is then I %. In nonpolar solvents, the emission from dioxetane 4 shows only one emission peak, which matches the fluorescence of the product. In polar solvents, a strong second peak at longer wavelengths indi- cates the formation of an intramolecular exciplex, conformationally stabilized by the polar solvent. Upon deprotonation, the lifetime of the dioxetane 5 decreases from years to seconds (in acetonitrile), and the emission becomes very intense, with a chemiexcitation yield of 45%.
less the process of bond breaking and nuclear rearrangement are nearly simultaneous with the first electron transfer, a drastic requirement. Therefore it is perhaps not surprising that such a dissociative electron transfer is a rare event.” However, here is a dilemma: the model peroxides are un- questionably decomposed by the activator, and the decom- position rates do depend on the activator oxidation poten- tial-just as predicted by Eq. 1 and CIEEL. Are two pro- cesses, both depending on the E,, of the activator, going on in parallel? Conceivably, as the 0-0 bond gets thermally elongated, charge transfer interactions with the activator could lower the transition state, causing the bond to break, without formation of radical ions. Some of the time, albeit rarely, the whole CIEEL sequence would occur, and it would be responsible for the weak emission. Or else, a polar mech- anism would by itself be responsible for excitation. These suggestions have none of the attractiveness of the concrete CIEEL steps, but it is important to keep in mind that electron transfer and development of charges are not a sine qua non condition for chemiexcitation.
The second possible cause of inefficiency in a CIEEL
0-0
Figure 6. The hypothetical dioxetane of firefly luciferin and its de- composition to produce the emitter.
mechanism, namely diffusion of the radical ions away from each other before the final charge annihilation takes place, was deemed unlikely in the case of diphenoyl peroxide. The basis for this conclusion was experimental: one would have expected considerable amounts of specific products of radi- cal reactions with solvent, while only traces were found.I4 However, recent photophysical results show that the rate constants for solvation of a contact radical ion pair to give a solvent-separated radical ion pair, and completely solvent- separated radical ions are fast, on the order of lo9 s-’.I8
If the key to efficiency was to prevent the electron transfer partners from getting away from each other, it may in part explain why some intramolecular reactions initiated by elec- tron or charge transfer can be extremely efficient, as the following examples illustrate.
The chemiluminescence yield from N-methylacridan diox- etane stabilized by an adamantyl group (3, Fig. 5) is ca lo%, and the dioxetane lifetime is estimated to be a year. How- ever, in the presence of alumina, which probably polarizes the 0-0 bond and induces a shift to an electron transfer mechanism, this lifetime is reduced to seconds, and most of the catalyzed decomposition yields excited states.I9
Another revealing example is provided by the very unsta- ble dioxetane 4, in Fig. 5. In nonpolar solvents, the emission matches the fluorescence of the product. But in polar sol- vents, a second strong emission peak appears at longer wavelengths, which is assigned to an intramolecular exciplex (an excited charge-transfer complex) stabilized by the sol- vent.20
Finally, the family of dioxetanes of type 5 (Fig. 5) pro- vides many fascinating examples. Protected by a sily] group, for example, the dioxetane is stable. But if the protective group is removed, the decomposition is extremely rapid and the chemiluminescence yield very high, ca 20%, which cor- responds to an astonishing 50% excitation yield.2’ When the protective groups can be enzymatically removed, the list of analytical applications based on this type of compounds is endless.2z
In these examples, there is no doubt that electron shifts or transfers (or other polar processes) play triggering roles, but precisely how is still the question. These efficient reac- tions are evidently much better models of bioluminescence; a comparison between the firefly case (Fig. 6) and the re- action of 5 in Fig. 5 makes that p ~ i n t . ~ ~ , ~ ~ Here, too, the phenolate ion is required; if the phenol is methylated, the reaction does not occur. However, because these reactions

S ymposium-in-Print
0 2 R RCHO
FMN+ H20
H EoFOOA E*F*
Scheme 1.
are intramolecular and very fast, the reaction pathways are less tractable than that of the intermolecular, activator-initi- ated reactions discussed above. Nonetheless, chemists still have to identify the intermediates and pathways involved in the Cyalume “light stick” reaction of a peroxyoxalate, H20, and a fluorescer, which is astonishingly efficient (30%)25 even though it is not an intramolecular reaction, nor one facilitated by an enzyme, whose role could be to hold the reactants together.
The most classic case of chemiluminescence, that of lu- minol, has not yet yielded all its secrets either, but it is very likely that a transient six membered cyclic peroxide is formed and that radical ion processes play a critical ro]e.2627,28
INTERMEDIACY OF LINEAR PEROXIDES
Bacterial bioluminescence is the foremost example of a bioluminescence that does not involve the intermediacy of a cyclic peroxide. Bacterial luciferase catalyzes the mixed function oxidation of a long-chain aldehyde and reduced fla- vin mononucleotide (FMNH,), Eq. 7. The quantum yield is -0.2. Much is now known of the mechanism, which is dis- cussed in detail e l s e ~ h e r e . ~ ~ , ~ ~ Several intermediates have been characterized, although the most critical, the postulated peroxyhemiacetal, E.FOOA in Scheme 1 (see above), has so far resisted isolation. The emitter is the enzyme-bound 4a- hydroxyflavin, E.F* Electrochemical studies,3’J2 as well as the effect of S-posi- tion substituents of various oxidation potentials on the flavin ring,33 suggest that the rate-detenning step is influenced by electron transfer (or charge redistribution) toward the per- oxide bond, which is estimated to be unusually weak: cleav- age of the 0-0 bond could follow either the transfer of an electron from the N5 lone pair or the shift of a proton from the aldehyde moiety to the 0-0 bond. The excitation step is presently regarded as a charge annihilation between two radical ion centers. All electron shifts and bond rearrange- ments are considered to take place within the constraints of the enzyme pocket, which is assumed to keep the reacting groups in contact and in proper orientation (the role tenta- tively attributed to the solvent cage in the examples of in- tramolecular CIEEL, discussed above). Because none of the postulated reaction intermediates are stable outside the en- zyme, their identities and therefore the sequence of reaction steps in which they partake remain obviously hypothetical.
Up to now, the chemiluminescence quantum yields of model systems have been disappointingly low.
CONCLUDING REMARKS
The intense focus on the role of electron transfer processes in chemi- and bioluminescences has had a remarkably in- vigorating effect on the field in the last two decades. Such processes are, of essence, extremely fast, whereas the chem- istry that precedes or follows them is, in most cases, much slower. When the overall efficiency of a chemiluminescence is low, it is evidently risky to try and assign the excitation step to a particular chemical process. McCapra recently ex- pressed a similar note of caution.34 The role of hypotheses is to suggest critical experiments, which may, hopefully, bring definitive results.
Acknowledgements-Support from the National Science Foundation (MCB93-06-879) is gratefully acknowledged. I thank J. W. Hastings and E. A. Chandross for enjoyable discussions and comments on the manuscript.
REFERENCES
1. Adam, W., M. Heil, T. Mosandl and C. R. Saha-Moller (1992) Dioxetanes and a-peroxylactones, 4-membered ring cyclic per- oxides. In Organic Peroxides (Edited by W. Ando), pp. 252- 254. Wiley & Sons, Chichester, England.
2. Baumstark, A. L. (1988) Thermolysis of “alkyl”-1,2-dioxe- tanes. In Advances in Oxygenated Processes, Vol. 1 (Edited by A. L. Baumstark), pp. 31-84. Jai Press Inc., Greenwich, CT.
3. Wilson, T. (1976) Chemiluminescence in the liquid state: ther- mal cleavage of dioxetanes. Znt. Rev. Sci. Phys. Chem. Ser. [21 9, 265-322.
4. Marcus, R. A. and N. Sutin (1985) Electron transfers in chem- istry and biology. Biochim. Biophys. Acta 811, 265-322.
5. Wilson, T. and A. M. Halpern (1995) Dioxetane decomposition revisited. A semiempirical study of the potential energy surface. J. Phys. Org. Chem. and references therein. 8, 359-363.
6. McCapra, F. and K. D. Perring (1985) Luciferin hiolumines- cence In Chemi- and Bioluminescence (Edited by J. Burr), pp. 359-386. Marcel Dekker, New York.
7. Adam, W. and G. Cilento (eds.) (1982) Chemical and Biological Generation of Excited States. Academic Press, New York.
8. Schmidt, S. P. and G. B. Schuster (1978) Kinetics of unimole- cular dioxetanone chemiluminescence. Competitive reaction paths. 1. Am. Chem. SOC. 100, 5559-5561.
9. Schmidt, S. P. and G. B. Schuster (1980) Chemiluminescence of dimethyldioxetanone. Unimolecular generation of excited singlet and triplet acetone. Chemically initiated electron-ex- change luminescence, the primary light generating reaction. J. Am. Chem. Soc. 102, 306-314.
10. Adam, W. and 0. Cueto (1979) Fluorescer-enhanced chemilu-

THERESE WILSON 606
minescence of a-peroxylactones via electron exchange. J. Am. Chem. SOC. 101, 6511-651s.
I 1. McCapra, F. (1973) Chemiluminescence of organic compounds. Prog. Org. Chem. 8, 231-277.
12. Schuster, G. B. (1979) Chemiluminescence of organic perox- ides, Conversion of ground-state reactants to excited-state prod- ucts by the chemically initiated electron-exchange luminescence mechanism. Acct. Chem. Rex 12, 366-373.
13. Faulkner, L. R. (1976) Chemiluminescence in the liquid phase: electron transfer. Int. Rev. Sci. Phys. Chem. Ser. [ 2 ] 9, 213- 263.
14. Koo, J.-Y. and G. B. Schuster (1978) Chemiluminescence of diphenoyl peroxide. Chemically initiated electron-exchange lu- minescence. A new general mechanism for chemical production of electronically excited states. J . Am. Chem. SOC. 100, 4496- 4503.
1s. Catalani, L. H. and T. Wilson (1989) Electron transfer and che- miluminescence. Two inefficient systems: 1,4-dimethoxy-9,10- diphenylanthracene peroxide and diphenoyl peroxide. J. Am. Chern. SOC. 111, 2633-2639.
16. Schmidt, S. P., M. A. Vincent, C. E. Dykstra and G. B. Schuster ( I 981) Chemiluminescence of dioxetanone investigated by self- consistent-field theory. J. Am. Chem. Soc. 103, 1292-1293.
17. Workentin, M. S., F. Maran and D. D. M. Wayner (1995) Re- duction of di-tert-butyl peroxide: evidence for nonadiabatic electron transfer. J. Am. Chem. Soc. 117, 2120-2121.
18. Arnold, B. R., D. Noukakis, S. Farid, J. L. Goodman and I. R. Gould (1995) Dynamics of interconversion of contact and sol- vent-separated radical ion pairs. J. Am. Chem. Soc. 117, 4399- 4400.
19. McCapra, F., 1. Begeshiti, A. Burford, R. A. Hann and K. A. Zaklika (1977) Singlet excited states from dioxetane decompo- sition. J. Chem. SOC. Chem. Commun., 944-946.
20. Nakamura, H. and T. Goto (1979) Studies on aminodioxetanes as model of bioluminescence intermediates. 1 -( I-Methyl-3-in- dolyl)-6-phenyl-2,5,7,8-tetraoxabicyclo[4,2,O]octane, and ami- nodioxetane resulting in efficient ultraviolet and exciplex che- miluminescence. Phofochem. Photobiol. 30, 27-33.
21. Schaap, A. P., T. S. Chen, R. S. Handley, R. DeSilva and B. P. Giri (1987) Chemical and enzymatic triggering of 1,2-diox- etanes. 2. Fluoride-induced chemiluminescence from terr-bu- tyldimethylsiloxy-substituted dioxetanes. Tetrahedron Lett. 28, 1155-3158.
22. Beck, S . and H. Koster (1990) Applications of dioxetane chem- iluminescent probes to molecular biology. Anal. Chem. 62, 2258-2270, and references therein.
23. White, E. H., M. G. Steinmetz, J. D. Miano, P. D. Wildes and R. M. Morland (1980) Chemi- and bioluminescence of firefly luciferin. J. Am. Chem. SOC. 102, 3199-3208.
24. Koo, J.-Y., S. P. Schmidt and G. B. Schuster (1978) Biolumi- nescence of the firefly: key steps in the formation of the elec- tronically excited state for model systems. Proc. Natl. Acad. Sci. USA 57, 30-33.
25. Mohan, A. G. (1985) Peroxyoxalate chemiluminescence. In Chemi- and Bioluminescence (Edited by J. Burr), pp. 245-258. Marcel Dekker, New York.
26. White, E. H. and D. F. Roswell (1985) Luminol chemilumines- cence. In Chemi- and Bioluminescence (Edited by J. G. Burr), pp. 215-244. Marcel Dekker, Inc., New York.
27. McCapra, F. and K. D. Perring (1985) General organic chemi- luminescence. In Chemi- and Bioluminescence (Edited by J. G. Burr), pp. 259-320. Marcel Dekker, Inc., New York.
28. Merenyi, G., J. Lind and T. E. Eriksen (1986) Nucleophilic addition to diazaquinones. Formation of tetrahedral intermedi- ates in relation to luminol chemiluminescence. J . Am. Chem. Soc. 108, 7716-7726.
29. Mager, I. X. and S.-C. Tu (1995) Chemical aspects of biolumi- nescence. Phorochem. Photobiol. 62, 607-6 14.
30. Baldwin, T. 0. and M. M. Ziegler (1992) In Chemistry and Biochemistry of Flavoenzymes, Vol. 111 (Edited by F. Muller), pp. 467-530. CRC Press, Boca Raton, FL.
31. Mager, H. I. X., D. Sazou, Y. H. Liu, S.-C. Tu and K. M. Kadish (1988) Reversible one-electron generation of 4a,5-substituted flavin radical cations: models for a postulated key intermediate in bacterial bioluminescence. J. Am. Chem. Soc. 110, 3759- 3762.
32. Merenyi, G., J. Lind, H. I. X. Mager and S.-C. Tu (1992) Prop- erties of 4a-hydroxy-4a.5-dihydroflavin radicals in relation to bacterial bioluminescence. J. Phys. Chem. 96, 10528-10533.
33. Eckstein, J. W., J. W. Hastings and S . Ghisla (1993) Mechanism of bacterial bioluminescence: 4a,5-dihydroflavin analogs as models for luciferase hydroperoxide intermediates and the effect of substituents at the 8-position of flavin on luciferase kinetics. Biochemistry 32, 4 0 4 4 1 1.
34. McCapra, F. (1990) Chemiluminescence and bioluminescence. J . Photochem. Photobiol. A Chem. 51, 21-28.