supplementary information - the royal society of … · supplementary information threading the...
TRANSCRIPT

S1
Supplementary Information
Threading the needle: guest transport in a versatile 0D porous molecular crystal.
V. I. Nikolayenko, A. Heyns and L. J. Barbour*
Department of Chemistry and Polymer Science, University of Stellenbosch, Stellenbosch 7600, South
Africa. [email protected]
All reagents employed in this study were purchased from commercial sources with no further
purification. The L1 ligand was synthesized according to a previously reported method.1
Ligand Synthesis
Solid 2,4-bis(chloromethyl)-1,3,5-trimethylbenzene (2.32 g, 10.7 mmol) was added to a
solution of imidazole (6.71 g, 98.6 mmol) in 100 mL methanol. The mixture was heated to
reflux. The reaction was monitored using TLC, and was found to be complete after 24 hrs.
Excess solvent was removed under reduced pressure and gentle heating before precipitation of
the product by adding excess 10% K2CO3 solution. After washing with ethyl acetate, the pure
white product was filtered and left to dry overnight. Yield: 1.532 g, 47%. H-NMR (CDCl3, 300
MHz): δ 2.18 (3H, s, CH3), 2.31 (6H, s, CH3), 5.14 (4H, s, CH2), 6.73 (2H, s, Him), 7.01 (2H,
s, Him), 7.03 (1H, s, HBz) 7.29 (2H, s, Him) ppm.
Crystallisation
Crystals of 1 were grown under solvothermal conditions in a programmable oven (Labotech,
Ecotherm) (heated at 80 °C for 24 hrs before cooling to room temperature over a period of 36
hrs) by adding a solution 15.2 mg (0.089 mmol) of CuCl2∙2H2O in 2 ml of methanol to a
Electronic Supplementary Material (ESI) for ChemComm.This journal is © The Royal Society of Chemistry 2017

S2
solution of 25 mg (0.089mmol) L1 in 3 ml of methanol. Green block-shaped crystals suitable
for single-crystal X-ray diffraction analysis (SCD) were obtained.
The solvent-free complex 1a was obtained by placing crystals of 1 at 100 °C under reduced
pressure for 24 hrs. Both products were characterised by means of PXRD, SCD and thermal
analysis. The PXRD patterns of 1 and 1a are representative of the powder patterns simulated
from the SCD structures (Figure S1(a) and S1(b)).
Figure S1 (a) PXRD patterns of 1 calculated from the SC structure (grey), and the
experimental pattern (black) of 1, showing that the SCD structure is representative of the bulk
material. (b) PXRD pattern of 1a calculated from the SC structure (grey), and the experimental
pattern (black) of 1a, showing that the SCD structure is representative of the bulk material.
5 10 15 20 25
2θ/°
a
5 10 15 20 25 30 35 40
2θ/°
b

S3
Guest Exchange
In order to determine whether 1 is porous a series of guest exchange experiments where
undertaken. Each experiment consisted of immersing several single crystals of 1 in a small vial
containing 1 ml of one of the solvents listed in Table S1. The crystals of the stable complex 1
are not soluble in any of the organic solvents typically used for crystallisation. The systems
were left overnight to equilibrate. Crystals were removed from the vial with a spatula, the tip
of which was covered in Paratone oil to prevent rapid desolvation during mounting prior to
data collection.
Table S1: Organic molecules employed in guest exchange experiments.
Sorption of sublimed naphthalene and p-dichlorobenzene
Single-crystals of 1a were sealed in the presence of freshly ground crystals of the targeted guest
(Table S2), taking special care to allow crystals of 1a to be in contact with the excess
naphthalene or p-dichlorobenzene. In both cases the crystals remained intact such that their
structures could be determined using single-crystal X-ray diffraction.
Table S2: Volatile solids employed in guest exchange experiments.
Solvate
Formula
Molecular
weight
(g mol-1)
Melting
point (°C)
Vapour
pressure
(mmHg)
Naphthalene C10H8 128.2 88.5 0.08
p-Dichlorobenzene C6H4Cl2 147.0 53.1 1.76
Solvate
Formula
Molecular
weight
(g mol-1)
Boiling
point
(°C)
Density
(g cm-3)
Density
conversion
factor
Vapour
pressure
(mmHg @
20 °C)
Methanol CH3OH 32.0 64.7 0.79 0.002 97.3
Water H2O 18.0 100.0 1.00 0.001 20.0
Acetone C3H6O 58.1 56.3 0.79 0.003 183.1
Acetonitrile CH3CN 41.1 81.6 0.79 0.002 72.0
Carbon disulphide CS2 76.1 46.3 1.26 0.003 500.0
Dichloromethane CH2Cl2 84.9 39.8 1.33 0.003 350.0
p-Difluorobenzene C6H4F2 114.3 114.3 1.17 0.004 72.0
Benzene C6H6 78.1 80.1 0.87 0.004 75.1
Chloroform CHCl3 119.4 61.2 1.48 0.004 157.1

S4
Single-crystal X-ray diffraction
X-ray intensity data were recorded on a Bruker SMART APEX II2 equipped with a Mo sealed
tube source. The diffractometer employs an Oxford Cryosystems 700+ Plus cryostat to control
the temperature of the sample. Data reduction was carried out by means of standard procedures
using the Bruker software package SAINT.3 Absorption corrections and correction of other
systematic errors were carried out using SADABS.4 All structures were solved by direct
methods using SHELXS-16 and refined using SHELXL-16.5 X-Seed6 was used as the graphical
interface for the SHELX program suite. Solvent-accessible voids can be visualised by
calculating Connolly surfaces using MS-ROLL,7 another program incorporated into X-Seed.
Hydrogen atoms were placed in calculated positions using riding models.
Selected crystallographic data are given in Table S3.

S5
Table S3 Crystallographic data for metallocycles 1-11.
1 1a 2 3 4 5
Guest methanol water acetone acetonitrile Carbon disulphide
Empirical formula C36 H46 Cl4 Cu2 N8 O2 C34 H40 Cl4 Cu2 N8 C34 H40Cl4Cu2N8O1.4 C37H46Cl4Cu2N8O1 C36H43Cl4Cu2N9 C35H40Cl4Cu2N8S2
Formula weight 891.71 829.64 851.40 888.88 869.95 905.77
Temperature (K) 100(0) 100(0) 100(0) 100(0) 100(0) 100(0)
Wavelength (Å) Monoclinic Monoclinic Monoclinic Monoclinic Monoclinic Monoclinic
Crystal system 0.71073 0.71073 0.71073 0.71073 0.71073 0.71073
a/Å 8.426(1) 8.327(2) 8.333(4) 8.386(3) 8.325(5) 8.348(11)
b/ Å 10.707(1) 10.489(3) 10.394(5) 10.778(4) 10.568(6) 10.598(14)
c/ Å 21.974(3) 22.225(5) 22.167(10) 22.282(8) 22.158(1) 22.200(3)
° 90.0 90.0 90.0 90.0 90.0 90.0
° 99.441(2) 98.546(3) 98.264(7) 99.205(4) 98.506(7) 98.700(2)
° 90.0 90.0 90.0 90.0 90.0 90.0
Space group P21/c P21/c P21/c P21/c P21/c P21/c
Volume (Å3) 1955.6(4) 1919.6(8) 1900.0(2) 1988.0(1) 1928.0(2) 1941.5(4)
Z 2 2 2 2 2 2

S6
Table S3 continued
6 7 8 9 10 11
Guest dichloromethane difluorobenzene benzene chloroform naphthalene dichlorobenzene
Empirical formula C35H42Cl6Cu2N8 C40H44Cl4Cu2F2N8 C40H46Cl4Cu2N8 C35H41Cl7Cu2N8 C38H44Cl4Cu2N8 C40H44Cl6Cu2N8
Formula weight 912.53 943.73 907.04 949.01 886.16 976.63
Temperature (K) 100(0) 100(0) 100(0) 100(0) 100(0) 100(0)
Wavelength (Å) 0.71073 0.71073 0.71073 0.71073 0.71073 0.71073
Crystal system Monoclinic Monoclinic Monoclinic Monoclinic Monoclinic Monoclinic
a/Å 8.349(1) 8.424(2) 8.368(4) 16.671(2) 8.297(2) 8.383(2)
b/ Å 10.646(1) 10.944(3) 10.906(5) 10.779(2) 10.771(2) 10.868(3)
c/ Å 22.378(3) 22.094(6) 22.082(11) 22.437(3) 22.520(4) 22.633(6)
° 90.0 90.0 90.0 90.0 90.0 90.0
° 99.387(2) 99.735(5) 99.017(7) 99.497(2) 99.655(2) 79.007(3)
° 90.0 90.0 90.0 90.0 90.0 90.0
Space group P21/c P21/c P21/c P21/c P21/c P21/c
Volume (Å3) 1962.5(5) 2007.6(9) 1990.3(2) 3976.8(9) 1984.0(6) 2024.2(9)
Z 2 2 2 4 2 2
S7
Table S4 Selected crystallographic and thermogravimetric parameters for 1-11.
Additional crystal structure diagrams and structure descriptions
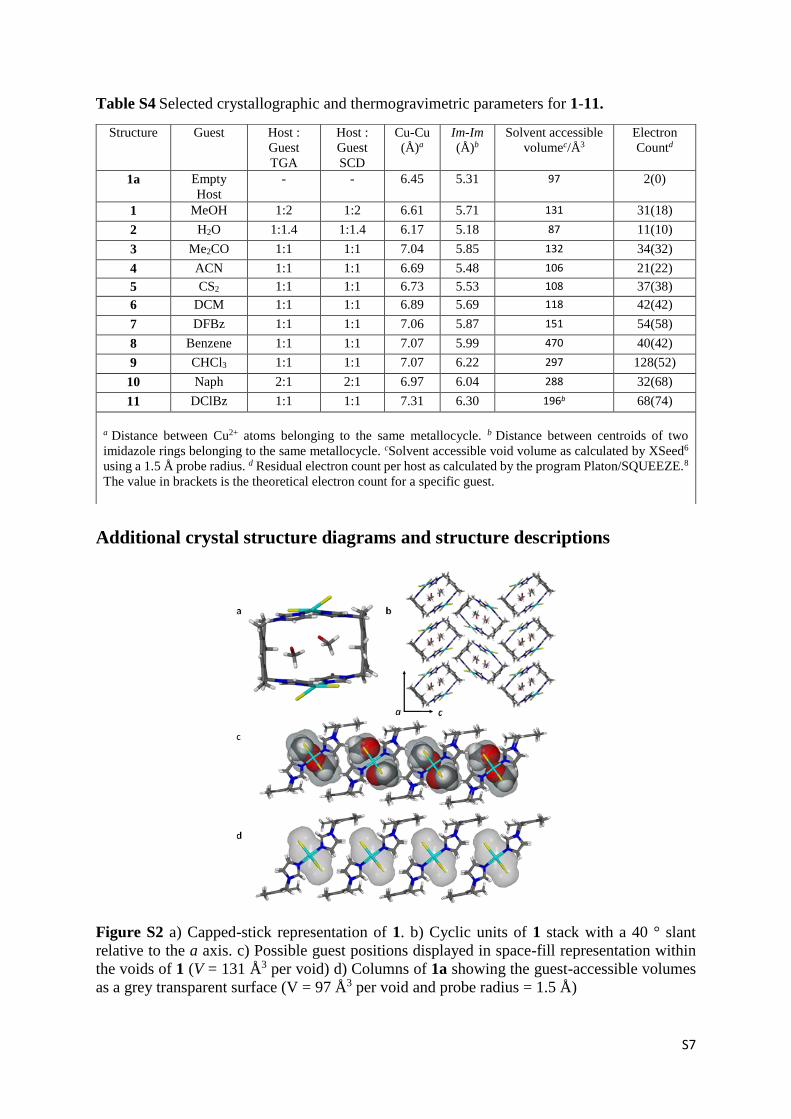
Figure S2 a) Capped-stick representation of 1. b) Cyclic units of 1 stack with a 40 ° slant
relative to the a axis. c) Possible guest positions displayed in space-fill representation within
the voids of 1 (V = 131 Å3 per void) d) Columns of 1a showing the guest-accessible volumes
as a grey transparent surface (V = 97 Å3 per void and probe radius = 1.5 Å)
Structure Guest Host :
Guest
TGA
Host :
Guest
SCD
Cu-Cu
(Å)a
Im-Im
(Å)b
Solvent accessible
volumec/Å3
Electron
Countd
1a Empty
Host
- - 6.45 5.31 97 2(0)
1 MeOH 1:2 1:2 6.61 5.71 131 31(18)
2 H2O 1:1.4 1:1.4 6.17 5.18 87 11(10)
3 Me2CO 1:1 1:1 7.04 5.85 132 34(32)
4 ACN 1:1 1:1 6.69 5.48 106 21(22)
5 CS2 1:1 1:1 6.73 5.53 108 37(38)
6 DCM 1:1 1:1 6.89 5.69 118 42(42)
7 DFBz 1:1 1:1 7.06 5.87 151 54(58)
8 Benzene 1:1 1:1 7.07 5.99 470 40(42)
9 CHCl3 1:1 1:1 7.07 6.22 297 128(52)
10 Naph 2:1 2:1 6.97 6.04 288 32(68)
11 DClBz 1:1 1:1 7.31 6.30 196b 68(74)
a Distance between Cu2+ atoms belonging to the same metallocycle. b Distance between centroids of two
imidazole rings belonging to the same metallocycle. cSolvent accessible void volume as calculated by XSeed6
using a 1.5 Å probe radius. d Residual electron count per host as calculated by the program Platon/SQUEEZE.8
The value in brackets is the theoretical electron count for a specific guest.

S8
Figure S3 a) The locations of the unique water guest molecules in 2. The weak coordination
of Cu2+∙∙∙O1A = 2.765(1) Å and is shown as a solid line. b) Possible placement of the water
guests in 2 within a stacked column of host complexes. Guest pockets are shown as semi-
transparent grey surfaces; volume = 87 Å3 per void. Hydrogen atoms of the water molecules
could not be located in difference electron density maps.
The ASU of 3 contains half an acetone guest molecule situated on a general position.
Careful inspection of the space-filled guest model shows that they are distributed
randomly throughout the structure (Fig S4).9 The host-guest ratio is 1:1, with weak
hydrogen bonds present between the carbonyl group of the guest and the inner methyl
group of the host (labelled C7 in Fig S4a). In the past, interactions between the
electron-rich carbonyl group of the guest and imidazole (Im) rings of the ligand would
have been overlooked. However, a report by Mooibroek et al. showed that the lone pair
(lp)∙∙∙π interaction is of supramolecular relevance.10 These interactions stabilise the
acetone guest between the two imidazole rings of each L1 ligand (Fig S4a,
O1A∙∙∙Im(1,2) = 3.12 Å and 3.15 Å, respectively). Relative to 1a, a noteworthy increase
in solvent accessible-volume, Cu2+∙∙∙Cu2+ and Im∙∙∙Im distances occurs upon saturation
with the larger acetone guest (Table S4, V = 132 Å3 per void, 7.04 Å and 5.85 Å,
respectively).

S9
Figure S4 a) Molecular structure of 3 with select atoms labelled. One complete host is shown
in CPK colours and the disordered guest is illustrated in green and orange. The lone pair (lp)∙∙∙π
interactions are indicated by red dashed lines. The imidazole rings are numbered (1 4). b) A
perspective view of a column of metallocycles showing possible positions of the acetone guests
in the cavities of 3 (V = 132 Å3 per void).
The ASU of 4 contains of half a host and one acetonitrile guest disordered over two
positions of equal occupancy. Despite the comparable size of acetonitrile (3.80 x 5.49
Å) to methanol (3.80 x 4.7 Å), only one acetonitrile molecule can occupy each pocket
in 4 (Fig S5a). We propose that it is the coordination of the methanol that allows two
molecules to be accommodated in the cavity. Furthermore, guest-guest interactions
enable the methanol molecules to be closer to each other. Weak attractive forces
between the imidazole rings of the host in 4 and the acetonitrile guest draw the Im-Im
moieties closer together preventing further guest inclusion. One half occupancy CS2,
together with half a host molecule, constitute the ASU in 5. The CS2 guest is disordered
over two symmetry-related sites, but the host-guest ratio is only 1:1. The disorder in 5
is similar to that of 4 (Fig S5b). Apart from a weak C–H∙∙∙S interaction (Cdonor–
Sacceptor = 3.792(4) Å), no strong interactions between the host and guest exist.

S10
Figure S5 A perspective view of a column of metallocycles showing possible positions of a)
the acetonitrile, V = 106 Å3 and b) carbon disulphide, V = 108 Å3 guests in the cavities of 4
and 5.
The ASU of 6 also consists of half a host and a disordered guest molecule with a site-
occupancy factor of 0.5. The only significant host-guest interaction is a C–H∙∙∙Cl
hydrogen bond (Fig S6, C1A donor∙∙∙Cl1 acceptor = 3.72(8) Å). The dichloromethane
guests do not occupy the centre of the cavity, but are situated on either side to take part
in this host-guest interaction.
Figure S6 a) The ASU of 6 with selected atoms labelled. b) A column of metallocycles in
6 showing possible positions of the DCM guests (V = 118 Å3 per void).

S11
Having determined that 1a can accommodate a benzene guest molecule, single-crystals of 1
were immersed in p-difluorobenzene. The ASU of 7 contains half a host and half a guest. The
Connolly surface confirmed discrete cavities with volumes of 151 Å3 per void. The fluorine
atoms protruding from the Connolly surface imply interactions between adjacent hosts within
a stacked column (Cdonor∙∙∙Facceptor = 3.085(9)Å).
Figure S7 A perspective view of a column of metallocycles showing positions of the DFBz
guests in the cavities of 7 (V = 151 Å3 per void).
The ASU of the benzene solvate 8 contains half a host with an Im moiety disordered
over two positions (site occupancies of 64 and 46 %). One of the two partially occupied
benzene guest molecules is situated about an inversion centre (Fig S7a). It is apparent
from the size of the cavity and the electron count that a maximum of only one benzene
guest can be accommodated per host (Table S4). Owing to the strong π-character of the
aromatic guest, two host-guest interactions influence the occupancy (Cu2+∙∙∙∙π
coordination and π∙∙∙π stacking with the Im rings). The slightly higher occupancy of
guest A is facilitated by coordination to the metal centre (Fig S8a, Cu2+∙∙∙centroid ~ 3.86
Å). The π∙∙∙π interactions between the benzene guest and the Im rings of the metallocycle
are of similar strength (centroid∙∙∙centroid ~ 4.11 Å). The inclusion of benzene is
accompanied by an elongation of the Cu2+∙∙∙Cu2+ distance (from 6.61 to 7.07 Å).
Connolly surface analysis reveals that benzene inclusion results in enlarged discrete
voids wherein one green and two orange or one green and two yellow benzene molecules
can be accommodated per column pocket, with a cavity volume of 470 Å3 per unit cell
(Fig S8b). Owing to the Im rings rotating about the Cu–N coordination bond, it is
proposed that the Im rings can adopt either an “open” or “closed” conformation thus
tuning the cavity size for a specific guest (Fig S8c).

S12
Figure S8 a) The ASU of 8 (left), showing the metallocycle and two benzene guests molecules,
one of which is disordered over two sites (shown on the right). b) Perspective view showing
the merged discrete voids in 8 with both position of A and B represented in space fill. c) A
column of host molecules showing the “gate open” (blue) and “gate closed” (red) orientations
of the imidazole moieties that facilitate void merging.
a
b
A
B
OPEN
CLOSED
c

S13
A noteworthy adjustment of unit cell parameters occurs upon saturation with
chloroform. Although the space group symmetry remains identical, 1a undergoes
distortion in order to accommodate the relatively large guest (Fig. 9a). Since the Im rings
can rotate it is proposed that similar to 8, L1 in 9 can also adopt either an “open” or
“closed” conformation (Im∙∙∙Im = 6.223 Å and 5.610 Å, respectively). This was shown
to be dependent on close contact distances between the host and guest (Fig S9b). The
resulting distorted host is acentric, but the overall crystal structure of 9 is
centrosymmetric. Metallocycle distortion is accompanied by doubling of the a-axis,
with the chloroform inclusion inducing a considerable elongation of the Cu2+∙∙∙Cu2+ and
Im∙∙∙Im distances (6.45 Å to 7.07 Å and from 5.31 Å to 6.22 Å respectively). The
presence of the guest provides a stimulus to open or close the imidazole “gate”, yielding
a complex that consists of an adaptable binding site. These pockets are sensitive to the
shape and size of the guest. Remarkably, they can adapt their shape without any
significant loss in single-crystallinity.
Figure S9 Comparison of the pockets in a) 1a and b) 9. The length of the a-axis is indicated
with an arrow. 9 experiences doubling of the a-axis with im rings in the “open” (1-2) or
“closed” (3-4) conformation. Enlarged cavities form between two adjacent metallocycles and
enclose two chloroform guests, V = 297 Å3 per void

S14
The ASU of 10 consists of half a metallocycle and half a naphthalene guest molecule, at half
occupancy (Fig S10a, 2:1 host-guest ratio). One of the Im moieties in the metallocycle is
disordered over two positions of equal occupancy. The guest is located between two
neighbouring host molecules of a stacked column, rather than within the complex ring.
Although a similar arrangement is observed in 9, the size of naphthalene only permits one guest
per pocket. The host-guest interactions can be mapped on the Hirshfeld11 surface of the host,
using the program CrystalExplorer12 (Fig 10c). Each metallocycle shares a guest with only one
neighbouring metallocycle in order to avoid repulsive van der Waals contacts between nearby
guests. Thus, an enlarged pocket is formed between two hosts by means of a similar mechanism
explained for the sorption of chloroform (Fig 1b-1c). Removal of naphthalene from 10 occurs
readily at room temperature (Fig S13). The percentage mass loss on the TGA thermogram also
corresponds to a host-guest ratio of 2:1. Furthermore, the DSC thermogram reveals two
endothermic peaks (Fig S13). The sharp peak at ±80°C correlates to the melting point of
naphthalene (Table S2). The broad peak at ±110 °C is thought to be a host distortion, a
consequence of naphthalene diffusing out of the cavities, reverting the structure back to that of
1a. Owing to its molecular volume and high vapour pressure at room temperature, the
permeability to p-dichlorobenzene was also tested (Table S2).
Figure S10 a) ASU of 10 showing one disordered imidazole group. b) Perspective view
showing the elongated pockets in 10. c) Hirshfeld surface analysis was carried out for the
metallocycle and the position of the naphthalene guest is illustrated; red surfaces reveal strong
interactions between host and guest.
a
c
b

S15
SCD analysis of 11 indicated an ASU comprised of half a metallocycle and half a p-
dichlorobenzene guest molecule. The solvent accessible space in 11 contains a void volume of
341 Å3 per unit cell. Fig 1d illustrates the tight fit of the guest molecules within the empty
space. Instead of forming a temporary channel between neighbouring metallocycles as
observed for 10, the hosts accommodate the large guests by creating a permanent p-
dichlorobenzene-filled channel. The reversibility of this host deformation is supported by the
broad endotherm in the DSC thermogram (Fig S14). We suspect that the strong guest-guest
interactions play a major role in both the tight fit in the channel and the hosts strong affinity
for p-dichlorobenzene (Cl∙∙∙Cl = 4.181 Å, ∟C–Cl∙∙∙Cl = 110.67°). Strong host-guest
interactions also contribute to the 1a’s affinity for p-dichlorobenzene (π∙∙∙π stacking:
centroid∙∙∙centroid ~ 3.642 Å; C–H∙∙∙π hydrogen bond: Cdonor∙∙∙centroid ~ 3.532 Å). Unlike
most of the guests investigated, p-dichlorobenzene does not desorb readily at room temperature
and can only be removed by heating above 70 °C. The permeability of these guests was further
investigated kinetically using an in-house constructed gravimetric system.13 Freshly ground
samples of 1a where exposed to volatile solids for 24 hours. The host-guest ratio of 2:1,
observed in the SCD of 10, is reached after 700 minutes .Kinetic sorption measurements for p-
dichlorobenzene showed an occupancy of 0.9 after approximately 1000 minutes. (Fig S15). A
1:1 host-guest ratio consistent with the crystallographic data was observed.
Thermogravimetric Analysis
Thermogravimetric analyses were carried out using a TA Instruments Q500 thermogravimetric
analyser to record the weight loss of samples 1-11 as a function of temperature. Sample
preparation was carried out using the powdered phase of 1 for 1-9 and 1a for 11-12. In each
case, the sample was exposed to the relevant liquid or vapour for one week before analysis.
The balance and sample were purged with dry N2 gas flowing at rates of 50 and 70 cm3 min-1,
respectively. Samples masses typically ranged from 2-7 mg and samples were heated at a
constant rate of 10 °C min-1 from 30 °C to 300 °C. Thermograms were analysed using the TA
Instruments Universal Analysis program13 and are summarised in Figure S12. TGA
thermograms indicate that in all cases the single-step desolvation process occurred readily,
even at room temperature, indicating that gentle heating is sufficient for breaking the host-guest
interactions present in complexes. Note that decomposition of the metallocycle framework
occurs above 250 °C in all cases.

S16
Figure S11 Thermogravimetric traces for 1 (blue) and 1a (green).
Figure S12 Thermogravimetric traces of nine organic guests encapsulated in 1-9. Since
metallocycle decomposition occurs at 250 °C, each sample was only heated to 250°C.
40
50
60
70
80
90
100
30 80 130 180 230 280
We
igh
t %
Temperature (°C)
84
86
88
90
92
94
96
98
100
30 80 130 180 230
We
igh
t %
Temperature/ °C
1 2 3 4 5 6 7 8 9

S17
Differential scanning calorimetry (DSC)
Differential scanning calorimetry was carried out using a TA Instruments Q100 differential
scanning calorimeter. Samples were prepared by crimping the sample pan and lid (a pin hole
was placed in the lid to prevent pressure build-up). A reference pan was prepared in the same
manner for each analysis. Analyses were generally carried out in the temperature range 20-200
ºC and a general experimental procedure consisted of heating/cooling the sample while the heat
flow into or out of the sample, relative to the reference, was measured as a function of time and
temperature in a controlled atmosphere. N2 gas, flowing at a rate of 50 ml. min-1 was used to
purge the furnace. The resulting thermograms were analysed using TA Instruments Universal
Analysis program and figures were prepared with Microsoft Excel (Figure S13-S14)
Figure S13 Thermoanalytical data for desorption of 10. The TGA thermogram is shown in
blue and the DSC thermogram in red. Desorption of naphthalene results in a mass loss of
approximately 6.41%.
-18
-16
-14
-12
-10
-8
-6
-4
-2
0
70
75
80
85
90
95
100
30 80 130 180 230 280
He
at
Flo
w (
W/g
)
We
igh
t %
Temperature (°C)

S18
Figure S14 Thermoanalytical data for desorption of 11. The TGA thermogram is shown in
blue and the DSC thermogram in red. Desorption of p-dichlorobenzene results in a mass loss
of 15.83%.
Volumetric sorption of volatile organic solids
The permeability to these guests was further investigated kinetically using an in-house
constructed gravimetric system based on an instrument that was previously used for such
measurements.14 Data obtained from the sorption experiments were plotted as the occupancy
(mol guest/mol host) versus the measurement time (Figure S15).
-6
-5.5
-5
-4.5
-4
-3.5
-3
-2.5
-2
-1.5
-1
75
80
85
90
95
100
30 80 130 180 230 280
Hea
t fl
ow
(W
/g)
We
igh
t %
Temperature (°C)

S19
Figure S15 Kinetic isotherms (measured at room temperature) for naphthalene (green) and
p-dichlorobenzene (yellow) at guest vapour pressures of 0.08 and 1.76 mmHg, respectively.
Sorption isotherms were measured using an in-house constructed gravimetric system.15 Full
occupancy for naphthalene uptake (host:guest ratio = 2:1) is reached after approximately 700
minutes. Full occupancy for p-dichlorobenzene uptake (host:guest ratio = 1:1) is not reached
in the experimental time allowed.
0.0
0.2
0.4
0.6
0.8
1.0
1.2
0 200 400 600 800 1000
Oc
cu
pa
nc
y (
mm
ol/
mm
ol)
Time (min)

S20
References 1. L. Dobrzańska, G. O. Lloyd, H. G. Raubenheimer, L. J. Barbour, J. Am. Chem. Soc.,
2006, 128, 698.
2. SMART Data Collection Software, Version 5.629, Bruker AXS Inc., Madison, WI, 2003.
3. SAINT Data Reduction Software, Version 6.45, Bruker AXS Inc., Madison, WI, 2003.
4. SADABS, Bruker AXS Inc., Madison, WI, 2014.
5. G. M. Sheldrick, Acta Crystallogr., Sect. C, 2015, 71, 3.
6. L. J. Barbour, J. Supramol. Chem., 2001, 1, 189.
7. a) M. L. Connolly, Science, 1983, 221, 709. ;b) M. L. Connolly, J. Mol. Graphics, 1993, 11,
139.
8. a) A. L. Spek, Acta Crystallogr., Sect. D, 2009, 65, 148.; b) P. Van Der Sluis, A. L. Spek,
Acta Crystallogr., Sect. A, 1990, 46, 194.
9. T. Jacobs, M. W. Bredenkamp, P. H. Neethling, E. G. Rohwer, L. J. Barbour, Chem.
Commun. 2010, 46, 8341.
10. T. J. Mooibroek, P. Gamez, J. Reedijk, CrystEngComm. 2008, 10, 1501.
11. F. L. Hirshfeld, Theor. Chim. Acta, 1977, 44, 129.
12. S. K. Wolff, D. J. Grimwood, J. J. McKinnon, D. Jayatilaka and M. A. Spackman, Crystal
Explorer 2.1 (381), University of Western Australia, Perth, 2007.
13. Universal Analysis 2000, Version 4.5A, TA instruments, Waters-LLC, 2007.
14. L. Dobrzańska, G. O. Lloyd, H. G. Raubenheimer, L. J. Barbour, J. Am. Chem. Soc. 2006,
128, 698.
15. L. J. Barbour, K. Achleitner, J. R. Greene, Thermochim. Acta 1992, 205, 171.