structure function relationship of the nucleocapsid
TRANSCRIPT

MOLECULAR CHARACTERIZATION OF THE NUCLEOCAPSID PROTEIN OF
ARTERIVIRUSES
A Thesis
Presented to
The Faculty of Graduate Studies
of
The University of Guelph
by
HAKIMEH MOHAMMADI
In partial fulfilment of requirements
for the degree
Doctor of Philosophy
January 2010
© Hakimeh Mohammadi, 2010

ABSTRACT
MOLECULAR CHARACTERIZATION OF THE NUCLEOCAPSID PROTEIN OF
ARTERIVIRUSES
Hakimeh Mohammadi Advisor:
University of Guelph, 2010 Dr. Dongwan Yoo
Co-advisor:
Dr. Shayan Sharif
Nuclear localization of the nucleocapsid (N) protein of arteriviruses and the
possible function of N in the nucleus for host cell function modulation were studied.
Subcellular localization of N of lactate dehydrogenase-elevating virus (LDV) was
determined by tagging N with enhanced green fluorescence protein (EGFP) on the N- and
C-termini. Both N-EGFP and EGFP-N fusion proteins were found to localize to the
nucleus and nucleolus of gene-transfected cells. A ‘pat4’ motif was identified in N as a
potential nuclear localization signal (NLS), and its functional significance was
determined by expressing a series of deletion constructs. The results showed that the
‘pat4’ NLS in LDV N was essential for nuclear translocation. LDV N interacted with
importin-α and -β proteins suggesting that its nuclear localization is mediated through the
importin-dependent nuclear transport pathway. This study was expanded to the equine
arteritis virus (EAV) N protein. The EAV N gene was fused with EGFP at the N- or C-
terminus, and its cellular distribution was investigated in gene-transfected cells. Both N-
EGFP and EGFP-N fusion proteins of EAV N accumulated in the nucleus and nucleolus
in addition to the cytoplasm. A series of deletion mutants were made by progressively

deleting amino acids from the C- and N-termini of EAV N to determine the functional
significance of the NLS-motif at amino acids positions 4-16. Studies using the mutant
genes showed that the EAV N nuclear localization required the entire amino acid
sequence from 4-20 position. EAV N interacted with both importin-α and -β proteins but
also with some other fusion proteins suggesting that factors other than the importin-
regulated nuclear transport pathway may be involved in this process. The simian
hemorrhagic fever virus (SHFV) N gene was cloned as a fusion protein with EGFP. The
functionality of the potential ‘pat7’ type NLS present at amino acid positions 22-28 of
SHFV N was determined by constructing a series of deletion mutants and individually
expressing them in cells. The results showed that the ‘pat7’ NLS was essential for nuclear
translocation of SHFV N. The interaction of SHFV N with importin-α suggests that N
recruits this chaperone protein for its nuclear localization. These finding confirm that the
nuclear localization of N is a common property of arteriviruses. Next, modulation of cell
cycle by arteriviruses was investigated. Replication of PRRSV and EAV in cells lead to
accumulation of cells at the G2/M phase. Furthermore, cells expressing N protein from
all four arteriviruses indicated that the number of cells in the G2/M phase was higher
compared to controls. The modulatory effect of N protein on G2/M arrest was similar for
all four N proteins, although PRRSV N and EAV N showed stronger effects on the cell
number increase at the G2/M phase than LDV N and SHFV N. The results obtained from
this research improve our understanding of N protein nuclear localization and its
biological role during infection by members of the Arteriviridae family.

i
ACKNOWLEDGEMENTS
This is an honor for me to thank those who made this thesis possible:
I would like to express my sincere gratitude to my advisor, Dr. Dongwan Yoo, for
providing me with the great opportunity to prepare my research under his supervision in
his laboratory. He has been of such a great help and support throughout in many aspects
including but not restricted to writing grant proposals, scholarship applications and this
thesis. I am grateful to his support, patience and guidance and will always be.
I would like to render my heartfelt appreciations to Dr. Shayan Sharif who
thoughtfully and kindly served as my co-advisor after Dr. Yoo left, providing unfaltering
support and learning opportunity in his lab. From the day one I started my PhD, I have
always relied on his sage hints and discernment. His enthusiasm, inspiration, and great
efforts in explaining complicated matters so perceivably have secured him one of my best
teachers and mentors ever. Throughout the ups and downs of my research and thesis-
writing period as well as the hard times in my life he, with his heart of gold has always
been one of a kind of encouragement, support, initiation, and morale. I would have been
lost without his help in those dire days of dismal doldrums. Moreover by allowing me to
be a part of his laboratory I was privileged to get to know with many nice and talented
people in the laboratory. I doubt that I could ever be able to fully convey my gratitude to
him the way I feel it and hence suffice to round up with saying that I am eternally grateful
to him.
I would also like to thank the other members of my committee, Dr. Dorothee
Bienzle, Dr. Ray Lu, and Dr. Sarah Wootton for the great succor they all provided me
with during this research project. Special thanks go to Dr. Bienzle’s extremely helpful
comments throughout the work, using DAPI in particular as well as creating learning and
training opportunities of professional software and instruments. I am very grateful to Dr.
Wootton who generously not only allowed me to complete my research project after
taking Dr. Yoo’s laboratory over but has also made an invaluable companion, both
professional and confidant.
I wish to thank the members of the Department of Pathobiology including
professors and staff who have helped me from the day one I started my PhD. I would
especially like to thank the Chair of the Department Dr. Robert Jacobs for his kindness
and help at the gloomy times of despair. Without his full support and management I
would have not been able to complete my degree. My especial thanks go to Betty-Anne
McBey who has been always helpful in FACS area and without her help and effort
nothing can be done there. Donna Kangas, Jean Bagg, Cathy Bernardi, Elizabeth
Gilbertson, and many others deserve special mention.
I would also like to thank my old colleagues in Dr. Yoo’s past laboratory
including Frances Lai, Paul Rosenfeld, Dr. Yanlong Pei, Cheng Song, Dr. Gang Li, Dr.

ii
Changhee Lee, Sheila Watson, and Maged Gomaa for their friendship, help and
willingness to participate in the scientific environment we had in the laboratory.
I would like to thank to all friends and colleagues in Dr. Sharif’s lab who so
warmly welcomed, supported and treated me that the lab time became an enjoyable span
of day: Leah Read, Hamid Haghighi, Payvand Parvizi, Niroshan Thanthrige, Kamran-Ul
Haq, Raveendra Kulkarni, Jen Brisbin, Amirul Mallick, and other members of this lab.
During my PhD at Guelph, I have met many wonderful people in the Department,
OVC, and university of Guelph who have offered their unrestrained friendship and help. I
am especially thankful for the International Advisor Mr. Benny Quey who has been a full
support whenever needed. I am so blessed to have so many friends and family members
who have been all help and for them I am always grateful.
I would also like to thank my and my husband’s families for the support they have
provided me through my entire life. Particularly I would like to thank my parents
Mohammadali Mohammadi and Sara Hosaini for their unconditional love and support
from the day I was born. Without their support and love I would not be able to stand at
the place I am in now. I would also like to thank my brothers and sisters, and my brother-
and sister- in law. They have always been inspiring me with love and support. I would
also very much like to remember the dear memories and supports of my departed brother
and mother-in-law who always loved and encouraged me to study and overcome the
difficulty that life might throw in.
And the last but most importantly, my great appreciation is for the greatest ocean
of affection, love and support; my loving husband, Alireza Omumi, who has been my
best friend and comrade in bad and good times. Without your passionate love, support,
encouragement and inspiring assistance, I would not be able to complete this thesis. I am
here now because you have always been inspiring, supportive and patient. I am forever
grateful to your kind heart.
In conclusion, I recognize that this research would not have been possible without
the financial assistance of “Iranian Ministry of Health and Medical Education” that
provided me the full scholarship for 4 years and Dr. Yoo’s funds (Natural Sciences and
Engineering Research Council, and US Department of Agriculture National Research
initiative), as well as the support of the department of pathobiology and university of
Guelph. I express my gratitude to those.

iii
I wish to dedicate this thesis to my departed brother, Abdolkarim Mohammadi.

iv
TABLE OF CONTENTS
ACKNOWLEDGEMENTS...…..…………………………………………………………i
TABLE OF CONTENTS……………………………………….………………………..iii
LIST OF TABLES…………………………………………………….………………….vi
LIST OF FIGURES……………………………………………………………………...vii
LIST OF ABBREVIATIONS……………………………………………………..…...…ix
INTRODUCTION……………………………...…………………………………………1
CHAPTER 1:
I. Review of Literature
1. The family Arteriviridae, Classification, and the Diseases………….…2
1.1 History and Taxonomy
1.2 Diseases Caused by Arteriviruses
1.3 Transmission
2. Virion Structure and Genome Organization of Arteriviruses…………..4
2.1 Virus Particle and Virion Composition
2.2 Genome Organization
2.3 Major Structural Proteins
2.3.1 M protein
2.3.2 Glycoprotein 5
2.3.3. Nucleocapsid (N) protein
2.4 Minor Structural Proteins
3. Life Cycle of Arteriviridae……………………………………………12
3.1 Cell Tropism
3.2 Attachment and Entry
3.3 Replication and Transcription
3.4 Virion Assembly and Release from Cell
4. Pathogenesis of Arteriviruses…………………………………………21
4.1 Natural Infection and Persistence
4.1.1 Equine Arteritis Virus (EAV)
4.1.2 Porcine Reproductive and Respiratory Syndrome
Virus (PRRSV)
4.1.3 Lactate Dehydrogenase Elevating Virus (LDV)
4.1.4 Simian Hemorrhagic Fever Virus (SHFV)
4.2 Immune Response to Arterivirus Infections.
4.2.1 Innate Immune Response to Arteriviruses
4.2.2 Adaptive Immune Response to Arteriviruses
5. Nuclear Localization of Viral Proteins in +ssRNA viruses……..…….28

v
5.1 Nucleolus and Nuclear Localization of Proteins
5.2 Nucleolar Localization of +ssRNA Virus Proteins
5.3 Function of Nuclear Localization of Viral Nucleocapsid
Proteins
5.4 Modulation of Cell Cycle by Viral Proteins
5.5 Nucleocapsid Protein in Arteriviridae
II. Objectives of the Thesis………………………………………………………44
CHAPTER 2: The Lactate Dehydrogenase-Elevating Virus Capsid Protein is a Nuclear-
Cytoplasmic Protein.
Abstract…………………………………………………………………………..47
Introduction………………………………………………………………………48
Materials and Methods…………………………………………………………...50
Results……………………………………………………………………………54
Discussion………………………………………………………………………..64
CHAPTER 3: Nuclear Localization of Equine Arteritis Virus and Simian Hemorrhagic
Fever Virus Nucleocapsid Proteins.
Abstract…………………………………………………………………………..69
Introduction………………………………………………………………………70
Materials and Methods………………………………………………………...…72
Results……………………………………………………………………………80
Discussion………………………………………………………………………..96
CHAPTER 4: The Role of Nucleocapsid Protein of Arteriviruses in Cell Cycle
Modulation.
Abstract…………………………………………………………………………102
Introduction……………………………………………………………………..103
Materials and Methods………………………………………………………….105
Results…………………………………………………………………………..108
Discussion………………………………………………………………………114
CHAPTER 5: General Conclusions……………………………...……………………..119
REFERENCES…………………………………………………………………………124

vi
LIST OF TABLES
CHAPTER 2: The lactate dehydrogenase-elevating virus capsid protein is a nuclear-
cytoplasmic protein.
Table 2.1. List of primers used for EGFP fusion constructions for LDV N……………..51
Table 2.2. Site-directed mutagenesis of LDV N NLS motif and subcellular localization of
NLS mutants……………………………………………………………………………..60
CHAPTER 3: Nuclear localization of equine arteritis virus and simian hemorrhagic
fever virus nucleocapsid proteins.
Table 3.1. List of primers used for EGFP fusion constructions for EAV N…………….74
Table 3.2. List of primers used for EGFP fusion constructions for SHFV N…………...75
Table 3.3. Site-directed mutagenesis of the SHFV N NLS motif and subcellular
localization of NLS mutants……………………………………………………………..93

vii
LIST OF FIGURES
CHAPTER 1: Literature review
Figure 1.1: Schematic representation and genome organization of arteriviruses…………5
Figure 1.2: Overview of the genomes of the arteriviruses………………………………...7
Figure 1.3: Schematic representation of the life cycle of arteriviruses…………………..16
Figure 1.4: The nested set of RNAs in arteriviruses and the discontinuous strategy for
mRNA transcription………...…..…………….………………………………………….19
Figure 1.5: The schematic illustration of the nuclear protein transport …………………30
Figure 1.6: Schematic presentation of a cell cycle……………………………...……….38
CHAPTER 2: The lactate dehydrogenase-elevating virus capsid protein is a nuclear-
cytoplasmic protein.
Figure 2.1: Subcellular localization of LDV-N protein………………………………….55
Figure 2.2: Identification of an NLS motif in the LDV-N protein and its functional
mapping…………………………………………………………………………………..58
Figure 2.3: Subcellular localization of NLS mutants…………………………………….61
Figure 2.4: Subcellular localization of the NLS-null LDV-N-EGFP mutants in HeLa and
NIH-3T3 murine cells…………………………………………………………………....62
Figure 2.5: Interactions of LDV-N with mouse importin proteins by GST-pull down
assays…………………………………………………………………………………….65
CHAPTER 3: Nuclear localization of equine arteritis virus and simian hemorrhagic
fever virus nucleocapsid proteins.
Figure 3.1: Confirmation of the EAV N nuclear localization ……………………….......81
Figure 3.2: Subcellular localization of EAV N protein……………………………….....82
Figure 3.3: Identification of NLS in EAV N protein and its functional mapping……….84
Figure 3.4: Subcellular localization of EAV N NLS mutants…………………………...86
Figure 3.5: Interaction of EAV-N with importin proteins by GST-pull down assay……87

viii
Figure 3.6: Subcellular localization of SHFV-N protein………………………………...89
Figure 3.7: Identification of NLS in SHFV N protein and its functional mapping……...90
Figure 3.8: Subcellular location of the NLS-null SHFVN-EGFP mutant……………….94
Figure 3.9: Interaction of SHFV N with importin proteins by GST-pull down assay…...95
CHAPTER 4: The role of nucleocapsid protein of arteriviruses in cell cycle modulation.
Figure 4.1: Experimental design for analysis of Arteriviruses and their nucleocapsid
protein effects on cell cycle…………………………………………………………….107
Figure 4.2: Co-localization of LDV N and EAV N with fibrillarin…………………….109
Figure 4.3: CPE of PRRSV infected MARC-145 cell at different times ………………111
Figure 4.4: Cell cycle profile of Mock/PRRSV infected MARC cells…………………112
Figure 4.5: Cell cycle profile of mock/EAV infected BHK-21 cells…………………...113
Figure 4.6: Cell cycle profile in arterivirus N transfected BHK-21cells…….………....115

ix
LIST OF ABBREVIATIONS
BHK Baby hamster kidney
BSA Bovine serum albumin
CAS Cellular Apoptosis Susceptibility protein
Ci Curie
CPE Cytopathogenic effects
DAPI 4',6-Diamidino-2-phenylindole dihydrochloride
DFC Dense fibrillar component
DMEM Dulbecco’s minimum essential medium
DNA Deoxyribonucleic acid
dpi Day post infection
EAV Equine arteritis virus
E.coli Escherichia coli
EDTA Ethylenediamine tetraacetic acid
EM Electron microscopy
ER Endoplasmic reticulum
FBS Fetal bovine serum
FACS Fluorescence activated cell scanning
FC Fibrillar center
G1 First gap
G2 Second gap
GC Granular component
GDP Guanosine diphosphate
GP Glycoprotein
RanGEF Ran Guanine nucleotide -Exchange Factor
GST Glutathione-S transferase
GTP Guanosine-5'-triphosphate
h Hour
HCV Hepatitis C virus
HDV Hepatitis delta virus
hpi Hours post infection
HIV Human immunodeficiency virus
IBV Infectious bronchitis virus
ID50 Infectious dose 50%
IF Immunofluorescence
IgG Immunoglobulin G
Impα Importin α
Impβ Importin β
IPTG Isopropyle-β-D-thiogalactopyranoside
IRES Internal ribosome entry site
kDa Kilodalton
JEV Japanese encephalitis virus
JSs Junction sites
LD50 Lethal dose 50%
LDV Lactate dehydrogenase-elevating virus

x
LV Lelystad virus
M Mitosis
M protein Matrix protein
μl Microliter
ml Mililiter
MAb Monoclonal antibody
MHV Mouse hepatitis virus
MOI Multiplicity of infection
mRNA Messenger RNA
N Nucleocapsid protein
NE Nuclear envelope
NES Nuclear export signal
NLS Nuclear localization signal
NoLS Nucleolar localization signal
NPC Nuclear pore complex
Nsp Nonstructural protein
ORF Open reading frame
PAGE Polyacrylamide gel electrophoresis
PAMS Porcine alveolar macrophages
PBS Phosphate buffered saline
PCR Polymerase chain reaction
PFU Plaque forming unit
PMSF Phenylmethylsulfonyl fluoride
PRRSV Porcine reproductive and respiratory syndrome virus
Ran Ras-related nuclear protein
RanGAP Ran GTPase activating protein
RIs Replicative intermediates
RNA Ribonucleic acid
rRNA Ribosomal RNA
RT Reverse transcriptase
SARS Severe acute respiratory syndrome
SDS Sodium dodecyl sulfate
sgmRNA Subgenomic mRNA
SHFV Simian hemorrhagic fever virus
ssRNA Single stranded RNA
tRNA Transfer RNA
TCID50 50% tissue culture infectious dose
TGEV Transmissible gastroenteritis virus
UBF upstream binding factor
TLRs Transcription regulating sequences
UTR Untranslated region

1
INTRODUCTION
Arteriviridae is a family of viruses comprising four viruses some of which cause
important diseases in animals with major economic losses. These viruses are: equine
arteritis virus (EAV), lactate dehydrogenase-elevating virus (LDV), porcine reproductive
and respiratory syndrome virus (PRRSV), and simian hemorrhagic fever virus (SHFV).
These viruses have a linear positive stranded RNA as genome and based on similarity of
their genome organization and replication strategy with Coronaviridae are categorized in
Nidovirales order of viruses. They replicate in the cytoplasm of infected cells but it has
been reported that the nucleocapsid protein of PRRSV and EAV, as one of the major
structural proteins can localize in the nucleus of infected cells and interact with nucleolar
proteins involved in cellular activities. A similar pattern of cellular distribution has been
reported for the nucleocapsid protein of coronaviruses. Understanding the nucleolar
localization of N protein of other members of arteriviruses demands further investigation
into its biological function and whether this is a common feature of members of this
family in the Nidovirales order. Moreover, previous studies of coronavirus N proteins and
their modulation of cell cycle have raised the possibility that the capsid protein of
Arteriviridae has a major role in cell cycle progression during virus replication. Thus,
elucidation of nuclear localization of capsid proteins of all four member viruses of the
Arteriviridae family and involvement in the cell cycle could open a window to
understand the importance of this feature in virus pathogenesis. Moreover, advancing our
understanding of viral N protein interactions with host proteins may lead to the design of
novel vaccines. The new strategies for vaccine against arteriviruses could target the N
protein as one of the most important viral proteins to achieve better prevention.

2
CHAPTER 1
I. REVIEW OF LITERATURE
1. Arteriviridae, Classification, and the Diseases
1.1 History and Taxonomy. The family Arteriviridae was established by the
International Committee on Txonomy of Viruses (ICTV) in 1996 (Cavanagh et al., 1994;
Cavanagh 1997). Arteriviridae is comprised of four member viruses: equine arteritis virus
(EAV), lactate dehydrogenase-elevating virus (LDV), porcine reproductive and
respiratory syndrome virus (PRRSV), and simian hemorrhagic fever virus (SHFV). Three
of these viruses, EAV, LDV, and SHFV have been known for the last 4-5 decades,
whereas PRRSV emerged in the late 1980s in Europe and early 1990s in North America
(Snijder and Meulenberg, 1998; Albina, 1997). These viruses cause different diseases in
susceptible animals, ranging from mild asymptomatic to persistent or severe clinical
syndromes. EAV is the prototype virus of the family and was first isolated in 1957 in
Bucyrus, Ohio (USA) from the lungs of horses suffering from abortion (Doll et al, 1957).
PRRSV is the causative agent of porcine reproductive and respiratory syndrome (PRRS)
(Collins et al., 1992; Rossow, 1998). LDV is the causative agent of an asymptomatic
persistent infection with lifelong viremia in mice. LDV has been mostly used as an in
vivo model in many investigations on persistent infections (Riley et al, 1960; Rowson and
Mahy, 1985). SHFV is the etiological agent of a fatal hemorrhagic fever of monkeys and
was first isolated in 1968 during several outbreaks of a febrile hemorrhagic disease in
colonies of macaque monkeys at the National Institute of Health (NIH) (Palmer at al,
1968; Allen et al, 1968; Tauraso et. al 1968). Although SHFV leads to 100% mortality in

3
macaque, it has been shown to cause an endemic asymptomatic infection in some other
genera of African monkeys (London et al, 1977; Gravell et al, 1986b).
A unique feature of the family Arteriviridae is the production of a nested set of
subgenomic (sg) mRNAs from their genome, a property shared with another family of
viruses, the Coronaviridae. Based on the similarity of their genome structure, mechanism
of gene expression, and replicase genes, Arteriviridae and Coronaviridae have been
placed in the new order of viruses, Nidovirales (the nidus in the Latin means the “nest’),
along with Roniviridae, a newly identified group of viruses in invertebrates (de Vries et
al., 1997).
1.2 Diseases caused by arteriviruses. The disease caused by EAV may be asymptomatic
or symptomatic with severe manifestations including edema, hemorrhage of small
arteries (hence the name EAV), and abortion in pregnant mares (Doll et al, 1957, and Del
Piero, 2000). PRRS is one of the most economically important viral diseases of the swine
industry. Manifestations of PRRSV-induced disease include severe reproductive failure
in sows and gilts with a high abortion rate and an increased number of stillborn and weak
piglets. The disease has also been related to respiratory problems due to interstitial
pneumonitis (Rossow, 1998). LDV infection in mice has no apparent clinical signs except
an increased activity of lactate dehydrogenase in plasma, from which the name of LDV is
derived (Plagemann et al., 1995). LDV has very limited in vitro replication with restricted
tropism to mouse primary macrophages (Brinton-Darnell et al., 1975; Brinton-Darnell
and Plagemann 1975; Stueckemann et al., 1982; Onyekaba et al., 1989a). LDV infection
is usually asymptomatic, but in specific strains of mice it has been shown to cause lethal
poliomyelitis due to cytocidal replication of the virus (Contag and Plagemann, 1989).

4
SHFV induces a lethal febrile hemorrhagic disease in different genera of Asian and
African monkeys (Palmer et al., 1968; Tauraso, et al., 1968).
1.3 Transmission. Both EAV and PRRSV can be spread mainly through the respiratory
system and both viruses can induce persistent infections in animals. Persistent infection
in male animals enables venereal transmission of the virus a common way for spread of
EAV and PRRSV (Timoney and McCollum 1993; Wensvoort et al., 1993). PRRSV has
been found in saliva, respiratory tract secretions, and urine. Also, the virus can be
transmitted via the placenta (Rossow, 1998). LDV is secreted in urine, feces, and saliva,
and horizontal transmission is not an effective route of LDV spread except in invasive
fighting in male mice. The transmission of LDV through placenta and milk is a more
effective route of virus transfer (Plagemann and Moennig, 1992). SHFV can transmit
through direct contact and aerosols, but unlike other members of the family, it is not
transmitted vertically via placenta to the fetus (Gravell et al., 1986a).
2. Virion Structure and Genome Organization of Arteriviruses
2.1 Virus Particle and Virion composition. Arteriviruses are a group of spherical
viruses with virion size of 45-60 nm in diameter (Fig. 1.1 a; Brinton, 1999; Snijder and
Meulenberg, 1998; Snijder and Spaan, 2007). Their core is an icosahedral structure of 25-
35 nm enclosed in a lipid envelope with small projections as viral proteins, which are
quite different from the long spikes seen in coronaviruses. These viruses are sensitive to
non-ionic detergents, pH > 7.5 and < 6, and storage at 4oC or higher temperature
(Horzinek et al., 1971; Brinton-Darnell and Plagemann, 1975; Sagripanti, 1984;
Meulenberg et al., 1995a; Burki, 1966; Tauraso et al, 1968; Snijder and Meulenberg,
1998).

5
Fig. 1.1. (A) Schematic representation of PRRSV particle. The virion of arteriviruses is
spherical surrounding an icosahedral capsid formed by the nucleocapsid protein (N)
which in turn encloses the positive sense RNA genome of the virus. The membrane is
composed of five proteins: the membrane protein (M), the major glycoprotein (GP)5, the
minor glycoproteins GP2a and GP4, and the envelope (E) protein. The GP3 is believed to
be structural for the European PRRSV and a secretory protein in case of the North
American type of this virus (Adapted from Dea et al, 2000). (B) The genome
organization of EAV, prototype of the Arteriviridae family of viruses, and transcription
of subgenomic mRNAs (adapted from Snijder and Meulenberg, 1998).

6
Arteriviruses have a buoyant density of 1.13 to 1.17 g/cm3 in sucrose and their
sedimentation coefficient varies from 214 S to 230 S.
A schematic figure of these viruses is shown in Fig. 1.1 a. The nucleocapsid of the
family Arteriviridae contains a positive-sense single-stranded RNA (ssRNA) genome of
12.7 to 15.7 kb enclosed by the nucleocapsid protein (N) (Brinton et al, 1999, Snijder and
Meulenberg, 1998; Snijder and Spaan, 2007). Studies on EAV and PRRSV have
suggested that there are six envelope proteins embedded in the lipid bilayer envelope of
arteriviruses. For all members of the family, the three major structural proteins that have
been described as virion components are the nucleocapsid protein N (12-15 kDa), the
non-glycosylated membrane protein M (16-20 kDa) and the major glycoprotein (GP) (22-
44 kDa, GP5 in PRRSV, LDV and EAV and GP7 in SHFV). M and GP5 of PRRSV, EAV
and LDV form a heterodimer via a disulfide linkage (de Vries et al., 1995a; Faaberg et
al., 1995; Snijder et al., 2003). The major structural proteins are encoded by open reading
frames (ORF) located at the 3’ end of the viral genome (Snijder and Spaan, 2007). The
minor structural proteins include GP2 (coded by ORF2a of PRRSV and ORF2b of
EAV/LDV), GP3, GP4, and the small non-glycosylated envelope protein E (Snijder and
Spaan, 2007). GP2, GP3, GP4 of PRRSV and EAV have been shown to form heterotrimers
in the virus particle (Wieringa et al., 2003; Wissink et al., 2005). Using reverse genetics,
it has been shown that all major and minor structural proteins are necessary in order to
produce infectious EAV (Wieringa et al., 2004).
2.2 Genome Organization. Viruses in the family Arteriviridae contain a single-stranded
positive-sense RNA genome with a length of 12.7 to 15.7 kb (Snijder and Swaan, 2007).

7
Fig. 1.2. Overview of the genomes of the arteriviruses. The genome organization of the
family prototype EAV is shown at the top. The genome includes the replicase open
reading frames (ORF) 1a and 1b which are followed by ORFs 2-7 (9 in SHFV) encoding
for structural proteins. The structural genes include genes for the E protein, three minor
glycoproteins (GP2a/b-4), major glycoprotein GP5 (GP7 in SHFV), and the genes for the
membrane (M) and nucleocapsid (ORF7) proteins. The 3’ end of the SHFV genome has a
large insertion which is assumed for the putative extra ORFs (Snijder and Spaan, 2007).

8
The genome of SHFV has been reported to have a type I cap structure (Sagripanti et al.,
1986). The necessity of cap structure for in vitro transcription of infectious viral RNA
from the full-length cDNA clones of PRRSV and EAV has been described. The full-
length genomes of all members including two European and North American isolates of
EAV, more than 20 European and North American isolates of PRRSV, LDV strains, and
one isolate of SHFV have been sequenced (Fig. 1.2) (Snijder and Spaan, 2007).
Approximately three-fourth of the 5’ end of the genome is comprised of two large ORFs,
called ORF1a and ORF1b, which encode non-structural proteins. The ORF1a-encoded
polyprotein is more variable in sequence and size among members of the family
compared to the ORF1b-encoded polypeptide (Snijder and Meulenberg, 1998). The 3’
part of the genome of EAV, LDV, and PRRSV includes a group of seven smaller
overlapping ORFs which encode structural proteins of these viruses. SHFV genome
contains three additional ORFs at the 3’ end. These ORFs are located between ORF1b
and ORF5 of the other three members of the Arteriviridae family (Fig. 1.2). The
significance of these extra ORFs in the SHFV genome is not clear. In the 3’ end of the
replicase gene and in the leader sequence of EAV, PRRSV, and SHFV, the presence of
small ORFs have been reported with no clear significance (Snijder and Meulenberg,
1998).
2.3 Major Structural Proteins. The three most abundant viral proteins in arteriviruses
are GP5 (GP7 in SHFV), M, and N. These proteins constitute 90-95% of the structural
proteins of the virion and are expressed from three ORFs located at the 3’ end of the
genome in the mentioned order (Snijder and Spaan, 2007).

9
2.3.1 M Protein. The M protein is a non-glycosylated structural protein of arteriviruses
and is known to be one of the most abundant structural proteins. The M protein is the
most conserved structural protein in this group of viruses. Its configuration and function
are similar to the M protein of Coronaviridae and is suggested to be a membrane protein
(de Vries et al., 1992; Kuo et al., 1992; Meulenberg et al., 1993b). The N-terminal half of
the arterivirus M protein is thought to span the membrane three times (de Vries et al,
1992; Faaberg and Plagemann, 1995; Meulenberg et al., 1993b) resulting in a
configuration consisting of the N-terminus of the M protein at the external and the C-
terminus of M towards the internal sides of the virion as well as exposure of a short
stretch of amino acids (10-18 residues) at the surface of the virion (de Vries et al., 1995a;
Faaberg et al., 1995a; Mardassi et al., 1996). It is proposed that the M protein of
Arteriviridae has an important role in assembly and budding of viruses (Wieringa et al.,
2004). The M protein is localized in the endoplasmic reticulum (ER) and through the Cys
residues of its N-terminal ectodomain binds to the Cys residues of GP5, leading to the
formation of heterodimer of M-GP linked by disulfide bonds (de Vries et al., 1995a;
Faaberg et al., 1995a; Mardassi et al., 1996). The disulfide linked M-GP5 heterodimers
have been detected in PRRSV and EAV infected cells (de Vries et al., 1995a). It has also
been reported that disruption of this disulfide bond by treating LDV virion with DTT
decreases the infectivity of this virus. This finding indicates the importance of M-GP5/7
heterodimer in virus infectivity likely at the beginning of the replication cycle (Faaberg et
al., 1995a).
2.3.2 Glycoprotein5/Glycoprotein7 (GP5/GP7). GP5/GP7 is the major GP of
Arteriviridae and is a hydrophobic protein. The internal part of GP5 is presumed to

10
traverse the viral membrane three times (Snijder and Meulenberg, 1998). The size of the
ectodomain is smaller in PRRSV and LDV compared to the EAV equivalent. However, a
common feature in all three viruses is the presence of an N-glycosylation site in
ectodomain. Glycosylation of GP5 of EAV and LDV, is the result of adding different
numbers of repeated lactosamines (de Vries et al., 1992; Li et al., 1998). Neutralizing
antibodies are mainly produced against GP5 in mice and horses infected with LDV and
EAV, respectively (Cafruny et al., 1986; Chirnside et al., 1995a, 1995b). Moreover,
monoclonal antibodies (MAbs) against PRRSV GP5 have been found to neutralize
PRRSV infection (Pirzadeh and Dea, 1997). Reports have shown that the ectodomain of
GP5 is the target for neutralizing antibodies in EAV-infected horses and for MAbs raised
against GP5 (Chirnside et al., 1995a; Balasuriya et al., 1997). The reports also show that
mutations or deletions of this portion of GP5 help EAV mutants escape the host immune
response without affecting their infectivity (Balasuriya et al., 1995; Glaser et al., 1995;
Balasuriya et al., 1997). A similar location of the epitope for neutralizing antibodies has
been shown in the ectodomain of the GP5 glycoprotein of LDV (Li et al., 1998). The
number of glycosylation sites at the ectodomain of GP5 is different in LDV strains
(Faaberg et al., 1995b). This difference is suggested to be involved in the persistence and
neurovirulence of LDV. Li et al. (1998) demonstrated that binding of neutralizing
antibodies to the highly glycosylated GP5 of LDV is decreased.
2.4 Minor Structural Proteins. Several minor structural proteins have been
characterized for arteriviruses. The ORF2a of PRRSV and ORF2b of EAV/LDV encode
GP2 (previously called GPs for EAV). GP2 is a part of the virion in PRRSV and EAV. It
is a type I integral membrane protein and has a signal peptide at the N-terminus and a

11
transmembrane domain located at the C-terminus. This protein is suggested to be N-
glycosylated (de Vries et al., 1992; Meulenberg and Petersen-den Besten, 1996). There
are cysteine residues of EAV GP2, which form not only intrachain disulfide bonds in
GP2-GP2 homodimers found in the virion, but also intermolecular disulfide bonds with
the ectodomain of another minor GP (GP4), resulting in the formation of GP2-GP4
disulfide-linked complexes (Wieringa et al., 2003). The GP2 protein of PRRSV and EAV
remains in the ER when expressed as a recombinant protein (de Vries et al., 1995b;
Meulenberg and Petersen-den Besten, 1996). This suggests that it needs other structural
proteins or assembly of the virion for translocation from the ER to the Golgi complex.
Recently, an ion channel function for the PRRSV E protein has been reported which
suggests that the E protein might have a function in fusion of the virus envelope with the
endosomal membrane leading to disassembly of the genome and nucleocapsid of PRRSV
after entry (Lee and Yoo, 2006). GP3 and GP4 are the glycoproteins encoded by ORF3
and ORF4 of PRRSV, EAV, and LDV, respectively. GP3 is an integral membrane protein
which is highly glycosylated. This glycoprotein is one of the minor structural proteins of
the PRRSV virion (van Nieuwstadt et al., 1996), but it has yet to be detected in EAV and
LDV virions or cells infected with these viruses. The GP3 and GP4 proteins of LDV
have been shown to be soluble and membrane-attached, respectively, in vitro (Faaberg
and Plagemann, 1997). GP4 is also a type I integral membrane protein. MAbs raised
against PRRSV ORF4 protein have been shown to possess neutralizing activity and the
MAb-binding site has been reported to be a highly heterogeneous region of this protein in
different strains of PRRSV (van Nieuwstadt et al., 1996; Meulenberg et al., 1997).
Heterotrimers of GP2, GP3, and GP4 have been reported for EAV (Wieringa et al., 2003).

12
There are three more ORFs located at the 3’ end of SHFV genome which are suggested to
be replicas of ORFs 2 to 4 (Godeny et al., 1998).
3. Life Cycle of Arteriviridae
3.1 Cell Tropism. Viruses in the Arteriviridae family have limited growth ability in
vitro. Primary macrophages from natural hosts are the select cells for their replication.
LDV has a very limited growth in murine macrophage, and it only replicates in specific
primary macrophages of mice (Plagemann and Moening, 1992). SHFV and PRRSV
replicate in simian and porcine macrophages, respectively. They both replicate in African
green monkey kidney cells (MA-104) and its derivatives (CL2621 and MARC-145)
(Gravell et al, 1986b; Benfield et al, 1992; Kim et al, 1993). Some strains of PRRSV
replicate better in porcine alveolar macrophages (PAMs) while some others grow better
in CL2621 (Bautista et al, 1993). Unlike other arteriviruses, EAV infects and replicates in
kidney cell lines such as baby hamster kidney (BHK-21; Hyllseth, 1969), rabbit kidney
(RK-13; Snijder and Meulenberg, 1998), and African green monkey kidney (Vero cells;
Konishi et al, 1975) as well as in primary macrophages. Using one-step growth
experiments, it has been shown that it takes 10 to 20 hrs to detect the first virus particles
released from virus-infected cells (Stueckemann et al., 1982; van Berlo et al., 1982;
Gravell et al., 1986b; Kim et al., 1993). High rates of cytolysis and cell death result from
infection of primary macrophages and cell lines by these viruses. The titer of PRRSV and
SHFV in the cell culture can be 106-10
7 of 50% tissue culture infectious dose per ml
(TCID50 /ml) but it can reach 108
TCID50/ml for EAV (Tauraso et al., 1968; Onyekaba et
al., 1989b; Snijder and Meulenberg, 1998). Replication of LDV in cell culture is very
limited and thus LDV is generally titrated in vivo in mice (Plagemann and Moennig,

13
1992). In cells infected with EAV, SHFV, and PRRSV, cytopathic effects (CPE) are
described as cell rounding and detachment from the surface of culture plates (Hyllseth,
1969; van Berlo et al., 1980; Gravell et al., 1986b; Benfield et al., 1992; Kim et al., 1993;
van Nieuwstadt et al., 1996). Presence of “double membrane vesicles” (DMVs) in
infected cells is common in Arteriviridae, and occurs 3-6 hr post-infection (Snijder and
Meulenberg, 1998; Wood et al., 1970; Stueckemann et al., 1982; Rossow, 1998; Pol et
al., 1997). The role of these structures in replication and viral RNA transcription is not
yet known, though it does not seem that they are involved in assembly of the virus.
3.2 Attachment and Entry. Transfection of non-permissive cells with LDV and PRRSV
genomic RNA leads to production of virus particlest (Inada et al, 1993; Meulenberg et al,
1998). When different cell lines derived from rats and mice were first infected with
murine leukemia virus, they supported later infection with LDV (Inada and Yamazaki,
1991). This suggests that one of the determinants in susceptibility of cell lines to
arteriviruses replication is the existence of specific receptors on the surface of these cells.
The cell tropism has been well investigated for PRRSV. It has been found that pre-
incubation of PRRSV or MARC-145 cells with heparin or cells with heparinase, prevents
PRRSV infection. This suggested that there is a heparin-like molecule on the surface of
MARC-145 cells which supports virus attachment and subsequent entry of the virus (Jusa
et al, 1997). Subsequently, sialoadhesin (or sialic acid-dependent lectin-like receptor 1;
Siglec-1), which is a macrophage-specific molecule, was found to mediate PRRSV
attachment to the surface of alveolar macrophages (Vanderheijden et al, 2003). In other
investigations, heparin sulfate on the surface of macrophages and sialic acid on the
PRRSV virion were shown to play a role in virus entry (Delputte et al., 2002;

14
Vanderheijden et al., 2001; Vanderheijden et al., 2003). In the present model of infection
of macrophages with PRRSV, it is suggested that the virus first binds to heparin sulfate.
This facilitates binding of viral ligands to Siglec-1 which happens via sialic acid present
on viral glycoproteins (Delputte et al, 2005). This is followed by internalization of virus
through endocytosis which is dependent on clathrin-coated vesicles (Fig. 1.3) (Delputte et
al., 2004, 2005, 2007; Delputte and Nauwynck, 2004). As mentioned, infection of the
permissive cells such as MARC-145 is partially mediated by the presence of a heparin-
like molecule on the surface (Jusa et al, 1997). However, the absence of sialoadhesin on
these cells and on cells belonging to non-macrophage lineages, suggested that there was
another molecule which mediated viral entry. It was shown that the micro-filament
vimentin in MARC-145 cells binds to the nucleocapsid protein of PRRSV. This
suggested that vimentin may transport PRRSV to host cells by its interactions with other
compartments of the cytoskeleton (Kim et al, 2006). CD151 has also been shown to
interact with PRRSV RNA and it is suggested to mediate fusion of viral membrane and
endosome (Shanmukhappa et al., 2007). Another investigation showed that scavenger
receptor CD163, a member of the scavenger receptor cysteine-rich family class B, is
necessary for infection of MARC-145 by PRRSV. Its expression in a non-permissive cell
line resulted in replication of PRRSV in these cells (Calvert et al., 2007). A recent study
showed that sialoadhesin is the cellular receptor involved in PRRSV internalization and
CD163 is needed for viral entry (Van Gorp, et al, 2008).
Studies using drugs affecting the pH of intracellular compartments have revealed
that low pH is needed for PRRSV entry. Electron microscopy studies have shown that
PRRSV and LDV particle are visible in clathrin coated vesicles (Kreutz and Ackermann,

15
1996, Kowalchyk and Plagemann, 1985). A recent investigation suggests that EAV also
enters cells by clathrin-dependent endocytosis and is delivered to acidic endosomal
compartments (Nitschke et al, 2008). All the above evidence indicates that arteriviruses
use the low pH-dependant endosomal pathway to enter cells.
3.3 Replication and Transcription. Replication of arteriviruses takes place in the
cytoplasm of infected cells (Fig. 1.3). After virus entry and uncoating, viral genome RNA
is released into the cytoplasm and translated into two large polyproteins, pp1a (1727 for
EAV and 2502 amino acids for PRRSV) and pp1ab (3175 for EAV 3959 amino acids for
PRRSV) which provide the replicase proteins needed for viral RNA synthesis
(Molenkamp et al, 2000b; Snijder and Spaan, 2007). ORF1b of the replicase gene starts
at the -1 position to the stop codon location of ORF1a. The translation of the replicase
polyproteins starts at the 5’ end non-translated region (NTR) of the genome after
“conventional” scanning of the genomic 5’ NTR by ribosome (Van den Born, 2005). The
translation of ORF1b requires two structural signals, located at the ORF1a/1b junction,
which mediate the ribosomal frameshift. One signal is a “slippery” sequence at the
location of the frameshift before the stop codon of ORF1a. The proposed slippery site for
EAV starts upstream of the stop codon of ORF1a (5’GUUAAAC 3’), but for LDV and
PRRSV, there is an additional codon between the slippery sequence and the stop codon.
The other element required for the ribosomal frameshift is a secondary structure
downstream of the slippery sequence, which is referred to as the “pseudoknot” (Jacks et
al, 1988; Brierley1995). The pseudoknot structure has been suggested to contain two
stems. The stems 1 and 2 at the frameshift sites of EAV, PRRSV, and LDV, are
suggested to be 11-12 and 6- 7 nucleotides, respectively (den Boon et al, 1991; Godney et

16
Fig 1.3. Schematic representation of the life cycle of arteriviruses. Entering of
arteriviruses into susceptible cells occurs through receptor mediated endocytosis. At first
the viruses bind to specific receptors on the cell, and then the virus is taken into the cell
by clathrin-coated vesicles. The low pH stimulates fusion of virion envelope with the
endosomal membrane leading to the release of the viral nucleocapsid into the cytoplasm
of the infected cell. Translation and transcription are the next steps of the life cycle
followed by the replication of the genome and assembly of virion which happens by
budding the nucleocapsid via ER and Golgi complex. The final step is the exit of virion
which happens by exocytosis from the infected cells (adapted from Snijder and Spaan,
2007).

17
al, 1993; Meulenburge et al., 1993b). The frameshifting for EAV is reported to be 15% to
20% efficient (den Boon et al, 1991). Proteolytic processing of the polyprotein after
translation forms a structure to copy a full genome length complementary strand
(negative genome RNA) which functions as a replication template for the genome of
arteriviruses.
A nested series of subgenomic (sg) minus-strand RNAs (complementary of
sgmRNAs) are produced during transcription which will be discussed later. Each of (–)
sgRNA and (+) sgRNA has similar 5’ and 3’ ends (Snijder and Spaan, 2007). The
presence of specific structures for RNA replication of arteriviruses has yet to be
discovered. There are two NTRs located at the 5’ and 3’ ends of the genome of 156 to
221 and 59 to 117 nucleotides in lengths, respectively (Molenkamp et al., 2000a; Tijms et
al, 2001). The 3’ NTR has 33-47% identity in sequence between members of the
Arteriviridae family. A conserved sequence is found among all of these viruses in the 3’
NTR, located just before the poly A sequence (Godeny et al., 1993). In the 5’ end of
LDV, PRRSV, and SHFV, the start codon of ORF1a is located immediately after the
common leader sequence but for EAV, it is located 12 nucleotides downstream of the
leader sequence. This results in the presence of the entire genomic 5’ NTR in all of sg
mRNAs in LDV, PRRSV, and SHFV. There are interactions between host cell proteins
and the 3’ end of the negative-sense genomic RNA for SHFV, LDV, and EAV (Hwang
and Brinton, 1998). This indicates that for initiation of the (+) RNA transcription, the 3’
end of the (–) RNA and the interactions with the host proteins are crucial (Snijder and
Spaan, 2007). Using defective interfering RNAs of EAV, it has been found that the
presence of a 300 nucleotide region at both ends of the genome is required for efficient

18
replication. This suggests that NTR’s of both termini and some regions of the coding
sequence may contain signals required for replication of EAV (Molenkamp et al., 2000b;
Tijms et al, 2001). Similarly, interaction between the RNA hairpin secondary structure in
the 3’ NTR and the N protein gene, which is called the “kissing interaction”, has been
reported for PRRSV as vital for synthesis of viral RNA (Verheije et al., 2002).
Nidovirus transcription for generation of a nested set of sgmRNAs is a unique
feature of the replication cycle of these viruses. sgmRNAs are synthesized to express the
structural genes located at the 3’ end of the genome of these viruses (Baric et al., 1983;
Spaan et al., 1983; van Berlo et al., 1982, 1986; de Vries et al., 1990, 1997; Kuo et al.,
1991; Meulenberg et al., 1993b). The mRNAs of arteriviruses have common 3’ terminal
and 5’ leader sequences (Snijder and Horzinek, 1993; de Vries et al., 1997). The 5’ leader
sequence of sgmRNAs is the 5’ end sequence of the genome. The process by which this
leader sequence is merged into the sgmRNAs is the common mechanism of transcription
for Arteriviridae and Coronaviridae and is called “discontinuous transcription” (Snijder
and Meulenberg, 1998). The presence of conserved sequences at the “leader-body”
junctions of sg mRNAs of arteriviruses has been reported (de Vries et al., 1990; Chen et
al., 1993; Meulenberg et al., 1993a; Zeng et al.,1995; den Boon et al., 1996; Godeny et
al., 1998). A series of negative-strand (-) sg replicative intermediates (RIs) of EAV has
been detected. These (-)sgRIs, which are complementary of (+)sg mRNAs, are thought to
be involved in mRNA synthesis of arteriviruses (den Boon et al., 1996; Chen et al.,
1994). Fig. 1.4 is an illustration of the nested set composition of genomic and (+) and (-)
subgenomic strands of RNAs in Arteriviridae, and two proposed models for the
transcription are depicted. As demonstrated in Fig. 1.1 b and 1.4 a, the sg mRNAs of

19
Fig. 1.4. The nested set of RNAs in arteriviruses and the discontinuous strategy for the
mRNA transcription. (a) The structure of arteriviruses nested series of RNA. The positive
and negative strand RNAs are shown in white and black respectively. Below the full
strands of genomic and its complementary full RNA, two examples of + and – strand
subgenomic (sg) RNAs are depicted. The small white and black boxes on the RNAs
indicate the (-) and (+) transcription-regulating sequences (TRSs) respectively. The
leader and anti leader sequences are shown as hatched boxes. The sgmRNAs contain a
leader sequence drived from the 5’ end of the genome which is connected to the specific
sequences from 3’ end of the genome through a discontinouse mechanism of RNA
transcription. (b) and (c) show two proposed mechanisms for the transcription of
arteriviruses. The initial model for transcription was the leader-jump. In this method plus
strand RNA synthesis happens by falling the + leader sequence and base-pairing with the
– TRS on the downstream of the (-) genome RNA (b). However by the discovery of the –
sg RNAs the second model as body-jump (c) was suggested. In the body-jump method,
the body TRS falls during the synthesis of the –RNA. Then it base pairs with the leader
sequence leading to the synthesis of the – sg RNA which can be used as template for the
+sg mRNAs synthesis. The second method is more favorable by researches (Adapted
from Snijder and Meulenberg, 1998).

20
Arteriviridae contain the “leader” and “body” sequences. These two parts of the sg
mRNAs are not located in a continuous position in the genome sequence rather they are
transcribed from the 3’ and 5’-termini of the genome. Conserved sequences called
transcription regulating sequences (TRSs) connect the leader and body sequences of each
mRNA. The TRSs are located at the 3’ end of the leader sequence as well as at the 5’ end
of the body of each mRNA (Snijder and Meulenberg, 1998).
Two models have been proposed for mRNA transcription of members of the order
Nidovirales. The first model is called “leader-primed transcription” in which the
synthesis of the (+) sg RNA is disrupted and the (+) TRS located at the 3’ end of the
leader sequence complements with the (-) TRS of the 5’ end of each transcription unit
(Fig. 1.4b; Baric et al., 1983; Spaan et al., 1983). After the (+) and (-) TRS base-pairing
starts, it moves toward extension of the leader resulting in a sg mRNA. This model leads
to continuous synthesis of the (+) genomic RNA and discontinuous synthesis of the (+)
sg RNA (Snijder and Meulenberg, 1998). However, subsequent studies showed the
presence of a nested series of RIs that included (-) sg RNAs containing an anti-leader
sequence, and another model was proposed for RNA transcription. This model involves
discontinuous synthesis of (-) strand RNAs from the genome template (Fig. 1.4c). Base-
pairing of (-) leader TRS and (+) TRS of the template is crucial in both transcription
models (Snijder and Spaan, 2007). The TRS sequence for all members of the
Arteriviridae family has been determined. The sequence of TRS in the EAV genome is
highly conserved (den Boon et al., 1991). However, the TRS sequence is not the sole
determinant by which the virus transcription machinery chooses the correct TRS
sequence as the interaction of viral and/or cellular proteins, and viral RNA has also been

21
suggested to be involved in this process (Snijder and Spaan, 2007; Pasternak et al., 2004).
In addition to the leader TRS, 17 other conserved TRS sequences are detected in the
genome of EAV, but six of them are known to play a role in the EAV transcription
(Snijder and Meulenberg, 1998). The 3’ side of TRS is more conserved than the 5’ side.
This may indicate that the 3’ side of TRS is more important than the 5’ side (Chen et al.,
1993; Meulenberg et al., 1993a; Godeny et al., 1998). This finding suggests that “the
leader-primed model” is less favorable for viral mRNA transcription has been more
widely accepted in recent years.
3.4 Virion Assembly and Release from the Cell. The subcellular localization of EAV N
has been reported (Tijms et al., 2002). Since there is no evidence that N protein is
required for genome replication and transcription of arteriviruses, this protein has been
suggested to be involved in virus assembly. Viruses of the Arteriviridae family replicate
in the cytoplasm of host cells and their assembled nucleocapsid buds from the
intracellular membranes where the viral membrane proteins are already synthesized and
located (Fig. 1.3). The preformed nucleocapsid buds through the smooth endoplasmic
reticulum followed by the Golgi system. Then, it is released from cells by exocytosis and
finally by lysis of infected cells (Magnusson et al., 1970; Wood et al., 1970;
Stueckemann et al., 1982; Pol and Wagenaar, 1992).
4. Pathogenesis of Arteriviruses
4.1. Natural Infection and Persistence
4.1.1 EAV. EAV infection occurs via the respiratory route which is followed by virus
replication in alveolar macrophages and endothelial cells. The virus then tranclocates to

22
lymph nodes, and through the lymphatic system, it can spread to other host tissues.
Viremia is developed 3 days post-infection and virus can be isolated from all tissues by
this time (Balasuria and Maclachlan, 2004). Infection of horses with EAV may remain
subclinical, but clinical signs can include flu-like signs, abortion in pregnant mares, and
interstitial pneumonia in newborns (Bryans et al 1957; Balasuria and Maclachlan, 2004).
Injuries of the endothelial system and small muscular arteries, along with high
permeability of the vascular system are some of the mechanisms for pathogenesis of
EAV (Balasuria and Maclachlan, 2004). The EAV infection naturally results in persistent
infection affecting about 35% of infected male horses (Timoney and McCollum, 1993).
These “carrier stallions” are persistently infected animals that keep the virus in the
reproductive tract and shed it into the semen which helps to spread the virus (Snijder and
Spaan, 2007). The exact mechanism of the persistence of EAV is not fully understood.
4.1.2 PRRSV. Infection of pigs with PRRSV occurs via the respiratory route and it is
also transmissible to the fetus across the placenta (Zimmerman et al, 1997). The primary
cells that become infected are alveolar macrophages and, subsequently, macrophages in
other tissues also become infected. Viremia is seen at 12 to 24 hours post-infection and
may last 1 to 2 weeks in older pigs and up to eight weeks in piglets (Snijder and Spaan,
2007). Clinical features of PRRSV infection include high temperature, anorexia, lesions
in the lung, as well as stillborn, dead or weak piglets due to the reproductive problems of
infected sows. Different factors influence the outcome of infection with PRRSV, which
include the age, gender, and immune condition of the host and the strain of virus
infecting the host (Wensvoort et al., 1993).

23
4.1.3 LDV. Infection of the host by LDV is asymptomatic but results in a permanent
viremia in the infected animals. Maintenance of LDV happens by constant replication of
the virus in a small group of peritoneal macrophages (Onyekaba et al., 1989b). The titer
of virus reaches 1010
lethal dose 50% (LD50) per ml of plasma at 24 hours post-infection,
but afterwards it declines to 104
ID50 to 106 ID50. The latter is the titer of LDV which
persists along with the increased amount of lactate dehydrogenase through the life of the
infected mouse. Virus can be detected in spleen, lymph nodes, thymus, and liver of
infected mice during the persistent period (Cafruny et al., 1986). In specific inbred lines
of mice, a deadly poliomyelitis resulting from infection by neurovirulent strains of LDV
can occur, which is age-related (Contag and Plagemann, 1989).
4.1.4 SHFV. SHFV leads to asymptomatic, acute or persistent infections in African
monkeys based on the strain of the virus which infects these animals (Gravell et al.,
1986a; London, 1977). However, the virus caused deadly hemorrhagic fever outbreaks in
captive macaques kept in the National Institutes of Health (NIH) quarantine which were
shipped from India (Tauraso et al., 1968). Pathogenesis of SHFV in macaques is largely
unknown. Fever, erythem face, edema, dehydration, and hemorrhages are the
manifestations of SHFV infection in macaques. Normally, the SHFV-infected macaques
die by two weeks post-infection and the rate of mortality is near 100%. The target cells
for SHFV in the host are macrophages, similar to other members of the family (Gravell et
al., 1986b).
4.2 Immune Response to Arteriviruses Infections.
4.2.1 Innate Immune Response to Arteriviruses. Characteristics of innate immunity to
arteriviruses include production and expression of pro-inflammatory cytokines and

24
interleukins, and activation of infiltrating natural killer (NK) cells and macrophages
(Snijder and Spaan, 2007). Of the pro-inflammatory cytokines, induction of tumor
necrosis factor-alpha (TNF-α) has been reported in vitro in macrophages infected with
the virulent and avirulent strains of EAV. In the case of infection by avirulent EAV
strains, production of TNF-α was lower (Balasuriya and Maclachlan, 2004; Moore et al.,
2003). The induction of innate immune mechanisms is believed to be poor in PRRSV
infection (Murtaugh et al., 2002). Lee et al. (2004) demonstrated that induction and
sensitivity to interferon-alpha (IFN-α) in porcine alveolar macrophages varies among
different field isolates of PRRSV. PRRSV was shown to suppress the activation of type I
IFN transcription factor, interferon regulatory factor 3 (IRF3), in MARC-145 infected
cells and it is suggested that PRRSV N is involved in the deactivation of IRF3 (Lai F.W.,
2006). Several studies indicate that non-structural (nsp) proteins of PRRSV have
important role in suppression of innate immune response. For example two auto-clevage
products of nsp1 (nsp1-alpha and nsp1-beta) are involved in inhibition of synthesis and
signaling pathway by IFN-beta (Chen et al., 2009a). Moreover, in cells expressing two
nsp1-alpha, nsp1-beta, and nsp11 of PRRSV, double-stranded RNA (dsRNA) signaling
pathway was inhibited. The correlation of nsp1-beta to inhibit gene induction by IRF3
and NF-kappaB mediated by dsRNA and Sendai virus is reported (Beura et al., 2010).
Also the down-regulated expression of IL-1 beta and TNF-alpha in cells infected with
virus carrying the mutant forms of nsp2, suggests a regulatory role for this protein in
innate immue respone for PRRSV (Chen et al., 2009b). LDV has been reported to
activate NK cells in the infected host, which results in elevated interferon-gamma (IFN-γ)

25
in the serum, although elevated levels of this cytokine cannot clear the infection
(Markine-Goriaynoff et al., 2002).
4.2.2 Adaptive Immune Response to Arteriviruses. As mentioned above, persistent
infection is a common feature of arteriviruses. It can be as long as 2-3 months in the case
of PRRSV-infected swine, or may be a life-long in LDV-, EAV-, and SHFV-infected
hosts (1992; Onyekaba et al., 1989a; Timoney and McCollum, 1993; Gravell et al.,
1986b). The mechanism of evasion from the host immune system by arteriviruses is not
known.
The involvement of antibody-mediated immune response in arterivirus infection
has been studied for different members of the family. One week post-infection, antibodies
against viral proteins can be detected (Coutelier et al., 1986; Gravell et al., 1986b;
McCollum, 1986; Nelson et al., 1994). In EAV-infected animals, antibodies are induced
against N, M, GP5, and GP2 proteins (Balasuriya et al., 2002; MacLachlan et al., 1998).
In PRRSV-infected pigs, serum antibodies are present against GP2 to GP5, N, and M. In
the serum of these animals, the most abundant antibody is against the N protein (Loemba
et al., 1996; Meulenberg et al., 1995b).
Most of the neutralizing antibodies in animals infected with arteriviruses are
induced against GP5 (Cafruny et al., 1986; Chirnside et al., 1995a, 1995b; Gonin et al.,
1999). In EAV-, LDV-, and PRRSV-infected animals, the presence of neutralizing
antibodies against GP5 has been reported with the neutralization epitopes located in the
ectodomain of GP5 (Balasuriya et al., 2004; Chirnside et al., 1995 a, 1995b; Li et al.,
1998; Plagemann, 2004). The ectodomain of GP4 of PRRSV seems to harbor the binding
domain for a second MAb (Meulenberg et al., 1997). Virus neutralization by MAbs

26
raised against GP5 is more efficient than by MAbs against GP4 (Wieland et al., 1999).
The immune response against structural proteins of SHFV has not been investigated
thoroughly (Snijder and Spaan, 2007).
In EAV-infected horses, highly neutralizing antibodies appear at the same time as
the virus is cleared. These antibodies remain in the host for a long time and they have
been suggested to be important for protection against EAV (Snijder and Meulenberg,
1998). Low titers of neutralizing antibodies against LDV and PRRSV have been
observed and it seems that these antibodies do not clear or lower virus burden in the
blood (Cafruny et al., 1986; Loemba et al., 1996; Yoon et al., 1996). It has been reported
that antibodies against LDV and PRRSV affect virus intake by macrophages in vitro.
This suggests that the complex of virus-antibody in infected mice and pigs may increase
the infection (Cafruny and Plagemann, 1982; Yoon et al., 1996). It has been shown that
maternal antibodies can protect progeny of LDV- and PRRSV-infected animals against
these infections (Broen and Cafruny, 1993; Snijder and Meulenberg, 1998). Antibodies
against LDV can inhibit the progression of infection towards age-dependant poliomyelitis
in mice (Harty et al., 1987; Harty and Plagemann, 1990; Plagemann and Moennig, 1992).
The factors involved in LDV persistence include infection of newly developed
macrophages leading to slow and consistent formation of virus-susceptible cells,
emergence of immune resistant virus variants during the persistent infection, and
presence of infectious virus-antibody immune complexes in LDV infected mice
(Onyekaba et al., 1989a; Rowland et al., 1994; Monteyne and Coutelier, 1994; Chen et
al., 1997). The antibody-mediated response against SHFV is dependent on the species of
the infected monkeys as well as virus variants. There is no efficient immune response in

27
infected macaque monkeys which die promptly after SHFV infection. However, in
infected Patas monkeys, two different antibody responses have been shown, depending
on SHFV strains causing different forms of the disease. The SHFV strains causing acute
infection induce high titer of neutralizing antibodies against the virus and this coincides
with the complete disappearance of virus from blood. However, the viral strains that
cause persistent infection of the patas monkeys induce only low titers of non-neutralizing
antibodies against the virus (Gravell et al., 1986b).
Cell-mediated immune response against Arteriviridae has not been well studied.
Cell-mediated immune (CMI) response seems to play an important role in the immune
response against SHFV. Subsequent clearance of super-infection in Patas monkeys which
had already been persistently infected with a different strain of SHFV is supportive of
CMI response to SHFV. Moreover the lack of cross- neutralization between the
antibodies raised against the two different strains of SHFV supports CMI involvement as
well (Gravell et al., 1986a, Snijder and Meulenberg, 1998). Experiments on EAV
infected ponies showed that cytotoxic CD8+T-cells are mediators of the cell-mediated
response against EAV and that response is specific for the virus (Balasuriya and
MacLachlan, 2004; Castillo-Olivares et al., 2003). The presence of cytotoxic T
lymphocytes (CTLs) and helper T cell responses stimulated by LDV infection has been
shown. These responses lasted for 30 and 250 days post-infection in different
investigations, but were not effective in prevention of LDV replication (Even et al., 1995;
van den Broek et al., 1997). Both CD4+ and CD8
+ responses have been detected 4 to 8
weeks post-PRRSV infection (Murtaugh et al., 2002; Xiao et al., 2004). T-cell
proliferation responses against M, GP2, and GP5 have been detected between 4-12 weeks

28
after PRRSV infection (Bautista et al., 1999). More investigations are needed to
determine if cell-mediated immunity is involved in protecting animals from infection
with arteriviruses.
5. Nuclear Localization of Viral Proteins in (+) ssRNA viruses
5.1 Nucleolar and Nuclear Localization of Proteins. The nucleolus is a part of
eukaryotic cell which is known for ribosome biogenesis (Melese and Xue, 1995; Sirri et
al., 2008). Transcription of rRNAs, cleavage of ribosomal RNA precursors, and
aggregation of ribosomal RNAs with ribosomal protein to construct the 40S and 60S
subunits of the ribosome all occur in the nucleolus (Sirri et al, 2008). Using electron
microscopy (EM), three distinct parts of the nucleolus have been observed. The “dense
fibrillar component” (DFC) and the “granular component” (GC) are two major
compartments of the nucleolus observed by conventional EM (Jordan 1984). The third
compartment is called the “fibrillar center” (FC) which comprises a small portion of the
nucleolus (Shaw and Jordan 1995). The FCs and DFCs are inside the GC which is
composed of granules that are 15-20 nm in size. When proteins are in nucleus, either
transferred by importins or diffused, some are localized to specific parts of the nucleus,
such as nucleoli, and others are seen all over the nucleus (Sirri at al, 2008; Andersen et al.
2002, 2005; Leung et al. 2003). All of the required proteins for ribosomal synthesis are
cycling between the nucleolus and the nucleoplasm in the interphase stage of the cell
cycle. Also, the diffusion of nucleolar proteins between these compartments of the
nucleus is a permanent feature of nucleus, which happens very rapidly (Dundr et al. 2004;
Sirri et al, 2008). Many factors are important in the nucleolar localization of a protein.
The most important factor for this activity is the localization of the protein to the nucleus

29
which is followed by other mechanisms that lead a protein to the nucleolus. The
mechanisms and signals for nuclear localization of a protein and their export to the
cytoplasm have been investigated thoroughly, but the molecular bases for nucleolar
localization of proteins are still to be discovered (Sirri et al, 2008). The cytoplasm and
nucleus are separated by the nuclear envelope (NE). The NE is a continuation of the
endoplasmic reticulum (ER). It contains many holes, named nuclear pore complexes
(NPCs), for transferring macromolecules between the nucleus and the cytoplasm. Some
of these macromolecules are imported from the cytoplasm including nuclear proteins
such as histones and transcription factors. Some molecules synthesized in the nucleus
must be exported to the cytoplasm, including transfer RNA (tRNA), ribosomal RNA
(rRNA), and messenger RNA (mRNA). The NPCs are the only path through which these
translocations happen. The numbers of NPCs differ depending on the size and activity of
each cell. For example, a human cell can contain 3,000-5,000 NPCs in the NE. NPCs
have octuple rotational symmetry and each of them contains a central core compartment
inserted in the NE with extended cytoplasmic and nuclear elongations to shape the
cytoplasmic extensions and the nuclear basket structures, respectively. The proteins
constituting the NPCs are called nucleoporins which also provide the place that nuclear
transport proteins and elements dock and interact for their activities (Gorlich and Kutay,
1999). NPCs shape the routes through which all nuclear trafficking processes occur
(Fedherr et al, 1984; Gorlich and Kutay, 1999). The trafficking of small molecules less
than 9 nm in diameter, such as metabolites, takes place by passive diffusion through these
channels (Paine et al, 1975; Gorlich and Kutay, 1999). On the other hand, the
translocation of larger macromolecules greater than ~40 kD through the NPCs happens

30
Fig. 1.5.Schematic illustration of nuclear protein transportation. The cargo protein
containing a nuclear localization signal (NLS) forms an import complex with importin-α
(α) and importin-β (β). This complex docks onto the nuclear pore complex (NPCs) and
enters into the nucleus where the binding of RanGTP to imp-β separates it from the
complex. This is followed by disassembly of the cargo and imp-α in the nucleus and
return of the imp-α to the cytoplasm by its nuclear export protein, CAS in association
with RanGTP. In the cytoplasm RanGAP induces the hydrolysis of GTP and releases
imp-α. (From Cook et al., 2007).

31
actively and is a signal-mediated process regulated by the presence of a particular signal
on the protein and its interaction with transportins and NPCs. This mechanism is also
involved in transferring some smaller proteins and RNAs into the nucleus (Breeuwer and
Goldfarb, 1990; Jäkel et al., 1999; Gorlich and Kutay, 1999, Stewart, 2007). The first
identification of nuclear translocation signal on a protein was reported for the
nucleoplasmin of Xenopus laevis oocytes (Dingwall et al 1982), and the simian virus 40
(SV40) large-T antigen was the first protein reported to contain the nuclear localization
signal (NLS) for import into the nucleus (Kalderon et al., 1984; Lanford and Butel, 1984;
Robbins et al., 1991; Gorlich and Kutay, 1999). Three types of NLSs have been reported
so far. “Pat 4” is a type that includes a stretch of four basic amino acids of mostly lysines
or arginines. “Pat 7” is a second type of NLS which contains a motif of seven amino
acids starting with a proline followed by two other amino acids which are followed by
four other amino acids and three of them being basic residues. The third type of NLS is
“bipartite” which begins with two basic amino acids followed by 10-12 residues as a
spacer and ends with five amino acids, among which at least three are basic (Robbins et
al., 1991; Nakai and Kanehisa,1992). It is now accepted that in NLS-related nuclear
import, four essential components are involved. Two of them are importin α (Impα),
importin β (Impβ), and two others are Ran and nuclear transport factor 2 (NTF2) which
belong to the RanGTPase system (Adam and Adam, 1994; Gorlich et al., 1994; Chi et al.,
1995; Gorlich et al., 1995; Melchior et al. 1993; Moore and Blobel, 1993; Moore and
Blobel, 1994; Paschal and Gerace, 1995). More investigation has shown that different
nuclear transfer pathways are involved in translocating proteins into the nucleus. Most of
these mechanisms include a super family of carrier proteins called β-karyopherins.

32
Among them are Impα and Impβ which mediate the nuclear import of many proteins.
Often the complex of Impβ-Impα transfers the protein of interest into the nucleus, in
which Impα acts as an adaptor for Impβ by recognizing the NLS on the cargo protein
(Stewart, 2007). Every minute, approximately 100-1000 cargo proteins are transported
through each NPC by the classical pathway of nuclear protein import. The import cycle
includes four phases: the cargo-carrier complex formation in the cytoplasm, transfer via
NPCs, dissociation of the import complex in the nucleus, and recovery of the importins
(Fig. 1.5). First in the cytoplasm, the cargo protein containing NLS binds to the Impβ-
Impα heterodimer via the NLS binding site on the Impα which is acting as an adaptor.
The import complex of cargo and Impβ-Impα-heterodimer then moves towards the NPCs
and docks there. By Ran-GDP, this complex passes via the NPCs. In the nucleus, the
present Ran-GTP binds to Impβ resulting in disassembly of the import complex and
liberation of the cargo protein into the nucleus in which it either diffuses to a specific site
or spreads evenly. The last step is recycling importins and RanGTP. Impβ returns to the
cytoplasm along with RanGTP and Impα leaves the nucleus along with the β-karyopherin
CAS (Cellular Apoptosis Susceptibility protein) and RanGTP. In the cytoplasm, the Ran
GTPase activating protein (RanGAP) prompts the Ran GTPase to produce RanGDP
leading to release of the importins (Stewart, 2007). Protein import is an orchestrated
cycle which involves groups of controlled interactions between cargoes and carriers.
These interactions are coordinated by the nucleotide condition of Ran which is cycling
between GTP- and GDP-bound forms of Ran controlled by its Guanine nucleotide-
exchange factor (RanGEF) and RanGAP (Ran GTPase activating protein). The RanGEF
resides in the nucleus and catalyses the recharging of RanGDP with GTP. RanGAP is

33
cytoplasmic and stimulates hydrolysis of GTP. The RanGTP is localized in the nucleus
and RanGAP is in the cytoplasm. The positioning of RanGTP in the nucleus mediates
export of Impβ protein from the nucleus to cytoplasm where GTP is hydrolysed leading
to the release of RanGDP to the cytoplasm preparing to be used in another cycle for
protein import. In addition to the nucleotide state of Ran, its conformational changes
during the nuclear import process are also determinants of its interaction with the
importin family members and the energy produced by RanGTPase for nuclear import
(Stewart, 2007). Hydrolysis of RanGTP in the cytoplasm and exchange of the nucleotide
state of Ran from GTP to GDP in the cytoplasm retains a gradient of RanGTP/RanGDP
which is supported by the presence of the RanGAP in the cytoplasm and RanGEF in the
nucleus. This gradient determines the direction of nuclear transport (Cook et al., 2007).
5.2 Nucleolar Localization of (+)ssRNA Virus Proteins. Many viruses including RNA
viruses, DNA viruses, and retroviruses have molecular interactions with the nucleus and
nucleolus. The nucleus is the place in which many DNA viruses, retroviruses, and a few
RNA viruses replicate. Positive-strand RNA viruses mostly replicate in the cytoplasm of
infected cells and do not require the function of the nucleus for their replication cycle.
Their essential proteins have been shown to localize in the nucleus which is not the place
for their replication (Hiscox, 2007; Sirri et al, 2008; Wilhelmsen, et al., 1981; Evans et
al., 1980). Nucleolar localization of many proteins of positive-strand RNA viruses,
particularly the nucleocapsid protein, has been a topic of interest for many researchers.
The nucleocapsid protein of coronaviruses such as IBV (infectious bronchitis virus),
SARS (severe acute respiratory syndrome) virus, and MHV (mouse hepatitis virus), the
capsid protein and nonstructural protein nsP2 of alphavirus, and the core protein of

34
dengue virus, are all examples of (+)ss RNA viruses which have been reported to localize
to the nucleus and nucleolus of infected cells (Hiscox et al 2001; Wurm et al., 2001; You
et al. 2005; Timani et al., 2005; Tijms et al., 2002; Rowland et al., 1999; Michel et al.,
1990; Rikkonen et al., 1992; Wang et al., 2002). In order to fulfill their cytoplasmic
activities such as their involvement in the structure and replication of viruses, these viral
proteins must translocate back from the nucleus to the cytoplasm. It is well known that
the presence of an NLS motif is a vital part of nuclear transport for many viral proteins.
However, the nucleolar localization signal (NoLS) is not well characterized for viral or
host proteins (Hiscox, 2007). In some viruses, NLS and NoLS work in harmony to
localize a protein to the nucleus. The presence of NoLS with a functional NLS motif in
the sequence and resemblance to host NoLSs has been well studied for PRRSV
nucleocapsid protein. Two NLS motifs have been identified in PRRSV N: the ‘pat4’ type
NLS-1 at positions 10-13 and the ‘pat7’ type NLS-2 located at 41-47. The cooperation of
NLS and NoLS in the case of PRRSV N results in translocation of this protein into the
nucleolus. By constructing truncated proteins fused to enhanced green fluorescent protein
(EGFP), the region of PRRSV N responsible for nucleolar localization has been mapped
to position 41-72 which contains the functional NLS-2 motif. The N protein also contains
a leucine rich motif located at 106-117, which is the nuclear export signal (NES)
responsible for transferring the protein back to the cytoplasm (Rowland et al., 2003;
Rowland and Yoo, 2003). The other way to transport a protein to the nucleolus is
through collaboration of the viral protein with cellular proteins. The presence of a
proposed NoLS in the hepatitis delta virus (HDV) delta antigen is responsible for both
nucleolar localization and binding to the nucleolar protein nucleolin. Mutation of the

35
NoLS of HDV delta antigen prevents this protein from either translocating into the
nucleus or binding to nucleolin. This implies that nucleolar translocation is correlated
with binding to the host protein nucleolin (Lee et al, 1998). Unlike NLS, there is no
available program to predict NoLS. Investigation on the IBV N protein nucleolar
localization has revealed that a motif of 8 basic amino acids on this protein functions as a
NoLS signal and indeed interacts with nucleolar protein leading the IBV N protein into
the nucleolus. This motif is also an interactive part of IBV N with nucleolar proteins
(Reed et al., 2006; Hiscox, 2007; Sirri et al, 2008).
5.3 Function of Nuclear Localization of Viral Nucleocapsid Proteins. The presence of
NoLS on viral proteins indicates that viruses use the nucleolus for specific reasons and
suggests that viruses have undergone evolution to manifest and maintain this function.
Evidence suggests that the nuclear or nucleolar localization of some (+) SS RNA viruses
influences the pathogenesis and virulence of these viruses. Examples of viral proteins
affecting the virus replication and pathogenesis are explained below.
The non-structural protein nsP2 of Semliki Forest virus has been shown to
localize to the nucleus and by changing one amino acid in this protein, its trafficking to
the nucleus was interfered, which resulted in lowered virulence of the virus (Peranen et
al., 1990; Fazakerley et al., 2002). Lee et al., (2006b) by infecting pigs with an infectious
clone of PRRSV which had mutations in its functional pat7 NLS, demonstrated that
viremia was shorter. This was the first in vivo report of the importance of nuclear
localization in the pathogenesis of (+)ssRNA viruses (Lee et al., 2006a, 2006b). Another
example is the core protein of Japanese encephalitis virus (JEV) which has been reported
to improve the replication of JEV. By eliminating the nuclear and nucleolar localization

36
activity of JEV core protein, the recombinant virus showed reduced replication compared
to the wild-type virus (Mori et al., 2005). Interaction of the JEV core protein with the
nucleolar protein B23 has been shown (Tsuda et al., 2006). Moreover, the newly
synthesized viral RNA was found in the nucleus along with the viral replicase in infected
cells. Nucleolar localization of the flavivirus core protein has also been suggested to be
important in viral RNA synthesis (Uchil et al., 2006). Interaction of many (+) ssRNA
viruses with the nucleus and nucleolus is believed to help viral replication.
Another example of RNA viruses that interact with nucleus is the retroviruses.
The best studied example is human immunodeficiency virus (HIV). The genome of HIV
includes two copies of positive-sense RNA and its replication takes place in two separate
phases: cytoplasm and nucleus of infected cells. In the cytoplasmic phase, the viral
genome is first reverse-transcribed by the viral RT and then transferred to the nucleus
where the viral genome is transcribed and is transported back to the cytoplasm (Hiscox,
2007).
One of the first assumptions for the nucleolar localization of capsid and other
viral proteins of RNA viruses was that they diffuse through NPCs to reside in the nucleus
without any impact on the virus life cycle. But the possible selective pressure on keeping
this phenomenon during RNA virus replication leads to the idea that this function is a
helpful strategy for efficient viral replication. This maybe achieved by occupying the
nucleolus of the cell and using the nucleolar proteins involved in cellular transcription
and ribosomal biogenesis for virus benefit (Hiscox, 2007). Disruption of the nucleus
functions seems to be one of the influences of nucleolar localization of viral proteins.
Such an effect can be related to the reduced or elevated levels of the nucleolar proteins in

37
the nucleus, nucleolus, and cytoplasm as a result of moving or reorganization of these
proteins by viral proteins (Hiscox, 2002). The result of nucleolar localization of viral
proteins could interfere with the structure and activities of the nucleolus in virus-infected
cells. For instance, the disruption of the nucleolar functions has been reported in
poliovirus-infected cells. In poliovirus-infected cells, nucleolin is relocated to the
cytoplasm from the nucleolus (Waggoner and Sarnow 1998). Also, inactivation of the
upstream binding factor (UBF) has been reported for poliovirus which leads to the
inhibition of the RNA polymerase I-mediated transcription in virus-infected cells
(Banerjee et al., 2005). The nucleolar structure has been shown to be disrupted in IBV-
infected cells (Dove et al., 2006a). Moreover, G2 arrest of the cell cycle and abnormal
cytokinesis have been reported in IBV-infected cells (Wurm et al., 2001; Dove et al.,
2006b). The nucleocapsid proteins of IBV and PRRSV have been shown to interact with
nucleolar proteins, nucleolin and fibrillarin (Chen et al., 2002; Yoo et al., 2003). HIV-I
Rev protein is another example of a viral protein from (+) ssRNA viruses is located in the
DFCs and GCs compartments of the nucleolus and disrupts the architecture of nucleolus
and interacts with B23.1 (Miyazaki et al., 1996). The high number of cells at the G2 and
M stages of cell cycle (Miyazaki et al., 1996) and failed cytokinesis (Cannavo et al.,
2001) are two changes in Rev expressing cells.
It has been suggested that RNA viruses hijack the nucleolus by recruiting its
proteins during their replication. These nucleolar proteins have vital functions in
synthesis, processing and translation of cellular RNA. For example, picornaviruses
interact with and use nucleolin during their replication. Nucleolin interacts with the
picornavirus internal ribosome entry site (IRES) located at the 5’ end of its genome. This

38
Fig 1.6. Schematic presentation of a normal cell cycle. The cell cycle has two major
divisions: mitosis (M) and interphase. The second starts after mitosis and include three
phases: G1 which is the stage of preparing cells for the next stage the S phase in which
the DNA synthesis happens and is followed by G2 phase which prepares cells for the
next M. Each stage is regulated by different cyclines and Cdks (adapted from Schafer,
1998).

39
induces the IRES mediated translation which is cap-independant (Izumi et al., 2001).
Nucleolin also has been shown to interact with the 3’ non-coding terminus of the genome
of picornaviruses. This region acts as a regulator of the negative-strand RNA synthesis at
the early stages of poliovirus infection (Waggoner and Sarnow, 1998). Nucleolin
interacts with IRES in hepatitis C virus (HCV) and its interaction with HCV NS5B
protein has been proposed to be important in NS5B function (Hirano et al. 2003).
Localization of viral proteins in the nucleolus can abolish the translation process in
infected cells. Investigations have shown that nucleolar localization of the precursors of
3C protease of human rhinovirus, 3CD` and 3CD, at the beginning of the infection
prevents cellular RNA transcription by their protease activity (Amineva et al., 2004).
Such a shut-down of cellular protein translation has been reported as a result of nucleolar
localization of encephalomyocarditis virus (EMCV) 2A protein (Aminev et al, 2003).
5.4 Modulation of the Cell Cycle by Viral Proteins. The cell cycle is a synchronized
and controlled process for the growth and extension of cells. It is involved in the growth
and development of the organism as well as other biological functions such as repairing
DNA damage, tissue growth in injuries and cancer development. This process is
regulated by several proteins which orchestrate a series of phases leading to the formation
of two daughter cells. The key regulatory proteins for the cell cycle include the cyclin-
dependent kinases (Cdks) and the cyclin proteins which control moving cells through
four phases: G1 (gap 1), S (synthesis), G2 (gap 2), and M (mitosis) (Fig. 1.6).
Morphological subdivisions of cell cycle include two distinct phases, interphase and
mitosis (M). The interphase includes the G1, S, and G2 phases of cell cycle and the M
phase comprises of four stages of prophase, metaphase, anaphase, and telophase.

40
Interphase and M are two clear stages of cell cycle involved in DNA synthesis and
division of cells into two daughter cells, respectively. These two are separated by “gaps”
called G1 and G2. In the G1 step, the cells provide enzymes and proteins involved in
DNA synthesis which happens in the S phase, followed by the G2 in which the cell
becomes ready for M phase. Therefore, the DNA content of the cell at the G1, S, and G2
are 2N, 4N, and 4N, respectively. G0 is the quiescent stage during which cells are not
active and do not cycle (Schafer, 1998).
The key regulatory factors of cell cycle are cyclin proteins and Cdks. Cdks are a
group of serine/ threonine kinases and are vital for the progression of cell cycle. Cdks
become activated mostly by phosphorylation of their threonine and tyrosine residues.
Each stage of cell cycle contains specific regulatory factors which are neither active nor
abundant in the other phases of the cell cycle (Schafer, 1998). Many pathogens including
viruses have been reported to modulate the cell cycle. Viruses intervene with the cell
cycle by different strategies including: destroying the host cell normal growth to replicate
more efficiently, providing a better situation for virus genome replication, and prolonging
the cell cycle at different phases to provide more time for virus assembly. Most of
investigations of viral effects on cell cycle have been for DNA viruses and retroviruses.
However, interest in the interference of RNA viruses with cell cycle has been increasing.
Cell cycle control by cyclines and CdKs has been studied intensively, though recent
investigations show that nucleolus has direct or indirect effects on cell cycle along with
the aforementioned regulatory factors. Interaction of RNA viruses and their proteins with
nucleolus has been of great interest for researchers in this area (Emmett et al., 2004).
There are two major mechanisms that DNA viruses use to interfere with cell cycle. The

41
first strategy is used largely by DNA viruses with a large genome, such as herpesviruses,
which stops cells at the G1 stage inhibiting their transfer to the S phase of cell cycle
(Flemington, 2001). The second strategy is mostly used by DNA viruses with a small
genome, such as adenoviruses, which encode proteins interfering with regulatory factors
of the cell cycle (Moran, 1993). Cell cycle interruption has been well studied for
retroviruses. HIV infection leads to accumulation of infected cells in the G2 phase of cell
cycle which increases virus propagation. The viral protein responsible for this
phenomenon has been identified as Vpr, which localizes to the nucleus (Braaten et al,
1995; Poon et al, 1998). Negative- and positive-stranded RNA viruses have been reported
to modify cell cycle at different stages. For instance, the G0 arrest of cell cycle in cells
infected with measles virus, a negative ssRNA virus, has been reported (Naniche et al.,
1999), and the paramyxovirus simian virus V protein has been shown to slow cell cycle at
G1 and G2 stages (Lin and Lamb, 2000). Cell cycle modifications by positive-sense
RNA viruses have also been investigated. IBV accumulates infected cells at the S/G2
stage and inhibits normal cytokinesis (Chen et al., 2002; Wurm et al., 2001). Replication
of coxsackievirus is increased in cells arrested in G1 or G1/S phases (Feuer et al., 2002).
The other mechanism by which viruses interact with cell cycle control is by targeting
sub-nuclear structures involved in cell cycle such as nucleoli and their associated proteins
(Hiscox, 2002; Hiscox 2003). The concentration and distribution of nucleolar proteins
such as nucleolin, B23, and fibrillarin have been shown to be involved in regulation of
the cell cycle process (Sirri et al., 1997; Azum-Gelade et al, 1994; Fomproix et al., 1998).
Investigations have indicated that both non-structural and structural proteins of different
viruses can influence cell cycle progression. Among non-structural proteins, the

42
following have been implicated: the Vpr protein of HIV-1 which arrest the cell cycle in
G2/M (Mahalingam et al, 1997; Matsuda et al, 2003); σ1S protein of alpha reovirus
stalls cells at the G2/M phase (Poggioli et al, 2000); the V protein of the paramyxovirus
simian virus 5 (SV5) slows the cell cycle progress (Lin and Lamb, 2000); the non-
structural proteins 5A and 2 of hepatitis C virus regulate cell cycle phases (Ghosh et al,
1999; Yang et al, 2006), and MHV NS protein p28 and SARS 7a protein halt the cell
cycle at G0/G1 (Chen et al, 2004, Yuan et al, 2006). Among the structural proteins of
viruses, studies indicate that transmissible gastroenteritis virus nucleocapsid protein
(TGEV N) and IBV N play a role in cell cycle regulation. TGEV N is suggested to
modify cell cycle by increasing the number of dividing cells (Wurm et al, 2001) and the
nuclear localization of IBV N has been shown to be influenced by the cell cycle phase. In
synchronized Vero cells at the G2/M phase of the cell cycle upon transfection by IBV N
protein, the number of cells which showed nuclear localization for the N protein were
increased (Cawood et al, 2007).
5.5 Nucleocapsid Protein of Arteriviridae. The nucleocapsid (N) protein is the most
abundant protein in the arterivirus particle, comprising up to 40% of the virion protein.
The N protein is encoded by the 3’ most ORF of the genome. It is a basic protein and has
a molecular weight of 12-15 kDa (de Vries et al., 1992; Faaberg and Plagemann, 1995;
Godeny et al., 1995; Bautista et al., 1996). EAV N and PRRSV N are phosphorylated
(Zeegers et al., 1976; Wootton et al, 2002). The N protein is present as disulfide-linked
homodimers in the PRRSV particles (Mardassi et al., 1996; Meulenberg and Petersen-den
Besten, 1996) but the N of EAV and LDV has been reported to be in a monomeric form
in viral particles (de Vries et al., 1995a; Faaberg et al., 1995a). The N protein is an RNA

43
binding protein and the RNA binding domain for PRRSV N has been shown to overlap
with the functional NLS of this protein (Rowland and Yoo, 2003). In the sera from
infected mice with LDV, the level of antibodies against N is very low (Coutelier et al.,
1986; Faaberg and Plagemann, 1995). But in infected animals with EAV and PRRSV,
antibodies against N are abundant, which makes the N protein a good candidate for
serodiagnosis (Chirnside et al., 1995c; Kheyar et al., 1997; Meulenberg et al., 1995a;
Denac et al., 1997; Rodriguez et al., 1997). The N proteins of EAV and PRRSV have
been shown to localize to the nucleus and nucleolus of virus-infected and gene-
transfected cells (Tijms et al., 2002; Rowland et al., 1999). The PRRSV N has been
shown to interact and colocalize with the nucleolar protein fibrillarin, bind to the viral
and ribosomal RNA, and partner with the inhibitor of MyoD family-a (I-mfa) domain-
containing protein (HIC) (Yoo et al., 2003; Song et al., 2009). It has been shown that HIC
interacts with a T1 type of cyclin which in turn is part of the transcription system and is
involved in cell cycle progression (Young et al., 2003). Further studies have shown that
PRRSV infection using a mutant virus with disabled NLS is clinically attenuated
compared to the wild-type virus, indicating the importance of nuclear localization in the
viral pathogenesis (Lee et al., 2006a, 2006b; Pei et al., 2008). The interaction and
colocalization of N with the nucleolar proteins involved in cellular activities, such as cell
cycle, suggests that N has a regulatory function to influence the cell cycle process, in
addition to the structural function for virion assembly.

44
II.OBJECTIVES OF THE THESIS
The nucleolar localization of N protein of arteriviruses demands further
investigation for its biological function and whether it is a common feature of this family
in the Nidovirales order. The interactions of PRRSV N with the nucleolar and cellular
proteins result in modulation of cellular activities. Previous studies of coronavirus N
proteins and their modulation of cell cycle provides a basis for the hypothesis that the
capsid protein of Arteriviridae has a major role in cell cycle progression during virus
replication. Thus, the main goal of this study was to elucidate the nucleolar localization
of capsid proteins of all four member viruses of the Arteriviridae family and involvement
in the cell cycle.
Outline of Specific Objectives. Investigation on N protein has been mainly
accomplished using PRRSV, and information on other member viruses within the family
Arteriviridae is limited. Therefore, the first objective of this study was to investigate the
nuclear and nucleolar localization activity for N proteins of LDV, EAV, and SHFV.
The N protein of LDV was cloned as a GFP fusion protein and examined for its
subcellular distribution. The LDV N nucleolar localization signal was identified and
structure-function studies were conducted using mutant constructs. Subsequently, key
amino acids for the nuclear localization of LDV N were determined. Based on
experimental data and information from PRRSV N, the conclusion was made that LDV N
uses an active nuclear transport mechanism (Chapter 2).
Following the study of LDV N, our investigation was extended to EAV N and
SHFV N proteins. Using EGFP-EAV N and EGFP-SHFV N genes, subcellular
localization of the EAV N and SHFN explored. A functional NLS was identified for

45
EAV N and SHFV N and mapped using a series of mutant constructs. This was followed
by investigating of the molecular basis for their nuclear localization which (Chapter 3).
PRRSV N interacts with nucleolar and cellular proteins which play a role in
regulating cell cycle. This led us to hypothesize that the capsid protein of Arteriviridae
has a regulatory role in the cell cycle during virus replication. Therefore, in Chapter 4,
cell cycle modification by arteriviruses was investigated using PRRSV and EAV in virus-
infected cells. The role of N in cell cycle modification was then studied using N genes of
all 4 member viruses within the family by over expression of N in gene transfected BHK-
21 cells (Chapter 4).

46
CHAPTER 2
The Lactate Dehydrogenase-Elevating Virus Capsid Protein is a Nuclear-
Cytoplasmic Protein
Hakimeh Mohammadi1, Shayan Sharif
1, Raymond R. Rowland
2, Dongwan Yoo
3,*
1Department of Pathobiology, University of Guelph, Guelph, Ontario N1G 2W1, Canada.
2Department of Diagnostic Medicine and Pathobiology, Kansas State University,
Manhattan, KS 66506, USA.
3Department of Pathobiology, University of Illinois at Urbana-Champaign, 2001 South
Lincoln Avenue, Urbana, IL 61802, USA.
Published in: Archives of Virology. 2009.154(7):1071-80.

47
ABSTRACT
Arteriviruses replicate in the cytoplasm and do not require the nucleus function of
cell for virus multiplication in vitro. However, nucleocapsid (N) protein of two
arteriviruses, porcine reproductive respiratory syndrome virus (PRRSV) and equine
arteritis virus (EAV), has been observed to localize in the nucleus and nucleolus of virus-
infected and N gene-transfected cells, in addition to their normal cytoplasmic
distribution. In the present study, the N protein of lactate dehydrogenase-elevating virus
(LDV) of mice was examined for nuclear localization. Subcellular localization of LDV-N
was determined by tagging N with enhanced green fluorescence protein (EGFP) on the
N- and C-terminus. Both N-EGFP and EGFP-N fusion proteins localized to the nucleus
and nucleolus of gene-transfected cells. Labeled N also accumulated in the perinuclear
region, the site of virus replication. LDV-N sequence contains a putative ‘pat4’ type
nuclear localization signal (NLS) consisting of KKKK. To determine its functional
significance, a series of deletion constructs of N were generated and individually
expressed in cells. The results showed that the ‘pat4’ NLS was essential for nuclear
translocation. In addition, the LDV-N interacted with the importin-α and -β proteins,
suggesting that the LDV-N nuclear localization occurs via the importin-mediated nuclear
transport pathway. These results provide further evidence for the nuclear localization of
N as a common feature within the arteriviruses.

48
INTRODUCTION
Lactate dehydrogenase-elevating virus (LDV) is a murine virus causing lifelong
persistent viremia in mice with no apparent clinical signs except an increased level of the
plasma enzyme lactate dehydrogenase (Plagemann et al., 1995). LDV shows a limited in
vitro replication which is restricted to only specific murine primary macrophages and
presents a low level of virus production in culture (Brinton-Darnell et al., 1975; Brinton-
Darnell and Plagemann 1975; Stueckemann et al., 1982; Onyekaba et al., 1989a). LDV is
an RNA virus of 62-80 nm in diameter which belongs to the family Arteriviridae. This
family of viruses contains a single-stranded positive-sense RNA as the genome and
includes three other member viruses: porcine reproductive and respiratory syndrome
virus (PRRSV), equine arteritis virus (EAV), and simian hemorrhagic fever virus (SHFV)
(Snijder et al, 1998). The family Arteriviridae is grouped in the order Nidovirales along
with the Coronaviridae family. Arteriviruses infect macrophages in the host animal and
are responsible for a number of important diseases such as abortions and respiratory
disorders in pigs by PRRSV, persistent viremia in mice by LDV, abortion and arteritis in
horses by EAV, and hemorrhagic fever in monkeys by SHFV (Snijder and Spann, 2007).
The LDV genome is 14.1 kb in length dominated by two large open reading
frames ORF1a and ORF1ab, in the 5’ three-fourths of the genome and code for two large
polyproteins PP1a and PP1ab. The polyproteins are predicted to be proteolytically
cleaved into 12 cleavage products designated Nsp1 (non-structural protein 1) through
Nsp12. Downstream from ORF1ab, are ORF2 through ORF7, which encode at least 7
structural proteins; GP2a, GP2b (E), GP3, GP4, GP5, matrix (M) and nucleocapsid (N)

49
proteins (Snijder and Spann, 2007). The nucleocapsid protein of LDV is 115 amino acids
in length and is the most abundant protein within the virion.
Nidoviruses replicate exclusively in the cytoplasm. However, the N protein of
PRRSV, EAV and the coronaviruses, avian infectious bronchitis virus (IBV) and
transmissible gastroenteritis virus (TGEV), localize to the nucleolus (Rowland et al.,
1999; Tijms et al., 2002, Wurm et al, 2001). An interesting exception is the N protein of
SARS-CoV. One study showed that SARS-CoV N protein is localized exclusively in the
cytoplasm (Rowland et al, 2005), while another study has reported the SARS-CoV N
protein is present in the both cytoplasm and the nucleus/nucleolus (You et al, 2007).
These findings suggest that localization of N to the nucleolus may not be a general
property for all nidovirus N proteins. Once in the nucleus, the PRRSV and IBV N
proteins interact with fibrillarin and nucleolin, where they may be involved in the cell
cycle regulation (Yoo et al., 2003; Chen et al., 2002; Wurm et al., 2001). For nuclear
proteins, trafficking between cytoplasm and the nucleus is through the nuclear pore
complexes (NPC). While small molecules enter the nucleus by diffusion through NPC,
large molecules enter the nucleus by an energy-dependant pathway using a nuclear
localization signal (NLS) (Golrich and Kutay, 1999). The NLS on the cargo protein
interacts with importin proteins in the cytoplasm and then are shuttled across the NPC
and into the nucleus. Three types of NLSs have been identified. A “pat4” type NLS is a
stretch of four basic amino acids such as ‘KKKK’. A second type is “pat7” which is a
string of amino acids starting with proline (P) followed by any other two amino acids
which is followed by four amino acids of which at least three are basic residues. The third
type is a bipartite form. The bipartite NLS has two areas of basic residues separated by a

50
10-12 amino acids. The first group of basic residues includes two basic amino acids and
the second group contains 5 amino acids consisting of at least three basic residues (Nakai
and Kanehisa, 1992; Rowland and Yoo, 2003). In this report, we examined the N protein
nuclear localization of LDV and identified a functional ‘pat-4’ signal sequence essential
for N protein translocation into the nucleus.
MATERIALS AND METHODS
Cells, viruses, and bacterial strains: MARC-145 (Kim et al, 1993), a derivative of MA-
104 cells, HeLa cells, and NIH-3T3 cells were used for this study. Cells were maintained
in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% heat
inactivated fetal bovine serum (FBS, Invitrogen, Carlsbad, CA) in a humidified incubator
with 5% CO2 at 37ºC. PRRSV strain PA8 (Wootton et al., 2000) was grown in MARC-
145 cells. E. coli strains DH5α and XL-1 Blue (Stratagene, La Jolla, CA) were used for
gene cloning and mutagenesis, and strain BL-21 (Stratagene) was used for protein
expression.
Plasmid constructions: The LDV-N gene was amplified by PCR from a plasmid
containing the LDV genomic fragment, and using the BamHI sequence at both ends of
the primers, cloned into pEGFP-N1 and pEGFP-C1 (Clontech, Mountain View, CA)
upstream and downstream of the enhanced green fluorescence protein (EGFP) gene,
respectively. LDV-N was also subcloned using BamHI in pCITE-2C (Novagen) for in
vitro transcription and translation.
Mutant construction for LDV-N: Deletion constructs of LDV-N were made by PCR
using respective primer sets (Table 2.1) under the following conditions: preincubation for

51
Table 2.1. List of primers used for EGFP fusion constructions for LDV N.
Construct Primer pair Sequence a
LDV N-EGFP N-EGFP-FWD
N-EGFP-REV
5’-ATggatccATGTCTCAAAATAAGAAGAA-3’
5’-ATggatccGAAGCAGAAGAATTAGCAGAAG-3’
EGFP-LDV N EGFP-N-FWD
EGFP-N-REV
5’-ATggatccATGTCTCAAAATAAGAAGAA-3’
5’-ATggatccTTAAGCAGAAGAATTAGCAG-3’
LDV N- ΔC40 N-ΔC40-FWD
N- ΔC40-REV
5’-ATggatccATGTCTCAAAATAAGAAGAA-3’
5’-ATggatccGACAGCTTGGGCTGCTTCTTCTT-3’
LDV N- ΔC65 N-ΔC65-FWD
N- ΔC65-REV
5’-ATggatccATGTCTCAAAATAAGAAGAA-3’
5’-ATggatccGACGCCATAGGGAAGTGCAGCTT-3’
LDV N- ΔC70 N-ΔC70-FWD
N- ΔC70-REV
5’-ATggatccATGTCTCAAAATAAGAAGAA-3’
5’-ATggatccGAGAACAAGGTTACCAATGAGGC-3’
LDV N- ΔC82 N-ΔC82-FWD
N- ΔC82-REV
5’-ATggatccATGTCTCAAAATAAGAAGAA-3’
5’-ATggatccGATTTATTCTGTCCGGCATTGCG-3’
a Primers were designed based on sequence information available for LDV.
b Lowercase letters indicate BamHI recognition sequence.

52
5 min at 37ºC, denaturation for 30 sec at 95ºC, annealing for 1 min at 56ºC, and extension
for 1 min at 72ºC per cycle for 35 cycles, and final extension for 10 min at 72ºC in the
GeneAmp 2400 thermocycler (Perkin Elmer). PCR products were digested with BamHI
and subcloned into pEGFP-N1. Each construct was verified by restriction digestion and
nucleotide sequencing of the junction between N and EGFP. Cloning and DNA
manipulations were conducted according to the standard protocols (Sambrook and
Russell, 2001). For mutagenesis, PCR-based site-directed mutagenesis was performed by
overlapping extension using mismatch primers as previously described (Table 2.2)
(Wootton et al., 2001). PCR was carried out using 15 ng of pLDV-N-EGFP plasmid
DNA, 300 ng of each forward and reverse primer, 1 mM dNTPs, 1X buffer [10 mM KCl,
10 mM (NH4)2SO4, 20 mM Tris-HCl (pH 8.8), 2 mM MgSO4, 0.1% Triton X-100], and
2.5 units of Pfu DNA polymerase (Stratagene) for 16 cycles as follows: denaturation at
94°C for 30 sec, primer annealing at 56°C for 1 min, and primer extension at 68°C for 8
min. The PCR products were digested with DpnI and transformed to E. coli XL1-Blue.
Transformants were randomly selected for plasmid DNA preparation using a QIAprep
spin miniprep kit (QIAGEN, Santa Clarita, CA), and mutagenized sequences were
verified by nucleotide sequencing in both directions.
Fluorescent microscopy: Expression of N-GFP fusion proteins was determined in HeLa
cells by transfection using Lipofectin (Invitrogen) according to the manufacturer’s
instruction. Cells were grown on microscope coverslips and transfected with GFP fusion
constructs of LDV-N. PRRSV N gene and PRRSV N-GFP fusion constructs were
included as a positive control. At 24 hrs post-transfection, cells were fixed with methanol,
blocked with 1% bovine serum albumin (BSA) in PBS (phosphate buffered saline) for 30

53
min, and visualized for fluorescence using a fluorescence microscope (model AX70,
Olympus). For PRRSV-infected cells, N protein was stained with MAb SDOW-17
(Nelson et al., 1993) at a dilution at 1:400 in 1% BSA-PBS, followed by goat anti-mouse
antibody conjugated with AlexaFluor 488 (Molecular Probes) in dilution of 1:100. After
5 washes with PBS, cells were stained with DAPI (4',6-Diamidino-2-phenylindole
dihydrochloride, Sigma) for 5 min, washed again, mounted on a slide using 40 μl of the
mounting buffer (60% glycerol, 0.1% sodium azide in PBS), and visualized for
fluorescence.
GST-fusion protein expression and coupling with Sepharose beads: Mouse importin-
α and importin-β genes were cloned in pGEX-3X (Amersham, Piscataway, NJ) and
expressed as GST (glutathione-S-transferase)-importin-α and GST-importin-β fusion
proteins. One ml of Luria-Bertani medium containing 100 g/ml of ampicillin was
inoculated with 1/100 of an overnight culture. When optical density of the culture reaches
0.6 at 600 nm, IPTG (isopropyl-β-D-thiogalactopyranoside) was added to the final
concentration of 1 mM and the culture was further incubated for 3 h. Bacterial cells were
collected by centrifugation at 6,000 RPM for 10 min 4oC (Avanti J30I, Beckman) and
resuspended in 10 ml of PBS. The bacterial suspension was sonicated on ice three times
(model W-385; Ultrasonics Inc.), each time for 30 sec with 2 sec intervals. The
suspension was incubated with 1% Triton X-100 for 30 min at 4oC with occasional
agitation and centrifuged at 10,000 RPM for 10 min at 4oC to remove the insoluble
fractions and cell debris. The supernatant which contained expressed GST-fusion proteins
was incubated with 100 l of glutathion-Sepharose 4B beads (Amersham) in 50% slurry
overnight at 4oC with constant agitation. GST-fusion proteins bond to beads were

54
centrifuged and washed 5 times in PBS containing 1% Triton X-100. The collected beads
were then resuspended in a final volume of 250 l of incubation buffer (20 mM Tris-HCl
[pH 7.5], 100 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 5 mM dithiothreitol, 0.5% NP-40, 1
mM PMSF, 5% glycerol). This resulted in 20% slurry of beads to use for GST pull-down
assay. For production of radiolabelled N protein, the LDV-N gene was cloned into pCITE
(Novogen) and translated using TNT® Quick Coupled in vitro Transcription/Translation
system (Promega, Madison, WI) in the presence of [35
S]methionine (EasyTag EXPRESS
protein-labeling mix of [35
S]methionine and [35
S]cysteine; specific activity 407 MBq/ml,
Perkin-Elmer) according to the manufacturer’s instruction. Based on the quantity as
measured by SDS-PAGE, approximately equal amounts of Sepharose-coupled GST
fusion proteins and LDV-N were incubated overnight at 4°C with constant agitation. The
Sepharose beads were washed five times with PBS, boiled for 5 min in SDS sample
buffer (10 mM Tris-HCl [pH 6.8], 25% glycerol, 10% SDS, 10% β-mercaptoethanol, and
0.12% [wt/vol] bromophenol blue), and analyzed by 12% SDS-PAGE. Gels were dried
and exposed for image analysis by Phosphorimager (Molecular Dynamics).
RESULTS
Subcellular localization of EGFP-tagged LDV-N: The LDV nucleocapsid protein was
expressed as an EGFP-N fusion protein in HeLa cells and examined for subcellular
distribution. Cells were counterstained with DAPI for nuclear staining. EGFP alone was
distributed throughout transfected cells (Fig. 2.1B). For the purpose of comparisons,
localization of the PRRSV N protein in virus-infected MARC-145 cells is shown (Fig.
2.1C). Detection of PRRSV N by N-specific MAb SDOW17 showed the accumulation of

55

56
Fig. 2. 1. Subcellular localization of LDV-N protein. HeLa cells grown on the coverslips
were transfected with EGFP-LDV-N fusion protein genes using Lipofectin. At 24 hrs
post-transfection, cells were fixed with methanol and stained with DAPI for nuclear
staining. PRRSV-infected MARC-145 cells were stained with SDOW-17 MAb, followed
by staining with goat anti-mouse Ab conjugated with AlexaFluor 488 and finally were
stained with DAPI for nuclear staining. Cells on the coverslips were visualized by
fluorescence microscopy (model AX70, Olympus). Panels: A, HeLa cells without
transfection; B, HeLa cells with the pEGFP-N1 empty vector; C, PRRSV-infected
MARC-145 cells stained with the N-specific MAb SDOW-17; D, PRRSV-infected
MARC-145 cells stained with bovine coronavirus spike protein-specific MAb; E, HeLa
cells with PRRSV-N gene stained with SDOW-17; F, HeLa cells with PRRSV-N-GFP;
G, HeLa cells with LDV-N-EGFP; H, HeLa cells with EGFP-LDV-N.

57
N in the nucleus and nucleolus in addition to the normal cytoplasmic distribution. In
PRRSV N and PRRSV N-GFP transfected cells, N accumulated in the nucleus (Fig. 2.1E
and 2.1F), and similarly, both LDV-N-EGFP and LDV-EGFP-N fusion proteins showed
the accumulation of fluorescence in the nucleus (Fig. 2.1G and 2.1H), with a distribution
pattern similar to PRRSV N, indicating that LDV-N localized both in the cytoplasm and
the nucleolus.
Identification and mapping of NLS in the LDV-N protein: The sequence analysis of
the LDV-N protein identified the presence of a single basic region of KKK which
resembled a ‘pat-4’ type NLS located between amino acid positions 38-41 (Fig. 2.2A).
The location of this lysine-rich region corresponded to the ‘pat-7’ motif of PRRSV N
protein. To determine the functional significance of the ‘pat-4’ motif of LDV-N, a series
of deletion mutants were made by progressively deleting from the C-terminus of N (Fig.
2.2B). Mutant constructs were then transfected into cells and their fluorescence was
examined. LDV-N-C40-EGFP, LDV-N-C65-EGFP, and LDV-N-C70-EGFP showed the
fluorescence in the nucleus and nucleolus (Fig. 2.2(C); panels B, C, D).
In contrast, LDV-N-C82-EGFP in which the putative motif was deleted showed the
presence of cytoplasmic fluorescence but the absence of nuclear fluorescence (panels E).
These results indicate that the predicted ‘pat-4’ motif was indeed a functional NLS that
translocates LDV-N to the nucleus and nucleolus in cells.
To further confirm the function of predicted NLS and to identify core amino acids
essential for this function, individual residues of ’38-KKKK-41’ were substituted with
glycine (G) at single or multiple sites (Table 2.2), and the individual mutants were then
expressed in HeLa cells (Fig. 2.4A). This study showed that two ‘K’s at positions 38 and

58

59
Fig. 2. 2. Identification of an NLS motif in the LDV-N protein and its functional
mapping. A, a predicted ‘pat-4’ type nuclear localization signal in the LDV-N protein
identified by the PSORT II program. The PRRSV N protein sequence is aligned for
comparison with LDV-N. Dashes indicate deletions. Bold letters with underlines indicate
‘pat-4’ (for LDV) and ‘pat-7’ (for PRRSV) type NLS sequences with their amino acids
positions. B, schematic presentation of LDV-N deletion mutants fused with EGFP.
Darkened areas represent predicted NLS at amino acid positions 38-41. C, subcellular
localization of LDV-N mutants, upper and lower images are GFP expression of the LDV
N constructs and the overlay of GFP and DAPI staining of the same area respectively.
Panels (A), wild type LDV-N-EGFP; (B), deletion constructs LDV-N-C40-EGFP; C,
LDV-N-C65-EGFP; D, LDV-N-C70-EGFP; E, LDV-N-C82-EGFP.

60
Table 2.2. Site-directed mutagenesis of LDV N NLS motif and subcellular localization of
NLS mutants.
Construct Sequence of NLSa
Localizationb
Cy Nu No
LDV-N 38-KKKK-41 ++++ ++ ++++
LDVN- K38G 38-GKKK-41 ++++ ++++ +++
LDV-N- K39G 38-KGKK-41 ++++ +++ +
LDV-N- K40G 38-KKGK-41 ++++ ++ +
LDV-N- K41G 38-KKKG-41 ++++ ++++ +++
LDV-N- K38,39G 38-GGKK-41 ++++ ++++ +
LDV-N- K38,40G 38-GKGK-41 ++++ ++ +
LDV-N- K38,41K 38-GKKG-41 ++++ +++ +
LDV-N- K39,40G 38-KGGK-41 ++++ +++ ++
LDV-N- K39,41G 38-KGKG-41 ++++ +++ +
LDV-N- K40,41G 38-KKGG-41 ++++ +++ +/-
LDV-N- K39,40,41G 38-KGGG-41 ++++ +++ +/-
LDV-N- K38,39,40G 38-GGGK-41 ++++ +++ +/-
LDV-N- K38,39,40,41G 38-GGGG-41 ++++ - -
aIndividual lysines (underlined) in the NLS were substituted with glycines.
bFor scoring of the fluorescence signal for each mutant construct, 100 transfected cells
were counted presented as + or -. Each one ‘+’ represents fluorescence of 25 transfected
cells either in the cytoplasm (Cy), nucleus (Nu), or nucleolus (No).

61
Fig. 2. 3. Subcellular localization of NLS mutants. Panels A through D, single
substitutions of LDV-N (LDV-N-K38G, LDV-N-K39G , LDV-N-K40G, and LDV-N-
K41G, respectively) panels E through J are double mutations in NLS (LDV-N-K38,39G ,
LDV-N-K38,40G, LDV-N-K38,41K , LDV-N-K39,40G , LDV-N-K39,41G , LDV-N-
K40,41G, respectively). Panels K and L are triple mutations in NLS (LDV-N-
K39,40,41G and LDV-N-K38,39,40G). (A), GFP staining; (B), the same cells in (A)
overlaid with DAPI staining.

62
Fig. 2. 4. Subcellular localization of the NLS-null LDV-N-EGFP mutants in HeLa cells
(A) and NIH-3T3 murine cells (B). The NLS (38-KKKK) of LDV-N was mutated to 38-
GGGG-41 to knock out the NLS function. The NLS-null LDV-N gene was transfected to
HeLa cells or NIH-3T3 cells followed by staining with DAPI for fluorescent microscopy.
LDV-N-wt, wild type LDV-N-EGFP; LDV-N-null, NLS-null mutant LDV-N-EGFP;
PRRSV-N, porcine reproductive respiratory syndrome virus wild type N protein.

63
40 function as core amino acids for nucleolar translocation of N and ‘K’s at positions 38,
40, and 41 play a crucial role for this function (Table 2.2 and Fig. 2.3). The mutant N in
which all four ‘K’s were substituted with ‘G’s was exclusively cytoplasmic and did not
localize to either nucleus or nucleolus (Fig. 2.4), confirming that the pat-4’ motif was
indeed the functional NLS for LDV-N protein for nuclear localization. Since LDV is a
murine virus, the function of NLS and the nuclear localization of LDV-N were confirmed
in mouse cells using the same constructs (Fig. 2.4B). NIH-3T3 cells were transfected
with either wild-type LDV-N or LDV-N NLS-null mutant construct and examined for
fluorescence. The wild-type LDV-N protein was clearly nuclear-localized, and the LDV-
N NLS-null mutant was cytoplasmic without distinct nuclear staining, indicating that the
LDV-N nuclear localization is not cell type-dependent but is rather the nature of the N
protein.
LDV-N interaction with importin proteins: To determine the basis for LDV-N nuclear
localization, interactions between N and importin proteins were determined by GST pull-
down assay. Importin-α and importin-β were individually expressed in E.coli as a GST-
fusion protein and coupled to glutathione-Sepharose beads. The radiolabelled LDV-N
protein was then synthesized in vitro by transcription translation, and importin-coupled
GST beads were incubated with the radiolabelled N protein. Unbond proteins were
washed off and the bond proteins were resolved by SDS-PAGE and autoradiography. As
with the PRRSV N protein which was previously shown to interact with both importin-α
and -β proteins (Rowland et al., 2003; Fig. 2.5, lanes 7 and 9), both importin-α and
importin-β specifically bond LDV-N (lanes 8 and 10), while GST alone did not bind to
both PRRSV-N and LDV-N proteins (lanes 5 and 6). This study demonstrates the specific

64
interaction of LDV-N with both importin proteins, and suggests that LDV-N nuclear
localization may be importin-mediated.
DISCUSSION
Nuclear localization of capsid protein has been studied for several RNA viruses
that restrict their replication in the cytoplasm, and capsid proteins of Semliki forest virus,
dengue fever virus, and hepatitis C virus have been shown to localize in the nucleus and
nucleolus (Favre et al., 1994; Yasui et al., 1998, Wang et al., 2002). Nidoviruses also
replicate exclusively in the cytoplasm of infected cells (Brayton et. al, 1981), but N
protein has been shown to localize in the nucleus and nucleolus for PRRSV, EAV, IBV,
MHV and TGEV (Hiscox et al, 2001; Wurm et al, 2001; Rowland et al., 1999; Tijms et
al., 2002). The N protein of PRRSV contains two putative NLS motifs, ‘pat 4’ and ‘pat
7’, and the “pat 7” located at positions 41-47 has been shown to be the active and primary
NLS for N (Rowland et al., 2003). In the present study, we showed that the LDV-N
protein localized to the nucleus and nucleolus in N-EGFP gene transfected cells, and
using mutant constructs showed that the nuclear localization of N was dependent on a
‘pat-4’ motif located at positions 38-41 (Fig. 2. 2A; 2C). The location of ‘pat-4’ motif in
LDV-N is similar to the region where the functional ‘pat 7’ motif is located in PRRSV N.
We further identified two lysine residues at positions 38 and 40 in LDV-N that form core
residues for nuclear transport of N. For PRRSV N, amino acids at position 43 and 44 are
critical residues for nuclear localization (Rowland et al., 2003; Lee et al., 2006a; Pei et
al., 2008). For a protein to localize in the nucleus, the cellular chaperone importin-α
recognizes the NLS on the cargo protein and subsequently, importin-β binds to the

65
Fig. 2. 5. Interactions of LDV-N with mouse importin proteins by GST-pull down assays.
Importin-α- and importin-β-glutathione-S-transferase (GST) fusion proteins were
expressed in E-coli and coupled to glutathion-Sepharose 4B beads. Beads-bound proteins
were incubated overnight at 4°C with in vitro-translated radiolabelled N protein. After
washing, bound proteins were visualized by 12% SDS-PAGE and autoradiography. Left
panel, in vitro translation of vector control (lane 2), PRRSV-N (lane 3), and LDV-N (lane
4). Middle and right panels, GST pull-down assays for LDV-N by importin-α (lane 8)
and importin-β (lane 10), respectively.

66
importin-α/cargo protein complex, transporting the complex in the presence of Ran-GDP
to NPC on the nuclear membrane. We found that LDV-N binds to importin-α (Fig. 2.5,
lane 8), which is consistent with the utilization of the classical pathway for nuclear
transport. It is interesting to note that LDV-N also binds to importin-β (Fig. 2.5, lane 10).
PRRSV N also binds to both importin-α and importin-β (Rowland et al, 2003) and
several other viral proteins have been reported to interact with both importin-α and -β.
The polyomavirus capsid protein requires the interaction with both importin-α and -β to
enter the nucleus (Qu et al., 2004), and SV40 VP3 also interacts with both importin
proteins for nuclear translocation (Nakanashi et al, 2002). The HIV Rev protein binds
both importin-α and -β, but its binding to importin-α is NLS-independent while the
importin- β binding is NLS-dependent (Henderson et al., 1997). For PRRSV, two
nucleolar proteins, nucleolin and B23, have been identified to bind to N (unpublished
data), and thus a possibility for the LDV-N nuclear transport is the incorporation of
importin-dependent pathway with a ‘piggy-backing’ mechanism, in which N binds to a
nuclear targeted cellular protein in addition to importin proteins.
Since N proteins of PRRSV, EAV, and LDV all localize to the nucleus and
nucleolus, the nuclear role of N during infection is of major interest. The core protein of
hepatitis C virus is believed to modify cellular function by preventing translocation of
host cell proteins to the nucleus (Isoyama et al., 2002; Suzuki et al., 2005), and the N
protein of TGEV has been suggested to play a role in the disruption of cell division
(Wurm et al., 2001). For PRRSV N, disruption of NLS does not affect virus replication in
cell culture, but in pigs, NLS-null viruses are attenuated in their virulence (Lee et al.,
2006a, 2006b; Pei et al., 2008). The NLS motif of PRRSV N was associated with strong

67
selective pressure for reacquisition of the nuclear function in vivo, suggesting the
involvement of N nuclear localization in the viral pathogenesis. N proteins of several
member viruses in the order Nidovirales colocalize and interact with fibrillarin and
nucleolin in the nucleolus, supporting the hypothesis that N modulates the host cell
ribosomal biogenesis and cell cycle progression (Chen et al., 2002; Hiscox, 2007). In
summary, we show that LDV-N contains a functional ‘pat-4’ type NLS at positions 38-41
and localizes in the nucleolus of cell via the importin-mediated pathway. The capsid
protein nuclear localization may be a common feature in the Arteriviridae family, which
may play a common role for replication and pathogenesis of this group of viruses.

68
CHAPTER 3
Nuclear Localization of Equine Arteritis Virus and Simian Hemorrhagic Fever Virus
Nucleocapsid Proteins

69
ABSTRACT
In the present study, the subcellular localization of nucleocapsid (N) proteins of
two member viruses in the Arteriviridae family, equine arteritis virus (EAV) and simian
hemorrhagic fever virus (SHFV) were examined by immunofluorescence microscopy.
EAV N and SHFV N proteins were fused with the enhanced green fluorescence protein
(EGFP) on the N- or C-terminus, and the cellular distribution of fusion proteins was
examined in gene-transfected cells. Both N-EGFP and EGFP-N fusion proteins of EAV
and SHFV showed accumulation of fluorescence in the nucleus and nucleolus of gene-
transfected cells. The EAV N gene contains a putative ‘bipartite’ type nuclear
localization signal (NLS) at position 4-20, and the SHFV N gene has a potential ‘pat-7’
type NLS at amino acid positions 22-28. To determine the functional significance of the
NLS-like sequences, a series of deletion mutants were made by progressively deleting
amino acids from the C- and N-termini of both EAV N and SHFV N proteins and the
mutant proteins were individually expressed in HeLa cells. The results show that the
‘pat7’ NLS was essential for nuclear translocation of SHFV N protein, while EAV N
protein required the entire motif containing 4-20 N-terminal residues for nuclear
localization. Furthermore, the interaction of SHFV N protein with importin-α indicates
the use of the importin-mediated nuclear transport pathway. EAV N protein interacted
with both importin-α and importin-β proteins, which indicates that the importin mediated
pathway is likely involved in its nuclear transfer. Taken together, this study indicates that
nucleolar localization of the N protein is a common feature of the Arteriviridae family
and possibly plays a significant role in the virus life cycle of this family.

70
INTRODUCTION
Arteriviruses include four members of enveloped RNA viruses containing a
single-stranded positive-sense RNA genome of 12.7 to 15.7 kilobases (kb) in size with
the 5` cap structure and 3` polyadenylated tail (Snijder and Swaan, 2007). Based on the
similarity of the mechanism for their genome replication, the Arteriviridae family has
been placed along with the Coronaviridae family, into the order Nidovirales (Snijder and
Spann, 2007; Gorbalenya et al, 2006).
Equine arteritis virus (EAV) and simian hemorrhagic fever virus (SHFV) are two
member viruses of the Arteriviridae family and cause viral arteritis in horses and
hemorrhagic fever in monkeys, respectively. EAV causes respiratory and reproductive
failures in horses, donkeys, and mules resulting in economic losses for the equine
industry in North America (Timoney and McCollum, 1993). The causative agent for viral
arteritis is EAV which was first identified in 1957 in Bucyrus, Ohio from lungs of sick
horses affected by this disease (Doll et al, 1957; Bryans et al 1957). SHFV causes acute,
febrile and fatal disease with a mortality rate of up to 100% in rhesus monkeys (Palmer et
al, 1968). It was first isolated from a fatal hemorrhagic fever of macaque monkeys in
1968 (Madden et al., 1978, Trousdale et. al 1975).
EAV is the prototype virus of the Arteriviridae family (Snijder and Meulenberg,
1998). Its genome is an infectious single-stranded RNA 12.7 kb in size which contains 9
open reading frames (ORF). ORF1a and ORF1ab are translated directly from the viral
genome, and the two polyproteins are cleaved to produce 18 non-structural proteins.
These proteins are involved in viral genome transcription and replication. ORFs 2a, 2b, 3,
4, 5, 6, and 7 code for structural proteins E, GP2 (GP2b/Gs), GP3, GP4, GP5 (GL), M, and N

71
respectively. The M and GP4 are two major envelope proteins, and E, GP2b, GP3, and
GP4 are the minor envelop proteins (Snijder and Spann, 2007; de Vries et al, 1992;
Wieringa et al, 2004; Molenkamp et al, 2000b). N protein is the most abundant protein of
EAV involved in virion RNA encapsidation, while being dispensable for viral genome
replication (de Vries et al, 1992; Wieringa et al., 2004; Molenkamp et al, 2000b). EAV N
protein is a phosphoprotein of 110 amino acids with the molecular weights of 12-14 kD
(Zeeger et al, 1976; de Vries et al, 1992).
The genome of SHFV is also an infectious RNA of 15.7 kb in size. It contains two
non-structural and 9 structural ORFs. Like other members of arteriviruses, non-structural
proteins of SHFV are also translated from the two largest ORFs; ORF1a and ORF1b.
ORFs 2a, 2b, 3, 4a, 4b, 5, 6, 7, 8, and 9 code for structural proteins Gp2a, Gp2b, Gp3, E,
Gp4, Gp5, Gp6, Gp7(p54), M, and N, respectively. ORF9 encodes nucleocapsid (N)
protein which is one of the most abundant structural proteins of SHFV with 115 amino
acids in length (Sagripanti 1984).
Previous studies have shown that the N protein of PRRSV localizes to the
cytoplasm and the nucleus/nucleolus of PRRSV-infected and N-gene transfected cells
(Rowland et al., 1999). EAV N also seems to localize in the nucleus and nucleolus of
infected cells (Tijms et al., 2002). A similar observation has been reported for the N
protein of lactate dehydrogenase-elevating virus (LDV) in human and mouse cells
expressing LDV N (Mohammadi et al., 2009). Similar cellular distribution of the N
proteins have been reported for coronaviruses infectious bronchitis virus
(IBV),
transmissible gastroenteritis virus (TGEV), and severe acute respiratory syndrome
(SARS) virus (Wurm et al, 2001, You et al, 2007). As in Coronaviridae, viruses in the

72
Arteriviridae family replicate in the cytoplasm of infected cells and do not need the
nuclear function of infected cells for their life cycle. The finding of the nucleolar
localization of N protein in some arteriviruses including PRRSV and EAV suggests that
the nucleolar localization of N protein may be a common feature in nidoviruses with an
important function for viral replication in addition to its structural role for virion
assembly of these viruses. Despite the investigation using PRRSV and LDV, the cellular
distribution of SHFV N and EAV N has yet to be investigated.
The objectives of this study were to determine the SHFV N transport to the
nucleus, to define the nuclear localization signal (NLS) for EAV and SHFV N proteins,
and to elucidate the mechanism of their transport to the nucleus. This study shows that
the nuclear localization of N is a conserved feature of arteriviruses and indicates that the
nucleolar localization of the N protein may play an important role in the pathogenesis of
diseases caused by these viruses.
MATERIALS AND METHODS
Cells, viruses, and bacterial strains. Baby hamster kidney cells (BHK-21), MARC-145
(Kim et al, 1993) which is a derivative of the MA104 cell line, COS-1, and HeLa cells
were used in this study. Cells were maintained in Dulbecco’s modified Eagle’s medium
(DMEM) supplemented with 10% heat inactivated fetal bovine serum (FBS; GIBCO
BRL) in a humidified incubator with 5% CO2 at 37ºC. EAV strain Bucyrus (Doll et al.,
1957) and PRRSV strain PA8 (Wootton et al., 2000) were grown in MARC-145 and
BHK-21 cells, respectively. Escherichia coli (E. coli) strains XL-1 Blue (Stratagene, La

73
Jolla, CA), and DH5α were used for gene cloning and mutagenesis, and E. coli strain BL-
21 (Novagen) was used for protein expression.
Antibodies. A mouse MAb SDOW-17 (IgG isotype) against the PRRSV N (Nelson et al.,
1993) was used to stain for PRRSV N protein. A mouse MAb 3E2 (IgG type) against
EAV N was a generous gift from Dr. Udeni Balasuriya (Gluck Equine Research Center,
University of Kentucky, Lexington, USA) and used to stain for EAV N protein. Goat
anti-mouse antibody conjugated with AlexaFluor 488 was purchased from Molecular
Probes and used as a secondary antibody.
Plasmid constructions. The EAV N gene was amplified from pEAV030 containing the
full genome sequence of EAV (van Dinten et al., 1997) by PCR using the specific
primers containing a BamHI recognition sequence at the 5’ end (Table 3.1). The PCR
product was subcloned in-frame into pEGFP-N1 and pEGFP-C1 (Clontech Inc.)
upstream and downstream of the enhanced green fluorescence protein (EGFP) gene,
respectively. In order to clone the EAV N gene upstream of EGFP, the reverse primer
was designed to delete the stop codon of EAV N and the PCR product was subcloned in
frame with EGFP. SHFV N gene was PCR-amplified from a cDNA library of SHFV
genome (provided by Dr. E. Snijder, Leiden University Medical Center, Leiden,
Netherlands) using specific primers (Table 3.2) and sub-cloned upstream and
downstream of EGFP in the pEGFP-N1 and pEGFP-C1 in-frame, respectively. In order
to construct the SHFV N-EGFP in frame, the reverse primer of N-EGFP was designed to
eliminate stop codon of the SHFV N gene. Both EAV N and SHFV N were subcloned
into BamHI site of pCITE-2C (Novogen) for in vitro transcription and translation.

74
Table 3.1. List of primers used for EGFP fusion constructions for EAV N.
Construct Primer pair Sequence a
EAV N-EGFP N-EGFP-FWD
N-EGFP-REV
5’-ATggatccATGGCGTCAAGACGATCACGT-3’
5’-ATggatccGACGGCCCTGCTGGAGGCGCAA-3’
EGFP-EAV N EGFP-N-FWD
EGFP-N-REV
5’-ATggatccATGGCGTCAAGACGATCACGT-3’
5’-ATggatccTTACGGCCCTGCTGGAGGCGCA-3’
EAV N- ΔC40 N-ΔC40-FWD
N- ΔC40-REV
5’-ATggatccATGGCGTCAAGACGATCACGT-3’
5’-GTggatccGATTGTACGTTCGACGAAAGGGT-3’
EAV N- ΔC70 N-ΔC70-FWD
N- ΔC70-REV
5’-ATggatccATGGCGTCAAGACGATCACGT-3’
5’-GTggatccGAGGGCGGTTTGCGGACCCGCAT-3’
EAV N- ΔC87 N-ΔC87-FWD
N- ΔC87-REV
5’-ATggatccATGGCGTCAAGACGATCACGT-3’
5’-TAggatccGAGCTTGTAGGCTGTCGCCGC-3’
EAV N- ΔN20 N-ΔN20-FWD
N- ΔN20-REV
5’-CGggatccATGCCTACAAGCTACAATGACCT-3’
5’-ATggatccGACGGCCCTGCTGGAGGCGCAA-3’
a Primers were designed based on sequence information available for EAV.
b Lowercase letters indicate BamHI recognition sequence.

75
Table 3.2. List of primers used for EGFP fusion constructions for SHFV N.
Construct Primer pair Sequence a
SHFV N-EGFP N-EGFP-FWD
N-EGFP-REV
5’-GGTggatccATGGCTGGCAAACCAAAAAC-3’
5’-GTggatccGAGGTGGAGGAGGTGACCTT-3’
EGFP-SHFV N EGFP-N-FWD
EGFP-N-REV
5’-GGTggatccATGGCTGGCAAACCAAAAAC-3’
5’-AGTggatccCTAGGTGGAGGAGGTGACCTTC-3’
SHFV N- ΔC20 N-ΔC20-FWD
N- ΔC20-REV
5’-CGggatccATGGCTGGCAAACCAAAA-3’
5’-GTggatccGAAGCAAAATTGATTCT-3’
SHFV N- ΔC40 N-ΔC40-FWD
N- ΔC40-REV
5’-CGggatccATGGCTGGCAAACCAAAA-3’
5’-GTggatccGAGATCAGCAGCTGTTT-3’
SHFV N- ΔC70 N-ΔC70-FWD
N- ΔC70-REV
5’-CGggatccATGGCTGGCAAACCAAAA-3’
5’-GTggatccGATAGAGGCTTGTGGAC-3’
SHFV N- ΔN30 N-ΔN30-FWD
N- ΔN30-REV
5’-CGggatccATGCGTAGAGCTGCTCCT-3’
5’-GTggatccGAGGTGGAGGAGGTGACCTT-3’
a Primers were designed based on sequence information available for SHFV.
b Lowercase letters indicate BamHI recognition sequence.

76
Deletion construction and site-directed mutagenesis. Deletion constructs of EAV N
and SHFV N were made by PCR using respective primer sets (Tables 3.1 and 3.2) under
the following amplification conditions: preincubation for 5 min at 37ºC followed by 30
sec at 95ºC for denaturation, 1 min at 56ºC for annealing, and 1 min at 72ºC for extension
for 35 cycles, and the final extension for 10 min at 72ºC (GeneAmp 2400 thermocycler,
Perkin Elmer). Individual PCR products were digested with BamHI and subcloned into
the BamHI site of pEGFP-N1. Each construct was verified by restriction digestion and
nucleotide sequencing of the junction between N and EGFP. Cloning and DNA
manipulations were conducted according to the standard protocols (Sambrook and
Russell, 2001). To introduce mutations to NLS, PCR-based site-directed mutagenesis was
performed by overlapping extension using primer sequences for each mutation (Wootton
et al., 2001). PCR was performed using 15 ng of pEAV N-EGFP and pSHFV N-EGFP
plasmid DNA, 300 ng of forward and reverse primers (Tables 3.2 and 3.4), 1 mM dNTPs,
1X buffer [10 mM KCl, 10 mM (NH4)2SO4, 20 mM Tris-HCl (pH 8.8), 2 mM MgSO4,
0.1% Triton X-100], and 2.5 units of Pfu DNA polymerase (Stratagene) for 16 cycles
under the following parameters: denaturation at 94°C for 30 sec, primer annealing at
56°C for 1 min, and primer extension at 68°C for 8 min. Then, DpnI was added to PCR
products for 30 min and the digested DNA was transformed to E. coli XL1-Blue.
Colonies were randomly selected for plasmid DNA preparation using a QIAprep spin
miniprep kit (QIAGEN, Santa Clarita, Calif.) and sequencing. Nucleotide sequencing
was done in both directions to verify introduced mutations.
Fluorescence microscopy. Expression of N-GFP fusion proteins was determined in
HeLa cells by transfection using Lipofectin (Invitrogen) according to the manufacturer’s

77
instruction. For SHFV N-GFP fusion constructs COS-1 and HeLa cells were used for
transfection. Cells were grown on microscope coverslips and transfected with EAV N-
GFP and SHFV N-GFP fusion constructs using Lipofectin according to the
manufacturer’s instructions. PRRSV N gene and PRRSV N-GFP fusion constructs were
included as a positive control. BHK-21 cells were infected with EAV at a MOI of 1. One
group of cells was treated with 10 µM of leptomycin-B (LMB). At 24 h post-transfection
and at 2, 4, 6, 8, 10, 12, 24, 36, and 48 h post-infection, cells were examined for EAV N
protein distribution. Briefly, cells were fixed with methanol, blocked with 1% bovine
serum albumin (BSA) in PBS (phosphate buffered saline) for 30 min, stained with DAPI
(4', 6-Diamidino-2-phenylindole dihydrochloride, Sigma) for 5 min, washed again,
mounted on a slide using 40 μl of the mounting buffer (60% glycerol, 0.1% sodium azide
in PBS), and visualized for GFP fluorescence by fluorescent microscope (model AX70,
Olympus). For PRRSV-infected and EAV-infected cells, they were stained with MAb
SDOW-17 (Nelson et al., 1993) and MAb 3E2 at a dilution at 1:400 in 1% BSA-PBS,
followed by goat anti-mouse antibody conjugated with AlexaFluor 488 (Molecular
Probes) at the dilution of 1:100. After 5 times washing with PBS, cells were stained with
DAPI for 5 min, washed again, mounted on a slide using 40 μl of the mounting buffer
and visualized for fluorescence by fluorescence microscope.
Protein expression in E. coli and coupling with Sepharose beads. Plasmids containing
the murine importin genes, mImp-α (PTAC58) and mImp-β (PTAC97), were used to
express the gutathione-S-transferase T-importin-α and -β fusion proteins (GST-Impα and
-β) in E. coli strain BL-21 (Rowland et al., 2003). One ml of Luria-Bertani medium
containing 100 g/ml of ampicillin was inoculated with 1/100 of an overnight culture.

78
When the optical density of bacterial culture reached 0.6 at 600 nm, IPTG (isopropyl-β-
D-thiogalactopyranoside) was added to the final concentration of 1 mM and the culture
was further incubated for 3 hours. The bacterial cells were collected by centrifugation at
6,000 RPM for 10 min 4oC (Beckman Avanti J30I centrifuge) and resuspended in 10 ml
of PBS. The bacterial suspension was sonicated (model W-385; Ultrasonics Inc.) three
times, each time for 30 sec with 2 sec intervals. The suspension was incubated with 1%
Triton X-100 for 30 min at 4oC with occasional agitation and centrifuged at 10,000 RPM
for 10 min at 4oC to remove the insoluble fractions and cell debris. The supernatant
which contained expressed GST-fusion proteins was incubated with 100 l of glutathion-
Sepharose 4B beads (Pharmacia) in 50% slurry overnight at 4oC with constant agitation.
GST-fusion proteins bound to beads were centrifuged and washed 5 times in PBS
containing 1% Triton X-100. The collected beads were then resuspended in a final
volume of 250 l of incubation buffer (20 mM Tris-HCl [pH 7.5], 100 mM KCl, 2 mM
CaCl2, 2 mM MgCl2, 5 mM dithiothreitol, 0.5% NP-40, 1 mM PMSF, 5% glycerol). This
resulted in 20% slurry of beads to use for GST pull-down assay.
GST pull-down assay. To produce radiolabeled EAV N and SHFV N proteins, each N
gene was cloned into pCITE (Novogen) and translated using TNT® Quick Coupled in
vitro Transcription/Translation system (Promega) in the presence of 50 μCi/ml
[35
S]methionine (EasyTag EXPRESS protein-labeling mix of [35
S]methionine and
[35
S]cysteine; specific activity 407 MBq/ml, Perkin-Elmer) according to the
manufacturer’s instruction. The quantity of GST fusion proteins and translated EAV N
and SHFV N was measured by SDS-PAGE. Equal amounts of radiolabeled EAV N and
SHFV proteins were incubated with the GST fusion proteins bound to glutathione-

79
Sepharose beads in 20% slurry in binding buffer separately (20 mM Tris-HCl [pH 7.5],
100mM KCl, 2 mM CaCl2, 2 mM MgCl2, 5 mM dithiothreitol, 0.5% NP-40, 1 mM
PMSF, 5% glycerol) to the final volume of 400 μl overnight at 4°C with constant
agitation. The beads were washed five times with PBS, boiled for 5 min in SDS sample
buffer (10 mM Tris-HCl [pH 6.8], 25% glycerol, 10% SDS, 10% β-mercaptoethanol, and
0.12% [wt/vol] bromophenol blue), and analyzed by 12% SDS-PAGE. Gels were dried
and exposed to Phosphorimager (Molecular Dynamics) for image analysis.
Radioimmunoprecipitation: BHK-21 cells, grown to 75% confluency, were infected
for 1 h at 37oC with EAV at a MOI of 1. Cells were washed once with PBS and incubated
at 37o
C in fresh DMEM. At 48 h post-infection, the culture medium was removed and
cells were starved for 1 h in DMEM without methionine. Then, cells were labeled for 5 h
with 50 μCi of EasyTag EXPRESS protein-labeling mix ([35S]methionine and
[35S]cysteine; specific activity, 407 MBq/ml; Perkin-Elmer). After 5 h, cells were
harvested, washed twice with cold PBS, and lysed in RIPA buffer (1% Triton X-100, 1%
sodium deoxycholate, 150 mM NaCl, 50 mM Tris-HCl [pH 7.4], 10 mM EDTA, 0.1%
sodium dodecyl sulfate [SDS]) containing 1 mM phenylmethylsulfonyl fluoride (PMSF).
Cell lysates were incubated on ice for 20 min, centrifuged at 14,000 RPM for 30 min in a
microcentrifuge and the supernatants were put aside for immunoprecipitation (IP). For IP,
cell lysates were adjusted with RIPA buffer to a final volume of 100 l, and incubated
with 1 l of 3E2 MAb against EAV N for 2 h at room temperature. The immune
complexes were incubated with 7 mg of protein A-Sepharose CL-4B beads (Amersham
Biosciences) for 16 h at 4°C with constant agitation. In the following day, the complexes
were centrifuged at 6,000 RPM for 2 min, and beads were washed with RIPA buffer two

80
times and resuspended in 30 l of SDS-polyacrylamide gel electrophoresis (PAGE)
sample buffer (10 mM Tris- HCl [pH 6.8], 25% glycerol, 10% SDS, 0.12% [wt/vol]
bromophenol blue) containing 10% -mercaptoethanol. The mixtures were boiled for 5
min and run on 12% SDS–PAGE gel. The gels were dried and analyzed by a
Phosphorimager (model SI; Molecular Dynamics).
RESULTS
Subcellular localization of EAV N. To study the nucleolar localization of EAV N,
BHK-21 cells were infected with EAV and treated with and without LMB. Cells were
fixed with methanol and stained with a monoclonal antibody against EAV N as well as
DAPI at different time points post LMB treatment. The results of time course study of
EAV infection in BHK-21 cells are shown in Fig. 3.1. As demonstrated in Fig. 3.1, the
EAV N protein localizes in the nucleolus as early as 8 h post-infection (p.i). In the
absence of LMB, many cells showed the nucleolar localization until 12 h p.i. However,
during later stages of infection, the nucleolar localization of EAV N was limited to only a
few cells. At 24 h p.i., the EAV N localization was observed mostly in the cytoplasm and
at 36 h p.i., most of the infected cells were positive for cytoplasmic staining of EAV N
and no nucleolar or nuclear localization was observed (Fig. 3.1, panel A). In the presence
of LMB, EAV N was localized into nucleolus at early and late phases of infection (Fig.
3.1, panel B).
The EAV nucleocapsid protein was expressed as an EGFP-N fusion protein in
HeLa cells and examined for subcellular distribution. Cells were counterstained with the
nuclear stain, DAPI (Fig 3.2). Localization of the PRRSV N protein in infected MARC-

81
Fig 3.1. Localization of EAV N in the nucleus and nucleolus of BHK-21 cells at different
time points. Cells were fixed at the indicated times and stained with EAV N specific
MAb followed by staining with goat anti-mouse Ab conjugated with AlexaFluor 488.
Cells were then visualized by fluorescence microscopy. Panels demonstrate EAV-
infected BHK in the absence (A) or presence (B) of leptomycin B (LMB).

82
Fig 3.2. Subcellular localization of EAV N protein. HeLa cells grown on the coverslips
were transfected with EGFP-EAV N fusion protein genes using Lipofectin. At 24 hrs
post-transfection, cells were fixed with methanol and stained with DAPI for nuclear
staining. PRRSV-infected MARC-145 cells were stained with MAb SDOW-17, followed
by staining with goat anti-mouse Ab conjugated with AlexaFluor 488 and DAPI for
nuclear staining. Cells were visualized by fluorescence microscopy. Panels A, HeLa
without transfection; B, PRRSV-infected MARC-145 cells stained with the N-specific
MAb SDOW-17; C and D, EAV-infected BHK-21 cells stained with EAV N protein
specific MAb in the absence (C) or presence (D) of leptomycin B respectively; E, HeLa
cells with EAV N-EGFP; F, HeLa cells with EGFP-EAVN. Arrows show the nucleolar
staining of the N protein.

83
145 cells, is shown for the purpose of comparison (Fig. 3.2B). Staining of PRRSV N
showed the accumulation of N in the nucleus. Similarly, both EAV N-EGFP and EGFP-
EAV N fusion proteins showed the accumulation of fluorescence in the nucleus with a
distribution pattern similar to PRRS N (Fig. 3.2E and F, respectively). These results
indicate that EAV N was localized both in the cytoplasm and the nucleolus.
Identification and mapping of NLS in the EAV N. Sequence analysis of the EAV N
protein identified the presence of a basic region, which resembles a ‘bipartite’ type NLS
in the EAV N protein predicted by the PSORT II program (Nakai and Kanehisa, 1992).
This motif was located at positions 4-20 as 4-RRSRPQAASFRNGRRRQ-20 (Fig. 3.3A).
To determine the functional significance of this NLS-like sequence, a series of deletion
mutants were made by progressively deleting amino acids from the C- and N-termini of
EAV N (Fig. 3.3B). The mutant constructs were then transfected into HeLa cells and
their fluorescence was examined. EAVN C40-EGFP, EAVN C70-EGFP, and EAVN
C87-EGFP showed fluorescence in the nucleus and nucleolus (Fig. 3.3C, panels B, C and
D). In contrast, EAVN N20-EGFP in which the putative motif was deleted showed the
absence of nuclear fluorescence but the presence of cytoplasmic fluorescence (Fig. 3.3C,
panel D). These results indicate that the predicted ‘bipartite’ sequence may be a
functional NLS that translocates EAV N to the nucleus and nucleolus in transfected cells.
To confirm the function of the predicted NLS and to identify core amino acids essential
for this function, individual arginine (R) residues of 4-RRSRPQAASFRNGRRRQ-20
were substituted with glycine (G). Double, triple, quintuple, sextuple, and septuple G
mutants of EAV N were constructed (Fig 3.4). Individual mutants were expressed in
HeLa cells and visualized by fluorescence microscopy (Fig. 3.4). All of mutants with R

84

85
Fig 3.3. Identification of NLS in EAV N protein and its functional mapping. (A)
identification of a putative ‘bipartite’ type NLS in the EAV N protein predicted by the
PSORT II program (Nakai and Kanehisa, 1992). The PRRSV N protein sequence is
aligned for comparisons. Dashes indicate amino acid deletions. Boxes indicate ‘bipartite’
(for EAV), ‘pat-4’ and ‘pat-7’ (for PRRSV) type NLS sequences. (B). Schematic
presentation of EAVN deletion mutants fused with EGFP. The darkened areas represent
predicted NLS at amino acid positions 4-20. (C). Subcellular localization of EAV N
mutants. Panels A, wild type EAVN-EGFP; B-E, deletion construct: EAVN-EGFP-C40
(B); EAVN-EGFP-C70 (C); EAVN-EGFP-C87 (D); EAVN-EGFP-N20 (E).

86
Fig 3.4. Subcellular localization of EAV N NLS mutants. Names of mutants and their
amino acid sequences are indicated. Panels show immunofluorescence of each mutant .
Overlay, superimposition with DAPI staining.

87
Fig 3.5. Interaction of EAV-N with importin proteins by GST-pull down assay. Panels A
represents GST pull-down assay using EAV-infected BHK-21 cell lysates and panel B
represents the assay using in vitro translated product. Lane 1 is the immunoprecipitation
(IP) of EAV-infected cells with EAVN MAb. Lanes: 2 and 8, GST; 3 and 9, GST-Jenv as
a negative control; 4 and 10, GST-Imp-α; 5 and 11, GST-Imp-β; 6 and 12, GST-EAVN;
7, uninfected BHK-21 cell lysates; 13 in vitro translated EAV N.

88
residues substituted with G showed nuclear localization (Fig. 3.4). This suggests that
these residues may not be the key amino acids to the nuclear localization of EAV N.
Molecular basis for EAV N nuclear localization. To further determine the basis for
EAV N nuclear localization, the interactions between importin proteins and EAV N were
examined. Importin-α and importin-β were expressed in E. coli as a GST-fusion protein
and coupled to glutathione-sepharose beads. The EAV N protein was synthesized either
in vitro by transcription and translation or prepared from EAV infected BHK-21 cells in
the presence of [35
S]methionine. Importin-coupled GST beads were then incubated with
the radiolabelled EAV N protein, and the bound proteins were resolved by SDS-PAGE
and visualized by autoradiography. Despite some degree of non-specific binding of EAV
N to GST, it seems that EAV N interacts with both GST-Imp-α, GST-Imp-β in both in
vitro and cells by pull down assay (Fig 3.5 A and B).
Subcellular localization of SHFV N. HeLa and Cos-1 cells were grown on coverslips
and transfected with genes encoding EGFP-SHFV N and SHFV N-EGFP fusion proteins
using Lipofectin. At 24 hrs post-transfection, cells were fixed with methanol and stained
with DAPI for nuclear staining followed by fluorescence microscopy. The results indicate
that in both cell types, SHFV N-EGFP and SHFV N-EGFP were localized in the nucleus
and nucleolus of transfected cells (Fig. 3.6).
Identification and mapping of NLS in the SHFV N protein. To identify an NLS in
SHFV N protein the PSORT II program (Nakai and Kanehisa, 1992) was used and a ‘pat-
7’ type NLS was predicted at amino acid positions 22-28 of the N protein (Fig. 3.7A). In
order to examine the functional significance of the ‘pat7’ NLS, a series of deletion
mutants of SHFV N were constructed as EGFP fusion proteins (Fig. 3.7B). HeLa cells

89
Fig. 3.6. Subcellular localization of SHFV-N protein in HeLa and Cos-1 cells. Cells
grown on the coverslips were transfected with EGFP-SHFV N fusion protein genes using
Lipofectin. At 24 hrs post-transfection, cells were fixed with methanol and stained with
DAPI for nuclear staining. Cells were then visualized by fluorescence microscopy (model
AX70, Olympus). Panels A, Cos-1 cells with SHFV N-EGFP; B, HeLa cells with SHFV
N-EGFP; C, Cos-1 cells with EGFP-SHFV N; D, HeLa with EGFP-SHFV N.
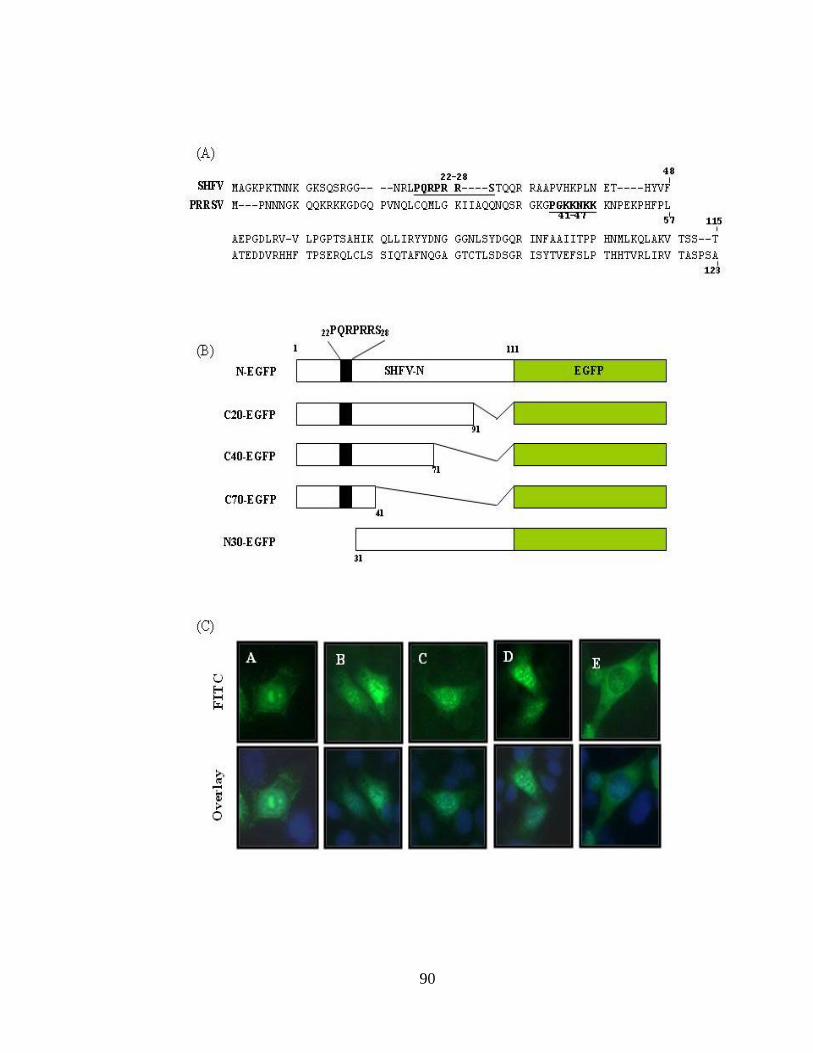
90

91
Fig 3.7. Identification of NLS motif in SHFV N protein. (A) a ‘pat-7’ type NLS in the
SHFV N protein predicted by the PSORT II program (Nakai and Kanehisa, 1992).
PRRSV N protein sequence is aligned for comparison. Dashes indicate amino acid
deletions. Boxes indicate ‘pat-7’ (for SHFV) and ‘pat-7’ (for PRRSV) type NLS
sequences. (B) Schematic presentation of SHFVN deletion mutants fused with EGFP.
Darkened areas represent predicted NLS at amino acid positions 22-28. (C) Subcellular
localization of SHFV N mutants. Panels A, wild type SHFVN-EGFP; B, Deletion
construct SHFVN-EGFP-C20; C, deletion construct SHFVN-EGFP-C40; D, deletion
construct SHFVN-EGFP-C70; E, deletion construct SHFVN-EGFP-N30.

92
were transfected with the mutant constructs and subcellular localization of SHFV N
mutant proteins was determined (Fig. 3.7C). The SHFV N constructs containing the
putative ‘pat7’ type of NLS localized in the both cytoplasm and nucleolus of gene-
transfected HeLa cells (Fig. 3.7C, panels B, C, and D), whereas the mutants that did not
contain the predicted NLS showed only cytoplasmic staining (Fig. 3.7C, panel E). These
observations indicate that the putative NLS identified at positions 22-28 was a functional
motif. To determine core amino acids essential for the function of ‘pat7’ type NLS of
SHFV N, the R residues in NLS (22-PQRPRRS) of SHFV N were individually mutated
to G (Table 3.3). The NLS-null SHFV N gene was transfected to HeLa cells followed by
staining with DAPI and fluorescence microscopy. Fig. 3.8 demonstrates the subcellular
location of the NLS-null SHFVN-EGFP mutant in the transfected HeLa cells. The NLS-
null mutant protein of SHFV N showed neither nuclear nor nucleolar localization, and
fluorescence was seen only in the cytoplasm of transfected cells. This indicates that all
arginine residues of SHFV N protein are important for its nuclear localization (Fig.
3.8A).
Molecular basis for SHFV N nuclear localization. To determine the molecular basis
for SHFV N nuclear localization, interactions between importin shuttle proteins and the
N protein were examined. The SHFV N protein was synthesized in vitro by transcription
and translation in the presence of [35
S] methionine. The GST fusion proteins of Imp-α
and Imp-β expressed in E. coli and coupled to glutathione-Sepharose beads were then
incubated with the radiolabelled SHFV N protein. After washing, bound proteins were
released by boiling for 5 min in SDS sample buffer and resolved by 12% SDS PAGE for
autoradiography using Phosphorimager. The result shown in Fig. 3.9 indicates that SHFV

93
Table 3.3. Site-directed mutagenesis of the SHFV N NLS motif and subcellular
localization of NLS mutants.
Construct Sequence of NLSa
Localizationb
Cy Nu No
SHFV-N 22-PQRPRRS ++++ ++ ++++
SHFV-N- R24,26,27G 22-PQGPGGS-28
++++ - -
aIndividual Arginines (underlined) in the NLS were substituted with glycines.
bFor scoring of the fluorescence signal for each mutant construct, 100 transfected cells
were counted presented as + or -. Each one ‘+’ represents fluorescence of 25 transfected
cells either in the cytoplasm (Cy), nucleus (Nu), or nucleolus (No).

94
Fig. 3.8. Subcellular localization of the NLS-null SHFVN-EGFP mutant. The basic
residues in the NLS motif (22-PQRPRRS) of SHFV N were mutated into 22-
PQGPGGS-28. The NLS-null SHFV N gene was transfected to HeLa cells followed by
staining with DAPI and fluorescent microscopy. A, wild type SHFVN-EGFP; B, NLS-
null SHFVN-EGFP.

95
Fig 3.9. Interaction of SHFV N with importin proteins by GST-pull down assay.
Importin-α and importin-β-glutathione-S-transferase (GST) fusion proteins were
expressed in E-coli and coupled to glutathion Sepharose 4B beads (Pharmacia). The
beads-bound proteins were incubated with radiolabelled in vitro-translated SHFV N
protein overnight at 4°C with constant agitation. After washing, bound proteins were
released by boiling for 5 min in SDS sample buffer and resolved by 12% SDS PAGE for
autoradiography using Phosphorimager. Lanes: M, protein marker; 1, in vitro translated
product of SHFV N; 2, GST pull-down assay for SHFV N and GST; 3, SHFV N and
GST-Jenv; 4, SHFV N using importin-α, 5, SHFV N and importin-β.

96
N interacts with both importin-α and importin-β, with a stronger affinity for importin-α
(Fig. 3.9, lanes 5 and 6).
DISCUSSION
Nuclear localization of capsid protein for cytoplasmic RNA viruses has been
investigated in this chapter using EAV and SHFV as a model. Nuclear localization of
capsid protein from cytoplasmic RNA viruses has been observed previously, and
examples include capsid proteins of Semliki Forest virus, dengue fever virus, and
hepatitis C virus, all of which are translocated to the nucleus and nucleolus (Favre et al.,
1994; Yasui et al., 1998, Wang et al., 2002). The exclusive replication of nidoviruses in
the cytoplasm of infected cells has been previously reported for MHV, which replicates
in enucleated cells (Brayton et. al, 1981). This indicates that these viruses do not need
nucleus for their replication. But, nuclear and nucleolar localization for nucleocapsid
proteins of PRRSV, EAV, IBV, MHV and TGEV has been observed (Hiscox et al, 2001;
Wurm et al, 2001; Rowland et al., 1999; Tijms et al., 2002). The nuclear localization of
proteins is mainly mediated by the nuclear localization signal (Golrich and Kutay, 1999).
In arteriviruses, PRRSV N contains two putative NLS motifs, ‘pat 7’ and ‘pat 4’
(Rowland et al., 2003). The location of “pat 7”, the primary and active NLS of the
protein, is between positions 41-47 (Rowland et al., 2003). The ‘pat-4’ NLS is also
functional but only in the absence of ‘pat7’ motif and leads PRRSV N to the nucleus
(Rowland et al., 2003). In addition to PRRSV N, we have shown study that the LDV N
protein is also localized in the nucleus and nucleolus of transfected cells (Mohammadi et
al., 2009). Our studies using mutant LDV N constructs showed that the nuclear

97
localization of LDV N was dependent on a ‘pat-4’ motif at positions 38-41 (Fig. 2.2B;
Fig. 2.3), indicating that this motif was indeed functional. The nuclear localization of
nucleocapsid proteins of two other members of the family Arteriviridae, EAV N and
SHFV N, had not been investigated in detail prior to the current study, even though one
report showed the nucleolar translocation of EAV N in virus- infected cells (Tijms et al.,
2002). No such information was available for SHFV N. Thus, we conducted detailed
studies and report the nuclear and nucleolar localization of nucleocapsid proteins for both
EAV and SHFV.
EAV N was previously observed to localize in the nucleolus of infected cells
(Tijms et al., 2002). We expanded the study to further examine its nuclear translocation
and identify the presence of an NLS. Based on our data, EAV N is localized in the
nucleolus of infected cells at the early stage of virus infection, as the fluorescence was
detected at 8 hr p.i (Fig. 3.1). EAV N did not translocate to the nucleus in the absence of
the first 20 residues at the N-terminus of the protein. This motif contains a putative
bipartite NLS motif (Fig. 3.3. B and C). Fine mapping studies of the putative NLS
showed that the substitution of basic amino acids in this motif did not prevent the nuclear
localization of EAV N (Fig. 3.4). This suggests that EAV N may require all of the N
terminal 20-amino-acids within the motif to be functional for nuclear translocation. A
bipartite NLS has been identified in many eukaryotic proteins such as nucleoplasmin,
human pRB, human p53, and human interleukin 5 (IL5) (Munoz-Fontedla et al, 2003).
Functional bipartite NLSs have also been identified in viral proteins. For example,
nucleoproteins of Thogoto virus and influenza A virus, phosphoprotein of Borna disease

98
virus, and the core protein of hepatitis C virus all contain functional bipartite NLSs
(Weber et. al, 1998; Schwemmle et. al, 1999; Suzuki et al., 2005).
We also investigated the nuclear localization of SHFV N. For this protein, a ‘pat-
7’ type NLS was identified at amino acid positions 22-28 (Fig. 3.7). Studies using a series
of deletion mutants of SHFV N showed that the putative ‘pat7’ type of NLS was
necessary to lead SHFV N to the nucleus in the N gene-transfected cells (Fig. 3.7, B and
C). The key amino acids for the NLS of SHFV N were further identified by substituting
arginines in the putative NLS with glycines (Table 3.2). The results showed that all of the
arginines within the motif were essential for nuclear localization of SHFV N (Fig. 3.8A).
The cellular mechanism for nuclear localization of a protein starts by recruiting a family
of cellular proteins, and among them importins are the most common proteins. The
process begins by binding of importin-α to NLS on the cargo protein followed by binding
of importin-β to the importin-α/cargo protein complex, which finally transports the entire
complex to nuclear pore complex (NPC) on the nuclear membrane in the presence of
Ran-GDP. We showed here that EAV N and SHFV N both could bind to importin-α and
importin β (Fig. 3.5 and Fig. 3.9). EAV N appeared to bind to GST as well, but perhaps
this interaction would be non-specific. PRRSV N and LDV N bind to importin-α and
importin-β (Rowland et al., 2003; Mohammadi et al., 2009). Several viral proteins
have been reported to interact with both importin-α and -β. For instance, the
polyomavirus capsid protein requires the interaction with both importin-α and -β through
its NLS to enter the nucleus (Qu et al., 2004), and SV40 VP3 also bins to importinα-α/β
complex to enter the nucleus (Nakanashi et al, 2002). The HIV Rev protein directly binds
to both importin-α and -β, but its binding to importin-α is NLS-independent while

99
binding to importin-β is NLS-dependent via its arginine-rich NLS (Henderson et al.,
1997). It is possible that these proteins use another cellular protein which mediates the
transport to the nucleus. Binding of EAV N to GST in addition to GST-importin fusion
proteins could be a source of non-specific binding of the N protein to these proteins.
Since N protein of all arteriviruses are localized to the nucleus and nucleolus, the
nuclear role of N during infection is of major interest. The core protein of hepatitis C
virus is believed to modify cellular functions by preventing translocation of host cell
proteins to the nucleus (Isoyama et al., 2002). The N protein of TGEV has been
suggested to play a role in the disruption of cell division (Wurm et al., 2001). For PRRSV
N, disruption of NLS does not affect virus replication in cell culture, but in pigs, NLS-
null viruses were attenuated (Lee et al., 2006a, 2006b; Pei et al., 2008). A recent study
showed that the nuclear import of PRRSV N from the cytoplasm was faster than the
export from the nucleus and the N protein was not kept in the nucleolus (You et al.,
2008). We also noticed that EAV N protein localizes in the nucleus at the early stage of
EAV infection. Our observation along with another report on PRRSV N (You et al.,
2008) suggest an important role for N during the early stage of PRRSV and EAV
replication in which N is localized predominantly in the nucleolus. N protein of several
member viruses in the order Nidovirales have been shown to co-localize and interact with
fibrillarin and nucleolin in the nucleolus, supporting the hypothesis that N plays a role
during host cell ribosomal biogenesis and cell cycle modulation (Yoo et al., 2003; Chen
et al., 2002).
In summary, we show that EAV N and SHFV N are both localized in the nucleus
and nucleolus of N gene transfected cells. EAV N localization is dependent on a part of

100
the protein which contains a bipartite NLS. Also SHFV N has a functional ‘pat7’ type
NLS at positions 22-28. The capsid protein nuclear localization is a common feature in
the Arteriviridae family, and this protein may play a role in replication and pathogenesis
of this group of viruses.

101
CHAPTER 4
The Role of Nucleocapsid Protein of Arteriviruses in Cell Cycle Modulation

102
ABSTRACT
Replication of porcine reproductive and respiratory syndrome virus (PRRSV) and
equine arteritis virus (EAV) in the dividing MARC-145 and BHK-21 cells, respectively,
led to accumulation of cells at G2/M phase of the cell cycle. The current study was
conducted to identify the viral component involved in this function. The N protein was
the most probable viral protein for this activity since it localizes to the nucleolus of virus-
infected and N gene transfected cells and interacts with nucleolar proteins including
fibrillarin which participates in the ribosome biogenesis of cells. In BHK-21 cells
expressing GFP-N of arteriviruses (GFP-PRRSV N, GFP-EAV N, GFP-LDV N, and
GFP-SHFV N), the number of cells in G2/M phase was higher compared to untransfected
cell controls. This suggests that the nucleocapsid protein of arteriviruses plays a
modulatory role during cell cycle. The effect of N protein was similar for the all four N
proteins but some difference were observed. PRRSV N and EAV N had more cells at
G2/M stage compared to LDV N and SHFV N. This study for the first time suggests that
N protein of arteriviruses affects cell cycle process in the N-expressing cells.

103
INTRODUCTION
The family Arteriviridae is a group of RNA viruses consisting of four different
members: porcine reproductive and respiratory syndrome virus (PRRSV), equine arteritis
virus (EAV), lactate dehydrogenase-elevating virus (LDV), and simian hemorrhagic
fever virus (SHFV) (Snijder and Meulenberg 1998). The Arteriviridae family, along with
the Coronaviridae family, is grouped in the Nidovirales order of viruses (Cavanagh et al,
1997). Despite their replication in the cytoplasm of infected cells, the nucleocapsid (N)
protein of PRRSV and EAV have been shown to localize in the nucleus and nucleolus of
virus-infected and gene-transfected cells (Rowland et al, 1999; Tijms et al, 2002). The N
protein of these viruses is encoded by the 3’ ultimate open reading frame (ORF) of viral
genome and varies in size between 14-15 kDa (Snijder et al, 1998). The N protein of
other member viruses localizes in the nucleus and nucleolus of infected and N-gene
transfected cells (Mohammadi et al., 2009; Chapter 2). Studies have also shown the
colocalization and interaction of PRRSV N with nucleolar proteins fibrillarin and
nucleolin. These proteins are known to be involved in a variety of cellular processes,
including cell cycle progression (Yoo et al., 2003; Chen et al., 2002; Wurm et al., 2001).
A recent study showed that PRRSV N interacts and co-localizes with human I-mfa
domain-containing protein (HIC) which is a cellular transcription factor (Song et al,
2009). HIC itself interacts with cyclin T1 which is part of a protein complex controlling
the progression of cell cycle (Young et al, 2003). This suggests that PRRSV N protein
may play a role in cell cycle modulation of the virus infected cells. A role for nuclear
localization of the N protein and the consequence of its interaction with cellular proteins
has not been investigated previously.

104
The cell cycle is a complex process controlled by many regulatory proteins which
leads to mitosis and generation of daughter cells. This process is regulated by specific
cellular proteins, namely cyclins and cyclin-dependent kinases (Cdks). The cell cycle is
divided into four phases: G0/G1 (quiescent phase/ Gap1), S (Synthesis phase), G2/M
(Gap2/ Mitosis), and M. Each stage of cell cycle is controlled by specific cyclins and
Cdks. Alteration in any of these regulatory factors can influence or disturb the normal
cell cycle (Schafer, 1998). Several studies have demonstrated the effect of viruses and
viral proteins on cell proliferation and cycle at different stages, mostly G2/M in the case
of RNA viruses. Examples of viruses arresting the cell cycle at G2/M include human
immunodeficiency virus type I (HIV-1), reoviruses, and coronaviruses (Emmet et al,
2005; Zhao et al, 2005 and Davy and Doorbar et al, 2007). Human Parvovirus B19 stops
the cell cycle at the G2 stage and increases the cellular regulatory factors of mitosis step
(Morita et al, 2001). For Nidovirales, several member viruses of the family
Coronaviridae, such as infectious bronchitis virus (IBV), murine hepatitis virus (MHV),
and severe acute respiratory syndrome (SARS) virus, affect cell cycle at different stages.
IBV induces cell cycle arrest at S and G2/M stage in infected cells (Dove et al, 2006b; Li
et al, 2007). However, investigations show that MHV and SARS virus arrest cell cycle at
the stage of G0/G1 (Chen and Makino 2004; Yuan et al, 2006). There is no report on
modulation of cell cycle by Arteriviridae and their viral proteins. Therefore, the
objectives of this study were firstly to investigate the effects of arteriviruses on cell cycle
and secondly to examine arterivirus N protein for possible involvement in this process.
Here we show that PRRSV and EAV also regulate cell cycle in MARC-145 and BHK-21

105
cells, respectively. Further, we demonstrate that N protein is the viral component
associated with this modulation in BHK-21 cells.
MATERIALS AND METHODS
Cells and viruses. Baby hamster kidney cells (BHK-21) and MARC-145 (Kim et al,
1993) which is a derivative line of MA104 cells were used in this study. Cells were
maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10%
heat inactivated fetal bovine serum (FBS, Gibco BRL) in a humidified incubator with 5%
CO2 at 37ºC. EAV strain Bucyrus (Doll et al., 1957) and PRRSV strain PA8 (Wootton et
al., 2000) were grown in MARC-145 cells and BHK-21 cells, respectively.
Plasmid constructs. The pEGFP-N1 (Clontech Inc.) was used in this study. EAV N,
LDV N, and SHFV N coding sequences were sub-cloned into Bam HI site of the
pEGFP-N1 upstream of the enhanced green fluorescence protein (EGFP) in-frame from
PCR products using specific primers (Mohammadi et al., 2009; Chapter 3 Table 3.1 and
3.3) and the fusion genes EAV N-GFP, LDV N-GFP, and SHFV N-GFP were
constructed. To amplify the EAV N gene, pEAV030 containing the full genome sequence
of EAV (van Dinten et al., 1997) was used as the template for PCR reaction. SHFV N
gene was cloned in pGEM-T using a cDNA library of SHFV genome. It was then
amplified by PCR from the plasmid using specific primers and sub-cloned upstream of
EGFP in the pEGFP-N1 and in frame. The LDV-N gene was amplified by PCR from a
plasmid containing the LDV genomic fragment, and using the Bam HI sequence at both
ends of the primers, cloned into pEGFP-N1 upstream of the EGFP gene, respectively.

106
The PRRSV N-EGFP fusion protein has been described previously (Rowland et al., 1999;
Rowland et al., 2003).
Co-staining of viral N and fibrillarin. For immunofluorescence purpose, HeLa cells
were used to express LDV N-EGFP fusion protein and BHK-21 cells were used for EAV
infection. Cells were seeded on coverslips in 35 mm diameter dishes. At 75% confluency,
cells were either transfected with 2 μg of a plasmid containing LDV N-EGFP fusion
construct and Lipofectin (Invitrogen) according to the manufacturer’s instruction or
infected with EAV at a multiplicity of infection (MOI) of 1. After 24 hours, cells were
fixed by methanol, and stained with a specific antibody for fibrillarin followed by
staining with goat anti-rabbit IgG conjugated with Texas red dye (Molecular Probes,
Eugene, Oreg.,) or Alexa fluor 488, and finally stained with DAPI (4',6-Diamidino-2-
phenylindole dihydrochloride). The EAV infected cells were co-stained with a mAb
against EAV N (MAb 3E2, IgG isotype) which was a generous gift from Dr. Udeni B.R.
Balasuriya (Department of Veterinary Science, Gluck Equine Research Center,
University of Kentucky, Lexington, USA) and fibrillarin followed by incubation with
goat anti-mouse immunoglobulin G conjugated with AlexaFluor 488 (Molecular Probes)
and goat anti-rabbit immunoglobulin G conjugated with Texas red dye (Molecular
Probes, Eugene, Oreg.). After washing, cells were finally stained with DAPI. After last
staining, cells were washed and mounted on a slide using 40 μl of the mounting buffer
(60% glycerol, 0.1% sodium azide in PBS), and visualized for fluorescence.
Flow cytometry of virus infected cells. Fig. 4.1 shows the experimental design used for
analysis of arteriviruses and their nucleocapsid protein on cell cycle. 06 cells of MARC-
145 or BHK-21 were seeded onto every 100 mm diameter cell culture dishes. The

107
Fig 4.1. Experimental design for analysis of arteriviruses and their nucleocapsid protein
on cell cycle.

108
following day, cells were either mock or virus-infected at a multiplicity of infection of 5,
with PRRSV or EAV for MARC-145 and BHK-21 cells, respectively. At different time
points post-infection (12, 24, and 48 hrs), cell monolayers were washed, trypsinized, and
fixed in 4% paraformaldehyde (PFA) for 10 min at room temperature. Then, cells were
washed twice with cold PBS and stained with propidium iodide (PI) (50g/ml propidium
iodide, 0.1% NP-40, 0.1% Sodium citrate, and 20 g/ml RNase A in PBS). After 30 min
incubation at room temperature, the DNA content of 30,000 cells was assessed using
FACScan flow cytometry instrument (Becton Dickinson). Then DNA content of the
acquired cells was analyzed using the ModFit LT 3.0 DNA analysis software. Each
experiment was performed in triplicate and repeated three times.
Flow cytometry of N gene transfected cells. Approximately 3x10
6 BHK-21 cells per
dish were seeded onto 100 mm diameter dishes. After 24 h, cells were transfected with
GFP-N gene using 60 l of 1 mg/ml of stock solution of PEI (Linear polyethylenimines;
Polysciences Inc.) and 20 ug of plasmid DNA in serum-free DMEM. At 16 hrs post-
transfection, the medium was replaced with DMEM containing 10% fetal bovine serum.
At 60 h post-transfection, cells were harvested, fixed in 4% paraformaldehyde and
stained with PI for 30 minutes at room temperature. Then the DNA content of 10,000
transfected cells was analyzed by FACScan.
RESULTS
Co-localization of LDV N and EAV N with fibrillarin. Previously, co-localization of
PRRSV N and the nucleolar protein fibrillarin was reported (Yoo et al., 2003). In this
study, this feature was examined for two other member viruses of the Arteriviridae
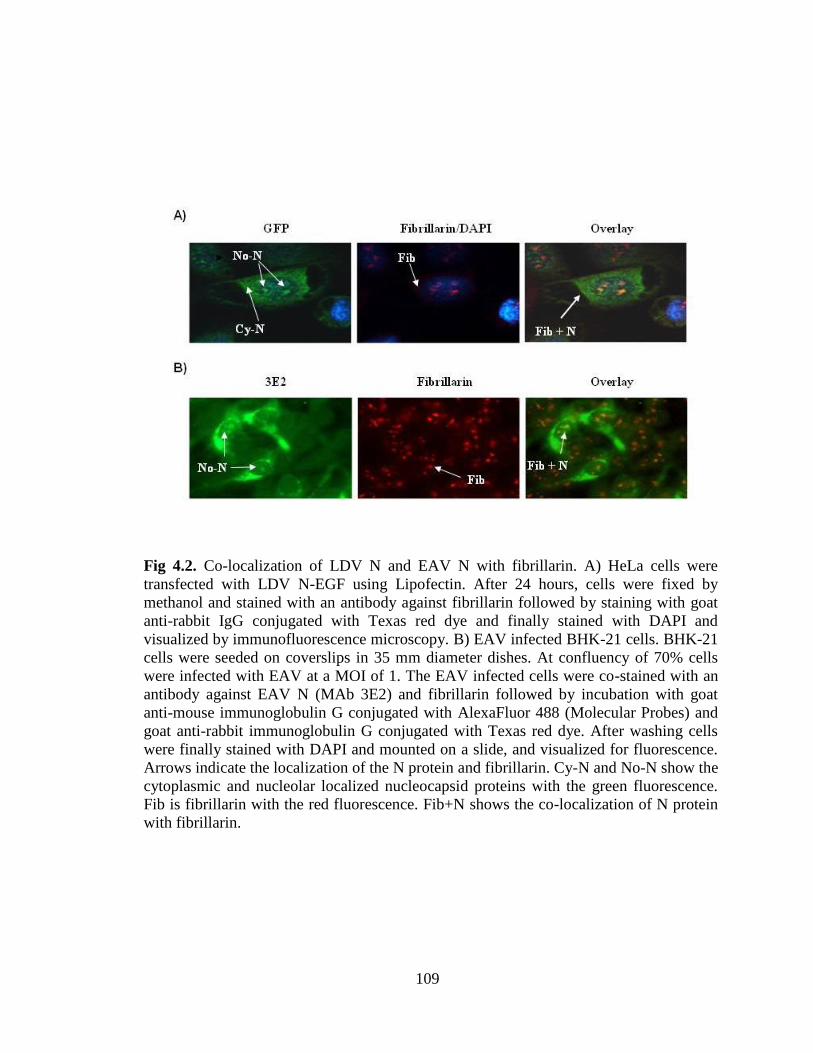
109
Fig 4.2. Co-localization of LDV N and EAV N with fibrillarin. A) HeLa cells were
transfected with LDV N-EGF using Lipofectin. After 24 hours, cells were fixed by
methanol and stained with an antibody against fibrillarin followed by staining with goat
anti-rabbit IgG conjugated with Texas red dye and finally stained with DAPI and
visualized by immunofluorescence microscopy. B) EAV infected BHK-21 cells. BHK-21
cells were seeded on coverslips in 35 mm diameter dishes. At confluency of 70% cells
were infected with EAV at a MOI of 1. The EAV infected cells were co-stained with an
antibody against EAV N (MAb 3E2) and fibrillarin followed by incubation with goat
anti-mouse immunoglobulin G conjugated with AlexaFluor 488 (Molecular Probes) and
goat anti-rabbit immunoglobulin G conjugated with Texas red dye. After washing cells
were finally stained with DAPI and mounted on a slide, and visualized for fluorescence.
Arrows indicate the localization of the N protein and fibrillarin. Cy-N and No-N show the
cytoplasmic and nucleolar localized nucleocapsid proteins with the green fluorescence.
Fib is fibrillarin with the red fluorescence. Fib+N shows the co-localization of N protein
with fibrillarin.

110
family to determine if it was a common feature of N protein in this group of viruses. LDV
N- EGFP fusion protein was expressed in HeLa cells and stained with a rabbit polyclonal
antibody against fibrillarin (Abcam, ab5821) as well as with DAPI. The result in Fig. 4.2
A shows that LDV N co-localizes with fibrillarin in the nucleolus of the N-gene
transfected cells. For EAV, BHK-21 cells were infected and the N protein was examined
by staining. It was apparent that EAV N also co-localized with fibrillarin in virus-infected
BHK-21 cells (Fig. 4.2B).
PRRSV infection of asynchronized MARC-145 cells increases the number of cells in
the G2/M phase of cell cycle. Co-localization of N protein with nucleolar proteins
suggests that N protein may modulate the normal cellular activities such as cell cycle
progression. This possibility was examined first using virus-infected MARC-145 cells
infected with PRRSV at an MOI of 5 and were harvested at 12, 24, and 48 hr p.i. Cells
were then stained with PI and DNA contents of cells were analyzed by flow cytometry.
To monitor the infection, cytopathic effects (CPEs) were also determined using
microscopy. CPE was increased in virus-infected cells and the increase was time-
dependent (Fig. 4.3). Fig. 4.4 shows the cell cycle profile of PRRSV-infected MARC-145
cells. The number of virus-infected cells was significantly (P 0.05) higher compared to
mock-infected (uninfected) cells in the G2/M phase at 12 and 24 h p.i. At 48 hrs, the
number of infected cells was higher in the S phase compared to mock-infected cells.
Accumulation of EAV infected BHK-21 cells in the G2/M phase. To examine if the
cell cycle modification was common for arteriviruses, EAV was chosen. BHK-21 cells
were either mock-infected or virus-infected with EAV (strain Bucyrus) at a MOI of 5. At
24 h p.i, cells were harvested and stained with PI as described for DNA quantification.

111
Fig 4.3. CPE of PRRSV-infected MARC-145 cell at different time points. Panels A, B,
and C are mock-infected cells at 12h, 24h, and 48h post infection respectively and panels
D, E, and F demonstrate the PRRSV infected MARC-145 cells (with an MOI of 5) at
12h, 24h, and 48h post-infection with PRRSV. Arrows show the CPE.

112
Fig. 4.4. Cell cycle profile of PRRSV-infected MARC-145 cells. Averaged values from
three independent experiments are presented. MARC-145 cells were infected with
PRRSV strain PA8 at an MOI of 5. At 12hr, 24hr, and 48hr post-infection, cells were
trypsinized, washed, fixed with 4% PFA, and stained with PI. After 30 min, the amount
of DNA of cells were analyzed using flow cytometry. The upper graph demonstrates the
comparison of mock- and PRRSV-infected cells at different stages of cell cycle. The
asterisks indicate significant difference between two groups (P-value less than 0.05) and
the error bars show the standard error. The lower graph shows the trend of cell cycle
analysis at different time points in mock- and PRRSV- infected cells separately.

113
Fig. 4.5. Cell cycle profile of EAV-infected BHK-21 cells. BHK-21 cells were infected
with EAV strain Bucyrus at an MOI of 5. At 24 hrs post-infection cells were trypsinized,
washed, fixed with 4% PFA, and stained with PI. After 30 min, the amount of DNA of
cells was analyzed using flow cytometry . Each experiment is an average of three
independent experiments. The upper graph demonstrates the comparison of mock- and
EAV-infected cells at different stage of cell cycle. Asterisks demonstrate a significant
difference between groups (the P-value less than 0.05) and the error bars are
representative of standard errors. The lower graph shows the trend of cell cycle analysis
at different time points in mock- and EAV-infected cells.

114
Fig. 4.5 demonstrates the cell cycle profile of from this experiment. The number of cells
was significantly (P 0.05) higher at the S and G2/M phases of cell cycle in EAV-
infected cells compared to mock-infected cells.
Nucleocapsid protein of arteriviruses arrests cell cycle in G2/M stage. Since PRRSV
and EAV appear to modulate the cell cycle during infection, it was of interest to
determine the viral component responsible for this function. Since the nucleocapsid
protein of these viruses co-localizes with nucleolar proteins, the N protein was examined
for this function. BHK-21 cells were either untransfected or transfected with GFP, GFP-
PRRSV N, GFP-EAV N, GFP-LDV N, GFP-SHFV N, and the patterns of cell cycle were
determined at 60 h post-transfection (Fig. 4.6). The cell cycle profile was similar in
untransfected and GFP-transfected cells. The number of cells in the G2/M phase was
higher in N-gene transfected cells for all members of the Arteriviridae family compared
to the negative controls (untransfected and GFP expressing BHK-21 cells). However, for
PRRSV N and EAV N gene-transfected cells the number of cells in the G2/M phase were
more than those for LDV N and SHFN transfected cells.
DISCUSSION
In this study we have demonstrated that viruses in the Arteriviridae family can
cause infected cells to accumulate in the G2/M stage of the cell cycle. We further show
that the nucleocapsid protein of arteriviruses is responsible for this G2/M accumulation.
PRRSV-infected MARC-145 cells were analyzed for DNA content at different
time points post-infection (p.i.). It was noticed that the number of cells at G2/M was
significantly higher in PRRSV-infected cells compared to uninfected cells at 12 and 24 h

115
Fig. 4.6. Cell cycle profiles in N-gene transfected BHK-21cells. BHK-21 cells were
transfected with GFP, GFP-PRRSV N, GFP-EAV N, GFP-LDV N, and GFP-SHFV N.
At 60 hrs post-transfection cells were trypsinized, washed, fixed with 4% PFA, and
stained with PI. After 30 min, the amount of DNA was analyzed using FACScan.

116
p.i. (Fig. 4.4). However at 48 h p.i at which the maximum CPE was observed (Fig 4.3F),
the number of cells at G2/M in the infected group was lower than that of uninfected cells.
At 48 h p.i. infected cells were significantly lower and higher at the G0/G1 phase and the
S phase of cell cycle respectively. It seems that infected cells move faster to G2/M at 24
hours and then to S phase at 48 h p.i. in comparison with uninfected cells. This indicates
that virus infected cells appear to cycle faster than uninfected ones, which can be
considered as favored for virus replication. EAV infection of BHK-21 cells led to a
significant increase in the number of cells at the S and G2/M phases and a significant
decrease in the number of cells at the G0/G1 phases of cell cycle at 24 h p.i (Fig. 4.5).
In order to determine which viral components are involved in cell cycle
modulation by arteriviruses, the most likely candidate was the N protein as it interacts
with nucleolar proteins involved in the cell cycle (Fig 4.2). As it is shown in Fig 4.2, both
LDV N and EAV N proteins colocalize with fibrillarin, a host cell nucleolar protein.
These data along with the previous findings of PRRSV N co-localization with fibrillarin
indicate that co-localization of N with fibrillarin is a common feature in arteriviruses,
suggesting that N protein may be involved in modulation of the cell cycle. We
determined if N had any impact on cell cycle modification. N proteins of arteriviruses
were transfected in to BHK-21 cells as GFP fusion proteins and the DNA content of the
transfected cells was analyzed. Our results show that the number of cells at the G2/M
stage was higher at 60 h post-transfection in the cells expressing N proteins of the
Arteriviridae family compared to untransfected and GFP transfected negative controls
(Fig 4.6). However, the PRRSV N and EAV N had stronger activity on the number of
cells at G2/M compared to LDV N and SHFV N at 60 h post transfection (Fig. 4.6). Our

117
results indicate that the N proteins of arteriviruses modulate the cell cycle in the similar
manner but they may function differently.
Cell cycle modification by RNA viruses has been reported, particularly with
regards to stalling at G2/M phase (Davy and Doorbar, 2007; Zhao and Elder 2005). The
stage of cell cycle might be a determinant of permissiveness and level of virus production
for some viruses. For instance, Dengue virus type 2 infects different types of permissive
cells based on the cell cycle stage. HepG2 cells are more permissive to Dengue virus
infection as well as virus production at the G2/M step of cell cycle (Phoolcharoen and
Smith. 2004).
The involvement of viral proteins in modulation of the cell cycle has been studied
for several viruses. In this regard, mostly non-structural (NS) viral proteins are involved
in cell cycle modulation. The Vpr protein of HIV-1, which is a nucleo-cytoplasmic
protein, has been found to arrest cells in the G2/M phase of the cell cycle (Mahalingam et
al, 1997; Matsuda et al, 2003). Furthermore, the σ1S protein of alpha reovirus also stops
cell cycle at the G2/M phase (Poggioli et al, 2000). The paramyxovirus simian virus 5
(SV5) V protein slows down the cell cycle process (Lin and Lamb 2000). Hepatitis C
virus non-structural proteins 5A and 2 have been shown to regulate cell cycle phases
(Ghosh et al, 1999; Yang et al, 2006). The NS protein p28 of MHV and NS protein 7a of
SARS coronavirus are shown to block the G0/G1 progress (Chen et al, 2004, Yuan et al,
2006). Structural proteins of RNA viruses can also play a role in cell cycle modulation.
Transmissible gastroenteritis virus nucleocapsid protein (TGEV N) has been shown to
modify the cell cycle. When cells were transfected with TGEV N, only 30% of
transfected cells were dividing, which suggested cell cycle arrest at the G2 phase (Wurm

118
et al, 2001). The nuclear localization of IBV N protein is dependant on stage of the cell
cycle. In G2/M synchronized Vero cells expressing IBV N, the nuclear localization of N
was monitored by live microscopy and the percentage of nuclear localization was found
to be higher. This indicates that at the G2/M stage of cell cycle is more suitable for
nuclear localization of IBV N (Cawood et al, 2007).
Here we showed that the N protein affects cell cycle by accumulation of cells at
G2/M stage. These findings suggest that the N protein plays a significant role in
pathogenesis of these viruses, and further studies are needed to scrutinize its mechanism
of function in host cells and pathogenesis of arteriviruses.

119
CHAPTER 5
General Discussion
Nuclear localization of the most abundant structural protein of arteriviruses,
which is N protein, in the virus infected cells has been of great interest to investigators
working on these viruses. Moreover, the exclusive cytoplasmic replication of
arteriviruses with no need for cell nucleus makes this phenomenon more interesting. The
primary objective of this research was to investigate the nuclear localization property of
the nucleocapsid (N) protein of the viruses in the family Arteriviridae. We also
investigated the possible role of N protein in modulation of the host cell function in virus-
infected cells and N gene-transfected cells.
The nucleolar localization for N protein in the family of Arteriviridae was first
reported for PRRSV N. The presence of a functional NLS in PRRSV N as well as the
mechanism for its nuclear translocation, have been investigated thoroughly. However,
there was little information available for N proteins of other members of the family
including LDV, SHFV, and EAV. Since LDV exhibits limited cell tropism and grows
only in a limited number of mouse primary macrophages, its subcellular localization was
investigated using the cloned LDV N gene. Subcellular localization of LDV-N was
determined by tagging N protein with enhanced green fluorescence protein (EGFP) on
the N- or C-terminus. The fusion proteins localized to the nucleus and nucleolus of gene-
transfected cells. Further studies using a series of mutant constructs of LDV N indicated
that a functional ‘pat4’ type NLS consisting of KKKK is involved in nuclear localization
of this protein and the key amino acids for this activity were identified. The location of
NLS for LDV N was in the similar region of PRRSV N functional NLS. Furthermore, the

120
LDV N interaction with the importin-α and -β proteins suggests that its nuclear
localization occurs through the importin-mediated nuclear transport pathway. These
findings show that the nuclear localization is the property of LDV N in addition to
PRRSV N.
Despite the report on nucleolar localization of EAV N in infected BHK-21 cells
(Tijms et al., 2001), little was known about the details of its nuclear localization. Thus,
we conducted further studies on EAV N nucleolar localization. EAV N fusion constructs
were made to fuse with EGFP on the N- and C-termini and their cellular distribution was
investigated in the gene-transfected cells. Both N-EGFP and EGFP-N fusion proteins of
EAV N showed accumulation of fluorescence in the nucleus and nucleolus of gene-
transfected cells. The presence of a putative ‘bipartite’ type NLS on EAV N sequence at
amino acids position 4-20 was found using the PSORT II program. Based on the cellular
distribution of a series of deletion mutants of EAV N , it appeared that EAV N needed the
entire motif of 4-20 residues for nuclear localization. Because of not clear interaction of
EAV N with importin-α and -β proteins, it is possible that it may not utilize the importin-
regulated nuclear transport pathway. Its interaction with GST itself and an unrelated
GST-fusion protein might be a nonspecific binding. The other possibility could be that it
uses other cellular protein to go to nucleus.
The N protein of SHFV is the least characterized N protein in the family so far.
The SHFV N gene was cloned as a fusion protein with EGFP on the N- or C-terminus.
Both SHFV N-EGFP and EGFP-SHFV N fusion proteins showed nuclear and nucleolar
localization. The significance of a functional ‘pat-7’ type NLS at amino acid positions
22-28 predicted by PSORT II, was determined by constructing a group of progressively

121
deleted mutants for SHFV N protein. Furthermore, the interaction of SHFV N protein
with importin-α suggests that it recruits this chaperon protein for its nuclear transport.
These findings resemble the data for the N proteins of other members of arteriviruses,
indicating that nucleolar localization of the N protein is a common and conserved feature
for Arteriviridae.
The conserved feature of nucleolar localization of N protein among all members
of the Arteriviridae family suggests that it plays an important accessory role during viral
replication in addition to its participation in virion assembly. The nuclear localization of
nucleocapsid protein of PRRSV was investigated in vitro intensively and only one in vivo
study was conducted using infectious clone harboring the PRRSV N with deleted NLS.
This study showed that the mutant protein had limited replication compared to the wild
type PRRSV which indicates that N protein is important in the pathogenesis of PRRSV
(Lee et al, 2006a). PRRSV N, like N protein of the member viruses of Coronaviridae,
interacts with nucleolar and cellular proteins involved in regulating cell cycle. The co-
localization of LDV N and EAV N proteins with fibrillarin indicates that it is also a
shared property between members of Arteriviridae. Since no data was available for the
role of N in cell cycle modulation by Arteriviridae, the research on this subject would
elucidate a very important aspect of arterivirus-cell correlation. Our results showed that
replication of PRRSV and EAV in growing cells leads to accumulation of cells at the
G2/M phase of the cell cycle. We further examined if N protein was the viral component
involved in this function. The N protein of all 4 member viruses of the Arteriviridae
family was so expressed in BHK-21 cells and analyzed for DNA contents. The results
indicated that the number of cells was higher in the G2/M phase in N gene transfected

122
cells compared to controls. This suggests that the nucleocapsid protein of arteriviruses
may be associated with cell cycle modulation in arterivirus infected cells. The N proteins
of all members of the family Arteriviridae affected cell cycle in a similar way but their
influence on the cell cycle modulation was different. In the BHK-21 cells expressing the
N protein, PRRSV N and EAV N resulted in more accumulation of cells at G2/M stage
compared to LDV N and SHFV N.
Taken together, it seems that the nucleocapsid protein of Arteriviridae is a
multifunctional protein. It is a nuclear-cytoplasmic protein, and binds to cellular proteins
which are involved in regulation of cell functions, such as transcription and cell cycle. N
protein also affects cell cycle by accumulation of cells at the G2/M stage. These findings
raise the possibility that N protein plays a role in pathogenesis of these viruses, and
further studies are needed to scrutinize its mechanism of function in host cells and
pathogenesis of arteriviruses.
One of the caveats of this research was the lack of an in vivo study for the N
protein nuclear localization and its behavior in primary cells of the natural host. Also, the
cell cycle modification by arteriviruses could have been more deeply investigated.
Therefore, future approaches to deciphering the role of arterivirus N in cell cycle could
be by focusing on each stage of cell cycle. This may be done by synchronizing cells at
G0/G1, S, and G2/M phases followed by virus infection and N protein transfection.
Furthermore, live cell microscopy for monitoring cellular distribution of N protein at
different stages of cell cycle could open another view on its behavior and function in host
cells.

123
In conclusion, the results obtained from the present study shed light on the
subcellular distribution of N protein of arteriviruses. The conserved property of nucleolar
localization of N protein suggests a significant role for this protein in virus pathogenesis.
The finding that N protein is involved in cell cycle modification, supports the assumption
that in addition to its structural role for virus assembly, N protein plays a non-structural
role during virus replication.

124
REFERENCES
Adam, E. J. and S. A. Adam. 1994. Identification of cytosolic factors required for
nuclear location sequence-mediated binding to the nuclear envelope. J. Cell Biol.
125:547-555.
Albina, E. 1997. Epidemiology of porcine reproductive and respiratory syndrome
(PRRS): an overview. Vet. Microbiol. 55:309-316.
Allen, A. M., A. E. Palmer, N. M. Tauraso, and A. Shelokov. 1968. Simian
hemorrhagic fever. II. Studies in pathology. Am. J. Trop. Med. Hyg. 17:413-421.
Aminev, A. G., S. P. Amineva, and A. C. Palmenberg. 2003. Encephalomyocarditis
virus (EMCV) proteins 2A and 3BCD localize to nuclei and inhibit cellular mRNA
transcription but not rRNA transcription. Virus Res. 95: 59–73.
Amineva S. P., A. G. Aminev, A. C. Palmenberg, and J. E. Gern. 2004. Rhinovirus
3C protease precursors 3CD and 3CD' localize to the nuclei of infected cells. J. Gen.
Virol. 85: 2969-2979.
Andersen, J. S., C. E. Lyon, A. H. Fox, A. K. Leung, Y. W. Lam, H. Steen, M. Mann,
and A. I. Lamond. 2002. Directed proteomic analysis of the human nucleolus. Curr.
Biol. 12:1-11.
Andersen, J. S., Y. W. Lam, A. K. Leung, S. E. Ong, C. E. Lyon, A. I. Lamond, and
M. Mann. 2005. Nucleolar proteome dynamics. Nature. 433:77-83.
Azum-Gelade, M. C., J. Noaillac-Depeyre, M. Caizergues-Ferrer, and N. Gas. 1994.
Cell cycle redistribution of U3 snRNA and fibrillarin. Presence in the cytoplasmic
nucleolus remnant and in the prenucleolar bodies at telophase. J. Cell. Sci. 107 ( Pt
2):463-475.
Balasuriya, U. B., N.J. MacLachlan, A.A.F. de Vries, P.V. Rossitto, and P.J.M.
Rottier. 1995. Identification of a neutralization site in the major envelope glycoprotein
(GL) of equine arteritis virus. Virology. 207: 518-527.
Balasuriya, U. B., J.F. Patton, P.V. Rossitto, P.J. Timoney, W.H. McCollum, and
N.J. MacLachlan. 1997. Neutralization determinants of laboratory strains and field
isolates of equine arteritis virus: identification of four neutralization sites in the amino-
terminal ectodomain of the GL envelope glycoprotein. Virology. 232: 114-128.
Balasuriya, U. B., H. W. Heidner, N. L. Davis, H. M. Wagner, P. J. Hullinger, J. F.
Hedges, J. C. Williams, R. E. Johnston, W. David Wilson, I. K. Liu, and N. James
MacLachlan. 2002. Alphavirus replicon particles expressing the two major envelope

125
proteins of equine arteritis virus induce high level protection against challenge with
virulent virus in vaccinated horses. Vaccine. 20:1609-1617.
Balasuriya, U. B., J. C. Dobbe, H. W. Heidner, V. L. Smalley, A. Navarrette, E. J.
Snijder, and N. J. MacLachlan. 2004. Characterization of the neutralization
determinants of equine arteritis virus using recombinant chimeric viruses and site-specific
mutagenesis of an infectious cDNA clone. Virology. 321:235-246.
Balasuriya, U. B. and N. J. MacLachlan. 2004. The immune response to equine
arteritis virus: potential lessons for other arteriviruses. Vet. Immunol. Immunopathol.
102:107-129.
Banerjee, R., M. K. Weidman, S. Navarro, L. Comai, and A. Dasgupta. 2005.
Modifications of both selectivity factor and upstream binding factor contribute to
poliovirus-mediated inhibition of RNA polymerase I transcription. J. Gen. Virol.
86:2315-2322.
Baric, R. S., S. A. Stohlman, and M. M. Lai. 1983. Characterization of replicative
intermediate RNA of mouse hepatitis virus: presence of leader RNA sequences on
nascent chains. J. Virol. 48:633-640.
Bautista, E. M., S.M. Goyal, I.J. Yoon, H.S. Joo, and J.E. Collins. 1993. Comparison
of porcine alveolar macrophages and CL 2621 for the detection of porcine reproductive
and respiratory syndrome (PRRS) virus and anti-PRRS antibody. J. Vet. Diagn. Invest. 5:
163-165.
Bautista, E. M., J. J. Meulenberg, C. S. Choi, and T. W. Molitor. 1996. Structural
polypeptides of the American (VR-2332) strain of porcine reproductive and respiratory
syndrome virus. Arch. Virol. 141:1357-1365.
Bautista, E. M., P. Suarez, and T. W. Molitor. 1999. T cell responses to the structural
polypeptides of porcine reproductive and respiratory syndrome virus. Arch. Virol.
144:117-134.
Benfield, D. A., E.A. Nelson, J.E. Collins, L. Harris, S.M. Goyal, D. Robison, W.T.
Christianson, R.B. Morrison, D. Gorcyca, and D.W. Chladek. 1992. Characterization
of swine infertility and respiratory syndrome (SIRS) virus (isolate ATCC VR-2332). J.
Vet. Diagn. Invest. 4: 127-133.
Beura, L. K., S. N. Sarkar, B. Kwon, S. Subramaniam, C. Jones, A. K. Pattnaik, and
F. A. Osorio. 2010. Porcine reproductive and respiratory syndrome virus nonstructural
protein 1beta modulates host innate immune response by antagonizing IRF3 activation. J.
Virol. 84:1574-1584.

126
Braaten Re F., D., E. K. Franke, and J. Luban. 1995. Human immunodeficiency virus
type 1 Vpr arrests the cell cycle in G2 by inhibiting the activation of p34cdc2-cyclin B. J.
Virology. 69:6859-6864.
Brayton, P. R., R. G. Ganges, and S. A. Stohlman. 1981. Host cell nuclear function
and murine hepatitis virus replication. J. Gen. Virol. 56:457-460.
Breeuwer, M. and D. S. Goldfarb. 1990. Facilitated nuclear transport of histone H1 and
other small nucleophilic proteins. Cell. 60:999-1008.
Brierley, I. 1995. Ribosomal frameshifting on viral RNAs. J. Gen. Virol. 76: 1885-1892.
Brinton M.A. 1999. Arteriviruses (Arteriviridae). In: Encyclopedia of Virology, 2nd
Edition (A. Granoff and R.G. Webster, eds.), pp. 89-97. Academic Press, San Diego.
Brinton-Darnell, M., J. K. Collins, and P. G. Plagemann. 1975. Lactate
dehydrogenase-elevating virus replication, maturation, and viral RNA synthesis in
primary mouse macrophage cultures. Virology. 65:187-195.
Brinton-Darnell, M. and P. G. Plagemann. 1975. Structure and chemical-physical
characteristics of lactate dehydrogenase-elevating virus and its RNA. J. Virol. 16:420-
433.
Broen, J. J. and W. A. Cafruny. 1993. Immunoglobulin transfer from immune-
reconstituted SCID mice to nursing neonates: blood distribution of antibody and
association with perinatal virus protection. Reg. Immunol. 5:44-52.
Bryans, J. T., M. E. Crowe, E. R. Doll, and W. H. Mccollum. 1957. The blood picture
and thermal reaction in experimental viral arteritis of horses. Cornell Vet. 47:43-52.
Burki, F. 1966. Further properties of equine arteritis virus. Archiv fur die Gesamte
Virusforschung. 19: 123-129.
Cafruny, W. A. and P. G. Plagemann. 1982. Immune response to lactate
dehydrogenase-elevating virus: serologically specific rabbit neutralizing antibody to the
virus. Infect. Immun. 37:1007-1012.
Cafruny, W. A., S. P. Chan, J. T. Harty, S. Yousefi, K. Kowalchyk, D. McDonald, B.
Foreman, G. Budweg, and P. G. Plagemann. 1986. Antibody response of mice to
lactate dehydrogenase-elevating virus during infection and immunization with inactivated
virus. Virus Res. 5:357-375.
Cannavo, G., M. Paiardini, D. Galati, B. Cervasi, M. Montroni, G. De Vico, D.
Guetard, M. L. Bocchino, I. Picerno, M. Magnani, G. Silvestri, and G. Piedimonte.
2001. Abnormal intracellular kinetics of cell-cycle-dependent proteins in lymphocytes
from patients infected with human immunodeficiency virus: a novel biologic link

127
between immune activation, accelerated T-cell turnover, and high levels of apoptosis.
Blood. 97:1756-1764.
Castillo-Olivares, J., J. P. Tearle, F. Montesso, D. Westcott, J. H. Kydd, N. J. Davis-
Poynter, and D. Hannant. 2003. Detection of equine arteritis virus (EAV)-specific
cytotoxic CD8+ T lymphocyte precursors from EAV-infected ponies. J. Gen. Virol.
84:2745-2753.
Calvert, J. G., D. E. Slade, S.L. Shields, R. Jolie, R.M. Mannan, R.G. Ankenbauer,
and S.K. Welch. (2007). CD163 expression confers susceptibility to porcine
reproductive and respiratory syndrome viruses. J. Virol. 81: 7371–7379.
Cavanagh D., Brien D. A., Brinton M., Enjuanes L., Holmes K. V., Horzinek M. C.,
Lai M. M. C., Laude H. , Plagemann P. G. W. , Siddell S., Spaan W. J. M., Taguchi
F. and Talbot P. J. 1994. Revision of the taxonomy of the Coronavirus, Torovirus and
Arterivirus genera. Arch. Virol. 135:227-237.
Cavanagh, D. 1997. Nidovirales: a new order comprising Coronaviridae and
Arteriviridae. Arch. Virol. 142:629-633.
Cawood, R., S. M. Harrison, B. K. Dove, M. L. Reed, and J. A. Hiscox. 2007. Cell
cycle dependent nucleolar localization of the coronavirus nucleocapsid protein. Cell
Cycle. 6:863-867.
Chen, C. J. and S. Makino. 2004. Murine coronavirus replication induces cell cycle
arrest in G0/G1 phase. J. Virol. 78:5658-5669.
Chen, C. J., K. Sugiyama, H. Kubo, C. Huang, and S. Makino. 2004. Murine
coronavirus nonstructural protein p28 arrests cell cycle in G0/G1 phase. J. Virol.
78:10410-10419.
Chen, H., T. Wurm, P. Britton, G. Brooks, and J. A. Hiscox. 2002. Interaction of the
coronavirus nucleoprotein with nucleolar antigens and the host cell. J. Virol. 76:5233-
5250.
Chen, Z., L. Kuo, R. R. Rowland, C. Even, K. S. Faaberg, and P. G. Plagemann.
1993. Sequences of 3' end of genome and of 5' end of open reading frame 1a of lactate
dehydrogenase-elevating virus and common junction motifs between 5' leader and bodies
of seven subgenomic mRNAs. J. Gen. Virol. 74:643-659.
Chen, Z., K. S. Faaberg, and P. G. Plagemann. 1994. Detection of negative-stranded
subgenomic RNAs but not of free leader in LDV-infected macrophages. Virus Res.
34:167-177.

128
Chen, Z., R. R. Rowland, G. W. Anderson, G. A. Palmer, and P. G. Plagemann.
1997. Coexistence in lactate dehydrogenase-elevating virus pools of variants that differ in
neuropathogenicity and ability to establish a persistent infection. J. Virol. 71:2913-2920.
Chen, Z., S. Lawson, Z. Sun, X. Zhou, X. Guan, J. Christopher-Hennings, E. A.
Nelson, and Y. Fang. 2009a. Identification of two auto-cleavage products of
nonstructural protein 1 (nsp1) in porcine reproductive and respiratory syndrome virus
infected cells: nsp1 function as interferon antagonist. Virol. [Epub ahead of print].
Chen, Z., X. Zhou, J. K. Lunney, S. Lawson, Z. Sun, E. Brown, J. Christopher-
Hennings, D. Knudsen, E. Nelson, and Y. Fang. 2009b. Immunodominant epitopes in
nsp2 of porcine reproductive and respiratory syndrome virus are dispensable for
replication but play an important role in modulation of host immune response. J. Gen.
Virol. 2009 Nov 18. [Epub ahead of print].
Chi, N. C., E. J. Adam, and S. A. Adam. 1995. Sequence and characterization of
cytoplasmic nuclear protein import factor p97. J. Cell Biol. 130:265-274.
Chirnside, E. D., A.A.F. de Vries, J.A. Mumford, and P.J.M. Rottier. 1995a. Equine
arteritis virus-neutralizing antibody in the horse is induced by a determinant on the large
envelope glycoprotein GL. J. Gen. Virol. 76: 1989-1998.
Chirnside, E. D., P. M. Francis, A. A. de Vries, R. Sinclair, and J. A. Mumford.
1995b. Development and evaluation of an ELISA using recombinant fusion protein to
detect the presence of host antibody to equine arteritis virus. J. Virol. Methods 54:1-13.
Chirnside, E. D., P. M. Francis, and J. A. Mumford. 1995c. Expression cloning and
antigenic analysis of the nucleocapsid protein of equine arteritis virus. Virus Res. 39:277-
288.
Collins, J. E., D. A. Benfield, W. T. Christianson, L. Harris, J. C. Hennings, D. P.
Shaw, S. M. Goyal, S. McCullough, R. B. Morrison, and H. S. Joo. 1992. Isolation of
swine infertility and respiratory syndrome virus (isolate ATCC VR-2332) in North
America and experimental reproduction of the disease in gnotobiotic pigs. J. Vet. Diagn.
Invest. 4:117-126.
Cook, A., F. Bono, M. Jinek, and E. Conti. 2007. Structural biology of
nucleocytoplasmic transport. Annu. Rev. Biochem. 76:647-671.
Contag, C. H. and Plagemann, P. G. W. 1989. Age-dependent poliomyelitis of mice:
expression of endogenous retrovirus correlates with cytocidal replication of lactate
dehydrogenase-elevating virus in motor neurons. J. Virol. 63: 4362-4369.
Coutelier, J. P., E. Van Roost, P. Lambotte, and J. Van Snick. 1986. The murine
antibody response to lactate dehydrogenase-elevating virus. J. Gen. Virol. 67:1099-1108.

129
Davy, C. and J. Doorbar. 2007. G2/M cell cycle arrest in the life cycle of viruses.
Virology. 368:219-226.
Del Piero, F. 2000. Equine viral arteritis. Vet. Pathol. 37:287-296.
Delputte, P. L., N. Vanderheijden, H. J. Nauwynck, and M. B. Pensaert. 2002.
Involvement of the matrix protein in attachment of porcine reproductive and respiratory
syndrome virus to a heparin like receptor on porcine alveolar macrophages. J. Virol.
76:4312–4320.
Delputte, P.L. and H.J. Nauwynck. 2004. Porcine arterivirus infection of alveolar
macrophages is mediated by sialic acid on the virus. J. Virol. 78: 8094–8101.
Delputte, P.L., P. Meerts, S. Costers, and H.J. Nauwynck, H. J. 2004. Effect of virus-
specific antibodies on attachment, internalization and infection of porcine reproductive
and respiratory syndrome virus in primary macrophages. Vet. Immunol. Immunopathol.
102: 179–188.
Delputte, P. L., S. Costers, and H. J. Nauwynck. 2005. Analysis of porcine
reproductive and respiratory syndrome virus attachment and internalization: distinctive
roles for heparan sulphate and sialoadhesin. J. Gen. Virol. 86:1441-1445.
Delputte, P.L., W. Van Breedam, I. Delrue, C. Oetke, P.R. Crocker, and H.J.
Nauwynck. 2007. Porcine arterivirus attachment to the macrophage-specific receptor
sialoadhesin is dependent on the sialic acid-binding activity of the N-terminal
immunoglobulin domain of sialoadhesin. J. Virol. 81: 9546–9550.
Denac, H., C. Moser, J. D. Tratschin, and M. A. Hofmann. 1997. An indirect ELISA
for the detection of antibodies against porcine reproductive and respiratory syndrome
virus using recombinant nucleocapsid protein as antigen. J. Virol. Methods 65:169-181.
den Boon, J. A., E. J. Snijder, E. D. Chirnside, A. A. de Vries, M. C. Horzinek, and
W. J. Spaan. 1991. Equine arteritis virus is not a togavirus but belongs to the
coronaviruslike superfamily. J. Virol. 65:2910-2920.
den Boon, J. A., M. F. Kleijnen, W. J. Spaan, and E. J. Snijder. 1996. Equine arteritis
virus subgenomic mRNA synthesis: analysis of leader-body junctions and replicative-
form RNAs. J. Virol. 70:4291-4298.
de Vries, A. A. F., E.D. Chirnside, P.J. Bredenbeek, L.A. Gravestein, M.C.
Horzinek, and W.J.M. Spaan. 1990. All subgenomic mRNAs of equine arteritis virus
contain a common leader sequence. Nucleic Acids Res. 18: 3241-3247.
de Vries, A. A., E.D. Chirnside, M.C. Horzinek, and P. J. Rottier. 1992. Structural
proteins of equine arteritis virus. J. Virol. 66:6294-6303.

130
de Vries, A. A., S.M. Post, M.J. Raamsman, M.C. Horzinek, and P.J. Rottier. 1995a.
The two major envelope proteins of equine arteritis virus associate into disulfide-linked
heterodimers. J. Virol. 69:4668-4674.
de Vries, A. A. F., Raamsman, M. J., van Dijk, H. A., Horzinek, M. C. and Rottier,
P. J. M. 1995b. The small envelope glycoprotein (Gs) of equine arteritis virus folds into
three distinct monomers and a disulfide-linked dimer. J. Virol. 69:3441-3448.
de Vries, A. A. F., Horzinek, M. C., Rottier, P. J. M. & de Groot, R. J. 1997. The
genome organization of the Nidovirales: similarities and differences between arteri-, toro,
and coronaviruses. Semin. Virol. 8: 33-47.
Doll, E. R., Bryans, J. T., McCollum, W. H. & Crowe, M. 1957. Isolation of a
filterable agent causing arteritis of horses and abortion by mares: its differentiation from
the equine abortion (influenza) virus. Cornell Vet. 47: 3-41.
Dove, B. K., J. H. You, M. L. Reed, S. R. Emmett, G. Brooks, and J. A. Hiscox.
2006a. Changes in nucleolar morphology and proteins during infection with the
coronavirus infectious bronchitis virus. Cell. Microbiol. 8:1147-1157.
Dove, B., G. Brooks, K. Bicknell, T. Wurm, and J. A. Hiscox. 2006b. Cell cycle
perturbations induced by infection with the coronavirus infectious bronchitis virus and
their effect on virus replication. J. Virol. 80:4147-4156.
Dundr, M., M. D. Hebert, T. S. Karpova, D. Stanek, H. Xu, K. B. Shpargel, U. T.
Meier, K. M. Neugebauer, A. G. Matera, and T. Misteli. 2004. In vivo kinetics of
Cajal body components. J. Cell Biol. 164:831-842.
Emmett, S. R., B. Dove, L. Mahoney, T. Wurm, and J. A. Hiscox. 2005. The cell
cycle and virus infection. Methods Mol. Biol. 296:197-218.
Evans, M. R. and R. W. Simpson. 1980. The coronavirus avian infectious bronchitis
virus requires the cell nucleus and host transcriptional factors. Virology. 105:582-591.
Even, C., R. R. Rowland, and P. G. Plagemann. 1995. Cytotoxic T cells are elicited
during acute infection of mice with lactate dehydrogenase-elevating virus but disappear
during the chronic phase of infection. J. Virol. 69:5666-5676.
Faaberg, K. S., C. Even, G. A. Palmer, and P. G. Plagemann. 1995a. Disulfide bonds
between two envelope proteins of lactate dehydrogenase-elevating virus are essential for
viral infectivity. J. Virol. 69:613-617.
Faaberg, K. S., G.A. Palmer, C. Even, G.W. Anderson, and P.G.W. Plagemann.
1995b. Differential glycosylation of the ectodomain of the primary envelope glycoprotein
of two strains of lactate dehydrogenase elevating virus that differ in neuropathogenicity.
Virus Res. 39: 331-340.

131
Faaberg, K. S. and P. G. Plagemann. 1995. The envelope proteins of lactate
dehydrogenase-elevating virus and their membrane topography. Virology 212:512-525.
Faaberg, K. S. and P. G. W. Plagemann. 1997. ORF3 of lactate dehydrogenase
elevating virus encodes a soluble, nonstructural, highly glycosylated, and antigenic
protein. Virology. 227: 245-251.
Favre, D., E. Studer, and M. R. Michel. 1994. Two nucleolar targeting signals present
in the N-terminal part of Semliki Forest virus capsid protein. Arch. Virol. 137:149-155.
Fazakerley, J. K., A. Boyd, M. L. Mikkola, and L. Kaariainen. 2002. A single amino
acid change in the nuclear localization sequence of the nsP2 protein affects the
neurovirulence of Semliki Forest virus. J. Virol. 76:392-396.
Feldherr, C. M., E. Kallenbach, and N. Schultz. 1984. Movement of a karyophilic
protein through the nuclear pores of oocytes. J. Cell Biol. 99:2216-2222.
Flemington, E. K. 2001. Herpesvirus lytic replication and the cell cycle: arresting new
developments. J. Virol. 75:4475-4481.
Fomproix, N., J. Gebrane-Younes, and D. Hernandez-Verdun. 1998. Effects of anti-
fibrillarin antibodies on building of functional nucleoli at the end of mitosis. J. Cell. Sci.
111:359-372.
Ghosh, A. K., R. Steele, K. Meyer, R. Ray, and R. B. Ray. 1999. Hepatitis C virus
NS5A protein modulates cell cycle regulatory genes and promotes cell growth. J. Gen.
Virol. 80:1179-1183.
Godeny, E. K., L. Chen, S.N. Kumar, S.L. Methven, E.V. Koonin, and M.A.
Brinton. 1993. Complete genomic sequence and phylogenetic analysis of the lactate
dehydrogenase-elevating virus (LDV). Virology.194: 585-596.
Godeny, E. K., L. Zeng, S.L. Smith, and M.A. Brinton. 1995. Molecular
characterization of the 3’ terminus of the simian hemorrhagic fever virus genome. J.
Virol. 69: 2679-2683.
Godeny, E. K., A.A.F. de Vries, X.C. Wang, S.L. Smith, and R.J. de Groot. 1998.
Identification of the leader-body junctions for the viral subgenomic mRNAs and
organization of the simian hemorrhagic fever virus genome: evidence for gene
duplication during arterivirus evolution. J. Virol. 72: 862-867.
Gonin, P., B. Pirzadeh, C. A. Gagnon, and S. Dea. 1999. Seroneutralization of porcine
reproductive and respiratory syndrome virus correlates with antibody response to the GP5
major envelope glycoprotein. J. Vet. Diagn. Invest. 11:20-26.

132
Gorbalenya, A. E., L. Enjuanes, J. Ziebuhr, and E. J. Snijder. 2006. Nidovirales:
evolving the largest RNA virus genome. Virus Res. 117:17-37.
Gorlich, D., S. Prehn, R. A. Laskey, and E. Hartmann. 1994. Isolation of a protein
that is essential for the first step of nuclear protein import. Cell 79:767-778.
Gorlich, D., S. Kostka, R. Kraft, C. Dingwall, R. A. Laskey, E. Hartmann, and S.
Prehn. 1995. Two different subunits of importin cooperate to recognize nuclear
localization signals and bind them to the nuclear envelope. Curr. Biol. 5:383-392.
Gorlich, D. and U. Kutay. 1999. Transport between the cell nucleus and the cytoplasm.
Annu. Rev. Cell Dev. Biol. 15:607-660.
Gravell, M., W. T. London, M. Leon, A. E. Palmer, and R. S. Hamilton. 1986a.
Elimination of persistent simian hemorrhagic fever (SHF) virus infection in patas
monkeys. Proc. Soc. Exp. Biol. Med. 181:219-225.
Gravell, M., W. T. London, M. E. Leon, A. E. Palmer, and R. S. Hamilton. 1986b.
Differences among isolates of simian hemorrhagic fever (SHF) virus. Proc. Soc. Exp.
Biol. Med. 181:112-119.
Harty, J. T., S. P. Chan, C. H. Contag, and P. G. Plagemann. 1987. Protection of C58
mice from lactate dehydrogenase-elevating virus-induced motor neuron disease by non-
neutralizing antiviral antibodies without interference with virus replication. J.
Neuroimmunol. 15:195-206.
Harty, J. T. and P. G. Plagemann. 1990. Monoclonal antibody protection from age-
dependent poliomyelitis: implications regarding the pathogenesis of lactate
dehydrogenase-elevating virus. J. Virol. 64:6257-6262.
Henderson, B. R. and P. Percipalle. 1997. Interactions between HIV Rev and nuclear
import and export factors: the Rev nuclear localisation signal mediates specific binding to
human importin-beta. J. Mol. Biol. 274:693-707.
Hirano, M., S. Kaneko, T. Yamashita, H. Luo, W. Qin, Y. Shirota, T. Nomura, K.
Kobayashi, and S. Murakami. 2003. Direct interaction between nucleolin and hepatitis
C virus NS5B. J. Biol. Chem. 278:5109-5115.
Hiscox, J. A., T. Wurm, L. Wilson, P. Britton, D. Cavanagh, and G. Brooks. 2001.
The coronavirus infectious bronchitis virus nucleoprotein localizes to the nucleolus. J.
Virol. 75:506-512.
Hiscox, J. A. 2002. The nucleolus--a gateway to viral infection? Arch. Virol. 147:1077-
1089.

133
Hiscox, J. A. 2003. The interaction of animal cytoplasmic RNA viruses with the nucleus
to facilitate replication. Virus Res. 95:13-22.
Hiscox, J. A. 2007. RNA viruses: hijacking the dynamic nucleolus. Nat. Rev. Microbiol.
5:119-127.
Horzinek, M. C., J. Maess, and R. Laufs. 1971. Studies on the substructure of
togaviruses. II. Analysis of equine arteritis, rubella, bovine viral diarrhea, and hog
cholera viruses. Arch Gesamte Virusforsch. 33: 306-318.
Hwang, Y. K. and M. A. Brinton. 1998. A 68-nucleotide sequence within the 3'
noncoding region of simian hemorrhagic fever virus negative-strand RNA binds to four
MA104 cell proteins. J. Virol. 72:4341-4351.
Hyllseth, B. 1969. A plaque assay of equine arteritis virus in BHK-21 cells. Arch
Gesamte Virusforsch. 28: 26-33.
Inada, T. and S. Yamazaki. 1991. Replication of lactate dehydrogenaseelevating virus
in cells infected with murine leukaemia viruses in vitro. J. Gen. Virol. 72: 2437-2444.
Inada, T., H. Kikuchi, and S. Yamazaki. 1993. Comparison of the ability of lactate
dehydrogenase-elevating virus and its virion RNA to infect murine leukemia virus-
infected or -uninfected cell lines. J. Virol. 67: 5698-5703.
Isoyama, T., S. Kuge, and A. Nomoto. 2002. The core protein of hepatitis C virus is
imported into the nucleus by transport receptor Kap123p but inhibits Kap121p-dependent
nuclear import of yeast AP1-like transcription factor in yeast cells. J. Biol. Chem.
277:39634-39641.
Izumi, R. E., B. Valdez, R. Banerjee, M. Srivastava, and A. Dasgupta. 2001.
Nucleolin stimulates viral internal ribosome entry site-mediated translation. Virus Res.
76:17-29.
Jäkel S., W. Albig, U. Kutay, F. R. Bischoff, K. Schwamborn, D. Doenecke, and D.
Görlich. (1999). The importin β/importin 7 heterodimer is a functional nuclear import
receptor for histone H1. EMBO J. 18: 2411–2423.
Jacks, T., H. D. Madhani, F. R. Masiarz, and H. E. Varmus. 1988. Signals for
ribosomal frameshifting in the Rous sarcoma virus gag-pol region. Cell. 55:447-458.
Jordan, E. G. 1984. Nucleolar nomenclature. J. Cell. Sci. 67:217-220.
Jusa, E. R., Y. Inaba, M. Kouno, and O. Hirose. 1997. Effect of heparin on infection
of cells by porcine reproductive and respiratory syndrome virus. Am. J. Vet. Res. 58:488-
491.

134
Kalderon, D., W. D. Richardson, A. F. Markham, and A. E. Smith. 1984. Sequence
requirements for nuclear location of simian virus 40 large-T antigen. Nature 311:33-38.
Kheyar, A., S. Martin, G. St-Laurent, P. J. Timoney, W. H. McCollum, and D.
Archambault. 1997. Expression cloning and humoral immune response to the
nucleocapsid and membrane proteins of equine arteritis virus. Clin. Diagn. Lab.
Immunol. 4:648-652.
Kim, H. S., J. Kwang, I. J. Yoon, H. S. Joo, and M. L. Frey. 1993. Enhanced
replication of porcine reproductive and respiratory syndrome (PRRS) virus in a
homogeneous subpopulation of MA-104 cell line. Arch. Virol. 133:477-483.
Kim, J. K., A. M. Fahad, K. Shanmukhappa, and S. Kapil. 2006. Defining the cellular
targets of porcine reproductive and respiratory syndrome virus blocking monoclonal
antibody 7G10. J. Virol. 80:689–696.
Konishi, S., H. Akashi, H., A. Sentsui, and M. Ogata. 1975. Studies on equine viral
arteritis Characterisation of the virus and trial survey on antibody with Vero cell culture.
J. Vet. Med. Sci. 37: 259-267.
Kowalchyk, K. and P. G. Plagemann. 1985. Cell surface receptors for lactate
dehydrogenase-elevating virus on subpopulation of macrophages. Virus Res. 2:211-229.
Kreutz, L. C. and M. R. Ackermann. 1996. Porcine reproductive and respiratory
syndrome virus enters cells through a low pH-dependent endocytic pathway. Virus Res.
42:137-147.
Kuo, L. L., J. T. Harty, L. Erickson, G. A. Palmer, and P. G. Plagemann. 1991. A
nested set of eight RNAs is formed in macrophages infected with lactate dehydrogenase-
elevating virus. J. Virol. 65:5118-5123.
Kuo, L. L., Chen, Z., Rowland, R. R. R., Faaberg, K. S. & Plagemann, P. G. W. 1992. Lactate dehydrogenase-elevating virus (LDV): subgenomic mRNAs, mRNA leader
and comparison of 3’-terminal sequences of two LDV isolates. Virus Res. 23: 55-72.
Lai, Frances W. 2006. Modulation of type 1 interferon by porcine reproductive and
respiratory syndrome virus. Thesis. University of Guelph.
Lanford, R. E. and J. S. Butel. 1984. Construction and characterization of an SV40
mutant defective in nuclear transport of T antigen. Cell. 37:801-813.
Lee, C. H., S. C. Chang, C. J. Chen, and M. F. Chang. 1998. The nucleolin binding
activity of hepatitis delta antigen is associated with nucleolus targeting. J. Biol. Chem.
273:7650-7656.

135
Lee, C., D. Hodgins, J. G. Calvert, S. K. Welch, R. Jolie, and D. Yoo. 2006a.
Mutations within the nuclear localization signal of the porcine reproductive and
respiratory syndrome virus nucleocapsid protein attenuate virus replication. Virology.
346:238-250.
Lee, C., D. C. Hodgins, J. G. Calvert, S. K. Welch, R. Jolie, and D. Yoo. 2006b. The
nuclear localization signal of the PRRS virus nucleocapsid protein viral replication in
vitro and antibody response in vivo. Adv. Exp. Med. Biol. 581:145-148.
Lee, C. and D. Yoo. 2006. The small envelope protein of porcine reproductive and
respiratory syndrome virus possesses ion channel protein-like properties. Virology.
355:30-43.
Lee, S. M., S. K. Schommer, and S. B. Kleiboeker. 2004. Porcine reproductive and
respiratory syndrome virus field isolates differ in in vitro interferon phenotypes. Vet.
Immunol. Immunopathol. 102:217-231.
Leung, A. K., J. S. Andersen, M. Mann, and A. I. Lamond. 2003. Bioinformatic
analysis of the nucleolus. Biochem. J. 376:553-569.
Li, K., Z. Chen, and P. Plagemann. 1998. The neutralization epitope of lactate
dehydrogenase-elevating virus is located on the short ectodomain of the primary
envelope glycoprotein. Virology. 242:239-245.
Li, F. Q., J. P. Tam, and D. X. Liu. 2007. Cell cycle arrest and apoptosis induced by the
coronavirus infectious bronchitis virus in the absence of p53. Virology .365:435-445.
Lin, G. Y. and R. A. Lamb. 2000. The paramyxovirus simian virus 5 V protein slows
progression of the cell cycle. J. Virol. 74:9152-9166.
Loemba, H. D., S. Mounir, H. Mardassi, D. Archambault, and S. Dea. 1996. Kinetics
of humoral immune response to the major structural proteins of the porcine reproductive
and respiratory syndrome virus. Arch. Virol. 141:751-761.
London, W. T. 1977. Epizootiology, transmission and approach to prevention of fatal
simian haemorrhagic fever in rhesus monkeys. Nature 268:344-345.
MacLachlan, N. J., U. B. Balasuriya, J. F. Hedges, T. M. Schweidler, W. H.
McCollum, P. J. Timoney, P. J. Hullinger, and J. F. Patton. 1998. Serologic response
of horses to the structural proteins of equine arteritis virus. J. Vet. Diagn. Invest. 10:229-
236.
Madden, D. L., D. A. Fuccillo, J. A. Dorosz, W. T. London, A. E. Palmer, and G. A.
Castellano. 1978. Antigenic relationship of two strains of simian hemorrhagic fever
virus. Lab. Anim. Sci. 28:422-427.

136
Magnusson, P., B. Hyllseth, and H. Marusyk. 1970. Morphological studies on equine
arteritis virus. Arch. Gesamte Virusforsch. 30:105-112.
Mardassi, H., B. Massie and S. Dea. 1996. Intracellular synthesis, processing and
transport of proteins encoded by ORFs 5 to 7 of porcine reproductive and respiratory
syndrome virus. Virology. 221: 98–112.
Mahalingam, S., V. Ayyavoo, M. Patel, T. Kieber-Emmons, and D. B. Weiner. 1997.
Nuclear import, virion incorporation, and cell cycle arrest/differentiation are mediated by
distinct functional domains of human immunodeficiency virus type 1 Vpr. J. Virol.
71:6339-6347.
Markine-Goriaynoff, D., X. Hulhoven, C. L. Cambiaso, P. Monteyne, T. Briet, M. D.
Gonzalez, P. Coulie, and J. P. Coutelier. 2002. Natural killer cell activation after
infection with lactate dehydrogenase-elevating virus. J. Gen. Virol. 83:2709-2716.
Matsuda, M., N. Matsuda, A. Watanabe, R. Fujisawa, K. Yamamoto, and M.
Masuda. 2003. Cell cycle arrest induction by an adenoviral vector expressing HIV-1 Vpr
in bovine and feline cells. Biochem. Biophys. Res. Commun. 311:748-753.
McCollum, W. H. 1986. Responses of horses vaccinated with avirulent modified-live
equine arteritis virus propagated in the E. Derm (NBL-6) cell line to nasal inoculation
with virulent virus. Am. J. Vet. Res. 47:1931-1934.
Melchior, F., B. Paschal, J. Evans, and L. Gerace. 1993. Inhibition of nuclear protein
import by nonhydrolyzable analogues of GTP and identification of the small GTPase
Ran/TC4 as an essential transport factor. J. Cell Biol. 123:1649-1659.
Melese, T. and Z. Xue. 1995. The nucleolus: an organelle formed by the act of building
a ribosome. Curr. Opin. Cell Biol. 7:319-324.
Meulenberg, J. J. M., E.J. de Meijer, and R.J.M. Moormann. 1993a. Subgenomic
RNAs of Lelystad virus contain a conserved leader-body junction sequence. J. Gen.
Virol. 74: 1697-1701.
Meulenberg, J. J. M., M.M. Hulst, E.J. de Meijer, P.L. Moonen, A. den Besten, E.P.
de Kluyver, G. Wensvoort, and R.J.M. Moormann. 1993b. Lelystad virus, the
causative agent of porcine epidemic abortion and respiratory syndrome (PEARS), is
related to LDV and EAV. Virology. 192: 62-72.
Meulenberg, J. J., A. Petersen-den Besten, E. P. De Kluyver, R. J. Moormann, W.
M. Schaaper, and G. Wensvoort. 1995a. Characterization of proteins encoded by ORFs
2 to 7 of Lelystad virus. Virology. 206:155-163.
Meulenberg, J. J., R. J. Bende, J. M. Pol, G. Wensvoort, and R. J. Moormann.
1995b. Nucleocapsid protein N of Lelystad virus: expression by recombinant baculovirus,

137
immunological properties, and suitability for detection of serum antibodies. Clin. Diagn.
Lab. Immunol. 2:652-656.
Meulenberg, J. J. M. and A. Petersen-den Besten. 1996. Identification and
characterization of a sixth structural protein of Lelystad virus: the glycoprotein GP (2)
encoded by ORF2 is incorporated in virus particles. Virology. 225: 44-51.
Meulenberg, J. J., A. P. van Nieuwstadt, A. van Essen-Zandbergen, and J. P.
Langeveld. 1997. Posttranslational processing and identification of a neutralization
domain of the GP4 protein encoded by ORF4 of Lelystad virus. J. Virol. 71:6061-6067.
Meulenberg, J. J., A. P. van Nieuwstadt, A. van Essen-Zandbergen, J. N. Bos-de
Ruijter, J. P. Langeveld, and R. H. Meloen. 1998. Localization and fine mapping of
antigenic sites on the nucleocapsid protein N of porcine reproductive and respiratory
syndrome virus with monoclonal antibodies. Virology. 252:106-114.
Miyazaki, Y., T. Nosaka, and M. Hatanaka. 1996. The post-transcriptional regulator
Rev of HIV: implications for its interaction with the nucleolar protein B23. Biochimie.
78:1081-1086.
Michel, M. R., M. Elgizoli, Y. Dai, R. Jakob, H. Koblet, and A. P. Arrigo. 1990.
Karyophilic properties of Semliki Forest virus nucleocapsid protein. J. Virol. 64:5123-
5131.
Mohammadi, H., S. Sharif, R. R. Rowland, and D. Yoo. 2009. The lactate
dehydrogenase-elevating virus capsid protein is a nuclear-cytoplasmic protein. Arch.
Virol. 154:1071-1080.
Molenkamp, R., B. C. Rozier, S. Greve, W. J. Spaan, and E. J. Snijder. 2000a.
Isolation and characterization of an arterivirus defective interfering RNA genome. J.
Virol. 74:3156-3165.
Molenkamp, R., H. van Tol, B. C. Rozier, Y. van der Meer, W. J. Spaan, and E. J.
Snijder. 2000b. The arterivirus replicase is the only viral protein required for genome
replication and subgenomic mRNA transcription. J. Gen. Virol. 81:2491-2496.
Monteyne, P. and J. P. Coutelier. 1994. Difference in neutralization between lactate
dehydrogenase-elevating virus isolated from acutely and chronically infected mice. J.
Gen. Virol. 75:1173-1176.
Moore, B. D., U. B. Balasuriya, J. L. Watson, C. M. Bosio, R. J. MacKay, and N. J.
MacLachlan. 2003. Virulent and avirulent strains of equine arteritis virus induce
different quantities of TNF-alpha and other proinflammatory cytokines in alveolar and
blood-derived equine macrophages. Virology. 314:662-670.

138
Moore, M. S. and G. Blobel. 1993. The GTP-binding protein Ran/TC4 is required for
protein import into the nucleus. Nature. 365:661-663.
Moore, M. S. and G. Blobel. 1994. Purification of a Ran-interacting protein that is
required for protein import into the nucleus. Proc. Natl. Acad. Sci. U. S. A. 91:10212-
10216.
Moran, E. 1993. Interaction of adenoviral proteins with pRB and p53. FASEB J. 7:880-
885.
Mori, Y., T. Okabayashi, T. Yamashita, Z. Zhao, T. Wakita, K. Yasui, F. Hasebe,
M. Tadano, E. Konishi, K. Moriishi, and Y. Matsuura. 2005. Nuclear localization of
Japanese encephalitis virus core protein enhances viral replication. J. Virol. 79:3448-
3458.
Morita, E., K. Tada, H. Chisaka, H. Asao, H. Sato, N. Yaegashi, and K. Sugamura.
2001. Human parvovirus B19 induces cell cycle arrest at G(2) phase with accumulation
of mitotic cyclins. J. Virol. 75:7555-7563.
Munoz-Fontela, C., E. Rodriguez, C. Nombela, J. Arroyo, and C. Rivas. 2003.
Characterization of the bipartite nuclear localization signal of protein LANA2 from
Kaposi's sarcoma-associated herpesvirus. Biochem. J. 374:545-550.
Murtaugh, M. P., Z. Xiao, and F. Zuckermann. 2002. Immunological responses of
swine to porcine reproductive and respiratory syndrome virus infection. Viral Immunol.
15:533-547.
Nakai, K. and M. Kanehisa. 1992. A knowledge base for predicting protein localization
sites in eukaryotic cells. Genomics. 14:897-911.
Nakanishi, A., D. Shum, H. Morioka, E. Otsuka, and H. Kasamatsu. 2002.
Interaction of the Vp3 nuclear localization signal with the importin alpha 2/beta
heterodimer directs nuclear entry of infecting simian virus 40. J. Virol. 76:9368-9377.
Naniche, D., S. I. Reed, and M. B. Oldstone. 1999. Cell cycle arrest during measles
virus infection: a G0-like block leads to suppression of retinoblastoma protein expression.
J. Virol. 73:1894-1901.
Nelson, E. A., J. Christopher-Hennings, T. Drew, G. Wensvoort, J. E. Collins, and
D. A. Benfield. 1993. Differentiation of U.S. and European isolates of porcine
reproductive and respiratory syndrome virus by monoclonal antibodies. J. Clin.
Microbiol. 31:3184-3189.
Nelson, E. A., J. Christopher-Hennings, and D. A. Benfield. 1994. Serum immune
responses to the proteins of porcine reproductive and respiratory syndrome (PRRS) virus.
J. Vet. Diagn. Invest. 6:410-415.

139
Nitschke, M., T. Korte, C. Tielesch, G. Ter-Avetisyan, G. Tunnemann, M. C.
Cardoso, M. Veit, and A. Herrmann. 2008. Equine arteritis virus is delivered to an
acidic compartment of host cells via clathrin-dependent endocytosis. Virology. 377:248-
254.
Onyekaba, C. O., J.T. Harty, C. Even, B.G. Hu, and P.G.W. Plagemann. 1989a.
Persistent infection of mice by lactate dehydrogenase elevating virus: effects of
immunosuppression on virus replication and antiviral immune responses. Virus Res. 14:
297-315.
Onyekaba, C. O., J. T. Harty, and P. G. Plagemann. 1989b. Extensive cytocidal
replication of lactate dehydrogenase-elevating virus in cultured peritoneal macrophages
from 1-2-week-old mice. Virus Res. 14:327-338.
Paine, P. L., L. C. Moore, and S. B. Horowitz. 1975. Nuclear envelope permeability.
Nature. 254:109-114.
Palmer, A. E., A. M. Allen, N. M. Tauraso, and A. Shelokov. 1968. Simian
hemorrhagic fever. I. Clinical and epizootiologic aspects of an outbreak among
quarantined monkeys. Am. J. Trop. Med. Hyg. 17:404-412.
Paschal, B. M. and L. Gerace. 1995. Identification of NTF2, a cytosolic factor for
nuclear import that interacts with nuclear pore complex protein p62. J. Cell Biol.
129:925-937.
Pei, Y., D. C. Hodgins, C. Lee, J. G. Calvert, S. K. Welch, R. Jolie, M. Keith, and D.
Yoo. 2008. Functional mapping of the porcine reproductive and respiratory syndrome
virus capsid protein nuclear localization signal and its pathogenic association. Virus Res.
135:107-114.
Peranen, J., M. Rikkonen, P. Liljestrom, and L. Kaariainen. 1990. Nuclear
localization of Semliki Forest virus-specific nonstructural protein nsP2. J. Virol.
64:1888-1896.
Phoolcharoen, W. and D. R. Smith. 2004. Internalization of the dengue virus is cell
cycle modulated in HepG2, but not Vero cells. J. Med. Virol. 74:434-441.
Pirzadeh B. and S. Dea. 1997. Monoclonal antibodies to the ORF5 product of porcine
reproductive and respiratory syndrome virus define linear neutralizing determinants. J.
Gen. Virol. 78: 1867–1873.
Plagemann, P. G. and V. Moennig. 1992. Lactate dehydrogenase-elevating virus,
equine arteritis virus, and simian hemorrhagic fever virus: a new group of positive-strand
RNA viruses. Adv. Virus Res. 41:99-192.

140
Plagemann, P. G. W., J.T. Harty, and C. Even. 1992. Mode of neutralization of lactate
dehydrogenase-elevating virus by polyclonal and monoclonal antibodies. Arch. Virol.
123: 89-100.
Plagemann, P. G., R. R. Rowland, C. Even, and K. S. Faaberg. 1995. Lactate
dehydrogenase-elevating virus: an ideal persistent virus? Springer Semin. Immunopathol.
17:167-186.
Poggioli, G. J., C. Keefer, J. L. Connolly, T. S. Dermody, and K. L. Tyler. 2000.
Reovirus-induced G(2)/M cell cycle arrest requires sigma1s and occurs in the absence of
apoptosis. J. Virol. 74:9562-9570.
Pol, J. M., F. Wagenaar, and J.E.G. Reus. 1997. Comparative morphogenesis of three
PRRS virus strains. Vet. Microbiol. 55: 203-208.
Poon, B., K. Grovit-Ferbas, S. A. Stewart, and I. S. Chen. 1998. Cell cycle arrest by
Vpr in HIV-1 virions and insensitivity to antiretroviral agents. Science. 281:266-269.
Qu, Q., H. Sawa, T. Suzuki, S. Semba, C. Henmi, Y. Okada, M. Tsuda, S. Tanaka,
W. J. Atwood, and K. Nagashima. 2004. Nuclear entry mechanism of the human
polyomavirus JC virus-like particle: role of importins and the nuclear pore complex. J.
Biol. Chem. 279:27735-27742.
Reed, M. L., B. K. Dove, R. M. Jackson, R. Collins, G. Brooks, and J. A. Hiscox.
2006. Delineation and modelling of nucleolar retention signal in the coronavirus
nucleocapsid protein. Traffic. 7:833-848.
Rikkonen, M., J. Peranen, and L. Kaariainen. 1992. Nuclear and nucleolar targeting
signals of Semliki Forest virus nonstructural protein nsP2. Virology. 189:462-473.
Riley, V., F. Lilly, E. Huerto, and D. Bardell. 1960. Transmissible Agent Associated
with 26 Types of Experimental Mouse Neoplasms. Science. 132:545-547.
Robbins, J., S. M. Dilworth, R. A. Laskey, and C. Dingwall. 1991. Two
interdependent basic domains in nucleoplasmin nuclear targeting sequence: identification
of a class of bipartite nuclear targeting sequence. Cell. 64:615-623.
Rodriguez, M. J., J. Sarraseca, J. Garcia, A. Sanz, J. Plana-Duran, and J. Ignacio
Casal. 1997. Epitope mapping of the nucleocapsid protein of European and North
American isolates of porcine reproductive and respiratory syndrome virus. J. Gen. Virol.
78:2269-2278.
Rossow, K. D. 1998. Porcine reproductive and respiratory syndrome. Vet. Pathol. 35:1-
20.

141
Rowland, R. R., C. Even, G. W. Anderson, Z. Chen, B. Hu, and P. G. Plagemann.
1994. Neonatal infection of mice with lactate dehydrogenase-elevating virus results in
suppression of humoral antiviral immune response but does not alter the course of
viraemia or the polyclonal activation of B cells and immune complex formation. J. Gen.
Virol. 75:1071-1081.
Rowland, R. R., R. Kervin, C. Kuckleburg, A. Sperlich, and D. A. Benfield. 1999.
The localization of porcine reproductive and respiratory syndrome virus nucleocapsid
protein to the nucleolus of infected cells and identification of a potential nucleolar
localization signal sequence. Virus Res. 64:1-12.
Rowland, R. R., P. Schneider, Y. Fang, S. Wootton, D. Yoo, and D. A. Benfield.
2003. Peptide domains involved in the localization of the porcine reproductive and
respiratory syndrome virus nucleocapsid protein to the nucleolus. Virology. 316:135-145.
Rowland, R. R. and D. Yoo. 2003. Nucleolar-cytoplasmic shuttling of PRRSV
nucleocapsid protein: a simple case of molecular mimicry or the complex regulation by
nuclear import, nucleolar localization and nuclear export signal sequences. Virus Res.
95:23-33.
Rowland, R. R., V. Chauhan, Y. Fang, A. Pekosz, M. Kerrigan, and M. D. Burton.
2005. Intracellular localization of the severe acute respiratory syndrome coronavirus
nucleocapsid protein: absence of nucleolar accumulation during infection and after
expression as a recombinant protein in vero cells. J. Virol. 79:11507-11512.
Rowson, K. E. and B. W. Mahy. 1985. Lactate dehydrogenase-elevating virus. J. Gen.
Virol. 66 ( Pt 11):2297-2312.
Sagripanti, J. L. 1984. Studies on simian hemorrhagic fever virus nucleocapsids.
Arch.Virol. 82: 49-59.
Sagripanti, J. L., Zandomeni, R. O. & Weinmann, R. 1986. The cap structure of
simian hemorrhagic fever virion RNA. Virology. 151: 146-150.
Sambrook, J., and D. W. Russell. 2001. Molecular cloning: a laboratory manual, 3rd ed.
Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
Schafer, K. A. 1998. The cell cycle: a review. Vet. Pathol. 35:461-478.
Schwemmle, M., C. Jehle, T. Shoemaker, and W. I. Lipkin. 1999. Characterization of
the major nuclear localization signal of the Borna disease virus phosphoprotein. J. Gen.
Virol. 80:97-100.
Shaw, P. J. and E. G. Jordan. 1995. The nucleolus. Annu. Rev. Cell Dev. Biol. 11:93-
121.

142
Shanmukhappa, K., J.K. Kim, and S. Kapil, S. 2007. Role of CD151, a tetraspanin, in
porcine reproductive and respiratory syndrome virus infection. Virol. J. 4: 62.
Sirri, V., P. Roussel, M. C. Gendron, and D. Hernandez-Verdun. 1997. Amount of
the two major Ag-NOR proteins, nucleolin, and protein B23 is cell-cycle dependent.
Cytometry. 28:147-156.
Sirri, V., S. Urcuqui-Inchima, P. Roussel, and D. Hernandez-Verdun. 2008.
Nucleolus: the fascinating nuclear body. Histochem. Cell Biol. 129:13-31.
Snijder, E. J. and M.C. Horzinek. 1993. Toroviruses: replication, evolution and
comparison with other members of the coronavirus-like superfamily. J Gen. Virol. 74:
2305-2316.
Snijder, E. J. and J. J. Meulenberg. 1998. The molecular biology of arteriviruses. J.
Gen. Virol. 79:961-979.
Snijder, E. J., J. C. Dobbe, and W. J. Spaan. 2003. Heterodimerization of the two
major envelope proteins is essential for arterivirus infectivity. J. Virol. 77:97-104.
Snijder, E. J., and W. J. M. Spaan. 2007. Arteriviruses. In: Fields Virology, 5th
Edition
(D.M. Knipe, P.M. Howley, D.E. Griffin, R.A. Lamb, M.A. Martin, B. Roizman, and
S.E. Straus, eds.), pp. 1338-1355. Lippincott, Williams, and Wilkins, Philadelphia.
Song, C., R. Lu, D. Bienzle, H. C. Liu, and D. Yoo. 2009. Interaction of the porcine
reproductive and respiratory syndrome virus nucleocapsid protein with the inhibitor of
MyoD family-a domain-containing protein. Biol. Chem. 390:215-223.
Spaan, W., H. Delius, M. Skinner, J. Armstrong, P. Rottier, S. Smeekens, B. A. van
der Zeijst, and S. G. Siddell. 1983. Coronavirus mRNA synthesis involves fusion of
non-contiguous sequences. EMBO J. 2:1839-1844.
Stewart M. 2007. Molecular mechanism of the nuclear protein import cycle. Nat. Rev.
Mol. Cell Biol. 8: 195-208.
Stokes, H. L., A. J. Easton, and A. C. Marriott. 2003. Chimeric pneumovirus
nucleocapsid (N) proteins allow identification of amino acids essential for the function of
the respiratory syncytial virus N protein. J. Gen. Virol. 84:2679-2683.
Stueckemann, J. A., M. Holth, W. J. Swart, K. Kowalchyk, M. S. Smith, A. J.
Wolstenholme, W. A. Cafruny, and P. G. Plagemann. 1982. Replication of lactate
dehydrogenase-elevating virus in macrophages. 2. Mechanism of persistent infection in
mice and cell culture. J. Gen. Virol. 59:263-272.
Suzuki, R., S. Sakamoto, T. Tsutsumi, A. Rikimaru, K. Tanaka, T. Shimoike, K.
Moriishi, T. Iwasaki, K. Mizumoto, Y. Matsuura, T. Miyamura, and T. Suzuki.

143
2005. Molecular determinants for subcellular localization of hepatitis C virus core
protein. J. Virol. 79:1271-1281.
Tauraso, N. M., A. Shelokov, A. E. Palmer, and A. M. Allen. 1968. Simian
hemorrhagic fever. 3. Isolation and characterization of a viral agent. Am. J. Trop. Med.
Hyg. 17:422-431.
Tijms, M. A., L. C. van Dinten, A. E. Gorbalenya, and E. J. Snijder. 2001. A zinc
finger-containing papain-like protease couples subgenomic mRNA synthesis to genome
translation in a positive-stranded RNA virus. Proc. Natl. Acad. Sci. U. S. A. 98:1889-
1894.
Tijms, M. A., Y. van der Meer, and E. J. Snijder. 2002. Nuclear localization of non-
structural protein 1 and nucleocapsid protein of equine arteritis virus. J. Gen. Virol.
83:795-800.
Timani, K. A., Q. Liao, L. Ye, Y. Zeng, J. Liu, Y. Zheng, L. Ye, X. Yang, K.
Lingbao, J. Gao, and Y. Zhu. 2005. Nuclear/nucleolar localization properties of C-
terminal nucleocapsid protein of SARS coronavirus. Virus Res. 114:23-34.
Timoney, P. J. and W. H. McCollum. 1993. Equine viral arteritis. Vet. Clin. North Am.
Equine Pract. 9:295-309.
Trousdale, M. D., D. W. Trent, and A. Shelokov. 1975. Simian hemorrhagic fever
virus: a new togavirus. Proc. Soc. Exp. Biol. Med. 150:707-711.
Tsuda, Y., Y. Mori, T. Abe, T. Yamashita, T. Okamoto, T. Ichimura, K. Moriishi,
and Y. Matsuura. 2006. Nucleolar protein B23 interacts with Japanese encephalitis virus
core protein and participates in viral replication. Microbiol. Immunol. 50:225-234.
Uchil, P. D., A. V. Kumar, and V. Satchidanandam. 2006. Nuclear localization of
flavivirus RNA synthesis in infected cells. J. Virol. 80:5451-5464.
van Berlo, M. F., M.C. Horzinek, and B.A.M. van der Zeijst. 1980. Temperature-
sensitive mutants of equine arteritis virus. . J. Gen. Virol. 49: 97-104.
van Berlo, M. F., M.C. Horzinek, and B.A.M. van der Zeijst. 1982. Equine arteritis
virus-infected cells contain six polyadenylated virus specific RNAs. Virology.118: 45-52.
van Berlo, M. F., P.J.M. Rottier,M.C. Horzinek, and B.A.M. van der Zeijst. 1986.
Intracellular equine arteritis virus (EAV)-specific RNAs contain common sequences.
Virology. 152: 492-496.
van den Born, E., C. C. Posthuma, A. P. Gultyaev, and E. J. Snijder. 2005.
Discontinuous subgenomic RNA synthesis in arteriviruses is guided by an RNA hairpin
structure located in the genomic leader region. J. Virol. 79:6312-6324.

144
van den Broek, M. F., R. Sporri, C. Even, P. G. Plagemann, E. Hanseler, H.
Hengartner, and R. M. Zinkernagel. 1997. Lactate dehydrogenase-elevating virus
(LDV): lifelong coexistence of virus and LDV-specific immunity. J. Immunol. 159:1585-
1588.
Vanderheijden, N., P. Delputte, H. Nauwynck, and M. Pensaert. 2001. Effects of
heparin on the entry of porcine reproductive and respiratory syndrome virus into alveolar
macrophages. Adv Exp Med Biol. 494: 683–689.
Vanderheijden, N., P. L. Delputte, H. W. Favoreel, J. Vandekerckhove, J. Van
Damme, P. A. van Woensel, and H. J. Nauwynck. 2003. Involvement of sialoadhesin
in entry of porcine reproductive and respiratory syndrome virus into porcine alveolar
macrophages. J. Virol. 77:8207-8215.
van Dinten, L. C., J. A. den Boon, A. L. Wassenaar, W. J. Spaan, and E. J. Snijder.
1997. An infectious arterivirus cDNA clone: identification of a replicase point mutation
that abolishes discontinuous mRNA transcription. Proc. Natl. Acad. Sci. U. S. A. 94:991-
996.
Van Gorp, H., W. Van Breedam, P. L. Delputte, and H. J. Nauwynck. 2008.
Sialoadhesin and CD163 join forces during entry of the porcine reproductive and
respiratory syndrome virus. J. Gen. Virol. 89:2943-2953.
van Nieuwstadt, A. P., J.J.M. Meulenberg, A. van Essen-Zanbergen, A. Petersen-
den Besten, R.J. Bende, R.J.M. Moormann, and G. Wensvoort. 1996. Proteins
encoded by open reading frames 3 and 4 of the genome of Lelystad virus (Arteriviridae)
are structural proteins of the virion. J. Virol. 70: 4767-4772.
Verheije, M. H., R. C. Olsthoorn, M. V. Kroese, P. J. Rottier, and J. J. Meulenberg.
2002. Kissing interaction between 3' noncoding and coding sequences is essential for
porcine arterivirus RNA replication. J. Virol. 76:1521-1526.
Waggoner, S. and Sarnow, P. 1998. Viral ribonucleoprotein complex formation and
nucleolar–cytoplasmic relocalization of nucleolin in poliovirus-infected cells. J. Virol.
72: 6699–6709 .
Wang, S. H., W. J. Syu, K. J. Huang, H. Y. Lei, C. W. Yao, C. C. King, and S. T. Hu.
2002. Intracellular localization and determination of a nuclear localization signal of the
core protein of dengue virus. J. Gen. Virol. 83:3093-3102.
Weber, F., G. Kochs, S. Gruber, and O. Haller. 1998. A classical bipartite nuclear
localization signal on Thogoto and influenza A virus nucleoproteins. Virology. 250:9-18.
Weiland, E., M. Wieczorek-Krohmer, D. Kohl, K. K. Conzelmann, and F. Weiland.
1999. Monoclonal antibodies to the GP5 of porcine reproductive and respiratory

145
syndrome virus are more effective in virus neutralization than monoclonal antibodies to
the GP4. Vet. Microbiol. 66:171-186.
Wensvoort, G. 1993. Lelystad virus and the porcine epidemic abortion and respiratory
syndrome. Vet. Res. 24:117-124.
Wilhelmsen, K. C., J. L. Leibowitz, C. W. Bond, and J. A. Robb. 1981. The
replication of murine coronaviruses in enucleated cells. Virology. 110:225-230.
Wieringa, R., A. A. de Vries, and P. J. Rottier. 2003. Formation of disulfide-linked
complexes between the three minor envelope glycoproteins (GP2b, GP3, and GP4) of
equine arteritis virus. J. Virol. 77:6216-6226.
Wieringa, R., A. A. de Vries, J. van der Meulen, G. J. Godeke, J. J. Onderwater, H.
van Tol, H. K. Koerten, A. M. Mommaas, E. J. Snijder, and P. J. Rottier. 2004.
Structural protein requirements in equine arteritis virus assembly. J. Virol. 78:13019-
13027.
Wissink, E. H., M. V. Kroese, H. A. van Wijk, F. A. Rijsewijk, J. J. Meulenberg, and
P. J. Rottier. 2005. Envelope protein requirements for the assembly of infectious virions
of porcine reproductive and respiratory syndrome virus. J. Virol. 79:12495-12506.
Wood, O., N. Tauraso, and H. Liebhaber. 1970. Electron microscopic study of tissue
cultures infected with simian haemorrhagic fever virus. J. Gen. Virol. 7: 129-136.
Wootton, S., D. Yoo, and D. Rogan. 2000. Full-length sequence of a Canadian porcine
reproductive and respiratory syndrome virus (PRRSV) isolate. Arch. Virol. 145:2297-
2323.
Wootton, S. K., R. R. Rowland, and D. Yoo. 2002. Phosphorylation of the porcine
reproductive and respiratory syndrome virus nucleocapsid protein. J. Virol. 76:10569-
10576.
Wurm, T., H. Chen, T. Hodgson, P. Britton, G. Brooks, and J. A. Hiscox. 2001.
Localization to the nucleolus is a common feature of coronavirus nucleoproteins, and the
protein may disrupt host cell division. J. Virol. 75:9345-9356.
Xiao, Z., L. Batista, S. Dee, P. Halbur, and M. P. Murtaugh. 2004. The level of virus-
specific T-cell and macrophage recruitment in porcine reproductive and respiratory
syndrome virus infection in pigs is independent of virus load. J. Virol. 78:5923-5933.
Yang, X. J., J. Liu, L. Ye, Q. J. Liao, J. G. Wu, J. R. Gao, Y. L. She, Z. H. Wu, and
L. B. Ye. 2006. HCV NS2 protein inhibits cell proliferation and induces cell cycle arrest
in the S-phase in mammalian cells through down-regulation of cyclin A expression. Virus
Res. 121:134-143.

146
Yasui, K., T. Wakita, K. Tsukiyama-Kohara, S. I. Funahashi, M. Ichikawa, T.
Kajita, D. Moradpour, J. R. Wands, and M. Kohara. 1998. The native form and
maturation process of hepatitis C virus core protein. J. Virol. 72:6048-6055.
Yoo, D., S. K. Wootton, G. Li, C. Song, and R. R. Rowland. 2003. Colocalization and
interaction of the porcine arterivirus nucleocapsid protein with the small nucleolar RNA-
associated protein fibrillarin. J. Virol. 77:12173-12183.
Yoon, K. J., L. L. Wu, J. J. Zimmerman, H. T. Hill, and K. B. Platt. 1996. Antibody-
dependent enhancement (ADE) of porcine reproductive and respiratory syndrome virus
(PRRSV) infection in pigs. Viral Immunol. 9:51-63.
You, J., B. K. Dove, L. Enjuanes, M. L. DeDiego, E. Alvarez, G. Howell, P. Heinen,
M. Zambon, and J. A. Hiscox. 2005. Subcellular localization of the severe acute
respiratory syndrome coronavirus nucleocapsid protein. J. Gen. Virol. 86:3303-3310.
You, J. H., M. L. Reed, and J. A. Hiscox. 2007. Trafficking motifs in the SARS-
coronavirus nucleocapsid protein. Biochem. Biophys. Res. Commun. 358:1015-1020.
You, J. H., G. Howell, A. K. Pattnaik, F. A. Osorio, and J. A. Hiscox. 2008. A model
for the dynamic nuclear/nucleolar/cytoplasmic trafficking of the porcine reproductive and
respiratory syndrome virus (PRRSV) nucleocapsid protein based on live cell imaging.
Virology. 378:34-47.
Young, T. M., Q. Wang, T. Pe'ery, and M. B. Mathews. 2003. The human I-mfa
domain-containing protein, HIC, interacts with cyclin T1 and modulates P-TEFb-
dependent transcription. Mol. Cell. Biol. 23:6373-6384.
Yuan, X., J. Wu, Y. Shan, Z. Yao, B. Dong, B. Chen, Z. Zhao, S. Wang, J. Chen, and
Y. Cong. 2006. SARS coronavirus 7a protein blocks cell cycle progression at G0/G1
phase via the cyclin D3/pRb pathway. Virology. 346:74-85.
Zeegers, J. J., B. A. Van der Zeijst, and M. C. Horzinek. 1976. The structural proteins
of equine arteritis virus. Virology. 73:200-205.
Zeng, L., E.K. Godeny, S.L. Methven, and M.A. Brinton. 1995. Analysis of simian
hemorrhagic fever virus (SHFV) subgenomic RNAs, junction sequences, and 5’ leader.
Virology. 207: 543-548.
Zhao, R. Y. and R. T. Elder. 2005. Viral infections and cell cycle G2/M regulation. Cell
Res. 15:143-149.
Zimmerman, J. J., K. J. Yoon, R. W. Wills, and S. L. Swenson. 1997. General
overview of PRRSV: a perspective from the United States. Vet. Microbiol. 55:187-196.