“some like it shallower” – p-type doping in sic
TRANSCRIPT

phys. stat. sol. (b) 235, No. 1, 139–145 (2003) / DOI 10.1002/pssb.200301522
© 2003 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim 0370-1972/03/23501-0139 $ 17.50+.50/0
“Some like it shallower” – p-type doping in SiC
Peter Deák*1, Bálint Aradi1, Adam Gali1, and Uwe Gerstmann2
1 Department of Atomic Physics, Budapest University of Technology and Economics, Budafoki út 8, Budapest, 1111 Hungary
2 Theoretical Physics, University of Paderborn, 33100 Paderborn, Germany
Received 24 July 2002, accepted 27 July 2002 Published online 30 December 2002
PACS 71.55.Ht, 71.55.Mb
The usual p-type dopants of SiC, B, and Al, do not produce really shallow levels. In fact, boron can give rise to a secondary very deep acceptor level as well. The picture is complicated by hydrogen which is rea-dily incorporated during in-growth doping into p-type material and can passivate both dopants but to a different degree. First principle calculations are reported regarding the interaction of hydrogen with B and Al in SiC. The results explain why hydrogen is incorporated in much higher amounts into B-doped than into Al-doped samples, and also reveal the influences of hydrogen on boron to produce the shallower acceptor. It will be shown that hydrogen incorporation during growth does not influence Al. Finally an Al–N–Al complex is proposed as a shallower acceptor in SiC.
Introduction Silicon carbide is a wide band gap semiconductor intended for use in high power, high temperature electronics. This fact increases somewhat the tolerance for what can be termed a shallow level, still the traditional p-type dopants, boron and aluminium, seem to behave rather unsatisfactorily in SiC. Boron can produce a deep acceptor state about 0.55 eV above the valence band, as well as a shal-lower one at 0.30 eV [1], and it is not clear why the former appears. Aluminium gives only one level at about 0.21 eV [2], which is still too high for planned high-frequency devices operating at moderate tem-peratures. (Note that SiC comes in many different polytypes, the most important ones being the cubic 3C and the hexagonal 4H and 6H. The numbers here denote the number of inequivalent Si–C bilayers stacked on each other within one period along the cubic [111] or the hexagonal [0001] direction. Unless otherwise noted, values quoted refer to the 4H polytype.) Doping of SiC is complicated by two facts. For one, in a compound semiconductor dopants can choose between the two sublattices where they can behave rather differently. Second, the strong bonds and tight structure of SiC precludes diffusion doping. Therefore, doping may be achieved in growth during chemical vapour deposition (CVD) of homoepitaxial layers, or by implantation. The former pro-cedure introduces hydrogen impurities, the latter causes intrinsic point defects – in both cases the dopants may form complexes. As for the site selection, the behaviour of Al is relatively predictable: it prefers the silicon site (AlSi), where it fits in more conveniently, and produces the “shallow” acceptor level mentioned above. Boron, on the other hand, has been reported to occupy both, the silicon and the carbon site, alas, with preference for the Si site [3]. If SiC is grown under Si-rich conditions in CVD (offer of empty C sites for B), the boron incorporation is substantially lower than in the case of C-rich conditions (offer of empty Si sites). The latter (C-rich growth) produces the “shallow” acceptors mentioned above while the “deep” ones were observed in the former (Si-rich growth). Therefore, although no conclusive proof has yet been
* Corresponding author: e-mail: [email protected], Phone: [36]-(1)-463-1393, Fax: [36]-(1)-463-4357

140 Peter Deák et al.: “Some like it shallower” – p-type doping in SiC
shown, BSi is generally associated with the shallow acceptor, and it has been suggested [4] that BC might be the deep acceptor. On the other hand, substantial amounts of the deep boron acceptor could only be produced by implantation [5], so, in addition (based on theoretical calculations [6]) an “isomer” of BC, BSi + SiC, as well as (based on paramagnetic resonance studies [7]) a complex with a carbon vacancy, BSi + VC, were also suggested as possible origins of the “deep” boron center. The latter assignment was, however, not supported by the calculations, while BSi + SiC was predicted considerably higher in energy than BC. Hydrogen can form passive complexes with both kind of p-type dopants in SiC. The measured disso-ciation energy of the B + H and Al + H complexes are, however, rather different, 2.51 and 1.61 eV, res-pectively, indicating a different kind of bonding configuration in the two cases [8]. In contrast to n-type, hydrogen is easily incorporated into p-type SiC during CVD growth, but to a different degree in B- and Al-doped samples. In as-grown material doped with boron, the incorporated hydrogen concentra-tion is proportional to the amount of boron [3, 9]. In contrast, in Al doped samples, the hydrogen concen-tration is about two–three orders of magnitude lower than the dopant concentration [10]. In the follow-ing it will be shown that hydrogen has a crucial effect in making boron a “shallow” acceptor, while it does not influence aluminium. Methods The acceptors of SiC, the incorporation of hydrogen and the complex formation among them has been studied by first principles calculations based on density-functional theory in the local-density approximation (LDA). The FHI96MD code [11] for plane-wave supercell LDA calculations was used with a 36 Ry cut-off and Troullier-Martins norm conserving soft-core pseudopotentials. 3C–SiC and 4H–SiC were modeled by 128 and 96 atom supercells, respectively, using 23 Monkhorst-Pack k-point sets for Brillouin-zone (BZ) summation. The defect geometry was determined by allowing three shells of host atoms to relax around the defect until the forces were less than 0.0005 hartree/bohr. The total energy was corrected for the LDA gap error and for dispersion due to the supercell approach. Details of the method and references are given in Ref. [12]. Hydrogen incorporation excluding complexes with dopants As mentioned already in the introduc-tion, hydrogen can be incorporated into SiC during CVD growth. This is no equilibrium process but under stationary growth condition the assumption of thermal equilibrium between the hydrogen content of the reactive gas mixture (SiH4, C3H8 and B2H6 or Al(CH4)3 in H2 carrier gas) and the growing crystal is acceptable. In typical CVD growth the substrate temperature is between 1400–1700 °C. At these tem-peratures an estimated 1% of the hydrogen is atomic (due to H2 dissociation but also as a product of near surface reactions). The total pressure in CVD is 1 atm, most of it coming from hydrogen. So, assuming 0.01 atm of atomic hydrogen sets the chemical potential of hydrogen to (see Ref. [12])
5 5 2H (H) 8.617386 10 ln(19.314058 )E T pTµ
−
= + × , (1)
where E(H) is the calculated total energy of atomic hydrogen. After calculating the total energy of the supercell containing the defect (in the relevant charge state q), the defect formation energy can be de-fined as
form Si C H Si C H F tot Si C H Si Si C C H H F( , , ; , , , ) ( , , )q qE n n n E E n n n n n n qEµ µ µ µ µ µ= − − − + , (2)
where ni and µi are the number and chemical potential of atoms i in the system, and EF is the Fermi en-ergy (the chemical potential of electrons) in the doped crystal at the temperature of growth. (Vibration contributions to Etot are neglected.) The chemical potentials of C and Si are connected, µSi + µC = bulk
SiCµ , and are limited, on the one hand, by bulk silicon formation (extreme Si-rich conditions, µSi = bulk
Siµ ) and, on the other hand, by bulk graphite formation (extreme C-rich conditions, µC = bulk
Cµ ). Neglecting entropy contributions within the crystal, the concentration of a hydrogen defect in SiC can be obtained as
0 formexpiq
i i
EN N
kT
−=
. (3)

phys. stat. sol. (b) 235, No. 1 (2003) 141
1019 1018 1017 1016 1017 1018 10191012
1014
1016
1018
1013
1015
1017
1019
[p] [n]
+
+
[Hi ] in p(Ha) = 0.01 atm[VC] in p(Ha) = 0.01 atm+
+
Of course, all possible defects involving hydrogen should be considered. In case any of these are elec-trically active (defect level in the gap), the charge state appropriate for the given Fermi energy has to be considered. Since the ionisation of the defect influences it, the position of the Fermi-level has to be de-termined from the charge neutrality condition self-consistently with the defect concentrations, assuming a given concentration of dopants. This is a rather ambitious task, so we applied some restrictions. First, since the formation energy of the important defects turns out to be rather independent of the polytype, we have carried out the calculations in 3C-SiC which has the smallest unit cell (two atoms), so a sufficiently large supercell (4 × 4 × 4) with 128 atoms could be applied. Second, we have excluded interaction between hydrogen and the dopants (i.e. complex formation) at the beginning. The following defects have been considered: interstitial H atom, interstitial dihydrogen complexes, vacancies (both, C and Si) and vacancy–hydrogen complexes contain-ing one and two hydrogen atoms. The calculations [12, 13] resulted in the diagram shown in Fig. 1. As can be seen, the only hydrogen defect appearing in the growing SiC crystal is interstitial atomic hydrogen in the positive charge state. Even that is only incorporated into p-type samples in appreciable amounts – in agreement with experimental observations. The calculated dependence of the concentration of hydrogen on the concentration of the p-type dopants nicely follows the ratio observed in Al-doped samples [10]. Apparently, the neglected complex formation with hydrogen had no effect in the case of aluminium. Since hydrogen is incorporated into boron-doped samples in equal amount to the boron con-centration, it is reasonable to assume that they are incorporated together [14]. The interaction of hydrogen with boron and aluminium At this point we switched to 4H-SiC, and calculated the interaction of interstitial hydrogen with boron and aluminium on the Si site and with boron on the C site [15, 16]. We found that the most sable configuration of H and B or Al is always an electri-cally inactive complex. H always binds to the more electronegative atom (to C in case of BSi + H and AlSi + H, to B in case of BC + H). Near the smaller B atom in BSi + H it remains near the bond-centre (BC) site, while it is forced to go to the antibonding site of the carbon atom (ABC) next to the Al atom in AlSi + H. In BC + H, it is again in the antibonding position (to B) for it is easier to pull boron back into an almost planar configuration with its neighbours than to push back an Si atom from the tetrahedrally co-ordinated lattice position. The binding energy of H+ to the “shallow” acceptors B–
Si and Al–Si have been
calculated to be 1.6 and 0.7 eV for BSi + HBC and AlSi + HAB(C), respectively. The dissociation energy is approximately the sum of the binding energy and the activation energy for H+ diffusion, which has been calculated to be 0.4 eV [17]. This gives values for the dissociation energy of 2.0 and 1.1 eV, to be com-pared to the observed 2.5 and 1.6 eV. The difference is due to the error committed by compensating the charge of the defects by a jellium charge of opposite sign to keep the supercell neutral. This error is often corrected by a Madelung term (see e.g. [4]) which is independent of the sign of the defect charge and in a 128 atom cell is about 0.2 eV. (In our case, i.e. for A + H → A– + H+, the error is committed twice, so a ~0.4 eV correction applies.)
Fig. 1 The calculated concentration [cm–3] of defects in SiC assuming equilibrium with 0.01 atm of atomic hydrogen at 1400 °C as a function of the dopant concentration [cm–3]. Extreme C-rich (Si-rich) conditions have been assumed for p- (n-) type doping.

142 Peter Deák et al.: “Some like it shallower” – p-type doping in SiC
1400 °C 1700 °C
0
-1
+1[eV]
Ef(BSi) - Ef(BSi + H)
Ef(AlSi) - Ef(AlSi + H)
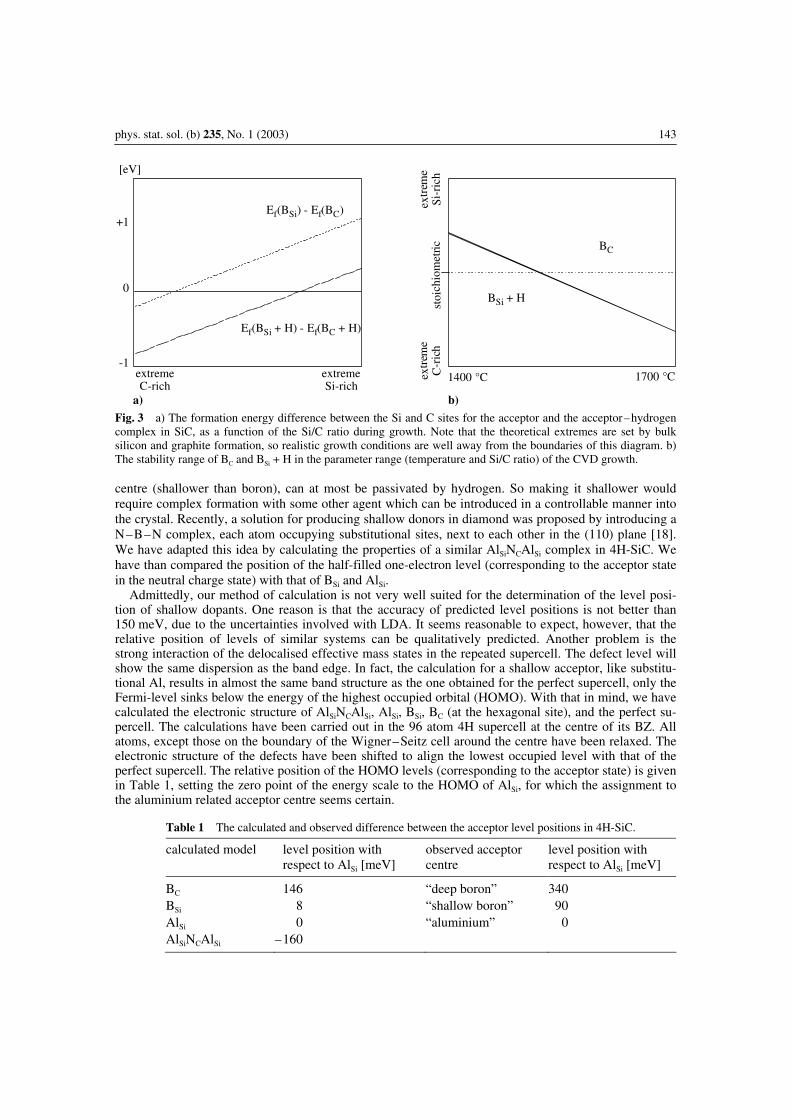
Our results explain the difference in the energy needed for reactivating the hydrogen-passivated ac-ceptor complexes but – by the same token (the stronger interaction of H with BSi) – sheds light onto the higher hydrogen concentration of H in boron doped samples. Let us take into account that in the gas phase the precursor molecules of the dopants dissociate. Using standard heat of formation data the disso-ciation leading to a BH (AlH) molecule requires 1.2 eV (0.7 eV) less energy than dissociation leading to free B (Al) atoms. Therefore, BH and AlH molecules are definitely present in the gas phase. If these molecules are incorporated into the crystal, the acceptor–hydrogen complex may form in the first place. Let us compare the formation energy of the isolated acceptor with that of the acceptor–hydrogen com-plex. For the temperature range of CVD growth, this is shown in Fig. 2. The negative value for Al means that isolated Al is more stable than the AlSi + H complex for any growth temperature, so Al atoms are incorporated as isolated substitutional acceptors, and the amount of incorporated hydrogen depends on the Al concentration only through the doping effect of the latter. The positive value for B, however, indicates that the BSi + H complex is favoured over the isolated B atom, i.e., it is more likely that boron and hydrogen gets incorporated together, in equal amounts. This is in agreement with the observations. How about boron at the C site? Figure 3a shows the formation energy difference between isolated B and the B + H complex at the C as well as at the Si site as a function of the Si/C ratio during growth. As can be seen, for realistic growth conditions, BC and BSi + H are favoured. Comparing their forma-tion energy in the 1400–1700 °C temperature range in Fig. 3b, it can be seen that except of strongly Si-rich conditions or very high temperatures, BSi + H is more likely, i.e., boron incorporation onto the Si site, together with hydrogen is preferred. How to make it shallower The results outlined above explain not only the higher amount of hydrogen in boron doped samples but also the observation that the attainable boron concentration is lower under Si-rich conditions. Accepting the proposal of Ref. [4] for assigning the deep boron acceptor to BC, it becomes clear why the concentration of deep boron acceptors was so low in the CVD layers grown even in Si-rich conditions at 1350 °C in Ref. [1], while it could be really high in implanted samples [5]. It is the presence of hydrogen in the as grown epilayers which make the boron atom produce the shallower acceptor! If hydrogen is present, boron is incorporated together with a hydrogen atom, preferentially onto the Si site. Since the binding energy of the complex is rather low, this passive complex readily dissoci-ates during further growth, leaving the shallower acceptor, BSi behind. Change of the site would require at least 4 eV! In case of implantation, additional boron is introduced without hydrogen in its neighbour-hood. Therefore, the following heat treatment activates boron as deep acceptor on the C site, i.e., as BC. So the recipe of making boron shallower is to have hydrogen around! Still, even the shallower boron acceptor is not really very shallow. On the other hand, aluminium, always producing just one acceptor
Fig. 2 The formation energy difference be-tween the acceptor and the acceptor–hydrogen complex in SiC, as a function of temperature, assuming 0.01 atm atomic hydrogen during CVD growth.
phys. stat. sol. (b) 235, No. 1 (2003) 143
+1
0
-1
[eV]
extremeC-rich
extremeSi-rich
Ef(BSi + H) - Ef(BC + H)
Ef(BSi) - Ef(BC)
a) b)
Fig. 3 a) The formation energy difference between the Si and C sites for the acceptor and the acceptor–hydrogen complex in SiC, as a function of the Si/C ratio during growth. Note that the theoretical extremes are set by bulk silicon and graphite formation, so realistic growth conditions are well away from the boundaries of this diagram. b) The stability range of BC and BSi + H in the parameter range (temperature and Si/C ratio) of the CVD growth. centre (shallower than boron), can at most be passivated by hydrogen. So making it shallower would require complex formation with some other agent which can be introduced in a controllable manner into the crystal. Recently, a solution for producing shallow donors in diamond was proposed by introducing a N–B–N complex, each atom occupying substitutional sites, next to each other in the (110) plane [18]. We have adapted this idea by calculating the properties of a similar AlSiNCAlSi complex in 4H-SiC. We have than compared the position of the half-filled one-electron level (corresponding to the acceptor state in the neutral charge state) with that of BSi and AlSi. Admittedly, our method of calculation is not very well suited for the determination of the level posi-tion of shallow dopants. One reason is that the accuracy of predicted level positions is not better than 150 meV, due to the uncertainties involved with LDA. It seems reasonable to expect, however, that the relative position of levels of similar systems can be qualitatively predicted. Another problem is the strong interaction of the delocalised effective mass states in the repeated supercell. The defect level will show the same dispersion as the band edge. In fact, the calculation for a shallow acceptor, like substitu-tional Al, results in almost the same band structure as the one obtained for the perfect supercell, only the Fermi-level sinks below the energy of the highest occupied orbital (HOMO). With that in mind, we have calculated the electronic structure of AlSiNCAlSi, AlSi, BSi, BC (at the hexagonal site), and the perfect su-percell. The calculations have been carried out in the 96 atom 4H supercell at the centre of its BZ. All atoms, except those on the boundary of the Wigner–Seitz cell around the centre have been relaxed. The electronic structure of the defects have been shifted to align the lowest occupied level with that of the perfect supercell. The relative position of the HOMO levels (corresponding to the acceptor state) is given in Table 1, setting the zero point of the energy scale to the HOMO of AlSi, for which the assignment to the aluminium related acceptor centre seems certain.
Table 1 The calculated and observed difference between the acceptor level positions in 4H-SiC.
calculated model –level position with –respect to AlSi [meV]
observed acceptor centre
level position with respect to AlSi [meV]
BC –146 “deep boron” 340 BSi – 8 “shallow boron” 90 AlSi – 0 “aluminium” 0 AlSiNCAlSi –160
1400 °C 1700 °Cextr
eme
C-r
ich
extr
eme
Si-r
ich
BSi + H
BC
stoi
chio
met
ric

144 Peter Deák et al.: “Some like it shallower” – p-type doping in SiC
4
2
0
--2
--4
BC
--4 --2 0 2 4
4
2
0
--2
--4
AlSi
--4 --2 0 2 4
4
2
0
--2
--4
BSi
--4 --2 0 2 4
4
2
0
--2
--4
AlNAl
--4 --2 0 2 4
Fig. 4 The spatial distribution of the acceptor wave function in the negative charge state of BC, BSi, AlSi, and AlSiNCAlSi. (The centres of the p-like distributions mark the carbon sublattice.)
The comparison with the observed difference among the acceptor level positions shows that – al-though with considerable error – the sequence is at least qualitatively reproduced. By the force of this comparison we expect that the AlSiNCAlSi complex will provide a shallower level than the others. A look at the spatial distribution of the acceptor wave functions in the negative charge state (Fig. 4) strengthens the confidence in this prediction. Generally, the more shallow the defect level, the less localized is the orbital. This trend can be followed on the acceptor states (composed of p-orbitals on the carbon sublat-tice) of SiC, from BC (rather strongly localized on the boron), through BSi and AlSi (decreasingly local-ized on a carbon atom next to the acceptor) to AlSiNCAlSi, for which the acceptor state is the least local-ized. According to the calculation, the (AlSiNCAlSi)
– complex is at least 0.7 eV lower in energy than two isolated Al–
Si and an NC+. Regarding the error committed in calculating the energy of charged defects, the
energy gain on complex formation can be estimated to be about 1.4 eV. Since Al and N are charged oppositely, this complex may easily form after coimplantation. Also, it could be produced in growth, using the Al2N molecule as precursor for doping. Why should the Al–N–Al complex give rise to a more shallow level than the isolated acceptor at-oms? The answer probably lies in geometrical factors. BSi is not really a shallow acceptor – not only because of the position of its level but also because the corresponding orbital is not an effective mass (EMT) state. In equilibrium at 0 K, the relatively small boron atom moves off-centre, binding stronger to three of its carbon neighbours. The acceptor state is, in fact, the dangling bond of the fourth carbon. (At finite temperature motional averaging occurs.) The carbon atoms reconstruct accordingly. Aluminium is larger, has less room to move and is forced to stay in a fourfold coordinated position pulling the carbon

phys. stat. sol. (b) 235, No. 1 (2003) 145
neighbours inwards. It gives rise to an EMT state. Generally speaking, the acceptor level is shallowest when the lattice is least disturbed, i.e., the central cell correction to EMT is smallest. In the three cases considered, the average relaxation of the carbon neighbours from their ideal position is 6.3, 4.6, and 3.5% for BSi, AlSi, and AlSiNCAlSi, respectively. The reason is that the nitrogen atom (“larger” than car-bon) compensates for the Al atoms (which are “smaller” than silicon). Accordingly, this complex gives rise to a geometrically least disturbed lattice and to an EMT state with small central cell correction.
Acknowledgment The authors thank W. J. Choyke, R. P. Devaty, E. Janzén, and N. T. Son for helpful discus-sions. Support by the Hungarian OTKA T32174 grant as well as the bilateral program between the Hungarian, Swedish and German Academies are appreciated. This work was also supported by grants from the Pittsburgh Su-percomputer Center (PHY970006P) and the Swedish National Supercomputer Center (2002002).
References [1] S. G. Sridhara, L. L. Clemen, R. P. Devaty, W. J. Choyke, D. J. Larkin, H. S. Kong, T. Troffer, and G. Pensl,
J. Appl. Phys. 83, 7909 (1998). [2] R. P. Devaty and W. J. Choyke, phys. stat. sol. (a) 162, 21 (1997). [3] D. J. Larkin, phys. stat. sol. (b) 202, 305 (1997). [4] M. Bockstedte, A. Mattausch, and O. Pankratov, Mater. Sci. Forum 353, 447 (2001). [5] M. Gong, C. V. Reddy, C. D. Beling, S. Fung, G. Brauer, H. Wirth, and W. Skorupa, Appl. Phys. Lett. 72,
2739 (1998). [6] B. Aradi, A. Gali, P. Deák, E. Rauls, Th. Frauenheim, and N. T. Son, Mater. Sci. Forum 353–356, 455 (2001). [7] A. Duijn-Arnold, T. Ikoma, O. G. Poluektov, P. G. Baranov, E. N. Mokhov, and J. Schmidt, Phys. Rev. B 57,
1607 (1998). [8] M. S. Janson, A. Hallén, M. K. Linnarsson, and B. G. Svensson, Phys. Rev. B 64, 195202 (2001). [9] A. Henry, B. Magnusson, M. K. Linnarsson, A. Ellison, M. Syväjärvi, R. Yakimova, and E. Janzén, Mater. Sci.
Forum 353, 373 (2001). [10] M. K. Linnarsson, U. Forsberg, M. S. Janson, E. Janzén, and B. G. Svensson, Mater. Sci. Forum 389–393, 565
(2002). [11] M. Bockstedte, A. Kley, J. Neugebauer, and M. Scheffler, Comput. Phys. Commun. 107, 187 (1997). [12] B. Aradi, A. Gali, P. Deák, J. E. Lowther, N. T. Son, E. Janzén, and W. J. Choyke, Phys. Rev. B 63, 245202
(2001). [13] P. Deák, A. Gali, B. Aradi, Mater. Sci. Forum 353–356, 421 (2001). [14] D. J. Larkin, phys. stat. sol. (b) 202, 305 (1997). [15] B. Aradi, P. Deák, N. T. Son, E. Janzén, R. P. Devaty and W. J. Choyke, Appl. Phys. Lett. 79, 2746 (2001). [16] P. Deák, B. Aradi, and A. Gali, J. Phys.: Condens. Matter 13, 9019 (2001). [17] B. Aradi, A. Gali, P. Deák, N. T. Son, and E. Janzén, to be published. [18] H. Katayama-Yoshida, T. Nishimatsu, T. Yamamoto, and N. Orita, J. Phys.: Condens. Matter 13, 8901 (2001).