smartstarttm your clinical drug drug devl... · manual/mop completed ... • glp and gcp is a must...
TRANSCRIPT

SMARTSTARTTM YOUR CLINICAL DRUG DEVELOPMENT

CLINICAL CONSULTING SERVICES OVERVIEW
Founded: 2005, Private LLC Employees: 80+
Our focus is getting needed drugs and devices to patients through a unique approach to:
• Strategic regulatory and clinical services from discovery through commercialization
• Clinical trial recruitment and retention • Patient support technology platform with
front-end automation and back-end data analytics

A TRUSTED PARTNER in LIFE SCIENCES Fully integrated pre-clinical, clinical, regulatory and site management service provider Founded 2005, with offices in Baltimore, MD, San Francisco, CA, London, UK, and Hyderabad, India Strategic, multi-faceted team: 80+ FTEs Global experience in 40+ countries and 20+ therapeutic areas 500+ new drugs, biologics and device products successfully commercialized

FDA is the Federal agency that is required by law to review and approve all new drugs in the US 1906: FDA Act prohibited mislabeling but did not require pre-market approval of drugs or devices 1938: Food, Drugs and Cosmetics Act required submission of evidence of a drug’s safety before it could be marketed
HISTORY of FEDERAL OVERSIGHT of the DRUG DEVELOPMENT PROCESS

Reviews and evaluates new drugs based on evidence presented from the clinical research studies performed by the sponsor Regulate interstate commerce Center for Drug Evaluation and Research (CDER) is the largest of the FDA’s five centers and is responsible for prescription and over-the-counter (OTC) drug safety and efficacy Center for Biologic Evaluation and Research (CBER) regulates biologics Primary goal: to ensure patient safety
ROLE of the FDA

• Development of new drugs is complex and costly
• Mean cost: $1.3 billion • Mean time: 12 years to bring one
drug from discovery to the patient • For every one drug that reaches
market, 10,000-30,000 compounds are screened
DRUG DEVELOPMENT PROCESS
PMA, 2015

1. What are the goals and objectives? o Early exit, commercialization vs. licensing
2. What are the desired product attributes that differentiate value?
o Indication, dosage, formulation, claims, PK/PD, safety & efficacy
3. How can you create an unmet need? o Why is it important, and what are the issues and value
4. What stakeholders and market channels are important?
o Patients, physicians, payers, VC, shareholders, etc.
5. Where will the drug be marketed? o United States, Europe, Asia, etc
6. What has been successful, or not, in the past? o What will work for product X and what issues must be
considered? Model for success!
STRATEGIC ROAD MAP FOR SUCCESS

“WHAT IF?” Contingency planning for highly probable events!
• What if API can only be made in small batches or the ship sinks from China? • What if formulation is not stable? • What if the FDA does not agree with your planned studies? • What if you fail to achieve your primary endpoint, or so what data? • What if a competitor’s product launches first?
Spend less time on the improbable and more on probable events (i.e., timelines are lagging) Create your Risk Management Plan or REMS apriori
• Ensure benefits outweigh the risks

Preclinical, nonclinical and animal studies
Investigational new drug (IND) application
Clinical studies (Phases 1-3)
New drug or biologic application (NDA/BLA)
FDA/CDER or CBER review
Approval
Post approval evaluation/Phase 4
DRUG APPROVAL PROCESS

New drug applications (NDAs) or Biologic License Applications (BLA) require clinical trials to demonstrate safety and efficacy Studies must be contacted under GLP and GCP conditions Federal law requires a drug to be FDA approved before transporting across state lines The FDA Investigational New Drug (IND) is the process by which an exemption to the law is obtained Human studies can only begin after IND is reviewed and approved by the FDA and an institutional review board (IRB)
DEFINITIONS – INVESTIGATIIONAL NEW DRUG (IND)

Submit IND protocols for review at least 90 days before anticipated start of study State in cover letter that review of protocol &/or study report is requested Provide adequate information / data
facilitates review & response to Questions
Describe role of study in overall development program and/or potential regulatory outcomes (e.g., label claim)
EFFECTIVE IND COMMUNICATION STRATEGY

Fully understand the full lifecycle of any clinical trial, regardless of the Phase (I-IV) or indication The process stays the same and here to stay! Love the process, not the compound under study
Compounds are a “dime a dozen” and come and go. (Quote: Michael Poole, MD Vice President, Wyeth)
Focus on SmartStudyTM (Study, site and patient feasibility) Most drugs fail due to the process (i.e., study)
THE PROCESS of CONDUCTING CLINICAL TRIALS

Life Cycle of a Clinical Trial
Protocol Synopsis finalized
Schedule of Activities finalized
Protocol finalized
Model ICF finalized
Sites selected
Operations Manual/MOP completed
CRFs finalized
IRB approvals obtained
Site subcontracts/ payment schedule in place
Finalize Contracts with third party vendors (labs, ECGs etc.)
DSMB established
Build database
Finalize Study drug packaging/labeling
Enroll subjects Distribution of study drug to sites Answer Protocol/CRF questions Take incident calls SAEs Dosage Adjustments Premature Withdrawals Drug Disclosure
Data query process Clean/Close database Transfer database to
Biostatistics
Perform primary/ secondary analysis Submit abstract Submit manuscript Submit CTR Post results on
www.clinicaltrials.gov Post-hoc analysis
Orientation or Initiation Meeting
Database Locked
Analysis
CONCEPTUAL PHASE
PLANNING PHASE
IMPLEMENTATION PHASE
ANALYSIS/ PUBLICATION
PHASE
Grant Award and/or Parent
contract established

IRB’s ensure the rights and welfare of subjects participating in clinical trials, both before and during trial participation Ensure subjects are fully informed and have given written, informed consent before participating in studies Risks, benefits and protection of human subjects and data IRB’s are located in hospitals, research centers and private (Central) Local vs. Central IRB’s IRB approval varies from days to > year, and often after contracting
ROLE of the INSTITUTIONAL REVIEW BOARD (IRB)

• Emphasis on drug safety, PK/PD, biomarker
• SAD, MAD, Asian bridging study • Identify major side-effects (NIH Grade
1-4), metabolism, routes of excretion • Sufficient information about PK/PD to
permit design of well-controlled Phase 2 studies
• 70% of drugs that make it to this phase will pass this phase
PHASE 1 CLINICAL STUDIES
Duration: 1-2 years
Cost:
$1.5-$6+ mil
20-80 healthy volunteers

• Emphasis on effectiveness, safety, dosage, biomarker
• Closely monitored, evaluate short-term side-effects and risks
• Often 2+ studies are conducted • Patients receiving the drug are
compared to similar patients receiving a placebo or another drug
• 33% of drugs will pass this phase • Adaptive designs may lead to
registration
PHASE 2 CLINICAL STUDIES
Duration: 2-4 years
Cost:
$6-20+ mil
100-300 people with target disease

• Emphasis on safety, effectiveness, QOL, Costs
• Investigates different populations and dosages as well as combination with other drugs, surrogate biomarkers
• Randomized, placebo controlled • Typically, 2 confirmatory studies
required • Extrapolation to a general target
population • Acquire data used for physician
labeling • 25-30% pass this phase
PHASE 3 CLINICAL STUDIES
Duration: 2-8 years
Cost:
$20 - $600+ mil ($30--$150+K per patient)
300-10,000+ people with target disease

• Post-market surveillance of the drug to continually assess safety of the drug
• Risk Evaluation Mitigation Safety surveillance (REMS) • Required under accelerated basis of approval • May include: incidence and severity of rare adverse
reactions, cost-effectiveness analyses, comparative trials, and quality of life studies
• Tool to leverage FDA with reporting requirements • May have a “Big Data Play”
PHASE 4 CLINICAL STUDIES
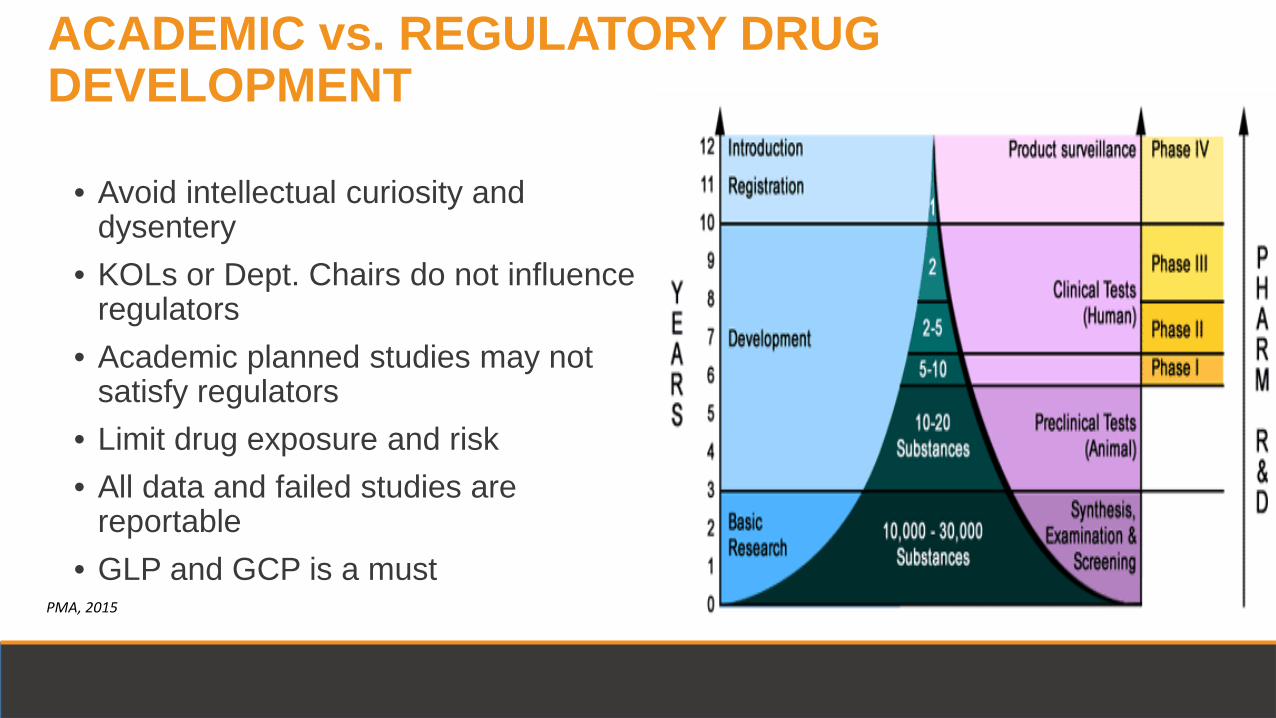
• Avoid intellectual curiosity and dysentery
• KOLs or Dept. Chairs do not influence regulators
• Academic planned studies may not satisfy regulators
• Limit drug exposure and risk • All data and failed studies are
reportable • GLP and GCP is a must
ACADEMIC vs. REGULATORY DRUG DEVELOPMENT
PMA, 2015

Orphan Well defined by FDA and OOPD, yet subject to agency interpretation Common EMEA/FDA Application with different Sponsor benefits Clinically superior to existing or previous unapproved or new orphan indication for same disease Establish rationale for indication(< 200K US cases) and Rx with medical plausibility Benefits: Patent protection (7 yrs.), protocol assistance, grants, contracts, tax incentives
FDA ORHPAN & FAST TRACK DESIGNATION
Fast Track Serious unmet medical need Preliminary safety and clinical activity superior to SOC Accelerated approval and priority review More frequent FDA meetings and engagement Sponsor requested after IND or EOP1

FDA Priority REVEIW and GAIN PROGRAMs Priority Review • FDA voucher program • Intended for Rx to treat rare and neglected diseases • Set to expire, Oct. 2016. Publically, FDA is not in favor • Priority review < 6 months • Transferable FDA GAIN Act • Generating Antibiotics Incentives Now (est. 2012) • Goal: generate new anti-microbial Rx for world pathogens • FDA qualified list of pathogens • 5 year market exclusivity, agency commitment and
engagement

Breakthrough Therapy Superior improvement over existing Rx. Fast Track benefits plus: intensive FDA guidance and commitment File no later than EOP2
Accelerated Approval Superior surrogate end point Post marketing pharmacovigilance and REMS commitment Agency commitment and engagement
FDA SPECIAL DESIGNATIONS

• NDA/BLA: Formal step asking FDA to consider approving a drug for marketing • Starting in 1938, all data; preclinical and clinical data of an IND are required to become
part of the NDA/BLA • Plan studies wisely, nothing out of FDA/EMA reach
• Electronic submission of all data, IND and NDA/BLA • e Publishing requirements
• Consist of as many as 15 different sections • Pre-NDA/BLA period: FDA and drug sponsors meet • FDA has 1 year to decide whether it will file the NDA/BLA for approval consideration • If filed, a FDA review team is assigned to evaluate the new drug • 2017, preclinical data must follow current electronic data and requirements for IND
NEW DRUG or Biologic APPLICATION (NDA/BLA)

• FDA review team evaluates the research on safety and effectiveness • Labeling information reviewed • Study audit • Inspection of GMP production facilities; methods and validation processes • Decision and justification letter:
Not approvable: Lists deficiencies in application and why it cannot be approved Approvable: Ultimately can be approved and lists deficiencies that can be corrected, including labeling changes and requests for post-approval studies Approval: Drug is approved
FDA APPROVAL

Developed in the conceptual and planning phase Develop a REALISTIC timeline and work scope Keep It Simple Stupid! whenever possible Plan for contingencies It always costs more and takes longer
KEYS to STAYING on SCHEDULE

Create a scope of work document clearly delineating who is responsible for what: sponsor, Project Team, External Vendors, Sites, Monitors
Create a detailed timeline of all activities that need to complete in each phase of the Project Lifecycle
Both documents will provide the roadmap for the overall project Tools for timeline development:
Excel Microsoft Project
Copyright © 2009 Clinical Trials Coordination Center/University of Rochester: All Rights Reserved
TIMELINE and WORK SCOPE

Select DSMB members Typically 3-5 independent scientists with no conflicts-of-interest and no other role in the study (should include: a biostatistican, disease expert, expert on drug under study, expert in body system with AE profile of greatest concern, ideally most have prior clinical trials experience)
Create/Finalize DSMB charter Specifies exactly what the DSMB is charged to monitor, stopping rules for efficacy/safety, major areas of concern (e.g., renal, hepatic etc.)
DMSB members and final charter should be in-place prior to FPFV
ESTABLISH DATA SAFETY MONITORING BOARD (DSMB) and CHARTER

Food and Drug Administration Amendments Act of 2007 or FDAAA), Title VIII, Section 801 mandates that a "responsible party" (i.e., the sponsor or designated principal investigator) register and report results of certain "applicable clinical trials":
Trials of Drugs and Biologics: Controlled, clinical investigations, other than Phase I investigations, of a product subject to FDA regulation; Trials of Devices: Controlled trials with health outcomes of a product subject to FDA regulation (other than small feasibility studies) and pediatric postmarket surveillance studies.
"Applicable clinical trials" generally include interventional studies (with one or more arms) of drugs, biological products, or devices that are subject to FDA regulation, meaning that the trial has one or more sites in the U.S, involves a drug, biologic, or device that is manufactured in the US (or its territories), or is conducted under an investigational new drug application (IND).
REGISTER TRIAL WITH WWW.CLINICALTRIALS.GOV BEFORE FPFV

21 CFR 320
ICH Guidance Documents (15+)
EMEA Guidance Documents (30+)
General BA/BE Guidance
Population PK Guidance
Exposure/Response Guidance
USDA (Animal Welfare Act)
Good Laboratory Practices
BCS Guidance
IVIVC Guidance
SUPAC Guidance documents (IR, MR and SS)
REGULATIONS and a FEW GUIDANCE DOCUMENTS
http://www.fda.gov/cder/guidance

Open, timely, exchange of ideas between Sponsor and regulatory agencies Obtain Agency’s views on questions / issues
Opportunity for specific questions to be addressed – (e.g., # of special population, interaction studies, biomarker) Request a SPA (Special Protocol Assessment) Minimize regulatory decision surprises Facilitates good review management principles
COMMUNICATION: WHY?

0
10
20
30
40
50
60
70
80
90
100
2008 2009 2010 2011 Source: Forbes, August 2015
2012 2013 2014 2015
Total new drug approvals Only new molecules
0
5
10
15
20
25
30
35
40
45
50
2004 2005 2006 2007 2008 2009 2010 2011 2012 2013 2014 2015
NME Approvals NME Applications Filed
Source: CDER 2015 Novel New Drugs Reports
• 2013/2014 had several biotech blockbuster launches (Sovaldi/Harvoni, Tecfidera, Imbruvica, Eyelea), but 2015 was relatively quiet (Praluent, Repatha)
• Streamlined FDA approval pathways in 2015 (31% Fast Track, 22% Breakthrough, 53% Priority Review, 13% Accelerated Approval)
Record 45 FDA drug approvals in 2015 Drug Approval Rate Reaches New High
• Orphan Drugs were 47% of all approvals in 2015; First-in-Class drugs were 36%
ROBUST RATE of DRUG APPROVALS

PAGE 32
• Immuno-oncology technologies
High valuations and hype – defensible given limited clinical data, growing competition, and demonstrated impact only in liquid tumors?
• Gene Therapy and Gene Editing
Early successes in narrow indications but failures in heart failure and back-of-eye diseases • Cell based therapies
• Companion Diagnostics
Tailored medicines based on a patient’s specific genetic profile or disease subtype • M&A
Buyer’s market among large pharmaceutical players with patent expirations and slow growth to acquire higher growth biotechnology products
• Pricing Pressure
Pharmacy benefit manufacturers, physician groups, politicians and media have been proactive with specialty drugs • Valuations and Financing Environment
What is fair value and is the financing window “open”? Greatest valuation is Phase 2 data.
VC “watch and wait” till after Phase 1 or 510(k) cleared
CURRENT “Hot” TOPICS IN Bio-Pharma Investing

SMARTSTARTTM YOUR CLINICAL DRUG DEVELOPMENT