size control of magnetic carbon nano particles for drug delivery
TRANSCRIPT

8/3/2019 Size Control of Magnetic Carbon Nano Particles for Drug Delivery
http://slidepdf.com/reader/full/size-control-of-magnetic-carbon-nano-particles-for-drug-delivery 1/7
Size control of magnetic carbon nanoparticles for drug delivery
W.-K. Oh, H. Yoon, J. Jang*
School of Chemical and Biological Engineering, Seoul National University, 599 Gwanangro, Sillim-dong, Gwanak-gu, Seoul 151-742, Republic of Korea
a r t i c l e i n f o
Article history:
Received 22 September 2009Accepted 8 October 2009Available online 29 October 2009
Keywords:
Drug deliveryNanoparticleMagnetismPorosityCell viablility
a b s t r a c t
Carbonized polypyrrole nanoparticles with controlled diameters were readily fabricated by the pyrolysisof polypyrrole nanoparticles. The carbonized polypyrrole nanoparticles showed narrow size distribution,
large micropore volume, and high surface area. Magnetic phases were introduced into the carbonnanoparticles during the pyrolysis without sophisticated process, which resulted in useful magneticproperties for selective nanoparticle separation. Field emission scanning electron microscopy, Ramanspectrometer, N2 adsorption/desorption, X-ray diffraction, and superconducting interference device wereemployed for characterizing the carbonized polypyrrole nanoparticles. Hydrophobic guest moleculeswere incorporated into the carbonized polypyrrole nanoparticles by surface adsorption, pore filling, andsurface covalent coupling. The carbonized polypyrrole nanoparticles exhibited embedding capabilityusing pyrene as a typical hydrophobic fluorescent molecule. In addition, ibuprofen was incorporated intothe carbon nanoparticles, and drug-loaded carbon nanoparticles sustained release property. In addition,the carbonized polypyrrole nanoparticles revealed low toxicity at concentrations below 100 mg mL À1 viacell viability test and were uptaken inside the cells. These results suggest a new platform for the drugdelivery using carbonized polypyrrole nanoparticles.
Ó 2009 Elsevier Ltd. All rights reserved.
1. Introduction
Recent progress in synthesis of nanomaterials has made itpossible to fabricate nanometer-sized materials with controlledstructures and functionalities [1–3]. In particular, versatile porousmaterials with nanometer feature sizes have emerged as promisingcandidates for applications in the fields of catalysis [4–6], energyconversion and storage [7,8], separation [9], and biomedical science[10,11]. For example, as a typical microporous material, zeolite hasbeen extensively utilized for molecular sieves, scaffolds, andtemplates for microporous replicas [12]. In addition, mesoporouscarbons have been obtained using sacrificial templates includingcolloidal silica nanoparticles and mesoporous silicas. Various
precursors, such as sucrose, phenol resin, polypyrrole, poly-acrylonitrile, and poly(furfuryl alcohol), were employed forobtaining porous carbon structures [13,14].
Carbon nanomaterials are of great interest in applicationsfor biological fields [15,16]. Typically, carbon nanotubes (CNTs) havea feature of endohedral filling of 2–10 nm in diameter leadingto encapsulation of small molecules. CNTs can be heterogeneouslysurface-functionalized and stained cytochemically with non-
quenching and non-photobleaching. Accordingly, CNTs may besuitable for bio–applications in biorecognition and drug deliverysystems [17–19]. However the biocompatibility of CNTs is stillcontroversial and the tedious functionalization process of CNTsurface remains major obstacles to practical applications [20,21].
The synthetic strategies towards carbon nanoparticles involve:i) pyrolysis of organic precursors under inert atmosphere [22] andii) physical and chemical vapor deposition techniques [23]. Whilethe pyrolysis approach is applicable to large-scale production, itoffers very limited control on the carbon nanostructure. It has beenwell-known that carbon black is a mass-product manufactured bythermal decomposition or incomplete combustion of carbonhydrogen compounds [24]. However, carbon black consists of
aggregates of spherical particles with the diameter of individualparticles commonly ranging from 10 nm to 200 nm. Although thevapor deposition method allows precise control on the carbonnanostructure, it has also significant drawbacks such as limitedyield, high cost, and complex equipment. In general, carbonencapsulated magnetic nanoparticles were obtained by depositionof carbon onto Fe, Co, and Ni nanocatalysts using arc-dischargetechniques [25]. However, it is known that the arc-dischargetechnique often gives an irreproducible and low yield due to the by-products such as graphitic flakes, carbon nanotubes, and carbides.
Recently, we have explored the fabrication of several types of carbon nanoparticles from polymer nanoparticles as carbon
* Corresponding author. Fax: þ82 2 8881604.E-mail address: [email protected] (J. Jang).
Contents lists available at ScienceDirect
Biomaterials
j o u r n a l h o m e p a g e : w w w . e l s e v i e r . c o m / l o c a t e / b i o m a t e r i a l s
0142-9612/$ – see front matter Ó 2009 Elsevier Ltd. All rights reserved.
doi:10.1016/j.biomaterials.2009.10.018
Biomaterials 31 (2010) 1342–1348

8/3/2019 Size Control of Magnetic Carbon Nano Particles for Drug Delivery
http://slidepdf.com/reader/full/size-control-of-magnetic-carbon-nano-particles-for-drug-delivery 2/7
precursors [26,27]. Direct carbonization of tailored polymer nano-particles is favorable to the formation of carbon nanoparticles withthe desired structure and composition. Furthermore, carbonizationin mild temperature such as from 700 to 900 C remained their ownbiocompatibility of polymers, resulting in applying for biologicalfields. Notably, magnetic carbon nanospheres with controlled sizeand shape were fabricated via carbonization of iron-doped polymernanomaterials [28,29]. Magnetic carbon nanostructures havearoused a great deal of interest due to their possible biologicalapplications, including drug/gene delivery, thermal tumor therapy,in–vitro cell separation, and magnetic resonance imaging [30,31].The geometry (size and shape) of the nanostructures is a key factordetermining cellular uptake rate and mechanism. It has been foundthat an optimum geometry for endocytotic uptake is ca. 50 nm andspherical shape [32,33].
Here we report the fabrication of carbonized polypyrrolenanoparticles (CPyNs) with controlled diameters and their texturalproperties. Furthermore, we investigate the potential capability of CPyNs as imaging probes and drug carriers based on their porosity,magnetic property and biocompatibility. The guest molecule-loading of CPyNs was conducted with pyrene as a typical hydro-phobic dye and the guest molecule-releasing test was performed
with ibuprofen as a typical hydrophobic drug.
2. Materials and methods
2.1. Materials
Dodecyltrimethylammonium bromide (DTAB, 99.0%), decyl alcohol (99.0%),pyrrole (98.0%), and ferric chloride (97.0%) were purchased from Aldrich. Ethanol(99.9%), tetrahydrofuran (THF, 99.9%), methanol (99.9%), and phosphate bufferedsolution (PBS: 0.1 M, pH 7.4) were also obtained from Aldrich. Pyrene (99%) andibuprofen(98%) were purchasedfrom Aldrich. A human fibroblastadherent cell line,which was derived from fetal lung tissue IMR90 (CCL-186), was purchased fromAmerican Type Culture Collection (ATCC).
2.2. Fabrication of CPyNs
Polypyrrole (PPy) nanoparticles as the carbon precursor were prepared by
micelle templating method and went through carbonization process in order togenerate CPyNs.First, to prepare 60 nm–diameterPPy nanoparticles, DTAB(8 g) wasdissolved in distilled water (160 mL) containing decyl alcohol (4.8 g) at 3 C. Pyrrole(1 g) was added dropwise into the DTAB/decyl alcohol solution and then ferricchloride (11.2 g) was added into the above solution. The chemical oxidation poly-merization of pyrrole monomer was carried out for 1 h at 3 C. To prepare 80 nm–/105 nm–diameter PPy nanoparticles, DTAB (8 g/8.4 g) was introduced into distilledwater (160 mL) containing decyl alcohol (4.4 g/3.2 g). Pyrrole (1.6 g/2 g) was addeddropwise into the surfactant solution, and ferric chloride (11.2 g) was added into thepyrrole/surfactant solution. The polymerization proceeded for 1 h at 3 C. Theresulting products were washed with ethanol to remove the surfactant and otherresidual reagents and subsequently dried in a vacuum oven at room temperature.Carbonization of the PPy nanoparticles was conducted in a quartz tubular furnaceunder nitrogen atmosphere. The nanoparticles were heated to 800 C at a heatingrate of 1 C minÀ1, held for 3 h, and cooled to room temperature. CPyNs withcontrolled diameters could be obtained from the PPy precursors at approximatelychar yields of ca. 50%. Field emission scanning electron microscopy (FE-SEM) was
performed with a JEOL 6330F at an acceleration voltage of 10 kV. The sizedistribution of CPyNs was calculated by software based on more than 100 particlesin the images. Raman spectra were obtained in the range of 800–1800 cmÀ1 usinga Jobin–Yvon T64000 spectrometer. The Brunauer–Emmett–Teller (BET) nitrogensorption experiments were conducted to calculate pore size distributions andcumulative pore volume with a Micromeritics ASAP 2020 at 77 K X-ray diffraction(XRD) measurement was performed using a M18XHF-SRA diffractometer (MACScience Co.) equipped with a CuKa radiation source (l¼1.5406 Å) at 40 kV and300 mA (12 kW). The magnetic property of CPyNs was measured using a super-conducting interference device (SQUID) magnetometer (Quantum Design MPMS5).
2.3. Pyrene loading into CPyNs
Pyrene was dissolved in THF solution at a concentration range of 5 Â10À2 to25 mM and then mixed with an aqueous solution containing CPyNs for 12 h. Thesteady-state fluorescence spectrum of each solution (lexc¼ 334 nm) was obtainedand were scanned in the range of 350–450 nm using a Quanta Master Fluorescence
Steady-State Spectrometer.
2.4. Surface modification with amino groups using plasma treatment
The plasma reactor (Korea Vacuum Co.) is the parallel–electrode type witha 13.56 MHz radio–frequency generator. The diameter of the powered electrode onwhich the sample is placed is 35 cm and the distance between the two electrodes is8 cm. The carbon nanoparticles were added into the plasma chamber, and thechamber was evacuated. When the chamber was evacuated below 10À3 Torr, theNH3 carrier gas was introduced into the chamber at rate of 30 cm3 minÀ1 (operatingpressure: 160 mTorr). The plasma output power was fixed at 80 W and the plasma
treatment was conducted in 2 min. This plasma treatment method and character-ization were previously reported in the literature [34].
2.5. Release profile of ibuprofen from CPyNs
Ibuprofen (2 mg) was dissolved in methanol (2 mL) and then mixed into anaqueous solution (20 mL) containing CPyN-1 (10 mg). The CPyN-1 solution wasstirred for 12 h, and subsequently it was separated by an external magnet andwashed three times with water to remove residual drug. The drug-loaded CPyN-1was dried in a vacuum oven at room temperature. To carry out time-dependentrelease tests, the following steps were employed: drug-loaded CPyN-1 wasintroduced into 0.1 M PBS (50 mL, pH) solution and incubated at 37 C. The samples(0.2 mL) were extracted at a constant time interval and dissolved in methanol(1.8 mL). The samples were quantitatively analyzed by HPLC (WatersÒ). Separationof the analytes was achieved with an acetonitrile/water (1:1) containing 0.1%phosphate buffer, flow 1 mLminÀ1, detection 228 nm and 270 nm, pressure10,35,000–1047000 psi, injected volume 50 mL, and C18 column. Linear regression
coefficients from six-point linear calibration curves (concentration range0.5–10 mg mL À1) were between 0.9982 and 0.9999 for all compounds.
2.6. Cell viability assay
IMR90 cells were cultured in Eagle’s Minimum Essential Medium (EMEM)containing sodium pyruvate (1 mM), L –glutamine (2 mM), 10% heat–inactivated FetalBovine Serum (FBS), penicillin (10,000 IUmL À1), and streptomycin (10 mg mL À1).The cells were grown in a humidified incubator at 37 C (95% air, 5% CO2). Cellviability of the CPyN-treated cells was investigatedusing CellTiter glow luminescentcell viability assay (Promega, Madison, WI). For estimating viable cells, 10,000 cellsperwell were platedand treatedwith different concentrations of CPyNs (10, 25,100,250, and 500 mg mL À1) for 24 h.
2.7. Transmission electron microscopy(TEM) of CPyNs-treated cells
The ultrastructural alterations of IMR90 cell lines induced by the CPyNs were
observed with TEM (JEM-2000EXII, JEOL). IMR 90 cells were incubated in Lab-Tek IIchamber slides until 80% confluence. After a 24 h exposure of CPyNs, IMR90 washarvested by a cell scraper and prefixed with 2% paraformaldehyde and 2% glutar-aldehyde at 4 C for4 h.After being washed with 0.05 M cacodylatebuffer, IMR90waspostfixed with 1% osmiumtetraoxide at 4 C for 2 h and washed with distilled waterand then stained with 0.5% uranyl acetate at 4 C. The cells were dehydrated througha series of ethyl alcoholconcentrations (30%, 50%, 70%, 80%, 90%,100%,100%,and dryalcohol). Then, the cells were treated with propylene oxide followed by 1:1propylene oxide: spurr’s resin for 2 h. The cells were infiltrated in spurr’s resin at70 C for 24 h and ultramicrotome was performed with diamond knife, collected oncarbon grids. Then, samples were observed with the TEM at 120 kV.
3. Results and discussion
Polypyrrole (PPy) nanoparticles with controlled diameters wereprepared by micelle templating in oil/water emulsions and used for
carbon precursors in order to fabricate CPyNs. PPy consists of five-membered heterocyclic rings with cross-linking structures (a,a- anda,b-links) and includes iron cations as the dopant from the oxidizingagent (ferric chloride), which can act as the catalyst to facilitate theformation graphite structure [35]. During heat-treatment processunder an inert atmosphere, thewell-definedPPy nanoparticlescan bereadily converted into carbon nanoparticles and the doped ironcationsare transformed into magnetic phases.As previouslyreported,spherical micelles were formed with DTAB in aqueous solution, anddecyl alcohol as a cosurfactant was used to minimize the destabili-zation phenomenon such as Ostwald ripening effect to fabricate PPynanoparticles with controlled diameters [2,25]. The size of micellesstrongly depends on the concentrations of surfactant. In general, thesize of nanoparticle decreases with increasing the surfactant
concentration [35]. Under our experimental conditions, the diameter
W.-K. Oh et al. / Biomaterials 31 (2010) 1342–1348 1343

8/3/2019 Size Control of Magnetic Carbon Nano Particles for Drug Delivery
http://slidepdf.com/reader/full/size-control-of-magnetic-carbon-nano-particles-for-drug-delivery 3/7
of PPy nanoparticle was effectively controlled with varying concen-trations of DTAB and decyl alcohol. DTAB was employed to formmicelles as a nanoreactor, and decyl alcohol was used as a cosurfac-tant. Decyl alcohol, a water-insoluble long chain alcohol, can retard orhinder the diffusion of pyrrole through the aqueous phase. Therefore,the size of the nanoparticles was controlled by the ratio betweenpyrrole and DTAB/decyl alcohol. When the monomer/surfactant ratiodecreases, the diameter of PPy nanoparticles decreases. PPy nano-particles were prepared with three different diameters (60, 80, and105 nm) and were successfully transformed to CPyNs with uniformsizes by the carbonization procedure (char yield: ca. 50%). Fig. 1represents FE–SEM images and size distributionhistograms of CPyNs.The FE–SEM images exhibited spherical nanoparticles with reason-ably uniform sizes and their diameters were 56 Æ 6 nm (CPyN–1),76Æ 9 nm (CPyN–2), and 99Æ 8 nm (CPyN–3), respectively.
The successful carbonization of CPyNs and the impregnation of iron oxides were confirmed by Raman spectroscopy. In Fig. 2a, theRaman spectrum of the CPyNs represented two distinct peaks at1595 cmÀ1 and 1350 cmÀ1. The band at 1595 cmÀ1 (G band) wasassigned to the E2g vibration of graphitic carbon with sp2 configu-ration. On the other hand, the peak at 1350 cmÀ1 (D band) wasattributable to the A1g mode of an imperfection in the crystalstructure of graphite (e.g., iron) with sp3 configuration. AlthoughCPyNs were carbonized at mild temperature (800 C), the CPyNsrepresented the crystal structure and could be categorized asa glassy carbon structure. In addition, the XRD pattern of CPyNsconfirmed the existence of graphite in the nanoparticles, origi-nating the characteristic 002 and 100 Bragg reflections of graphe-nes (Fig. 2b). Furthermore, the XRD pattern showed the presence of g-Fe2O3 and a-Fe (labeled F and A, respectively) as magnetic phases
Fig. 1. FE-SEM images and size distribution histograms of CPyNs with different diameters: CPyN-1(56 Æ 6 nm) (a), CPyN-2(76Æ 9 nm) (b), and CPyN-3(99 Æ 8 nm) (c).
W.-K. Oh et al. / Biomaterials 31 (2010) 1342–13481344

8/3/2019 Size Control of Magnetic Carbon Nano Particles for Drug Delivery
http://slidepdf.com/reader/full/size-control-of-magnetic-carbon-nano-particles-for-drug-delivery 4/7
in the CPyNs. It is considered that iron-based complexes encapsu-lated in PPy nanoparticles formed iron oxides at 800 C.
Nitrogen sorption experiments were performed to characterizethe textural properties of CPyNs. The nitrogen adsorption–desorption isotherms and the cumulative pore volume curves of CPyNs are displayed in Fig. 3. The isotherms represent an increasein the volume adsorbed up to the relative pressure in the range of 0.01–0.02, depending on the particular sample, and then leveledoff. At low relative pressures, micropore filling mainly occurs due tothe strong adsorbent–adsorbate interactions and the amount
adsorbed in the mesopores is negligible. Accordingly, this behavioris associated with adsorption in micropores and may also originatefrom adsorption in mesopores with sizes close to the microporerange. The CPyNs displayed a steep increase in the volumeadsorbed at pressures close to the saturation vapor pressure. Thisphenomenon is due to the capillary condensation of nitrogen ininterparticle pores with some contribution of multilayer adsorptionon the surface of these pores [36]. BET surface areas were ca.408 m2 gÀ1, 235 m2 gÀ1, and 164 m2 gÀ1 for CPyN–1, CPyN–2, andCPyN–3, respectively, which was in inverse proportion to thediameter of CPyNs. The pore size distributions of CPyNs derivedfrom nitrogen adsorption isotherms was close to the microporerange of 0.0–2.5 nm in size. The total pore volumes of CPyNs werecalculated to be ca. 0.62 cm3 gÀ1, 0.39 cm3 gÀ1, and 0.22 cm3 gÀ1 for
CPyN–1, CPyN–2, and CPyN–3, respectively. The micropore volumes
calculated from the Horvath–Kawazoe (HK) formalism were ca.0.17 cm3 gÀ1, 0.13 cm3 gÀ1, and 0.10 cm3 gÀ1, respectively (Fig. 3insets). It is considered that the micropore volume increased ininverse proportion to the size of CPyNs because the heat conduc-tion and mass transfer of CPyNs werevaried with respect to the sizeof CPyNs, where the heat conduction proceeded from outside toinside of CPyNs and the mass transfer proceeded from inside to
outside of CPyNs during carbonization process. Interestingly, the
Fig. 2. Raman spectrum of CPyN-1 measured between 800 and 1800 cm-1 a), powder
XRD pattern of CPyN-1 (G: graphite, F: g–Fe2O3, A: a–Fe) b).
Fig. 3. Nitrogen adsorption/desorption isotherms of CPyNs (insets: cumulative pore
volumes of CPyNs at a pore diameter of less than 3 nm calculated by HK formalism):
CPyN-1 (a), CPyN-2 (b), and CPyN-3 (c).
W.-K. Oh et al. / Biomaterials 31 (2010) 1342–1348 1345

8/3/2019 Size Control of Magnetic Carbon Nano Particles for Drug Delivery
http://slidepdf.com/reader/full/size-control-of-magnetic-carbon-nano-particles-for-drug-delivery 5/7
micropore volume of CPyNs was comparable to that of ZSM–5zeolite (micropore volume 0.14 cm3 gÀ1, mesopore volume0.02 cm3 gÀ1), as a well–known microporous material [37].
Magnetic properties of the CPyNs were investigated with anSQUID magnetometer. In Fig. 4, the hysteresis loop of the CPyNs at300 K represented typical ferromagnetic behavior. Moreover,saturation of the magnetization from the hysteresis loop was foundto be 12.5 emu gÀ1. The coercivity (H c) was observed at a field of 25 Oe. Compared to the value of Hc for bulk iron (z1 Oe), a signif-icant increase of coercivity was observed in the CPyNs. Themagnetic property of CPyNs is useful for efficient targeting andseparating of drug carriers.
In general, carbon nanoparticles could load functional guestmolecules by surface adsorption, pore filling, and surface covalentcoupling [38]. As the diameter of a nanoparticle becomes smaller, ithas more populations to contact with guest species due to theenhanced surface area. When the guest molecules reach to thenanoparticle surface, they can be incorporated into the nano-particle by physical interactions such as van der Waals force andhydrophobic interaction. In order to gain a better insight into theadsorption behavior of guest molecules in CPyNs, pyrene wasloaded into CPyNs by a phase separation method. Emission spec-
trum of pyrene shows characteristic the intensity ratio between thefirst (369.5 nm) and the third (380 nm) monomeric peaks (I m1/I m3).The I m1/I m3 value is susceptible to the micro-environmentalpolarity of the solubilized pyrene molecules [39,40]. In general, theI m1/I m3 value increases as the solvent polarity increases. Accord-ingly, the location of pyrene in CPyNs could be traced on themolecular level by monitoring the I m1/I m3 value. Fig. 5a exhibitsrepresentative fluorescence spectrum of pyrene/CPyN-1/watersolution. The I m1/I m3 ratio was systematically investigated andnormalized by dividing it by the initial value (the concentration of pyrene¼ 5Â10À2 mM). Fig. 5b displays changes in the normalizedI m1/I m3 value for pyrene/CPyN/water systems as a function of pyrene concentration. The normalized I m1/I m3 value did not changeup to a critical concentration (C crit), which was dependent on the
size of CPyNs, and then gradually increased with further increasingpyrene concentration. At low concentrations, most pyrene mole-cules can existin the hydrophobicporeor surface of CPyNs. The C crit
increased in the order of CPyN-3< CPyN-2< CPyN-1. Because thepyrene amount in CPyN was determined by the pore volume andsurface area of CPyNs, the C crit was strongly affected by the size of CPyNs. The increase in Im1/Im3 value at higher concentrationsreflects that pyrene locates a more hydrophilic microenvironment.
In other words, it is considered that an excess of pyrene moleculesover the C crit exists in aqueous phase. These results confirm the factthat CPyNs have high affinity with hydrophobic molecules and canreadily load a certain amount of guest molecules.
Potential capability of CPyNs to load guest molecules providesthe application to a drug carrier. Ibuprofen is a well knownnonsteroidal antiinflammatory drug and was investigated toreleasing molecules of CPyNs in aqueous medium. Furthermore,a surface-modified CPyN-1 (termed CPyN-1-NH2) was prepared byfurther functionalization with ammonia plasma treatment for
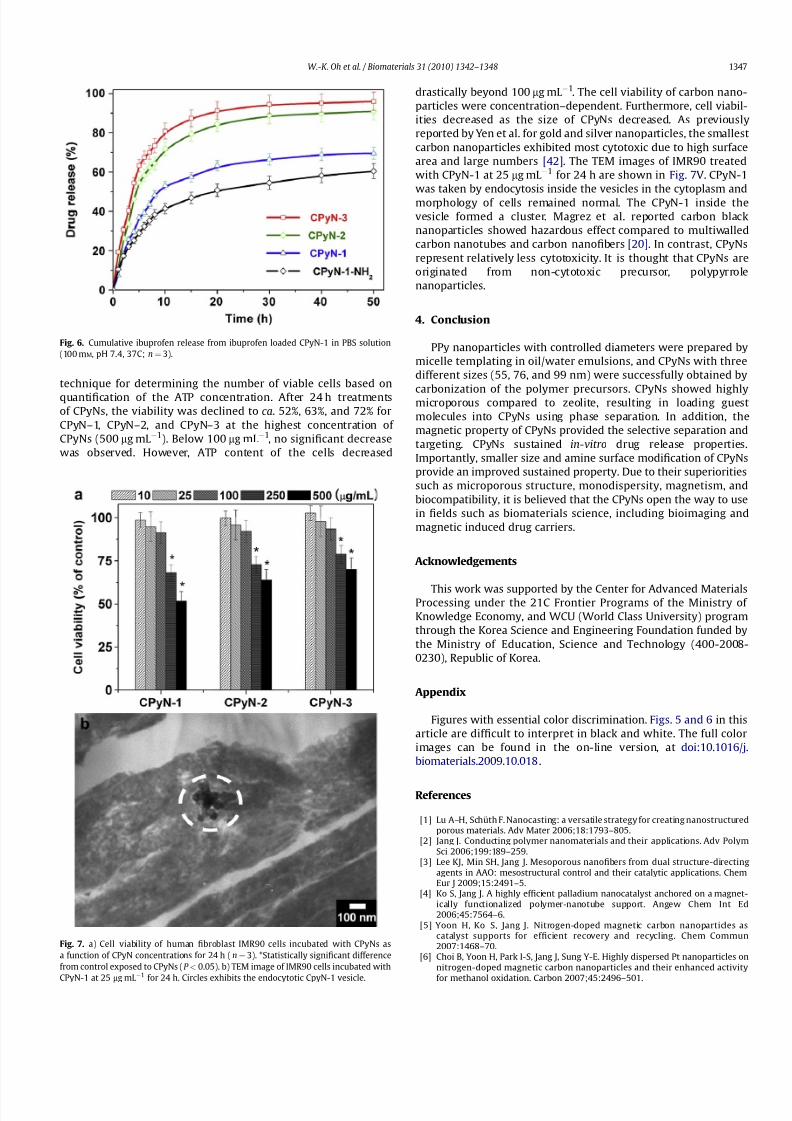
preparing the CPyN-1 with amino groups. Fig. 6 exhibits the in-vitrorelease profiles of CPyNs for ibuprofen. CPyN-2 and CPyN-3 releasedover 70% of naproxen during 10 h, and more than 90% of naproxenwas released within 50 h. In the case of CPyN-1, about 53% of ibuprofen released within 10 h, and CPyN-1 was sustained ca.70%of ibuprofen during 50 h. CPyN-1 exhibits the relatively slow releasebecause of higher surface area and pore volume. Notably, CPyN-1-NH2 delayed the release of ibuprofen such as about 60% within50 h, indicating that the ionic interaction between the amino groupof the CPyN-1-NH2 and the carboxyl group of ibuprofen playsa crucial role of sustaining release property [41].
The biocompatibility of CPyNs is a key factor in their biologicalapplications. The CPyNs were incubated with human lung fibro-blast cell line (IMR90), and ATP assay was used for the study on the
cell viability of CPyNs (Fig. 7a). The ATP assay is a homogeneous
Fig. 4. Magnetic hysteresis loop of CPyN-1 measured at 300 K between À15 and
þ
15 kOe (inset: magnified area betweenÀ
50 andþ
50 Oe).
Fig. 5. Adsoption behavior of pyrene-loaded CPyN-1; Fluorescence spectrum of pyr-
ene/CPyN-1/aqueous solution (a), and relative hydrophobic index of pyrene-loaded
CPyN-1 as a function of pyrene concentration (b) ( I m1/I m3 value: micro-environmental
polarity of the solubilized pyrene molecules).
W.-K. Oh et al. / Biomaterials 31 (2010) 1342–13481346
8/3/2019 Size Control of Magnetic Carbon Nano Particles for Drug Delivery
http://slidepdf.com/reader/full/size-control-of-magnetic-carbon-nano-particles-for-drug-delivery 6/7
technique for determining the number of viable cells based on
quantification of the ATP concentration. After 24 h treatmentsof CPyNs, the viability was declined to ca. 52%, 63%, and 72% forCPyN–1, CPyN–2, and CPyN–3 at the highest concentration of CPyNs (500 mg mL À1). Below 100 mg mL À1, no significant decreasewas observed. However, ATP content of the cells decreased
drastically beyond 100 mg mL À1. The cell viability of carbon nano-particles were concentration–dependent. Furthermore, cell viabil-ities decreased as the size of CPyNs decreased. As previouslyreported by Yen et al. for gold and silver nanoparticles, the smallestcarbon nanoparticles exhibited most cytotoxic due to high surfacearea and large numbers [42]. The TEM images of IMR90 treatedwith CPyN-1 at 25 mg mL À1 for 24 h are shown in Fig. 7V. CPyN-1was taken by endocytosis inside the vesicles in the cytoplasm andmorphology of cells remained normal. The CPyN-1 inside thevesicle formed a cluster. Magrez et al. reported carbon blacknanoparticles showed hazardous effect compared to multiwalledcarbon nanotubes and carbon nanofibers [20]. In contrast, CPyNsrepresent relatively less cytotoxicity. It is thought that CPyNs areoriginated from non-cytotoxic precursor, polypyrrolenanoparticles.
4. Conclusion
PPy nanoparticles with controlled diameters were prepared bymicelle templating in oil/water emulsions, and CPyNs with threedifferent sizes (55, 76, and 99 nm) were successfully obtained by
carbonization of the polymer precursors. CPyNs showed highlymicroporous compared to zeolite, resulting in loading guestmolecules into CPyNs using phase separation. In addition, themagnetic property of CPyNs provided the selective separation andtargeting. CPyNs sustained in-vitro drug release properties.Importantly, smaller size and amine surface modification of CPyNsprovide an improved sustained property. Due to their superioritiessuch as microporous structure, monodispersity, magnetism, andbiocompatibility, it is believed that the CPyNs open the way to usein fields such as biomaterials science, including bioimaging andmagnetic induced drug carriers.
Acknowledgements
This work was supported by the Center for Advanced MaterialsProcessing under the 21C Frontier Programs of the Ministry of Knowledge Economy, and WCU (World Class University) programthrough the Korea Science and Engineering Foundation funded bythe Ministry of Education, Science and Technology (400-2008-0230), Republic of Korea.
Appendix
Figures with essential color discrimination. Figs. 5 and 6 in thisarticle are difficult to interpret in black and white. The full colorimages can be found in the on-line version, at doi:10.1016/j.biomaterials.2009.10.018 .
References
[1] Lu A–H, Schu th F. Nanocasting: a versatile strategy for creating nanostructuredporous materials. Adv Mater 2006;18:1793–805.
[2] Jang J. Conducting polymer nanomaterials and their applications. Adv PolymSci 2006;199:189–259.
[3] Lee KJ, Min SH, Jang J. Mesoporous nanofibers from dual structure-directingagents in AAO: mesostructural control and their catalytic applications. ChemEur J 2009;15:2491–5.
[4] Ko S, Jang J. A highly efficient palladium nanocatalyst anchored on a magnet-ically functionalized polymer-nanotube support. Angew Chem Int Ed2006;45:7564–6.
[5] Yoon H, Ko S, Jang J. Nitrogen-doped magnetic carbon nanoparticles ascatalyst supports for efficient recovery and recycling. Chem Commun2007:1468–70.
[6] Choi B, Yoon H, Park I-S, Jang J, Sung Y-E. Highly dispersed Pt nanoparticles onnitrogen-doped magnetic carbon nanoparticles and their enhanced activity
for methanol oxidation. Carbon 2007;45:2496–501.
Fig. 6. Cumulative ibuprofen release from ibuprofen loaded CPyN-1 in PBS solution
(100 mM, pH 7.4, 37C; n¼ 3).
Fig. 7. a) Cell viability of human fibroblast IMR90 cells incubated with CPyNs as
a function of CPyN concentrations for 24 h (n¼ 3). *Statistically significant difference
from control exposed to CPyNs (P < 0.05). b) TEM image of IMR90 cells incubated with
CPyN-1 at 25 mg mL
À1
for 24 h. Circles exhibits the endocytotic CpyN-1 vesicle.
W.-K. Oh et al. / Biomaterials 31 (2010) 1342–1348 1347

8/3/2019 Size Control of Magnetic Carbon Nano Particles for Drug Delivery
http://slidepdf.com/reader/full/size-control-of-magnetic-carbon-nano-particles-for-drug-delivery 7/7
[7] Moon S-H, Jin W-J, Kim T-R, Hahm H-S, Cho B-W, Kim M-S. Performance of graphite electrode modified with carbon nanofibers for lithium ion secondarybattery. J Ind Eng Chem 2005;11:594–602.
[8] Choi M, Jang J. Heavy metal ion adsorption on polypyrrole-impregnatedporous carbon. J Colloid Interface Sci 2008;325:287–9.
[9] Giri S, Trewyn BG, Stellmaker MP, Lin VS–Y. Stimuli-responsive controlled-release delivery system based on mesoporous silica nanorods capped withmagnetic nanoparticles. Angew Chem Int Ed 2005;44:5038–44.
[10] Yang P, Quan Z, Lu L, Huang S, Lin J. Luminescence functionalization of mes-oporous silica with different morphologies and applications as drug delivery
systems. Biomaterials 2008;29:692–702.[11] Guo S, Li D, Zhang L, Li J, Wang E. Monodisperse mesoporous super-
paramagnetic single-crystal magnetite nanoparticles for drug delivery.Biomaterials 2009;30:1881–9.
[12] Pelster SA, Kalamajka R, Schrader W, Schu th F. Monitoring the nucleation of zeolites by mass spectrometry. Angew Chem Int Ed 2007;46:2299–302.
[13] Jang J, Bae J. Fabrication of polymer nanofibers and carbon nanofibers by usinga salt-assisted microemulsion polymerization. Angew Chem Int Ed2004;43:3803–6.
[14] Vinu A. Two-dimensional hexagonally-ordered mesoporous carbon nitrideswith tunable pore diameter, surface area and nitrogen content. Adv FunctMater 2008;18:816–27.
[15] Kelly S, Regan EM, Uney JB, Dick AD, McGeehan JP, Mayer EJ, et al. Patternedgrowth of neuronal cells on modified diamond-like carbon substrates.Biomaterials 2008;29:2573–80.
[16] Yushin G, Hoffman EN, Barsoum MW, Gogotsi Y, Howell CA, Sandeman SR,et al. Mesoporous carbide-derived carbon with porosity tuned for efficientadsorption of cytokines. Biomaterials 2006;27:5755–62.
[17] Hayashi T, Kim YA, Natsuki T, Endo M. Mechanical properties of carbonnanomaterials. ChemPhysChem 2007;8:999–1004.
[18] Jang J, Yoon H. Fabrication of magnetic carbon nanotubes using a metal-impregnated polymer precursor. Adv Mater 2003;15:2088–91.
[19] Peckys DB, Melechko AV, Simpson ML, McKnight TE. Immobilization andrelease strategies for DNA delivery using carbon nanofiber arrays and self-assembled monolayers. Nanotechnology 2009;20:145304–11.
[20] Magrez A, Kasas S, Salicio V, Pasquier N, Seo JW, Celio M, et al. Cellular toxicityof carbon-based nanomaterials. Nano Lett 2006;6:1121–5.
[21] Cheng C, Mu ller KH, Koziol KKK, Skepper JN, Midgley PA, Welland ME, et al.Toxicity and imaging of multi-walled carbon nanotubes in human macrophagecells. Biomaterials 2009;30:4152–60.
[22] Sun X, Li Y. Colloidal carbon spheres and their core/shell structures withnoble-metal nanoparticles. Angew Chem Int Ed 2004;43:597–601.
[23] Qiao WM,Song Y,Lim SY, HongSH, Yoon SH,Mochida I, et al.Carbonnanospheresproduced in an arc-discharge process. Carbon 2006;44:187–90.
[24] HouP-X,OrikasaH, YamazakiT,MatsuokaK, TomitaA, Setoyama N,et al.Synthesisof nitrogen-containing microporous carbon with a highly ordered structure andeffectof nitrogen doping on H2O adsorption. Chem Mater 2005;17:5187–93.
[25] Mirabile Gattia D, Vittori Antisari M, Marazzi R. AC arc discharge synthesis of single-walled nanohorns and highly convoluted grapheme sheets. Nano-technology 2002;18:255604–10.
[26] Jang J, Yoon H. Multigram-scale fabrication of monodisperse conductingpolymer and magnetic carbon nanoparticles. Small 2005;1:1195–9.
[27] Jang J, Oh JH. A top-down approach to fullerene fabrication using a polymernanoparticle precursor. Adv Mater 2004;16:1650–3.
[28] Bystrzejewski M, Cudzi1o S, Huczko A, Lange H, Soucy G, Cota-Sanchez G, et al.Carbon encapsulated magnetic nanoparticles for biomedical applications:thermal stability studies. Biomole Eng 2007;24:555–8.
[29] Kim S, Shibata E, Sergiienko R, Nakamura T. Purification and separation of carbon nanocapsules as a magnetic carrier for drug delivery systems. Carbon2008;46:1523–9.
[30] Miyawaki J, Yudasaka M, Imai H, Yorimitsu H, Isobe H, Nakamura E, et al. Invivo magnetic resonance imaging of single-walled carbon nanohorns bylabeling with magnetite nanoparticles. Adv Mater 2006;18:1010–4.
[31] Lu A–H, Salabas EL, Schu th F. Magnetic nanoparticles: synthesis, protec-tion, functionalization, and application. Angew Chem Int Ed 2007;46:1222–44.
[32] Decuzzi P, Pasqualini R, Arap W, Ferrari M. Intravascular delivery of particulatesystems: does geometry really matter? Pharm Res 2009;26:235–43.
[33] Jiang W, Kim BYS, Rutka JT, Chan WCW. Nanoparticle-mediated cellularresponse is size-dependent. Nat Nanotechnol 2008;3:145–50.
[34] Oh W-K, Yoon H, Jang J. Characterization of surface modified carbon nano-particles by low temperature plasma treatment. Diam Relat Mater2009;18:1316–20.
[35] Jang J, Oh JH, Stucky GD. Fabrication of ultrafine conducting polymer andgraphite nanoparticles. Angew Chem Int Ed 2002;41:4016–9.
[36] Kyotani T. Synthesis of nitrogen-containing microporous carbon with a highlyordered structure and effect of nitrogen doping on H2O adsorption. ChemMater 2005;17:5187–93.
[37] Pelster SA, Kalamajka R, Schrader W, Schu th F. Monitoring the nucleation of zeolites by mass spectrometry. Angew Chem Int Ed 2007;46:2299–302.
[38] Schu th F. Non-siliceous mesostructured and mesoporous materials. ChemMater 2001;13:3184–95.
[39] Evertsson H, Nilsson S, Holmberg C, Sundelof L–O. Temperature effects on theinteractions between EHEC and SDS in dilute aqueous solutions. Steady-stateflurescence quenching and equilibrium dialysis investigations. Langmuir1996;12:5781–9.
[40] Sun Q, Ren B, Liu X, Zeng F, Liu P, Tong Z. Fluorescence study for the elec-trostatic interaction and aggregation in dilute polar solution of poly-electrolytes. Macromol Symp 2003;192:251–64.
[41] Yang P, Quan Z, Hou Z, Li C, Kang X, Cheng Z, et al. A magnetic, luminescentand mesoporous core-shell structured composite material as drug carrier.Biomaterials 2009;30:4786–95.
[42] Yen H-J, Hsu S-H, Tsai C-L. Cytotoxicity and immunological response of goldand silver nanoparticles of different sizes. Small 2009;5:1553–61.
W.-K. Oh et al. / Biomaterials 31 (2010) 1342–13481348