sickle celled aneamia
TRANSCRIPT

8/3/2019 Sickle Celled Aneamia
http://slidepdf.com/reader/full/sickle-celled-aneamia 1/13
INTRODUCTION
Sickle-cell disease (SCD), or sickle-cell anaemia (or anemia, SCA)or drepanocytosis, is an autosomal recessive genetic blood
disorder with overdominance , characterized by red blood cells that assume anabnormal, rigid, sickle shape. Sickling decreases the cells' flexibility andresults in a risk of various complications. The sickling occurs because of a mutation in the haemoglobin gene. Life expectancy is shortened, withstudies reporting an average life expectancy of 42 in males and 48 infemales.[1]
Sickle-cell disease, usually presenting in childhood, occurs more commonly inpeople (or their descendants) from parts of tropical and sub-tropical regionswhere malaria is or was common. One-third of all indigenous inhabitants
of Sub-Saharan Africa carry the gene,[2] because in areas where malaria iscommon, there is a fitness benefit in carrying only a single sickle-cell gene(sickle cell trait). Those with only one of the two alleles of the sickle-celldisease, while not totally resistant, are more tolerant to the infection and thusshow less severe symptoms when infected.[3]
The prevalence of the disease in the United States is approximately 1 in5,000, mostly affecting Americans of Sub-Saharan African descent, accordingto the National Institutes of Health.[4] In the United States, about 1 out of 500African-American children born will have sickle-cell anaemia.[5]
Sickle-cell anaemia is the name of a specific form of sickle-cell disease inwhich there is homozygosity for the mutation that causes HbS. Sickle-cellanaemia is also referred to as "HbSS", "SS disease", "haemoglobin S" or permutations thereof. In heterozygous people, who have only one sickle geneand one normal adult haemoglobin gene, it is referred to as "HbAS" or "sicklecell trait". Other, rarer forms of sickle-cell disease include sickle-haemoglobinC disease (HbSC), sickle beta-plus-thalassaemia (HbS/β+) and sickle beta-zero-thalassaemia (HbS/β0). These other forms of sickle-cell diseaseare compound heterozygous states in which the person has only one copy of
the mutation that causes HbS and one copy of another abnormal haemoglobin allele.
The term disease is applied, because the inherited abnormality causes a
pathological condition that can lead to death and severe complications. Not all
inherited variants of haemoglobin are detrimental, a concept known as genetic
polymorphism.
8/3/2019 Sickle Celled Aneamia
http://slidepdf.com/reader/full/sickle-celled-aneamia 2/13
STRUCTURE OF SICKLE CELL
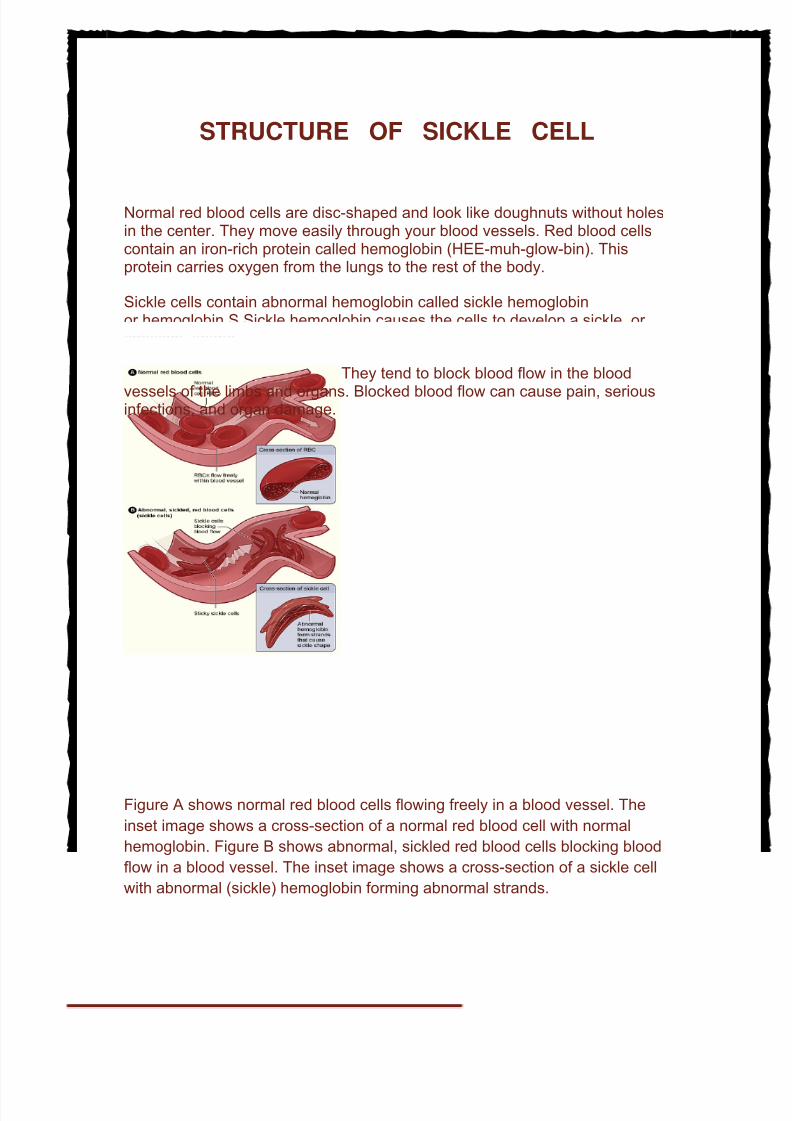
Normal red blood cells are disc-shaped and look like doughnuts without holesin the center. They move easily through your blood vessels. Red blood cellscontain an iron-rich protein called hemoglobin (HEE-muh-glow-bin). Thisprotein carries oxygen from the lungs to the rest of the body.
Sickle cells contain abnormal hemoglobin called sickle hemoglobinor hemoglobin S.Sickle hemoglobin causes the cells to develop a sickle, or crescent, shape.
Sickle cells are stiff and sticky. They tend to block blood flow in the blood
vessels of the limbs and organs. Blocked blood flow can cause pain, seriousinfections, and organ damage.
Figure A shows normal red blood cells flowing freely in a blood vessel. The
inset image shows a cross-section of a normal red blood cell with normal
hemoglobin. Figure B shows abnormal, sickled red blood cells blocking blood
flow in a blood vessel. The inset image shows a cross-section of a sickle cell
with abnormal (sickle) hemoglobin forming abnormal strands.

8/3/2019 Sickle Celled Aneamia
http://slidepdf.com/reader/full/sickle-celled-aneamia 3/13
SYMPTOMS
Signs and symptoms of sickle cell anemia usually show up after an infant is 4
months old and may include:
• Anemia. Sickle cells are fragile. They break apart easily and die,leaving you chronically short on red blood cells. Red blood cells usually live for
about 120 days before they die and need to be replaced. However, sickle cells
die after only 10 to 20 days. The result is a chronic shortage of red blood cells,
known as anemia. Without enough red blood cells in circulation, your body
can't get the oxygen it needs to feel energized. That's why anemia causes
fatigue.
• Episodes of pain. Periodic episodes of pain, called crises, are a major
symptom of sickle cell anemia. Pain develops when sickle-shaped red blood
cells block blood flow through tiny blood vessels to your chest, abdomen and joints. Pain can also occur in your bones. The pain may vary in intensity and
can last for a few hours to a few weeks. Some people experience only a few
episodes of pain. Others experience a dozen or more crises a year. If a crisis
is severe enough, you may need to be hospitalized.
• Hand-foot syndrome. Swollen hands and feet may be the first signs of
sickle cell anemia in babies. The swelling is caused by sickle-shaped red
blood cells blocking blood flow out of their hands and feet.
•
Frequent infections. Sickle cells can damage your spleen, an organthat fights infection. This may make you more vulnerable to infections. Doctors
commonly give infants and children with sickle cell anemia antibiotics to
prevent potentially life-threatening infections, such as pneumonia.
• Delayed growth. Red blood cells provide your body with the oxygen
and nutrients you need for growth. A shortage of healthy red blood cells can
slow growth in infants and children and delay puberty in teenagers.
• Vision problems. Some people with sickle cell anemia experience
vision problems. Tiny blood vessels that supply your eyes may become
plugged with sickle cells. This can damage the retina — the portion of the eye
that processes visual images.
When to see a doctor
Although sickle cell anemia is usually diagnosed in infancy, if you or your child
develops any of the following problems, see your doctor right away or seek
emergency medical care:

8/3/2019 Sickle Celled Aneamia
http://slidepdf.com/reader/full/sickle-celled-aneamia 4/13
• Unexplained episodes of severe pain, such as pain in abdomen,
chest, bones or joints.
• Swelling in the hands or feet.
• Abdominal swelling, especially if the area is tender to touch.
• Fever. People with sickle cell anemia have an increased risk of
infection, and fever can be the first sign of an illness.
• Pale skin or nail beds.
• Yellow tint to the skin or the whites of the eyes.
• Any signs or symptoms of stroke. If you notice any one-sided
paralysis or weakness in the face, arms or legs, confusion, trouble walking or
talking, sudden vision problems or numbness, or a headache, call 911 or your
local emergency number right away.
Symptoms of sickle cell crisis include:
• Severe pain• Anemia• Chest pain and difficulty breathing• Strokes• Joint pain and arthritis• Blockage of blood flow in the spleen or liver • Severe infections
Patients with sickle cell disease develop severe pain in the chest, back,arms, legs, and abdomen. Pain can occur anywhere in the body. Sicklered blood cells in the lungs can cause severe illness with chest pain,fever, and difficulty breathing. Sickle cell disease can also causepermanent damage to the brain, heart, kidneys, liver, spleen, andbones. The severity and symptoms vary greatly from person to person,even within the same family.

8/3/2019 Sickle Celled Aneamia
http://slidepdf.com/reader/full/sickle-celled-aneamia 5/13
CAUSES
Sickle cell disease is caused by a genetic abnormality in the gene for hemoglobin, which results in the production of sickle hemoglobin. Whenoxygen is released from sickle hemoglobin, it sticks together and forms longrods, which damage and change the shape of the red blood cell. The sicklered blood cells causes the symptoms of sickle cell disease.
Children are born with sickle cell disease; it is not contagious. It occurs whena child inherits two sickle hemoglobin genes, one from each parent. About2,000 babies are born with sickle cell disease each year in the United States.People who inherit only one sickle hemoglobin gene are carriers (sickle celltrait) and do not have anemia or painful sickle cell crises. About 2 millionAmericans have sickle cell trait.

8/3/2019 Sickle Celled Aneamia
http://slidepdf.com/reader/full/sickle-celled-aneamia 6/13
TREATMENT
The goal of treatment is to manage and control symptoms, and to limit the number of
crises.
Patients with sickle cell disease need ongoing treatment, even when they are not
having a painful crisis.
Folic acid supplements should be taken. Folic acid is needed to make red blood cells.
T rea t m en t f o r a s i ck l e ce ll cr i s is i n c lu d es :
• Blood transfusions (may also be given regularly to prevent stroke)• Pain medicines• Plenty of fluids
O th e r t r e a t m e n t s f o r s ick le c ell a n e m ia m a y in c lu d e :• Hydroxyurea (Hydrea), a medicine that may help reduce the number of pain
episodes (including chest pain and difficulty breathing) in some people• Antibiotics to prevent bacterial infections, which are common in children with
sickle cell disease
Treatments for complications of sickle cell anemia may include:
• Kidney dialysis or kidney transplant for kidney disease• Drug rehabilitation and counseling for psychological complications•
Gallbladder removal in those with gallstone disease• Hip replacement for avascular necrosis of the hip• Treatments, including surgery, for persistent, painful erections (priapism)• Surgery for eye problems• Wound care, zinc oxide, or surgery for leg ulcers
Bone marrow or stem cell transplants can cure sickle cell anemia. However, they are
current not an option for most patients. Sickle cell anemia patients are often unable
to find well-matched donors.

8/3/2019 Sickle Celled Aneamia
http://slidepdf.com/reader/full/sickle-celled-aneamia 7/13
Possible Complications
Acute chest syndrome
•
Anemia
• Blindness/vision impairment
• Brain and nervous system (neurologic) symptoms andstroke
• Death
•
Disease of many body systems (kidney, liver, lung)
• Drug (narcotic) abuse
• Erectile dysfunction (as a result of priapism)
• Gallstones
• Hemolytic crisis
• Infection, including pneumonia, gallbladder inflammation(cholecystitis), bone infection (osteomyelitis), and urinary tractinfection
• Joint destruction
• Leg sores (ulcers)
•
Loss of function in the spleen
• Parvovirus B19 infection, leading to low red blood cellproduction (aplastic crisis)
• Splenic sequestration syndromeTissue death in the kidney

8/3/2019 Sickle Celled Aneamia
http://slidepdf.com/reader/full/sickle-celled-aneamia 8/13
PREVENTION
Sickle cell anemia can only occur when two people who carry sickle cell trait have a
child together. Genetic counseling is recommended for all carriers of sickle cell trait.
About 1 in 12 African Americans has sickle cell trait.
It is possible to diagnose sickle cell anemia during pregnancy.
I f y o u h a v e s ic k l e ce ll a n a e m i a , y o u c a n p r e v e n t t h e c h a n g e in r e d b l o o d
ce ll shap e by :
• Getting enough fluids• Getting enough oxygen
• Quickly treating infections
• Have a physical exam every 3 - 6 months to make sure that you are getting
enough nutrition and activity, and that you are receiving the proper vaccinations.
Regular eye exams are recommended.
P R E V E NTI N G CR I SE S
It is important to maintain good oxygen levels and to preventdehydration. The
following steps can help prevent a sickle cell crisis:
• Avoid strenous activities, stress, smoking, high-altitudes, nonpressurizedflights, and other events that reduce your oxygen level
• Always have plenty of fluids with you• Avoid too much sun exposure
Consider having the child with sickle cell anemia wear a Medic Alert bracelet. Sharethe above information with teachers and other caretakers, when necessary.
PREV ENTIN G INFECTIONS
• People with sickle cell anemia need to keep their immunizations up to date toprevent illness.
• Some patients may receive antibiotics to prevent infections

8/3/2019 Sickle Celled Aneamia
http://slidepdf.com/reader/full/sickle-celled-aneamia 9/13
Bahrain 'winning war on sickle celldisease'SICKLE cell patients in Bahrain have one of the longest life expectancies inthe world, it has been declared.
The average age of patients is 45, which compares favourably to 40 in theUS and only around two years in some parts of Africa. Bahrain'sachievements in preventing the disease have helped it win globalrecognition, said consultant geneticist and head of the Salmaniya MedicalComplex (SMC) genetics department Dr Shaikha Al Arrayed.
However, she said the management of patients still needed to be seriouslylooked into.
"Facilities for such patients have already been augmented at the SMC whilemore are on the way," she said.
"A new centre is already coming up at the hospital, which will further enhance treatment and management facilities."
Speaking following the conclusion of two weeks of activities to mark WorldSickle Cell Diseases Day, Dr Al Arrayed said the ministry was aware of the"immense problems" being faced by patients.
But she said everyone should understand that the government was takingsteps to solve the problems.
"The new centre, for example, will be one of the very few specialisedfacilities in this region," said Dr Al Arrayed.
"It will have its own entry and exit as well as a dedicated emergency room,among other facilities. We are hoping it will be ready in the next 18 to 24months."
Bahrain Society for Sickle Cell Anaemia Patient Care chairman ZakareyaEbrahim Al Kadhem said the government plans were welcomed, but neededto be finished quickly.
Facilities
He said it was important to increase facilities at the SMC even temporarily,
until the centre was built. "We cannot have people suffering in pain and dyingin the absence of a specialised centre," he said.
According to the ministry, congenital and genetic disorders are responsiblefor a major proportion of infant mortality, morbidity and handicap cases inBahrain.
"We can easily avoid this situation," claimed Mr Al Kadhem. He earlier said11 of the 23 patients who died of the disease last year were aged less than

8/3/2019 Sickle Celled Aneamia
http://slidepdf.com/reader/full/sickle-celled-aneamia 10/13
30, including a 10-year-old boy.
Mr Al Kadhem said the society was concerned many of the deaths wereamong the younger age group. He said the average age was 40 in 1980 andcalled for greater awareness and action.
The GDN reported last month that Bahrain was close to eradicating thepotentially deadly sickle cell disease in newborn babies, with less than one
per cent of all children now born with the agonising blood disorder. Thereport said education and awareness campaigns have helped it achieve a 70per cent drop in cases in the last 25 years and the genetic disease nowaccounted for only 0.6 per cent of births.
The rate fell considerably after Bahrain started the compulsory screeningprocess for couples intending to get married five years ago, according toSMC genetic diseases consultant Dr Amani Al Hajeri.
Around 18,000 people in Bahrain are living with the disease, which hasalready killed nine people this year.
Features of sickle-cell disease in Bahrain
Genetic disorders of haemoglobin are prevalent in Bahrain. In a study of the hospital populationcovering 56 198 Bahrainis, we found that 2% of newborns have sickle-cell disease (SCD) and 18%have sickle-cell trait, while 24% are carriers of the (-thalassaemia gene. In a study of the presentation
of SCD among Bahrainis it was found that the mild form of the disease predominates, but a wideclinical variability is apparent. It was also found that their haematological values are similar to those of patients from Eastern Province, Saudi Arabia, where the mild form of the disease predominates. Introduction The state of Bahrain is an archipelago of 33 islands, with the kingdom of Saudi Arabia to the west andQatar to the east. The 1991 population was 500 000, one third non-Bahraini. Falciparum malaria wasendemic in Bahrain until 1970 and so the malaria-associated genetic defects of red cells (sickle-celldisease [SCD], thalassaemia and glucose 6 phosphate dehydrogenase deficiency) were found to becommon [1]. In 1990 it was found that hereditary anaemias were the third most frequent diagnosis at the
Salmaniya Medical Centre, which is the main hospital in the country [ 1].
Sickle-cell disease (SCD) drains a country's health resources and dramatically affects family andpersonal life. Accordingly we decided to study sickle-cell disease among Bahrainis. The aims of these studies were to: 1. ascertain the incidence of genetic disorders of haemoglobin in the hospital population in Bahrain 2. ascertain the natural history of sickle-cell disease among Bahrainis

8/3/2019 Sickle Celled Aneamia
http://slidepdf.com/reader/full/sickle-celled-aneamia 11/13
3. investigate the haematological characteristics of the Bahraini SCD patient 4. identify the haplotype associated with SCD mutation among Bahrainis. We present here a summary of four studies performed on sickle-cell disease among our population. 1. Prevalence of genetic disorders of haemoglobins in the hospital population of Bahrain Blood samples of 56 198 Bahraini nationals were analysed over a six-year period (1982- 1987). Of the total, 5 503 were neonatal samples (see Table 1) and the rest non-neonatal. Abnormalhaemoglobin was detected in 44.35% of neonatal samples (24.2% were a-thalassaemia cases, 18.1%showed sickle-cell trait [SCT] and 2.1% had SCD). Hb Barts was the most common abnormalhaemoglobin seen. In the non-neonatal cases, the overall frequency of SCD was found to be 10.44%, and the frequencyof those with SCD and Hb F present was 8.75%, which means that nearly 84 % of the SCD patientshad Hb F present. Table 2 shows the distribution of quantitation of fetal haemoglobin (Hb F) in SCDpatients with Hb S/F. Hb F varied between 2% and 40%. The majority of cases (about 76%) had Hb Fin the range between 4.1% and 20%. The favourable protective role played by Hb F in sickle-celldisease is well-recognized by several workers [2-8], with the severity of the disease being inverselyproportional to the quantity of Hb F. The high incidence among the non-neonatal cases is due to the fact that a good number of caseswere referred for Hb electrophoresis from outpatient clinics and hospital wards, and from healthcentres after getting positive results from a sickling test. 2. The nature of sickle-cell disease in Bahrain Sickle-cell disease in Bahrain and Saudi Arabia presents special features. SCD in this area ishaematologically and clinically mild, and mortality is low in both children and adults [ 9]. This benignpicture results in part from very high levels of fetal Hb in the community and also from a highprevalence of a-thalassaemia. However in this environment clinical variability is apparent, with somecases dying from septicaemia and serious morbidity resulting from salmonella osteomyelitis. This study was conducted with a view to ascertaining the nature of SCD in the Bahraini population,helping us to formulate certain palliative and corrective measures. The study was community based; aquestionnaire was sent to and completed by 100 school children aged between 8 and 12 and their parents. From this study we found that the most frequent factor cited as precipitating a crisis was exposure tocold (45% of cases). Other factors included fever or elevated body temperature (35%), exhaustionand severe physical activity (35%), hot humid weather (10%), stuffy and crowded places (10%)[10,11] (Table 3). Regarding the clinical picture, fever was mentioned as the most frequent symptom (69% of responders). Other symptoms mentioned were pain in the hands (59%), pain in the limbs (58%),abdominal pain (56%) and pain in the knees (55%). Of the sample, 36% had chest pain and only 18%had urinary problems (Table 4). Almost 55% of respondents mention fava beans as a precipitating cause of a crisis. Although notdocumented, an explanation for this may be the high incidence of glucose 6 phosphatedehydrogenase (G6PD) deficiency in the area [1,12,14]. An improvement in the patient's conditionwas noted with increased intake of fluids, fruits, vegetables and milk (Tables 5 and 6). The study found that 19% of respondents suffered a painful crisis (which might last from a few hoursto a few weeks) once a week, 48% once a month, and 33% between one and four times a year. Onemight expect that school absenteeism would echo the above data: we found that 43% of thoseresponding had experienced irregular schooling due to frequent crisis and 2% had had to discontinue

8/3/2019 Sickle Celled Aneamia
http://slidepdf.com/reader/full/sickle-celled-aneamia 12/13
schooling as a result of the severity of SCD. Of those surveyed, 10% had experienced the death of some family member due to SCD. The need of patients for qualified advice was clearly indicated by70% being in favour of premarital counselling and 62% in favour of specialized sickling clinics (Table7). 3. Haematological characteristics of Bahraini sickle-cell disease patients It is well known that the three major types of haemoglobinopathy are found in Bahrain [ 1], and manydifferent combinations of haemoglobinopathies genes occur. All may happen with or without the
coincidental G6PD deficiency. These complex interactions produce a continuous spectrum of severity,both clinical and haematological [14]. This study was of the haematological picture of Bahraini sickle-cell disease patients. A total of 50such cases was sampled. The ages of these patients ranged from 15 to 50 years. We found that 60% of the patients had Hb lower than 10 g/dl and that only 8.8% had Hb above 12g/dl. The normal Hb for an adult is 12 g/dl or higher [15]. Of these patients, 57% had HCT below 30.MCH was below 25 pg in 64%; the normal level of MCH is 28 pg or above. The low level of MCH inthese patients is partly due to the presence of the thalassaemia gene. MCV was also shown to be onthe low side_62% had MCV below 76 fl, indicating microcytosis, which is partly due to the coexistenceof the a-thalassaemia gene with SCD [7] (Table 8). A study was done in Saudi Arabia comparing the haematological values in SCD patients from EasternProvince and those from Western Province [16] (Table 8). These two groups were found to havedifferent haplotypes [17,18]. The Asian haplotypes predominated in the patients from EasternProvince while the African haplotype, benign type or S1 predominated in the patients from WesternProvince. There were significant differences in the total haemoglobin, red blood cell and haematocritvalues, but the red cell indices (mean cell volume), mean cell haemoglobin concentration and thepercentage of Hb F did not show any significant difference. If we compare the patient values from our study with these two groups (Table 9), we find that the Bahraini numbers are similar to those fromEastern Province, Saudi Arabia. This is consistent with the results of a molecular study presentedlater. 4. Beta globin gene haplotypes in Bahraini sickle-cell disease patients Molecular genetic studies were undertaken to determine the haplotypes of chromosomes carrying the
sickle-cell allele in Bahraini patients. A total of 59 individuals from 19 families were studied. Of these,35 were carriers. Haplotypes were investigated by PCR amplification of globin target sequencesfollowed by restriction digestion using Hind III, Ava II, Hind II, and Hinf l polymorphism [19,20] In the 19 families the Bs gene was found to be linked to the Asian haplotype in 33 chromosomes(90%), to the S2 haplotype in two chromosomes (5%), to the haplotype S1 in one chromosome(2.5%) and to the haplotype found in association with b-thalassaemia in one family (2.5%). Fig. 1 shows the pedigree of a family with sickle-cell disease exhibiting the Asian haplotype,while Table 10 shows the different haplotypes reported in Africa, Saudi Arabia and Bahrain. The present study shows that all Bahraini patients with sickle-cell disease studied to date have onehaplotype in common_the Asian haplotype. It is present in all the 19 families studied. Of the affectedindividuals in the 19 families, 27 were homozygous with the Asian haplotypes, five were heterozygous(Asian, S2), two were heterozygous (Asian, S1) and two were heterozygous (Asian, b-thalassaemia). In Saudi Arabia, four haplotypes were found: the Asian, S2 and S1, together with a rare Saudihaplotype (Kulozik 1986). Kulozik suggests that a West African population carrying the S1 haplotypemigrated to North Africa, to the Mediterranean and to the southwest of Saudi Arabia. The Asian Bsmutation may have originated in east Saudi Arabia, spreading to India with the Arab expansion in thefirst millennium AD, perhaps along the Indian-Arab trade route [9,20,21]. This study indicates that there are at least three different Bs haplotypes in the small islands of Bahrain, and that the Asian haplotype is predominant. The sickle-cell alleles in Bahrain probably

8/3/2019 Sickle Celled Aneamia
http://slidepdf.com/reader/full/sickle-celled-aneamia 13/13
derive from different sources, mainly Asian and partly African reflecting the migrating populations thathave passed through the country in the past. References 1. Mohammed AM et al. Haemoglobinopathies and glucose 6 phosphate dehydrogenase in hospitalpopulation in Bahrain. Annals of Saudi Medicine, 1992, 12:536-9. 2. Gelpi AP. Glucose-6-phosphate dehydrogenase deficiency, the sickling trait and malaria. Saudi
Arabia Journal of Pediatrics, 1967, 71:138-146. 3. Perrine RP et al. Natural history of sickle cell anaemia in Saudi Arabs; a study of 270 subjects.Annals of Internal Medicine, 1978, 88:1-16. 4. Pembrey ME et al. Foetal haemoglobin production and sickle gene in the oases of Eastern SaudiArabia. British Journal of Haematology, 1978, 40:415. 5. Powars DR et al. The natural history stroke in sickle cell disease. American Journal of Medicine,1978, 65:461-472.