sean k. wang may 2014 - stanford universityfx203jc8736/wang_sean_bio... · sean k. wang . may 2014....
TRANSCRIPT

Cas9-Mediated Genome Editing in Mouse Primary Cells
Sean K. Wang
May 2014

CAS9-MEDIATED GENOME EDITING IN MOUSE PRIMARY CELLS
An Honors Thesis Submitted to the Department of Biology
in partial fulfillment of the Honors Program STANFORD UNIVERSITY
by
Sean K. Wang May 2014

Cas9-Mediated Genome Editing in Mouse Primary Cells
by
Sean K. Wang
Approved for submittal to the Department of Biology for consideration of granting graduation with honors:
Research Sponsor Irving L. Weissman ______________________ Date ________________________
(signature) Second Reader Or Gozani ______________________ Date ________________________
(signature)

ACKNOWLEDGEMENTS
Thank you to James Y. Chen and Masanori Miyanishi for your day-to-day guidance,
patience, and enthusiasm throughout this project. I have learned so much about science
from you two and will continue to keep your snippets of advice with me wherever I go.
Thank you to Jonathan Tsai and Roy Louis Maute for offering reagents and your
expertise. Thank you to Mingye Feng and Ingrid Ibarra for your regular words of
encouragement and willingness to help. Thank you to Stephen Willingham for assistance
with ultracentrifugation and always friendly attitude. Thank you to Kyle M. Loh for your
endless knowledge and occasional inquiries into my social life.
Thank you to Professors Jamie Inam and Jennifer Wolf for your mentorship during Bing
Honors College. Thank you to Dr. Russ Carpenter for all your helpful feedback during
Bio 199W.
Thank you to Professor Or Gozani for agreeing to review my honors thesis despite your
hectic schedule. Thank you to Dr. Irving L. Weissman for training me as an aspiring
physician-scientist, making me feel welcome in the lab, pushing me to be critical of
reported findings, and serving as my researcher sponsor and esteemed mentor during my
time at Stanford.

Table of Contents
Abstract 1
Introduction 2
Methods 5
Construct Design and Synthesis 5
Vector Purification and Verification 6
CRISPR Design and Synthesis 6
Cell Culture Conditions 6
Embryonic Stem Cell Targeting and Validation 7
Lentivirus Production and Infection 8
Flow Cytometry and Fluorescence-Activated Cell Sorting 8
Results 9
Homology-Directed Integration of Cas9 in Mouse Embryonic Stem Cells 9
Toxicity of Cas9 Nucleases in Cultured Mouse Cells 10
Genome Editing in Cas9 Mouse Cells via Stable Guide RNA Integration 12
Discussion 14
Supplementary Figures and Tables 17
References 20

List of Figures
Figure 1. Homologous Recombination Schematic 2
Figure 2. Targeting Construct Design and Process 9
Figure 3. Integration of Cas9 in Mouse Embryonic Stem Cells 10
Figure 4. Toxicity of Constitutive Cas9 Expression in Mouse Cells 11
Figure 5. Genome Editing in Cas9 Mouse Cells via Guide RNA Delivery 13
Figure 6. Applications of a Cas9 Transgenic Mouse 16
Figure S1. Efficiency of Cas9- and Cas9n-Mediated Genome Editing 17

List of Tables
Table S1. PCR Primers 18
Table S2. Sequencing Primers 18
Table S3. CRISPR Guide RNA Sequences 19

1
Abstract
Gene targeting through Clustered Regularly Interspaced Short Palindromic
Repeats (CRISPR)/Cas9-mediated homologous recombination is a novel and powerful
technique with promising applications in human disease modeling and therapeutic
development. However, one major factor limiting genome editing efficiency is the time
and difficulty of obtaining a transgenic animal, which may require several generations of
breeding and expensive maintenance. While the establishment of an animal line with
endogenous Cas9 nuclease expression could potentially accelerate the study of both gene
knock-out and overexpression models, particularly in primary cells, previous work has
yet to test the feasibility of such a system. This project provided the first in vitro proof of
concept for a Cas9 transgenic mouse line as a research tool through the generation and
characterization of Cas9-expressing mouse cells. In NIH3T3 cells, constitutive
expression of either the Cas9 nuclease or nickase resulted in toxicity, with high levels of
wild-type Cas9 expression appearing to be lethal over time. Following delivery of a
CRISPR guide RNA sequence into cultured Cas9-expressing mouse cells via lentivirus,
efficient genome editing was also observed. These findings demonstrate the prospect of
high-speed multiplex genome engineering in mouse primary cells with future clinical
implications such as cancer modeling and gene therapy. Moving forward, the generation
of an inducible Cas9 transgenic animal should be pursued.

2
Introduction
For the past forty years, scientists have utilized an approach called genome
editing to investigate the roles of individual genes.1 Through the ability to add, alter, or
delete genetic information from living cells, this process has enabled an unequaled level
of precision in studies of gene function, allowing researchers and clinicians today to
better understand the mechanisms behind various genetic abnormalities.2 In animal
research, one of the leading applications of genome editing has been the generation of
accurate mouse models, which may subsequently be used to represent human diseases
during experimentation.3 Moreover, correction of deleterious point mutations in the
human genome through gene targeting may provide the basis to treat monogenic
disorders such as cystic fibrosis, sickle-cell anemia, Duchenne muscular dystrophy,
Huntington's disease, and hemophilia.4,5
Modification of endogenous genes in eukaryotic cells is typically achieved
through homologous recombination, one of two primary mechanisms by which inevitable
DNA breaks are repaired.6 Defined as using a homologous DNA template to controllably
restore breaks in the genome, homologous recombination makes possible the transfer of
modifications of a cloned gene into the genome of a living cell and is thus central to the
practice of genome editing.7 By introducing a cloned DNA sequence with “homologous
arms” identical to a specific chromosomal locus, the phenomenon allows researchers to
insert exogenous sequences at virtually any induced genomic break site with the outcome
of precise genome targeting (Fig. 1).8
Figure 1. Schematic of homologous recombination as a genome-editing strategy. Following the induction a
DNA break at a specific genomic locus, dividing cells which recognize homologous regions (blue) on an
introduced template or targeting vector may incorporate the exogenous sequence (green) during the DNA
repair process, resulting in a modified target gene.

3
In the absence of mutagenic agents, homologous recombination is an
exceptionally rare process, with an estimated frequency of 10-6 spontaneous occurrences
per mammalian cell gene or roughly 1-10 per cell division.9,10 To mediate site-specific
insertion of clinically or scientifically relevant DNA sequences into the genome, a
number of genome editing techniques have been developed to induce DNA cleavage,
including Zinc Finger Nucleases (ZFNs), Transcription Activator-Like Effector
Nucleases (TALENs), and more recently, the Clustered Regularly Interspaced Short
Palindromic Repeats (CRISPR)/Cas9 system.11 Consisting of the Cas9 nuclease and two
noncoding RNAs (crRNA and tracrRNA) which can be fused into a single guide RNA
(gRNA) transcript to recognize the target sequence, the CRISPR/Cas9 system exhibits
higher cutting efficiency and targeting frequency of mammalian cells compared to ZFNs
and TALENs.12 In fact, CRISPR/Cas9 nucleases have been able to improve the rate of
homologous recombination by up to 130-fold in yeast models, confirming their
designation as revolutionary tools for genome engineering.13
Several versions of the Cas9 nuclease have previously been characterized for
eukaryotic genome editing including wild-type Cas9, a D10A mutant nickase (Cas9n),
and various Cas9-transcription factor complexes (Cas9-VP16, Cas9-VP64, Cas9-
VP160).14 Whereas wild-type Cas9 normally represses a gene by producing a blunt-
ended, double-stranded break within the gRNA recognition sequence,15 Cas9n is thought
to cut only one of the two strands, resulting in a “nick.” In contrast, Cas9-transcription
factor complexes have been altered to act as transcriptional activators instead of
nucleases, allowing for gene-specific modulation and overexpression.14 For all three of
these varieties of Cas9, a single construct may be able to introduce several short gRNAs
each targeting a separate chromosomal locus, thereby facilitating the simultaneous
modification of multiple genes in a cell.12
While the CRISPR/Cas9 system has undoubtedly improved the process of gene
targeting, obtaining a transgenic animal under this method may still require two or three
successive generations of breeding and years of maintenance. Considering the versatility
of Cas9 nucleases, it may be advantageous to establish a mouse line with constitutive
Cas9 expression in order to accelerate the study of knock-out and overexpression models
pertinent to genetic disorders. If the generation of such a transgenic mouse is ultimately

4
feasible, primary cells from the animal could easily be edited by using lentivirus,
adenovirus, or other DNA delivery vehicles to introduce gRNA sequences corresponding
to each gene of interest. Consequently, the time and cost necessary to study how a gene
functions in particular cell populations would be greatly reduced.
With this broader goal in mind, my project aims to provide the first in vitro proof
of concept for a Cas9 transgenic mouse system through the generation and
characterization of Cas9-expressing mouse cells. By assessing the toxicity and genome-
editing efficiency of constitutive Cas9 expression as a research tool, I plan to demonstrate
the prospect of high-speed multiplex genome engineering in mouse primary cells with
clinical implications in disease modeling and gene therapy.

5
Methods
Construct Design and Synthesis
All constructs were first designed in silico using ApE plasmid editor and
synthesized according to the Golden Gate cloning strategy as detailed by Sanjana et al.16
In this approach, type IIS restriction endonuclease sites with unique overhangs were
introduced onto the ends of individual DNA fragments by primers and polymerase chain
reaction (PCR). Following digestion with the appropriate restriction enzymes, these
fragments were ligated into a vector backbone to generate each construct. Primers for
PCR and subsequent sequencing reactions were designed using online Primer-BLAST
(National Center for Biotechnology Information) and synthesized by Elim
Biopharmaceuticals (Table S1). To determine PCR conditions, annealing temperatures
and elongation times were estimated using IDT Oligo Analyzer (Integrated DNA
Technologies).
In the majority of cases, amplification by PCR was performed using Phusion
High-Fidelity DNA Polymerase (Thermo Scientific) in a 50 µL reaction. For regions with
high guanine-cytosine content (>70%) or longer sequences (>4 kb), respectively,
AmpliTaq Gold with GC Enhancer (Life Technologies) or TaKaRa LA Taq (Clontech)
was selected as the polymerase. To obtain fragments corresponding to elements of the
CRISPR/Cas9 system such as the Cas9 nucleases or tracrRNA, restriction enzymes (New
England Biolabs) were used to cut out the desired sequences from Addgene plasmids
pX330, pX335, pX260, and pLentiCRISPR. Template sequences to clone fluorescent
proteins (Zsgreen, TdTomato, mCerulean) and the neomycin resistance gene were
provided by collaborators in the lab.
To isolate individual fragments after PCR and restriction enzyme digest,
electrophoresis was performed using 0.5-2.0% agarose gels depending on the size of the
desired band. For all gels, DNA was stained using a 1:10000 dilution of SYBR Green
(Life Technologies) and visualized under ultraviolet illumination in a Gel Doc XR+
System (Bio-Rad). Upon excision from the gel with a clean scalpel, fragments were
purified using a gel extraction kit (Qiagen) and ligated into the appropriate vector
backbone containing ampicillin resistance, an origin of replication, and nucleotide
overhangs specific for the enzyme-digested product. Ligation for all constructs was

6
conducted in a 10 µL reaction with T7 ligase (New England Biolabs) at 23oC for at least
two hours.
Vector Purification and Verification
Ligated plasmids were transformed into competent E. coli cells (Invitrogen) via
electroporation if the vector size was over 9 kb, or chemical transformation in all other
cases. Electroporation was performed using the Nucleofector II (Amaxa Lonza) and
sterile 1 mm cuvettes according to the manufacturer’s protocol. For each construct, 3-5
individual colonies were picked from ampicillin plates and grown in LB ampicillin media
to shake at 37oC overnight. Plasmid from each colony was then isolated using a DNA
minipreparation kit (Qiagen).
To verify the sequence of each construct, DNA sequencing was performed by
Elim Biopharmaceuticals using multiple primer sets (Table S2), from which one colony
without mutations for each vector was expanded overnight in 200 mL of LB ampicillin
media. After 12-16 hours of shaking at 37oC, plasmid from the selected colony was
isolated using an endotoxin-free DNA maxipreparation kit (Qiagen).
CRISPR Design and Synthesis
To induce homology-directed integration of the Cas9 targeting construct into the
genome, the CRISPR/Cas9 genome editing system was employed. Targeting was
performed using six CRISPR guide RNAs which bound to specific regions of the Rosa26
locus (Table S3). Guide RNAs were designed using the online CRISPR Design Tool
(crispr.mit.edu) and generated by the slow annealing and phosphorylation of two
complementary oligonucleotides (Elim Biopharmaceuticals) with T4 polynucleotide
kinase (New England Biolabs) in a 10 µL reaction. A 1:250 dilution of the annealed
product was subsequently ligated into a pre-cut Cas9-vector backbone as described by
Cong et al.12 Plasmid purification and endotoxin removal for CRISPR constructs were
performed by an outside company, Transcriptic.
Cell Culture Conditions

7
C57BL/6 mouse embryonic stem cells (mESCs) were cultured in ESGRO
Complete Clonal Grade medium (Millipore) under the ‘1i’ method in the presence of the
leukemia inhibitory factor (LIF) cytokine. Following treatment with Accutase cell
detachment solution (Millipore) to dissociate cells from the plate, mESC colonies were
seeded at 10% confluence and incubated in 7.5% CO2 at 37oC. For all wells, culture
media was exchanged every 24 hours.
HEK293T and NIH3T3 cells were grown in Dulbecco's Modified Eagle Medium
supplemented with 10% fetal bovine serum and 1% penicillin streptomycin (Life
Technologies). Both cell lines were passaged every 48 hours using 0.25% trypsin-EDTA
(Gibco) and were maintained under humidified conditions in 5% CO2 at 37°C.
Embryonic Stem Cell Targeting and Validation
To mediate site-specific integration of the Cas9 targeting vector into the genome,
mESCs were transfected with a 1:4 ratio of the targeting construct and associated
CRISPR/Cas9 plasmid in a six-well plate using Effectene transfection reagent (Qiagen).
Two days following transfection, each well was treated with 100 µg/mL neomycin for a
week in order to positively select for drug-resistant integrants. Next, mESC colonies from
the well with the highest survival rate were passaged to remove dead cells and subjected
to Effectene-assisted transfection with a plasmid containing both Cre-recombinase and
puromycin resistance. After 24 hours, cells were treated with 0.5 µg/mL puromycin for
another two days to isolate colonies which had undergone Cre recombination and excised
the neomycin resistance cassette. Following targeting, 48 mESC colonies were picked
and moved to individual wells in 24-well plates to allow for clonal expansion.
To screen for correctly targeted mESCs exhibiting homology-directed integration,
genomic DNA was collected from single colonies using a DNA micropurification kit
(Qiagen). To validate whether or not integration occurred, colony DNA was subjected to
long-range PCR at the targeted locus using primers specific for both the homologous
arms and integrated sequence. All genomic DNA PCR was performed using TaKaRa Taq
DNA polymerase (Clontech) with product sizes estimated by gel electrophoresis. Any
colony failing to display an expected band was excluded from further analysis.

8
Lentivirus Production and Infection
Lentiviral particles were packaged by the co-transfection of HEK293T cells with
the viral vector and packaging plasmids pCAG-HIVgp, pRSV-Rev, and pENV-IRES-
puro. Transfection was achieved using X-tremeGENE HP DNA transfection reagent
(Roche) according to the manufacturer’s instructions, after which the cells were
incubated in six-well plates at 37oC for 48 hours. Supernatant from each well was then
harvested, passed through a 0.22 µm filter to remove cell debris, and frozen as 1 mL
aliquots at -80oC.
Virus titering was performed on NIH3T3 cells with a desired multiplicity of
infection (MOI) of 0.1. In cases of low titer, virus particles were concentrated via
ultracentrifugation using an SW32 Ti rotor (Beckman Coulter) at 28000 rpm for 90
minutes. Following aspiration of the supernatant, the resulting pellet was resuspended in
1 mL of culture media, passed again through a 0.22 µm filter, and frozen as 200 µL
aliquots at -80oC.
NIH3T3 cells were infected with lentivirus at approximately 15% confluence in
the presence of 8 µg/mL polybrene (Millipore). After five hours of incubation at 37oC,
infected media from each well was aspirated and exchanged for sterile culture media.
Cells were maintained for at least 48 hours prior to analysis.
Flow Cytometry and Fluorescence-Activated Cell Sorting
Flow cytometric analysis and cell sorting were performed on a BD FACSAria II
instrument using the FACSDiva software (BD Biosciences). To assess levels of
fluorescence, cells from each specimen were trypsinized and centrifuged at 1200 g for 5
minutes, with the resulting pellet resuspended in 200 µL FACS buffer (phosphate
buffered saline with 2% fetal bovine serum). Single cell suspensions were sampled with a
low flow rate until at least 50000 events had been recorded. Data were analyzed using
FlowJo 9 software by gating on a polygonal region containing approximately 60% of the
cell population in the forward scatter-side scatter plot. A 1:1000 dilution of Sytox Red
(Life Technologies) was used exclude dead cells and debris. For each experiment,
triplicate cultures were measured.
9
Results
Homology-Directed Integration of Cas9 in Mouse Embryonic Stem Cells
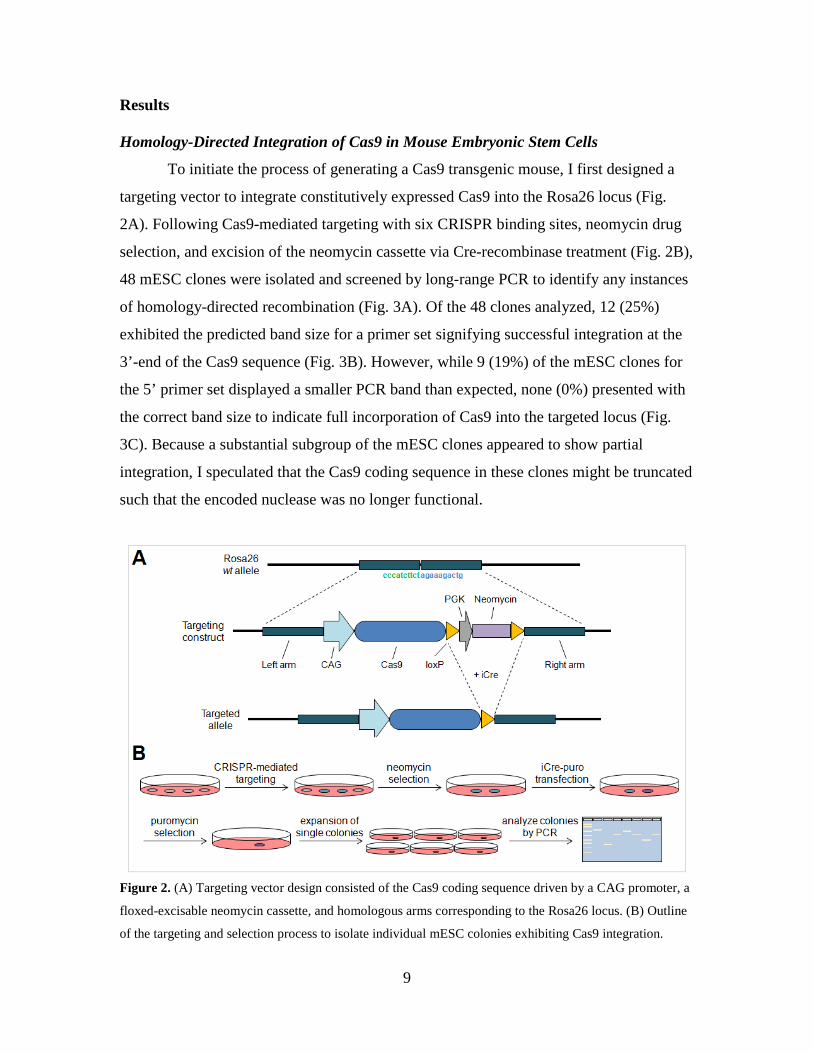
To initiate the process of generating a Cas9 transgenic mouse, I first designed a
targeting vector to integrate constitutively expressed Cas9 into the Rosa26 locus (Fig.
2A). Following Cas9-mediated targeting with six CRISPR binding sites, neomycin drug
selection, and excision of the neomycin cassette via Cre-recombinase treatment (Fig. 2B),
48 mESC clones were isolated and screened by long-range PCR to identify any instances
of homology-directed recombination (Fig. 3A). Of the 48 clones analyzed, 12 (25%)
exhibited the predicted band size for a primer set signifying successful integration at the
3’-end of the Cas9 sequence (Fig. 3B). However, while 9 (19%) of the mESC clones for
the 5’ primer set displayed a smaller PCR band than expected, none (0%) presented with
the correct band size to indicate full incorporation of Cas9 into the targeted locus (Fig.
3C). Because a substantial subgroup of the mESC clones appeared to show partial
integration, I speculated that the Cas9 coding sequence in these clones might be truncated
such that the encoded nuclease was no longer functional.
Figure 2. (A) Targeting vector design consisted of the Cas9 coding sequence driven by a CAG promoter, a
floxed-excisable neomycin cassette, and homologous arms corresponding to the Rosa26 locus. (B) Outline
of the targeting and selection process to isolate individual mESC colonies exhibiting Cas9 integration.

10
Figure 3. (A) Long-range PCR primer design to analyze the 48 selected mESC colonies for specific
integration of the Cas9 targeting construct. (B) Integration of the 3’-coding sequence at the Rosa26 locus
was indicated by a 4882-bp band. (C) Integration of the 5’-coding sequence from the same set of mESC
colonies was indicated by a 5235-bp band.
Toxicity of Cas9 Nucleases in Cultured Mouse Cells
In an earlier experiment, several members of the lab and I estimated the efficiency
of Cas9- and Cas9n-mediated homology-directed recombination in mESCs to be 67%
and 17%, respectively (Fig. S1A, S1B). Considering the lack of successful integrants for
the Cas9 sequence despite earlier demonstrations of feasible genome editing in mESCs, I
suspected that constitutive expression of particular elements from the CRISPR/Cas9
system might in fact be toxic to mouse cells. In order to investigate the potential toxicity
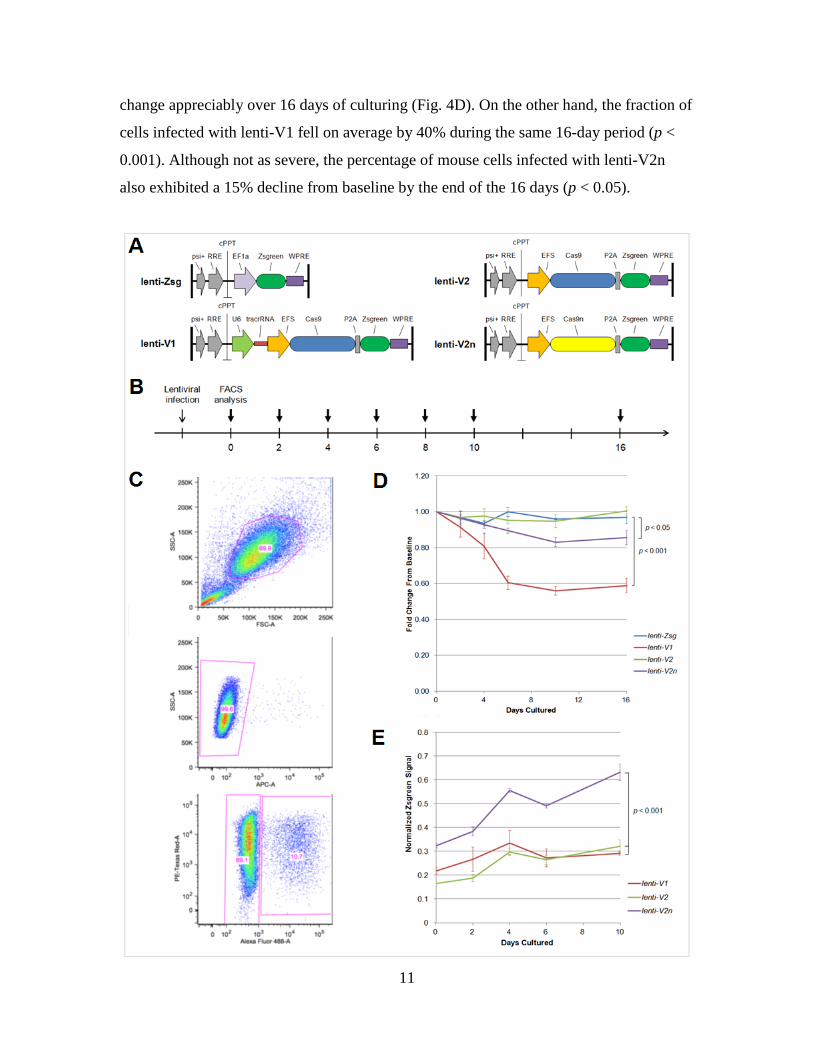
of continuous Cas9 expression, I constructed a set of lentiviral vectors to stably integrate
Cas9 with or without the tracrRNA sequence into the genome of TdTomato-expressing
mouse NIH3T3 cells (Fig. 4A). As measured by Zsgreen expression in flow cytometry
(Fig. 4B, C), the percentage of infected cells for lenti-Zsg and lenti-V2 samples did not
11
change appreciably over 16 days of culturing (Fig. 4D). On the other hand, the fraction of
cells infected with lenti-V1 fell on average by 40% during the same 16-day period (p <
0.001). Although not as severe, the percentage of mouse cells infected with lenti-V2n
also exhibited a 15% decline from baseline by the end of the 16 days (p < 0.05).

12
Figure 4. (A) Lentiviral constructs to assess the toxicity of stable Cas9 expression. Lenti-V1 consisted of
the Cas9 nuclease coupled to a Zsgreen fluorescent protein sequence with expression of the tracrRNA but
not crRNA recognition sequence. Lenti-V2 and lenti-V2n consisted of the Cas9 nuclease and nickase,
respectively, coupled to a Zsgreen fluorescent protein sequence without expression of the tracrRNA or
chRNA. Lenti-Zsg was designed as a Zsgreen-only control. (B) Timeline depicting time points of flow
cytometry analysis following initial lentiviral infection. (C) Representative flow cytometry gating for
NIH3T3 cells. Following exclusion of doublet events and Sytox Red-positive cells, samples were analyzed
for TdTomato and ZsGreen expression using the PE-Texas Red and Alexa Fluor 488 channels,
respectively. (D) Fold change of Zsgreen-positive cells over 16 days of culturing. Baseline was set as two
days after initial lentiviral infection. P-values were calculated using a two-tailed, unpaired Student’s t-test.
(E) Mean ZsGreen intensity of infected cells as measured by flow cytometry. Signal strength at each time
point was normalized with respect to the lenti-Zsg control.
Because lentiviral vectors for lenti-V1, lenti-V2, and lenti-V2n were designed
using the identical EFS promoter sequence and P2A self-cleaving peptide, the relative
expression level of Cas9 in each infected population could be roughly estimated by the
average Zsgreen signal strength.17 Based on this approximation, mean Zsgreen signal
intensity and accordingly mean Cas9 expression level of infected cells at every time point
were higher in lenti-V2n samples compared to lenti-V1 and lenti-V2 cultures (p < 0.001,
Fig. 4E).
Genome Editing in Cas9-Expressing Mouse Cells via Stable Guide RNA Integration
Having obtained mouse cells with constitutive Cas9 expression, I lastly was
curious to test whether a separately introduced vector containing the gRNA sequence
could still be used to induce efficient genome editing. To assess this approach and hence
the feasibility of modifying primary cells via lentivirus in the proposed Cas9 transgenic
mouse, I synthesized another lentiviral construct labeled with the mCerulean fluorescent
protein (Fig. 5A) and stably integrated a gRNA sequence targeting the TdTomato gene
into Cas9- and TdTomato-expressing NIH3T3 cells.
Within 48 hours of infection, over 20% of cells expressing both wild-type Cas9
and the gRNA were defined as negative for the TdTomato signal by flow cytometry (Fig.
5B, 5C, 5D). In contrast, less than 3% of cells expressing wild-type Cas9 but not the
gRNA were found to be TdTomato-negative. For cultures subjected to Cas9n-mediated

13
knock-out, only 5.52% of cells infected with both the nickase and gRNA were negative
for TdTomato compared to 3.94% with the nickase alone after 48 hours.
Figure 5. (A) Lentiviral construct to stably integrate a guide RNA sequence targeting the TdTomato gene.
Infected NIH3T3 cells were identified via flow cytometry based on expression of the mCerulean
fluorescent protein in the AmCyan channel. (B) Representative flow cytometry gating to analyze the
Zsgreen-positive and Zsgreen/mCerulean double-positive subpopulations. Expression levels of the
TdTomato fluorophore were assessed using the PE-Texas Red channel. (C) Histograms detailing
population-level expression of TdTomato in Cas9-expressing cells with (blue) and without (green) stable
guide RNA integration. Values indicate mean TdTomato signal intensity. (D) Percentage of TdTomato-
negative events in each culture of Cas9-expressing NIH3T3 cells with and without stable integration of the
guide RNA. Cells were analyzed 48 hours after lentiviral infection.

14
Discussion
Within the past three years, CRISPR/Cas9 technology has become one of the
most widely utilized research tools to date, revolutionizing the speed, cost, and precision
of eukaryotic genome editing and disease modeling.18,19 Consistent with the published
literature, earlier experiments from our lab found wild-type Cas9 to be highly efficient in
inducing homology-directed recombination, producing complete and site-specific
integration of the exogenous sequence in up to 67% of mESC clones after neomycin
treatment. While our estimated efficiency of isolating homologous recombinants with the
Cas9 nickase was lower at 17%, this targeting frequency was still favorable and may
have additionally benefited from a reduced likelihood of mutagenesis near the CRISPR
target site.20 Overall, compared to previous ZFN- or TALEN-based methods for genome
engineering,11 CRISPR/Cas9 thus represents a significant scientific advancement through
its immediate acceleration of genetic studies across a spectrum of research fields.
Given the many advantages of the Cas9 nucleases in achieving genomic
modifications, it is evident that the generation of a Cas9 transgenic mouse would present
a powerful means to expedite the study of important biological questions. Nonetheless,
my thesis reveals several unexpected obstacles which may compromise the feasibility of
such a system. First, as suggested by the declining fractions of cells infected with lenti-
V1 and lenti-V2n, constitutive expression of either Cas9 or Cas9n may be toxic to mouse
cells in as little as one to two weeks. Based on flow cytometry analysis for lenti-V1 in
particular, continuous expression of the noncoding tracrRNA sequence may exacerbate
Cas9 toxicity even if the chRNA recognition site is absent. Furthermore, as shown by the
weaker Zsgreen intensity in lenti-V1 and lenti-V2 cultures with respect to lenti-V2n,
there appears to be a lower tolerance for wild-type Cas9 expression in mouse cells
compared to the Cas9 nickase. In fact, accounting for the failure to integrate Cas9 into the
Rosa26 locus despite analyzing 48 mESC clones, I conclude that high levels of Cas9
expression in mouse cells are likely lethal over time, thereby precluding the prospect of
generating a constitutive Cas9 transgenic mouse. In agreement with this notion, only
mESC clones which failed to integrate the entire Cas9 coding sequence were observed by
long-range PCR, hinting that any clone which did succeed in homology-directed
recombination presumably did not survive.

15
Recently with the advent of lentiviral CRISPR/Cas9 plasmids, several groups
have proposed using stable integration of Cas9 to perform genome-scale loss-of-function
screening in humans.21,22 In these assays, candidate genes for further study are
characterized by the depletion or enrichment of cells expressing both Cas9 and a specific
gRNA library sequence, generally for durations of up to two weeks. Notably, the data
presented here suggest that although the majority of cells in these genomic screens may
experience comparable levels of Cas9 expression, there is a chance that Cas9-related
toxicity may lead to false results by biasing the cells toward depletion. The duration of
constitutive Cas9 use should therefore be considered when conducting such a screen to
identify novel targets.
Despite foreseeable issues with constitutive Cas9 expression, it may still be
possible to generate an inducible Cas9 transgenic mouse. However, while the genome-
editing efficiency of wild-type Cas9 appears preferable to that of the nickase, there is a
risk that even transient Cas9 expression might be toxic in a transgenic animal. To address
these concerns, one option moving forward could be to combine a tightly controlled
inducible promoter with the paired nickase strategy, in which two gRNAs on opposite
strands of the same locus ‘nick’ adjacent regions to mimic a double stranded break.23
Because I expect an inducible Cas9 nickase to be more tolerable than either an inducible
Cas9 nuclease or a constitutively expressed nickase, this is the version I plan to pursue in
generating the Cas9 transgenic mouse line.
From a broader perspective, the ultimate advantage of a Cas9 transgenic animal is
a higher-throughput method to rapidly modify candidate genes in primary cells. As an
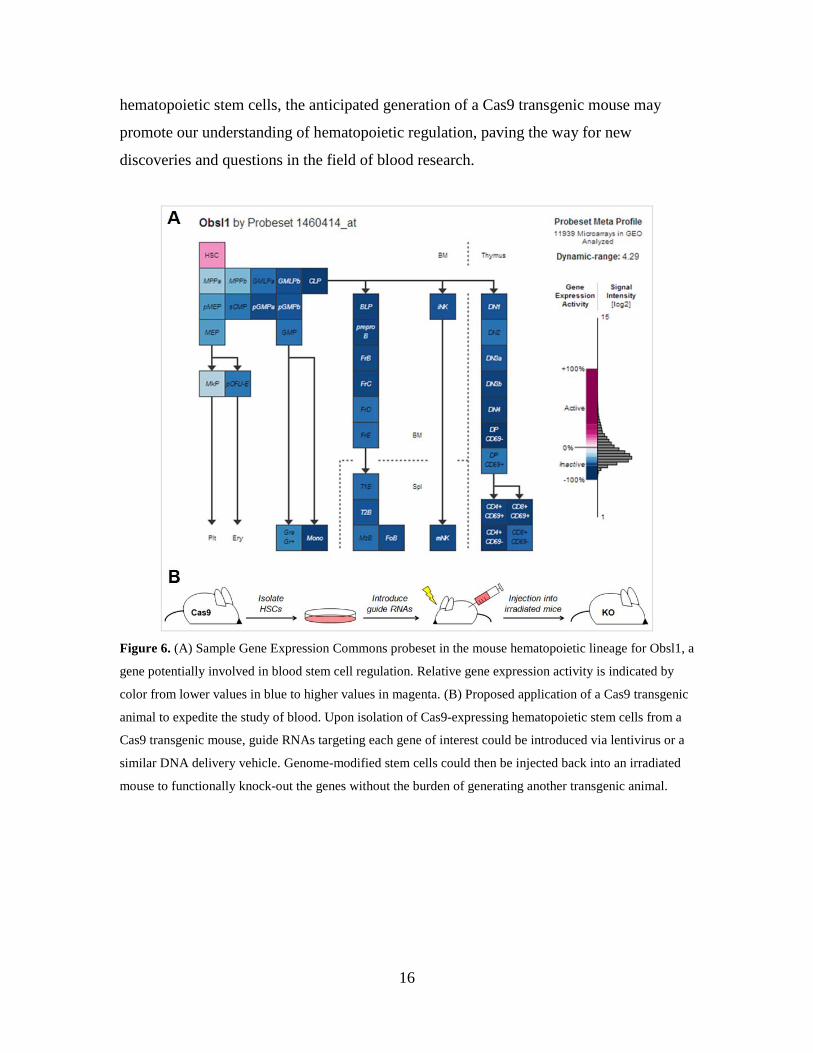
aspiring researcher interested in hematopoietic regulation, I intend to first use gene
profiling resources such as the Gene Expression Commons to identify a list of genes
potentially involved in self-renewal of the blood stem cell (Fig. 6A).24 Then, utilizing the
genome-editing ability of a Cas9 transgenic mouse, I plan to isolate Cas9-expressing
hematopoietic stem cells and introduce gRNAs corresponding to each candidate gene to
obtain precisely modified blood stem cells in vitro (Fig. 6B). Upon injection of these
stem cells into irradiated mice, I will be able to assess the likely role of each gene in
relation to hematopoiesis, all without having to generate a new knock-in or knock-out
animal. Finally, because there currently exists no efficient method to edit the genome of
16
hematopoietic stem cells, the anticipated generation of a Cas9 transgenic mouse may
promote our understanding of hematopoietic regulation, paving the way for new
discoveries and questions in the field of blood research.
Figure 6. (A) Sample Gene Expression Commons probeset in the mouse hematopoietic lineage for Obsl1, a
gene potentially involved in blood stem cell regulation. Relative gene expression activity is indicated by
color from lower values in blue to higher values in magenta. (B) Proposed application of a Cas9 transgenic
animal to expedite the study of blood. Upon isolation of Cas9-expressing hematopoietic stem cells from a
Cas9 transgenic mouse, guide RNAs targeting each gene of interest could be introduced via lentivirus or a
similar DNA delivery vehicle. Genome-modified stem cells could then be injected back into an irradiated
mouse to functionally knock-out the genes without the burden of generating another transgenic animal.

17
Supplementary Figures and Tables
Figure S1. (A) Efficiency of Cas9-mediated targeting at the Obsl1 locus as estimated by long-range PCR.
Knock-in and wild-type were indicated by 5225-bp and 2813-bp bands, respectively. Of the 12 mESC
clones examined, 8 (67%) exhibited the knock-in band size while 3 (25%) showed a smaller band
indicating incomplete integration. For one clone (8%), PCR failed to detect a knock-in band, suggesting
random integration of the knock-in sequence at some other region of the genome. (B) Analogous PCR
result of mESC clones targeted with the Cas9n nickase. Of the 12 mESC clones, 2 (17%) displayed the
knock-in band size while 5 (42%) appeared to be instances of incomplete integration. The remaining 5
(42%) clones did not present a knock-in band and were likely cases of random integration.

18
Table S1. PCR Primers
Primer Set Forward and Reverse Sequence Rosa26 left arm
F: ATTGACGGTCTCTTGACAAAGGAACCACCTTTTACAGATGT R: CAGAGTGGTCTCAAGCTAGAAGATGGGCGGGAGTC
Rosa26 right arm
F: ATTGACGGTCTCTCTTCAGAAAGACTGGAGTTGCAGATCAC R: CAGAGTGGTCTCAGAGTGGTGGGCAGGAATGCG
CAG promoter F: ATTGACGGTCTCTTTCTTAGTTATTAATAGTAATCAATTACGGGGTCAT R: CAGAGTGGTCTCACCGGGCCGCCGGTCACA
Cas9 adapter F: ATTGACCGTCTCACCGGAGAGACCATGGCCCAGTCCAAGCA R: CAAAGTCGTCTCTCTTCTGAGACCTCAGGGCAAGGCGGAG
2a-Zsgreen F: TACATTGCTAGCGCCACCAACTTCAGCCTGCT R: ATAGATGGTCTCACGCGTTTAGGGCAAGGCGGAGCC
lentiCRISPR adapter
F: GTTACATCGCGGCCGCTGATCTTCAG R: ATATCTGAATTCTCTGGGTCCCCTCGGGG
Cas9n adapter F: CACCGTCTCGAATTCTAGATCTTGAGACAAATGGC R: ATTTATCGTCTCAGCCAGGCCGATGCTGTACT
mCerulean F: ATAAATGGATCCGCCACCATGGATTACAAAGACGATGACGATAAGAT R: GCTCGCCGAACGCGTTTACTTGTACAGCTCGTCCATG
Neomycin cassette
F: TGCGTCCGTCTCTCTTCAGAGACCGGCAGAATGCTTAATGAATT R: AGTCAGCGTCTCAGAGTCTTGAGCAACTGATAGCTGTC
3’ Rosa26 long range PCR
F: CAAGAAAATCGAGTGCTTCGACTCC R: GAGGAGAGGCGTTCAGGAAGATTAT
5’ Rosa26 long range PCR
F: TGGTCTTCTGTATCCCACAAGTCTG R: CGTTTTCCTCATTGTCCAGGAAGTC
Table S2. Sequencing Primers
Primer Sequence Rosa26 left-F CGAGGCGGATCACAAGCAAT Rosa26 right-R CGTTTCCGACTTGAGTTGCC gRNA-F ACATGTGAGGGCCTATTTCCC D10A mut-F AAGGGATGGTTGGTTGGTGG Cas9-F GTGATCACCGACGAGTACAAG Cas9-R GCCCACAGAGTTGGTGCCGAT EFS-F TTTTCGCAACGGGTTTGCC EFS-R AGCAGCGTATCCACATAGCG Zsgreen-F TTCCTGTTCGAGGACGGCGC lentiCRISPR-F GTTAGGCAGGGATATTCACCA Puromycin-F GCGCACCTGGTGCATGACCC Neomycin-R GACTGCCTTGGGAAAAGCGCC

19
Table S3. CRISPR Guide RNA Sequences
gRNA Sequence Rosa26-1 GACUGGAGUUGCAGAUCACGAGG Rosa26-2 ACUGGAGUUGCAGAUCACGAGGG Rosa26-3 UGGGCGGGAGUCUUCUGGGCAGG Rosa26-4 AAGAUGGGCGGGAGUCUUCUGGG Rosa26-5 GGAGUGUUGCAAUACCUUUCUGG Rosa26-6 CGACAAAACCGAAAAUCUGUGGG TdTomato CGCCCUCGAUCUCGAACUCGUGG

20
References 1. Smith HW, Muller WJ. Transgenic mouse models--a seminal breakthrough in
oncogene research. Cold Spring Harb Protoc. 2013;2013(12).
2. Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. Genome editing with engineered zinc finger nucleases. Nat Rev Genet. 2010 Sep;11(9):636–46.
3. Park K-E, Telugu BPVL. Role of stem cells in large animal genetic engineering in the TALENs-CRISPR era. Reprod Fertil Dev. 2013 Dec;26(1):65–73.
4. Holmes MC, Urnov FD, Lee Y-L, Miller JC, Gregory PD. 713. Gene Correction Therapy Using Designed Zinc Finger-Based Endonucleases. Mol Ther. 2004 May;9(S1):S272–S272.
5. Campeau PM, Scriver CR, Mitchell JJ. A 25-year longitudinal analysis of treatment efficacy in inborn errors of metabolism. Mol Genet Metab. 2008 Sep;95(1–2):11–6.
6. Wei C, Liu J, Yu Z, Zhang B, Gao G, Jiao R. TALEN or Cas9 – Rapid, Efficient and Specific Choices for Genome Modifications. J Genet Genomics. 2013 Jun 20;40(6):281–9.
7. Capecchi MR. Altering the genome by homologous recombination. Science. 1989;244(4910):1288–92.
8. Zu Y, Tong X, Wang Z, Liu D, Pan R, Li Z, et al. TALEN-mediated precise genome modification by homologous recombination in zebrafish. Nat Methods. 2013 Apr;10(4):329–31.
9. Lin FL, Sternberg N. Homologous recombination between overlapping thymidine kinase gene fragments stably inserted into a mouse cell genome. Mol Cell Biol. 1984 May;4(5):852–61.
10. Hendricks CA, Almeida KH, Stitt MS, Jonnalagadda VS, Rugo RE, Kerrison GF, et al. Spontaneous mitotic homologous recombination at an enhanced yellow fluorescent protein (EYFP) cDNA direct repeat in transgenic mice. Proc Natl Acad Sci. 2003 May 27;100(11):6325–30.
11. Gaj T, Gersbach CA, Barbas CF 3rd. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013 Jul;31(7):397–405.
12. Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013 Feb 15;339(6121):819–23.
13. DiCarlo JE, Norville JE, Mali P, Rios X, Aach J, Church GM. Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res. 2013 Mar 4;4336–43.

21
14. Mali P, Aach J, Stranges PB, Esvelt KM, Moosburner M, Kosuri S, et al. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol. 2013 Sep;31(9):833–8.
15. Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, et al. Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression. Cell. 2013 Feb 28;152(5):1173–83.
16. Sanjana NE, Cong L, Zhou Y, Cunniff MM, Feng G, Zhang F. A transcription activator-like effector toolbox for genome engineering. Nat Protoc. 2012 Jan;7(1):171–92.
17. Kim JH, Lee S-R, Li L-H, Park H-J, Park J-H, Lee KY, et al. High Cleavage Efficiency of a 2A Peptide Derived from Porcine Teschovirus-1 in Human Cell Lines, Zebrafish and Mice. PLoS ONE. 2011 Apr 29;6(4):e18556.
18. Zhang F, Wen Y, Guo X. CRISPR/Cas9 for genome editing: progress, implications and challenges. Hum Mol Genet. 2014 Mar 20;ddu125.
19. Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013 Nov;8(11):2281–308.
20. Shen B, Zhang W, Zhang J, Zhou J, Wang J, Chen L, et al. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat Methods. 2014 Apr;11(4):399–402.
21. Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014 Jan 3;343(6166):84–7.
22. Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014 Jan 3;343(6166):80–4.
23. Ran FA, Hsu PD, Lin C-Y, Gootenberg JS, Konermann S, Trevino AE, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013 Sep 12;154(6):1380–9.
24. Seita J, Sahoo D, Rossi DJ, Bhattacharya D, Serwold T, Inlay MA, et al. Gene Expression Commons: an open platform for absolute gene expression profiling. PloS One. 2012;7(7):e40321.