role of protein kinase c α in endothelin-1 stimulation of cytosolic phospholipase a2 and...
TRANSCRIPT

Ž .Biochimica et Biophysica Acta 1392 1998 127–144
Role of protein kinase C a in endothelin-1 stimulation of cytosolicphospholipase A and arachidonic acid release in cultured cat iris2
sphincter smooth muscle cells
Shahid Husain, Ata A. Abdel-Latif )
Department of Biochemistry and Molecular Biology, Medical College of Georgia, Augusta, GA 30912-2100, USA
Received 1 October 1997; revised 9 December 1997; accepted 6 January 1998
Abstract
Ž . Ž .We have investigated the role and mechanism of protein kinase C PKC isoforms in endothelin-1 ET-1 -inducedŽ . Ž .arachidonic acid AA release in cat iris sphincter smooth muscle CISM cells. ET-1 increased AA release in a
Ž . Ž . Ž .concentration EC s8 nM and time-dependent t s1.2 min manner. Cytosolic phospholipase A cPLA , but not50 1r2 2 2Ž .phospholipase C PLC , is involved in the liberation of AA in the stimulated cells. This conclusion is supported by the
findings that ET-1-induced AA release is inhibited by AACOCF , quinacrine and manoalide, PLA inhibitors, but not by3 2
U-73122, a PLC inhibitor, or by RHC-80267, a diacylglycerol lipase inhibitor. A role for PKC in ET-1-induced AA releaseis supported by the findings that the phorbol ester, PDBu, increased AA release by 96%, that prolonged treatment of thecells with PDBu resulted in the selective down regulation of PKCa and the complete inhibition of ET-1-induced AArelease, and that pretreatment of the cells with staurosporine or RO 31-8220, PKC inhibitors, blocked the ET-1-induced AArelease. Go-6976, a compound that inhibits PKCa and b specifically, blocked ET-1-induced AA release in a¨
Ž .concentration-dependent manner with an IC value of 8 nM. Thymeatoxin 0.1 mM , a specific activator of PKCa , b , and50
g induced a 150% increase in AA release. Treatment of the cells with ET-1 caused significant translocation of PKCa , butnot PKCb , from cytosol to the particulate fraction. These results suggest that PKCa plays a critical role in ET-1-inducedAA release in these cells. Immunochemical analysis revealed the presence of cPLA , p42mapk and p44mapk in the CISM2
cells. The data presented are consistent with a role for PKCa , but not for p42rp44 mitogen-activated protein kinaseŽ . Ž .MAPK , in cPLA activation and AA release in ET-1-stimulated CISM cells since: i the PKC inhibitor, RO 31-8220,2
inhibited ET-1-induced AA release, cPLA phosphorylation and cPLA activity, but had no inhibitory effect on p42rp442 2Ž .MAPK activation, ii genistein, a tyrosine kinase inhibitor, inhibited ET-1-stimulated MAPK activity but had no inhibitory
Abbreviations: ET, endothelin; cPLA , cytosolic phospholipase A ; AA, arachidonic acid; PG, prostaglandin; PLC, phospholipase C;2 2
PKC, protein kinase C; CISM cells, cat iris sphincter smooth muscle cells; DAG, 1,2-diacylglycerol; PDBu, phorbol 12,13-dibutyrate;MBP, myelin basic protein
) Corresponding author. Fax: q1-706-721-6608; E-mail: [email protected]
0005-2760r98r$19.00 q 1998 Elsevier Science B.V. All rights reserved.Ž .PII S0005-2760 98 00011-3

( )S. Husain, A.A. Abdel-LatifrBiochimica et Biophysica Acta 1392 1998 127–144128
effect on AA release in the ET-1-stimulated cells. We conclude that in CISM cells, ET-1 activates PKCa , which activatescPLA , which liberates AA for prostaglandin synthesis. q 1998 Elsevier Science B.V. All rights reserved.2
Keywords: Endothelin-1; Protein kinase C; Cytosolic phospholipase A ; Arachidonic acid; Iris sphincter smooth muscle cell2
1. Introduction
Ž .Endothelin-1 ET-1 is a 21-amino acid peptide,w xinitially purified from vascular endothelial cells 1 .
The ETs are a family of three peptides, termed ET-1,ET-2, and ET-3, which have a variety of biologicaleffects. These include the contraction of varioussmooth muscles, cardiac inotropism, modulation ofother hormone and cytokine production, regulation oftransport in the renal tubule, and stimulation of mito-
Ž w x.genesis reviewed in Ref. 2 . The ETs trigger theiractions by activating specific cell-surface receptorsthat belong to the heptahelical G-protein-coupled su-
w xperfamily of receptors 2,3 . In mammalian tissues,two major ET receptors have been identified, namely
w xET and ET 2 . In smooth muscle, the ET receptorA B AŽ .is coupled to the activation of phospholipase C PLCŽ .and to the generation of 1,2-diacylglycerol DAG
Ž . w xand inositol trisphosphate IP and contraction 1–4 .3
In earlier studies from this laboratory, we reportedthat in rabbit iris sphincter smooth muscle, ET-1 is apotent agonist for phosphoinositide hydrolysis and
w xmuscle contraction 5 , and furthermore, in this tis-Ž .sue, the peptide induces arachidonic acid AA re-
lease, through activation of phospholipase A 2Ž . Ž . w xPLA , for prostaglandin PG synthesis 6 , and2
phosphatidic acid via activation of phospholipase Dw x7 . Moreover, in immortalized cat iris sphinctersmooth muscle cells, ET-1 markedly stimulated the
Ž .activities of PLA , PLC and phospholipase D PLD2w x8 . More recently, we reported that PGE could2
mediate the stimulatory effects of ET-1 on cAMPw xaccumulation in both the sphincter 9 and ciliary
w xsmooth muscles 10 . It was concluded from thesefindings that ET-1 binds to the ET receptor subtypeA
to activate PLA and to release AA for PG synthesis.2
PGs, such as PGE , may then interact with the EP2 2
receptor to stimulate the adenylate cyclase system.Ž .While the protein kinase C PKC activator, phorbol
Ž .12,13-dibutyrate PDBu , markedly increases AA re-w xlease and PG synthesis in the iris sphincter 6 , the
role of PKC in activating PLA in the ET-1-stimu-2
lated AA release in this smooth muscle remainsunexplored. Phorbol esters have previously been re-ported to stimulate AA release in vascular smooth
w xmuscle cells 4 , human pericardial smooth musclew x w xcells 11 and rat aortic rings 12 .
There is accumulating evidence which indicatesthat in a wide variety of tissues, the key enzymeresponsible for AA release for eicosanoid biosynthe-
w xsis is cPLA 13–15 . The different isoforms of2
PLA have been subdivided into two main classes:2Ž .the 14-kDa secreted forms include type I pancreatic
Ž .and type II secretory, sPLA and the 85-kDa non-2Ž . w xsecreted form, namely, type IV cPLA 16 . cPLA2 2
is unique among the PLA s known to date since it is2
a receptor-regulated, Ca2q-dependent PLA that as-2
sociates with its phospholipid substrate when theconcentration of free Ca2q is raised from basal levelsŽ .100 nM to concentrations typically found in acti-
Ž . w xvated cells G300 nM 15 . Despite numerous inves-tigations, the regulation of cPLA is not completely2
understood. There is evidence that this regulationinvolves an increase in intracellular calcium concen-tration, mediated by activation of PLC, and phospho-rylation of the cPLA polypeptide by a kinase acti-2
w xvated during cell stimulation, such as PKC 17 .Thus, phorbol esters, the PKC activators, and calciumionophores A-23187 and ionomycin have been shownto increase AA release and PG synthesis in a wide
w xvariety of isolated cells 18–28 . Involvement of mi-Ž .togen-activated protein kinase MAPK in enhancing
cPLA activation was reported in Chinese hamster2w xovary cells 29 . In addition, ET-1 stimulated MAPK
w xactivity in cardiac myocytes 30 and bovine trachealw xsmooth muscle cells 31 .
There is little information about the role and regu-lation of cPLA in agonist-induced AA release in2
smooth muscle. The purpose of the present study wasto determine the role of PKC and its isoforms inET-1-induced AA release in cultured cat iris sphinc-
Ž .ter smooth muscle CISM cells. The results obtaineddemonstrate that ET-1 is a potent agonist for AArelease in these cells, that the peptide induces a

( )S. Husain, A.A. Abdel-LatifrBiochimica et Biophysica Acta 1392 1998 127–144 129
PKC-dependent phosphorylation and activation ofcPLA , that PKCa is the isoform mainly involved in2
the stimulated AA release. Furthermore, the datapresented do not support a role for p42rp44 MAPKin cPLA activation and AA release in ET-1-stimu-2
lated CISM cells. This is the first report on theinvolvement of PKCa isoform in agonist-inducedAA release in smooth muscle.
2. Materials and methods
2.1. Materials
ŽAffinity purified polyclonal anti-PKC isoforms a ,.b , g , d , ´ , h and z antibodies were purchased
Ž .from Gibco BRL Gaithersburg, MD . PolyclonalŽ .anti-MAPK R2 Erk1-CT antibodies, recognizing
p42mapk, p43mapk and p44mapk, and polyclonal 14-kDasPLA antibodies were purchased from Upstate2
Ž .Biotechnology Lake Placid, NY . Cytosolic PLA 2Ž .85-kDa polyclonal antibodies were obtained through
Žthe courtesy of the Genetics Institute Cambridge,. w 32 x ŽMA . g -P ATP Specific Activity, 3000 Ci
y1. w3 x Žmmol and H arachidonic acid Specific Activ-y1.ity, 184.6 Ci mmol and L-3-phosphatidylcholine
w 14 x Ž1-stearoyl-2- 1- C arachidonyl specific activity 53y1.m Ci mmol were obtained from Amersham Life
Ž .Science Arlington Heights, IL and Dupont NewŽ .England Nuclear Boston, MA , respectively. ET-1
Žwas obtained from Peptide International Louisville,.KY . RO 31-8220, staurosporine, thymeatoxin, Go-¨
6976 and arachidonyltrifluoromethyl ketoneŽ . ŽAACOCF were purchased from Calbiochem La3
. Ž .Jolla, CA . PDBu, Myelin Basic Protein MBP , Pro-tein A-Agarose, quinacrine dihydrochloride and
Žmanoalide were purchased from Sigma Chemical St..Louis, MO . U-73122 was purchased from Research
Ž .Biochemical International Natick, MA and RHC-Ž80267 from Biomol Research Laboratories Plymouth
.Meeting, PA . Fetal Bovine Serum was obtainedfrom Atlanta Biological and all other Cell Culture
Žsupplies were obtained from Cell Gro Herndon,.VA .
2.2. Methods
2.2.1. Cell cultureIris sphincter smooth muscle cells were isolated
from 4–6 months old cats as described previously
w x32 . Cat eyes were obtained through the courtesy ofRichmond County Animal Control, Augusta, GA.Briefly, the eyes were enucleated immediately afterdeath and transported to the laboratory packed in ice.The sphincter muscle was dissected out, furthercleaned and cut into 1–2 mm2 pieces. The explantswere placed in Dulbecco’s modified eagle mediumŽ . y1DMEM containing 2 mg ml collagenase type IA,
Ž . y110% fetal bovine serum FBS and 50 mg mlgentamicin and then incubated for 1–2 h at 378C withoccasional gentle shaking. The major parts of theexplants were then dispersed into single cells orgroups of 2–5 cells. The cell suspension was cen-trifuged at 200=g for 7 min and resuspended inequal parts of DMEM and Ham’s F-12 supplemented
Ž .with 10% vrv heat inactivated FBS and 100 unitsmly1 penicillin, 100 mg mly1 streptomycin and 0.25mg mly1 amphotericin B in 5% CO humidified2
atmosphere. To selectively remove contaminating fi-broblasts which are more adhesive than smooth mus-cle cells in primary culture, the cell suspension wasincubated in a 25 cm2 tissue culture flask for 30 min.The cells that were still in suspension were trans-ferred to another flask and cultured at 378C in 5%CO , humidified air. After 3 days, one third of the2
culture medium was replaced with fresh medium andafter 1 week, the cells were confluent. The smoothmuscle cells were subcultured at a split ratio of 1:4using 0.05% trypsin and 0.02% EDTA.
[3 ]2.2.2. Measurement of H arachidonic acid releaseŽ .CISM cells passages 3–10 were grown to a
confluence in 12-well plates and incubated with 0.75y1 w3 xmCi ml H AA for 24 h at 378C in DMEM
containing 0.1 mg mly1 bovine serum albumin. Afterlabeling, the cells were washed three times withnon-radioactive DMEM to remove unincorporatedAA and then incubated in the absence or presence ofagonists in 1 ml serum free DMEM as described
w xpreviously 8 . In experiments where protein kinaseinhibitors or phospholipase inhibitors were employed,the cells were pre-incubated with the inhibitor for 15min prior to the addition of the agonist. At the end ofincubation, the medium was centrifuged at 1400=gfor 10 min at 48C, and radioactivity in the supernatantdetermined by liquid scintillation spectrometry. Todetermine the radioactivity recovered in the free AA

( )S. Husain, A.A. Abdel-LatifrBiochimica et Biophysica Acta 1392 1998 127–144130
released into the medium as percentage of the totalradioactivity in the medium, the radioactivity in themedium was analyzed by means of thin-layer chro-
Ž . w xmatography TLC as described previously 6 .Briefly, the medium was acidified with 10% formicacid to pH 3.5 and extracted three times with 3 ml ofethyl acetate. The solvent was evaporated under N .2
The residue was dissolved in chloroformrmethanolŽ .2:1, vrv , spotted on Whatman pre-coated silica gelLK6DF plates and developed in the organic phase ofa solvent system of ethyl acetateracetic acidrtri-
Ž .methylpentanerwater 11:2:5:10, by volume . Aftervisualization of AA and PG standards by exposure toI vapor, the radioactive spots were scraped and2
measured by counting in a Beckman liquid-scintilla-tion counter.
2.2.3. Phospholipase A assay2
Cells grown in 25 cm2 flask were arrested for 16 hand stimulated with or without agonists and lysed in50 mM HEPES buffer, pH 7.4 containing 350 mMsucrose, 1 mM EGTA, 10 mg mly1 phenylmethysul-fonyl fluoride, 10 mg mly1 leupeptin, 10 mg mly1
aprotinin, 200 mM sodium orthovanadate, and 50mM sodium fluoride. PLA activity in lysates or in2
Ž100 000=g cytosolic fraction of CISM cells 50–100.mg of proteinrassay was measured using
w14 xC arachidonyl-phosphatidylcholine as substrate asw xdescribed 33 with slight modifications. A total of 11
ml of radiolabeled phospholipid stock were driedunder N and added to 0.5 ml of reaction mixture2Ž25-mM HEPES, pH 7.4, 150 mM NaCl, 5 mM
y1 .CaCl , 1 mM DTT, 1 mg ml BSA and sonicated2Ž .on ice. The reaction mixture 50 ml containing 25
ml of protein was incubated at 378C for 30 min. Thereaction was stopped by first adding 2.5 ml of Dole’s
Ž .reagent 2-propanol, heptane, 0.5 M H SO , 20:5:12 4
and then 1.5 ml of heptane and 1 ml of water andmixed thoroughly. The heptane phase containing theradioactive fatty acid was passed through a silicicacid chromatography column. The eluates were col-lected and the radioactivity was measured by liquidscintillation spectrometry.
2.2.4. Labeling of cells with 32P and immunoprecip-i
itation of 32P -labeled cPLAi 2
CISM cells were grown in 25 cm2 flasks to con-fluency and starved for 24 h in serum-free medium.
w32 xPrior to the labeling with P , the cells were pre-in-i
cubated in phosphate-free medium for 30 min at378C. The cells were then incubated for 6 h in 3 mlof phosphate-free DMEM containing carrier freew32 x Ž y1.P orthophosphate 150 mCi ml and then incu-i
bated in the absence or presence of 100 nM ET-1 or200 nM PDBu for 5 and 30 min, respectively. ThePKC inhibitors were added 15 min prior to theaddition of the agonists. The labeled cells were first
Ž 2q 2q .washed with ice-cold PBS Ca rMg free , lysedŽin 1 ml of lysis buffer 50 mM Tris–HCl, pH 7.5, 1%
Triton X-100, 0.5% deoxycholate, 10 mM EDTA, 1mM phenylmethyl-sulfonyl fluoride, 10 mM sodiumpyrophosphate, 50-mM NaF, 200 mM sodium ortho-vanadate, and 10 mg mly1 leupeptin and 50 mg mly1
.aprotinin , then placed on ice for 30 min and soni-cated. The cells were sonicated and centrifuged at13 000=g for 15 min at 48C and the cPLA isolated2
from the supernatant by immunoprecipitation usingrabbit anti-cPLA polyclonal antibodies for 2 h, fol-2
lowed by incubation for 1 h with protein A-agarosew xas described by Bosch-Haubold et al. 34 . The im-¨
munoprecipitates were collected and washed threetimes with 1 ml of 10 mM Tris–HCl buffer, pH 7.4,containing 0.5% Triton X-100 and 150 mM NaClŽ .wash buffer-A , followed by two washes with washbuffer-A containing 750 mM NaCl, and finally, twicewith 1 ml wash buffer A.
The immunoprecipitates were collected and ex-tracted with Laemmli buffer for 15 min at 608C and
w xresolved on 10% SDS-PAGE 35 . Proteins weretransferred to nitrocellulose membranes and blockedwith 3% BSA for 2 h. Membranes were exposed toAmersham Hyperfilm at y708C. Phosphorylation wasquantitated by densitometry.
2.2.5. Densitometric quantitationThe densities of the bands were determined by
scanning with Ultrascan XL enhanced laser densito-w xmeter as described previously 36 . Each band was
Ž .expressed as arbitrary units AU , which was calcu-lated from the area of peak of a selected bandscanned by a densitometer.
2.2.6. SDS-PAGE and immunoblottingCISM cells were lysed in 50-mM Tris–HCl buffer,
pH 8.0, containing 100 mM NaCl, 1-mM EDTA, 1%Nonidet P-40, 0.1% SDS, 0.5% deoxycholate, 50

( )S. Husain, A.A. Abdel-LatifrBiochimica et Biophysica Acta 1392 1998 127–144 131
mM NaF, 1 mM Na VO , 5 mM PMSF, 10 mg mly13 4
Leupeptin and 50 mg mly1 aprotinin for 20 min onice. Cell lysates were centrifuged at 2500=g for 10min and the supernatant was used for immuno-detec-
w xtion of PKC isoforms 28 . The cytosolic and mem-brane fractions were prepared as previously describedw x36 . Equal amounts of proteins were then resolved
w xon 10% SDS–polyacrylamide gels 35 . PrestainedŽ .Kaleidoscope range 7.1–208 kDa and SDS-PAGE
Ž .low range 20.5–112 kDa standards were run inparallel as protein molecular weight markers. Ratbrain protein extract, purified cPLA and Swiss 3T32
cell lysates were used as positive controls for thedetection of PKC isoforms, cPLA and MAPK, re-2
spectively. Proteins were then transferred to nitro-cellulose membranes. After blocking with 3% BSA in
ŽTTBS 20 mM Tris–HCl, 155 mM NaCl, 0.05%.Tween-20, pH 7.5 , the nitrocellulose membrane was
Žincubated with primary antibodies i.e., anti-PKCisoforms polyclonal antibodies at a dilution of 1:1000,anti-cPLA polyclonal antibodies at a dilution of2
1:1500, and anti-MAPK polyclonal antibodies at a.dilution of 1:1000 for 2 h at 208C with gentle
Žshaking. After washing with TTBS two washes for.15 min followed by three washes for 5 min , the
membrane was incubated with secondary antibodiesŽHRP conjugated goat anti-rabbit IgG at 1:3000 dilu-
.tion for 1 h at 208C. For each isoform of PKC, anŽ y1.inhibitory peptide 1–5 mg ml was added to the
primary antibody solution prior to addition to theblots, in order to selectively block the binding ofantibodies to an antigen. For chemiluminescent detec-tion, the membranes were treated with ECL reagentfor 1 min and subsequently exposed to ECL hyper-film for 1–2 min.
2.2.7. Measurement of MAPK actiÕation in CISMcells
MAPK activity was characterized by the in situŽ .myelin basic protein MBP phosphorylation assay
adapted from the method of Kameshita and Fujisawaw x37 . Briefly, quiescent cells were stimulated with100 nM ET-1 for 5 min. After treatment, the cellswere washed three times in ice-cold Ca2qrMg2q-freeDulbecco’s PBS and scraped into ice-cold extraction
Žbuffer 20 mM 2-Phosphoglycerol, 20 mM NaF, 2mM EDTA, 0.2 mM sodium vanadate, 1 mM PMSF,25 mg mly1 Leupeptin, 10 mg mly1 Aprotinin and
Ž . .0.3% vrv b-mercaptoethanol, pH 7.5 as describedw xby Bogoyevitch et al. 30 . The cell extracts were
centrifuged at 10 000=g for 10 min at 48C, and thesupernatant was resolved on a 10% SDS polyacryl-amide gel co-polymerized with 0.5 mg mly1 MBP.After electrophoresis, the gels were washed with 50
Ž .mM Tris–HCl buffer, pH 8.0, containing 20% vrvpropanol to remove SDS, then washed with denatur-
Žing buffer 50 mM Tris–HCl, pH 8.0, containing 6Mguanidine hydrochloride and 5 mM b-mercapto-
.ethanol . The enzymes on the gel were then renaturedby washing with 50-mM Tris–HCl buffer, pH 8.0,
Ž .containing 0.04% vrv Tween-40 and 5 mM b-mer-captoethanol at 48C for 21 h. The gel was thenpre-incubated with assay buffer containing 40 mMHepes, pH 8.0, 10 mM MgCl , 2 mM dithiothreitol2
and 0.1 mM EGTA at 308C for 30 min. The kinaseactivity was determined by incubating the gel with 20ml of the assay buffer, which contained 20 mM ATP
w 32 xand 100 mCi g- P ATP, at 308C for 1 h. AfterŽ .extensive washing in 5% wrv trichloroacetic acid
containing 10-mM sodium pyrophosphate, the gelwas dried and autoradiographed at y708C.
2.2.8. Protein estimationProteins were measured by the method of Lowry etw xal. 38 using bovine serum albumin as standard.
3. Results
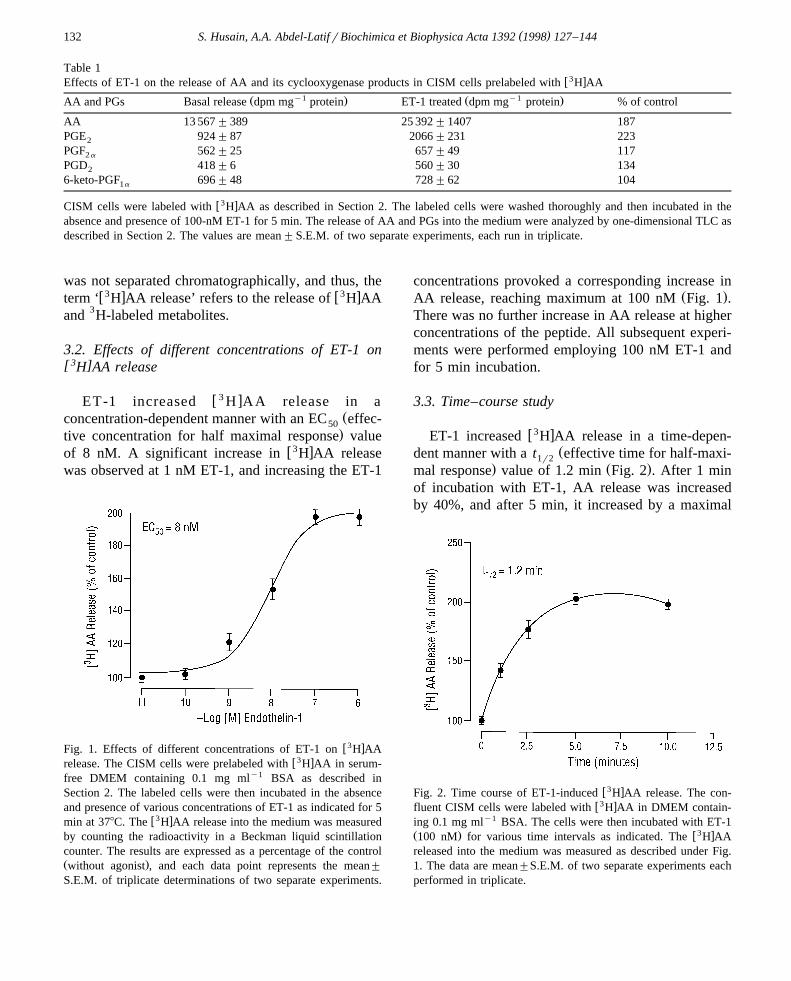
3.1. Effects of ET-1 on the release of AA and itscyclooxygenase products in CISM cells prelabeled
[ 3 ]with H AA
In order to estimate the radioactivity recovered inthe free AA released into the medium as percentageof the total radioactivity in the medium, the labeledcells were incubated in the absence and presence ofET-1 for 5 min and the radioactivity in AA and itsmetabolites released into the medium was analyzedby TLC as described in Section 2. As shown in Table1, greater than 80% of the counts released into themedium were associated with the AA fraction, theremaining 20% of the counts were cyclooxygenaseproducts and other unidentified metabolites. In thefollowing experiments, radioactivity in the medium
( )S. Husain, A.A. Abdel-LatifrBiochimica et Biophysica Acta 1392 1998 127–144132
Table 1w3 xEffects of ET-1 on the release of AA and its cyclooxygenase products in CISM cells prelabeled with H AA
y1 y1Ž . Ž .AA and PGs Basal release dpm mg protein ET-1 treated dpm mg protein % of control
AA 13 567"389 25 392"1407 187PGE 924"87 2066"231 2232
PGF 562"25 657"49 1172a
PGD 418"6 560"30 1342
6-keto-PGF 696"48 728"62 1041a
w3 xCISM cells were labeled with H AA as described in Section 2. The labeled cells were washed thoroughly and then incubated in theabsence and presence of 100-nM ET-1 for 5 min. The release of AA and PGs into the medium were analyzed by one-dimensional TLC asdescribed in Section 2. The values are mean"S.E.M. of two separate experiments, each run in triplicate.
was not separated chromatographically, and thus, thew3 x w3 xterm ‘ H AA release’ refers to the release of H AA
and 3H-labeled metabolites.
3.2. Effects of different concentrations of ET-1 on[3 ]H AA release
w 3 xET-1 increased H AA release in aŽconcentration-dependent manner with an EC effec-50.tive concentration for half maximal response value
w3 xof 8 nM. A significant increase in H AA releasewas observed at 1 nM ET-1, and increasing the ET-1
w3 xFig. 1. Effects of different concentrations of ET-1 on H AAw3 xrelease. The CISM cells were prelabeled with H AA in serum-
free DMEM containing 0.1 mg mly1 BSA as described inSection 2. The labeled cells were then incubated in the absenceand presence of various concentrations of ET-1 as indicated for 5
w3 xmin at 378C. The H AA release into the medium was measuredby counting the radioactivity in a Beckman liquid scintillationcounter. The results are expressed as a percentage of the controlŽ .without agonist , and each data point represents the mean"
S.E.M. of triplicate determinations of two separate experiments.
concentrations provoked a corresponding increase inŽ .AA release, reaching maximum at 100 nM Fig. 1 .
There was no further increase in AA release at higherconcentrations of the peptide. All subsequent experi-ments were performed employing 100 nM ET-1 andfor 5 min incubation.
3.3. Time–course study
w3 xET-1 increased H AA release in a time-depen-Ždent manner with a t effective time for half-maxi-1r2
. Ž .mal response value of 1.2 min Fig. 2 . After 1 minof incubation with ET-1, AA release was increasedby 40%, and after 5 min, it increased by a maximal
w3 xFig. 2. Time course of ET-1-induced H AA release. The con-w3 xfluent CISM cells were labeled with H AA in DMEM contain-
ing 0.1 mg mly1 BSA. The cells were then incubated with ET-1Ž . w3 x100 nM for various time intervals as indicated. The H AAreleased into the medium was measured as described under Fig.1. The data are mean"S.E.M. of two separate experiments eachperformed in triplicate.

( )S. Husain, A.A. Abdel-LatifrBiochimica et Biophysica Acta 1392 1998 127–144 133
Table 2Effects of inhibitors of PLA , PLC and diacylglycerol lipase on2w3 xH AA release
3w xAdditions H AA releasey1dpm mg % of
protein control
None 9 800"480 100100 nM ET-1 19796"743 202100 nM ET-1q100 nM AACOCF 13524"600 1383
100 nM ET-1q1 mM quinacrine 10486"250 107100 nM ET-1q1 mM manoalide 10094"650 103100 nM ET-1q1 mM U-73122 20090"850 205100 nM ET-1q1 mM RHC-80267 19502"789 199
w3 xConfluent CISM cells were labeled with 0.75 mCi H AArwellfor 24 h at 378C. The cells were then washed three times withDMEM and treated with or without AACOCF , quinacrine,3
manoalide, U-73122 or RHC-80267 for 15 min before the addi-tion of 100 nM ET-1 for 5 min at 378C. After incubation, the
w3 xrelease of H AA into the medium was determined as describedin Section 2.Each value represents the mean " S.E.M. of triplicate determina-tions of two separate experiments.
value of 97%. There was no further increase in AArelease at longer time intervals.
3.4. Effects of inhibitors of PLA , PLC and diacyl-2[3 ]glycerol lipase on ET-1-induced H AA release
Arachidonic acid can be released from membranephospholipids either through the direct action of PLA 2w x39 or through the indirect actions of PLC and DAG
w xlipase 40 . To determine whether the ET-1-inducedAA release was the result of PLA activation or PLC2
and DAG lipase activations, cells were pre-incubatedŽ .with quinacrine or manoalide PLA inhibitors ,2
w Ž .xAACOCF a selective cPLA inhibitor , U-731223 2Ž . ŽPLC inhibitor or RHC-80267 DAG lipase in-
.hibitor . As shown in Table 2, quinacrine andmanoalide, PLA inhibitors, completely blocked the2
w3 xET-1-induced H AA release, however, AACOCF3w3 xinhibited the H AA release by 64%. In contrast,
U-73122 and RHC-80267 failed to show any in-hibitory effect on the stimulated AA release. Thesedata demonstrate that PLA , but not PLC and DAG2
lipase, is involved in the ET-1-induced AA release inthese cells.
3.5. Effects of PKC inhibitors on ET-1- and PDBu-[ 3 ]induced H AA release
Previously, we reported that ET-1 increased AAw xrelease both in rabbit iris sphincter smooth muscle 6
and in transformed cat iris sphincter smooth muscleŽ . w xSV-CISM-2 cells 8 , and that PDBu, an activatorof PKC, potentiated the stimulated AA release in the
w xsphincter muscle 6 . The involvement of PKC inagonist-induced AA release has been reported in
Fig. 3. Effects of PKC inhibitors on ET-1- and PDBu-inducedw3 x w3 xH AA release. The confluent cells were prelabeled with H AAin serum-free DMEM in the presence of 0.1 mg mly1 BSA. The
Ž .cells were then pretreated with either staurosporine 1 mM orŽ .RO 31-8220 1 mM for 15 min followed by incubation with 100
nM ET-1 or 200 nM PDBu for 5 and 30 min, respectively. Afterw3 xincubation, the H AA release into the medium was estimated as
described in Section 2. The data are "S.E.M. of four experi-ments each performed in triplicate.

( )S. Husain, A.A. Abdel-LatifrBiochimica et Biophysica Acta 1392 1998 127–144134
smooth muscle as well as in other tissues. In thesestudies, pharmacological inhibitors of PKCw x6,27,28,34 and prolonged treatment of the tissue
w xwith phorbol esters 20 , designed to downregulatethe enzyme, were employed. To demonstrate theinvolvement of PKC in AA release in the CISM cells,
w3 xthe cells were labeled with H AA and then treatedwith either ET-1 or PDBu for 5 and 30 min, respec-tively. As shown in Fig. 3, ET-1 increased AA
Ž .release by 97% Fig. 3A and PDBu increased it byŽ .96% Fig. 3B . In addition, the stimulatory effects of
Ž .PDBu and ET-1 were not additive data not shown .To confirm the involvement of PKC in the stimulatedrelease of AA in these cells, we employed PKC
Ž .inhibitors. Staurosporine 1 mM completely blockedŽ .ET-1- and PDBu-induced AA release Fig. 3A,B .
Since staurosporine is a non-specific PKC inhibitor,we have employed the staurosporine analogue RO31-8220, which is a potent and more selective in-
w xhibitor of PKC 41 . Pre-incubation of the cells withRO 31-8220 inhibited ET-1- and PDBu-induced AA
Ž .release by 82 and 100%, respectively Fig. 3A,B .These data demonstrate that PKC is involved inET-1-induced AA release in the CISM cells.
3.6. Effect of AACOCF , on ET-1-stimulated3
phospholipase A actiÕity in CISM cells2
Ž .As shown in Fig. 4, ET-1 100 nM increasedcPLA catalytic activity by 70% when phosphatidyl-2
w 14 xcholine 1-stearoyl-2- 1- C arachidonyl was used assubstrate. To confirm the involvement of cPLA , we2
have used AACOCF , a selective cell permeable3
cPLA inhibitor, which has no inhibitory effect on2w xsecretory and other forms of PLA 42 . As shown in2
Fig. 4, ET-1-induced cPLA activation is completely2
inhibited by 1-mM AACOCF . These data show that3
most, if not all, PLA activity is contributed by the2
cytosolic form of PLA .2
3.7. Western blot analysis of PKC isoforms in theCISM cells
Ž .To determine which isoform s of PKC is involvedin ET-1-induced AA release in the CISM cells, it wasnecessary to demonstrate the type of isoforms ex-
Fig. 4. Effect of ET-1 on PLA activity in CISM cells. Cells2Ž .were incubated with or without ET-1 100 nM for 5 min then
homogenized and fractionated as described in Section 2. Todetermine the effect of the cPLA inhibitor, the cells were2
Ž .pre-incubated with AACOCF 1 mM for 30 min prior to ET-13
treatment. PLA activity in the 100000= g cytosolic fraction2w 14 xwas determined using phosphatidylcholine 1-stearoyl-2 1- C -
arachidonyl as described in Section 2.
pressed in these cells. As shown in Fig. 5, whole cellextracts were subjected to SDS-PAGE and immuno-blotted with antibodies specific for PKCa , b , g , ´ ,
w xd , h, and z isoforms as described previously 36 .All of the isoforms examined were detected in ratbrain homogenate which was employed as a positive
Ž .control lane 3 , suggesting that the antibodies hadsufficient titer and binding affinity. Antibodies di-rected against PKCa , b , ´ , d , h, and z revealed
Ž .immunoreactive bands in the CISM cells lanes 1 ,indicating the presence of these isoforms in thesecells. In contrast, antibodies against PKCg failed todetect immunoreactive bands in extracts from CISMcells, indicating that this isoform is not expressed to a
Ž .significantly detectable level lane 1 . The specificityof each isoform was confirmed by deletion of the
Ž .immunoreactive band lane 2 upon incubation withŽthe appropriate inhibitory peptide which was used to
.raise that particular antibody . The apparent molecu-lar weight of PKCa , b , and d is about 80 kDa,whereas those of PKCh and z are slightly lower.PKC´ showed a slightly higher molecular weight,about 90 kDa. These observations are consistent withthe known tissue-specific distribution of the PKC
w xisoforms in the cat iris sphincter muscle 36 .

( )S. Husain, A.A. Abdel-LatifrBiochimica et Biophysica Acta 1392 1998 127–144 135
Fig. 5. Expression of PKC isoforms in CISM cells. The CISMŽcells were lysed and 12 mg protein of whole cell lysate lanes 1
. Ž .and 2 or rat brain homogenate lanes 3 , were subjected to 10%SDS-PAGE. Proteins were transferred to nitrocellulose mem-branes and immunoblotted with antibodies specific to PKC a , b ,g , ´ , d , h, and z as described in Section 2. Specificity of PKCisoforms was determined in the presence of immunizing peptidesŽ .lanes 2 . The immunoreactive bands were detected using ECLchemiluminescent reagent after exposure to ECL Hyperfilm for1–2 min.
[3 ]3.8. Effect of PKC downregulation on H AArelease
Tumor-promoting phorbol esters such as phorbolŽ .12-myristate 13-acetate PMA and PDBu can substi-
tute for DAG as activators of PKC. The cellularresponse to phorbol esters is biphasic: the initialresponse involves translocation and activation of PKCw x43 , however, prolonged activation of the enzymeresults in its downregulation, therefore, the mem-brane form of the enzyme is more susceptible to
w xproteolysis 44 . Both activation and downregulationof PKC by phorbol esters are widely used experimen-tal paradigms for implicating the enzyme in a biolog-ical response. It has been demonstrated that atypical
Ž .PKC isoforms z , l are insensitive to PMA-induced
w3 xFig. 6. Effect of PKC downregulation on H AA release inŽ . w3 xCISM cells. A CISM cells were labeled with H AA in the
presence of 0.1 mg mly1 BSA and incubated in the absence andpresence of 500 nM PDBu for 16 h. The cells were thenstimulated with either 100 nM ET-1 or 200 nM PDBu for 5 and
w3 x30 min, respectively. The H AA released into the medium wasdetermined as described in Section 2. The results are expressed asmean"S.E.M. of two separate experiments each performed in
Ž . Ž .triplicate. B CISM cells were incubated without lanes 1 orŽ . Žwith 500 nM PDBu lanes 2 for 16 h. The cell lysates 12
.mgrlane were then separated on 10% SDS-PAGE, followed byimmunoblotting with anti-PKCa , b , ´ , d , h, and z antibodiesas described in Section 2. Rat brain homogenate was used as a
Ž .positive control lanes 3 . Specificity of each PKC isoform wasdetermined by deletion of the band in the presence of theimmunizing peptide as shown in Fig. 5.

( )S. Husain, A.A. Abdel-LatifrBiochimica et Biophysica Acta 1392 1998 127–144136
Fig. 7. Effect of ET-1 on translocation of PKCa in CISM cells.Cells were treated with 100-nM ET-1 for 5 min followed byisolation of cytosolic and membrane fractions. PKCa and b
isoforms were detected in these fractions as described in Section 2.
Ždownregulation and the classical PKC isoforms a ,.b , b , g are relatively more sensitive to PMA as1 11
Ž .compared to the novel PKC isoforms d , e , h, u , i .
Since CISM cells express both PDBu-sensitive andPDBu insensitive PKC isoforms, studies were under-taken in order to determine the effects of long-termPDBu treatment on the expression of the variousisoforms and on the ET-1-induced AA release. Cellswere treated with 500-nM PDBu for 16 h and thenthe ET-1-stimulated AA release was investigated. Ascan be seen from Fig. 6A, ET-1 and PDBu did notenhance AA release in cells pretreated with PDBu,
Žbut did in the control cells without PDBu pretreat-.ment . Under similar experimental conditions, PDBu
pretreated cells were investigated for the expressionof the various PKC isoforms. As shown in Fig. 6B,prolonged treatment of the cells with 500 nM PDBuresulted in selective downregulation of PKCa , how-ever, there was no depletion of any other PKC iso-form by the phorbol ester treatment. Furthermore,
Ž .higher concentrations of PDBu 1 mM and pro-
w3 x Ž .Fig. 8. Effects of different concentrations of PDBu on PKCa down-regulation and on agonist-induced H AA release. A PBDu-in-Ž .duced PKCa downregulation. CISM cells were incubated with or without different concentrations of PDBu 1–500 nM for 16 h. The
Ž .cell lysates 12 mgrlane were separated on 10% SDS-PAGE and then immunoblotted with anti-PKCa antibody as described in SectionŽ .2. The specificity of PKCa was determined as described in Fig. 5. Rat brain homogenate was employed as a positive control. B Effects
w3 xof PKCa downregulation on AA release. The confluent cells were labeled with H AA and incubated in the absence and presence ofŽ .different concentrations of PDBu 1–500 nM for 16 h. The labeled cells were then stimulated with 100 nM ET-1 or 200 nM PDBu for 5
w3 xand 30 min, respectively. Release of H AA into the medium was determined as described in Fig. 6. The data are mean"S.E.M. of twoseparate experiments each performed in triplicate.

( )S. Husain, A.A. Abdel-LatifrBiochimica et Biophysica Acta 1392 1998 127–144 137
longed incubations with the PKC inhibitor, up to 24h, had no effect on PKCb and other isoforms of
Ž .PKC data not shown . These data are compatiblewith involvement of the PKCa isoform in the ET-1-induced AA release in the CISM cells.
3.9. Translocation of PKC isoforms
Treatment of CISM cells with 100 nM ET-1 orwith 200 nM PDBu caused significant translocationof PKCa , but not PKCb , from cytosol to the partic-
Ž .ulate fraction Fig. 7 . This is in support of theconclusion that PKCa is involved in the agonist-in-duced AA release in these cells.
3.10. Effects of different concentrations of PDBu onPKCa downregulation and on agonist-induced[3 ]H AA release
To further investigate the involvement of PKCa
in AA release, the CISM cells were pre-incubatedwith different concentrations of PDBu for 16 h. Asshown in Fig. 8A, prolonged exposure to PDBuresulted in a concentration-dependent downregulationof PKCa in these cells. PKCa was partially de-
Fig. 9. Concentration-dependent effect of Go-6976 on ET-1-in-¨w3 xduced H AA release. The CISM cells were prelabeled with
w3 xH AA as described in Fig. 1, then incubated with differentŽ .concentrations of Go-6976 0.01–100 nM for 15 min. The cells¨
were then stimulated with 100-nM ET-1 for 5 min and the releasew3 xof H AA into the medium was determined as described in
Section 2. Each data point represents the mean"S.E.M. of twoseparate experiments, each run in triplicate.
Table 3w3 xEffect of thymeatoxin on H AA release in CISM cells3w xAdditions H AA release
y1dpm mg protein % of control
None 12 332"580 100100-nM ET-1 25 218"1021 204100-nM thymeatoxin 30 830"1561 250
w3 xConfluent cells were prelabeled with 0.75 mCi H AArwell for24 h at 378C. The cells were washed three times with DMEM andthen incubated with 100 nM ET-1 and 100 nM thymeatoxin for 5
w3 xand 30 min, respectively. H AA release into the medium wasdetermined as described in Section 2.Each value represents mean " S.E.M. of triplicate determinationof two separate experiments.
pleted in the presence of 100-nM PDBu and com-pletely depleted in the presence of 500 nM. Similarly,the PDBu pretreatment inhibited ET-1- and PDBu-in-duced AA release in a concentration-dependent man-
Ž .ner Fig. 8B . These data strongly support the in-volvement of PKCa in the agonist-induced AA re-lease in the CISM cells.
3.11. Effect of the PKC inhibitor Go-6976 on ET-1-¨[ 3 ]induced H AA release
To further demonstrate the type of PKC isoforminvolved in ET-1-induced AA release, we used theisoform-specific inhibitor Go-6976, which inhibits¨PKCa , b , and g isoforms. As shown in Fig. 9,pretreatment of the cells with Go-6976 inhibited ET-¨
w3 x1-induced H AA in a concentration-dependent man-ner with an IC value of 8 nM.50
[3 ]3.12. Effect of thymeatoxin on H AA release
Thymeatoxin, an activator of PKCa , b and g
w x45 , induced AA release from the CISM cells andŽthis was comparable to that obtained with ET-1 Ta-
.ble 3 . These data suggest the involvement of PKC inAA release in these cells.
3.13. Effects of RO 31-8220 on ET-1- and PDBu-induced cPLA phosphorylation2
There is accumulating evidence to indicate thatcPLA plays a critical role in the receptor-mediated2
mobilization of AA and eicosanoid biosynthesis. We

( )S. Husain, A.A. Abdel-LatifrBiochimica et Biophysica Acta 1392 1998 127–144138
have demonstrated the presence of an 85-kDa cPLA 2
in the CISM cells by immunoblotting analysis em-ploying rabbit polyclonal antibodies raised againstpurified cPLA . The immunoreactive protein band2
had an apparent molecular weight of 100 kDa andco-migrated with purified 85-kDa cPLA used as a2
Ž .positive control in 10% SDS-PAGE Fig. 10A .To provide additional support for the conclusion
that PKC mediates the effect of ET-1 on AA releasein the CISM cells via activation of cPLA , we have2
investigated the effects of RO 31-8220, a PKC in-hibitor, on ET-1- and PDBu-induced cPLA phos-2
phorylation. The CISM cells were labeled with 32PiŽ y1.150 mCi ml for 6 h, and then incubated in theabsence or presence of ET-1 and PDBu for 5 min and
30 min, respectively. The cPLA was then immuno-2
precipitated from the 32P -labeled cells using poly-i
clonal rabbit anti-cPLA IgG. As shown in Fig. 10B,2
the extent of cPLA phosphorylation was markedly2
increased following ET-1 and PDBu stimulation. Toconfirm the involvement of PKC in cPLA phospho-2
rylation, the cells were pre-incubated for 15 min withRO 31-8220 prior to the addition of ET-1 and PDBu.As shown in Fig. 10B, the ET-1- and PDBu-inducedcPLA phosphorylation was significantly inhibited2
by the PKC inhibitor, thus implicating the involve-ment of PKC in the phosphorylation and activation ofcPLA in the CISM cells. The extent of cPLA2 2
phosphorylation was also measured by densitometry.As shown in Fig. 10C, ET-1 and PDBu increased thephosphorylation by 221 and 137%, respectively, andin the presence of RO 31-8220, the phosphorylationwas inhibited by 72 and 100%, respectively. We havealso carried out Western blotting of the cPLA in2
order to demonstrate that comparable amounts ofprotein were immunoprecipitated in all of the sam-
Ž .ples Fig. 10D . All lanes show the presence of thesame amounts of cPLA . To rule out the possibility2
of the involvement of another isoform of PLA which2
is capable of cross-reacting with the cPLA antibody,2
we have used the 100 000=g supernatant, instead ofthe cell lysate for immunoprecipitation. The data
Fig. 10. Effects of RO 31-8220 on ET-1- and PDBu-inducedŽ .cPLA phosphorylation. A Immunochemical identification of2
Ž .cPLA . Cell lysate 16 mg protein, lane 1 analyzed by 10%2
SDS-PAGE and immunoblotted with specific polyclonal cPLA 2
antibody as described in Section 2. Lane 2 contained purifiedŽ .cPLA used as a positive control. B Autoradiogram of im-2
munoprecipitated cPLA . Cells were labeled with 32P for 6 h,2 iŽ .then incubated with or without RO 31-8220 1 mM for 15 min.
The cells were then stimulated with 100 nM ET-1 or 200 nMPDBu for 5 and 30 min, respectively. The immunoprecipitate wasresolved on 10% SDS-PAGE as described in Section 2. Theradioactive bands were detected by autoradiography. Lane 1Ž . Ž . Žcontrol , lane 2 100-nM ET-1 , lane 3 100 nM ET-1q1 mM
. Ž . ŽRO 31-8220 , lane 4 200 nM PDBu and lane 5 200 nM. Ž .PDBuq1 mM RO 31-8220 . C Quantitation of cPLA phos-2
phorylation. Phosphorylation of cPLA in the CISM cells was2
quantitated by densitometry. The autordiographs were quantitatedby scanning with Ultrascan XL-enhanced laser densitometer and
Ž . Ž .expressed as arbitrary units AU , as in Section 2. D Westernblot analysis of the immunoprecipitated cPLA . After auto-2
radiography, the nitrocellulose membrane was used for cPLA 2
detection by Western blotting analysis as described in Section 2.

( )S. Husain, A.A. Abdel-LatifrBiochimica et Biophysica Acta 1392 1998 127–144 139
Fig. 11. Effect of RO 31-8220 on ET-1 and PDBu-induced PLA 2
activity. Confluent CISM cells were starved for 16 h, then treatedwith or without 100 nM ET-1 or 200 nM PDBu for 5 and 30 min,
Žrespectively. The cells were pre-incubated with RO 31-8220 1.mM for 15 min prior to addition of the agonist. cPLA activity2
was determined as described in Fig. 4.
obtained were identical with those observed in theŽ .cell lysate shown in Fig. 10 data not shown . This
confirms our conclusion that the 85-kDa band corre-sponds to cPLA in CISM cells. These data demon-2
Fig. 12. Effect of ET-1 on p42mapk and p44mapk activation inŽ .CISM cells. A Immunochemical identification of MAPK. TheŽ .cell lysate 10 mg protein, lane 1 was separated on 10%
SDS-PAGE, then transferred to nitrocellulose membranes. Themembrane was probed with polyclonal anti-MAPK antibodies asdescribed in Section 2. Lane 2 contained 3T3 cell lysate which
Ž .was used as positive control. B Effect of RO 31-8220 andgenistein on ET-1-induced activation of MAPK. The cell lysateswere subjected to electrophoresis on 10% SDS-PAGE containing0.5 mg mly1 MBP. Proteins resolved in the gel were denaturedand renatured as described in Section 2 and were incubated with
w 32 x20 mM ATP and 100 mCi g- P ATP in the kinase assay bufferat 308C for 1 h. After drying the gel, the phosphorylated bands
Ž . Žwere visualized by autoradiography. Lane 1 control , lane 2 100. Ž .nM ET-1 , lane 3 100 nM ET-1q1 mM RO 31-8220 and lane
Ž . Ž .4 100 nM ET-1q1 mM genistein . C Quantitation of MAPKphosphorylation. p42mapk and p44mapk bands were cut from thegel and quantitated by liquid scintillation counting as described inSection 2. The data represent phosphorylation of both p42mapk
and p44mapk bands. The results represent the mean of twoŽ .separate experiments. D Western blot analysis of MAPK
Ž mapk. Žp42rp44 in cell lysates: a portion of the samples exactlyŽ ..the same amount as was used for MBP phosphorylation in B
were resolved on 10% SDS-PAGE, transferred to nitrocellulosemembranes and immunoblotted using polyclonal anti-p42rp44mapk antibodies as described in Section 2.
strate that phosphorylation of cPLA is required for2
its activation and that it is mediated through a PKC-dependent pathway in these cells.
3.14. Effects of ET-1 and PDBu on phospholipase A2
actiÕity
To confirm whether or not the observed phospho-rylation of cPLA correlates with an increase in the2
catalytic activity of the enzyme, the CISM cells wereŽ . Ž .treated with ET-1 100 nM or PDBu 200 nM for 5
and 30 min, respectively, and the enzyme activitywas measured. As shown in Fig. 11, ET-1 and PDBu

( )S. Husain, A.A. Abdel-LatifrBiochimica et Biophysica Acta 1392 1998 127–144140
increased cPLA activity by 70 and 100%, respec-2
tively, and in the presence of RO 31-8220, the activ-ity of the enzyme was almost completely inhibited.These data demonstrate that ET-1 and PDBu inducephosphorylation of the enzyme which increases itscatalytic activity and AA release. Furthermore, thedata show that the effects of these agonists on cPLA 2
activity in these cells are acute.
3.15. ET-1-induced actiÕation of MAPK in CISMcells
There is evidence that MAPK are involved inw xphosphorylation of cPLA in stimulated cells 15 .2
The CISM cells expressed both p42mapk and p44mapk
isoforms when polyclonal anti-MAPK antibodies wereŽ .used Fig. 12A . CISM cells were stimulated with
100 nM ET-1 for 5 min and the MAPK activity wasdetermined using an in-gel renaturation kinase assay.Both isoforms of MAPK were significantly stimu-
Ž .lated in the presence of ET-1 Fig. 12B, lane 2 .From the in situ phosphorylation of MBP after SDS-PAGE, the molecular masses for MAPK in the ET-1-stimulated cells were 42 and 44 kDa. Incorporationof 32P into MBP in each treatment was estimated bycutting the gels, followed by counting the radioactiv-ity. As shown in Fig. 12C, ET-1 increased MAPKphosphorylation by 165%.
To investigate whether PKC is involved in theactivation of MAPK, the PKC inhibitor, RO 31-8220,was used. Pre-incubation of the cells with RO 31-8220had no inhibitory effect on ET-1-induced MAPK
Ž .activity Fig. 12B,C, lane 3 . However, genistein, apotent protein tyrosine kinase inhibitor, had signifi-cant inhibitory effect on ET-1-induced MAPK activ-
Ž .ity Fig. 12B, lane 4 . In the presence of 1-mMgenistein, the ET-1-induced phosphorylation was re-
Ž .duced to 15% Fig. 12C, lane 4 , suggesting a poten-tial role for tyrosine phosphorylation in ET-1-inducedMAPK activation. To rule out the possibility ofvariation in the levels of p42mapk and p44mapk, equalamounts of all samples were loaded on SDS-PAGEand immunoblotted with anti-MAPK polyclonal anti-bodies. As can be seen from Fig. 12D, all samplescontained comparable amounts of p42mapk andp44mapk. To further confirm the non-involvement ofPKC in ET-1-induced MAPK activation the cells
Ž .were exposed to PDBu 500 nM for 16 h, to deplete
Table 4w3 xEffect of genistein on ET-1-induced H AA release in CISM
cells3w xAdditions H AA release
y1dpm mg protein % of control
None 9 500"450 100100-nM ET-1 19665"793 207100-nM ET-1q1-mM 21660"1153 228genistein
w3 xConfluent cells were prelabeled with 0.75 mCi H AArwell for24 h at 378C. After washing, the cells were treated with orwithout genistein for 15 min before the addition of 100 nM ET-1
w3 xfor 5 min at 378C. The release of H AA into the medium wasdetermined as described in Section 2. Each value represents themean"S.E.M. of two separate experiments, run in triplicate.
PKC, and then the MAPK activity was measured. Wefound that downregulation of PKC has no significantinhibitory effect on ET-1-induced MAPK activationŽ .data not shown . These data provide evidence thatPKC is not involved in MAPK activation in CISMcells.
[3 ]3.16. Effect of genistein on ET-1-induced H AArelease
To investigate whether or not tyrosine phosphory-lation is involved in ET-1-induced AA release, wehave investigated the effect of genistein on the re-lease of AA in the CISM cells. As shown in Table 4,the tyrosine kinase inhibitor had no effect on theET-1-induced AA release.
4. Discussion
In this study, we employed biochemical and phar-macological tools to investigate how ET-1 binding toET receptors in CISM cells stimulates AA release.We found that in these cells, ET-1 is a potent agonistfor AA release. Greater than 80% of the countsreleased into the medium were associated with the
Ž .AA fraction Table 1 . It stimulated AA release in aconcentration- and time-dependent manner with anEC value of 8 nM and a t value of 1.2 min50 1r2Ž .Figs. 1 and 2 . These values are comparable to thosewe reported previously on the effects of the peptide
w xon AA release in the intact iris sphincter 6 . The

( )S. Husain, A.A. Abdel-LatifrBiochimica et Biophysica Acta 1392 1998 127–144 141
stimulated release of AA from membrane phospho-lipids occurs either through a direct release by PLA 2
or through an indirect release by the action of PLCŽfollowed by DAG lipases for reviews, see Refs.
w x.46,47 . The data presented here suggest that inCISM cells, ET-1-induced AA release occurs throughthe action of PLA . This conclusion is supported by2
the findings that the PLA inhibitors, quinacrine and2
manoalide, blocked completely ET-1-stimulated AArelease, whereas AACOCF blocked 64% of the AA3
release. Since AACOCF is a selective cPLA in-3 2w xhibitor 42 , other PLA s may be involved in the2
mechanism of ET-1-induced AA release. In contrast,U-73122 and RHC-80267, inhibitors of PLC andDAG lipase, respectively, had no effects on the ET-
Ž .1-induced AA release Table 2 . Therefore, PLC andmonoacylglycerolrDAG lipases do not play a signifi-cant role in the ET-1-stimulated AA release in thesecells. Evidence against a major role for DAG lipasein the liberation of AA has already been reported in
w xplatelets 18 . These authors demonstrated that inthese cells, Ca2q ionophore induces substantialamounts of AA release in the absence of PLC activa-tion.
The data presented here show that in CISM cellsET-1 stimulation activates PKCa to phosphorylateand activate cPLA to liberate AA for PG synthesis.2
This conclusion is supported by the following find-Ž .ings: 1 using immunochemical techniques, we have
demonstrated the presence of cPLA which has an2Ž . Ž .apparent molecular weight of 100 kDa Fig. 10A . 2
To address the question as to whether PKC is in-volved in the activation of cPLA , experiments were2
Ž .carried out using agents that either stimulate PDBuŽ .or inhibit staurosporine, RO 31-8220 the PKC activ-
ity. ET-1 and PDBu increased AA release by 97 and96%, respectively, and these effects were blocked by
Ž .the PKC inhibitors Fig. 3A,B . The CISM cellsŽexpressed PKCa , b , ´ , d , h and z isoforms Fig.
.5 . PKCg was not detected in these cells which isconsistent with our previous finding in the cat iris
w x Ž .sphincter 36 . 3 Downregulation of PKC with PDBuresulted in complete inhibition of ET-1- and PDBu-
Ž .induced AA release Fig. 6A and in the specific lossŽ .of the PKCa isoform Fig. 6B . This finding was
confirmed through the prolonged exposure of theCISM cells to different concentrations of PDBu.These treatments resulted in downregulation of PKCa
Ž .in a concentration-dependent manner Fig. 8A and ina corresponding suppression of ET-1- and PDBu-in-
Ž .duced AA release Fig. 8B . Chronic exposure ofcells to PDBu depletes some of the PKC isoforms inmany cells. This approach has been used to investi-gate the involvement of PKC in the mechanism of
w xaction of a wide variety of agonists 20,28 . Sincetreatment of the CISM cells with PDBu could lead toreceptor downregulation, reduction in receptor num-ber andror affinity, or receptor desensitization, wehave investigated the effects of PKC downregulationon ET-1-induced inositol phosphates production. Wefound that comparable amounts of inositol phos-phates were released by ET-1 in both untreated and
Ž . ŽPDBu-treated 500 nM PDBu for 16 h cells S..Husain, A.A. Abdel-Latif, unpublished results . This
finding could suggest that chronic treatment of theCISM cells with PDBu had no effect on the ETreceptors. Thus, inhibition of ET-1-induced AA re-lease in the PDBu-treated cells is not due to a loss inthe ET receptor number or affinity but it is due todownregulation of the PKCa isoform in these cells.Ž .4 ET-1 and PDBu also increased cPLA activity by2
70 and 100%, respectively, which was inhibited bythe selective cPLA inhibitor, AACOCF , and by the2 3
Ž . Ž .PKC inhibitor RO 31-8220 Figs. 4 and 11 . 5 Thestudies on the effects of the PKC isoform-specificinhibitor Go-6976, an inhibitor of PKCa , b and g¨
w xisoforms 48,49 , and thymeatoxin, an activator ofw xthese isoforms 45 , on AA release also supports the
involvement of PKCa in ET-1-induced AA releasein the CISM cells. Go-6976 inhibited ET-1-induced¨AA release in a concentration-dependent manner with
Ž .an IC value of 8 nM Fig. 9 . Go-6976 has been¨50
reported to be the most potent inhibitor of calcium-Ž .dependent PKC isoforms a , b , and g and it in-
hibits these isoforms in nanomolar concentrations,whereas micromolar concentrations of this inhibitorhad no effect on the activities of the Ca2q-indepen-
Ž .dent PKC isoforms d , ´ and z . Since PKCg is notŽ .expressed in the CISM cells Fig. 5 , and since
Ž .PKCb is not downregulated by PDBu Fig. 6B norŽ .translocated to the membrane fraction Fig. 7 , we
can conclude that PKCa is the isoform involved inET-1-induced cPLA activation and AA release.2
Thymeatoxin also increased AA release in the CISMcells at a rate which is comparable to that observed
Ž .with ET-1 Table 3 . To the best of our knowledge,

( )S. Husain, A.A. Abdel-LatifrBiochimica et Biophysica Acta 1392 1998 127–144142
the present study constitutes the first report on theinvolvement of PKCa isoform in agonist-inducedAA release in smooth muscle. Phorbol esters havebeen previously reported to stimulate AA release in
w xvascular smooth muscle cells 4 , rabbit iris sphincterw x w xsmooth muscle 6 and rat aortic rings 12 . In addi-
tion, PKCa has been reported to be involved in AArelease in various cell lines, including PC12 cellsw x Ž . w x28 , Madin–Darby canine kidney MDCK cells 20 ,
w xChinese hamster ovary cells 25,26 and mouse peri-w xtoneal macrophages 24 . Phosphorylation of cPLA 2
by PKCa could play an important role in ET-1-in-duced activation of cPLA and subsequently leads to2
an increase in AA release and PG synthesis.cPLA plays an important role in receptor-media-2
ted mobilization of AA and production of eicosanoids.Activation of cPLA in intact cells is associated with2
w xits phosphorylation 21,50 , which increases its activ-w xity in a reconstitution assay 50 . In Chinese hamster
ovary cells, cPLA is phosphorylated on serine2
residues after treatment with phorbol esters, ATP orw xthrombin 21,29 . In contrast, phosphorylation of
cPLA is not necessary for thrombin-stimulated AA2w xrelease in platelets 34 , since staurosporine abolished
cPLA phosphorylation without reducing the libera-2
tion of AA release. In the present work, ET-1 andPDBu increased cPLA phosphorylation by 221 and2
137%, respectively, and in the presence of RO 31-8220 cPLA phosphorylation was inhibited by 722
Ž .and 100%, respectively Fig. 10C . Under similarexperimental conditions, ET-1 and PDBu increasedcPLA activity by 70 and 100%, respectively, and2
these stimulatory effects were inhibited by RO 31-Ž .8220 Fig. 11 . These observations suggest that in
CISM cells, ET-1- and PDBu-induced phosphoryla-tion is necessary for cPLA activation and that the2
catalytic activity of this enzyme is mediated througha PKC-dependent pathway.
There is accumulating evidence which indicatesthat the MAPK ERK1 and ERK2 are involved in theactivating phosphorylation of cPLA in stimulated2
w xcells 15 . Interestingly, the cPLA sequence contains2
potential recognition sites for various kinases includ-w xing PKC, PKA, MAPK and tyrosine kinases 51 . It
is known that cPLA is phosphorylated on Ser-5052w xduring expression in SF9 cells 52 , however, it was
recently reported that three additional serine residuesare phosphorylated on cPLA during expression in2
w xinsect cells 53 . Stimulation with Okadaic acid re-sulted in an increase in phosphorylation of Ser-727
w xon cPLA 53 . This site may have a regulatory role2
in activation of cPLA and its phosphorylation may2
occur by protein kinases other than MAPK. Purifiedhuman cPLA has been reported to be phosphory-2
w xlated by both PKC and MAPK 23 . Thus, to deter-mine whether or not MAPK are involved in ET-1-in-duced AA release, we investigated the effect of ET-1on p42rp44 MAPK activation in the CISM cells. Wefound that p42mapk and p44mapk are present in these
Ž .cells Fig. 12A , and that both kinases are activatedŽ .upon stimulation with ET-1 Fig. 12B,C . Treatment
of the cells with 100-nM ET-1 increased MAPKŽ .activity by 165% Fig. 12C , and pre-incubation of
the cells with RO 31-8220 had no effect on the ET-1Ž .stimulation Fig. 12B,C . Furthermore, treatment of
the cells with PDBu for 16 h, to deplete PKC, had noŽeffect on ET-1-induced MAPK activity S. Husain,
.A.A. Abdel-Latif, unpublished results . These find-ings suggest that ET-1 may stimulate p42rp44 MAPKvia a PKC-independent pathway. In cardiac my-
Žocytes, treatment with the phorbol ester, TPA 1.mM , for 24 h decreased ET-1-stimulated MAPK
activation by 62%, but it had no effect on aFGFw xstimulation of the kinase 30 . Prolonged exposure to
the phorbol ester, PMA, did not suppress agonist-in-w xduced MAPK activation in macrophages 27 .
MAPK are also regulated by phosphorylation onw xtyrosine residues 51 . Thus, pre-incubation of the
CISM cells with the tyrosine kinase inhibitor, genis-Ž .tein 1 mM , significantly inhibited ET-1-induced
Ž .MAPK activation Fig. 12B,C , however, it had now3 xinhibitory effect on ET-1-induced H AA release in
Ž .these cells Table 4 . These observations suggest thatin these cells tyrosine phosphorylation is involved inET-1-induced MAPK activation, but not in ET-1-in-duced cPLA phosphorylation and AA release.2
Therefore, in CISM cells, ET-1-induced MAPK acti-vation is mediated through a PKC-independent path-way.
In conclusion, the data presented here are consis-tent with a role for PKCa , but not for p42rp44MAPK, in cPLA activation and AA release in ET-2
Ž .1-stimulated CISM cells since: i the PKC inhibitor,w3 xRO 31-8220, inhibited ET-1-induced H AA release,
cPLA phosphorylation and cPLA activity, but had2 2Ž .no inhibitory effect on MAPK activation, ii pro-

( )S. Husain, A.A. Abdel-LatifrBiochimica et Biophysica Acta 1392 1998 127–144 143
longed treatment of the cells with PDBu resulted in aselective downregulation of PKCa which correlatedwell with the suppression of ET-1-induced AA re-
Ž .lease, iii Go-6976, a specific inhibitor of PKCa ,¨inhibited ET-1-induced AA release in a concentra-tion-dependent manner with an IC value of 8 nM,50
Ž .and iv genistein, a tyrosine kinase inhibitor, inhib-ited ET-1-induced MAPK activation but had no in-hibitory effect on AA release in the ET-1-stimulatedcells. ET-1 may bind to ET receptor subtype andA
activate PLC to hydrolyze polyphosphoinositides intoDAG and IP . DAG and Ca2q, generated through3
IP -induced Ca2q release, may then activate PKCa3
to phosphorylate and activate cPLA to liberate AA,2
from membrane phospholipids, for PG synthesis.Studies on ET-1-induced AA release and PG synthe-sis in the smooth muscles of the iris could throwmore light on the mechanism of the intraocular pres-
Ž .sure IOP -lowering effect of this peptide in the eye.ET-1 has recently been reported to lower IOP in
w xrabbit eyes 54 , and the PGF analogue, la-2a
tanoprost, is now routinely used to lower IOP inw xglaucoma patients 55 .
Acknowledgements
This work was supported by NIH grants RO1-EY-04387 and R37-EY04171 from US Public HealthService. The authors wish to thank Mr. Justin M.Verner for technical assistance, the Genetic Institutefor supplying us with cPLA polyclonal antibodies,2
and Ms. Jacqueline Negron for typing the manuscript.
References
w x1 M. Yanagisawa, H. Kurihara, S. Kimura, Y. Tomobe, M.Kobayashi, Y. Mitsui, Y. Yazaki, K. Goto, T. Masaki,
Ž .Nature 332 1988 411–415.w x Ž .2 G.M. Rubanyi, M.A. Polokoff, Pharmacol. Rev. 46 1994
325–415.w x3 T. Sakurai, M. Yanagisawa, T. Masaki, Trends Pharmacol.
Ž .Sci. 13 1992 103–108.w x4 E.E. Reynolds, L.L.S. Mok, S. Kurokawa, Biochem. Bio-
Ž .phys. Res. Commun. 160 1989 868–873.w x5 A.A. Abdel-Latif, Y. Zhang, Invest. Ophthalmol. Vis. Sci.
Ž .32 1991 2432–2438.w x6 A.A. Abdel-Latif, Y. Zhang, S.Y.K. Yousufzai, Curr. Eye
Ž .Res. 10 1991 259–265.
w x Ž .7 Y. Zhang, A.A. Abdel-Latif, Cell. Signal. 4 1992 777–788.w x8 A.A. Abdel-Latif, K.-H. Ding, R.A. Akhtar, S.Y.K.
Ž .Yousufzai, J. Lipid Mediators Cell Signal. 14 1996 147–155.
w x9 S.Y.K. Yousufzai, Z. Ye, A.A. Abdel-Latif, J. Pharmacol.Ž .Exp. Ther. 275 1995 1280–1287.
w x10 A.A. Abdel-Latif, S.Y.K. Yousufzai, A.M. El-Mowafy, Z.Ž .Ye, Invest. Ophthalmol. Vis. Sci. 37 1996 328–338.
w x11 J.R. Wu-Wong, B.D. Dayton, T.J. Opgenorth, Am. J. Phys-Ž .iol. 271 1996 c869–c877, Cell Physiol. 40.
w x12 G.K. Oriji, J.E. Tate, H.R. Keiser, ProstaglandinsŽ .Leukotriens Essent. Fatty Acids 55 1996 309–313.
w x Ž .13 R.F. Irvine, Biochem. J. 204 1982 3–16.w x14 E.A. Dennis, S.G. Rhee, M.M. Billah, Y.A. Hannun, FASEB
Ž .J. 5 1991 2068–2077.w x15 R.M. Kramer, D.T. Stephenson, E.F. Roberts, J.A. Clemens,
Ž .J. Lipid Mediators Cell Signal. 14 1996 3–7.w x Ž .16 R.J. Mayer, L.A. Marshall, FASEB J. 7 1993 339–348.w x Ž .17 J.H. Exton, Biochim. Biophys. Acta 1212 1994 26–42.w x18 S.P. Halenda, G.B. Zavoico, M.B. Feinstein, J. Biol. Chem.
Ž .260 1985 12484–12491.w x Ž .19 J. Parker, L.W. Daniel, M. Waite, J. Biol. Chem. 262 1987
5385–5393.w x20 C. Godson, B.A. Weiss, P.A. Insel, J. Biol. Chem. 265
Ž .1990 8369–8372.w x21 L.-L. Lin, A.Y. Lin, J.L. Knopf, Proc. Natl. Acad. Sci.
Ž .USA. 89 1992 6147–6151.w x Ž .22 C. Godson, K.S. Bell, P.A. Insel, J. Biol. Chem. 268 1993
11946–11950.w x23 R.A. Nemenoff, S. Winitz, N.-X. Qian, V. Van Putten, G.L.
Ž .Johnson, L.E. Heasley, J. Biol. Chem. 268 1993 1960–1964.
w x Ž .24 A. Huwiler, J. Pfeilschifter, Eur. J. Biochem. 217 199369–75.
w x25 S. Arkinstall, I. Emergy, D. Church, A. Chollet, E.Ž .Kawashima, FEBS. Lett. 338 1994 75–80.
w x26 S. Clark, R. Keogh, M. Dunlop, Biochim. Biophys. ActaŽ .1224 1994 221–227.
w x Ž .27 Z.-H. Qiu, C.C. Leslie, J. Biol. Chem. 269 1994 19480–19487.
w x28 W.-H. Zheng, D.W. Fink, G. Guroff, J. Neurochem. 66Ž .1996 1868–1875.
w x29 L.-L. Lin, M. Wartmann, A.Y. Lin, J.L. Knopf, A. Seth,Ž .R.J. Davis, Cell 72 1993 269–278.
w x30 M.A. Bogoyevitch, P.E. Glennon, M.B. Andersson, A. Clark,A. Lazou, C.J. Marshall, P.J. Parker, P.H. Sugden, J. Biol.
Ž .Chem. 269 1994 1110–1119.w x31 K. Malarkey, E.R. Chilvers, M.F. Lawson, R. Plevin, Br. J.
Ž .Pharmacol. 116 1995 2267–2273.w x32 A. Ocklind, S.Y.K. Yousufzai, S. Ghosh, M. Coca-Prados,
Ž .J. Stjernschantz, A.A. Abdel-Latif, Exp. Eye Res. 61 1995535–545.
w x Ž .33 C.C. Leslie, Methods Enzymol. 187 1990 216–225.w x34 A.G. Bosch-Haubold, R.M. Kramer, S.P. Watson, J. Biol.¨
Ž .Chem. 270 1995 25885–25892.w x Ž .35 U.K. Laemmli, Nature 227 1970 680–685.

( )S. Husain, A.A. Abdel-LatifrBiochimica et Biophysica Acta 1392 1998 127–144144
w x Ž .36 S. Husain, A.A. Abdel-Latif, Curr. Eye Res. 15 1996329–334.
w x Ž .37 I. Kameshita, H. Fujisawa, Anal. Biochem. 183 1989139–143.
w x38 O.H. Lowry, N.J. Rosebrough, A.L. Farr, R.J. Randall, J.Ž .Biol. Chem. 193 1951 265–275.
w x Ž .39 R.J. Flower, G.J. Blackwell, Biochem. Pharmacol. 25 1976285–291.
w x40 R.L. Bell, D.A. Kennerly, N. Stanford, P.W. Majerus, Proc.Ž .Natl. Acad. Sci. USA. 76 1979 3238–3241.
w x41 S.E. Wilkinson, P.J. Parker, J.S. Nixon, Biochem. J. 294Ž .1993 335–337.
w x42 F. Bartoli, H.-K. Lin, F. Ghomashchi, M.H. Gelb, M.K.Ž .Jain, R. Apitz-Castro, J. Biol. Chem. 269 1994 15625–
15630.w x Ž .43 A.S. Kraft, W.B. Anderson, Nature 301 1983 621–623.w x44 S. Young, P.J. Parker, A. Ullrich, S. Stabel, Biochem. J. 244
Ž .1987 775–779.w x45 W.J. Ryves, A.T. Evans, A.R. Olivier, P.J. Parker, F.J.
Ž .Evans, FEBS. Lett. 288 1991 5–9.w x46 A.A. Abdel-Latif, Prostaglandins Leukotrienes Essent. Fatty
Ž .Acids 44 1991 71–82.
w x47 A.A. Abdel-Latif, Progress in Retinal and Eye Research, in:Ž .N.N. Osborne, G.J. Chader Eds. , Pergamon, Oxford, 14,
1995, pp. 75–107.w x48 G. Martiny-Barou, M.G. Kazanietz, H. Mischak, P.M.
Blumberg, G. Kochs, H. Hug, D. Marme, C. Schachtele, J.¨Ž .Biol. Chem. 268 1993 9194–9197.
w x49 K.A. Qatsha, C. Rudolph, D. Marme, C. Schachtele, W.S.¨Ž .May, Proc. Natl. Acad. Sci. USA 90 1993 4674–4678.
w x50 R.M. Kramer, E.F. Roberts, J.V. Manetta, P.A. Hyslop, J.A.Ž .Jakubowski, J. Biol. Chem. 268 1993 26796–26804.
w x Ž .51 B.E. Kemp, R.B. Pearson, Trends Biochem. Sci. 15 1990342–346.
w x52 K. Abdullah, W.A. Cromlish, P. Payette, F. Laliberte, Z.Huang, I. Street, B.P. Kennedy, Biochim. Biophys. Acta
Ž .1244 1995 157–164.w x53 M.G.S. de Carvalho, A.L. Mccormack, E. Olson, F.
Ghomashchi, M.H. Gelb, J.R. Yates III, C.C. Leslie, J. Biol.Ž .Chem. 271 1996 6987–6997.
w x54 T. Taniguchi, K. Okada, M.S.R. Haque, K. Sugiyama, Y.Ž .Kitazawa, Curr. Eye Res. 13 1994 461–464.
w x Ž .55 J. Stjernschantz, B. Resul, Drugs Future 17 1992 691–704.