role of immunocompetent cells in nonimmune renal diseases
TRANSCRIPT

Kidney International, Vol. 59 (2001), pp. 1626–1640
PERSPECTIVES IN BASIC SCIENCE
Role of immunocompetent cells in nonimmune renal diseases
BERNARDO RODRIGUEZ-ITURBE, HECTOR PONS, JAIME HERRERA-ACOSTA,and RICHARD J. JOHNSON1
Renal Service Hospital Universitario de Maracaibo, Venezuela; Department of Nephrology, Instituto Nacional de Cardiologıa“Ignacio Chavez,” Mexico City, Mexico; and Renal Division, Department of Medicine, University of Washington MedicalCenter, Seattle, Washington, USA
Role of immunocompetent cells in nonimmune renal diseases. changes that lead to end-stage renal disease indepen-Renal infiltration with macrophages and monocytes is a well- dently of the initial insult, but are also conspicuouslyrecognized feature of not only immune, but also nonimmune present, sometimes in early stages, in a variety of diseaseskidney disease. This review focuses on the investigations that
of the kidney that do not have an immune etiopathogen-have shown accumulation of immunocompetent cells in experi-esis.mental models of acute and chronic ischemia, protein overload,
hypercholesterolemia, renal ablation, obstructive uropathy, We do not discuss the participation of lymphocytespolycystic kidney disease, diabetes, aging, murine hyperten- and macrophages in the development of renal fibrosission, and nephrotoxicity. We examine the mechanisms of infil-
since these aspects have been well covered by authorita-tration of immunocompetent cells and their participation in thetive reviews [3–5]; rather, we examine investigations thatself-perpetuating cycle of activation of the angiotensin system,
generation of reactive oxygen species, and further recruitment have shown accumulation of immunocompetent cells inof monocytes and lymphocytes. We also discuss the possibility nonimmune diseases and the mechanisms for infiltrationof antigen-dependent and antigen-independent mechanisms of and activation of these cells. Finally, we summarize re-immune cell activation in these animal models. Finally, we
cent research that indicates that significant improvementreview the recent studies in which suppression of cellular immu-in histologic and functional damage in nonimmune dis-nity with mycophenolate mofetil has proven beneficial in atten-
uating or preventing the progression of renal functional and eases of the kidney may result from treatment aimed athistologic damage in experimental conditions of nonimmune suppressing cellular immunity.nature.
TUBULOINTERSTITIAL INFILTRATION OFIn 1970, Rocklin, Lewis, and David reported that lym- MACROPHAGES AND LYMPHOCYTES IN
phocytes from some patients with glomerulonephritis NONIMMUNE RENAL DAMAGEshowed in vitro reactivity against glomerular basement Infiltration of macrophages and lymphocytes is a con-membrane [1]. Since the prevailing view at the time stant feature in conditions associated with chronic renalwas that humoral immunity was responsible for immune- damage. Whatever the mechanism for their accumula-mediated renal disease, this finding merited an editorial tion, these cells play a central role in most, if not all,with this title: “What are sensitized cells doing in glomer- pathways leading to progressive scarring of the kidney.ulonephritis?” [2]. Now, three decades later, it may be Immunocompetent cells are particularly conspicuous inworthwhile to ask what immunocompetent cells are do- areas of active tubulointerstitial injury, which since theing in nonimmune renal diseases. This review focuses
landmark observations of Risdon, Sloper, and de Warde-on the presence and role of macrophages and lympho-
ner [6] and Schainuck et al [7], is generally recognizedcytes that not only are associated with the sclerotic
to correlate with the severity of renal failure [8–10].Tubulointerstitial inflammatory injury may result fromantigen-specific stimulation of dendritic cells, infiltratingKey words: macrophages, lymphocytes, cell activation, tubulointersti-
tial injury, proteinuria, progressive renal disease, hypertension. macrophages, and resident endothelial and tubular cells,but cell-mediated injury may also occur in the absence1 Present address is Division of Nephrology, Department of Medicine,
Baylor University, Houston, Texas, USA. of antigen stimulation [11–15]. Table 1 shows a partiallist of experimental conditions that are assumed to have
Received for publication June 20, 2000a pathogenesis unrelated to antigen-specific immune re-and in revised form September 26, 2000
Accepted for publication November 15, 2000 sponses.In the ischemic models of renal damage, there is a 2001 by the International Society of Nephrology
1626

Rodrıguez-Iturbe et al: Immunocompetent cells 1627
Table 1. Experimental nonimmune renal diseases with this model of renal disease has a nonimmune pathogene-tubulointerstitial infiltration of immunocompetent cells
sis, despite the fact that the induction requires daily intra-Experimental condition References peritoneal injections of bovine serum albumin (BSA)Ischemia into Lewis rats. In fact, humoral immune mechanisms
Acute [16] are unlikely to play a role since proteinuria developsChronic [17]
within 24 to 48 hours of the initiation of the albuminProtein overload [19, 40]Hypercholesterolemia [40] injections, and there are no immune deposits in the kid-Reduction of renal mass [46, 52–54, 56, 57] ney. However, the possibility of participation of immuno-Obstructive uropathy [63, 65]
competent cells in the pathogenesis of this model is notPolycystic kidney disease [67, 68]Diabetic nephropathy [75, 78] ruled out. Studies by Eddy have shown that there is anAging [81, 86] early (,7 days) and prominent macrophage infiltrationPyelonephritis [88, 89]
that remains unabated throughout the duration of theHypertensionDahl sensitive [90, 91] overload proteinuria; furthermore, after two weeks,SHR [92, 93] T-helper and T-cytotoxic cells are also observed in theDOCA-salt sensitive
tubulointerstitial infiltate [19]. While the T-helper infil-Catecholamine infusion [94]Angiotensin II infusion [95] trate tends to decrease after the third week, the T-cyto-Two-kidney one-clip [96, 97] toxic cells remain for at least seven weeks. T-cell de-Nitric oxide synthetase inhibition [98, 103]
pletion with intraperitoneal anti–T-cell monoclonalNephrotoxicityPuromycin aminonucleoside [20, 109, 110, 112, 113] antibody (mAb) administration did not modify the mac-Mercuric chloride [107, 108] rophage infiltration, indicating that the influx of theseCyclosporine A [21, 118, 119]
cells was not dependent on the lymphocytes [19], butTacrolimus [117]more likely resulting from locally expressed osteopontin,References correspond to investigations reporting specific details about the
lymphocyte and macrophage infiltration. monocyte chemoattractant protein-1 (MCP-1), andadhesion molecules vascular cell adhesion molecule(VCAM) and intracellular adhesion molecule (ICAM)[24] and a chemotactic urinary lipid possibly generated
prominent infiltration of macrophages and lymphocytes. by the tubular catabolism of albumin-borne fatty acidsIn the acute ischemia induced in mice by temporal occlu- [25]. Nevertheless, structural changes in the tubules aresion of the renal flow to one kidney, Goes et al found an present in this experimental model [26, 27], and the toxicincreased steady-state mRNA for a number of cytokines damage could result in the expression of autoantigensinvolved in macrophage and T-cell activation, including that could elicit a delayed cellular response. For instance,interferon-g (IFN-g), interleukin (IL)-2, IL-10, and gran- vimentin filaments found in the tubules of rats with over-ulocyte-macrophage colony-stimulating factor (CSF), as load proteinuria [19] are not expressed in normal tubuleswell as overexpression of major histocompatibility com- and neoantigens expressed after activation of the C5b-9plex (MHC) class I and class II molecules [16]. There was attack complex, likely resulting from stimulation of thean intrarenal accumulation of T cells and macrophages complement cascade by proximal tubular cells [28], haveassociated with this cytokine response. been demonstrated in the brush border of tubular cells
In experimental chronic ischemia induced by unilat- [19, 28].eral renal arterial stenosis in the rat, Truong et al found In 1971, Wellman and Volk published one of the earli-that the ischemic kidney showed marked tubulointersti- est investigations linking hypercholesterolemia with re-tial damage, including an abundant inflammatory infil- nal injury [29], but widespread interest in the field wastrate and tubulitis in 42% of the tubular cross sections fueled by Moorhead et al’s article suggesting that in-[17]. The infiltrating cells were mostly macrophages, B creased lipoprotein synthesis associated with proteinuriacells, and T-helper lymphocytes, and there were impor- contributes to the progression to chronic renal failuretant changes in the antigenic constitution of the tubules. [30]. Since then, it has been established that high choles-The authors noted similarities between the phenotypic terol feeding causes experimental renal disease [31, 32]characteristics of the infiltrate in chronic ischemia and and that reduction in lipid levels reduces the renal dam-the infiltrate in autoimmune tubulointerstitial nephritis age of not only hyperlipidemic nephrotic rats [33], but[18], overload proteinuria [19], aminonucleoside nephro- also in a variety of hyperlipidemic renal diseases. Notsis [20], and cyclosporine nephrotoxicity [21]. surprisingly, the initial investigations focused on the
It has been known for more than half a century that pathogenesis of lipid-induced glomerulosclerosis, indi-parenteral administration of proteins induces proteinuria cating that oxidized lipoproteins in the glomeruli would[22, 23], and recent models of overload proteinuria have follow similar pathogenetic pathways, as observed in ath-emphasized the importance of tubulointerstitial infiltra- erosclerosis, including recruitment of macrophages with
scavenger functions, thromboxane-induced glomerulartion with mononuclear cells. It is generally assumed that

Rodrıguez-Iturbe et al: Immunocompetent cells1628
vasoconstriction, and mesangial cell proliferation [31, The histologic changes that follow ureteral obstructionwere initially described almost three decades ago [58].34–40]. Primary tubulointerstitial damage caused by hy-
percholesterolemia has also been documented in the rig- Tubular and interstitial injury is accompanied by cellproliferation and extracellular matrix deposition [59]. Anorous investigation of Eddy, Liu, and McCulloch who
showed that ingestion of a 4% cholesterol diet resulting increased production of angiotensin and thromboxane[60, 61] is responsible for well-defined hemodynamicin a threefold to fourfold increment in plasma cholesterol
with the deposition of neutral lipids and oxidized lipids changes [62]. Careful studies by Schreiner et al detectedan influx of macrophages and lymphocytes in the ob-in both glomerular and tubulointerstitial areas [40]. By 12
weeks, there was a prominent infiltration of macrophages structed kidney as early as 4 hours after obstruction, andthe infiltration of these cells reached maximal intensitythat was preceded by or accompanied with the increment
in mRNA levels for MCP-1, osteopontin, and VCAM. after 24 hours and remained at the same levels until theobstruction was released [63]. Since macrophages are aTransforming growth factor-b1 (TGF-b1) was present
before and during the fibrotic phase of the disease. No known source of angiotensin and thromboxane [64], itis not surprising that the intensity of the mononuclearreference was made to lymphocytes in this work [40], but
T cells are known to be present in the tubulointerstitial cell infiltrate varied in parallel with the levels of thesevasoconstrictors and the fall in glomerular filtration. Ininfiltrate of kidneys with arteriolosclerosis that resem-
bles, at times, that found in pyelonephritis [41], and lym- addition to the participation in the pathogenesis of he-modynamic and renal functional changes, the infiltratingphocytes are a constant finding in atherosclerotic plaques
(Fig. 4A) [42] and in ischemic kidneys [17]. macrophages are also involved in the late fibrogenicchanges resulting from increased synthesis of TGF-b1A reduction of renal mass is a well-defined nonim-
mune model of progressive renal failure studied at length [65]. After release of obstruction, the glomerular macro-phages remain significantly increased, a finding that hasby Hostetter et al after the original description of glomer-
ulosclerosis following 5/6 nephrectomy by Shimamura been postulated to be related to postobstruction loss offunctional glomeruli [63]. Lymphocyte infiltration fol-and Morrison [43, 44]. The development of glomerulo-
sclerosis is related to the hemodynamic disturbances lows the same pattern as the macrophage infiltration.Interestingly, in contrast with most other models of non-present in the remnant nephrons as a result of the reduc-
tion in functioning kidney mass [45]. Mesangial cell acti- immune renal disease, the suppressor-cytotoxic subsetof infiltrating T lymphocytes is five times more promi-vation (a-smooth muscle actin expression) and prolifera-
tion occur within days after renal ablation in association nent than the helper phenotype [63].Polycystic kidney disease is a hereditary disorder thatwith platelet-derived growth factor (PDGF) expression
and precede the mononuclear cell infiltrate and the glo- is the cause of a rapidly fatal form, with uremia shortlyafter birth in the C57BL/6 J-cpk mouse [66], or a moremerular and interstitial sclerosis [46]. Activation of the
renin-angiotensin system by the intraglomerular hemo- slowly progressive form in the DBA/2FG-pcy mouse[67], the latter resembling the autosomic dominant poly-dynamic changes may result in increased TGF-b and
subsequent glomerular sclerosis [47–49], a process that cystic kidney disease in the human. The DBA/2FG-pcymice develop an autosomal recessive form of polycysticis accelerated by a high-protein diet [50, 51]. Macrophage
infiltration in glomeruli and tubulointerstitium is already kidney, manifested at birth by segmental dilation of distaltubules and collecting ducts that progress to cysts, whichfound after 10 days, increases after 4 weeks, and reaches
a maximum after 12 weeks’ postablation [52–54]. The are numerous and dilated by eight weeks of age, andconstitute the dominant feature in the late stages of theinfiltration of macrophages is facilitated by the expres-
sion of MCP-1 [55] and stimulated by angiotensin II [56]. disease [67]. In this model, a prominent tubulointerstitialinfiltrate consisting predominantly of lymphocytes andApproximately one third of the infiltrating macrophages
are proliferating in the remnant kidney [52], and the total macrophages appears between the 18 and 30 weeks ofage. Increased expressions of PDGF, insulin-like growthnumber of macrophages correlate with the functional
parameters of renal failure [56]. T lymphocytes are also factor-1 (IGF-1), and TGF-b have been demonstratedand are thought to play a role in the cyst formationconspicuously present in the tubulointerstitial infiltrate
after seven days [53] and remain in significant numbers [68]. Since macrophages are one of the cells capable ofproducing PDGF [69, 70] and IGF-1 [71], as well asfor the following weeks [53, 54]. The disease is associated
with an overexpression of MHC class II antigens, and TGF-b [72], Nakamura et al have suggested that macro-phages could have a role in the pathogenesis of the dis-the infiltrating T cells are predominantly of the T-helper
phenotype [57]. The pathogenetic importance of immu- ease [68].Diabetic nephropathy is a complex and progressivenocompetent cells in this hemodynamically-induced
chronic renal damage has been dramatically shown by the process that in its early phase is associated with nephronhypertrophy [73] and later with the development of dif-improvement obtained with immunosuppressive treat-
ment [53, 54, 57]; this is discussed later. fuse glomerulosclerosis that sometimes assumes a nodu-

Rodrıguez-Iturbe et al: Immunocompetent cells 1629
lar appearance [74] often associated with widespread rats, there is intense accumulation of macrophages inbrain and kidney. In the brain, they are scattered aroundinterstitial fibrosis [75]. The increase in kidney size is
mostly the result of hypertrophy, even though there is the lesions and appear to act only as scavengers; how-ever, in the kidney, macrophages expressing class II-an early component of hyperplasia. Mesangial expansion
accompanies the global hypertrophic changes and is as- MHC antigens in association with T lymphocytes arelocated in perivascular areas, and their infiltration pre-sociated with an increase TGF-b1 in both glomerular
and the tubulointerstitium. The severity of glomerular cedes the fibrocellular proliferative lesions [92]. The ad-ministration of an angiotensin-converting enzyme inhibi-[76] and tubulointerstitial damage [77] correlates with
functional deterioration. Rarely mentioned is the fact tor or angiotensin II-receptor blocker was associatedwith a significant reduction on macrophage and T-helperthat streptozotocin-treated rats have an important inter-
stitial inflammatory infiltrate [75]. Characteristics of this lymphocyte infiltration and, in parallel, with reduced tu-bulointerstitial proliferation and sclerosis [93]. An infil-infiltrate are undefined but, in humans with diabetic ne-
phropathy, the inflammatory cells are predominantly T tration of macrophages in the interstitium has also beennoted in catecholamine-induced hypertension [94], an-lymphocytes and to a lesser degree macrophages [78].
Clearly, the pathogenesis of the sclerotic transformation giotensin II-induced hypertension [95], and two-kidneyone-clip hypertension [96, 97], and in these models, theof the kidney in diabetes is multifactorial. Hyperglycemia
and nonenzymatically glycated proteins are known to interstitial injury correlates with the blood pressure. Fur-thermore, a reanalysis of the catecholamine infusionstimulate mesangial and proximal tubular cell produc-
tion of TGF-b1 as well as production and gene expression study [94] shows that the macrophage interstitial infiltra-tion present at the end of the catecholamine infusionof fibronectin and type IV collagen [79], but in addition,
immunocompetent cells may participate in the diabetic also correlates with the subsequent blood pressure re-sponse to a high-salt diet (Fig. 2).injury. Macrophage recruitment may be induced by high
serum lipid levels, as discussed earlier, and with advanc- Chronic administration of nitro-l-arginine-methyl es-ther (L-NAME) produces arterial hypertension as a con-ing arteriosclerotic disease, chronic ischemia becomes
an increasingly important mechanism of generation of sequence of suppression of nitric oxide biosynthesis. Thisexperimental model, first described by Oliveira-Ribeiroreactive oxygen species (ROS) and infiltration of mono-
nuclear cells (discussed later in this article). et al [98], induces activation of the renin-angiotensinsystem [99–101] and sympathetic nervous system [102],Aging is associated with a variety of functional and
structural changes [80, 81]. Glomerulosclerosis is the causing intense vasoconstriction. Monocyte/macrophageinfiltration is intense in the intima and media of preglo-classic finding that has attracted most of the research
effort [82–85], but tubulointerstitial injury is also an im- merular arteries and arterioles as well as in tubulointer-stitial areas, where these cells are two to three timesportant characteristic of the senescent kidney. Tubular
dilation and atrophy, widening and fibrosis of intersti- increased in number [103]. Nitric oxide suppresses theendothelial expression of several cytokines and cell ad-tium, phenotypic changes consisting in cellular expres-
sion of a-smooth muscle actin, and imbalance between hesion molecules, particularly VCAM-1 and P-selectin;in addition, L-NAME administration augments the ex-proliferation and apoptosis have all been demonstrated
in the aging rat [86]. TGF-b1 is increased in parallel with pression of VCAM-1. These findings suggest a mecha-nism for the mononuclear cell infiltration associated withthe fibrotic tubulointerstitial changes [87]. Importantly,
a prominent infiltration with macrophages and lympho- inhibition of nitric oxide synthase [104–106].Not surprisingly, experimental models of acute renalcytes in association with increased osteopontin and
ICAM-1 expression has been observed in the intersti- failure are also associated with intense inflammatory in-filtrate in association with acute tubular necrosis. Mercu-tium in aging animals [81].
Experimental pyelonephritis induced by intrarenal ric chloride nephrotoxicity is characterized by proximaltubular necrosis and intense mononuclear cell infiltrationbacterial inoculation is characterized by interstitial
edema, inflammation, and scarring. Contraction and col- that precedes growth factor expression [107]. The macro-phage and lymphocyte infiltration may be stimulatedlapse of the tubulointerstitial parenchyma with fibrosis
are major pathophysiologic mechanisms for new colla- by the generated reactive oxygen species (ROS), sincerecent work from our group found that it decreases withgen production [88]. Mononuclear cell infiltration has
been noted since early descriptions of this model [89]. antioxidant therapy [108].The model of puromycin aminonucleoside nephrosisIn practically every experimental model of hyperten-
sion, there is a prominent tubulointerstitial infiltrate of is characterized by focal glomerulosclerosis and diffuseinterstitial fibrosis [109]. However, several reports haveimmunocompetent cells. In Dahl-sensitive rats, mononu-
clear cell infiltration is prominent in the tubulointersti- emphasized the existence of important interstitial in-flammatory infiltrate [110–112] constituted mainly bytium, especially around sclerosed glomeruli and arteri-
oles [90, 91]. In stroke-prone spontaneously hypertensive macrophages but also by lymphocytes [111]. Macro-

Rodrıguez-Iturbe et al: Immunocompetent cells1630
phages are known to have an important pathogenetic both the functions of a chemokine and the functions ofadhesion molecules.role in the model induced by a single intravenous injec-
tion of puromycin aminonucleoside, since inhibition of Adhesion molecules are responsible for the rolling,adhesion, and eventually penetration of these cells fromthe macrophage infiltration in the acute phase results
in a reduction of the late glomerular injury [113, 114]. circulation into the interstitial spaces [reviewed in 122].Of the various adhesion molecules, ICAM-1 (CD54) andOxidative stress [112, 115] and increased TGF-b1 expres-
sion [111] may result from macrophage activity and pro- VCAM1 (CD106) appear to be particularly importantin experimental models of renal disease [123, 124].mote further macrophage recruitment.
Cyclosporine A and tracolimus induce renal damage Table 2 examines the characteristics of antigen-inde-pendent renal injuries that are capable of inducing thethat has similar histologic features. Increased TGF-b1
production and extracellular matrix deposition [116, 117] production of chemokines and expression of adhesionmolecules.are present in association with intense macrophage infil-
tration and stimulation of the intrarenal renin-angioten- A final pathway of most injuries leading to chronickidney damage includes the participation of proteinuria,sin system [118]. Macrophage accumulation and renal
damage are potentiated by a low-salt diet [119], which oxidized lipids, and chronic ischemia, the cross-talk ofwhich results in a self-perpetuating cycle of activationstimulates angiotensin activity, and injury is reduced by
angiotensin-receptor blockade [118]. Therefore, intrare- of the renin-angiotensin system, generation of ROS, andrecruitment and activation of macrophages and lympho-nal ischemia, angiotensin II generation, and macro-
phage-dependent cytokines are pathogenetically linked cytes (Fig. 1).The role of angiotensin II (Ang II) in the generationin the development of the striped interstitial fibrosis,
tubular atrophy, and hyalinosis of the afferent arteriole, of sclerotic changes is beyond the scope of this articleand has been the subject of excellent reviews [125, 126].which are typical of both cyclosporine and tacrolimus
nephrotoxicity [120]. Pertinent to the present discussion, it should be empha-sized that fibrosis increases chronic ischemia in the rem-nant kidney tissue since the fibrous transformation re-
MECHANISMS OF INFILTRATION ANDduces the interstitial vascular network. Fine, Orphanides,
ACTIVATION OF IMMUNOCOMPETENT CELLS and Norman have emphasized that chronic tubulointer-IN NONIMMUNE RENAL DISEASE stitial hypoxia is capable of promoting progressive renal
The infiltration of immunocompetent cells requires scarring [127]. A variety of growth factors and fibrogenicboth the attraction exerted by the local expression of cytokines is produced by renal cells in vitro in responsechemotactic factors and the expression of adhesion mole- to hypoxia [128].cules. The chemokines are one of the most important Angiotensin II may promote the accumulation of im-chemotactic compounds. They are proteins of 8 to 10 munocompetent cells by its hemodynamic effects, bykD secreted by activated immune cells as well as resident stimulating synthesis of chemokines and adhesion mole-renal cells. Chemokines stimulate only migration but, cules and by direct actions on resident and infiltratingunder certain conditions, may also stimulate cell prolifer- cells. The hemodynamic effects of angiotensin promoteation, angiogenesis, and leukocyte activation. The che- macrophage accumulation by at least two mechanismsmokine superfamily now consists of four subfamilies with contemplated in Figure 1. (1) Generalized vasoconstric-40 members and 17 different receptors [reviewed in 121]. tion resulting from intrarenal angiotensin activity pro-The two more numerous subfamilies are the C-C subfam- motes chronic ischemia and ischemia-derived generationily, which has two cysteines adjacent to each other, and of ROS [129], and (2) angiotensin exerts preferentialthe C-X-C subfamily, which has an additional amino efferent vasoconstriction in the glomerular capillary,acid interposed between the cysteines. Most of C-X-C thereby increasing intraglomerular filtration pressurechemokines are chemoattractant for neutrophils, while and proteinuria [130, 131]. Prevention of these hemody-the C-C chemokines attract monocytes, T cells, and natu- namic effects is largely responsible for the reduction inral killer cells. Two of the cytokines most commonly proteinuria observed with administration of angiotensin-implicated in renal disease, MCP-1 (which is primarily converting enzyme inhibitors [132, 133]. In turn, protein-chemotactic for macrophages) and RANTES (which is uria induces enhanced angiotensinogen mRNA expres-primarily chemotactic for lymphocytes), belong to C-C sion in the proximal tubular cells [134].subfamily. Lymphotactin, a member of the third subfam- In addition to its vasoactive properties, Ang II inducesily (C family), lacks two of the four cysteine residues, the expression of adhesion molecules [135–137], acti-but is homologous to the CC chemokines in the carboxyl vates nuclear factor kappa-B (NF-kB), and stimulatesend. Lymphotactin is a powerful attractor of T lympho- the synthesis of MCP-1 [138], and osteopontin [139],cytes. The final subfamily (CX3C subfamily) is consti- which is assumed to be a macrophage attractor. Further-
more, amino and carboxy-terminal Ang II tetrapeptidestuted by Fractalkine, which has been shown to have

Rodrıguez-Iturbe et al: Immunocompetent cells 1631
Table 2. Expression of chemoattractants and adhesion molecules in pathophysiologic conditions associated withinfiltration of cells of the immune system in the kidney
Chemoattractants Adhesion molecules References
Proteinuria Chemokines, complement (C5a) growth factors, VCAM, ICAM [19, 24, 25, 204, 205]vasoactive mediators, urinary lipid chemoattrac-tant, osteopontin
Ischemia Chemokines, growth factors, osteopontin VCAM, ICAM [16]Oxidized lipids Chemokines, vasoactive mediators, osteopontin VCAM, ICAM [40, 159–161] [206]Obstruction to ICAM-1, VCAM [207–209]
urine flow Chemokines, vasoactive mediators, osteopontinShear stress Chemokines, growth factors/vasoactive mediators ICAM-1, VCAM-1 [210–214]
Chemokines include MCP-1, RANTES and growth factors/vasoactive mediators include PDGF, colony stimulating factors, endothelin-1 and angiotensin II.
are chemotactic for monocytes [140]. In addition to pro- production of ROS with inhibition of the membrane-bound nicotinamide adenine dinucleotide phosphatemoting macrophage recruitment, Ang II is capable of
activating them [141]. (NADPH)-dependent oxidase system, or using ROSscavengers, such as dimethyl- and trimethyl-thio-urea,The role of angiotensin in the accumulation and activa-
tion of immunocompetent cells in vivo is increasingly reduces the expression of these same chemoattractantsand adhesion molecules. ROS may function as secondrecognized by the beneficial effects of angiotensin recep-
tor blockade in several models of immune-mediated dis- messengers [156] in the generation of MCP-1, RANTES,and ICAM-1 by a mechanism that may involve an inter-eases [142–145]. While these findings are largely indirect,
immune modulation by Ang II is strongly suggested by mediate step of activation of the transcription NF-kB[157].the existence of angiotensin II type 1 (AT1) receptor in
macrophages and T cells [146], and more recently by the Oxidized lipids have monocyte chemoattractant activ-finding that proliferation of T cells is induced by Ang II ity [158] and induce endothelial cell expression of adhe-through the AT1 receptor by a calcineurin-dependent sion molecules ICAM-1 and VCAM [159–161]. Highpathway [147]. Finally, Ang II may stimulate indirectly lipid diets induce mRNA of MCP-1, osteopontin, andthe lipid-induced macrophage activation since it has a VCAM in the rat kidney [40]. A reduction of serumstimulatory effect in low-density lipoprotein (LDL) oxi- lipids reduces injury in models of kidney disease thatdation [148]. do not cause increased serum lipids, such as models of
Reactive oxygen species are mediators of tissue injury ureteral obstruction [162, 163]. However, the beneficialthat are pathogenetically involved in a variety of experi- effects of HMG-CoA reductase inhibitors used in manymental models of immune, toxic, and ischemic renal dam- of such studies may result from a direct antiproliferativeage. Resident renal cells and infiltrating leukocytes are effect of these drugs, rather than from its lipid-loweringknown sources of ROS [149]. Macrophages produce su- properties, since they inhibit the mevalonate pathwayperoxide (O2
2), hydrogen peroxide (H2O2) and hydroxyl that generates metabolites that stimulate mesangial pro-radical (·OH) in inflammatory immune-mediated glo- liferation [164] and type IV collagen secretion [165].merulonephritis [149]. ROS have been reported to cause Finally, recruitment of immunocompetent cells by oxi-mesangiolysis and endothelial damage in some [150] but dized lipoproteins may also be indirectly mediated bynot all species [151]. If injected in the renal artery, H2O2 the generation of ROS, which these compounds inducecauses a massive, abrupt, and reversible proteinuria re- in endothelial and mesangial cells (Fig. 1) [166].sulting from a size-selective defect caused by iron-depen-dent metabolites of hydrogen peroxide [151]. Studies by
ARE IMMUNOCOMPETENT CELLSJohnson et al have shown that glomerular interactionACTIVATED BY ANTIGEN-DEPENDENT ORbetween myeloperoxidase, H2O2, and a halide causedANTIGEN-INDEPENDENT MECHANISMS INproteinuria and a damage consisting of necrosis of endo-NONIMMUNE RENAL DISEASES?thelial cells with platelet activation and proliferation of
While the infiltration of neutrophils and macrophagesresident glomerular cells [152, 153].is part of a nonspecific inflammatory reaction, the pres-In addition, ROS are actively involved in the activationence of lymphocytes and dendritic cells within lesionsof chemotactic factor and adhesion molecules (Fig. 1).raises questions about their role in the maintenance andIn a series of elegant experiments, Satriano et al haveamplification of the inflammatory response and whethershown that generation of superoxide anions by the addi-or not their recruitment and activation is mediated bytion of xanthine oxidase and hypoxanthine to mesangialan antigen-specific immune response. These questionscells increase the mRNA levels of MCP-1, RANTES,
and ICAM-1 [154, 155]. Furthermore, suppressing remain largely unanswered at the present time.
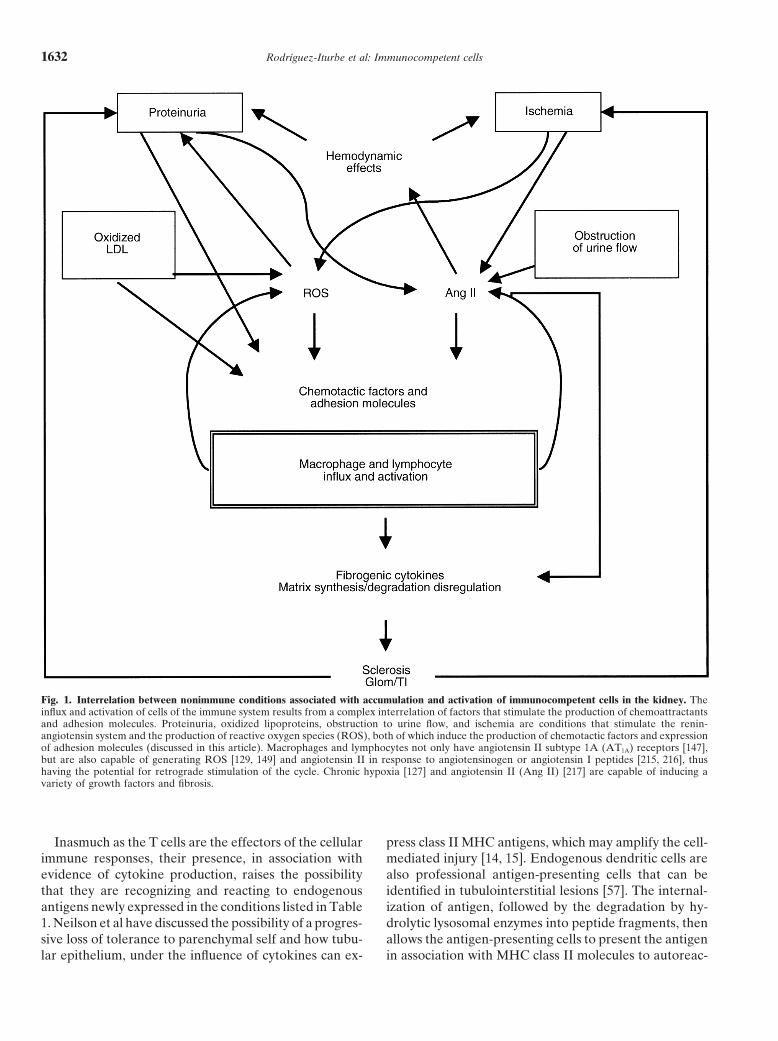
Rodrıguez-Iturbe et al: Immunocompetent cells1632
Fig. 1. Interrelation between nonimmune conditions associated with accumulation and activation of immunocompetent cells in the kidney. Theinflux and activation of cells of the immune system results from a complex interrelation of factors that stimulate the production of chemoattractantsand adhesion molecules. Proteinuria, oxidized lipoproteins, obstruction to urine flow, and ischemia are conditions that stimulate the renin-angiotensin system and the production of reactive oxygen species (ROS), both of which induce the production of chemotactic factors and expressionof adhesion molecules (discussed in this article). Macrophages and lymphocytes not only have angiotensin II subtype 1A (AT1A) receptors [147],but are also capable of generating ROS [129, 149] and angiotensin II in response to angiotensinogen or angiotensin I peptides [215, 216], thushaving the potential for retrograde stimulation of the cycle. Chronic hypoxia [127] and angiotensin II (Ang II) [217] are capable of inducing avariety of growth factors and fibrosis.
Inasmuch as the T cells are the effectors of the cellular press class II MHC antigens, which may amplify the cell-mediated injury [14, 15]. Endogenous dendritic cells areimmune responses, their presence, in association with
evidence of cytokine production, raises the possibility also professional antigen-presenting cells that can beidentified in tubulointerstitial lesions [57]. The internal-that they are recognizing and reacting to endogenous
antigens newly expressed in the conditions listed in Table ization of antigen, followed by the degradation by hy-drolytic lysosomal enzymes into peptide fragments, then1. Neilson et al have discussed the possibility of a progres-
sive loss of tolerance to parenchymal self and how tubu- allows the antigen-presenting cells to present the antigenin association with MHC class II molecules to autoreac-lar epithelium, under the influence of cytokines can ex-

Rodrıguez-Iturbe et al: Immunocompetent cells 1633
in the initiation of autoimmunity [172]. T lymphocyteswith ab TCR have a very wide distribution, while lym-phocytes with gd TCR are mostly restricted to specificareas such as epidermis, intestinal epithelium, uterus,and tongue, but they have also been found in the kidneyin IgA nephropathy, where their presence is associatedwith progressive disease [173]. It is therefore conceivablethat T cells with gd TCR could play a role in renalautoimmunity recognizing antigenic stress proteins de-rived from the tubulointerstitial cells injured by varietyof pathogenetic influences.
Antigen-independent pathways may also result inT-lymphocyte activation (Table 1). This possibility hasbeen raised in chronic hepatitis C and in multiple sclero-sis, because in these diseases, there is a discrepancy be-tween the large number of infiltrating T cells and theFig. 2. Relationship of interstitial macrophage infiltration to systolic
blood pressure in catecholamine-induced hypertension [94]. In this small number of them that are specifically reactive forstudy, rats were administered phenylephrine for eight weeks. During hepatitis C virus (HCV) proteins or central nervous sys-this time, blood pressure was episodically elevated. At the end of the
tem antigens, respectively. One explanation for this ob-infusion one kidney was removed and the number of ED-1–positivemacrophages in the interstitium was determined (semiquantitative score servation is the possibility of antigen-independent mech-0 to 5). Rats were subsequently placed on a high (8% NaCl) salt diet anisms that could activate bystander T cells [174]; severalfor four to eight weeks. Shown is the systolic blood pressure at the end
studies have focused on the possibility that T cells couldof the study. (, vehicle-perfused rats; , phenylephrine-infused rats; N 517, r2 5 0.48, P , 0.003). proliferate and display effector functions in response
to cytokines in the absence of TCR occupancy. Infact, Unutmaz et al have found that a combination ofIL-2, tumor necrosis factor (TNF)-a, and IL-6 could in-
tive T cells via specific T-cell receptor (TCR). Peptide duce highly purified naive (CD45RA1) and memoryfragments bound to class II MHC molecules are recog- (CD45RO1) T cells to proliferate and resting memorynized predominantly in T cells expressing CD4 molecules T cells to synthesize cytokines and promote IgG produc-and require a costimulatory signal that is antigen inde- tion by B cells [175, 176]. Bacon et al found thatpendent. RANTES, in addition to its chemotactic properties, in-
Truong et al found that chronic ischemia induces pro- duces antigen-independent cytokine release, IL-2 recep-found changes in the antigens expressed by the renal tor expression, and T-cell proliferation [177], and similartubules [17]. These changes included a loss of cell mem- results were obtained with IL-7 by Fukui et al [178].brane glycoproteins, characterized by a loss of binding Antigen-independent production of mRNA for IL-2properties for the lectins Dolichos biflorus and glycine and expression of IL-2 receptor may also be inducedmax, which are specific markers for D-galactose-N-acetyl by the binding of specific costimulatory molecules, inglucosamine. In addition, there was exposure of cytoskel- particular CD28 [179, 180] and the vitronectin receptor
(VNR), which if engaged by extracellular matrix pro-eton filament antigens that are normally expressed onlyteins, independently of occupancy of the TCR, may in-in embryonic cells, such as vimentin and keratin. Of note,duce secretion of IL-2 in gd TCR T cells [181].these changes are not specific for ischemic injury since
At present, it is not known to what extent, if any,they also occur in other conditions, such as puromycinautoreactive cellular immunity is responsible for the[20] and protein overload [18] nephropathies, raisingmononuclear cell infiltration clearly present in “nonim-the possibility that these alterations in tubular antigenicmune” renal diseases and whether nonspecific activationprofile are common to many, if not all, models of severeof these cells plays a role in their proliferation and ef-tubulointerstitial injury.fector activity.In addition, the early events of injury in the tubuloin-
terstitial cells likely include the release of heat shock(stress) proteins, which are found in all prokaryotic and IS THE PRESENCE OF IMMUNE CELLS
PATHOGENETICALLY RELEVANT INeukaryotic cells and are assumed to be transporters ofNONIMMUNE RENAL DISEASES?target antigens in the initial protective response againstRESULTS OBTAINED WITHmany infectious organisms [167]. These stress proteinsMYCOPHENOLATE MOFETILhave essential roles in the assembly and transport of
other molecules, can be recognized by the gamma delta Several studies have investigated the role of the cellu-lar immune system in some experimental conditionsT cell receptor (gd TCR) [168–171], and may be involved
Rodrıguez-Iturbe et al: Immunocompetent cells1634
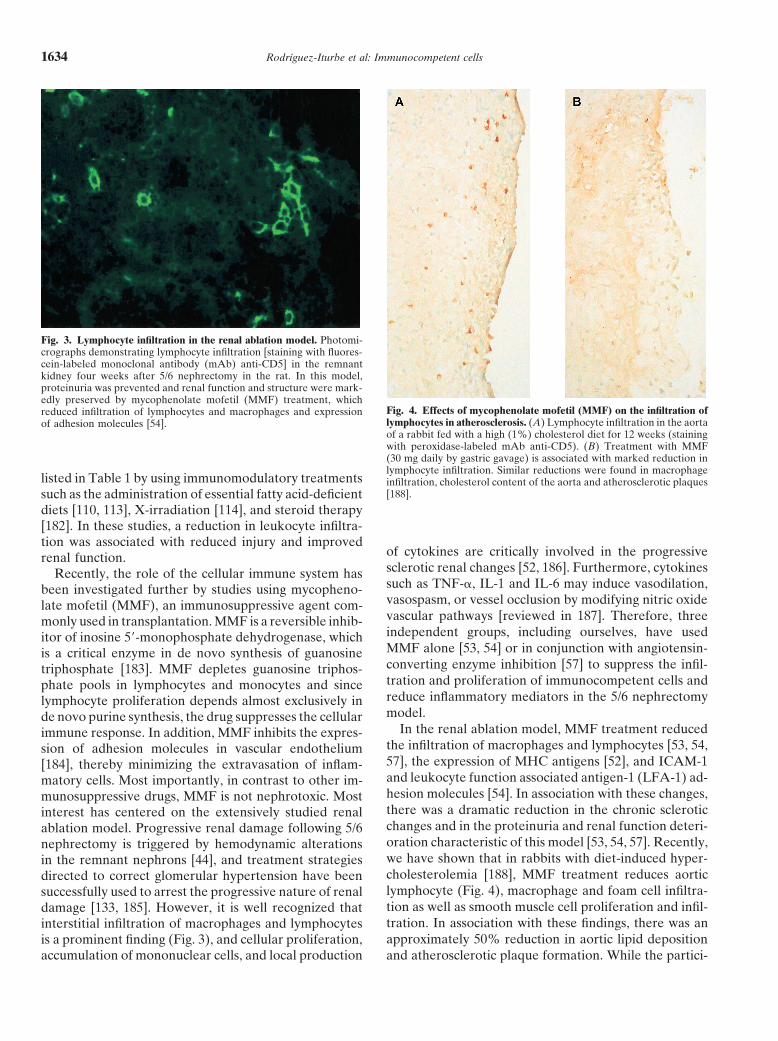
Fig. 3. Lymphocyte infiltration in the renal ablation model. Photomi-crographs demonstrating lymphocyte infiltration [staining with fluores-cein-labeled monoclonal antibody (mAb) anti-CD5] in the remnantkidney four weeks after 5/6 nephrectomy in the rat. In this model,proteinuria was prevented and renal function and structure were mark-edly preserved by mycophenolate mofetil (MMF) treatment, which
Fig. 4. Effects of mycophenolate mofetil (MMF) on the infiltration ofreduced infiltration of lymphocytes and macrophages and expressionlymphocytes in atherosclerosis. (A) Lymphocyte infiltration in the aortaof adhesion molecules [54].of a rabbit fed with a high (1%) cholesterol diet for 12 weeks (stainingwith peroxidase-labeled mAb anti-CD5). (B) Treatment with MMF(30 mg daily by gastric gavage) is associated with marked reduction inlymphocyte infiltration. Similar reductions were found in macrophage
listed in Table 1 by using immunomodulatory treatments infiltration, cholesterol content of the aorta and atherosclerotic plaques[188].such as the administration of essential fatty acid-deficient
diets [110, 113], X-irradiation [114], and steroid therapy[182]. In these studies, a reduction in leukocyte infiltra-tion was associated with reduced injury and improved
of cytokines are critically involved in the progressiverenal function.sclerotic renal changes [52, 186]. Furthermore, cytokinesRecently, the role of the cellular immune system hassuch as TNF-a, IL-1 and IL-6 may induce vasodilation,been investigated further by studies using mycopheno-vasospasm, or vessel occlusion by modifying nitric oxidelate mofetil (MMF), an immunosuppressive agent com-vascular pathways [reviewed in 187]. Therefore, threemonly used in transplantation. MMF is a reversible inhib-independent groups, including ourselves, have useditor of inosine 59-monophosphate dehydrogenase, whichMMF alone [53, 54] or in conjunction with angiotensin-is a critical enzyme in de novo synthesis of guanosineconverting enzyme inhibition [57] to suppress the infil-triphosphate [183]. MMF depletes guanosine triphos-tration and proliferation of immunocompetent cells andphate pools in lymphocytes and monocytes and sincereduce inflammatory mediators in the 5/6 nephrectomylymphocyte proliferation depends almost exclusively inmodel.de novo purine synthesis, the drug suppresses the cellular
In the renal ablation model, MMF treatment reducedimmune response. In addition, MMF inhibits the expres-the infiltration of macrophages and lymphocytes [53, 54,sion of adhesion molecules in vascular endothelium57], the expression of MHC antigens [52], and ICAM-1[184], thereby minimizing the extravasation of inflam-and leukocyte function associated antigen-1 (LFA-1) ad-matory cells. Most importantly, in contrast to other im-hesion molecules [54]. In association with these changes,munosuppressive drugs, MMF is not nephrotoxic. Mostthere was a dramatic reduction in the chronic scleroticinterest has centered on the extensively studied renalchanges and in the proteinuria and renal function deteri-ablation model. Progressive renal damage following 5/6oration characteristic of this model [53, 54, 57]. Recently,nephrectomy is triggered by hemodynamic alterationswe have shown that in rabbits with diet-induced hyper-in the remnant nephrons [44], and treatment strategiescholesterolemia [188], MMF treatment reduces aorticdirected to correct glomerular hypertension have beenlymphocyte (Fig. 4), macrophage and foam cell infiltra-successfully used to arrest the progressive nature of renaltion as well as smooth muscle cell proliferation and infil-damage [133, 185]. However, it is well recognized thattration. In association with these findings, there was aninterstitial infiltration of macrophages and lymphocytesapproximately 50% reduction in aortic lipid depositionis a prominent finding (Fig. 3), and cellular proliferation,
accumulation of mononuclear cells, and local production and atherosclerotic plaque formation. While the partici-

Rodrıguez-Iturbe et al: Immunocompetent cells 1635
pation of immune cells in atherosclerosis is well recog- by which MMF prevents salt-sensitive hypertension, thereduction of angiotensin II-producing infiltrate couldnized [42], the treatment with other immunosuppressive
drugs has given conflicting results [189–192]; therefore, certainly play a role. Taken together, the data suggestthat immunocompetent cells play an important role inthe usefulness of MMF in the rabbit model of atheroscle-
rosis is of considerable interest. Clearly, the suppression several mechanisms that are implicated in the pathogen-esis of nonimmune renal damage. Gaining insight intoof the infiltration and proliferation of immunocompetent
cells in response to oxidized lipoproteins is of potential these mechanisms has obvious clinical relevance sincehuman conditions commonly associated with chronic re-benefit in renal conditions in which atherogenic lipopro-
teins play a primary role or participate as a secondary nal failure, such as severe essential hypertension [201],chronic pyelonephritis [202], and diabetes [203] haverisk factor.
More recently, we have used MMF treatment in well- similar infiltration of immunocompetent cells. Immuno-suppressive drugs that lack nephrotoxicity and have spe-established models of salt-sensitive hypertension that,
as discussed earlier, are known to be associated with cific antilymphocytic action as well as a reasonably widetherapeutic window may represent an additional optiontubulointerstitial infiltration of immunocompetent cells.
Preliminary data in the salt-sensitive hypertension that in the treatment of progressive renal damage and shouldbe investigated further.follows short-term angiotensin infusion [95] indicate that
while the hypertension induced by angiotensin infusionis unchanged by MMF, the increase in blood pressure ACKNOWLEDGMENTSobserved when a high salt intake is administered after Research in the authors’ laboratories is supported by Asociacion
de Amigos del Rinon, Maracaibo and Consejo Nacional de Investigaci-angiotensin infusion is prevented by MMF. A pathogenicones Cientıficas (CONICIT), Venezuela; Consejo de Investigacionesrelationship between salt-sensitive hypertension and im-Cientıficas, Mexico; and U.S. Public Health Service Grants DK-43422,
munocompetent cells is suggested by the concomitant DK-52121, and DK-47659.reduction of lymphocyte infiltration and activation, oxi-
Reprint requests to Bernardo Rodrıguez-Iturbe, M.D., Apartadodative stress, and tubulointerstitial damage in the kidney.Postal 1430, Maracaibo 4001-A, Venezuela.
Macrophage infiltration was not significantly reduced E-mail: [email protected][218]. Similar studies were done in the model of hyper-tension induced by inhibition of nitric oxide (NO) syn- REFERENCESthesis [98]. In this model, the administration of L-NAME
1. Rocklin RE, Lewis EJ, David JR: In vitro evidence for cellularinduces infiltration with macrophages and lymphocytes hypersensibility to glomerular basement-membrane antigens in
human glomerulonephritis. N Engl J Med 283:497–501, 1970and hypertension that subsides after stopping the drug.2. Dixon F: What are sensitized cells doing in glomerulonephritis?However, three weeks of NO synthesis inhibition causes
N Engl J Med 283:536–537, 1970the subsequent development of hypertension in rats 3. Strutz F, Neilson EG: The role of lymphocytes in the progres-given a high-salt diet for several weeks. Also in this sion of interstitial disease. Kidney Int 45(Suppl 45):S106–S116,
1994model, MMF treatment prevents the infiltration of im-4. Schena FP, Gesualdo L, Grandaliano G, Montinaro V: Pro-mune cells and salt-sensitive hypertension in [219]. Inter- gression of renal damage in human glomerulonephritides: Is there
estingly, in both models of salt-sensitive hypertension, a sleight of hand in winning the game? Kidney Int 52:1439–1457,1997we have found interstitial accumulation of angiotensin
5. Muller GA, Schettler V, Muller CA, Strutz F: PreventionII-producing cells. Double staining studies demonstrated of progression of renal fibrosis: How far are we? Kidney Intthat about one third of infiltrating lymphocytes in the 49:S75–S82, 1996
6. Risdon RA, Sloper JC, de Wardener HE: Relationship betweenrats with salt-sensitive hypertension stain positive forrenal function and histologic changes found in renal biopsy speci-angiotensin II and that MMF treatment was associated mens from patients with persistent glomerulonephritis. Lancet
with reduction of this infiltrate. To our knowledge, there 2:363–366, 19687. Schainuck LI, Stricker GE, Cutler RE, Benditt EP: Structural-are no studies evaluating specifically the renal hemody-
functional correlations in renal disease. II. The correlations. Humnamic alterations associated with immune infiltrates in Pathol 1:631–641, 1970nonimmune renal disease. However, a constant finding 8. Strutz F, Muller GA: On the progression of chronic renal
disease. Nephron 69:371–379, 1995in almost every condition discussed earlier has been in-9. Bohle A, Muller GA, Whermannn M, et al: Pathogenesis ofcreased glomerular capillary pressure and therapeutic
chronic renal failure in the primary glomerulopathies, renal vas-regimes that directly or indirectly suppress angiotensin culopathies and chronic interstitial nephritides. Kidney Int
49(Suppl 54):S2–S9, 1997II activity, usually prevent or retard the progression of10. Nath KA: Tubulointerstitial changes as a major determinant inrenal disease. Such an approach has been used in diabe-
the progression of renal damage. Am J Kidney Dis 20:1–17, 1992tes [133, 193], hypertensive renal disease [194, 195], aging 11. Main IW, Nikolic-Paterson DJ, Atkins RC: T cells and macro-
phages and their role in renal injury. Semin Nephrol 12:395–407,[132], obesity [196], hyperlipidemia [32], puromycin1992aminonucleoside nephrosis [197, 198], and extensive re-
12. Lan HY, Bacher M, Yang N, et al: The pathogenic role ofnal ablation [43, 132, 199, 200]. macrophage migration inhibitory factor in immunologically in-
duced kidney disease in the rat. J Exp Med 185:1455–1465, 1997While our observations do not define the mechanism

Rodrıguez-Iturbe et al: Immunocompetent cells1636
13. Miyazaki K, Isbel NM, Lan HY, et al: Up-regulation of macro- 37. Magil AB, Cohen AH: Monocytes and focal glomerulosclerosis.Lab Invest 61:404–409, 1989phage colony-stimulating factor (M-CSF) and migration inhibi-
38. Schonholzer KW, Waldron M, Magil AB: Intraglomerulartory factor (MIF) expression and monocyte recruitment duringfoam cells and human focal glomerulosclerosis. Nephron 62:130–lipid-induced glomerular injury in the exogenous hypercholes-135, 1992terolaemic (ExHC) rat. Clin Exp Immunol 108:318–323, 1997
39. Keane WF: Lipids and the kidney. Kidney Int 46:910–920, 199414. Neilson EG: Pathogenesis and therapy of interstitial nephritis.40. Eddy AA, Liu A, McCulloch L: Interstitial inflammation andKidney Int 35:1257–1270, 1989
fibrosis in rats with diet induced hypercholesterolemia. Kidney15. Yee J, Kuncio GS, Neilson EG: Tubulointerstitial injury follow-Int 50:1139–1149, 1996ing glomerulonephritis. Semin Nephrol 11:361–366, 1991
41. Heptinstall RH: Pyelonephritis: Pathologic features, in Pathol-16. Goes N, Urmson J, Ramassar V, Halloran PF: Ischemic acuteogy of the Kidney (4th ed), edited by Heptinstall RH, Boston,tubular necrosis induces and extensive local cytokine response.Little, Brown and Co., 1992, pp 1540–1541Transplantation 59:565–572, 1995
42. Ross R: Atherosclerosis: An inflammatory disease. N Engl J Med17. Truong LD, Farhood A, Tasby J, Gillum D: Experimental340:115–126, 1999chronic renal ischemia: Morphologic and immunologic studies.
43. Hostetter TH, Olson JL, Venkatachalam MA, Brenner BM:Kidney Int 41:1676–1689, 1992Hyperfiltration of remnant nephrons: A potentially adverse re-18. Mampaso FM, Wilson CB: Characterization of inflammatory cellssponse to renal ablation. Am J Physiol 241:F85–F93, 1981in autoimmune tubulointerstitial nephritis in rats. Kidney Int
44. Shimamura T, Morrison AB: A progressive glomerulosclerosis23:448–457, 1983occurring in partial five-sixth nephrectomy. Am J Pathol 79:95101,19. Eddy AA: Interstitial nephritis induced by protein overload pro-1975teinuria. Am J Pathol 135:719–733, 1989
45. Zatz R: Haemodynamically mediated glomerular injury: The end20. Eddy AA, Michael AF: Acute tubulointerstitial nephritis associ-of a 15-year-old controversy? Curr Opin Nephrol Hypertensated by aminonucleoside nephrosis. Kidney Int 33:14–23, 19885:468–475, 199621. Gillum DM, Truong L, Tasby J: Characterization of the intersti-
46. Floege J, Burns MW, Alpers CE, et al: Glomerular cell prolifera-tial cellular infiltrate in experimental chronic cyclosporine ne-tion and PDGF expression precede glomerulosclerosis in the rem-phropathy. Transplantation 49:793–797, 1990nant kidney model. Kidney Int 41:297–309, 199222. Lippman RW: Mechanism of proteinuria: Effect of parenteral
47. Wolf G, Mueller E, Stahl RA, Zyyadeh FN: Angiotensin IIbovine albumin injections on hemoglobin excretion in rats. Aminduced hypertrophy of cultured murine proximal tubular cells isJ Physiol 154:532–536, 1948mediated by endogenous transforming growth factor-beta. J Clin23. Terry R, Hawkins DR, Church EH, Whipple GH: ProteinuriaInvest 92:1366–1372, 1993related to hyperproteinemia in dogs following plasma given par-
48. Kagami S, Border WA, Miller DE, Noble NA: Angiotensin IIenterally: A renal threshold for plasma proteins. J Exp Medstimulates extracellular matrix protein synthesis through induc-87:561–573, 1948tion of transforming growth factor-beta expression in rat glomeru-24. Eddy AA, Giachelli CM: Renal expression of genes that pro-lar mesangial cells. J Clin Invest 93:2431–2437, 1994mote interstitial inflammation and fibrosis in rats with protein 49. Wu L, Cox A, Roe CJ, et al: Transforming growth factor b1 andoverload proteinuria. Kidney Int 47:1546–1557, 1995 the renal injury following subtotal nephrectomy in the rat: Role
25. Kees-Folts D, Sadow JL, Schreiner GF: Tubular catabolism of of the renin-angiotensin system. Kidney Int 51:1553–1567, 1997albumin is associated with the release of an inflammatory lipid. 50. Hostetter TH, Meyer TW, Rennke HG, Brenner BM: ChronicKidney Int 45:1697–1709, 1994 effects of dietary protein in the rat with intact and reduced renal
26. Anderson MS, Recant L: Fine structural alterations in the rat mass. Kidney Int 30:509–517, 1986following intraperitoneal bovine albumin. Am J Pathol 40:555– 51. Correa-Rotter R, Hostetter T, Rosenberg M: Effect of dietary560, 1962 protein on renin and angiotensinogen gene expression after renal
27. Andrews PM: A scanning and transmission electron microscopic ablation. Am J Physiol 262:F631–F638, 1992comparison of puromycin aminonucleoside-induced nephrosis to 52. Yang N, Wu LL, Nikolic-Paterson DJ, et al: Local macrophagehyperalbuminemia-induced proteinuria with emphasis on kidney and myofibroblast proliferation in progressive renal injury in thepodocyte pedicel loss. Lab Invest 36:183–197, 1977 rat remnant kidney. Nephrol Dial Transplant 13:1967–1974, 1998
28. Camussi G, Rotunno M, Segoloni G, et al: In vitro alternative 53. Fuhihara CK, Avancini DA, Zatz R, Noronha IL: Mycopheno-pathway activation of complement by the brush border of proxi- late mofetil attenuates renal injury in the rat remnant kidney.mal tubules of normal rat kidney. J Immunol 128:1659–1663, 1982 Kidney Int 54:1510–1519, 1998
29. Wellmann KF, Volk BW: Renal changes in experimental hyper- 54. Romero F, Rodrıguez-Iturbe B, Parra G, et al: Mycophenolatecholesterolemia in normal and subdiabetic rabbits. II. Long-term mofetil prevents the progressive renal failure induced by 5/6 renalstudies. Lab Invest 24:144–155, 1971 ablation in rats. Kidney Int 55:945–955, 1999
30. Moorhead JF, Chan MK, El Nahas M, Varghese Z: Lipid 55. Schiller B, Moran J: Focal glomerulosclerosis in the remnantnephrotoxicity in chronic progressive glomerular and tubulointer- kidney model—An inflammatory disease mediated by cytokines.stitial disease. Lancet 2:1309–1311, 1982 Nephrol Dial Transplant 12:430–437, 1997
31. Grone H-J, Walli A, Grone E, et al: Induction of glomeruloscle- 56. Wu LL, Yang N, Roe CJ, et al: Macrophage and myofibroblastrosis by dietary lipids: A functional and morphologic study in the proliferation in remnant kidney. Role of angiotensin II. Kidneyrat. Lab Invest 60:433–446, 1989 Int 52(Suppl 63):S221–S225, 1997
32. Kasiske BL, O’Donnell MP, Schmitz PG, et al: Renal injury of 57. Remuzzi G, Zoja G, Gagliardini E, et al: Combining an antipro-diet-induced hypercholestemia in rats. Kidney Int 37:880–891, teinuric approach with mycophenolate mofetil suppresses pro-1990 gressive nephropathy of experimental animals. J Am Soc Nephrol
33. Diamond JR, Hanchak NA, McCarter MD, Karnovsky MJ: 10:1542–1549, 1999Cholestyramine resin ameliorates chronic aminonucleoside ne- 58. Nagle RB, Bulger RB, Cutter RB, et al: Unilateral obstructivephrosis. Am J Clin Nutr 51:606–611, 1990 uropathy in the rabbit. I. Early morphologic, physiologic and
34. Diamond JR, Karnovsky MJ: A putative role of hypercholesterol- histochemical changes. Lab Invest 28:456–467, 1971emia in progressive glomerular injury. Annu Rev Med 43:83–92, 59. Nagle RB, Johnson ME, Jervis HR: Proliferation of renal inter-1992 stitial cells following injury induced by ureteral obstruction. Lab
35. Keane WF, Mulcahy WS, Kasiske BL, et al: Hyperlipemia and Invest 35:18–22, 1976progressive renal disease. Kidney Int 39(Suppl 31):S41–S48, 1991 60. Okegawa T, Jonas PE, Deschryver K, et al: Metabolic and cellu-
36. Diamond JR: Effect of lipid abnormalities on the progression of lar alterations underlying the exaggerated renal prostaglandinrenal damage: Analogous pathobiologic mechanisms in glomeru- and thromboxane synthesis in ureter obstruction in rabbits. J Clinlosclerosis and atherosclerosis. Kidney Int 39(Suppl 31):S29–S34, Invest 71:61–90, 1983
61. Lefkowitz JB, Okegawa T, Deschryver-Kecskemeti K, Nee-1991

Rodrıguez-Iturbe et al: Immunocompetent cells 1637
dleman P: Macrophage-dependent arachidonate metabolism in The effect of chronic active pyelonephrhitis on renal function.Kidney Int 9:23–29, 1976hydronephrosis. Kidney Int 26:10–17, 1984
62. Klahr S: Pathophysiology of obstructive nephropathy. Kidney 90. Tamaki K, Okuda S, Nakayama M, et al: Transforming growthfactor-b1 in hypertensive renal injury in Dahl salt-sensitive rats.Int 23:414–426, 1983
63. Schreiner GF, Harris KPG, Pukerson MI, Klahr S: Immuno- J Am Soc Nephrol 7:2578–2589, 199691. Johnson RJ, Gordon KL, Giachelli C, et al: Tubulointerstitiallogical aspects of acute ureteral obstruction: Immune cell infiltrate
in the kidney. Kidney Int 34:487–493, 1988 injury and loss of nitric oxide synthases parallel the developmentof hypertension in the Dahl-S rat. J Hypertens 18:1497–1505, 200064. Halushka PV, Cook JA, Wise WC: Thromboxane A2 and prosta-
cyclin production by lipopolysaccharide-stimulated peritoneal 92. Abumiya T, Masuda J, Kawai J, et al: Heterogeneity in theappearance and distribution of macrophage subsets and theirmacrophages. J Reticulonedothel Soc 30:445–450, 1981
65. Kaneto H, Morrissey J, Klahr S: Increased expression of possible involvement in hypertensive vascular lesions in rats. LabInvest 75:125–136, 1966TGF-b1 mRNA in the obstructed kidney of rats with unilateral
ureteral ligation. Kidney Int 44:313–321, 1993 93. Geiger H, Fierlbeck W, Mai M, et al: Effects of early and lateantihypertensive treatment on extracellular matrix proteins and66. Mandell J, Koch WK, Nidess R, et al: Congenital polycystic
kidney disease. Am J Pathol 113:112–114, 1983 mononuclear cells in uninephrectomized SHR. Kidney Int 51:750–761, 199767. Takahashi H, Calvet JP, Dittemore-Hoover D, et al: A heredi-
tary model of slowly progressive polycystic kidney disease in the 94. Johnson RJ, Gordon KL, Suga S, et al: Renal injury and salt-sensitive hypertension after exposure to catecholamines. Hyper-mouse. J Am Soc Nephrol 1:980–989, 1991
68. Nakamura T, Ebihara I, Nagaoka I, et al: Growth factor gene tension 34:151–159, 199995. Lombardi D, Gordon KL, Polinski P, et al: Salt-sensitive hyper-expression in kidney of murine polycystic kidney disease. J Am
Soc Nephrol 3:1378–1386, 1993 tension develops after short-term exposure to angiotensin II. Hy-pertension 33:1013–1019, 199969. Kagaoka I, Trapnell BC, Crystal RG: Upregulation of platelet-
derived growth factor A and B gene expression in alveolar macro- 96. Mai M, Geiger H, Hilgers KF, et al: Early interstitial changesin hypertension-induced renal injury. Hypertension 22:754–765,phages in individuals with idiopathic pulmonary fibrosis. J Clin
Invest 85:2023–2027, 1990 199397. Eng E, Veniant M, Floege J, et al: Renal proliferation and70. Nakamura T, Ebihara I, Nagaoka I, et al: Activated peripheral
blood mononuclear cells in IgA nephropathy express platelet- phenotypic changes in rats with two-kidneys one-clip Goldblatthypertension. Am J Hypertens 7:177–185, 1994derived growth factor. J Lab Clin Med 120:212–221, 1992
71. Kagaoka I, Trapnell BC, Crystal RG: Regulation of insulin- 98. Oliveira-Ribeiro M, Antunes E, de Nucci G, et al: Chronicinhibition of nitric oxide synthesis: A new model of arterial hyper-like growth factor I gene expression in the human macrophage-
like cell line U937. J Clin Invest 85:448–455, 1990 tension. Hypertension 20:298303, 199299. Jover B, Herizi A, Ventre F, et al: Sodium and angiotensin in72. Eddy AA: Molecular insights into renal interstitial fibrosis. J Am
Soc Nephrol 7:2495–2508, 1996 hypertension induced by long-term nitric oxide blockade. Hyper-tension 21:944–948, 199373. Wolf G, Ziyadeh FN: Molecular mechanisms of diabetic renal
hypertrophy. Kidney Int 56:393–405, 1999 100. Pollock DM, Polakowski JS, Divish BJ, Opgenorth TJ: Angio-tensin blockade reverses hypertension during long-term nitric74. Kimmelstiel P, Wilson C: Intercapillary lesions in glomeruli of
the kidney. Am J Pathol 12:83–97, 1936 oxide synthase inhibition. Hypertension 21:660–666, 1993101. Melaragno MG, Fink GD: Role of angiotensin II in hypertension75. Ziyadeh FN, Goldfarb S: The renal tubulointerstitium in diabe-
tes mellitus. Kidney Int 39:464–475, 1991 produced by chronic inhibition of nitric oxide synthase in con-scious rats. Am J Physiol 271:H806–H811, 199676. Mauer S, Steffes M, Ellis E, et al: Structural-functional relation-
ships in diabetic nephropathy. J Clin Invest 74:1143–1155, 1984 102. Cunha RS, Cabral AM, Vasquez EC: Evidence that the auto-nomic nervous system plays a major role in the L-NAME-induced77. Lane PH, Steffes MW, Fioretto P, Mauer MS: Renal interstitial
expansion in insulin-dependent diabetes mellitus. Kidney Int hypertension in conscious rats. Am J Hypertens 6:806–809, 1993103. Verhagen AMG, Rabelink TJ, Braam B, et al: Endothelin A43:661–667, 1993
78. Hooke DH, Gee DC, Atkins RC: Leukocyte analysis using mono- receptor blockade alleviates hypertension and renal lesions asso-ciated with chronic nitric oxide synthase inhibition. J Am Socclonal antibodies in human glomerulonephritis. Kidney Int 31:
964–973, 1987 Nephrol 9:755–762, 1998104. De Caterina R, Libby P, Peng HB, et al: Nitric oxide decreases79. Cohen MP, Zyyadeh FN: Role of Amadori-modified nonenzy-
matically glycated serum proteins in the pathogenesis of diabetic cytokine induced endothelial activation: Nitric oxide selectivelyreduces endothelial expression of adhesion molecules and proin-nephropathy. J Am Soc Nephrol 7:183–190, 1996
80. Lindeman RD: Overview: Renal physiology and pathophysiology flammatory cytokines. J Clin Invest 96:60–68, 1995105. Spiecker M, Darius H, Kaboth K, et al: Differential regulationof aging. Am J Kidney Dis 4:275–282, 1990
81. Baylis C, Corman B: The aging kidney: Insights from experimen- of endothelial cell adhesion molecule expression by nitric oxidedonors and antioxidants. J Leukoc Biol 63:732–739, 1998tal studies. J Am Soc Nephrol 9:699–709, 1998
82. Epstein M: Aging and the kidney. J Am Soc Nephrol 7:1106–1122, 106. Lefer AM, Lefer DJ: The role of nitric oxide and cell adhesionmolecules on the microcirculation in ischaemia-reperfusion. Car-1996
83. Couser WG, Stilmant MM: Mesangial lesion and focal glomeru- diovasc Res 32:743–751, 1996107. Verstrepen WA, Nouwen EJ, Zhu MQ, et al: Time course oflosclerosis in the aging rat. Lab Invest 33:493–501, 1975
84. Goldstein RS, Tarloff JB, Hook JB: Age-related nephropathy growth factor expression in mercuric chloride acute renal failure.Nephrol Dial Transplant 10:1361–1371, 1995in laboratory rats. FASEB J 2:2241–2251, 1988
85. Abrass C, Adcox MJ, Raugi GJ: Aging-associated change in 108. Nava M, Romero F, Quiroz Y, et al: Melatonin attenuates theacute renal failure and the oxidative stress induced by mercuricextracellular matrix. Am J Pathol 146:742–752, 1995
86. Thomas SA, Anderson S, Gordon KL, et al: Tubulointerstitial chloride in rats. Am J Physiol (Renal ) F910–F918, 2000109. Glassock RJ, Velosa JA, Michael AF: Experimental model ofdisease in aging: Evidence for underlying peritubular capillary
damage, a potential role for renal ischemia. J Am Soc Nephrol focal sclerosis. I. Relationship to protein excretion in aminonucle-oside nephrosis. Lab Invest 36:519–526, 19779:231–242, 1998
87. Ruiz-Torres MP, Bosch RJ, O’Valle F, et al: Age-related in- 110. Harris KPG, Lefkowitz JB, Klahr S, Schreiner GF: Essentialfatty acid deficiency ameliorates acute renal dysfunction in thecrease in expression of TGF-b1 in the rat kidney: Relation to
morphologic changes. J Am Soc Nephrol 9:782–791, 1998 rat after the administration of aminonucleoside of puromycin.J Clin Invest 86:1115–1123, 199088. Hewitson TD, Darby IA, Bisucci T, et al: Evolution of tubuloin-
terstitial fibrosis in experimental renal infection and scarring. 111. Jones CL, Buch S, Post M, et al: Pathogenesis of interstitialfibrosis in chronic purine aminonucleoside nephrosis. Kidney IntJ Am Soc Nephrol 9:632–642, 1998
89. Miller TE, Layzel D, Stewart E: Experimental pyelonephritis: 40:1020–1031, 1991

Rodrıguez-Iturbe et al: Immunocompetent cells1638
112. Magil A: Inhibition of progression of chronic puromycin amino- 136. Grafe M, Auch-Schwelk W, Zakrzewicz A, et al: Angiotensinnucleoside nephrosis by probucol, an antioxidant. J Am Soc II-induced leukocyte adhesion on human coronary endothelialNephrol 7:2340–2347, 1996 cells is mediated by E-selectin. Circ Res 81:804–811, 1997
113. Diamond JR, Pesek I, Ruggieri S, Karnovsky MJ: Essential fatty 137. Krejcy K, Eichler HG, Jilma B, et al: Influence of angiotensinacid deficiency during acute puromycin nephrosis ameliorates late II on circulating adhesion molecules and blood leukocyte countrenal injury. Am J Physiol 257:F798–F807, 1989 in vivo. Can J Physiol Pharmacol 74:9–14, 1996
114. Diamond JR, Pesek-Diamond I: Sublethal X-irradiation during 138. Ruiz-Ortega M, Bustos C, Hernandez-Presa MA, et al: Angio-the acute puromycin nephrosis prevents the late renal injury: Role tensin II participates in mononuclear cell recruitment in experi-of macrophages. Am J Physiol 260:F779–F786, 1991 mental immune complex nephritis through nuclear factor-kappa
115. Diamond JR: The role of reactive oxygen species in animal models B activation and monocyte chemoattractant protein-1 synthesis.of glomerular disease. Am J Kidney Dis 19:292–300, 1992 J Immunol 161:430–439, 1998
116. Shihab FS, Andoh TF, Tanner AM, et al: Role of transforming 139. Johnson RJ, Alpers CE, Yoshimura A, et al: Renal injury fromgrowth factor-b1 in experimental chronic cyclosporine nephropa- angiotensin II-mediated hypertension. Hypertension 19:464–474,thy. Kidney Int 49:1141–1151, 1996 1992
117. Shihab FS, Bennett WM, Tanner AM, Andoh TF: Mechanism 140. Goetzl EJ, Klickstein LB, Watt KWK, Wintroub BU: Theof fibrosis in experimental tracolimus nephrotoxicity. Transplan- preferential human mononuclear leukocyte chemotactic activitytation 64:1829–1837, 1997 of the substituent tetrapeptides of angiotensin II. Biochem Bio-
118. Pichler R, Franceschini N, Young BA, et al: Pathogenesis of phys Res Commun 97:1097–1102, 1980cyclosporine nephropathy: Roles of angiotensin II and osteopon- 141. Hahn AWA, Jonas U, Buhler FR, Resink TJ: Activation oftin. J Am Soc Nephrol 6:1186–1196, 1995 human peripheral monocytes by angiotensin II. FEBS Lett
119. Elzinga LW, Rosen S, Bennett WM: Dissociation of glomerular 347:178–180, 1994filtration rate from tubulointerstitial fibrosis in experimental 142. Furukawa Y, Matsumori A, Hirozane T, Sasayama S: Angio-chronic cyclosporine nephropathy: Role of sodium intake. J Am tensin II receptor antagonist reduces graft coronary artery diseaseSoc Nephrol 4:214–221, 1993 and preserves graft status in a murine model. Circulation 93:333–
120. Bennett WM, Burdmann EA, Andoh TF, et al: Nephrotoxicity 339, 1996of immunosuppressive drugs. Nephrol Dial Transplant 9(Suppl 4): 143. Tanaka A, Matsumori A, Wang W, Sasayama S: An angiotensin141–145, 1994 II receptor antagonist reduces myocardial damage in an animal
121. Segerer S, Nelson PJ, Scholondorf D: Chemokines, chemokine model of myocarditis. Circulation 90:2051–2055, 1994receptors and renal disease: From basic science to pathophysio- 144. Amuchastegui S, Azzolini SC, Mister M, et al: Chronic allograftlogic and therapeutic studies. J Am Soc Nephrol 11:152–176, 2000 nephropathy in the rat is improved by angiotensin II receptor
122. Brady HR: Leukocyte adhesion molecules and kidney diseases. blockade but not by calcium channel antagonism. J Am SocKidney Int 45:1285–1300, 1994 Nephrol 9:1948–1955, 1998
123. Hill PA, Lan HY, Nikolic-Paterson DJ, Atkins RC: ICAM-1 145. Benedikrsson H, Chea R, Davidoff A, Paul L: Antihyperten-directs migration and localization of interstitial leukocytes in ex- sive drug treatment in chronic renal allograft rejection in the rat.perimental glomerulonephritis. Kidney Int 45:32–42, 1994 Effect on structure. Transplantation 62:1634–1642, 1996124. Tang WW, Feng L, Mathison JC, Wilson CB: Cytokine expres-
146. Shimada K, Yazaki Y: Binding site for angiotensin II in humansion, upregulation of intracellular adhesion molecule-1 and leuko-mononuclear leukocytes. J Biochem 84:1013–1015, 1978cyte infiltration in tubulointerstitial nephritis. Lab Invest 70:631–
147. Nataraj C, Oliveiro MJ, Mannon RB, et al: Angiotensin regu-638, 1994lates cellular immune responses through a calcineurin-dependent125. Matsusaka T, Hymes J, Ichikawa I: Angiotensin in progressivepathway. J Clin Invest 104:1693–1701, 1999renal diseases: Theory and practice. J Am Soc Nephrol 7:2025–
148. Keldar S, Kaplan M, Shapira C, et al: Low density lipoprotein2043, 1996isolated from patients with essential hypertension exhibits in-126. Fogo A: The role of plasminogen II and plasma plasminogencreased propensity for oxidation and enhanced uptake by macro-activator inhibitor-1 in progressive glomerulonephritis. Am J Kid-phages: A possible role for angiotensin II. Atherosclerosis 107:71–ney Dis 36:179–188, 200084, 1994127. Fine LG, Orphanides C, Norman JT: Progressive renal disease:
149. Boyce NW, Tipping PG, Holdsworth SR: Glomerular macro-The chronic hypoxia hypothesis. Kidney Int 53(Suppl 65):S74–S78,phages produce reactive oxygen species in experimental glomeru-1998lonephritis. Kidney Int 35:778–782, 1989128. Fine LG, Bandyopadhay D, Norman JT: Is there a mechanism
150. Stratta P, Canavese C, Mazzucco G, et al: Mesangiolysis andfor the progression of different types of renal diseases other thanendothelial lesions due to peroxidative damage in the rabbit.proteinuria? Towards the unifying theme of chronic hypoxia.Nephron 51:250–256, 1989Kidney Int 57(Suppl 75):S22–S26, 2000
151. Yoshioka T, Ichikawa I, Fogo A: Reactive oxygen metabolites129. Laurent B, Ardaillou R: Reactive oxygen species: Productioncause massive, reversible proteinuria and a glomerular sievingand role in the kidney. Am J Physiol 251:F765–F776, 1986defect without apparent ultrastructural abnormality. J Am Soc130. Blantz RC, Konnen KS, Tucker BJ: Angiotensin II effects uponNephrol 2:902–912, 1991the glomerular microcirculation and ultrafiltration coefficient of
152. Johnson RJ, Couser WG, Chi EY, et al: New mechanism ofthe rat. J Clin Invest 57:419–434, 1976glomerular injury: Myeloperoxidase-hydrogen peroxide-halide131. Bohrer MP, Deen WM, Robertson CR, Brenner BM: Mecha-system. J Clin Invest 79:1379–1387, 1987nism of angiotensin-induced proteinuria in the rat. Am J Physiol
153. Johnson RJ, Gugenheim SJ, Klebanoff SJ, et al: Morphological233:F13–F21, 1977correlates of glomerular oxidant injury induced by the myeloper-132. Anderson S, Rennke HG, Brenner BM: Therapeutic advantageoxidase-hydrogen peroxide-halide system of he neutrophil. Labof converting enzyme inhibitors in arresting progressive renalInvest 58:294–301, 1988disease associated with systemic hypertension. J Clin Invest 77:
154. Satriano JA, Shuldiner M, Hora K, et al: Oxygen radicals as1993–2000, 1986second messengers for expression of monocyte chemoattractant133. Zatz R, Dunn BR, Meyer TW, et al: Prevention of diabeticprotein, JE/MCP-1, and the monocyte colony stimulating factor,glomerulopathy by pharmacological amelioration of glomerularCSF-1, in response to tumor necrosis factor-a and immunoglobu-capillary hypertension. J Clin Invest 77:1925–1930, 1986lin G. J Clin Invest 92:1564–1571, 1993134. Largo R, Gomez-Garre D, Soto K, et al: Angiotensin-converting
155. Satriano JA, Schlondorf D: Regulation of RANTES and ICAMenzyme is upregulated in the proximal tubules of rats with intenseexpression in mesangial cells. J Am Soc Nephrol 8:596–603, 1997proteinuria. Hypertension 33:732–739, 1999
156. Schreck R, Baeuerle PA: A role for oxygen radicals as second135. Kim J, Berliner J, Nadler J: Angiotensin II increases monocytemessengers. Trend Cell Biol 1:39–42, 1991binding to endothelial cells. Biochem Byophys Res Commun
226:862–868, 1996 157. Schlondorff D: The role of chemokines in the initiation and

Rodrıguez-Iturbe et al: Immunocompetent cells 1639
progression of renal disease. Kidney Int 47(Suppl 49):S44–S47, tial requirements of naıve and memory T cells for CD28 costimula-tion in autoimmune pathogenesis. Histol Histopathol 14:1269–1995
158. Quinn MT, Parthasarathy S, Fong LG, Steinberg D: Oxida- 1276, 1999181. Sturmhofel K, Brando C, Martinon F, et al: Antigen-indepen-tively modified low-density lipoproteins: A potential role in re-
cruitment and retention of monocyte/macrophages during athero- dent, integrin-mediated T cell activation. J Immunol 154:2104–2111, 1995genesis. Proc Soc Natl Acad Sci USA 84:2995–2998, 1987
159. Witzum JL, Steinberg D: Role of oxidized low-density lipopro- 182. Saito T, Atkins RC: Contribution of mononuclear leukocytes tothe progression of experimental focal glomerular sclerosis. Kidneyteins in atherosclerosis. J Clin Invest 88:1785–1792, 1991
160. Kume N, Cybulsky MI, Gimbrine MAJ: Lisophosphatidylcholine, Int 37:1076–1083, 1990183. Sollinger HW: Mycophenolate mofetil. Kidney Int 48(Suppla component of atherogenic lipoproteins, induces mononuclear
leukocytes in cultured human and rabbit arterial endothelial cells. 52):S14–S17, 1995184. Hauser IA, Johnson DR, Thevenod F, Gopplet-Strube M: Ef-J Clin Invest 90:1138–1144, 1992
161. Khan BV, Parthasarathy SS, Alexander RW, Medford RM: fect of mycophenolic acid on TNF alpha-induced expression ofadhesion molecules in human endothelial cells in vitro. Br J Phar-Modified low-density lipoproteins and its constituents augment
cytokine-activated vascular cell adhesion molecule-1 gene expres- macol 122:1315–1322, 1995185. Remuzzi A, Imberti G, Puntorieri S, et al: Dissociation betweension in human vascular endothelial cells. J Clin Invest 95:1262–
1270, 1995 anti-proteinuric and anti-hypertensive effects of angiotensin-con-verting enzyme inhibition in rats. Am J Physiol 267:F1034–F1044,162. Modi KS, Morrissey J, Shah SV, et al: Effects of probucol on
renal function in rats with bilateral ureteral obstruction. Kidney 1994186. Kliem V, Johnson RJ, Alpers CE, et al: Mechanisms involvedInt 38:843–850, 1990
163. Modi KS, Schreiner GF, Pukerson ML, Klahr S: Effect of in the pathogenesis of tubulointerstitial fibrosis in 5/6-nephrecto-mized rats. Kidney Int 49:666–678, 1996probucol in the renal function and structure in rats with subtotal
kidney ablation. J Lab Clin Med 120:310–317, 1992 187. Bhagat K, Vallance P: Effects of cytokines on nitric oxidepathways in human vasculature. Curr Opin Nephrol Hypertens164. Yoshimura A, Nemoto T, Sugenova Y, et al: Effect of simvastatin
of proliferative nephritis and cell-cycle protein expression. Kidney 8:89–96, 1999188. Romero F, Rodrıguez-Iturbe B, Pons H, et al: MycophenolateInt 56(Suppl 71):S84–S87, 1999
165. Nishimura M, Tanaka T, Yasuda T, et al: Effect of pravastatin mofetil treatment reduces cholesterol-induced atherosclerosis inrabbits. Atherosclerosis 152:127–133, 2000on type IV collagen secretion and mesangial cell proliferation.
Kidney Int 56(Suppl 71):S97–S100, 1999 189. Emeson EE, Shen M-L: Accelerated atherosclerosis in hyperlipid-emic C57BL mice treated with cyclosporin A. Am J Pathol 142:166. Galle J, Heermeier K, Wanner C: Atherogenic lipoproteins,
oxidative stress and cell death. Kidney Int 56(Suppl 71):S62–S65, 1906–1915, 1993190. Roselaar SE, Schoenfeld G, Daugherty A: Enhanced develop-1999
167. Rook G: Immunity to bacteria and fungi, in Immunology (5th ment of atherosclerosis in cholesterol-fed rabbits by suppressionof cell-mediated immunity. J Clin Invest 96:1389–1394, 1995ed), edited by Roitt I, Brostoff J, Male D, London, Mosby,
1998, pp 239–240 191. Matsumoto T, Saito E, Watanabe H, et al: Influence of FK506on the experimental atherosclerosis in cholesterol-fed rabbits.168. Heng MK, Hneg MC: Heat-shock protein 65 and activated
gamma/delta T cells in injured arteries. Lancet 2:921–923, 1994 Atherosclerosis 139:95–106, 1998192. Drew AF, Tipping PG: Cyclosporin treatment reduces early ath-169. Fu X, Cranfill R, Vollmer M, et al: In vivo response of murine
gamma delta T cells to heat shock protein-derived peptide. Proc erosclerosis in the cholesterol-fed rabbits. Atherosclerosis 116:181–189, 1995Natl Acad Sci USA 90:322–324, 1993
170. Haas W, Pereira P, Tonegawa S: Gamma/delta cells. Annu Rev 193. Fujihara CK, Padilha RM, Zatz R: Glomerular abnormalitiesin long-term experimental diabetes: Role of hemodynamic andImmunol 11:637–685, 1993
171. Rajagopalan S, Zordan T, Tsokos GC, Datta SK: Pathogenetic nonhemodynamic factors and effects of antihypertensive therapy.Diabetes 41:286–293, 1992anti-DNA autoantibody-inducing T helper cell lines from patients
with active lupus nephritis: Isolation of CD4-T helper cell lines 194. Tapia E, Gabbai FB, Calleja C, et al: Determinants of renaldamage in rats with systemic hypertension. Kidney Int 38:642–648,that express gamma delta T-cell antigen receptors. Proc Natl Acad
Sci USA 87:7020–7024, 1990 1990195. Simons JL, Provoost AP, Anderson S, et al: Pathogenesis of172. Hohlfeld R, Engel AG: The role of gamma-delta T lymphocytes
in inflammatory muscle disease. Chem Imunol 53:75–85, 1992 glomerular injury in the fawn-hooded rat: Early glomerular hyper-tension predicts glomerular sclerosis. Am J Nephrol 3:1775–1782,173. Falk MC, Ng G, Zhang GY, et al: Infiltration of the kidney by
alpha beta and gamma delta T cells: Effect on progression of IgA 1993196. Schmitz PG, O’Donnell MP, Kasiske BL, et al: Renal injurynephropathy. Kidney Int 47:177–185, 1995
174. Abrignani S: Antigen-independent activation of resting T cells in obese Zucker rats: Glomerular hemodynamic alterations andeffects of enalapril. Am J Physiol 263:F496–F502, 1992in the liver of patients with chronic hepatitis. Dev Biol Stand
92:191–191, 1998 197. Anderson S, Diamond JR, Karnovsky MJ, Brenner BM: Mech-anisms underlying transition from acute glomerular injury to late175. Unutmaz D, Pileri P, Abrignani S: Antigen-independent activa-
tion of naive and memory resting T cells by a cytokine combina- glomerular sclerosis in a rat model of nephrotic syndrome. J ClinInvest 82:1757–1766, 1988tion. J Exp Med 180:1159–1164, 1994
176. Unutmaz D, Baldoni F, Abrignani S: Human naıve T cells 198. Amato D, Tapia E, Bobadilla N, et al: Mechanisms involvedin the progression to glomerular sclerosis induced by systemicactivated by cytokine differentiate into split phenotype with func-
tional features intermediate between naıve and memory T cells. hypertension during mild puromycin aminonucleoside nephrosis.Am J Hypertens 5:629–636, 1992Int Immunol 7:1417–1424, 1995
177. Bacon KB, Premack BA, Gardner P, Schall TJ: Activation of 199. Lafayette RA, Mayer T, Park SK, Meyer TW: Angiotensin IIreceptor blockade limits glomerular injury in rats with reduceddual T cell signaling pathways by the chemokine RANTES. Sci-
ence 269:1727–1730, 1995 renal mass. J Clin Invest 90:766–771, 1992200. Mackenzie HS, Troy JL, Rennke HG, Brenner BM: TCV116178. Fukui T, Katamura K, Abe N, et al: IL-7 induces proliferation,
variable cytokine-producing ability and IL-2 responsiveness in prevents the progressive renal injury in rats with extensive renalmass ablation. J Hypertens 12(Suppl 9):S11–S16, 1994naıve CD41 T cells from human cord blood. Immunol Lett 59:21–
28, 1997 201. Fogo A, Breyere JA, Smith MC, et al: Accuracy of the diagnosisof hypertensive nephrosclerosis in African Americans: A report179. Siefken R, Kurrie R, Schwinzer R: CD28-mediated activation
of resting human T cells without costimulation of the CD3/TCR from the African American Study of Kidney Disease (AASK)trial. Kidney Int 51:244–252, 1997complex. Cell Immunol 176:59–65, 1997
180. Perrin PJ, Lovett-Racke A, Phillips SM, Racke MK: Differen- 202. Weiss S, Parker F: Pyelonephritis: its relation to vascular lesions

Rodrıguez-Iturbe et al: Immunocompetent cells1640
and to arterial hypertension. Medicine (Baltimore) 18:221–316, lial platelet-derived growth factor messenger RNA levels. Am JPhysiol 260:H642–H646, 19911939
212. Gimbrone MA, Resnick N, Nagel T, et al: Hemodynamics, endo-203. Gilbert ARE, Cooper ME: The tubulointerstitium in progressivethelial gene expression and atherogenesis. Ann NY Acad Scidiabetic kidney disease: More than an aftermath of glomerular811:1–11, 1997injury? Kidney Int 56:1627–1637, 1999
213. Wampola PL, Gotlieb AI, Cybulsky MI, Langille BL: Expres-204. Wang YP, Rangan GP, Tay YC, et al: Induction of monocytesion of ICAM-1 and VCAM-1 and monocyte adherence in arterieschemoattractant protein-1 by albumin is mediated by nuclearexposed to altered shear stress. Arterioscler Thromb Vasc Biolfactor kappa B activation. J Am Soc Nephrol 10:1204–1213, 199915:2–10, 1995205. Zoja C, Donadelli R, Colleoni S, et al: Protein overload stimu-
214. Ishida T, Takahashi M, Corson MA, Bark BC: Fluid shearlates RANTES production by proximal tubular cells dependingstress-mediated signal transduction: How do endothelial cellson NF-kappa B activation. Kidney Int 53:1608–1615, 1998transduce mechanical force into biological responses. Ann NY206. Lynn EG, Show YL, Karmin O: Very low-density lipoproteinAcad Sci 811:12–24, 1997stimulates the expression of monocyte chemoatttractant protein-1
215. Dezso B, Jacobsen J, Poulsen K: Evidence for the presence ofin mesangial cells. Kidney Int 57:1483–1472, 2000angiotensins in normal, unstimulated alveolar macrophages and207. Ricardo SD, Levinson ME, Dejoseph MR, et al: Expressionmonocytes. J Hypertens 7:5–11, 1989of adhesion molecules in rat renal cortex during experimental
216. Weinstock J, Blum A: Granuloma macrophages in murine Schis-hydronephrosis. Kidney Int 50:2002–2010, 1996 tosomiasis mansoni generate components of the angiotensin sys-208. Klahr S, Morrisey JJ: The role of growth factors, cytokines tem. Cell Immunol 89:39–45, 1984
and vasoactive compounds in obstructive nephropathy. Semin 217. Klahr S, Morrisey JJ: The role of vasoactive compounds, growthNephrol 18:622–632, 1998 factors and cytokines in the progression of renal disease. Kidney
209. Morrisey JJ, Klahr S: Differential effects of ACE and AT1 Int 57(Suppl 75):S7–S14, 2000receptor inhibition on chemoattractant and adhesion molecule 218. Rodrıguez-Iturbe B, Pons H, Quiroz Y, et al: Mycophenolatesynthesis. Am J Physiol 274:F580–F586, 1998 mofetil prevents salt-sensitive hypertension resulting from angio-
210. Shyy H-J, Hsieh H-J, Usami S, Chien S: Fluid shear stress induces tensin II exposure. Kidney Int (in press)a biphasic response of human monocyte chemotactic protein-1 219. Quiroz Y, Pons H, Gordon KL, et al: Mycophenolate mofetilgene expression in vascular endothelium. Proc Natl Acad Sci USA prevents the salt-sensitive hypertension resulting from short-term91:4678–4682, 1994 nitric oxide synthesis inhibition. A role for immune cells and
microvascular injury. Am J Physiol (in press)211. Hsieh H-J, Li N-Q, Frangos JA: Shear stress increases endothe-