rights / license: research collection in copyright - …33111/... · vonweizenstroh-lignin imlaufe...
TRANSCRIPT

Research Collection
Doctoral Thesis
Chemische Untersuchungen über die Umwandlung vonWeizenstroh-Lignin im Laufe der Verrottung
Author(s): Schobinger, Ulrich
Publication Date: 1958
Permanent Link: https://doi.org/10.3929/ethz-a-000092016
Rights / License: In Copyright - Non-Commercial Use Permitted
This page was generated automatically upon download from the ETH Zurich Research Collection. For moreinformation please consult the Terms of use.
ETH Library

Prom. Nr. 2728
Chemische Untersuchungen über die
Umwandlung von Weizenstroh-Ligninim Laufe der Verrottung
Von der
EIDGENÖSSISCHEN TECHNISCHEN
HOCHSCHULE IN ZÜRICH
zur Erlangung der Würde eines
Doktors der technischen Wissenschaften
genehmigte
PROMOTIONSARBEIT
vorgelegt von
ULRICH SCHOBINGER
Dipl. Ing.-Agr. E.T.H.
von Luzern
Referent: Herr Prof. Dr. H. Deuel
Korreferent: Herr Prof. Dr. E. Crasemann
VERLAG HANS SCHELLENBERG • WINTERTHUR 1958

Leer - Vide - Empty

DEM ANDENKEN MEINES LIEBEN VATERS
MEINER LIEBEN MUTTER GEWIDMET

Diese Arbeit wurde am Instititut für Biochemie des Bodens der Forschungsanstaltfür Landwirtschaft in Braunschweig-Völkenrode durchgeführt. Sie kam durch ein
freundliches Übereinkommen zwischen Herrn Prof. Dr. H. Deuel, E.T.H., Zürich,und Herrn Prof. Dr. W. Flaig, Braunschweig, zustande. Für die wertvollen Rat¬
schläge und die vielen Anregungen sowie die mir zuteil gewordene Förderung bin
ich Herrn Professor Flaig besonders dankbar.
An dieser Stelle sei auch dem Deutschen Akademischen Austauschdienst für ein
Austausch-Stipendium, dem Bundesministerium für Ernährung, Landwirtschaft
und Forsten für eine Beihilfe, der E.T.H., Zürich, für die mir zur Verfügunggestellten Mittel und dem Kuratorium des Laur-Fonds für einen Beitrag an die
Druckkosten der Promotionsarbeit vielmals gedankt.
Ferner möchte ich den Herren Dr. Th. Breyhan vom Chemischen Untersuchungs¬laboratorium für Beratung bei analytischen Problemen und Dr. O. Graff vom
Institut für Humuswirtschaft der Forschungsanstalt für die Ratschläge zur techni¬
schen Durchführung der Verrottungsversuche meinen herzlichen Dank aussprechen.

INHALTSVERZEICHNIS
1. Einleitung und Problemstellung 1
2. Literaturbesprechung 2
21. Heutiger Stand der Ligninforschung 2
211. Allgemeines 2
212. Definition und Isolierung des Lignins 2
213. Physikalische Eigenschaften 3
214. Chemische Eigenschaften 4
215. Biosynthese des Lignins 6
22. Theorie der Humusbildung 8
221. Allgemeines 8
222. Biologische Zersetzung des Lignins 9
3. Experimenteller Teil 10
31. Versuchsanordnung und Beschreibung des Materials. .
10
32. Analysenmethoden 11
321. Bestimmung des pH-Wertes, der Trockensubstanzund der Asche 12
322. Bestimmung der Extraktstoffe 13
323. Bestimmung des Lignins 14
324. Bestimmung der Holozellulose 16
325. Bestimmung der a-Zellulose 17
326. Bestimmung der Hemizellulosen als Furfurol...
18
327. Decarboxylierung der Strohproben mit 12 °/oigerSalzsäure 20
328. Bestimmung des Ammoniak- und Nitratstickstoffes 22
329. Bestimmung der Elementarzusammensetzung ... 23
33. Ergebnisse des Abbauversuches 24
34. Isolierung von Lignin und Huminsäuren nach verschiede¬
nen Methoden 30
341. Isolierung von Nativlignin nach Brauns ....31
342. Isolierung von Lignin nach Björkman 31
3421. Björkman-Ligrim aus frischem Weizenstroh.
32
3422. Björkman-Lignin aus verrottetem Weizenstroh 33
343. Isolierung von Thioglykolsäure-Lignin nach
Holmberg 35
344. Isolierung von Schwefelsäurelignin 36
345. Isolierung von Huminsäuren 37
35. Enzymatische Oxydation von Ligninen 39
V

351. Isolierung des Enzyms 40
352. Aktivitätsbestimmungen der Champignonoxydase 41
353. Enzymatische Oxydation von Björkman-Lignin . .42
354. Enzymatische Oxydation von Ligninen in der
Warburg-Apparatur ....43
36. Charakterisierung der Rotteprodukte im Vergleich zum
Ausgangsmaterial 44
361. Elementarzusammensetzung der isolierten Lignineund Huminsäuren 44
362. Hydrolyse von Stroh, Ligninen und Huminsäuren 46
363. Alkalischmelze von Ligninen und Huminsäuren . .49
364. Decarboxylierung von Ligninen und Huminsäuren
mit 12 °/oiger Salzsäure 52
365. Potentiometrische Titration von Ligninen .... 53
366. Methylierung von Ligninen 56
367. Reaktion mit 2,4-Dinitrophenylhydrazin ....57
368. Spektrophotometrische Messungen von Ligninen undHuminsäuren im ultravioletten Licht 58
369. Infrarotspektrographische Studien an Ligninen und
Huminsäuren 59
3691. Zuordnung der Banden 60
3692. Diskussion der Spektren der frischen, unver-
rotteten Lignine 61
3693. Diskussion der Spektren der aus verrottetem
Stroh isolierten Lignine 64
3694. Diskussion der Huminsäure-Spektren ...66
4. Diskussion der Ergebnisse 70
41. Abbau der verschiedenen Zellwandkomponenten ...70
42. Chemische Änderungen im Ligninmolekül 72
5. Zusammenfassung 75
6. Literaturverzeichnis 77
VI

i. Einleitung und Problemstellung
Lignin ist einer der widerstandsfähigsten organischen Naturstoffe in den
Pflanzen und bildet somit eines der wichtigsten Ausgangsmaterialien für die
Humusbildung. Die Umformung des Lignins zu Huminsäuren ist schon in
einer Reihe von Arbeiten diskutiert und untersucht worden12 3 * 5 6. Eines
der wesentlichen Probleme ist dabei die Entmethylierung der Ligninbau-steine. Durch die Spaltung von Aetherbindungen können o-Diphenole ent¬
stehen7. Das polymère Lignin wird somit wenigstens teilweise zu phenoli¬schen Substanzen abgebaut. Diese können auf rein chemischem Wege oder
auch unter Einwirkung von Enzymen nach Dehydrierungsreaktionen zu
fulvosäure- und huminsäureähnlichen Polymerisaten aufgebaut werden.
Trotzdem herrscht noch keine klare Vorstellung über die Umwandlungvon Lignin zu Fulvo- bzw. Huminsäuren. Vor allem ist noch nicht abge¬klärt, wieweit die mit verdünnten Alkalilösungen oder Natriumsalzen kalk¬
fällender Säuren durchgeführte Extraktion von Huminsäuren wirklich eine
neugebildete Stoffgruppe erfasst, die eine von der restlichen organischenSubstanz völlig verschiedene Zusammensetzung aufweist, oder ob es sich
dabei um ein niedermolekulares Spaltprodukt des gesamten Huminkom-
plexes handelt. Durch Anwendung alkalischer Extraktionsmittel werden
trotz mehrfacher Reinigungsoperationen ausser Huminstoffen noch andere
Pflanzenstoffe, vor allem Lignin und Hemizellulosen, gelöst, was zu einer
mehr oder weniger starken Verunreinigung der Huminsäuren führt. Ob die
in den Huminsäuren festgestellten niederen Methoxylgehalte auf eine Ver¬
unreinigung zurückzuführen oder aber charakteristisch für das Huminsäure-
molekül sind, konnte bisher noch nicht mit Sicherheit entschieden werden.
In der vorliegenden Arbeit ist daher der Frage nachgegangen worden,welche Zusammenhänge zwischen dem Abbau von Lignin und der Bildungvon Huminsäuren bestehen. Von verschiedenen Autoren8 9 10 ist versucht
worden, schonend isoliertes Lignin auf chemischem oder biologischem Wegein Huminsäuren umzuwandeln. In keinem Falle wurden die Ligninpräpa-rate so stark verändert, dass von einer eigentlichen Huminsäurebildung ge¬
sprochen werden kann. Auch schonend isoliertes Lignin scheint schon soweit
verändert zu sein, dass es nicht mehr so reaktionsfähig ist, wie das in den
Pflanzen vorliegende genuine Lignin8. Aus diesem Grunde wurde versucht,die durch biochemische Prozesse verursachten Veränderungen des in der
Pflanze vorliegenden Lignins im Verlaufe der biologischen Zersetzung zu
studieren. Zu diesem Zwecke ist Weizenstrohlignin zu verschiedenen Zeit¬
punkten der Verrottung nach einem schonenden Verfahren isoliert und mit
den neugebildeten Huminsäuren verglichen worden. Dabei war es natürlich
auch notwendig, die Veränderungen zu verfolgen, denen die gesamte
Pflanzenmasse im Laufe des Rotteprozesses unterworfen ist.
1

2. Literaturbesprechung
21. Heutiger Stand der Ligninforschung
Die in diesem Kapitel dargelegten Ausführungen sollen in Kürze einen
Überblick über die wichigsten Erkenntnisse der Ligninforschung der letzten
Jahre geben. Im übrigen sei auf die in jüngster Zeit erschienenen Zusammen¬
fassungen auf diesem Gebiet verwiesen111213.
211. Allgemeines
Das Lignin ist im Pflanzenreich weit verbreitet und stellt vor allem unter
den höheren Pflanzen einen Hauptbestandteil der Holzfaser dar. Seine
Funktion besteht darin, das Gefüge der aus Polysacchariden bestehenden
Zellwand und ihre Zwischenräume auszufüllen und zu versteifen. Seine
Gegenwart verursacht den physiologischen Tod des Gewebes.
Coniferenholz enthält im allgemeinen 26—30 °/o, Laubholz 20—22 °/o
Lignin. Alle übrigen Pflanzen, insbesondere die krautartigen, weisen einen
niedrigeren Ligningehalt auf.
Die Frage, ob Lignin in einer Bindung mit den Kohlenhydraten vorliegt,ist immer noch umstritten. Schubert und Nord1* sind der Ansicht, dass
Lignin nur physikalisch mit der Zellulose verbunden ist. Sie basieren ihre
Behauptungen auf der Tatsache, dass nach einem teilweise enzymatischenAbbau der Kohlenhydrate ein zusätzlicher Teil an «enzymatisch freigeleg¬tem» Lignin mit organischen Lösungsmitteln herausgelöst werden kann,das sich in seinen Eigenschaften nicht von dem direkt aus Holz extrahier¬
baren Nativlignin unterscheidet. Neuere Arbeiten15 16 zeigen jedoch, dass
eine chemische Verbindung von Lignin mit den Kohlenhydraten, vor allem
mit Pentosen, sehr wohl möglich ist. Björkman1'1 gelang es in jüngster Zeit,aus sehr fein gemahlenem Fichtenholz mit Dimethylformamid einen Lignin-Kohlenhydratkomplex zu isolieren, wobei nach der Hydrolyse dieselben
Zucker nachgewiesen werden konnten, die auch in der Hemizellulose vor¬
handen sind.
212. Definition und Isolierung des Lignins
Freudenbergls definiert das Lignin folgendermassen: «Lignin ist eine poly¬mère, in Wasser und den üblichen organischen Lösungsmitteln grösstenteils
2

unlösliche Substanz, die thermoplastisch und im verholzten Gewebe der
Pflanzen abgelagert ist, ein charakteristisches UV-Spektrum besitzt, 59—
67 °/o Kohlenstoff enthält, bei geeigneter Oxydation etwa 25 "/• aromatische
Aldehyde liefert, mehr oder weniger Methoxyl enthält, sowie mit Bisulfit
und Thioglykolsäure unter den bekannten Bedingungen reagiert. In starker
Schwefelsäure ist es ganz oder grösstenteils unlöslich.»
Nach Schrameckla gibt es zur Isolierung von Lignin nach Entfernungder akzessorischen Bestandteile (Fette, Harze, Proteine) grundsätzlich drei
Möglichkeiten:a) Lösen des Kohlenhydratanteils durch Kupferoxydammoniak oder durch
Hydrolyse mit starken Mineralsäuren (72 °/oige Schwefelsäure oder 42
°/oige Salzsäure), wobei das Lignin als Lösungsrückstand zurückbleibt.
b) Lösen oder Dispergieren des Lignins mit geeigneten Peptisationsmittelnwie Natronlauge, Phenolen, Alkoholen, organischen Säuren, Dioxan,
Bisulfit, Chlordioxyd; die hochpolymeren Kohlenhydrate verbleiben
zum grössten Teil als Rückstand.
c) Herstellen von Acetyl-, Benzyl- und anderen Derivaten von Kohlen¬
hydraten und Lignin mit nachfolgender Trennung auf Grund der ver¬
schiedenen Löslichkeiten in organischen Lösungsmitteln.Bis heute gibt es noch kein Verfahren, das gestattet, die Gesamtmenge des
Lignins mit indifferenten Lösungsmitteln ohne Anwendung von Hitze oder
von Katalysatoren zu extrahieren20. Es ist zwar möglich, einen kleinen
Teil durch Extraktion mit organischen Lösungsmitteln (Dioxan, Aceton)ohne Anwendung eines Katalysators zu isolieren. Dieser lösliche Anteil
beträgt 1—2 %> des Gesamt-Lignins und wird als «lösliches Lignin» oder
Brauns-Uign'm bezeichnet21. Alle übrigen Isolierungsmethoden scheinen trotz
gegenteiliger Ansicht22 vor allem die physikalischen und auch die chemischen
Eigenschaften des Lignins zu verändern.
213. Physikalische Eigenschaften
Die exakte Form und Grösse des Ligninmoleküls ist noch nicht festgelegt.Das durchschnittliche Molekulargewicht beträgt 1000—10 000. Nach neu¬
esten röntgenographischen Untersuchungen ist Lignin eine dreidimensional
vernetzte Kettensubstanz, die als amorphes Paket die Zellulosefibrillen um¬
gibt23. Bei weitem der grösste Teil des Lignins ist, so lange er unverändert
bleibt, in Wasser und organischen Lösungsmitteln unlöslich. Lignine, die in
löslicher Form isoliert werden und deshalb bestimmten Abbaureaktionen un¬
terworfen worden sind, haben niedere spezifische Viskositäten. Sie bestehen
wahrscheinlich aus 5—6 Phenylpropaneinheiten24. Gralén25 fand bei Fich-
tenholzlignin (Thioglykolsäurelignin) mit der Svedberg-Ultrazentrifuge ein
Molekulargewicht von 6—7000. Durch Vorbehandlung des Holzes mit
3

Salzsäure in der Hitze stieg das Molekulargewicht auf ca. 34 000 an. Osmo-
metrische Messungen ergaben Werte von 3800—450024. Die nach der kryo-skopischen und ebullioskopischen Methode erhaltenen, sowie die aus der
Bruttoformel errechneten «Molekulargewichte» liegen meist zwischen 8—
900. Man darf deshalb annehmen, dass dieser Wert das Gewicht einer Ein¬
heit darstellt, wovon das Ligninmolekül ein Vielfaches ist.
Lignin zeigt im UV ein typisches Maximum zwischen 270 und 280 mu.,
das in Übereinstimmung mit anderen phenolischen Substanzen bei Messun¬
gen in alkalischem Medium nach dem langwelligen Gebiet verschoben wird.
Diese Tatsache ist von Aulin-Erdtman26 zur quantitativen Bestimmung der
phenolischen Hydroxylgruppen im Lignin ausgenutzt worden. Die Lignineweisen zudem ein differenziertes IR-Spektrum auf, mit dessen Hilfe Nähe¬
res über die peripheren Gruppen im Molekül und die Verknüpfung der
einzelnen Ligninbausteine ausgesagt werden kann.
214. Chemische Eigenschaften
Auf Grund einer grossen Zahl von Arbeiten27 28 29 steht fest, dass Ligninin der Pflanze aromatische Grundstruktur besitzt, wobei Guajacylpropan-körper wesentlich am Aufbau beteiligt sind. Diese Phenylpropan-Monome-ren können nach Adler und Mitarb.30 miteinander verknüpft sein durch
Aetherbindungen (I), sowohl durch Aether- wie auch C-C-Bindungen (II)und teilweise nur durch C-C-Bindungen (III).
i* c
*V*OCH,
Da Lignin nur wenige phenolische Hydroxylgruppen enthält31 32, wirdder Typ III nur selten vorkommen, sodass der grösste Teil der Lignin-Mono-meren durch Aryl-Aetherbindungen verknüpft sein muss (I + II). Die Ver¬
knüpfung der Aryl-Aetherbindungen über das a-C-Atom der Seitenkette
4

ist auch möglich (vergl. Dehydrodiconiferylalkohol), obwohl in vielen Fäl¬
len an diesem C-Atom entweder eine Alkoxyl- oder eine Hydroxylgruppesitzt33.
Eine der wichtigsten Reaktionen zur Aufklärung der Konstitution des
Lignins stellt der Abbau zu aromatischen Aldehyden durch alkalische Oxy¬dation mit Nitrobenzol dar. Die verschiedenen Pflanzenlignine weisen jenach ihrer Herkunft eine charakteristische Zusammensetzung an diesen
Spaltprodukten auf34. So liefert Fichtenlignin vor allem Vanillin (IV), Laub-
holzlignin dagegen ein Gemisch von Vanillin und Syringa-Aldehyd (V).Zu diesen Aldehyden tritt bei gewissen Ligninarten, insbesondere der Gra¬
mineen, der p-Oxybenzaldehyd (VI) hinzu35. Nach Behandlung von Fich¬
tenlignin mit starkem Alkali in der Hitze und anschliessender Methylierungerhielt Richtzenhain36 bei der Oxydation neben Veratrumsäure (VII) einigeProzente Isohemipinsäure (VIII); das lässt auf eine C-C-Bindung zwischen
zwei Phenylpropaneinheiten (Ca—C3) schliessen.
ji vu r=h
Vi R=COOH
Neben diesen aromatischen Spaltprodukten erhält man noch eine Reihe
aliphatischer Verbindungen wie Formaldehyd, Acetaldehyd, Ameisensäure,
Essigsäure, Oxalsäure und andere mehr37 38, die jedoch wenig zur Struktur¬
aufklärung des Ligninmoleküls beitragen.Während bei den bisher genannten Oxydationsprodukten des Lignins
die Seitenkette abgebaut ist, konnten durch geeignete Abbaureaktionen
Ce—C3-Bruchstücke isoliert werden. So hat Schorygina39 durch Behand¬
lung von Fichtenholz in flüssigem Ammoniak mit Natrium bis zu 13 °/o
l-Guajacylpropanol-2 (IX) erhalten. Hibbert und Mitarb.40 behandelten
Lignin oder Holz mit Alkoholen in der Wärme unter Zusatz einer geringenMenge Mineralsäure. Es treten dabei einige Prozente monomerer Bausteine,die sogenannten Hibbert'schen Ketone auf (X bis XIII). Die Druck hydrie-rung des Lignins führt zu verschiedenen Derivaten des Propylcyclohexans41.
Trotz dieser grossen Zahl von Spaltprodukten konnte bis vor kurzer
Zeit kein genaues Schema über die Art der Verknüpfung dieser Bausteine
entworfen werden. Im folgenden Abschnitt sind einige weitere Verknüp¬fungsmöglichkeiten dieser monomeren Bausteine zu höhermolekularen Ge¬
bilden aufgezeigt.
5
5

T*3 T* r r
pHCOH
C=0
1C*0
lHÇOC^
1c=o
1
O0CH3OH
J
HCOC2H5
R
R=Ouajaeyl -
fR
od Syringylrest
C-0
1R
c«o
1R
Ü I S a üi
215. Biosynthese des Lignins
Erdtman42 vermutet schon im Jahre 1933, dass Lignine wie auch Lignaneim Prinzip durch Dehydrogenierung von einfachen Phenylpropanbausteinengebildet werden. Diesen Vorstufen wird in der Biosynthese einer grossen
Gruppe von aromatischen Verbindungen im Pflanzenreich eine zentrale
Stellung zugeschrieben43.Prinzipiell werden Derivate vom p-Oxyphenylpropan als Ligninvor-
stufen benötigt. Diese können durch Peroxydasen oder andere phenoloxy¬dierende Enzyme zu Polymeren mit den chemischen und physikalischenEigenschaften der Lignine kondensiert werden24.
Am besten ist bisher die enzymathische Dehydrierung des Coniferylalko-hols (XV) untersucht worden, der bei den Coniferen das Hauptausgangs¬material der Ligninbildung darstellt44. Die dabei erhaltenen Dehydrierungs¬polymerisate (DHP) zeigen im C-, H- und OCH3-Gehalt keine wesent¬
lichen Unterschiede gegenüber dem aus Fichtenholz isolierten Acetonligninnach Brauns. Desgleichen stimmen auch die UV- und IR-Spektren des DHP
und der entsprechenden Ligninpräparate gut überein45. Kratzl und
Schweers*6 konnten in diesem DHP neben Vanillin durch Aethanolyse die
gleichen Hibbert'schen Ketone nachweisen, die auch aus Fichtenlignin erhal¬
ten werden.
Auf Grund der bisherigen Arbeiten von Freudenberg und Mitarb.44 4T 48
kann man den Vorgang der Verholzung bei den Coniferen wie folgt deuten:
Aus seinen Bildungsstellen wird Coniferin (XIV) an die Innenseite des Cam¬
biums geleitet, wo aus ihm durch ß-Glukosidase Coniferylalkohol (XV) frei
gemacht wird. Die reichlich vorhandenen Phenoldehydrasen und Pero¬
xydasen verwandeln diesen unter Entzug von Wasserstoff zunächst in min¬
destens drei dimere Dehydrierungsprodukte, Dehydro-diconiferylalkohol(XVI), DL-Pinoresinol (XVII) sowie Guajacylglycerin-coniferyläther(XVIII), und in Coniferylaldehyd. Diese Zwischenprodukte werden im
6

Gegensatz zum primären Baustein Coniferylalkohol als sekundäre Bausteine
bezeichnet. Sie werden ihrerseits im Gemisch miteinander durch weitere
Dehydrierung kondensiert und bilden so das Lignin der Coniferen. Bei den
Laubhölzern tritt als weitere Komponente der Sinapinalkohol und bei den
Gramineen der p-Cumaralkohol hinzu.
Coniferin
Coniferylalkohol
Die Bildung der genannten Substanzen ist nach Freudenberg*9 so zu deu¬
ten, dass die Phenolredoxasen dem Conyferylalkohol zunächst das Phenol¬
wasserstoffatom entziehen, wobei das entstandene Radikal 4 mesomere
Formen (Resonanzformen) bilden kann (XlXa bis d).
XIX a XIX b XIX c XIX d
7

Aus diesen zahlreichen Formen erklärt sich auch die Mannigfaltigkeitder Kondensationsprodukte. Die Wirkung der Fermente besteht demnach im
Entzug des Phenolwasserstoffes, während deren Mitwirkung für die weite¬
ren Reaktionen nicht mehr notwendig ist.
22. Theorie der Humusbildung
221. Allgemeines
Die organische Substanz des Bodens stellt ein Produkt mikrobiologischerund chemischer Umformung von Ausgangsmaterial dar, das fast ausschliess¬
lich pflanzlichen Ursprungs ist. Zahlreiche Zersetzungsversuche mit den
verschiedensten Pflanzenmaterialien ergeben übereinstimmend, dass die
Kohlenhydrate relativ schnell abgebaut werden, während sich Lignin in
Verbindung mit gewissen stickstoffhaltigen Komplexen im Rückstand anrei¬
chert50 51 52 B3 54. Obwohl das im Pflanzenverband vorliegende Lignin zum
Teil beträchtlich abgebaut wird55, muss ihm bei der Humusbildung doch
eine zentrale Stellung zugeschrieben werden. Trotzdem gibt es noch eine
Reihe anderer Naturstoffe, die bei der Bildung von Huminstoffen eine
Rolle spielen. Die verschiedenen Entstehungsmöglichkeiten von Huminsäu¬
ren aus Lignin und Gerbstoffen, Kohlenhydraten, Eiweiss und gewissenStoffwechselprodukten von Bodenorganismen sind von Weite58 eingehenddiskutiert worden.
Die durch ihre Dunkelfärbung gekennzeichnete Gruppe der Huminstoffe
lässt sich auf Grund der verschiedenen Löslichkeitseigenschaften weiter un¬
terteilen57. Wegen der Unlöslichkeit in starken Alkalien lassen sich zunächst
die Humine und Humuskohlen als spezielle Untergruppe leicht abtrennen.
Die wichtigste Gruppe der Huminstoffe stellen die Huminsäuren dar. Sie
lassen sich durch verdünnte Alkalien und Alkalisalzlösungen kalkfällender
Säuren (Natriumfluorid bzw. Natriumoxalat) aus dem Boden extrahieren
und durch Mineralsäuren fällen. Diese amorphen, höhermolekularen, dun¬
kelbraunen Substanzen stellen ein verhältnismässig stabiles Produkt biolo¬
gischer und chemischer Umsetzungen der organischen Substanz im Boden
dar. Zahlreiche Arbeiten berichten von einer Förderung des Pflanzenwachs¬
tums durch Huminsäuren58 59 60. Zur Aufklärung der Konstitution und der
Bildungsweise dieser Stoffe sind auch synthetische Huminsäuren auf Chinon-
oder Phenolbasis studiert worden81 62 83.
Die auf Grund verschiedener Abbaureaktionen erhaltenen Spaltproduktenatürlicher Huminsäuren lassen auf einen gewissen Zusammenhang im
Molekülaufbau der Huminsäuren und des Lignins schliessen. So sind aus
der Alkalischmelze von Huminsäure Protokatechusäure und Brenzcatechin
isoliert worden84, die Oxydation mit Nitrobenzol in Alkali ergibt geringe
8

Mengen Vanillin65 und die Hydrierung mit Molybdänsulfid als Katalysator
Propylcyclohexanolderivate66.
222. Biologische Zersetzung des Lignins
Das in der Pflanze vorliegende Lignin kann durch eine Reihe von Mikro¬
organismen zum Teil sehr weitgehend abgebaut werden55 67 68, während es
dem Angriff dieser Organismen in isoliertem und gereinigtem Zustand
vermehrten Widerstand entgegensetzt. Erst in jüngster Zeit ist es durch
Adaptationsversuche gelungen, mit holzzerstörenden Pilzen auf isoliertem
Nativlignin als alleinige C-Quelle gutes Wachstum zu erreichen69. Wie beim
Abbau der Kohlenhydrate, Eiweisse, Fette etc. so spielen auch beim Abbau
des Lignins Enzyme eine wichtige Rolle70. Nach Untersuchungen von
Lindeberg11 scheiden sämtliche Lignizersetzer Phenoloxydasen in das
Nähr-Medium aus. Lindeberg und Fahraeus72 haben Versuche mit den
Weissfäulcpilzen Polyporus versicolor und Polyporus zonatus angestellt.Die dabei ausgeschiedenen Phenolasen sind vom Typ der Laccase.
Die Gruppe der Phenoloxydasen umfasst wenigstens zwei verschiedene
Enzym-Typen, nämlich Tyrosinase und Laccase73. Di- und Polyphenolemit zwei Hydroxylgruppen in Ortho-Stellung werden von beiden Enzymenoxydiert. Tyrosinase katalysiert dagegen auch die Oxydation von Mono-
phenolen. Laccase ist solchen Stoffen gegenüber inaktiv, oxydiert aber
Hydrochinon und p-Phenylendiamin.Mehrere Autoren berichten von Warburg-Versuchen, bei denen Brauns-
Lignin als Substrat benutzt wurde. So oxydiert Dion''* verschiedene Lig-ninpräparate mittels der Kulturflüssigkeit von Polyporus versicolor. Gott¬
lieb und Geller76 zeigen, dass rohes Enzympulver aus dem Pressaft von
Agaricus campestris die Oxydation von Nativlignin katalysiert. Ähnliche
Versuche sind auch von Vliet durchgeführt worden.
Die durch die Wirkung der Enzyme und Mikroorganismen verursachten
chemischen Veränderungen im Ligninmolekül beruhen in erster Linie auf
der Abspaltung von Methoxylgruppen" 7T 78 79. Ferner ist eine leichte Ab¬
nahme der C- und H-Werte beobachtet worden78. Bartlett und Norman80
berichten von einer starken Zunahme der Austauschkapazität und des N-
Gehaltes im Lignin. Diese Zunahme der Austauschkapazität führen sie zum
grössten Teil auf die Bildung von Carboxylgruppen durch Oxydation der
Seitenketten zurück, da die Entmethylierung von Hydroxylgruppen unge¬
nügend war, um diesen Anstieg zu erklären. Die Löslichkeit der Rückstände
in Alkohol-Benzol, Heisswasser und Alkali nehmen mit steigendem Ver-
rottungsgrad zu54.
Bei der Oxydation von verrottetem Holz mit Nitrobenzol in Alkali wer¬
den im allgemeinen die gleichen Oxydationsprodukte erhalten wie aus fri-
9

schem Holz. Mit zunehmender Zersetzung nehmen jedoch die auf den Lig-
ningehalt berechneten Ausbeuten beträchtlich ab81 82. Diese Befunde dürften
somit auch die bei der alkalischen Nitrobenzoloxydation von Humin-
säuren65 oder der organischen Substanz des Bodens83 erhaltene geringe Aus¬
beute an aromatischen Aldehyden erklären.
3. Experimenteller Teil
31. Versuchsanordnung und Beschreibung des Materials
Weizenstroh wurde in Mitscherlichgefässen unter aeroben Bedingungen bei
28° C und 95—98 %> relativer Luftfeuchtigkeit der Verrottung unterworfen.
Es handelte sich dabei um ein Sommerweizenstroh der Sorte «Peko» aus dem
Versuchsgut Salzdahlum der Forschungsanstalt für Landwirtschaft, Braun¬
schweig-Völkenrode. Das Stroh stammte aus einer Parzelle mit schwerem
Boden mit einem pH von 6,8 bis 7,3.Durch Häckseln wurde das Stroh auf eine Halmlänge von ca. 3 bis 5 cm
gebracht. Je Mitscherlichgefäss wurden 300 g lufttrockenes, gehäckseltesStroh eingewogen und mit 900 ml Wasser, dem folgende Nährsalze beige¬geben wurden, gut durchfeuchtet:
Je Gefäss: KH2PO4 2,05 g
NaNOs 8,50 g
MgSOé : 7 H2O 0,50 gKCl 0,30 g
% dieser Nährsalzmenge wurde gleich zu Anfang verabreicht, der Rest nach
einem Monat. Das lufttrockene Stroh wies einen Wassergehalt von 5,8 %
auf; somit ergab sich für die gesamte Trockenmasse je Gefäss folgende Zu¬
sammensetzung:
Stroh 282,48 gNährsalzc und Bodensuspension 11,92 g
Gesamttrockengewicht je Gefäss: 294,40 g
Um die Verdunstung in den Gefässen auf ein Minimum herabzusetzen,wurden sie mit einer Kunststoffhaube überzogen, wodurch ein schwacher
Luftaustausch gewährleistet wird. Zum Auffangen des Sickerwassers wur¬
den die Mitscherlichgefässe auf Weckgläser von 1 Liter Fassungsvermögen
10

gestellt, wobei eine unten am Gefäss angebrachte Schwammgummidichtungdie Verdunstung verhinderte. Das Sickerwasser wurde in bestimmten Zeit¬
abständen immer wieder in die Gefässe zurückgegossen, wobei es mit zuneh¬
mender Zersetzung vom Stroh meist völlig absorbiert wurde. Von Zeit zu
Zeit war es notwendig, etwas destilliertes Wasser nachzugiessen, um geringeVerdunstungsverluste auszugleichen. Folgende Variation in der Versuchs¬
anordnung wurde vorgenommen:
25 Gefässe wurden mit Nährlösung von pH 4,5 angesetzt und
25 mit Nährlösung von pH 7,5, eingestellt durch zufügen von NaOH.
Nachdem das Stroh mit der Nährlösung gut durchfeuchtet war, wurde mit
einer Bodensuspension geimpft. Zu deren Herstellung wurde eine bestimmte
Menge Komposterde mit Wasser aufgeschlämmt und mit einem Homogeni¬sator fein verteilt. Nach 3 Minuten Sedimentationszeit wurde je Gefäss 10ml
der Suspension beigegeben. Das Material wurde nach 70,180, 260, 340 und
410 Tagen Rottezeit analysiert auf:
pHTrockensubstanz
Organische Substanz (Glühverlust)LigninHolozellulose und a-ZelluloseHemizellulosen (Furfurolbildende Substanzen)Decarboxylierbare Substanzen (Erhitzen mit 12%iger HCl)Nitrat- und Ammoniakstickstoff
C, H, N, OCH3
Nach 180 Tagen trat sowohl bei der Trockensubstanz wie auch bei den
übrigen Komponenten keine wesentliche Änderung mehr ein. Da diese
Erscheinung eventuell eine Folge des Fehlens von Nährsalzen, vor allem
von Stickstoff, sein konnte, wurden drei Gefässe der letzten Serie zusätz¬
lich mit je einem Drittel der Normalmenge an Nährsalzen (pH 7,5) versetzt
und neu mit Bodensuspension beimpft, da die mikrobielle Tätigkeit auf ein
Minimum gesunken zu sein schien. Ferner wurden die bei pH 4,5 angesetz¬ten Proben nach 70 Tagen Rottedauer nicht mehr weiter untersucht, da der
pH-Wert im Verlaufe dieser Zeitpanne auf ca. pH 8 angestiegen war und
somit kein wesentlicher Unterschied mehr zwischen den beiden Varianten
bestand.
32. Analysenmethoden
Von der ersten Rottestufe wurde der Inhalt von je 5 Gefässen einzeln un¬
tersucht, während von den nachfolgenden Rottestufen je 1 Gefäss einzeln
und 4 im Gemisch auf die Zusammensetzung analysiert wurden.
11
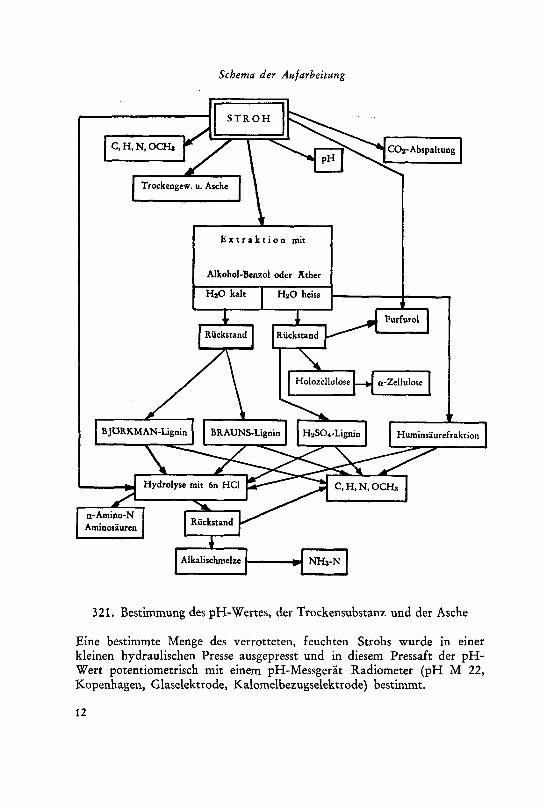
Schema der Aufarbeitung
QH.N.OCHstA
STROH
Trockengew. u. Asche
C02-Abspaltung
Extraktion mit
Alkohol-Benzol oder Äther
H20 kalt H2O heiss
Rückstand
a-Amino-N
Aminosäuren
BJÖRKMAN-Lignin Huminsäurefraktion
Rückstand
*
NH3-N
321. Bestimmung des pH-Wertes, der Trockensubstanz und der Asche
Eine bestimmte Menge des verrotteten, feuchten Strohs wurde in einer
kleinen hydraulischen Presse ausgepresst und in diesem Pressaft der pH-Wert potentiometrisch mit einem pH-Messgerät Radiometer (pH M 22,
Kopenhagen, Glaselektrode, Kalomelbezugselektrode) bestimmt.
12

Der Inhalt der Mitscherlichgefässe wurde auf Packpapier ausgebreitetund an der Luft getrocknet. Das lufttrockene Material wurde nach vorhe¬
rigem Abwiegen in einer Condux-Mühle fein zermahlen. Durch Trocknen
einer eingewogenen Probe bei 105° C bis zur Gewichtskonstanz wurde der
Feuchtigkeitsgehalt der Probe bestimmt. Das lufttrockene Strohmehl wurde
hierauf in einen Kolben von 2 Liter Inhalt eingefüllt, der an die Wasser¬
strahlpumpe unter Zwischenschaltung einer Kühlfalle mit Aceton-Kohlen-
säure angeschlossen wurde. Der Kolben stand in einem Gefäss mit Wasser,
das durch einen Heizthermostaten auf 45° C gehalten wurde. Nach 6—7
Stunden betrug der Wassergehalt meist nur noch 2—3 °/o, worauf das Stroh¬
mehl in Flaschen abgefüllt und bis zur Analyse auf Lignin, Holozellulose
und furfurolbildende Substanzen gut verschlossen aufbewahrt wurde.
Die Bestimmung der Asche erfolgte durch Glühen im Muffelofen bei
600° C während 5 Stunden.
322. Bestimmung der Extraktstoffe
Da für die Bestimmung von Lignin, Holozellulose und Furfurol allgemeindie Anwesenheit von Fetten, Harzen, Wachsen und heisswasserlöslichen
Substanzen wie Zucker, Stärke, Aminosäuren, Pektin und Huminsäuren
störend wirkt, wurden je Probe ca. 10 g Strohmehl zunächst während 35
Stunden im Soxhlet mit Äther extrahiert. Parallel dazu wurde auch eine
Extraktion mit Alkohol-Benzol 1:1 vorgenommen. Die Extraktlösungenwurden hierauf eingedampft und der Rückstand nach Trocknung bei 105° C
gravimetrisch bestimmt. Zur Bestimmung der Heisswasserextrakte, die mit
zunehmender Verrottung dunkel bis schwarz gefärbt waren, wurde das mit
Äther vorextrahierte Strohmehl (ca. 10 g) mit 500 ml dest. Wasser versetzt
und in einem Becherglas 3 Stunden auf siedendem Wasserbad unter gele¬gentlichen Umrühren stehen gelassen. Die Temperatur des Wassers zur Ex¬
traktion betrug ca. 60—70° C. Hernach wurde 15 Minuten bei 3000 T/Min.
zentrifugiert und nochmals mit dest. Wasser gewaschen. Eine aliquoteMenge des Extraktes wurde auf dem Wasserbad in einem kleinen Kölbchen
eingedampft, bei 105° C getrocknet und gewogen.
Vergleichsweise wurde auch der Kaltwasserextrakt durch Schütteln von
1 g Strohmehl in 1 1 dest. Wasser auf der Schwingmühle während 2 Stunden
bestimmt. Hernach wurde durch ein Papierfilter filtriert und analog wie
beim Heisswasserextrakt die in Lösung gegangene Menge als Trockensub¬
stanz besimmt.
13

323. Bestimmung des Lignins
Bei der Bestimmung des Lignins wurde diejenige Methode bevorzugt, die
eine möglichst quantitative Erfassung des Lignins gewährleistet. Adams
und Castagne** haben den Ligningehalt mehrerer Stroharten mit Schwefel¬
säure verschiedener Konzentration bestimmt und gezeigt, dass die Lignin-
bestimmung mit 72 %>iger Schwefelsäure die Forderungen nach einem «wirk¬
lichen» totalen Ligningehalt besser befriedigt als die Bestimmung mit 42
°/oiger HCl. Sie haben mit 71 °/«iger Schwefelsäure ein Minimum an Ligninmit einem Maximum an Methoxyl erhalten, was nach der Ansicht von
Freudenberg und Ploetzm das Kriterium einer brauchbaren Ligninbestim-
mung darstellt. Die Schwefelsäuremethode ergab allgemein etwas höhere
Ligninwerte. Die genannten Autoren zeigen jedoch, dass diese Erhöhungnicht dem Auftreten von Inkohlungsprodukten zugeschrieben werden kann.
Die Ligninbestimmung mit 72 Voiger Schwefelsäure erfolgte nach einer
Vorschrift, die an der Nederlands Proefstation voor Stroverwerking te
Groningen86 mit Erfolg auf frische und verrottete Strohproben angewendetwird. Diese Vorschrift wurde in Anlehnung an die Methode von Ritter,
Seborg und Mitchell87 ausgearbeitet. An Stelle der vorgeschriebenen Alko¬
hol-Benzolextraktion wurde nur mit Äther extrahiert, neben einer nach¬
folgenden Extraktion mit warmem Wasser.
Ausführung:
In einem Becherglas von 100 ml wird 0,5 g fein gemahlenes, bei 105° C getrocknetesStrohmehl, das zuvor einer Äther- und Wasserextraktion unterzogen wurde, mit
25 ml 72°/oiger Schwefelsäure von 20° C (Gewichtsprozent = 1180 g Schwefel¬
säure/Liter) übergössen und nachher gut miteinander vermischt. Diese Mischunglässt man 2 Stunden im Thermostaten bei 20° C stehen, wobei von Zeit zu Zeit
gut gerührt wird, damit das Material von der Schwefelsäure völlig durchfeuchtetwird. Hernach wird der Inhalt des Becherglases mit 575 ml dest. Wasser in einen
Rundkolben von 1 Liter überspült. Die Schwefelsäurekonzentration ist hiermit
auf 5 °/o gebracht. Nun wird der Inhalt des Kolbens am Rückfluss auf einem Sand¬
bad 4 Stunden zu leichtem Sieden erhitzt, anschliessend über einen Filtertiegelnach Gooch mit einer dicken Asbestdecke abgesaugt, wobei darauf zu achten ist,dass immer eine Schicht Flüssigkeit im Tiegel bleibt und dass der Inhalt des Kolbens
sich nicht abkühlt. Deshalb stellt man den Kolben jedesmal, nachdem der Tiegelnachgefüllt ist, in ein siedendes Wasserbad. Man wäscht mit Wasser bis zu neu¬
traler Reaktion auf Dimethylgelb aus, saugt ab, trocknet den Tiegel bei 105° C
während 14 Stunden und wiegt. Nachher wird der Inhalt des Tiegels im Muffel¬
ofen bei 680° C verascht und nach dem Abkühlen erneut gewogen. Der Gewichts¬unterschied stellt das aschefreie Lignin dar.
Die Methode wurde an frischem Weizenstroh geprüft.
Die Analysenergebnisse sind in Tabelle 1 zusammengestellt.
14

Tabelle 1: Ligninbestimmung an frischem Weizenstroh (in Prozent des extrakt¬freien Materials).
Lignin und Asche
in %>
aschefreies Ligninin %
23,12
22,25
22,03
22,50
20,19
20,28
19,93
20,32
im Durchschnitt: 20,18
Die Ergebnisse von Adams und Castagne8* liegen um 1 %> tiefer, was
nach Brauns11* der Entfernung eines Teils des Lignins durch die Alkohol-
Benzol-Vorbehandlung zugeschrieben werden muss. Eigene Untersuchungenan mit Alkohol-Benzol 1:1 extrahiertem Strohmehl lieferten ebenfalls nur
19,13 °/o aschefreies Lignin. Als Grundlage für die Berechnungen des Lig-ningehaltes wurden jedoch die Werte des mit Äther extrahierten Materials
benutzt.
Da mit zunehmender Zersetzung die Lignine auch einen steigenden Gehalt
an Stickstoff aufwiesen, wurde untersucht, wie sich eine 2-stündige Vor¬
hydrolyse mit 5 Voiger Salzsäure vor der Ligninbestimmung mit 72 VoigerSchwefelsäure auf die Elementarzusammensetzung auswirkt, im Vergleichzu einem Schwefelsäurelignin, das ohne diese Vorbehandlung hergestelltwurde. Für diesen Vergleich wurde eine Strohprobe untersucht, die 340
Tage dem Verrottungsprozess ausgesetzt gewesen war (Tabelle 2).
Tabelle 2: Elementarzusammensetzung eines Schwefelsäurelignins (Prozente bezo¬
gen auf aschefreie Substanz).
Material %> C °/o H °/o O °/o N %> S °/o OCH3
H2S04-Ligninohne Voihydrolysemit 5 "/oiger HCl
54,61 5,96 32,45 3,08 3,9 S,61
H2SU4-Ligninmit 5 Voiger HCl
vorhydrolysiert55,53 5,73 32,32 2,42 4,0 9,04
Der N-Gehalt stickstoffreicher Lignine wird oft als Protein (N x 6,25)in Abzug gebracht88. Nach Norman und Jenkins89 ist eine solche Korrektur
falsch, denn in einem Säurelignin könne der Stickstoff nicht mehr in Form
15

von Proteinen, sondern nur als Proteinspaltstücke vorliegen, so dass der
Fehler durch eine solche Korrektur oft grösser wird als die Störung durch
N-haltiges Material.
Wie man aus Tabelle 2 ersieht, konnte der N-Gehalt durch die Vorbe¬
handlung mit 5 °/oiger Salzsäure nur zu einem geringen Teil erniedrigt wer¬
den. Die leichte Abnahme der Wasserstoffwerte deutet darauf hin, dass der
Stickstoff zum Teil als Ammoniak abgespalten wurde. Diese Vorhydrolysewurde bei der Bestimmung der übrigen Proben nicht mehr durchgeführt.
324. Bestimmung der Holozellulose
Die Holozellulosefraktion von extraktfreien Hölzern oder anderen Pflanzen-
materialien stellt nahezu quantitativ den gesamten Gehalt an wasserunlös¬
lichen Polysacchariden der Zellwand dar. Die Bestimmung beruht darauf,dass das Lignin durch Natriumchlorit (NaClOa) unter möglichster Erhal¬
tung der übrigen Zellwandkomponenten in Lösung gebracht wird. Diese
Holozellulose kann als Ausgangsmaterial zur Bestimmung der Cross- und
5et>!*«-Zellulose, a-Zellulose und der Hemizellulosen benutzt werden. Im
folgenden wurde unter Einführung geringfügiger Variationen die Vorschrift
von Adams und Castagne^0 benutzt, die die Anwendbarkeit dieser Methode
zur Bestimmung der Holozellulose in Stroh untersucht haben.
Ausführung:
2 g extrahiertes Strohmehl werden in eine weithalsige Flasche von 0,51 Inhalt
eingefüllt und mit 100 ml dest. Wasser versetzt. Die so gefüllte Flasche wird in ein
heisses Wasserbad gestellt, das durch einen Thermostaten genau auf 75° C (± 0,5° C)gehalten wird, da zur Erzielung reproduzierbarer Resultate eine konstante Tem¬
peratur unbedingt erforderlich ist. Wenn die Mischung die notwendige Temperaturerreicht hat, werden 0,12 ml Eisessig und 3 ml einer 50 Voigen Natriumchlorit-
Lösung beigegeben. Die Flasche wird mit einem Uhrglas zugedeckt und von Zeit
zu Zeit geschüttelt. Nach einer Stunde werden nochmals die gleichen Reagenzienbeigegeben. Drei solche Behandlungen zu je einer Stunde genügen, um eine Aus¬
beute an Holozellulose zu erhalten, die dem theoretischen Wert sehr nahe kommt.Es wird nun durch einen Glasfiltertiegel 1 G 1 filtriert, mehrmals mit Eiswasser
und schliesslich mit Aceton gewaschen und anschliessend im Trockenschrank bei105° C bis zur Gewichtskonstanz getrocknet. Von der so erhaltenen rohen Holo¬zellulose wird darauf eine Aschebestimmung, eine Ligninbestimmung und eine
N-Bestimmung zur Korrektur auf vorhandene Proteine durchgeführt. Es ist anzu¬
nehmen, dass noch andere N-haltige Verbindungen ausser Proteinen anwesend
waren, die in diesem Zusammenhang nicht speziell berücksichtigt wurden.
Die zur Prüfung der Methode an frischem Weizenstroh durchgeführtenAnalysen sind in Tabelle 3 zusammengestellt. Etwa 2—3 °/o Lignin werden
von der Holozellulose festgehalten und können auch durch mehrere Natri-
umchloritbehandlungen nicht völlig entfernt werden. Bei mehr als 3 Chlorit-
behandlungen tritt auch ein Verlust an Polysacchariden ein.
16

Bei einem durchschnittlichen N-Gehalt der rohen Holozellulose aus fri¬
schem Stroh von 0,2 °/o ergibt sich unter Anwendung des Faktors 6,25 x °/o N
ein Abzug von 1,25 %> der Holozellulose für Proteine. Die dadurch erhal¬
tene Ausbeute von 75,9 °/o reiner Holozellulose deckt sich ziemlich genau
mit den Ergebnissen von Adams und Castagne90, die aus Weizenstroh
75,5 °/o reine Holozellulose erhielten.
Tabelle 3: Holozellulose-Bestimmung an frischem Weizenstroh
Rohe Holozell.
in °/o des extrakt¬freien Strohs
Aschegehaltder Holozell.
in °/o
Ligningehaltder Holozell.
in °/o
Rohe Holozell.
(korr. auf Ascheund Lignin) in °/o
des extrafreienStrohs
83,1
83,9
84,1
5,2
5,9
5,1
2,49
3,60
2,57
76,7
75,8
77,6
M* 83,7 M 5,4 M 2,88 M 76,8
Während mit obiger Vorschrift beim frischen Stroh gut reproduzierbareResultate erhalten wurden, traten mit zunehmender Verrottung Schwierig¬keiten bei der Filtration ein. Die Ligninlösung wurde deshalb vorsichtigabdekantiert, worauf das zurückgebliebene Material mit Eiswasser gewa¬
schen und hernach abzentrifugiert wurde. Nach Abgiessen des Wassers
wurde der Inhalt des Zentrifugenglases mit wässrigem Aceton (90 °/o) quan¬
titativ in den Filtertiegel überführt. Der schleimige Rückstand der Holo¬
zellulose wurde durch diese Behandlung mit Aceton körnig und konnte ohne
Schwierigkeit filtriert werden.
325. Bestimmung der a-Zellulose
Als a-Zellulose bezeichnet man den Teil der Zellulose, der in 24 %>iger KOHoder 17,5 %>iger NaOH unlöslich ist. DerBegriff a-Zellulose deckt sich nicht
mit dem Begriff Baumwollzellulose, die nur aus polymerer Glukose besteht,sondern sie enthält noch geringe Mengen an Pentosanen und Asche und un¬
terscheidet sich von der Baumwollzellulose durch den Polymerisationsgrad.Wise, Murphy und d'Addieco91 fraktionierten die Holozellulose mit 5- und
24 "/oiger KOH in Stickstoffatmosphäre, um eine Oxydation zu vermeiden.
Im vorliegenden Falle wurde lediglich beabsichtigt, den bei 20° C in 24
"/oiger KOH unlöslichen Teil eines hauptsächlich aus Kohlenhydraten beste-
*M = Arithmetisches Mittel.
17

henden Materials (a-Zellulose) zu bestimmen. Die von frischem Weizen¬
stroh erhaltenen Analysenergebnisse sind in Tabelle 4 aufgeführt. Die a-
Zellulose wurde auf Asche, jedoch nicht auf Pentosane korrigiert.
Ausführung:
Auf 1 g getrocknete Holozellulose werden 40 ml 24°/oige KOH bei 20° C (imThermostaten) unter Umrühren beigegeben. Den Inhalt des Becherglases lässt man
2 Stunden bei dieser Temperatur unter gelegentlichem Umrühren stehen. Dann wird
durch ein Glasfiltertiegel 1 Gl filtriert, mit 25 ml KOH der gleichen Konzentra¬
tion, dann mit 100 ml Wasser, 10 ml 10 Voiger Essigsäure, fünfmal mit Eiswasser
und fünfmal mit Aceton gewaschen und hernach bei 105° C bis zur Gewichtskon¬
stanz getrocknet.
Tabelle 4: a-Zellulose-Bestimmung an frischem Weizenstroh.
a-Zellulose korr. auf Asche
i. °/o d. Holozellulosea-Zellulose i. %> d.
unextrahierten Strohs
47,2
49,4
48,4
36,6
38,3
37,5
M 48,3 M 37,5
Adams und Castagne92 erhielten 38,3 bis 38,7 °/o a-Zellulose bezogen auf
unextrahiertes Weizenstroh.
326. Bestimmung der Hemizellulosen als Furfurol
Die allgemeine Methode zur Bestimmung der Hemizellulosen (furfurolbil-dende Substanzen) beruht auf der Umwandlung von Pentosanen in Pentose¬
zucker durch Säurehydrolyse. Diese Pentosezucker werden gleichzeitig zu
Furfurol dehydratisiert, zu dessen Bestimmung gravimetrische, volumetri-
sche und kolorimetrische Verfahren benutzt werden. Die in den Hemizellu¬
losen vorhandenen Uronsäuren spalten unter diesen Bedingungen CO2 ab
und bilden in 35—40 Voiger Ausbeute ebenfalls Furfurol. Nachdem Adamsund Castagne93 in einer Untersuchung gezeigt haben, dass die kolorimetri¬sche Bestimmung mit Anilinacetat bessere Resultate ergibt als die übrigenMethoden, wurde dieses Verfahren mit einigen kleinen Abänderungen in
Anwendung gebracht. Zur Bestimmung der Eichkurve wurde handelsübli¬
ches Furfurol durch Destillation im Vakuum gereinigt und davon eine Ver¬
dünnungsreihe hergestellt. Von den einzelnen Konzentrationsreihen wurde
18

je 1 ml in ein Reagensglas pipettiert, 9 ml Anilinreagens zugefügt, kurz
geschüttelt und im Thermostaten bei 20° C in der Dunkelheit 1 Stunde ste¬
hen gelassen; dann wurde im Unicam Spectrophotometer Sp 600 bei 518 mu,
die Absorption gemessen. Da Methyl- und Oxymethylfurfurol aus Methyl-pentosen, Hexosezuckern, Stärke und Zellulose bei dieser Wellenlängekeine Absorption zeigen93, ist es möglich, Furfurol in Anwesenheit dieser
sehr verwandten Verbindungen zu bestimmen- Um die Stabilität der Fur-
furol-Anilinfarbe zu erhöhen, testeten Adams und Castagne98 verschiedene
Substanzen durch und fanden, dass eine Mischung von NaCl, Oxalsäure
und Dinatriumphosphat die beste stabilisierende Wirkung hatte.
Anilinacetatreagens: •
5 ml frisch destilliertes Anilin
50 ml Eisessig10 ml 20°/oige NaCl-Lösung5 ml 5 °/oige Oxalsäurelösung5 ml 10 °/oiges Dinatriumphosphat
Blindwert: Summe aller Reagenzien — Furfurol = 100%
Ausführung:
Bei frischem Material werden ca. 0,5 g, mit zunehmender Verrottung 1—2 g, mit
Äther und Wasser extrahiertes Strohmehl in einen Zweihalskolben von 600 ml
Inhalt eingefüllt. Dann werden 150 ml 12 Voige HCl und 20 g NaCl zugefügt,worauf der Inhalt durchgeschüttelt wird. Das Niveau im Kolben wird bei der
Destillation auf gleicher Höhe gehalten durch Zufügen von frischer HCl aus dem
mit einem Seitenarm durch Glasschliff verbundenen Tropftrichter. Der andere
Seitenarm ist durch einen Destillieraufsatz mit Tropfenfänger nach Stutzer und
darüber mit einem Destillieraufsatz mit angeschmolzenem Liebig-Kühler verbunden,der am Ende in einen Messkolben von 1 Liter Inhalt einmündet. Mit Hilfe einer
Pilzheizhaube wird der Inhalt soweit erhitzt, dass bei 110° C die Destillations¬
menge in 10 Minuten ca. 22 ml beträgt. Die Destillation ist beendet, wenn im
Destillat mit Anilinreagens nach 10—15 Minuten keine Farbreaktion mehr auf¬
tritt. Zu diesem Zweck wird 1 ml Destillat mit NaOH neutralisiert und mit 1 ml
frisch destilliertem Anilin, 10 ml Wasser und 9 ml Eisessig vermischt.
Die erforderliche Destillationsmenge beträgt in allgemeinen ca. 600 ml, wozu
eine Destillationszeit von ca. 4—5 Stunden notwendig ist. Das Destillat wird
mit Wasser bis zur Marke aufgefüllt und gut gemischt. 2 ml davon werden in einen
Kolben von 50 ml Inhalt pipettiert, 9 ml Anilinreagens beigefügt, kurz geschütteltund in der Dunkelheit bei 20° C im Thermostaten 1 Stunde stehen gelassen. Dann
wird im Unicam bei 518 mu, die Extinktion gemessen und mit Hilfe der aufgenom¬menen Eichkurve die Menge Furfurol bestimmt.
Von den Analysen wurden jeweils Doppelbestimmungen durchgeführt.Bekanntlich werden durch die Heisswasserextraktion unter anderem auch
furfurolbildende Substanzen (Uronsäuren, Pektine) extrahiert. Deshalb
wurde zudem noch eine Furfurolbestimmung im Doppel an unextrahiertem
Material vorgenommen. Theoretisch müsste die Differenz zwischen den
19

Furfurolwerten des extrahierten und des unextrahierten Materials dem
Gehalt an furfurolbildenden Substanzen im Heisswasserextrakt entsprechen.In Tabelle 5 sind die Werte, die aus frischem Stroh erhalten wurden, zusam¬
mengestellt.
Tabelle 5: Furfurolbestimmung an extrahiertem und unextrahiertem, frischem
Weizenstroh.
Stroh extrahiert mit Äther
und H2OStroh unextrahiert
Fur
in °/o des
Trockengew.*
fur 0 1
in °/o der
org. Substanz
Fur
in °/o des
Trockengew.
f u r 0 1
in °/o der
org. Substanz
21,72
21,66
21,87
21,43
23,14
23,07
23,30
22,83
21,19
20,72
21,08
20,80
22,57
22,07
22,46
22,16
Mittel: 21,67 23,08 20,95 22,31
* Unter Berücksichtigung der Extraktstoffe auf Ausgangsmaterial umgerechnet.
Wie man aus der Tabelle ersieht, erfasst man bei der Bestimmung in unex¬
trahiertem Material annähernd 1 °/o Furfurol weniger. Diese Erscheinungkonnte zum Teil auch bei den verrotteten Proben beobachtet werden. Die
heisswasserlösliche Extrakte scheinen bei der Furfurolbestimmung störend
zu wirken. Verschiedene Autoren94 95 berichten, dass Lignin und Gerbstoffe
mit Furfurol Kondensationsprodukte bilden und so dessen Ausbeute ver¬
mindern. Der Furfurolgehalt der Heisswasserextrakte konnte deshalb auf
die beschriebene Weise nicht ermittelt werden. Um die erhaltenen Furfurol-
werte auf Pentosane umzurechnen, müsste noch ein Abzug für Uronsäuren
gemacht werden.
327. Decarboxylierung der Strohproben mit 12 %>iger HCl
Viele Hemizellulosen und andere Polysaccharide enthalten Uronsäuren,deren Bestimmung auf der Tatsache beruht, dass sie durch Erhitzen mit
genügend konzentrierten Mineralsäuren mehr oder weniger quantitativdecarboxyliert werden98. Das dabei freigewordene CO2 wird zur Berechnungder freien und gebundenen Uronsäuren benutzt. Auf diese Weise wurde von
einer Reihe von Forschern 97 98 " 10° 1M der Uronsäuregehalt in Böden und
20

Komposten bestimmt. Dabei wurden Uronsäuremengen von 10—38 "/• der
gesamten organischen Bodensubstanz erhalten9710°. Diese Ergebnisse werden
jedoch von einigen Autoren angezweifelt102 103. Bei der Isolierung von Poly¬sacchariden aus verschiedenen Böden nach der Methode von Forsyth konn¬
ten Deuel und Mitarb.104 nur eine geringe Menge an decarboxylierbarenPolysacchariden erfassen, die in keiner Weise mit den durch saure Decar-
boxylierung ermittelten «Bodenuronsäuren» übereinstimmt. Sie nehmen da¬
her an, dass im Boden neben Uronsäuren noch andere decarboxylierbareSubstanzen vorhanden sind. Es war deshalb im Rahmen dieser Arbeit von
besonderem Interesse, das Verhältnis von abspaltbarem CO2 zu Furfurol
im Verlaufe der Verrottung zu verfolgen. Die Decarboxylierung der Stroh¬
proben erfolgte im Prinzip in der von Dubach*105 beschriebenen Apparatur.Die Apparatur wurde zuvor mit reinem Na2C03 (100 mg) getestet. Gefun¬
den: 41,58 und 41,60 %> CO2. Theorie: 41,51 %> CO2.
Ausführung:
In den Reaktionskolben A (Abb. 1) von 500 ml Inhalt werden 2—5 g unextrahiertesStrohmehl eingewogen. Um eine Störung durch eventuell vorhandene Carbonate
auszuschalten, wird das Strohmehl aus dem angesetzten Tropftrichter mit kalter12 °/oiger HCl Übergossen und für ca. 10 Minuten ein Stickstoffstrom durchgeleitet,
Abb. 1: Apparatur zur Bestimmung von Uronsäuren.
* Herrn P. Dubach, Agrikulturchemisches Institut der E. T. H., Zürich, möchte ich
herzlich für die Durchführung von C02-Bestimmungen an einigen Strohprobendanken.
21

ohne die beiden Absorptionsgefasse E und F anzuschliessen. Dann werden die unter
Stickstoffstrom mit einer bestimmten Menge 0,02 n Ba(OH)2 beschickten Absorp¬tionsgefasse E und F angeschlossen. Nun wird das Ölbad allmählich auf 135—
140° C erhitzt und fur 4% Stunden auf dieser Temperatur gehalten. Der Stickstoff-
ström wird so eingestellt, dass in Gefass B pro sec. 1 Blase durchperlt. Die aufstei¬
genden HCl-Dampfe kondensieren im Ruckflusskuhler. Das Waschgefass B enthalt
12 °/oige mit Phloroglucin gesattigte HCl zur Absorption von übergetriebenemFurfurol. Das Waschgefass C enthalt CC>2-gesattigtes Wasser und Gefass D 10 "/oigesSilbernitrat zur Absorption von eventuell übergetriebenen HCl-Dampfen. Die m
den Absorptionsgefassen E und F eingeschmolzenen Fritten lassen das Gasgemisch(N2 + CO2) als ganz feine Bläschen hochperlen.
Nach beendeter Reaktionszeit werden die Absorptionsgefasse E und F abgehängt;der Rest an Ba(OH)2 wird mit 0,1 n HCl gegen Phenolphtalein als Indikator zu¬
rücktitriert. Die Duichmischung der Flüssigkeit wahrend der Titration erfolgtdurch einen kraftigen Stickstoffstrom.
328. Bestimmung des Ammoniak- und Nitratstickstoffes
Um zu erfahren, bis zu welchem Grade der Gesamtstickstoff organischgebunden vorliegt, ist es notwendig, den Anteil an Ammoniak- und Nitrat¬
stickstoff zu bestimmen. Den organisch gebundenen Stickstoff erhält man
durch Substraktion des Ammoniak- und Nitratstickstoffes vom Gesamt¬
stickstoff.
Bremner und Shawiw bestimmten den Ammoniak- und Nitratstickstoff im
Boden mit Hilfe der Mikrodiffusionsanalyse nach Conway. Bremner101
wandte diese Methode mit Erfolg auch bei Strohkomposten an. Sie beruht
darauf, dass Ammoniak bei niederer Temperatur aus einer alkalischen Lö¬
sung freigesetzt und von einer geeigneten Saure wieder absorbiert wird.
Der gesamte Vorgang spielt sich in einem geschlossenen Spezialgefäss ab.
Zur Bestimmung des Nitratstickstoffes wird analog vorgegangen, nur
wird der Nitratstickstoff vorher zu Ammoniak reduziert.
Während nach dieser Methode für Ammoniakstickstoff gute und repro¬duzierbare Resultate erhalten wurden, schwankten die Werte beim Nitrat¬
stickstoff stark. Dies ist darauf zurückzuführen, dass die Magnesiumoxyd¬suspension mit der Titanylsulfatlösung eine unbewegliche, gallertartigeMasse bildet, wodurch die Reaktion gestört wird. Um dies zu vermeiden,wurde die Methode nach einer Vorschrift von Conway108 abgeändert, wel¬
cher für stark saure Lösungen Kaliummetaborat oder 50 %ige KOH emp¬fiehlt. Die Reduktion des Nitratstickstoffes erfolgte mit feingepulverterDevarda'scher Legierung. Die Überprüfung der Methode mit (NHé^SOéund KNO3 als Testsubstanzen lieferte unter Anwendung von 50 Voiger KOHsehr gute und reproduzierbare Resultate.
22

Reagenzien:
1 n K.2SO4 eingestellt auf pH 1—1,5 mit H2SO4.
2 Voige Borsäure enthaltend eine Indikatorlösung (20 ml/1) nach Conway und
Malleyim. Das Reagens wird bereitet, indem 20 g p. a. Borsäure in einer Mischungvon 200 ml abs. Alkohol und 700 ml dest. Wasser gelöst werden. Dieser Lösungwerden 200 ml Mischindikator beigefügt. Dieser letztere wird hergestellt durch
Auflösen von 0,033 g Bromkresolgrün und 0,066 g Methylrot in 100 ml abs. Alkohol.
Nach dem Mischen wird die Lösung durch sorgfältiges Zufügen von 0,05 n NaOH
zu einer schwach gelblich-grünen Färbung gebracht und dann mit dest. Wasser
auf 11 verdünnt.
Standard 0,005 n H2SO4, (1 ml entspricht 0,07 mg N), fein pulverisierte Devar-
da'sche Legierung, 50 °/oige KOH.
Ausführung:
0,3—0,5 g feingemahlenes Material wird in einem verschlossenem Erlenmeyerkolbenvon 100 ml Inhalt zusammen mit 10 ml K.2SC>4-Lösung von pH 1,1 während 2
Stunden auf der Schüttelmaschine geschüttelt. Dann wird durch einen Faltenfilter
filtriert. Zur Bestimmung des NH3-N pipettiert man 1 ml von diesem Filtrat in
den Aussenraum des Mikrodiffusionsgefässes und in den Innenraum 1 ml Borsäure¬
lösung, die den Mischindikator enthält. Nach Zufügen von 1 ml 50 °/oiger KOHin den Aussenraum wird das Gefäss sofort mit einem Deckel verschlossen und mit
einem Klebestreifen gut zugeklebt. Das Gefäss bleibt nun 2 Stunden bei Zimmer¬
temperatur auf dem Labortisch stehen. Dann wird der Inhalt des Innenraumes
mit einer Pipette entnommen und mehrmals mit dest. Wasser ausgewaschen. Die
Titration erfolgt mit 0,005 n H2SO4 aus einer Mikrobürette, bis der Umschlag nach
rotgelb erfolgt. In einer parallelen Reihe werden dem Aussenraum vor dem Zufü¬
gen von KOH einige Spatelspitzen Devarda'sche Legierung beigeben und gut ver¬
teilt. Die Bestimmung, die sonst analog verläuft wie vorher, ergibt die Menge an
vorhandenem Nitrat- und Ammoniak-Stickstoff. Durch Subtraktion des vorher
gefundenen Wertes an Ammoniakstickstoff von diesem letzteren Wert erhält man
die vorhandene Menge an Nitratstickstoff. Die Analysen wurden stets im Doppelausgeführt.
329. Bestimmung der Elementarzusammensetzung
Die verschiedenen Strohproben sowie auch die übrigen Präparate wurden
im Achatmörser staubfein pulverisiert, in der Trockenpistole bei 65° C über
P2O5 getrocknet und hernach auf C, H und Gesamtstickstoff nach Dumas*
untersucht.
Zur Bestimmung der Methoxylwerte wurde die Mikromethode von Vie-
böck und Brecher110 in Anwendung gebracht mit der Abänderung, dass
statt 0,5 °/oiger Natriumthiosulfatlösung 0,1 n NaOH als Waschflüssigkeitverwendet wurde. Mit Natriumthiosulfat resultierten oft zu niedrige Werte,
was wahrscheinlich auf die Bildung des Methylesters der Thioschwefelsäure
* Die Analysen wurden im Chemischen Untersuchungslaboratorium der Forschungs¬anstalt für Landwirtschaft, Braunschweig-Völkenrode, durchgeführt.
23

zurückgeführt werden muss. Die Apparatur wurde von Zeit zu Zeit mit
Vanillin als Testsubstanz geprüft Die Analysen wurden stets im Doppel
ausgeführt. Die Einwaage betrug 4—8 mg.
33. Ergebnisse des Abbauversuches
In den folgenden Tabellen 6—10 sind die während der Rotte von Weizen¬
stroh eingetretenen Veränderungen in der Zusammensetzung kurz darge¬stellt.
Tabelle 6: Menge an Extraktstoffen zu verschiedenen Zeitpunkten der Verrottung(absolut in g und in Prozent des Trockengewichtes).
Rottezeit Tr. Gew. Ätherlöslidi Heisswasserlöslidi Alk.-r}eiaoUöslidi Heisswasserlösl."^ '
(anorg. + org.) (aschefrei) (anorg. + org.)in m
abs. i. % d. abs. i. %d. abs. i.%d. abs. i. % à.
Tagen g in g Tr. Gew. in g Tr. Gew. in g Tr. Gew. in g Tr. Gew.
0 282,5+ 2,17 0,77 20,1 7,1 10,8 3,8 12,1 4,3
70 148,5 1,42 0,96 24,2 16,3 2,4 1,6 23,3 15,7180 68,9 0,46 0,66 18,6 26,9 1,1 1,5 18,1 26,2260 73,4 0,32 0,43 22,1 31,4 1,0 1,4 19,7 26,8340 71,1 0,28 0,39 20,8 29,3 1,1 1,7 17,8 25,0410 70,5 0,30 0,43 18,6 26,5 0,7 1,0 18,2 25,8410+N" 72,1 0,25 0,35 28,7 39,8 0,4 0,6 28,3 39,3
* Bezieht sich auf die Serie mit zusätzlicher Nährsalzmenge.+ Strohtrockengewicht ohne Nährsalze.++ Nach vorheriger Extraktion mit Alkohol-Benzol 1:1.
Aus der Tabelle 6 geht hervor, dass mit zunehmender Verrottung der in
Äther und Alkohol-Benzol lösliche Anteil stetig abnimmt. Würde man zu
den heisswasserlöslichen Stoffen aus dem frischen Stroh noch die zugefügteDüngermenge von 11,3 g hinzuzählen, so wäre auch in der absoluten Mengedes heisswasserlöslichen Anteils eine Abnahme zu verzeichnen, während bei
vorliegender Betrachtungsweise eine leichte Zunahme oder zum mindesten
nur eine geringfügige Abnahme resultiert. Die in dieser Tabelle angegebenenWerte für die heisswasserlöslichen Stoffe stellen den gesamten Anteil an
anorganischen und organischen Bestandteilen dar, der unter den genannten
Bedingungen in Lösung geht. Der Grund für die relative hohe Ausbeute an
heisswasserlöslichen, organischen Stoffen dürfte vor allem auf das in be¬
trächtlicher Menge als Stickstoffquelle zugegebene Na-Nitrat zurückzufüh¬
ren sein, wobei nach der bakteriellen Verwertung des Nitrat-Ions als Zwi¬
schenprodukt Na-Carbonat entsteht, welches seinerseits mit den im Verlauf
des Rotteprozesses entstandenen sauren Gruppen wasserlösliche Na-Humate
bildet.
24

Tabelle 7: Trockengewicht, Lignin, Holozellulose und a-Zellulose nach 70 TagenRotte in den bei unterschiedlichem pH angesetzten Strohproben (absolut in g undAbnahme in Prozent des Ausgangsmaterials).
Nû ix
1 in V—(
o.-, 1 On oo oo -^ in
**no nû su oo m
d
S 2 C_o
1nû Ö vfH \0 On +1 CS* i-Î Ö Ö *n +1
3 'S < vO N su sfi vfi^H
N N K N N00
•=> < 1-t4) (J
a
NO IV.
Su v* es im so oo m O fNj cnj m ,-h es o
.s VO m' h ö « n in C» O H H OÔ dO r^ m -*t m m m es m <n m es m
^v»- *•
«i .S a e ON v~î
.ellulosf
Lign Protei•s 1 On nû 00 •<$ On
r"tOO co i-; p OO
v-1
c 1 00* r-ï SU OÔ iH +1 Ö K O Ö Tt" +15 NÛ hv NÛ \0 N.
m
Ov
N nû N N N
d
Holoi korr.au
Asche,°
vO IV.
»fi
.5 m_ On »t; On Is k in q o * <û h Cvj
00 i-* vo »n es in* d oô -^ On On O oôOv \û m nû nû m VO in nû m m m m
V"H
o
1G
• m
•S00 N.
'S ... 1 00 00 ON nO 00es
nO f*% O p fsjv
<x> S 1 oô on tn *-* o* +1 rj \d in on in* +1
•i 1es m es m m
en
m es m es mvO
o sc/5 ni
m m
0Û OO *û 00 h h M tn \o on t-o *n es r-*
K .s fi K l-î ON \û -r-* tn in oô ^»- hv Tt" vÖ«n m m m f*ï m tn m f» en en m («")
i 4>
E
tsin
Iv.
p
eng 9 1 CS eS On On OO+1 p in nû i-; T-H
*-<
C l» tfj -t oô ri en oô «-* es in* +1
* < ** m tc -^j- m vO m *** m m m^
:troc # OvVf
esm
sam* v*; \û on rn m y-* in t N 't ff; r- 1-H
60•«t es is* es* ö o* oô oô *-" es" ci es H
.S Ov in en no m rn Vf <n m ^- * m <<*-4> rvi vpH v?-« vp-I r-H ,-« T-4 T-^ 1—( 1—* 1—t T-<
—2 o m m
« :8 V—< > >
S S
O
T5 1 fl^ OO C IS OO Is O IN O ho ,-H
X «1 oô oô cô oô oô oô 00 00 00 00 00 oô
a
< 1 in in
*-»l< «f
'8 a
2 Ö 60*j •- rt
<â H o o Ois f^
25

£en
Mittelwert
arithmetischer
=M
Nährsalzen.
zusätzlichen
mit
2o
S*
»
5-
croc
aSi
C
N2
i^R
-i<û.SC
*i
-.
rt
vi
oa
E.
n
Ng
SI
o2.^2
l^aa
-'
c«sisL.
fer
CIO
_s
<=>5tFg
Nn
8-
TO
g2.2
•?
'
5"a
?»
n"
Nf&
3rt
££
nET*
^«a
pis'il-
96.2
3.9
94.5
11.0
65.7
18.1
75.6
72.1
7.2
3N*
+410
99.3
0.7
95.9
9.7
63.0
19.5
76.1
70.5
7.1
3410
97.1
3.1
96.3
7.4
64.6
18.7
75.8
71.1
7.1
M
97.7
2.5
95.4
4.9
95.9
8.1
97.3
5.4
65.6
18.2
61.7
20.2
76.0
70.7
75.4
72.2
7.2
6.8
31340
97.8
2.3
95.0
9.9
64.2
18.9
75.1
73.4
7.3
M
98.0
2.1
97.1
3.1
95.0
9.9
95.1
9.8
64.0
19.0
65.0
18.5
75.0
73.8
75.6
72.0
7.3
7.5
41260
95.2
5.1
93.8
12.4
68.0
16.9
76.6
68.9
7.5
M
95.4
4.9
94.5
5.9
93.8
12.4
93.7
12.5
68.2
16.8
67.0
17.4
77.1
67.3
74.4
75.4
7.5
7.6
41180
67.1
35.0
69.5
60.5
33.2
35.3
49.6
148.5
8.7
v.5
M70
—106.4
—198.5
—52.8
—
294.4
—1
0
Abnahme
°/o
gin
asch
efre
i
(X-Z
ellu
lose
Abnahme
"/o
gin
Protein
Asche,
Lign
in,
auf
korr.
Holozellulose
Abnahme
°/o
gin
aschefrei
U2SOi-Lignin
Abnahme
°/o
gin
Gefäss
je
Gesamttrockengew.
pH
End-
Gefässe
Anzahl
Tagen
in
Rottezeit

Id
1
O"8
aS2.çg.g.
cre
n3gcre3
«-t
ss
re
n=
<
oro
Co¬s)
a°
2pro_
a
a.T22.S3
33
eo
Cl¬
3nO-Q.a
n>STcp
>-t
ft-
<00
SS.
l-t
_
3o"
'S
o•
fr»*
rttna.
re
sS-
"•
N
^S
S
Stroh.
unextrahiertem
in
Bestimmt
*
2.84
4.06
2.04
3.02
4.31
2.16
3.49
4.99
2.51
50.4
N+
410
2.21
3.31
1.55
2.85
4.27
2.02
3.12
4.70
2.20
47.0
410
2.14
3.10
1.52
2.95
4.35
2.11
3.90
5.75
2.83
49.3
340
2.07
3.07
1.51
3.33
4.93
2.41
3.21
4.76
2.34
49.2
260
1.56
2.20
1.11
4.99
7.06
3.52
5.87
8.31
4.18
50.4
180
0.88
1.05
1.30
12.92
15.43
19.14
13.81
16.49
20.50
124.3
70
0.85
0.90
2.38
20.95
22.32
59.14
21.67
23.09
60.97
265.1
0
Gew.
Tr.
d.
°/o
in
S.org.
d.%>
in
gin
absol.
Gew.
Tr.
S.org.
d.°/
oin
d.%>
in
gin
absol.
Gew.
Tr.
d.°/
oin
S.org.
d.°/o
in
gin
abso
l.g
Tagen
CO2*
Furfurol
unextrahiert
Stroh
1
Äther
0fur
Fur
H2O
+
mit
extrah.
Stroh
Subst.
Organ.
zeit
Rotte¬

Serie.
angesetzten
4,5
pH
bei
der
Analysen
5von
Mittel
=B**
Serie.
angesetzten
7,5
pH
bei
der
Analysen
5von
Mittel
=A*
NaNÛ3.
zugefügtem
.aus
N=
Rest
"/«,
0,74
=Stroh
im
Total-N
=+
8.8
23.3
9.6
31.8
4.34
5.17
4.36
5.58
45.54
4.29
5.03
4.51
5.87
48.32
30.16
83.1
33.30
89.5
3.05
3.00
0.56
0.05
3.61
3.01
2.91
0.41
0.04
3.36
3N
+410
3410
10.7
36.1
6.22
47.93
3.82
4.51
4.93
Î1Î
Toi
32.24
84'8
29.91
„o.
3.15
3.40
2.64
0.43
0.04
3'08
X340
10.6
34.1
5.99
49.60
6.18
50.22
32.62
85'°
32.56
~o,
3.00
2.67
0.44
0.03
3.08
4\260
12.6
43.1
«3.«
«-
22
28.68
85'9
30.09
„„„
3.38
2.38
0.31
0.08
'78
2
4\180
26.2
33.4
24.3
28.6
1.73
1.84
4.24
6.17
48.16
1.88
1.99
4.26
6.21
48.34
16.44
94.1
16.29
94.6
3.55
1.45
0.03
0.06
1.54
3.57
1.58
0.04
0.05
1.67
5B-
70
5A*
,A
60.2
—0.71
1.29
4.04
6.47
46.36
6.15
55.4
3.80
0.67
0.49
0.05
1.21+
10
NGesamt
des
N:
C0/0
in
N-Ver!ust
org.N
Total-N
OCH3
HC
Substanz
org.
der
°/o
in
Total-N
°/o
in
von
Asche
°/>
inOrg.N
OCHs
Norg.
NOs-N
NNHs
Total-N
Troc
keng
ewic
htes
des
%in
fasse
Tagen
Ge¬
in
zeit
Anzahl
Rotte-

In Tabelle 7 sind die Ergebnisse über den Abbau der bei unterschiedlichem
pH angesetzten Versuchs-Serien zusammengestellt. Bei beiden Serien ist ein
starker Anstieg des pH-Wertes festzustellen, so dass nach 70 Tagen auch die
bei pH 4,5 angesetzten Gefässe alkalische Reaktion aufweisen. Dieser
starke Anstieg darf dem in beträchtlicher Menge als Stickstoffquelle zuge¬fügtem Na-Nitrat zugeschrieben werden, denn auch Jansson und Clarklilhaben bei Verwendung von Na-Nitrat in gepufferter Nährlösung gleicheEffekte beobachtet. Ein Anstieg des pH-Wertes zu Beginn der aeroben Zer¬
setzung von Pflanzen verschiedenster Herkunft ist jedoch auch bei Ab¬wesenheit von physiologisch alkalisch wirkenden Nährsalzen festgestelltworden112.
Infolge dieser Angleichung der Reaktionsverhältnisse sind im Verlaufevon 70 Tagen in der Zusammensetzung der verschiedenen Komponentenzwischen den beiden Serien keine wesentlichen Unterschiede eingetreten.Während die Werte der Trockensubstanz, der Holozellulose und der a-Zel-lulose auf einen vermehrten Abbau in der bei pH 4,5 angesetzten Serie
schliessen lassen, ist jedoch infolge der erheblichen Streuung der Einzelwerte
nur der vermehrte Abbau der a-Zellulose im sauren Milieu gegenüber demAbbau im alkalischen gesichert.
Die in Tabelle 8 zusammengestellten Ergebnisse über den Abbau der ver¬
schiedenen Komponenten zeigen eindeutig, dass die Werte des Gesamttrok-
kengewichts, des Lignins und der Holozellulose sich nach 180 Tagen Rotte¬
zeit kaum mehr ändern. Die schwache Abnahme der Werte für die a-Zellu-lose im Zeitraum von 180 bis 410 Tagen Rottedauer deutet jedoch auf einen
geringen, zusätzlichen Abbau hin. Daraus lässt sich ableiten, dass sich die
Holozellulose mit zunehmender Verrottung zum überwiegenden Teil aus
Hemizellulosen (= Holozellulose minus a-Zellulose) zusammensetzt. Dasich die Werte der Serie mit nachträglich beigefügten Nährsalzen (410 + N)kaum von denjenigen der parallelen Serie (410) unterscheiden, kann man
den Schluss ziehen, dass die Ursache des Stillstandes im Abbau nicht dem
Mangel an Mineralsalzen, sondern einem anderen Umstand zugeschriebenwerden muss.
Aus Tabelle 9 geht hervor, dass beim Abbau der furfurolbildenden Sub¬
stanzen in Übereinstimmung mit den Ergebnissen der Tabelle 8 eine ähnliche
Tendenz festgestellt werden kann. Im Gegensatz zu den übrigen Kompo¬nenten tritt beim Abbau der Hemizellulosen erst nach 260 Rottetagen ein
Stillstand ein. Die Furfurolwerte der unextrahierten Strohproben zeigenjedoch nach diesem Zeitpunkt noch eine geringe Abnahme. Die Erscheinung,dass unextrahiertes Stroh geringere Furfurolwerte ergibt, ist an andererStelle schon diskutiert worden (siehe Abschnitt 326).
Die durch saure Decarboxylierung erhaltenen C02-Mengen nehmen inden ersten 180 Rottetagen um mehr als die Hälfte ab, um hernach langsam
29

wieder anzusteigen. Da dieser Erhöhung der CCb-Menge keine entsprechendeZunahme an Furfurol gegenübersteht, kann dieser Anstieg nicht mit der
Neubildung von Uronsäurekomplexen (mikrobielle Polysaccharide) erklärt
werden, sondern dürfte auf die durch oxydative Prozesse bedingte Entste¬
hung anderer decarboxylierbarer Substanzen zurückgeführt werden.
Aus Tabelle 10 kann man ersehen, dass der prozentuale Anteil des Total-
und des organischen Stickstoffs trotz eines N-Verlustes von 30 bis 40 °/o
in Form von Ammoniak im Verlauf der Verrottung ständig zunimmt. Diese
Zunahme an Stickstoff kommt auch deutlich in der Abnahme des C/N-Quo-tienten zum Ausdruck, der nach 260 Tagen den für stabile Humusformen
charakteristischen Wert von ungefähr 10 aufweist. In Übereinstimmungmit den nach 70 Tagen Rottezeit festgestellten, geringen Werten für den
Nitrat-Stickstoff darf man schliessen, dass in diesem Zeitpunkt der als N-
Quelle zugegebene Nitrat-Stickstoff von den Mikroorganismen nahezu völ¬
lig ausgenützt und zu einem grossen Teil in organische Stickstoffverbin¬
dungen umgewandelt wurde. Darauf weist auch der hohe Anteil an organi¬schem Stickstoff in Prozent des Gesamt-N hin. Während die Werte für den
Ammoniak-Stickstoff stets gering und mehr oder weniger konstant bleiben,nimmt der prozentuale Anteil an Nitrat-Stickstoff nach 70 Tagen wieder
zu. Diese Zunahme des Nitrat-Stickstoffes ist auf den durch nitrifizierende
Bakterien verursachten Mineralisierungsprozess zurückzuführen, wobei das
aus der Eiweisszersetzung stammende Ammoniak zu Nitrit und Nitrat
oxydiert wird.
Die Kohlenstoffwerte zeigen einen ständigen Anstieg und erreichen mit
180 Rottetagen ihr Maximum, um hernach auf gleicher Höhe zu bleiben
oder teilweise leicht abzufallen. Stark erniedrigte Kohlenstoffwerte weist
die Serie mit nachträglich zugefügten Nährsalzen auf. Die Wasserstoffwerte
zeigen eine ständige geringfügige Abnahme. Die auf Trockensubstanz be¬
zogenen Methoxylwerte nehmen infolge des starken Anstiegs des Aschege¬haltes ab, während die auf organische Substanz berechneten Werte leicht
zunehmen. Die Zunahme der C- und OCH3-Werte müsste jedoch viel be¬
deutender sein, wenn die Kohlenstoff- und Methoxylwerte im Lignin nicht
auch abgenommen hätten.
34. Isolierung von Lignin und Huminsäuren nach
verschiedenen Methoden
Braunsuh benennt das Lignin, so wie es in der Pflanze vorliegt, als «natives»
oder Protolignin. Dieses erhält man durch Extraktion von pflanzlichemMaterial mit Alkohol, Dioxan oder mit irgendeinem anderen indifferenten
Lösungsmittel bei Zimmertemperatur.
30

Um die im Verlaufe der Zersetzung von Stroh eingetretenen Änderungender Ligninstruktur zu verfolgen, musste ein Verfahren angewendet werden,das ein möglichst schonendes Herauslösen gewährleistet. Nur auf diese
Weise war es möglich, sich zu vergewissern, ob das nach der Schwefelsäure¬
methode hergestellte Lignin tatsächlich noch dem «wirklichen» Lignin ent¬
spricht.
341. Isolierung von Nativlignin nach Brauns21
Frisches, in der Conduxmühle gemahlenes Stroh wird mit peroxydfreiem Äther
35 Stunden lang im Soxhlet extrahiert und mit kaltem destilliertem Wasser gründ¬lich gewaschen. 900 g dieses feingemahlenen, extrahierten Strohmehls werden in
ein Glasrohr mit Abflusshahn eingefüllt. 12 Tage lang wird bei Zimmertemperaturmit reinem 96 Voigem Alkohol extrahiert. Das in Lösung gegangene Lignin wird
täglich unten abgezapft und oben wieder frischer Alkohol nachgeschüttet, bis der
Alkohol praktisch farblos abfliesst (10—12 Tage). Die alkoholische Lösung wird
nun im Vakuum vorsichtig auf kleines Volumen eingedampft, zuletzt dest. Wasser
beigegeben und der Rest des Alkohols völlig abgedampft. Dabei fällt ein schwach¬
gelber Niederschlag aus. Dieser wird gut mit destilliertem Wasser und Äther ge¬
waschen, an der Luft und schliesslich im Vakuumexsikkator getrocknet. Nach einem
Tag erhält man durch Zerreiben ein gelbbraunes Pulver. Davon wird mit gereinig¬tem Dioxan (11c) eine 10°/oige Lösung hergestellt und bei mittlerer Geschwindig¬keit kurz zentrifugiert. Die braune Lösung wird in feinem Strahl unter starkemRühren in die 15-fache Menge dest. Wasser eingetragen, worauf ein käsiger, creme¬
farbener Niederschlag ausfällt, der zentrifugiert und im Vakuumexsikkator ge¬trocknet wird. Dann wird erneut in wasserfreiem, gereinigtem Dioxan gelöst (10-Voige Lösung), zentrifugiert, filtriert und in feinem Strahl unter starkem Um¬
rühren langsam in die 15-fache Menge absoluten Äther eingegossen. Dabei fällt ein
hellbraunes Pulver aus. Dieses wird abzentrifugiert, zweimal mit frischem Äther
gewaschen, kurz getrocknet und erneut in Dioxan gelöst, abermals in absolutem
Äther ausgefällt, abzentrifugiert, im Zentrifugenglas einmal mit frischem Äther,zweimal mit reinem Benzol, zweimal mit Petroläther (40°) gewaschen und im
Vakuumexsikkator über P2O5 getrocknet.
Die Ausbeute an gereinigtem Lignin betrug 208 mg = 0,12 °/o vom Lig-ninanteil. Obwohl Brauns21 aus Fichtenholz eine viel bessere Ausbeute er¬
hielt, scheint bei Stroharten der lösliche Ligninanteil unter den von ihm
angewandten Bedingungen viel geringer zu sein. So erhielten Stone und
Tanner113 aus 563 g Weizenstroh nur 50 mg gereinigtes Nativlignin.Das aus frischem Strohmehl erhaltene Nativlignin nach Brauns wurde
auf die Elementarzusammensetzung untersucht und zu Infrarotstudien ver¬
wendet. Eine Extraktion von Lignin nach dieser Methode aus verrottetem
Stroh wurde nicht vorgenommen.
342. Isolierung von Lignin nach Björkman
Björkman11 U4 konnte auf Grund eingehender Untersuchungen zeigen, dass
aus Fichtenholz, das in genügend kleiner Menge einer Mahlung in der
31

Schwingmühle in einer das Holz nicht quellenden Flüssigkeit unterworfen
wird, ein grosser Teil (50—60 %>) des Lignins mit organischen Lösungs¬mitteln extrahiert werden kann. Es sollte deshalb versucht werden, ob sich
bei Weizenstroh die Ausbeute an schonend herausgelöstem Lignin nach die¬
sem Verfahren erhöhen lässt.
Björkman hat das Holzmehl in einer speziell konstruierten Schwingmühlemit rostfreien Stahlkugeln staubfein zermahlen. Das dabei erhaltene Ligninnennt er «milled wood lignin». Im vorliegenden Falle wurde mit einer
Schwingmühle (Fabrikat Vibratom) unter Verwendung von Porzellanku¬
geln gearbeitet. Es ist anzunehmen, dass der Mahlungsgrad dabei nicht so
hoch war. Im übrigen wurden genau die Vorschriften von Björkman115eingehalten. Das auf diese Weise extrahierte Lignin wird im folgenden als
«Björkman-lÄgwln-» bezeichnet.
3421. Björkman-Lign'm aus frischem Weizenstroh
Zur Verwendung gelangte das nach der Nativligninbestimmung nach
Brauns verwendete zurückgebliebene Strohmehl. Die Schwingmahlung er¬
folgte in 2 Porzellangefässen zu je 4 1 Fassungsvermögen. Je Gefäss wurden
60 g Strohmehl eingefüllt und so viel reinstes Toluol beigegeben, dass das
Strohmehl gut durchfeuchtet war. Nach Zugabe einer grösseren Anzahl von
Porzellankugeln wurden die Gefässe gut verschlossen und während 65 Stun¬
den auf der Schwingmühle geschüttelt. Nach Abtrennung des Toluols durch
Zentrifugieren wurde das staubfeine Material 3 Tage bei Zimmertemperaturmit wässrigem 90 Voigem Dioxan extrahiert. Die Dioxanlösung wurde im
Vakuum eingeengt, im 10-fachen Volumen Äther gefällt, das abzentrifu-
gierte Lignin in 90 Voiger Essigsäure gelöst und die Lösung unter starkem
Umrühren in das 10-fache Volumen Wasser eingetragen. Durch Zusatz einer
Spur Na2SOé konnte besseres Ausflocken erreicht werden. Die abzentrifu-
gierte Fällung wurde nach vorherigem Ausbreiten in einem Luftstrom (Föhn)getrocknet. Dann wurde wieder in einer Mischung von Aethylenchlorid undAethylalkohol (2:1) gelöst und das Lignin aus der Lösung schliesslich nach
kurzem Abzentrifugieren durch Eingiessen in absoluten Äther gefällt. Die
abzentrifugierte Fällung wurde im Zentrifugenglas dreimal mit frischem
Äther, einmal mit Petroläther gewaschen und im Vakuumexsikkator über
PïOb getrocknet. Der Rest des verbliebenen Mehles wurde nochmals 4 Tagemit Dioxan extrahiert.
Erste Ausbeute: 0,44 gZweite Ausbeute: 0,05 g
Gesamt-Ausbeute: 0,49 g
Im ganzen wurden durch diese Behandlungen rund 2,5 %> des Lignins aus
frischem Weizenstroh auf schonende Art isoliert.
32

Durch Erwärmen des verbliebenen Strohmehls in 90 °/oigem Dioxan wäh¬
rend 24 Stunden auf dem Wasserbad konnten nochmals ungefähr 500 mg
Lignin extrahiert werden. Die Temparatur im Kolben betrug dabei 60—
70° C.
Da ein Zusatz einer geringen Menge Mineralsäure zum organischen Lö¬
sungsmittel die nahezu quantitative Herauslösung des Lignins gestattet116,wurde je 100 ml 90 Voiges Dioxan 1 ml konz. HCl beigegeben und das vor¬
her nur mit Dioxan extrahierte Material eine Nacht lang auf dem Wasser-
bad am Rückflusskühler gekocht. Die Aufarbeitung des so isolierten Lignins
erfolgte wie vorher. Das unter Säurezusatz herausgelöste Lignin wies eine
dunklere Farbe als die beiden vorhergehenden Präparate auf.
Von diesen drei unter verschiedenen Bedingungen isolierten Ligninenwurde die Elementarzusammensetzung bestimmt (Tabelle 11) und eine Un¬
tersuchung im Infrarotspektographen vorgenommen. Die IR-Spektren der
beiden ersten Lignine waren praktisch völlig identisch, während bei dem
durch HCl-Zusatz erhaltenen Lignin Änderungen im Bereich von 7,25—
7,57 (i und 6,06—6,17 u, festgestellt wurden (vergl. hierzu IR-SpektrumNo. IV, Seite 62).
Tabelle 11: Elementarzusammensetzung von Björkman-Lignin aus frischem Stroh
isoliert unter verschiedenen Bedingungen (Prozente bezogen auf aschefreie Sub¬
stanz).
Extraktionsbedingungen C%> HVo N%> OCHs %> Asche °/o
Zimmertemperatur 60,68 5,79 0,42 16,76 1,11
60—70° C 60,46 5,72 0,42 16,70 4,03
80» C + HCl 61,08 5,97 0,19 16,86 0,76
Aus Tabelle 11 ersieht man, dass durch den HCl-Zusatz die C- und H-
Werte leicht erhöht werden. Wahrscheinlich wurde ein beträchtlicher Teil
des Stickstoffes in Form von Aminosäuren abhydrolysiert. Der noch vor¬
handene Stickstoff muss ziemlich fest an das Molekül gebunden sein (siehehierzu Abschnitt 362).
3422. Björkman-Lignin aus verrottetem Weizenstroh
Die Isolierung von Björkman-Lignm aus den verrotteten Proben erfolgteunter den gleichen Bedingungen wie beim frischen Material, nur dass an
Stelle der Äther- und Kaltwasserextraktion vor der Schwingmahlung mit
33
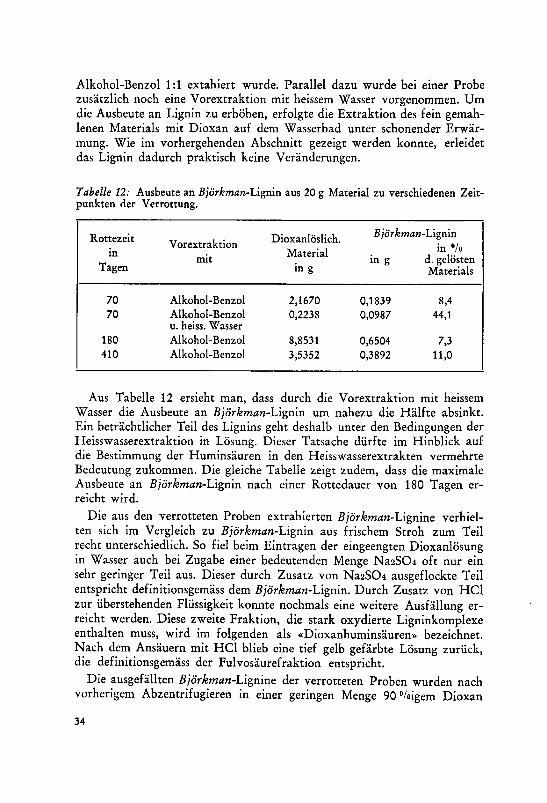
Alkohol-Benzol 1:1 extahiert wurde. Parallel dazu wurde bei einer Probe
zusätzlich noch eine Vorextraktion mit heissem Wasser vorgenommen. Um
die Ausbeute an Lignin zu erhöhen, erfolgte die Extraktion des fein gemah¬lenen Materials mit Dioxan auf dem Wasserbad unter schonender Erwär¬
mung. Wie im vorhergehenden Abschnitt gezeigt werden konnte, erleidet
das Lignin dadurch praktisch keine Veränderungen.
Tabelle 12: Ausbeute an B}örkman-\A%a\n aus 20 g Material zu verschiedenen Zeit¬
punkten der Verrottung.
Rottezeit
in
Tagen
Vorextraktion
mit
Dioxanlöslich.
Material
in g
Björkman
in g
-Ligninin °/o
d. gelöstenMaterials
70
70
180
410
Alkohol-Benzol
Alkohol-Benzol
u. heiss. Wasser
Alkohol-Benzol
Alkohol-Benzol
2,1670
0,2238
8,8531
3,5352
0,1839
0,0987
0,6504
0,3892
8,4
44,1
7,3
11,0
Aus Tabelle 12 ersieht man, dass durch die Vorextraktion mit heissem
Wasser die Ausbeute an Björkman-Lignin um nahezu die Hälfte absinkt.
Ein beträchtlicher Teil des Lignins geht deshalb unter den Bedingungen der
Heisswasserextraktion in Lösung. Dieser Tatsache dürfte im Hinblick auf
die Bestimmung der Huminsäuren in den Heisswasserextrakten vermehrte
Bedeutung zukommen. Die gleiche Tabelle zeigt zudem, dass die maximale
Ausbeute an Björkman-Lignin nach einer Rottedauer von 180 Tagen er¬
reicht wird.
Die aus den verrotteten Proben extrahierten Björkman-Lignine verhiel¬
ten sich im Vergleich zu Björkman-Lignin aus frischem Stroh zum Teil
recht unterschiedlich. So fiel beim Eintragen der eingeengten Dioxanlösungin Wasser auch bei Zugabe einer bedeutenden Menge Na2SOé oft nur ein
sehr geringer Teil aus. Dieser durch Zusatz von Na2SOé ausgeflockte Teil
entspricht definitionsgemäss dem Björkman-Lignin. Durch Zusatz von HCl
zur überstehenden Flüssigkeit konnte nochmals eine weitere Ausfällung er¬
reicht werden. Diese zweite Fraktion, die stark oxydierte Ligninkomplexeenthalten muss, wird im folgenden als «Dioxanhuminsäuren» bezeichnet.
Nach dem Ansäuern mit HCl blieb eine tief gelb gefärbte Lösung zurück,die definitionsgemäss der Fulvosäurefraktion entspricht.
Die ausgefällten Björkman-Lignine der verrotteten Proben wurden nach
vorherigem Abzentrifugieren in einer geringen Menge 90 %>igem Dioxan
34

gelöst und in dünnem Strahl unter Umrühren in absol. Äther eingegossen.Dabei konnte keine richtige Ausflockung mehr erreicht werden, sondern auf
dem Boden des Kolbens blieb eine schmierige Masse zurück, die bei längeremStehenlassen an der Luft nachdunkelte und verharzte. Die Lignine wurden
deshalb nach dem Ausfällen der eingeengten Dioxanlösungen in Wasser
nach dem Zentrifugieren mit Benzol und Petroläther gewaschen und dann
durch Gefriertrocknung im Hochvakuum getrocknet. Die so erhaltenen Lig¬nine waren orangebraun gefärbt, etwas dunkler als die aus frischem Material
extrahierten Björkman-Lignine.Die «Dioxanhuminsäuren» wurden nach dem Ausfällen mit HCl (pH 1)
in der Zentrifuge kurz mit destilliertem Wasser gewaschen und hernach wie
die Björkman-Lignine getrocknet. Die Farbe dieser «Dioxanhuminsäuren»
war ebenfalls orangebraun.
343. Isolierung von Thioglykolsäure-Lignin nach Holmberg117
Ein weiteres Lösungsmittel, das eine relativ schonende Extraktion gestattet,ist die erstmals von Holmberg angewandte Thioglykolsäure. Da die Thio-
glykolsäure mit dem Lignin sehr schnell reagiert, werden die reaktionsfähi¬
gen Gruppen geschützt und das Auftreten von sekundären Reaktionen ver¬
hindert. Zum Vergleich mit den bisher isolierten Ligninen wurden deshalb
einige Präparate nach dieser Methode hergestellt.
Ausfährung:
10 g mit Alkohol-Benzol 1:1 und heissem Wasser vorextrahiertes Weizenstrohmehlwerden mit 6 g 80 Voiger Thioglykolsäure in 50 ml 2 n HCl während 4 Stundenauf dem Wasserbad digeriert, ab und zu umgerührt, nach dem Erkalten durcheinen Büchnertrichter filtriert, mit 100 ml Wasser gewaschen und im Trocken¬
schrank bei 45° C getrocknet. Das durch die Thioglykolsäure-Salzsäure-Mischungnicht gelöste Material wird nun während 48 Sid. bei Zimmertemperatur mit 100 mlreinem Alkohol behandelt, dann durch einen Büchnertrichter abgesaugt, mit dem
gleichen Volumen Alkohol gewaschen und an der Luft getrocknet. Das Alkohol-filtrat wird bei schwach erhöhter Temperatur eingedampft, wobei als Rückstandein erstes Rohprodukt an Lignothioglykolsäure = Li-thj erhalten wird.
Der getrocknete Rückstand nach der Alkoholextraktion wird nun während24 Stunden mit 100 ml 0,5 n NaOH behandelt. Dieser Prozess wurde im Gegensatzzu den Vorschriften von Holmberg in Stickstoff-Atmosphäre durchgeführt, um
eine Oxydation zu verhindern. Hierauf wird abgesaugt und mit 100 ml H2O
gewaschen. Das braungefärbte Laugenfiltrat wird mit 15 ml 5n HCl angesäuert,wobei ein käsiger Niederschlag entsteht, der über Nacht stehen gelassen wird. Die
ganze Mischung wird am anderen Tag während 15 Min. auf dem Wasserbad
erwärmt, wodurch die Fällung in eine halbfeste, teigige Masse übergeht. Diese
wird beim Erkalten spröde und harzig. Dann wird sie unter Wasser pulverisiert,abfiltriert, mit H2O gewaschen, getrocknet und nach dem Trocknen im Mörser
fein gerieben. Es entsteht ein braunes Pulver = Li-thjj.
35

Die Ausbeute aus frischem Stroh betrug für Li—thj = 29 %> und für
Li—thn = 52,6 °/o. Durch diese beiden Behandlungen wurden also rund
82 V« des Lignins herausgelöst. Auf eine zweite Behandlung mit Lauge wurde
verzichtet.
Analog wurde aus 10 g extraktfreiem Stroh, das 180 Tage dem Verrot-
tungsprozess ausgesetzt gewesen war, 362 mg eines Rohproduktes an Li—
thj erhalten = 11,1 V» des Ligninanteils.
Dieses Präparat, wie auch Li—tht aus frischem Stroh, wurde durch mehr¬
maliges, abwechselndes Ausfällen des in Dioxan gelösten Lignins in H2O
und Äther gereinigt und bei 60° C über P2O5 getrocknet.
344. Isolierung von Schwefelsäurelignin
Bei der Herstellung der Schwefelsäurelignine zu analytischen Zwecken
wurde das feingemahlene Strohmehl im Gegensatz zu den früheren Bestim¬
mungen (siehe S. 13), bei denen mit Äther extrahiert wurde, mit Alkohol-
Benzol 1:1 während 40 Std. extrahiert. Die anschliessende Extraktion mit
heissem Wasser wurde nicht abgeändert.
Das mit heissem Wasser behandelte Material wurde 15 Min. bei 3000 T/Min
zentrifugiert, einmal im Zentrifugenglas mit dest. Wasser, dann auf einem Filter¬
tiegel Gi mit warmem Wasser und schliesslich mit Äther gewaschen, um ein bes¬
seres Trocknen zu erreichen. Beim Trocknen ist darauf zu achten, dass das Material
während der Trocknung im Trockenschrank zuerst nicht zu stark erhitzt (55° C)und dabei ständig zerteilt wird, da sonst steinharte Klumpen entstehen, die im
Mörser nur mit Mühe zerkleinert werden können. Dieses so extrahierte Material
wird nun wie üblich der Schwefelsäurebehandlung unterworfen mit dem Unter¬
schied, dass das Lignin nach der Säurehydrolyse nicht durch Goochtiegel filtriert,sondern zentrifugiert wird. Der Niederschlag wird im Zentrifugenglas solangegewaschen, bis das Waschwasser neutral auf Dimethylgelb ist. Dann wird im Trok-
kenschrank bei 60° C vorgetrocknet, hernach im Achatmörser fein zerkleinert und
anschliessend in der Trockenpistole im Vakuum bei 90" C über P2O5 getrocknet.
Da die durch die Behandlung mit Schwefelsäure erhaltenen Lignine meist
3—50/0 Schwefel enthalten (11 d), wird zu dessen Entfernung oft eine
Nachhydrolyse mit 0,5 °/oiger Salzsäure während 11 Stunden empfohlen.Um durch den Kontakt mit heisser Salzsäure die Schwefelsäurelignine nicht
zu stark zu denaturieren, wurde im vorliegenden Falle die Bestimmung des
Schwefelgehaltes* vorgezogen.
* Durchgeführt vom Chemischen Untersuchungslaboratorium der Forschungsan¬stalt für Landwirtschaft, Braunschweig-Völkenrode.
36

345. Isolierung von Huminsäuren
Durch die Heisswasserbehandlung der verrotteten Strohproben wurden be¬
deutende Mengen wasserlöslicher Stoffe extrahiert, die beim Ansäuern mit
HCl ausflockten. Da diese säurefällbaren Stoffe mit Wasser herausgelöstwerden, fallen sie definitionsgemäss nicht mehr unter den Begriff Lignin,weshalb sie im folgenden als «Huminsäuren» bezeichnet werden. Wahr¬
scheinlich enthält diese Fraktion hauptsächlich oxydierte Ligninkomplexe.Im Vergleich zu den im Abschnitt 3422 beschriebenen «Dioxanhumin¬
säuren» wurden auch die schwarzbraunen Heisswasserextrakte aus den ver¬
schiedenen Strohproben nach vorherigem Zentrifugieren mit HCl auf pH 1
angesäuert, zentrifugiert, mit dest. Wasser gewaschen und hierauf elektro-
dialysiert, bis keine Cl-Ionen mehr nachgewiesen werden konnten. Die
Trocknung der dialysierten Präparate erfolgte unter schonendem Erwär¬
men auf dem Wasserbad, wobei schwarze, glänzende, amorphe Plättchen,
analog den aus Boden isolierten Huminsäuren erhalten wurden- Durch Ge¬
friertrocknung im Hochvakuum erhält man ein braunes Pulver, etwas dunk¬
ler als bei den «Dioxanhuminsäuren». Beim Eintrocknen auf dem Wasserbad
scheint demnach ein Oxydations- und Kondensationsprozess stattgefundenzu haben.
Da zur Gewinnung von Huminsäuren aus dem Boden oft Natronlaugeverwendet wird, sollte geprüft werden, ob mit 0,5 Voiger NaOH eine wesent¬
lich grössere Menge an Huminsäuren erfasst würde als durch die Heisswasser¬
behandlung. Zu diesem Zwecke wurde eine bestimmte Menge Stroh (18,6 g),das 70 Tage dem Verrottungsprozess ausgesetzt gewesen war, während 20
Stunden bei Zimmertemperatur mit 1 Liter 0,5 Voiger NaOH behandelt.
Die ungelösten Bestandteile wurden abzentrifugiert, die überstehende Lö¬
sung mit HCl auf pH 1 angesäuert, der Niederschlag nach dem Zentrifu¬
gieren mit dest. Wasser gut ausgewaschen und gefriergetrocknet. Die beiden
folgenden Tabellen 13 und 14 geben einen Überblick über die aus den ver¬
schiedenen Rottestufen erhaltenen Mengen an Kalt- und Heisswasserex-
traktstoffen und die .darin enthaltenen Anteile an Fulvo- und Huminsäuren.
Die kalt- und heisswasserlöslichen Extraktstoffe wurden an getrennten
Proben bestimmt.
37

Tabelle 13: Kalt- und heisswasserlösliche Extraktstoffe zu verschiedenen Zeit¬
punkten der Verrottung. Prozentualer Anteil säurefällbarer, organischer Substanz
in den Heisswasserextrakten (= Huminsäurefraktion).
Rottezeit Tr. Gew.
in in
Tagen g
Kaltwasserlösl.
Anteil (anorg. + org.)
abs. i. °/o des
in g Tr. Gew.
Heisswasserlösl.*
Anteil (aschefrei)
abs. i °/o des
in g Tr. Gew.
Vo säurefällbare
org. Substanz
(Heisswasser-
extrakt) ++
0 282,5 20,1 7,1 5,9 2,1 3,5
70 148,5 23,7 16,0 15,0 10,1 33,2
180 68,9 20,8 30,2 10,6 15,4 38,5
260 73,4 18,3 24,9 11,5 15,7 35,7
340 71,1 15,2 21,4 10,2 14,3 40,3
410 70,5 10,6 15,1 10,7 15,2 37,5
410+N 72,1 13,2 18,3 12,8 17,7 31,9
* Vorextraktion mit Alkohol-Benzol 1:1.
++—"h der säurefällbaren org. Subst. bezogen auf den org. Anteil des Heisswasser-extraktes.
Aus der Tabelle 13 geht hervor, dass die absolute Menge an kaltwasser¬
löslichen Stoffen nach einem geringen Anstieg zu Beginn der Rottezeit nach¬
her stetig abnimmt. Anfänglich wird durch die Kaltwasserextraktion nahezu
die gleiche Menge erfasst wie durch die Heisswasserextraktion (vergl. Tab.
6), während mit zunehmender Rottezeit die Kaltwasserlöslichkeit ziemlich
stark abnimmt. Beim heisswasserlöslichen organischen Anteil ist gleich zu
Beginn des Rotteprozesses ein starker Anstieg zu beobachten, während mit
zunehmender Verrottung auch hier eine Abnahme zu verzeichnen ist. Nur
in der Serie mit nachträglich zugesetzten Nährsalzen ist eine Ausnahme von
dieser Tendenz festzustellen. Der säurefällbare organische Anteil in den
Heisswasserextrakten bleibt dagegen während der ganzen Rotteperiodemehr oder weniger konstant. Interessant ist in diesem Zusammenhang, dass
auch aus frischem Stroh in geringer Ausbeute ein heisswasserlöslicher, säure¬
fällbarer Anteil extrahiert werden konnte, dessen IR-Spektrum grosse Ähn¬
lichkeit mit der Huminsäurefraktion der verrotteten Proben aufwies (sieheAbschnitt. 369). Wahrscheinlich handelt es sich dabei um einen ähnlichen
Polysaccharid-Ligninkomplex, wie ihn Kratzl und Silbemagellis bei der
thermischen Behandlung von Fichtenholz mit Wasser erhielten; der Lignin-anteil liess sich dabei durch Oxydation mit Nitrobenzol-Alkali in guterAusbeute zu Vanillin abbauen.
38

Tabelle 14: Ausbeute an Fulvosäuren ( = mit Säuren nicht fällbare organischeSubstanz) und an Huminsäuren zu verschiedenen Zeitpunkten der Verrottung(Werte für Fulvo- und Huminsäuren auf aschefreie Substanz umgerechnet).
Rottezeit Organ.Subst.
in g
Extraktions¬
Fulvosäuren
(aschefrei)
Huminsäuren
(aschefrei)in
Tagenmittel i. °/o der
org. S.
abs.
in g
i. 9/o der
org. S.
abs.
in g
70 124,3Wasser 70° C 8,0 10,0 4,0 5,0
0,5 °/oige NaOH 10,6 13,2 8,8 11,0
180 50,4 Wasser 70° C 12,8 6,5 8,1 4,1
260 49,2 Wasser 70° C 15,0 7,4 8,3 4,1
340 49,3 Wasser 70° C 12,3 6,1 8,3 4,1
410 47,0 Wasser 70° C 14,2 6,7 8,5 4,0
410+N 50,4 Wasser 70° C 17,3 8,7 8,1 4,1
Aus Tabelle 14 ersieht man, dass im Gegensatz zur Heisswasserextrak-
tion durch die Behandlung mit 0,5 Voiger NaOH mehr als die doppelteMenge an Huminsäuren erfasst wird. Die Ausbeute an Fulvosäuren wird
dagegen nur unbedeutend erhöht. Diese stark erhöhte Ausbeute an Humin¬
säuren ist sehr wahrscheinlich der Tatsache zuzuschreiben, dass durch die
Behandlung mit Natronlauge ausser Hemizellulosen in vermehrtem Masse
weniger stark oxydiertes Lignin erfasst wurde, worauf auch die erhöhten
Methoxylwerte und das IR-Spektrum dieser Huminsäure hindeuten (sieheTab. 17). Ferner ersieht man, dass sowohl bei der Fulvo- wie bei der Hu-
minsäurefraktion im Verlaufe des Rotteprozesses keine nennenswerten Än¬
derungen mehr eingetreten sind. Die etwas höhere Ausbeute an Fulvosäuren
in der Serie mit zusätzlicher Nährsalzbeigabe dürfte zum Teil dem Wieder¬
aufleben der Mikroorganismentätigkeit zuzuschreiben sein. Die erhöhten
Furfurolwerte dieser Serie deuten darauf hin, dass durch die Extraktion mit
heissem Wasser in vermehrtem Masse mikrobielle Polysaccharide erfasst
wurden. Es ist zudem interessant, dass der Anteil der Fulvosäurefraktion
auch bei verschiedener Extraktionsart stets grösser ist als der Anteil der
entsprechenden Huminsäurefraktion. Dieser Tatsache dürfte im Hinblick
auf die pflanzenphysiologische Wirksamkeit dieser wasserlöslichen Rotte¬
produkte vermehrte Bedeutung zukommen.
35. Enzymatische Oxydation von Ligninen
Im Abschnitt über die biologische Zersetzung des Lignins ist gezeigt worden,welch grosse Bedeutung den verschiedenen Enzymen beim Prozess der Hu-
39

musbildung zukommt. Bei der enzymatischen Oxydation von Lignin schei¬
nen vor allem die Polyphenoloxydasen eine bedeutende Rolle zu spielen.Es sollte deshalb untersucht werden, bis zu welchem Grade solche Enzymedie Übertragung von Sauerstoff auf Lignin aus frischem und verrottetem
Stroh zu katalysieren vermögen.
351. Isolierung des Enzyms
Die Darstellung der Polyphenoloxydasen erfolgte aus Abfällen des Speise¬champignons (Psalliota campestris) nach einer Methode, die in Anlehnungan Arbeiten von Freudenberg und Richtzenhain119 am hiesigen Institut
ausgearbeitet worden ist120. Diese Methode deckt sich im Prinzip mit den
kürzlich von Freudenberg121 veröffentlichten Angaben zur Gewinnung von
Champignonoxydase.
Ausführung:
Die Champignonabfälle, bestehend aus 1—2 Tage alten Stengelteilen des Frucht¬
körpers und der Rhizomorphen, werden in einer Fleischhackmaschine zerkleinert
und in einer hydraulischen Presse bei 400 Atü abgepresst. Der braunrot gefärbtePilzsaft wird im Eiskochsalzgemisch auf 3° C abgekühlt, dann mit der gleichenMenge vorgekühltem Methylalkohol unter Umrühren versetzt und die gefälltenAnteile abzentrifugiert. Die Flockung hat eine dunkelbraune Färbung. Der Nie¬
derschlag wird mit Methanol und Äther nachgewaschen und im Vakuumexsikkator
über CaCl2 bei Zimmertemperatur getrocknet.Zur Gewinnung einer zweiten Fraktion wird die zentrifugierte und gekühlte
Lösung nochmals mit der gleichen Menge Methylalkohol versetzt, worauf man
wie bei der ersten Fällung verfährt.
Die Ausbeute aus 2 kg Champignons betrug in der ersten Fraktion 2,49 g,
aus der zweiten Fraktion 1,24 g. Beide Enzymfällungen wurden im Vakuum-
Exsikkator über Blaugel bei einer Temperatur von 3° C aufbewahrt. Wäh¬
rend das Enzym in wässriger Lösung auch in der Kälte seine Aktivität in
kurzer Zeit verliert121, ist es als Trockenpräparat über mehrere Monate halt¬
bar, allerdings auch unter langsamer Einbusse an Aktivität. Bei den beschrie¬
benen Versuchen zur enzymatischen Oxydation von Ligninen ist das Trok-
kenpulver der zweiten Fraktion der Champignonoxydase verwendet wor¬
den, da diese eine höhere Aktivität aufwies.
Auf einen genauen Test der Substratspezifität wurde verzichtet, weil
Champignonoxydase-Präparate von verschiedenen Autoren119 la2 auf ihre
Eigenschaften geprüft worden sind. Danach muss angenommen werden, dass
gezüchtete Champignons in den Myzelen Laccase, in den Rhizomorphenund Fruchtkörperteilen Tyrosinase und Brenzcatechinoxydase enthalten.
40

352. Aktivitätsbestimmung der Champignonoxydase
Die bei der Oxydation mit einer Phenoloxydase während einer bestimmten
Zeit verbrauchte Menge Sauerstoff ist ein Mass für die Aktivität des En¬
zyms. Nach Freudenberg121 ist eine Enzymeinheit diejenige Menge Enzym,die bei pH 5,5 und 25° C in einer wässrigen Lösung von 4 cm3 in 40 Min.
auf 3 mg Dihydro-ferulasäure 120 mm3 Sauerstoff überträgt.Als Grundlage für die eigenen Versuche zur Bestimmung von Phenol-
oxydase-Aktivitäten ist die von Steinmetz120 ausgearbeitete Purpurogallin-methode benutzt worden. Das Prinzip ist dabei folgendes:Das Oxydationsenzym katalysiert die Bildung einer seiner Aktivität ent¬
sprechenden Menge Purpurogallin aus Pyrogallol. Die Menge des gebildetenFarbstoffes wird kolorimetrisch bestimmt. Die Aktivität der Champignon¬oxydase wird charakterisiert durch die Purpurogallinzahl = PN, die folgen-dermassen definiert ist: 1 PN = mg Purpurogallin pro mg Trockengewichtdes Enzympräparates gebildet innerhalb von 5 Minuten bei 20° C, pH 6,3und einem Sauerstoffstrom von 400 ccm/Min.
Ausführung:
Zur Bestimmung der Purpurogallin¬zahl hat sich das in Abbildung 2 dar¬
gestellte Glasgefäss bewährt. In das
etwa 20 cm lange, 200 ml fassende
zylindrische Gefäss K mit eingeschlif¬fenem Glaskopf pipettiert man 20 ml
1/15 mol Phosphatpuffer von pH 8,0.Dies kann durch das EntlüftungsrohrM geschehen. Die Apparatur wird bis
zum Kolbenhals in einen Thermosta¬
ten gehängt. Dann stellt man den
Sauerstoffstrom, der vorher durch ein
Strömungsmanometer fliesst, auf etwa
400 ccm pro Minute ein. Alsdann wer¬
den durch das Entlüftungsrohr M
20 ml einer schwefelsauren 0,25 °/oigenPyrogallol-Lösung (5 ml 0,2n Schwe¬
felsäure/100 ml 0.25 %ige Pyrogallol-Lösung) hinzugefügt. Das Reaktions-
gemisch stellt sich darauf auf pH 6,3ein. Nun lässt man aus der einen
Bürette eine bestimmte Menge Enzym¬lösung zulaufen und bestimmt die Zeit
mit der Stoppuhr. Nach 5 Minuten
wird die Reaktion durch Zugabe von
2,5 ml einer 2n Schwefelsäure aus einer
zweiten Bürette unterbrochen. Das Abb. 2: Apparatur zur Bestimmung der
Reaktionsgemisch wird in einen Schei- Purpurogallinzahlendetrichter gebracht, das Gefäss K
41

zweimal mit 10 ml dest. Wasser gewaschen und die vereinigte wässrige Lösung drei¬
mal mit 25 ml Äther ausgeschüttelt. Die Ätherauszüge werden über wasserfreiem
Natriumsulfat getrocknet und dann in einen 100 ml Messkolben filtriert. Das Na¬
triumsulfat wird mit Äther nachgewaschen und damit bis zur Marke aufgefüllt.Die gebildete Purpurogallinmenge ergibt sich aus der Extinktion im Eppendorf-Kolorimeter bei 10 mm Schichtdicke und dem Filter Hg 366 mm mit Hilfe einer
Eichkurve.
Beim Blindversuch mit hitzeinaktiviertem Enzym wird wie beim Haupt¬versuch verfahren. Die wahre Enzymaktivität ergibt sich aus der Differenz
der gefundenen Purpurogallinwerte.Die Aktivität der ersten Fraktion nach 3-monatiger Lagerung als Trok-
kenpräparat bei 3° C betrug 0,65 PN/mg Gesamttrockengewicht, diejenigeder zweiten Fraktion noch 2,70 PN. Verglichen mit den Anfangsaktivitätenvon 2,17 PN resp. 8,28 PN/mg hatte das Enzym demnach in dieser Zeit¬
spanne rund % seiner ursprünglichen Aktivität verloren.
353. Enzymatische Oxydation von Björkman-Lignin
Auf einer Schüttelapparatur werden zwei 1 Liter Dreihalsrundkolben be¬
festigt. Auf dem mittleren Hals wird mittels eines durchbohrten Gummi¬
stopfens ein Thermometer befestigt. In beiden Kolben werden nacheinander
430 ml 1/15 mol Phosphatpuffer pH 6,5 und 20 ml einer Ligninsuspension(1 g Lignin) zugegeben. Zu deren Herstellung werden 2 g Lignin in einer
geringen Menge wässrigem Aceton (90 °/oig) gelöst und unter starkem Um¬
rühren in 35 ml Wasser eingetragen. Das Aceton wird unter vermindertem
Druck abgedampft, worauf eine milchig-trübe Ligninsuspension zurück¬
bleibt. In dem einen Kolben werden zur Feststellung des Blindwertes statt
der Fermentlösung lediglich 50 ml dest. Wasser und in den anderen 50 ml
zentrifugierte, wässrige Champignonoxydaselösung (250 mg der feinpulveri¬sierten zweiten Fraktion) zugegeben. Die Temperatur der Gesamtlösungsoll 20° C betragen. Die Leeratmung des Ferments wurde in einem getrenn¬ten Versuch bestimmt.
Der Raum oberhalb der Flüssigkeit in den Kolben wird mit kräftigemSauerstoffstrom aus einer Flasche gefüllt. Die Kolben werden darauf ver¬
schlossen. Sie sind durch einen Gummischlauch mit einem mit Sauerstoff
gefüllten, graduierten Gasometergefäss verbunden. Es ist wichtig, die Tem¬
peratur des Gemisches auf 1/10" C zu bestimmen und bei den Ablesungenkonstant zu halten. Alsdann werden die Kolben mit einer Frequenz von
100/min. geschüttelt. In bestimmten Zeitintervallen wird die Sauerstoffauf¬
nahme abgelesen.Nach einer Reaktionszeit von 15 Stunden wurden von 1 g Björkman-
Lignin aus frischem Weizenstroh nach Abzug eines Blindwertes von 0,6 ml
insgesamt 2,0 ml und nach 2 Tagen 5 ml Sauerstoff aufgenommen. Bei einem
42

zweiten Versuch, in dem das Lignin in einer geringen Menge n-Oktylalko-hol unter Zugabe von wenig Äthanol suspendiert wurde, konnte keine
wesentlich höhere Sauerstoffaufnahme festgestellt werden. Dieses oxydierteLignin wurde nachher wieder aufgearbeitet und zu infrarotspektrographi-schen Studien verwendet (siehe hierzu Abschnitt 369).
354. Enzymatische Oxydation von Ligninen in der
Warburg-Apparatur
Zur genaueren Überprüfung der vorher erhaltenen Ergebnisse mit Björkman-Lignin aus frischem Weizenstroh ist auch ein Versuch in der Warbttrg-Apparatur durchgeführt worden. Da es in diesem Zusammenhang von In¬
teresse war, ob Lignin durch den Rotteprozess dem oxydativen Angriff der
Champignonoxydase zugänglicher geworden war, wurde ein Ligninpräpa-rat (Björkman-IAgmn) der ältesten Rottestufe (410 + N) in die Untersu¬
chungen mit einbezogen.Die Suspension des frischen Lignins wurde analog wie vorher zubereitet.
Vom verrotteten Lignin konnte ohne Mühe eine wässrige Suspension herge¬stellt werden. Die Oxydation beider Lignine erfolgte im Doppel. Ferner
wurde in je einer Blindprobe die Leeratmung des Enzyms und der Substrate
bestimmt.
Versuchsbedingungen :
Die Temperatur betrug 27° C. In den Hauptraum des Warburg-Gefässes wurden
1,9 ml Ligninsuspension, enthaltend 30 mg Lignin und Phosphatpufferlösung nach
Sörensen (m/15, pH 6,5), gegeben,
mi
X»
,in den Innenraum 0,2 ml 10 °/oige
ir[^02 KOH zur Absorption von even¬
tuell gebildetem CO2, und in den
Seitenarm 0,4 ml wässrige Enzym¬lösung (10 mg der feinpulverisier¬ten zweiten Fraktion).
/
/,/
50t /
//
//.
/
Abb. 3: Sauerstoff-Aufnahme von
Ligninen in der
-
BJORKMAN-Lignm aus tnschem We.zenstroh W'ar&arg-Apparatur.
-BJORKMAN-Lgnin 410 Tage verrottet
43

Die in Abbildung 3 dargestellten Ergebnisse stellen den Mittelwert aus
den beiden Parallelbestimungen nach Abzug der Blindwerte dar. Durch
Verdoppelung der Enzymmenge beim frischen Lignin konnte noch eine ge¬
ringe Erhöhung der Sauerstoffaufnahme erreicht werden.
Aus dieser Abbildung kann man deutlich ersehen, dass die durch Poly¬phenoloxydasen katalysierte Sauerstoffaufnahme der Lignine mit zuneh¬
mendem Zersetzungsgrad stark ansteigt.
36. Charakterisierung der Rotteprodukte im Vergleichzum Ausgangsmaterial
Die in diesem Abschnitt dargelegten Untersuchungen dienen zur näheren
Charakterisierung der im Verlaufe des Rotteprozesses eingetretenen Verän¬
derungen der als Lignin und Huminsäuren definierten Produkte durch che¬
mische und physikalische Methoden-
361. Elementarzusammensetzung der isolierten Lignineund Huminsäuren
Die in Tabelle 15 angeführten Gehalte an Schwefel sind zum grössten Teil
auf die Einführung von Sulfo-Gruppen in das Ligninmolekül durch Schwe¬
felsäurebehandlung zurückzuführen.
Tabelle IS: Elementarzusammensetzung der zu verschiedenen Zeitpunkten iso¬lierten Schwefelsäurelignine (Prozente bezogen auf aschefreie Substanz).
Rottezeit
in TagenC°/o H°/o 0»/o N°/o S°/o OCH3%> Asche %>
0 58,59 5,60 31,37 0,54 3,9 15,33 14,2270 58,78 5,93 29,48 2,01 3,8 11,14 26,73
180 58,67 5,91 29,15 2,37 3,9 9,06 40,62260 58,00 5,83 29,93 3,14 3,9 7,84 37,40340 54,97 5,96 32,09 3,08 3,9 8,57 38,64410 54,29 5,86 32,88 2,97 4,0 7,32 32,71410+N 56,71 5,58 30,66 2,85 4,2 7,16 32,35
Aus Tabelle 15 kann man ersehen, dass die Kohlenstoffwerte nach 180
Tagen Rottedauer mit Ausnahme der Serie 410+N ziemlich stark abneh¬
men, während die Wasserstoffwerte nur innerhalb enger Grenzen schwan¬ken. Die Sauerstoffwerte zeigen nach anfänglich abnehmender Tendenz mit
44

steigender Rottezeit wieder eine leichte Zunahme. Auffallend stark ist die
Zunahme des Stickstoffgehaltes, der gegen Ende der Rotte wieder leicht
abnimmt. Interessant ist, dass trotz zusätzlicher Stickstoffgabe in der Serie
410+N die N-Werte tiefer liegen. Während die Schwefelwerte mehr oder
weniger konstant bleiben, ist bei den Methoxylwerten eine starke Abnahme
zu verzeichnen. Die hohen Aschengehalte dürften vor allem der starken
Anreicherung an Kieselsäure (S1O2) zuzuschreiben sein.
Tabelle 16: Elementarzusammensetzung von Nativlignin nach Brauns und von
Björkman-Ligninen (= Bj.-Lig.) isoliert zu verschiedenen Zeitpunkten der Ver¬
rottung (Prozente bezogen auf aschefreie Substanz).
Lignine + Zeit i. T. C/o H %> OVo N»/o OCH3 °/o Asche °/o
Nativlig. n. Brauns 60,87 6,11 32,58 0,44 14,29 0,42
Bj.-Lig. frisch 60,68 5,79 33,11 0,42 16,76 1,11
Bj.-Lig. 70 Tage* 59,75 7,35 30,93 1,97 10,84 21,91
Bj.-Lig. 180 Tage 58,81 6,56 31,27 3,36 8,19 9,32
Bj.-Lig. 340 Tage* 57,37 6,95 31,14 4,54 7,50 49,36
Bj.-Lig. 410+N 64,67 7,87 24,99 2,47 7,01 4,98
* bei höherer Temperatur extrahiert.
Aus Tabelle 16 geht hervor, dass in der Elementarzusammensetzung der
Björkman-Lignint prinzipiell die gleiche Tendenz festgestellt werden kann
wie bei den Schwefelsäureligninen. Gegenüber den letzteren zeigen jedochdie Björkman-Lignine eine Zunahme der Wasserstoffwerte und eine ste¬
tige Abnahme der Sauerstoffwerte. Interessant ist ferner der schon von
Björkman114 beobachtete höhere Methoxylgehalt des Björkman-Lignlns im
Vergleich zu Branns-Llgmn.Aus Tabelle 17 (siehe hierzu Seite 46) kann man ersehen, dass die Humin-
säuren gegenüber den Ligninen in der Elementarzusammensetzung doch be¬
trächtliche Unterschiede aufweisen. So sind zum Beispiel die Kohlenstoff-
und Wasserstoffwerte niedriger als bei den schonend isolierten Ligninen.Die Rottedauer scheint jedoch auf die Zusammensetzung der entstandenen
Huminsäuren einen ähnlichen Einfluss auszuüben wie auf die der Lignine.Die Kohlenstoff- und Methoxylwerte nehmen dabei ab und die Sauerstoff-
und Stickstoffwerte zu. Wie bei den Schwefelsäureligninen nimmt der N-
Gehalt der Huminsäuren mit 340 Rottetagen wieder ab. Der hohe Methoxyl¬gehalt der mit Natronlauge isolierten Huminsäure dürfte auf die Heraus¬
lösung weniger stark abgebauter Ligninkomplexe zurückzuführen sein.
45

Tabelle 17: Elementarzusammensetzung der mit heissem Wasser extrahierten Hu¬
minsäuren (Prozente bezogen auf aschefreie Substanz).
Rottezeit
in TagenC°/o H «/o OVo N°/o OCH3V0 Asche Vo
70 55,02 5,26 36,97 2,75 8,84 2,16
70* 56,20 5,59 36,30 1,91 10,80 3,19
180 55,65 5,22 35,75 3,38 8,47 2,10
260 53,74 5,19 37,63 3,44 7,66 2,89
340 52,92 5,21 38,93 2,94 8,31 3,87
410+N 53,55 5,11 38,57 2,77 6,80 6,80
* Isoliert mit 0,5 »/oiger NaOH.
362. Hydrolyse von Stroh, Ligninen und Huminsäuren
Um zu untersuchen, ob die in den Ligninen und Huminsäuren beobachtete
Anreicherung an Stickstoff auf Proteine oder Proteinspaltstücke zurückzu¬
führen ist, wurden die verschiedenen Präparate einer Hydrolyse mit 6n
HCl unterworfen. Zu diesem Zweck wurden die Präparate (20 ml 6n HCl/
g Trockensubstanz) auf dem Sandbad während 16 Stunden am Rückfluss
auf 116° C erhitzt. Dabei war nach der Aufarbeitung teilweise eine bedeu¬
tende Abnahme an Trockengewicht zu beobachten.
Zur Feststellung des Gewichtsverlustes wurde der Hydrolysenrückstanddurch einen Filtertiegel 1 G 4 filtriert, bei 105° C getrocknet und gewogen.
Von diesen hydrolysierten Strohproben, Ligninen und Huminsäuren wurde
in einigen Fällen die Elementarzusammensetzung bestimmt.
Zur Entfernung der in den Filtraten enthaltenen Salzsäure wurden diese
im Vakuum dreimal fast bis zur Trockene eingedampft, hernach mit dest.
Wasser auf 10 ml aufgefüllt und auf a-Aminostickstoff nach der titrimetri-
schen Ninhydrinmethode von Van Slyke, Mac Fadyen und Hamilton123
analysiert. Zur Feststellung der durch die Hydrolyse freigewordenen Menge
an Aminosäuren wurde diese Methode bevorzugt, weil die quantitative,papierchromatographische Methode für Serienuntersuchungen zu langwierigund nicht genügend genau ist.
Um ein Bild über die Anzahl Aminosäuren in den Björkman-Ligainender verschiedenen Rottestufen zu erhalten, wurden die Hydrolysate einiger
Präparate papierchromatographisch untersucht. Die Auftrennung erfolgtemit n-Butanol-Eisessig-Wasser (70:20:10) nach dem aufsteigenden Verfah¬
ren auf Chromatographiepapier Schleicher und Schüll 2043 b. Nach dem
46

Durchlaufen des Lösungsmittels wurden die Chromatogramme getrocknetund hernach auf diese Weise noch dreimal in der gleichen Richtung laufen
gelassen. Die Entwicklung geschah durch Besprühen der Chromatogrammemit einer 0,4 %>igen Lösung von Ninhydrin in Methanol. Bei iJr^ww-Ligninkonnten dabei 4, bei Björkman-Lignm aus frischem Stroh 5 Aminosäuren
festgestellt werden. Nach 70 Tagen Rottedauer wies Björkman-Lignm 13
Aminosäuren auf. Diese Zahl veränderte sich auch nach 180 Tagen Rotte
nicht. Nach 410 Tagen wurden noch 12 Aminosäuren festgestellt.Ob die in den schonend gewonnenen Ligninpräparaten aus frischem
Stroh festgestellten Aminosäuren als Verunreinigungen anzusehen sind oder
tatsächlich mit dem Ligninmolekül verknüpft sind, kann nicht sicher ent¬
schieden werden. Da jedoch der Stickstoffgehalt dieser schonend isolierten
Lignine auch nach mehrmaligem Reinigen durch Auflösen in Dioxan und
Ausfällen in Wasser und Äther bei allen Präparaten immer einen konstanten
Wert erreichte, muss man annehmen, dass Aminosäuren am Molekülaufbau
teilnehmen. Dass aromatische Aminosäuren (z. B. Phenylalanin u. a. m.)am Aufbau des Ligninmoleküls beteiligt sein können, zeigt die Arbeit von
Brown und Neish124:
Aus Tabelle 18 ersieht man im Gegensatz zu den nicht hydrolysiertenSchwefelsäureligninen der Tabelle 15 viel deutlicher, dass der Kohlenstoff¬
gehalt der Lignine bedeutend abgenommen hat und sich somit stark dem
Wert nähert, der bei den Huminsäuren erhalten wurde. Dagegen ist eine
bedeutende Zunahme an säureresistentem Stickstoff und an Sauerstoff fest¬
zustellen, während die Wasserstoffwerte ziemlich gleich bleiben.
Tabelle 18: Elementarzusammensetzung der Schwefelsäurelignine nach der Hydro¬lyse (Prozente, bezogen auf aschefreie Substanz).
Rottezeit
in TagenC°/o H»/o O"/. N%> Asche %>
0 63,39 5,41 30,98 0,22 14,85
70 60,15 5,68 33,20 0,97 28,55
180 60,40 5,66 32,86 1,08 43,70
260 58,86 5,61 34,15 1,38 40,15
340 56,66 5,62 36,05 1,67 42,08
410+N 59,17 5,19 33,87 1,77 35,26
Auf eine Korrektur bezüglich des Schwefelgehaltes konnte hier verzichtet
werden, da durch die Hydrolyse der Schwefel praktisch entfernt wird (11 d).
47

Tabelle 19: Elementarzusammensetzung von Björkman-Ligninen, Huminsäuren
und Strohproben, jeweils nach der Hydrolyse (Prozente, bezogen auf aschefreie
Substanz).
Substanz O/o H%» o%> N%> Asche %>
Bj.-Lig. frisch
Bj.-Lig. 70 Tage
Bj.-Lig. 180 Tage
Bj.-Lig. 410 Tage (+N)
Huminsäure 260 TageHuminsäure 340 Tage
69,35
68,45
67,36
70,89
57,53
56,70
62,36
59,78
58,12
5,88
7,45
6,37
8,18
4,58
4,56
5,33
5,30
4,91
24,59
22,97
24,90
19,71
36,94
37,78
32,05
33,63
35,68
0,18
1,13
1,37
1,22
0,95
0,96
0,26
1,29
1,29
2,80
11,28
2,57
3,97
2,89
3,98
9,45
30,38
31,99
Stroh frisch
Stroh 180 TageStroh 410 Tage (+N)
Tabelle 19 zeigt, dass in den hydrolysierten Strohproben die gleiche Ten¬
denz wie in den Ligninen und Huminsäuren festgestellt werden kann. Wäh¬
rend die Kohlenstoffwerte der hydrolysierten Huminsäuren praktisch gleichsind wie in den hydrolysierten Schwefelsäureligninen, sind die Wasserstoff-
und Stickstoffwerte geringer und die Sauerstoffwerte höher. Dies deutet
auf eine stärkere Oxydation hin und könnte damit auch die Wasserlöslich¬
keit dieser Produkte erklären. Die hohen Kohlenstoffwerte der Björkman-
Lignine dürfen wohl den durch die Hydrolyse mit 6n HCl verursachten
Kondensationsprozessen zugeschrieben werden.
Aus Tabelle 20 (siehe hierzu Seite 49) geht eindeutig hervor, dass mit
zunehmender Rottezeit der prozentuale Anteil des a-Aminostickstoffes am
Gesamtstickstoff sowohl in den Ligninen, Huminsäuren wie auch in den
Strohproben ständig abnimmt. Wenn auch eine gewisse Zerstörung von Ami¬
nosäuren unter diesen Bedingungen der Säurehydrolyse stattfinden kann107,so dürfte die mit zunehmender Rottedauer auftretende Diskrepanz zwischen
dem Gehalt an Gesamtstickstoff und a-Aminostickstoff doch einer anderen
Ursache zugeschrieben werden.
Bremner101 hat festgestellt, dass bei Zersetzung von Stroh Aminozucker-
komplexe gebildet werden. Nach Untersuchungen von Bremner und Shaw106
kann 5—10% des Gesamtstickstoffes im Boden solchen Aminozucker-
komplexen zugeschrieben werden. Diese können durch Säurehydrolyse unter
Abspaltung von Ammoniak zu einem beträchtlichen Teil zerstört werden.
Andererseits besteht auch die Möglichkeit, dass im Verlauf des Abbaupro¬zesses ein Teil der Eiweissbausteine durch Einwirkung von Decarboxylasenaus den Bakterien zu Aminen decarboxyliert und so durch die titrimetrische
Ninhydrinmethode nicht mehr erfasst werden128. Bekanntlich wird bei der
48

Zersetzung von pflanzlichem und tierischem Eiweiss eine Reihe von Dia¬
minen, wie z. B. Putrescin und Cadaverin, gebildet126. Man kann annehmen,dass die beobachtete Abnahme an a-Aminostickstoff zu einem grossen Teil
solchen Prozessen zugeschrieben werden darf.
Tabelle 20: a-Aminostickstoff in den Ligninen, Huminsäuren und Strohproben(Prozente, bezogen auf aschefreie Substanz).
Substanz
°/o*N
vor der
Hydrol.
a-Aminostickstoffim Hydrolysat
in °/o des
in»/. 8es.org.N vor der
Hydrol.
Gew. Verlust
°/o*N durch die
nach der Hydrol.
Hydrol. in °/o
H2S04-Lig. frisch 0,55 0,32 58,1 0,22 17
H2S04-Lig. 70 Tage 2,09 0,82 39,2 0,97 20
H2S04-Lig. 180 Tage 2,46 0,52 21,1 1,08 24
H2S04-Lig. 260 Tage 3,26 0,68 20,8 1,38 24
H2S04-Lig. 340 Tage 3,20 0,65 20,3 1,67 32
H2S04-Lig. 410+N 2,97 0,52 17,5 1,77 11
Huminsäure 260 Tage 3,44 0,72 20,7 0,95 46
Huminsäure 340 Tage 2,94 0,50 17,0 0,96
0,18
47
24Bj.-Lig. frisch 0,42 0,20 47,6
Bj.-Lig. 70 Tage 1,97 0,81 41,1 1,13 46
Bj.-Lig. 180 Tage 3,36 1,14 33,9 1,37 43
Bj.-Lig. 410+N 2,47 0,52 21,0
52,7
1,22
0,26
30
Stroh frisch 0,74 0,39 72
Stroh 180 Tage 3,37 0,68 20,1 1,29 65
Stroh 410+N 4,34 0,61 14,0 1,29 66
* (N in °/o der org. Substanz, bestimmt nach Dumas).
363. Alkalischmelze von Ligninen und Huminsäuren
Um zu untersuchen, wie sich der nach der Säurehydrolyse in den Ligninenund Huminsäuren verbliebene Stickstoff bei der alkalischen Hydrolyse ver¬
hält, wurden die Proben anschliessend der Alkalischmelze* nach der Me¬
thode von Breyban121 unterworfen. Diese Mikromethode zur Stickstoff¬
bestimmung in pflanzlichem Material basiert auf der Erscheinung, dass
* Diese Untersuchung wurde in dankenswerter Weise von Herrn Dr. Th. Breyhan,Chemisches Untersuchungslaboratorium der Forschungsanstalt für Landwirt¬
schaft, Braunschweig-Völkenrode, ausgeführt.
49

trockenes, stickstoffhaltiges Material bei der Schmelze mit AlkalihydroxydAmmoniak abspaltet, das in einem Stickstoffstrom übergetrieben und in
einer Vorlage mit Schwefelsäure bestimmter Konzentration absorbiert wird.
Der heterocyclisch im Indol-Imidazol- und Chinolinring festgelegte Stick¬
stoff wird dabei nicht erfasst. Phenylglycin, das als Ausgangsmaterial zur
Indigosynthese Verwendung findet, lieferte unter diesen reduktiven Bedin¬
gungen (Stickstoffatmosphäre und Eisentiegel) keine nachweisbaren Indol-
körper.Zur Bestimmung werden 30—50 mg hydrolysiertes Material in ein kleines Eisen-
kölbchen eingewogen und mit 0,7 g fein zerriebener KOH und 0,1 g krist. Natrium-
acetat gut miteinander vermischt. Hernach wird 7 Minuten auf ca. 270° C erhitzt.
Das im Stickstoff übergetriebene Ammoniak wird in 10 ml 0,02n Schwefelsäure
aufgefangen und mit 0,02n NaOH unter Verwendung von Tashiro-Indikator
zurücktitriert.
Tabelle 21: Prozentuale Menge an Ammoniakstickstoff, erhalten durch Alkali¬
schmelze der hydrolysierten Lignine und Huminsäuren (bezogen auf organischeSubstanz).
Substanz N'/o NHs-N %> «Rest-N» in °/o
Björkm.-Lig frisch 0,18 0,19 0
H2S04-Lig. frisch 0,22 0,22 0
H2S04-Lig. 70 Tage 0,97 0,58 0,39
H2S04-Lig. 180 Tage 1,08 0,62 0,46
H2S04-Lig. 260 Tage 1,38 0,56 0,82
H2S04-Lig. 340 Tage 1,67 0,82 0,85
H2S04-Lig. 410+N 1,77 0,37 1,40
Huminsäure 260 Tage 0,95 0,51 0,44
Huminsäure 340 Tage 0,96 0,64 0,32
Gleichzeitig wurde an 3 nicht-hydrolysierten Präparaten der «Rest-N»
durch Alkalischmelze bestimmt. Dieser betrug für:
Björkman-Lignm aus frischem Stroh: 0,02 °/o N
Sehwefelsäurelignin aus frischem Stroh: 0,04 °/o N
Huminsäure aus Stroh 260 T. verrottet: 1,16 °/o N
Aus Tabelle 21 geht eindeutig hervor, dass der «Rest-N» in den Ligninenim Verlauf der Verrottung zunimmt. Man darf annehmen, dass dieser «Rest-
Stickstoff» zum grössten Teil heterocyclisch gebundenem Stickstoff ent¬
spricht. Der höhere Gehalt an «Rest-N» in den nicht hydrolysierten Proben
kann dadurch erklärt werden, dass Tyrosin und Indolverbindungen wie z. B.
Tryptophan etc. empfindlich gegen Mineralsäuren sind und durch die Säure¬
hydrolyse wohl zu einem grossen Teil zerstört worden sind.
50

Um diese Befunde eingehender zu prüfen, wurden Rückstände nach durchgeführ¬ter Alkalischmelze mit Wasser aufgenommen, mit 2n HCl auf pH 4 angesäuertund mit Äther extrahiert. Diese Extrakte wurden eingeengt und mit Isopropanol-Ammoniak-Wasser (10:1:1) auf Schleicher und Schüll-Papier 2043 b nach der
aufsteigenden Methode chromatographiert. Zur Untersuchung auf Indolverbindun¬
gen wurden die Chromatogramme mit einer 1 °/oigen Lösung von p-Dimethylamino-benzaldehyd in n HCl (Ehrlich's Reagens) besprüht und die Färbung bei 65° Centwickelt.
Abb. 3: Papierchromatographischer Nachweis von Indolverbindungen in Ligninenund Huminsäuren.
0.9t
044
033
021
D o O O O o
c O o o o o o
B O O O
A o
12 3 4 5 6 7
1 = H2SU4-Lignin aus Stroh, 70 Tage verrottet
2 = H2S04-Lignin aus frischem Stroh3 = Huminsäure aus Stroh, 340 Tage verrottet
4 = H2SU4-Lignin aus Stroh, 260 Tage verrottet
5 = Björkman-Lignin aus frischem Stroh6 = Björkman-Lignin aus frischem Stroh, hydrolysiert7 = Huminsäure aus Schwarzerde
Reaktion mit Ehrlichs Reagens: Die Flecken zeigen eine mehr oder weni¬
ger intensive blau-violette Färbung.
51

Reaktion mit Phosphormolybdänsäure: Die Flecken vom Rf-Wert 0,91
(D) weisen eine blau-graue Färbung auf. Dies deutet darauf hin, dass es sich
bei diesen Verbindungen wahrscheinlich um Oxy-Indole handelt.
Flaig und Breyhan12S haben inzwischen bei der Alkalischmelze von Tryp¬
tophan und Schwarzerde-Huminsäure im Papierchromatogramm beim Be¬
sprühen mit Ehrlichs Reagens blau-violette Flecken erhalten, die mit den
Flecken vom Rf-Wert 0,41 identisch sind und als Indolkörper angespro¬
chen werden.
364. Decarboxylierung von Ligninen und Huminsäuren
mit 12 Voiger Salzsäure
In der Literatur findet man zahlreiche Arbeiten, die darauf hinweisen, dass
Pentosane und auch Polyuronide eng mit dem Ligninmolekül verknüpftsindlle 17. Unterwirft man Nativlignin, isoliert aus Fichtenholz, der Be¬
handlung mit starker Salz- oder Schwefelsäure (72 °/o), so werden nach
Brauns11* 90 bis 92 %> als Säurelignin zurückgewonnen. Dabei wird ein
Ansteigen des Methoxylgehaltes von 14,8 auf 15,5 °/o beobachtet. Dieser Ge¬
wichtsverlust des Lignins von 8—10 °/o und das gleichzeitige Ansteigen des
Methoxylgehaltes kann dadurch erklärt werden, dass durch die Säurebe¬
handlung ein nicht methoxylhaltiges Bruchstück abgespalten wird. Sicher¬
lich kann ein Teil dieses Gewichtsverlustes auch dem Verlust von Konsti¬
tutionswasser durch Kondensation zugeschrieben werden.
Da die im Verlauf der Verrottung beobachtete Zunahme an decarboxy-lierbaren Substanzen von einer konstanten Abnahme an Furfurol begleitetwar, durfte man annehmen, dass durch den Zersetzungsprozess neue decar-
boxylierbare Substanzen entstanden sind, oder dass schon vorhandene Ver-
dindungen (z. B. Lignin) durch den Oxydationsprozess so verändert wur¬
den, dass sie unter diesen Bedingungen CO2 abgeben. Zunächst sollte des¬
halb überprüft werden, ob auch ein schonend hergestelltes Lignin (Björkman-Lignin) unter diesen Bedingungen CO2 abspaltet. Daneben wurden 2 Björk-man-Lignine aus verrottetem Stroh und eine mit kaltem Wasser herausge¬löste Huminsäurefraktion auf gleiche Weise untersucht. Die Herstellungder Huminsäurefraktion erfolgte durch 2-stündige Extraktion einer mit
Wasser versetzten Strohprobe auf der Schüttelmaschine und nachherigesAusfällen der zentrifugierten Lösung mit HCl. Die Ergebnisse bezogenauf organische Substanz sind in der folgenden Aufstellung dargelegt:
Björkman-Lign'm aus frischem Stroh: 0,47 °/o CO2
Björkman-Lign'm aus Stroh 70 Tage verrottet: 0,64 °/o CO2
Björkman-Lignin aus Stroh 180 Tage verrottet: 1,34 °/o CO2
Huminsäure aus Stroh 340 Tage verrottet: 2,90 °/o CO2
52

Bekanntlich wird beim Erhitzen von Uronsäuren in 12 Voiger Salzsäure
auch Furfurol gebildet. Diese Reaktion verläuft jedoch nicht quantitativ.Aus Glucuronsäure wird auf diese Weise 39,4 %> und aus Galacturonsäure
43,1 °/o der theoretischen Furfurolausbeute erhalten129.
Die an dieser Huminsäure durchgeführte Furfurolbestimmung ergab0,95 °/o Furfurol bezogen auf organische Substanz. Die abgespaltene CO2-
Menge würde theoretisch 11,60 %> Uronsäureanhydrid entsprechen. Bei
einer durchschnittlichen Furfurolausbeute von 40 °/o müssten bei die¬
ser Uronsäuremenge 2,63 °/o Furfurol anstatt der erhaltenen 0,95 °/o gebildetwerden. Diese Berechnung zeigt, dass die durch Decarboxylierung mit Salz¬
säure ermittelte Menge an CO2 nicht nur von Uronsäuren stammen kann.
Die im Verlauf des Rotteprozesses von Mikroorganismen gebildeten Zucker¬
und Schleimsäuren können an dieser vermehrten C02-Bildung kaum betei¬
ligt sein, denn sie liefern unter diesen Bedingungen Dehydroschleimsäure130.Sicher darf man einen bedeutenden Teil des erhaltenen CO2 der Decar¬
boxylierung anoxydierter Ligninkomplexe zuschreiben. Da von den Björk-man-Lignmen nur noch geringe Mengen zur Verfügung standen, konnten
damit keine Furfurolbestimmungen durchgeführt werden.
365. Potentiometrische Titration von Ligninen
Zur Bestimung der phenolischen Hydroxylgruppen im Lignin lässt sich eine
in jüngster Zeit von Brockmarin und Meyer131 ausgearbeitete potentiome¬trische Mikrotitrationsmethode anwenden. Die Titration in nicht wässrigenLösungsmitteln, wie z. B. Äthylendiamin oder Dimethylformamid mit Cola-
min gestattet daher, auch bei Anwesenheit von Carboxylgruppen, die
schwach sauren phenolischen Oxygruppen zu erfassen. Jeder Stufe der
Titrationskurve kann nur eine saure Gruppe zugeordnet werden, und aus
&0H
„Blindwert
Abb. S: Titration von Benzoesäure
und Vanillin
Abb. 6: Titration von
p-Oxybenzoesäure
53

der zwischen zwei Wendepunkten verbrauchten Basenmenge lässt sich der
Gehalt einer funktionellen Gruppe berechnen, die nur einmal in der Molekel
vorhanden ist.
Die Titration der Lignine in Äthylendiamin erfolgte in der von Brockman und
Meyer1&1 beschriebenen Apparatur. Als Potentiometer diente ein pH-Meter 22
(Fa. Radiometer Kopenhagen). Als Elektroden wurden 3 mm breite und 6 mm
lange Streifen aus Platinblech verwendet. Wasserfreies Äthylendiamin wurde aus
handelsüblichem Monohydrat durch Destillieren über NaOH und schliesslich über
Natrium gewonnen. Äthanolamin (Colamin) wurde zweimal durch eine 60 cm
Vigreux-Kolonne destilliert132. Die Titerstellung erfolgte mit Benzoesäure. Die
Einwaagen wurden auf organische Substanz berechnet und lagen in der Grössen-
ordnung von 3 mg.
Während die Titration von Modellsubstanzen, wie o-Oxybenoesäure, p-
Oxybenzoesäure, Vanillin (siehe Abb. 5 und 6), befriedigende Umschlag¬
punkte ergibt, wurde bei 3,4-Dioxybenzoesäure eine unspezifische Titra¬
tionskurve erhalten. Dies muss darauf zurückgeführt werden, dass Stoffe,
die in alkalischer Lösung durch Luft leicht oxydiert werden, depolarisierendwirken und daher die potentiometrische Messung in dieser Apparatur un¬
möglich machen131.
Aus der bis zum Wendepunkt verbrauchten Menge Colaminat ergaben sich
im Durchschnitt von zwei Messungen für die verschiedenen Lignine fol¬
gende Äquivalentgewichte:
Björkman-Lignia frisch: 560 ±13
Bra»»5-Lignin frisch: 405 ±10
Björkman-Lignin 70 Tage verrottet: 577 ±11
Björkman-lAgain 180 Tage verrottet: 429 ± 7
Björkman-Lignin 340 Tage verrottet: 412 ± 9
Björkman-Lignin 410 (+N) Tage verrottet: 564 ± 11
Basierend auf einem Molekulargewicht von 180—190 für eine Phenyl-propaneinheit ergibt sich für frisches Björkman-Lignin aus Stroh auf drei
Einheiten eine phenolische Hydroxylgruppe, d. h. 0,33 Phenolhydroxyl jeEinheit. Brauns Lignin enthält wesentlich mehr, im Durchschnitt 0,5 Phe¬
nolhydroxyl je Einheit. Diese Befunde stimmen mit den Ergebnissen von
Freudenberg133 überein, der für Björkman-Lignin aus Fichte 0,33 und für
Brauns-Lignin 0,5—0,7 phenolische Hydroxylgruppen (bezogen auf C6—
Oxymatairesinol
54

C3) fand. Diesen erhöhten Gehalt an Phenolhydroxyl in Brauns-Lignin ist
nach Freudenberg133 darauf zurückzuführen, dass dieses «lösliche Lignin»zur Hauptsache aus niedermolekularen, dem Coniferylalkohol verwandten
Anteilen besteht und daneben erhebliche Mengen eines als Substanz und
in Derivaten sehr schwer kristallisierbaren Lignans enthält, dem die For¬
mel eines Oxymatairesinols zugeschrieben wird (siehe S. 54).Der Verlauf der Titrationskurven der unverrotteten Lignine (Abb. 7)
lässt darauf schliessen, dass nach der Neutralisation der phenolischen Hy¬droxylgruppe noch eine schwach saure Gruppe neutralisiert wurde, die
keinen deutlichen Wendepunkt mehr zeigt. Es darf angenommen werden
(siehe Abschnitt 3692), dass es sich dabei wahrscheinlich um enolische Hy¬droxylgruppen in der Phenylpropanseitenkette handelt. Björkman-Ligninaus Weizenstroh würde demnach unter den genannten Bedingungen zwei
titrierbare saure Gruppen enthalten, woraus sich ein Molekulargewicht von
1120 errechnen lässt.
Die bei den verrotteten Ligninen (Abb. 8) auftretenden Änderungen des
Verlaufes der Titrationskurven lassen sich durch eine steigende Zunahme an
sauren Gruppen im Ligninmolekül erklären. Mit zunehmender Rottedauer
nähert sich das Aussehen der Titrationskurven immer mehr derjenigen der
Benzoesäure. Diese Erscheinung kann dem Auftreten von Carboxylgruppenoder aber auch von Oxy-Gruppen in Chinonen zugeschrieben werden, die
zum Teil saurer sind als die Carboxylgruppe der Essigsäure (134). Deshalb
lassen sich auf diese Weise Oxy-Gruppen in Chinonen nicht mehr von Car¬
boxylgruppen unterscheiden. Dies kommt sehr deutlich zum Ausdruck,wenn man die Titrationskurve von Chinizarin, ein 1,4-Dioxy-Anthrachinon(131), mit derjenigen der Benzoesäure vergleicht. Für das Vorhandensein
von Oxychinonen spricht auch die Tatsache, dass mit zunehmender Verrot¬
tung der Lignine (siehe Abschn. 366), z. B. nach 70 Tagen, mit Diazomethan
weniger OH-Gruppen erfasst werden können. Dies kann damit begründetwerden, dass zwar durch die biologische Entmethylierung (Phenoläther¬spaltung) neue OH-Gruppen entstehen, diese aber sofort zu Chinonen auf¬
oxydiert werden.
Die Titration von Thioglykolsäurelignin ergab zwei Wendepunkte, einen
deutlichen für die Carboxylgruppe und einen schwächeren für das Phenol¬
hydroxyl.Beim Betrachten der Titrationskurven der verrotteten Björkman-Lignine
(Abb. 8) konstatiert man, dass die Anzahl saurer Gruppen im Laufe des
Rotteprozesses zunimmt (siehe hierzu auch Aequivalentgewichte S. 54).Björkman-L'igmn aus der 410 Tage alten Probe zeigt ein von dieser Ten¬
denz abweichendes Verhalten. Die erhöhten Kohlenstoffwerte dieses Ligninsdeuten darauf hin, dass wahrscheinlich durch weitere Oxydation zu Keto-
säuren Decarboxylierungs- und anschliessende Kondensationsprozesse statt¬
gefunden haben.
55
Äquivalente CA BÄquivalente
Abb. 7: Titration von Ligninen Abb. 8: Titration von Ligninenaus frischem Stroh aus verrottetem Stroh
366. Methylierung von Ligninen
Die Zunahme des Methoxylgehaltes bei der Methylierung von Ligninengestattet Rückschlüsse auf die Anzahl vorhandener Hydroxylgruppen.Die Tatsache, dass Nativlignin je nach Methylierungsmittel zu verschie¬
denem Grade methyliert werden kann, deutet auf die Anwesenheit verschie¬
dener Arten von Hydroxylgruppen hin. Mit Diazomethan wird nur ein
Teil der Hydroxylgruppen erfasst. In wasserfreien Lösungsmitteln (Äther)reagiert es vor allem mit sauren Hydroxylgruppen, wie Carboxylgruppen,phenolischen und enolischen, aber auch aliphatischen Hydroxylgruppen.Brauns (11 g) erhöhte durch Methylieren von Nativlignin aus Fichtenholz
mit Diazomethan in Äthersuspension den Methoxylgehalt von 14,8 %> auf
18,3 °/o und bei Durchführung der Methylierung in Dioxanlösung auf 21,4 °/o
OCH3. Während das erste Produkt noch in Natronlauge löslich war, gingdas Lignin mit dem höheren Methoxylgehalt darin nicht mehr in Lösung und
gab auch die typischen Farbreaktionen des Lignins nicht mehr. Brauns führt
dies darauf zurück, dass im ersten Falle eine phenolische Hydroxylgruppeund im zweiten Fall eine enolisierte Carbonylgruppe methyliert wurde.
Durch Methylieren mit Methanol-Salzsäure und mit Dimethylsulfat-NaOHerhielt er ein Lignin mit 32,3 %> OCH3. Durch solche Behandlungen kann
das Ligninmolekül schon tiefgreifenden Veränderungen unterliegen.In diesem Zusammenhang interessierte lediglich das Vorhandensein von
freien Hydroxylgruppen, weshalb die Methylierung mit Diazomethan be¬
vorzugt wurde. Das Lignin wurde zu diesem Zwecke in Aceton unter Zu¬
gabe einer Spur Wasser gelöst und an der Kälte 12 Stunden mit einem
56

Überschuss an Diazomethan behandelt. Die methylierten Präparate wiesen
eine nahezu weisse Farbe auf. Es wurden dabei folgende Resultate erhalten:
Björkman-Ligmn aus frischem Stroh: 16,76 °/o OCH3
Björkman-Ligmn aus frischem Stroh methyliert: 21,76 °/o OCH3
Björkman-Lignin 70 Tage verrottet: 10,84 °/o OCH3
Björkman-Lignin 70 Tage verrottet methyliert: 13,97 °/o OCH3
Basierend auf einem Molekulargewicht (Teilchengewicht) von rund 1120
(siehe hierzu Abschnitt 365) erhöht sich durch Methylieren mit Diazomethan
die Zahl der Methoxylgruppen für Björkman-Lignm aus frischem Stroh von
6,06 auf 8,25. Durch diese Behandlung müssen demnach rund 2,2 Hydroxyl¬gruppen erfasst worden sein. Bei einem Teilchengewicht von 1150 für
Björkman-Ligmn aus verrottetem Stroh (70 Tage) werden auf diese Weise
nur noch rund 1,3 Hydroxylgruppen erfasst. Man sieht daraus, dass trotz
der Spaltung von Methylätherbindungen die Anzahl freier Hydroxylgrup¬pen nicht erhöht wurde.
367. Reaktion mit 2,4-Dinitrophenylhydrazin
Zur Feststellung von Carbonylgruppen im Lignin wurde eine von Traynardund Eymery135 mit Erfolg angewandte Methode benutzt.
Ausführung:
Eine gesättigte Lösung von 2,4-Dinitrophenylhydrazin in 1 °/oiger HCl wird ineine 2 %ige alkoholische Ligninlösung eingegossen. Der dabei entstandene rot¬
braune Niederschlag wird filtriert und hernach gut mit Wasser gewaschen. Die
Trocknung erfolgt im Vakuum über P2O5 bei 45° C.
Der Carbongehalt der verschiedenen Lignine wurde auf Grund der Stick¬
stoffzunahme, resultierend aus der Reaktion mit 2,4-Dinitrophenylhydra¬zin, berechnet (Tabelle 22).
Tabelle 22: Carbonylgehalt von Ligninen verschiedenen Rottegrades.
N-Gehalt in %>
Carbonyl¬gehaltin°/o
in der Ausgangs¬substanz im 2,4-Dinitro-
(M v. 2 Analysen) phenylhydrazonZunahme
Björkman-IAgnin frisch 0,42 1,12
Björkman-Ugnin 70 Tage 1,97 2,91
Björkman-Lignin 410+N 2,47 3,62
0,70
0,94
1,15
0,36
0,47
0,59
57

Der für frisches Björkman-Lignm gefundene Carbonylgehalt von 0,36 %>
kann zum Teil auf die für die Farbreaktion des Lignins verantwortliche
Zimtaldehydgruppe136137 zurückgeführt werden. Die im Verlauf der Ver¬
rottung festgestellte Zunahme an Carbonylsauerstoff dürfte mit der durch
oxydative Prozesse verursachten Bildung von CO-Gruppen in der Seiten¬
kette oder aber auch mit der Oxydation phenolischer OH-Gruppen zu
Chinonen zusammenhängen.
368. Spektrophotometrische Messungen von Ligninenund Huminsäuren im ultravioletten Licht
Das nicht sehr spezifische Absorptionsspektrum von Lignin im UV ist von
einer Reihe von Autoren sehr unterschiedlich gedeutet worden. Eine Zusam¬
menfassung all dieser Arbeiten liegt von Jones* vor. In Übereinstimmungmit den Ergebnissen anderer Forscher und auf Grund von eigenen Unter¬
suchungen schreibt er das im Bereich von 275—285 m|x liegende Maximum
dem Sauerstoff-substituierten Benzolkern im Lignin und die starke Ab¬
sorption zwischen 300 und 350 mjx der Anwesenheit chromophorer Gruppen(Carbonylgruppen oder konjugierte Doppelbindungen) in der Seitenkette zu.
Scheffer und Welte1S9 schliessen aus ihren UV-spektroskopischen Unter¬
suchungen an verschiedenen Ligninen und Huminsäuren auf eine strukturelle
Verwandtschaft.
Da die Huminsäuren in UV im allgemeinen kein Maximum aufweisen,sollte untersucht werden, ob die UV-Kurve des Lignins im Verlaufe der
Verrottung eine entsprechende Änderung erfährt. Die UV-Kurven der Hu¬
minsäuren werden im allgemeinen meist in alkalischer Lösung aufgenom¬men. Bekanntlich wird das bei 280 mu, liegende Maximum des Lignins bei
Messungen in alkalischer Lösung nach dem langwelligen Bereich verscho¬
ben und zugleich stark abgeschwächt26 lm. Aus diesem Grunde wurden die
in Abbildung 9 und 10 dargestellten Kurven in 90 °/oigem Dioxan aufge¬nommen (Unicam-Spektrophotometer Sp. 500). Die Einwaagen wurden auf
organische Substanz berechnet und betrugen 1 mg pro ml 90 '/oiges Dioxan.
Von den Huminsäuren löste sich jedoch nur ca. ein Viertel der eingewoge¬nen Menge, weshalb die erhaltenen Werte auf die entsprechende Konzen¬
tration umgerechnet wurden.
Die in Abbildung 9 dargestellte UV-Kurve von frischem Weizenstroh-
lignin (I) stimmt in ihrem Verlauf gut mit derjenigen von nativem Fichten-
holzlignin überein138. Das Absorptionsmaximum liegt bei 276 mu. Nach
410 Tagen Rottezeit ist das Maximum zum Teil nur noch sehr schwach
(III) oder leicht nach dem langwelligen Bereich verschoben (II). Auch die
bei frischem Lignin (I) vorhandene Absorption im Bereich von 350 mfx ist
bei verrotteten Ligninen stark abgeschwächt, was durch oxydative Verän¬
derungen in der Phenylpropanseitenkette erklärt werden dürfte.
58

—WWM*y* A
000 500 *00 330 SOO ISO
5 OTiW#fflVlVi'MiSV|'li'l'iVM^
* U-./,
hi,i,M,
v
I.U,l,l,l,l,l,Uil,liUK
- Wdknzahl ? -
l.liUil.MiLl
tMnXi
•«fern'
Abb. 9: UV-Aufnahme von LigninenI Lignin nach Bjorkman frisch
II Lignin nach Bjorkman 410 Tage (+N)III Lignm nach Bjorkman 410 Tage
Abb. 10: UV-Aufnahmen von
Hummsäuren
I Huminsäure aus Stroh nach 70 T. Rottezeit
II Hummsaure aus Stroh nach 410 T Rottezeit
Aus dem Kurvenverlauf der in Abbildung 10 dargestellten Huminsäuren
kann man ersehen, dass die Übereinstimmung zwischen diesen Kurven und
derjenigen der verrotteten Lignine sehr gross ist. Dies muss wohl darauf
zurückgeführt werden, dass sich in 90 Voigem Dioxan vor allem die in den
Huminsäuren enthaltenen Ligninkomplexe gelöst haben. Die Huminsäure I
wurde durch schonendes Erwärmen auf dem Wasserbade getrocknet, wäh¬
rend die Huminsäure II gefriergetrocknet wurde. Diese unterschiedliche
Behandlung hat jedoch auf den Kurvenverlauf kaum einen merklichen
Einfluss.
369. Infrarotspektrographische Studien an Ligninenund Huminsäuren
Das differenzierte IR-Spektrum des Lignins (speziell aus Holz) wird in
jüngster Zeit von einer grossen Anzahl Autoren140 141142143zu Fragen der
Identifizierung und Konstitutionsaufklärung herangezogen. Ceh und
Hadziui berichten von infrarotspektrographischen Studien an natürlichen
Huminsäuren aus Kohle und an synthetischen Huminsäuren. BregerU5 hat
das IR-Spektrum von Nativlignin aus Fichtenholz mit dem von Torfhumin-
säure verglichen und ist zum Schluss gekommen, dass Huminsäure und
Lignin strukturell verwandt sein müssen.
59

Die auf den folgenden Seiten abgebildeten IR-Spektren wurden mit einem
vollautomatischen Infrarot-Spektrographen der Firma Leitz (Wetzlar)aufgenommen. Die Substanzen wurden vorerst im Mörser fein zermahlen
(z. T. in Alkoholsuspension), worauf eine bestimmte Menge davon mit
Kaliumbromid vermischt wurde (1 mg Substanz auf 400 mg KBr). Die
Proben wurden vorher im Hochvakuum bei 30—40 ° C getrocknet.
3691. Zuordnung der Banden
Als Grundlage zur Interpretation der IR-Spektren dienten neben Angabender Literatur, die beiden Werke von Bellamy^*- und Brügelxm°. Die disku¬
tierten Lignine zeigen im Bereich zwischen 2,8 bis 12,5 jj. (3571—800 K)
Banden, die, verglichen mit verschiedenen, dem Lignin verwandten Ver¬
bindungen und mit den in der Literatur141 146 147 beschriebenen Spektrenvon Ligninen, wie folgt gedeutet werden können:
2,74—2,82 ]x (3650—3546 K): freie OH-Gruppen Bei Aufnahmen in festem Zu¬
stand (KBr) treten oft Frequenzverschiebungen nach niederen
Frequenzen ein, was auf Assoziationserscheinungen zurückzuführen
ist.
2,90—3,15 ix (3448—3175 K): wasserstoffbrückengebundene Hydroxylgruppen.3,35—3,50 m (2985—2857 K): aliphatische CH-Valenzfrequenz.5,80—5,87 n (1724—1704 K): C=0 aus Keto-, Säure- oder Estergruppen.5,95—6,10 \i (1681—1639 K): Aryl-Aldehyde (in KBr), et, ß-ungesättigte Ketone.
6,25—6,60 u (1600—1515 K): substituierte Benzolkerne, C=C-Ringschwingun-gen.
6,80—6,90 und 7,30 jx (1470—1449 und 1370 K): CH-Deformationsfrequenzen,wobei die Intensität der letzten Bande ein Mass für die Anzahl
der Gruppen mit CH-Deformations-Schwingung ist, die von CH3-
Gruppen herrühren147.
7,00 u, (1429 K): zeigt die Anwesenheit aliphatischer Gruppen im Lignin-komplex an.
7,50—8,85 ix (1333—1130 K): Über die Bedeutung der Banden in diesem Bereich
können keine sichern Angaben gemacht werden. Da die Banden
in dieser Gegend aus Schwingungen des Moleküls als Ganzes ent¬
stehen können, kann die Deutung nicht sehr spezifisch sein147. In
diesem Gebiet liegen auch Banden von OH-Gruppen, substituierten
Benzolkernen, Ätherbindungen, Säuren und Säure-Estern. Lage und
Intensität dieser Banden sind stark abhängig von der Art, in der
diese Gruppen an den Rest des Moleküls gebunden sind.
9,35—10 n (1070—1000 K):
11,50—12,50 [x ( 860— 800 F
ierten Benzolkernen her.
) KV ^
)XV IB3110^11 (CH-Waggingschwingungen) in diesen
'' * Bereichen rühren von verschieden substitu-
Die zitierten Banden beziehen sich vorwiegend auf Untersuchungen in Nujol.In der vorliegenden Arbeit wurde hauptsächlich mit KBr-Presslingen gearbeitet,wobei bekanntlich einige Banden des öfteren nach dem langwelligen Bereich ver¬
schoben werden. Von den Spektren wird nur der Bereich von 5—13,5 u. wieder¬
gegeben.
60

1700 1500 1300 1100ji.,t.ji|...i..|,i,.i,i.,,. i.. i , i. . i.j
1000 900 800 cm'1
11 12 O yu
3692. Diskussion der Spektren der frischen, unverrotteten Lignine
Die Spektren der aus frischem Weizenstroh nach verschiedenen Methoden
isolierten Lignine (I—VII) zeigen, abgesehen von wenigen Ausnahmen,
alle sehr ähnliche Banden. Auch das aus Fichtenholz isolierte Cuproxam-lignin weist eine weitgehende Ähnlichkeit mit den Strohligninen auf. Ver¬
gleichende Betrachtungen mit den dem Lignin nahestehenden Verbindungen(speziell Vanillin) zeigen die grosse Übereinstimmung, die zwischen einigenvon diesen Modellsubstanzen und dem Lignin besteht. Sämtliche Ligninezeigen eine breite Bande in der Gegend von 3jj, (3400 K), die auf wasser-
stoffbrückengebundene OH-Gruppen zurückzuführen ist. Auch bei Aufnah¬
men in Nujol konnten freie OH-Gruppen nicht beobachtet werden. Dabei
wurde in Übereinstimmung mit den Untersuchungen von Jones148 an Nativ-
61

1700 1500 1300 1100Il I Ol.I ..Il, III. !.. I I
, I I l,|_1000 900 800 cm"'
' '1
'r \ ' • ', ' I
IV —VI
lignin aus Fichtenholz im Bereich von 2,98 \i (3350 K.) eine breite Bande
erhalten. Die Spektren aller Lignine weisen auch bei 3,4 u. (2941 K) eine
Bande auf, die auf aliphatische CH-Valenz-Schwingung zurückzuführen
ist.
Sämtliche Spektren der Strohlignine zeigen eine Schulter oder schwache
Bande bei 5,85 \i (1710 K). Smithli% schreibt auf Grund von Untersuchun¬
gen am Weizenstrohlignin eine Bande in diesem Bereich (1697 K) Estergrup¬
pierungen zu. Während beim Cuproxamlignin diese Schulter nur ganz
schwach angedeutet ist, weist das Thioglykolsäurelignin in diesem Bereich
(5,8 |i) eine deutliche Bande auf. Diese Erscheinung dürfte auf die bei der
Isolierung in das Ligninmolekül eingeführte Thioglykolsäuregruppe zurück¬
zuführen sein. Mit Ausnahme des Cuproxamlignins zeigen alle Lignineeine Bande bei 6,0—6,16 u. (1667—1650 K), die im allgemeinen aromati-
62

sehen Aldehydgruppen zugeschrieben wird146. Diese Bande kann jedochebensogut mit C-C-Doppelbindungen oder konjugierten Keto-Gruppen in
der Phenylpropanseitenkette erklärt werden. Rasmussen und Mitarb.150
schreiben für enolisierbare ß-Ketoester und ß-Diketone die Bande bei 6,06 \x
(1650 K) dem C = O eines «konjugierten Chelatringes» folgender Grund¬
struktur zu: m / \
O-H O O-H O
I II II IR'-C = CR" — C-R"' und R'-C — CR" = C-R'"
Im vorliegenden Falle R' = PhenylrestR" =H
R"' — aliphatischer Rest oder -O-R
Ein Vergleich der Spektren von Björkman-Lignin aus frischem Stroh und
methyliertem Lignin legt den Schluss nahe, dass im Björkman-Lignin aus
Stroh zum Teil solche Resonanzstrukturen vorliegen. So zeigt frisches
Björkman-Lignin neben der typischen Aromatenbande bei 6,25 \i (1600 K),die hier allerdings nur schwach angedeutet ist, noch eine deutliche zweite
Bande bei 6,31 u. (1585 K), die charakteristisch für BenzoylVerbindungenist150. So zeigt z. B. auch Acetophenon ein Dublett in diesem Bereich150.
Nach Methylieren des Lignins mit Diazomethan verschwindet die Bande
bei 6,31 \i. Offenbar wird der grösste Teil dieser leicht enolisierbaren Keto-
gruppe methyliert. Dafür spricht auch die Tatsache, dass das methylierteLignin in Natronlauge fast unlöslich ist, während es in Nitrobenzol gutlöslich ist. Die bei Methylierung deutlicher in Erscheinung tretende Bande
(Schulter) bei 5,8 \i (1724 K), die beim nicht methylierten Lignin nur
schwach angedeutet ist, dürfte in diesem Falle auf die Entfernung des an
der Chelatbildung beteiligten Wasserstoffatoms zurückzuführen sein.
Die bei 6,25 ^ (1600 K) und 6,6 y, (1515 K) auftretenden C=C-Ring-schwingungen sind bei sämtlichen Ligninen deutlich wahrzunehmen. Ferner
zeigen alle Lignine in Übereinstimmung mit vielen Modellsubstanzen (z. B.
Vanillin, Eugenol) eine Bande bei 6,8 bis 6,9 p, (1450—1480 K), und eine
andere bei 7 |x (1430 K), die in diesem Fall wohl CH-Deformationsschwin-
gungen zugeschrieben werden müssen. Die Banden im Bereich von 8 (i, (1250K), 8,9 (x (1120 K) und 9,75 ^ (1025 K) sind auf C-O-Valenz- und OH-
Deformationsschwingungen zurückzuführen. Ob diese Banden jedoch auf
Aryläther, Phenole, Ester, Säuren oder andere Verbindungen mit der Stuk-
tur = C—O— zurückzuführen sind, kann nicht entschieden werden. Die
bei 12 n (835 K) liegende Bande liegt im gleichen Bereich wie jene von einem
1,2,4- oder 1,2,3,4- substituierten Benzolkern. Für die ortho-Substitution
spricht auch die bei 13,1 |x (765 K) liegende, schwach angedeutete Bande.
Interessant ist auch die Tatsache, dass bei der Säurehydrolyse von Björk¬man-Lignin die gleichen Änderungen der Banden im Bereich von 7,5—10 |x
(1350—1000 K) eintreten wie bei der Herstellung eines Säurelignins.
63

1800 1600 1400 BOO 1200 1100,,.... i. .. i
, . . . i .
1000 900 800 cm-1
3693. Diskussion der Spektren der aus verrottetem Stroh isolierten Lignine
Nach 70 Tagen Rottezeit sind im IR-Spektrum des Björkman-Lignim
(VIII) noch keine sehr grossen Veränderungen eingetreten. Lediglich die
Bande bei 6,31 ^ (1585 K) ist verschwunden. Die starke Absorption im
Bereich von 8,7 \i (C-O-Valenzschwingungen von Sauren, Alkoholen,Äthern und Estern) wird durch Methylierung mit Diazomethan stark ver¬
mindert, so dass das Spektrum dieses methylierten Präparates (IX) nahezu
identisch ist mit demjenigen von frischem Björkman-Lignin (VII). Mit
Zunahme der Rottezeit weist die Bande bei 5,8 \l (1729 K) eine stetige Zu¬
nahme der Absorption (Zunahme an COOH) auf, während die Bande bei
6,06 |x (1650 K) immer mehr verschwindet. Die Verbreiterung der Bande im
Bereich von 6,1—6,2 \i (1639 bis 1613 K) kann eine Folge des Stickstoff¬
einbaues in das Ligninmolekül sein, denn in diesem Bereich liegen C=N-
64

100
80
60
40
20
0
1700 1500 1300,r..li.i.i....liitf |V. ,\ . . . I . . . i . . |
£ 80
5 60
2
ifcfc^
J.)kcl\
1100 1000 900 800 cm"'11 • » I ! i
BJÖRKMAN - Lignin180 Toge verrottet
H
X —XII
Valenzschwingungen heterocyclischer Aromaten, NH-Deformationsschwin-
gungen von primären und sekundären Aminen und auch NH2-Deforma-
tionsschwingungen primärer Amide. Während die Gerüstschwingungen(C=C im Ring) auch nach längerer Rottedauer trotz Verminderung der
Intensität noch deutlich zu erkennen sind, werden jedoch die Banden im
Bereich von 8—14 \i (1250—713 K) immer undeutlicher. Diese Erscheinungkann wohl auf die in der peripheren Molekülgruppierung eingetretenen Än¬
derungen zurückgeführt werden.
In Übereinstimmung mit den vorherigen Befunden ist auch durch die
enzymatische Oxydation von Lignin (XIII) die Bande bei 6,1 u, (1650 K)verschwunden und eine Änderung im Bereich von 7,4 u, (1350 K) einge¬treten. Interessanterweise werden durch die Behandlung von Björkman-Lignin mit 0,1 n NaOH (XIV) während 35 Stunden bei Zimmertemperatur
65

1700 1500 1300 1100 1000 900 800 cm'1]l-'lt.M.ll.|l I HI . I I I . I I ! I I , . I | II I , . I I , I , I . | ,
,I .
XIII — XV
und nachfolgendes Ausfällen mit HCl genau die gleichen Veränderungen
hervorgerufen. Man darf deshalb annehmen, dass die enzymatische Oxy¬dation von Lignin sich auf die gleiche Gruppierung auswirkt, die auch durch
eine Behandlung mit NaOH angegriffen wird. Dass Ligninpräparate durch
die Behandlung mit NaOH Veränderungen erfahren, kann man auch aus
dem IR-Spektrum der Ligninthioglykolsäure n = Li-th n (XV) ersehen, die
im Gegensatz zu Li-thj (II) mit 0,5 n NaOH in Stickstoff-Atmosphäreextrahiert worden ist.
3694. Diskussion der Huminsäure-Spektren
In Übereinstimmung mit den Ergebnissen anderer Autoren144 Ub ,51 konnten
bei den Huminsäuren nur wenige und unscharfe Banden wahrgenommen
66

1700 1500 1300 1100 1000 900 800 cm-1100
10
...!..• i. ,.!.. .1... .i.. . i.. .. i 11. 111 , , 1 ' ' 1,., 1 i ' . i i i i
~>
\\ A
\/ m
Huminsäure aus
frischem Stroh
0r
\ ^
X
\ •
m 50
lA VJ tof^ 'X M
.° on
i^J Vd *SHuminsäure 70 T
extrah m Natronlauge
â20
0
\•
60
40
/r*~-
f\/^
'
sm
iJ V / "Di oxan huminsäure" au s
Stroh, 410 läge werrott
o
•
5 S 7 J
>
<
:vi
>
-X
i
VIII
0 1 1 2 1'/*
werden. Präparat XVI ist eine «Huminsäure», die durch Ansäuern des
Heisswasserextraktes erhalten wurde. Es ist möglich, dass es sich dabei um
eine Ligninvorstufe handelt, die wasserlöslich ist. Bei der mit Natronlaugeaus 70 Tage verrottetem Stroh extrahierten Huminsäure (XVII) sieht man
deutlich, dass das Präparat einen beträchtlichen Teil an nicht oxydiertemLignin enthält. Dieser Befund deckt sich auch mit den bei der Elementar¬
zusammensetzung festgestellten hohen Methoxylwerten des genannten Prä¬
parates. Die sehr starke Ähnlichkeit zwischen Huminsäuren und verrotte¬
tem Lignin geht auch aus den IR-Spektren der beiden Präparate XVIII und
XIX hervor. Bei der «Dioxanhuminsäure» (XVIII), deren Herstellung in
Abschnitt 345 beschrieben wurde, ist die Ähnlichkeit mit verrottetem Ligninviel stärker als bei der mit heissem Wasser extrahierten Huminsäure (XIX).Durch Behandlung einer mit heissem Wasser extrahierten Huminsäure mit
67

1700 1500 1300 1100 1000 900 800 cm-'JQQ jiiiIiiiiIiiiiIijiiIiiiiIii. I i i ii I i ,i i I .... I i i i , J , , . , I . i i tl i i | , I , i
68

72 %>iger H2SO4 (gleiche Bedingungen wie bei der Ligninbestimmung) wer¬
den die Banden im Bereich zwischen 6 und 7 n stark verändert und abge¬schwächt (XX). Beim Methylieren der «Dioxanhuminsäure» (XXI) mit
Diazomethan verschwindet die Bande bei 6,15 ji (1635 K) und bei 5,75 \i
(1739 K) tritt eine neue Bande auf. Dies deutet darauf hin, dass die Bande
bei 6,15 u, auf eine konjugierte C=0-Gruppe einer Carbonsäure zurück¬
zuführen ist. Als Vergleich zu diesen Huminsäuren aus Stroh wurde auch
eine mit einer Mischung von Natriumfluorid und Natronlauge aus Moor¬
boden extrahierte Huminsäure untersucht (XXII). Sie weist neben den
Banden bei 3 und 3,4 [x (3356 und 2941 K) die fast bei allen Huminsäuren
beobachteten zwei Banden bei 5,8 \i und 6,10—6,15 \i (1724 K und 1639—
1653 K) auf. Die C=C-Ringschwingungen bei 6,25 \i (1600 K) und 6,65 \i
(1504 K) sind jedoch in diesem Präparat nicht vorhanden. Das Lignin die¬
ses Präparates ist offenbar noch viel stärker zersetzt als dasjenige der Stroh-
Huminsäuren.
69

4- Diskussion der Ergebnisse
41. Abbau der verschiedenen Zellwandkomponenten
Die durch den Rotteversuch erhaltenen Ergebnisse stimmen im wesentlichen
mit den Befunden anderer Autoren50 5152 53 54 55 überein. Aus Abbildung 11
kann man ersehen, dass im Verlauf von 410 Tagen rund 65 °/o des Ligninsund 96 °/o der Holozellulose abgebaut werden. Interessant ist, dass nach
180 Tagen praktisch keine weitere Gewichtsabnahme erfolgte.
Abbau
In Gew.- V,
''2.0 098 0.32 0.13 0.19 0.11
Lignin
-»• Rotte-Quotient = Qp
a Zellulose in V. der org Subst.
in % der org Subst.
Lignin
70 180 260 340 410
410 *H
org Subst
,0 sHolozellulose
Rottezeit
in Tagen
Abb. 11: Abnahme an organischer Substanz, Lignin und Holozellulose im Ver¬lauf der Verrottung (Prozente bezogen auf Ausgangsmaterial).
Mit dieser Erscheinung steht auch das viel diskutierte Problem der Aus¬
nutzung von Lignin durch Mikroorganismen (als alleinige C-Quelle) im
engsten Zusammenhang69. Da auch die nachträgliche Zugabe anorganischerNährstoffe keinen merklichen Einfluss auf den Abbau der organischen Sub¬
stanz und das Lignin ausübte, darf man schliessen, dass der Abbau nahezuzum Stillstand kommt, wenn der Rotte-Quotient QR unter den Wert von
0,30 abfällt. Der Rottequotient Qj^ ist folgendermassen definiert:
qq-Zellulose in °/o der organischen Substanz
Lignin in °/o der organischen Substanz
70

Zur Berechnung des Rotte-Quotienten ist deshalb die a-Zellulose benutzt
worden, weil sich die Holozellulose mit zunehmender Verrottung aus mi-
krobiellen Polysaccariden zusammensetzt, die nur sehr schwer abgebautwerden können152.
Dieser Rotte-Quotient ist zugleich ein Mass für die Stabilität eines Hu¬
muskomplexes. Man darf somit annehmen, dass bei einem Wert für Qr =
0,20 bis 0,30 die organische Substanz in mehr oder weniger stabiler Form
vorliegt und keine nennenswerten Änderungen mehr erfährt.
Trotz der bedeutenden Abnahme reichert sich das Lignin im Rückstand
relativ stark an (siehe hierzu Abbildung 12). Gleichzeitig damit findet auch
eine Anreicherung an heisswasserlöslichen Extraktstoffen statt.
Die am hiesigen Institute durchgeführte Überprüfung der pflanzenphy¬siologischen Wirksamkeit der kaltwasserlöslichen Extrakte ergab eine Zu¬
nahme der fördernden Wirkung dieser Stoffe bis zum Zeitpunkt von 180
Rotte-Tagen, um hernach wieder leicht abzusinken153. Es scheint demnach,dass die Förderung des Pflanzenwachstums von kaltwasserlöslichen, orga¬
nischen Substanzen abhängig ist. Da diese Stoffe schnell in die tieferen
Bodenschichten eingewaschen werden, ist zur Erzielung einer langdauern¬den Förderwirkung ein kontinuierlicher Abbau organischer Substanz (Lig¬nin) notwendig. Dies ist nur möglich, wenn den Mikroorganismen zum
Abbau des Lignins eine geeignete C-Quelle (Zellulose) zur Verfügung steht.
Lignin
Heisswasserlosl org S
Holozellulose
«- Zellulose
180 260 340 410Rottezeit
in Togen
Abb. 12: Lignin, Holozellulose, a-Zellulose und heisswasserlösliche Extraktstoffe
zu verschiedenen Zeitpunkten der Verrottung (in Prozent der organischen Substanz)
Wenn man die in Abbildung 12 angegebenen prozentualen Werte für
Lignin, Holozellulose und heisswasserlösliche Extraktstoffe addiert, erhält
man mit Ausnahme des Ausgangsmaterials stets nur einen Wert von 75—
71

90 °/o. Diese fehlenden 10—25 °/o müssen zum Teil der Anwesenheit von
Proteinen zugeschrieben werden. Sicher ist ein beträchtlicher Teil dieser
Diskrepanz darauf zurückzuführen, dass grössere Mengen des anoxydiertenLignins bei der Säureligninbestimmung in Lösung gegangen sind.
42. Chemische Änderungen im Ligninmolekiil
Obwohl die organische Substanz nach 180 Tagen praktisch keine Gewichts¬
abnahme mehr zeigt, finden im Rückstand auch weiterhin enzymatischeund chemische Umwandlungsprozesse statt. Nach Broadbent nimmt die
Sauerstoffaufnahme von Lignin in alkalischer Suspension in der Warburg-Apparatur mit steigendem Rottegrad zu. Nach den Befunden der IR-
Studien bewirkt der erste Angriff der Enzyme (Polyphenoloxydasen) im
Ligninmolekül die gleichen Veränderungen, die auch bei der Behandlungvon Lignin mit Alkali an der Luft beobachtet werden. Dass die Angreif¬barkeit des Lignins durch Polyphenoloxydasen im Verlauf der Verrottungebenfalls zunimmt, zeigen die in der Warburg-Apparatur durchgeführtenVersuche.
Die auf Grund der infrarotspektrographischen Untersuchungen erhalte¬
nen Befunde deuten darauf hin, dass im Weizenstrohlignin zum Teil ß-Ketoestergruppierungen vorliegen. Daraus können im Verlauf der Zer¬
setzung ß-Ketosäuren gebildet werden, die unter den Bedingungen der Decar-
boxylierung mit 12°/oiger HCl sehr leicht CO2 abspalten. Aus Abbildung13 kann man ersehen, dass nur ein Teil des dabei abgespaltenen CO2 aus
Uronsäuren stammen kann, denn der Erhöhung der CO2-Werte nach 180
Rottetagen steht keine vermehrte Ausbeute an Furfurol gegenüber. Die
starke Abnahme der Furfurolausbeute nach 70 und 180 Tagen Rottezeit
kann jdoch zum Teil auch auf die durch die Zunahme der Heisswasser-
extrakte verursachte erhöhte Störung der Furfurolbestimmung zurückge¬führt werden (siehe hierzu Abschnitt 326).Neben der Abnahme des Methoxylgehaltes ist die Zunahme des Stick¬
stoffgehaltes im Lignin eines der wesentlichsten Merkmale der Verrottung.Nach Ansicht von Waksman und Iyer15i liegt der grösste Teil des Humin-
stickstoffs in Form von Proteinen vor die durch chemische Bindung mit
dem Lignin zum sog. «Lignin-Proteinkomplex» stabilisiert werden. Mattson
und Koutler-Andersson155 haben diese Theorie zurückgewiesen und gezeigt,dass die Huminsäuren mit einiger Wahrscheinlichkeit durch Autoxydationvon Lignin und gleichzeitige NH3-Fixierung entstehen. Forsyth156 konnte
durch Hydrolysieren von Huminsäuren mit 2 "/oiger HCl während 4 Stun¬
den nur einen geringen Teil des Stickstoffs entfernen, woraus er den Schluss
zieht, dass der Huminsäure-Stickstoff nicht in Form von Proteinen vor¬
liegen kann.
72

Furfurol
180 260 340 410Rottezeit
in Tagen
Abb. 13: Änderungen der Furfurol- und CC>2-Werte (durch saure Decarboxylie-rung). Gehalt im Ausgangsmaterial = 100 °/o.
Eigene Ergebnisse deuten darauf hin, dass zu Beginn der Rottezeit ein
grosser Teil des Stickstoffs in Form von Polypeptiden oder als Aminosäuren
im Lignin fixiert ist. Diese Peptide können nicht als lose Beimengung oder
Verunreinigung vorliegen, denn selbst die Schwefelsäure-Lignine zeigenerstaunlich hohe a-Aminostickstoffwerte. Mit zunehmender Verrottungwird jedoch der Stickstoff durch oxydative Prozesse in heterocyclischerForm in das Molekül eingebaut. Dies geht einesteils aus der bei der Alkali¬
schmelze erhaltenen Zunahme an «Reststickstoff» und andernteils aus dem
papierchromatographischen Nachweis von Indolverbindungen hervor. Es
wäre zu prüfen, ob diese Zunahme an heterocyclischen Stickstoffverbindun¬
gen ein Grund für die erhöhte Widerstandsfähigkeit oxydierter Lignin-komplexe gegenüber Acetylbromid ist.
Der Einbau stickstoffhaltiger Verbindungen ist jedoch erst dann mög¬
lich, wenn das Lignin durch die Spaltung von Phenolätherbindungen in
einen reaktionsfähigeren Zustand versetzt wird. Die Untersuchungen haben
gezeigt, dass der Methoxylgehalt der aus dem verrotteten Stroh isolierten
Lignine im Laufe der Zersetzung abnimmt, wobei Polyphenole entstehen
können. Da die Phenole nicht in der Lage sind, stickstoffhaltige Verbindun¬
gen zu addieren, muss der Einbau von Stickstoff in die Oxydationsproduktedes Lignins über die Stufe des a-Oxy-o-Chinons verlaufen7. Die Untersu¬
chungen von Steinmetz120 haben den Beweis erbracht, dass bei der enzyma-
tischen Oxydation von möglichen Abbauprodukten des Lignins, wie Proto-
catechusäure, Gallussäure und deren Ester, die Zwischenstufe eines a-Oxy-o-Benzochinons tatsächlich durchlaufen wird. Die Reaktion mit Anilin und
73

Methylanilin ergab definierte Verbindungen und Hinweise über den Einbau
des Stickstoffes. Dabei sind auch Stickstoffheterocyclen gefunden worden,die als Phenacinderivate angesprochen werden.
Es erhebt sich nun die Frage, ob man diese stark entmethylierten und
mit Stickstoff angereicherten Komplexe noch als Lignin bezeichnen darf.
Streng genommen ist die Bezeichnung «Lignin» nicht mehr gerechtfertigt.Die vorliegenden Untersuchungen haben jedoch gezeigt, dass bei der Zer¬
setzung von pflanzlichem Material eine mit heissem Wasser extrahierbare
Stoffgruppe entsteht, die sich schon zu Beginn der Rotte in mancher Hin¬
sicht von dem zurückbleibenden, weniger stark veränderten Lignin unter¬
scheidet. Diese als «Huminsäuren» bezeichnete Stoffgruppe weist nahezu
die gleichen Methoxylwerte und N-Gehalte auf, wie das restliche Lignin.Die Kohlenstoff- und Wasserstoffwerte liegen jedoch bedeutend tiefer und
die erhöhten Sauerstoffwerte deuten auf einen stärkeren Oxydationsgradhin. Am deutlichsten kommen diese Unterschiede im IR-Spektrum zum Aus¬
druck. Speziell zu Beginn der Rottezeit sind zwischen den Huminsäuren
und dem verrotteten Lignin bedeutende Unterschiede wahrzunehmen, wenn
auch die infrarotspektrographischen Befunde die Herkunft der Huminsäuren
vom Lignin noch deutlicher erkennen lassen. Neben den relativ hohen Me-
thoxylgehalten kommt dies auch in der Tatsache zum Ausdruck, dass diese
Huminsäuren unter den Bedingungen der Säureligninbestimmung einen
«Ligningehalt» von 60—75°/o aufweisen. Der «Ligningehalt» der aus Moor¬
boden isolierten Huminsäure liegt bei 75 °/o.
Der Methoxylgehalt der aus dem Boden isolierten Huminsäuren ist im
allgemeinen niedriger; wahrscheinlich findet während der längeren Ver-
rottungszeit im Boden eine stärkere Entmethylierung statt.
Mit zunehmender Rottedauer kann infrarotspektrographisch ein lang¬samer Übergang von verändertem Lignin zu Huminsäuren festgestellt wer¬
den. Abschliessend kann man sagen, dass die Huminsäuren im Gegensatzzum restlichen Lignin neben Proteinen und Polysacchariden zum grösstenTeil aus stark modifizierten Ligninkomplexen bestehen. Inwieweit sich diese
Erkenntnisse auf die Verhältnisse im Boden übertragen lassen, müsste noch
untersucht werden. Es ist jedoch anzunehmen, dass die im Boden stattfin¬
denden Umwandlungsprozesse einen sehr ähnlichen Verlauf nehmen.
74

5. Zusammenfassung
In der vorliegenden Arbeit ist versucht worden, die bei der Humifizierungpflanzlichen Materials eintretenden Veränderungen in möglichst umfassen¬
der Weise zu verfolgen. In erster Linie wurde dabei der Abbau des Ligninsund dessen Umwandlung zu Huminstoffen untersucht. Zu diesem Zwecke
wurde Weizenstroh in Mitscherlichgefässen bei 28° C unter aeroben Bedin¬
gungen während 410 Tagen der Verrottung unterworfen.
Nach bestimmten Zeitintervallen wurde das Material auf Lignin, Holo¬
zellulose, Hemizellulosen, a-Zellulose und die Elementarzusammensetzunguntersucht. Lignin wurde mittels der Schwefelsäuremethode von Ritter,
Seborg und Mitchell, die Holozellulose mit Natriumchlorit nach Adams und
Castagne, die Hemizellulosen als Furfurol und die a-Zellulose durch Behan¬
deln der Holozellulose mit 24 %>iger Kalilauge bestimmt. Im Vergleich zu
den Schwefelsäureligninen wurde das Lignin im Laufe der Rotte auch auf
schonende Weise nach der Methode von Björkman isoliert.
Die Versuchsergebnisse lassen sich wie folgt zusammenfassen:
1. Das Lignin wird nur solange abgebaut, als eine leichtverwertbare C-
Quelle (Zellulose) zur Verfügung steht. Nach 180 Tagen hat das Ligninum 65 %>, die Holozellulose sogar um 95 °/o abgenommen. Nach diesem
Zeitpunkt treten in der gesamten organischen Substanz nur noch geringeVeränderungen ein. Auch durch eine erneute Beigabe anorganischerNährsalze ist kein zusätzlicher Abbau erreicht worden.
2. In der organischen Substanz der verrotteten Strohproben steigen der
Kohlenstoffgehalt und der Methoxylgehalt leicht an, die Wasserstoff¬
werte dagegen bleiben praktisch gleich. Der organisch gebundene Stick¬
stoff erhöht sich auf den sechsfachen Wert.
3. In den nach den obenstehenden Methoden isolierten Ligninen nimmt der
Methoxylgehalt im Laufe der Zersetzung um mehr als die Hälfte ab.
Auch beim Kohlenstoffgehalt ist eine Abnahme zu verzeichnen, während
die Wasserstoffwerte konstant bleiben. Der Sauerstoffgehalt nimmt an¬
fänglich ab, um mit steigendem Zersetzungsgrad wieder leicht zuzuneh¬
men. Der Stickstoffgehalt nimmt um mehr als das Fünffache zu.
4. Die mit heissem Wasser extrahierten Huminsäuren weisen einen höheren
Sauerstoff- und einen tieferen Kohlenstoff- und Wasserstoffgehalt auf
als die schonend isolierten Björkman-Lignine. Mit steigender Rottezeit
nimmt der Stickstoff- und der Sauerstoffgehalt zu, der Kohlenstoff- und
Methoxylgehalt ab.
75

5. Die Menge des nach der Hydrolyse der Lignine mit 6n Salzsäure zurück¬
bleibenden Stickstoffes nimmt mit steigender Zersetzung zu, der Anteil
an a-Amino-Stickstoff in Prozent des Gesamt-N ab. Der nach der Alka¬
lischmelze der hydrolysierten Lignine im Rückstand verbleibende, hetero-
cyclische Stickstoff nimmt im Laufe der Rotte ständig zu. Im ätherischen
Extrakt der angesäuerten Lösung der Alkalischmelze sind auf dem Pa-
pierchromatogramm mit Ehrlichs Reagens Indolkörper nachgewiesenworden.
6. Der stetigen Abnahme des Furfurolgehaltes steht eine zunehmende Ab¬
spaltung an CO2 bei der Erhitzung der Proben mit 12 Voiger Salzsäure
gegenüber. Es wird gezeigt, dass ein bedeutender Teil dieser Zunahme
an CO2 oxydierten Ligninen zugeschrieben werden kann.
7. Die durch Polyphenoloxydasen katalysierte Sauerstoffaufnahme von
schonend isolierten Ligninen in der Warburg-Apparatur wird nach 410
Tagen Rottezeit bedeutend erhöht. An Hand von infrarotspektrographi-schen Studien wird gezeigt, dass der erste Angriff ligninoxydierenderEnzyme die gleichen Veränderungen im Ligninmolekül bewirkt wie die
Behandlung von Lignin mit 0,1 n Natronlauge während 35 Stunden bei
Zimmertemperatur.8. Zu Beginn der Rotte ist infrarotspektrographisch zwischen Huminsäuren
und Lignin ein bedeutender Unterschied festzustellen. Mit zunehmender
Zersetzung gleichen sich jedoch die Ligninspektren immer mehr denjeni¬gen der Huminsäuren an.
76

6. LITERATURVERZEICHNIS
1. W. Grosskopf, Brennstoff-Chem. 7, 293 (1926).2. F. Fischer und R. Lieske, Biochem. Z. 203, 351 (1928).3. H. Schrader, Brennstoff-Chem. 3, 161 (1922).4. H. Pringsheim und W. Fuchs, Ber. dtsch. ehem. Ges. 56, 2095 (1923).5. W. Fuchs, Brennstoff-Chem. 9, 298 (1928).6. T. A. Kukharenko, ref. in Chem. Abstr. 44, 9147 g (1950).7. W. Flaig, Holzforschung 9, 1 (1955).8. E. Junker, Kolloid-Z. 95, 213 (1941).9. H. Perrenoud, Diss. E. T. H. Zürich 1944.
10. J. Wehrmann, Z. Pflanzenernähr., Düng., Bodenkunde 70, 34 (1955).11. F.E.Brauns, The Chemistry of Lignin, New York: Acad.Press 1952.
IIa. F.E.Brauns, The Chemistry of Lignin, p. 49.
IIb. F.E.Brauns, The Chemistry of Lignin, p. 3.
11c. F.E.Brauns, The Chemistry of Lignin, p. 745.
lid. F.E.Brauns, The Chemistry of Lignin, p. 56.
lie. F.E.Brauns, The Chemistry of Lignin, p. 436.
llf. F.E.Brauns, The Chemistry of Lignin, p. 52.
llg. F.E.Brauns, The Chemistry of Lignin, p. 295.
12. L. E. Wise und E. C. Jahn, Wood Chemistry, New York: Reinhold Publ.
CO 1952.
13. K. Freudenberg, Fortschr. Chem. org. Naturstoffe 11, 43 (1954).14. W.J.Schubert und F.F.Nord, J. Amer. chem. Soc. 72, 977 (1950).15. I. Kawamura und T. Higuchi, J. Soc. Textile und Cellulose Ind. Japan 9,
157 (1953).ibid: 9, 454 (1953).ibid: 10, 15 (1954).
16. P. H. Traynard und A. M. Ayroud, Bull. Soc. chim. Fr. 1954, 345.
17. A. Björkman, Svensk. Papperstidn. 59, 477 (1956).18. K. Freudenberg, in Moderne Methoden der Pflanzenanalyse. Abschnitt Lignin.
(Herausgegeben K. Paech und M. V. Tracey) Berlin-Göttingen-Heidelberg,Springer-Verlag 1954.
19. H. Schrameck, Diss. E. T. H. Zürich 1944.
20. F. E. Brauns, Papier 7, 446 (1953).21. F.E.Brauns, J. Amer. chem. Soc. 61, 2120 (1939).22. E. Hägglund und H. Richtzenhain, Tappi 35, 281 (1952).23. G. Becherer und G. Voigtländer-Tetzner, Naturwiss. 42, 577 (1955).24. S.M.Siegel, Quart. Rev. Biol. 31, 1 (1956).25. N. Gratén, J. Colloid Sei. /, 453 (1946).26. G. Aulin-Erdtman, Tappi 32, 160 (1949).
Svensk. Papperstidn. 57, 745 (1954).27. K. Freudenberg, Cellulosechemie 22, 117 (1944).28. K. Kratzl, Internat. Holzmarkt 2, 7 (1950).29. A. v. Wacek und K. Kratzl, Ber. dtsch. ehem. Ges. 76, 891 (1943).30. E. Adler, B. O. Lindgren und U. Saeden, Svensk. Papperstidn. 55, 245 (1952).31. G. Aulin-Erdtman, Svensk. Papperstidn. 55, 745 (1952).32. E. Adler und S. Hernestam, Acta chem. Scand. 9, 319 (1955).33. B. O. Lindgren, Svensk. Papperstidn. 55, 78 (1952).
77

34. R. H. J. Creighton, R. D. Gibbs und H. Hibbert, J. Amer. chem. Soc. 66, 32
(1944).35. R. H. J. Creighton und H. Hibbert, J. Amer. chem. Soc. 66, 37 (1944).36. H. Richtzenhain, Svensk. Papperstidn. 35, 644 (1950).37. H. Richtzenhain, Ber. dtsch. ehem. Ges. 75, 269 (1942).38. K.Kratzl, Mh. Chem. 80, 314 (1949).39. N. N. Schorygina und T. Ja. Kefeli, J. Obschtschei Chimii 20, (82) 1199 (1950),
réf. in Chem. Zbl. 1951 II, 233.
N. N. Schorygina, T. Ja. Kefeli und A. F. Ssemetschkina, J. Obschtschei Chimii
19, (81) 1558 (1949), réf. in Chem. Zbl. 1950 I, 728.
40. H. Hibbert, réf. in F. E. Brauns, Fortschr.Chem.org.Naturstoffe 5, 175 (1948).41. H. Adkins, R.L.Frank und E.S.Bloom, J. Amer. chem. Soc. 63, 549 (1941).
E.E. Harris, J.D'Ianni und H. Adkins, J. Amer. chem. Soc. 60, 1467 (1938).42. H.Erdtman, Biochem. Z. 258, 172 (1933).43. T. Geissman und E. Hinreiner, Bot. Rev. 18, 77 (1952).44. K. Freudenberg, H. Reznik, H. Boesenberg und D. Rasenack, Chem. Ber. 58,
641 (1952).45. K. Freudenberg und W. Heimberger, Chem. Ber. 83, 519 (1950).46. K.Kratzl und W. Schweers, Mh. Chem. 85, 1166 (1954).47. K. Freudenberg, Z. Pflanzenernähr., Düng., Bodenkunde 69, 1 (1955).48. K. Freudenberg, H. Reznik, W.Fuchs und M. Reichert, Naturwiss. 42, 29
(1955).49. K. Freudenberg, Angew. Chem. 68, 84 (1956).50. F. G.Tenney und S. A. Waksman, Soil Sei. 30, 143 (1930).51. S. A. Waksman und F. C. Gerretsen, Ecology 12, 33 (1931).52. M.Phillips, H.D.Weihe und N.R.Smith, Soil Sei. 30, 383 (1930).53. U. Springer, Praktische Blätter für Pflanzenbau und Pflanzenschutz 21/22,
1 (1944/45).54. A. Apenitis, H.Erdtman und B.Leopold, Svensk. kern. Tidskr. 63, 195 (1951).55. A. Stöckli, Diss. E. T.H. Zürich 1952.
56. E. Weite, Z. Pflanzenernähr., Düng., Bodenkunde 56, 105 (1952).57. F. Scheffer und E. Weite, Naturwiss. 37, 321 (1950).58. W. Flaig, Landwirtschaftl. Forschung 8, Heft 2, 133 (1955).59. E. Saalbach, VI Inter. Congr. Sei. du Sol, Paris IV, 17, 107 (1956).60. R. Chaminade und R. Blanchet, C. R. Acad. Sei. 233, 1486 (1951).61. W. Eller und K.Koch, Ber. dtsch. chem. Ges. 53, 1469 (1920).62. H. Erdtman, Liebigs Ann. Chem. 513, 240 (1934).63. W. Flaig, H. Schulze, E. Küster und H. Biergans, Landbouwkund. Tijdschr.
66, 392 (1954).64. S. S. Dragunow, N. N. Schelochowtzewa und E. T. Strelkowa, Pedologija 7,
409 (1948), réf. n. W. Flaig, Landwirtsch. Forsch., Sonderheft 6, 94 (1955).65. H. Stach, Brennstoff-Chem. 22, 170 (1941).67. G. Lindeberg, Symb. Bot. Upsala VIII: 2 (1944).
Arkiv f. Bot. Upsala 331, Nr. 10 (1946).68. G. Fahraeus, R. Nilsson und G. Nilsson, Svensk. bot. Tidskr. 43, 343 (1949).69. M.L.Pelczar, S. Gottlieb und W. O. Day, Arch. Biochem. 25, 449 (1950).70. E. Hofmann, Z. Pflanzenernähr., Düng., Bodenkunde 69, 165 (1955).71. G. Lindeberg, Physiol. Plant. /, 196 (1948).72. G. Lindeberg und G. Fahraeus, Physiol. Plant. 5, 277 (1952).73. G. Lindeberg, Z. Pflanzenernähr., Düng., Bodenkunde 69, 142 (1955).74. W. M. Dion, Canad. J. Bot. 3D. 9 (1952).75. S. Gottlieb und S. H. Geller, Science 110, 189 (1949).76. W. F. van Vliet, Biochim. Biophys. Acta 15, 211 (1954).77. O. Fernandez und B. Regueiro, Farm, nueva 11, 57 (1946).
78

78. F. E. Broadbent, Proc. Soil Sei. Soc. America 18, 165 (1954).79. J. B. Bartlett, Iowa State Coll. J. Sei. 14, 11 (1939).80. J. B. Bartlett und A. G. Norman, Proc. Soil Sei. Soc. America 3, 210 (1939).81. B.Leopold, Svensk. kern.Tidskr. 62, 260 (1950).82. T. Enkvist, E. Solin, U. Maunula, Paperi ja Puu 36, 65 (1954).83. S. Gottlieb und S.B.Hendricks, Proc. Soil Sei. Soc. America 10, 117 (1946).84. G. A. Adams und A. E. Castagne, Canad. J. Res. Sect. B. 27, 915 (1949).85. K. Freudenberg und Th. Ploetz, Ber. dtsch. ehem. Ges. 73, 754 1940).86. Privatmitteilung.87. G. J. Ritter, R. M. Seborg und R. L. Mitchell, Ind. Engng. Chem., analyt.
Edit. 4, 202 (1932).88. Th. J. de Man und J. G. de Heus, Recueil Trav. chim. Pays-Bas 69, 271 (1950).89. A. G. Norman und S. H. Jenkins, Biochem.J. 28, 2160 (1934).90. G. A. Adams und A. E. Castagne, Canad. J. Res. Sect. B. 26, 325 (1948).91. L.E.Wise, M.Murphy und A. A. D'Addieco, Paper Trade J. 122, 35 (1946).92. G. A. Adams und A. E. Castagne, Canad. J. Res. Sect. B. 27, 907 (1949).93. G. A. Adams und A. E. Castagne, Canad. J. Res. Sect. B. 26, 314 (1948).94. R. S. Hilpert und H. Meybier, Ber. dtsch. ehem. Ges. B. 71, 1962 (1938).95. A. P. Sakostschikoff, V. T. Ivanowa und A. M. Kurenowa, Ind. Engng. Chem.
analyt. Edit. 6, 205 (1934).96. K. V. Lefère und B. Tollens, Ber. dtsch. ehem. Ges. 40, 4513 (1907).97. E. C. Shorey und J. B. Martin, J. Amer. chem. Soc. 52, 4907 (1930).98. S. A.Waksman und H. W. Reuszer, Soil Sei. 33, 135 (1932).99. U.Springer, Z.Pflanzenernähr.,Düng.,Bodenkunde 18, 129 (1940).
100. A.G.Norman und W.V.Bartholomew, Soil Sei. 56, 143 (1943).101. W. H. Fuller, Soil Sei. 64, 143 (1947).102. S. Mattson und E. Koutler-Andersson, Ann. agric. Coll. Sweden 12, 70 (1944).103. M. J.Bremner, J. Soil Sei. 2, 67 (1951).104. P. Dubach, G. Zweifel, R. Bach und H. Deuel, Z. Pflanzenernähr., Düng.,
Bodenkunde 69, 97 (1955).105. P. Dubach, Diss. E. T. H. Zürich (im Druck).106. J.M.Bremner und K.Shaw, J. agric. Sei. 44, 152 (1954).107. J. M. Bremner, J. agric. Sei. 45, 469 (1955).108. Ë. J. Conway, «Microdiffusion Analysis and Volumetrie Error» p. 90, Crosby
Lockwood & Son Ltd., London 1950, 3rd Revised Ed.109. E. J. Conway und E. O'Malley, Biochem. J. 36, 655 (1942).110. F. Vieböck und C.Brecher in F. Pregl (S. 266). «Quantitative organische
Microanalyse» neubearbeitet von H. Roth, Springer Verlag Wien 1949.111. S. L. Jansson undF. E. Clark, Proc. Soil. Soc. America 16, 330 (1952).112. S. Mattson und E. Koutler-Andersson, Ann. aric. Coll. Sweden 9, 1 (1941).113. J.E.Stone und K. G. Tanner, Canad. J. Chem. 30, 166 (1952).114. A.Björkman, Nature (London) 174, 1057 (1954).115. Privatmitteilung, Prof. E. Adler, Göteborg (Schweden).116. W. Stumpf und K. Freudenberg, Angew. Chem. 62, 537 (1950).117. B. Holmberg, Ing. Vetensk.-Akad. Handl., No. 131, 5 (1934).118. K.Kratzl und H. Silbernagel, Mh. Chem. 83, 1022 (1952).119. K. Freudenberg und H. Richtzenhain, Ber. dtsch. chem. Ges. 76, 937 (1943).120. A. Steinmetz, Diss. T. H. Braunschweig 1956.
121. K. Freudenberg, Angew. Chem. 68, 508 (1956).122. G. Lindeberg, Nature (London) 166, 739 (1950).123. D. D. Van Slyke, D. A. Mac Fadyen und P. Hamilton, J. biol. Chemistry 141,
671 (1941).124. S. A. Brown und A. C. Neish, Nature (London) 175, 688 (1955).125. E. F. Gale, Advances in Enzymol. 6, 1 (1946).
79

126. K. Paech und M. V. Tracey, «Moderne Methoden der Pflanzenanalyse». Vol
IV, S. 517, Berlin-Göttingen-Heidelberg: Springer-Verlag 1954.
127. Th. Breyhan, Z. analyt. Chem. 152, 412 (1956).128. W. Flaig und Th. Breyhan, Z. Pflanzenernähr., Düng., Bodenkunde 75, 132
(1956).129. G. Jayme und P. Sarten, Biochem. Z. 310, 1 (1941).130. P. A. Yoder und B. Tollens, Ber. dtsch. chem. Ges. 34, 3446 (1901).131. H. Brockmann und E.Meyer, Chem. Ber. 86, 1514 (1953).132. M.L.Moss, J.H.Elliott und R.T.Hall, Analytic, chem. 20, 784 (1948).133. K. Freudenberg, Angew. Chem. 68, 84 (1956).134. W. Flaig, H. Beutelspacher und H. Söchtig, «Kalium Symposium» 1954, 81—
107.
135. Ph.Traynard und A. Eymery, Holzforschung 10, 6 (1956).136. E.Adler und L. Ellmer, Acta chem. scand. 2, 93 (1948).137. J. C.Pew, J. Amer. chem. Soc. 74, 2850 (1952).138. E.J.Jones, jr., Tappi 32, 311 (1949).139. F. Scheffer und E.Weite, Z. Pflanzenernähr., Düng., Bodenkunde 48, 250
(1950).140. W. J. Schubert und F.F.Nord, J. Amer. chem. Soc. 72, 3835 (1950).141. S. F.Kudzin und F.F.Nord, J. Amer. chem. Soc. 73, 690 (1951).142. R. M. de Baun und F. F. Nord, J. Amer. chem. Soc. 73, 1358 (1951).143. K. Kratzl und H. Tschamler, Mh. Chem. 83, 786 (1952).144. M. Ceh und D. Hadzi, Fuel 35, 77 (1956).145. I. A.Breger, Fuel 30, 204 (1951).145a. L. J. Bellamy, «Ultrarotspektrum und chemische Konstitution»; Verlag
D. Steinkopff, Darmstadt 1955.
145b. W. Brügel, «Einführung in die Ultrarotspektroskopie», Verlag D. Steinkopff,Darmstadt (1954).
146. K. Freudenberg, W. Siebert und W. Heimberger, Chem. Ber. 83, 533 (1950).147. K. Freudenberg, H.Dietrich und W. Siebert, Chem. Ber. 84, 961 (1951).148. E. J. Jones, J. Amer. chem. Soc. 70, 1984 (1948).149. D. C. C. Smith, Nature (London) 176, 267 (1955).150. R. S. Rasmussen, D. D. Tunnicliff und R. R. Brattain, J. Amer. chem. Soc. 71,
1068 (1949).R. S. Rasmussen und R. R. Brattain, J. Amer. chem. Soc. 71, 1073 (1949).
151. M. M. Kononowa und N. P. Beltschikowa, VI Int. Congr. Sei. du Sol, Paris,Vol.B. 557 (1956).
152. R.L. Whistler und K. W. Kirby, J. Amer. chem. Soc. 78, 1755 (1956).153. W. Flaig, E. Saalbach und U. Schobinger, unveröffentlichte Versuche.154. S. A. Waksman und K. R. N. Iyer, Soil Sei. 34, 43 (1932).
S. A. Waksman und K. R. N. Iyer, Soil Sei. 34, 71 (1932).S. A. Waksman und K. R. N. Iyer, Soil Sei. 36, 57 (1933).S. A. Waksman und K. R. N. Iyer, Soil Sei. 36, 69 (1933).
155. S. Mattson und E. Koutler-Andersson, Ann. agric. Coll. Sweden 11, 107 (1943).156. W. G. C. Forsyth, J. agric. Sei. 37, 132 (1947).

CURRICULUM VITAE
Am 24. Januar 1929 wurde ich in Menzingen (Kt. Zug) als Sohndes Arztes Josef Schobinger und dessen Ehefrau Stefanie Wyssgeboren. In Menzingen besuchte ich auch die erste Klasse der
Primarschule.
Nach der Umsiedlung nach Zug absolvierte ich dort von 1937—
1942 die Primarschule und anschliessend während einem Jahre die
Sekundärschule, um hernach ins Gymnasium der Kantonsschule
Zug einzutreten. Die Maturitätsprüfung legte ich im Jahre 1949 ab.
Während 8 Monaten besuchte ich die landwirtschaftliche Schule
in Grangeneuve bei Fribourg. Den Rest meiner landwirtschaftli¬
chen Praxis absolvierte ich auf verschiedenen Betrieben des In-
und Auslandes. Im Herbst 1950 begann ich das Studium der Land¬
wirtschaft an der Eidg. Technischen Hochschule in Zürich. Nach
bestandenem Diplom im Frühjahr 1954 war ich während 4 Mona¬
ten an der Eidgenössischen Versuchsanstalt für Obst-, Wein- und
Gartenbau in Wädenswil tätig.Im Herbst 1954 erhielt ich von der Eidg. Technischen Hoch¬
schule ein Austauschstipendium zu wissenschaftlicher Weiterbil¬
dung in Deutschland, welches mir ermöglichte, die vorliegendeArbeit am Institut für Biochemie des Bodens der Forschungsanstaltfür Landwirtschaft in Braunschweig-Völkenrode vom Oktober
1954 bis März 1957 durchzuführen.