rheology of peptide
TRANSCRIPT

UNCORRECTED PROOFS
Advanced Review
Rheology of peptide- andprotein-based physical hydrogels:Are everyday measurements justscratching the surface?Sameer Sathaye,1 Armstrong Mbi,2 Cem Sonmez,3,4 Yingchao Chen,1
Daniel L. Blair,2 Joel P. Schneider4 and Darrin J. Pochan1∗
Rheological characterization of physically crosslinked peptide- and protein-basedhydrogels is widely reported in the literature. In this review, we focus on solidinjectable hydrogels, which are commonly referred to as ‘shear-thinning andrehealing’ materials. This class of what sometimes also are called ‘yield-stress’materials holds exciting promise for biomedical applications that requirewell-defined morphological and mechanical properties after delivery to a desiredsite through a shearing process (e.g., syringe or catheter injection). In addition tothe review of recent studies using common rheometric measurements on peptide-and protein-based, physically crosslinked hydrogels, we provide experimentallyobtained visual evidence, using a rheo-confocal microscope, of the fracture andsubsequent flow of physically crosslinked 𝛽-hairpin peptide hydrogels understeady-state shear mimicking commonly conducted experimental conditions usingbench-top rheometers. The observed fracture demonstrates that the supposed bulkshear-thinning and rehealing behavior of physical gels can be limited to the yield-ing of a hydrogel layer close to the shearing surface with the bulk of the hydro-gel below experiencing negligible shear. We suggest some measures to be takenwhile acquiring and interpreting data using bench-top rheometers with a particularfocus on physical hydrogels. In particular, the use of confocal-rheometer assem-bly is intended to inspire studies on yielding behavior of hydrogels perceived asshear-thinning and rehealing materials. A deeper insight into their yielding behav-ior will lead to the development of yield-stress, injectable, solid biomaterials, andhopefully inspire the design of new shear-thinning and rehealing hydrogels andmore thorough physical characterization of such systems. Finally, more examplesof bulk fracture in some physical hydrogels based on peptides and proteins areexplored in the light of their behavior as yield-stress materials. © 2014 Wiley Periodicals,Inc.
How to cite this article:WIREs Nanomed Nanobiotechnol 2014. doi: 10.1002/wnan.1299
∗Correspondence to: [email protected] of Materials Science and Engineering and Delaware Biotechnology Institute, University of Delaware, Newark, DE, USA2Department of Physics, Georgetown University, Washington, DC, USA3Department of Chemistry, University of Delaware, Newark, DE, USA4Chemical Biology Laboratory, National Cancer Institute, Frederick National Laboratory for Cancer Research, Frederick, MD, USA
Conflict of interest: The authors have declared no conflicts of interest for this article.Additional Supporting Information may be found in the online version of this article.
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
Volume 2, 2014 © 2014 Wiley Per iodica ls, Inc. 1

UNCORRECTED PROOFS
Advanced Review wires.wiley.com/nanomed
INTRODUCTION
‘Hydrogel’ is a general term used for water swollenand porous materials of polymeric, protein,
peptidic, colloidal, surfactant, or lipid origin. Hydro-gels are a mainstay in the food and pharmaceuticalindustries but are also increasingly finding applica-tions in areas such as biosensing,1,2 microfluidics,3–6
drug delivery,7–10 and tissue engineering.7,9,11,12 Abroad classification of hydrogels based on the type ofcrosslinking is commonly made, i.e., chemically (cova-lently) crosslinked or physically crosslinked (basedon secondary interactions such as hydrogen bond-ing, hydrophobic and electrostatic interactions). Thehighly hydrated and porous nature of hydrogels ingeneral can be leveraged for their utility as encapsulat-ing agents and delivery devices for therapeutic agentssuch as cells,13–18 growth factors,19–21 DNA,22–25
peptides and proteins,26–28 and drugs.29–33
A very important property of some hydrogels rel-evant to their applications as drug delivery vectors,34
tissue engineering scaffolds,35 or biomedical implantsand biosealants36–38 is that they can be injected usinga simple device such as a syringe or a catheter. A largenumber of studies have been focused on injection ofliquid precursors that are injected at a desired site intothe body and then undergo designed sol to gel tran-sition (via physical or chemical mechanisms) at thesite of delivery and, thus, solidify postinjection. Thesematerials have the capacity to encapsulate therapeuticagent and cell payloads as a consequence of gelation atthe site of delivery. Although injectable delivery of liq-uid precursors is a widely studied strategy, the strategycan suffer from a number of setbacks. Liquid precur-sors injected at a desired site of delivery in the bodyare subject to dilution with bodily fluids or leakageinto neighboring tissue.16,39 Owing to these factors,mechanical properties of the hydrogels formed fromthese precursors ex vivo might be compromised,40,41
and toxicity concerns might arise because of the pres-ence of unreacted initiators, crosslinking agents, anduncrosslinked polymers. Injection of liquid precur-sors offers little control over flow properties, spatiallocation, and biological response of uncrosslinked liq-uid precursors immediately after injection. This lackof control can lead to potential ambiguity in desiredperformance of in vivo hydrogels in terms of estab-lished ex vivo parameters such as specific morphology,drug/protein release profile, and encapsulated cell bio-logical behavior.
In this tutorial review, we focus on the solid,injectable, ‘shear-thinning, and rehealing’ hydrogels.Ex vivo, physically crosslinked, solid hydrogels thatare formed prior to injection can undergo flow duringinjection and reheal into percolated, solid networks
after injection are of particular interest in biomedicalapplications.16–18,26,27,30,40,42–52 These materials sim-ply will be referred to as solid injectable hydrogelsin this review. Drugs, cell, and growth factor pay-loads (or combinations thereof) can be encapsulatedin preformed solid hydrogels and injected directly ata specific desired site owing to the shear-thinningnature of injectable solid hydrogels. They offer sev-eral advantages over injectable liquid precursor mate-rials that are usually crosslinked at the site of deliv-ery and solidify postinjection. Preformed solids canmaintain their structural integrity even immediatelyafter injection, allowing the encapsulated therapeu-tics to be localized effectively and largely eliminat-ing limitations posed by injectable liquid precursors.53
Because they are already physically crosslinked, pre-formed solids eliminate chemical effects of the hydro-gel crosslinking process such as UV irradiation andthermal crosslinking on the underlying body tissue.The ex vivo crosslinked nature of injectable solidhydrogels offers reliable predictability of hydrogelmorphology, flow behavior, and encapsulated payloadrelease profile in vivo as compared with liquid precur-sors, thus overcoming some of the major limitations ofinjectable liquid precursors.16,17 Thus, injectable, pre-formed solid hydrogels demonstrate great potential indelivery of encapsulated therapies over injectable pre-cursor liquids. Yield-stress materials exhibit solid-likeresponse of resistance to flow when subject to smallerstress values but liquid-like response of yielding andflowing above a threshold stress value.54 On a com-mercial scale, these materials are everywhere aroundus used in products such as paints, mayonnaise,cement, toothpaste, and concrete.55 Injectable solidhydrogels demonstrate a rheological response simi-lar to yield-stress materials, and thus are amenableas injectable materials that flow when subjected tosyringe-induced shear.
Physically crosslinked, solid injectable hydrogelscan be developed from peptides, proteins, or poly-mers. With rapid advances in peptide,56,57 protein,58,59
polymer,60,61 and protein–polymer conjugate syn-thesis62,63 and materials development, rheologicalcharacterization has become an increasingly impor-tant tool to obtain more information about the vis-coelastic and flow properties of hydrogels based onpeptides, proteins, and polymers. Common rheolog-ical studies conducted on hydrogel materials includemeasurement of shear storage modulus (G′) (qualita-tively the material stiffness), loss modulus (G′′) (qual-itatively the material liquid-like flow properties), andloss factor tan (𝛿) (the ratio of liquid-like behavior tosolid-like behavior), measured as functions of time,oscillatory frequency, and oscillatory strain. Such
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
2 © 2014 Wiley Per iodica ls, Inc. Volume 2, 2014

UNCORRECTED PROOFS
WIREs Nanomedicine and Nanobiotechnology Rheology of peptide- and protein-based physical hydrogels
studies can provide insight about gelation kinetics,linear viscoelastic regions, and relaxation timescalesrelevant to the studied hydrogels. A commonly con-ducted measurement for studying the shear-thinningand rehealing behavior of physical hydrogels is thestorage modulus evolution of a hydrogel immedi-ately after it has been subject to steady-state shearof large amplitude by the upper plate of a bench-toprheometer. Such a measurement often shows a signif-icant reduction in the value of the storage modulusupon shearing and subsequent gradual storage modu-lus evolution post-shear cessation.16,26,41,44,64–68 Solidinjectable hydrogels are subject to shear treatmentusually using a rheometer and also, but less often, asyringe or capillary to study their under shear andpost-shear behavior.
Rheological characterization is a widely usedand convenient strategy for investigating shearresponse of solid injectable hydrogels. Studying theshear response of solid injectable hydrogels in terms ofexact flow profile and potential changes in hydrogelbulk structural characteristics is a step forward inunderstanding the shear behavior of solid injectablehydrogels. Physically crosslinked hydrogels, whichform the hydrogel class representing most solidinjectable hydrogels, have been reported to demon-strate diverse behavior such as bulk fracture,69–72
strain stiffening,73–76 and shear banding77 when sub-jected to large amplitude oscillatory or steady-stateshear. Rheological characterization provides infor-mation about the changes in mechanical behavior ofphysically crosslinked, solid, injectable hydrogels. Itis often not possible to obtain correlations betweenmechanical and morphological behavior of physicallycrosslinked hydrogels when subject to shear solely bythe use of rheometric experiments. In this review, wediscuss the use of a confocal-rheometer compoundassembly that provides a direct visual evidence of theflow pattern of a solid injectable peptide hydrogel sam-ple under steady-state shear. The rheometer-inducedshear treatment in the experiment reported hereinis very similar to that reported in citations in theprevious paragraph that discuss shear-thinning andrehealing behavior of physical hydrogels. Thus, usinga combined assembly of a rheometer and a confocalmicroscope, we subject the hydrogel to steady-stateshear as described by reports studying shear-thinningand rehealing hydrogels with a traditional bench-toprheometer while being able to simultaneously obtaina visual understanding of the exact flow pattern of thehydrogel. Upon being subjected to certain amplitudeof shear, the peptide hydrogel displays bulk fracture athin layer away from the shearing upper plate of therheometer. Preliminarily, the thickness of the fractured
layer depends upon the amplitude of applied shear.
AQ1
The phenomenon of bulk fracture demonstrates thatthe perceived shear-thinning and rehealing behaviorof physical gels might, perhaps, be due to yieldingbehavior of a thin layer of the hydrogel very closeto the shearing surface that is the top plate of therheometer, with a bulk of the hydrogel below thatlayer experiencing negligible shear. Additionally,upper plate-induced shear produced in standardrheometry might not be the best representation of theshear treatment via syringe injection for a variety ofreasons. Unless carefully conducted, a measurementinvolving rheometer-induced shear is also prone toartifacts such as wall slip, gap effects, time effects, andshear history. Measurements carried out in a mannersimilar to the one described above might literally andfiguratively be ‘scratching the surface’ of what exactphenomena might be occurring across the entire gapheight in a standard rheometer.
In the next section, we suggest some proto-cols and precautionary measures to be observedwhile acquiring and interpreting data using com-mon bench-top rheometers with a particular focuson physical hydrogels. To put the phenomenon ofbulk fracture observed by the experiments reportedhere into perspective, we review some of the recentaccounts of bulk fracture observed in physicallycrosslinked hydrogel systems. Correlation of changesin rheological behavior to bulk structural changes inphysically crosslinked proteins and peptides hydro-gels is a crucial step in understanding the behaviorof these hydrogels in various mechanical and bio-logical environments. The following section alsoincludes a review of rheological characterization ofpeptide- and protein-based physically crosslinkedhydrogels. These studies demonstrate that rheolog-ical characterization is critical to understanding ofhydrogel characteristics such as gelation kinetics, gelstiffness, solid-like character, and yield-stress behav-ior. The study of these and related hydrogels canpotentially provide interesting correlations betweenshear-induced structural changes and rheologicalproperties.
RHEOLOGICAL MEASUREMENTS OFPHYSICAL HYDROGELS: PROTOCOLSAND PRECAUTIONS
Some measurements common to all rheometric exper-iments, such as oscillatory time sweep, frequencysweep and strain sweep measurements, are carriedout using common bench-top rheometers while study-ing rheological properties of physical hydrogels. Someexperimental protocols, for example, the order in
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
Volume 2, 2014 © 2014 Wiley Per iodica ls, Inc. 3
UNCORRECTED PROOFS
Advanced Review wires.wiley.com/nanomed
which these measurements should be carried outalong with any specific precautions that should betaken while studying different physical hydrogels,are discussed in this section. For detailed explana-tion of measurements of raw, physical parameters byrheometers (for example, torque, rotational speed, anddeflection angle) and their conversion to correspond-ing rheological parameters such as shear stress, shearrate, and shear deformation, the reader is referredto Refs 78 and 79 In the case of controlled-strainrheometers, shear strain is applied to the sample in asinusoidal oscillation, 𝛾(t)= 𝛾o (sin 𝜔t), and the mea-sured shear stress is a phase-shifted sine wave with𝜏(t)= 𝜏o (sin 𝜔t+ 𝛿) in which 𝜔 is the applied angularfrequency and 𝛿 is the phase difference between thetwo waves. For stress-controlled rheometers, the shearstress is applied as 𝜏(t)= 𝜏o (sin 𝜔t) and the resultingshear strain is measured as 𝛾(t)= 𝛾o (sin 𝜔t+ 𝛿). Theequation relating shear stress and shear strain is givenby 𝜏(𝜔, t)=G′.𝛾o. (sin 𝜔t)+G′′.𝛾o. (cos 𝜔t), withthe coefficients G′ and G′′, respectively, indicatingthe energy stored elastically and energy lost throughflow per cycle when oscillatory shear is applied toa viscoelastic material. For a purely elastic material,the strain and stress waves are in phase (𝛿 =0∘) whilea purely viscous response has the two waves out ofphase by 90∘ (𝛿 = 90∘). Most viscoelastic materials,if not all, demonstrate an intermediate phase angle0∘ <𝛿 < 90∘.80 Since the G′ and G′′ are ratios of theoscillatory stress measured in Pascal to the oscillatorystrain which is a dimensionless quantity, G′ and G′′
are measured in the unit of stress, which is Pascal(Pa).78,81
In a common oscillatory rheological measure-ment, the storage modulus, G′, and loss modulus,G′′, are the most common parameters that are mea-sured for a hydrogel. G′ (Pa) and G′′ (Pa) are usuallymonitored as a function of time, applied angular fre-quency, and applied oscillatory strain. In a viscous solstate, G′′ is greater than G′. Therefore, for a solid,physical gel the storage modulus is greater than theloss modulus (G′ ≫G′′), particularly at low frequen-cies as shown in Figure 1. An extensive account ofthe theory and practical techniques concerning accu-rate measurement of a gel point of polymers hasbeen provided by Winter and Mours.82–84 Microrhe-ology has also been used to monitor the gelationof physical hydrogel systems through use of themotion of dispersed colloidal probe particles to mea-sure viscoelastic properties.85–87 Schultz and Furst88
have described multiple particle-tracking microrheol-ogy as a technique useful for precise determinationof hydrogel sol–gel transitions in a comprehensivereview.
1000
100
G′,
G′′
(Pa)
Strain %
10
100.1 100 1000
1
1
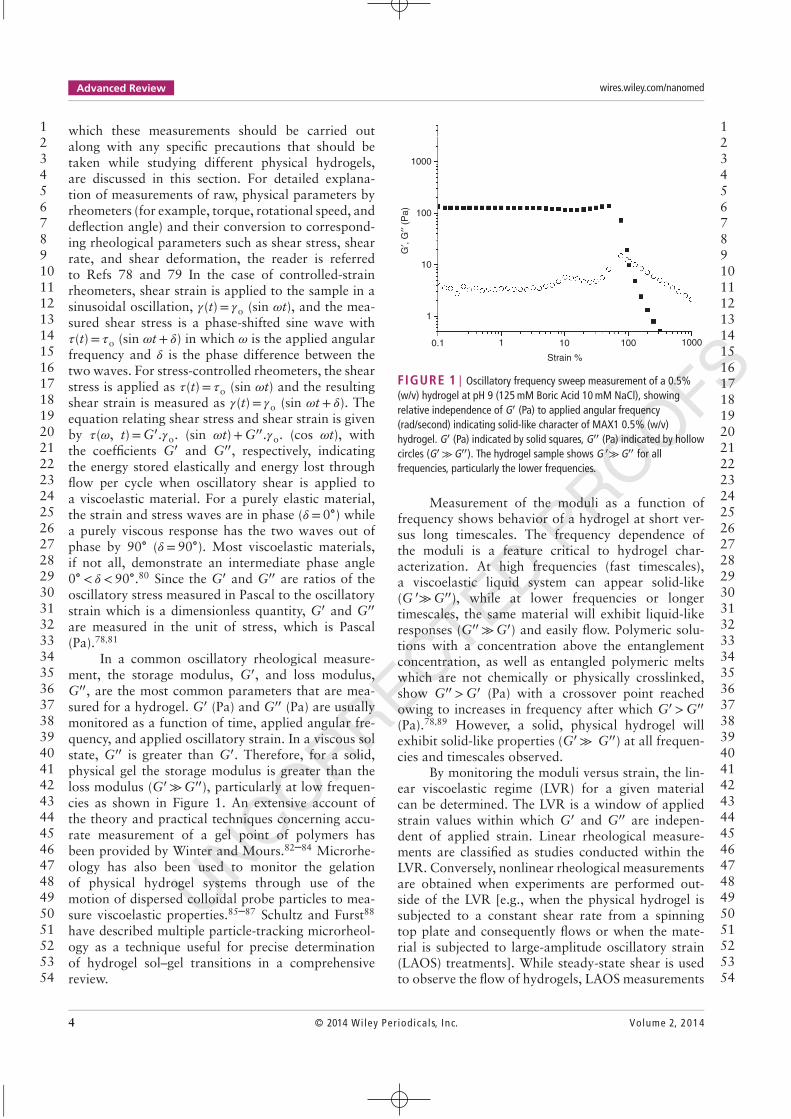
FIGURE 1 | Oscillatory frequency sweep measurement of a 0.5%(w/v) hydrogel at pH 9 (125 mM Boric Acid 10 mM NaCl), showingrelative independence of G′ (Pa) to applied angular frequency(rad/second) indicating solid-like character of MAX1 0.5% (w/v)hydrogel. G′ (Pa) indicated by solid squares, G′′ (Pa) indicated by hollowcircles (G′ ≫G′′). The hydrogel sample shows G ′≫G′′ for allfrequencies, particularly the lower frequencies.
Measurement of the moduli as a function offrequency shows behavior of a hydrogel at short ver-sus long timescales. The frequency dependence ofthe moduli is a feature critical to hydrogel char-acterization. At high frequencies (fast timescales),a viscoelastic liquid system can appear solid-like(G ′≫G′′), while at lower frequencies or longertimescales, the same material will exhibit liquid-likeresponses (G′′ ≫G′) and easily flow. Polymeric solu-tions with a concentration above the entanglementconcentration, as well as entangled polymeric meltswhich are not chemically or physically crosslinked,show G′′ >G′ (Pa) with a crossover point reachedowing to increases in frequency after which G′ >G′′
(Pa).78,89 However, a solid, physical hydrogel willexhibit solid-like properties (G′ ≫ G′′) at all frequen-cies and timescales observed.
By monitoring the moduli versus strain, the lin-ear viscoelastic regime (LVR) for a given materialcan be determined. The LVR is a window of appliedstrain values within which G′ and G′′ are indepen-dent of applied strain. Linear rheological measure-ments are classified as studies conducted within theLVR. Conversely, nonlinear rheological measurementsare obtained when experiments are performed out-side of the LVR [e.g., when the physical hydrogel issubjected to a constant shear rate from a spinningtop plate and consequently flows or when the mate-rial is subjected to large-amplitude oscillatory strain(LAOS) treatments]. While steady-state shear is usedto observe the flow of hydrogels, LAOS measurements
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
4 © 2014 Wiley Per iodica ls, Inc. Volume 2, 2014

UNCORRECTED PROOFS
WIREs Nanomedicine and Nanobiotechnology Rheology of peptide- and protein-based physical hydrogels
are instrumental in characterizing large, rapid materialdeformations and help offer a more complete perspec-tive of soft material responses to processing. LAOSmeasurements and some typical rheological responsesfrom complex fluids have been reviewed by Hyunet al.90 and Deshpande et al.91 In order to ascertainthe strain and frequency windows in which the stud-ied hydrogel behaves like a solid (i.e., measurementsare performed in the LVR), the oscillatory strain sweepmeasurement and the frequency sweep measurementshould be the first measurements to be carried out ona hydrogel during its rheological characterization. Aprecise idea of frequency and strain response is impor-tant since the values of G′ and G′′ measured by therheometer during an oscillatory time sweep measure-ment (e.g., during initial gelation or during rehealingafter shear-thin flow) will be at constant frequency andneed to be at strains within the LVR.
Common rheological measurements such as evo-lution of G′ and G′′ over time or frequency canhelp the researcher gain knowledge about the mech-anism and kinetics of physical gel network forma-tion. In particular, various solution stimuli such aspH, temperature, specific metal ions, enzymes, andlight are instrumental in triggering the assembly ofpeptides and polypeptides. Rheology provides a directview of the effects of different types and magni-tudes of assembly stimuli on peptides designed tobe stimulus-dependent materials. Extensive reviews ofstimuli-dependent (pH, salt, cations/anions, tempera-ture, enzyme) peptide-based, self-assembled materialshave been provided by Lowik et al.92 and Raeburnet al.93 For example, zinc ion-triggered assembly ofa 𝛽-hairpin peptide containing a zinc binding non-natural amino acid residue has been reported toform hydrogels by Micklitsch et al.94 Cation-inducedhydrogelation of napthylalanine dipeptides at high pHhas been reported by Chen et al.95 Similarly, a seriesof functionalized dipeptide systems were reported toundergo salt-induced gelation in solution to formshear-thinning and rehealing hydrogels.67 In anotheraccount, cysteine-containing 𝛽-hairpin peptides under-going metal-triggered assembly by binding to metalssuch as arsenic have been reported by Knerr et al.96
Toledano et al.97 have discussed protease-triggeredself-assembly of peptides into nanofibrous hydrogelsvia reversed hydrolysis. In a vast majority of thesereports and the reports reviewed by Lowik et al.,92
the magnitude of triggering stimulus greatly influ-ences peptide and polypeptide self-assembly pathwaysand, in some cases, ultimate mechanical propertiesof the hydrogels formed. For some peptide-basedgels, the rate of peptide assembly strongly influ-ences bulk mechanical hydrogel properties such as
stiffness.40,46,98–100 For these systems, the faster thekinetics of self-assembly, the stiffer the formed hydro-gels are. An example is shown in Figure 2, where thedependence of gel formation over time relative to dif-ferent solution ionic strengths was demonstrated usingan oscillatory time sweep measurement for G′, withthe value of G′ ≫G′′ for all samples. The assembling𝛽-hairpin peptide samples at weaker stimulus, which isthe lower ionic-strength buffer solution conditions inthis case, show a characteristic lag time while devel-oping into fully percolated gels depending upon theionic strength. The sample with 20 mM NaCl concen-tration shows the highest lag time of about 45 minand leads to the least stiff hydrogels, while the samplewith 400 mM NaCl concentration assembles fastestwith almost no lag time and leads to the stiffest hydro-gels. The hydrogel sample triggered using a buffer with150 mM NaCl concentration shows an intermediatelag time of ∼15 min as compared to the 20 mM and400 mM NaCl before it shows a sharp increase in G′
values. Interestingly, the final hydrogels all have thesame peptide concentration despite having moduli thatdiffer over an order of magnitude, a clear indicationof differences in network structure due to the differ-ent assembly stimuli magnitude and resultant networkstructures. A final example of stimuli-triggered assem-bly is the specially designed 𝛽-hairpin peptide that con-tains a photocage that is released from the sequencewhen the peptide solution is irradiated by UV radi-ation as discussed by Haines et al.101 The UV irra-diation produced consequent peptide assembly andhydrogelation as evidenced by oscillatory rheology.The stimuli-triggered assembly behavior and hydro-gel rheological characterization of peptides demon-strate the diversity of the types of stimuli that can beused to assemble different peptide designs. Many moreexamples of peptide and polypeptide physical assem-bly into hydrogels will be discussed in the remainderof the paper.
In addition to the study of gelation kineticsand the effects of different stimuli on assembly, rhe-ological measurements have a particular relevancein studying mechanical properties of biomaterialsintended as tissue engineering scaffolds and cellculturing substrates. For example, the mechanicalproperties of hydrogels are important in applica-tions as scaffolds until formation of extracellularmatrix (ECM) by cells is complete.102,103 For appli-cations of hydrogels as therapeutic cell deliveryvectors, cellular viability and phenotype depend onhydrogel mechanical properties.104 When hydrogelsare used as substrates for tissue engineering, cel-lular differentiation, proliferation, and migrationare influenced by hydrogel mechanical properties
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
Volume 2, 2014 © 2014 Wiley Per iodica ls, Inc. 5

UNCORRECTED PROOFS
Advanced Review wires.wiley.com/nanomed
10,000
1000
100
G′ (
Pa)
10
0 30 60
Time (min)
90 120 1501
FIGURE 2 | Dynamic Time Sweep (1% strain, 6 rad/second) of 2 wt% MAX1 [(VK)4-VDPPT-(KV)4] solution with 20 mM (G′, filled circles),150 mM (G′, filled triangles and 400 mM (G′, filled squares) NaCl at20∘C. For all three samples (G ′≫G′′) indicating gel-like rheologicalbehavior from all samples at all times. (Reprinted •with permissionAQ2from Ref 46)
as discussed by Discher et al.105 A seminal reportfrom the Discher group106 discusses the influenceof hydrogel matrix elasticity on stem cell lineagespecification. Oda et al.107 have reported dependenceof proliferation rate and cell cycles of pluripotentstem cells on the storage moduli of the hydrogelswhen the cells are encapsulated within chemicallycrosslinked hydrogels. The hydrogels were based onpoly(2-methacryloyloxyethyl phosphorylcholine-co-n-butyl methacrylate-co-p-vinylphenylboronic acid)(PMBV) and poly(vinyl alcohol) (PVA) polymers withfinely tunable storage modulus values. The conver-sion of mechanical signals from their environmentto biochemical signals by cells due to cell-surfacereceptor-mediated connection between the ECM andcytoskeleton has also been suggested. These examplesunderscore the importance of mechanical propertiesof hydrogels that can be potentially used as ECMmimetic materials.108,109
An important choice relevant to rheological mea-surement pertains to the instrument geometry usedin the rheological measurement. Different types ofrheometer geometries are available for making rheo-logical measurements namely parallel plate, cone andplate, concentric cylinder, double walled concentriccylinder, and combined cone and plate with cylinder(Mooney/Ewart measuring systems).79 For rheologi-cal measurements on physical hydrogels, parallel plateand cone and plate geometries are most popular. Thecone and plate geometry consists of a flat plate anda low-angle cone 𝜃 (1∘ <𝜃 <4∘) that rotates againstthe flat plate. A truncated cone may also be used toavoid damage to the apex of the cone and preventing
trapping of sample particles between the apex and theflat plate. The main advantage of the cone and plategeometry is that the shear rate does not vary fromthe axis of rotation because both the linear velocityand the gap between the cone and plate increase withincrease of distance from the rotational axis. Paral-lel plate geometry consists of two parallel plates withadjustable gap for holding the sample between theplates. The advantage of this geometry is the variablegap that can be adjusted to accommodate dispersionswith large particles or domain sizes as well as samplesthat have large heterogeneities in structure (e.g., largevariation in pore sizes). For parallel plates, variationsin shear rate across the gap height and instabilities inshear field at high rates are observed.110 Thus, in caseof parallel plates, calculation of the actual shear ratefrom the shear rates measured at different gap must becarried out. Synthetic limitations often constrain theamount of peptide or protein available for the formu-lation of a peptidic or protein-based hydrogel. Giventhis constraint, the cone and plate geometry is typicallyimplemented for rheological studies, as it requires theleast amount of sample volume for a measurement.Cone and plate geometries are not the best choice forparticulate hydrogels or hydrogels with large domainsizes like hierarchically assembled fibers, when the par-ticle size can be on the order of the gap size.110 Samplesedimentation in the case of heterogeneous hydrogelsover time that results in a layer depleted of particlesnear the cone has also been reported to skew the rhe-ological measurement.110 Thus, for particulate hydro-gels and gels containing large domain sizes, parallelplate geometries should be used.
Gap effects are commonly observed during rheo-logical characterization of soft, physically crosslinkedhydrogels. A precise calibration of the gap height, i.e.,the distance between the upper plate and a lower platein parallel plate geometries, must be obtained. Beforeconducting any measurement, the gap measurementat which two surfaces are in physical contact withone another should be calibrated as the ‘zero gap’.This zero gap calibration helps as reference in settinga desired gap height for the experiments. When mea-surements of G′ and G′′ are carried out for the samesample at different gap heights, different values of themeasured parameters may be obtained because of theeffects of heterogeneity. Thus, multiple measurementsat various gap heights should be carried out and a suit-able gap height should be fixed such that values ofmeasured parameters stay constant at heights abovethis gap height.
Similarly, flow-induced heterogeneity with orwithout shear and specific physical or chemical inter-actions between the sample and the plate walls can
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
6 © 2014 Wiley Per iodica ls, Inc. Volume 2, 2014

UNCORRECTED PROOFS
WIREs Nanomedicine and Nanobiotechnology Rheology of peptide- and protein-based physical hydrogels
lead to wall slippage and distort the measurement.111
Multiple measurements carried out at various gapheights can also help in analyzing the presence ofpotential wall slip, which, if present, can lead to dif-ferences in measurements at different gap heights. Wallslip can be minimized in case of measurements usingparallel plate geometries by roughening of the geome-try surface, ensuring adequate momentum exchangewith the gel thereby avoiding slip. A simple solu-tion toward eliminating wall slip is the attachmentof a piece of high grit sand paper to either or bothplates of a bench-top rheometer. Detailed accountsof attempts made at determination and eliminationof wall slip have been given by Yoshimura et al.112
and Carotenuto et al.,113 who specifically discuss theimplementation of rough tool walls in order to avoidslip in their rheological measurements. Walls et al.114
reported a significant reduction in wall slip during rhe-ological characterization of fumed silica gels by usinggeometries with rough surfaces as opposed to geome-tries with smooth surfaces. Clasen et al.115 and Keles-sidis et al.116 have presented specific examples of thedetermination wall slip in yield-stress fluids.
Another important consideration for rheologi-cal measurements on peptide- and polypeptide-basedhydrogels is the material used to fabricate the rheome-ter geometries. Some examples of materials usedto fabricate rheometer geometries include stainlesssteel and acrylate resins. Peptides and polypeptidesdemonstrate a multitude of secondary interactionssuch as hydrophobic interactions, hydrogen bonding,and electrostatic interactions. Taking such interactionsinto account, a careful choice of geometry based onthe material used to fabricate it should be made. Formost experimental setups, the geometries used forthe characterization of hydrogels from these systemsshould be, ideally, chemically inert to these systems.For samples that demonstrate wall slip at the tool sur-face, it is sometimes beneficial to ensure appropriateadhesive interactions between the sample and the toolsurface via secondary interactions to avoid wall slip.Any potential physical bonding interactions betweenthe materials under study and the rheometer geome-try should be carefully accounted for while interpret-ing rheological data using the concerned geometry.For example, if hydrophobic interactions dominatethe self-assembly and hydrogel formation from cer-tain peptide molecules and a stainless steel or acrylicrheometer geometry is used to probe the gelationkinetics, hydrophobic interactions between the pep-tide molecules and the geometry must be considered.Hydrophilic-functionalized geometries might help inminimization of hydrophobic interactions betweenthe geometry and the sample in such a case. As
an experimental illustration of sample–plate interac-tions, Walls et al.114 have reported that in case of ahydrophobic sample, a fumed silica gel, hydropho-bic plates undergo hydrophobic interactions leading todecrease in wall slip. When hydrophilic plates are usedthey repel the hydrophobic silica sample leading to aparticle-lean layer near the plate leading to occurrenceof wall slip.
The next section describes solid injectable hydro-gels based on 𝛽-hairpin peptides that undergo hierar-chical self-assembly into fibrillar nanostructures. Therheological characterization of the hydrogels basedon this family of peptides has been conducted tak-ing the above-described protocols and precautionsinto consideration. While these hydrogels demonstrateshear-thinning and rehealing behavior when investi-gated with a bench-top rheometer, they undergo bulkfracture under steady-state shear as evidenced by theconfocal-rheometry experimental study described inthe next section.
BIOMEDICALLY PROMISING SOLIDINJECTABLE HYDROGELS FROMSELF-ASSEMBLING 𝜷-HAIRPINPEPTIDES
Hydrogels from 𝜷-Hairpin Peptides Basedon the Parent Sequence MAX1[VKVKVKVK-(VDPPT)-KVKVKVKV-NH2]Solid, injectable hydrogels are formed by the hier-archical self-assembly of 𝛽-hairpin forming peptides.The 𝛽-hairpin peptide assembly process leading tohydrogel formation, and the importance of rheolog-ical characterization in studying hydrogel mechanicalproperties, gelation kinetics, and yield-stress materialbehavior is discussed in this section. The rheologicalcharacterization of these hydrogels has been carriedout in accordance to the protocols and precautions dis-cussed in Section Rheological Measurements of Phys-ical Hydrogels: Protocols and Precautions.
The Pochan and Schneider groups have studiedextensively the ‘MAX’ family of peptides, whichis based on a parent sequence MAX1.45,46,117–119
MAX1 is a 20 amino acid residue amphiphilic peptidesequence VKVKVKVK-(VDPPT)-KVKVKVKV-NH2,with alternating hydrophobic valine (V) andhydrophilic lysine (K) residues.45 The type II’ turnsequence -VDPPT-; where DP is the nonnatural,right-handed enantiomer of proline, P is the natural,left-handed proline amino acid, and T is threo-nine; in the center is responsible for chain reversal and𝛽-hairpin formation at elevated pH, ionic strength andtemperature solution conditions from a random coil
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
Volume 2, 2014 © 2014 Wiley Per iodica ls, Inc. 7

UNCORRECTED PROOFS
Advanced Review wires.wiley.com/nanomed
(d)
(f)(e)50 nm
(c)(b)(a)
FIGURE 3 | Schematic of MAX1 self-assembly (a) MAX1 random coil conformation at low to neutral pH and low temperature, the pink sidechains represent lysine side chains and the blue side chains represent valine side chains. (b) 𝛽-Hairpin conformation induced by rise in the pH,temperature, and/or ionic strength of the peptide solution. (c) Subsequent to intramolecular folding, facial hydrophobic collapse of two hairpinsleading to formation of a bilayer type structure. (d) Direction of lateral hydrophobic interactions among multiple bilayer type structures (e)hierarchically assembled branched fibril of MAX1. (f) Cryogenic transmission electron microscopy showing fibrillar structure of MAX1.
conformation in aqueous solution of low to neutralpH and low ionic strength (Figure 3(b)). An increasein pH of an acidic/neutral peptide solution serves todeprotonate some of the lysine side chain primaryamino groups and allows folding and self-assemblyto occur. In addition, increasing the ionic strength ofa neutral peptide solution can also initiate peptidefolding and assembly by screening the positive chargeson the lysine side chains, both leading to 𝛽-hairpinformation. The 𝛽-hairpin conformation has the twoamphiphilic arms of MAX1 parallel to each otherstabilized by intramolecular hydrogen bonding. Anincrease in temperature emphasizes hydrophobic inter-actions primarily between the valine side chains andserves as another factor promoting the peptide foldingand self-assembly. Facial hydrophobic interactions(Figure 3(c)) at the valine face between two 𝛽-hairpinsform the core of the growing nanofibrils.117 Lateralintermolecular hydrophobic interactions (Figure 3(d))along the axis of the growing fibrils, along with thefacial hydrophobic interactions, lead to hierarchicalassembly of these 𝛽-hairpins into uniform fibrils(Figure 3(e)).118 Lateral intermolecular hydrogenbonding interactions also play a significant role in theassembly. Sometimes, during the facial hydrophobiccollapse, two hairpins undergo an incomplete burial
of the hydrophobic face. This incomplete burial mani-fests itself as a defect that is responsible for nucleationof two fibrils emanating from the defect junction.Thus, these junctions of fibrils act as branching pointsand contribute to physical crosslinking of the fibrilsin addition to fibrillar entanglement leading to forma-tion of self-standing, solid hydrogels.120 The cryogenictransmission electron micrograph (Figure 3(f)) showsthe uniform fibrillar structure of the MAX1 net-work. The fibrils have a uniform thickness ∼3 nm,corresponding to the strand length of each 𝛽-hairpinof MAX1.
In case of derivatives of MAX1 with slightlydifferent primary structures, the specific pH, ionicstrength, and temperature conditions, or suitablecombination of these solution parameters, used for𝛽-hairpin formation and consequent intermolecularassembly and gelation are dependent on the specificpeptide primary sequence. As an example, the peptideMAX8 is obtained by point substitution of a posi-tively charged lysine residue in MAX1 with a glutamicacid residue with a negative charge. At the same pep-tide concentration and solution conditions, MAX8demonstrates faster assembly kinetics owing to lessoverall positive charge (+7 as compared with +9 incase of MAX1) and additional attractive electrostatic
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
8 © 2014 Wiley Per iodica ls, Inc. Volume 2, 2014
UNCORRECTED PROOFS
WIREs Nanomedicine and Nanobiotechnology Rheology of peptide- and protein-based physical hydrogels
interactions between the added glutamic acid andlysine residues.40,121 MAX8 peptides can undergoself-assembly to form hydrogel materials under phys-iological pH (∼7.4),121 ionic strength30 (∼150 mMsalt), and temperature conditions (37∘C).30,52,122
MAX8 allows uniform three-dimensional (3D) livingmesenchymal cell or drug payload encapsulationdue to the fast gelation time (<1 min vs ∼30 minfor MAX1) at physiological solution conditions.122
This expedited gelation leading to ultimately stifferhydrogels from MAX8, as compared to those fromMAX1 at the same peptide concentration and solu-tion conditions, has been shown clearly by oscillatoryrheological measurements.123 Thus, MAX8 hasparticular relevance to homogeneous 3D cell encap-sulation and potential tissue engineering applications.Leonard et al.124 have provided solid-state NMRspectroscopy evidence of the 𝛽-hairpin conformationbeing the dominant conformation within assembledfibrils of MAX8 peptide. MAX8 also has been shownto have a very similar fibrillar nanostructure as MAX1in terms of fibril width (∼3.2 nm).120 Owing to fastergelation kinetics than MAX1, MAX8 fibrils formnetworks with smaller pore sizes indicating morecrosslink density of MAX8 fibrils as compared toMAX1 as evidenced by cryogenic transmission elec-tron microscopy (cryo-TEM) and small-angle neutronscattering (SANS).125 Hydrogels based on MAX1and MAX8 have demonstrated cytocompatibility,126
non-inflammatory properties122 and in some casesbiologically effective properties such as antibacte-rial activity (MAX1127 and its derivatives47). Thesepeptide systems provide flexibility of peptide struc-ture through introduction of specific biochemicalfunctionalities such as post-self-assembly chemicalcrosslinking to yield stiffer hydrogels.51 Macro-molecule self-diffusion and bulk release studies withMAX1 and MAX8 hydrogels have shown macro-molecule mobility within, and release out of, thegels.27,29 This demonstrates the ability of the porous𝛽-hairpin peptide hydrogels with tunable mesh sizesas viable candidates for tissue engineering and drugdelivery scaffolds since they allow transport of nutri-ents and metabolites. Thus, owing to the favorabletherapeutic encapsulation, initial biocompatibilityand, in some cases, bioactivity, MAX1 and MAX8demonstrate significant potential for their use inbiomedical applications, particularly in light of theinjectable solid attributes displayed as discussedbelow.
A well-defined linear viscoelastic region andyield strain value (∼10–40%) dependent upon the spe-cific peptide sequence and solution conditions usedto trigger self-assembly are demonstrated by these
2500
2000
1500
1000G´, G
´´ (
Pa)
500
00 20 40 60 80
Time (min)100 120 140 160 180
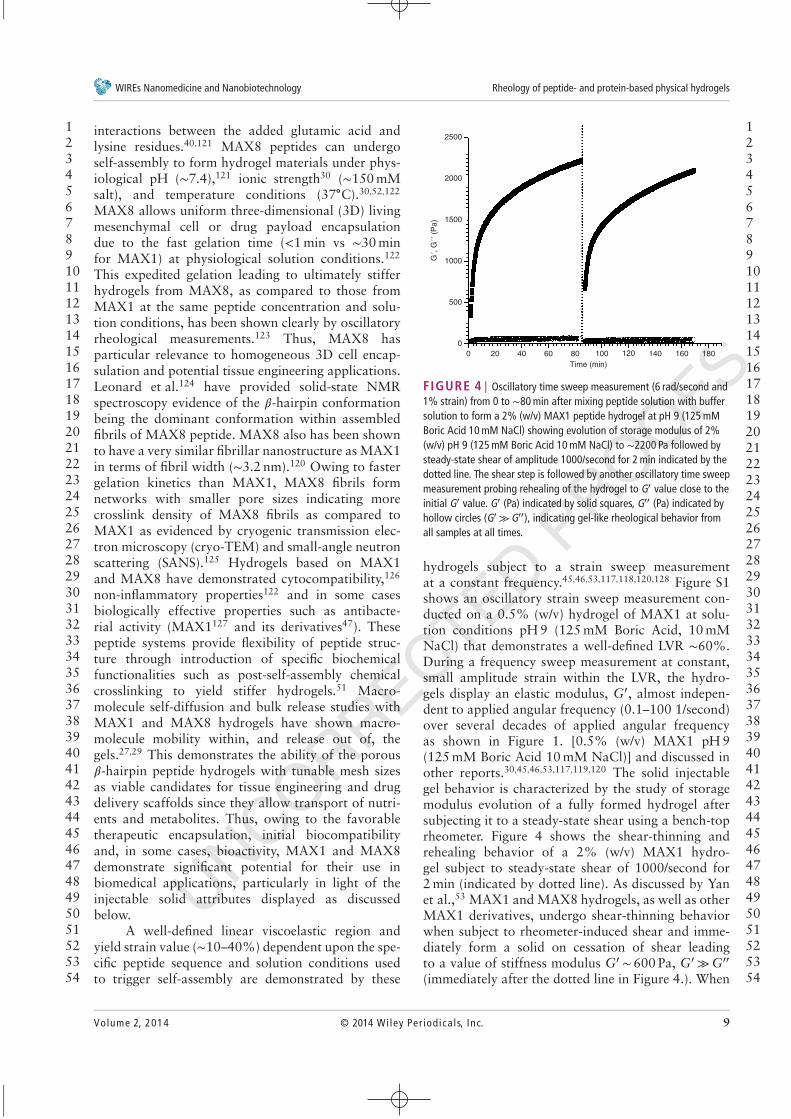
FIGURE 4 | Oscillatory time sweep measurement (6 rad/second and1% strain) from 0 to ∼80 min after mixing peptide solution with buffersolution to form a 2% (w/v) MAX1 peptide hydrogel at pH 9 (125 mMBoric Acid 10 mM NaCl) showing evolution of storage modulus of 2%(w/v) pH 9 (125 mM Boric Acid 10 mM NaCl) to ∼2200 Pa followed bysteady-state shear of amplitude 1000/second for 2 min indicated by thedotted line. The shear step is followed by another oscillatory time sweepmeasurement probing rehealing of the hydrogel to G′ value close to theinitial G′ value. G′ (Pa) indicated by solid squares, G′′ (Pa) indicated byhollow circles (G′ ≫G′′), indicating gel-like rheological behavior fromall samples at all times.
hydrogels subject to a strain sweep measurementat a constant frequency.45,46,53,117,118,120,128 Figure S1shows an oscillatory strain sweep measurement con-ducted on a 0.5% (w/v) hydrogel of MAX1 at solu-tion conditions pH 9 (125 mM Boric Acid, 10 mMNaCl) that demonstrates a well-defined LVR ∼60%.During a frequency sweep measurement at constant,small amplitude strain within the LVR, the hydro-gels display an elastic modulus, G′, almost indepen-dent to applied angular frequency (0.1–100 1/second)over several decades of applied angular frequencyas shown in Figure 1. [0.5% (w/v) MAX1 pH 9(125 mM Boric Acid 10 mM NaCl)] and discussed inother reports.30,45,46,53,117,119,120 The solid injectablegel behavior is characterized by the study of storagemodulus evolution of a fully formed hydrogel aftersubjecting it to a steady-state shear using a bench-toprheometer. Figure 4 shows the shear-thinning andrehealing behavior of a 2% (w/v) MAX1 hydro-gel subject to steady-state shear of 1000/second for2 min (indicated by dotted line). As discussed by Yanet al.,53 MAX1 and MAX8 hydrogels, as well as otherMAX1 derivatives, undergo shear-thinning behaviorwhen subject to rheometer-induced shear and imme-diately form a solid on cessation of shear leadingto a value of stiffness modulus G′ ∼ 600 Pa, G′ ≫G′′
(immediately after the dotted line in Figure 4.). When
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
Volume 2, 2014 © 2014 Wiley Per iodica ls, Inc. 9

UNCORRECTED PROOFS
Advanced Review wires.wiley.com/nanomed
allowed to age, these hydrogels heal into networkswith modulus G′ comparable to that of the net-work pre-shear (∼2000 Pa). The significant observa-tion from the results shown in Figure 4 is that thehydrogels have G′ (∼600 Pa)≫G′′ (∼20 Pa) immedi-ately after cessation of shear, indicating a solid perco-lated network nature. This can be considered as the‘recovery’ of the sheared materials under flow to asolid network immediately after shear cessation. Theincrease of the G′ (Pa) values to pre-shear values overseveral hours is indicative of further healing/stiffeningof the hydrogels over time, with a decrease in thetan 𝛿 and a corresponding increase in elastic natureof the hydrogels. The rheological characterization ofthe MAX1/MAX8 hydrogels demonstrates the utilityof these materials as preformed solid hydrogels thatcan retain their solid nature when injected in vivo.This shear-thinning and rehealing behavior will bediscussed in more detail later in Section RheologicalCharacterization of the Shear-Thinning and RehealingBehavior of 𝛽-Hairpin Hydrogels.
Along with MAX1 and MAX8, different pri-mary structures of MAX1 derivative peptides leadto differences in the assembly stimulus requirement,assembly kinetics, the nanostructure, and mechani-cal properties of hydrogels formed from the peptides.The examples discussed below demonstrate differ-ences in physical properties of hydrogels from pep-tides with different primary structures based on theMAX1 peptide sequence. For example, the MAX3peptide is obtained by substitution of two hydropho-bic side chain valine residues with hydrophilic sidechain threonine residues. Owing to these point sub-stitutions, the MAX3 peptide under the same solutionconditions as MAX1 forms hydrogels only at a muchhigher temperature (∼70∘C) than MAX1 at the samesolution conditions (∼30∘C). When cooled to temper-atures significantly below 70∘C (∼5∘C), gelation andassembly are reversed and the material becomes a lowviscosity solution.117 MAX3 hydrogels can undergomultiple cycles of sol to gel and gel to sol transi-tions. [Such reversibility of hydrogel like propertiesis observed also in case of MAX1 when temperatureis used as the dominant stimulus for self-assemblyalong with appropriate pH/ionic-strength solutionconditions (i.e., low pH, low ionic strength).] Thesecyclic changes in MAX3 mechanical properties wereobserved using oscillatory rheological measurements.Other examples of MAX1 derivatives include pep-tides with varying properties such as twisted fib-rils, non-twisted and laminated fibrils all formingstiff, shear-thinning and rehealing hydrogels.129–131
Owing to the designs of the appropriately named‘strand swapping peptides’, SSP1, SSP2, and SSP3
undergo strand swapping. Hydrophobic associationof strand-swapped dimers drives core fibril structureformation. But, instead of two hairpins forming a fib-ril cross section in the case of MAX1, four hairpinsare required to form a fibril structure cross sectionby burial of valine side chains in the SSP peptides.Thus, effectively twice the concentration of peptideswas required to form fibrils and hydrogels with SSPpeptides as compared to MAX1 as shown via rhe-ology. The strand swapping incurred during peptidefolding results in the formation of different nanostruc-tures are formed from SSP1 (singular twisted fibrillar),SSP2 (singular non-twisted fibrillar), and SSP3 (lami-nated non-twisted fibrillar).
Rheological measurements also were usedto probe the controlled biodegradation of peptidehydrogels based on a series of degrading peptides(DP) through interaction with metalloproteinase-13(MMP-13).132 Oscillatory rheological characteriza-tion was used to measure stiffness values of hydrogelsas they were subject to degradation using MMP-13and thus helped directly validate that the enzymeswere degrading the peptide fibrils that constituted thehydrogel. Hydrogel based on enantiomeric mixturesof self-assembling 𝛽-hairpins (MAX1 and D-MAX1)showing non-additive, synergistic, enhancement inmaterial rigidity, compared to gels prepared fromeither pure enantiomer, have been reported by Nagyet al.133 The fibrillar morphology of the hydrogelformed from the enantiomeric mixture is negligiblydifferent from that based on either pure enantiomer.Results from rheometeric experiments are useful indemonstrating the non-additive mechanical synergis-tic effects when peptide enantiomers are mixed.
Similar to 𝛽-hairpin peptides, hydrogels fromamphiphilic linear peptides that form hydrogels basedon tapes and laminates have been reported.49 Theabsence of a 𝛽-hairpin conformation affects the nanos-tructure formed from linear peptides, but the hydro-gels can be studied rheologically. Many examples oflinear peptides are discussed later in the paper.
Rheological Characterization of theShear-Thinning and Rehealing Behaviorof 𝜷-Hairpin HydrogelsAs discussed above, rheological conditions thatemploy small amplitude oscillatory strain conditionshave been used to determine various mechanicalattributes of hydrogels based on MAX1, MAX8, andderivative peptide sequences. Results from rheologicalmeasurements help in the understanding of variousproperties such as hydrogel stiffness, linear viscoelas-tic region windows, and yield-stress values of these
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
10 © 2014 Wiley Per iodica ls, Inc. Volume 2, 2014
UNCORRECTED PROOFS
WIREs Nanomedicine and Nanobiotechnology Rheology of peptide- and protein-based physical hydrogels
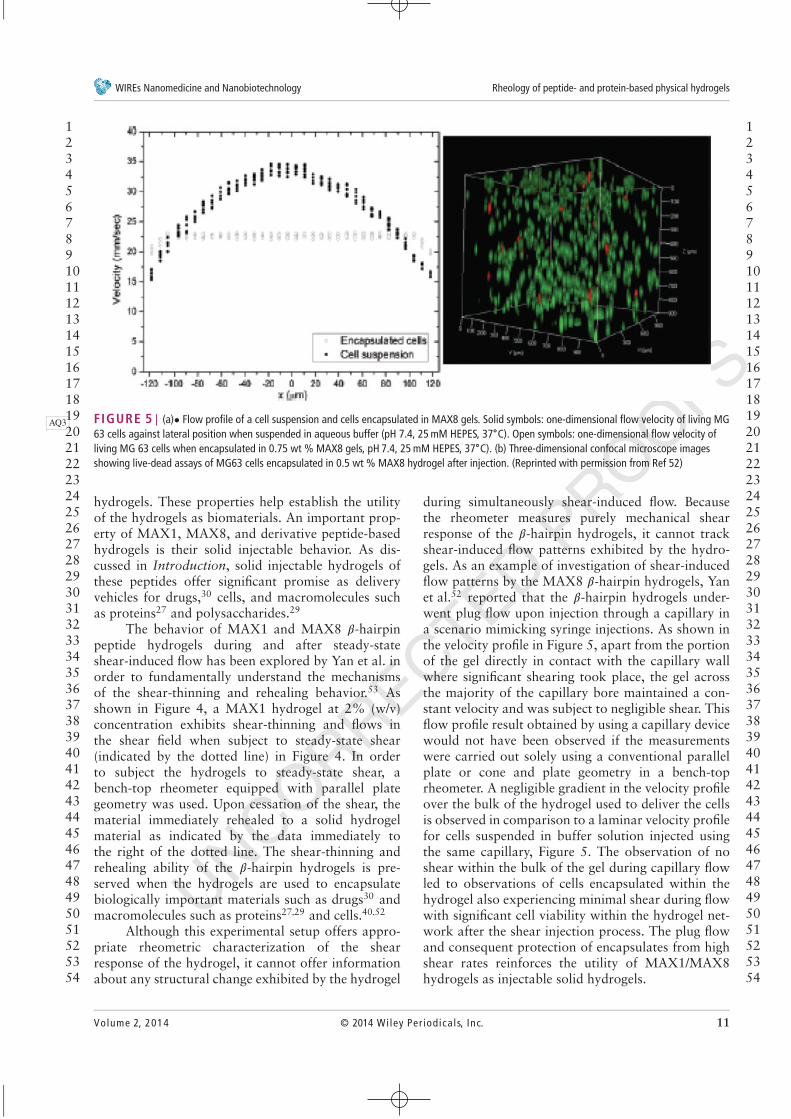
FIGURE 5 | (a)• Flow profile of a cell suspension and cells encapsulated in MAX8 gels. Solid symbols: one-dimensional flow velocity of living MGAQ363 cells against lateral position when suspended in aqueous buffer (pH 7.4, 25 mM HEPES, 37∘C). Open symbols: one-dimensional flow velocity ofliving MG 63 cells when encapsulated in 0.75 wt % MAX8 gels, pH 7.4, 25 mM HEPES, 37∘C). (b) Three-dimensional confocal microscope imagesshowing live-dead assays of MG63 cells encapsulated in 0.5 wt % MAX8 hydrogel after injection. (Reprinted with permission from Ref 52)
hydrogels. These properties help establish the utilityof the hydrogels as biomaterials. An important prop-erty of MAX1, MAX8, and derivative peptide-basedhydrogels is their solid injectable behavior. As dis-cussed in Introduction, solid injectable hydrogels ofthese peptides offer significant promise as deliveryvehicles for drugs,30 cells, and macromolecules suchas proteins27 and polysaccharides.29
The behavior of MAX1 and MAX8 𝛽-hairpinpeptide hydrogels during and after steady-stateshear-induced flow has been explored by Yan et al. inorder to fundamentally understand the mechanismsof the shear-thinning and rehealing behavior.53 Asshown in Figure 4, a MAX1 hydrogel at 2% (w/v)concentration exhibits shear-thinning and flows inthe shear field when subject to steady-state shear(indicated by the dotted line) in Figure 4. In orderto subject the hydrogels to steady-state shear, abench-top rheometer equipped with parallel plategeometry was used. Upon cessation of the shear, thematerial immediately rehealed to a solid hydrogelmaterial as indicated by the data immediately tothe right of the dotted line. The shear-thinning andrehealing ability of the 𝛽-hairpin hydrogels is pre-served when the hydrogels are used to encapsulatebiologically important materials such as drugs30 andmacromolecules such as proteins27,29 and cells.40,52
Although this experimental setup offers appro-priate rheometric characterization of the shearresponse of the hydrogel, it cannot offer informationabout any structural change exhibited by the hydrogel
during simultaneously shear-induced flow. Becausethe rheometer measures purely mechanical shearresponse of the 𝛽-hairpin hydrogels, it cannot trackshear-induced flow patterns exhibited by the hydro-gels. As an example of investigation of shear-inducedflow patterns by the MAX8 𝛽-hairpin hydrogels, Yanet al.52 reported that the 𝛽-hairpin hydrogels under-went plug flow upon injection through a capillary ina scenario mimicking syringe injections. As shown inthe velocity profile in Figure 5, apart from the portionof the gel directly in contact with the capillary wallwhere significant shearing took place, the gel acrossthe majority of the capillary bore maintained a con-stant velocity and was subject to negligible shear. Thisflow profile result obtained by using a capillary devicewould not have been observed if the measurementswere carried out solely using a conventional parallelplate or cone and plate geometry in a bench-toprheometer. A negligible gradient in the velocity profileover the bulk of the hydrogel used to deliver the cellsis observed in comparison to a laminar velocity profilefor cells suspended in buffer solution injected usingthe same capillary, Figure 5. The observation of noshear within the bulk of the gel during capillary flowled to observations of cells encapsulated within thehydrogel also experiencing minimal shear during flowwith significant cell viability within the hydrogel net-work after the shear injection process. The plug flowand consequent protection of encapsulates from highshear rates reinforces the utility of MAX1/MAX8hydrogels as injectable solid hydrogels.
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
Volume 2, 2014 © 2014 Wiley Per iodica ls, Inc. 11
UNCORRECTED PROOFS
Advanced Review wires.wiley.com/nanomed
0.20
0.25
0.15
0.10
Vel
ocity
(m
m/s
)
0.05
Average velocity = 0.151 mm/s
Capillary wall
Centerline
t
x
200 μm
0.000.0 0.1 0.2
Radial position (mm)
0.3 0.4
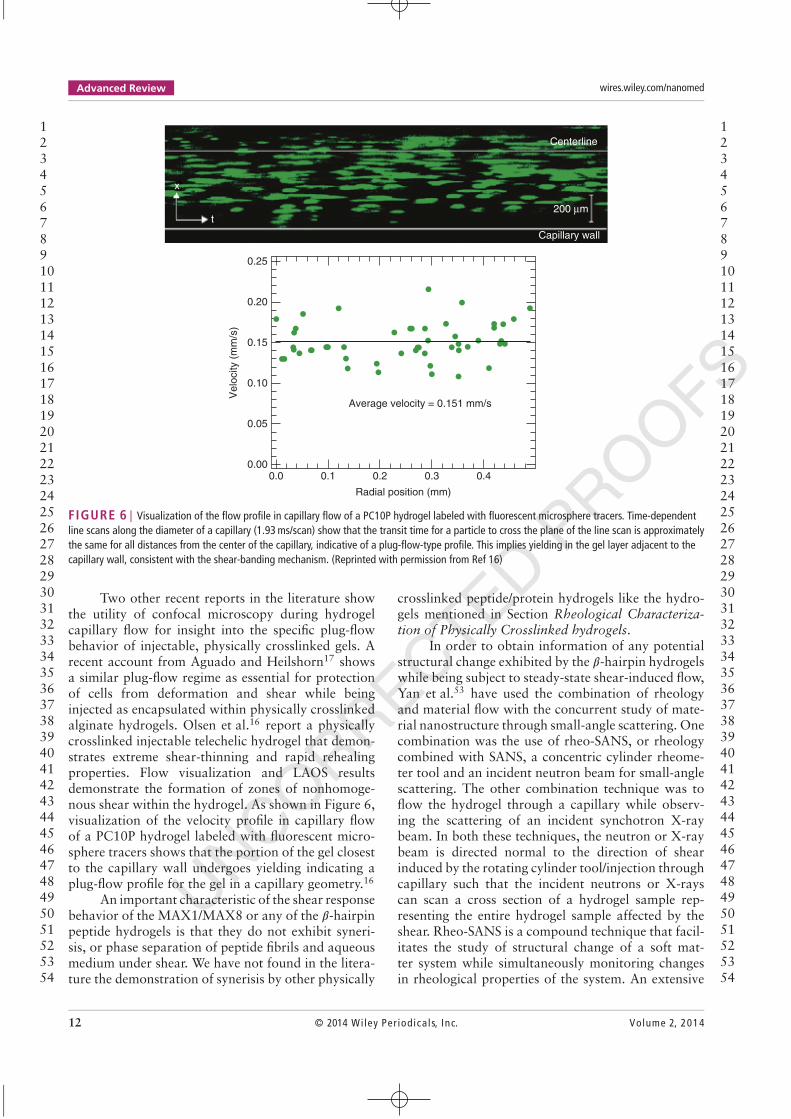
FIGURE 6 | Visualization of the flow profile in capillary flow of a PC10P hydrogel labeled with fluorescent microsphere tracers. Time-dependentline scans along the diameter of a capillary (1.93 ms/scan) show that the transit time for a particle to cross the plane of the line scan is approximatelythe same for all distances from the center of the capillary, indicative of a plug-flow-type profile. This implies yielding in the gel layer adjacent to thecapillary wall, consistent with the shear-banding mechanism. (Reprinted with permission from Ref 16)
Two other recent reports in the literature showthe utility of confocal microscopy during hydrogelcapillary flow for insight into the specific plug-flowbehavior of injectable, physically crosslinked gels. Arecent account from Aguado and Heilshorn17 showsa similar plug-flow regime as essential for protectionof cells from deformation and shear while beinginjected as encapsulated within physically crosslinkedalginate hydrogels. Olsen et al.16 report a physicallycrosslinked injectable telechelic hydrogel that demon-strates extreme shear-thinning and rapid rehealingproperties. Flow visualization and LAOS resultsdemonstrate the formation of zones of nonhomoge-nous shear within the hydrogel. As shown in Figure 6,visualization of the velocity profile in capillary flowof a PC10P hydrogel labeled with fluorescent micro-sphere tracers shows that the portion of the gel closestto the capillary wall undergoes yielding indicating aplug-flow profile for the gel in a capillary geometry.16
An important characteristic of the shear responsebehavior of the MAX1/MAX8 or any of the 𝛽-hairpinpeptide hydrogels is that they do not exhibit syneri-sis, or phase separation of peptide fibrils and aqueousmedium under shear. We have not found in the litera-ture the demonstration of synerisis by other physically
crosslinked peptide/protein hydrogels like the hydro-gels mentioned in Section Rheological Characteriza-tion of Physically Crosslinked hydrogels.
In order to obtain information of any potentialstructural change exhibited by the 𝛽-hairpin hydrogelswhile being subject to steady-state shear-induced flow,Yan et al.53 have used the combination of rheologyand material flow with the concurrent study of mate-rial nanostructure through small-angle scattering. Onecombination was the use of rheo-SANS, or rheologycombined with SANS, a concentric cylinder rheome-ter tool and an incident neutron beam for small-anglescattering. The other combination technique was toflow the hydrogel through a capillary while observ-ing the scattering of an incident synchotron X-raybeam. In both these techniques, the neutron or X-raybeam is directed normal to the direction of shearinduced by the rotating cylinder tool/injection throughcapillary such that the incident neutrons or X-rayscan scan a cross section of a hydrogel sample rep-resenting the entire hydrogel sample affected by theshear. Rheo-SANS is a compound technique that facil-itates the study of structural change of a soft mat-ter system while simultaneously monitoring changesin rheological properties of the system. An extensive
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
12 © 2014 Wiley Per iodica ls, Inc. Volume 2, 2014

UNCORRECTED PROOFS
WIREs Nanomedicine and Nanobiotechnology Rheology of peptide- and protein-based physical hydrogels
review of Rheo-SANS study of soft matter systemshas been provided by Eberle et al.134 The constructionof a modified commercial rheometer used to conductrheometric measurements while simultaneously beingable to monitor structural changes has been discussedby Porcar et al.135 SANS and small-angle X-ray scat-tering (SAXS) data from the hydrogels under shearhelp to elucidate any changes in hydrogel nanos-tructure during the process of rheometer-induced orcapillary-induced shear. Results from the combina-tion techniques demonstrate that there is no noticeablechange in hydrogel morphology during rheometer- orcapillary-induced shear. Based on these results andresults from bench-top rheology of 𝛽-hairpin peptide(MAX1/MAX8) hydrogels, a model has been pro-posed to explain how the gel network is fracturedinto domains during shear-thinning and flow.53 Thedomains can flow during shear but can immediatelyre-percolate into a solid hydrogel when the shearis stopped. Within the fractured domains exists thesame nanostructure, same fibrillar thickness and sameporosity as the original bulk network. The lack ofchanges in hydrogel nanostructure during rheometeror capillary-induced shear may also point to bulk frac-ture within a certain thickness of the hydrogel sampleaway from the shearing surface as explained by Yanet al.53 Instead of fracturing into domains within thebulk of the gel thus allowing the material to flow,a hydrogel layer near the stationary rheometer toolmay fracture from the bulk material and consequentlyflow. In this case, the bulk of the hydrogel wouldremain stationary and thus would appear as the orig-inal bulk network by scattering. Given the possibil-ity of bulk fracture of the hydrogel throughout thesample cross section or fracture limited to a certainlayer of the hydrogel sample, obtaining visual evidenceof the exact, possible fracture characteristics of theMAX1/MAX8 hydrogels via imaging techniques suchas confocal microscopy is an important step forwardin understanding the shear response from these solidinjectable hydrogels.
Given the plug-flow behavior of the injectablesolid MAX1 or MAX8 hydrogels, one could expectfracture of a surface layer next to the top rheome-ter plate in the hydrogels when subjected to sheartreatment using a rotating upper plate of a bench-toprheometer. Intuitively, it can be expected that the shearinduced by the rheometer upper plate might be frac-turing only a localized layer of the hydrogel very nearthe upper plate, while the bulk of the gel might benegligibly affected by shear. Thus, direct visualiza-tion of the flow pattern of the hydrogel when subjectto shear using a rheometer upper plate would revealthe exact flow patterns obtained using a bench-top
rheometer. Rehealing behavior of MAX1 or MAX8hydrogels after being subject to steady-state shear hasalready been observed to be affected by the differ-ences in amplitude and duration of the applied shear.53
For example, a hydrogel subject to 1000/second ofsteady-state shear for 2 min recovers to a lower valueof stiffness modulus, G′ (Pa), as compared to a hydro-gel subject to 1000/second of steady-state shear for5 seconds. Is this difference due to differences in bulkshear of the gels or to differences in the fracturedlayer experiencing shear? Thus, imaging during shearapplication would help verify hydrogel flow patternsand any fracture properties as functions of variousparameters associated with the shear treatment such asapplied stress amplitude, duration, and shear history.
In the experiments reported herein, a confocalrheometer136 was used to obtain direct visualizationof the structural response of 𝛽-hairpin hydrogel whensubject to steady-state shear using a bench-top par-allel plate rheometer. Experimental details and theinstrumental configuration have been described inSection Experimental. Carboxyl-functionalized, fluo-rescent microparticles were encapsulated within theMAX8 hydrogels used for the study. The fluorescentmicroparticles indicated the presence of the hydrogelacross the gap height when imaged using the confocalmicroscope in the device. Video data obtained usingthe confocal microscope are included in the Support-ing Information. These data show a sheared hydro-gel sample at various steady-state shear rates whileit is being viewed from the bottom of the hydrogelsample looking upwards at the rheometer upper plateas shown in Figure 7. Specifically, each video showsimages of multiple layers of the hydrogels at increasingheights within the sample away from the rheometerlower plate. These increasing heights are indicated byan increasing z value in the videos and the constant xand y values indicate the position of the layers relativeto the lower plate. The total thickness of the hydrogelscanned (μm), thickness of fractured layers (μm), thecorresponding rates of steady shear (second−1), andthe thickness of the fractured layers as a percentageof the total hydrogel thickness scanned are reported inTable 1. The video data obtained on the MAX8 hydro-gel at the different steady-state shear rates (5/second,50/second, 250/second, 400/second, and 1000/sec-ond) show fractured layers of different thicknesses.The total hydrogel thickness between rheometer platesfor the shear rate 5/second was 980 μm, while for theshear rates of 50/second, 250/second, 400/second, and1000/second the total gel thickness was 790 μm. Understeady-state shear of 5/second, the bulk of the encapsu-lated microparticles, i.e., the bulk of the MAX8 hydro-gel, undergoes zero or negligible flow. Only a layer
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
Volume 2, 2014 © 2014 Wiley Per iodica ls, Inc. 13

UNCORRECTED PROOFS
Advanced Review wires.wiley.com/nanomed
Rheometerupper plate
Hydrogelsampleat rest
Incident laserfrom confocalmicroscope 400/s
Tf~400 μm
50/sTf~
64 μm
5/sTf~
50 μm
Shear amplitude&
thickness offractured layer
Fracturedlayer
StationarylayerGlass window
for confocalmicroscope
FIGURE 7 | Schematic showing results obtained from confocal-rheometer compound assembly. The hydrogel undergoes shear rate-dependentfracture in a layer of certain thickness away from the upper plate of the rheometer geometry, while the rest of the hydrogel undergoes no or negligibleflow. The square schematics indicate volumes across the cross section of the hydrogel sample between the rheometer plates when the sample issubject to 5/second, 50/second, 250/second, 400/second, and 1000/second rate of steady-state shear. The light blue and dark blue layers indicate thefractured and consequently flowing layers and stationary layers of the MAX8 hydrogel, respectively. T f indicates the % thickness of the fractured layer.
of average thickness ∼23 μm near the upper plate (atthe top of the bulk hydrogel) shows flow of encapsu-lated microparticles. This result clearly shows evidenceof bulk fracture of the physically assembled MAX8hydrogel. When the same hydrogel is subject to asteady-state shear of 50/second the layer of micropar-ticles undergoing flow stays approximately the sameat an average of 25 μm. When the rate of steady-stateshear is increased to 250/second the fracture betweenstationary and flowing gel next to the upper plateoccurs at an average thickness of 307 μm from theupper plate. At a steady-state shear of 400/second,the microparticles appear to be in motion at an aver-age thickness of ∼625 μm. At a steady-state shear of1000/second the microparticles appear to be in motionat thickness of ∼650 μm. These results provide evi-dence of the bulk fracture and yielding behavior ofthe MAX8 hydrogel that is commonly considered asa shear-thinning and rehealing material. The experi-mental results acquired using the confocal-rheomterstudying the MAX8 hydrogel clearly indicate shearrate-dependent fracture of the hydrogel.
The results from the confocal-rheometer assem-bly provide important direct visualization of the frac-ture and flow behavior of the 𝛽-hairpin hydrogelsunder commonly employed shear treatment using arheometer. The data clearly show that the hydrogelsdemonstrate bulk fracture under steady-state shear inlayers with thicknesses dependent on the shear rate. Asshown in Table 1, the thickness of the fractured layerincreases with increased shear rate.
TABLE 1 Results Obtained from Confocal-Rheometer Assembly
Shear
Rate
(second−1)
Total Gel
Thickness
(Gap Height)
(μm)
Fractured
Layer
Thickness
(μm)
% Thickness
Fractured
5 980 23± 2 2.4± 0.2
50 790 25± 4 2.8± 0.9
250 790 307± 9 38.8± 1.2
400 790 624.6± 11 79.0± 1.4
1000 790 652.0± 5 82.5± 0.7
The thickness of the hydrogel undergoing shear rate-dependent fracture awayfrom the upper plate of the rheometer geometry is obtained by recordingthe z values from the videos at which the encapsulated microparticlesstart demonstrating motion. The z values are then converted to thicknessof hydrogel layer in which negligible microparticle motion takes placeby multiplying by the factor (2.37) fixed during the confocal microscopymeasurement. The thickness of the fractured layer is obtained by subtractingthe thickness of the stationary microparticle layer from the total hydrogelthickness. Measurements performed at each shear rate were performed intriplicate.
According to the model proposed by Yanet al.,53 MAX1/MAX8 hydrogels undergo fractureinto smaller domains under shear indicated by yield-ing and flowing of the hydrogel within the shear field.Upon termination of rheometer or syringe-inducedshear, these fractured domains re-percolate to form asolid hydrogel network, indicated by bench-top rhe-ology with G′ >G′′ (Pa). The bulk fracture indicatedby the confocal rheometer serves to reinforce thehydrogel fracture model proposed by Yan et al. The
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
14 © 2014 Wiley Per iodica ls, Inc. Volume 2, 2014

UNCORRECTED PROOFS
WIREs Nanomedicine and Nanobiotechnology Rheology of peptide- and protein-based physical hydrogels
data from the compound assembly prove that yieldingand flow of the MAX1 or MAX8 hydrogels take placebut only within a defined layer parallel to the shearingplate of the rheometer. These data are also consistentwith the yielding and shearing of MAX8 hydrogelsin a layer close to the walls of a capillary duringcapillary flow of MAX8 hydrogels, while the bulkof the material within the capillary flows as a plugand experiences no shear. The experimental resultsfrom the capillary geometry and the compound con-focal rheometer clearly indicate shear rate-dependentfracture exhibited by the 𝛽-hairpin peptide hydro-gels. At higher amplitudes of steady-state shear, asignificant majority of the hydrogel bulk away fromthe rheometer upper plate undergoes fracture andshear-thinning. Upon cessation of this shear, it alsodemonstrates rehealing behavior into a bulk solidhydrogel. Importantly, even if one is confined touse only of a bench-top rheometer, which does notyield a direct correlation between hydrogel structuralchanges under flow and the corresponding rheologicalshear response, the rheometer still faithfully repre-sents shear-thinning and rehealing behavior within acertain layer thickness. As long as a shear-fracturedlayer constitutes a significant thickness of a bulkphysical hydrogel, the rheological behavior of thefractured layer can be considered qualitatively repre-sentative of that of the bulk hydrogel sample whenunder shear. While difficult to determine what layerthickness is considered a ‘significant thickness’, in allmeasurements performed herein, at least 20 μm ofhydrogel experienced fracture and shear flow at thelowest shear rate of 5/second. When subject to highrates of steady-state shear (1000/second), the hydrogelexhibits fracture and flow of a layer with thickness∼82% of the entire hydrogel sample thickness, a largemajority of the bulk of the hydrogel. Therefore, highershear rates are desired if one desires flow of as muchhydrogel volume as possible within the bench-toprheometer. Importantly, both low and high shearrate data exhibit qualitatively similar shear-thinningand immediate rehealing behavior. Almost identical,immediate rehealing behavior of a bulk hydrogelwhen subject to syringe injection-induced shear hasbeen reported by Yan et al.53 This syringe observa-tion also helps to validate the utility of exclusivelyrheometer-induced flow for studying shear-thinningand rehealing behavior of 𝛽-hairpin peptide-basedhydrogels and other physical hydrogels that show thisbehavior. Therefore, a combination of a visual tech-nique such as a confocal microscope and a rheometerhelps elucidate the correlation between the structuralchanges and corresponding rheological behavior fromphysical hydrogels.
The characteristic of 𝛽-hairpin peptide-basedhydrogels of undergoing plug flow when injected usinga syringe has been studied in depth by Yan et al.and the confocal-rheometer assembly. The plug-flowbehavior of the hydrogels can confer a specific benefitto the use of these hydrogels in therapeutic applica-tions like syringe delivery of pharmaceutical activesand cells encapsulated in the hydrogels. This specificbenefit is the protection of the encapsulates in thebulk of the hydrogels from syringe-induced shear. Theencapsulates in the hydrogel bulk undergo negligiblestress as only the hydrogel material at the walls of thesyringe undergoes yielding and syringe-induced shear.As is inferred from the results of the confocal-rheomterassembly, higher is the magnitude of shear to whichthe hydrogels are subjected, larger is the portion of thehydrogels that yields and flows. This shear-dependentfracturing of the hydrogels is an important propertythat allows injection of these hydrogels in cavities ofdifferent sizes. The gels can re-percolate immediatelyinto bulk networks at the injection sites, thus beingparticularly viable from a therapeutic standpoint.
Rheological characterization of 𝛽-hairpin hydro-gels as discussed in Section Hydrogels from 𝛽-HairpinPeptides Based on the Parent Sequence MAX1 [VKVKVKVK-(VDPPT)-KVKVKVKV-NH2] and otherassembled physical hydrogels as discussed inSection Rheological Characterization of PhysicallyCrosslinked Hydrogels has been carried out usinga bench-top rheometer using parallel plate or coneand plate geometries. These oscillatory rheologicalmeasurements have been carried out with appliedstrain amplitudes within the LVR of the hydro-gels and correctly represent the hydrogel behavior.These results help in understanding parameters suchas hydrogel stiffness and frequency dependencebehavior from the hydrogels. This utility of a purerheometer-induced flow can be reinforced by theuse of simultaneous visual investigation of potentialstructural changes of hydrogels under flow. The com-pound confocal-rheometer assembly is an exampleof an instrument with such reinforced utility. Effortstoward obtaining a complete perspective about thestructural as well as mechanical changes exhibited byvarious types of soft materials have been the focusof research well over a decade now. Visual confirma-tion of flow behavior of polymers solutions, melts,and networks has been investigated using particletracking in a variety of reports.137–140 Wang andcolleagues describe the development and implica-tions of the method of particle tracking velocimetry(PTV). This technique has been instrumental in directvisualization of flow behavior in case of rheologicalresponse from a variety of materials such as DNA
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
Volume 2, 2014 © 2014 Wiley Per iodica ls, Inc. 15

UNCORRECTED PROOFS
Advanced Review wires.wiley.com/nanomed
Glass cell
Rotating bob
Focusing lenses
Laser
Camera
FIGURE 8 | Schematic representation of a particle-trackingvelocimetry apparatus. (Reprinted with permission from Ref 147)
solutions,141–144 polymeric melts,142,145 and wormlikemicelles146 from surfactants. A basic setup of a PTVinstrument as reported by Skrzeszewska et al.147 isshown in Figure 8. The principle of the PTV methodis to optically track the fluorescing or diffractingparticles embedded into the sample under study whenilluminated by a laser. This method provides thevisual proof of phenomena such as wall slip and/orinhomogeneous shear in case of polymer melts, andthis method has been suggested by the authors as an‘indispensable part of nonlinear rheological studies ofhighly viscoelastic materials’.148
In light of the popularity of oscillatory rheom-etry as a characterization technique for studyingself-assembly pathways, mechanical properties andflow behavior of physical hydrogels based on pep-tides and polypeptides, it is important that there isawareness regarding these protocols and precautionsdiscussed in Section Rheological Measurements ofPhysical Hydrogels: Protocols and Precautions.Additionally, combination techniques like theconfocal-rheometer assembly and other visualizationtechniques such as PTV would help in elucidationof the flow behavior of various ‘shear-thinning andrehealing’ hydrogels under rheometer-induced shear, acommonly used technique for characterization of suchgels. It could also help in easy determination of wallslip at the surface of hydrogels being studied using abench-top rheometer which is capable to distorting arheological measurement.
An overwhelming majority of rheological stud-ies on physical hydrogels as described later in SectionsHydrogels Based on 𝛽-Sheet Structures, HydrogelMaterials Based on Specific Molecular Recogni-tion, Elastin and Related Hydrogels, Gelatin Gels,
Globular Protein Hydrogels, Fibrous Protein Hydro-gels, Hydrogels Based on Peptide Amphiphiles, andHydrogel Based on Low Molecular Weight PeptideGelators of this review are focused on the linearrheological properties, i.e., the properties within theLVR. Representation of shear environments duringflow of physical hydrogels (e.g., syringe injection)cannot be obtained by restriction to oscillatoryrheological measurements only within small strainamplitude oscillatory conditions. Exposing hydrogelsto steady-state shear such as injection through asyringe is a good strategy to understand the abilityof the hydrogel to be processed and/or shaped into adesired morphology. For example, fully formed solidMAX8 hydrogels injected using a syringe into a cavityor crevice can easily adopt the shape of the cavity orcrevice. In case of steady-state shear treatment, infor-mation about elastic relaxation of hydrogels subjectto shear cannot be acquired. Sometimes, a thoroughunderstanding of the nonlinear rheological behaviorof these materials is interesting and beneficial to fur-ther explore their use as injectable drug/cell deliveryvehicles and load bearing biomaterials. Reviews oflarge amplitude oscillatory shear (LAOS) measure-ments (i.e., oscillatory application of strain outside ofthe LVR) and their utility in probing nonlinear (i.e.,outside the LVR) viscoelastic properties of complexfluids such as polymer solutions and melts, emulsions,biological gels, and micellar solutions has been pro-vided by Hyun et al.,90 and Deshpande.91 Steady-stateshear treatment, the non-oscillatory ‘spinning’ of therheometer top plate at a specified rate, consequentlyapplying strain that is far outside the LVR, is the otherexperimental rheological technique used to subject ahydrogel to shear treatment. While carrying out SAOS(small amplitude oscillatory stress) measurements likethe ones being reviewed herein, a linear viscoelasticresponse is obtained when oscillatory strain amplitudeof a small value is applied to viscoleastic materialssuch as a physically crosslinked hydrogel. A linearviscoelastic response is characterized by independenceof G′ (storage modulus) and G′′ (loss modulus) to theapplied strain and amplitude at a constant angular fre-quency, and the resulting stress is sinusoidal in nature.In the nonlinear region, however, applied strain is ofa large amplitude and the G′ (storage modulus) andG′′ (loss modulus) become dependent on the appliedstrain amplitude at a constant angular frequency. Theresulting stress waveform deviates from a sinusoidalform. On the basis of the characteristic deviations ofdifferent types of complex fluids, nonlinear propertiesof different materials can be studied using correla-tions and comparisons to the already known typicalresponses. Thus, steady-state and large amplitude
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
16 © 2014 Wiley Per iodica ls, Inc. Volume 2, 2014

UNCORRECTED PROOFS
WIREs Nanomedicine and Nanobiotechnology Rheology of peptide- and protein-based physical hydrogels
oscillatory shear measurements can be utilized tostudy rheological response of physical hydrogels inpractical environments.
Bulk fracture as observed in case of MAX1 orMAX8 hydrogels is an example of many importanthigh steady-state shear responses demonstrated by dif-ferent physically crosslinked systems. The next sectionreviews some of the latest studies of bulk fractureobserved in physically crosslinked materials such asnetworks from telechelic polymers and crosslinkedemulsions. Following this section, rheological char-acterization of physically crosslinked peptide- andprotein-based hydrogels is discussed. These hydrogelscan be used as model systems of diverse molecular ori-gins to gather in-depth understanding of relationshipsbetween shear-induced structural changes and rheo-logical behavior.
ACCOUNTS OF FRACTURING INPHYSICALLY CROSSLINKEDHYDROGELS
Solids, when subject to stress undergo a finite defor-mation while fluids under the same stress, flow,or continuously deform. Viscoelastic materials suchas physical hydrogels, display properties intermedi-ate to both these classes of materials beyond theirrespective LVR by showing shear responses such asmacroscopic fracture,69–72 strain stiffening,73–76 andshear banding.77 Macroscopic fracture in physicallycrosslinked hydrogels is an effect that can interferewith rheological measurements carried out using con-ventional geometries. As discussed in this review, aphysical hydrogel based on self-assembled peptidescan undergo macroscopic fracture at a certain distancealong the gel thickness from the shearing rheometerplate. Thus, only the portion of the gel within thefractured volume might yield and flow while a cer-tain layer of the gel might be negligibly affected byshear. Thus, an advanced study of macroscopic frac-ture behavior of physical hydrogels can enable deeperunderstanding of their deformation behavior underlarge amplitude shear. Physical hydrogels are excellentcandidates for potential biomaterial applications suchas developing extracellular matrix mimetic materials.Thus, such studies are a step in the direction of sim-ulating real-life mechanical and morphological envi-ronments that might be encountered by these physicalhydrogels used as biomaterials. An interesting perspec-tive about fracture of elastomers and gels has been pro-vided by Shull.70 A comprehensive review focusing onfracture behavior in transient networks has been pro-vided by Ligoure et al.72 A few studies related closely
to the fracture of physical hydrogels are highlightedbelow.
Tabuteau et al.69 have reported a study of frac-ture nucleation in a model viscoelastic fluid and pro-posed that like real solids, viscoelastic fluids can failin a brittle manner because of lowering of the overallstrength by the presence of micro-cracks. These tran-sient viscoelastic networks are telechelic polymers con-necting oil-in-water droplets in a microemulsion. Thetelechelic polymers have a hydrophilic •PEO back- AQ4
bone with a hydrophobic group at both ends. Theseend chains stick reversibly into the hydrophobic coreof the oil droplets and either loop or link the droplets,leading to a self-assembled structure. Under shear,these ‘links’ can escape and rebound to droplets whenshear is removed. The shear stress increases linearlywith shear rate up to a critical stress after whichthe flow curve shows a sharp discontinuity and thenetwork fractures instantaneously with cracks open-ing up all around the sample that grow rapidly. Thefracture is proposed to take place because of alreadypresent micro-cracks in the material. The tensile stressrather than the shear stress is reported to cause thefracturing of the networks. The critical stress for rup-ture is experimentally and theoretically verified to beon the order of magnitude of the Young’s modulusof the networks. Tixier et al.149 report on interestingnew materials that, topologically, are self-assembledtransient networks comprised of surfactant micelleslinked reversibly by telechelic polymers. Morpholo-gies of the micelles help define three distinct domains,a domain of isolated but not entangled micelles, par-tially entangled and fully entangled micelles. Thesedomains correspond to three distinguishable failuremodes in the nonlinear rheology: a brittle mode,an intermediate mode, and a ductile/shear-bandingmode, respectively. These networks serve to combinethe properties of wormlike micelles with those ofthe bridged micro emulsions discussed earlier. Ramoset al.71 report on tunable transient networks madeof telechelic PEO based polymer linked cetylpyrid-inuim chloride micelles that align under shear. Theauthors have proposed that fluctuations in the degreeof micelle alignment can be a structural probe ofthe fracture behavior under shear. The critical shearrate for fracture was reported to be either very closeto the shear rate at the onset of nonlinear behavior(for ‘brittle’ samples) or significantly larger than it (for‘ductile’ samples).
Berret et al.150 have reported a study of nonlin-ear rheological behavior of physical networks fromtelechelic polymers with a poly(ethylene oxide) mid-dle block and semiperfluorinated end caps. Thisis one of the first reports on shear-induced brittle
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
Volume 2, 2014 © 2014 Wiley Per iodica ls, Inc. 17

UNCORRECTED PROOFS
Advanced Review wires.wiley.com/nanomed
solid-like fracture in a physical network. A flowvisualization technique, very similar to the PTV setupas discussed in Section Accounts of Fracturing in Phys-ically Crosslinked Hydrogels in this review, was used.The flow visualization technique indicates the pres-ence of a clear fracture zone when the networks aresubject to strains above a certain threshold strain.Another study conducted by Berret et al.151 using verysimilar polymers discussed the existence of a discon-tinuity in the stress versus shear rate curve due toinhomogeneous flow conditions created by the ruptureof the network. As the steady shear stress for thesenetworks was studied in response to the shear rate,two stress regimes stood out with increasing shear rateseparated by a discontinuity. The first regime is shearthickening while the second is shear-thinning. Theshear-thickening regime was reported to display anenhancement of the elastic modulus of the networks.This discontinuous behavior was attributed to ruptureor fracture inside the networks. An example of hydro-gels that fracture under shear and reheal upon removalof shear has been discussed by Skrzeszewska et al.147
They report the study of macroscopic fracture of phys-ical networks formed by collagen-inspired telechelicpolypeptides under shear by particle image velocime-try (PIV), which is principally similar to the PTVtechnique discussed in Section Accounts of Fracturingin Physically Crosslinked Hydrogels. The stress andstrain at which failure occurs depend on the appliedshear rate. The width of the fracture zone (zone inwhich almost all the physical crosslink points are rup-tured) as studied by PIV was reported to be muchgreater than the distance between two crosslinks andincreased with magnitude of applied shear. Detailedstudy by the authors of the dependence of the rup-turing of the gel at constant stress and different shearrates has also been reported.
In the next section, we will review recent workthat reports basic rheological characterization onphysically crosslinked hydrogels. The oscillatory rheo-metric measurements carried out in a majority of thestudies reported herein are similar to the measure-ments described in Section Rheological Measurementsof Physical Hydrogels: Protocols and Precautions,e.g., oscillatory time sweep, frequency sweep, andstrain sweep measurements. In all these accounts ofphysically crosslinked hydrogels from various peptidesand proteins, rheological characterization was carriedout by conducting time sweep, oscillatory strain, fre-quency sweep measurements, or any combination ofthese measurements, with the applied shear stress val-ues and resultant strains within the linear viscoelas-tic regions of these hydrogels. The shear-thinningand rehealing behavior of some of the hydrogels has
been studied by applying steady shear, or oscillatoryshear at large strains outside of the LVR using therheometer upper plate as described in Section Rheo-logical Measurements of Physical Hydrogels: Proto-cols and Precautions.
RHEOLOGICAL CHARACTERIZATIONOF PHYSICALLY CROSSLINKEDHYDROGELS
This section describes rheological characterization ofphysically crosslinked hydrogels based on peptide𝛽-sheet structures, proteins, and peptides undergoingspecific molecular interactions, elastin, gelatin, glob-ular proteins, fibrous proteins, peptide amphiphiles(PAs), and low molecular weight peptide gelators. Anoverwhelming majority of measurements carried outon samples of various hydrogels from these materi-als have been conducted using a bench-top rheome-ter using either a parallel plate or cone and plategeometry. Many of these measurements can be car-ried out with greater reliability and reproducibilitywhen precautions and protocols for measurementsas described in Section Rheological Measurements ofPhysical Hydrogels: Protocols and Precautions areobserved. These precautions include evaluation ofwall slip potential of hydrogel sample in contact withgeometry, adjustment of appropriate gap height andstudy of different gap heights for conducting mea-surements and an appropriate choice of geometry andmaterial of fabricating the geometry depending uponthe hydrogel material under study.
Hydrogels Based on 𝜷-Sheet StructuresAulisia et al.44 have reported on shear-thinning andrehealing hydrogels based on multi-domain peptides;(a) K2(QL)6 K2, (b) E(QL)6E, (c) K2(SL)6 K2, (d)E(SL)6E, and (e) E(CLSL)3E. These multi-domain pep-tides have an ABA structure with the middle B domaincontaining alternating hydrophilic and hydrophobicamino acids. The A block at the end contains chargedamino acids that at neutral pH counters the tendencyof the central block to associate. These peptides areshown to form nanofibers that, in turn, form viscoelas-tic hydrogels. These gels display varying abilities ofrehealing on removal of shear treatment. Upon forma-tion, the charged nanofibers are crosslinked ionicallywith either phosphate or magnesium ions. Cryogenictransmission electron microscopy was used to con-firm the fibrillar identity of the self-assembled struc-tures. Fibers formed by peptides containing serine ascompared to glutamine in their central blocks werereported to form much longer and more entangled
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
18 © 2014 Wiley Per iodica ls, Inc. Volume 2, 2014

UNCORRECTED PROOFS
WIREs Nanomedicine and Nanobiotechnology Rheology of peptide- and protein-based physical hydrogels
fibers leading to stiffer hydrogels. The nanofibers frompeptides containing serines were reported to show amuch larger rise in storage modulus when crosslinkedwith an oppositely charged crosslinker (Mg+2 ions) ascompared to those containing glutamines crosslinkedwith phosphate ions. The hydrogels from serinecontaining peptides were shown to demonstrate anincreased propensity to reheal after being subjectedto shear treatment by flowing through a narrowbore syringe. When the nanofibers containing cys-tine residues were oxidized using hydrogen peroxide,forming covalent disulphide bonds along or normalto the fiber axis, the stiffness of the formed hydro-gels was dramatically increased (∼60×). This increasewas reported to be larger than that obtained when thenanofibers were crosslinked with Mg+2 ions. As sug-gested by the authors, this approach shows the versa-tility of the multi-domain peptide hydrogel mechanicalproperties as a function of the peptide sequence.
Further work by Bakota et al.152 employedextensive rheological strain sweep and time sweepmeasurements to demonstrate the enzymaticcrosslinking of nanofibers from multi-domain pep-tides. This phenomenon of exogenous enzymaticoxidative crosslinking has been suggested as a facilestrategy for tuning nanofibrillar hydrogel stiffnessfrom multi-domain peptides. Multi-domain peptidescontaining the cell attaching peptide sequence RGDhave also been reported by the same group to formmechanically stable nanofibrous, shear-thinning,and rehealing hydrogels, which makes their syringedelivery possible.26
Bowerman et al.153 have studied the relativeimportance of hydrophobicity of the non-aromaticversus aromatic amino acids in the (XEXE)2 peptidesduring amyloid-like self-assembly into helical fibrils.The authors have correlated the hydrogelation prop-erties of a set of peptides, inferred from the rheologicalbehavior of their hydrogels, to the hydrophobic char-acter of the individual amino acids (alanine, valine,leucine, phenylalanine, and cyclohexylalanine) in the‘X’ position of (XEXE)2. The rheological measure-ments provide a pivotal piece of information thatindicates an increase in the generic hydrophobicity,and not aromaticity, contributes to the non-covalentsupramolecular cross-linking of the fibrils. TEM hasplayed an important role in determining morphologyof the fibrils from different peptides. Some peptidesin this series form stable left-handed helices or uni-formly thick fibrils while some form transient helicesthat undergo morphological changes into mature fib-rils. TEM provides structural information comple-mentary to rheological characterization to understandself-assembly processes of these peptides. The Nilsson
group also reports the rheological characterization of𝛽-sheet peptide hydrogels based on a reductive triggerwhich induces a conformational change from a pep-tide macrocycle to a 𝛽-sheet.154
Ramachandran et al.66,155 report on co-assemblyof amphiphilic peptides into hydrogels with potentialbiomedical applications.43,153,155 The authors discusspeptides KAW10, KAW15, EAW10, and EAW15. Pep-tide molecules having the same charged residue (lysineor glutamic acid) do not undergo self-assembly witheach other but do so when blended with moleculescontaining an opposite charge (glutamic acid or lysine,correspondingly). The assembly strategy discussed bythe authors involved making hydrogels by blendingpairs of peptides from the four peptides generatingfour pairs, two with sticky ends (with unmatchedlengths of amino acids in peptide pair), and two withblunt ends (with exactly matched lengths of aminoacids in peptide pair). All four pairs were reportedto undergo co-assembly with very different kineticsbut form shear-thinning and rehealing fibrillar hydro-gels, as confirmed by TEM. The KAW:EAW blunt-endpair was found to form the most stable hydrogelspost-cessation of shear, negating the importance of thesticky ends for stiffer network formation.
Guilbaud et al.156 have reported on the corre-lation between enzyme concentration and hydrogelmorphology and properties of enzymatically triggered𝛽-sheet forming tetrapeptide hydrogels. They haveshown formation of heterogeneous hydrogel morphol-ogy due to denser hydrogel domains in the vicin-ity of the enzyme. These denser domains act as areinforcement phase for the hydrogels as determinedby rheometeric experiments. Peptide hydrogels basedon mussel inspired iron crosslinking and formationof shear-thinning and rehealing hydrogels have beenreported by Ceylan et al.157
Hydrogel Materials Based on SpecificMolecular RecognitionSpecific interactions between peptide, protein, andpolymer molecules have been used widely for design-ing smart responsive materials. This section discussesthe use of rheological characterization to probevarious assembly parameters such as optimum gela-tion concentration, temperature, pH, and solutionionic strength and their effects on bulk hydrogelmechanical properties formed as a result of specificmolecular recognition interactions in peptides andproteins. The Tirrell group put forward one of the firstaccounts of materials based on specific interactions,that of coiled-coil formation.158 These materials aremulti-domain proteins with a water-soluble polyelec-trolyte block flanked on both ends by leucine zipper
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
Volume 2, 2014 © 2014 Wiley Per iodica ls, Inc. 19

UNCORRECTED PROOFS
Advanced Review wires.wiley.com/nanomed
domains. The zipper domains formed coiled-coildimers because of interhelical hydrophobic intr-eractions. These dimers act as crosslink junctionsthat are shown to help in formation of transienthydrogels.158 The Kopecek group and other groupshave used the strategy of incorporating leucine zip-per domains.159–163 Susceptibility of these leucinezipper domains to denaturation at high pH or hightemperature leads to reversal of these gels into solu-tions. Plateau moduli of the hydrogels formed asa function of peptide concentration, pH, and ionicstrength were studied using rheological measure-ments and were attributed to network topology.164
Another study from the same group shows that thepH-dependent viscosity transition of such networkscan be attributed to the dynamic properties of thenetworks.165 Further, disulfide covalent bondingdue to cysteine incorporation in the leucine zipperdomain has been reported to show enhancementof hydrogel rheological properties.166,167 Xu andKopecek have presented coiled-coil-containing blockcopolypeptides in which the gelation process wasmonitored by microrheology.161 The authors report areversible sol–gel transition observed on addition orremoval of guanidine hydrochloride that denaturesthe coiled-coil domains. Hybrid hydrogels based on𝛽-sheet peptides grafted onto HPMA copolymersspecifically for the purpose of nucleation of hydrox-yapetite, ultimately intended for bone regeneration,have been reported by Wu et al.168 The bulk mechani-cal properties of the hybrid gels probed by rheologicalmeasurements were shown to be dependent on thepeptide concentration and incubation time. Woolf-son and colleagues discuss development of hybridpeptides by incorporation of 𝛼-helix and 𝛽-hairpincharacteristics into one peptide sequence that couldtransform from a 𝛼-helix into a 𝛽-hairpin upon heat-ing. The ultimately fibrous gel could be reversedinto a solution at lower pH values.169 The samegroup has reported on a 𝛼-helical, peptide-based,fibrillar hydrogel based on coiled-coil interactionswith the exception of the amino acids at certainpositions on the coiled-coil surfaces replaced withthree alanine residues for enhanced intermolecularinteraction. Mixing of complementary peptides underchilled conditions or room temperature was reportedto form self-supporting, thermally stable physicalgels.170,171 Deming and colleagues172–174 have usedbulk rheological measurements to study the extentof hydrogel formation from polypeptide diblockcopolymers comprised of hydrophilic polyelectrolytecharged segments and a corresponding hydropho-bic segment. The charged polyelectrolyte block wasshown to determine the sol–gel transition. The final
gel modulus was dependent on the hydrophilic tohydrophobic ratio and block length. Two factors,the conformational differences in the hydrophobicsegment in the diblock and solution ionic strength,were shown to affect the critical gelling concentra-tion. These hydrogels demonstrated shear-thinningand rehealing properties (∼80–90% of their pre-shearrigidity value after a large-amplitude oscillatory shearof 1000% strain). Grove at al.92,93 have describedthe fabrication of ionic-strength-responsive hydrogelsby using modular, bottom-up protein design basedon the 34 amino acid tetratricopeptide (TPR) repeat.Specifically, non-covalent interactions between repeatprotein modules and their partner peptide •PEG AQ5
ligands act as crosslink junctions for these hydrogels.Both bulk rheology and microrheology have beenused to study the kinetics and viscoelastic propertiesof these hydrogels. The authors discuss the potentialof hydrogels based on these modular protein designsfor tissue engineering applications. Foo et al.65 havereported a similar simple mixing strategy to inducea sol–gel phase transition without any extensivechanges in environmental conditions by using molec-ular recognition interactions between proline-richand di-tryptophan (WW)-rich domains in engineeredprotein materials. The transient physical cross-linkjunctions in such gels yield relatively weak (∼50 Pa)gels (as studied by microrheology), but provide thema shear-thinning and rehealing nature. Lu et al.18
report on a ’Dock and Lock’ physically crosslinkedhydrogel in which the crosslink junctions are theinteractions between the Docking and DimerizationDomain (DDD) of cAMP-dependent protein kinaseA (PKA) and the anchoring domain (AD) of A-kinaseanchoring proteins (AKAP). These hydrogels werefound to be relatively stiff (∼1000 Pa) as comparedto similar protein-based molecular recognition gelsand to demonstrate shear-thinning and much fasterrehealing properties due to inherently expeditedkinetics of the DDD and AD binding. Extensiveoscillatory time, frequency, and strain sweep mea-surements were carried out to characterize thesegels rheologically. Liu et al.175 reported on a physi-cally crosslinked hydrogel based on thiol-containingpolypeptide macromer assembled via coiled-coilinteractions. These coiled-coil interactions act aspoints of physical crosslinking, imparting a physicallycrosslinked nature to the hydrogel network formedfrom these macromers. These polypeptide macromersare covalently bound to vinyl sulfone-containing PEGmolecules through a biologically benign Michaeltype addition reaction. These unique gels were alsoshown to be benign toward epithelial cells encap-sulated within them. Oscillatory bulk rheology was
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
20 © 2014 Wiley Per iodica ls, Inc. Volume 2, 2014

UNCORRECTED PROOFS
WIREs Nanomedicine and Nanobiotechnology Rheology of peptide- and protein-based physical hydrogels
used to characterize the mechanical properties ofthese gels.
Elastin and Related HydrogelsElastin is a natural protein found abundantly inelastic tissues in the human body. Elasticity and themechanical reversible character of elastin allow tissuessuch as arteries, skin, and vocal folds to extend uponstress addition and recover upon stress removal. Theelastomeric properties of this protein provide majormotivation for the design of mimetic molecules,elastin-like polypeptides (ELPs).176 The pentapeptiderepeat unit valine-proline-glycine-X-glycine (where Xis any residue but proline) is the signature sequenceof elastin. The critical temperature of inverse phasetransition, above which ELP solubility decreases andELP hydrogelation occurs, can be varied by changingthe ’X’ amino acid.177,178 Thermal reversible gellinghydrogels for cartilage repair were developed usingthe thermal reversibility of the elastin molecule.179,180
Cytocompatible ELP coacervates, which mechani-cally do not measure up to natural connective tissue,have also been reported.179–182 A step up fromthese coacervates, crosslinked ELP hydrogels thatdisplayed enhanced mechanical properties over ELPcoacervates have been reported.182–186 ELP hydrogelswith improved mechanical properties, developed bycontrolling the phase separation characteristics inELP solutions, have also been reported.183,184,187–189
These efforts demonstrate control of rheological prop-erties by variation in polypeptide sequence and ionicstrength. Hybrid ELPs obtained by incorporating silkresidue repeat sequences into ELP sequences for drugand DNA delivery have also been reported.190,191
These proteins, with temperature-controlled kineticsof gelation, have been shown to be used for in situgelation application after syringe injection.192,193 Therheological characterization of cysteine-containingelastin-like proteins, which undergo thermal as well asoxidative gelation, has been reported by the Chilkotigroup.194,195
Gelatin GelsGelatin gels find utility in biomedical applicationssuch as protein delivery due to biocompatibilityand temperature sensitive gelation.196,197 Gelatinis produced by denaturation of naturally derivedcollagen in solution through an acidic or alkalineprocess that leads to separation of the triple-helicaltropocollagen into three single-stranded gelatinmolecules. These single-stranded molecules undergoa coil–helix transformation when cooled in aque-ous solutions.198,199 Above a critical protein
concentration, thermoreversible gelation takes placefrom helix association.198–200 Dependence of gelmodulus and gel melting temperature on gelatin con-centration and molecular weight, both experimentallyand theoretically, has been reported by Gilsenanand Ross-Murphy201,202 Creep and creep recoverymeasurements used to observe long-term viscoelasticbehavior of gelatin gels were also reported by the sameauthors.203 Comparison of creep compliance with theinverse of the final gel modulus, which providedpotential proof of elastic solid-like behavior of gelatingels, has also been reported. According to the authors,an observed non-recovery of the deformation at theend of the recovery phase provides an indication ofviscoelastic behavior of gelatin gels.
Globular Protein Hydrogels‘Globular proteins’ is another class of proteins thatdemonstrate gel-forming capabilities. Gels from glob-ular proteins are not only relevant industrially forcommercial applications in foods, but have also showngreat promise in emerging areas such as tissue engi-neering. Lysozome-based gels204–206 are a good, recentexample. Unfolding of globular proteins at highertemperatures allows aggregation of individual pro-tein molecules above a certain critical gelling concen-tration. This aggregation has been attributed to sec-ondary interactions such as hydrogen bonding andhydrophobic effects. Depending on whether solutionpH of the proteins is closer or further from the pI,globular proteins can undergo aggregation to formparticulate or fine stranded hydrogels. Extensive rheo-logical studies focused on gels from globular proteinssuch as bovine serum albumin, 𝛽-lactoglobulin, andovalbumin, and the correlation of their rheologicalproperties with their structural and molecular charac-teristics, have been reported in the literature200,206–212
Theoretical modeling has also been reported for bet-ter understanding of the gelation mechanisms. Thesemodels attempt to explain the dependence of gelationtime, critical gelation concentration, and equilibriumgel modulus on variables such as protein concentra-tion, pH, temperature, and ionic strength. In a series ofpublications, Ross-Murphy and colleagues190,210–214
have reported on several models to describe the gellingbehavior of fibrillar 𝛽-lactoglobulin gels formed atboth acidic and basic pH. Van der Linden andcolleagues215–218 have described an adjusted randomcontact model that correlates the critical percola-tion concentration and ionic strength for fine-strandedgels. Studies involving developments of models thatuse parameters obtained from data on the aggrega-tion process, hydrogel rheology, and complementarytechniques such as light scattering have also been
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
Volume 2, 2014 © 2014 Wiley Per iodica ls, Inc. 21

UNCORRECTED PROOFS
Advanced Review wires.wiley.com/nanomed
reported.207,219–223 Miller et al. have reported on ther-moreversible lysozyme gels formed in mixtures ofwater and dithiothreitol as solvents. The lysozymegelation was reported to be achieved by heating theprotein solution up to 85∘C and then slowly coolingback to room temperature, denaturing the lysozymeproteins and forming entangled 𝛽-sheet-rich fibrilsforming a gel network.204,205 Dynamic oscillatorymeasurements were reported to probe the gelationbehavior and mechanical properties.224 The plateauelastic modulus of the gels was reported to be depen-dent on lysozyme concentration in a power lawrelation.
Rheological characterization of whey proteingels has been reported by Eissa et al. Heating acidicwhey protein solutions (pH<4.6) produces weakand brittle gels. Eissa et al.225 have described anenzyme-catalyzed two-step process of producing pro-tein gels with substantially higher elastic moduli andhigher yield-stress and strain values. The enzyme treat-ment was reported to be carried out under alka-line conditions. In these studies, oscillatory rheologi-cal characterization was utilized as a critical methodin analyzing materials with significant commercialpotential in the food industry. The enzyme (transg-lutaminase) treatment allowed for crosslinking of theproteins. The same authors have also described animprovement on the above-described method226 byusing neutral solution conditions instead of alkalinesolution conditions for the enzymatic crosslinking ofwhy proteins. In this report, in addition to stiffnessvalues of the gels, rheological characterization wasalso used to study fractal dimensions of the gels, whichwere compared to the fractal dimensions on the samesamples studied using confocal microscopy. As analternative to forming whey protein gels at pH<4.5which leads to brittle and weak gels, whey proteingels with strong elastic and textural properties werereported by Vardhanabhuti et al.227 These gels wereformed at higher pH (7.5) and equilibrated at differ-ent acid solutions. The elastic properties of the gelswere reported to be affected by the acid type, acid con-centration, equilibration time, and equilibrating solu-tion pH.
Fibrous Protein HydrogelsSilks form a class of highly studied fibrous proteinsthat can form fibers with mechanical properties(Young’s modulus and tensile strength228,229) unsur-passed by almost any other polymer of biologicalor synthetic origin. The biocompatible and degrad-able silk proteins230–232 serve as motivation forvarious studies to explore silk gels as tissue engineer-ing scaffolds233–235 and drug delivery agents when
blended with gelatin.236,237 Silk fibroin is mainlyfound in protein from natural silkworm fibers.238
Above a critical concentration silk fibroin assem-bles in solution leading to a physical hydrogel of𝛽-sheet-rich fibrils. Cytocompatibility and biodegrad-ability of silk fibroin have been established by variousaccounts in the literature.233,235,239–243 These fibersare distinct from self-assembled fibrillar nanostruc-tures from 𝛽-sheet peptides in terms of dimensionsof fibrillar structures formed. The hydrogels formedfrom silk proteins are composed of micron-scalemacromolecular clusters of 𝛽-sheets244 as com-pared to the well-defined nanosized 𝛽-sheets fibrillarstructures from synthetic amphiphilic peptides asdiscussed in Sections Hydrogels from 𝛽-HairpinPeptides Based on the Parent Sequence MAX1[VKVKVKVK-(VDPPT)-KVKVKVKV-NH2] andHydrogels Based on Peptide Amphiphiles.
Kang• et al.158 and Yoo et al.159 report that the AQ6
stability and rigidity of silk fibroin hydrogel wereaffected by the amount of an added polymer, polox-amer 407. Wang et al245,246 has reported on expedit-ing gelation kinetics of silk fibroin under physiologi-cal conditions facilitating homogeneous 3D encapsu-lation of living mesenchymal stem cells. Yucel et al.244
report mechanical stimuli (vortexing) as a means ofexpediting gelation kinetics of fibroin protein solution.Moreover, these mechanically stimulated silk hydro-gels were also shown to be capable of shear-thinningby injection and rehealing to pre-shear rigidity imme-diately after shear cessation. Vollrath et al.247 discusspH-dependent gelation of spidroin proteins at lowerpH (∼5.5) and reversal to solution at pH 7. Fibrillarnetworks from synthetic spidroins stabilized by chem-ical or physical crosslinks have also been reported. Thephysical spidroin hydrogels were reported to be eas-ily disrupted because of the purely topological fibrillarentanglement acting as crosslinks, in contrast to muchstiffer chemically crosslinked gels with elastic moduliup to around 1000 Pa.248–250
Fibrin is another natural, fibrous protein withpotential biomaterial applications for wound heal-ing and growth factor delivery.251 In the event of aninjury fibrinogen is covalently crosslinked by throm-bin, thus forming fibrin. When further crosslinked byfactor XIII, fibrin forms a clot and forms a bloodcoagulating network.252 Fukada and Kaibara pro-vide early reports on various techniques to studydynamic rheological properties of fibrin clots alongwith mechanism of blood coagulation.253–256 Ryanet al.257,258 have reported a structure–property rela-tionship study relating rheological behavior of fibrinclots to their structural characteristics. They suggest adirect dependence of mechanical rigidity of fibrin clots
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
22 © 2014 Wiley Per iodica ls, Inc. Volume 2, 2014

UNCORRECTED PROOFS
WIREs Nanomedicine and Nanobiotechnology Rheology of peptide- and protein-based physical hydrogels
on the concentration of thrombin, calcium, and fib-rinogen and network cross-link densities.
Hydrogels Based on Peptide AmphiphilesPAs are an interesting family of functionally diversemolecules whose assembly properties have beenextensively studied both fundamentally and withrespect to biomedical applications.39,100,259–268 Someapplications targeted for PAs are biomineraliza-tion, scaffolding for therapeutic delivery and tissueregeneration.259–261,269 Some of the first studies ofPA self-assembly were put forward by Hartgerinket al.259,269 A PA molecule is a hydrophilic pep-tide sequence covalently bonded to a hydrophobicaliphatic segment. PAs are particularly suited totissue repair and the controlled therapeutic releaseas they offer flexibility of design. For example, abiological ligand can be used as a head group of thepeptide sequence of an individual PA molecule forbiofunctionalization.39 Additionally, kinetics of PAself-assembly and the assembled nanostructure canbe controlled by controlling the individual aminoacid sequences in the peptide sequence.268 Molec-ular amphiphilicity leads PAs to self-assemble intonanofibers39,259,269 or nanobelts268 and further intofibrillar networks above certain concentrations. Stuppand colleagues demonstrated that the sol–gel transi-tion was triggered upon mixing acidic and basic PAsat physiological pH.262 With the cell-binding ligandIKVAV as the head group, a PA solution mixed withneural cells could form a solid hydrogel when injectedin vivo.39 The authors also discussed the gelationbehavior and viscoelastic properties of PA-based gelsas studied via dynamic oscillatory measurements.Stendahl et al.100 performed a detailed investigationon effects of pH, ionic strength, and type of metalions on mechanical stiffness of PA gels. Mechan-ical stiffness of PA-based hydrogels as a functionof increasing calcium concentration in the solutionenvironment has been reported by Jun et al.270 Theauthors have attributed the decrease of storage mod-ulus after a threshold value of calcium concentrationto possible phase separation that occurs within thegel. In a separate study, it was found that stabilityand mechanical strength of PA-based hydrogels weredependent on the number and position of glycineresidue that can hydrogen bond.44 Pashuck et al.271
have reported the control of mechanical properties ofnanofibrillar PA hydrogels based on number and posi-tions of hydrophobic amino acid residues in the PAs.Greenfield et al.272 have reported tuning rheologicalproperties such as stiffness and strain response ofself-assembled fibrillar PA networks by changing the
types of interfibrillar interactions such as hydrogenbonding and ionic bridges. Toksoz et al.273 reportthe use of oscillatory rheological measurements toprobe the self-assembly and gelation properties ofPA molecules, specifically upon isolation and neu-tralization of ionic charges on the molecules. Chargeneutralization was reported by the use of counter-ionslike calcium and also macromolecules such as heparinand DNA.
Hydrogels Based on Low Molecular WeightPeptidic GelatorsIn addition to peptides and polypeptides from aminoacids, oligopeptides of low molecular weight, suchas fluorenylmethoxycarbonyl (Fmoc)-protected aminoacids, can self-assemble into supramolecular hydro-gels that can be studied using oscillatory rheol-ogy measurements. Biological studies have demon-strated potential biomedical applications for thesehydrogels such as tissue regeneration274–276 and drugdelivery.277–279 These short peptides form hydro-gels when using pH and temperature as a gelationtrigger275,277,280–282 or in the presence of a naturalenzyme.97,277,279,283–286
The Ulijn group has reported hydrogelationof Fmoc-functionalized amino acids initiated underphysiological conditions. Fmoc–diphenylalanine(Fmoc–F2) molecules were reported to undergoself-assembly into fibrous hydrogels that support 3Dcell culture of living cells.274–276 Functionalizationof the network with different chemical moieties hasbeen explored as an approach to tune mechanicalrigidity of Fmoc–F2 gels. The resulting gel networkswere shown to display optimized compatibility withvarious cells.275 Xu and coworkers have presented aseries of Fmoc–dipeptides, which show a reversiblesol–gel transition in response to shift in temper-ature and pH.281,282 In addition, the same groupreported hydrogelation in response to the bindingof vancomycin where gel stiffness was observed toincrease significantly.287 Mahler et al.284 have demon-strated sol–gel transition from Fmoc–F2 solution inwater at an appropriate concentration, leading toshear-thinning Fmoc–F2 gels. Adams et al.283 havereported the gelation of Fmoc–dipeptides induced byhydrolysis of glucono-𝛿-lactone. The resulting hydro-gels were homogeneous with reproducible mechanicalproperties irrespective of the pre-shear history expe-rienced by the hydrogels. The Adams group hasalso reported self-assembly of different dipeptidestriggered by various stimuli such as temperature andsolvent composition68 and salts.67 They have alsoprovided a report of rheological characterization of
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
Volume 2, 2014 © 2014 Wiley Per iodica ls, Inc. 23

UNCORRECTED PROOFS
Advanced Review wires.wiley.com/nanomed
dipeptide-based hydrogels showing differences in gelproperties with different characteristics such as overallhydrophobicity.288 Several groups have also studiedenzyme-induced gelation of low molecular weightpeptides. Specifically, the Ulijn group has discussedthe hydrogelation of Fmoc–tyrosine–OH triggeredunder physiological conditions by the presence ofalkaline phosphatase. The concentration of alkalinephosphatase was crucial in determining the gelationkinetics and stiffness of hydrogels formed.285 Xu andcoworkers discuss hydrogelation of Fmoc–tyrosine at37∘ C at pH 6.0 and pH 9.6281 induced by alkalinephosphate. The final gels were reversed to a solutionwith kinase.282 Other enzymes such as thermolysin,97
𝛽-lactamase,289 and MMP-9290 have been used totrigger hydrogelation of various hydrogelators.
CONCLUSION
Physically crosslinked hydrogels demonstrate signif-icant potential as biomaterials for drug/cell/growthfactor delivery agents and tissue engineeringscaffolds.291 Such hydrogels exhibit a rich diver-sity in hydrogelation stimuli such as pH, temperature,ionic strength, enzymes, and metal ions. In particular,preformed, solid hydrogels, such as the 𝛽-hairpinpeptide-based hydrogels, can be injected as a solidhydrogel using a syringe to a desired site in the bodyand reheal into solid hydrogels immediately afterinjection. These hydrogels, along with some othersimilar physical hydrogels, demonstrate a plug-flowpattern when injected using a syringe that resultsfrom the yielding of a specific layer of hydrogel nearthe syringe wall while the bulk of the hydrogel expe-riences minimal shear. This plug-flow behavior ofthe hydrogels offers a particular advantage towardsyringe delivery of cells encapsulated in the hydrogelsby subjecting them to minimal syringe-induced shear.When subject to steady-state shear using a bench-toprheometer upper plate, these hydrogels undergo bulkfracture at a specific distance away from the shearingupper plate depending on the amplitude of appliedshear. The complex flow patterns observed in theseyield-stress hydrogels under shear, when studied withvarious rheological geometries, present challengestoward physical characterization. Various other phys-ically crosslinked hydrogels that inherently behave asyield-stress, shear-thinning materials intuitively canbe expected to demonstrate such complex flow pat-terns when characterized using an everyday bench-toprheometer assembly. A large number of physicallycrosslinked hydrogel systems demonstrate the phe-nomenon of bulk fracture as described in the review.Thus, this tutorial review attempts to bring forth a
helpful set of precautions and protocols that warrantcareful consideration during rheological characteriza-tion of physically crosslinked yield-stress hydrogels. Alarge variety of physically crosslinked hydrogels basedon peptide, protein, PA, and small molecule peptidehydrogelator systems clearly reinforce the tremendousbiomedical potential of such hydrogels.
EXPERIMENTAL
Confocal-Rheometer Compound AssemblyThe assembly is constructed by mounting an MCR301 Anton Paar rheometer unto a Leica TCS SP5high-speed confocal microscope. The base plate of therheometer is replaced by a homemade cup-like basethat has an aperture to allow light from the objectiveto reach the sample. A glass coverslip is placed overthe aperture of the cup-like base. In order to reducevibrations from the rheometer to the microscope, aplastic brace is used to couple both devices throughthe cup-like base. In this configuration, both devicesoperate independently of each other. Details of thedevice are described by Dutta et al.136 Specifically forthis measurement, both the surface of the rheometergeometry which acts as the upper plate and the coverglass which functions as the lower plate in the rheo-logical measurement have been covered with pieces of600 grit sand paper (Mc-Master Carr, Elmhurst, IL,USA) to ensure as much elimination of wall slip effectsas possible. The confocal microscope is allowed toimage the in situ sample between the rheometer platesvia an opening created by cutting out a little portionof the sandpaper, thus uncovering the aperture.
Analysis of Video Data Acquired fromConfocal-Rheometer Compound AssemblyVideo data obtained using the confocal microscopeare included as Supporting Information. These datashow a sheared MAX8 hydrogel sample at varioussteady-state shear rates while it is being viewed fromthe bottom of the hydrogel sample looking upwardsat the rheometer upper plate as shown in Figure 7.Specifically, each video shows images of multiple lay-ers of the hydrogels at increasing heights within thesample away from the rheometer lower plate. Theseincreasing heights are indicated by an increasing zvalue in the videos. The constant x and y valuesthat indicate the position of the layers relative tothe lower plate remain the same. The thickness ofthe hydrogel undergoing shear rate-dependent frac-ture away from the upper plate of the rheometergeometry is obtained by recording the z values fromthe videos at which the encapsulated microparticles
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
24 © 2014 Wiley Per iodica ls, Inc. Volume 2, 2014

UNCORRECTED PROOFS
WIREs Nanomedicine and Nanobiotechnology Rheology of peptide- and protein-based physical hydrogels
start demonstrating motion. The z values are then con-verted to thickness of hydrogel layer in which neg-ligible microparticle motion takes place by multiply-ing by the factor (2.37) fixed during the confocalmicroscopy measurement. The thickness of the frac-tured layer is obtained by subtracting the thicknessof the stationary microparticle layer from the totalhydrogel thickness. The total thickness of the hydro-gel scanned (μm), thickness of fractured layers (μm),the corresponding rates of steady-shear (second−1),and the thickness of the fractured layers as a per-centage of the total hydrogel thickness scanned havebeen reported in Table 1. The video data obtainedon the MAX8 hydrogel at the different steady-stateshear rates (5/second, 50/second, 250/second, 400/sec-ond, 1000/second) show fractured layers of differ-ent thicknesses corresponding to the shear rates. Therange of the z variable for the shear rate 5/secondis 0< z< 414, corresponding to a minimum thicknessof 0 μm and maximum thickness scanned ∼980 μm.For the shear rates of 50/second, 250/second, 400/sec-ond, and 1000/second, the range of the z variable is0< z< 333, corresponding to a minimum thicknessof 0 μm and maximum thickness scanned ∼790 μm.Under steady-state shear rate of 5/second, the major-ity of the encapsulated microparticles, i.e., the bulk ofthe MAX8 hydrogel, undergo zero or negligible flowwith only ∼20 μm, or ∼2.5% of the gel, experiencingfracture and flow. When the same hydrogel is subjectto a steady-state shear rate of 50/second, the layer ofmicroparticles undergoing flow stays approximatelythe same at an average of 25 μm (z= 325/333). Whenthe rate of steady-state shear is increased to 250/sec-ond, the fracture between stationary and flowing gelnext to the upper plate occurs at an average thicknessof 307 μm (z= 125/333) from the upper plate. At asteady-state shear rate of 400/second, the micropar-ticles appear to be in motion at a z value of ∼75/333,corresponding to an average thickness of fracturedMAX8 hydrogel ∼625 μm. At a steady-state shear rateof 1000/second, which is the most widely used shearrate to apply rheometer-induced shear in the literature,the microparticles appear to be in motion at a z valueof ∼53/333, corresponding to an average thickness offractured MAX8 hydrogel ∼650 μm.
Sample PreparationThe zero gap height calibration for the rheometerwas performed. A 2-mg aqueous solution of MAX8peptide in 100 μL of HEPES (50 mM) is preparedleading to a 2% (w/v) solution. Fluorescent carboxyl-functionalized polystyrene microspheres (0.5–0.99 μmin diameter) from Bangs Laboratories (Fishers, IN,USA) are dispersed in DMEM (Dulbecco’s ModifiedEagle Medium, with 25 mM HEPES) An equal volumeof the DMEM is added to the HEPES peptide solutionto give a buffered solution of MAX8. The solution isquickly transferred to the lower plate of the assembly,which is the microscopy cover glass. A 25-mm parallelplate tool is slowly lowered at a speed of 5 μm/secondto a measuring gap H= 680 μm. The solvent trap isput in place to prevent drying of the sample. A 10×objective is used on the microscope. The objective ispositioned so that it is at a distance of ∼r/2 of theradius of the tool. A 488 nm laser is used for imaging.Using the rheometer a steady-state shear of variousshear rates 5/second, 50/second, and 100/second isapplied to the hydrogel. At the onset of shear, 3Dimages of the gel using the confocal microscope areacquired.
Cryogenic Transmission ElectronMicroscopyCryo-TEM was performed on a Tecnai G2-12 TwinTransmission Electron Microscope (FEI Inc., Hills-boro, OR, USA) operating at voltage of 120 kVlocated in the W.M.Keck Electron Microscopy Facilityat the University of Delaware. For sample prepara-tion, a copper TEM grid, 300 mesh pre-coated withlacey carbon film (Ted Pella, Redding, CA, USA). Adroplet of 2 μL solution was placed onto the coppergrid loaded in an FEI Vitrobot. Each grid was blottedtwice in order to obtain suitable specimen thicknessfor imaging. The blotting time was set as 2 seconds.The sample was quickly plunged into liquefied ethane(∼90 K) cooled by a reservoir of liquid nitrogen toensure the vitrification of water. The vitrified sampleswere transferred to a single tilt cryo transfer holderin a cryo transfer stage immersed in liquid nitrogen.During the imaging, the cryo-holder was kept below−170∘C to prevent sublimation of vitreous solvent.
ACKNOWLEDGMENTS
This manuscript was prepared under cooperative agreement 70NANB12H239 from NIST, U.S. Department ofCommerce. The statements, findings, conclusions, and recommendations are those of the author(s) and do notnecessarily reflect the view of NIST or the U.S. Department of Commerce.
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
Volume 2, 2014 © 2014 Wiley Per iodica ls, Inc. 25

UNCORRECTED PROOFS
Advanced Review wires.wiley.com/nanomed
REFERENCES1. Buenger D, Topuz F, Groll J. Hydrogels in sensing
applications. Prog Polym Sci 2012, 37:1678–1719.•AQ7
2. Miyata T, Uragami T, Nakamae K. Biomolecule-sensitive hydrogels. Adv Drug Deliv Rev 2002,54:79–98.
3. Bassetti MJ, Chatterjee AN, Aluru NR, Beebe DJ.Development and modeling of electrically triggeredhydrogels for microfluidic applications. J Microelec-tromech Syst 2005, 14:1198–1207.
4. Burdick JA, Khademhosseini A, Langer R. Fab-rication of gradient hydrogels using a microflu-idics/photopolymerization process. Langmuir 2004,20:5153–5156.
5. Domachuk P, Tsioris K, Omenetto FG, KaplanDL. Bio-microfluidics: biomaterials and biomimeticdesigns. Adv Mater 2010, 22:249–260.
6. Dong L, Jiang H. Autonomous microfluidics withstimuli-responsive hydrogels. Soft Matter 2007,3:1223–1230.
7. Hoffman AS. Hydrogels for biomedical applications.Adv Drug Deliv Rev 2002, 54:3–12.
8. Peppas NA, Bures P, Leobandung W, Ichikawa H.Hydrogels in pharmaceutical formulations. Eur JPharm Biopharm 2000, 50:27–46.
9. Peppas NA, Hilt JZ, Khademhosseini A, Langer R.Hydrogels in biology and medicine: from molecularprinciples to bionanotechnology. Adv Mater 2006,18:1345–1360.
10. Qiu Y, Park K. Environment-sensitive hydrogels fordrug delivery. Adv Drug Deliv Rev 2001, 53:321–339.
11. Langer R, Tirrell DA. Designing materials for biologyand medicine. Nature 2004, 428:487–492.
12. Lee KY, Mooney DJ. Hydrogels for tissue engineering.Chem Rev 2001, 101:1869–1879.
13. Kloxin AM, Kasko AM, Salinas CN, Anseth KS. Pho-todegradable hydrogels for dynamic tuning of physicaland chemical properties. Science 2009, 324:59–63.
14. Godier-Furnemont AFG, Martens TP, Koeckert MS,Wan L, Parks J, Arai K, Zhang G, Hudson B, HommaS, Vunjak-Novakovic G. Composite scaffold providesa cell delivery platform for cardiovascular repair. ProcNatl Acad Sci U S A 2011, 108:7974–7979.
15. Su J, Hu B-H, Lowe WL Jr, Kaufman DB, MessersmithPB. Anti-inflammatory peptide-functionalized hydro-gels for insulin-secreting cell encapsulation. Biomate-rials 2010, 31:308–314.
16. Olsen BD, Kornfield JA, Tirrell DA. Yielding behav-ior in injectable hydrogels from telechelic proteins.Macromolecules 2010, 43:9094–9099.
17. Aguado BA, Mulyasasmita W, Su J, Lampe KJ, Heil-shorn SC. Improving viability of stem cells duringsyringe needle flow through the design of hydrogel cellcarriers. Tissue Eng Part A 2012, 18:806–815.
18. Lu HD, Charati MB, Kim IL, Burdick JA.Injectable shear-thinning hydrogels engineeredwith a self-assembling dock-and-lock mechanism.Biomaterials 2012, 33:2145–2153.
19. Borselli C, Storrie H, Benesch-Lee F, Shvartsman D,Cezar C, Lichtman JW, Vandenburgh HH, MooneyDJ. Functional muscle regeneration with combineddelivery of angiogenesis and myogenesis factors. ProcNatl Acad Sci U S A 2010, 107:3287–3292.
20. McCall JD, Lin C-C, Anseth KS. Affinity peptidesprotect transforming growth factor 𝛽 during encap-sulation in poly(ethylene glycol) hydrogels. Biomacro-molecules 2011, 12:1051–1057.• AQ8
21. Vulic K, Shoichet MS. Tunable growth factor deliveryfrom injectable hydrogels for tissue engineering. J AmChem Soc 2012, 134:882–885.
22. Segura T, Chung PH, Shea LD. DNA deliveryfrom hyaluronic acid-collagen hydrogels via asubstrate-mediated approach. Biomaterials 2005,26:1575–1584.
23. Ma D, Zhang H-B, Chen D-H, Zhang L-M. Novelsupramolecular gelation route to in situ entrapmentand sustained delivery of plasmid DNA. J ColloidInterface Sci 2011, 364:566–573.
24. Shepard JA, Wesson PJ, Wang CE, Stevans AC, Hol-land SJ, Shikanov A, Grzybowski BA, Shea LD. Genetherapy vectors with enhanced transfection based onhydrogels modified with affinity peptides. Biomaterials2011, 32:5092–5099.
25. Dadsetan M, Szatkowski JP, Shogren KL, YaszemskiMJ, Maran A. Hydrogel-mediated DNA deliv-ery confers estrogenic response in nonresponsiveosteoblast cells. J Biomed Mater Res A 2009, 91A:1170–1177.
26. Bakota EL, Wang Y, Danesh FR, Hartgerink JD.Injectable multidomain peptide nanofiber hydrogel asa delivery agent for stem cell secretome. Biomacro-molecules 2011, 12:1651–1657.
27. Branco MC, Pochan DJ, Wagner NJ, Schneider JP. Theeffect of protein structure on their controlled releasefrom an injectable peptide hydrogel. Biomaterials2010, 31:9527–9534.
28. Aimetti AA, Machen AJ, Anseth KS. Poly(ethylene gly-col) hydrogels formed by thiol-ene photopolymeriza-tion for enzyme-responsive protein delivery. Biomate-rials 2009, 30:6048–6054.
29. Branco MC, Pochan DJ, Wagner NJ, SchneiderJP. Macromolecular diffusion and release fromself-assembled 𝛽-hairpin peptide hydrogels. Biomate-rials 2009, 30:1339–1347.
30. Altunbas A, Lee SJ, Rajasekaran SA, SchneiderJP, Pochan DJ. Encapsulation of curcumin inself-assembling peptide hydrogels as injectable drugdelivery vehicles. Biomaterials 2011, 32:5906–5914.
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
26 © 2014 Wiley Per iodica ls, Inc. Volume 2, 2014

UNCORRECTED PROOFS
WIREs Nanomedicine and Nanobiotechnology Rheology of peptide- and protein-based physical hydrogels
31. Branco MC, Schneider JP. Self-assembling materi-als for therapeutic delivery. Acta Biomater 2009,5:817–831.
32. Hudson SP, Langer R, Fink GR, Kohane DS. Injectablein situ cross-linking hydrogels for local antifungaltherapy. Biomaterials 2010, 31:1444–1452.
33. Sivakumaran D, Maitland D, Hoare T. Injectablemicrogel-hydrogel composites for prolongedsmall-molecule drug delivery. Biomacromolecules2011, 12:4112–4120.
34. Bae KH, Wang L-S, Kurisawa M. Injectable biodegrad-able hydrogels: progress and challenges. Journal ofMaterials Chemistry B 2013, 1:5371–5388.
35. Cong Truc H, Minh Khanh N, Lee DS. Injectable blockcopolymer hydrogels: achievements and future chal-lenges for biomedical applications. Macromolecules2011, 44:6629–6636.
36. Guvendiren M, Messersmith PB, Shull KR. Self-assembly and adhesion of DOPA-modified methacrylictriblock hydrogels. Biomacromolecules 2008,9:122–128.
37. Guvendiren M, Brass DA, Messersmith PB, Shull KR.Adhesion of DOPA-functionalized model membranesto hard and soft surfaces. J Adhes 2009, 85:631–645.
38. Artzi N, Shazly T, Baker AB, Bon A, EdelmanER. Aldehyde-amine chemistry enables modulatedbiosealants with tissue-specific adhesion. Adv Mater2009, 21:3399–3403.
39. Silva GA, Czeisler C, Niece KL, Beniash E, HarringtonDA, Kessler JA, Stupp SI. Selective differentiationof neural progenitor cells by high-epitope densitynanofibers. Science 2004, 303:1352–1355.
40. Haines-Butterick L, Rajagopal K, Branco M, SalickD, Rughani R, Pilarz M, Lamm MS, Pochan DJ,Schneider JP. Controlling hydrogelation kinetics bypeptide design for three-dimensional encapsulationand injectable delivery of cells. Proc Natl Acad Sci US A 2007, 104:7791–7796.
41. Chiu YL, Chen SC, Su CJ, Hsiao CW, Chen YM,Chen HL, Sung HW. pH-triggered injectable hydro-gels prepared from aqueous N-palmitoyl chitosan: invitro characteristics and in vivo biocompatibility. Bio-materials 2009, 30:4877–4888.
42. Yan CQ, Altunbas A, Yucel T, Nagarkar RP, Schnei-der JP, Pochan DJ. Injectable solid hydrogel: mech-anism of shear-thinning and immediate recovery ofinjectable 𝛽-hairpin peptide hydrogels. Soft Matter2010, 6:5143–5156.
43. Ramachandran S, Flynn P, Tseng Y, Yu YB. Elec-trostatically controlled hydrogelation of oligopep-tides and protein entrapment. Chem Mater 2005,17:6583–6588.
44. Aulisa L, Dong H, Hartgerink JD. Self-assembly ofmultidomain peptides: sequence variation allows con-trol over cross-linking and viscoelasticity. Biomacro-molecules 2009, 10:2694–2698.
45. Schneider JP, Pochan DJ, Ozbas B, Rajagopal K,Pakstis L, Kretsinger J. Responsive hydrogels from theintramolecular folding and self-assembly of a designedpeptide. J Am Chem Soc 2002, 124:15030–15037.
46. Ozbas B, Kretsinger J, Rajagopal K, Schneider JP,Pochan DJ. Salt-triggered peptide folding and conse-quent self-assembly into hydrogels with tunable mod-ulus. Macromolecules 2004, 37:7331–7337.
47. Veiga AS, Sinthuvanich C, Gaspar D, Franque-lim HG, Castanho M, Schneider JP. Arginine-richself-assembling peptides as potent antibacterial gels.Biomaterials 2012, 33:8907–8916.
48. Sinthuvanich C, Haines-Butterick LA, Nagy KJ,Schneider JP. Iterative design of peptide-based hydro-gels and the effect of network electrostatics onprimary chondrocyte behavior. Biomaterials 2012,33:7478–7488.
49. Geisler IM, Schneider JP. Evolution-based designof an injectable hydrogel. Adv Funct Mater 2012,22:529–537.
50. Rughani RV, Salick DA, Lamm MS, Yucel T,Pochan DJ, Schneider JP. Folding, self-assembly,and bulk material properties of a De novo designedthree-stranded 𝛽-sheet hydrogel. Biomacromolecules2009, 10:1295–1304.
51. Rughani RV, Branco MC, Pochan D, Schneider JP. Denovo design of a shear-thin recoverable peptide-basedhydrogel capable of intrafibrillar photopolymeriza-tion. Macromolecules 2010, 43:7924–7930.
52. Yan CQ, Mackay ME, Czymmek K, Nagarkar RP,Schneider JP, Pochan DJ. Injectable solid peptidehydrogel as a cell carrier: effects of shear flowon hydrogels and cell payload. Langmuir 2012,28:6076–6087.
53. Yan CQ, Pochan DJ. Rheological properties ofpeptide-based hydrogels for biomedical and otherapplications. Chem Soc Rev 2010, 39:3528–3540.
54. Bonn D, Denn MM. Yield stress fluids slowly yield toanalysis. Science 2009, 324:1401–1402.
55. Barnes HA. The yield stress - a review or ‘pi alphanu tau alpha rho epsilon iota’-everything flows? J Non-Newton Fluid Mech 1999, 81:133–178.
56. Aboye TL, Camarero JA. Biological synthesis ofcircular polypeptides. J Biol Chem 2012, 287:27026–27032.
57. Aimoto S. Contemporary methods for peptide andprotein synthesis. Curr Org Chem 2001, 5:45–87.
58. Papas S, Strongyis C, Tsikaris V. Synthetic approachesfor total chemical synthesis of proteins and protein-likemacromolecules of branched architecture. Curr OrgChem 2006, 10:1727–1744.
59. Kyle S, Aggeli A, Ingham E, McPherson MJ. Produc-tion of self-assembling biomaterials for tissue engineer-ing. Trends Biotechnol 2009, 27:423–433.
60. Badi N, Lutz JF. Sequence control in polymer synthe-sis. Chem Soc Rev 2009, 38:3383–3390.
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
Volume 2, 2014 © 2014 Wiley Per iodica ls, Inc. 27

UNCORRECTED PROOFS
Advanced Review wires.wiley.com/nanomed
61. Qin AJ, Lam JWY, Tang BZ. Click polymerization.Chem Soc Rev 2010, 39:2522–2544.
62. Dehn S, Chapman R, Jolliffe KA, Perrier S. Syntheticstrategies for the design of peptide/polymer conjugates.Polym Rev 2011, 51:214–234.
63. Canalle LA, Lowik D, van Hest JCM.Polypeptide-polymer bioconjugates. Chem Soc Rev2010, 39:329–353.
64. Dong H, Paramonov SE, Aulisa L, Bakota EL, Hart-gerink JD. Self-assembly of multidomain peptides:balancing molecular frustration controls confor-mation and nanostructure. J Am Chem Soc 2007,129:12468–12472.
65. Foo C, Lee JS, Mulyasasmita W, Parisi-Amon A, Heil-shorn SC. Two-component protein-engineered physi-cal hydrogels for cell encapsulation. Proc Natl AcadSci U S A 2009, 106:22067–22072.
66. Ramachandran S, Tseng Y, Yu YB. Repeated rapidshear-responsiveness of peptide hydrogels withtunable shear modulus. Biomacromolecules 2005,6:1316–1321.
67. Chen L, McDonald TO, Adams DJ. Salt-inducedhydrogels from functionalised-dipeptides. RSC Adv2013, 3:8714–8720.
68. Chen L, Raeburn J, Sutton S, Spiller DG, Williams J,Sharp JS, Griffiths PC, Heenan RK, King SM, Paul A,et al. Tuneable mechanical properties in low molecularweight gels. Soft Matter 2011, 7:9721–9727.
69. Tabuteau H, Mora S, Porte G, Abkarian M, LigoureC. Microscopic mechanisms of the brittleness of vis-coelastic fluids. Phys Rev Lett 2009, 102.
70. Shull KR. Fracture and adhesion of elastomers andgels: large strains at small length scales. J Polym Sci[B] 2006, 44:3436–3439.
71. Ramos L, Laperrousaz A, Dieudonne P, Ligoure C.Structural signature of a brittle-to-ductile transition inself-assembled networks. Phys Rev Lett 2011, 107.
72. Ligoure C, Mora S. Fractures in complex fluids:the case of transient networks. Rheol Acta 2013,52:91–114.
73. Erk KA, Henderson KJ, Shull KR. Strain stiffen-ing in synthetic and biopolymer networks. Biomacro-molecules 2010, 11:1358–1363.
74. Motte S, Kaufman LJ. Strain stiffening in collagen Inetworks. Biopolymers 2013, 99:35–46.
75. Giesa T, Pugno NM, Buehler MJ. Natural stiffeningincreases flaw tolerance of biological fibers. Phys RevE 2012, 86.
76. Kurniawan NA, Wong LH, Rajagopalan R. Earlystiffening and softening of collagen: interplay ofdeformation mechanisms in biopolymer networks.Biomacromolecules 2012, 13:691–698.
77. Olmsted PD. Perspectives on shear banding in complexfluids. Rheol Acta 2008, 47:283–300.
78. Macosko CW. Rheology: Principles, Measurementsand Applications. New York: Wiley-VCH, Inc.; 1994.
79. Mezger TG. The Rheology Handbook: For Usersof Rotational and Oscillatory Rheometers. 2nd ed.Hannover: xxxx; 2006.• AQ9
80. Kavanagh GM, Ross-Murphy SB. Rheological char-acterisation of polymer gels. Prog Polym Sci 1998,23:533–562.
81. Goodwin JW, Hughes RW. Rheology for Chemists.xxxxx: RSC Publishing; 2008.• AQ10
82. Mours M, Winter HH. Relaxation patterns of nearlycritical gels. Macromolecules 1996, 29:7221–7229.
83. Winter HH. Gel point. In: The Encyclopedia of Poly-mer Science and Technology. New York: John Wiley& Sons; 2003.
84. Winter HH, Mours M. Rheology of polymers nearsolid liquid transition. In: Advances in Polymer Sci-ence, vol. 134. Berlin: Springer Verlag; 1997.
85. Cicuta P, Donald AM. Microrheology: a reviewof the method and applications. Soft Matter 2007,3:1449–1455.
86. MacKintosh FC, Schmidt CF. Microrheology. CurrOpin Colloid Interface Sci 1999, 4:300–307.
87. Waigh TA. Microrheology of complex fluids. Rep ProgPhys 2005, 68:685–742.
88. Schultz KM, Furst EM. Microrheology of biomaterialhydrogelators. Soft Matter 2012, 8:6198–6205.
89. Larson RG. The Structure and Rheology of ComplexFluids. New York: Oxford University Press; 1998.
90. Hyun K, Wilhelm M, Klein CO, Cho KS, Nam JG,Ahn KH, Lee SJ, Ewoldt RH, McKinley GH. Areview of nonlinear oscillatory shear tests: analysisAnd application of large amplitude oscillatory shear(LAOS). Prog Polym Sci 2011, 36:1697–1753.
91. Deshpande A. Oscillatory shear rheology for prob-ing nonlinear viscoelasticity of complex fluids: largeamplitude oscillatory shear. In: Krishnan JM, Desh-pande AP, Kumar PBS, eds. Rheology of Complex Flu-ids. New York: Springer; 2010, 87–110.
92. Lowik D, Leunissen EHP, van den Heuvel M, HansenMB, van Hest JCM. Stimulus responsive peptide basedmaterials. Chem Soc Rev 2010, 39:3394–3412.
93. Raeburn J, Cardoso AZ, Adams DJ. The importanceof the self-assembly process to control mechanicalproperties of low molecular weight hydrogels. ChemSoc Rev 2013, 42:5143–5156.
94. Micklitsch CM, Knerr PJ, Branco MC, Nagarkar R,Pochan DJ, Schneider JP. Zinc-triggered hydrogelationof a self-assembling 𝛽-hairpin peptide. Angew ChemInt Ed 2011, 50:1577–1579.
95. Chen L, Revel S, Morris K, Adams DJ. Energy transferin self-assembled dipeptide hydrogels. Chem Commun2010, 46:4267–4269.
96. Knerr PJ, Branco MC, Nagarkar R, Pochan DJ,Schneider JP. Heavy metal ion hydrogelation of a
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
28 © 2014 Wiley Per iodica ls, Inc. Volume 2, 2014

UNCORRECTED PROOFS
WIREs Nanomedicine and Nanobiotechnology Rheology of peptide- and protein-based physical hydrogels
self-assembling peptide via cysteinyl chelation. J MaterChem 2012, 22:1352–1357.
97. Toledano S, Williams RJ, Jayawarna V, Ulijn RV.Enzyme-triggered self-assembly of peptide hydro-gels via reversed hydrolysis. J Am Chem Soc 2006,128:1070–1071.
98. Zamith Cardoso A, Alvarez Alvarez AE, Cattoz BN,Griffiths PC, King SM, Frithe WJ, Adams DJ. Theinfluence of the kinetics of self-assembly on the prop-erties of dipeptide hydrogels. Faraday Discuss 2013,XX:XX.•AQ11
99. Veerman C, Rajagopal K, Palla CS, Pochan DJ, Schnei-der JP, Furst EM. Gelation kinetics of 𝛽-hairpinpeptide hydrogel networks. Macromolecules 2006,39:6608–6614.
100. Stendahl JC, Rao MS, Guler MO, Stupp SI. Inter-molecular forces in the self-assembly of peptideamphiphile nanofibers. Adv Funct Mater 2006,16:499–508.
101. Haines LA, Rajagopal K, Ozbas B, Salick DA, PochanDJ, Schneider JP. Light-activated hydrogel forma-tion via the triggered folding and self-assemblyof a designed peptide. J Am Chem Soc 2005,127:17025–17029.
102. Sundelacruz S, Kaplan DL. Stem cell- andscaffold-based tissue engineering approaches toosteochondral regenerative medicine. Semin Cell DevBiol 2009, 20:646–655.
103. Martins AM, Alves CM, Kasper FK, Mikos AG, ReisRL. Responsive and in situ-forming chitosan scaffoldsfor bone tissue engineering applications: an overviewof the last decade. J Mater Chem 2010, 20:1638–1645.
104. Schmidt JJ, Rowley J, Kong HJ. Hydrogels used forcell-based drug delivery. J Biomed Mater Res A 2008,87A:1113–1122.
105. Discher DE, Janmey P, Wang YL. Tissue cells feeland respond to the stiffness of their substrate. Science2005, 310:1139–1143.
106. Engler AJ, Sen S, Sweeney HL, Discher DE. Matrixelasticity directs stem cell lineage specification. Cell2006, 126:677–689.
107. Oda H, Konno T, Ishihara K. The use of the mechan-ical microenvironment of phospholipid polymerhydrogels to control cell behavior. Biomaterials 2013,34:5891–5896.
108. Galbraith CG, Sheetz MP. Forces on adhesive con-tacts affect cell function. Curr Opin Cell Biol 1998,10:566–571.
109. Geiger B, Bershadsky A, Pankov R, Yamada KM.Transmembrane extracellular matrix-cytoskeletoncrosstalk. Nat Rev Mol Cell Biol 2001, 2:793–805.
110. Mewis J, Moldenaers P. xxxxx. Korea Aust Rheol J1999, 11:313–320.•AQ12
111. Tabilo-Munizaga G, Barbosa-Ca ́novas GV. Rheologyfor the food industry. J Food Eng 2005, 67:147–156.
112. Yoshimura A, Prudhomme RK. Wall slip correctionsfor Couette and parallel disk viscometers. J Rheol1988, 32:53–67.
113. Carotenuto C, Minale M. On the use of rough geome-tries in rheometry. J Non-Newton Fluid Mech 2013,198:39–47.
114. Walls HJ, Caines SB, Sanchez AM, Khan SA. Yieldstress and wall slip phenomena in colloidal silica gels.J Rheol (1978-present) 2003, 47:847–868.
115. Clasen C. Determining the true slip of a yield stressmaterial with a sliding plate rheometer. RheologicaActa 2012, 51:883–890.
116. Kelessidis VC, Hatzistamou V, Maglione R. Wall slipphenomenon assessment of yield stress pseudoplas-tic fluids in Couette geometry. Appl Rheol 2010,20:U29–U36.
117. Ozbas B, Rajagopal K, Schneider JP, Pochan DJ.Semiflexible chain networks formed via self-assemblyof 𝛽-hairpin molecules. Phys Rev Lett 2004, 93.
118. Rajagopal K, Ozbas B, Pochan DJ, Schneider JP. Prob-ing the importance of lateral hydrophobic associationin self-assembling peptide hydrogelators. Europ Bio-phys J Biophys Lett 2006, 35:162–169.
119. Pochan DJ, Schneider JP, Kretsinger J, Ozbas B,Rajagopal K, Haines L. Thermally reversible hydro-gels via intramolecular folding and consequentself-assembly of a de Novo designed peptide. J AmChem Soc 2003, 125:11802–11803.
120. Yucel T, Micklitsch CM, Schneider JP, Pochan DJ.Direct observation of early-time hydrogelation in𝛽-hairpin peptide self-assembly. Macromolecules2008, 41:5763–5772.
121. Rajagopal K, Lamm MS, Haines-Butterick LA, PochanDJ, Schneider JP. Tuning the pH responsiveness of𝛽-hairpin peptide folding, self-assembly, and hydro-gel material formation. Biomacromolecules 2009,10:2619–2625.
122. Haines-Butterick LA, Salick DA, Pochan DJ, SchneiderJP. In vitro assessment of the pro-inflammatory poten-tial of 𝛽-hairpin peptide hydrogels. Biomaterials 2008,29:4164–4169.
123. Haines-Butterick LA, Rajagopal K, Lamm M, PochanDJ, Schnieder JP. Controlling hydrogelation kineticsvia peptide design for three-dimensional encapsulationand injectable delivery of cells. Biopolymers 2007,88:518.
124. Leonard SR, Cormier AR, Pang XD, Zimmerman MI,Zhou HX, Paravastu AK. Solid-state NMR Evidencefor 𝛽-hairpin structure within MAX8 designer peptidenanofibers. Biophys J 2013, 105:222–230.
125. Hule RA, Nagarkar RP, Altunbas A, Ramay HR,Branco MC, Schneider JP, Pochan DJ. Correlationsbetween structure, material properties and bioprop-erties in self-assembled 𝛽-hairpin peptide hydrogels.Faraday Discuss 2008, 139:251–264.
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
Volume 2, 2014 © 2014 Wiley Per iodica ls, Inc. 29

UNCORRECTED PROOFS
Advanced Review wires.wiley.com/nanomed
126. Kretsinger JK, Haines LA, Ozbas B, Pochan DJ,Schneider JP. Cytocompatibility of self-assembledss-hairpin peptide hydrogel surfaces. Biomaterials2005, 26:5177–5186.
127. Salick DA, Kretsinger JK, Pochan DJ, Schneider JP.Inherent antibacterial activity of a peptide-based𝛽-hairpin hydrogel. J Am Chem Soc 2007, 129:14793–14799.
128. Ozbas B, Rajagopal K, Haines-Butterick L, Schnei-der JP, Pochan DJ. Reversible stiffening transition in𝛽-hairpin hydrogels induced by ion complexation. JPhys Chem B 2007, 111:13901–13908.
129. Nagarkar RP, Hule RA, Pochan DJ, SchneiderJP. Strand swapping controls the nanostructure of𝛽-hairpin peptide hydrogels. Biopolymers 2007,88:614.
130. Hule RA, Nagarkar RP, Hammouda B, SchneiderJP, Pochan DJ. Dependence of self-assembled peptidehydrogel network structure on local fibril nanostruc-ture. Macromolecules 2009, 42:7137–7145.
131. Nagarkar RP, Hule RA, Pochan DJ, Schneider JP. Denovo design of strand-swapped 𝛽-hairpin hydrogels. JAm Chem Soc 2008, 130:4466–4474.
132. Giano MC, Pochan DJ, Schneider JP. Con-trolled biodegradation of self-assembling 𝛽-hairpinpeptide hydrogels by proteolysis with matrixmetalloproteinase-13. Biomaterials 2011, 32:6471–6477.
133. Nagy KJ, Giano MC, Jin A, Pochan DJ, SchneiderJP. Enhanced mechanical rigidity of hydrogels formedfrom enantiomeric peptide assemblies. J Am Chem Soc2011, 133:14975–14977.
134. Eberle APR, Porcar L. Flow-SANS and Rheo-SANSapplied to soft matter. Curr Opin Colloid Interface Sci2012, 17:33–43.
135. Porcar L, Pozzo D, Langenbucher G, Moyer J, But-ler PD. Rheo-small-angle neutron scattering at theNational Institute of Standards and Technology Cen-ter for Neutron Research. Rev Sci Instrum 2011,82:XX.•AQ13
136. Dutta SK, Mbi A, Arevalo RC, Blair DL. Developmentof a confocal rheometer for soft and biological mate-rials. Rev Sci Instrum 2013, 84:XX.
137. Adrian RJ. Twenty years of particle image velocimetry.Exp Fluids 2005, 39:159–169.
138. Cowen EA, Monismith SG. A hybrid digital parti-cle tracking velocimetry technique. Exp Fluids 1997,22:199–211.
139. Maas HG, Gruen A, Papantoniou D. Particle trackingvelocimetry in 3-dimensional flows .1. photogrammet-ric determination of particle coordinates. Exp Fluids1993, 15:133–146.
140. Melling A. Tracer particles and seeding for par-ticle image velocimetry. Meas Sci Technol 1997,8:1406–1416.
141. Boukany PE, Hu YT, Wang SQ. Observations of wallslip and shear banding in an entangled DNA solution.Macromolecules 2008, 41:2644–2650.
142. Boukany PE, Wang SQ. Exploring the transition fromwall slip to bulk shearing banding in well-entangledDNA solutions. Soft Matter 2009, 5:780–789.
143. Boukany PE, Wang SQ. Shear banding or not inentangled DNA solutions depending on the level ofentanglement. J Rheol 2009, 53:73–83.
144. Hemminger OL, Boukany PE, Wang SQ, Lee LJ.Flow pattern and molecular visualization of DNAsolutions through a 4:1 planar micro-contraction. JNon-Newton Fluid Mech 2010, 165:1613–1624.
145. Wang SQ, Ravindranath S, Boukany P, OlechnowiczM, Quirk RP, Halasa A, Mays J. Nonquiescent relax-ation in entangled polymer liquids after step shear.Phys Rev Lett 2006, 97.
146. Boukany PE, Wang SQ. Use of particle-trackingvelocimetry and flow birefringence to study nonlinearflow behavior of entangled wormlike micellar solu-tion: from wall slip, bulk disentanglement to chainscission. Macromolecules 2008, 41:1455–1464.
147. Skrzeszewska PJ, Sprakel J, de Wolf FA, Fokkink R,Stuart MAC, van der Gucht J. Fracture and self-healingin a well-defined self-assembled polymer network.Macromolecules 2010, 43:3542–3548.
148. Wang SQ, Ravindranath S, Boukany PE. Homoge-neous shear, wall slip, and shear banding of entan-gled polymeric liquids in simple-shear rheometry:a roadmap of nonlinear rheology. Macromolecules2011, 44:183–190.
149. Tixier T, Tabuteau H, Carriere A, Ramos L, LigoureC. Transition from "brittle" to "ductile" rheologicalbehavior by tuning the morphology of self-assemblednetworks. Soft Matter 2010, 6:2699–2707.
150. Berret JF, Serero Y. Evidence of shear-induced fluidfracture in telechelic polymer networks. Phys Rev Lett2001, 87.
151. Berret JF, Sereo Y, Winkelman B, Calvet D, Collet A,Viguier M. Nonlinear rheology of telechelic polymernetworks. J Rheol 2001, 45:477–492.
152. Bakota EL, Aulisa L, Galler KM, Hartgerink JD. Enzy-matic cross-linking of a nanofibrous peptide hydrogel.Biomacromolecules 2011, 12:82–87.
153. Bowerman CJ, Ryan DM, Nissan DA, NilssonBL. The effect of increasing hydrophobicity on theself-assembly of amphipathic 𝛽-sheet peptides. MolBiosyst 2009, 5:1058–1069.
154. Bowerman CJ, Nilsson BL. A reductive trigger forpeptide self-assembly and hydrogelation. J Am ChemSoc 2010, 132:9526–9527.
155. Ramachandran S, Trewhella J, Tseng Y, Yu YB.Coassembling peptide-based biomaterials: effects ofpairing equal and unequal chain length oligopeptides.Chem Mater 2006, 18:6157–6162.
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
30 © 2014 Wiley Per iodica ls, Inc. Volume 2, 2014

UNCORRECTED PROOFS
WIREs Nanomedicine and Nanobiotechnology Rheology of peptide- and protein-based physical hydrogels
156. Guilbaud JB, Rochas C, Miller AF, Saiani A. Effect ofenzyme concentration of the morphology and prop-erties of enzymatically triggered peptide hydrogels.Biomacromolecules 2013, 14:1403–1411.
157. Ceylan H, Urel M, Erkal TS, Tekinay AB, Dana A,Guler MO. Mussel inspired dynamic cross-linking ofself-healing peptide nanofiber network. Adv FunctMater 2013, 23:2081–2090.
158. Petka WA, Harden JL, McGrath KP, Wirtz D, TirrellDA. Reversible hydrogels from self-assembling artifi-cial proteins. Science 1998, 281:389–392.
159. Wang C, Kopecek J, Stewart RJ. Hybrid hydrogelscross-linked by genetically engineered coiled-coil blockproteins. Biomacromolecules 2001, 2:912–920.
160. Wang C, Stewart RJ, Kopecek J. Hybrid hydrogelsassembled from synthetic polymers and coiled-coilprotein domains. Nature 1999, 397:417–420.
161. Xu C, Kopecek J. Genetically engineered block copoly-mers: influence of the length and structure of thecoiled-coil blocks on hydrogel self-assembly. PharmRes 2008, 25:674–682.
162. Wu K, Yang J, Konak C, Kopeckova P, KopecekJ. Novel synthesis of HPMA copolymers containingpeptide grafts and their self-assembly into hybridhydrogels. Macromol Chem Phys 2008, 209:467–475.
163. Klok H-A. Peptide/protein-synthetic polymer con-jugates: Quo Vadis. Macromolecules 2009, 42:7990–8000.
164. Shen W, Zhang KC, Kornfield JA, Tirrell DA. Tun-ing the erosion rate of artificial protein hydrogelsthrough control of network topology. Nat Mater 2006,5:153–158.
165. Shen W, Kornfield JA, Tirrell DA. Dynamic proper-ties of artificial protein hydrogels assembled throughaggregation of leucine zipper peptide domains. Macro-molecules 2007, 40:689–692.
166. Shen W, Lammertink RGH, Sakata JK, Kornfield JA,Tirrell DA. Assembly of an artificial protein hydrogelthrough leucine zipper aggregation and disulfide bondformation. Macromolecules 2005, 38:3909–3916.
167. Shen W, Kornfield JA, Tirrell DA. Structure andmechanical properties of artificial protein hydrogelsassembled through aggregation of leucine zipper pep-tide domains. Soft Matter 2007, 3:99–107.
168. Wu LC, Yang JY, Kopecek J. Hybrid hydrogelsself-assembled from graft copolymers containing com-plementary 𝛽-sheets as hydroxyapatite nucleation scaf-folds. Biomaterials 2011, 32:5341–5353.
169. Ciani B, Hutchinson EG, Sessions RB, Woolfson DN.A designed system for assessing how sequence affects𝛼 to 𝛽 conformational transitions in proteins. J BiolChem 2002, 277:10150–10155.
170. Pandya MJ, Spooner GM, Sunde M, Thorpe JR,Rodger A, Woolfson DN. Sticky-end assembly of adesigned peptide fiber provides insight into proteinfibrillogenesis. Biochemistry 2000, 39:8728–8734.
171. Banwell EF, Abelardo ES, Adams DJ, Birchall MA,Corrigan A, Donald AM, Kirkland M, Serpell LC, But-ler MF, Woolfson DN. Rational design and applicationof responsive 𝛼-helical peptide hydrogels. Nat Mater2009, 8:596–600.
172. Nowak AP, Breedveld V, Pakstis L, Ozbas B, Pine DJ,Pochan D, Deming TJ. Rapidly recovering hydrogelscaffolds from self-assembling diblock copolypeptideamphiphiles. Nature 2002, 417:424–428.
173. Pochan DJ, Pakstis L, Ozbas B, Nowak AP, DemingTJ. SANS and cryo-TEM study of self-assembleddiblock copolypeptide hydrogels with rich nano-through microscale morphology. Macromolecules2002, 35:5358–5360.
174. Breedveld V, Nowak AP, Sato J, Deming TJ, PineDJ. Rheology of block copolypeptide solutions: hydro-gels with tunable properties. Macromolecules 2004,37:3943–3953.
175. Liu Y, Liu B, Riesberg JJ, Shen W. In situ formingphysical hydrogels for three-dimensional tissue mor-phogenesis. Macromol Biosci 2011, 11:1325–1330.
176. Jia X, Kiick KL. Hybrid multicomponent hydrogels fortissue engineering. Macromol Biosci 2009, 9:140–156.
177. Urry DW. Physical chemistry of biological free energytransduction as demonstrated by elastic protein-basedpolymers. J Phys Chem B 1997, 101:11007–11028.
178. Meyer DE, Chilkoti A. Quantification of the effects ofchain length and concentration on the thermal behav-ior of elastin-like polypeptides. Biomacromolecules2004, 5:846–851.
179. Betre H, Setton LA, Meyer DE, Chilkoti A. Character-ization of a genetically engineered elastin-like polypep-tide for cartilaginous tissue repair. Biomacromolecules2002, 3:910–916.
180. Betre H, Ong SR, Guilak F, Chilkoti A, FermorB, Setton LA. Chondrocytic differentiation ofhuman adipose-derived adult stem cells in elastin-likepolypeptide. Biomaterials 2006, 27:91–99.
181. Setton LA, Mow VC, Howell DS. Mechanical-behavior of articular-cartilage in shear is altered bytransection of the anterior cruciate ligament. J OrthopRes 1995, 13:473–482.
182. McHale MK, Setton LA, Chilkoti A. Synthesis andin vitro evaluation of enzymatically cross-linkedelastin-like polypeptide gels for cartilaginous tissuerepair. Tissue Eng 2005, 11:1768–1779.
183. Lee J, Macosko CW, Urry DW. Mechanical prop-erties of cross-linked synthetic elastomeric polypen-tapeptides. Macromolecules 2001, 34:5968–5974.
184. Lim DW, Nettles DL, Setton LA, Chilkoti A. Rapidcross-linking of elastin-like polypeptides with (hydrox-ymethyl)phosphines in aqueous solution. Biomacro-molecules 2007, 8:1463–1470.
185. Annabi N, Mithieux SM, Boughton EA, Ruys AJ,Weiss AS, Dehghani F. Synthesis of highly porous
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
Volume 2, 2014 © 2014 Wiley Per iodica ls, Inc. 31

UNCORRECTED PROOFS
Advanced Review wires.wiley.com/nanomed
crosslinked elastin hydrogels and their interaction withfibroblasts in vitro. Biomaterials 2009, 30:4550–4557.
186. Trabbic-Carlson K, Setton LA, Chilkoti A. Swellingand mechanical behaviors of chemically cross-linkedhydrogels of elastin-like polypeptides. Biomacro-molecules 2003, 4:572–580.
187. Wright ER, McMillan RA, Cooper A, Apkarian RP,Conticello VP. Thermoplastic elastomer hydrogels viaself-assembly of an elastin-mimetic triblock polypep-tide. Adv Funct Mater 2002, 12:149–154.
188. Nagapudi K, Brinkman WT, Thomas BS, ParkJO, Srinivasarao M, Wright E, Conticello VP,Chaikof EL. Viscoelastic and mechanical behav-ior of recombinant protein elastomers. Biomaterials2005, 26:4695–4706.
189. Chow D, Nunalee ML, Lim DW, Simnick AJ, ChilkotiA. Peptide-based biopolymers in biomedicine andbiotechnology. Mater Sci Eng R 2008, 62:125–155.
190. Dinerman AA, Cappello J, Ghandehari H, HoagSW. Solute diffusion in genetically engineeredsilk-elastinlike protein polymer hydrogels. J ControlRelease 2002, 82:277–287.
191. Megeed Z, Haider M, Li DQ, O’Malley BW, CappelloJ, Ghandehari H. In vitro and in vivo evaluation ofrecombinant silk-elastinlike hydrogels for cancer genetherapy. J Control Release 2004, 94:433–445.
192. Megeed Z, Cappello J, Ghandehari H. Controlledrelease of plasmid DNA from a genetically engi-neered silk-elastinlike hydrogel. Pharm Res 2002,19:954–959.
193. Hatefi A, Cappello J, Ghandehari H. Adenoviral genedelivery to solid tumors by recombinant silk-elastinlikeprotein polymers. Pharm Res 2007, 24:773–779.
194. Asai D, Xu DH, Liu WG, Quiroz FG, Callahan DJ,Zalutsky MR, Craig SL, Chilkoti A. Protein polymerhydrogels by in situ, rapid and reversible self-gelation.Biomaterials 2012, 33:5451–5458.
195. Xu DH, Asai D, Chilkoti A, Craig SL. Rheologicalproperties of cysteine-containing elastin-like polypep-tide solutions and hydrogels. Biomacromolecules2012, 13:2315–2321.
196. Young S, Wong M, Tabata Y, Mikos AG. Gelatin as adelivery vehicle for the controlled release of bioactivemolecules. J Control Release 2005, 109:256–274.
197. Yamamoto M, Tabata Y, Ikada Y. Growth factorrelease from gelatin hydrogel for tissue engineering. JBioact Compat Polym 1999, 14:474–489.
198. Rossmurphy SB. Structure and rheology of gelatin gels- recent progress. Polymer 1992, 33:2622–2627.
199. Kt N. On the nature of crosslinks in thermoreversiblegels. Polym Bull 2007, 58:27–42.
200. Clark AH, Rossmurphy SB. Structural andmechanical-properties of bio-polymer gels. AdvPolym Sci 1987, 83:57–192.
201. Gilsenan PM, Ross-Murphy SB. Rheological charac-terisation of gelatins from mammalian and marinesources. Food Hydrocolloids 2000, 14:191–195.
202. Gilsenan PM, Ross-Murphy SB. Viscoelasticity ofthermoreversible gelatin gels from mammalian andpiscine collagens. J Rheol 2000, 44:871–883.
203. Gilsenan PM, Ross-Murphy SB. Shear creep of gelatingels from mammalian and piscine collagens. Int J BiolMacromol 2001, 29:53–61.
204. Yan H, Saiani A, Gough JE, Miller AF. Thermore-versible protein hydrogel as cell scaffold. Biomacro-molecules 2006, 7:2776–2782.
205. Yan H, Frielinghaus H, Nykanen A, RuokolainenJ, Saiani A, Miller AF. Thermoreversible lysozymehydrogels: properties and an insight into the gelationpathway. Soft Matter 2008, 4:1313–1325.
206. Foegeding EA. Food biophysics of protein gels: achallenge of nano and macroscopic proportions. FoodBiophys 2006, 1:41–50.
207. Durand D, Gimel JC, Nicolai T. Aggregation, gelationand phase separation of heat denatured globular pro-teins. Physica A 2002, 304:253–265.
208. Totosaus A, Montejano JG, Salazar JA, Guerrero I. Areview of physical and chemical protein-gel induction.Int J Food Sci Technol 2002, 37:589–601.
209. Gosal WS, Ross-Murphy SB. Globular proteingelation. Curr Opin Colloid Interface Sci 2000,5:188–194.
210. Gosal WS, Clark AH, Pudney PDA, Ross-MurphySB. Novel amyloid fibrillar networks derived froma globular protein: 𝛽-lactoglobulin. Langmuir 2002,18:7174–7181.
211. Gosal WS, Clark AH, Ross-Murphy SB. Fibrillar𝛽-lactoglobulin gels: Part 2. Dynamic mechanical char-acterization of heat-set systems. Biomacromolecules2004, 5:2420–2429.
212. Gosal WS, Clark AH, Ross-Murphy SB. Fibrillar𝛽-lactoglobulin gels: Part 3. Dynamic mechanical ofsolvent-induced systems. Biomacromolecules 2004,5:2430–2438.
213. Kavanagh GM, Clark AH, Ross-Murphy SB.Heat-induced gelation of globular proteins: 4. Gela-tion kinetics of low pH 𝛽-lactoglobulin gels. Langmuir2000, 16:9584–9594.
214. Tobitani A, RossMurphy SB. Heat-induced gelation ofglobular proteins. 2. Effect of environmental factorson single-component and mixed-protein gels. Macro-molecules 1997, 30:4855–4862.
215. Sagis LMC, Veerman C, van der Linden E. Mesoscopicproperties of semiflexible amyloid fibrils. Langmuir2004, 20:924–927.
216. Veerman C, Ruis H, Sagis LMC, van der LindenE. Effect of electrostatic interactions on the perco-lation concentration of fibrillar 𝛽-lactoglobulin gels.Biomacromolecules 2002, 3:869–873.
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
32 © 2014 Wiley Per iodica ls, Inc. Volume 2, 2014

UNCORRECTED PROOFS
WIREs Nanomedicine and Nanobiotechnology Rheology of peptide- and protein-based physical hydrogels
217. Veerman C, de Schiffart G, Sagis LMC, van derLinden E. Irreversible self-assembly of ovalbumin intofibrils and the resulting network rheology. Int J BiolMacromol 2003, 33:121–127.
218. Veerman C, Sagis LMC, Heck J, van der Linden E.Mesostructure of fibrillar bovine serum albumin gels.Int J Biol Macromol 2003, 31:139–146.
219. Pouzot M, Benyahia L, Nicolai T. Dynamic mechan-ical characterization of the heat-induced formationof fractal globular protein gels. J Rheol 2004,48:1123–1134.
220. Pouzot M, Durand D, Nicolai T. Influence of the ionicstrength on the structure of heat-set globular proteingels at pH 7. 𝛽-Lactoglobulin. Macromolecules 2004,37:8703–8708.
221. Pouzot M, Nicolai T, Durand D, Benyahia L. Structurefactor and elasticity of a heat-set globular protein gel.Macromolecules 2004, 37:614–620.
222. Mehalebi S, Nicolai T, Durand D. Light scatteringstudy of heat-denatured globular protein aggregates.Int J Biol Macromol 2008, 43:129–135.
223. Mehalebi S, Nicolai T, Durand D. The influence ofelectrostatic interaction on the structure and the shearmodulus of heat-set globular protein gels. Soft Matter2008, 4:893–900.
224. Yan H, Nykanen A, Ruokolainen J, Farrar D, GoughJE, Saiani A, Miller AF. Thermo-reversible proteinfibrillar hydrogels as cell scaffolds. Faraday Discuss2008, 139:71–84.
225. Eissa AS, Bisram S, Khan SA. Polymerization andgelation of whey protein isolates at Low pH usingtransglutaminase enzyme. J Agric Food Chem 2004,52:4456–4464.
226. Eissa AS, Khan SA. Acid-induced gelation of enzymat-ically modified, preheated whey proteins. J Agric FoodChem 2005, 53:5010–5017.
227. Vardhanabhuti B, Khayankan W, Foegeding EA. For-mation of elastic whey protein gels at low pH by acidequilibration. J Food Sci 2010, 75:E305–E313.
228. Pins GD, Christiansen DL, Patel R, Silver FH.Self-assembly of collagen fibers. Influence of fibrillaralignment and decorin on mechanical properties.Biophys J 1997, 73:2164–2172.
229. Perez-Rigueiro J, Viney C, Llorca J, Elices M. Mechan-ical properties of single-brin silkworm silk. J ApplPolym Sci 2000, 75:1270–1277.
230. Dal Pra I, Freddi G, Minic J, Chiarini A, Armato U.De novo engineering of reticular connective tissue invivo by silk fibroin nonwoven materials. Biomaterials2005, 26:1987–1999.
231. Meinel L, Hofmann S, Karageorgiou V, Kirker-HeadC, McCool J, Gronowicz G, Zichner L, Langer R,Vunjak-Novakovic G, Kaplan DL. The inflammatoryresponses to silk films in vitro and in vivo. Biomaterials2005, 26:147–155.
232. Arai T, Freddi G, Innocenti R, Tsukada M. Biodegra-dation of Bombyx mori silk fibroin fibers and films. JAppl Polym Sci 2004, 91:2383–2390.
233. Fini M, Motta A, Torricelli P, Glavaresi G, AldiniNN, Tschon M, Giardino R, Migliaresi C. The heal-ing of confined critical size cancellous defects in thepresence of silk fibroin hydrogel. Biomaterials 2005,26:3527–3536.
234. Aoki H, Tomita N, Morita Y, Hattori K, Harada Y,Sonobe M, Wakitani S, Tamada Y. Culture of chondro-cytes in fibroin-hydrogel sponge. Biomed Mater Eng2003, 13:309–316.
235. Motta A, Migliaresi C, Faccioni F, Torricelli P, FiniM, Giardino R. Fibroin hydrogels for biomedicalapplications: preparation, characterization and in vitrocell culture studies. J Biomater Sci Polym Ed 2004,15:851–864.
236. Gil ES, Frankowski DJ, Spontak RJ, Hudson SM.Swelling behavior and morphological evolutionof mixed gelatin/silk fibroin hydrogels. Biomacro-molecules 2005, 6:3079–3087.
237. Gil ES, Spontak RJ, Hudson SM. Effect of 𝛽-sheetcrystals on the thermal and rheological behavior ofprotein-based hydrogels derived from gelatin and silkfibroin. Macromol Biosci 2005, 5:702–709.
238. Inoue S, Tanaka K, Arisaka F, Kimura S, Ohtomo K,Mizuno S. Silk fibroin of Bombyx mori is secreted,assembling a high molecular mass elementary unitconsisting of H-chain, L-chain, and P25, with a 6 : 6 :1 molar ratio. J Biol Chem 2000, 275:40517–40528.
239. Altman GH, Diaz F, Jakuba C, Calabro T, Horan RL,Chen JS, Lu H, Richmond J, Kaplan DL. Silk-basedbiomaterials. Biomaterials 2003, 24:401–416.
240. Horan RL, Antle K, Collette AL, Huang YZ, HuangJ, Moreau JE, Volloch V, Kaplan DL, Altman GH. Invitro degradation of silk fibroin. Biomaterials 2005,26:3385–3393.
241. Kim UJ, Park JY, Li CM, Jin HJ, Valluzzi R, KaplanDL. Structure and properties of silk hydrogels.Biomacromolecules 2004, 5:786–792.
242. Kim UJ, Park J, Kim HJ, Wada M, Kaplan DL.Three-dimensional aqueous-derived biomaterialscaffolds from silk fibroin. Biomaterials 2005,26:2775–2785.
243. Nazarov R, Jin HJ, Kaplan DL. Porous 3-D scaffoldsfrom regenerated silk fibroin. Biomacromolecules2004, 5:718–726.
244. Yucel T, Cebe P, Kaplan DL. Vortex-induced injectablesilk fibroin hydrogels. Biophys J 2009, 97:2044–2050.
245. Wang H, Zhang YP, Shao HL, Hu XC. A study onthe flow stability of regenerated silk fibroin aqueoussolution. Int J Biol Macromol 2005, 36:66–70.
246. Wang X, Kluge JA, Leisk GG, Kaplan DL.Sonication-induced gelation of silk fibroin for cellencapsulation. Biomaterials 2008, 29:1054–1064.
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
Volume 2, 2014 © 2014 Wiley Per iodica ls, Inc. 33

UNCORRECTED PROOFS
Advanced Review wires.wiley.com/nanomed
247. Vollrath F, Knight DP, Hu XW. Silk production in aspider involves acid bath treatment. Proc R Soc B BiolSci 1998, 265:817–820.
248. Slotta U, Hess S, Spiess K, Stromer T, Serpell L,Scheibel T. Spider silk and amyloid fibrils: a structuralcomparison. Macromol Biosci 2007, 7:183–188.
249. Rammensee S, Huemmerich D, Hermanson KD,Scheibel T, Bausch AR. Rheological characterizationof hydrogels formed by recombinantly producedspider silk. Appl Phys A 2006, 82:261–264.
250. Huemmerich D, Scheibel T, Vollrath F, Cohen S, GatU, Ittah S. Novel assembly properties of recombi-nant spider dragline silk proteins. Curr Biol 2004,14:2070–2074.
251. Zisch AH, Lutolf MP, Hubbell JA. Biopolymeric deliv-ery matrices for angiogenic growth factors. CardiovascPathol 2003, 12:295–310.
252. Yoo MK, Kweon HY, Lee KG, Lee HC, Cho CS.Preparation of semi-interpenetrating polymer net-works composed of silk fibroin and poloxamermacromer. Int J Biol Macromol 2004, 34:263–270.
253. Fukada E, Kaibara M. Rheological measurements offibrin gels during clotting. Thromb Res 1976, 8:49–58.
254. Kaibara M. Fukada E. Effect of temperature ondynamic viscoelasticity during clotting reaction of fib-rin. Biochim Biophys Acta 1977, 499:352–361.
255. Kaibara M, Fukada E. An analysis of the clottingcurves of complex dynamic rigidity for fibrinogen-thrombin solutions. Biorheology 1980, 17:255–259.
256. Kaibara M, Date M. A new rheological method tomeasure fluidity change of blood during coagulation- application to invitro evaluation of anticoagulabilityof artificial materials. Biorheology 1985, 22:197–208.
257. Ryan EA, Mockros LF, Weisel JW, Lorand L. Struc-tural origins of fibrin clot rheology. Biophys J 1999,77:2813–2826.
258. Ryan EA, Mockros LF, Stern AM, Lorand L. Influenceof a natural and a synthetic inhibitor of factor XIIIa onfibrin clot rheology. Biophys J 1999, 77:2827–2836.
259. Hartgerink JD, Beniash E, Stupp SI. Self-assemblyand mineralization of peptide-amphiphile nanofibers.Science 2001, 294:1684–1688.
260. Rajangam K, Behanna HA, Hui MJ, Han X, HulvatJF, Lomasney JW, Stupp SI. Heparin binding nanos-tructures to promote growth of blood vessels. NanoLett 2006, 6:2086–2090.
261. Webber MJ, Tongers J, Renault M-A, Roncalli JG,Losordo DW, Stupp SI. Development of bioactivepeptide amphiphiles for therapeutic cell delivery. ActaBiomater 2010, 6:3–11.
262. Niece KL, Hartgerink JD, Donners J, Stupp SI. Self-assembly combining two bioactive peptide-amphiphilemolecules into nanofibers by electrostatic attraction. JAm Chem Soc 2003, 125:7146–7147.
263. Behanna HA, Donners J, Gordon AC, Stupp SI.Coassembly of amphiphiles with opposite peptidepolarities into nanofibers. J Am Chem Soc 2005,127:1193–1200.
264. Beniash E, Hartgerink JD, Storrie H, StendahlJC, Stupp SI. Self-assembling peptide amphiphilenanofiber matrices for cell entrapment. Acta Biomater2005, 1:387–397.
265. Bull SR, Guler MO, Bras RE, Meade TJ, Stupp SI.Self-assembled peptide amphiphile nanofibers conju-gated to MRI contrast agents. Nano Lett 2005, 5:1–4.
266. Rajangam K, Arnold MS, Rocco MA, Stupp SI. Peptideamphiphile nanostructure-heparin interactions andtheir relationship to bioactivity. Biomaterials 2008,29:3298–3305.
267. Tysseling-Mattiace VM, Sahni V, Niece KL, BirchD, Czeisler C, Fehlings MG, Stupp SI, Kessler JA.Self-assembling nanofibers inhibit glial scar formationand promote axon elongation after spinal cord injury.J Neurosci 2008, 28:3814–3823.
268. Cui H, Muraoka T, Cheetham AG, Stupp SI. Self-assembly of giant peptide nanobelts. Nano Lett 2009,9:945–951.
269. Hartgerink JD, Beniash E, Stupp SI.Peptide-amphiphile nanofibers: a versatile scaffoldfor the preparation of self-assembling materials. ProcNatl Acad Sci U S A 2002, 99:5133–5138.
270. Jun HW, Yuwono V, Paramonov SE, Hartgerink JD.Enzyme-mediated degradation of peptide-amphiphilenanofiber networks. Adv Mater 2005, 17:2612–2617.
271. Pashuck ET, Cui HG, Stupp SI. Tuning supramolecularrigidity of peptide fibers through molecular structure.J Am Chem Soc 2010, 132:6041–6046.
272. Greenfield MA, Hoffman JR, de la Cruz MO, StuppSI. Tunable mechanics of peptide nanofiber gels. Lang-muir 2010, 26:3641–3647.
273. Toksoz S, Mammadov R, Tekinay AB, GulerMO. Electrostatic effects on nanofiber formationof self-assembling peptide amphiphiles. J ColloidInterface Sci 2011, 356:131–137.
274. Jayawarna V, Ali M, Jowitt TA, Miller AE, Saiani A,Gough JE, Ulijn RV. Nanostructured hydrogels forthree-dimensional cell culture through self-assemblyof fluorenylmethoxycarbonyl-dipeptides. Adv Mater2006, 18:611–614.
275. Jayawarna V, Richardson SM, Hirst AR, Hodson NW,Saiani A, Gough JE, Ulijn RV. Introducing chemicalfunctionality in Fmoc-peptide gels for cell culture. ActaBiomater 2009, 5:934–943.
276. Zhou M, Smith AM, Das AK, Hodson NW, CollinsRF, Ulijn RV, Gough JE. Self-assembled peptide-basedhydrogels as scaffolds for anchorage-dependent cells.Biomaterials 2009, 30:2523–2530.
277. Yang Z, Liang G, Ma M, Gao Y, Xu B. In vitro and invivo enzymatic formation of supramolecular hydrogels
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
34 © 2014 Wiley Per iodica ls, Inc. Volume 2, 2014

UNCORRECTED PROOFS
WIREs Nanomedicine and Nanobiotechnology Rheology of peptide- and protein-based physical hydrogels
based on self-assembled nanofibers of a 𝛽-amino acidderivative. Small 2007, 3:558–562.
278. Yang ZM, Xu KM, Wang L, Gu HW, Wei H, ZhangMJ, Xu B. Self-assembly of small molecules affordsmultifunctional supramolecular hydrogels for topi-cally treating simulated uranium wounds. Chem Com-mun 2005, XX:4414–4416.
279. Sutton S, Campbell NL, Cooper AI, Kirkland M,Frith WJ, Adams DJ. Controlled release from modifiedamino acid hydrogels governed by molecular size ornetwork dynamics. Langmuir 2009, 25:10285–10291.
280. Smith AM, Williams RJ, Tang C, Coppo P, Collins RF,Turner ML, Saiani A, Ulijn RV. Fmoc-diphenylalanineself assembles to a hydrogel via a novel architecturebased on 𝜋-𝜋 interlocked 𝛽-sheets. Adv Mater 2008,20:37–41.
281. Yang ZM, Gu HW, Fu DG, Gao P, Lam JK, XuB. Enzymatic formation of supramolecular hydrogels.Adv Mater 2004, 16:1440–1444.
282. Yang ZM, Liang GL, Wang L, Xu B. Using akinase/phosphatase switch to regulate a supramolecu-lar hydrogel and forming the supramoleclar hydrogelin vivo. J Am Chem Soc 2006, 128:3038–3043.
283. Adams DJ, Butler MF, Frith WJ, Kirkland M, MullenL, Sanderson P. A new method for maintaining homo-geneity during liquid-hydrogel transitions using lowmolecular weight hydrogelators. Soft Matter 2009,5:1856–1862.
284. Mahler A, Reches M, Rechter M, Cohen S, GazitE. Rigid, self-assembled hydrogel composed of a
modified aromatic dipeptide. Adv Mater 2006, 18:1365–1370.
285. Thornton K, Smith AM, Merry CLR, Ulijn RV. Con-trolling stiffness in nanostructured hydrogels producedby enzymatic dephosphorylation. Biochem Soc Trans2009, 37:660–664.
286. Zhang Y, Gu HW, Yang ZM, Xu B. Supramolecularhydrogels respond to ligand-receptor interaction. J AmChem Soc 2003, 125:13680–13681.
287. Zhang Y, Yang ZM, Yuan F, Gu HW, Gao P, XuB. Molecular recognition remolds the self-assemblyof hydrogelators and increases the elasticity of thehydrogel by 10(6)-fold. J Am Chem Soc 2004,126:15028–15029.
288. Adams DJ, Mullen LM, Berta M, Chen L, Frith WJ.Relationship between molecular structure, gelationbehaviour and gel properties of Fmoc-dipeptides. SoftMatter 2010, 6:1971–1980.
289. Yang Z, Ho P-L, Liang G, Chow KH, Wang Q,Cao Y, Guo Z, Xu B. Using 𝛽-lactamase to triggersupramolecular hydrogelation. J Am Chem Soc 2007,129:266–267.
290. Yang Z, Ma M, Xu B. Using matrix metalloprotease-9(MMP-9) to trigger supramolecular hydrogelation.Soft Matter 2009, 5:2546–2548.
291. Altunbas A, Pochan DJ. Peptide-based andpolypeptide-based hydrogels for drug delivery andtissue engineering. In: Deming T, ed. Peptide-BasedMaterials, vol. 310. 2012, 135–167.
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
Volume 2, 2014 © 2014 Wiley Per iodica ls, Inc. 35

UNCORRECTED PROOFS
QUERIES TO BE ANSWERED BY AUTHOR
IMPORTANT NOTE: Please mark your corrections and answers to these queries directly onto the proof at therelevant place. DO NOT mark your corrections on this query sheet.
Queries from the Copyeditor:
AQ1. We have provided “Rheology of peptide- and protein-based physical hydrogels” as the running head forthis article. Please confirm if this is appropriate.
AQ2. Please provide the copyright year and publisher name for the sources mentioned in the captions of Figures2, 5, 6, and 8.
AQ3. These figures (5, 6) are poor quality. Kindly resupply.
AQ4. Please spell out “PEO” at the first instance.
AQ5. Please spell out “PEG” at the first instance.
AQ6. For citations 159 and 160, the author names Kang et al and Yoo et al do not match with the respectivereferences. Please confirm whether the author names can be changed here.
AQ7. Please note that as refs 41 and 65 were duplicate references, we have deleted the duplicate reference andrenumbered accordingly.
AQ8. We have changed all Greek charecters to Greek symbols including references as per journal style. Pleasecheck and confirm.
AQ9. Please provide the publisher name for ref 79.
AQ10. Please provide the place of publication for ref 81.
AQ11. Please provide the volume number and page range for ref 98.
AQ12. Please provide the article title for ref 110.
AQ13. Please provide the page range for refs 135 and 136

UNCORRECTED PROOFS

UNCORRECTED PROOFS