review sample preparation - unamdepa.fquim.unam.mx/amyd/archivero/preparaciondemuestras_6120.pdf ·...
TRANSCRIPT

A
dcps©
K
C
0d
Available online at www.sciencedirect.com
Journal of Chromatography A, 1184 (2008) 191–219
Review
Sample preparation
Yi Chen a,∗, Zhenpeng Guo a,b, Xiaoyu Wang a,b, Changgui Qiu a,b
a Beijing National Laboratory of Molecular Science; Laboratory of Analytical Chemistry for Life Science, Institute of Chemistry,Chinese Academy of Sciences, Beijing 100080, China
b Graduate School, Chinese Academy of Sciences, Beijing 100039, China
Available online 16 October 2007
bstract
A panorama of sample preparation methods has been composed from 481 references, with a highlight of some promising methods fast developeduring recent years and a somewhat brief introduction on most of the well-developed methods. All the samples were commonly referred to molecular
omposition, being extendable to particles including cells but not to organs, tissues and larger bodies. Some criteria to evaluate or validate a samplereparation method were proposed for reference. Strategy for integration of several methods to prepare complicated protein samples for proteomictudies was illustrated and discussed. 2007 Elsevier B.V. All rights reserved.eywords: Sample preparation; Highlighted method; Method survey; Protein; Criteria for method evaluation
ontents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1922. A brief historical retrospect . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1923. A survey of methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 193
3.1. Chemical processing . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1933.2. Non-chemical processing . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 195
3.2.1. Liquid partition methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1953.2.2. Adsorptive methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1953.2.3. Filtration . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1963.2.4. Speed-dependent methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 196
4. Highlighted methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1974.1. Environment friendly methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 197
4.1.1. Supercritical fluids . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1974.1.2. Room temperature ionic liquids . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 198
4.2. Acceleration techniques . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1984.2.1. Pressurized liquid extraction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1984.2.2. Microwave- or sonication-assisted extraction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 200
4.3. Scale down . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2014.3.1. Liquid-phase microextraction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 201
4.4. Adsorptive methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2024.4.1. Multifunctional sorbents . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 202
4.4.2. Selective sorbents . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .4.4.3. Solid-phase microextraction . . . . . . . . . . . . . . . . . . . . . .4.4.4. Stir bar sorptive extraction . . . . . . . . . . . . . . . . . . . . . . . .4.4.5. Matrix solid-phase dispersion . . . . . . . . . . . . . . . . . . . . .∗ Corresponding author. Tel.: +86 10 62618240; fax: +86 10 62559373.E-mail address: [email protected] (Y. Chen).
021-9673/$ – see front matter © 2007 Elsevier B.V. All rights reserved.oi:10.1016/j.chroma.2007.10.026
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 203. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 203. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 206. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 206

192 Y. Chen et al. / J. Chromatogr. A 1184 (2008) 191–219
4.5. Microdialysis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2074.6. On-line stacking . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 207
4.6.1. Isotachophoresis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2074.6.2. Capillary isoelectric focusing . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2074.6.3. Field amplification . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2074.6.4. pH regulation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2084.6.5. Sweeping . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 208
4.7. Derivatization . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2094.8. Miniaturization and integration . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 210
5. Criteria for method validation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2106. Integration of sample preparation methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 211
6.1. Preparation of proteomics-oriented proteins . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2127. Conclusions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 213
Acknowledgements . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 213. . . . .
1
mtvratg
aatsst
sb[(nsmsmhrstes
tsipov
htoo
2
vpadcsypl(ia
bocpsy
dtttapmg
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. Introduction
In this review, sample preparation is in most caseseant to be the isolation and/or (on-line or off-line) concen-
ration of some components of interest or target analytes fromarious matrices, making the analytes more suitable for sepa-ation and detection. Chemical modification of the interestednalytes could be involved in the procedure of sample prepara-ion for an easy isolation, facile later separation and/or detection,roup protection or molecular structure elucidation.
Sample preparation impacts nearly all the later assayed stepsnd is hence critical for unequivocal identification, confirmationnd quantification of analytes. In common, a clean sample assistso improve separation and detection, while a poorly treatedample may invalidate the whole assay. Use of ideally cleanedamples also reduces the time to maintain instruments and inurn the cost of assay.
It is because of the importance of sample preparation thatome excellent reviews on this topic have appeared in 2002 inooks [1,2] and special journal issues [3,4]. In 2003, Pawliszyn3] summarized the fundamental aspects of sample extractionequilibrium and kinetics related to mass transfer), which wereecessary for the development of methods, with special con-iderations of on-site and in situ extractions. Smith [4] providedany examples on extraction and concentration of analytes from
olid, liquid and gas matrices. Selective extraction methods witholecularly imprinted polymers (MIPs) and affinity columns
ave also been considered. In 2004 and 2006, Raynie [5,6]eviewed the experimental and fundamental developments onample extraction, and related methodology appeared duringhe calendar years of 2002–2005, with an exclusion of gen-ral application articles but an inclusion of some individualteps.
This review thus aims at systemizing the sample prepara-ion methods to have a panorama on this field, with a stress onome promising methods appeared and/or fast developed dur-
ng recent years. The matured methods towards such cell andarticle preparation are only briefly discussed, while tissues andrgans are commonly excluded. Some criteria to evaluate oralidate a method for better preparation of complicated samplesq
tc
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 213
ave been proposed for reference. The strategy on total prepara-ion of complicated protein samples (used in proteomic study)r polysaccharides from dried plant materials (for comparisonnly) is illustrated.
. A brief historical retrospect
Study of sample preparation might be traced back to theery beginning of analytical chemistry when complex sam-les were first touched. Following the rapid development ofnalytical techniques in the post-World War II era, increasingemands were placed on sample quality because the samplesollected from natural environment, living body and many otherources had very complex matrices, and their subsequent anal-sis was undoubtedly difficult or even impossible without anyretreatment. As known, sample matrix remains a serious chal-enge in conducting liquid chromatography–mass spectrometryLC–MS), not only reducing the resolution of LC and the ion-zation efficiency of MS but also increasing the detection noisend ultimately the limits of detection.
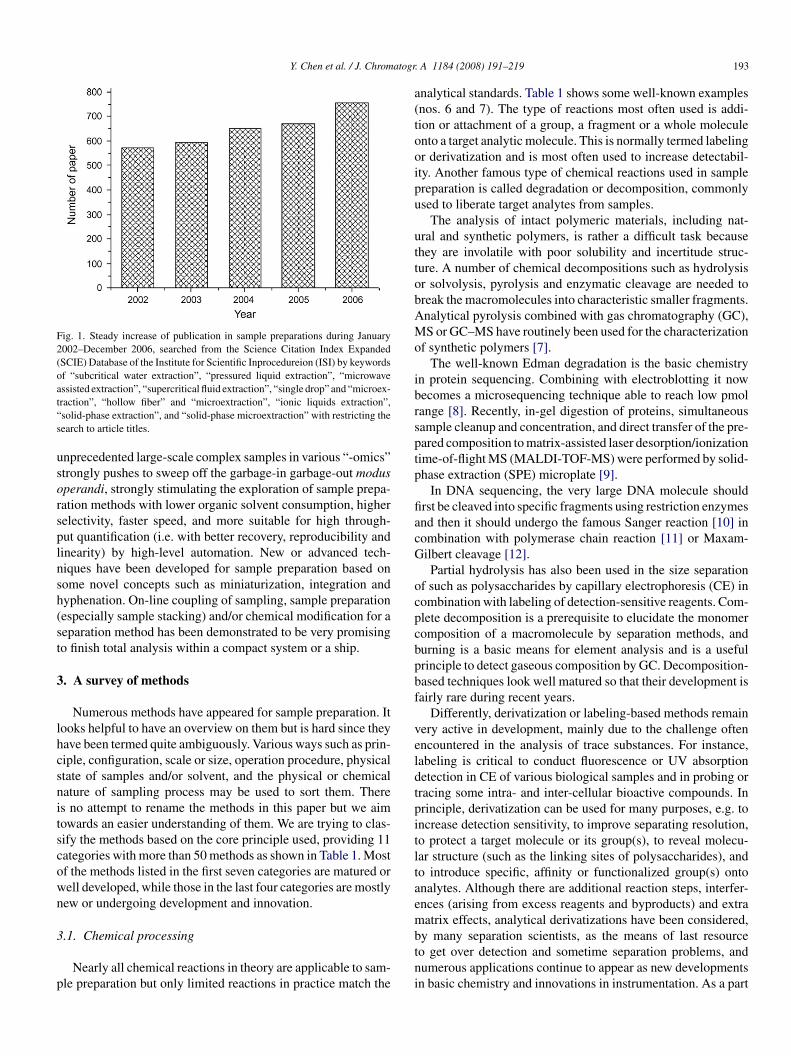
As analytical chemistry grows, sample preparation graduallyecomes a major part of analysis, capable of taking up to 80%f the total time of a complete separation-based analytical pro-ess, which typically includes five steps, that is sampling, samplereparation, separation, detection and data analysis. Since then,ample preparation has developed increasingly during recentears (Fig. 1).
Environmental application is a main cause driving theevelopment of many procedures of sample preparation dueo the increased public awareness that environmental con-aminants are a health risk. The increased demands inhe analysis of foods and natural products have broughtnother pressure to develop the technologies of samplereparation. The appearance of more sensitive and reliableethodology to monitor environment is also impelled by
overnmental necessity to elevate public living standard and
uality.During the past decade, active research on sample prepara-ion has also been fueled by pressure to analyze combinatorialhemistry and biological samples. The urge to analyze the
Y. Chen et al. / J. Chromatogr
Fig. 1. Steady increase of publication in sample preparations during January2002–December 2006, searched from the Science Citation Index Expanded(SCIE) Database of the Institute for Scientific Inprocedureion (ISI) by keywordsof “subcritical water extraction”, “pressured liquid extraction”, “microwaveassisted extraction”, “supercritical fluid extraction”, “single drop” and “microex-t“s
usorsplnsh(st
3
lhcsnitscown
3
p
a(tooipu
uttobAMo
ibrsptp
fiacG
ocpcbpbf
veldtpitltaem
raction”, “hollow fiber” and “microextraction”, “ionic liquids extraction”,solid-phase extraction”, and “solid-phase microextraction” with restricting theearch to article titles.
nprecedented large-scale complex samples in various “-omics”trongly pushes to sweep off the garbage-in garbage-out modusperandi, strongly stimulating the exploration of sample prepa-ation methods with lower organic solvent consumption, higherelectivity, faster speed, and more suitable for high through-ut quantification (i.e. with better recovery, reproducibility andinearity) by high-level automation. New or advanced tech-iques have been developed for sample preparation based onome novel concepts such as miniaturization, integration andyphenation. On-line coupling of sampling, sample preparationespecially sample stacking) and/or chemical modification for aeparation method has been demonstrated to be very promisingo finish total analysis within a compact system or a ship.
. A survey of methods
Numerous methods have appeared for sample preparation. Itooks helpful to have an overview on them but is hard since theyave been termed quite ambiguously. Various ways such as prin-iple, configuration, scale or size, operation procedure, physicaltate of samples and/or solvent, and the physical or chemicalature of sampling process may be used to sort them. Theres no attempt to rename the methods in this paper but we aimowards an easier understanding of them. We are trying to clas-ify the methods based on the core principle used, providing 11ategories with more than 50 methods as shown in Table 1. Mostf the methods listed in the first seven categories are matured orell developed, while those in the last four categories are mostlyew or undergoing development and innovation.
.1. Chemical processing
Nearly all chemical reactions in theory are applicable to sam-le preparation but only limited reactions in practice match the
btni
. A 1184 (2008) 191–219 193
nalytical standards. Table 1 shows some well-known examplesnos. 6 and 7). The type of reactions most often used is addi-ion or attachment of a group, a fragment or a whole moleculento a target analytic molecule. This is normally termed labelingr derivatization and is most often used to increase detectabil-ty. Another famous type of chemical reactions used in samplereparation is called degradation or decomposition, commonlysed to liberate target analytes from samples.
The analysis of intact polymeric materials, including nat-ral and synthetic polymers, is rather a difficult task becausehey are involatile with poor solubility and incertitude struc-ure. A number of chemical decompositions such as hydrolysisr solvolysis, pyrolysis and enzymatic cleavage are needed toreak the macromolecules into characteristic smaller fragments.nalytical pyrolysis combined with gas chromatography (GC),S or GC–MS have routinely been used for the characterization
f synthetic polymers [7].The well-known Edman degradation is the basic chemistry
n protein sequencing. Combining with electroblotting it nowecomes a microsequencing technique able to reach low pmolange [8]. Recently, in-gel digestion of proteins, simultaneousample cleanup and concentration, and direct transfer of the pre-ared composition to matrix-assisted laser desorption/ionizationime-of-flight MS (MALDI-TOF-MS) were performed by solid-hase extraction (SPE) microplate [9].
In DNA sequencing, the very large DNA molecule shouldrst be cleaved into specific fragments using restriction enzymesnd then it should undergo the famous Sanger reaction [10] inombination with polymerase chain reaction [11] or Maxam-ilbert cleavage [12].Partial hydrolysis has also been used in the size separation
f such as polysaccharides by capillary electrophoresis (CE) inombination with labeling of detection-sensitive reagents. Com-lete decomposition is a prerequisite to elucidate the monomeromposition of a macromolecule by separation methods, andurning is a basic means for element analysis and is a usefulrinciple to detect gaseous composition by GC. Decomposition-ased techniques look well matured so that their development isairly rare during recent years.
Differently, derivatization or labeling-based methods remainery active in development, mainly due to the challenge oftenncountered in the analysis of trace substances. For instance,abeling is critical to conduct fluorescence or UV absorptionetection in CE of various biological samples and in probing orracing some intra- and inter-cellular bioactive compounds. Inrinciple, derivatization can be used for many purposes, e.g. toncrease detection sensitivity, to improve separating resolution,o protect a target molecule or its group(s), to reveal molecu-ar structure (such as the linking sites of polysaccharides), ando introduce specific, affinity or functionalized group(s) ontonalytes. Although there are additional reaction steps, interfer-nces (arising from excess reagents and byproducts) and extraatrix effects, analytical derivatizations have been considered,
y many separation scientists, as the means of last resourceo get over detection and sometime separation problems, andumerous applications continue to appear as new developmentsn basic chemistry and innovations in instrumentation. As a part

194 Y. Chen et al. / J. Chromatogr. A 1184 (2008) 191–219
Table 1Classification of sample preparation methods by principleNo. Core principle Method Common usage
1 Mechanics Grinding Powdering, alloyingBlending Alloying, mixingSieving Solid particle sieving
2 Gravity Sedimentation Solid material isolationCentrifugation Phase/density separationUltracentrifugation Macromolecule isolation/purification
3 Magnetic field Magnetic sedimentation Particle-based isolation4 Size exclusion Size-exclusion chromatography Molecular sieving5 Electrochemistry Electro-sedimentation or dissolution Electric active sample preparation
6 Derivatization or labeling Methylation Increasing volatility, identifying linking or branching sites, group protectionMethoxylationSilylation. . .
Stravidinylation, avidinylation, biotinylation, etc. Conjugation of protein, DNA, etc.Aldehyde addition Immobilizing/fixing proteins. . .
Chiral addition Chirality eliminationIn situ labeling Selective probing or sensingPre-, off or after column labeling Detection by absorption, emission or radiation, etc.On-column labeling
7 Degradation Edman degradation Protein sequencingSanger reaction DNA sequencingMaxam-Gilbert cleavagePartial hydrolysis SequencingTotal hydrolysis Composition determinationBurning Detection or Elements analysis
8 Filtration and membrane separation Dialysis Special types of liquid–liquid extraction used for purificationof or cutting off macromolecules
MicrodialysisMembrane-separated liquid extractionElectrodialysisFiltration (frit, paper or membrane filter) Cleanup, particle isolationUltra-filtration, microfiltration Macromolecule isolation/purificationGel filtration Sieving/purification
9 Phase partition Liquid-phase extraction Isolation of soluble analytesLiquid–liquid extractionLiquid-phase microextractionAqueous two-phase extractionCloud point extractionLiquid–solid extraction Analyte isolation from solid matricesSoxhlet extractionSub-critical water extractionPressurized liquid extractionSupercritical fluid extractionMicrowave/sonication-assisted extractionSolid matrix-supported liquid film extraction Extracting analytes from solution or gasesGas–liquid extraction Extraction of gaseous analytesLiquid absorbingHeadspace liquid-phase microextractionMembrane extraction with sorbent interface
10 Sorption or equilibration Solid-phase extraction Collection or concentration of analytes from gaseous and liquid matricesSolid-phase microextractionMatrix solid-phase dispersionStir bar sorptive extractionPolystyrene surface adsorption Immobilazation of antibody or antigen for such as enzyme-
linked immunosorbent assay (ELISA)11 Speed variation Preparative chromatography Thin-layer chromatography General preparation
Ion-exchange chromatography Desalting, purificationCounter-current chromatography Productive preparation and purificationColumn chromatography General preparation
Electrophoresis (EP) Isoelectric focusing For preparing zwitterionsFree flow electrophoresis Large scale preparation of proteins, DNA, Cells and other
charged particlesGel EP (including disc electrophoresis) Preparing proteins and DNAField flow fraction For soluble species, it can better be sorted in chromatography
On-line stacking Field amplification For charged analytesIsotachophoresis For charged analytesSweeping For chargeable substancespH regulation For dissociable samplesAt column capture DependsBarrage Depends

atogr
otb
tpsthtSs
cbCtmopsos[uawc[
lrp
fspsad
3
TTatatesbfipio
3
sel
atfbpivvmc(flcaoteurtlt
apbcmtt(Fittcatl
ppRi
3
Y. Chen et al. / J. Chrom
f sample preparation, various derivatization protocols, deriva-izing reagents and reaction conditions for many analytes haveeen reported in several excellent books and reviews [1,13–15].
In GC analysis, most of the derivatizations aim at increasinghe volatility and stability of the analytes by following princi-al reactions: (1) formation of trimethylsilyl ethers from sugars,teroids and alkaloids, (2) methylation of fatty acids, (3) transes-erification of lipids, and (4) acylation of amines. Derivatizationas also been used for structure confirmation in MS, to obtainhe spectra of derivatives containing molecular ion signals [16].ome derivatizations are adapted in concentrating process orample cleanup.
Much more frequently, the derivatization is adopted tohange the analyte properties for sensitive detection or/andetter separation (by GC, LC, CE, etc.). Derivatization forE is mainly conducted to enhance the detectability, partially
o modify both the detectability and polarity (or charge-to-ass ratio) to increase resolution [17,18]. Pre-, on-, post- or
ff-column labeling has extensively been explored for high-erformance LC (HPLC) and especially for capillary-basedeparation techniques such as nano-LC and CE, thereinto then- or in-column labeling is the most promising which con-umes very limited (down to sub-nanoliter) reagents and samples19]. This is highly favored in single cell analysis or otherltra-trace analysis. Derivatization is also used to introducen energy exchanging structure into a fluorescent molecule,hich is at present a novel technique to prepare molecular bea-
ons [20] or measure fluorescence resonance energy transfer21].
Other types of chemical reactions may also be consideredike electrochemistry-, heat-, sonication-, or microwave-assistedeaction. Enzymatic reactions are gentle and favorable for thereparation of biomolecules.
It should be noted that surface modification is inseverablerom many processes of sample preparation. To improve theelectivity or to get off the non-specific adsorption of sam-le preparation materials like cartridge, channel(s) or packings,urface modifications by chemical (and sometimes physical)dsorption of some affinity or inert substances have to be con-ucted.
.2. Non-chemical processing
Most of the methods listed in nos. 1–4 and nos. 8–11 inable 1 can roughly be considered as non-chemical techniques.he mechanical processes such as grinding, blending and sievingre mainly used to prepare sized particles. They are suitable forhe preparation of solid samples. Gravity-based methods suchs centrifugation and sedimentation are often used for the isola-ion of heterogeneous samples and liquid layers in liquid–liquidxtraction. Centrifugation is known as a robust technique forample preparation routinely used in chemical and especiallyiological laboratories. When filtration is incorporated, ultra-
ltration can be obtained. The methods based on filtration, phaseartitioning, adsorption and equilibration or speed variation aren most cases non-chemical approaches remaining in fast devel-pment.baae
. A 1184 (2008) 191–219 195
.2.1. Liquid partition methodsLiquid partitioning means the transfer and distribution of
oluble analytes in a liquid-containing phase system. Variousxtraction methods have been created based on the liquid–liquid,iquid–solid or sometimes liquid–gas partitioning systems.
Liquid–liquid extraction (LLE) transfers target analytes fromliquid matrix into another immiscible liquid-phase according
o solubility difference. The most classical extraction is per-ormed in separating funnels to extract analytes from an aqueousiological or environmental solution into a non-polar or lessolar organic solvent. Classical LLE operates manually, requir-ng repetitive coalescence and phase separation, which can beery slow when emulsions form and in turn produces a largeolume of organic waste. Some alternative techniques includeembrane-separated liquid extraction (MSLE) [22], counter-
urrent chromatography (CCC), single drop microextractionSDME) [23], and laminar flow techniques [24], etc. The laminarow methods are commonly performed in a miniaturized devicealled microfluidic system, allowing extraction of analytes fromliquid into another miscible liquid-phase [25]. Based on mixingf two incompatible polymers or polymer-salt in water, extrac-ion of analytes can be performed using aqueous two-phasextraction without the conditions of laminar flow [26]. In case ofsing aqueous micellar solutions, cloud point extraction can beealized by adjusting temperature and pressure [27]. In additiono water and common organic solvents, room temperature ioniciquids (RTILs) are drawing more and more attention as a newype of extraction solvents.
Liquid–solid extraction (LSE) is used to isolate analytes fromsolid or semi-solid matrix into solvents by liquid–solid-phaseartitioning and/or desorption, which can simply be performedy stirring a solid sample in a hot or cold solvent, classically, in alosed vessel like the well-known Soxhlet extraction. Sometimesicrowave [28–30] or sonication [31–37] is used to accelerate
he dissolution of analytes and the penetration of solvent(s) intohe solid matrix, they are termed microwave-assisted extractionMAE) and sonication-assisted extraction (SAE) respectively.urther development of the extraction strategies based on new
nstrumentation and new fluids have been achieved, allowingo reduce the consumption of solvent, sample and extractionime, and to enhance the selectivity of extraction. Two typi-al examples are the supercritical fluids and sub-critical fluids,nd the corresponding techniques are supercritical fluid extrac-ion (SFE), sub-critical water extraction (SWE), and pressurizediquid extraction (PLE).
Liquid–gas extraction is widely used to capture atmosphericollutants by dynamic or passive techniques. Sampling andreconcentration of analytes are often integrated in one step.ecently, liquids have been used as a sorptive collecting phase
n some headspace techniques.
.2.2. Adsorptive methodsAdsorptive extraction methods first trap analytes onto immo-
ilized phases and the adsorbed analytes are eluted by anppropriate solvent or desorbed thermally. These methods canlso be considered as special solid–liquid or gas–liquid-phasextraction techniques but are considerably faster and consume

1 togr.
shpsm
ctmamrhmm
esmtaa
3
wmffiwsimtwb
tducasdtGpc
frgf
owc
scShsfi
3
tscpTistdt
dtca
LrtrTsnosnHpmtf
ltat
apmwait
96 Y. Chen et al. / J. Chroma
ignificantly smaller volumes of solvents and samples. Theyave hence a wide use in a further treatment of the samplesrepared by other methods and can also be used for gaseousampling/extraction via either dynamic (forced flow) or passiveode.There is another type of adsorptive extraction humorously
alled “poor man’s chromatography” or rigorously SPE. Thisechnique likely starts off with the conventional column chro-
atographic cleanup and fractionation, and later develops inton extraction mode with various formats. The desire for evenore selective phases is the current driving force in SPE
esearch, and restricted-access materials (RAMs) and MIPs haveence been explored. Innovation of adsorptive extraction for-ats is the most recent effort that has brought solid-phaseicroextraction (SPME) in this world.Magnetic separation may be included in solid–liquid-phase
xtraction in combination with affinity techniques. Solution sub-tances can be extracted by affinity reagents immobilized onagnetic particles and separated from the solution by attracting
he particles with a magnetic field. Since strong magnets are nowvailable in a small sizes, magnet-assisted extraction is space-nd cost-effective and easy in operation.
.2.3. FiltrationNearly all filtration methods are membrane-based techniques
orking with the size-exclusion mechanism via the through-embrane pores which allow small molecules to pass through
reely but stop the flow of large molecules. More accurately, alter with a certain size of pores can “cut-off” the moleculesith a size larger than the pores, making them free from the
maller molecules or reversely. The molecular size cut-off values thus a key factor to characterize a filter or a membrane. Since
acromolecules are often identified by their molecular weight,he characterizing thus uses the value of molecular weight cut-offhich is the molecular mass of the smallest compound retainedy a membrane to an extent larger than 90%.
There are several different forces available to drive filtra-ions. The most common filtrations are operated under pressureifferences simply caused by gravity so that they can easily besed in many common laboratories and even in industrial pro-esses. The more advanced micro- and ultra-filtrations need thessistance of vacuum or centrifugation. Dialysis, microdialy-is and other MSLE are induced by concentration difference oriffusion. Electrodialysis should be conducted under an elec-ric potential difference coupled with a concentration gradient.el filtration is achieved by retaining small molecules in gelores and eluting macromolecules from the gel body or packedolumns.
Filtration is frequently used to separate suspended particlesrom dissolved substances, supposing the particles meet the sizeequirement of a specific filter or membrane. Ultra-filtration andel filtration are often used for purifying, concentrating andractionating macromolecules or colloidal suspensions.
Filters are essential in filtration. They can be a membranef fibers (paper), glass, celluloses or other plastic substancesith various porosities, of which paper and glass frit filters are
lassical and remain in use in various laboratories. Cellulose and
aiei
A 1184 (2008) 191–219
ome plastic membrane filters are better alternations for theirlean feature with pore size and scale selectable or variable.yringe filters with different diameters and filter thicknessesave been used for microfiltration. Filters incorporated into aample vial or into a centrifuge tube are now often used for theltration of viscous samples at a very small volume [38].
.2.4. Speed-dependent methodsSpeed variation happens in nearly all the sample prepara-
ion methods but is the core of separation-based techniquesuch as electrophoresis, chromatography and on-line or in-olumn stacking. Chromatography is frequently used for samplereparation in various laboratories and industrial processes.hin-layer chromatography is a very common technique used
n organic synthetic laboratories, it prepares target analytes byeparating the sample dots printed on one side of a silica gel slabhrough development using one or mixed solvents. The separatedots are cut-off from the silica gel slab and eluted by solvent(s)o collect the required analytes.
Ion-exchange chromatography is commonly used to isolate,esalt or purify neutral molecules and the analytes chargedhe same sign as the exchanger. They are not retained as theounter-ionic impurity does. Reverse operation is also possiblend adoptable.
CCC pioneered by Ito et al. [39] can also be included inLE since it is an all-liquid method without solid-phases. CCC
elies on the partition of a sample of two immiscible solventso achieve separation, and has been the subject of numerousesearch papers, review articles [40–43] and books [44–50].his technique is now developing rapidly from its time con-uming formats into droplet CCC and rotation locular CCC andew generations of CCC instruments called high-speed CCCr high-performance CCC have also appeared, of which high-peed CCC has been widely used in preparative separation ofatural products [51–54]. Although CCC is not as efficient asPLC, it is an excellent alternative for other large-scaled sampleretreating approaches able to preserve the chemical integrity ofixtures. With these advantages, CCC is a preferred purifica-
ion tool for natural products, especially for the bioassay-guidedractionation of plant-derived compounds.
In addition, all column chromatographic methods more oress suit for sample isolation (by exploring their separation func-ion), of which analytically reversed phase HPLC is often used assemi-preparative tool in various laboratories for it is a common
ool at hand.Field flow fraction (FFF) proposed by Giddings [63–65] can
lso be considered a special class of chromatographic methodsarticularly suitable for the purification and characterization ofacromolecules, colloids and particles [66,67]. In FFF, samplesith different molar mass, size and/or other physical properties
re separated, under some fields, into different velocity regionsn a parabolic flow of mobile phase across the channel, andhen exit the channel at different retention times. Different FFF
pproaches can be available by varying the applied fields, fornstance, electrical field flow fraction is obtained when usinglectric field as the driving force and has been shown usefuln the separation of proteins [63], DNA [68] and polystyrene
atogr
sctmtimeprfin(tii
tmatuf
iptp
ttstatnesbm[
togrotpttfepopi
(ibrapIaalr
4
nulclp
4
4
mat
gvsafltcevoehtippnmsra
Y. Chen et al. / J. Chrom
ulfonates [69] but is hindered by the unavailability of commer-ial instruments. Crossflow or flow FFF is the most universalechnique in FFF family, applicable to macromolecules with
olecular weight of 103–109 Da and particles with diameter upo 50 �m [70]. Sedimentation FFF uses a centrifugal field ands a high-resolution separation technique for submicrometer- to
icrometer-sized particles [71]. Thermal or temperature gradi-nt FFF has been used mainly for separating the organosolubleolymers [72] and to a lesser extent the particles. The mostecent appealing applications of FFF include: (1) sorting andngerprinting of bacteria for whole-cell vaccine production; (2)on-invasive and tagless sorting of immature and stem cells;3) separation of intact proteins and enzymes in top-down pro-eomics; and (4) the development of flow-assisted, multianalytemmunoassays using nano- and micrometer-sized particles withmmobilized biomolecules [73,74].
Similar to chromatography, electrophoresis can be used forhe preparation of various charged samples especially biologicacromolecules such as proteins, glycoconjugates and nucleic
cids. It has hence been adopted more often in biological studieshan chromatography. The powerful methods of electrophoresissed for sample preparation include isoelectric focusing (IEF),ree flow electrophoresis (FFE), and gel electrophoresis.
IEF is a known format of electrophoresis. It achieves samplesolation and concentration by forcing analytes to migrate in aH gradient medium (either free solutions or gels) and stop athe isoelectric points (pI), and is commonly adopted to focusroteins at their pI.
FFE, introduced more than 40 years ago [55], allows a con-inuous injection of samples into a carrier solution flowing as ahin laminar film (0.3–1.0 mm wide) between two plates, and theamples are separated while flowing down with the carrier solu-ion and colleted at the bottom outlets for subsequent analysis bypplying an electric field along the liquid film perpendicular tohe direction of flow. Either IEF or zone electrophoretic mecha-ism can be adopted for the preparation of proteins, organelles,tc. In 2004, Mortiz et al. [56,57] described a two-dimensionaleparation system that used FFE to fractionate protein mixturesy IEF into 96 well pools. A recent development of FFE isiniaturization to improve its performance and heat dissipation
58–61].Gel electrophoresis is termed not according to principle but to
he medium. For sample preparation the most often used meth-ds are (1) sodium dodecyl sulfate or sulfonate-polyacrylamideel electrophoresis (SDS-PAGE) which is a zone electrophoresisetarded by gel pores, and (2) disc electrophoresis which is a typef isotachophoresis performed on gel column with leading anderminating electrolytes (or discontinuous buffers). Samples pre-ared by gel media are collected by cutting off sample dots fromhe gel and eluted or dissolved in solvents. Electroblotting pro-ein onto chemically inert membranes is probably a better wayor micro-sample preparation. In the case of two-dimensionallectrophoresis (2-DE) which is currently the workhorse for
roteomics when combing with MS [62], the first dimensionf separation can actually be considered as a means of samplereparation and the prepared samples were “on-line” transferrednto the second analytical dimension by electric field.pawm
. A 1184 (2008) 191–219 197
Possibly enlightened by IEF, in-column sample stackingICSS) techniques have been explored in CE since 1990s. Orig-nally ICSS was developed to improve the detection sensitivityy increasing sample load. However, an on-line sample prepa-ation way is able to integrate sampling, purifying, desaltingnd concentrating into one step and is thus especially useful forretreating samples with a limited volume. There are variousCSS methods developed during the last 20 years. The methodsre distinguishable according to their principle including fieldmplification (FA), isotachophoresis (ITP), sweeping, pH regu-ation at column capture and barrage. For more discussion pleaseefer to Section 4.6.
. Highlighted methods
During recent years, many modified, innovated and evenovel sample preparation methods have been proposed andnderwent various evaluations or validations to meet the chal-enges constantly appearing in life sciences. This sectionollects some advanced and promising methods to hit and high-ight the new trends in developing the methods for samplereparation.
.1. Environment friendly methods
.1.1. Supercritical fluidsAn environment friendly method of sample preparation
ainly means the use of environment friendly solvents suchs water, supercritical fluids, RTILs, etc. SFE and SWE are twoypical environment friendly methods.
Supercritical fluids, the intervening physical state betweenas and liquids, possess unique properties. In particular, theiriscosity is lower than that of liquids, allowing faster diffu-ion and more efficient extractions. By such fluids, SFE obtainsbility to perform selective extractions through adjusting theuid properties by regulating pressure, temperature and the con-
ent of modifiers. Carbon dioxide is currently the solvent ofhoice. Non-polar supercritical CO2 produces high extractionfficiency for compounds from non-polar to low polarity. Cosol-ent systems combining CO2 with one or more small amountsf modifiers (≤15%) extend the utility of CO2 to polar andven ionic compounds. The numerous adjustable parametersave not only made SFE flexible but also tedious in optimiza-ion and difficult in use. Important points in favor of SFE arets relatively short extraction time, mild pressures and tem-eratures used, which minimize the risks of losing activity toreserve the integrity of functional compounds of food andatural products, and to extract labile compounds from environ-ental samples [75–79]. SFE works the best for finely powdered
olids with good permeability, such as soils and dried plant mate-ials. Extraction of wet or liquid samples and solutions can bechieved by SFE but with somewhat difficulty [80,81].
SFE can be accomplished in a static mode in which sam-
le and solvent are mixed and kept for a user-specified timet a constant pressure and temperature, or in a dynamic modehere the solvent flows through the sample in a continuousanner. The extracted analytes can be collected into an off-line
198 Y. Chen et al. / J. Chromatogr.
Fig. 2. Schematic configuration of SFE–LC–GC–MS system. C, position ofbel
ddssiddmmnS
ihimr
atbhmpsapcaoatbr
dcSse
llpt[
eptecb
4
ooefet
tqatic[o[aaff
stpIup[
cRia
4
4
ack pressure regulator operated during elution; CR, capillary restrictor; EV,xtraction vessel; R, pressure restrictor; V1–V7, multi-port valves (all at theoading position) [93]. From ref. [93], with permission.
evice or transferred to a on-line chromatographic system forirect analysis. The off-line trapping is performed by depres-urizing the supercritical fluid and absorbing analytes into aolvent or onto a solid sorbent. For on-line coupling, supercrit-cal fluid chromatography, GC or LC can be chosen but withifferent interfaces. With SFE–GC the analytes can be trappedirectly in the GC injector with a restrictor, or first on a solidaterial and then transferred to GC column [82]. SFE–LC isost commonly conducted through solid-phase trapping tech-
iques [83–85]. Fig. 2 shows an example of on-line coupledFE–LC–GC–MS [86,87].
Although various advancements have been achieved includ-ng techniques, instrumentation and applications [88–91], SFEas turned more or less to analytical aspects [92]. Although SFEs inherently superior to many other techniques, it is not a facile
eans that can be used widely. How to facilitate the use of SFEemains a challenge.
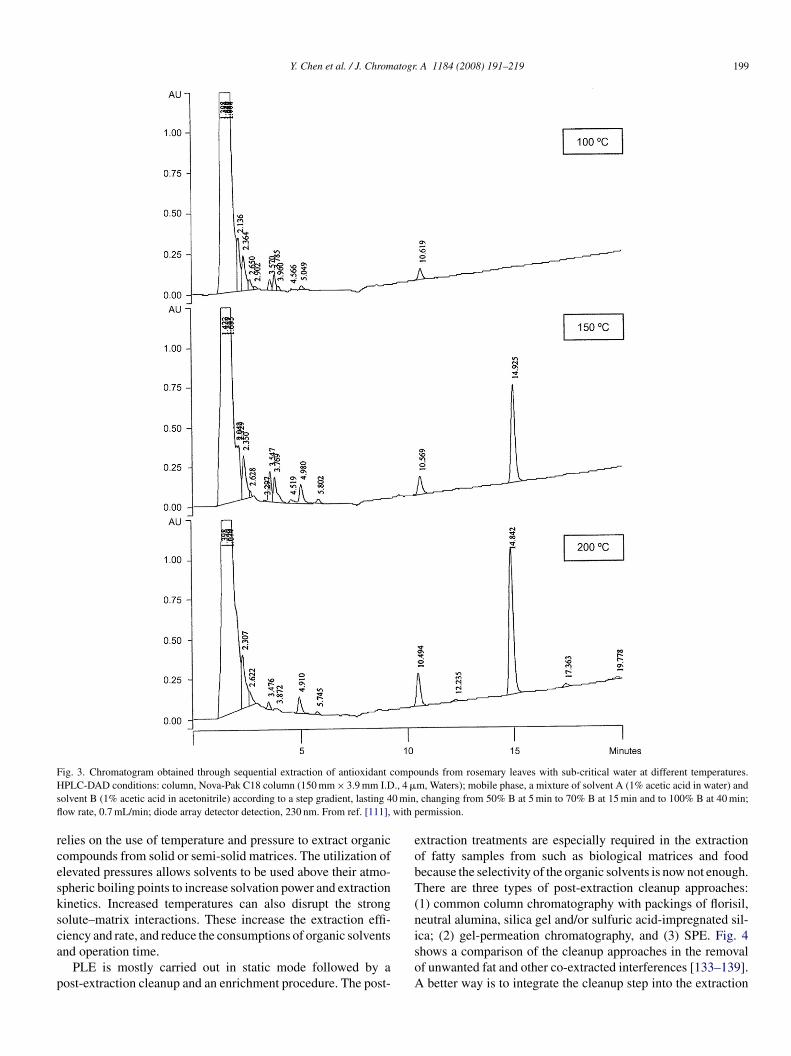
Water is much too polar to be used for extracting non-nd moderate-polar organic compounds at a room tempera-ure. However, when water is brought to its sub-critical statey increasing temperature up to 100–374 ◦C at a sufficientlyigh pressure, its polarity, viscosity and surface tension decreasearkedly [94,95]. Water is thus able to extract low-polar com-
ounds at a higher temperature and polar compounds at auitably lower temperature (Fig. 3). SWE has been shownpplicable to the extraction of organic pollutants such asolychlorinated biphenyls (PCBs), polycyclic aromatic hydro-arbons (PAHs), pesticides, herbicides, phenolic compoundsnd others from soils and plant materials [96,97]. The extractionf volatile components such as essential oils in plant materi-ls has also been reported [98,99]. The application of SWEo the extraction of bioactive compounds or biomarkers fromotanicals and medicinal plant materials has been well reviewedecently [100].
SWE is generally performed in a flowing mode, giving fairlyiluted aqueous extracts which has to be further extracted and
oncentrated with a small volume of organic solvents, SPE orPME [101,102]. Alternatively, the analytes can be trapped initu by addition of a SPE sorbent disk or cartridge into the SWExtraction vessel [103,104]. Another interesting alternative is toshps
A 1184 (2008) 191–219
ink the extraction system directly to LC or it is even better toink it to a superheated water chromatography through a solid-hase trapping interface [105–108]. SWE has also been coupledo GC using a membrane or a solid-phase trap as an interface109,110].
Nevertheless, SWE suffers from two disadvantages of lowxtraction efficiency and impassability for thermal instable com-osition. A way to kill two birds with one stone is modifyinghe water with organic solvents or surfactants to reduce thextraction temperature and improve extraction efficiencies. Inonclusion, SWE is waiting for further investigation before itecomes widely adaptable.
.1.2. Room temperature ionic liquidsRTILs are absorbing new type of liquids having at least
ne organic cation or anion, integrating both of the advantagesf water and organic solvents into one molecule. Because ofxtremely low vapor pressure, they are safe to use and possiblyriendly to environment, and have since been considered to benvironment friendly solvents as an alternative to the conven-ional solvents.
RTILs can be hydrophilic or hydrophobic depending onhe structures of their skeletal cations and anions. Conse-uently, their extraction selectivity and efficiency are somewhatdjustable with the assistance of other additives or extrac-ants [112,113]. For instance, in the extraction of special metalons from aqueous solutions, RTILs are used together with 18-rown-6 family crown ethers and some synthesized extractants114–116]. For better extraction of metal ions from aque-us solutions, some task-specific RTILs have been synthesized117,118], combining both functions of hydrophobic solventnd extractant in one molecule. RTILs extraction has also beenpplied to the removal of organic environmental contaminantsrom water [119,120] and to the deep desulfurization of dieseluels [121].
Importantly, extraction of biomolecules using RTILs hasignificantly increased during recent years [122]. The extrac-ion of proteins or double-stranded DNA from an aqueoushase into RTILs has been reported quite recently [123–125].n SPME 1-octyl-3-methylimidazolium (OMIM) PF6 wassed as a disposable liquid absorbent [126] and in liquid-hase microextraction (LPME) as an extraction solvent127,128].
Some reviews emphasizing the use of RTILs in analyti-al chemistry can be found in references [129–131]. AlthoughTILs themselves are not a new type of chemicals, they are
ndeed a novel and promising extraction solvents worth of tryingnd exploring with great efforts.
.2. Acceleration techniques
.2.1. Pressurized liquid extractionPLE is also known as pressurized fluid extraction, pressurized
olvent extraction, accelerated solvent extraction, pressurizedot solvent extraction, high-pressure solvent extraction, high-ressure and high-temperature solve extraction or sub-criticalolvent extraction [132], possibly rooting in SWE. Clearly PLE
Y. Chen et al. / J. Chromatogr. A 1184 (2008) 191–219 199
F ompoH ., 4 �
s 0 minfl with p
rcesksca
p
eobT(n
ig. 3. Chromatogram obtained through sequential extraction of antioxidant cPLC-DAD conditions: column, Nova-Pak C18 column (150 mm × 3.9 mm I.D
olvent B (1% acetic acid in acetonitrile) according to a step gradient, lasting 4ow rate, 0.7 mL/min; diode array detector detection, 230 nm. From ref. [111],
elies on the use of temperature and pressure to extract organicompounds from solid or semi-solid matrices. The utilization oflevated pressures allows solvents to be used above their atmo-pheric boiling points to increase solvation power and extractioninetics. Increased temperatures can also disrupt the strongolute–matrix interactions. These increase the extraction effi-
iency and rate, and reduce the consumptions of organic solventsnd operation time.PLE is mostly carried out in static mode followed by aost-extraction cleanup and an enrichment procedure. The post-
isoA
unds from rosemary leaves with sub-critical water at different temperatures.m, Waters); mobile phase, a mixture of solvent A (1% acetic acid in water) and, changing from 50% B at 5 min to 70% B at 15 min and to 100% B at 40 min;ermission.
xtraction treatments are especially required in the extractionf fatty samples from such as biological matrices and foodecause the selectivity of the organic solvents is now not enough.here are three types of post-extraction cleanup approaches:
1) common column chromatography with packings of florisil,eutral alumina, silica gel and/or sulfuric acid-impregnated sil-
ca; (2) gel-permeation chromatography, and (3) SPE. Fig. 4hows a comparison of the cleanup approaches in the removalf unwanted fat and other co-extracted interferences [133–139].better way is to integrate the cleanup step into the extraction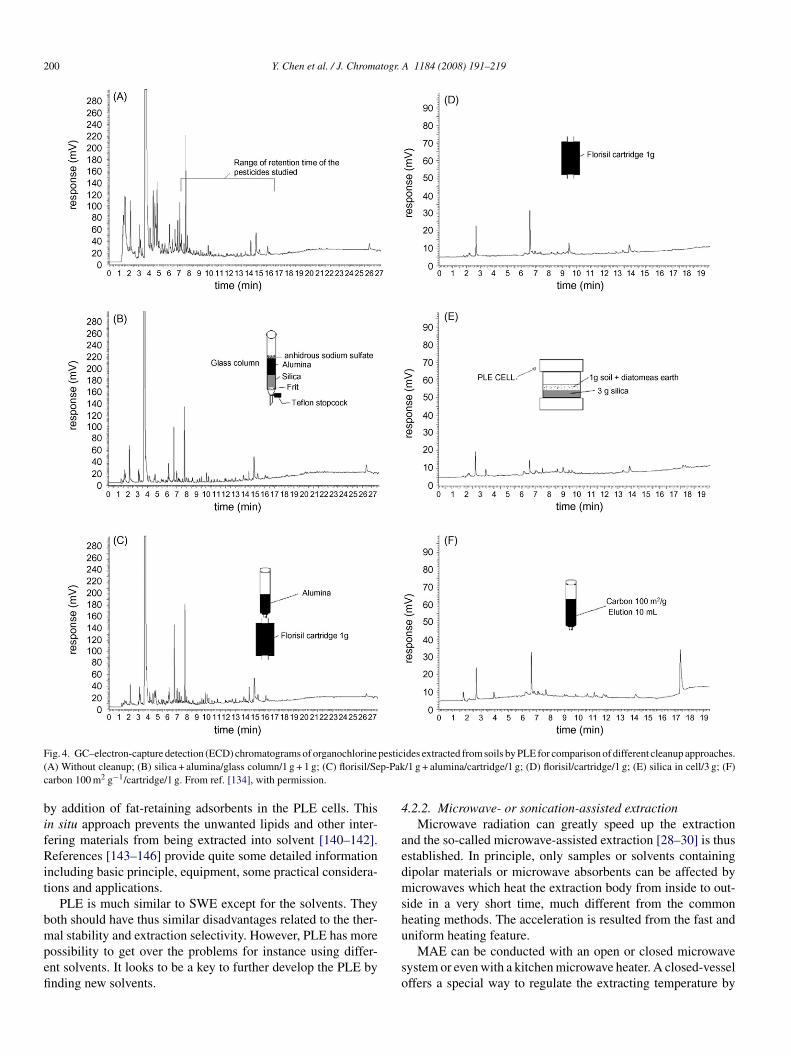
200 Y. Chen et al. / J. Chromatogr. A 1184 (2008) 191–219
F pestic( p-Pakc
bifRit
bmpefi
4
aedmsh
ig. 4. GC–electron-capture detection (ECD) chromatograms of organochlorineA) Without cleanup; (B) silica + alumina/glass column/1 g + 1 g; (C) florisil/Searbon 100 m2 g−1/cartridge/1 g. From ref. [134], with permission.
y addition of fat-retaining adsorbents in the PLE cells. Thisn situ approach prevents the unwanted lipids and other inter-ering materials from being extracted into solvent [140–142].eferences [143–146] provide quite some detailed information
ncluding basic principle, equipment, some practical considera-ions and applications.
PLE is much similar to SWE except for the solvents. Theyoth should have thus similar disadvantages related to the ther-
al stability and extraction selectivity. However, PLE has moreossibility to get over the problems for instance using differ-nt solvents. It looks to be a key to further develop the PLE bynding new solvents.
u
so
ides extracted from soils by PLE for comparison of different cleanup approaches./1 g + alumina/cartridge/1 g; (D) florisil/cartridge/1 g; (E) silica in cell/3 g; (F)
.2.2. Microwave- or sonication-assisted extractionMicrowave radiation can greatly speed up the extraction
nd the so-called microwave-assisted extraction [28–30] is thusstablished. In principle, only samples or solvents containingipolar materials or microwave absorbents can be affected byicrowaves which heat the extraction body from inside to out-
ide in a very short time, much different from the commoneating methods. The acceleration is resulted from the fast and
niform heating feature.MAE can be conducted with an open or closed microwaveystem or even with a kitchen microwave heater. A closed-vesselffers a special way to regulate the extracting temperature by
atogr. A 1184 (2008) 191–219 201
sifrcp
fssal
twtpcace
4
4
pflM
onaId[iamtcoee
oent[
ndsIa
Fdw
gc
(ci((or
tt
Y. Chen et al. / J. Chrom
imply adjusting the vessel pressure in a way somewhat sim-lar to PLE technology [147]. The use of open-vessel mainlyocused on Soxhlet extraction has been reported and reviewedecently [148]. Although almost all reported MAE methods wereonducted off-line, on-line approaches have been shown to beossible [149–151].
The main advantage of MAE is its wide applicability forast extractions of analytes including some thermal instable sub-tances. Its main disadvantage is less incorporable into a flowystem. It should be noted that MAE is better conducted withthermostatic microwave oven. Microwave can also accelerate
abeling reaction [152].Sonication is an alternative means to enhance the extraction
hrough induced cavitations which creates microenvironmentsith high temperatures and high pressures, and in turn speed up
he removal of analytes from sample matrices. SAE [33,36] areerformed mostly in static modes, some in dynamic or on-lineombinations with analytical systems [31–35,37]. Sonication isn expeditious, inexpensive and efficient means to innovate someonventional extraction techniques such as SFE, PLE, Soxhletxtraction and LLE [153–157].
.3. Scale down
.3.1. Liquid-phase microextractionFor analytical purposes, scaling down the size of sample
reparation is more applicable than scaling-up which is criticalor productive preparation. There are two important scale-downiquid-phase extraction approaches, i.e. SDME [23,158] and
SLE pioneered by Audunsson [159].SDME normally extracts analytes into a (1–10 �L) droplet
f water immiscible extracting solvent attached at a syringeeedle. The droplet can either be immersed into a stirredqueous solution or hung over a sample (Fig. 5A and B).n common, the liquid drop varies its volume regularly orynamically (Fig. 5C) to improve the extraction efficiency160] by simulating the traditional separatory funnel work-ng manner. Further improvement of the performance can bechieved by automation [161–163]. Since 2000, a new for-at of SDME appeared which captures analytes by inserting
he droplet in a flowing sample stream and hence termedontinuous-flow microextraction [164,165]. Due to the continu-us contact with the flowing fresh sample solution, the extractionfficiency and concentration factor are higher than the staticxtraction.
The hanging-over or headspace SDME allows the use of bothrganic and aqueous [166,167] solvents as receiving phase toxtract volatile or semivolatile compounds since the droplet doesot contact with the sample solution directly. This type of extrac-ion is mostly preferred by GC [168–171] and somewhat by LC172], CE [167,173]) and MS [174,175].
Clearly SDME features simplicity, cost-effectiveness andegligible solvent consumption. However, it inherits some
rawbacks from LLE such as the formation of emulsion and dis-olution of the liquid droplet in dealing with some dirty samples.n most cases the micro liquid drop is not as stable as desired,nd dedicated operators may be a prerequisite to conduct an ele-bpta
ig. 5. Schematic diagram of (A and B) direct immersion and headspace single-rop microextraction [23] and (C) automatic dynamic LPME. From ref. [160],ith permission.
ant extraction. To improve the stability of the droplet is thus ahallenging topic needed to get over.
MSLE uses a porous membrane to separate the sample phasedonor) from liquid extractant (acceptor) and microextractionan be achieved by using hollow fibers. Its most absorb-ng advantages include (1) very low consumption of solvent,2) remarkable cleanup efficiency, (3) high enrichment factors>100-fold) for inorganic and organic analytes in a wide rangef polarity, and (4) capability of on-line coupling to chromatog-aphy and other instrumental systems [22,176–180].
MSLE may work across more than two phases. In most ofwo-phase systems, the donor and acceptor contact each otherhrough the membrane pores [181] and the mass transfer is driven
y the concentration gradient or diffusion. When the pores arere-filled with an organic solvent, the two-phase system changeso a three-phase system, where the donor and acceptor (both arequeous phase) are separated by the organic filled hydrophobic
202 Y. Chen et al. / J. Chromatogr. A 1184 (2008) 191–219
F 97], a(
msaitpTpt
ftTpduu
sotti[
4
4
mA
ig. 6. Liquid-phase microextraction systems using (A and B) flat-membrane [1E) and without automatic flow (reprinted with permission).
embrane which extracts analytes from one aqueous sampleolution and is back-extracted by the other aqueous phase. Suchthree-phase system can suit the extraction of polar and even
onic compounds like organic acids, bases and metal ions. If thewo aqueous phases at the both sides of membrane have differentH, higher selectivity and enrichment factors can be obtained.he separating membrane can be made of silicone rubber orolyethylene that provides a mechanically stable system but athe cost of losing extraction speed [179].
These two systems can both be performed in a flow-throughormat, suitable for on-line combination with chromatographicechniques [22,179] such as LC [182,183] and GC [184–188].he flow-through cells are formed by intercalating a flat micro-
orous membrane in between donor and acceptor solvents withifferent designs (Fig. 6A and B). Hollow fibers (HF) are alsosed in flow-through format to largely reduce the channel vol-mes [189–191], but the handing of the hollow fibers tends to beuhut
nd (C [198] and E [196]) rod-shaped and (D) U-shaped [199] hollow fiber with
omewhat difficult [179]. HF-LPME has recently been focusedn in-vial format either in a rod-like or U-shaped configura-ion (Fig. 6C and D) [192], and the moving phase, normallyhe acceptor, can be automated (Fig. 6E) [193–196], whichmproves the extraction speed with higher enrichment factor194].
.4. Adsorptive methods
.4.1. Multifunctional sorbentsThe principal goals of SPE should be trace enrichment,
atrix simplification (sample cleanup) and medium exchange.lthough SPE disks have been developed, since 1990s, to scale
p the sample preparation, miniaturized techniques and devicesave been causing more and more concerns to handle small vol-me of samples. Thin disk and small column have enabled SPEo largely increase its throughput by automation in combina-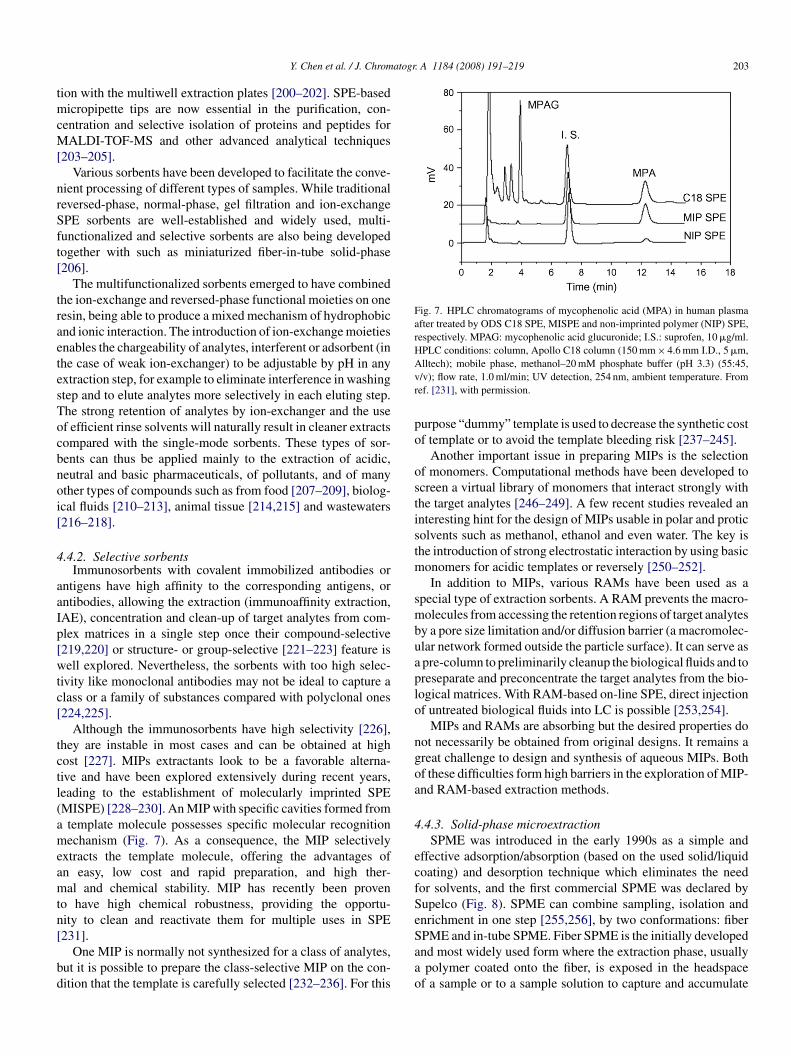
atogr. A 1184 (2008) 191–219 203
tmcM[
nrSft[
traetesTocbnoi[
4
aaIp[wtc[
tctl(ameamtn[
bd
Fig. 7. HPLC chromatograms of mycophenolic acid (MPA) in human plasmaafter treated by ODS C18 SPE, MISPE and non-imprinted polymer (NIP) SPE,respectively. MPAG: mycophenolic acid glucuronide; I.S.: suprofen, 10 �g/ml.HPLC conditions: column, Apollo C18 column (150 mm × 4.6 mm I.D., 5 �m,Avr
po
ostistm
smbuaplo
ngoa
4
ecfSe
Y. Chen et al. / J. Chrom
ion with the multiwell extraction plates [200–202]. SPE-basedicropipette tips are now essential in the purification, con-
entration and selective isolation of proteins and peptides forALDI-TOF-MS and other advanced analytical techniques
203–205].Various sorbents have been developed to facilitate the conve-
ient processing of different types of samples. While traditionaleversed-phase, normal-phase, gel filtration and ion-exchangePE sorbents are well-established and widely used, multi-unctionalized and selective sorbents are also being developedogether with such as miniaturized fiber-in-tube solid-phase206].
The multifunctionalized sorbents emerged to have combinedhe ion-exchange and reversed-phase functional moieties on oneesin, being able to produce a mixed mechanism of hydrophobicnd ionic interaction. The introduction of ion-exchange moietiesnables the chargeability of analytes, interferent or adsorbent (inhe case of weak ion-exchanger) to be adjustable by pH in anyxtraction step, for example to eliminate interference in washingtep and to elute analytes more selectively in each eluting step.he strong retention of analytes by ion-exchanger and the usef efficient rinse solvents will naturally result in cleaner extractsompared with the single-mode sorbents. These types of sor-ents can thus be applied mainly to the extraction of acidic,eutral and basic pharmaceuticals, of pollutants, and of manyther types of compounds such as from food [207–209], biolog-cal fluids [210–213], animal tissue [214,215] and wastewaters216–218].
.4.2. Selective sorbentsImmunosorbents with covalent immobilized antibodies or
ntigens have high affinity to the corresponding antigens, orntibodies, allowing the extraction (immunoaffinity extraction,AE), concentration and clean-up of target analytes from com-lex matrices in a single step once their compound-selective219,220] or structure- or group-selective [221–223] feature isell explored. Nevertheless, the sorbents with too high selec-
ivity like monoclonal antibodies may not be ideal to capture alass or a family of substances compared with polyclonal ones224,225].
Although the immunosorbents have high selectivity [226],hey are instable in most cases and can be obtained at highost [227]. MIPs extractants look to be a favorable alterna-ive and have been explored extensively during recent years,eading to the establishment of molecularly imprinted SPEMISPE) [228–230]. An MIP with specific cavities formed fromtemplate molecule possesses specific molecular recognitionechanism (Fig. 7). As a consequence, the MIP selectively
xtracts the template molecule, offering the advantages ofn easy, low cost and rapid preparation, and high ther-al and chemical stability. MIP has recently been proven
o have high chemical robustness, providing the opportu-ity to clean and reactivate them for multiple uses in SPE
231].One MIP is normally not synthesized for a class of analytes,ut it is possible to prepare the class-selective MIP on the con-ition that the template is carefully selected [232–236]. For this
Saao
lltech); mobile phase, methanol–20 mM phosphate buffer (pH 3.3) (55:45,/v); flow rate, 1.0 ml/min; UV detection, 254 nm, ambient temperature. Fromef. [231], with permission.
urpose “dummy” template is used to decrease the synthetic costf template or to avoid the template bleeding risk [237–245].
Another important issue in preparing MIPs is the selectionf monomers. Computational methods have been developed tocreen a virtual library of monomers that interact strongly withhe target analytes [246–249]. A few recent studies revealed annteresting hint for the design of MIPs usable in polar and proticolvents such as methanol, ethanol and even water. The key ishe introduction of strong electrostatic interaction by using basic
onomers for acidic templates or reversely [250–252].In addition to MIPs, various RAMs have been used as a
pecial type of extraction sorbents. A RAM prevents the macro-olecules from accessing the retention regions of target analytes
y a pore size limitation and/or diffusion barrier (a macromolec-lar network formed outside the particle surface). It can serve aspre-column to preliminarily cleanup the biological fluids and toreseparate and preconcentrate the target analytes from the bio-ogical matrices. With RAM-based on-line SPE, direct injectionf untreated biological fluids into LC is possible [253,254].
MIPs and RAMs are absorbing but the desired properties doot necessarily be obtained from original designs. It remains areat challenge to design and synthesis of aqueous MIPs. Bothf these difficulties form high barriers in the exploration of MIP-nd RAM-based extraction methods.
.4.3. Solid-phase microextractionSPME was introduced in the early 1990s as a simple and
ffective adsorption/absorption (based on the used solid/liquidoating) and desorption technique which eliminates the needor solvents, and the first commercial SPME was declared byupelco (Fig. 8). SPME can combine sampling, isolation andnrichment in one step [255,256], by two conformations: fiber
PME and in-tube SPME. Fiber SPME is the initially developednd most widely used form where the extraction phase, usuallypolymer coated onto the fiber, is exposed in the headspacef a sample or to a sample solution to capture and accumulate

204 Y. Chen et al. / J. Chromatogr. A 1184 (2008) 191–219
ce ma
anfi
iwiTt
mtscFtubc
clBft
mstr
swe
Stmfmflha
wpiabo
lcico(m
Fig. 8. Design of the first commercial SPME devi
nalytes. After a certain time to reach equilibration (sometimesear-equilibrium [257]), the loaded extraction phase is trans-erred to the injection port of chromatographic or other analyticalnstrument in use.
However, fiber SPME has the drawbacks of limited capac-ty and complexity in coupling with LC. The in-tube SPMEas thus explored which uses an open tubular fused-silica cap-
llary instead of the fiber [259] to suit on-line hyphenation.he column length and the thickness of extractant coating are
unable.The study of stationary phases for SPME assists the develop-
ent of applications [260–262]. Several coatings from non-polaro polar are commercially available, including polydimethyl-iloxane (PDMS), polyacrylate, divinylbenzene, Carboxen (aarbon molecular sieve) and Carbowax (polyethylene glycol).or coating, sol–gel technique provides an efficient incorpora-
ion of organic components into inorganic polymeric structuresnder extraordinarily mild thermal conditions [263], and haseen applied to the preparation of coated fibers [264–267] andapillary columns [268–270].
Fibers are available in different film thicknesses with singleoatings, combined coatings or co-polymers but remain veryimited, which restricts the wide application of SPME [260].esides coatings, monolithic sorbents with different kinds of
unctional groups [271–275], including MIPs [276], have showno be a promising alternative.
SPME may integrate sampling with sample preparation,
aking it suitable for on-site sampling and analysis. Corre-ponding passive sampling devices have been reported for theime-weighted average air/water sampling in which the fiber isetracted a known distance into its needle housing during the
oa
s
de by Supelco. From ref. [258], with permission.
ampling period [277–280]. Recently PDMS-rod and membraneere used as the passive samplers of SPME to improve extraction
fficiency and sensitivity [281–283].The small dimension and nearly solvent-free feature of
PME enable in vivo sampling without severe damage tohe live organisms. The reported in vivo methods include
onitoring the biogenic volatile organic compounds emittedrom plants, isolating the insect semiochemicals and othericrobiological inspections [284,285]. Direct extraction fromowing blood [286] and sampling of volatiles emitted byumans [287–289] and insects [290,291] have also beenchieved.
Most fiber SPME methods have been used in combinationith GC and LC (Fig. 9) [292–294]. A fiber SPME is incor-orated with HPLC through a desorption chamber as a part ofnjection loop (Fig. 9D) [295–298]. In-tube SPME can be fullyutomated [259,299], its capillary column is generally placed inetween the HPLC autosampler needle and the injection valver inserted in the injection loop [300].
On-line coupling of SPE with GC is similar to those used forarge volume injection and on-line LC–GC. On-column, inter-alated loop and programmable temperature vaporiser (PTV)nterfaces are the basic choice (Fig. 10) [302,303]. SPE-HPLCan be built by “column switching” that is inserting a piecef SPE material as a small pre-column into the injection loopFig. 11) [304]. By setting robust and reliable chromatographicethods and cartridge exchangeable modules, large increase
f the sample throughput is possible with a saving of the totalnalysis time [305–307].
Micro SPE can be on-line coupled to CE through a Tee-haped interface [308,309] or in-line coupled by placing
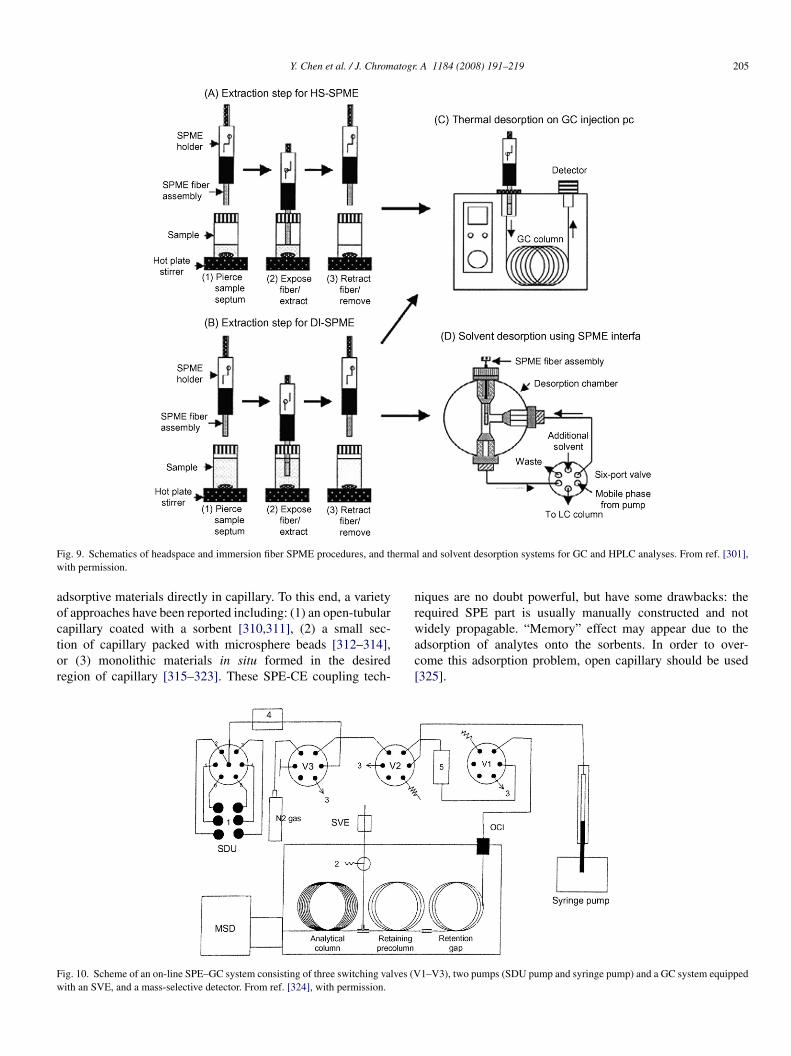
Y. Chen et al. / J. Chromatogr. A 1184 (2008) 191–219 205
F ermaw
aoctor
nr
Fw
ig. 9. Schematics of headspace and immersion fiber SPME procedures, and thith permission.
dsorptive materials directly in capillary. To this end, a varietyf approaches have been reported including: (1) an open-tubular
apillary coated with a sorbent [310,311], (2) a small sec-ion of capillary packed with microsphere beads [312–314],r (3) monolithic materials in situ formed in the desiredegion of capillary [315–323]. These SPE-CE coupling tech-wac[
ig. 10. Scheme of an on-line SPE–GC system consisting of three switching valves (Vith an SVE, and a mass-selective detector. From ref. [324], with permission.
l and solvent desorption systems for GC and HPLC analyses. From ref. [301],
iques are no doubt powerful, but have some drawbacks: theequired SPE part is usually manually constructed and not
idely propagable. “Memory” effect may appear due to thedsorption of analytes onto the sorbents. In order to over-ome this adsorption problem, open capillary should be used325].
1–V3), two pumps (SDU pump and syringe pump) and a GC system equipped
206 Y. Chen et al. / J. Chromatogr. A 1184 (2008) 191–219
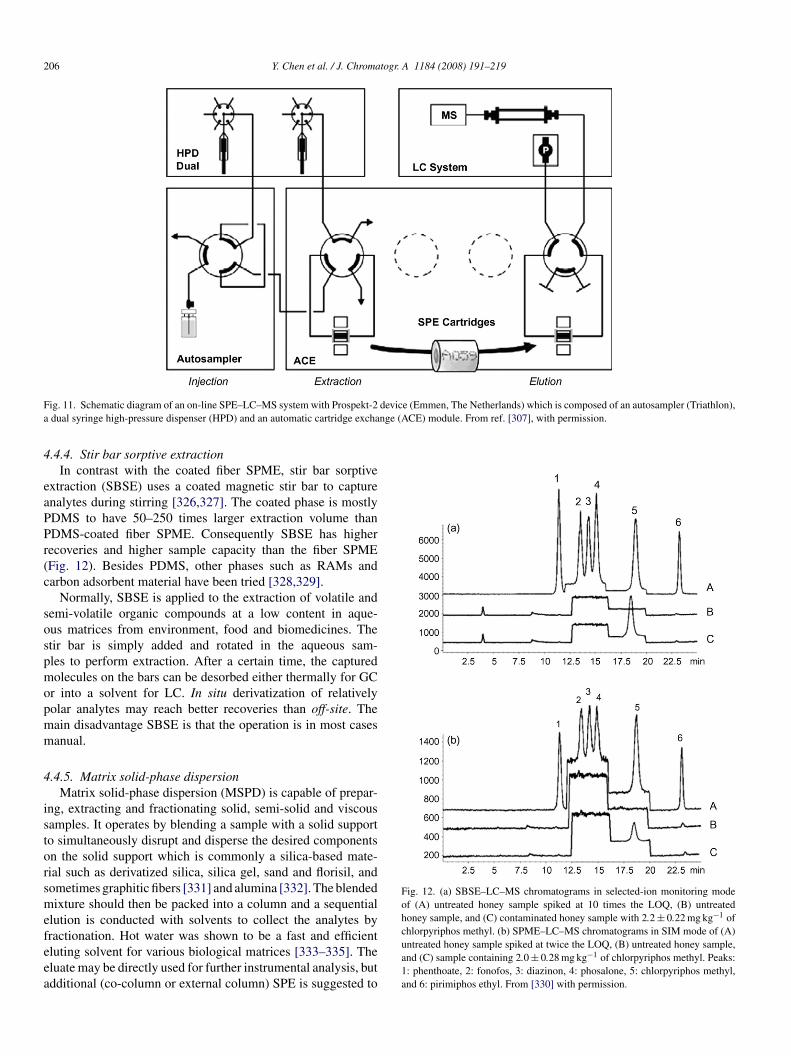
F device (Emmen, The Netherlands) which is composed of an autosampler (Triathlon),a nge (ACE) module. From ref. [307], with permission.
4
eaPPr(c
sospmopmm
4
istorsmefeea
Fig. 12. (a) SBSE–LC–MS chromatograms in selected-ion monitoring modeof (A) untreated honey sample spiked at 10 times the LOQ, (B) untreatedhoney sample, and (C) contaminated honey sample with 2.2 ± 0.22 mg kg−1 of
ig. 11. Schematic diagram of an on-line SPE–LC–MS system with Prospekt-2dual syringe high-pressure dispenser (HPD) and an automatic cartridge excha
.4.4. Stir bar sorptive extractionIn contrast with the coated fiber SPME, stir bar sorptive
xtraction (SBSE) uses a coated magnetic stir bar to capturenalytes during stirring [326,327]. The coated phase is mostlyDMS to have 50–250 times larger extraction volume thanDMS-coated fiber SPME. Consequently SBSE has higherecoveries and higher sample capacity than the fiber SPMEFig. 12). Besides PDMS, other phases such as RAMs andarbon adsorbent material have been tried [328,329].
Normally, SBSE is applied to the extraction of volatile andemi-volatile organic compounds at a low content in aque-us matrices from environment, food and biomedicines. Thetir bar is simply added and rotated in the aqueous sam-les to perform extraction. After a certain time, the capturedolecules on the bars can be desorbed either thermally for GC
r into a solvent for LC. In situ derivatization of relativelyolar analytes may reach better recoveries than off-site. Theain disadvantage SBSE is that the operation is in most casesanual.
.4.5. Matrix solid-phase dispersionMatrix solid-phase dispersion (MSPD) is capable of prepar-
ng, extracting and fractionating solid, semi-solid and viscousamples. It operates by blending a sample with a solid supporto simultaneously disrupt and disperse the desired componentsn the solid support which is commonly a silica-based mate-ial such as derivatized silica, silica gel, sand and florisil, andometimes graphitic fibers [331] and alumina [332]. The blendedixture should then be packed into a column and a sequential
lution is conducted with solvents to collect the analytes by
ractionation. Hot water was shown to be a fast and efficientluting solvent for various biological matrices [333–335]. Theluate may be directly used for further instrumental analysis, butdditional (co-column or external column) SPE is suggested tochlorpyriphos methyl. (b) SPME–LC–MS chromatograms in SIM mode of (A)untreated honey sample spiked at twice the LOQ, (B) untreated honey sample,and (C) sample containing 2.0 ± 0.28 mg kg−1 of chlorpyriphos methyl. Peaks:1: phenthoate, 2: fonofos, 3: diazinon, 4: phosalone, 5: chlorpyriphos methyl,and 6: pirimiphos ethyl. From [330] with permission.

atogr
rf
arom
4
elomDpiC
mRScAh
evihpbbccts
p[b
4
sas
4
ewsat
lltsttt
sbcIIbt
temsewpttictha[
4
a[pciemcpts
4
wcist
Y. Chen et al. / J. Chrom
emove the co-eluted interferent or to cleanup the analytes byurther fractionation [336,337].
MSPD can eliminate many complicated steps in classical LSEnd/or SPE [338–340] and is useful for the isolation of a wideange of drugs, pesticides, naturally occurring constituents andther compounds from a wide variety of complex biologicalatrices. This method is however fairly labour intensive.
.5. Microdialysis
Microdialysis is inherently an in vivo sampling techniquextensively used in clinical research, medicine development andife sciences. A microdialysis system is essentially composedf a micropump, a microdialytic probe with a semipermeableembrane at the tip and liquid delivery and collection devices.uring sampling, the probe is implanted into a living being anderfused with buffered solutions, and the flowing-out dialysates collected into microvials or directly transferred into a LC orE separation system.
Initially, microdialysis sampling is used to collect smallolecules such as pharmaceuticals and neurotransmitters.ecently, its application has extended to macromolecules.chutte et al. [341] used polyethersulfone microdialysis probe toollect proteins and dextrans ranging from 3000 to 150,000 ando et al. [342] obtained inflammatory cytokines with relativelyigh recovery using a similar method.
In order to increase the recovery of microdialysis, annhanced technique has been developed by chemically con-erting the target species to other forms once they diffusento the receiving solution. This conversion can maintain theighest driving force of diffusion required for analyte trans-ortation. For example, metal ions can efficiently be collectedy converting them into complexes with chelating agents and/oriopolymers added in the receiving solutions [343]. Similarly, �-yclodextrins are used to extract some drugs through host–guestomplexation [344]. By introducing affinity solid particles intohe receiving solution, Pettersson et al. [345] has developed aolid-support-enhanced microdialysis method.
Compared with others, microdialytic system is ready to cou-le with column separation systems such as CE [346], HPLC347] and microchip electrophoresis [348]. This creates a broadridge to link an analytic system to a living body.
.6. On-line stacking
Stacking is originally explored to increase the detection sen-itivity of CE by increasing sample loading, but it is actuallynew type of sample preparation route waiting for exploration
ince it can tremendously concentrate analytes into a tiny zone.
.6.1. IsotachophoresisWhen an analyte plug is sandwiched in between a leading
lectrolyte having the fastest ion and a terminating electrolyte
ith a slowest co-ion, the analyte co-ions can only migrate andeparate in between the leading and terminating ions. The sep-rated analyte zones will line up one after another accordingo their apparent mobility or speed, neither isolating nor over-
ptas
. A 1184 (2008) 191–219 207
apping each other, and all migrate at the same speed as theeading ion. Their concentration should be adjusted to a bit lesshan that of the leading ion by largely reducing their zone lengthince they are generally at a trace level. The enrichment factorhus depends on the content of the leading ion which can be upo more than 0.1 M. High stacking factor is expected by ITP inheory and has been achieved in practice.
There are two distinguishable approaches to conduct an ITPtacking, i.e. two dimensional coupling and in-capillary com-ination called transient-ITP (t-ITP) [349,350]. The formeronducts first a step of sample clean-up and concentration byTP with a wide bore capillary and second a step of separatingTP (ITP-ITP) [351,352] or CE (ITP-CE) [352,353] in a narrowore capillary. Samples in urea can directly be analyzed by eitherhe ITP-ITP or ITP-CE.
t-ITP allows sample stacking at the beginning of CE separa-ion. The stacking is achieved by introducing a plug of leadinglectrolytes followed by a section of sample solution with ter-inating ions, or reversely, by introducing a plug of sample
olution with leading ion followed by a section of terminatinglectrolyte. As early as in 1993, Shihabi [354] suggested an easyay to perform the t-ITP by introducing a plug of sample pre-ared in acetonitrile and NaCl. The acetonitrile largely reduceshe viscosity of sample solution to accelerate Cl− or Na+ movingo the right leading position to fast build up an ITP environmentn a short sample plug [355]. This type of t-ITP has been suc-essfully applied to the analysis of physiological samples, ableo stack analytes by factors of 10- to 30-fold [356–358]. t-ITPave been shown to be useful for the preconcentration of tracenalytes in the matrices containing surplus ionic components359–362].
.6.2. Capillary isoelectric focusingCapillary isoelectric focusing (cIEF) works the same way
s IEF but in a capillary filled with free solution ampholytes363]. Proteins can be separated and focused or stacked at theirI position. cIEF is suitable for large volume (one column)oncentration of zwitterions and can be coupled with other cap-llary or column separation techniques such as capillary zonelectrophoresis (CZE) [364], ITP [365], capillary electrochro-atography [366], capillary gel electrophoresis (CGE) [367],
apillary non-gel sieving electrophoresis [368] and reversed-hase liquid chromatography [369,370]. Compared with otherechniques, cIEF is in theory an interesting candidate for on-lineample preparation but requires further intensive exploration.
.6.3. Field amplificationFA stacking technique first introduced by Mikkers et al. [371]
orks under a discontinuous electric field distribution to have aharged analyte migrate from high to low electric fields to losets speed suddenly, causing an accumulation of the analyte at thepeed dropping edge. FA can simply be realized in CE by dilutinghe sample zone with a pure solvent or by placing a section of
ure solvent in front of the sample in capillary. The solvent orhe low ionic strength sample zone draws higher electric fieldcross it than other parts in the capillary once the voltage iswitched on. However, the solvent plug can only persist in a
2 togr.
shfitz>cT11a[
popieTtpswtma[
anbdrcbAwloes
seaimt
4
midfica
tibmafsMfm0eata
mosHl
[tabspyGstpCTht
mw(pDnzmrm1
4
o
08 Y. Chen et al. / J. Chroma
hort time because the ions outside will soon fill in under theigh electric field. As the ionic strength increases, the electriceld strength decreases and so does the FA efficiency. To prolong
he stacking time, the conductivity difference between sampleone and background should be as greater as possible (normally10). This can be achieved by using the running buffers of highonductivity in addition to the use of pure solvent plug [372].he injected sample volume should also be optimized at about0–30% of capillary length. After optimization, FA can obtain0- to 10,000-fold enrichments in the analysis of opioids [373],lkaloids [374], heroin metabolites [375] and arsenic species376].
Large-volume sample stacking (LVSS) is possible by FArinciple and was first tried by Chien and Burgi [377]. A samplef 90% capillary volume can be concentrated by a voltage witholarity reversing to separation. It is very important that, dur-ng stacking, the background electrolyte for building up a lowlectric field is pumped into the capillary against the analytes.his can be achieved by making an electroosmotic flow opposite
o the direction of analyte migration or by completely sup-ressing the electroosmosis at a low pH condition [378]. Moreimply, LVSS can be conducted under normal voltage polarityith a back pressure [379]. This method has been applied to
he analysis of various substances such as 3-nitrotyrosine [380],ercaptopurine and its metabolites [381], modified aromatic
mino acids [382] and non-steroidal anti-inflammatory drugs383], with 20- to 100-fold enhancements.
Feng reported a modified approach called constant pressure-ssisted electrokinetic injection [384] for the analysis ofegatively charged nucleotides. The pressure is used to counter-alance the reverse electroosmotic flow in the capillary columnuring sample injection under FA conditions. At balance, theunning buffer in capillary is stationary and the injection timean extend up to 1200 s in CZE/MS and 3600 s in CZE/UV. Ait complicated method is in combination with LPME [385].water-immiscible sample solution was covered by a layer of
ater and electrokinetically injected into CE system. When theow conductive water is modified with a moderate content ofrganic solvent and a small amount of H+, it provides the high-st sensitivity for analyzing positively chargeable compounds,uch as cocaine and thebaine.
Erny and Cifuentes [386] introduced a type of field amplifiedeparation in CE. A capillary was coated to provide near-zerolectroosmotic flow at the required pH and the separation wasllowed to happen in the high field zone before stacking, lead-ng to 7-fold reduction of the total analysis time (40 s). This
ight open an avenue to create novel analytical methods fromhe existed sample preparation methods.
.6.4. pH regulationFor weak electrolytic analytes, pH can be a very excellent
eans to adjust their effective mobility and various stack-ng approaches have been explored by this principle, such as
ynamic pH junction [387–393] and pH-mediated field ampli-ed sample stacking [394–397]. The same as FA, pH regulationreates a discontinuous acidic–basic boundary to make weaknalytes lose their speed suddenly during stacking. Since most ofbamh
A 1184 (2008) 191–219
he biological samples are not strong electrolytes, pH regulations especially preferred. “Dynamic pH junction”, first mentionedy Britz-Mckibbin and Chen [388], makes analytes focus at theoving boundary of H+/OH−. Two electrolytes are required
t different pH values to form a sharp pH junction boundary,or example, to stack weak acidic analytes, the sample solutionhould be acidified while the running buffer should be basified.
onton et al. [392] used this method to concentrate peptides by aactor of 124-fold. Hsieh and Chang [398] employed it to deter-ine biologically active amines and acids with 5200- and 14
00-fold improvements of detection sensitivity. Over 1000-foldnrichment has been obtained in the preconcentration of proteinst their pI [399], and about 100-fold enrichment in the separa-ion of a group of steroids (including androgens, corticosteroidsnd estrogens) under alkaline conditions [400].
Importantly, dynamic pH junction is a selective stackingethod because its stacking efficiency depends on the pKa value
f analytes and the pH values of background electrolytes andample matrix. Dynamic pH junction is tolerant of ionic strength.owever, since the sample is introduced by pressure, the capil-
ary volume restricts the increase of total sample volume.“pH-mediated stacking” was first introduced by Lunte
401–403] to preconcentrate samples in highly conductive solu-ions. To form a zone of low conductivity to stack the ionicnalytes, the weak counter-ions in the sample zone are titratedy a plug of OH− or H+ electrokinetically injected following theample. Hoque et al. [404] used this pH-mediated technique toreconcentrate glutathione and glutathione disulfide in the anal-sis of microdialysis samples with about 26-fold enrichment.illogly and Lunte (Fig. 13) [394] used the same technique to
tack acidic composition with reverse pressure to push out theitrated neutral zone, increasing the sample loading for six times.H-mediated sample stacking has also been used in microchipGE to achieve high sensitive DNA fragment analysis [405].his pH-mediated method is adaptable to samples containingigh salt due the creation of a low conductive zone during titra-ion.
Our group has developed another type of pH-mediatedethod named acid barrage stacking (ABS) [406]. This methodas validated by determining either standard amino acids
Fig. 14) in Ringer’s solution or trace Glu and Asp in real sam-les of rat brain microdialysate, rat serum and human saliva.ifferent from the mentioned approaches, ABS is performed onormal polar CE by sucking in a plug of acid following a sampleone. The acid plug serves as a barrage to block the backwardigration of the weak anionic analytes due to a sudden mobility
eduction via acid–base reaction. It has been proven that thisethod can stand up to 500 mM NaCl and stack analytes by
03-fold increase of UV detection limits.
.6.5. SweepingSweeping is a technique for in-column sample concentration
f non-polar molecules with an 80- to 5000-fold enhancement
ased on the partitioning capacity of analytes between the waternd pseudo-stationary phase in micellar electrokinetic chro-atography. Similar to FA, the stacking happens at the boundaryaving a sudden slowdown of sample ions. A zone of a sam-
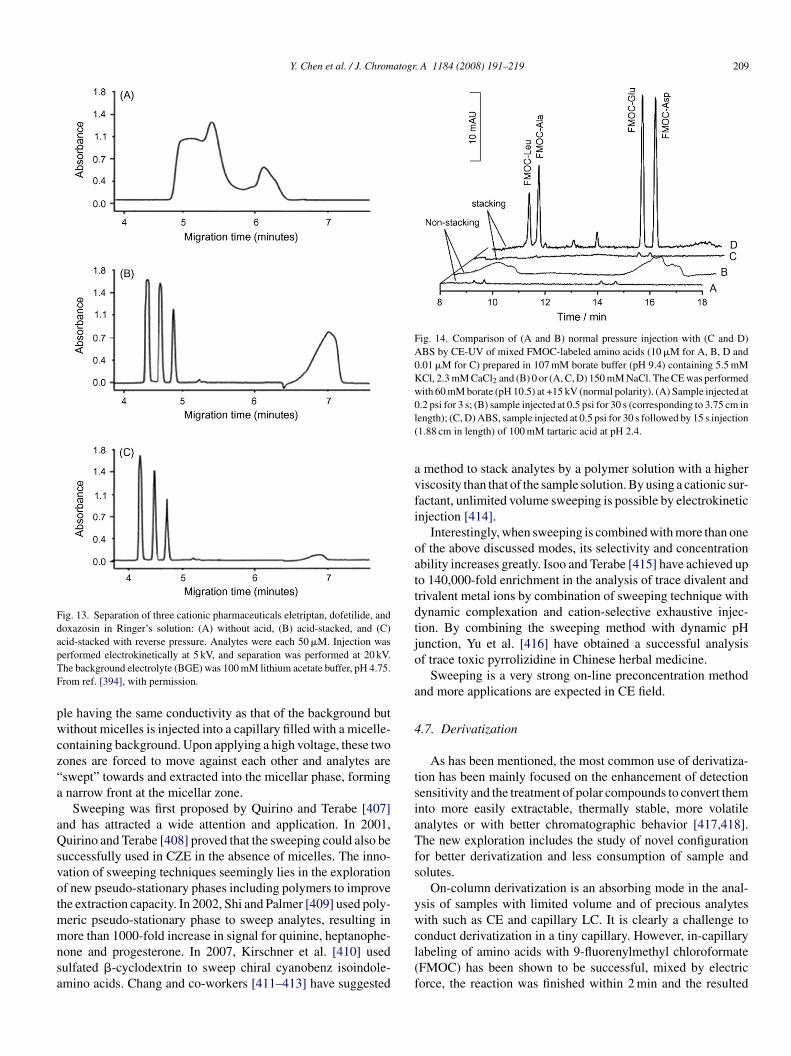
Y. Chen et al. / J. Chromatogr. A 1184 (2008) 191–219 209
Fig. 13. Separation of three cationic pharmaceuticals eletriptan, dofetilide, anddoxazosin in Ringer’s solution: (A) without acid, (B) acid-stacked, and (C)acid-stacked with reverse pressure. Analytes were each 50 �M. Injection waspTF
pwcz“a
aQsvotmmnsa
Fig. 14. Comparison of (A and B) normal pressure injection with (C and D)ABS by CE-UV of mixed FMOC-labeled amino acids (10 �M for A, B, D and0.01 �M for C) prepared in 107 mM borate buffer (pH 9.4) containing 5.5 mMKCl, 2.3 mM CaCl2 and (B) 0 or (A, C, D) 150 mM NaCl. The CE was performedwith 60 mM borate (pH 10.5) at +15 kV (normal polarity). (A) Sample injected at0l(
avfi
oattdtjo
a
4
tsiaTfs
yw
erformed electrokinetically at 5 kV, and separation was performed at 20 kV.he background electrolyte (BGE) was 100 mM lithium acetate buffer, pH 4.75.rom ref. [394], with permission.
le having the same conductivity as that of the background butithout micelles is injected into a capillary filled with a micelle-
ontaining background. Upon applying a high voltage, these twoones are forced to move against each other and analytes areswept” towards and extracted into the micellar phase, formingnarrow front at the micellar zone.
Sweeping was first proposed by Quirino and Terabe [407]nd has attracted a wide attention and application. In 2001,uirino and Terabe [408] proved that the sweeping could also be
uccessfully used in CZE in the absence of micelles. The inno-ation of sweeping techniques seemingly lies in the explorationf new pseudo-stationary phases including polymers to improvehe extraction capacity. In 2002, Shi and Palmer [409] used poly-eric pseudo-stationary phase to sweep analytes, resulting in
ore than 1000-fold increase in signal for quinine, heptanophe-one and progesterone. In 2007, Kirschner et al. [410] usedulfated �-cyclodextrin to sweep chiral cyanobenz isoindole-mino acids. Chang and co-workers [411–413] have suggested
cl(f
.2 psi for 3 s; (B) sample injected at 0.5 psi for 30 s (corresponding to 3.75 cm inength); (C, D) ABS, sample injected at 0.5 psi for 30 s followed by 15 s injection1.88 cm in length) of 100 mM tartaric acid at pH 2.4.
method to stack analytes by a polymer solution with a higheriscosity than that of the sample solution. By using a cationic sur-actant, unlimited volume sweeping is possible by electrokineticnjection [414].
Interestingly, when sweeping is combined with more than onef the above discussed modes, its selectivity and concentrationbility increases greatly. Isoo and Terabe [415] have achieved upo 140,000-fold enrichment in the analysis of trace divalent andrivalent metal ions by combination of sweeping technique withynamic complexation and cation-selective exhaustive injec-ion. By combining the sweeping method with dynamic pHunction, Yu et al. [416] have obtained a successful analysisf trace toxic pyrrolizidine in Chinese herbal medicine.
Sweeping is a very strong on-line preconcentration methodnd more applications are expected in CE field.
.7. Derivatization
As has been mentioned, the most common use of derivatiza-ion has been mainly focused on the enhancement of detectionensitivity and the treatment of polar compounds to convert themnto more easily extractable, thermally stable, more volatilenalytes or with better chromatographic behavior [417,418].he new exploration includes the study of novel configuration
or better derivatization and less consumption of sample andolutes.
On-column derivatization is an absorbing mode in the anal-sis of samples with limited volume and of precious analytesith such as CE and capillary LC. It is clearly a challenge to
onduct derivatization in a tiny capillary. However, in-capillaryabeling of amino acids with 9-fluorenylmethyl chloroformateFMOC) has been shown to be successful, mixed by electricorce, the reaction was finished within 2 min and the resulted

2 togr.
zc
oueifioidcSlt
4
paodmptosts
asfgmLfmtatbtcf
thnsCve1
o
fsh(ptbstaos[
5
piAtuaseaac
pbegspttareomeisac
thcnb
10 Y. Chen et al. / J. Chroma
one produced an excellent CE resolution of chiral amino acids,omparable with that of pre-column labeling [19].
Derivatization can also be carried out in sample matrix beforer simultaneously with extraction. This is a simple approachsed widely but is susceptible due to side reactions and interfer-nces. An improved approach is implementing the derivizationn the receiving or collection phase, mostly on the SPMEber. It can be performed by preloading the labeling agentn SPE cartridges [419,420], SPME fibers [260,418,421] orn the SDME micro liquid drops [422,423]. In this case, theerivatization occurs only when analytes are extracted into theollection phase, without any interference of sample matrices.uch sequential operation can be enlarged or stepped by first col-
ecting and concentrating the analytes and then exposing themo the labeling reagent [424,425].
.8. Miniaturization and integration
Micro total analysis system (�TAS) enables effective cou-ling of separation/detection processes with sample preparationpproaches. Although the capability for handling real samplesn microfluidic or lab-on-a-chip devices is a challenging hur-le which is currently restricting the advancement of �TAS,any techniques have been tried for on-line purification and/or
reconcentration of analytes, and some of them were showno be useful for coupling with separation approaches. Then-line preconcentration in �TAS can be achieved by usingolid sorbents, membranes, solvent microextraction, and elec-romigration focusing mechanisms, or sometimes by a uniquetructural design of the �TAS hardware.
Electrokinetic concentration of charged biomolecules suchs proteins and DNA has been conducted by allowing the pas-age of buffer ions but excluded larger migrating moleculesrom semipermeable interfaces [426–430]. LPME has been inte-rated into microchips utilizing co-current or counter-currenticroflow systems [25,431,432], producing microchip-basedPME. Wilson and Konermann [433] introduced an approach
or on-line desalting of macromolecule solutions in tens ofilliseconds by utilizing a two-layered laminar flow geome-
ry that exploits the differential diffusion of macromolecularnalytes and low molecular weight contaminants between thewo flow layers. In addition, droplet LPME can be achievedy trapping organic solvent droplets in recesses fabricated inhe channel walls, and delivering aqueous samples through thehannel [434]. Miniaturized MSLE has also been designed andabricated for sample enrichment [435].
Several sample preparation methods via electric field applica-ion including IEF [436–438], FA [439,440], and ITP [441–445]ave been reported. Ross and Locascio described a new tech-ique, temperature gradient focusing, for the concentration andeparation of ionic species within microchannels or capillaries.oncentration was achieved by balancing the electrophoreticelocity of analyte against the bulk flow of solution in the pres-
nce of a temperature gradient, with an enrichment factor up to04-fold [446].In addition to miniaturization, integration of different meth-ds into one microfluidic device is a new trend. SPE well suited
rclc
A 1184 (2008) 191–219
or integration into microfluidic devices for cleaning of unde-ired compounds and preconcentration of desired analytes. SPEas been performed on microchannels using packed particlesusually coated silica beads) or integrated monolithic porousolymers for DNA purification [447–451]. On-chip preconcen-ration of peptides and proteins followed by separation has alsoeen reported [452,453]. Broyles et al. showed that it was pos-ible to integrate on-chip filtration (by an array of thin channelso exclude particulates), sample concentration (employing C18s a stationary phase) and electrochromatographic separationn a microfabricated device [454]. On-chip filtration and dialy-is can be achieved with the aid of porous membrane materials455–459].
. Criteria for method validation
It is necessary to have some criteria to estimate a samplereparation method for selection over the numerous exist-ng techniques or to develop or innovate on a new approach.lthough numerous requirements may be encountered or need
o be considered, a sample preparation method should measurep first to the analytical purpose (for qualitative or quantitativenalysis, or a bit in detail, such as separation, detection, sensing,elective assaying, group protection, or structural elucidation,tc.), then to the regulation of safety, and to the cost-effectivenessnd simplicity. Of course, one has to consider what is at handnd potentially available in determining the importance ofriteria.
In other words, if there are no special limitations, a samplereparation method to be adopted or to be developed shoulde effective enough selectivity and throughput to produce highxtraction efficiency and concentration factor in extracting a tar-et analyte from any matrix of interest, the manageable sampleize and cleanness, which will depend on the sample matrix, theroperties and level of the analyte to be determined, should meethe demands of separation and detection, and it must be tailoredo the final analysis, considering the instrumentation to be usednd the degree of accuracy (or recovery), precision and linearityequired. Especially, the method should be safe for operators andnvironment, producing and discarding no pollutants. For mostf the users, the method should be cost-effective, consuming asinimum reagents and chemicals as possible and with as low
xpenses as possible on instrumentation and facility. A methods always preferred which is very easy to use, has the minimumteps and uses only simple devices or systems capable of fullutomation. Table 2 collects most of the items that need to beonsidered in sample preparation.
It is highly significant to minimize sample preparation stepso reduce the sources of error. A sample preparation method canave more than one step, such as homogenization, extraction,leanup, preconcentration and/or derivatization. The more theumber of steps are involved, the more will be uncertainty wille introduced into the assaying. Minimizing the sample prepa-
ation steps is also an effective way to save time and operationost. Using the minimum sample preparation step(s) is particu-arly favored in measuring the trace and ultra-trace analytes inomplex matrices.
Y. Chen et al. / J. Chromatogr. A 1184 (2008) 191–219 211
Table 2Some reference criteria for evaluating a sample preparation method
Sort Item Preference
Effectiveness Selectivity The higher the betterEfficiencyConcentrating factorThroughputSample size Depending on latter analysisQuantitation
Recovery Around 100%Linearity >2 orders, the wider the betterPrecision CV < 5%, the smaller the betterAccuracy Normally with >95% confidence limit
Separation demand As clean as possibleDetection limitations Clean background or detector-dependent
Safety Chemical hazardToxicity No or negligibleExplosivity No or
avoidableFlammabilityVolatility As low as possible or dependsRadiation Excluded whenever possiblePollution Measuring up legal regulations
Device security Safe in use, little garbage or freeOperation risk Naught or preventable
Cost Materials consumptionLowDevice or system price
Running costMaintenance Scarcely
Simplicity Steps MinimumConvenience HighIntegrating degree High
o(siitopib
tsaw
csptba
fi
Table 3Sample-state-based selection of a sample preparation method
Sample state Trappingmedium
Techniques
Solid/semi-solid
Liquid-phase
Soxhlet extractionSupercritical fluid extractionSub-critical water extractionPressurized liquid extractionMicrowave/Sonication assisted extraction
Solid-phase Matrix solid-phase dispersionHeadspacerelated
Static headspaceDynamic headspace (Purge and Trap)
Liquid
Liquid-phaseLiquid–liquid extractionLiquid-phase microextractionMembrane-separated liquid extraction
Solid-phase
Solid-phase extractionSolid-phase microextractionStir bar sorptive extractionMembrane extraction with sorbent interface
HeadspaceStatic headspaceDynamic headspace (Purge and Trap)
Gas-phaseLiquid-phase
Dynamic sampling/extractionPassive sampling/extraction
Solid-phaseDynamic sampling/extraction
ssmsfPtnr
6
pfitrmmfep
cat
Automation High or fullPreparation Free or rare
Increasing selectivity is a useful means to reduce the numberf sample preparation steps. Extraction with selective sorbentse.g. immunosorbents, MIPs, etc.) may eliminate or reduce manyteps like repeating separation and cleanup. Miniaturization andntegration open a novel way to shorten the sample extract-ng route. Two prominent examples are the SPME and SBSE,hey both may integrate sampling, isolation and enrichment intonly a single step. Introducing automatic techniques into sam-le preparation is also highly effective in saving time and inmproving reproducibility compared with the manual methodsut involves some cost.
For quantitative analysis, consideration must also be giveno the most appropriate preparation of calibration standards. Inome cases matrix-matched standards or the method of standarddditions is necessary. The use of a suitable internal standard isidely adopted to eliminate some effects of matrices.The choice of an appropriate sample preparation technique
an be based on the chemical and physical properties of analytes,uch as molecular weight, charge, solubility (hydrophobicity),olarity and volatility. Some selective methods utilize the selec-ivity for specific structural groupings, like IAE, or mimic a
iological selectivity such as MIP-based extraction. Volatilenalytes are often determined through headspace techniques.In general, a sample preparation method is better selected byrst considering the physical state of the sample and then the
rmtd
Passive sampling/extraction
olubility, polarity and particular recognition forces. A gaseousample or aerosol is better concentrated by some absorptiveethods, while liquid or solid samples are better extracted by
olvents or similar techniques. Tables 2 and 3 show two strategiesor the selection or evaluation of a sample preparation method.articularly, when a strong goal has been assigned such as in
he case of preparation of “-omics”-oriented samples, there willot be too much space for rotation. But the four sorting criteriaemain effective.
. Integration of sample preparation methods
Nearly all the molecular related analysis concerns the sam-le preparation. By surveying most of the complicated researchelds such as environment, food and life sciences, we have found
hat there are two types of samples that may serve as the rep-esentative to guide the establishment of a total or integratedethod for the preparation of samples from very complicatedatrices. The first case is the preparation of protein samples
rom tissues or cells for proteomic studies and the second is thextraction of polysaccharides from the raw materials of driedlants.
There is no attempt in this review to go into a detailed dis-ussion on the preparation of such a colligated method, but forn integrality of this review, route maps as to how to preparehese two types of samples are illustrated in Figs. 15 and 16,espectively. As the preparation of polysaccharides is not at this
oment an urgent topic, it will not be further discussed. Instead,he preparation of proteins is discussed, to some extent, inetail.

212 Y. Chen et al. / J. Chromatogr. A 1184 (2008) 191–219
F
6
tprotrpoimtstpptims(tsa
(
F
(
ig. 15. A strategy for the preparation of proteomic oriented protein samples.
.1. Preparation of proteomics-oriented proteins
There are different purposes to prepare proteins but at presenthe most urgent task is to prepare better protein samples forroteomic investigations. Considering that the most possibleesource to prepare this type of protein samples is tissues (orrgans) and cells, the preparation method design should neverurn away the sight from these tender materials. Since suchaw materials contain various types of biochemicals, the com-lete extraction of all the target proteins may not be possibler necessary. Actually in proteomic studies, the key at presents to uncover all the “hidden” protein fractions which are nor-
ally the low-abundance and membrane proteins. In addition,he availability of separation and identification methodologyhould seriously be taken into consideration in protein prepara-ion. Over all, the consistency and standardization in proteomicsrotocols for sample preparation are essential and under the aus-ices of the Human Proteome Organization, the laboratories ofhe Sample Processing Working Group are working out researchnitiatives to develop new procedures. However, at present, the
ost powerful tools remain to be the 2-DE and multidimen-ional chromatography in combination with the biological MSbio-MS). Under these considerations, a route map to preparehe complex protein samples should be more or less like the onehown in Fig. 15, where some specifications should be furtherddressed as follows:
1) The preliminary steps should include tissue disruption, celllysis, protein extraction and pre-fractionation. Many tech-niques are available for tissue disruption and cell lysis
ig. 16. A reference route for the preparation of polysaccharides from raw plant.
such as by detergents and mechanical methodologies. Pre-fractionation procedures constitute a valuable tool to findthe “hidden” protein fraction [460]. Although many poten-tial methods can be found, the centrifugation, affinity-and immune-based methods, chromatography and elec-trophoresis are at present the most often used tools forthe pre-fractionation purpose [461]. The selection of thetechniques strongly depends on the nature of samples andthe objects of study. Protein micro arrays are a goodalternative for pre-fractionation, but they remain very expen-sive [462]. Over all, the 2-DE should be considered andoptimized.
2) Sample cleanup for electrophoresis should carefully be con-sidered. 2-DE is the first step in the classical proteomicsstrategy to resolve the inherently complex nature of cellularproteomes but the samples should be as clean as possible sothat the interfering compounds, such as salts, nucleic acids,
polysaccharides, lipids and particulate materials and somedetergents can be removed prior to analysis. The salts canbe removed by precipitating the proteins with trichloroacetic
atogr
(
(
(
odnm
bccshcSpacsds
7
atrivicveoSsrcpsvlb
A
(eo2
R
Y. Chen et al. / J. Chrom
acid (TCA) or TCA/acetone [463]. Dialysis is another alter-native frequently used but it is time consuming.
3) The relative abundance of the protein(s) of interest mustbe considered when choosing the solubilization approach[464]. The presence of high abundant proteins causes enor-mous problems for the detection and analysis of the lessabundant proteins. Multi-component immuno-precipitationand affinity chromatography are now the methods of choicefor the removal of abundant proteins. When dealing with lowabundant proteins like transcription factors, crude extractsmust be enriched to recover enough amounts of proteinsfor visualization in the electrophoretic gels [465]. Themain enrichment strategies are (1) subcellular organellefractionation using density-gradient centrifugation andFFE, and (2) the selective fractionation and enrichmentof proteins by sequential solubilization, selective precip-itation, affinity- and immunocapture-based purification,electrophoretic techniques or various chromatographic pro-cedures.
4) Protein digestion is performed using either specificenzymes, or chemically, using peptide bond specificreagents, such as cyanogen bromide [466]. Chemical andenzymatic digestions may be sequentially applied [467]through in-gel, in-solution or in-column format. The in-column digestion reduces the reaction time from hours tominutes due to the high concentration of enzyme able toreach inside the column. By this in-column approach, theprotein digestion, peptide separation and MS identifica-tion can be on-line coupled, making possible whole-lineautomation. There is hence a growing interest in the use ofimmobilized enzymes to perform digestions for proteomicsstudies [468,469]. It is also widely used to digest proteinson nitrocellulose [470] or polyvinylidene fluoride [471]membranes after electroblotting. Electroelution [472] is aninstrumental approach to isolate intact proteins separated byPAGE.
5) MS has driven a significant improvement in sam-ple preparation. For MALDI-MS home-made disposablemicro-columns are available as a fast cleanup and con-centrating step prior to MS [473]. More recently, it hasbeen further simplified by using commercial prepackedZipTips (Millipore, Bedford, MA, USA). Apart fromstainless-steel MALDI target plates, the use of matrix-precoated targets [474] for the MALDI-TOF-MS analysisof peptides and proteins have been investigated fairlywidely. Many publications during the last 10 years refer toeither on-line procedures (coupling capillary reversed phaseHPLC separation with electrospray ionization MS [475]) oroff-line procedures using home-made or commercial micro-columns in which samples can be concentrated and elutedin few microliters [476].
Recent advances in protein preparation feature the use
f miniaturized devices that have an integration of proteinigestion, one- or two-dimensional LC or CE separation withano-electrospray ionization tandem MS. Li et al. designed aicro device with nanospray interface to MS and the possi-. A 1184 (2008) 191–219 213
ility to preconcentrate 10 �L samples makes the separationonsume only 1/10 of the concentrated sample [477]. With thiship a throughput of 12 samples per hour was obtained, with aensitivity of 25 fmol peptides on-chip digested. Paterson et al.ave introduced an integrated device which has a 40-nL micro-olumn with immobilized trypsin for protein digestion and aPE micro cartridge for desalting/concentration of the digestedeptides [478]. Ramsey et al. presented a two-dimensional sep-ration chip in which micellar electrokinetic chromatography isombined with CE [479]. A peak capacity of 4200 was demon-trated. However, the microfluidic technology is still underevelopment and is not presently adoptable to the proteomictudies [480,481].
. Conclusions
In conclusion, sample preparation is an inseverable step innalytical chemistry but it has not been fueled until environmen-al and life sciences became hot research topics which urgentlyequire automatic sample preparation methods of high sensitiv-ty, high throughput and high selectivity (H3 method) to treat theery complicated samples. Although many other techniques (fornstance, probing or sensor) may be used to analyze some targetomposition of interest, sample preparation is at present an uni-ersal, cost-effective and facile way to conquer the difficultiesncountered in many scientific researches, allowing various lab-ratories able to manipulate their complicated samples in situ.ample preparation is hence expected to further develop at a highpeed or to open a vast research field. High recovery and envi-onment friendly sample preparation methods for finding andoncentrating the trace analytes enshrouded by abundant com-osition will be the focus of research for 10 years. Chip-basedample preparation methods which can easily be integrated intoarious analytical systems will be highlighted. Flexible and on-ine-integrable sample preparation methods and devices tend toe the center of research and commercialization from now on.
cknowledgements
We gratefully acknowledge the financial support from NSFCNo. 20435030 & No. 20628507), Chinese Academy of Sci-nces (KJCX2-YW-H11), Ministry of Science and Technologyf China (No. 2006BAK03A09, No. 2007CB714504 & No.002CB713803).
eferences
[1] S.C. Moldoveanu, V. David, Sample Preparation in Chromatography,Elsevier, Amsterdam, 2002.
[2] J. Pawliszyn, Sampling and Sample Preparation for Field and Laboratory,Elsevier, Amsterdam, 2002.
[3] J. Pawliszyn, Anal. Chem. 75 (2003) 2543.[4] R.M. Smith, J. Chromatogr. A 1000 (2003) 3.[5] D.E. Raynie, Anal. Chem. 76 (2004) 4659.
[6] D.E. Raynie, Anal. Chem. 78 (2006) 3997.[7] T.P. Wampler, J. Anal. Appl. Pyrol. 71 (2004) 1.[8] K. Gevaert, J. Vandekerckhove, Electrophoresis 21 (2000) 1145.[9] M. Nissum, U. Schneider, S. Kuhfuss, C. Obermaier, R. Wildgruber, A.Posch, C. Eckerskorn, Anal. Chem. 76 (2004) 2040.

2 togr.
14 Y. Chen et al. / J. Chroma[10] F. Sanger, A.R. Coulson, J. Mol. Biol. 94 (1975) 441.[11] K. Mullis, F. Faloona, S. Scharf, R. Saiki, G. Horn, H. Erlich, Cold Spring
Harbor Symp. Quant. Biol. 51 (1986) 263.[12] A.M. Maxam, W. Gilbert, Proc. Natl. Acad. Sci. U.S.A. 74 (1977)
560.[13] K. Blau, J. Halket (Eds.), Handbook of Derivatives for Chromatography,
Wiley, New York, 1993.[14] T. Toyooka, Modern Derivatization Methods for Separation Science,
Wiley, New York, 2002.[15] J.M. Rosenfeld, Trends Anal. Chem. 22 (2003) 785.[16] G. Vas, K. Vekey, J. Mass Spectrom. 39 (2004) 233.[17] C. Kempter, W. Potter, N. Binding, H. Klaning, U. Witting, U. Karst,
Anal. Chim. Acta 410 (2000) 47.[18] W.N. Vreeland, C. Desruisseaux, A.E. Karger, G. Drouin, G.W. Slater,
A.E. Barron, Anal. Chem. 73 (2001) 1795.[19] Y. Han, Y. Chen, Electrophoresis 28 (2007) 2765.[20] W. Tan, K. Wang, T.J. Drake, Curr. Opin. Chem. Biol. 8 (2004) 547.[21] X. Duan, Z. Zhao, J. Ye, H. Ma, A. Xia, G. Yang, C. Wang, Angew. Chem.
Int. Ed. 43 (2004) 4216.[22] J.A. Jonsson, L. Mathiasson, J. Chromatogr. A 902 (2000) 205.[23] L. Xu, C. Basheer, H.K. Lee, J. Chromatogr. A 1152 (2007) 184.[24] Y. Wang, S. Yue, D. Li, M. Jin, C. Li, Solvent Extr. Ion Exc. 20 (2002)
345.[25] H. Miyaguchi, M. Tokeshi, Y. Kikutani, A. Hibara, H. Inoue, T. Kitamori,
J. Chromatogr. A 1129 (2006) 105.[26] K. Mondal, M.N. Gupta, Biomol. Eng. 23 (2006) 59.[27] C.D. Stalikas, Trends Anal. Chem. 21 (2002) 343.[28] J.M.R. Belanger, J.R.J. Pare, Anal. Bioanal. Chem. 386 (2006)
1049.[29] J.L. Luque-Garcıa, M.D. Luque de Castro, Trends Anal. Chem. 22 (2003)
90.[30] J.A. Nobrega, L.C. Trevizan, G.C.L. Araujo, A.R.A. Nogueira, Spec-
trochim. Acta B 57 (2002) 1855.[31] A. Caballo-Lopez, M.D. Luque de Castro, Anal. Chem. 78 (2006)
2297.[32] R. Japon-Lujan, J.M. Luque-Rodrıguez, M.D. Luque de Castro, J. Chro-
matogr. A 1108 (2006) 76.[33] J.L. Luque-Garcıa, M.D. Luque de Castro, Trends Anal. Chem. 22 (2003)
41.[34] S. Morales-Munoz, M.D. Luque de Castro, J. Chromatogr. A 1066 (2005)
1.[35] S. Morales-Munoz, R.J.J. Vreuls, M.D. Luque de Castro, J. Chromatogr.
A 1086 (2005) 122.[36] F. Priego-Capote, M.D. Luque de Castro, Trends Anal. Chem. 23 (2004)
644.[37] C. Sanchez, M. Ericsson, H. Carlsson, A. Colmsjo, J. Chromatogr. A 993
(2003) 103.[38] R. Lombardi, LC–GC (Suppl.) (1998) S47.[39] Y. Ito, M. Weinstein, I. Aoki, R. Harada, E. Kimura, K. Nunogaki, Nature
212 (1966) 985.[40] N. Fischer, B. Weinreich, S. Nitz, F. Drawert, J. Chromatogr. 538 (1991)
193.[41] A.P. Foucault, L. Chevolot, J. Chromatogr. A 808 (1998) 3.[42] K. Hostettmann, A. Marston, Anal. Chim. Acta 236 (1990) 63.[43] A. Marston, K. Hostettmann, J. Chromatogr. A 658 (1994) 315.[44] A. Berthod (Ed.), Countercurrent Chromatography: The Support-Free
Liquid Stationary Phase Comprehensive Analytical Chemistry, vol. 38,Elsevier, Amsterdam, 2003.
[45] W.D. Conway, Countercurrent Chromatography: Apparatus, Theory andApplication, VCH, New York, 1990.
[46] W.D. Conway, R.J. Petroski (Eds.), Modern Countercurrent Chro-matography (ACS Symposium, No. 593), American Chemical Society,Washington, DC, 1995.
[47] A.P. Foucault (Ed.), Centrifugal Partition Chromatography (Chromato-graphic Science), vol. 68, Marcel Dekker, New York, 1994.
[48] K. Hostettmann, A. Marston, M. Hostettmann, Preparative Chromatog-raphy Techniques: Applications in Natural Product Isolation, second ed.,Springer, Berlin, 1998.
A 1184 (2008) 191–219
[49] Y. Ito, W.D. Conway (Eds.), High-Speed Countercurrent Chromatography(Chemical Analysis), vol. 132, Wiley-Interscience, New York, 1996.
[50] N.B. Mandava, Y. Ito (Eds.), Countercurrent Chromatography: Theoryand Practice (Chromatographic Science), vol. 44, Marcel Dekker, NewYork, 1988.
[51] C. Juberta, G. Bailey, J. Chromatogr. A 1140 (2007) 95.[52] W. Remus-Borel, N. Shallow, D.J. McNally, C. Labbe, R.R. Belanger, J.
Chromatogr. A 1121 (2006) 200.[53] W. Zhao, C. Gao, X. Ma, X. Bai, Y. Zhang, J. Chromatogr. B 850 (2007)
523.[54] Q. Tang, C. Yang, W. Ye, J. Liu, S. Zhao, J. Chromatogr. A 1144 (2007)
203.[55] K. Hannig, Fresenius Zeitschr. Anal. Chem. 181 (1961) 244.[56] R.L. Moritz, A.B. Clippingdale, E.A. Kapp, J.S. Eddes, H. Ji, S. Gilbert,
L.M. Connolly, R.J. Simpson, Proteomics 5 (2005) 3402.[57] R.L. Moritz, H. Ji, F. Schutz, L.M. Connolly, E.A. Kapp, T.P. Speed, R.J.
Simpson, Anal. Chem. 76 (2004) 4811.[58] D.P. de Jesus, L. Blanes, C.L. do Lago, Electrophoresis 27 (2006) 4935.[59] B.R. Fonslow, V.H. Barocas, M.T. Bowser, Anal. Chem. 78 (2006) 5369.[60] B.R. Fonslow, M.T. Bowser, Anal. Chem. 77 (2005) 5706.[61] N. Tajima, H. Suzuki, K. Kano, E. Shinohara, Proceedings of the 22nd
International Symposium on Capillary Chromatography, Gifu, ChemicalSociety of Japan, 1999, p. 2.
[62] A. Gorg, W. Weiss, M.J. Dunn, Proteomics 4 (2004) 3665.[63] K.D. Caldwell, J.C. Giddings, M.N. Myers, L.F. Kesner, Science 176
(1972) 296.[64] J.C. Giddings, M.N. Myers, K.D. Caldwell, S.R. Fisher, Methods
Biochem. Anal. 26 (1980) 79.[65] J.C. Giddings, Anal. Chem. 58 (1986) 2052.[66] J.C. Giddings, Anal. Chem. 67 (1995) 592A.[67] J.C. Giddings, Science 260 (1993) 1456.[68] A.I.K. Lao, I.M. Hsing, Lab Chip 5 (2005) 687.[69] J.M. Davis, F.R.F. Fan, A.J. Bard, Anal. Chem. 59 (1987) 1339.[70] S.K. Ratanathanawongs, I. Lee, J.C. Giddings, ACS Symp. Ser. 472
(1991) 229.[71] B.N. Barman, J.C. Giddings, Langmuir 8 (1992) 51.[72] M.E. Schimpf, J. Liq. Chromatogr. Rel. Technol. 25 (2002) 2101.[73] T. Chianea, N.E. Assidjo, P.J.P. Cardot, Talanta 51 (2000) 835.[74] S.K.R. Williams, D. Lee, J. Sep. Sci. 29 (2006) 1720.[75] R. Batlle, C. Nerın, C. Crescenzi, H. Carlsson, Anal. Chem. 77 (2005)
4241.[76] A. Halasz, C. Groom, E. Zhou, L. Paquet, C. Beaulieu, S. Deschamps, A.
Corriveau, S. Thiboutot, G. Ampleman, C. Dubois, J. Hawari, J. Chro-matogr. A 963 (2002) 411.
[77] E.E. Stashenko, B.E. Jaramillo, J.R. Martınez, J. Chromatogr. A 1025(2004) 105.
[78] M. Sun, F. Temelli, J. Supercrit. Fluids 37 (2006) 397.[79] Z. Wang, M. Ashraf-Khorassani, L.T. Taylor, Anal. Chem. 76 (2004)
6771.[80] J. Pol, B.W. Wenclawiak, Anal. Chem. 75 (2003) 1430.[81] Z. Wang, M. Ashraf-Khorassani, L.T. Taylor, J. Chromatogr. A 1033
(2004) 221.[82] L. Chiappini, E. Perraudin, R. Durand-Jolibois, J.F. Doussin, Anal.
Bioanal. Chem. 386 (2006) 1749.[83] Z. Wang, M. Ashraf-Khorassani, L.T. Taylor, Anal. Chem. 75 (2003)
3979.[84] J. Zhang, L. Zhang, J. Duan, Z. Liang, W. Zhang, Y. Huo, Y. Zhang, J.
Sep. Sci. 29 (2006) 2514.[85] M. Zougagh, A. Rıos, M. Valcarcel, Anal. Chim. Acta 524 (2004) 279.[86] T. Aro, C. Brede, P. Manninen, H. Kallio, J. Agric. Food Chem. 50 (2002)
1970.[87] M. Shimmo, P. Anttila, K. Hartonen, T. Hyotylainen, J. Paatero, M. Kul-
mala, M.-L. Riekkola, J. Chromatogr. A 1022 (2004) 151.
[88] G. Anitescu, L.L. Tavlarides, J. Supercrit. Fluids 38 (2006) 167.[89] M.C. Henry, C.R. Yonker, Anal. Chem. 78 (2006) 3909.[90] M. Herrero, A. Cifuentes, E. Ibanez, Food Chem. 98 (2006) 136.[91] J.W. King, Adv. Chromatogr. 43 (2005) 109.[92] M. Zougagh, M. Valcarcel, A. Rıos, Trends Anal. Chem. 23 (2004) 399.
atogr
Y. Chen et al. / J. Chrom[93] M. Shimmo, H. Adler, T. Hyotylainen, K. Hartonen, M. Kulmala, M.-L.Riekkola, Atmos. Environ. 36 (2002) 2985.
[94] S.B. Hawthorne, Y. Yang, D.J. Miller, Anal. Chem. 66 (1994) 2912.[95] Y. Yang, M. Belghazi, A. Lagadec, D.J. Miller, S.B. Hawthorne, J. Chro-
matogr. A 810 (1998) 149.[96] R.M. Smith, Anal. Bioanal. Chem. 385 (2006) 419.[97] R.M. Smith, J. Chromatogr. A 975 (2002) 31.[98] C. Deng, A. Wang, S. Shen, D. Fu, J. Chen, X. Zhang, J. Pharm. Biomed.
Anal. 38 (2005) 326.[99] M.Z. Ozel, H. Kaymaz, Anal. Bioanal. Chem. 379 (2004) 1127.
[100] E.S. Ong, J.S.H. Cheong, D. Goh, J. Chromatogr. A 1112 (2006) 92.[101] C. Deng, J. Ji, X. Wang, X. Zhang, J. Sep. Sci. 28 (2005) 1237.[102] K. luthje, T. Hyotylainen, M.-L. Riekkola, J. Chromatogr. A 1025 (2004)
41.[103] X. Lou, D.J. Miller, S.B. Hawthorne, Anal. Chem. 72 (2000) 481.[104] A.E. McGowin, K.K. Adom, A.K. Obubuafo, Chemosphere 45 (2001)
857.[105] M.C. Herrera, R.C. Prados-Rosales, J.L. Luque-Garcıa, M.D. Luque de
Castro, Anal. Chim. Acta 463 (2002) 189.[106] M. Papagiannopoulos, B. Zimmermann, A. Mellenthin, M. Krappe, G.
Maio, R. Galensa, J. Chromatogr. A 958 (2002) 9.[107] R. Tajuddin, R.M. Smith, Analyst 127 (2002) 883.[108] R. Tajuddin, R.M. Smith, J. Chromatogr. A 1084 (2005) 194.[109] K. Kuosmanen, T. Hyotylainen, K. Hartonen, M.-L. Riekkola, J. Chro-
matogr. A 943 (2001) 113.[110] K. Luthje, T. Hyotylainen, M. Rautiainen-Rama, M.-L. Riekkola, Analyst
130 (2005) 52.[111] E. Ibanez, A. Kubatova, F.J. Senorans, S. Cavero, G. Reglero, S.B.
Hawthoran, J. Agric. Food Chem. 51 (2003) 375.[112] S. Chun, S.V. Dzyuba, R.A. Bartsch, Anal. Chem. 73 (2001) 3737.[113] A.E. Visser, R.P. Swatloski, W.M. Reichert, S.T. Griffin, R.D. Rogers,
Ind. Eng. Chem. Res. 39 (2000) 3596.[114] H. Luo, S. Dai, P.V. Bonnesen, Anal. Chem. 76 (2004) 2773.[115] H. Luo, S. Dai, P.V. Bonnesen, A.C. Buchanan III, J.D. Holbrey, N.J.
Bridges, R.D. Rogers, Anal. Chem. 76 (2004) 3078.[116] K. Shimojo, M. Goto, Anal. Chem. 76 (2004) 5039.[117] A.E. Visser, R.P. Swatloski, W.M. Reichert, R. Mayton, S. Sheff,
A. Wierzbicki, J.H. Davis, R.D. Rogers, Chem. Commun. (2001)135.
[118] A.E. Visser, R.P. Swatloski, W.M. Reichert, R. Mayton, S. Sheff, A.Wierzbicki, J.H. Davis, R.D. Rogers, Environ. Sci. Technol. 36 (2002)2523.
[119] J.G. Huddleston, H.D. Willauer, R.P. Swatloski, A.E. Visser, R.D. Rogers,Chem. Commun. (1998) 1765.
[120] S.T.M. Vidal, M.J. Neiva Correia, M.M. Marques, M.R. Ismael, M.T.A.Reis, Sep. Sci. Technol. 39 (2004) 2155.
[121] A. Boesmann, L. Datsevich, A. Jess, A. Lauter, C. Schmitz, P. Wasser-scheid, Chem. Commun. (2001) 2494.
[122] F. Kubota, M. Goto, Solvent Extr. Res. Dev. 13 (2006) 23.[123] K. Shimojo, N. Kamiya, F. Tani, H. Naganawa, Y. Naruta, M. Goto, Anal.
Chem. 78 (2006) 7735.[124] K. Shimojo, K. Nakashima, N. Kamiya, M. Goto, Biomacromolecules 7
(2006) 2.[125] J. Wang, D. Cheng, X. Chen, Z. Du, Z. Fang, Anal. Chem. 79 (2007) 620.[126] J. Liu, N. Li, G. Jiang, J. Liu, J.A. Josson, M. Wen, J. Chromatogr. A
1066 (2005) 27.[127] J. Liu, Y. Chi, G. Jiang, C. Tai, J. Peng, J. Hu, J. Chromatogr. A 1026
(2004) 143.[128] J. Liu, G. Jiang, Y. Chi, Y. Cai, Q. Zhou, J. Hu, Anal. Chem. 75 (2003)
5870.[129] J.L. Anderson, D.W. Armstrong, G.-T. Wei, Anal. Chem. 78 (2006)
2892.[130] J. Liu, J.A. Jonsson, G. Jiang, Trends Anal. Chem. 24 (2005) 20.
[131] H. Zhao, S. Xia, P. Ma, J. Chem. Technol. Biotechnol. 80 (2005) 1089.[132] B.E. Richter, B.A. Jones, J.L. Ezzell, N.L. Porter, N. Avdalovic, C. Pohl,Anal. Chem. 68 (1996) 1033.[133] Y.G. Ahn, J. Seo, J.H. Shin, J. Khim, J. Hong, Anal. Chim. Acta 576
(2006) 31.
. A 1184 (2008) 191–219 215
[134] E. Concha-Grana, M.I. Turnes-Carou, S. Muniategui-Lorenzo, P. Lopez-Mahıa, E. Fernandez-Fernandez, D. Prada-Rodrıguez, J. Chromatogr. A1047 (2004) 147.
[135] M.I.H. Helaleh, A. Al-Omair, A. Nisar, B. Gevao, J. Chromatogr. A 1083(2005) 153.
[136] A.M. Jacobsen, B. Halling-Sørensen, F. Ingerslev, S.H. Hansen, J. Chro-matogr. A 1038 (2004) 157.
[137] M. Janska, M. Tomaniova, J. Hajslova, V. Kocourek, Anal. Chim. Acta520 (2004) 93.
[138] K. Kawata, T. Asada, K. Oikawa, J. Chromatogr. A 1090 (2005) 10.[139] O. Pardo, V. Yusa, N. Leon, A. Pastor, J. Chromatogr. A 1107 (2006) 70.[140] A. de la Cal, E. Eljarrat, D. Barcelo, J. Chromatogr. A 1021 (2003)
165.[141] J. Poerschmann, R. Carlson, J. Chromatogr. A 1127 (2006) 18.[142] S. Sporring, E. Bjorklund, J. Chromatogr. A 1040 (2004) 155.[143] R. Carabias-Martınez, E. Rodrıguez-Gonzalo, P. Revilla-Ruiz, J.
Hernandez-Mendez, J. Chromatogr. A 1089 (2005) 1.[144] H. Giergielewicz-Mozajska, L. Dabrowski, J. Namiesnik, Crit. Rev. Anal.
Chem. 31 (2001) 149.[145] L. Ramos, E.M. Kristenson, U.A.Th. Brinkman, J. Chromatogr. A 975
(2002) 3.[146] M.M. Schantz, Anal. Bioanal. Chem. 386 (2006) 1043.[147] B.W. Renoe, Am. Lab. 26 (1994) 34.[148] J.L. Luque-Garcıa, M.D. Luque de Castro, Talanta 64 (2004) 571.[149] M. Ericsson, A. Colmsjo, Anal. Chem. 75 (2003) 1713.[150] M. Ericsson, A. Colmsjo, J. Chromatogr. A 964 (2002) 11.[151] A. Serrano, M. Gallego, J. Chromatogr. A 1104 (2006) 323.[152] L. Qi, S. Zhang, M. Zuo, Y. Chen, J. Pharm. Biomed. Anal. 41 (2006)
1620.[153] S. Balachandran, S.E. Kentish, R. Mawson, M. Ashokkumar, Ultrason.
Sonochem. 13 (2006) 471.[154] P. Richter, M. Jimenez, R. Salazar, A. Marican, J. Chromatogr. A 1132
(2006) 15.[155] M.D. Luque de Castro, F. Priego-Capote, Anal. Chim. Acta 583 (2007)
2.[156] J.L. Luque-Garcıa, M.D. Luque de Castro, J. Chromatogr. A 1034 (2004)
237.[157] E. Riera, Y. Golas, A. Blanco, J.A. Gallego, M. Blasco, A. Mulet, Ultra-
son. Sonochem. 11 (2004) 241.[158] E. Psillakis, N. Kalogerakis, Trends Anal. Chem. 21 (2002) 53.[159] G. Audunsson, Anal. Chem. 58 (1986) 2714.[160] L. Hou, H.K. Lee, J. Chromatogr. A 976 (2002) 377.[161] S.W. Myung, S.H. Yoon, M. Kim, Analyst 128 (2003) 1443.[162] G. Ouyang, W. Zhao, J. Pawliszyn, Anal. Chem. 77 (2005) 8122.[163] M. Saraji, J. Chromatogr. A 1062 (2005) 15.[164] L. Xia, B. Hu, Z. Jiang, Y. Wu, Y. Liang, Anal. Chem. 76 (2004) 2910.[165] W. Liu, H.K. Lee, Anal. Chem. 72 (2000) 4462.[166] S. Fragueiro, I. Lavilla, C. Bendicho, Talanta 68 (2006) 1096.[167] J. Zhang, T. Su, H.K. Lee, Anal. Chem. 77 (2005) 1988.[168] M.A. Jeannot, F.F. Cantwell, Anal. Chem. 68 (1996) 2236.[169] M. Kawaguchi, R. Ito, N. Endo, N. Okanouchi, N. Sakui, K. Saito, H.
Nakazawa, J. Chromatogr. A 1110 (2006) 1.[170] M. Saraji, M. Bakhshi, J. Chromatogr. A 1098 (2005) 30.[171] A. Tor, J. Chromatogr. A 1125 (2006) 129.[172] E.M. Gioti, D.C. Skalkos, Y.C. Fiamegos, C.D. Stalikas, J. Chromatogr.
A 1093 (2005) 1.[173] S. Jermak, B. Pranaityte, A. Padarauskas, Electrophoresis 27 (2006) 4538.[174] R. Sekar, H. Wu, Anal. Chem. 78 (2006) 6306.[175] P.R. Sudhir, H. Wu, Z. Zhou, Anal. Chem. 77 (2005) 7380.[176] L. Chimuka, R.E. Majors, LC–GC N. Am. 22 (2004) 102.[177] N. Jakubowska, Z. Polkowska, J. Namiesnik, A. Przyjazny, Crit. Rev.
Anal. Chem. 35 (2005) 217.[178] J.A. Jonsson, L. Mathiasson, J. Sep. Sci. 24 (2001) 495.
[179] R.E. Majors, J.A. Jonsson, L. Mathiasson, LC–GC N. Am. 21 (2003) 424.[180] E. Psillakis, N. Kalogerakis, Trends Anal. Chem. 22 (2003) 565.[181] Y. Shen, J.A. Jonsson, L. Mathiasson, Anal. Chem. 70 (1998) 946.[182] J. Liu, J. Chao, G. Jiang, Y. Cai, J. Liu, J. Chromatogr. A 995 (2003)21.

2 togr.
16 Y. Chen et al. / J. Chroma[183] A. Drapała, J.A. Jonsson, P. Wieczorek, Anal. Chim. Acta 553 (2005) 9.[184] T. Barri, S. Bergstrom, A. Hussen, J. Norberg, J.A. Jonsson, J. Chro-
matogr. A 1111 (2006) 11.[185] T. Barri, S. Bergstrom, J. Norberg, J.A. Jonsson, Anal. Chem. 76 (2004)
1928.[186] T. Hyotylainen, K. Luthje, M. Rautiainen-Rama, M.-L. Riekkola, J. Chro-
matogr. A 1056 (2004) 267.[187] K. Luthje, T. Hyotylainen, M.-L. Riekkola, Anal. Bioanal. Chem. 378
(2004) 1991.[188] J. Norberg, E. Thordarson, Analyst 125 (2000) 673.[189] N. Fontanals, T. Barri, S. Bergstrom, J.A. Jonsson, J. Chromatogr. A 1133
(2006) 41.[190] D. Kou, S. Mitra, Anal. Chem. 75 (2003) 6355.[191] X. Wang, D. Kou, S. Mitra, J. Chromatogr. A 1089 (2005) 39.[192] K.E. Rasmussen, S. Pedersen-Bjergaard, Trends Anal. Chem. 23 (2004)
1.[193] K.J. Chia, S.D. Huang, J. Chromatogr. A 1103 (2006) 158.[194] L. Hou, G. Shen, H.K. Lee, J. Chromatogr. A 985 (2003) 107.[195] X. Jiang, C. Basheer, J. Zhang, H.K. Lee, J. Chromatogr. A 1087 (2005)
289.[196] X. Jiang, S.Y. Oh, H.K. Lee, Anal. Chem. 77 (2005) 1689.[197] M. Sandahl, L. Mathiasson, J.A. Jonsson, J. Chromatogr. A 975 (2002)
211.[198] L. Zhu, L. Zhu, H.K. Lee, J. Chromatogr. A 924 (2001) 407.[199] K.E. Rasmussen, S. Pedersen-Bjergaard, M. Krogh, H.G. Ugland, T.
Grønhaug, J. Chromatogr. A 873 (2000) 3.[200] B. Kaye, W.J. Herron, P.V. Macrae, S. Robinson, D.A. Stopher, R.F. Venn,
W. Wild, Anal. Chem. 68 (1996) 1658.[201] G. Rule, M. Chapple, J. Henion, Anal. Chem. 73 (2001) 439.[202] R.F. Venn, J. Merson, S. Cole, P. Macrae, J. Chromatogr. B 817 (2005)
77.[203] S. Ekstrom, L. Wallman, D. Hok, G. Marko-Varga, T. Laurell, J. Proteome
Res. 5 (2006) 1071.[204] R.E. Majors, LC–GC N. Am. 23 (2005) 358.[205] E. Stephens, S.L. Maslen, L.G. Green, D.H. Williams, Anal. Chem. 76
(2004) 2343.[206] Y. Saito, M. Imaizumi, T. Takeichi, K. Jinno, Anal. Bioanal. Chem. 372
(2002) 164.[207] L. Ge, J.W.Y. Yong, N.K. Goh, L.S. Chia, S.N. Tan, E.S. Org, J. Chro-
matogr. B 829 (2005) 26.[208] K.F. Nielsen, P.W. Dalsgaard, J. Smedsgaard, T.O. Larsen, J. Agric. Food
Chem. 53 (2005) 2908.[209] N. Rosales-Conrado, M.E. Leon-Gonzalez, L.V. Perez-Arribas, L.M.
Polo-Dıez, J. Chromatogr. A 1076 (2005) 202.[210] S. Huq, A. Dixon, K. Kelly, K.M.R. Kallury, J. Chromatogr. A 1073
(2005) 355.[211] R. Kahlich, C.H. Gleiter, S. Laufer, B. Kammerer, Rapid Commun. Mass
Spectrom. 20 (2006) 275.[212] C.P.W.G.M. Verweij-van Wissen, R.E. Aarnoutse, D.M. Burger, J. Chro-
matogr. B 816 (2005) 121.[213] M.J. Rose, C. Fernandez-Metzler, B.A. Johns, G.R. Sitko, J.J. Cook, J.
Yergey, J. Pharm. Biomed. Anal. 38 (2005) 695.[214] E. Jimenez-Lozano, D. Roy, D. Barron, J. Barbosa, Electrophoresis 25
(2004) 65.[215] D. Richard, B. Ling, N. Authier, T.W. Faict, A. Eschalier, F. Coudore,
Anal. Chem. 77 (2005) 1354.[216] E. Benito-Pena, A.I. Partal-Rodera, M.E. Leon-Gonzalez, M.C. Moreno-
Bondi, Anal. Chim. Acta 556 (2006) 415.[217] N. Fontanals, B.C. Trammell, M. Galia, R.M. Marce, P.C. Iraneta, F.
Borrull, U.D. Neue, J. Sep. Sci. 29 (2006) 1622.[218] S. Weigel, R. Kallenborn, H. Huhnerfuss, J. Chromatogr. A 1023 (2004)
183.[219] C. Maisonnette, P. Simon, M.-C. Hennion, V. Pichon, J. Chromatogr. A
1120 (2006) 185.[220] X. Zhang, D. Martens, P.M. Kramer, A.A. Kettrup, X. Liang, J. Chro-
matogr. A 1102 (2006) 84.[221] N. Delaunay-Bertoncini, V. Pichon, M.-C. Hennion, J. Chromatogr. A
999 (2003) 3.
A 1184 (2008) 191–219
[222] N. Sanvicens, E.J. Moore, G.G. Guilbault, M.-P. Marco, J. Agric. FoodChem. 54 (2006) 9176.
[223] P. Su, X. Zhang, W. Chang, J. Chromatogr. B 816 (2005) 7.[224] N. Delaunay-Bertoncini, M.-C. Hennion, J. Pharm. Biomed. Anal. 34
(2004) 717.[225] M.-C. Hennion, V. Pichon, J. Chromatogr. A 1000 (2003) 29.[226] N. Delaunay, V. Pichon, M.-C. Hennion, J. Chromatogr. B 745 (2000) 15.[227] S. Hu, L. Li, X. He, Prog. Chem. 17 (2005) 531.[228] E. Caro, R.M. Marce, F. Borrull, P.A.G. Cormack, D.C. Sherrington,
Trends Anal. Chem. 25 (2006) 143.[229] V.B. Kandimalla, H. Ju, Anal. Bioanal. Chem. 380 (2004) 587.[230] F. Qiao, H. Sun, H. Yan, K.H. Row, Chromatographia 64 (2006) 625.[231] J. Yin, S. Wang, G. Yang, G. Yang, Y. Chen, J. Chromatogr. B 844 (2006)
142.[232] R. Carabias-Martınez, E. Rodrıguez-Gonzalo, E. Herrero-Hernandez,
M.E. Dıaz-Garcıa, J. Sep. Sci. 28 (2005) 453.[233] E. Caro, R.M. Marce, P.A.G. Cormack, D.C. Sherrington, F. Borrull, J.
Sep. Sci. 28 (2005) 2080.[234] F. Chapuis, V. Pichon, F. Lanza, B. Sellergren, M.-C. Hennion, J. Chro-
matogr. B 804 (2004) 93.[235] G. Karasova, J. Lehotay, J. Sadecka, I. Skacani, M. Lachova, J. Sep. Sci.
28 (2005) 2468.[236] L. Zhu, L. Chen, X. Xu, Anal. Chem. 75 (2003) 6381.[237] B. Dirion, F. Lanza, B. Sellergren, C. Chassaing, R. Venn, C. Berggren,
Chromatographia 56 (2002) 237.[238] J. Haginaka, H. Sanbe, Anal. Chem. 72 (2000) 5206.[239] S. Hu, S. Wang, X. He, Analyst 128 (2003) 1485.[240] T. Kubo, K. Hosoya, Y. Watabe, T. Ikegami, N. Tanaka, T. Sano, K. Kaya,
J. Chromatogr. A 987 (2003) 389.[241] T. Kubo, M. Nomachi, K. Nemoto, T. Sano, K. Hosoya, N. Tanaka, K.
Kaya, Anal. Chim. Acta 577 (2006) 1.[242] J. Matsui, K. Fujiwara, T. Takeuchi, Anal. Chem. 72 (2000) 1810.[243] J. Ou, L. Kong, C. Pan, X. Su, X. Lei, H. Zou, J. Chromatogr. A 1117
(2006) 163.[244] G. Theodoridis, A. Kantifes, P. Manesiotis, N. Raikos, H. Tsoukali-
Papadopoulou, J. Chromatogr. A 987 (2003) 103.[245] J.L. Urraca, M.D. Marazuela, E.R. Merino, G. Orellana, M.C. Moreno-
Bondi, J. Chromatogr. A 1116 (2006) 127.[246] I. Chianella, K. Karim, E.V. Piletska, C. Preston, S.A. Piletsky, Anal.
Chim. Acta 559 (2006) 73.[247] I. Chianella, S.A. Piletsky, I.E. Tothill, B. Chen, A.P.F. Turner, Biosens.
Bioelectron. 18 (2003) 119.[248] S. Piletsky, E. Piletska, K. Karim, G. Foster, C. Legge, A. Turner, Anal.
Chim. Acta 504 (2004) 123.[249] L. Wu, K. Zhu, M. Zhao, Y. Li, Anal. Chim. Acta 549 (2005) 39.[250] C. Baggiani, C. Giovannoli, L. Anfossi, C. Tozzi, J. Chromatogr. A 938
(2001) 35.[251] E. Caro, R.M. Marce, P.A.G. Cormack, D.C. Sherrington, F. Borrull, J.
Chromatogr. A 1047 (2004) 175.[252] R.G.C. Silva, F. Augusto, J. Chromatogr. A 1114 (2006) 216.[253] N.M. Cassiano, V.V. Lima, R.V. Oliveira, A.C. de Pietro, Q.B. Cass, Anal.
Bioanal. Chem. 384 (2006) 1462.[254] S. Souverain, S. Rudaz, J.-L. Veuthey, J. Chromatogr. B 801 (2004) 141.[255] C.L. Arthur, J. Pawliszyn, Anal. Chem. 62 (1990) 2145.[256] J. Pawliszyn, Solid-Phase Microextraction: Theory and Practice, Wiley-
VCH, New York, 1997.[257] J. Ai, Anal. Chem. 69 (1997) 1230.[258] H. Lord, J. Pawliszyn, J. Chromatogr. A 885 (2000) 153.[259] R. Eisert, J. Pawliszyn, Anal. Chem. 69 (1997) 3140.[260] C. Dietz, J. Sanz, C. Camara, J. Chromatogr. A 1103 (2006) 183.[261] W.M. Mullett, J. Pawliszyn, J. Sep. Sci. 26 (2003) 251.[262] J.B. Quintana, I. Rodrıguez, Anal. Bioanal. Chem. 384 (2006) 1447.[263] S.L. Chong, D. Wang, J.D. Hayes, B.W. Wilhite, A. Malik, Anal. Chem.
69 (1997) 3889.[264] M. Azenha, C. Malheiro, A.F. Silva, J. Chromatogr. A 1069 (2005)
163.[265] C. Basheer, S. Jegadesan, S. Valiyaveettil, H.K. Lee, J. Chromatogr. A
1087 (2005) 252.

atogr
Y. Chen et al. / J. Chrom[266] X. Li, Z. Zeng, S. Gao, H. Li, J. Chromatogr. A 1023 (2004) 15.[267] Z. Zeng, W. Qiu, Z. Huang, Anal. Chem. 73 (2001) 2429.[268] K. Alhooshani, T.Y. Kim, A. Kabir, A. Malik, J. Chromatogr. A 1062
(2005) 1.[269] T.Y. Kim, K. Alhooshani, A. Kahir, D.P. Fries, A. Malik, J. Chromatogr.
A 1047 (2004) 165.[270] S. Kulkarni, L. Fang, K. Alhooshani, A. Malik, J. Chromatogr. A 1124
(2006) 205.[271] Y. Fan, Y. Feng, J. Zhang, S. Da, M. Zhang, J. Chromatogr. A 1074 (2005)
9.[272] Y. Fan, M. Zhang, Y. Feng, J. Chromatogr. A 1099 (2005) 84.[273] J. Huang, B. Lin, Q. Yu, Y. Feng, Anal. Bioanal. Chem. 384 (2006)
1228.[274] H. Zhang, J. Huang, H. Wang, Y. Feng, Anal. Chim. Acta 565 (2006) 129.[275] M. Zhang, F. Wei, Y. Zhang, J. Nie, Y. Feng, J. Chromatogr. A 1102
(2006) 294.[276] J. Courtois, G. Fischer, B. Sellergren, K. Irgum, J. Chromatogr. A 1109
(2006) 92.[277] Y. Chen, J. Pawliszyn, Anal. Chem. 75 (2003) 2004.[278] Y. Chen, J. Pawliszyn, Anal. Chem. 76 (2004) 6823.[279] G. Ouyang, Y. Chen, J. Pawliszyn, Anal. Chem. 77 (2005) 7319.[280] B. Vrana, G.A. Mills, E. Dominiak, R. Greenwood, Environ. Pollut. 142
(2006) 333.[281] L. Bragg, Z. Qin, M. Alaee, J. Pawliszyn, J. Chromatogr. Sci. 44 (2006)
317.[282] I. Bruheim, X. Liu, J. Pawliszyn, Anal. Chem. 75 (2003) 1002.[283] W. Zhao, G. Ouyang, M. Alaee, J. Pawliszyn, J. Chromatogr. A 1124
(2006) 112.[284] F. Augusto, A.L.P. Valente, Trends Anal. Chem. 21 (2002) 428.[285] J. Pawliszyn, Aust. J. Chem. 56 (2003) 155.[286] H.L. Lord, R.P. Grant, M. Walles, B. Incledon, B. Fahie, J.B. Pawliszyn,
Anal. Chem. 75 (2003) 5103.[287] E. Pionnier, C. Chabanet, L. Mioche, J.-L. Le Quere, C. Salles, J. Agric.
Food Chem. 52 (2004) 557.[288] E. Pionnier, E. Semon, C. Chabanet, C. Salles, Sci. Aliment 25 (2005)
193.[289] Z. Zhang, J. Cai, G. Ruan, G. Li, J. Chromatogr. B 822 (2005) 244.[290] D. Djozan, T. Baheri, R. Farshbaf, S. Azhari, Anal. Chim. Acta 554 (2005)
197.[291] D.C. Gilley, G. DeGrandi-Hoffman, J.E. Hooper, J. Insect. Physiol. 52
(2006) 520.[292] T. Kumazawa, X.P. Lee, K. Sato, O. Suzuki, Anal. Chim. Acta 492 (2003)
49.[293] A.K. Malik, V. Kaur, N. Verma, Talanta 68 (2006) 842.[294] C.G. Zambonin, Anal. Bioanal. Chem. 375 (2003) 73.[295] A. Aresta, F. Palmisano, R. Vatinno, C.G. Zambonin, J. Agric. Food
Chem. 54 (2006) 1594.[296] A. Aresta, F. Palmisano, C.G. Zambonin, J. Pharm. Biomed. Anal. 39
(2005) 643.[297] J. Chen, J.B. Pawliszyn, Anal. Chem. 67 (1995) 2530.[298] K. Jinno, T. Muramatsu, Y. Saito, Y. Kiso, S. Magdic, J. Pawliszyn, J.
Chromatogr. A 754 (1996) 137.[299] H. Kataoka, Anal. Bioanal. Chem. 373 (2002) 31.[300] J. O’Reilly, Q. Wang, L. Setkova, J.P. Hutchinson, Y. Chen, H.L. Lord,
C.M. Linton, J. Pawliszyn, J. Sep. Sci. 28 (2005) 2010.[301] H. Kataoka, H.L. Lord, J. Pawliszyn, J. Chromatogr. A 880 (2000) 35.[302] T. Hyotylainen, M.-L. Riekkola, J. Chromatogr. A 1000 (2003) 357.[303] T. Hyotylainen, M.-L. Riekkola, J. Chromatogr. B 817 (2005) 13.[304] K. Pyrzynska, E. Pobozy, Crit. Rev. Anal. Chem. 32 (2002) 227.[305] Y. Alnouti, K. Srinivasan, D. Waddell, H. Bi, O. Kavetskaia, A.I. Gusev,
J. Chromatogr. A 1080 (2005) 99.[306] S. Rodriguez-Mozaz, M.J. Lopez de Alda, D. Barcelo, Anal. Chem. 76
(2004) 6998.
[307] A. Schellen, B. Ooms, D. van de Lagemaat, R. Vreeken, W.D. van Don-gen, J. Chromatogr. B 788 (2003) 251.[308] F.W.A. Tempels, J. Teeuwsen, I.K. Kyriakou, G. Theodoridis, W.J.M.
Underberg, G.W. Somsen, G.J. de Jong, J. Chromatogr. A 1053 (2004)263.
. A 1184 (2008) 191–219 217
[309] Z. Zhang, Y. He, J. Chromatogr. A 1066 (2005) 211.[310] M.C. Breadmore, A.S. Palmer, M. Curran, M. Macka, N. Avdalovic, P.R.
Haddad, Anal. Chem. 74 (2002) 2112.[311] S. Zhang, M. Macka, P.R. Haddad, Electrophoresis 27 (2006) 1069.[312] N.A. Guzman, R.J. Stubbs, Electrophoresis 22 (2001) 3602.[313] K. Sandra, F. Lynen, B. Devreese, J. van Beeumen, P. Sandra, Anal.
Bioanal. Chem. 385 (2006) 671.[314] N.M. Vizioli, M.L. Rusell, C.N. Carducci, Anal. Chim. Acta 514 (2004)
167.[315] J.M. Armenta, B. Gu, P.H. Humble, C.D. Thulin, M.L. Lee, J. Chromatogr.
A 1097 (2005) 171.[316] N.E. Baryla, N.P. Toltl, Analyst 128 (2003) 1009.[317] J.P. Hutchinson, M. Macka, N. Avdalovic, P.R. Haddad, J. Chromatogr.
A 1106 (2006) 43.[318] J.P. Hutchinson, P. Zakaria, A.R. Bowie, M. Macka, N. Avdalovic, P.R.
Haddad, Anal. Chem. 77 (2005) 407.[319] S. Oguri, H. Tanagaki, M. Hamaya, M. Kato, T. Toyooka, Anal. Chem.
75 (2003) 5240.[320] J.P. Quirino, M.T. Dulay, R.N. Zare, Anal. Chem. 73 (2001) 5557.[321] G. Ping, Y. Zhang, W. Zhang, L. Zhang, L. Zhang, P. Schmitt-Kopplin,
A. Kettrup, Electrophoresis 25 (2004) 421.[322] D. Schaller, E.F. Hilder, P.R. Haddad, Anal. Chim. Acta 556 (2006) 104.[323] J.A. Starkey, Y. Mechref, C.K. Byun, R. Steinmetz, J.S. Fuqua, O.H.
Pescovitz, M.V. Novotny, Anal. Chem. 74 (2002) 5998.[324] A.J.H. Louter, C.A. van Beekvelt, P. Cid Montanes, J. Slobodnik, J.J.
Vreuls, U.A.Th. Brinkman, J. Chromatogr. A 725 (1996) 67.[325] L. Zhang, X. Wu, Anal. Chem. 79 (2007) 2562.[326] F. David, P. Sandra, J. Chromatogr. A 1152 (2007) 54.[327] M. Kawaguchi, R. Ito, K. Saito, H. Nakazawa, J. Pharm. Biomed. Anal.
40 (2006) 500.[328] C. Bicchi, C. Cordero, E. Liberto, P. Rubiolo, B. Sgorbini, F. David, P.
Sandra, J. Chromatogr. A 1094 (2005) 9.[329] J.P. Lambert, W.M. Mullett, E. Kwong, D. Lubda, J. Chromatogr. A 1075
(2005) 43.[330] C. Blasco, M. Fernandez, Y. Pico, G. Font, J. Chromatogr. A 1030 (2004)
77.[331] C. Blasco, G. Font, Y. Pico, J. Chromatogr. A 1028 (2004) 267.[332] N. Furusawa, Anal. Bioanal. Chem. 378 (2004) 2004.[333] G. Berardi, S. Bogialli, R. Curini, A. Di Corcia, A. Lagana, J. Agric. Food
Chem. 54 (2006) 4537.[334] S. Bogialli, R. Curini, A. Di Corcia, A. Lagana, M. Mele, M. Nazzari, J.
Chromatogr. A 1067 (2005) 93.[335] S. Bogialli, R. Curini, A. Di Corcia, A. Lagana, M. Nazzari, M. Tonci, J.
Chromatogr. A 1054 (2004) 351.[336] J.J. Ramos, M.J. Gonzalez, L. Ramos, J. Sep. Sci. 27 (2004) 595.[337] H.B. Xiao, M. Krucker, K. Albert, X.M. Liang, J. Chromatogr. A 1032
(2004) 117.[338] S.A. Barker, J. Biochem. Biophys. Methods 70 (2007) 151.[339] S. Bogialli, A. Di Corcia, J. Biochem. Biophys. Methods 70 (2007) 163.[340] E.M. Kristenson, L. Ramos, U.A.Th. Brinkman, Trends Anal. Chem. 25
(2006) 96.[341] R.J. Schutte, S.A. Oshodi, W.M. Reichert, Anal. Chem. 76 (2004) 6058.[342] X. Ao, T.J. Sellati, J.A. Stenken, Anal. Chem. 76 (2004) 3777.[343] N. Torto, D. Mogopodi, Trends Anal. Chem. 23 (2004) 109.[344] A.N. Khramov, J.A. Stenken, Anal. Chem. 71 (1999) 1257.[345] A. Petterson, A. Amirkhani, B. Arvidsson, K. Markides, J. Bergquist,
Anal. Chem. 76 (2004) 1678.[346] J. Ruiz-Jimenez, M.D. Luque de Castro, Trends Anal. Chem. 25 (2006)
563.[347] W.C. Tseng, G.W. Cheng, C.F. Lee, L.H. Wu, Y.L. Huang, Anal. Chim.
Acta 543 (2005) 38.[348] B.H. Huynh, B.A. Fogarty, R.S. Martin, S.M. Lunte, Anal. Chem. 76
(2004) 6440.
[349] A.R. Timerbaev, T. Hirokawa, Electrophoresis 27 (2006) 323.[350] J. Petr, V. Maier, J. Horakova, J. Sevcık, Z. Stransky, J. Sep. Sci. 29 (2006)2705.[351] D. Flottmann, J. Hins, C. Rettenmaier, N. Schnell, Z. Kuci, G. Merkel,
G. Seitz, G. Bruchelt, Microchim. Acta 154 (2006) 49.

2 togr.
18 Y. Chen et al. / J. Chroma[352] D. Bexheti, E.I. Anderson, A.J. Hutt, M. Hanna-Brown, J. Chromatogr.A 1130 (2006) 137.
[353] S. Chen, M.L. Lee, Anal. Chem. 72 (2000) 816.[354] Z.K. Shihabi, J. Chromatogr. 652 (1993) 471.[355] K. Yu, Liq. J. Liq. Chromatogr. Rel. Technol. 29 (2006) 1561.[356] Z.K. Shihabi, Electrophoresis 23 (2002) 1612.[357] Z.K. Shihabi, Electrophoresis 23 (2002) 1628.[358] Z.K. Shihabi, M.E. Hinsdale, C.P. Cheng, Electrophoresis 22 (2001) 2351.[359] K. Fukushi, N. Ishio, M. Sumida, S. Takeda, S.I. Wakida, K. Hiiro,
Electrophoresis 21 (2000) 2866.[360] T. Hirokawa, T. Ichihara, K. Ito, A.R. Timerbaev, Electrophoresis 24
(2003) 2328.[361] T. Hirokawa, M. Yoshioka, H. Okamoto, A.R. Timerbaev, G. Blaschke,
J. Chromatogr. B 811 (2004) 165.[362] K. Yokota, K. Fukushi, S. Takeda, S.I. Wakida, J. Chromatogr. A 1035
(2004) 145.[363] R. Rodriquez-Diaz, T. Wehr, M. Zhu, V. Levi, J.P. Landers (Eds.), Hand-
book of Electrophoresis, second ed., CRC Press, Boca Raton, FL, 1997,p. 101.
[364] C. Yang, L. Zhang, H. Liu, W. Zhang, Y. Zhang, J. Chromatogr. A 1018(2003) 97.
[365] M. Deepa, S.L. Cheng, Electrophoresis 23 (2002) 3160.[366] M. Zhang, Z. El Rassi, J. Proteome Res. 5 (2006) 2001.[367] C. Yang, H. Liu, Q. Yang, L. Zhang, W. Zhang, Y. Zhang, Anal. Chem.
75 (2003) 215.[368] H. Liu, C. Yang, Q. Yang, W. Zhang, Y. Zhang, Chin. J. Anal. Chem. 32
(2004) 273.[369] J. Chen, B.M. Balgley, D.L. DeVoe, C.S. Lee, Anal. Chem. 75 (2003)
3145.[370] T. Guo, P.A. Rudnick, W. Wang, C.S. Lee, D.L. Devoe, B.M. Balgley, J.
Proteome Res. 5 (2006) 1469.[371] F.E.P. Mikkers, F.M. Everaerts, Th.P.E.M. Verheggen, J. Chromatogr. 169
(1979) 11.[372] W. Ding, M.J. Thornpson, J.S. Fritz, Electrophoresis 19 (1998) 2133.[373] A.B. Wey, W. Thormann, J. Chromatogr. A 924 (2001) 507.[374] S. Liu, Q. Li, X. Chen, Z. Hu, Electrophoresis 23 (2002) 3392.[375] A. Alnajjar, B. McCord, J. Pharmaceut. Biomed. 33 (2003) 463.[376] Z. Hen, J. Lin, R. Naidu, Anal. Bioanal. Chem. 375 (2003) 679.[377] R.L. Chien, D.S. Burgi, Anal. Chem. 64 (1992) 1046.[378] J.P. Quirino, S. Terabe, J. Chromatogr. A 850 (1999) 339.[379] S. Palmarsdottir, L. Mathiasson, J.A. Josson, L.E. Edholm, J. Chromatogr.
B 688 (1997) 127.[380] N. Maeso, A. Cifuentes, C. Barbas, J. Chromatogr. B 809 (2004) 147.[381] C.-C. Wang, S.-S. Chiou, S.-M. Wu, Electrophoresis 26 (2005) 2637.[382] T. Tabi, K. Magyar, E. Szoko, Electrophoresis 26 (2005) 1940.[383] A. Macia, F. Borrull, C. Aguilar, M. Calull, Electrophoresis 24 (2003)
2779.[384] Y. Feng, J. Zhu, Anal. Chem. 78 (2006) 6608.[385] H. Fang, Z. Zeng, L. Liu, D. Pang, Anal. Chem. 78 (2006) 1257.[386] G.L. Erny, A. Cifuentes, Anal. Chem. 78 (2006) 7557.[387] P. Britz-McKibbin, G.M. Bebault, D.D.Y. Chen, Anal. Chem. 72 (2000)
1729.[388] P. Britz-McKibbin, D.D.Y. Chen, Anal. Chem. 72 (2000) 1242.[389] P. Britz-McKibbin, S. Terabe, J. Chromatogr. A 1000 (2003) 917.[390] J.-B. Kim, P. Britz-McKibbin, T. Hirokawa, S. Terabe, Anal. Chem. 75
(2003) 3986.[391] J.-B. Kim, Y. Okamoto, S. Terabe, J. Chromatogr. A 1018 (2003) 251.[392] M.R.N. Monton, K. Imami, M. Nakanishi, J.-B. Kim, S. Terabe, J. Chro-
matogr. A 1079 (2005) 266.[393] W. Wei, G. Xue, E.S. Yeung, Anal. Chem. 74 (2002) 934.[394] J.A. Gillogly, C.E. Lunte, Electrophoresis 26 (2005) 633.[395] Z. Mala, L. Krivankova, P. Gebauer, P. Bocek, Electrophoresis 28 (2007)
243.
[396] D.J. Weiss, K. Saunders, C.E. Lunte, Electrophoresis 22 (2001) 59.[397] S.D. Arnett, C.E. Lunte, Electrophoresis 24 (2003) 1745.[398] M.M. Hsieh, H.T. Chang, Electrophoresis 26 (2005) 187.[399] C.A. Nesbitt, J.T.-M. Lo, K.K.C. Yeung, J. Chromatogr. A 1073 (2005)175.
A 1184 (2008) 191–219
[400] P. Britz-McKibbin, T. Ichihashi, K. Tsubota, D.D.Y. Chen, S. Terabe, J.Chromatogr. A 1013 (2003) 65.
[401] S. Park, C.E. Lunte, J. Microcol. Sep. 10 (1998) 511.[402] Y. Zhao, C.E. Lunte, Anal. Chem. 71 (1999) 3985.[403] Y. Zhao, K. Mclaughin, C.E. Lunte, Anal. Chem. 70 (1998) 4578.[404] M.E. Hoque, S.D. Arnett, C.E. Lunte, J. Chromatogr. B 827 (2005)
51.[405] D.-K. Kim, S.H. Kang, J. Chromatogr. A 1064 (2005) 121.[406] Y. Han, M. Zuo, L. Qi, K. Liu, L. Mao, Y. Chen, Electrophoresis 27 (2006)
4240.[407] J.P. Quirino, S. Terabe, Science 282 (1998) 465.[408] J.P. Quirino, S. Terabe, Chromatographia 53 (2001) 285.[409] W. Shi, C.P. Palmer, J. Sep. Sci. 25 (2002) 215.[410] D.L. Kirschner, M. Jaramillo, T.K. Green, Anal. Chem. 79 (2007) 736.[411] T.C. Chiu, Y. Lin, C.C. Hung, A. Chrambach, H.T. Chang, Electrophoresis
24 (2003) 1730.[412] M.M. Hsieh, C.E. Hsu, W.L. Tseng, H.T. Chang, Electrophoresis 23
(2002) 1633.[413] M.M. Hsieh, W.L. Tseng, H.T. Chang, Electrophoresis 21 (2000) 2904.[414] M. Gong, K.R. Wehmeyer, P.A. Limbach, W.R. Heineman, Anal. Chem.
78 (2006) 6035.[415] K. Isoo, S. Terabe, Anal. Chem. 75 (2003) 6789.[416] L. Yu, S. Fong, Y. Li, Electrophoresis 26 (2005) 4360.[417] Y.C. Fiamegos, C.G. Nanos, C.D. Stalikas, J. Chromatogr. B 813 (2004)
89.[418] E.E. Stashenko, J.R. Martınez, Trends Anal. Chem. 23 (2004) 553.[419] Z. Kuklenyik, J. Ekong, C.D. Cutchins, L.L. Needham, A.M. Calafat,
Anal. Chem. 75 (2003) 6820.[420] J.M. Rosenfeld, J. Chromatogr. A 843 (1999) 19.[421] L. Pan, J. Pawliszyn, Anal. Chem. 69 (1997) 196.[422] C. Deng, N. Yao, N. Li, X. Zhang, J. Sep. Sci. 28 (2005) 2301.[423] N. Li, C. Deng, X. Yin, N. Yao, X. Shen, X. Zhang, Anal. Biochem. 342
(2005) 318.[424] M. Kojima, N. Matsui, S. Tsunoi, M. Tanaka, J. Chromatogr. A 1078
(2005) 1.[425] L. Yu, M.J.M. Wells, J. Chromatogr. A 1143 (2007) 16.[426] R. Dhopeshwarkar, L. Sun, R.M. Crooks, Lab Chip 5 (2005) 1148.[427] R.S. Foote, J. Khandurina, S.C. Jacobson, J.M. Ramsey, Anal. Chem. 77
(2005) 57.[428] S. Song, A.K. Singh, B.J. Kirby, Anal. Chem. 76 (2004) 4589.[429] Y. Wang, A.L. Stevens, J. Han, Anal. Chem. 77 (2005) 4293.[430] Y. Zhang, A.T. Timperman, Analyst 128 (2003) 537.[431] A. Smirnova, K. Mawatari, A. Hibara, M.A. Proskurnin, T. Kitamori,
Anal. Chim. Acta 558 (2006) 69.[432] M. Tokeshi, T. Minagawa, K. Uchiyama, A. Hibara, K. Sato, H. Hisamoto,
T. Kitamori, Anal. Chem. 74 (2002) 1565.[433] D.J. Wilson, L. Konermann, Anal. Chem. 77 (2005) 6887.[434] H. Chen, Q. Fang, X. Yin, Z. Fang, Lab Chip 5 (2005) 719.[435] X. Wang, C. Saridara, S. Mitra, Anal. Chim. Acta 543 (2005) 92.[436] Y. Li, D.L. DeVoe, C.S. Lee, Electrophoresis 24 (2003) 193.[437] W. Tan, Z.H. Fan, C.X. Qiu, A.J. Ricco, I. Gibbons, Electrophoresis 23
(2002) 3638.[438] Y. Xu, C. Zhang, D. Janasek, A. Manz, Lab Chip 3 (2003) 224.[439] B. Jung, R. Bharadwaj, J.G. Santiago, Electrophoresis 24 (2003) 3476.[440] Y. Liu, R.S. Foote, S.C. Jacobson, J.M. Ramsey, Lab Chip 5 (2005) 457.[441] B. Grass, R. Hergenroder, A. Neyer, D. Siepe, J. Sep. Sci. 25 (2002) 135.[442] W.N. Vreeland, S.J. Williams, A.E. Barron, A.P. Sassi, Anal. Chem. 75
(2003) 3059.[443] A. Wainright, S.J. Williams, G. Ciambrone, Q. Xue, J. Wei, D. Harris, J.
Chromatogr. A 979 (2002) 69.[444] Z. Xu, T. Ando, T. Nishine, A. Arai, T. Hirokawa, Electrophoresis 24
(2003) 3821.[445] P.K. Wong, C. Chen, T. Wang, C.M. Ho, Anal. Chem. 76 (2004) 6908.
[446] D. Ross, L.E. Locascio, Anal. Chem. 74 (2002) 2556.[447] A. Bhattacharyya, C.M. Klapperich, Anal. Chem. 78 (2006) 788.[448] M.C. Breadmore, K.A. Wolfe, I.G. Arcibal, W.K. Leung, D. Dickson,B.C. Giordano, M.E. Power, J.P. Ferrance, S.H. Feldman, P.M. Norris,J.P. Landers, Anal. Chem. 75 (2003) 1880.

atogr
Y. Chen et al. / J. Chrom[449] T.N. Chiesl, W. Shi, A.E. Barron, Anal. Chem. 77 (2005) 772.[450] L.A. Legendre, J.M. Bienvenue, M.G. Roper, J.P. Ferrance, J.P. Landers,
Anal. Chem. 78 (2006) 1444.[451] Y. Xu, B. Vaidya, A.B. Patel, S.M. Ford, R.L. McCarley, S.A. Soper,
Anal. Chem. 75 (2003) 2975.[452] A.B. Jemere, R.D. Oleschuk, F. Ouchen, F. Fajuyigbe, D.J. Harrison,
Electrophoresis 23 (2002) 3537.[453] J.D. Ramsey, G.E. Collins, Anal. Chem. 77 (2005) 6664.[454] B.S. Broyles, S.C. Jacobson, J.M. Ramsey, Anal. Chem. 75 (2003) 2761.[455] J.E. Kim, J.H. Cho, S.H. Paek, Anal. Chem. 77 (2005) 7901.[456] P.N. Floriano, N. Christodoulides, D. Romanovicz, B. Bernard, G.W.
Simmons, M. Cavell, J.T. McDevitt, Biosens. Bioelectron. 20 (2005)2079.
[457] P. Myers, K.D. Bartle, J. Chromatogr. A 1044 (2004) 253.[458] S. Song, A.K. Singh, T.J. Shepodd, B.J. Kirby, Anal. Chem. 76 (2004)
2367.[459] Q. Wang, S. Lin, K.F. Warnick, H.D. Tolley, M.L. Lee, J. Chromatogr. A
985 (2003) 455.[460] S.K. Pedersen, J.L. Harrys, L. Sebastian, J. Baker, M.D. Traini, J.T.
McCarthy, A. Manoharan, M.R. Wilkins, A.A. Gooley, P. Rhighetti, N.H.Parcker, K.L. Williams, B. Herbert, J. Proteome Res. 2 (2003) 303.
[461] P.G. Righetti, A. Castagna, B. Herbert, G. Candiano, Biosci. Rep. 25(2005) 3.
[462] J. Ingvarsson, M. Lindstedt, C.A.K. Borrebaeck, C. Wingren, J. Proteome
Res. 5 (2006) 170.[463] T. Rabilloud, M. Chevallet, T. Rabilloud (Eds.), Proteome Research:Two-Dimensional Electrophoresis and Identification Methods, Springer,Heidelberg, 1999, p. 9.
[464] B. Herbert, Electrophoresis 20 (1999) 660.
. A 1184 (2008) 191–219 219
[465] S. Garbis, G. Lubec, M. Fountoulakis, J. Chromatogr. A 1077 (2005) 1.[466] R.G. Krishna, F. Wold, R.H. Angeletti (Eds.), Proteins: Analysis and
Design, Academic Press, Oxford, 1998 (Chapter 2).[467] B.A. van Montfort, M.K. Doeven, B. Canas, L.M. Veenhoff, B. Poolman,
G.T. Robillard, BBA Bioenergetics 1555 (2002) 111.[468] S. Wang, F.E. Regnier, J. Chromatogr. A 913 (2001) 429.[469] G.W. Slysz, D.F. Lewis, D.C. Schriemer, J. Proteome Res. 5 (2006) 1959.[470] J.L. Luque-Garcıa, G. Zhou, T. Sun, T.A. Neubert, Anal. Chem. 78 (2006)
5102.[471] R.M. Methogo, G. Dufresne-Martin, P. Leclerc, R. Leduc, K. Klarskov,
J. Proteome Res. 4 (2005) 2216.[472] S.P. Radko, H.-T. Chen, S.F. Zakharov, L. Bezrukov, A.L. Yergey, N.E.
Vieira, A. Chrambrach, Electrophoresis 23 (2002) 985.[473] M. Wilm, A. Shevchenko, T. Houthaeve, S. Breit, L. Schweigerer, T.
Fotsis, M. Mann, Nature 379 (1996) 466.[474] C. pan, S.X.H. Zhou, Y. Fu, M. Ye, H. Zou, Anal. Bioanal. Chem. 387
(2007) 193.[475] M.T. Davis, D.C. Stahl, S.A. Hefta, T.D. Lee, Anal. Chem. 67 (1995)
4549.[476] I.A. Papayannopoulos, Mass Spectrom. Rev. 14 (1995) 49.[477] J. Li, T. LeRiche, T.L. Tremblay, C. Wang, E. Bonneil, D.J. Harrison, P.
Thibault, Mol. Cell. Proteomics 1 (2002) 157.[478] D.S. Paterson, T. Rohr, F. Svec, J.M.J. Frechet, Anal. Chem. 75 (2003)
5328.
[479] J.D. Ramsey, S.C. Jacobson, C.T. Culbertson, J.M. Ramsey, Anal. Chem.75 (2003) 3758.[480] B. Canas, C. Pineiro, E. Calvo, D. Lopez-Ferrer, J.M. Gallardo, J. Chro-
matogr. A 1153 (2007) 235.[481] S.L.S. Freire, A.R. Wheeler, Lab Chip 6 (2006) 1415.