resveratrol induces p53-independent, xiap-mediated · pdf fileresveratrol induces...
TRANSCRIPT

1
RESVERATROL INDUCES p53-INDEPENDENT, XIAP-MEDIATED BAX OLIGOMERIZATION ON MITOCHONDRIA TO INITIATE CYTOCHROME C RELEASE AND CASPASE
ACTIVATION Raghu Gogada1, Varun Prabhu1, Michael Amadori, Rachael Scott, Sana Hashmi, and
Dhyan Chandra*
From the Department of Pharmacology and Therapeutics, Roswell Park Cancer Institute, Elm and Carlton Streets, Buffalo, NY 14263
Running head: Resveratrol induces Bax-dependent apoptosis Key words: Resveratrol, apoptosis, mitochondria, cytochrome c, Bax, Bak, XIAP, and p53
1These two authors have contributed equally to this work *Corresponding author: Department of Pharmacology and Therapeutics, Roswell Park Cancer Institute,
Elm and Carlton Streets, Buffalo, NY 14263. Tel: (716) 845-4882; Fax: (716) 845-8857;
email: [email protected]
Resveratrol, a naturally occurring phytoalexin, is known to induce apoptosis in multiple cancer cell types, but the underlying molecular mechanisms remain unclear. Here, we show that resveratrol induces p53-independent, X-linked inhibitor of apoptosis protein (XIAP)-mediated translocation of Bax to mitochondria, where it undergoes oligomerization to initiate apoptosis. Resveratrol treatment promotes interaction between Bax and XIAP in the cytosol and on mitochondria, suggesting that XIAP plays a critical role in the activation and translocation of Bax to mitochondria. This process does not involve p53, but requires accumulation of Bim and t-Bid on mitochondria. Bax primarily undergoes homo-oligomerization on mitochondria and plays a major role in release of cytochrome c to the cytosol. Bak, another key protein that regulates the mitochondrial membrane permeabilization, does not interact with p53 but continues to associate with Bcl-xL. Thus, the pro-apoptotic function of Bak remains suppressed during resveratrol-induced apoptosis. Caspase-9 silencing inhibits resveratrol-induced caspase activation, whereas caspase-8 knockdown does not affect caspase activity, suggesting that resveratrol induces caspase-9-dependent apoptosis. Together, our findings characterize the molecular mechanisms of resveratrol-induced caspase activation, and subsequent apoptosis in cancer cells.
Anticancer agents induce cell death in cancer and normal cells via mechanisms including apoptosis or autophagy (1-4). Therefore, there is a need for alternative anticancer agents that can promote cancer cell death while avoiding killing of normal, non-cancerous cells. Resveratrol (trans-3,5,4’-trihydroxystilbene) is a naturally occurring polyphenolic phytoalexin, found at high levels in the skin of grapes and in red wine. It is also present in peanuts and other plant products. Resveratrol has been shown to possess an apoptosis-dependent anticancer activity and minimal toxicity to normal cells (5-11). How resveratrol induces apoptosis or cancer cell death is not clearly known, but available evidence indicates that resveratrol induces p53-dependent signaling, which leads to cell-cycle arrest and apoptosis induction (10,12,13). Additionally, resveratrol targets mitochondria to induce cytochrome c release, and thereby, triggers caspase-dependent apoptotic cell death in multiple types of cancer cells (14-18). How resveratrol induces cytochrome c release and caspase activation to execute apoptosis remains unclear.
Caspases are activated by proteolytic processing and are broadly divided into initiator caspases (e.g., procaspase-8, and -9); and executioner caspases such as procaspase-3, and -7 (19-22). During apoptosis, the released cytochrome c from mitochondria triggers caspase-9 activation whereas ligation of death receptors on the plasma membrane activates caspase-8. Active caspase-8 generated upon death receptor ligation requires Bid-mediated cytochrome c release to
http://www.jbc.org/cgi/doi/10.1074/jbc.M110.202440The latest version is at JBC Papers in Press. Published on June 28, 2011 as Manuscript M110.202440
Copyright 2011 by The American Society for Biochemistry and Molecular Biology, Inc.
by guest on April 26, 2018
http://ww
w.jbc.org/
Dow
nloaded from

2
execute apoptotic cell death in epithelial cancer cells (22-25). Proapoptotic BH3-only proteins such as activated Bid/Bim translocate to mitochondria to initiate Bax and/or Bak activation leading to the channel formation on the outer mitochondrial membrane (OMM) and permeabilization of mitochondria (22,26,27). Bak localizes on the OMM whereas Bax mostly exist in the cytosol. Although resveratrol has been shown to modulate the levels of Bax/Bak or other Bcl-2 family proteins (14,28), it is unclear whether and how resveratrol activates Bax/Bak to permeabilize the mitochondrial membrane.
Here we demonstrate that resveratrol induces caspase-dependent apoptosis by targeting mitochondria. Our findings indicate that resveratrol may induce Bax oligomerization in the cytosol. Bax activation and its translocation to mitochondria seem to be regulated by XIAP, however, p53 does not directly participate in the activation of Bax/Bak. Bax recruitment to and its oligomerization on mitochondria are associated with cytochrome c release, caspase activation, and apoptosis. These findings suggest that resveratrol may be a novel inducer of Bax-mediated caspase activation and apoptosis in cancer cells, which normally lack p53 activity or harbor mutant p53.
EXPERIMENTAL PROCEDURES
Cells and reagents- Colon cancer cells (HCT116, HCT116-Bax-KO, and HCT-p53-KO) were kindly provided by Dr. B. Vogelstein (29,30) and cultured in McCoy’s 5A media supplemented with 10% FBS. Prostate cancer (PC3 and LNCaP), breast cancer (MDA-MB231, MCF-7, and MDA-MB435), immortalized normal human fibroblast (GM701), Jurkat WT, Jurkat caspase-8-/-, MEFs WT, and MEFs Apaf-1-/- cells were obtained from the ATCC or from various investigators and were subcultured as described previously (31-36). The primary antibodies were against: cytochrome c (mAb, monoclonal antibody), Apaf-1 and Bax (Rb pAb, rabbit polyclonal antibody), Bid, caspase-8, XIAP, and Bcl-xL were purchased from BD Pharmingen. Bax N-terminus or NT (Rb pAb; Upstate), Bak (Rb pAb; Santa Cruz), p53 (Santa Cruz), COX II (Mito Sciences), Hsp60 (Millipore), Hsp70 (Stressgen), Bak NT (Rb pAb; Upstate), VDAC-1 and Bim (Calbiochem),
caspase-3 (Rb pAbs; Biomol), caspase-9 (Cell Signaling), poly (ADP-ribose) polymerase (PARP), lactate dehydrogenase (LDH), and actin (mAb; ICN). Secondary antibodies and ECL reagents were acquired from GE Healthcare. AlexaFluor 594 or 488 conjugated goat anti-mouse or rabbit IgG (H+L) and mitochondrial dye (i.e., MitoTracker Orange, CMTMRos) were purchased from Molecular Probes (Invitrogen). The fluorogenic caspase substrates DEVD-AFC, LEHD-AFC, general caspase inhibitor z-VAD-fmk, and crosslinkers were bought from Enzo Life Sciences. All other chemicals were purchased from Sigma unless specified otherwise.
Subcellular fractionation and Western blotting- The preparation of whole cell lysates, mitochondrial and cytosolic fractions, and Western blotting were preformed as previously detailed (35).
Quantification of apoptosis and caspase activity measurement- Apoptotic cells were counted based on live cell staining with DAPI to label apoptotic nuclei (35). In addition, both live and dead cells were counted using Trypan blue dye. DEVDase and LEHDase activities were measured as previously described (35).
Establishment of cancer cells stably expressing caspase-9 or caspase-8 siRNA using shRNA lentiviral vectors- Green fluorescence protein (GFP)-tagged short hairpin RNAs (shRNAs) specific to caspase-9, caspase-8, and negative control shRNA were cloned into the pGIPZ (Open Biosystems) lentiviral vector to generate lentiviral particles. The shRNA sequences were: caspase-8 (5’-GACTTCAGCAGAAATCTTT-3’), and caspase-9 (5’- CCAGGCAGCTGATCATAGA-3'). Lentiviral particles specific for caspase-9, caspase-8, and control shRNAs were obtained from the Roswell Park Cancer Institute (RPCI) shRNA core resource and were directly utilized to infect cells at a multiplicity of infection (MOI) of 3. After 48 h, puromycin (1 µg/ml) was added to the medium to select caspase-8 or caspase-9 knockdown cells (37).
Immunofluorescence- Cells grown on coverslips were treated with resveratrol and, 15 min prior to the end of treatment, were incubated live with either DAPI alone (to label nucleus) or MitoTracker Orange (CMTMRos) and DAPI (to
by guest on April 26, 2018
http://ww
w.jbc.org/
Dow
nloaded from

3
label mitochondria and nuclei, respectively). Cells were then fixed, permeabilized, and immunolabeled for cytochrome c (32,35).
Chemical crosslinking and oligomerization assays- Freshly harvested cells or freshly purified mitochondria or cytosol (50 µg) were suspended in 45 µl of HIM buffer (200 mM mannitol, 70 mM sucrose, 10 mM HEPES-KOH, 1mM EGTA, pH 7.5) followed by addition of freshly prepared BMH (bismaleimidohexane) or EGS (Ethylene glycolbis[succinimidylsuccinate]) to a final concentration of 2 mM and incubated at room temperature for 30 min. Mitochondria were then mixed with protein sample buffer and subjected to Western blotting (32).
Gel filtration analysis- Various types of cancer cells were treated with resveratrol or vehicle (DMSO), and washed twice in cold phosphate-buffered saline. Cytosolic and mitochondrial fractions were purified as described previously (31,32,34) and were loaded onto a Superdex 200 HR10/30 column (GE Healthcare). Proteins were eluted at 0.5 ml/min and fractions (0.5 ml) were collected for Western analyses (38).
Immunoprecipitation- Purified mitochondrial lysates or cytosols were precleared with mouse or rabbit (depending on the primary antibodies used) IgG-conjugated agarose beads and incubated with primary antibodies against Bax or Bak or p53 or XIAP or Rb-IgG (as control), followed by addition of rabbit or mouse IgG beads. Finally, the beads were washed thoroughly and analyzed by TrueBlot (eBioscience) Western blotting system (34).
Cell-free reconstitution experiments- Purified mitochondria were incubated with cytosol or homogenizing buffer in a total reaction mixture of 50 µl at 37oC for 60 min followed by Western blotting to detect XIAP translocation (36).
Statistical analysis- Results are presented as mean ± standard deviation (SD) of data from at least three independent experiments. Statistical analysis was performed by ANOVA using Sigma Stat. Significant changes (p<0.01) are represented by *.
RESULTS
Resveratrol induces caspase-dependent apoptosis. Resveratrol induces apoptotic cell death
in multiple types of cancer cells, but the molecular mechanism is still unclear. To understand the involvement of apoptosis in resveratrol-induced cell death, we treated MDA-MB231, LNCaP, PC3, and GM701 cells with increasing doses (20-120 µM) of resveratrol and observed a dose-dependent apoptosis and caspase-3 activation (Fig. 1A, data not shown). Caspase-3 is a 34-kDa protein and processed during apoptosis to generate p20, p19, and p17 fragments (39,40). To investigate if cleaved caspase fragments are functionally active, substrate cleavage assay i.e., DEVDase assay, which represents caspase-3/7 activity were performed after treating cells with resveratrol for 4 and 24 h time periods. As shown in Fig. 1B, resveratrol induced 4, 6, 13, and 5 fold increase in caspase-3 activity as compared to DMSO in MDA-MD231, GM701, LNCaP, and PC3 cells, respectively, at 24 h time period. To further demonstrate that apoptosis induced by resveratrol is caspase-dependent, we pretreated MDA-MB231 and GM701 cells with pan caspase inhibitor (z-VAD), and observed that resveratrol-induced caspase-3 processing (i.e., p17) and DEVDase activity was inhibited by z-VAD (Fig. 1C). Similarly, resveratrol-induced cell death was inhibited in the presence of caspase inhibitor (Fig. 1D). Altogether, these findings demonstrate that resveratrol induces caspase-dependent apoptosis.
Resveratrol treatment leads to cytochrome c release from mitochondria. To explore the molecular mechanism of resveratrol-induced caspase-dependent apoptosis, we purified mitochondrial and cytosolic fractions from untreated or resveratrol-treated MDA-MB231 cells, followed by Western blotting to detect cytochrome c release from mitochondria, a critical step to trigger Apaf-1 dependent caspase activation. As shown in Fig. 2A, resveratrol treatment induced a low level of cytochrome c release in the cytosol as early as 12 h in MDA-MB231 cells prior to caspase activation, which happened around 24 h after resveratrol treatment. The levels of mitochondrial cytochrome c did not decrease, which support previous findings that treatment of cells with apoptotic inducers upregulate mitochondrial respiratory chain proteins such as cytochrome c (35,41-44). We did not detect cytochrome c oxidase subunit II (COX II, a marker for mitochondria) in the cytosol, and
by guest on April 26, 2018
http://ww
w.jbc.org/
Dow
nloaded from

4
lactate dehydrogenase (LDH, a marker for cytosol) in mitochondria, indicating that cytosolic fractions were not contaminated with mitochondrial proteins and vice versa. The released cytochrome c was followed by caspase activation as caspase-3 was processed to p20/17 in the cytosol and mitochondria at 24 h and onwards after resveratrol treatment (Fig. 2A). The substrate cleavage assay (i.e., DEVDase) also showed an increase in caspase-3 activity at 24 h onward upon resveratrol treatment (Fig. 1B, and data not shown). Similarly, resveratrol induced cytochrome c release in MDA-MB435, HCT116, and LNCaP cells (see Fig. 4F, 7A, and data not shown).
Cytochrome c is encoded by the nuclear genome and is synthesized in the cytosol. This newly synthesized cytochrome c (i.e., apo-cytochrome c) then translocates to mitochondria, where a heme moiety is attached to generate holo-cytochrome c (i.e., mitochondrial cytochrome c), which participates in the electron transport chain (45-47). Once released from mitochondria, holo-cytochrome c triggers apoptosome-dependent caspase activation (20,35). To further demonstrate that the cytosolic increase in cytochrome c levels was not due to the upregulation of newly synthesized cytochrome c (i.e., apo-cytochrome c), we performed immunolabeling to detect holo-cytochrome c. As shown in Fig. 2B, cytochrome c was mostly detected in mitochondria of control cells in MDA-MB231 and GM701 cells. Resveratrol treatment led to diffused cytochrome c staining suggesting that cytochrome c was released in response to resveratrol treatment. As expected, the released cytochrome c induces apoptosis as evidenced by the fragmentation of nucleus (Fig. 2B, panels c and g). Altogether, resveratrol treatment triggers cytochrome c release in multiple types of cells.
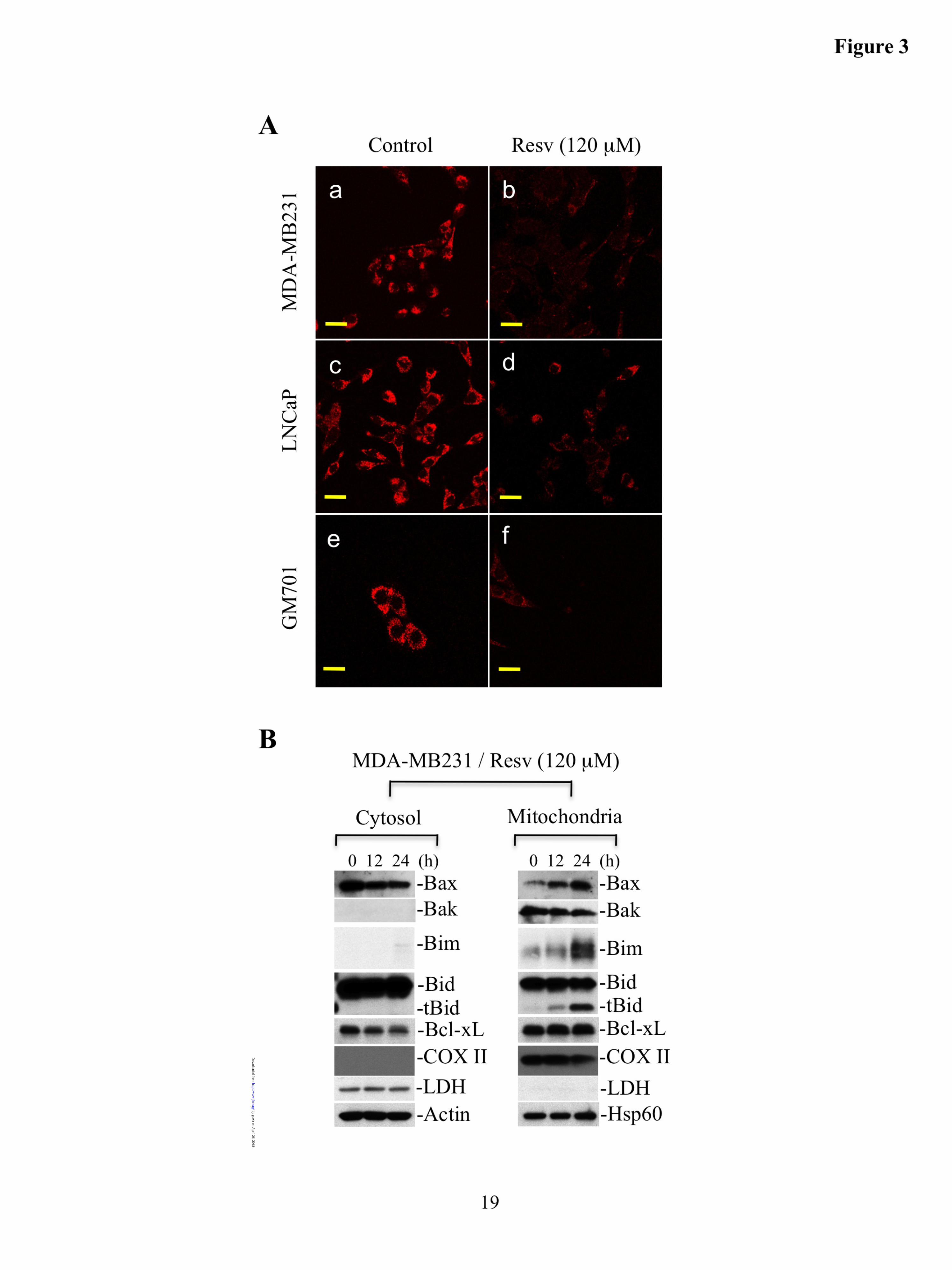
Resveratrol treatment causes loss of mitochondrial membrane potential. To understand the mechanism underlying cytochrome c release from mitochondria in response to resveratrol, we first evaluated the membrane potential of mitochondria by labeling MDA-MB231, LNCaP, and GM701 cells with MitoTracker Orange, which labels mitochondria in a membrane potential-dependent manner. Our data demonstrated that MitoTracker was accumulated in mitochondria of control cells. Upon resveratrol treatment,
MitoTracker was not observed in mitochondria suggesting the loss of membrane potential during apoptosis (Fig. 3A). These findings led us to conclude that resveratrol induces mitochondria dysfunction, which would account for the observed release of cytochrome c from mitochondria.
Resveratrol induces Bax translocation to mitochondria, and accumulation of Bim/t-Bid on mitochondria. Since Bax and/or Bak have been shown to form channels on the OMM during apoptosis (48,49), we investigated the subcellular localization of Bax/Bak by Western blotting using cytosolic and mitochondrial fractions. We found that during resveratrol-induced apoptosis, Bax translocated to mitochondria, whereas the level of Bak was not altered (Fig. 3B). Bax oligomerization on mitochondria is known to require the presence of activated proapoptotic proteins such as Bim and/or Bid (i.e., t-Bid) on mitochondria (48,49). Indeed, we observed accumulation of Bim and t-Bid on mitochondria upon resveratrol treatment. Additionally, the level of antiapoptotic protein, Bcl-xL was not modulated on mitochondria upon resveratrol treatment (Fig. 3B). These findings suggest that resveratrol induces Bim and t-Bid accumulation on mitochondria, contributing to the activation and oligomerization of Bax and/or Bak, and leading to the OMM permeabilization.
Bax oligomerizes on mitochondria upon resveratrol treatment. Translocation of Bax to mitochondria is followed by its oligomerization on the mitochondrial membrane (48,49). To investigate whether Bax undergoes oligomerization to form channels on mitochondria, freshly prepared mitochondria from untreated or resveratrol treated MDA-MB231 cells were incubated with BMH, a noncleavable, membrane-permeable homobifunctional maleimide that covalently and irreversibly cross-links sulfhydryl groups. We observed Bax oligomers on mitochondria (Fig. 4A). Similarly, BMH crosslinking of mitochondria isolated from HCT116 wild type (WT) cells treated with resveratrol also demonstrated Bax oligomerization, whereas Bax oligomers were not detected in HCT116 Bax-/- cells (Fig. 4B).
Bax has been shown to be expressed as
by guest on April 26, 2018
http://ww
w.jbc.org/
Dow
nloaded from

5
multiple isoforms with molecular weights ranging from 19-24 kDa (50-52). Additionally, Bax is cleaved during apoptosis to generate p18, which has also been shown to oligomerize with full length Bax (53). We have also detected multiple bands using Bax antibody by Western blotting upon 24 h of resveratrol treatment (Fig. 4A and B). These findings support that multiple Bax oligomers could be detected by Western analysis, which is consistent with previous findings (54,55).
To further demonstrate that resveratrol induces Bax oligomerization to execute apoptosis, resveratrol-treated or unstimulated MDA-MB231 and HCT116 cells were directly crosslinked with BMH and samples were analyzed by Western blotting. As shown in Fig. 4C and D, expected Bax oligomers (e.g., dimers and multimers) were detected in resveratrol-treated cells. To further confirm that resveratrol induces Bax oligomerization, we crosslinked unstimulated or resveratrol treated MDA-MB231 and HCT116 cells using another crosslinker, EGS. We observed Bax oligomerization mostly as multimers upon resveratrol treatment (Fig. 4C and D). Altogether, we demonstrate that resveratrol induces Bax oligomerization in order to permeabilize the OMM.
Bax oligomerization during resveratrol-induced apoptosis was further demonstrated by gel filtration analysis using MDA-MB231 cells. Bax was mostly eluted as monomers in mitochondria obtained from untreated cells (Fig. 5A, fractions 22-25). Resveratrol-treatment resulted in the elution of Bax (Fig. 5A, fractions 7-11 indicated by arrows) in higher molecular weight protein complexes (~400-700 kDa) in mitochondria.
To determine whether Bak also undergoes oligomerization, mitochondrial lysates were subjected to gel filtration analysis. Bak was eluted (Fig. 5A, fractions, 12-17) as higher molecular weight protein complexes (~158-440 kDa) in both untreated and resveratrol-treated MDA-MB231 cells. This suggests that Bak is already a part of larger protein complexes, which is consistent with our previous findings (32).
Bax forms homo-oligomers and Bak continues to associate with Bcl-xL upon resveratrol treatment. To understand if Bax hetero-oligomerizes with Bak to form Bax-Bak channel,
co-immunoprecipitation (co-IP) of Bax was performed in freshly isolated mitochondria of MDA-MB231 cells treated with resveratrol (120 µM for 36 hours). We observed very low levels of Bak by Western blotting, suggesting that Bax primarily undergoes homo-oligomerization to form Bax channels (Fig 4E, lane 5). Reciprocal IP with Bak in mitochondria of resveratrol-treated MDA-MB231 cells also did not significantly precipitate Bax (Fig. 4E, lane 6). Interestingly, Bcl-xL was detected upon co-IP using Bak but not with Bax, suggesting that proapoptotic functions of Bak may be inhibited by Bcl-xL (Fig. 4E, lanes 5 and 6). As expected, both Bax and Bak did not interact with each other under unstimulated conditions (Fig. 4E, lanes 1 and 2). Co-IP experiments indicated Bak sequesteration by Bcl-xL in unstimulated cells (Fig. 4E, lane 2). These findings suggest that resveratrol induces the formation of Bax channels, however, a very small amount of Bax and Bak may also hetero-oligomerize to form Bax-Bak channel on mitochondria.
Since Bak continues to associate with Bcl-xL during resveratrol-induced apoptosis on mitochondria, we examined if Bax oligomerization is required for cytochrome c release from mitochondria. To accomplish this, we treated MDA-MB231 and MDA-MB435 cells with lower dose (50 µM) of resveratrol. We observed that at 18 h treatment, a low level of cytochrome c release in MDA-MB231 cells is accompanied by slight Bax oligomerization (Fig. 4F). In MDA-MB435 cells, Bax oligomerization was observed with concomitant release of cytochrome c upon treatment with lower dose (50 µM) of resveratrol (Fig. 4F).
Bax translocation to and its oligomerization on mitochondria do not involve p53. Having established that Bax translocates to mitochondria, we next asked question how this is accomplished. Does Bax translocation to mitochondria require association with some cytosolic or mitochondrial proteins such as p53 (56) in addition to Bim or Bid? To investigate this possibility, we examined Bax co-elution with p53 by gel filtration analysis. As shown in Fig. 5B, p53 (fractions 17-21) and Bax (fractions 22-26) did not co-elute in the fractionated cytosols obtained from unstimulated or resveratrol-treated MDA-MB231 cells. To
by guest on April 26, 2018
http://ww
w.jbc.org/
Dow
nloaded from

6
further determine the interaction of p53 with Bax, we performed co-IP with p53 using cytosol obtained from MDA-MB231 cells treated with resveratrol, and observed that Bax was not precipitated with p53 co-IP (Fig. 5C, lane 3). Reciprocal IP using Bax antibody, which detects only activated Bax, as expected, a very little amount of Bax was precipitated without significant pull down of p53 despite abundant amounts of p53 in the cytosol (Fig. 5C, lane 2).
To understand if mitochondrially-localized Bax exists in p53-containing complexes, we performed gel filtration analysis using mitochondrial lysates obtained from untreated or resveratrol-treated MDA-MB231 cells. We observed that Bax was oligomerized and eluted in higher molecular protein complexes (Fig. 5A, fractions 7-11 indicated by arrows), which did not co-elute with p53 (Fig. 5A, fractions 11-16). These findings were further supported by co-IP experiments utilizing mitochondria isolated from untreated or resveratrol-treated MDA-MB231 cells. As shown in Fig. 4E (lanes 3 and 7), we did not observe Bax interaction with p53. These findings suggest that Bax and p53 interaction may not be essential for Bax recruitment and oligomerization on mitochondria in response to resveratrol treatment.
To further demonstrate that Bax oligomerization is not modulated by p53, we treated MDA-MB231 cells with pifithrin (PFT), a pharmacological inhibitor of p53, followed by crosslinking. Cyclic pifithrin-α (PFT-α) inhibits transactivation of p53 (57), whereas pifithrin-µ (PFT-µ) inhibits translocation of p53 to mitochondria (58). We observed that resveratrol-induced Bax oligomerization was not modulated by pretreatment of MDA-MB231 cells with PFT-α (Fig. 5D). Similarly, resveratrol-induced Bax oligomerization was not altered by pretreatment of MDA-MB435 cells with PFT-α or PFT-µ (Fig. 5E, and data not shown).
Bak does not interact with p53 on mitochondria during resveratrol-induced apoptosis. To investigate if an association between p53 and Bak plays a role in channel formation or if p53 activates Bak (59), gel filtration analysis using mitochondrial lysates was performed. We observed that Bak and p53 were co-eluted upon
fractionation of mitochondria obtained from untreated and resveratrol-treated MDA-MB231 cells (Fig. 5A, fractions 13-17). To examine whether p53 interacts with Bak to form p53-mediated Bak channels on mitochondria, we performed co-IP experiment using either Bak or p53. We observed that p53 did not interact with Bak suggesting that p53 may not play critical role in Bak oligomerization and channel formation to induce cytochrome c release (Fig. 4E, lanes 2, 3, 6, and 7). Similarly, gel filtration and IP analyses demonstrated that p53 did not interact with Bak during resveratrol-induced apoptosis in HCT116 and LNCaP cells (data not shown). Altogether, our findings demonstrate that p53 does not interact with Bak to permeabilize the mitochondrial membrane.
Resveratrol induces Bax oligomerization in the cytosol. BH3 only proteins such as Bid play an important role in the activation and recruitment of Bax on the OMM. How Bax translocates to mitochondria during apoptosis induction is not completely defined. However, current evidence suggests that the integration of cleaved Bid (t-Bid) on mitochondria recruits monomeric Bax, which undergoes oligomerization on the OMM to form the Bax channels (60-62). To investigate how Bax translocates to mitochondria during resveratrol-induced apoptosis, we crosslinked the cytosol obtained from untreated or resveratrol-treated MDA-MB231 cells. Surprisingly, we observed Bax oligomers in the cytosol as early as 12 hour after resveratrol treatment (Fig. 6A). Gel filtration analyses demonstrated Bax elution in fractions 22-26, which represents molecular weight in the range of 14-43 kDa suggesting that a small amount of Bax may exist as dimers in the unstimulated cytosols. Upon resveratrol treatment, Bax was eluted in fractions 22-25 suggesting that some amounts of Bax might have been dimerized (Fig. 5B). Similar to MDA-MB231 cells, increased Bax oligomers were also observed in the cytosol of HCT116 cells treated with resveratrol (Fig. 6B). Altogether, our findings suggest that resveratrol induces Bax activation in the cytosol, which translocates to the mitochondrial membrane to undergo a higher order of oligomerization to form the Bax channels.
XIAP regulates Bax activation and translocation to mitochondria. Since Bax does not
by guest on April 26, 2018
http://ww
w.jbc.org/
Dow
nloaded from

7
interact with p53, we asked how Bax is activated in the cytosol and how it is translocated to mitochondria. Recent findings demonstrate that XIAP translocates to mitochondria and promotes Bax-dependent permeabilization of the mitochondrial membrane (63). To determine whether XIAP translocates to mitochondria during resveratrol-induced apoptosis, we determined the levels of XIAP by Western blotting upon resveratrol treatment. As shown in Fig. 6C, XIAP translocation to mitochondria is also accompanied by a higher amount of Bax on mitochondria. To further demonstrate that XIAP may associate with Bax to promote Bax translocation to mitochondria during resveratrol-induced apoptosis, we performed reconstitution experiments using purified mitochondria and cytosol. We observed XIAP translocation to mitochondria when resveratrol-treated cytosol and mitochrondria were used in the reconstitution experiments (Fig. 6D). To determine XIAP interaction with Bax, we performed co-IP experiments using Bax or XIAP in mitochondria from MDA-MB231 cells treated with resveratrol (50 µM for 24h). Our findings clearly demonstrated that XIAP interacts with Bax as XIAP was co-precipitated with Bax IP (Fig. 6E, lane 1). However, reciprocal IP using XIAP could not precipitate Bax (Fig. 6E, lane 2) suggesting that Bax might stably associate with XIAP. This Bax-XIAP complex could not be precipitated by XIAP antibody. If XIAP interacts with Bax in mitochondria, it is possible that XIAP may associate with Bax in the cytosol to help translocate Bax to mitochondria. Indeed, we observed XIAP precipitation by Bax IP in the cytosol (Fig. 6F, lane 1) obtained from resveratrol (50 µM for 24 h) treated MDA-MB231 cells suggesting that XIAP associates with Bax to promote its translocation to mitochondria. Additionally, similar to a higher dose of resveratrol (i.e., in Fig. 4E and 5C), low dose of resveratrol further demonstrated that p53 does not interact with Bax in the cytosol or in mitochondria (Fig. 6E and F).
Bax plays a prominent role in resveratrol-induced cytochrome c release and caspase activation. Since Bak is sequestered by VDAC-2 (32,64) leading to the inhibition of its proapoptotic function, Bax translocation and oligomerization might play a dominant role to induce cytochrome c
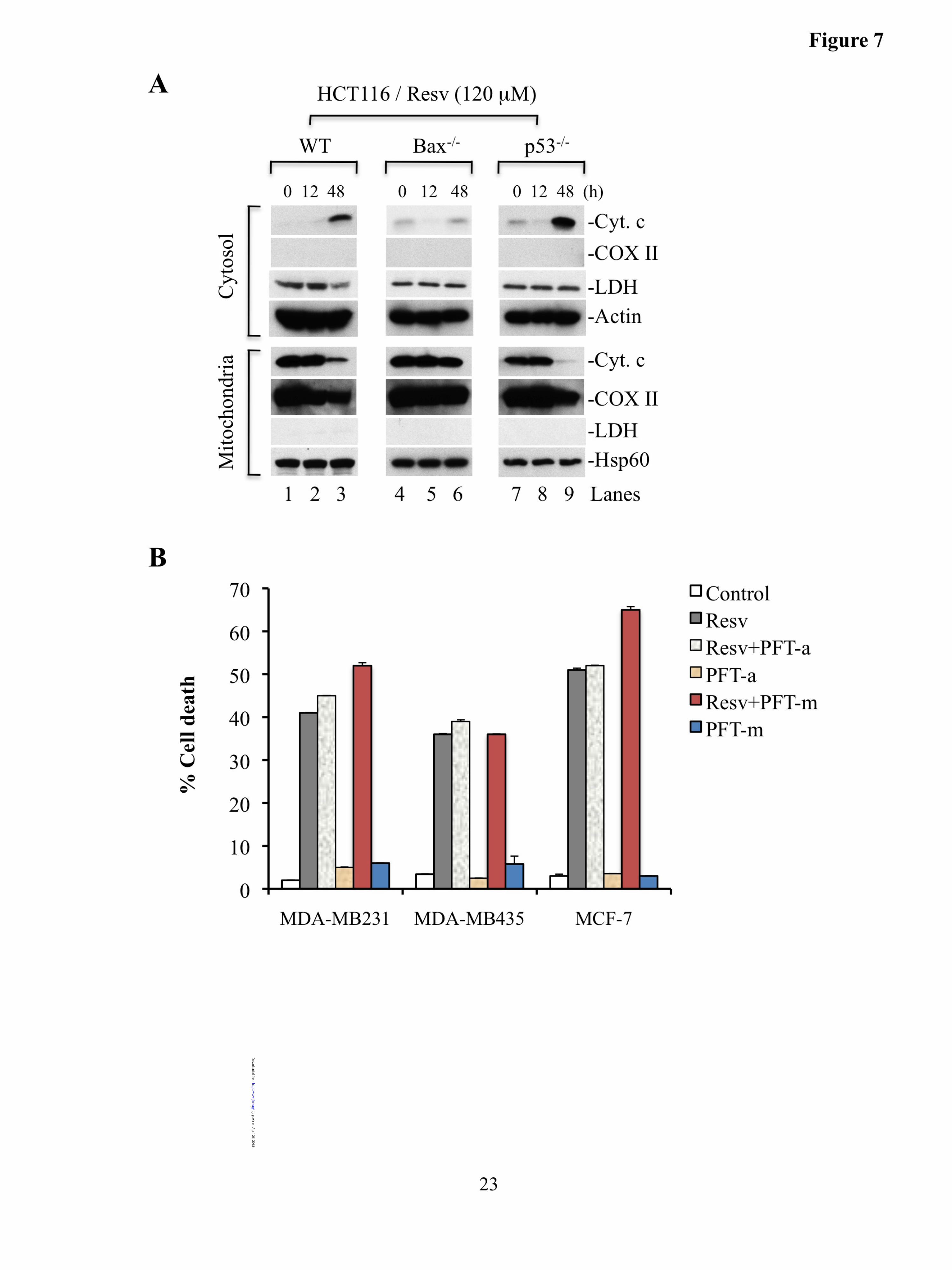
release. Additionally, Bak continues to associate with Bcl-xL on mitochondria during resveratrol-induced apoptosis (Fig. 4E). We have used MDA-MB231, HCT116, and LNCaP cells to study the molecular mechanisms of cytochrome c release from mitochondria and have observed similar results with all three types of cells. Therefore, to study the importance of Bax or p53 on the cytochrome c release, we utilized HCT116 isogenic cell lines deficient for Bax or p53. We treated HCT116 WT and Bax-deficient HCT116 cells with resveratrol and observed that cytochrome c release was significantly inhibited in Bax-deficient cells (Fig. 7A, lanes 3 and 6). Since our data support that p53 does not directly associate with Bax or Bak, the proapoptotic functions of p53 should be inhibited. Thus, we evaluated whether p53 plays a role in the outer membrane permeabilization. Cytosolic and mitochondrial fractions obtained from p53-deficient cells treated with resveratrol or DMSO were subjected to Western blotting. We observed that p53-deficiency did not inhibit cytochrome c release (Fig. 7A, lane 9), whereas cytochrome c release was blocked in Bax-deficient cells suggesting that Bax plays a critical role in resveratrol-induced cytochrome c release, and thus, apoptosis in a p53-independent manner.
If p53 does not regulate cytochrome c release from mitochondria in multiple types of cancer cells upon resveratrol treatment, then inhibition of p53 function using pharmacologic inhibitors should not affect the levels of resveratrol-induced apoptosis. Indeed, we observed that pretreatment of MDA-MB231, MDA-MB435, and MCF-7 cells with PFT-α or PFT-µ did not alter resveratrol-induced apoptosis (Fig. 7B).
Resveratrol induces caspase-9-mediated apoptotic cell death. The experiments described above clearly suggest that resveratrol treatment leads to Bax-dependent cytochrome c release from mitochondria, which activates caspase-9 with subsequent activation of caspase-3 and execution of apoptosis. We first determined whether caspase-9 is required for resveratrol-induced apoptosis. To accomplish this, we silenced caspase-9 in LNCaP (expressing wild type p53) cells using shRNA (Fig. 8A). These caspase-9 silenced stable cells were then treated with resveratrol for 24 hour. We observed that silencing
by guest on April 26, 2018
http://ww
w.jbc.org/
Dow
nloaded from

8
of caspase-9 inhibited cell death in LNCaP cells (data not shown). To further validate that activation of caspase-9 is required for resveratrol-induced cell death, we treated caspase-9 silenced cells with resveratrol to measure caspase-3 (DEVDase) activity. As shown in Fig. 8B, resveratrol-induced caspase-3 activity in control LNCaP cells, whereas in caspase-9 silenced LNCaP cells, caspase-3 activity was inhibited. In contrast, caspase-8 silenced LNCaP cells (Fig. 8B) showed enhanced caspase-3 activity similar to control LNCaP cells upon resveratrol treatment. Similarly, resveratrol-induced caspase activation was inhibited in caspase-9 silenced MDA-MB231 cells (expressing mutant p53) but not in caspase-8 silenced MDA-MB231 cells (Fig. 8C and D). Altogether, these findings demonstrate that resveratrol induces the intrinsic pathway to trigger caspase-9 dependent cell death in cancer cells.
DISCUSSION
A majority of epithelial cancer cells become resistant to known anticancer agents, making it necessary to apply such drugs at higher doses or to combine different anticancer drugs. These approaches are often associated with severe side effects in cancer patients. Therefore, anticancer agents that are minimally toxic to normal cells are highly sought for use in cancer therapy. Resveratrol has been shown to selectively kill cancer cells or possess anticancer properties, but how resveratrol induces apoptotic cell death is not clearly defined. Here we show that the mitochondrion is a critical target organelle for resveratrol-induced apoptosis in epithelial cancer cells. The induction of apoptosis is initiated by caspase-9 activation. The salient features of resveratrol-induced apoptosis are as follows: resveratrol induces mitochondrial dysfunction leading to the loss of mitochondrial membrane potential and cytochrome c release. The release of cytochrome c from mitochondria is dependent on Bax. Translocation and oligomerization of Bax on mitochondria are not facilitated by p53. Similarly, p53 does not interact with Bak on mitochondria during resveratrol-induced apoptosis. Most importantly, resveratrol induces Bax oligomerization in the cytosol, and XIAP interacts with Bax in the cytosol and on mitochondria
suggesting that XIAP regulates Bax-mediated release of cytochrome c from mitochondria. The oligomerization of Bax on mitochondria coincides with the release of mitochondrial cytochrome c, which triggers caspase-9 dependent apoptosis. These findings suggest that resveratrol activates Bax in a p53-independent mechanism. Furthermore, Bax recruitment and a high level of oligomerization to form Bax channels on the OMM are mediated by Bim/Bid-dependent mechanisms to induce cytochrome c release, caspase activation, and apoptosis.
We have demonstrated that resveratrol induces caspase-dependent apoptosis as evidenced by the presence of catalytically active caspase-3. Caspase activity and cell death were inhibited by pre-treatment of cells with the pan caspase-inhibitor (i.e., z-VAD) further demonstrating caspase-dependent apoptosis upon resveratrol treatment. Since we did not observe a release of AIF or Endo-G from the mitochondrial compartment (Prabhu et al., unpublished data), caspase-independent cell death does not seem to play a critical role in initiating cancer cell death upon resveratrol treatment. However, at later stages of apoptosis, the release of mitochondrial contents including AIF or Endo-G could amplify the process of apoptotic cell death.
How is the caspase cascade initiated? We have provided the comprehensive evidence that resveratrol induces caspase-9-dependent but caspase-8-independent apoptosis. This is supported by the fact that in the absence of Apaf-1 (by utilizing Apaf-1-/- MEFs, data not shown) or caspase-9 (silencing of caspase-9), caspase-3 activation was inhibited, whereas this was not the case in caspase-8-deficient Jurkat cells (data not shown) or caspase-8 silenced LNCaP and MDA-MB231 cells. Similarly, Western blot analysis clearly demonstrated caspase-9 activation but not caspase-8 activation in MDA-MB231 cells suggesting that caspase-9 is the initiator caspase for caspase-dependent apoptosis in response to resveratrol in cancer cells (data not shown). Our results are consistent with previous findings that resveratrol induces mitochondria-dependent but death-receptor independent apoptosis (65,66). Since resveratrol primarily targets mitochondria to induce apoptosis, resveratrol could be used as a synergistic agent to enhance death-receptor
by guest on April 26, 2018
http://ww
w.jbc.org/
Dow
nloaded from

9
mediated cancer cell death. Indeed, elegant works of other colleagues have demonstrated that resveratrol enhances TRAIL-induced apoptosis in cancer cells (67-70).
Since activation of caspase-9 requires cytochrome c release from mitochondria, resveratrol should permeabilize mitochondria to induce cytochrome c release. Indeed, we observed robust cytochrome c release from mitochondria into the cytosol upon resveratrol treatment in multiple types of cells. How is cytochrome c released? The release of cytochrome c from mitochondria requires multiple mechanisms such as Bax/Bak activation, a loss of mitochondrial membrane potential, and permeability transition pores (19,26,49). Our data demonstrate that Bax activation and translocation to mitochondria seems to play a prominent role in cytochrome c release. Active Bax on mitochondria undergoes oligomerization to form primarily Bax homo-oligomers. Although Bak exists in higher molecular protein complexes on mitochondria, Bax-deficient HCT116 cells (which still contain Bak) do not release cytochrome c from mitochondria. These data support previous observations that Bak is sequestered by VDAC-2 in the presence of Bax. Bak does not play a significant role in inducing cytochrome c release even if Bak is activated or oligomerized, as activated Bak remains in a complex with VDAC-2 (32,64) and Bak continues to associate with Bcl-xL (Fig. 4E). We did not observe VDAC-1 homo-oligomers, suggesting that resveratrol does not induce the formation of VDAC only channels (data not shown). The permeability transition pore (PTP) mechanism requires rupturing of the OMM leading to the release of proteins localized in the intermembrane space. However, we observed that AIF, and endonuclease G were not released from mitochondria suggesting that PTP may not be involved in resveratrol-induced cytochrome c release and apoptosis.
How is Bax translocated to mitochondria, and how is it activated? We observed a novel mechanism of Bax activation. For example, Bax is normally activated in a t-Bid-dependent manner in order to translocate to and oligomerize on mitochondria (22,23,48,49,60,61). We observed the presence of full-length Bid and t-Bid on mitochondria suggesting that resveratrol induces t-
Bid accumulation on mitochondria. Another direct activator of Bax could be activation/upregulation of Bim (48,49), which accumulates on mitochondria to participate in the activation/oligomerization of Bax leading to the formation of Bax-dependent channels on mitochondria. More importantly, we have demonstrated that XIAP interacts with Bax in the cytosol and on mitochondria suggesting that XIAP regulates activation/translocation of Bax to mitochondria. XIAP generally performs prosurvival functions by inhibiting caspase activation (71), however, recent study demonstrates that XIAP translocates to mitochondria and promotes Bax-dependent permeabilization of the mitochondrial membrane (63). Our findings further demonstrate the proapoptotic function of XIAP in resveratrol-induced apoptosis.
Is Bax only activated on the OMM? We provide the first evidence that Bax could be activated in the cytosol upon resveratrol treatment. How this could be achieved is still under investigation, but available data support that Bax could be activated by XIAP or by BH3-only proteins (Bim and Bid) in the cytosol. We observed that XIAP associates with Bax in the cytosol and on mitochondria, and translocation of XIAP to mitochondria is accompanied by increased level of Bax. These findings suggest that XIAP may regulate activation/translocation of Bax to mitochondria in order to promote Bax-dependent permeabilization of the mitochondria membrane. Since only transient interaction between BH3-only proteins and Bax is required for Bax activation (72), it is quite possible that small amounts of activated Bim or t-Bid or any other BH3-only proteins in the cytosolic compartment may transiently interact with the cytosolic Bax leading to its activation in the cytosol. Activated cytosolic Bax then undergoes oligomerization, mostly dimerization, which then translocates to mitochondria to undergo a higher order of oligomerization in a t-Bid/Bim-dependent mechanism or through association with XIAP to form Bax channels. How transient interaction of Bim or Bid with Bax induces Bax oligomerization is under investigation but dimerization of Bax in the cytosol may be explained by a recent model by Andrews and colleagues (60,61). For example,
by guest on April 26, 2018
http://ww
w.jbc.org/
Dow
nloaded from

10
BH3 activated domain of Bax by Bid or Bim can interact with another activated Bax in the cytosol to form Bax-dimer, which may then be recruited by mitochondrially-localized Bid or Bim. Subsequently, the recruited Bax-dimer may undergo a higher order of oligomerization to form Bax channels on the OMM in a t-Bid dependent manner. Since intracellular glutathione (GSH) depletion promotes Bax translocation to mitochondria (73,74), it is possible that GSH might interfere with Bax activation and oligomerization in the cytosol and its subsequent translocation to mitochondria. During resveratrol-induced apoptosis, depletion of GSH may facilitate Bax oligomerization in the cytosol leading to Bax channel formation on the mitochondrial membrane.
Bax could also be activated by cytosolic p53 (56), but gel filtration and immunoprecipitation analysis demonstrated that Bax does not associate with p53 upon resveratrol treatment suggesting that p53 is not critical for resveratrol-induced outer membrane permeabilization. These findings are consistent with the earlier report that resveratrol induces p53-independent apoptosis in cancer cells (65,67,73-76). Although Bak can be activated by p53 (59), we did not observe p53 involvement in Bak activation. It has also been reported that p53 could also suppress the anti-apoptotic function of Bcl-2/Bcl-xL/Mcl-1 (77-79), and thus, may cause an increase in cytochrome c release and apoptosis. Since the level of Bcl-2 is very low in MDA-MB231 cells, we tested whether levels of Bcl-xL were modified during resveratrol-induced cell death. We observed that the level of Bcl-xL was not modified during resveratrol-induced apoptosis (Fig. 3B). Additionally, we did not observe Bcl-xL interaction with p53 on mitochondria further supporting that p53 does not play a critical role in cytochrome c release upon resveratrol treatment. We are further investigating whether resveratrol modifies the anti-apoptotic function of Bcl-2/Bcl-xL/Mcl-1 through p53, but available evidence indicates that resveratrol induces caspase activation and apoptosis both in LNCaP (expressing wild type p53) and MDA-MB231 (expressing mutant p53) cells. Bax-deficient cells showed significantly reduced levels
of cytochrome c release, whereas p53-deficient cells showed equal or even slightly higher levels of cytochrome c release upon resveratrol treatment (Fig. 7A). These findings support that p53 does not interact with Bax/Bak, and that, p53-Bax or p53-Bak interaction may not be critical for resveratrol-induced cytochrome c release in cancer cells. It has been reported that wild type p53 as well as mutant p53 can promote Bax activation on mitochondria (80,81), whereas other studies suggest that p53 translocation to mitochondria does not induce Bax activation and apoptosis (82,83). Expression of mutant p53 in cancer cells confers selective advantage and resistant to apoptosis (84-86). Our findings suggest that resveratrol induces cytochrome c release, caspase activation, and apoptosis in cancer cells such as in MDA-MB231 and MDA-MB435 cells that harbor mutant p53.
Altogether, our findings provide comprehensive evidence that resveratrol induces Bax-dependent but p53-independent apoptosis in epithelial cancer cells, and further analysis on how resveratrol induces Bax activation may lead to the foundation of resveratrol-based anticancer agents. It is generally believed that for chemoprevention or cancer therapy, doses of resveratrol used to induce apoptotic cell death may not easily be achieved under physiological conditions through regular diets. However, various lines of evidence indicate that in vitro doses that have been shown to induce apoptotic cell death could be achieved in cancer cells through higher intake of resveratrol (87-91). For example, resveratrol at daily doses of up to 5 g for 29 days is not toxic to humans, and daily doses of 0.5 or 1 g produce levels of resveratrol in tumor cells that are sufficient to elicit anticancer effects such as induction of apoptosis in cancer cells (88). Additionally, further research on resveratrol modifications may increase the bioavailability of resveratrol in cancer tissues. Since we have demonstrated that resveratrol induces Bax activation in the cytosol and that XIAP interacts with Bax, further analysis on how to enhance Bax oligomerization on mitochondria in a p53-independent manner may provide a novel approach to enhance apoptotic cell death with lower doses of resveratrol.
by guest on April 26, 2018
http://ww
w.jbc.org/
Dow
nloaded from

11
REFERENCES
1. Mathew, R., and White, E. (2011) Curr Opin Genet Dev 21, 113-119 2. Zois, C. E., and Koukourakis, M. I. (2009) Autophagy 5, 442-450 3. Blagosklonny, M. V. (2002) Int J Cancer 98, 161-166 4. Lowe, S. W., and Lin, A. W. (2000) Carcinogenesis 21, 485-495 5. Delmas, D., Rebe, C., Lacour, S., Filomenko, R., Athias, A., Gambert, P., Cherkaoui-Malki, M.,
Jannin, B., Dubrez-Daloz, L., Latruffe, N., and Solary, E. (2003) J Biol Chem 278, 41482-41490 6. Fulda, S., and Debatin, K. M. (2004) Cancer Res 64, 337-346 7. Gill, C., Walsh, S. E., Morrissey, C., Fitzpatrick, J. M., and Watson, R. W. (2007) Prostate 67,
1641-1653 8. Niles, R. M., McFarland, M., Weimer, M. B., Redkar, A., Fu, Y. M., and Meadows, G. G. (2003)
Cancer Lett 190, 157-163 9. Pervaiz, S. (2003) FASEB J 17, 1975-1985 10. She, Q. B., Bode, A. M., Ma, W. Y., Chen, N. Y., and Dong, Z. (2001) Cancer Res 61, 1604-
1610 11. Estrov, Z., Shishodia, S., Faderl, S., Harris, D., Van, Q., Kantarjian, H. M., Talpaz, M., and
Aggarwal, B. B. (2003) Blood 102, 987-995 12. Joe, A. K., Liu, H., Suzui, M., Vural, M. E., Xiao, D., and Weinstein, I. B. (2002) Clin Cancer
Res 8, 893-903 13. Hsieh, T. C., Juan, G., Darzynkiewicz, Z., and Wu, J. M. (1999) Cancer Res 59, 2596-2601 14. Mohan, J., Gandhi, A. A., Bhavya, B. C., Rashmi, R., Karunagaran, D., Indu, R., and
Santhoshkumar, T. R. (2006) J Biol Chem 281, 17599-17611 15. Sareen, D., Darjatmoko, S. R., Albert, D. M., and Polans, A. S. (2007) Mol Pharmacol 72, 1466-
1475 16. Tinhofer, I., Bernhard, D., Senfter, M., Anether, G., Loeffler, M., Kroemer, G., Kofler, R.,
Csordas, A., and Greil, R. (2001) FASEB J 15, 1613-1615 17. van Ginkel, P. R., Sareen, D., Subramanian, L., Walker, Q., Darjatmoko, S. R., Lindstrom, M. J.,
Kulkarni, A., Albert, D. M., and Polans, A. S. (2007) Clin Cancer Res 13, 5162-5169 18. Huang, T. T., Lin, H. C., Chen, C. C., Lu, C. C., Wei, C. F., Wu, T. S., Liu, F. G., and Lai, H. C.
(2011) J Cell Physiol 226, 720-728 19. Boatright, K. M., Renatus, M., Scott, F. L., Sperandio, S., Shin, H., Pedersen, I. M., Ricci, J. E.,
Edris, W. A., Sutherlin, D. P., Green, D. R., and Salvesen, G. S. (2003) Mol Cell 11, 529-541 20. Jiang, X., and Wang, X. (2004) Annu Rev Biochem 73, 87-106 21. Salvesen, G. S., and Dixit, V. M. (1997) Cell 91, 443-446 22. Shi, Y. (2004) Cell 117, 855-858 23. Ashkenazi, A., and Dixit, V. M. (1998) Science 281, 1305-1308 24. Carrington, P. E., Sandu, C., Wei, Y., Hill, J. M., Morisawa, G., Huang, T., Gavathiotis, E., and
Werner, M. H. (2006) Mol Cell 22, 599-610 25. Scaffidi, C., Fulda, S., Srinivasan, A., Friesen, C., Li, F., Tomaselli, K. J., Debatin, K. M.,
Krammer, P. H., and Peter, M. E. (1998) EMBO J 17, 1675-1687 26. Garrido, C., Galluzzi, L., Brunet, M., Puig, P. E., Didelot, C., and Kroemer, G. (2006) Cell Death
Differ 13, 1423-1433 27. Kuwana, T., Mackey, M. R., Perkins, G., Ellisman, M. H., Latterich, M., Schneiter, R., Green, D.
R., and Newmeyer, D. D. (2002) Cell 111, 331-342 28. Aziz, M. H., Nihal, M., Fu, V. X., Jarrard, D. F., and Ahmad, N. (2006) Mol Cancer Ther 5,
1335-1341 29. Zhang, L., Yu, J., Park, B. H., Kinzler, K. W., and Vogelstein, B. (2000) Science 290, 989-992 30. Bunz, F., Dutriaux, A., Lengauer, C., Waldman, T., Zhou, S., Brown, J. P., Sedivy, J. M.,
Kinzler, K. W., and Vogelstein, B. (1998) Science 282, 1497-1501
by guest on April 26, 2018
http://ww
w.jbc.org/
Dow
nloaded from

12
31. Chandra, D., Bratton, S. B., Person, M. D., Tian, Y., Martin, A. G., Ayres, M., Fearnhead, H. O., Gandhi, V., and Tang, D. G. (2006) Cell 125, 1333-1346
32. Chandra, D., Choy, G., Daniel, P. T., and Tang, D. G. (2005) J Biol Chem 280, 19051-19061 33. Chandra, D., Choy, G., Deng, X., Bhatia, B., Daniel, P., and Tang, D. G. (2004) Mol Cell Biol 24,
6592-6607 34. Chandra, D., Choy, G., and Tang, D. G. (2007) J Biol Chem 282, 31289-31301 35. Chandra, D., Liu, J. W., and Tang, D. G. (2002) J Biol Chem 277, 50842-50854 36. Chandra, D., and Tang, D. G. (2003) J Biol Chem 278, 17408-17420 37. Jeter, C. R., Badeaux, M., Choy, G., Chandra, D., Patrawala, L., Liu, C., Calhoun-Davis, T.,
Zaehres, H., Daley, G. Q., and Tang, D. G. (2009) Stem Cells 27, 993-1005 38. Malladi, S., Challa-Malladi, M., Fearnhead, H. O., and Bratton, S. B. (2009) EMBO J 28, 1916-
1925 39. Fernandes-Alnemri, T., Armstrong, R. C., Krebs, J., Srinivasula, S. M., Wang, L., Bullrich, F.,
Fritz, L. C., Trapani, J. A., Tomaselli, K. J., Litwack, G., and Alnemri, E. S. (1996) Proc Natl Acad Sci U S A 93, 7464-7469
40. Nicholson, D. W., Ali, A., Thornberry, N. A., Vaillancourt, J. P., Ding, C. K., Gallant, M., Gareau, Y., Griffin, P. R., Labelle, M., Lazebnik, Y. A., and et al. (1995) Nature 376, 37-43
41. Heerdt, B. G., Houston, M. A., and Augenlicht, L. H. (1997) Cell Growth Differ 8, 523-532 42. Joshi, B., Li, L., Taffe, B. G., Zhu, Z., Wahl, S., Tian, H., Ben-Josef, E., Taylor, J. D., Porter, A.
T., and Tang, D. G. (1999) Cancer Res 59, 4343-4355 43. Sanchez-Alcazar, J. A., Ault, J. G., Khodjakov, A., and Schneider, E. (2000) Cell Death Differ 7,
1090-1100 44. Sanchez-Alcazar, J. A., Khodjakov, A., and Schneider, E. (2001) Cancer Res 61, 1038-1044 45. Poyton, R. O., and McEwen, J. E. (1996) Annu Rev Biochem 65, 563-607 46. Stuart, R. A., and Neupert, W. (1990) Biochimie 72, 115-121 47. Stuart, R. A., Nicholson, D. W., and Neupert, W. (1990) Cell 60, 31-43 48. Danial, N. N. (2007) Clin Cancer Res 13, 7254-7263 49. Brunelle, J. K., and Letai, A. (2009) J Cell Sci 122, 437-441 50. Oltvai, Z. N., Milliman, C. L., and Korsmeyer, S. J. (1993) Cell 74, 609-619 51. Cartron, P. F., Oliver, L., Martin, S., Moreau, C., LeCabellec, M. T., Jezequel, P., Meflah, K., and
Vallette, F. M. (2002) Hum Mol Genet 11, 675-687 52. Antonsson, B., Montessuit, S., Sanchez, B., and Martinou, J. C. (2001) J Biol Chem 276, 11615-
11623 53. Cao, X., Deng, X., and May, W. S. (2003) Blood 102, 2605-2614 54. Zhang, H., Kim, J. K., Edwards, C. A., Xu, Z., Taichman, R., and Wang, C. Y. (2005) Nat Cell
Biol 7, 909-915 55. Wolff, S., Erster, S., Palacios, G., and Moll, U. M. (2008) Cell Res 18, 733-744 56. Chipuk, J. E., Kuwana, T., Bouchier-Hayes, L., Droin, N. M., Newmeyer, D. D., Schuler, M., and
Green, D. R. (2004) Science 303, 1010-1014 57. Komarov, P. G., Komarova, E. A., Kondratov, R. V., Christov-Tselkov, K., Coon, J. S., Chernov,
M. V., and Gudkov, A. V. (1999) Science 285, 1733-1737 58. Strom, E., Sathe, S., Komarov, P. G., Chernova, O. B., Pavlovska, I., Shyshynova, I., Bosykh, D.
A., Burdelya, L. G., Macklis, R. M., Skaliter, R., Komarova, E. A., and Gudkov, A. V. (2006) Nat Chem Biol 2, 474-479
59. Pietsch, E. C., Perchiniak, E., Canutescu, A. A., Wang, G., Dunbrack, R. L., and Murphy, M. E. (2008) J Biol Chem 283, 21294-21304
60. Zhang, Z., Zhu, W., Lapolla, S. M., Miao, Y., Shao, Y., Falcone, M., Boreham, D., McFarlane, N., Ding, J., Johnson, A. E., Zhang, X. C., Andrews, D. W., and Lin, J. (2010) J Biol Chem 285, 17614-17627
61. Bogner, C., Leber, B., and Andrews, D. W. (2010) Curr Opin Cell Biol 22, 845-851
by guest on April 26, 2018
http://ww
w.jbc.org/
Dow
nloaded from

13
62. Kim, H., Tu, H. C., Ren, D., Takeuchi, O., Jeffers, J. R., Zambetti, G. P., Hsieh, J. J., and Cheng, E. H. (2009) Mol Cell 36, 487-499
63. Owens, T. W., Foster, F. M., Valentijn, A., Gilmore, A. P., and Streuli, C. H. (2010) J Biol Chem 285, 1081-1088
64. Cheng, E. H., Sheiko, T. V., Fisher, J. K., Craigen, W. J., and Korsmeyer, S. J. (2003) Science 301, 513-517
65. Mahyar-Roemer, M., Katsen, A., Mestres, P., and Roemer, K. (2001) Int J Cancer 94, 615-622 66. Dorrie, J., Gerauer, H., Wachter, Y., and Zunino, S. J. (2001) Cancer Res 61, 4731-4739 67. Pavet, V., Beyrath, J., Pardin, C., Morizot, A., Lechner, M. C., Briand, J. P., Wendland, M.,
Maison, W., Fournel, S., Micheau, O., Guichard, G., and Gronemeyer, H. (2010) Cancer Res 70, 1101-1110
68. Mader, I., Wabitsch, M., Debatin, K. M., Fischer-Posovszky, P., and Fulda, S. (2010) FASEB J 24, 1997-2009
69. Rushworth, S. A., and Micheau, O. (2009) Br J Pharmacol 157, 1186-1188 70. Ganapathy, S., Chen, Q., Singh, K. P., Shankar, S., and Srivastava, R. K. (2010) PLoS One 5,
e15627 71. Shi, Y. (2002) Mol Cell 9, 459-470 72. Perez, D., and White, E. (2000) Mol Cell 6, 53-63 73. Guha, P., Dey, A., Sen, R., Chatterjee, M., Chattopadhyay, S., and Bandyopadhyay, S. K. (2011)
J Pharmacol Exp Ther 336, 206-214 74. Honda, T., Coppola, S., Ghibelli, L., Cho, S. H., Kagawa, S., Spurgers, K. B., Brisbay, S. M.,
Roth, J. A., Meyn, R. E., Fang, B., and McDonnell, T. J. (2004) Cancer Gene Ther 11, 249-255 75. Chow, S. E., Wang, J. S., Chuang, S. F., Chang, Y. L., Chu, W. K., Chen, W. S., and Chen, Y. W.
(2010) Cancer Gene Ther 17, 872-882 76. Kim, M. Y., Trudel, L. J., and Wogan, G. N. (2009) Anticancer Res 29, 3733-3740 77. Mihara, M., Erster, S., Zaika, A., Petrenko, O., Chittenden, T., Pancoska, P., and Moll, U. M.
(2003) Mol Cell 11, 577-590 78. Chipuk, J. E., Bouchier-Hayes, L., Kuwana, T., Newmeyer, D. D., and Green, D. R. (2005)
Science 309, 1732-1735 79. Leu, J. I., Dumont, P., Hafey, M., Murphy, M. E., and George, D. L. (2004) Nat Cell Biol 6, 443-
450 80. Yamaguchi, H., Woods, N. T., Piluso, L. G., Lee, H. H., Chen, J., Bhalla, K. N., Monteiro, A.,
Liu, X., Hung, M. C., and Wang, H. G. (2009) J Biol Chem 284, 11171-11183 81. Yamaguchi, H., Chen, J., Bhalla, K., and Wang, H. G. (2004) J Biol Chem 279, 39431-39437 82. Mahyar-Roemer, M., Fritzsche, C., Wagner, S., Laue, M., and Roemer, K. (2004) Oncogene 23,
6226-6236 83. Essmann, F., Pohlmann, S., Gillissen, B., Daniel, P. T., Schulze-Osthoff, K., and Janicke, R. U.
(2005) J Biol Chem 280, 37169-37177 84. Di, X., Gennings, C., Bear, H. D., Graham, L. J., Sheth, C. M., White, K. L., Jr., and Gewirtz, D.
A. (2010) Breast Cancer Res Treat 124, 349-360 85. Forrester, K., Lupold, S. E., Ott, V. L., Chay, C. H., Band, V., Wang, X. W., and Harris, C. C.
(1995) Oncogene 10, 2103-2111 86. Oren, M., and Rotter, V. (2010) Cold Spring Harb Perspect Biol 2, a001107 87. Bishayee, A. (2009) Cancer Prev Res (Phila) 2, 409-418 88. Patel, K. R., Brown, V. A., Jones, D. J., Britton, R. G., Hemingway, D., Miller, A. S., West, K.
P., Booth, T. D., Perloff, M., Crowell, J. A., Brenner, D. E., Steward, W. P., Gescher, A. J., and Brown, K. (2010) Cancer Res 70, 7392-7399
89. Chow, H. H., Garland, L. L., Hsu, C. H., Vining, D. R., Chew, W. M., Miller, J. A., Perloff, M., Crowell, J. A., and Alberts, D. S. (2010) Cancer Prev Res (Phila) 3, 1168-1175
90. Baur, J. A., and Sinclair, D. A. (2006) Nat Rev Drug Discov 5, 493-506
by guest on April 26, 2018
http://ww
w.jbc.org/
Dow
nloaded from

14
91. Boocock, D. J., Faust, G. E., Patel, K. R., Schinas, A. M., Brown, V. A., Ducharme, M. P., Booth, T. D., Crowell, J. A., Perloff, M., Gescher, A. J., Steward, W. P., and Brenner, D. E. (2007) Cancer Epidemiol Biomarkers Prev 16, 1246-1252
FOOTNOTES
We thank Drs. B. Vogelstein and Terry Beerman for providing reagents. We are thankful to Drs. Jennifer Black, Dean Tang, and Terry Beerman for critical reading of this manuscript. We also thank Dr. Adrian Black for help with analysis of micrographs. This work was supported in part by a National Institutes of Health K01 Award CA123142 to DC and National Cancer Institute Center Support Grant CA016056 to the Roswell Park Cancer Institute. We apologize to those colleagues whose publications could not be cited due to space constraints.
FIGURE LEGENDS
Fig. 1. Resveratrol induces caspase-dependent apoptosis. A. MDA-MB231 cells were treated with resveratrol (Resv) for 36 h and percentage cell death (mean ± S.D. from four independent experiments) was determined using Trypan blue dye. *p<0.01. B. MDA-MB231, GM701 (GM), LNCaP, and PC3 cells were treated with resveratrol for the indicated time intervals and equal amounts of protein (50 µg) were subjected to caspase-3 activity measurements. Caspase activities are presented as values (mean ± S.D. from at least 3 independent experiments) relative to those in controls. *p<0.01. C. GM701 and MDA-MB231 cells were pretreated with pan caspase inhibitor z-VAD (50 µM) for 1 h, and then treated with resveratrol for 36 h. At the end of treatment, equal amounts of protein were used for Western blotting and DEVDase activity. The number in DEVDase activity represents fold change against untreated control (for example, lane 1 is control for GM701 cells, and lane 5 is control for MDA-MB231 cells). Data are the representative of four independent experiments. D. MDA-MB231 and GM701 cells were pretreated with pan caspase inhibitor z-VAD (50 µM) for 1 h, and then treated with resveratrol for 36 h. At the end of treatment, percentage cell death was determined and data are represented as mean ± S.D. from four independent experiments. *p<0.01 as compared with resveratrol treated cells. Resv, resveratrol; MDA, MDA-MB231; Procasp-3, procaspase-3; p17 is a cleaved caspase-3 fragment.
Fig. 2. Resveratrol induces cytochrome c release from mitochondria into the cytosol and is accompanied by fragmentation of the nucleus. A. MDA-MB231 cells were treated with resveratrol (Resv, 120 µM) for the indicated time intervals. At the end of the treatment, cytosolic and mitochondrial fractions were isolated and equal amounts of protein were subjected to Western blotting for the detection of the indicated molecules. Cyt. c, cytochrome c; procasp-3, procaspase-3; p20/p17 represent cleaved products of caspase-3; Actin or heat shock protein 60 (Hsp60) serves as loading controls; COX II, cytochrome c oxidase subunit II; and LDH, lactate dehydrogenase. B. Cells were treated with resveratrol (Resv, 120 µM) for 24 h. At the end of the treatment, cells were labeled live with DAPI and MitoTracker Orange to detect the nucleus and mitochondria (data not shown). Cells were then immunolabeled for cytochrome c (Cyt. c). Representative micrographs are shown and magnification bars represent 20 µM. Consistent with the Western blot analysis, the diffused staining for cytochrome c in individual cells reveals that it was released from mitochondria. Apoptotic cells show fragmented/shiny nucleus with DAPI staining in the panels c and g. Data are representative of at least three independent experiments.
Fig. 3. Resveratrol causes loss of mitochondrial membrane potential and induces Bax translocation to mitochondria, upregulates Bim and t-Bid on mitochondria. A. MDA-MB231, LNCaP, and GM701 cells were
by guest on April 26, 2018
http://ww
w.jbc.org/
Dow
nloaded from

15
treated with resveratrol (120 µM for 24 h), and just prior to fixation, were labeled live with MitoTracker Orange, which accumulates in mitochondria in a membrane potential-dependent manner. The images were captured by microscope, representative micrographs are shown, and magnification bars represent 20 µM. B. MDA-MB231 cells were treated with resveratrol (Resv, 120 µM) for the indicated time intervals. Cytosolic and mitochondrial fractions were purified followed by Western blotting for the indicated molecules by loading equal amounts of protein. COX II, cytochrome c oxidase subunit II. Actin or Hsp60 serves as loading controls. Data are representative of three independent experiments.
Fig. 4. Bax undergo homo-oligomerization on mitochondria and Bax oligomerization on mitochondria is accompanied by cytochrome c release. A-B. MDA-MB231 (A) cells were treated with resveratrol (Resv, 120 µM) for the indicated time intervals, HCT116 WT or HCT116 Bax-/- cells (B) were treated with resveratrol (Resv, 50 µM). Purified mitochondria were crosslinked with BMH, and subjected to Western blotting for Bax to detect its oligomers. Hsp60 serves as loading controls. C-D. MDA-MB231 and HCT116 cells were treated with resveratrol (50 µM) for indicated time periods. At the end of treatments, cells were crosslinked with BMH or EGS followed by Western blotting for Bax or Hsp60. E. Mitochondrial lysates isolated from MDA-MB231 treated with resveratrol (120 µM, 36 h) or from unstimulated were subjected to immunoprecipitation followed by Western blotting for indicated molecules. F. MDA-MB231 and MDA-MB435 were treated with resveratrol (50 µM) followed by mitochondria crosslinking and cytochrome c release assay by Western blotting. Hsp60, heat shock protein 60; MDA, MDA-MB231; MB435, MDA-MB435; Cyt. c, cytochrome c; LDH, lactate dehydrogenase. The asterisks in A, B, and F indicate a non-specific band. Data are representative of at least three independent experiments. Fig. 5. Bax undergoes oligomerization on mitochondria; Bax translocation and activation of Bax/Bak do not involve p53. A-B. Mitochondrial (A) and cytosolic (B) fractions isolated from MDA-MB231 cells treated with resveratrol (120 µM, 36 h) or unstimulated were fractionated on Superdex 200 column. Fractions (0.5 ml) were collected and a portion (20 µl) of fractions 6-28 was analyzed by Western blotting for Bax, Bak, and p53. C. Cytosol isolated from MDA-MB231 treated with resveratrol (120 µM, 36 h) was subjected to immunoprecipitation with p53 or Bax followed by Western blotting for indicated proteins. D-E. MDA-MB231 (D) and MDA-MB435 (E) cells were pretreated with PFT-α (30 µM) followed by resveratrol (50 µM) treatment. Cells were crosslinked and subjected to Western blotting to detect Bax oligomerization. Resv, resveratrol; Data are representative of at least three independent experiments. Fig. 6. Resveratrol induces Bax activation/oligomerization in the cytosol and XIAP interacts with Bax to regulate its activation and translocation to mitochondria. A-B. Equal amounts of cytosols obtained from MDA-MB231 cells (A) treated with resveratrol (120 µM for 0, 12, 24 and 36 h) or from HCT116 cells (B) treated with resveratrol (50 µM for 0, 12 and 24 h) were crosslinked with BMH followed by Western blotting for Bax. C. Purified mitochondria from resveratrol treated (50 µM for 24 h) or untreated cells were subjected to Western blotting to detect XIAP and Bax translocation to mitochondria. D. Purified cytosol and mitochondria obtained from resveratrol treated or untreated MDA-MB231 cells were reconstituted to examine XIAP translocation to mitochondria. E-F. IP was performed using mitochondria (E) or cytosol (F) obtained from MDA-MB231 cells treated with resveratrol (50 µM for 18 h) followed by Western blotting for indicated proteins. MDA, MDA-MB231; MB435, MDA-MB435; HCT, HCT116 WT; Resv, resveratrol; Cont, control; Cyto., cytosol; Mito., mitochondria. The asterisks in A and B indicate a non-specific band. Data are the representative of at least three independent experiments. Fig. 7. Bax plays a prominent role to induce cytochrome c release upon resveratrol treatment. A. HCT116 WT, Bax-/-, and p53-/- were treated with resveratrol (120 µM for 12 or 48 h). Cytosolic and mitochondrial fractions were subjected to Western blotting to detect cytochrome c release. Resv, resveratrol; Cyt. c, cytochrome c; COX II, cytochrome c oxidase subunit II. Actin or Hsp60 serves as loading controls. Data are
by guest on April 26, 2018
http://ww
w.jbc.org/
Dow
nloaded from

16
representative of three independent experiments. B. MDA-MB231, MDA-MB435, and MCF-7 cells were pretreated for 2 h with PFT-α (30 µM) or PFT-µ (10 µM) followed by resveratrol treatment (50 µM for 24 h). At the end of treatment, percentage cell death was quantified. Data are mean ± SD, n=3. PFT-a, PFT-α; PFT-m, PFT-µ. Fig. 8. Activation of caspase-9 is an initiating event in resveratrol-induced apoptosis. A. LNCaP cells were infected with caspase-9 or caspase-8 or control shRNA lentiviral particle at a MOI of 3. Stable cells were used for Western blotting to detect caspase-9 or caspase-8 silencing. B. Caspase-8 and caspase-9 silenced or control (non-targeting) cells were treated with resveratrol (120 µM for 24 h). C. Caspase-8 or caspase-9 was silenced in MDA-MB231 cells. D. Cells were treated with resveratrol (50 µM for 24 h). At the end, caspase-3 activity was determined. Resv, resveratrol; casp-9, caspase-9; casp-8, caspase-8. Actin or Hsp60 serves as loading controls. Data are mean ± SD, n=3, *p<0.01 as compared with resveratrol treated control cells.
by guest on April 26, 2018
http://ww
w.jbc.org/
Dow
nloaded from







ChandraRaghu Gogada, Varun Prabhu, Michael Amadori, Rachael Scott, Sana Hashmi and Dhyan
Mitochondria to Initiate Cytochrome c Release and Caspase ActivationResveratrol Induces p53-Independent, XIAP-Mediated Bax Oligomerization on
published online June 28, 2011J. Biol. Chem.
10.1074/jbc.M110.202440Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
by guest on April 26, 2018
http://ww
w.jbc.org/
Dow
nloaded from