resumen -...
TRANSCRIPT

1
RESUMEN
Introducción: El síndrome de West (SW) constituye un tipo de epilepsia con
un pronóstico evolutivo generalmente sombrío. Los estudios evolutivos en
series cubanas son escasos.
Objetivo: Determinar la evolución clínica de niños con SW, atendidos en el
Instituto de Neurología y Neurocirugía (INN) entre abril del 2010 y mayo del
2017.
Diseño Metodológico: Se trata de un estudio observacional, descriptivo,
retrospectivo, revisaron las historias clínicas de los pacientes con SW,
atendidos en el INN entre abril de 2010 y mayo de 2017. Se recopilaron
variables sociodemográficas: sexo y edad al ingreso; de la epilepsia: tipo de
espasmo epiléptico (EE), desarrollo psicomotor previo al debut de las crisis,
examen físico neurológico, otro tipo de crisis previo a los EE, edad de debut de
EE, presencia de patrón hipsarrítmico, etiología; y evolutivas: control de las
crisis, aparición de otro tipo de crisis, desaparición de la hipsarritmia, presencia
de retraso del neurodesarrollo.
Resultados: De los 31 pacientes con SW, 20 varones (64,5%), edad media de
debut (5,5 +/- 4,3 meses; rango, 1 a 25 meses); y edad media de última
evaluación 29,8 +/- 21,8 meses (rango de 6 a 84 meses). Predominaron los
casos sintomáticos, 19 (61%). El control de los EE se logró en 6 de 11 casos
criptogénicos (54%), y en 4 de 15 casos sintomáticos (27%). El retraso del
neurodesarrollo se constató en 23 de 26 pacientes (88,5%).
Conclusiones: Se evidenció una relación entre la etiología y la evolución, con
más tendencia al control de los EE y la desaparición de la hipsarritmia en los
casos criptogénicos.
Palabras clave: epilepsia, encefalopatía epiléptica, espasmos epilépticos,
niños, síndrome de West.

2
INTRODUCCIÓN
Los espasmos epilépticos (EE) son un tipo específico de crisis de la infancia y
la niñez temprana. Es un desorden significativo debido a su fuerte asociación
con el retraso de neurodesarrollo o su regresión que afecta al 75 - 93 % de los
niños afectados, la proporción de mortalidad es alta, la epilepsia es refractaria
al tratamiento convencional, y presenta sensibilidad a la terapia hormonal. 1, 2
Una de las formas de presentación de la epilepsia de comienzo en la infancia
temprana, cuya aparición se relaciona con la edad y que en sí misma se
considera un síndrome electroclínico es el síndrome de West (SW);
constituyendo un grupo heterogéneo de condiciones que comparten la triada
clínica de espasmos epilépticos, deterioro del neurodesarrollo y patrón de
hipsarritmia en el electroencefalograma (EEG). 3
El SW se presenta en niños entre 6 y 12 meses de edad con un pico de inicio
de los EE entre los 4-6 meses de edad. La incidencia promedio del SW es de
aproximadamente 0,31 (rango 0,05–0,60) por cada 1.000 nacidos vivos. 4
Las crisis de EE se caracterizan por una contracción brusca —más frecuente
bilateral y simétrica— de los músculos del cuello, tronco y miembros. Se
acompañan de una breve afectación de la conciencia. Pueden ser en flexión,
extensión o mixtos. Usualmente se presentan en series, grupos o salvas. 5
A veces ocurren espasmos aislados. Las series o salvas se manifiestan varias
veces al día, y son características al despertar o antes de dormir. Son raras
durante el sueño. Es posible observar manifestaciones autonómicas asociadas
a la crisis: rubicundez, dilatación pupilar y sudoración. 2

3
Numerosos niños muestran retardo del neurodesarrollo, aunque en algunos no
se detecta afectación del mismo previo al comienzo de las crisis. La etapa de
comienzo de los EE ocurre en la mayoría de los pacientes entre los 3 y 6
meses de edad, y un segundo pico entre los 6 y 8 meses. Es menos frecuente
la aparición de las crisis después del año de edad. Los espasmos se
acompañan a veces de risa y una expresión de susto. Pueden ser precedidos o
acompañados por crisis focales, atónicas o tónicas. 6, 7
La discapacidad cognitiva que se presenta en el 90% de los casos afectados
puede ser de diversos grados. Uno de sus aspectos clínicos más
sobresalientes se centra en la magnitud de las consecuencias que produce, en
el orden del neurodesarrollo, este tipo peculiar de síndrome epiléptico. 8
El pronóstico, aunque generalmente grave, resulta incierto dada las
peculiaridades evolutivas de esta entidad y debido a la multiplicidad de sus
bases patológicas. 8
El tratamiento del SW ha sido evaluado en el 2004 y el 2012 por la Academia
Americana de Neurología (AAN), y en una revisión sistemática de Cochrane en
el 2013. Las conclusiones estaban limitadas por la metodología global de los
estudios disponibles. La falta de adhesión a las definiciones de caso
estandarizadas y las medidas del resultado eran un problema con muchos
estudios. 9
Como resultado, aún quedan muchas preguntas con respecto al mecanismo, el
fármaco óptimo, la dosificación, la duración de terapia, y la importancia de
iniciación puntual de tratamiento después de la aparición de los EE. 9

4
EL SW es una encefalopatía epiléptica considerada como una enfermedad
compleja, pudiendo llegar a una discapacidad, lo que conlleva a una enorme
repercusión social y económica. 10
En Cuba los estudios sobre el este tipo de epilepsia son escasos y no se
disponen de estimados seguros de la frecuencia de la enfermedad y se
carecen de datos confiables sobre la misma.
El incremento de la calidad de vida en la población, el desarrollo de
herramientas para el asesoramiento genético, el desarrollo de las
especialidades médicas, el mejoramiento de los servicios de salud y educación,
que incluyen la rehabilitación, motivan al continuo esfuerzo por el dominio de la
enfermedad y la posibilidad de incorporar a los afectados a las actividades
estudiantiles, laborales y sociales. 3
Por todo lo expuesto con anterioridad, se hace imprescindible llevar a cabo una
investigación para determinar el comportamiento del SW en un grupo de niños
con este síndrome atendidos en el Instituto de Neurología y Neurocirugía, con
vistas a poder planificar mejor los recursos humanos y materiales destinados a
la atención de estos pacientes.

5
Problema Científico
¿Cuáles son las principales características clínicas y cómo evolucionan los
pacientes con Síndrome de West atendidos en el Servicio de Neurología Infantil
del Instituto de Neurología y Neurocirugía (INN)?

6
MARCO TEÓRICO
I. Concepto
El síndrome de West (SW) es uno de los tipos de encefalopatías epilépticas
reconocidas. Es definida por una tríada de espasmos epilépticos, hipsarritmia y
retardo o regresión del desarrollo psicomotor. 11, 12
Los espasmos epilépticos fueron originalmente descritos por el Dr. West en el
año 1841, cuando escribió una carta a Lancet sobre eventos clínicos que
observó en su hijo de 4 meses de edad, a los que se refirió como “una forma
peculiar de convulsión infantil”. 13
El síndrome de West es una epilepsia dependiente de la edad, que
frecuentemente se presenta en el primer año de vida con episodios de
espasmos que normalmente ocurren en racimos. Presenta características
típicas en el electroencefalograma, con ondas caóticas y de alto voltaje,
interictales, patrón conocido como hipsarritmia. Cuando debutan estos
espasmos en los niños, ya algunos casos presentan un desorden neurológico
cerebral con repercusión en el neurodesarrollo. 14
El grupo de estudio “West Delphi” siguiendo el consenso de 31 médicos de 15
países, elaboraron criterios diagnósticos para los espasmos epilépticos basado
en las signos clínicos.12, 15 El grupo concluyó que el resultado clínico primario,
nombrado libre de espasmos, debía definirse por la ausencia de espasmos
dentro de los 14 días de comienzo de tratamiento, y 28 días consecutivos, o
más, del último espasmo. El resultado electroclínico primario fue definido por el
cese de los espasmos con resolución de la hipsarritmia. 12,15

7
El grupo definió a los espasmos epilépticos como una característica
fundamental dentro de la triada que conforma al SW. Su reporte mantiene un
estándar para reportes modificados y rasgos atípicos de hipsarritmia. También
sugieren un grupo de características básicas y resultados que deben
informarse en los ensayos clínicos de pacientes con espasmos epilépticos, y
proponen una definición estándar de recaída. Sin embargo, no han alcanzado
un consenso para definir la hipsarritmia. 12-15
El patrón electroencefalográfico de hipsarritmia fue descrito por Gibbs y Gibbs
en el año 1952 y se caracteriza por: “puntas y ondas lentas de gran amplitud,
desordenadas, que varían de un momento a otro tanto en duración como en
localización”.16, 17
II. Epidemiología
La incidencia de SW varía de 2 a 3,5 por 10.000 nacidos vivos y representa el
13 - 45,5 % de las epilepsias de la infancia. 18 La mayoría de los casos se
presenta a una edad pico de ataque entre 3 y 7 meses; y el 90% se presenta
en el primer año de vida. La aparición de los EE después de 18 meses es rara,
aunque se han reportado hasta 4 años de edad. 19, 20, 21
En Eslovenia en periodo de estudio de 11 años, se reporta una incidencia de
2,06 por cada 10.000 nacidos vivos, con una incidencia anual que oscila entre
1,0 y 3,2 por cada 10.000 nacidos vivos. 22
En Georgia, la incidencia era 2,9 por 10.000 nacidos vivos entre 1975 a 1977.
La prevalencia entre niños de 10 años es de 2,0 por 10.000; la más baja

8
prevalencia que incidencia probablemente refleja la mortalidad sustancial en
este tipo de epilepsia. 23
En un estudio en Canadá que incluyó a 75 pacientes, los rangos de incidencia
anual y a los 5 años variaron. Durante dos períodos consecutivos (1979 a 1992
y 1993 a 1998), la incidencia media anual estimada disminuyó de 3,31 a 1,73
por cada 10.000 nacidos vivos. 24
El síndrome de West aparece en niños de todos los grupos étnicos, y los
varones son ligeramente más afectados que las niñas con una proporción de
60:40. Mientras otros estudios encuentran que afecta por igual a ambos sexos.
25
III. Etiología
La clasificación del SW está evolucionando. Tradicionalmente, se clasifica
como sintomáticos y criptogénicos: 26, 27
Pacientes con SW sintomáticos tienen una etiología conocida y/o retraso
significativo en el neurodesarrollo.
Pacientes con SW criptogénicos tienen una etiología desconocida y un
desarrollo normal del neurodesarrollo en el momento de la aparición del
espasmo.
El grupo de trabajo para la Clasificación y Terminología de la Liga Internacional
contra la epilepsia (ILAE) propuso agregar un grupo idiopático. Esta categoría
incluye a los pacientes con desarrollo normal a la aparición de los espasmos,

9
examen físico y neuroimágenes normales; y un modelo de hipsarritmia sin las
anormalidades epileptiformes focales. 26, 27
La proporción de categorías etiológicas del SW varía entre los estudios. Esta
amplia gama es el resultado del uso de diferentes definiciones, la edad
temprana al diagnóstico cuando los estados del neurodesarrollo pueden ser
difíciles de evaluar, y el aumento en el uso de estudios de neuroimágen que
revelan una causa subyacente en la mayoría de los casos. 28
En un informe de las provincias canadienses de Nova Scotia y Prince Edward
Island de 1978 a 1998, la proporción de categorías etiológicas era 68 % para
los sintomáticos, 24 % criptogénicos y 8 % para los casos idiopáticos. 20
Sin embargo, en una serie de 140 pacientes afectados cuya evaluación incluía
la tomografía computarizada (TAC), la imagen por resonancia magnética (IMR),
y/o la tomografía de emisión de positrón (PET), el 96 % de los casos eran
clasificados como sintomáticos. La evaluación médica de estos pacientes era
algo más extensa que lo que habitualmente se realiza en hospitales generales,
por lo que es poco probable que los casos sintomáticos sean tan
abrumadoramente altos en la mayoría de las series estudiadas. 28
En un estudio prospectivo y multicéntrico del Consorcio Nacional de los
Espasmos epilépticos, se estudiaron 251 infantes, y se identificó una causa en
161 pacientes (64 %). Las causas más comunes identificadas fueron
estructurales-adquiridas (22 %), genéticas (14 %), estructurales-congénitas (11
%), genética-estructural (10 %), metabólica (5 %), e infecciosa (2 %). 29

10
La evaluación clínica y la IMR proporcionaron un diagnóstico específico en 55
% de los infantes. Estos resultados dan énfasis a la importancia de la
evaluación clínica inicial y a la IRM en la evaluación de casos nuevos con SW,
seguido por la comprobación genética y metabólica, si ninguna etiología se
identifica. 29
Grupo Sintomático
Constituye el grupo más frecuente. Kurokawa y cols. 30, lo encontraron en el
45,7 % de su serie, Lombroso 31 en el 59 % de sus pacientes, Ohtahara y cols.
32, en el 63,4 % y Matsumoto y cols. 33, en el 67 % de sus casos.
El SW sintomático es el resultado de un trastorno demostrado del sistema
nervioso central. Uno o más factores etiológicos pueden identificarse en la
mayoría de casos (60 a 70 %). La evaluación continua con neuroimágen y PET
puede aumentar esta proporción hasta el 90 %. 34, 35
El tiempo crítico del daño al sistema nervioso central ocurre típicamente en el
período prenatal, perinatal, o postnatal temprano. La anormalidad prenatal más
común es la malformación del SNC. Otros transtornos incluyen desórdenes
genéticos, neurocutáneos, errores innatos del metabolismo, e infecciones
intrauterinas. 36
Período Prenatal
Malformaciones del SNC: la displasia cortical es el desorden más común, se
estima que constituye el 30 % de las causas prenatales con SW26. Otras
malformaciones de SNC que pueden producir EE son: la disgenesia cerebral
(ej: el síndrome de Aicardi), la lisencefalia (ej: el síndrome de Miller-Dieker), la

11
holoprosencefalia, y la hemimegalencefalia. Casi todos los pacientes con el
síndrome de Miller-Dieker y el síndrome de Aicardi presentan EE. 37
Síndrome de Miller-Dieker: Se produce como consecuencia de un
transtorno de la migración neuroblástica durante el desarrollo del SNC,
produciendo la lisencefalia. Otros rasgos característicos de este
síndrome incluyen retraso del crecimiento, microcefalia, anormalidades
craneofaciales como el surco bitemporal, micrognatia, anomalías de la
oreja y la nariz; defectos renales y cardíacos, así como anomalías
neurológicas como hipotonía, espasmos epilépticos, y retraso mental. La
mayoría de los pacientes con este desorden tiene una delección del gen
localizado en el cromosoma 17p13.3. 38, 39, 40
Síndrome de Aicardi: Es un trastorno con una herencia dominante ligada
al cromosoma X, solo observado en niñas, dado que en los varones es
una condición letal. El síndrome se caracteriza por espasmos epilépticos
(usualmente la forma de presentación), agenesia del cuerpo calloso,
corioretinopatía, anomalías vertebrales y retraso mental severo. Los
pacientes típicamente desarrollan crisis focales antes los 3 meses de
edad; cuando aparecen los EE, estos suelen ser asimétricos. El
electroencefalograma muestra a menudo una hipsarritmia asimétrica. 41,
42
Hemimegalencefalia: Se caracteriza por el aumento de volumen en un
hemisferio cerebral, con una estructura cortical anormal en el lado
afectado. Más de la mitad de los pacientes desarrollan a menudo EE. 43

12
Complejo Esclerosis Tuberosa (CET): Es un trastorno neurocutáneo
frecuentemente asociado con EE y representa entre el 10 y 30% de todas las
causas prenatales de SW, que se presentan en hasta el 68% de los casos con
CET. La incidencia real de CET en los niños con SW puede ser aún más alta
porque las características clínicas de esta enfermedad pueden aun no estar
presentes al momento del debut de los EE. 44, 45
En general, los pacientes con CET y SW tienen una evolución desfavorable
caracterizada por epilepsia fármaco-resistente y trastorno cognoscitivo severo.
En un reporte, el desarrollo era normal en sólo el 16 % de 24 niños con CET y
SW. 45 Otra serie de 50 niños reporta discapacidad intelectual en el 64 % de los
casos. 46
Los factores de riesgo para el retraso mental incluyen, una duración prolongada
del SW, un tiempo prolongado desde el inicio del tratamiento hasta el cese de
los EE, y pobre control de las crisis después del inicio del tratamiento.46
Otros trastornos neurocutáneos: El síndrome del nevus linear sebáceo,
incontinencia pigmenti, síndrome de Ito, y la neurofibromatosis tipo I (NF1)
frecuentemente presentan SW. Los pacientes con NF1 y EE generalmente
tienen espasmos simétricos, la hipsarritmia es típica en el EEG y ningún rasgo
focal. Comparado con la combinación de CET y EE, los pacientes con NF1
presentan una mejor evolución, con resolución de los EE tras el tratamiento en
el 87 % de los casos. 47
Anormalidades cromosómicas: Representan el 15% de las etiologías
prenatales con SW. El Síndrome de Down es la más frecuente. 47 En una serie

13
de 350 niños con síndrome de Down, 28 (8 %) tenían epilepsia; 13 de ellos con
SW. 48 En general, en estos pacientes existe afectación del neurodesarrollo. 49
Otras anormalidades cromosómicas asociadas a SW incluyen la duplicación
18q, duplicación 7q, así como la delección del gen de MAGI2 en el cromosoma
7q11.23-q21.11, y la trisomía parcial 2p. 50
Otras mutaciones genéticas: Se han descubierto genes específicos asociados
a los EE, que incluyen aquellos que codifican para las proteínas FOXG1, la
proteína STXBP1, la CASK, ALG13, PNPO, y ADSL. 51,52,53,54
Errores innatos del metabolismo: Pueden asociarse a SW. En los países donde
la fenilcetonuria (PKU) no se detecta por pesquisa recién nacidos, los EE son
una manifestación común de este desorden. En un informe de Beijing, China, el
SW ocurrió en 62 de 503 pacientes con PKU (12 %). 55 En esta serie, el
diagnóstico de PKU se realizó en los pacientes con edades entre 4 meses a 7
años de edad, los EE ocurrían antes del diagnóstico en la mayoría de los
casos. La intervención dietética redujo la frecuencia de espasmos y mejoró el
resultado cognoscitivo. 56
El SW ha sido asociado con más de 25 desórdenes metabólicos. 57 Éstos
incluyen la deficiencia de dihydropteridine reductasa 58, Enfermedad de Menkes
59, 60, la deficiencia de Piruvato Deshidrogenasa 61, Deficiencia de Citocromo C
oxidasa (el síndrome de Leigh) 62, histidinemia 63, la deficiencia de piridoxina, y
los trastornos del ciclo de la urea. 64

14
Infecciones congénitas: El SW pueden ocurrir en los infantes con infecciones
congénitas que afectan el SNC. Entre las infecciones prenatales más
frecuentes se encuentran la toxoplasmosis, la sífilis, y el citomegalovirus. 65
Período perinatal
Los EE pueden asociarse con los daños perinatales, incluidos la encefalopatía
hipóxico-isquémica y la hipoglicemia neonatal. 66 En una serie de 32 pacientes
con EE y una historia de daño perinatal, 15 tenían lesiones porencefálicas, 12
tenían leucomalacia periventricular, y 5 atrofia cerebral bilateral difusa. 67
La relación entre EE y el nacimiento pretérmino es incierta. En los niños con
EE, la proporción de bajo peso al nacer es aproximadamente tres a cuatro
veces mayor que en la población general, aunque la proporción de prematuros
es similar. Esto puede ser porque los EE son más probables que ocurran en
infantes pequeños para su edad gestacional, no así con los pretérminos de
peso adecuado. 68
Período postnatal
El SW se ha atribuido a injurias postnatales entre el 15 % y 67 % de los casos
de series con este tipo de crisis. Las causas más frecuentes en este período
incluyen: lesión traumática, asfixia, tumores, e infecciones del SNC, incluyendo
meningitis bacteriana y la encefalitis viral. En una revisión bibliográfica, estos
eventos ocurrieron típicamente dentro del primer año de vida, pero se reportó a
edades tardías como a los 31 meses. La latencia entre el daño y la aparición de
los EE va desde 1,5 a 11 meses. 68

15
Sin embargo, existen casos poco frecuentes en los que los EE aparecieron tras
un evento más tardío, como los casos de dos niños con desarrollo previamente
normal que desarrollaron EE a los 20 y 34 meses de edad respectivamente
después estar cerca de ahogarse.69
Grupo Criptogénico
Una propuesta de la ILAE en el 2001 recomendó el uso del término
"probablemente sintomático" en lugar del muy utilizado “criptogénico”. 27 Con
estos términos sinónimos se clasifican a los pacientes con SW en quienes se
presumen que puedan tener una causa sintomática, pero la misma no puede
ser demostrada. Tradicionalmente, los criterios para SW criptogénicos son:70
o Desarrollo normal antes del inicio de espasmos simétricos.
o Ningún otro tipo de crisis.
o Examen físico normal.
o TAC e IRM normales.
o Recurrencia de hipsarritmia entre los espasmos consecutivos de un
racimo.
o Falta de cualquier anormalidad focal en el EEG interictal o ictal.
La proporción de pacientes afectados con SW criptogénicos oscila entre 4 y 42
% 71. Los pacientes con este tipo de SW parecen tener un mejor pronóstico que
los casos con etiología sintomática.
Grupo Idiopático
Un Taller de la ILAE propuso la adición de este grupo. Esta categoría incluye a
los pacientes con un desarrollo normal, examen físico y estudios de

16
neuroimágenes normales; y un patrón de hipsarritmia sin anormalidades
epileptiformes focales. 27 Se presume que estos síndromes tienen una base
genética. Los estudios que han clasificado al SW en sintomáticos y
criptogénicos, incluyen este grupo de pacientes en la última categoría.
IV. Manifestaciones Clínicas
El síndrome de West se inicia en la mayoría de los niños durante el primer año
de vida, lo cual ocurre en hasta el 90 % de los casos. La incidencia máxima de
debut de las crisis (50 a 77%) está entre los tres y siete meses de edad; el
inicio después de 18 meses es raro.72, 73
Los espasmos epilépticos pueden involucrar los músculos del cuello, tronco, y
extremidades. Son simétricos y sincrónicos, pero puede haber variantes
clínicas. Ocurren típicamente dos fases de actividad muscular. La primera fase
consiste en contracciones musculares súbitas y breves de uno o más grupos
musculares. 74
La fase inicial se sigue por una fase tónica más larga. La contracción inicial
dura menos de dos segundos. La fase tónica que sigue es menos intensa,
normalmente dura de dos a diez segundos. A menudo, la contracción dura
menos de 0.5 segundos y ocurre sin una fase tónica. 74
Se han caracterizado tres tipos clínicos de espasmos. En un estudio de 5042
crisis de EE, en 24 pacientes, los espasmos fueron clasificados de flexores en
el 33,9 %, extensores en el 22,5 %, y mixtos (flexor-extensor) en el 42,0% de
los casos. La mayoría de los pacientes tiene más de un tipo de espasmo. 75

17
Los espasmos flexores consisten en la flexión súbita del cuello, tronco,
brazos, y piernas, y contracción de los músculos abdominales. Esta
última puede ser tan severa como para causar el movimiento del torso
hacia la cintura. La intensidad de las contracciones y el número de
grupos de músculos involucrados varía en los diferentes ataques.
Los espasmos extensores consisten en extensión abrupta del cuello y
tronco, con abducción de los brazos o piernas.
Los espasmos mixtos flexor-extensores normalmente consisten en
flexión del cuello, tronco, y brazos, y extensión de las piernas. Menos
frecuentemente, involucran flexión de las piernas y extensión de los
brazos.
V. Pronóstico
El pronóstico global de los niños con SW es malo. Los rangos de mortalidad
varían desde un 3 a 33%. 76, 77 Otros resultados adversos incluyen epilepsia
intratable y retraso del neurodesarrollo, desde moderado a severo.
Los espasmos clínicos desaparecen a los tres años de edad en
aproximadamente la mitad de los niños con EE y raramente persisten después
de los cinco años de edad. La hipsarritmia también tiende a resolverse con el
crecimiento. 78, 79
En conjunto, el 50 a 90 % de los pacientes desarrollan otro tipo de crisis. En
general, los pacientes con SW sintomáticos tienen más probabilidad de
desarrollar otro tipo de crisis que los pacientes con SW criptogénico en una
relación de 57,5 % vs 35,3%. 78, 79

18
Se ha observado que el control de las crisis es más favorable en los casos
criptogénicos que en los sintomáticos, y en aquéllos con un inicio más
temprano de los espasmos (<4 meses vs 4 a 8 meses vs 8 meses). 80
Un factor importante que influye en el pronóstico, es la etiología. El pronóstico
es mejor en los casos idiopáticos y criptogénicos. El pronóstico del SW
idiopático es favorable con desaparición de las crisis y un desarrollo psicomotor
normal. 81, 82
Se ha descrito, que en pacientes en los cuales se evidenció, mediante la
tomografía por emisión de positrones (PET), disminución del metabolismo de la
glucosa en ambas regiones temporales, presentaron un pronóstico
desfavorable a largo plazo y la mayoría manifestaron signos de autismo. 83
Se ha señalado que zonas de hipoperfusión multifocal, evidenciados mediante
la tomografía computadorizada por emisión de fotón único (SPECT), en
pacientes con síndrome de West sintomático, pueden indicar un pronóstico
desfavorable. 84
Hattori 85 ha referido la remisión espontánea de los espasmos en el síndrome
de West. Este autor señaló que de los pacientes que mostraron remisión
espontánea, en el 86 % de los casos, ésta fue precedida por infecciones virales
entre las cuales predominó el exantema súbito.
VI. Tratamiento
Las principales opciones de tratamiento del SW incluyen la terapia hormonal,
principalmente la Corticotropina (ACTH), y los fármacos antiepilépticos,
principalmente la Vigabatrina. La Piridoxina se ha usado a menudo como

19
primera línea de tratamiento en Japón, aunque no hay ningún ensayo
controlado aleatorizado de este medicamento. 86
Terapia Hormonal
El mecanismo de acción de la ACTH y los glucocorticoides no es conocido. La
administración de ACTH puede controlar los espasmos en los pacientes con
supresión suprarrenal, sugiriendo que el efecto es independiente de la
descarga de corticosteroides suprarrenales.
ACTH: La ACTH se recomienda como el tratamiento de primera línea en la
mayoría de los pacientes. En cinco ensayos controlados aleatorizados de
terapia de ACTH, la cesación de espasmos ocurrió en 42 - 87 % de los casos, y
el tiempo entre el inicio del tratamiento y la cesación de espasmos era 7 a 12
días. 87,88,89 En la mayoría de estos estudios, la respuesta a ACTH era mejor en
los pacientes con etiología criptogénica. Las proporciones de la recaída fueron
del 15 al 33 %. 90
Dos ensayos aleatorizados reportaron que la respuesta a bajas dosis era
comparable a las dosis más altas y producía menos efectos colaterales. 91 En
el más grande de los ensayos, 50 pacientes con reciente diagnóstico de SW
fueron asignados para recibir, al azar, altas o bajas dosis de ACTH.
Los grupos obtuvieron similar proporción de recaída y respuesta al tratamiento,
que fue evaluada por el cese de los espasmos y la desaparición de la
hipsarritmia, encontrándose una proporción de 50 y 58 % para el grupo de altas
dosis y bajas dosis respectivamente. 91

20
La hipertensión ocurrió más a menudo en el grupo de alta dosis con una
proporción de 31% contra 4 % del grupo de bajas dosis; otros efectos
colaterales fueron similares entre los grupos. Otros estudios retrospectivos
también confirman la eficacia similar y menos eventos adversos en los que
usaron dosis más baja. 92, 93
Glucocorticoides: Menos costoso, más fácil de administrar, se han sugerido los
regímenes de la terapia hormonales como las alternativas eficaces potenciales
a la ACTH. Los estudios han reportado otras terapias hormonales como
prednisona, prednisolona, metilprednisolona, o dexametasona, que parecen
controlar los espasmos en algunos pacientes. 94, 95
Varios pequeños ensayos controlados aleatorizados y otras investigaciones no
randomizadas han comparado la ACTH y la prednisona (cada uno a diferentes
dosis) para el tratamiento de EE. Los resultados han sido mixtos, con
variaciones en la dosis, formulación, y resultados entre los ensayos, lo que
hace difícil de tomar conclusiones firmes. 94, 95
Fármacos Antiepilépticos
Vigabatrina: Puede ser eficaz como tratamiento inicial. 96, 97 Es un inhibidor
irreversible de la GABA-transaminasa, que incrementa la concentración de
GABA en el sistema nervioso central. 98 Fue aceptado por la FDA en el 2009
para el tratamiento de los espasmos epilépticos. La mayor experiencia con el
uso de la vigabatrina como monoterapia inicial, proviene de un estudio
retrospectivo de 250 niños con SW. 99

21
En una serie de casos retrospectiva de 56 pacientes consecutivos, se trataron
21 casos con vigabatrina y 21 casos con ACTH. Se observó una proporción
similar de pacientes libres de espasmos, con desaparición de la hipsarritmia.
Las proporciones de recaída eran algo más altas en el grupo tratado con
ACTH. Los resultados del neurodesarrollo al año eran similares. 100
Ácido Valproico: Los informes de tratamiento con el ácido valproico tienen
resultados incoherentes. El ácido valproico puede beneficiar del 40 al 70 % de
los pacientes que no responden a la ACTH. Sin embargo, la respuesta clara
puede reflejar la historia natural de los EE, en lugar del efecto de tratamiento.
101
No existe ningún ensayo controlado del uso del ácido valproico para el
tratamiento de los EE. En el estudio de Siemes, 102 la cesación de espasmos y
resolución de hipsarritmia ocurrió entre el 73 y el 91 % de 22 niños, a seis
meses del inicio del tratamiento. La mayoría respondió en dos semanas, pero
el 23 % recayó, y un tercio desarrolló trombocitopenia. En otro estudio, el cese
de los espasmos ocurrió en el 72 % de los niños a los tres meses de iniciado el
tratamiento. 103
Zonisamida: Es un derivado de la sulfonamida. Su mecanismo de acción está
relacionado con el bloqueo de los canales de sodio voltaje-dependiente y de
calcio tipo C. 104 En una serie de casos, la zonisamida era eficaz en los
pacientes sin respuesta a la piridoxina o ácido valproico. 105
Topiramato: Se probó en un estudio de 11 pacientes con EE refractarios. 106 Se
utilizó una dosis inicial de 25 mg por día, además de su terapia actual. La dosis

22
se aumentó por 25 mg cada dos a tres días a un máximo de 25 mg/kg por día
hasta que se controlaran los espasmos o la dosis tolerada máxima fuera
alcanzada. Los espasmos se detuvieron y la hipsarritmia se resolvió en 5 de 11
pacientes. 107
Benzodiacepinas: Se incluye el nitrazepam, clonazepam, y diazepam, se han
usado anecdóticamente. 9
Otros tratamientos
Piridoxina: No existe ningún ensayo controlado aleatorizado de piridoxina para
el tratamiento de espasmos epilépticos. Aunque se han informado infantes con
el SW que responden al tratamiento con piridoxina a altas dosis (la vitamina B6,
30 a 400 mg diariamente). 9
Tratamiento Quirúrgico
Aunque muchos pacientes con SW tienen anormalidades cerebrales difusas,
algunos tienen una anormalidad estructural cortical focal que puede responder
a la escisión quirúrgica. 108
El tratamiento quirúrgico es considerado en pacientes con SW refractarios al
tratamiento médico, cuando hay regresión o detención del neurodesarrollo, y
cuando no hay evidencia de daño cerebral difuso o de enfermedad
degenerativa o metabólica. La cirugía no se debe considerar si la resección
cortical crearía un nuevo déficit neurológico. 108

23
OBJETIVOS
Objetivo General
Describir la evolución clínica de los pacientes con diagnóstico de
Síndrome de West atendidos en el Servicio de Neurología Infantil del
Instituto de Neurología y Neurocirugía (INN) en el periodo de abril del
2010 a mayo del 2017.
Objetivos Específicos
Describir las principales características sociodemográficas y de la
epilepsia en un grupo de niños con Síndrome de West.
Determinar las causas más frecuentes de Síndrome de West en la
serie estudiada
Precisar la evolución clínica y electroencefalográfica en el grupo de
pacientes estudiados.

24
DISEÑO METODOLÓGICO
Tipo de Estudio
Se realizó un estudio observacional, descriptivo, retrospectivo, de todos los
niños ingresados en el Servicio de Neurología Infantil del Instituto Neurología y
Neurocirugía (INN), durante el período comprendido entre abril del 2010 y
mayo del 2017, con el diagnóstico al egreso de Síndrome de West (SW).
UNIVERSO Y MUESTRA
El universo estuvo constituido por los 31 pacientes que ingresaron en el
servicio de neuropediatría del Instituto Neurología y Neurocirugía de Cuba, con
diagnóstico de Síndrome de West al momento del alta, en el período de abril
del 2010 a mayo del 2017. Se trabajó con todos los casos que cumplieron con
los criterios de inclusión.
Criterios de inclusión
Se utilizaron los siguientes criterios de inclusión.
Todos los niños hospitalizados en el Servicio de Neurología Infantil del
INN entre abril del 2010 y mayo del 2017, con diagnóstico al egreso de
SW.

25
OPERACIONALIZACIÓN DE VARIABLES
Variable Tipo Operacionalización Medida
Resumen Escala Definición
Edad Cuantitativa
Continua En números
Tiempo vivido en años
cumplidos
Media y desviación estándar
Sexo Cualitativa
nominal dicotómica
Masculino Femenino
Según el sexo biológico
determinado. Porciento
Color de la piel Cualitativa
nominal politómica
Blanca Negra
Mestiza
Según color de la piel
Porciento
Tipo de crisis precediendo a
los EE
Cualitativa nominal
politómica
Focal Generalizada Desconocida
Según tipo de crisis epiléptica.
Porciento
Desarrollo psicomotor previo a EE
Cualitativa nominal
dicotómica
Normal Anormal
Según desarrollo psicomotor
Porciento
Etiología Cualitativa
nominal politómica
Criptogénico Sintomático a:
Esclerosis Tuberosa Encefalopatía HI Malformaciones
Cerebrales Infecciones
Anomalías Genéticas Errores Congénitos
Metabólicos Otras
Según origen de la enfermedad
Porciento
Examen físico neurológico
Cualitativa nominal
dicotómica
Normal Anormal
Según resultado a la exploración
del paciente Porciento
EEG Interictal Cualitativa
nominal politómica
Si No
No precisado
Según la presencia de hipsarritmia al momento del
ingreso o diagnóstico.
Porciento
Estudio de Neuroimágen
Cualitativa nominal
dicotómica
Normal Anormal
Según resultados
imagenológicos. Porciento
Evolución Cualitativa
nominal dicotómica
Supresión de espasmos epilépticos
No supresión
Según la evolución clínica
del paciente Porciento
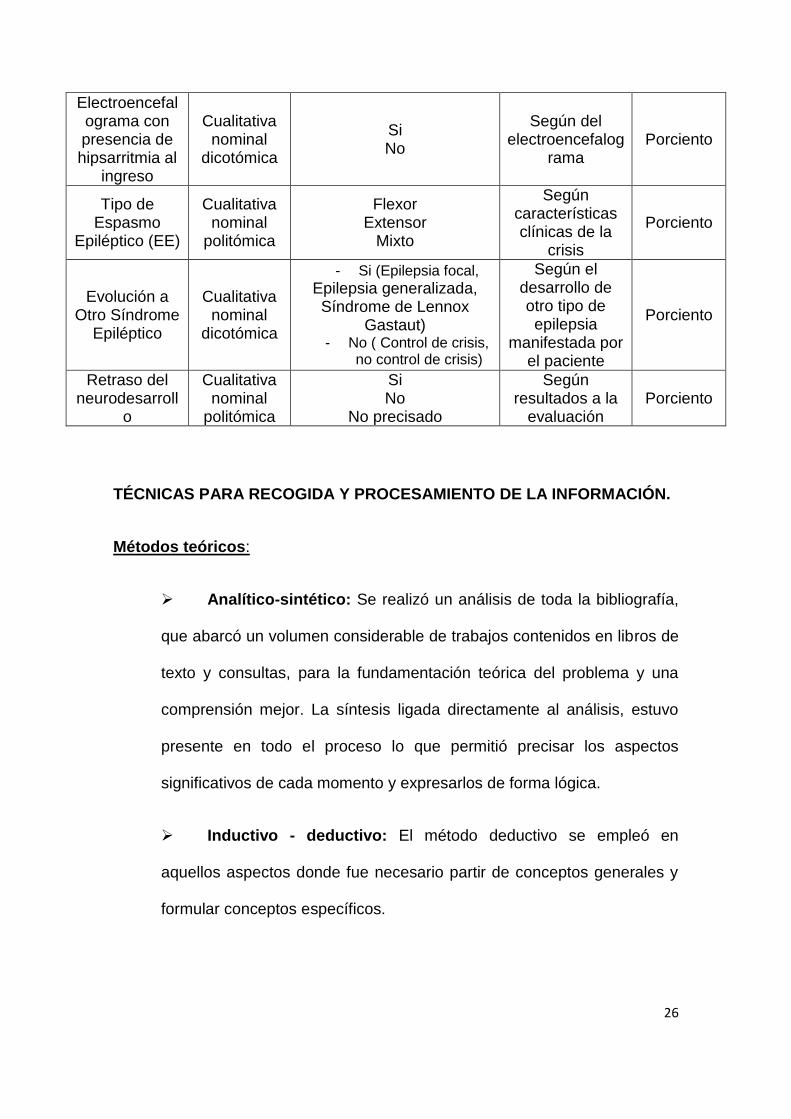
26
Electroencefalograma con presencia de hipsarritmia al
ingreso
Cualitativa nominal
dicotómica
Si No
Según del electroencefalog
rama Porciento
Tipo de Espasmo
Epiléptico (EE)
Cualitativa nominal
politómica
Flexor Extensor
Mixto
Según características clínicas de la
crisis
Porciento
Evolución a Otro Síndrome
Epiléptico
Cualitativa nominal
dicotómica
- Si (Epilepsia focal,
Epilepsia generalizada, Síndrome de Lennox
Gastaut) - No ( Control de crisis,
no control de crisis)
Según el desarrollo de otro tipo de epilepsia
manifestada por el paciente
Porciento
Retraso del neurodesarroll
o
Cualitativa nominal
politómica
Si No
No precisado
Según resultados a la
evaluación Porciento
TÉCNICAS PARA RECOGIDA Y PROCESAMIENTO DE LA INFORMACIÓN.
Métodos teóricos:
Analítico-sintético: Se realizó un análisis de toda la bibliografía,
que abarcó un volumen considerable de trabajos contenidos en libros de
texto y consultas, para la fundamentación teórica del problema y una
comprensión mejor. La síntesis ligada directamente al análisis, estuvo
presente en todo el proceso lo que permitió precisar los aspectos
significativos de cada momento y expresarlos de forma lógica.
Inductivo - deductivo: El método deductivo se empleó en
aquellos aspectos donde fue necesario partir de conceptos generales y
formular conceptos específicos.

27
Histórico - lógico: Se desarrolló a partir del análisis de
documentos, artículos, tesis y bibliografías, de forma general en la
sistematización de los antecedentes del problema de la investigación.
Métodos empíricos:
Planilla de recolección de datos: Para obtener información
relacionada con las variables definidas en el estudio.
RECOLECCIÓN DE LOS DATOS
Para la recolección de la información, se trabajó con las historias clínicas de los
pacientes. Se confeccionó la ficha de recolección de datos (anexo 1) y se
procedió a revisar las historias clínicas de los pacientes incluidos, de la cual se
extrajo la información referente a las variables seleccionadas para el estudio.
Para precisar la evolución de los niños con SW se tuvo en cuenta el control o
no de las crisis de espasmos epilépticos, el mantenimiento o desaparición del
patrón de hipsarritmia, y la evolución o no hacia otro tipo de epilepsia, así como
la presencia posterior de retraso global del neurodesarrollo, según los datos
recogidos en la historia clínica al momento de la última evaluación o consulta.
PROCESAMIENTO DE LA INFORMACIÓN
Para el procesamiento de la información, se creó una base de datos
automatizada con la hoja de cálculo electrónica Excel 2003. Los datos primarios
se procesaron en el programa estadístico SPSS 22.0.

28
Fuentes de información:
- Historias clínicas de los pacientes hospitalizados.
Procesamiento estadístico
Los datos del formulario, fueron vaciados en una base de datos creada en el
programa estadístico SPSS versión 22.0, donde se realizaron distribuciones de
frecuencias, cálculos porcentuales, cálculo de desviación estándar y medidas de
tendencia central. Se utilizó los métodos de la estadística descriptiva.
ÉTICA
En todo momento, la investigación se procedió de acuerdo con las normas
éticas imperantes en el Sistema de Salud del que disfrutamos en nuestra
sociedad. La información obtenida se utilizó solamente con fines científicos y
docentes.

29
RESULTADOS
Se detectaron un total de 31 niños ingresados en el Servicio de Neurología
Infantil del INN, en el periodo de abril del 2010 a mayo del 2017, con el
diagnóstico al egreso de Síndrome de West.
Las principales características sociodemográficas del grupo de niños
estudiados con SW, se pueden observar en la tabla 1.
Tabla 1. Principales características sociodemográficas y de la epilepsia del
grupo de niños con Síndrome de West.
Variables No. %
Género Masculino 20 64,5
Femenino 11 35,5
Color de la piel
Blanca 25 80,6
Negra 2 6,5
Mestiza 4 12,9
Tipo de Espasmo Epiléptico (EE)
Flexor 11 35,5
Extensor 2 6,4
Mixto 18 58,1
Desarrollo Psicomotor previo al inicio de los EE
Normal 8 25,8
Anormal 23 74,2
Examen neurológico al momento del ingreso
Normal 2 6,4
Anormal 29 93,6
Crisis Epilépticas Previas a los EE
Si 11 35,5
No 20 64,5
Electroencefalograma presencia de
hipsarritmia al ingreso
Si 31 100
No 0 0
Etiología Sintomática 19 61,3
Criptogénica 12 38,7
Edad media debut 5,5 +/- 4,3 meses
(rango, 1 a 25 meses)
Edad media en la última evaluación 29,8 +/- 21,8 meses
(rango de 6 a 84 meses)
Fuente: Historia Clínica
30
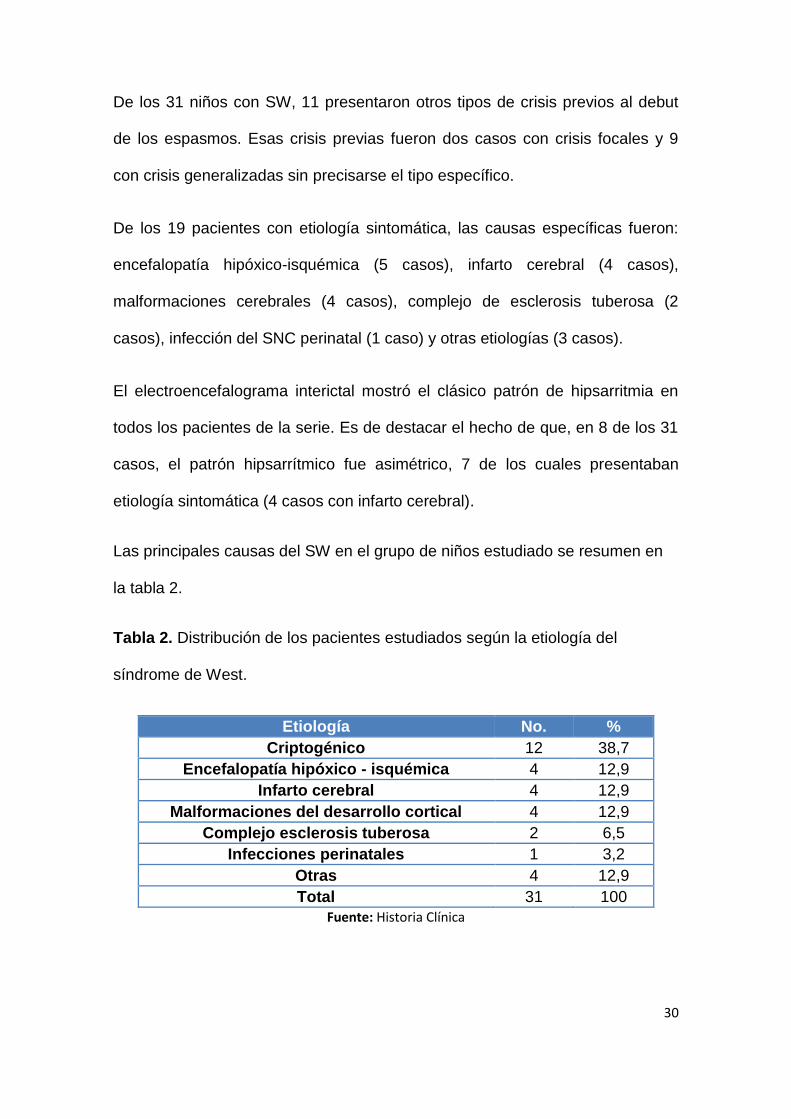
De los 31 niños con SW, 11 presentaron otros tipos de crisis previos al debut
de los espasmos. Esas crisis previas fueron dos casos con crisis focales y 9
con crisis generalizadas sin precisarse el tipo específico.
De los 19 pacientes con etiología sintomática, las causas específicas fueron:
encefalopatía hipóxico-isquémica (5 casos), infarto cerebral (4 casos),
malformaciones cerebrales (4 casos), complejo de esclerosis tuberosa (2
casos), infección del SNC perinatal (1 caso) y otras etiologías (3 casos).
El electroencefalograma interictal mostró el clásico patrón de hipsarritmia en
todos los pacientes de la serie. Es de destacar el hecho de que, en 8 de los 31
casos, el patrón hipsarrítmico fue asimétrico, 7 de los cuales presentaban
etiología sintomática (4 casos con infarto cerebral).
Las principales causas del SW en el grupo de niños estudiado se resumen en
la tabla 2.
Tabla 2. Distribución de los pacientes estudiados según la etiología del
síndrome de West.
Etiología No. %
Criptogénico 12 38,7
Encefalopatía hipóxico - isquémica 4 12,9
Infarto cerebral 4 12,9
Malformaciones del desarrollo cortical 4 12,9
Complejo esclerosis tuberosa 2 6,5
Infecciones perinatales 1 3,2
Otras 4 12,9
Total 31 100
Fuente: Historia Clínica

31
Los SW criptogénicos representaron el 38,7% de la serie, el resto (61,3%)
fueron sintomáticos, siendo la encefalopatía hipóxico-isquémica, el infarto
cerebral y las malformaciones del desarrollo cortical, las más frecuentes.
Se pudieron precisar datos evolutivos en 26 de los 31 pacientes lo que
representó el 83,9 % del total de la serie.
La evolución de los EE en los niños estudiados se puede apreciar en la figura
1.
Figura 1. Distribución de los pacientes según evolución de las crisis de
espasmos epilépticos.
Fuente: Historia Clínica
Se destaca que no se logró el control de las crisis en casi dos de cada tres
pacientes.
Al realizar un análisis del control de las crisis según la etiología del SW, se
encontró que, en el grupo con desaparición de las crisis, hubo más casos con
etiología criptogénica, a diferencia del grupo en que persistieron las crisis,

32
donde hubo un predominio de casos con etiología sintomática, aunque esta
diferencia no fue estadísticamente significativa. Ver tabla 3.
Tabla 3. Distribución de los pacientes según etiología y evolución de los
espasmos epilépticos.
Etiología
Evolución de Espasmos Epilépticos Total
Desaparición Persistencia
No. % No. % No. %
Sintomático 4 15,4 11 42,3 15 57,7
Criptogénico 6 23,1 5 19,2 11 42,3
Total 10 38,5 16 61,5 26 100
x2= 2,1 p ˃ 0,05
Fuente: Historia Clínica
Se logró precisar la evolución del EEG en 26 pacientes, en 9 de los cuales
ocurrió una desaparición del patrón de hipsarritmia. La relación entre la
evolución del patrón electroencefalográfico y la etiología del SW, se muestra en
la tabla 4.
Tabla 4. Distribución de los pacientes según etiología y evolución de
hipsarritmia.
Etiología
Evolución de Hipsarritmia Total
Desaparición Persistencia
No. % No. % No. %
Sintomático 4 15,4 11 42,3 15 57,7
Criptogénico 5 19,2 6 23,1 11 42,3
Total 9 34,6 17 65,4 26 100
x2= 0,9 p ˃ 0,05
Fuente: Historia Clínica
Se puede precisar que prácticamente la mitad de los casos con SW
criptogénicos tuvieron una desaparición del patrón de hipsarritmia, que solo fue

33
posible en menos de un tercio de los casos con SW sintomáticos. Estas
diferencias no fueron estadísticamente significativas (x2= 0,9 p ˃ 0,05).
De los 26 casos en que se pudieron precisar datos evolutivos, 6 (23%) tuvieron
un cambio de síndrome epiléptico, tal como se puede apreciar en la tabla 5. De
los 20 casos restantes, 13 mantenían las crisis de EE, al momento de la última
evaluación, y 7 habían alcanzado el estado de control de crisis.
Tabla 5. Distribución de los pacientes según evolución a otro síndrome
epiléptico.
Evolución a otro síndrome epiléptico No. %
Sí
Epilepsia focal 2 7,7
Epilepsia generalizada 3 11,5
Síndrome de Lennox Gastaut 1 3,8
No
Control de Crisis 7 26,9
No control de Crisis a 13 50,0
Etiología Sintomático 8 30,8
Criptogénico 5 19,2
n= 26
Fuente: Historia Clínica
a: continúan con espasmos epilépticos
En la tabla 6, se muestra la distribución de los pacientes estudiados según la
presencia evolutiva de retraso global del neurodesarrollo (RGND), que se
presentó en la mayoría de los casos (88,5%).
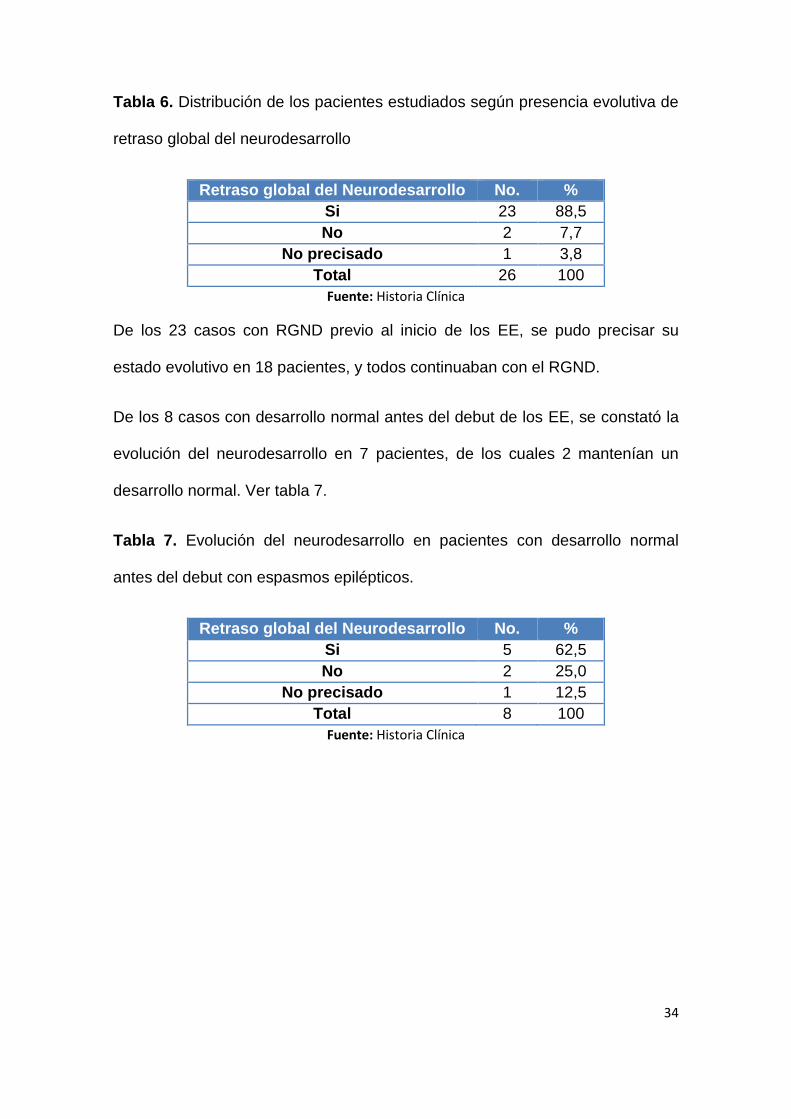
34
Tabla 6. Distribución de los pacientes estudiados según presencia evolutiva de
retraso global del neurodesarrollo
Retraso global del Neurodesarrollo No. %
Si 23 88,5
No 2 7,7
No precisado 1 3,8
Total 26 100
Fuente: Historia Clínica
De los 23 casos con RGND previo al inicio de los EE, se pudo precisar su
estado evolutivo en 18 pacientes, y todos continuaban con el RGND.
De los 8 casos con desarrollo normal antes del debut de los EE, se constató la
evolución del neurodesarrollo en 7 pacientes, de los cuales 2 mantenían un
desarrollo normal. Ver tabla 7.
Tabla 7. Evolución del neurodesarrollo en pacientes con desarrollo normal
antes del debut con espasmos epilépticos.
Retraso global del Neurodesarrollo No. %
Si 5 62,5
No 2 25,0
No precisado 1 12,5
Total 8 100
Fuente: Historia Clínica

35
DISCUSIÓN
El comportamiento de nuestra serie de casos de acuerdo a la distribución por
sexo mostró un predominio de los varones, con 20 casos (64,5%). Algunos
estudios han reportado que los niños y niñas se afectan por igual; mientras que
otros sugieren que el SW se presenta con más frecuencia en los varones, tal
como ocurrió en nuestra serie. 23-25, 109
Una de las principales características clínicas de los niños con SW lo
constituyen los EE, estas crisis suelen debutar en más del 90% de los casos
antes del año de edad, con un rango desde un día hasta los 4,5 años de edad.
11, 110
El pico de incidencia del debut de las crisis es entre los tres y siete meses de
edad; el debut después de los 18 meses de edad es raro. En nuestra serie solo
se presentó un caso con debut después del año de edad, una niña con inicio de
las crisis de espasmos epilépticos con 25 meses. Al analizar la edad media de
debut de los pacientes de nuestra serie, que fue de 5,5 meses de edad, se
evidencia un comportamiento similar al de otras series. 110
Las crisis de espasmos epilépticos han sido caracterizadas en tres tipos
clínicos, gracias a la revisión de las crisis por los registros video-
electroencefalográficos. En el estudio de Kellaway y cols. 111, reportaron 5042
crisis registradas en 24 lactantes, las cuales fueron clasificadas como flexoras,
extensoras o mixtas en 33,9; 22,5 y 42 % respectivamente.
En nuestra serie esta distribución fue de 35,5; 6,4 y 25,8 % respectivamente;
un comportamiento bastante parecido. Hay que destacar que la clasificación

36
del tipo de EE en nuestros casos fue realizada por juicio clínico, de acuerdo a
los resultados de la anamnesis, y no basados en la evaluación de registros
video-electroencefalográficos. 111
El carácter exacto de la crisis depende de si los músculos flexores o extensores
son predominantemente afectados y de la distribución de los grupos
musculares involucrados. 111
Las crisis de espasmos epilépticos (EE) pueden coexistir con otro tipo de crisis
en el mismo paciente. Las crisis focales pueden aparecer antes del debut de
los espasmos, coincidir con los EE, o aparecer una vez que los EE han cesado.
31, 111
El reconocimiento y la efectividad en el reporte de la cantidad y características
de los espasmos pueden ser difíciles tanto para padres como para el personal
médico. 111
Hay estudios realizados en los que se ha comparado los reportes de los padres
sobre las crisis de los niños y se han comparado con lo registrado en la
monitorización de video EEG, comprobándose que la mayoría de los padres
reportan de 5 a 10 veces menos crisis que lo evidenciado por el video EEG.
76,111
Este mismo análisis podría realizarse al evaluar la presencia de otro tipo de
crisis, que pueda preceder, coexistir o seguir a los EE, y que, al hacerse el
diagnóstico en base a los reportes de los familiares del niño, se corre el riesgo
de una observación no tan precisa como la que podría obtenerse a través del
video EEG. 76

37
Sin embargo, a pesar de esta limitación, la información obtenida a través de la
anamnesis es importante. Se demuestra en nuestra serie, como en los niños
con EE pueden presentarse otros tipos de crisis, incluso precediendo al debut
de los espasmos. 76
Otra de las principales características de los niños con SW, y que define su
tríada diagnóstica, lo constituye el patrón electroencefalográfico interictal de
hipsarritmia. Dicho patrón estuvo presente en el 100% de los casos al momento
la primera evaluación. Este patrón ha sido reportado entre el 7% y el 75% de
los pacientes con SW. 112
Hrachovy y cols. 113 han identificado cinco variaciones del patrón originalmente
descrito, pero en nuestro estudio no se entró en detalles sobre un subtipo en
específico de hipsarritmia.
La clasificación del SW está evolucionando. Tradicionalmente el SW se
clasifica como sintomático o criptogénico. Los pacientes con SW sintomáticos
tienen una etiología identificada y/o un retraso del neurodesarrollo al momento
del debut de los espasmos. Los casos con SW criptogénicos no tienen una
etiología conocida y presentan un neurodesarrollo normal al momento del inicio
de las crisis. 26, 70
Un grupo de trabajo sobre SW de la Liga Internacional Contra la Epilepsia
(ILAE) adicionó un grupo idiopático. En esta última categoría se incluyen
pacientes con neurodesarrollo normal al debut, examen físico y neuroimágenes
normales y patrón de hipsarritmia en el EEG, sin anomalías epileptiformes
focales. 26, 27

38
Las proporciones de las categorías etiológicas varían según diferentes
reportes. En uno de ellos, proveniente de estudios en las provincias
canadienses de las islas de Nueva Escocia y Prince Edward desde 1978 a
1998, la proporción de las categorías etiológicas fue 68%, 24% y 8% para
sintomática, criptogénica e idiopática, respectivamente. 24
En nuestra serie no se encontraron casos idiopáticos, pero si predominaron los
casos sintomáticos, que es lo reportado en diferentes series. En una serie de
140 niños afectados cuya evaluación incluyó estudios de neuroimágenes de
tomografía computarizada, resonancia magnética y/o tomografía por emisión
de positrón, el 96% de los casos fueron clasificados como sintomáticos. 28
La evaluación de estos pacientes fue más extensa y exhaustiva que lo habitual
en nuestro medio; es muy poco probable que los pacientes evaluados en la
comunidad puedan tener tan alta proporción de casos sintomáticos como lo
referido en el anterior estudio. 28
De forma general se puede considerar que el 20% de los casos con SW son
clasificados como criptogénicos, y el 80% restante como sintomáticos. 114 En
un estudio multicéntrico, prospectivo, que reunió 251 casos con SW, se
identificó la causa (sintomáticos) en 161 pacientes (64%). 29
El tiempo crítico del insulto típicamente ocurre en el período prenatal, perinatal
o postnatal temprano. Los factores prenatales representan la mayor proporción
de casos del SW, 43% en una serie 14, 32 similar resultado encontramos en
nuestra serie, con 10 de los 19 casos sintomáticos con factores prenatales.

39
La anormalidad prenatal más frecuente es la malformación del sistema
nervioso central (SNC); también se incluyen en este grupo los trastornos
genéticos y cromosómicos, los síndromes neurocutáneos, los errores
congénitos del metabolismo y las infecciones intrauterinas. 14, 32
En un estudio realizado en Reino Unido por Osborne y colaboradores 14, se
demostró la etiología en el (61%) de los pacientes con SW, de los cuales la
etiología más común fue la encefalopatía hipóxico-isquémica (10%),
cromosómicas (8%), malformaciones del desarrollo cortical (8%), el ictus
isquémico (8%), complejo esclerosis tuberosa (7%) y las enfermedades
metabólicas. 115,116.
En nuestro estudio, no se logró el control de las crisis en 16 pacientes. La
etiología estuvo asociada a la evolución del SW, predominó la persistencia de
las crisis y la hipsarritmia en los casos con etiología sintomática, mientras que
en los casos con etiología criptogénica fue más frecuente la desaparición de las
crisis y la hipsarritmia. Se ha descrito que la hipsarritmia tiende a desaparecer
con la maduración118, 119. Jeavons plantea que las crisis se detienen a los tres
años de edad en aproximadamente la mitad de los niños con EE y raramente
persiste después de que ellos son cinco años de edad. 117, 118
Se ha reportado una mejor evolución de las crisis en pacientes con EE
criptogénicos y en aquéllos con inicio temprano con EE, aunque estas
relaciones no se han encontrado en otros estudios. 80, 119 En los casos
sintomáticos, el resultado es más favorable en algunas etiologías como los

40
controles de crisis relativamente buenos reportados en pacientes con
neurofibromatosis tipo 1 o síndrome de Down. 34, 47, 120
En nuestro estudio, aproximadamente, uno de cada cuatro pacientes
evolucionó a otro síndrome epiléptico. En general, los pacientes con SW
sintomático tienen más probabilidad de desarrollar otro síndrome epiléptico que
aquéllos con SW criptogénico. Esta evolución se ha reportado que ocurre en el
57,5 % de los casos sintomáticos y el 35,3 % de los criptogénicos. 121, 122
Aproximadamente del 27 al 50 % de los pacientes con SW desarrollan una
forma severa de epilepsia conocido como síndrome de Lennox-Gastaut. 30, 81,
123 Rantala y cols. 124 reportan que el síndrome de Lennox-Gastaut se precedió
por EE en 10 de 25 casos. En la literatura publicada, sólo niños con SW
sintomático han evolucionado al síndrome de Lennox-Gastaut. 30, 81, 124
El retraso del neurodesarrollo ocurre en aproximadamente 85 % de los
pacientes; la mayoría tiene un déficit neurológico mayor (50 %) o menor (40 %).
31, 71, 78, 124 Resultados similares encontramos en nuestra serie, donde la
mayoría de los casos presentaron evolutivamente retraso global del
neurodesarrollo, incluso en aquellos con desarrollo normal al momento del
debut de las crisis de EE.
El pronóstico del neurodesarrollo a largo plazo parece depender de la etiología
subyacente. 31, 71, 125 Koo y cols. 123 estudiaron a 57 niños seguidos durante 12
a 60 meses y encontraron que el 44 % presentaron deterioro cognoscitivo
severo; mientras que en el 28 % tenían deterioro ligero o eran normales.

41
En otra serie casos de 68 niños, todos con SW sintomático, el retraso del
desarrollo se observó en el 100 % de los pacientes. 122 En otro estudio de 95
niños con SW, se reportó que el desarrollo de trastornos del espectro autista
era más frecuente en los casos sintomáticos que en los criptogénicos (8,73 %
vs 1,55 %, respectivamente). 125
Los trastornos cognitivos son comunes en niños con SW criptogénico, incluso
cuando los espasmos cesan. En un estudio, 126 el neurodesarrollo se evaluó,
por cuatro a seis años, en 15 niños en los cuales desaparecieron los espasmos
antes del primer año de edad; y se encontró una inteligencia normal en 12
niños. Sin embargo, el 41,7 % de los pacientes tuvieron trastornos cognitivos
específicos como problemas de atención, idioma, memoria, y las habilidades
viso-motoras.
Berg y cols. en un estudio prospectivo en niños menores de 3 años con
epilepsia de inicio reciente, encontraron tres factores que se asociaron de
forma independiente con una disminución del cociente de desarrollo a lo largo
del tiempo: presencia de síndromes considerados como encefalopatías
epilépticas, la etiología sintomática y la epilepsia refractaria. 128
En los niños sin ninguno de estos factores de riesgo no se observó disminución
del cociente de desarrollo. No obstante, este tipo de estudios no permiten
separar claramente el efecto de las crisis epilépticas del de la causa
subyacente. 127

42
La mejoría de los déficits cognitivos tras la resolución de las crisis epilépticas
en respuesta a la cirugía sugiere también un efecto deletéreo de ellas. Un
estudio de cohortes multicéntrico comparó los pacientes con encefalopatías
epilépticas tratados con fármacos y aquellos sometidos a cirugía resectiva o
paliativa. 129
Debe tenerse en cuenta que, de entrada, ambos grupos no son comparables
porque los pacientes intervenidos quirúrgicamente tenían obviamente alguna
indicación para ello. Como era de esperar, se observó una mayor proporción de
pacientes libres de crisis en aquellos tratados quirúrgicamente, que sólo fue
estadísticamente significativa para el subgrupo de cirugía resectiva. 128
Algunos estudios en niños con esclerosis tuberosa sugieren una relación entre
las crisis epilépticas y el deterioro neurológico. En un estudio prospectivo en
niños seguidos desde antes del inicio de las crisis, se observó una caída en el
cociente de inteligencia tras el inicio de los espasmos epilépticos, que fue
mayor en los casos con una mayor duración de los espasmos. En cambio, no
se observó este efecto en los niños que desarrollaron otros tipos de crisis. 117,
129
Otro estudio de cohortes prospectivo no aleatorizado, con un diseño antes-
después, mostró que la realización de electroencefalogramas periódicos y el
inicio del tratamiento preventivo con vigabatrina, en el caso de que se
detectaran alteraciones epileptiformes, e iniciado antes de la aparición de crisis
epilépticas, mejoró el control de las crisis y el cociente de desarrollo a los 24

43
meses de edad en comparación con los niños tratados tras el inicio de las
crisis. 130
No obstante, la contribución relativa de las crisis epilépticas y de la activación
de la vía mTOR en el origen de los déficits cognitivos en la esclerosis tuberosa
es todavía incierto. 131
El retraso mental ocurre en el 90 % de los casos y con frecuencia se asocia
con déficit motor, trastornos de conducta y rasgos autistas. La mortalidad es
del 5%; y del 55 a 60% de los niños con síndrome de West desarrollan
posteriormente otros tipos de epilepsia como el síndrome de Lennox-Gastaut y
epilepsias con crisis parciales complejas. 82
Un factor importante que contribuye a emitir un pronóstico, es si el paciente
inicialmente se clasifica como criptogénico, idiopático o sintomático. El
pronóstico es mejor en los casos idiopáticos y criptogénicos. 82
El pronóstico del síndrome de West idiopático es favorable con desaparición de
las crisis y un desarrollo psicomotor normal. En los casos criptogénicos la
demora en el inicio del tratamiento puede asociarse con un peor pronóstico
desde el punto de vista cognitivo. 82

44
CONCLUSIONES
En la serie estudiada de niños con SW se encontró un predominio de
varones. La edad promedio de debut de los espasmos epilépticos fue
similar a lo reportado en otras series.
La etiología más frecuente del síndrome de West fue la sintomática,
dentro de estas predominaron la encefalopatía hipóxico-isquémica, el
infarto cerebral y las malformaciones del desarrollo cortical.
Predominó la persistencia de las crisis y la hipsarritmia en los casos con
etiología sintomática, mientras que en los criptogénicos existió mayor
tendencia al control de las crisis y la desaparición de la hipsarritmia.
La evolución a otro síndrome epiléptico se presentó solo en casos
sintomáticos.
En la mayoría de los casos, evolutivamente presentaron retraso global
del neurodesarrollo, incluso los que iniciaron con desarrollo normal.
Se confirma en nuestra serie la pobre evolución de los pacientes con
SW, tanto por el control de las crisis, como por el retraso del
neurodesarrollo asociado, que hace de este tipo de epilepsia una de las
formas con mayor afectación a la salud del niño.

45
REFERENCIAS BIBLIOGRÁFICAS
1 Van der Berg BJ, Yerushalmy J. Studies on convulsive disorders in young
children. I. Incidence of febrile and nonfebrile convulsions by age and other
factors. Pediatr Res 1969; 3:298.
2 Nelson, K. Discussion: The Epidemiology of Epilepsy: A Workshop 1972.
3 Licourt Otero D, Travieso Tellez A. A strategy of genetic assessment in West’s
síndrome. Rev. Ciencias Médicas. 2013; 17(1):63-72
4 Okumura A, Watanabe K. Clinico-electrical evolution in pre-hypsarrhythmic
stage: towards prediction and prevention of West syndrome. Brain Dev.
2001;23(7):482–487.
5 Hrachovy RA, Frost J. Infantile Spasms. In: Dam l, editor. Comprehensive
epileptology. New York: Raven Press; 1990. p. 113-21.
6 Hamano S, Tanaka M, Mochizuki M. Long term follow up study of West
syndrome: differences of outcome among symptomatic etiologies. J Pediatr.
2003; 143:231-235.
7 Hino-Fukuyo N, Haginoya K, Iinuma K. Epidemiological and clinical studies of
West syndrome in Miyagi Prefecture, Japan. No To Hattatsu]; 39(4):257-61.
8 Oguni H, Otsuki T, Kobayashi K, Inoue Y, Watanabe E, Sugai K, et al. Clinical
analysis of catastrophic epilepsy in infancy and early childhood: results of the
Far-East Asia Catastrophic Epilepsy (FACE) study group. Brain Dev 2013; 35:
786-92.
9 Glaze DG. Management and prognosis of infantile spasms. UPTODATE.
2017.

46
10 Gurnet CA, Hedera P. New ideas in epilepsy genetics: novel epilepsy genes,
copy number alterations, and gene regulation. Arch Neurol 2007; 64(3): 324-8
11 Pellock JM, Hrachovy R, Shinnar S, Baram TZ, Bettis D, Dlugos DJ, et al.
Infantile spasms: A U.S. consensus report. Epilepsia. 2010;51(10):2175–2189.
12 Lux AL, Osborne JP. A proposal for case definitions and outcome measures
in studies of infantile spasms and West syndrome: consensus statement of the
West Delphi Group. Epilepsia. 2004;45(11):1416–1428.
13 Cowan LD, Hudson LS. The Epidemiology and Natural History of Infantile
Spasms. J Child Neurol. 1991;6:355-364.
14 Osborne JP, Lux AL, Edwards SW, Hancock E, Johnson AL, Kennedy CR, et
al. The underlying etiology of infantile spasms (West syndrome): Information
from the United Kingdom Infantile Spasms Study (UKISS) on contemporary
causes and their classification. Epilepsia. 2010;51(10):2168–2174.
15Wilmshurst JM , Ibekwe RC, O’ Callaghan FJK. Epileptic spasms - 175 years
on: Trying to teach an old dog new tricks. Seizure: Eur J Epilepsy. 2016.
16 Frost JD, Hrachovy RA, Kellaway P, Zion T. Quantitative analysis and
characterization of infantile spasms. Epilepsia. 1978;19:273–282.
17 Pozo Lauzán DR, Pozo Alonso AJ, Sayú Stewart JM. El
electroencefalograma en el síndrome de West y otras entidades clínicas
relacionadas. Revista Cubana de Pediatría. 2015;87(3):362-370.
18 Wilmshurst JM, Gaillard WD, Vinayan KP, Tsuchida TN, Plouin P, Van
Bogaert P, et al. Summary of recommendations for the management of infantile
seizures: Task Force Report for the ILAE Commission of Pediatrics. Epilepsia
2015;56:1185–97

47
19 Riikonen R. Epidemiological data of West syndrome in Finland. Brain Dev.
2001;23:539–541.
20 Ludvigsson P, Olafsson E, Sigurthardottir S, Hauser WA. Epidemiologic
features of infantile spasms in Iceland. Epilepsia. 1994;35:802–805.
21 Trevathan E, Murphy CC, Yeargin-Allsopp M. The Descriptive Epidemiology
of Infantile Spasms Among Atlanta Children. Epilepsia. 1999;40(6):748-751.
22 Primec ZR, Kopac S, Neubauer D. Epidemiologic Features of Infantile
Spasms in Slovenia. Epilepsia. 2002;43(2):183–187.
23 Lúthvígsson P, Olafsson E, Sigurthardóttir S, Hauser WA. Epidemiologic
features of infantile spasms in Iceland. Epilepsia 1994; 35:802.
24 Brna PM, Gordon KE, Dooley JM, Wood EP. The epidemiology of infantile
spasms. Can J Neurol Sci 2001; 28:309.
25 Sidenvall R, Eeg-Olofsson O. Epidemiology of infantile spasms in Sweden.
Epilepsia 1995; 36:572.
26 Commission on Classification and Terminology of the International League
Against Epilepsy: Workshop on infantile spasms. Epilepsia 1992; 33:195.
27 Lux AL, Osborne JP. A proposal for case definitions and outcome measures
in studies of infantile spasms and West syndrome: consensus statement of the
West Delphi group. Epilepsia 2004; 45:1416.
28 Chugani HT, Conti JR. Etiologic classification of infantile spasms in 140
cases: role of positron emission tomography. J Child Neurol 1996; 11:44.
29 Wirrell EC, Shellhaas RA, Joshi C, et al. How should children with West
syndrome be efficiently and accurately investigated? Results from the National
Infantile Spasms Consortium. Epilepsia 2015; 56:617.

48
30 Kurokawa T, Goya N, Fukuyama Y, Susuki M, Seki T, Ohtahara S. West
syndrome and Lennox-Gastaut syndrome: a survery of natural history.
Pediatrics 1980;65:81-8.
31 Lombroso CT. A prospective study of infantile spasms: clinical and
therapeutic correlations. Epilepsia 1983;24:135-58.
32 Ohtahara S, Ohtsuka Y, Yamatogi Y, Oka E, Yoshinaga H, Sato M. Prenatal
etiologies of West syndrome. Epilepsia 1993;34:716-22.
33 Matsumoto A, Watanabe K, Negoro T, Sugiura M, Iwase K, Hara K, et al.
Infantile spasms: etiological factors, clinical aspects, and long term prognosis in
200 cases. Eur J Pediatr 1981;135:239-44
34 Watanabe K. West syndrome: etiological and prognostic aspects. Brain Dev
1998; 20:1.
35 van Bogaert P, Chiron C, Adamsbaum C, et al. Value of magnetic resonance
imaging in West syndrome of unknown etiology. Epilepsia 1993; 34:701.
36 Aydinli N, Calişkan M, Ozmen M, Tonguç E. Neuroradiologic aspects of West
syndrome. Pediatr Neurol 1998; 19:211.
37 Van Allen M, Clarren SK. A spectrum of gyral anomalies in Miller-Dieker
(lissencephaly) syndrome. J Pediatr 2008; 102:559.
38 Willis J, Rosman NP. The Aicardi syndrome versus congenital infection:
diagnostic considerations. J Pediatr 2009; 96:235.
39 Singh R, Gardner RJ, Crossland KM, et al. Chromosomal abnormalities and
epilepsy: a review for clinicians and gene hunters. Epilepsia 2014; 43:127.
40 Dobyns WB, Stratton RF, Parke JT, et al. Miller-Diekersyndrome:
lissencephaly and monosomy 17p. J Pediatr 1983; 102:552.

49
41 Aicardi J, Chevrie JJ, Rousselie F. [Spasma-in-flexion syndrome, callosal
agenesis, chorioretinal abnormalities]. Arch Fr Pediatr 1969; 26:1103.
42 Bertoni JM, von Loh S, Allen RJ. The Aicardi syndrome: report of 4 cases and
review of the literature. Ann Neurol 1979; 5:475.
43 Guerrini R, Sicca F, Parmeggiani L. Epilepsy and malformations of the
cerebral cortex. EpilepticDisord 2003; 5 Suppl 2:S9.
44 Rizzuto N, Ferrari G. Familial infantile myoclonic epilepsy in a family suffering
from tuberous sclerosis. Epilepsia 1968; 9:117.
45 Roth JC, Epstein CJ. Infantile spasms and hypopigmented macules. Early
manifestations of tuberous sclerosis. Arch Neurol 1971; 25:547.
46 Goh S, Kwiatkowski DJ, Dorer DJ, Thiele EA. Infantile spasms and
intellectual outcomes in children with tuberous sclerosis complex. Neurology
2010; 65:235.
47 Motte J, Billard C, Fejerman N, et al. Neurofibromatosis type one and West
syndrome: a relatively benign association. Epilepsia 1993; 34:723.
48 Goldberg-Stern H, Strawsburg RH, Patterson B, et al. Seizure frequency and
characteristics in children with Down syndrome. Brain Dev 2001; 23:375.
49 Silva ML, Cieuta C, Guerrini R, et al. Early clinical and EEG features of
infantile spasms in Down syndrome. Epilepsia 1996; 37:977.
50 Marshall CR, Young EJ, Pani AM, et al. Infantile spasms is associated with
deletion of the MAGI2 gene on chromosome 7q11.23-q21.11. Am J Hum Genet
2008; 83:106.
51 Striano P, Paravidino R, Sicca F, et al. West syndrome associated with 14q12
duplication sharboring FOXG1. Neurology 2014; 76:1600.

50
52 Tohyama J, Yamamoto T, Hosoki K, et al. West syndrome associated with
mosaic duplication of FOXG1 in a patient with maternal uniparentaldisomy of
chromosome 14. Am J Med Genet A 2011; 155A:2584.
53 Mignot C, Moutard ML, Trouillard O, et al. STXBP1-related encephalopathy
presenting as infantile spasms and generalized tremor in three patients.
Epilepsia 2015; 52:1820.
54 Michaud JL, Lachance M, Hamdan FF, et al. The geneticlandscape of
infantilespasms. Hum Mol Genet 2014; 23:4846.
55 Zhongshu Z, Weiming Y, Yukio F, et al. Clinical analysis of West syndrome
associated with phenylketonuria. Brain Dev 2001; 23:552.
56 Low Nl, Bosma JF, Armstrong MD, Madsen JA. Infantile spasms with mental
retardation. I. Clinical observations and dietary experiments. Pediatrics 1958;
22:1153.
57 Gkampeta A, Pavlou E. Infantile spasms (West syndrome) in children with
inborn errors of metabolism: a review of the literature. J Child Neurol 2012;
27:1295.
58 Mikaeloff Y, Plouin P, Dhondt JL, et al. Clinical and EEG video-polygraphic
features of epileptic spasms in a child with dihydropteridine reductase
deficiency. Efficiency of hydrocortisone. EpilepticDisord 2015; 2:213.
59 Sfaello I, Castelnau P, Blanc N, et al. Infantile spasms and Menkes disease.
EpilepticDisord 2000; 2:227.
60 Bahi-Buisson N, Kaminska A, Nabbout R, et al. Epilepsy in Menkes disease:
analysis of clinical stages. Epilepsia 2014; 47:380

51
61 Naito E, Ito M, Yokota I, et al. Gender-specific occurrence of West syndrome
in patients with pyruvate dehydrogenase complex deficiency. Neuropediatrics
2013; 32:295.
62 Tsao CY, Luquette M, Rusin JA, et al. Leigh syndrome, cytochrome C
oxidase deficiency and hypsarrhythmia with infantile spasms.
ClinElectroencephalogr 1997; 28:214.
63 Duffner PK, Cohen ME. Infantile spasms associated with histidinemia.
Neurology 2013; 25:195.
64 Shih VE, Efron ML, Moser HW. Hyperornithinemia, hyperammonemia, and
homocitrullinuria. A new disorder of amino acid metabolism associated with
myoclonic seizures and mental retardation. Am J DisChild 1969; 117:83.
65 Feldman, RA, Schwartz, JF. Possible association between cytomegalovirus
infection and infantile spasms. Lancet 1968; 1:180.
66 Guggenheim MA, Frost JD Jr, Hrachovy RA. Time interval from a brain insult
to the onset of infantile spasms. Pediatr Neurol 2008; 38:34.
67 Cusmai R, Ricci S, Pinard JM, et al. West syndrome due to perinatal insults.
Epilepsia 1993; 34:738.
68 Riikonen R. Decreasing perinatal mortality: unchanged infantile spasm
morbidity. DevMedChild Neurol 1995; 37:232.
69 Hrachovy RA, Frost JD Jr, Gospe SM Jr, Glaze DG. Infantile spasms
following near-drowning: a report of two cases. Epilepsia 1987; 28:45.
70 Dimassi S, Labalme A, Ville D, et al. Whole-exome sequencing improves the
diagnosis yield in sporadic infantile spasm syndrome. Clin Genet 2016; 89:198.

52
71 Glaze DG, Hrachovy RA, Frost JD, et al. Prospective study of outcome of
infants with infantile spasms treated during controlled studies of ACTH and
prednisone. J Pediatr 1988; 112:389.
72 Hancock EC, Osborne JP, Edwards SW. Treatment of infantile spasms.
Cochrane Database Syst Rev 2013; :CD001770.
73 Mackay MT, Weiss SK, Adams-Webber T, et al. Practice parameter: medical
treatment of infantile spasms: report of the American Academy of Neurology
and the Child Neurology Society. Neurology 2004; 62:1668.
74 Brunson KL, Khan N, Eghbal-Ahmadi M, Baram TZ. Corticotropin (ACTH)
acts directly on amygdala neurons to down-regulate corticotropin-releasing
hormone gene expression. Ann Neurol 2001; 49:304.
75 Baram TZ, Mitchell WG, Brunson K, Haden E. Infantile spasms: hypothesis-
driven therapy and pilot human infant experiments using corticotropin-releasing
hormone receptor antagonists. Dev Neurosci 1999; 21:281.
76 Glaze DG, Zion TE. Infantile spasms. Curr Probl Pediatr 1985; 15:1.
77 Appleton RE. West syndrome: long-term prognosis and social aspects. Brain
Dev 2001; 23:688.
78 Lacy JR, Penry JK.. Infantile spasms, Raven Press, New York 2014.
79 Gibbs EL, Fleming MM, Gibbs FA. Diagnosis and prognosis of hypsarhythmia
and infantile spasms. Pediatrics 1954; 13:66.
80 Hamano S, Yamashita S, Tanaka M, et al. Therapeutic efficacy and adverse
effects of adrenocorticotropic hormone therapy in west syndrome: differences in
dosage of adrenocorticotropic hormone, onset of age, and cause. J Pediatr
2006; 148:485.

53
81 Riikonen R.A long-term follow-up study of 214 children with the syndrome of
infantile spasms. Neuropediatrics 1982; 13:14-23.
82 Pozo Alonso AJ, Pozo Lauzán D, Pozo Alonso D. Síndrome de West:
Etiología, Fisiopatología, Aspectos Clínicos y Pronósticos. Rev Cubana Pediatr
2002;74(2):151-61.
83 Chugani HT, Da Silva E, Chugani DC. Infantile spasms: III. Prognostic
implications of bitemporal hypometabolism on positron emission tomography.
Ann Neurol 1996;39:643-49
84 Karagol U, Deda G, Uysal S, Kabakus N, Ibis E, Gencoglu A, et al. Cerebral
blood flow abnormalities in symptomatic West syndrome: a single photon
emission computed tomography study. Pediatr Int 2001;43;66-70.
85 Hattori H. Spontaneous remission of spasms in West syndrome-implications
of viral infection. Brain Dev 2001;23:705-7
86 Go CY, Mackay MT, Weiss SK, et al. Evidence-based guideline update:
medical treatment of infantile spasms. Report of the Guideline Development
Subcommittee of the American Academy of Neurology and the Practice
Committee of the Child Neurology Society. Neurology 2012; 78:1974.
87 Yanagaki S, Oguni H, Hayashi K, et al. A comparative study of high-dose and
low-dose ACTH therapy for West syndrome. Brain Dev 2015; 21:461.
88 Vigevano F, Cilio MR. Vigabatrin versus ACTH as first-line treatment for
infantile spasms: a randomized, prospective study. Epilepsia 1997; 38:1270.
89 Baram TZ, Mitchell WG, Tournay A, et al. High-dose corticotropin (ACTH)
versus prednisone for infantile spasms: a prospective, randomized, blinded
study. Pediatrics 1996; 97:375.

54
90 Liu ZL, He B, Fang F, et al. Analysis of single nucleotide polymorphisms in
the melanocortin-4 receptor promoter in infantile spasms. Neuropediatrics 2007;
38:304.
91 Hrachovy RA, Frost JD Jr, Glaze DG. High-dose, long-duration versus low-
dose, short-duration corticotropin therapy for infantile spasms. J Pediatr 1994;
124:803.
92 Hamano S, Yamashita S, Tanaka M, et al. Therapeutic efficacy and adverse
effects of adrenocorticotropic hormone therapy in west syndrome: differences in
dosage of adrenocorticotropic hormone, onset of age, and cause. J Pediatr
2006; 148:485.
93 Oguni H, Yanagaki S, Hayashi K, et al. Extremely low-dose ACTH step-up
protocol for West syndrome: maximum therapeutic effect with minimal side
effects. Brain Dev 2006; 28:8.
94 Mytinger JR, Quigg M, Taft WC, et al. Outcomes in treatment of infantile
spasms with pulse methylprednisolone. J Child Neurol 2014; 25:948.
95 Kossoff EH, Hartman AL, Rubenstein JE, Vining EP. High-dose oral
prednisolone for infantile spasms: an effective and less expensive alternative to
ACTH. Epilepsy Behav 2009; 14:674.
96 Fejerman N, Cersósimo R, Caraballo R, et al. Vigabatrin as a first-choice
drug in the treatment of West syndrome. J Child Neurol 2012; 15:161.
97 Lux AL, Edwards SW, Hancock E, et al. The United Kingdom Infantile
Spasms Study comparing vigabatrin with prednisolone or tetracosactide at 14
days: a multicentre, randomised controlled trial. Lancet 2004; 364:1773.

55
98 Petroff OA, Rothman DL, Behar KL, et al. Human brain GABA levels rise
rapidly after initiation of vigabatrin therapy. Neurology 1996; 47:1567.
99 Aicardi J, Mumford JP, Dumas C, Wood S. Vigabatrin as initial therapy for
infantile spasms: a European retrospective survey. Sabril IS Investigator and
Peer Review Groups. Epilepsia 1996; 37:638.
100 Cossette P, Riviello JJ, Carmant L. ACTH versus vigabatrin therapy in
infantile spasms: a retrospective study. Neurology 2012; 52:1691.
101 Bachman DS. Use of valproic acid in treatment of infantile spasms. Arch
Neurol 1982; 39:49.
102 Siemes H, Spohr HL, Michael T, Nau H. Therapy of infantile spasms with
valproate: results of a prospective study. Epilepsia 1988; 29:553.
103 Fisher E, Siemes H, Pund R, et al. Valproate metabolites in serum and urine
during antiepileptic therapy in children with infantile spasms: abnormal
metabolite pattern associated with reversible hepatotoxicity. Epilepsia 1992;
33:165.
104 Schachter SC. The next wave of anticonvulsants. Focus on levetiracetam,
oxcarbazepine and zonisamide. CNS Drugs 2000; 14:229.
105 Yum SW, Zhang J, Mo K, et al. A novel recessive Nefl mutation causes a
severe, early-onset axonal neuropathy. Ann Neurol 2009; 66:759.
106 Glauser TA, Clark PO, Strawsburg R. A pilot study of topiramate in the
treatment of infantile spasms. Epilepsia 2014; 39:1324
107 Weber A, Cole JW, Mytinger JR. Infantile Spasms Respond Poorly to
Topiramate. Pediatr Neurol 2015; 53:130.

56
108 Moseley BD, Nickels K, Wirrell EC. Surgical outcomes for intractable
epilepsy in children with epileptic spasms. J Child Neurol 2012; 27:713.
109 Trevathan E, Murphy CC, Yeargin-Allsopp M. The descriptive epidemiology
of infantile spasms among Atlanta children. Epilepsia 1999; 40:748.
110 Vigevano F, Fusco L, Cusmai R, et al. The idiopathic form of West
syndrome. Epilepsia 1993; 34:743.
111 Kellaway P, Hrachovy RA, Frost JD Jr, Zion T. Precise characterization and
quantification of infantile spasms. Ann Neurol 1979; 6:214.
112 Alva-Moncayo E, Díaz-Leal MC, Olmos-García De Alba G.
Electroencephalographic discoveries in children with infantile massive spasms
in Mexico. Rev Neurol. 2002; 34: 928-932.
113 Hrachovy RA, Frost JD Jr, Kellaway P. Hypsarrhythmia: variations on the
theme. Epilepsia 1984; 25:317.
114 Hussain SA, Kwong G, Millichap JJ, et al. Hypsarrhythmia assessment
exhibits poor interrater reliability: a threat to clinical trial validity. Epilepsia 2015;
56:77.
115 Yu JY, Pearl PL. Metabolic causes of epileptic encephalopathy. Epilepsy
Res Treat 2013; 2013: 124934.
116 Ramos-Lizana J. Encefalopatías epilépticas. Rev Neurol 2017; 64 (Supl 3):
S45-8.
117 Jeavons PM, Bower BD, Dimitrakoudi M. Long-term prognosis of 150 cases
of "West syndrome". Epilepsia 1973; 14:153.
118 Jeavons PM, Bower BD. The natural history of infantile spasms. Arch Dis
Child 1961; 36:17.

57
119 Partikian A, Mitchell WG. Neurodevelopmental and epilepsy outcomes in a
North American cohort of patients with infantile spasms. J Child Neurol 2010;
25:423.
120 Sanmaneechai O, Sogawa Y, Silver W, et al. Treatment outcomes of West
syndrome in infants with Down syndrome. Pediatr Neurol 2013; 48:42.
121 Montenegro MA, Eck K, Jacob S, et al. Long-term outcome of symptomatic
infantile spasms established by video-electroencephalography (EEG)
monitoring. J Child Neurol 2014; 23:1288.
122 Koo B, Hwang PA, Logan WJ. Infantile spasms: outcome and prognostic
factors of cryptogenic and symptomatic groups. Neurology 1993; 43:2322.
123 Rantala H, Putkonen T. Occurrence, outcome, and prognostic factors of
infantile spasms and Lennox-Gastaut syndrome. Epilepsia 1999; 40:286.
124 Chevrie JJ, Aicardi J. Convulsive disorders in the first year of life:
neurological and mental outcome and mortality. Epilepsia 2011; 19:67.
125 Saemundsen E, Ludvigsson P, Rafnsson V. Risk of autism spectrum
disorders after infantile spasms: a population-based study nested in a cohort
with seizures in the first year of life. Epilepsia 2008; 49:1865.
126 Gaily E, Appelqvist K, Kantola-Sorsa E, et al. Cognitive deficits after
cryptogenic infantile spasms with benign seizure evolution. Dev Med Child
Neurol 1999; 41:660.
127 Berg AT, Smith SN, Frobish D, Beckerman B, Levy SR, Testa FM, et al.
Longitudinal assessment of adaptive behavior in infants and young children with
newly diagnosed epilepsy: influences of etiology, syndrome, and seizure
control. Pediatrics 2004; 114: 645-50.

58
128 Otsuki T, Kim HD, Luan G, Inoue Y, Baba H, Oguni H, et al. Surgical versus
medical treatment for children with epileptic encephalopathy in infancy and early
childhood: results of an international multicenter cohort study in Far-East Asia
(the FACE study). Brain Dev 2016; 38: 449-60.
129 Humphrey A, MacLean C, Ploubidis GB, Granader Y, Clifford M, Haslop M,
et al; Tuberous Sclerosis 2000 Study Group. Intellectual development before
and after the onset of infantile spasms: a controlled prospective longitudinal
study in tuberous sclerosis. Epilepsia 2014; 55: 108-16.
130 Józwiak S, Kotulska K, Domanska-Pakiela D, Lojszczyk B, Syczewska M,
Chmielewski D, et al. Antiepileptic treatment before the onset of seizures
reduces epilepsy severity and risk of mental retardation in infants with tuberous
sclerosis complex. Eur J Paediatr Neurol 2011; 15: 424-31.
131 Curatolo P, Aronica E, Jansen A, Jansen F, Kotulska K, Lagae L, et al. Early
onset epileptic encephalopathy or genetically determined encephalopathy with
early onset epilepsy? Lessons learned from TSC. Eur J Paediatr Neurol 2016;
20: 203-11.

59
ANEXOS
Anexo 1.
TARJETA DE RECOLECCION DE DATOS
Instituto de Neurología y Neurocirugía Dr Rafael Estrada Gonzales Servicio de Neuropediatria Investigación para optar por el Título de Especialista de 1er Grado en Neurología
Título: Comportamiento evolutivo de una serie de pacientes con síndrome
de west desde abril del 2010 hasta mayo del 2017.
De la base de datos de los pacientes ingresados en la sala de neuropediatria del Instituto de Neurologia y Neurocirugia se extraen los siguientes datos HISTORIA CLINICA_________ DATOS SOCIODEMOGRAFICOS Nombres y apellidos:____________________________________________________________ Telefono:______________________________________________________________________ Direccion:_____________________________________________________________________ Genero
1. masculino 2. femenino
Raza 1. blanca 2. mestiza 3. negra 4. no se encuentran datos
DATOS CLINICOS:
Edad de ingreso__________
Edad debut de las crisis de espasmos epilépticos______
Edad última evaluación______
Presencia de otras crisis epilépticas previas a los EE. (1. Si 2.
No 3. No precisado): _____
Tipo de crisis: _____ 1. Focal 2. Generalizada
3. Desconocida___
Desarrollo psicomotor previo al inicio de los EE. Normal o
Anormal
Etiología SW (1. Sintomático 2. Criptogénico): ______

60
SW sintomático a: _______
1. Complejo de Esclerosis Tuberosa (CET)
2. Encefalopatía HI pre, peri y postnatal
3. Malformaciones cerebrales
4. Infecciones congénitas o adquiridas
5. Anomalías genéticas (cromosómicas y génicas)
6. Errores congénitos del metabolismo
7. Otra. Cuál?_______
Tipo de EI: Flexión, Extensión, Mixto
Examen Físico neurológico (normal o anormal):
____________________
EEG interictal: presencia de hipsarritmia al momento del
ingreso o diagnóstico de SW: (1. Si 2. No 3. No precisado):
_____
Estudio de neuroimágen: Normal, Anormal. Si anormal
alteración, describir._______
1. Simétrico o asimétrico_______
Evolución (según datos de última consulta o ingreso)
1. Supresión espasmos (1. Sí 2. No) ____
2. Edad al suprimirse los espasmos: _______ (en meses)
3. Si control de crisis, qué tiempo después del debut y del
inicio del tratamiento_______
4. Desaparición del patrón de hipsarritmia (1. Si 2. No 3.
No precisado) ___
5. Edad al desaparecer la hipsarritmia: _______ (en
meses)
6. Evolución a otro tipo de epilepsia (1. Sí 2. No) ____
7. Cuál tipo de epilepsia evolutiva. 1. SLG 2. Focal
sintomática
8. Comorbilidad neuropsiquiátrica:
Retraso del Neurodesarrollo o RM (Sí, No)____
Trastorno del aprendizaje (1. Si 2. No 3. No
precisado) ___
TDAH (1. Si 2. No 3. No precisado) ___
Trastorno de conducta (1. Si 2. No 3. No
precisado) ___
Espectro autista (1. Si 2. No 3. No precisado) ___
9. Mortalidad: _____ 1. Si 2. No
10. De haber fallecido: Causa:_____________
Número de FAE utilizados: _______
FAE usados: _________

61
1. ACTH
2. Vigabatrina
3. Valproato
4. Topiramato
5. Clobazam
6. Vit B6
7. Otro
1. Si logró control de crisis con qué
fármaco(s)_________________