resonance fluorescence and resonance raman … · raman spectroscopy of bromine and iodine vapor c....
TRANSCRIPT
Resonance Fluorescence and Resonance Raman Spectroscopy of Bromine and Iodine Vapor C. Frank Shaw, Ill University of Wisconsin-Milwaukee, Milwaukee, WI 53201
Resonance Raman (RR) spectroscopy and resonance fluo- rescence (RF) spectroscopy are two techniques for which the advent of lasers has generated renewed interest (2-11). In resonance Raman and resonance fluorescence, a molecule interacts with a photon which corresponds to an electronic transition. then re-emits liaht which has changed in energs - - -. by one or more vibrational and rotational quanta. Thus the soectrum of adiatomic eas such as bromine or iodine in either - process consists of an evenly spaced vibrational progression (overtones) as shown in Figure 1.
'rhe;e re( hnique; prohe the \,ihrational and rurarional en- err\' levels ofthe excited ~ n d r rwnd electronic states uigns- eo& molecules. The basic experimental results, can h e ra- tionalized in terms of the molecular orbital, anharmonic os- cillator, and rigid rotator quantum mechanical results of a diatomic molecule as developed in an undergraduate physical chemistrv course. Useful information about the s~ectroscopic constants and molecular properties of the molecule in various electronic states can be obtained by detailed analysis of the fine structure of the overtone bands. Although a theoretical understanding of RR and RF and the relationship between them is still heing developed, two competing interpretations of their relationship have been proposed and will he summa- . -
rized briefly. Resonance Raman spectroscopy (5) is an inelastic scattering
process in which the incident (laser) photon "excites" the molecule to a virtual state in the vibrational-rotational con- tinuum of the excited state, accompanied by instantaneous re-emission of a Raman scattered photon, that has lost several quanta of energy because the molecule returns to a ground electronic, but vibrationally excited, state (Fig. 2a). The res- onance fluorescence ~henomena (6) involves excitation to a discrete vibrational-rotational level of an excited electronic state, which after a finite time interval fluoresces, returning the molecule to a vibrationally excited level of the ground electronic state (Fig. 2b). The different modes of absorption and re-emission for RF and RR spectroscopy result in dif- ferent spectral characteristics which are summarized in Table 1. Although the two techniques are experimentally distinct under most conditions, some workers, have suggested a common origin for them. However, there remain serious questions about their relationship, which are still heing in- vestigated theoretically and experimentally E l l , 12,17).
The selection rules for the RF and RR effects are very dif- ferent from those for infrared and ordinary Raman spec- troscopy, where only the fundamental vihratkmal tran>>iun (u" = O - t:" = 11' is allowed t o i n the harnlunic nscillntnr approximation, and ovenonei and rumhination hands appear weakly usually less than 1'; of the fundamental intcnsity~ due u, anharmonicitvefferts. In contrast. the o\.erronesof rhr RF and RR spectraare of comparable intensity to the funda- mental, decreasing gradually for higher quantum numbers (Fig. 1).
' Double primes are used to indicate the vibrational and rotational quantum numbers of the electronic ground state; single primes, those of the electronic excited states.
Comparison of Resonance Raman and Resonance Fluorescence Eflects
Resonance Characteristic Fluorescence Resonance Raman
Excited state Discrete Cont8nwm of excited state vibrational- rotational quantum level
Excited state Relatively long Instantaneous decay ( < l o " lifetime (-lo-a set) sec)
Observed bands Vibrational Vibrational overtones OYerlOnes
Band intensities Irregular Regular decrease with variation vibrational quantum number
Fine structure Doublets Q branch and rotational wings from each thermally populated vibrational State
Polarization Depoiarized Totally symmetric vibrations are polarized
Effect of increased intensity intensity increases gas pressure decreases (concentration effect)
(quenching) Effect of foreign Intensity Unaffected
gases decreases iauenchind
RESONANCE FLUORESCENCE 514.5nn 8on4 slils
RESONANCE RAMIN 4 8 8 . 0 nrn 80.-8 s l i t s
Figure 1. (a) RF spectrum of Br*: 700 mwans of 514.5 nm excitation; 8 cm-' slits: 25 cm-'Imin scan rate: 3000 cps full scale, 5 sec period. (b) RR spectrum of Ere: 500 mwanr of 488.0 cm-' excitation: 8 cm-' slits: 5 cm-'lmin scan rate: 300 cps full scale, 2 second perid.
Because of their simole structure and well understood quantum levels, the hacogen gases, especially bromine and iodine, which have intense visible absorptions, have been used extensively for developing and testing. rhe theoretical harei fur H H and RF sDectroscuDv ( 2 , 3.6-10). Hromine is a svm- metrical diatomii m~lecul~ipoint group D-h) and, therefore, has only one fundamental frequency (322 cm-1) and one-set of rotational energy levels, which means that the vibrational and rotational spectra have simple, easily interpreted features. Figure 3 shows the Morse potential curves (18) for the ground
Volume 58 Number 4 April 1981 343

F g.re 7 trc ut on .m re-emm on of ognt n RF ilnd RR enoclr tal RF re-, MS lrom exc tauon lo a 0 screle Lmron L 61itle o RR re% ls from e r c !allon lntu the continuum of an excited state.
Figure 3. Morse potential curves for 'Z,+ and %.+ states of Br, (atter Ref 11). The ueflical arrows show me relationships of 514.5 and 488.0 nm exciting lines to the convergence limit of excited state.
state, I&+, and second excited state, 3IIo,+, of Brz, which are involved in the RR and RF phenomena discussed below. Figure 4 shows the orbital occupation (molecular orhital schemes) for the two states.
Each electronic state has associated with it a characteristic series of vihrational enerev levels. which are overtones of the fundamental frequency f& that electronic state. The spacing of the vihrational enerw levels. G(u) , are not exact multi~les of the fundamental frequency, hut become progressively clker according to the relationship below, where w, is the harmonic frequency and wax, is the inharmonicity constant (typically less than 1% of we), u is the vibrational quantum number (0, 1 , 2 . . .), h is Planck's constant, and c is the speed of light:
As u increases, the spacing eventually becomes so small that a continuum of vibrational states is present. This point is re- ferred to as the convergence limit.
In BIZ the 3110,+ excited state lies at 15,891 cm-' ahove the ground state. The green (514.5 nm) line of the argon ion laser, which has an energy of 19,429.5 cm-', excites a bromine molecule into one of the vibrational-rotational levels of the sou+ state giving rise to the RF spectrum. In contrast, the
- - (G+ 'no+"
G R O U N D S T A T E E X C I T E D STATE
Fiaure 4. MO diaarams shawfno the orbital occupations of '&+and3IIo,+ states of Br2.
- E m i t t e d
+ L i g h t *
Figure 5. Cell lor RR and RF spectra of gases
higher energy 488.0 nm exciting line, 20,486.5 cm-1, is ahove the convergence limit of the 3&,+ state, 19,585 cm-', and causes a ";irtual excitation" in;; the continuum of vibra- tional-rotational levels thereby stimulating the RR effect (3,6). For molecular iodine, the convergence limit of the 3&,+ state is 20,162 cm-1 and laser wavelengths longer than 496.5 nm fall below it in the reeion of discrete vibronic states. and shorter wavelengths cause excitation into the continuum of states above the limit (8). The details of the excitation and re-emission processes of RR and RF are discussed.
The RR and RF Experiments Resonance Raman and Resonance Fluorescence Spectra
can be recorded routinely on a conventional laser Raman spectrometer. The laser serves as an intense. coherent. narrow band width excitation source, and the high resolution spec- trometer is necessarv to resolve the scattered lieht into the vihmtional l~ands with associated rotational fine structure. The light is ~~~~~~~~d wirh a sensitive ph~,tomulriplier nil*. m d the resulting elecrronic signal is ampliiied and displavcd by conventional means. A sample of the gas to be examined is illuminated in a simple glasibr quartz cell. The main experi- mental limitation is that the path lengthof the scattered light throurrh the cell must he verv short to avoid reabsorntioihv othermolecules (Fig. 5). o he only differenceb&een thk experimental designs for RF and RR experiments is the re-
344 Journal of Chemical Education
vihrational states of the molecule (19). Since only one vibra- tional state of the excited molecule is populated, a progression of bands differing by a vihrational quantum is observed. An- other conseauenie is that the rotational selection rule for
I I
F R E Q U E N C Y (crn-l)
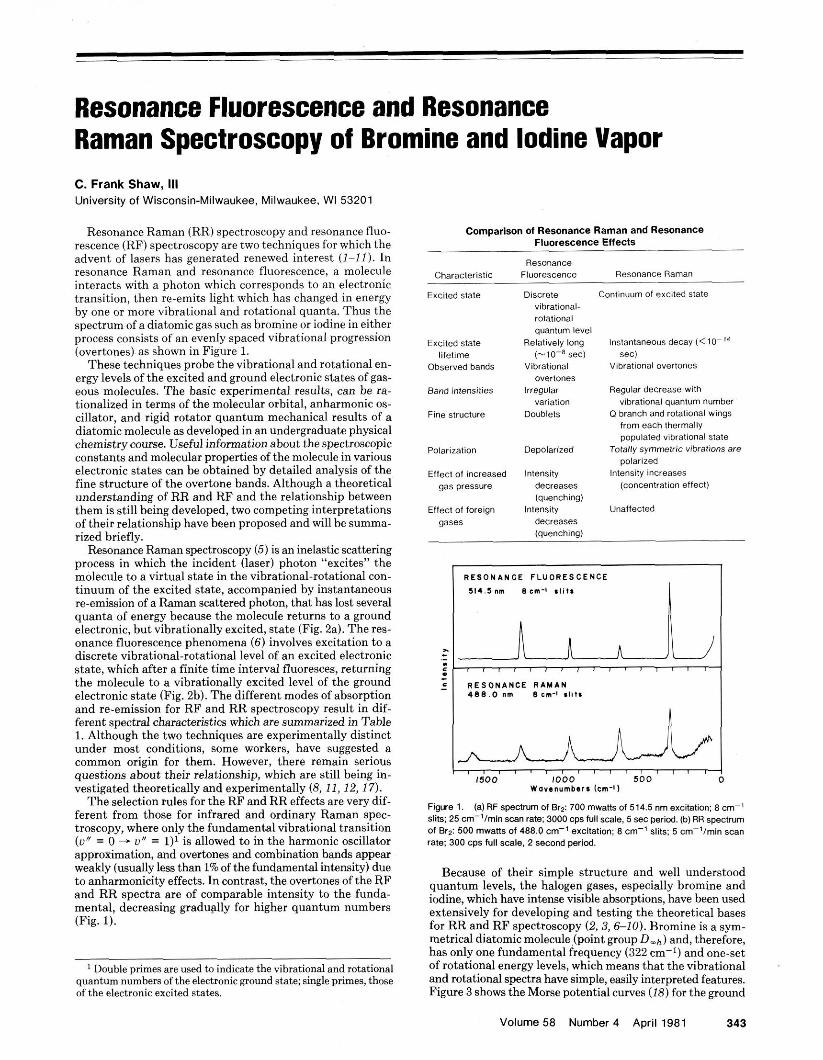
Figure 6. The doublet structure of the Av" = 1 band in the high resolution RF spectrum of Br2. excited with 514.5 nm laser line. Used with permission from Reference 3.
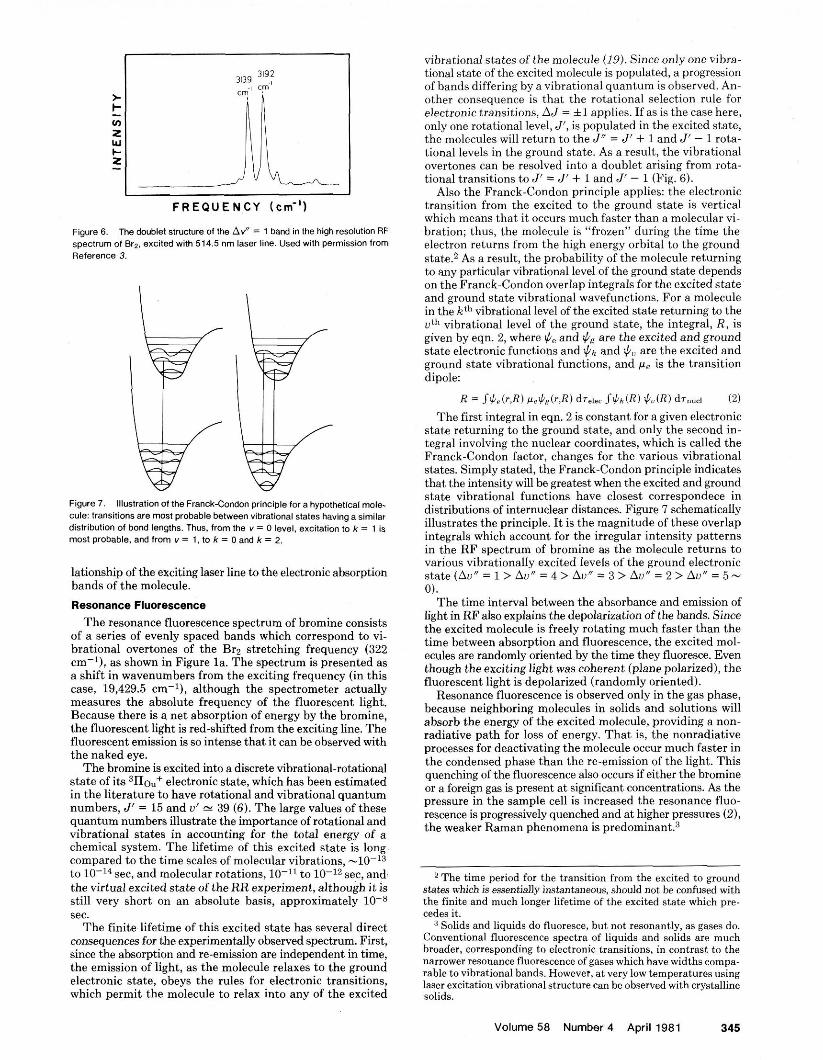
Figure 7. Illustration of the Franck-Condon principle for a hypothetical mole- cule: transitions are most probable between vibrational states having a similar distribution of bond lengths. Thus, from me v = 0 level, excitation to k = 1 is most probable, and from v = 1. to k = 0 and k = 2.
lationship of the exciting laser line to the electronic absorption bands of the molecule.
Resonance Fluorescence The resonance fluorescence spectrum of bromine consists
of a series of evenly spaced bands which correspond to vi- brational overtones of the Brz stretching frequency (322 cn-'), as shown in Figure la. The spectrum is presented as a shift in wavenumbers from the exciting frequency (in this case, 19,429.5 cm-'1, although the spectrometer actually measures the absolute frequency of the fluorescent light. Because there is a net absor~tion of enerev bv the bromine. -- - the fluorescent light is red-shifted from the exciting line. ~ h d fluorescent emission is so intense that it can be observed with the naked eye.
The bromine is excited into a discrete vibrational-rotational sratr of its ,'lloU7 electronic stale, which ha6 been estimated in the literature to have rotational and vibratimal quantum numl~era. .J' = 15 and I,' - 19 16). The Iar'le IWIUQS 01 thrsc quantuknnmbers illustrate the importanclof rotational and vibrational states in accountine for the total enerev of a chemical system. The lifetime i f this excited state$ long compared to the time scales of molecular vibrations. -10-'3 to 10-lP see, and molecular rotations, lo-" to 10-'2sec, and the virtual excited state of the RR experiment, altboueh i t is still very short on an absolute basis, appro&natel; 10-8 sec.
The finite lifetime of this excited state has several direct consequences for the experimentally observed spectrum. First. since the ahsorption and re-emission are independent in time, the emission of light, as the molecule relaxes to the ground electronic state, obeys the rules for electronic transitions, which permit the molecule to relax into any of the excited
electronic transitions, A J = f 1 applies. If as is the case here. only one rotational level, J', is populated in the excited state, the molecules will return to the J" = J' + 1 and J' - 1 rota- tional levels in the erouud state. As a result, the vihrational overtones can be r&lved into a doublet arising from rota- tional transitions to J' = J' + 1 and J' - 1 (Fig. 6 ) .
Also the Franck-Cundon principle applies: the electronic transition from the excited to the ground state is vertical which means that it occurs much faster than a molecular vi- bration; thus, the molecule is "frozen" during the time the electron returns from the hieh enerev orbital to the around state.2 As a result, the probability ofthe molecule returning to any particular vihrational level of the ground state depends on the Franck-Condon overlap integrals for the excited state and ground state vihrational wavefunctions. For a molecule in the kth vihrational level of the excited state returning to the uth vibrational level of the around state, the integral, R , is given by eqn. 2, where il., and fi8 are the excited a i d ground state electronic functions and $k and $, are the excited and ground state vibrational functions, and is the transition dipole:
R = J+,(r,R) &(r,R) dr,~,, S$a(R) v U R ) drnve~ (2)
The first inteeral in em. 2 is constant for a eiven electronic state returning& the ground state, and onl i the second in- t e ~ r a l involvine the nuclear coordinates, which is called the ~rinck-Condo; factor, changes for the various vihrational states. Simply stated, the Franck-Condon principle indicates that the intensity will he greatest when the excited and ground state vihrational functions have closest correspondece in distributions of internuclear distances. Figure 7 schematically illustrates the principle. It is the magnitude of these overlap integrals which account for the irreeular intensitv oatterns - u . . in the RF spectrum of bromine as the molecule returns to various vibrationallv excited levels of the around electronic state(Au" = 1 >A;,' = 4 > Au" = 3 > AU"= 2 > Au" = 5 - tll.
'The time interval hetween the ahsrrhanrr and emi-rim id lirht in HF also exl~lains thr d ~ ~ ~ ~ ~ r k ~ d f i ~ l l c t f the hnndr. :Sinre t i e excited molec>le is freelyktating much faster than the time between absorption and fluorescence, the excited mol- ecules are randomlymiented by the time they fluoresce. Even though the exciting light was coherent (plane polarized), the fluorescent light is depolarized (randomly oriented).
Resonance fluorescence is observed only in the gas phase, because neiehborine molecules in solids and solutions will absorb the energy of the excited molecule, providing anon- radiative Dath for loss of enerw. That is, the nonradiative ... processes fur deartivsting the mtdecnle #)(cur mu Ii fuarer in the cundens~d rlhase 1h.m lhr re.cmi.;iiun r , i thr lieht. 'l'hir quenching of thk fluorescence also occurs if either thebromine or a foreign pas is ureseut at significant concentrations. As the - - . pressure in the sample cell is increased the resonance fluo- rescence is promessively quenched and at higher pressures (21, the weaker-~a&an phenbmena is
cedes it. "Sods and liquids do fluoresce, but not resonantly, as gases do.
Conventional fluorescence spectra of liquids and solids are much broader, corresponding to electronic transitions, in contrast to the narrower resonance fluorescence of gases which have widths compa- rable to vibrational bands. However, at very low temperatures using h e r excitation vibrational structure can be observed with crystalline solids.
Volume 58 Number 4 April 1981 345
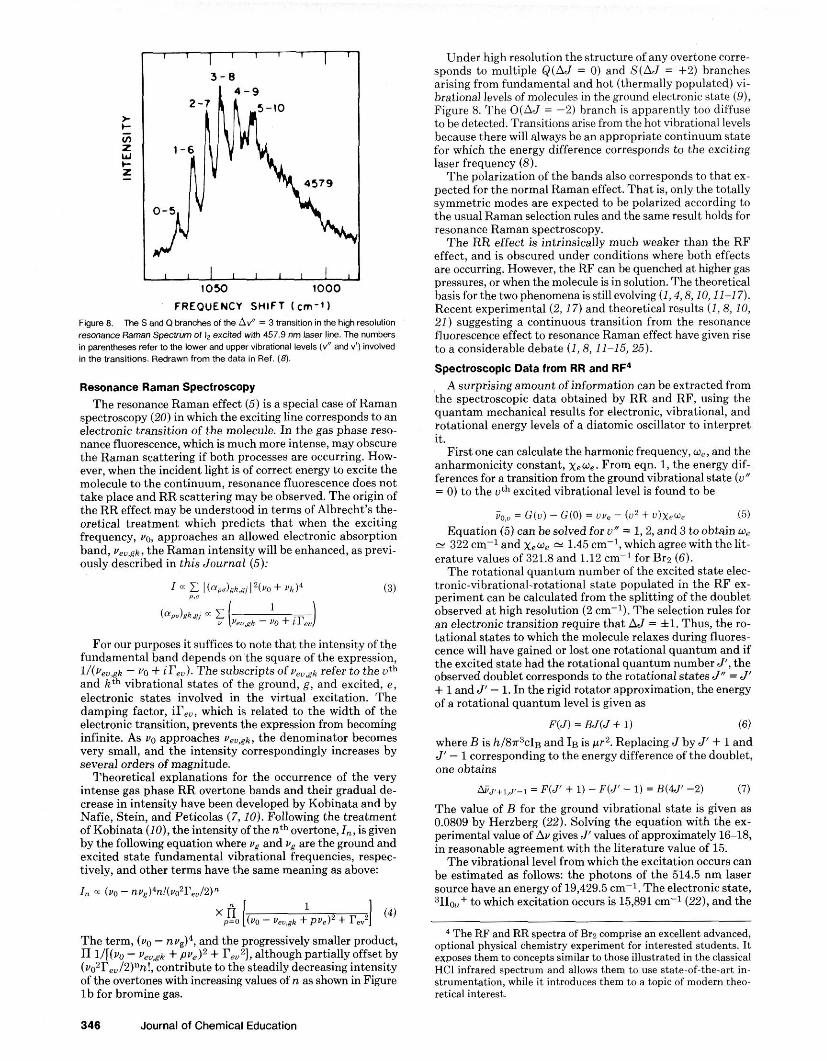
FREQUENCY SHIFT lcm-91 Figure 8. The S and Q branches of the Av" = 3 Vansition in the high resolution resonam R a m Spearurn of i, excited wim 457.9 nm laser line. The numbers in parentheses refw to me lower and upper vibrational levels (8 and v'l involved in the transitions. Redrawn from the data in Ref. (8) .
Resonance Raman Spectroscopy The resonance Raman effect (5) is a special case of Raman
spectroscopy (20) in which the exciting line corresponds to an electronic transition of the molecule. In the gas phase reso- nance fluorescence, which is much more intense, may obscure the Raman scattering if both processes are occurring. How- ever, when the incident light is of correct energy to excite the molecule to the continuum, resonance fluorescence does not take place and RR scattering may he observed. The origin of the RR effect may be understood in terms of Albrecht's the- oretical treatment which oredicts that when the excitinr - frequency, uo, approaches an allowed electronic absorption hand. u,,. ,b. the Raman intensitv will be enhanced. as orevi- , . . ously described in this Journal i5):
For our purposes it suffices to note that the intensity of the fundamental hand depends on the square of the expression, l/(v,,, k - vo + I P ~ " ) . The subscripts of ueUak refer to the uth 5 , . and k t vibrational states of the ground, g, and excited, e , electronic states involved in the virtual excitation. The damnine factor. ir,,,. which is related to the width of the . - , electronic transition, prevents the expression from becoming infinite. As un auoroaches u,,, .b. the denominator becomes - .. very small, and the intensity correspondingly increases by several orders of magnitude.
Theoretical explanations for the occurrence of the very intense gas phase RR overtone hands and their gradual de- crease in intensity have been developed by Kobinata and by Nafie, Stein, and Peticolas (7,101. Following the treatment of Kobinata (lo), the intensity of the nth overtone, I,,, is given by the following equation where v, and u, are the ground and excited state fundamental vibrational frequencies, respec- tively, and other terms have the same meaning as above:
I, 4 ("0 - n~~)'n!(vo~rJ2)"
The term, (uo - nu,)4, and the progressively smaller product, II l/[(uo - uegb + pue)2 + PJ, although partially offset by (uo2reU/2)"n!, contribute to the steadily decreasing intensity of the overtones with increasing values of n as shown in Figure Ih for bromine gas.
Under high resolution the structure of any overtone corre- sponds to multiple Q(AJ = 0) and S(AJ = +2) branches arising from fundamental and hot (thermally populated) vi- brational levels of molecules in the ground electronic state (91, Figure 8. The O(AJ = -2) branch is apparently too diffuse to he detected. Transitions arise from the hot vihrational levels hecause there will always be an appropriate continuum state for which the energy difference corresponds to the exciting laser freauencv (8). . . I he pdarizatism of the I~andi nlsu currhponds to !hat e x - iwcted fur the nmmal Rmanefiect. That i - , onis the to~alls . . Bymmetric modes are expected to be polarized according to the usual Raman selection rules and the same result holds for resonance Raman spectroscopy.
The RR effect is intrinsically much weaker than the RF effect. and is obscured under conditions where both effects ~~~ ~~, ~ ~ ~ ~
nre uccurring. Hcwe\.~r, the RF can t,c. quencl~cd nr h~gher yns ure>iures. or when the nit~lecule is in irhtion. The thwrrriml basis for the two phenomenais still evolving (1,4,8,10,11-17). Recent exoerimental(2. 17) and theoretical results (1.8.10. . . . . . 21) suggesting a continuous transition from the resonance fluorescence effect to resonance Raman effect have riven rise . to a considerable debate (1,8,11-15,25).
Spectroscopic Data from RR and RF4 A surwrisineamount of information can he extracted from
the spectroscopic data obtained by RR and RF, using the ouantam mechanical results for electronic, vibrational, and rotational energy levels of a diatomic oscillator to interpret it.
First one can calculate the harmonic frequency, we, and the anharmonicity constant, x,w,. From eqn. 1, the energy dif- ferences for a transition from the ground vibrational state (u" = 0) to the uth excited vibrational level is found to be
Fo,. = G(u) - G(0) = uu. - (u2 + u ) x , w , (5)
Equation (5) can he solved for u" = 1,2, and 3 toobtain w, -- 322 em-' and x,w, -- 1.45 em-', which agree with the lit- erature values of 321.8 and 1.12 cm-' for Brz (6 ) .
The rotational quantum number of the excited state elec- tronic-vibrational-rotational state populated in the R F ex- periment can he calculated from the splitting of the doublet observed at high resolution (2 cm-I). The selection rules for an electronic transition require that AJ = +I. Thus, the ro- tational states to which the molecule relaxes during fluores- cence will have gained ur lost unr rotarionJ qunntbm and it' the extitud srote had the rutntimal quantum numl~er J ' , rhe ~obier\.t-d rloulder arrresuonds ro rhe mraricmdl sratc..JW = J' + 1 and J' - 1. In the rigid rotator approximation, the energy of a rotational quantum level is given as
F(J) = BJ(J + I) ( 6 )
where B is h/8a3cIB and IB is prZ. Replacing J by J' + 1 and J' - 1 corresponding to the energy difference of the doublet, one obtains
AiJ,+lJ-l = F(J' + 1) - F(J' - 1) = B(4J' -2) (7)
The value of B for the ground vihrational state is given as 0.0809 bv Herzbere (22). Solvine the eauation with the ex- perimen& value or^" gives J' vzues of ~pproximately 1618, in reasonable agreement with the literature value of 15.
The vibrational level from which the excitation occurs can be estimated as follows: the photons of the 514.5 nm laser source have an energy of 19,429.5 cm-'. The electronic state, 3110u+ to which excitation occurs is 15,891 cm-' (22), and the - - - -
The RF and RR spectra of Br2 comprise an excellent advanced, optional physical chemistry experiment for interested students. It exnoses them to concepts similar to those illustrated in the classical HC1 infrared spectrum and allows them to use state-of-the-art in- strumentation, while it introduces them to a topic of modern theo- retical interest.
346 Journal of Chemical Education

additional energy of the exciting photons must correspond to the vibrational state of the excited molecule. The quantum number, u', can he estimated using eqn. 1 and the literature values for w, (169.7cm-l) and ,yew, (1.9cm-') of the "lou+ state as tabulated by Herzherg (221, a value for u' of 38 is oh- tained. This calculation ignoring higher order corrections is in good agreement with the literature estimate of u' = 39 ( 6 ) .
The harmonic frequencies for the I&.+ (ground) and 3110,+
(excited) states can be used to calculate force constants. and hence the curvature of the potential wells, for each electronic state. usine the eauation (which aodied onlv to diatomic . . molecules),
f = 4rr2,2u12fi/N (8)
where fi = reduced mass (glmole), c = speed of light (cmlsec), w, = harmonic vibrational frequency (em-'), f = force con- stant (Nlm ), and N = Avogadro's Number. The constants in eqn. 8. 4n2cLIN.have a value of 5.892 X loL5 kg-cm2-mole/ g-8ec" The calculated force constants, 244 Nlm for the ground state and 67.7 Nlm for the excited state, can be related to the curvature of the potential wells in ~ i g u r e 3. These numerical values clearlv emphasize the theoretical meanine of the force " . constant:
aZE a2E /=-=- aQZ arBrBIZ
(9)
E is the potential energy and r is the internuclear distance (bond stretching coordinate). The molecular orbital scheme of Figure 4 indicates that an electron has been promoted to a more antihonding level in the 3&,+ state. Thus, its poten- tial well is shallower, resulting in the smaller force con- stant. Time-Resolved Spectroscopy
An important advance during the past decade has been the develonment of time-resolved snectroscoov. TRS (8. 17). .", . , .. which itilizes a pulsed laser sourEe and very fast light detec- tion and amplification svstems to measure Raman and fluo- rescent emiision as a function of time following excitation of the sample by a pulsed laser source. Using a mode-locked. cavity d k p e d laser, which utilizes a c o u s t ~ ~ o ~ t i c a l coupling techniques, very short (less than a nanosecond) hut extremelv intm>e l , ~ i i ~ pulses can he rrp(~titivt4~ gmeruted, since a large I'ractiun or the rndiahn in the ln;er cavity is releainl in a very short time span. In a conventional or continuous-wave laser: a small amount of the radiation in the cavity (1-2%) is con- tinuously bled through an "imperfect" mirror: while in the pulsed laser, a burstbf intense light travelling in the laser cavity is periodically "dumped" to the exterior (23). Time resolved RF and RR spectra are obtained by monitoring the light emitted a t a single frequency as a function of time after the sample is excited by the laser pulse. The details of the sophisticated electronics required to carry out such mea- surements are beyond the scope of this article and can he found in the literature (24). However, the basic principle of the system is straightforward: the numbers of photons reaching the detector a t given time intervals after the laser is pulsed are accumulated in a multichannel analyzer (each channel corresponding to a different time interval). After a predetermined number of laser pulses. the data are disnlaved
displayed in Figure 9D. Time-resolved soectroscoov has been used to examine the
it shbnld be possible to explore the two phenomena by this method.
Figure 9 shows the TRS of molecular iodine (redrawn from the data of Ref. (8)), when the laser is in exact resonance with
T I M E ( " , . < I
Figure 9. Time-resolved spectra of l2 vapor excited with various modes of the 514.5 nm laser line. The intensity scale (photon counts) is arbitrary and varies for each spectrum. The laser excites lhe molecule from the v" = 0, J" = 15 vibronic level of the 'zg+ lo the d = 43, ,' = 16 vtbronlc level of the 3110u* excited state. The intensity is monitored at 216 cm-' which corresponds to emission which returns the molecule to the k" = 1. S = 17 level of the ground state. A. exact resonance of the laser with the initial ground state and excited statetransitionlAw =01.8. off resonance bv 0.040cm-'. C. off resonance bv 0.073 cm-'. D. he ti&dependenceof the &citing laser pulse (100 nsec width) for comparison. Redrawn from the data in Ref. 8.
and then slightly off-resonance from a electronic-vihra- tional-rotational transition from the u" = 0, J" = 15 vihronic level of the 'ZK+ ground state to the u' = 43, J' = 16 vihronic level of the 3110,+ electronic state, excited by the 514.5 nm argon ion laser line. This excited state vihronic level lies below the convergence limit, and when it is exactly excited, results in an RF-like spectrum in which the excited state has a lorg lifetime (Fie. 9A). . - .
Very fine tuning of the laser can be achieved using various modes of the laser (which result from slight changes in the geometry of the laser cavity). Each mode of the 514.5 nm line varies bv 130 MHz or 0.0043 cm-'. Using different modes of this line-spectra were obtained with the laser slightly offres- onance ( 4 w = 0.040 and 0.073 cm-I). I t can he seen in Figures 9B and C that the time dependence of the emitted light is dramatically changed, becoming progressively closer to the pattern of the exciting laser pulse (Fig. 9D). Although the spectra are plotted on an arbitrary intensity scale, the off- resonance spectra are actually much weaker than the exact resonance spectrum. Several hours are required to obtain the 4 w = 0.073 cm-I spectrum a t intensities comparable to the 4 w = 0.00 em-' sn&trum. Chanees in the slit width can also alter the appearance of the spectrum, since narrow slits dis- criminate against the slowly decaying emission, as the excited molecules diffuse away from the slit opening.
The interpretation of time-resolved spectra of Iz and BIZ has led to two opposing points of view which are descrihed briefly in the next section.
Theoretical Treatments The extensive use of resonance Raman spectroscopy in in-
organic and biological chemistry has stimulated the devel- opment and refinement of the theoretical bases for the in- terpretation of experimental data. The humble diatomic molecules Br? and I? have laved an extraordinarv role in this . , developmrnt. Their '2*' 11, 'n, .' transitima Iiaveaimtui- trru.5 r~litIim.ihip to thv iw.itinl: lines tof the krvpt1>11 nnd argon l,+sers, p+.rm~tting hurh l<F :tnd Rl< t u be n l~awred Anpl) hv retuning the Iw+ra, and t l ~ ~ ~ r ele* t r m ~ , ~ , ~ ~ l ~ r n t i ~ m d , and ro- t i l t i~ni~l energy It.veI-. .(re wc.1 I I I I ~ ~ ( . T A I ~ ~ U ~ 'l'hur. tlwy ar(. ~.xvellent mole,.de.; fu r testing thwwetic.a. ~~t~ve~oplrlellri.
.Although n deriled the~lretical disn~asion ut the tu,t# efiects and thr rdation;hip helwiw Illern is I*.pnd t l l c i c u p ~ o f t h ~ i nrta le, rwu ol~l)osiny point' ul riew have arisen rt.g;irdinn the Interpret:rtion uf 1ime.rr.dwd .pectr8.iccIpY :ind the qurdim d s h e t h e r rherei-.ac~,ntintl~rur tr:#n.~tion irum 1(1{ to RFor two discrete phenomena which occur simultaneously but to
Volume 58 Number 4 Aoril 1981 347

differine extents as the laser freauencv is tuned awav from
(25). Rousseau and Williams have cogently summarized the ev-
idence for a common origin of the two effects, based on a theoretical derivation in which RR and RF are the limitine cases. In their treatment, which effect is predominant depends on the relationship of the exciting laser line to the vibronic transitions of the molecule in question. Three terms, the decay time of the laser ~ u l s e . the decav time of the excited state vi- hronic level, and; term dependent on difference between the exciting frequency and the vibronic transition are necessary to describe the time-resolved spectrum. In fact, Roussean and Williams prefer the nomenclature, "discrete resonance Ramaa" for RF, which involves excitation into a single vi- hronic state, and "continuum resonance Raman" for RR, which involves excitation into the continuum. A contrary viewpoint, again derived from theory, is offered by Jortner and co-workers (12-14). In their treatment the two orocesses are physically distinct:'^^ scattering, which is instantaneous and therefore follows the time dependence of the exciting laser pulse and RF emission, which occurs with a longer decay time characteristic of the excited electronic-vibrational-rotational state which resulte from the absorption of a laser photon. Both nrocesses mav occur for anv eiven freauencv of excitation and - . - the time-resolved spectra can be analyzed as varying contri- butions of the two ~henomena. When there is exact resonance with a single vibronic level of the ground state, the RF effect predominates, but for off resonance excitation or excitation into the continuum, RR which follows the laser signal is ob- served. As the exciting frequency moves further off-resonance (e.g. Fig. 9), the RF contribution becomes progressively weaker and the RReffect makes aproportionately larger contribution to the ohserved time-resolved spectrum, even though it is intrinsically much weaker than RF. Testing and evaluation of the theoretical relationship between RF and RR is con- tinuing a t the present time by the researchers cited in this article and many other contrihuters to the field (8, 13, 16, 25).
Practical Applications Although the major interest in RF and RR spectroscopy of
vapor-phase molecules has been the development of the theory for the RF and RR effects, some practical applications
have emerged (23, 26-29). For example, continuous moni- toring of air for NO2 and S02, two of the most pernicious air ~ollutants, is Dossible with RF. NOn has a red-brown color. and its RF spectrum, which can be st&ulated by the 488.0 nm (blue) line of an argon ion laser or the 422 nm line of a Cd/Ne . laser, can he used to monitor the NO2 concentrations in am- bient air over a long period of time. Similar devices are being developed for SO2 monitoring. However, because SO2 is a colorless gas with an electronic absorption a t 180-230 nm, it is necessary to use UV excitation. Zn, Cd, and Sb vapor lamps which emit a t 213.8,217.5, and 228.8 nm have been success- fully employed in the laboratory, although more powerful sources are required for field use. Another exciting application is Raman LIDAR (an acronym for Light Detection and Ranging, in which a pulsed laser source is used to examine remote gases (exhaust stack plumes, the stratosphere, etc.). The time delay of the RR, RF, and ordinary Raman scattered light gives distance information, while the wavelengths of the scattered light provides chemical information about the scattering species (23,26-29).
Literature Cited (1) Behringer, J., J . Komon Spictroscopy.2.275 (1974). 12) B n W M.. Json,M..and Bernard L.,Con. J . Spectry., 17,60 (19721. GI Hulrer. W.. Murphy. W. F..andBernstein,H. J., J Chsm. Phyr., 52.399 (1970). (41 Clark,R. J. H..in"Advancesin Infraredand RarnanSpecboscu~y: !EditomClark,
R. J. H.. and Hester, H. E.) Vol 1, Heydan and Sons, London, 1975. (61 Strommen. D..and Nakarnntn, K , d CHEM. EDUC.54.474 119771. (6) Holrer. W.. Mmphy, W. F.,sndBarnstein, H. J.. J. Chrm. Phys.52.469 (19701. 17) Nafie. L stem P. and ~et icolar , w.L.. Chrm. phyr Lett.. 12,131 (1971). (81 Housseeu.D. L a n d Willisms, P. F.. J. Chem Phyr.. 64.3519 (1976). 19) Kiefer, W.. and B~rnstein, H.J .J . Moi. Spectry., 43, 366 (1972).
(10) Kobinafa, S., Ruli Chrm. S o c J a g , 46,3636 (1973). (11) Behringei, J., m "Mulecular Spectroscopy (Specialist Periodical Reports)." Vol. 2,The
Chemical Society, London, 1974, p. 1W. (121 Mukame1.S. and Jortner. J., J . Chrm. Phys.. 62,3609 (1975). cia1 M U ~ ~ ~ ~ I , S . , B ~ ~ ~ R ~ ~ V ~ ~ , A . , and ~ ~ ~ t ~ ~ ~ , J.. J c h r m ~hy,.. G ~ , , W (19761. (141 Mukamel,S.,Ben-Reuuen,A.. and Jortnei, J..Phyr. Rev. A , 12,947 (1975). (161 Riedman,H.and Wi1son.A. D., Chom. Phys. Lett., 46,307 (19771. (161 Mingardi. M. andsiehrand. W., J. Chem. Phys., 82.1074 (1975). (17) Williams, P. F., Rourseau, D. L., and Dwaretsky, S. H.. Phys. Re". Lett., 32. 196
(19761 ,~~ .,. (13) Hercberg. G;'SpectrsofDiatumie Moleculps,'. Van Nustrand ReinhddC~.. NPW Y M ~ .
1950. 0.456. 119) Ref. 11. p. 158. (201 Tobias. R. S., J CHEM EDUC., 44.1(1967) and 14.70 (1967). (211 Penny, C. M.,in ''Laser Rarnan GscDispostics," (Editors. Lapp, M.,andPenny.C.
MlPlenumPmss, NeuYork. 1974, o. 191. Ref. I l , p 612. Ready, J . F.."Industrisl Applicstions of Lasers." Academic Press, NOW York, 1978.
Z.Re0. Sci. Insfrmntr . 41.794 119771 (23) (24) Bachrach, R. ~, ~. ~ . -,
125) Nouak. F, A,, Friedman, J . M., and Hochstrasser, R. M., in '"Laser and Coherence Spectroscopy." (Editor, Steinfeld, J. I.) Plenum Press, New York, 1918.
(26) Birnbaurn. M., in "Modern Fluoreacenee Speelmscnpy." Yo1 1. Plenum Publishing Corp., New York. 1976.p. 121.
(27) Helfi. S. H., in " h e r Raman Gar Diagnostics," (Editors: Lapp, M.,andPenny, C. M.. and Penny, C. M.) Plenum Pr-, New York. 1974. p. 231.
(28) capub, 6.. ~ e o n s r d , D. A , and ~ungiierdo, J., Opiiroi Specria, l4(2), 57 (19801. (29) Gold rna~ L.. "Appiiostion of the Laser," CRC Press, Cleveland (1978).
348 Journal of Chemical Education