republica bolivariana de venezuela la … · anemias hemolÍticas dra. olga briceño republica...
TRANSCRIPT

ANEMIAS HEMOLÍTICAS
Dra. Olga Briceño
REPUBLICA BOLIVARIANA DE VENEZUELA
LA UNIVERSIDAD DEL ZULIA.
FACULTAD DE MEDICIA.
ESCUELA DE BIOANÁLISIS.

ANEMIAS HEMOLÍTICAS
Definición: Un grupo heterogéneo de
Anemias en donde hay un aumento en
la velocidad de destrucción eritrocitaria.

ANEMIAS HEMOLÍTICAS
Dx. de Laboratorio:
* Hb, Hto y GR:↓
* Reticulocitos:↑
* Sobrevida Eritrocitaria: Acortada
* Bilirrubina Total: ↑ a expensas de BI
* Urobilinógeno Fecal e Índice Hemolítico: ↑
* Producción Endógena de Monóxido de Carbono: ↑
* Imagen Roja: Variable.

ANEMIAS HEMOLÍTICAS
Pruebas Especiales:
* FO: Esferocitosis Congénita
* F. Falciforme: A. Falciforme
* Electroforésis de Hb: Hemoglobinopatías y Talasemia
* Coombs directo: A.H. Inmunes
* Test de HAM y Sucrosa: HPN
* Cuerpos de Heinz y Dosificación de enzimas:
Enzimopatías
* Hemoglobina Fetal.

ANEMIAS HEMOLÍTICAS
1.-Anemia hemolíticas por defecto intrínseco
1.1.-Defectos en la membrana eritrocitaria
1.1.1.-Esferocitosis Hereditaria
1.1.2.-Ovalocitosis Hereditaria
1.1.3.-Acantocitosis Hereditaria
1.1.4.-Estomatocitosis Hereditaria
1.2.- Hemoglobinopatías
1.2.1.-Anemia Falciforme
1.2.2.-Otras

ANEMIAS HEMOLÍTICAS
1.3.- Talasemia
1.3.1.- β Talasemia
1.3.2.- α Talasemia
1.4.-HPN (Hemoglobinuria Paroxística Nocturna)
1.5.- Deficiencia Enzimática
1.5.1.-Deficiencia de la G6PD
1.5.2.-Deficiencia de la PK
1.5.3.- Otras: Porfiria

ANEMIAS HEMOLÍTICAS
2.- Anemias Hemolíticas por Defectos
Extrínsecos.
2.1.- Anemia Hemolítica Inmune
2.1.1.- Anemia Autoinmune por Anticuerpos Calientes
2.1.2.- Anemia Autoinmune por Anticuerpos Fríos
2.1.3.- Anemia Hemolítica Inducida por Drogas.

ANEMIAS HEMOLÍTICAS
2.2.- Daño Mecánico al Glóbulo Rojo
2.2.1.-Anemia Hemolítica Microangiopática
2.2.2.-Hemoglobinuria de la Marcha
2.2.3.- Prótesis
2.2.4.- Quemaduras
2.3.-Secuestración Reticulo-Endotelial
2.4.-Infecciones y Tóxicos
2.5.-Enfermedad Hemolítica del Recién Nacido

ANEMIAS HEMOLÍTICAS
Principales Manifestaciones Clínicas
Palidez con perdida de coloración en la piel
Debilidad
Mareos
Fatiga
Intolerancia a la Actividad Física

ANEMIAS HEMOLÍTICAS
Ictericia, presencia color amarillento en piel y
mucosas
Hepatomegalia y esplenomegalia
Orina color oscuro
Afecciones cardiacas

ESFEROCITOSIS HEREDITARIA
AH por Defecto Intrínseco
Etiopatogenia:
↓15 - 20% de Colesterol y Fosfolípido
↑Permeabilidad Na y K.
↓Niveles de 2,3 DPG
Deficiencia de Espectrina, Anquirina, proteína 4.2 y banda 3.
Fisiopatología:
Poca Flexibilidad


FISIOPATOLOGÍA DE LA ESFEROCITOSIS HEREDITARIA

ESFEROCITOSIS HEREDITARIA
AH por Defecto Intrínseco
Manifestaciones Clínicas:
Anemias Discretas
Ictericia Acolúrica
Litiasis Biliar
Ulceras Crónicas
Esplenomegalia Moderada
Crisis Aplásica asociada a Infecciones por Parvovirus
Litiasis Vesicular

ESFEROCITOSIS HEREDITARIA
AH por Defecto Intrínseco
Laboratorio:
Hb: 8 y 12 gr/dL
Hto: ↓
GB: N o Lig↑
Plaquetas: N (↑ en Esplenectomía)
Frotis de SP: Esferocitosis,
Policromatofilia,
Basofilia Difusa.
Reticulocitos: ↑ > 6%

ESFEROCITOSIS HEREDITARIA
AH por Defecto Intrínseco
Laboratorio:
Índice Corpusculares:
CHCM: ↑ 36-0% de los casos
HCM: N
VCM: N o ↓
MO: Hiperplasia Eritroide
FO: (incubada): ↑ (aún en Hemólisis ligera)
(no Incubada) 20 – 25 % de los pacientes son normales
Bilirrubina Total: ↑ a expensas de BI
Urobilinógeno Fecal: ↑

ESFEROCITOSIS HEREDITARIA
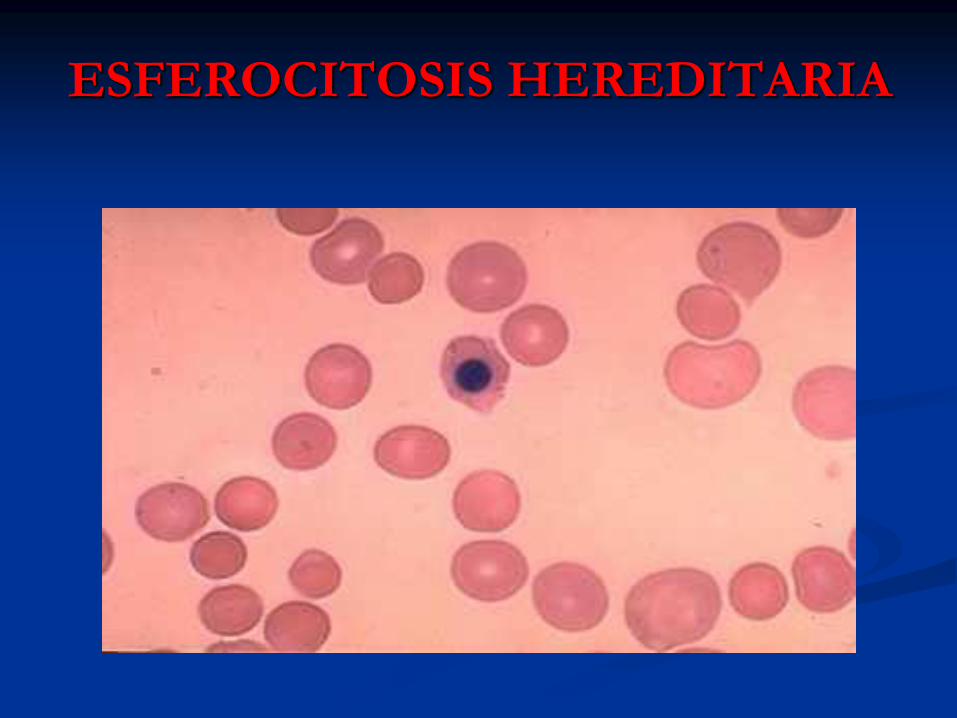
ESFEROCITOSIS HEREDITARIA

ESFEROCITOSIS HEREDITARIA

OVALOCITOSIS HEREDITARIA

ACANTOCITOSIS HEREDITARIA
AH por Defecto Intrínseco
Existen 2 condiciones clínicas:
Enfermedad Hepatocelular Severa
Abetalipoproteinemia
Caracterizado por Crecimiento retardado
Esteatorrea
Ataxia Progresiva
Rinitis pigmentosa
Acantocitosis

ACANTOCITOSIS HEREDITARIA

ESTOMATOCITOSIS HEREDITARIA
AH por Defecto Intrínseco
Etiopatogenia:
* ↓Defecto en la Membrana:
* Deficiencia de la Bomba de Na+y K+.
* ↑ Agua al medio intracelular. * Deficiencia de Estomatina o banda 7.2b.
Enfermedad por Rh Nulo

ESTOMATOCITOSIS HEREDITARIA
AH por Defecto Intrínseco
Condiciones Clínicas:
Alcoholismo (enfermedad)
Cirrosis Hepática
Deficiencia de la Glutatión Peróxidasa
Esferocitosis Hereditaria
Mononucleosis Infecciosa
Talasemia
Enfermedades Malignas en Niños y Adultos

ESTOMATOCITOSIS HEREDITARIA

HEMOGLOBINA
HUMANA
α
α
β
β
Grupo Hemo PM= 64,458

Hemoglobinopatías
•Enfermedades caracterizadas por presentar una
cadena de globina estructuralmente anómala.
•Causadas por mutaciones en la secuencia
codificante de un gen de globina que alteran su
estructura primaria.
•Conduce a la formación de variantes de Hb anormal
•Se han descrito en la actualidad aprox. 700 tipos
diferentes.
ALTERACIONES DE ORIGEN GENÉTICO EN LA
SÍNTESIS Y SECUENCIAS DE LAS GLOBINAS

ANEMIA FALCIFORME
AH por Hemoglobina Anormal
Etiopatogenia:
Hb S: 70 - 90% (Ac glutámico → Valina )
PO2: ↓ 45 mmHg (GR de media luna)
Células drepanocíticas irreversibles
Microcirculación (GR destruídos)
Hb: F (Efecto protector)


ANEMIA FALCIFORME

ANEMIA FALCIFORME
AH por Hemoglobina Anormal
Manifestaciones Clínicas:
Crecimiento y Desarrollo Retardado
Cambios Óseos
Ulceras en Piernas
Susceptibilidad a las Infecciones
Dactilitis
Cardiomegalia, Hepatomegalia
Litiasis Biliar
Manifestaciones Neurólogicas: Cefaleas, Convulsiones, afasia, entre otros. Manifestaciones Oculares: Tortuasidad de los vasos retinianos y hemorragias vitreas.

ANEMIA FALCIFORME
AH por Hemoglobina Anormal
Laboratorio:
Hb: 5 -11 gr.% Hto: ↓
Cb: ↑ (Neutrofilia)
Plaquetas: ↑
Frotis de SP:
Poiquilocitosis (drepanocitos)
Anisocitosis con predominio de macrocitosis
Corpúsculos de Howell Jolly
Eritroblastos ortocromático presente
Puntillado basófilo
Reticulocitos: ↑

ANEMIA FALCIFORME
AH por Hemoglobina Anormal
Laboratorio:
Índice Corpúsculares:
VCM:↑
CHCM: N
Médula Ósea: Hiperplasia eritroide
FO: ↓ Ferritina: ↑
Fenómeno Falciforme: +
Electrofóresis de Hb: Hb S
Hb F
B I: ↑ U F: ↑ I H: ↑


ANEMIA FALCIFORME

ANEMIA FALCIFORME

ANEMIA FALCIFORME

TARA FALCIFORME
AH por Hemoglobina Anormal
Laboratorio:
Hb: N Hto: N
CB: N
Contaje de Plaquetas: N
Frotis de SP: N Médula Ósea: N
Fenómeno Falciforme: +
Electrofóresis de Hb:
Hb A: 60 -80%
Hb S: 30 %
Hb F: 0 - 2 %

TARA FALCIFORME

HEMOGLOBINOPATÍA S C
AH por Hemoglobina Anormal
Manifestaciones Clínicas:
Dolores Abdominales, óseos o Toráxicos, a veces severos
Hepatomegalia: Ictericia discreta
Ulceras en las Piernas
Esplenomegalia
Cambios Cardiovasculares: Mínimos
Laboratorio:
Hb y Hto: ↓
Frotis de SP: Drepanocitos y dianocitos

HEMOGLOBINOPATÍA C
HOMOCIGOTA
AH por Hemoglobina Anormal
Etiopatogenia: Herencia Codominante Autosómica
Hb C (ác glutáminico → lisina)
↓ poco soluble
Cristaliza fácilmente
↓ más rígido
Esferocito
Manifestaciones Clínicas:
Anemia Moderada
Esplenomegalia
Molestias Abdominales Intermitentes
Ictericia Discreta
Artralgias
Laboratorio:
Frotis SP: Dianocitos abundante
Esferocitos
Médula ósea: Hiperplasia
Normoblástica
Electroforésis de Hb: HB C

HEMOGLOBINOPATÍA C

Fórmula:
N: 65%
L: 30%
M: 5%
Glóbulos
blancos:
Normales en
número y
morfología
Glóbulos Rojos:
Poiquilocitosis:
Drepanocitos ++,
Dianocitos ++,
esquistocitos +,
dacriocitos +
Marcada Policromatofília
Punteado basófilo ++,
Cuerpos de Howell Jolly +
Eritroblastos 26%
Plaquetas:
Normales en número y
morfología
VALIDACIÓN
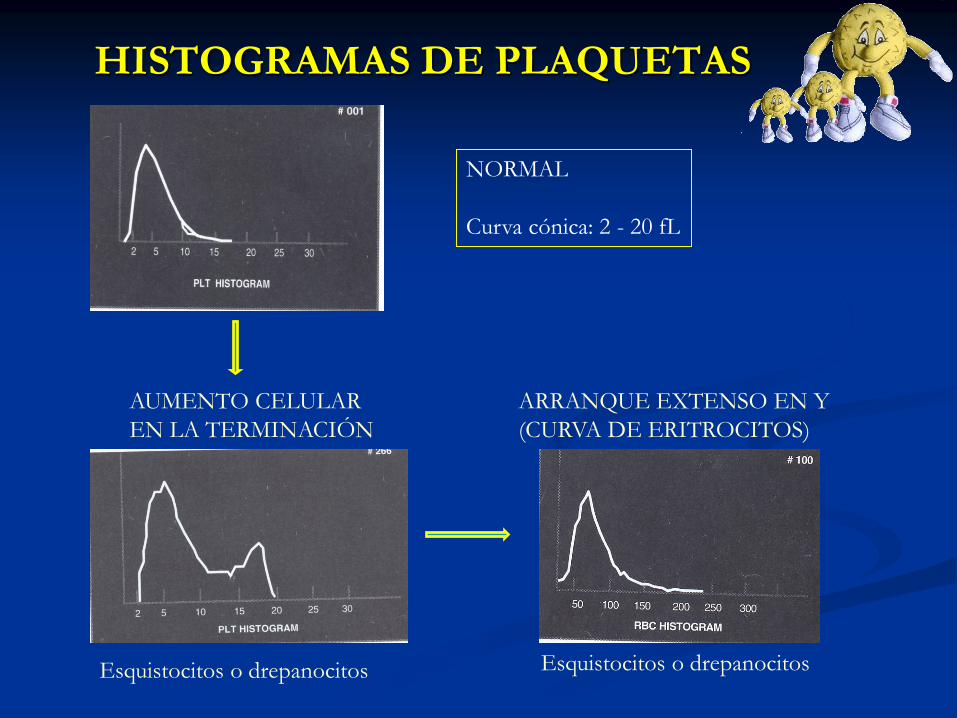
HISTOGRAMAS DE PLAQUETAS
NORMAL
Curva cónica: 2 - 20 fL
AUMENTO CELULAR
EN LA TERMINACIÓN
Esquistocitos o drepanocitos Esquistocitos o drepanocitos
ARRANQUE EXTENSO EN Y
(CURVA DE ERITROCITOS)

HISTOGRAMAS DE
ERITROCITOS
CURVA DESPLAZADA A LA IZQUIERDA
NORMAL
Curva cónica: 60 – 110 fl
Curva cóncava: 110 – 200 fl
Poiquilocitosis
(esquistocitos,dacriocitos,drepanocitos)

ALTERACIONES DE ORIGEN GENÉTICO EN LA
SÍNTESIS Y SECUENCIAS DE LAS GLOBINAS
Talasemias
•Grupo complejo de enfermedades congénitas que afectan la
expresión de los genes de las globinas y se heredan con carácter
autosómico dominante.
•Clasificadas según la cadena afectada, α, β,γ ,δ,ε talasemias.
• Mayor: Homocigoto Menor: Heterocigoto
•Caracterizadas por anemias hemolíticas microcíticas, debidas a una
disminución o ausencia de una o más cadenas polipeptídicas que
componen la hemoglobina.

Manifestaciones Clínicas:
Trastornos en el crecimiento y desarrollo
Retraso en la madurez sexual
Alteraciones óseas, dentarias y articulares
(faciales talasémicas, cráneo de cepillo)
Esplenomegalia e hiperesplenismo
Aumento del Hierro en órganos
Alteraciones Endoccrinas
Daño Cardiaco
Alteraciones Hepáticas
(αTalasemia : Hydrops fetalis)
β – TALASEMIA MAYOR

Laboratorio:
* Hb: 2 -4gr% * Hto: ↓
* CB: ↑ * Plaquetas: ↑
* Reticulocitos: Moderado↑
* Frotis de SP:
Hipocromía severa
Anisocitosis
Poiquilocitosis: Dianocitos moderados
Puntillado Basófilo
*Frotis Escandaloso
-VCM:↓
-HCM:↓
β – TALASEMIA MAYOR

Laboratorio:
- Médula Osea: Hiperplasia eritroide
Fe en deposito: ↑
- Fe Sérico:↑
- Ferritina: ↑
- Bilirrubina Total:↑ a expensas de BI
- Deshidrogenasa Láctica: ↑
- Electrofóresis de Hb: Hb F. 30 – 90%
Hb A2
Hb A : Si es Leve
β – TALASEMIA MAYOR

TALASEMIA

TALASEMIA
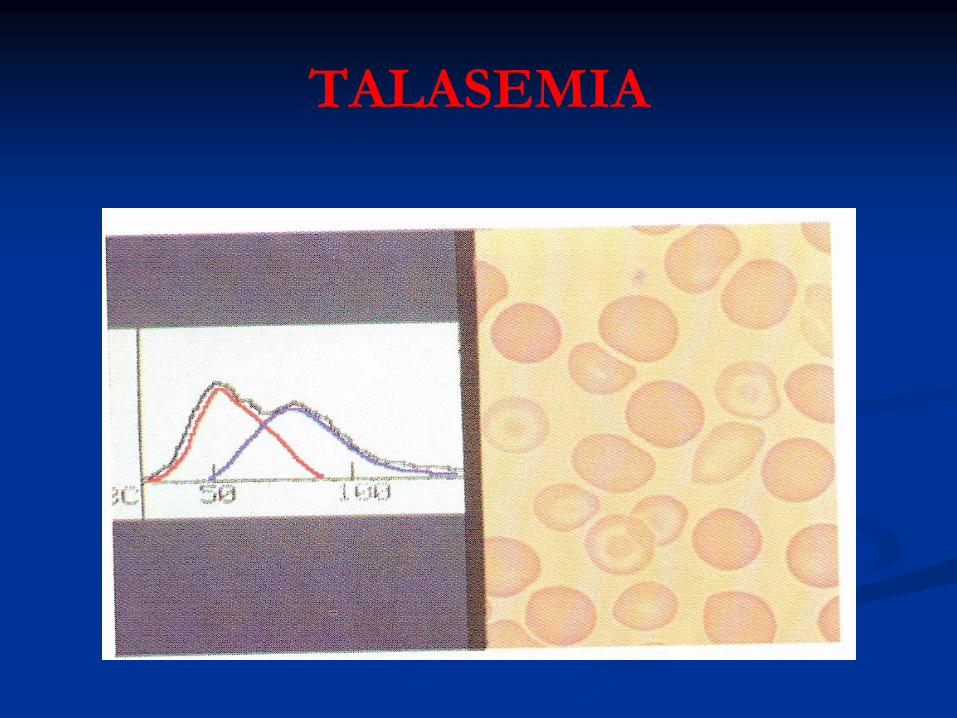
TALASEMIA
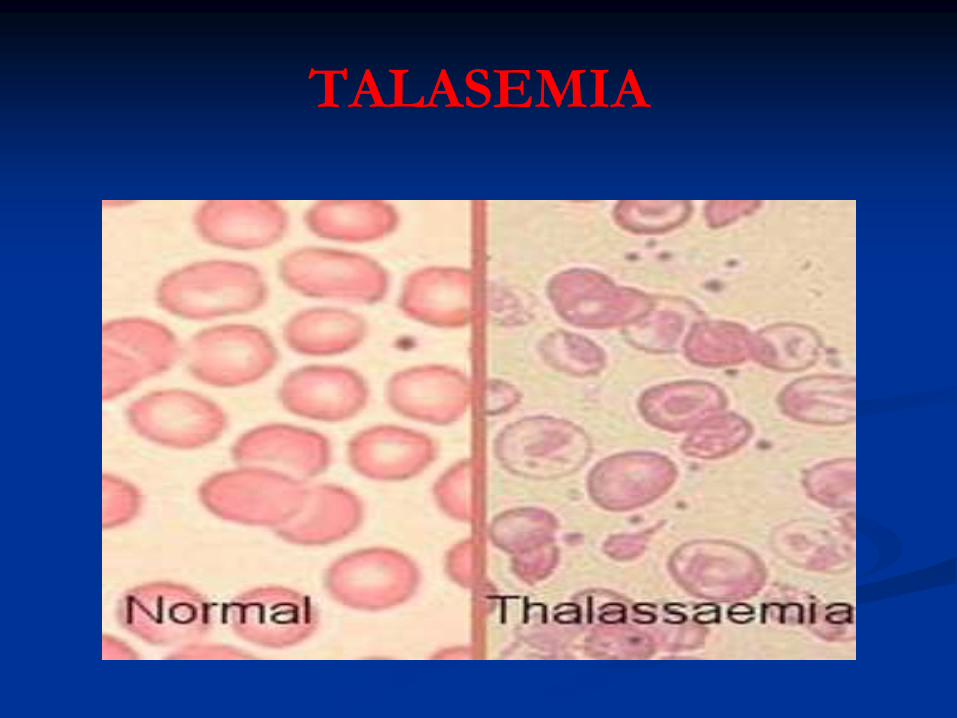
TALASEMIA

β – TALASEMIA MENOR
AH por Hemoglobina Anormal
Manifestaciones Clínicas:
Anemia Crónica
Varían en función de la coherencia con de α o δ Talasemia o deficiencia de Fe.
Laboratorio:
HB y HTO: ↓
Frotis de Sp:
Hipocromía
Microcitosis
Electroforesis de Hb:
Hb A : ↓
Hb A2 : ↑
HB F : ↑

ALFA TALASEMIAS Grupo de enfermedades de origen genético que resultan de una disminución o ausencia
de la síntesis de las cadenas α de globina. Se produce tanto en la vida fetal como en la
del adulto.
Talasemia α0
•Disminución en la síntesis de cadena α en un alelo.
Debido a:
• La deleción de uno de los genes de cada par (-α/) siendo este tipo
de mutación las mas frecuentes en el mundo.
• Mutaciones puntuales que hacen a los genes alfa inactivos parcial
o totalmente (NO DELECIÓN)
Talasemia α+
• Ausencia total de la síntesis de cadena α en un alelo.
Debido a:
• La pérdida o deleción de ambos genes de globina del
cromosoma 16, cuyo haplotipo es (--/).
• O bien por deleción del elemento regulatorio HS-40.
Clasificación

Normal
αα/ αα
Heterocigoto α+
αα/ α-
Heterocigoto α0
αα/ --
Homocigoto α +
α -/ α-
Enfermedad Hb H (β4)
α -/ --
Hidrops fetalis
Hb Bart (γ4)
--/--

DIAGNÓSTICO DE α TALASEMIA
1.- Estudio hemocitométrico convencional.
2.- Recuento de reticulocitos.
3.- Isoelectroenfoque de hemoglobinas (IEF).
4.- HPLC de cadenas de globina
5.- Determinación de Hb A2 y Hb F. (Para descartar β o δβ talasemia)
6.- Cuantificación de los Cuerpos de Inclusión.

Hemoglobinuria
Paroxística Nocturna
La enfermedad es un raro trastorno adquirido,
caracterizado por brotes intermitentes de hemólisis
intravascular y hemoglobinuria nocturna.
Etiopatogenia:
Defectos en la Adhesión de las proteínas Reguladoras:
•Factor acelerador del Decaimiento. (FAD)
•Proteína fijadora de C8. (C8bp)
Ausencia de un glucolípido de la Membrana Glucosil-fosfatidil-inositol (GPI)

Hemoglobinuria
Paroxística Nocturna
Mutación somática clonal de un gen del
cromosoma X denominado fosfatidilinositol
glucano clase A
Defectos en la Adhesión de las proteínas
Reguladoras: CD55 factor acelerador de la
degradación y CD59 inhibidor de membrana de
la lisis reactiva.

Hemoglobinuria
Paroxística Nocturna
CD55
CD59

Hemoglobinuria
Paroxística Nocturna
MANIFESTACIONES CLÍNICAS
Hemolísis acentuada
Trombosis venosa
Infección
Hipoplasia de la MO
Hemoglobinuria

HALLAZGOS DE LABORATORIO
EN LA HPN
Anemia Hb 8 - 10 gr/dl
Hto ↓
Glóbulos blancos: ↓
Reticulocitos: ↑ (5 a 10%)
Imagen Roja: Anisocitosis, hipocromía y eritroblastos ortocromáticos presente.
Hemoglobinuria: +
Hemosiderinuria: +
Test de Sucrosa: +
Test de HAM: +
