recycling and refurbishing old antitubercular drugs: the encouraging case of inhibitors of mycolic...
TRANSCRIPT

10.1586/ERI.13.24 429ISSN 1478-7210© 2013 Expert Reviews Ltdwww.expert-reviews.com
Review
Resistance to antitubercular drugs: the simplicity of the dispensableIn the last 20 years, the overwhelming advances in the understanding of mycobacterial physi-ology and genetics, driven by the availability of tools for genetic manipulation, provided a wealth of information that was quickly taken by research groups, attacking different aspects of the Mycobacterium tuberculosis infection, mechanisms of pathogenesis and, importantly, the battle between the pathogen and the drugs used to kill it. The information started to surge under the pressure of the burden of tuberculosis worldwide and quickly led to the start of lines of research; the one of interest for this review is that regarding the mechanisms of action of antitubercular drugs and antitubercular drug design. In this specific area, the flow of infor-mation was enormous and, as in a pole vault competition, the bar has steadily been set higher year after year [1–9].
There is little doubt that most of the relevant specific antitubercular drugs (thus not active on other Gram-positive or -negative bacteria) in clinical use for the treatment of tuberculosis
at any given time over the last 60 years, have the singularity of sharing a common target: the synthesis of mycolic acids. These α-alkyl, β-hydroxylated long-chain fatty acids are essen-tial components of the cell envelope structure and they also play a major role in modulating the interplay of the pathogen and the host’s immune system [10,11]. One of the many surprises provided by the analysis of the M. tuberculosis genome sequencing was the finding of two different sys-tems for the synthesis of fatty acids [12]. One of them, designated fatty acid synthase I system, is in charge of providing ‘medium’ chain length fatty acids (C16–C24), while a second one, fatty acid synthase type II (FASII), is responsible of elongating C18 fatty acyl primers until they reach a length of C60 (known as meromycolic acids). After that, other enzymatic systems con-densate the C24–C26 alkyl branch and form the mature mycolic acid [11,13].
Since its introduction as an antitubercular drug in 1952, isoniazid (INH; 2-isonicotinyl acid hydrazide) has been the most valuable weapon for treatment of tuberculosis [14], for this reason it seemed logical that this drug was
Juan M Belardinelli1,2 and Héctor R Morbidoni*1
1Cátedra de Microbiología, Facultad de Ciencias Médicas, Universidad Nacional de Rosario, Santa Fe 3100 (2000), Rosario, Argentina2Department of Microbiology, Immunology and Pathology, College of Veterinary Medicine and Biomedical Sciences, Colorado State University, Fort Collins, CO, USA*Author for correspondence: Tel.: +54 341 568 9618 Fax: +54 341 439 0465 [email protected]
One of the first approaches undertaken in the quest for antitubercular compounds was that of understanding the mechanism of action of old drugs and proposing chemical modifications or other strategies to improve their activity, generally lost to the mechanisms of resistance developed by Mycobacterium tuberculosis. A leading case was the work carried out on a set of compounds with proven activity on the essential pathway of the synthesis of mycolic acids. As a result, different solutions were presented, improving the activity of those inhibitors or producing novel compounds acting on the same molecular target(s), but avoiding the most common resistance strategies developed by the tubercle bacilli. This review focuses on the activity of those compounds, developed following the completion of the studies on several of the classic antitubercular drugs.
Keywords: antitubercular prodrugs • avoiding drug resistance • drug development • fatty acid synthase type II inhibitors • Mycobacterium tuberculosis • mycolic acid biosynthesis
Recycling and refurbishing old antitubercular drugs: the encouraging case of inhibitors of mycolic acid biosynthesisExpert Rev. Anti Infect. Ther. 11(4), 429–440 (2013)
Expert Review of Anti-infective Therapy
© 2013 Expert Reviews Ltd
10.1586/ERI.13.24
1478-7210
1744-8336
Review
For reprint orders, please contact [email protected]

Expert Rev. Anti Infect. Ther. 11(4), (2013)430
Review
the first choice to start the study of mechanisms of action and resistance of antitubercular drugs. In a brief account of the developments on INH (Figure 1A), it is necessary to pay hom-age to the early work by Gardner Middlebrook, who realized that M. tuberculosis clinical strains developed resistance to INH while losing catalase activity [15–17]. Almost 40 years later, this assertion was corroborated by Heym et al. by identifying the first genetic trait linked to resistance to this antitubercular drug [18]. Thus, their report demonstrated that katG, encoding a catalase-peroxidase, was crucial in resistance to INH and that several clinical strains displaying resistance to INH contained mutations in this gene [18,19]. Thus, those early reports dem-onstrated that KatG activity was essential to achieve killing activity, as shown by conferring Escherichia coli with suscepti-bility to INH simply by expressing Mycobacterium smegmatis katG in this otherwise INH-resistant microorganism [18]; it also generated the concept that antitubercular drugs may require an activation step, being in the strict sense of the meaning ‘prodrugs’. This was not at all a minor point, as demonstrated by a large body of evidence showing that katG mutations are sufficient to render M. tuberculosis resistant to high concentra-tions of INH, and as expected, they are present in most of the INH-resistant clinical strains worldwide [20–23]. In fact, several different mutations affecting catalase-peroxidase activity were identified over the years, contributing to the development of nucleic acid amplification or hybridization methods for the rapid testing of resistance to INH in clinical settings [23–25].
However, it is of interest to comment that different mutations and deletions in katG are predominant in clinical versus in vitro isolated INH-resistant mutants; as an example, the muta-tion most frequently reported in clinical isolates but hardly found in in vitro screenings (KatG S315-T) retains a fraction of its catalase-peroxidase activity while conferring resistance to intermediate levels (1–5 µg/ml) of this drug [26]. Conversely, deletions spanning katG (and sometimes adjacent chromosomal regions), are frequent in laboratory generated INH-resistant mutants, which in all cases displayed a high level of resistance (10–256 µg/ml) [26]. These results underscore the importance of the environment in which the strains are selected for and the comparison between their level of resistance. Intriguingly, although KatG-deficient strains were impaired to some extent in virulence, growth was comparable with the wild-type strain, suggesting that the catalase-peroxidase function was not essen-tial for normal growth except when exposed to an oxidative stress [27–30]. Thus, a sensible way to overcome the loss of action in those mutants was by designing molecules that would not need an activation step.
Subsequently, other drugs such as ethionamide (ETH), isoxyl (ISO) and thiacetazone (TAC) (Figure 1B–D), showed activity on the mycolic acid synthesis [31,32], although the understand-ing of the mechanism of action began to emerge only recently. Surprisingly, as shown in Figure 2, these last three drugs share a common activator, a flavin monooxygenase encoded by the gene ethA, for which a physiological role is still unknown [33,34].
Figure 1. Isoniazid, ethionamide, thiacetazone and isoxyl, and novel molecules proposed as antitubercular drugs.
Cl OH
Cl
O
O
NH
NNH
S
NH2
NH2
O
NH
S
NH
O
N
O NH
NH2
C l
O
Cl
C l OH
N
NH2S
O
5
OH
NN
ON
S
ON
NH
S
NH
O
O
NNH
S
TriclosanIsoniazid
Ethionamide
Isoxyl
ThiacetazoneSRI-224
BDM31343
Compound 19Compound 26
Compound 10
Belardinelli & Morbidoni

431www.expert-reviews.com
Review
Genetic analysis proved that ethA transcription was negatively regulated by an adjacent gene, ethR [33]. In a way comparable with what happened to INH, resistance to ETH, ISO and TAC has been demonstrated in strains containing mutations lying in ethA, reinforcing the notion that the activating enzymes are the key to the antitubercular effect of those compounds [34]. Moreover, the astonishing activity of INH is puzzling; with a MIC: 0.02–0.05 µg/ml, compared with the lower activity dis-played by ETH (MIC: 5 µg/ml). One of the explanations offered was that the level of activation of INH was much higher than that of ETH. Therefore, in the case of ETH, the efforts have focused on the design of compounds that would act on the regula-tor–activator pair, increasing the amount of activating enzyme available [35].
Finally, in the same string, both TAC and ISO are prodrugs used in the 1970s and 1980s especially in Africa and Latin America due to their low cost, their use being discontinued because of several side effects and a surprisingly high frequency of mutant strains arising during treatment [36,37]. Importantly, use of TAC in the antitubercular treatment of HIV patients was highly linked to a lethal condition, the Stevens–Johnson syndrome. This syndrome, causing severe cutaneous hypersensitivity reactions in HIV patients, especially at early ages, is also seen when other antitubercular drugs are given, but much less frequently than when TAC is used [38,39]. There is no clear understanding of the reasons for this toxicity until now, most likely due to the discon-tinuation of use of TAC, that in turn decreased the interest in this topic. Although an inhibitory effect by some of the mentioned drugs on mycolic acid synthesis was reported 50 years ago by the inspirational work of Winder’s group [31], there was little interest in them until recently, when two groups confirmed an enzyme of FASII as one of the enzymes involved in resistance to both drugs and, because of its essential role, and its potential as a molecular target [40,41].
Thus, the knowledge gathered on the mechanisms of resist-ance and actions of four prodrugs that are or have been used in treatment of tuberculosis (INH, ETH, TAC and ISO) allows the proposal of novel strategies aimed at the development of better compounds with more potency, fewer side effects and impor-tantly, avoiding activation steps. In the next sections, the authors will review the advances for each of the drugs mentioned above.
INH: how it works, how the tubercle bacilli avoids the killing & how to kill it anyway; tricking the tricksterEarly studies on the elucidation of the molecular mechanism of action of INH started almost 20 years ago when the isolation and characterization of M. smegmatis mutants resistant to this drug led to the identification of an enoyl-acyl carrier protein (ACP) reductase [42]. This enzyme, encoded by a gene designated as inhA, was located adjacent to a gene with homology to β-ketoacyl ACP reductases; thus, it was clear that both genes were probably part of a fatty acid biosynthetic pathway [43]. Taken together with the fact that INH treatment abrogated the synthesis of mycolic acids, both genes had to be part of the biosynthetic pathway responsible for that synthesis. Biochemical, genetic and crystallographic studies
led to the understanding of the mechanism responsible for the inhibition of the enzyme, demonstrating that once activated, one of the INH reactive derivatives formed (a hypothetical isonico-tinic acyl radical), reacted with NADH, forming a competitive inhibitor of the InhA enzyme [44–46]. This reaction led to the irreversible inactivation of this critical enzyme, shutting down the synthesis of mycolic acids in slow and fast-growing mycobacteria.
Although two different lines of research postulated either InhA or the β-ketoacyl ACP synthase KasA as the target of INH [42,47], the isolation and characterization of a M. smegmatis thermosensitive mutant resistant to INH led to the confirmation that InhA, not KasA, was the true target for INH [48]. Recently, the genetic replacement of a wild-type inhA gene by an allele carrying a specific point mutation in inhA conferred a high level of resistance to INH in a M. tuberculosis INH susceptible strain, ending the dispute of the target of this drug [49]. Thus, there has been a genuine interest in addressing a key issue: how can we inhibit InhA using the information gathered in the study of the mechanisms of action of INH? There are many publications reporting different scaffolds that are able to inhibit InhA, but perhaps the most promising studies arose from the observation that triclosan (TRC) (Figure 1A), a trichlorodiaryl ether, was active on bacterial and parasite enoyl-ACP reduc-tases, including the mycobacterial InhA [50–54]. Several studies on TRC revealed that it acted as a slow binding irreversible inhibitor that did not need any activation [53,55–57]. Thus, it seemed to be a logical candidate to replace INH; avoiding the problems caused by the loss of function of the INH activator, KatG. TRC has been in use for several years as an additive into healthcare products, such as mouth rinses, toothpastes and other applications, producing a large body of information on its safety [58]. However, TRC revealed itself as a molecule with low solubility and poor bioavailability [59,60]; thus, although useful as a primary scaffold, it was clear that changes had to be intro-duced to the molecule to improve its pharmacokinetics. Two laboratories pushed this issue, proposing different molecules
Figure 2. Activation of the antitubercular prodrugs isoniazid, ethionamide, isoxyl and thiacetazone. DesA3: Stearic acid desaturase; ETH: Ethionamide; HadABC: FAS II dehyadratase complex; INH: Isoniazid; InhA: FASII enoyl-ACP reductase; ISO: Isoxyl; MAMTs: Mycolic acid methyl transferases; TAC: Thiacetazone; TRC: Triclosan.
INH
TRC
KatG
EthA
ETH
TAC
ISO
InhA
MAMTs
HadABC
DesA3
Recycling & refurbishing old antitubercular drugs

Expert Rev. Anti Infect. Ther. 11(4), (2013)432
Review
with convenient chemical modifications that resulted in the appropriate changes in activity [56,61].
As a matter of fact, the rising interest on FabI (the bacterial analog of mycobacterial InhA) as a target for drug design gener-ated various drug discovery efforts focused on this enzyme of fatty acid biosynthesis [62]. Likewise, Sacchettini and cowork-ers extended results obtained on the Plasmodium falciparum FabI enzyme to InhA, where high-throughput screening helped identify two novel classes of InhA inhibitors [54]. One of those molecules, a piperazine-based compound, was highly active on the purified enzyme but had relatively poor in vitro antibacterial activity, while the second class, a pyrazole-based molecule acted as an InhA inhibitor with good antibacterial activity against both drug-sensitive and INH-resistant strains of M. tuberculosis [54].
While analyzing the transcriptional pattern of gene expression in M. tuberculosis treated with INH, TRC or the KasA/KasB inhibi-tor thiolactomycin, Betts et al. described a specific set of genes that were induced/repressed upon TRC treatment. Those genes, a signature of TRC treatment, were different to those affected by INH treatment [63]. Interestingly, genes encoding oxidoreductases and membrane proteins were among those upregulated by TRC treatment. Also, an ABC transporter (encoded by genes Rv1686c and Rv1687c) was induced in response to TRC treatment, suggest-ing a role in the efflux of the compound. Similarly, expression of two other genes, Rv3160c and Rv3161c (encoding a hypothetical transcriptional regulator of the TetR-AcrR family, and a possible dioxygenase related to bacterial aromatic dioxygenases involved in the degradation of arenes), were also induced [63]. Since TRC is a diphenylether derivative, it is possible that this pair of genes may be involved in the degradation of TRC. These results would explain to some extent the differences between the activity of TRC on purified InhA versus whole cell-based assays [64]. Work carried out by Tonge’s group focused on the development of a small library of diphenyl ethers with modifications (alkyl chains) in one of the rings; the activity displayed by those compounds showed that when the alkyl chain is extended from one to eight carbons, the affinity of the compounds for InhA increased, with the C8 compound (5-octyl-2-phenoxyphenol; nicknamed as ‘8PP’) binding to InhA about 200-fold more tightly than TRC itself (compound 19) (Figure 1A) [64]. The increase in affinity had a length limit since the C14 diphenylether (‘14PP’) binds much less tightly to InhA than 8PP. Importantly, the mentioned compounds did not induce expression of the detoxification mechanisms act-ing upon TRC, thus, increasing the activity by increasing their intracellular concentration and, of the outmost importance, being active not only on INH-sensitive M. tuberculosis as expected but also as predicted on KatG-deficient, INH-resistant strains [65,66]. Biological evaluation of those compounds demonstrated that they had longer half-life in serum, less cytotoxicity and good tolerance when administered orally [61].
In a similar way, Sacchetini’s and Jacobs’ groups pursued the same approach of chemical modification of the TRC scaffold; using a structure-based drug design approach, they produced a series of 5-substituted TRC derivatives (compound 26) (Figure 1A) containing either alkyl or aryl substituents [67]. Those new
molecules displayed enhanced potency against purified InhA enzyme, with the best candidate being 50-fold more active than the parental TRC molecule. Those compounds, based on the chlorine-containing structure of TRC, displayed higher activity when compared with the nonchlorine-containing diphenyl ethers developed by Tonge’s and Slayden’s groups, leading to the pro-posal that chlorine atoms in the B ring may contribute to binding to the enzyme and thus, to high activity [67]. Based on struc-ture–activity relationship analyses, refinement of the chemical modifications led to the development of a series of compounds, of which those containing a phenyl group linked by C1–C3 linkers to the A ring in position 5 displayed the highest level of activity known for TRC-related molecules [67]. Although those novel TRC derivatives were active on strains lacking the KatG activa-tor, they showed a four- to eight-fold decrease in activity against the two most common mutations affecting InhA; in this way, InhA (S94A) and InhA P(C-15T) mutants are less affected by the new compounds but at the same time confirm that InhA is their molecular target [67]. Considering that 15–25% of the clinical INH-resistant strains contain mutations affecting InhA, discovery of molecules capable of inhibiting this enzyme is a field worth of pursuing; from this point of view, further modifica-tion of the diaryl ether derivatives described could increase their potency as InhA inhibitors.
Increased activity of ethionamide on M. tuberculosis: with a little help from my friendsETH has been a second-line antitubercular prodrug for years, mostly being used after failure of INH due to M. tuberculosis resistance. Early work by Schwartz demonstrated that ETH was as good as INH in therapeutic effectiveness when combined with streptomycin, as measured by x-ray radiography as means of evalu-ation of cavity formation [68]. However, the dosage of ETH is higher than the dosage of INH; thus, although with less potency, ETH has come to the rescue in the countless times in which the therapeutic value of INH was lost due to KatG mutations, as explained in the past section. The mechanism of action of this compound and its prodrug nature were discovered in parallel to the studies on INH. Thus, previous reports suggested that ETH acted on the mycolic acid biosynthetic pathway [32], however, its molecular target and mechanism of action became clear only when the mechanisms of action of INH were studied. The fact that both INH and ETH shared InhA, the enoyl-ACP reductase of the mycolic acid biosynthetic pathway, as a target had profound implications in the understanding of the value of ETH as an INH back-up compound, as mutations in inhA could render M. tuber-culosis resistant to both prodrugs [42]. However, the fact that the activation step was fortunately catalyzed by a different enzyme, the mono-oxygenase EthA, strongly indicated that ETH could still be used in cases where the M. tuberculosis resistance to INH was due to KatG mutations [69]. Therefore, research interest was diverted into the understanding of the activation of ETH. This step is catalyzed by the flavin adenine dinucleotide-containing Baeyer–Villiger monooxygenase EthA [33,69]. Surprisingly, there is no known natural substrate for this enzyme in mycobacteria
Belardinelli & Morbidoni

433www.expert-reviews.com
Review
and its physiological function is still lacking. Early studies demon-strated that overexpression of the M. tuberculosis gene Rv3855 (latter designated as ethR) generated ETH resistance [33], and that the encoded protein displayed homology to members of the TetR family of transcriptional repressors [70]. The adjacent gene, Rv3854c, divergently transcribed, encodes the mentioned EthA enzyme and its role in the activation of ETH was clearly shown by overexpression, resulting in a large increase in susceptibility to this prodrug. Some years ago, an in vitro study showed that EthA is a bifunctional enzyme capable of converting ETH (although with low efficiency) to the S-oxide derivative and subsequently into the 2-ethyl-4-amidopyridine derivative (ETH-amide) [69]. As it was already known that this amide was inactive, secondary trans-formations were proposed. In this regard, DeBarber et al. used [14C]-ETH to identify the different metabolites formed inside the living bacilli; among those metabolites, S-oxide derivative, 2-ethyl-4-cyanopyridine, 2-ethyl-4-amido pyridine derivative, 2-ethyl-4-carboxypyridine and 2-ethyl-4-hydroxymethylpyri-dine were observed [34]. However, Baulard’s group raised con-cerns about the possibility that chemical modification (probably through oxidation) during extraction of the metabolites could yield erroneous results. Thus, this group proceeded to study ETH metabolism by high resolution magic angle spinning-nuclear mag-netic resonance, followed by sorting of the produced metabolites [71,72]. They found that the most relevant of such metabolites produced, 2-ethyl-4-hydroxymethylpyridine, is only present out-side of the mycobacterial cell. Intriguingly, an unidentified ETH metabolite was the only one detected inside the bacilli, being accumulated in the cytoplasm and thus giving support to a role as the molecular species leading to InhA inhibition. No other intermediates matching the ones reported by DeBarber et al. were identified, stressing their reactivity and the importance of the chosen method of analysis in order to preserve their chemical features [72].
The approaches undertaken to overcome the mycobacterial resistance to INH and ETH show interesting similarities and divergences. Both are useful antitubercular prodrugs; however, while INH is exquisitely active, ETH is less active due to a low activation, which leads to the use of larger doses and side effects [73]. As it has been described in the previous sections, both require specific activators in spite of hitting the same enzymatic target. However, in the case of INH, the strategy applied to obtain new InhA inhibitors was centered in using the large amount of biological and biophysical information available to design mol-ecules not requiring an activation step [64,67]. By contrast, in the case of ETH, an equally useful and original approach was based on the development of small molecules capable of inhibiting EthR, the negative regulator of EthA expression. This approach was based on the reports showing that overexpression of EthA in mycobacteria increases ETH sensitivity [33] and an ethR null mutation in bacille Calmette–Guerin also displayed increased ETH sensitivity compared with the parent strain [33]; therefore it is highly possible that ethA is usually partially repressed in myco-bacteria. Consequently, interference with EthR functionality may increase mycobacterial susceptibility to ETH. To this end,
crystal structures of EthR were developed and analyzed [70,74]. Those reports were compared by Frénois et al., who showed that in both cases, the crystal structure contained bound molecules; in one case a hexadecyl-octanoate molecule had fortuitously been included, in the second one, some impurities (dioxane, a component of the crystallization buffer) formed part of the crystal structure. Nonetheless, both structures allowed the demonstration that the crystal with bound ligands displayed a conformational state incompatible with binding of DNA, thereby repressing transcription, giving the opportunity to act on chemical molecules causing EthR to mimic that structure, and in consequence, relieving the inhibition of expression of EthA. The generation of a metabolic state in which over activation of ETH could be achieved would reduce the dosage necessary for its antitubercular effect, thus decreasing its toxicity effects. Such an outcome would be extended to other thioamides such as isoxyl and thiacetazone, especially since due to the severe side effects displayed by those two compounds, a strategy leading to lower doses would be of great benefit for the patient (see below). In the quest for such molecules, Frénois et al., anticipated that the structural features would include two aromatic cycles joined by a 6 Å linker; possible hydrogen bonding to stabilize the molecular interactions and generate a full induction of EthR were also pre-sented [75]. Such compounds were recently described by Willand et al. [35]. From a library of compounds, 131 were selected on the basis of their chemical structure and docking studies. Later on, this set was refined by analyzing their ability to prevent the binding of EthR to a double-stranded biotinylated probe encom-passing the operator region of EthR. This screening yielded two families of compounds; one of them (the most active) had a 1,2,4-oxadiazole linker and a second one (less active) contained a 1,2,4-triazole linker. Cocrystallization studies of EthR with two selected compounds (named BDM31381 and BDM31343) (see Figure 1B) confirmed that the conformational state of the repressor was not fit for binding, and microbiological analy-sis of their activity demonstrated that the activity of ETH was indeed increased 100-fold. Both compounds were also tested in ‘in vivo’ studies in a murine model of M. tuberculosis infec-tion, results of which showed that the mycobacterial counts were reduced at lower (threefold) doses of ETH when these activity ‘boosters’ were given simultaneously with ETH [35]. The struc-ture of BDM31343 was further refined in order to improve its oral availability in mice, leading to the discovery of a compound (BDM41906) that was able to boost the activity of ETH ten-fold on M. tuberculosis infected macrophages, and at the same time, had an increased availability in mice after oral administra-tion [76]. Recently, the screening of a 14,640-compound library identified a novel N-phenylphenoxyacetamide family of com-pounds inhibiting EthR [77], opening the way to the design of new boosters of ETH.
In summary, the strategy of increasing the potency of ETH through a chemical approach leading to a more activating enzyme has proven to be a valuable concept that can be further extended to other antitubercular prodrugs. Those booster molecules are potential companion drugs to be added to existing prodrugs,
Recycling & refurbishing old antitubercular drugs

Expert Rev. Anti Infect. Ther. 11(4), (2013)434
Review
which help to decrease the daily doses required to achieve killing activity during the treatment, thus potentially reducing toxicity effects to the patient.
TAC & ISO, two antitubercular drugs in search for their molecular target(s)Both INH and ETH are still therapeutic compounds in clinical use; however, other agents such as the thioureas TAC and ISO, have been withdrawn from the antitubercular treatment as a result of serious side effects and the relatively high frequency of resistant mutant species generation during treatment [37]. In spite of being known for a long time, it was only recently that studies on the activation of ETH revealed that these cheap prodrugs were also activated by EthA [78,79]. However, their molecular target was clearly different from InhA, the enzyme inhibited by activated ETH and INH, as overexpression of this enoyl-ACP reductase could not confer resistance to these drugs. Several reports helped the understanding of partial information on the mode of action of both prodrugs. In the first place, Phetsuksiri et al. demonstrated that ISO not only inhibited the synthesis of mycolic acids but also that of short chain fatty acids [80]. Analysis of these fatty acids showed that ISO specifically inhibited the synthesis of oleic acid, catalyzed by the stearoyl-ACP desaturase DesA3 (encoded by Rv3229c) [81]. As expected from the analysis of fatty acid contents, overexpression of DesA3 partially restored synthesis of oleic acid and conferred a low-level resistance to ISO. These results led the authors to hypothesize that ISO could inhibit other desatu-rases involved in mycolic acid synthesis [81]. The M. tuberculosis genome contains two other genes, apart from desA3, coding for additional desaturases DesA1 and DesA2, with homology to acyl-ACP desaturases, which led to the thinking that these desaturases were involved in the introduction of the two unsaturations pre-sent in meromycolic acid precursors. Despite this, DesA1 and DesA2 overexpression did not confer resistance to ISO, proving this hypothesis wrong [Buchieri MV, Morbidoni HR, Unpublished data] [81]. Without much information on a precise mode of action, sev-eral compounds were synthesized and tested in M. tuberculosis, displaying an inhibitory activity that, depending on the structural features, was more specific on the synthesis of oleic acid or on the synthesis of mycolic acids (Figure 1C) [82]. Those results, added to the ones previously described, strongly suggested that the mode of action of ISO could be far more complex than expected, involving steps in mycolic acid biosynthesis that were still not known.
Kremer’s group, working on the mechanism(s) of resistance to TAC, found that the treatment of Mycobacterium bovis var bacille Calmette–Guerin with this compound generated an evi-dent accumulation of a mycolic acid precursor which, after puri-fication and chemical analysis, was shown to contain one or two unsaturations at the expense of missing the cyclopropane and oxygenated groups [83]. Thus, it was proposed that TAC acted upon the mycolic acid methyltransferases (MAMTs), catalyz-ing the reactions leading to those modifications. Isolation and characterization of TAC-resistant M. bovis spontaneous mutants produced ample proof that the gene encoding one of the MAMTs, mmaA4, contained mutations in most (but not all) of the isolated
mutants [84]. The evidence was complemented by analysis of a M. tuberculosis mutant containing a transposon insertion in the mmaA4 gene; as expected, that mutant displayed resistance to TAC. Nevertheless, those results posed an unexpected problem, as after several meticulous genetic manipulations, Barkan et al. elegantly demonstrated that M. tuberculosis can be stripped of all the MAMT genes without losing viability although at the cost of severe cell envelope alterations and loss of virulence [85]. Thus, the current available information indicates that the MAMT genes are not essential, although they are necessary for a balanced growth and virulence of the tubercle bacilli. Moreover, overexpression of MmaA4 failed to confer resistance to TAC, adding more doubts on the role of this enzyme [84]. Taken together, those results indi-cate that although one of the mechanisms of action of TAC is the loss of mycolic acid cyclopropanation through the inhibition of MAMTs, the essential target for this drug is still unrevealed and lies buried in the M. tuberculosis chromosome. However, sev-eral compounds were designed as structural variants of TAC and tested for antitubercular activity (Figure 1D); some of those com-pounds exerted very good activity and hold promise as possible scaffolds for drug improvement but the lack of information on their mechanisms of action and molecular targets dampens that possibility to some extent [82,86].
New kids on the block: TAC & ISO put a novel FASII enzyme under the spotlightA remarkable feature of the M. tuberculosis genome is the abun-dance of genes involved in fatty acid metabolism (degradation, modification and synthesis); moreover, the high number of orthologs for any given gene makes the assignment of function complex [12,87]. In the specific pathway leading to the synthesis of mycolic acids, an important factor that increases this complexity is the mere nature of the products (very long modified fatty acids) and the number of steps of the pathway. Thus, it is quite difficult to demonstrate function, as no commercial substrates are available for this biochemical pathway that makes such complex products, and model substrates do not always reflect the true nature of the enzyme under scrutiny.
The recent efforts in the understanding of the pathway respon-sible for mycolic acid biosynthesis led to the identification of novel enzymes, one of which was the long-sought β-hydroxyacyl-ACP dehydratase [88,89]. This enzyme is, in reality, a complex formed by two heterodimers, HadAB and HadBC, encoded in a single operon (hadABC). Although the enzymatic reaction was char-acterized, there is still a void in the knowledge of some features, such as their genetic regulation and crystal structure. Although only hadAB has been proven to be essential in nature by TraSH analysis, defined deletions have not been made, thus the nones-sentiality of hadBC is still arguable [90]. However, compounds inhibiting these reactions are actively being sought [91,92].
In this scenario, a recent report by our group indicated the role of HadABC in conferring high resistance to TAC in M. tuberculosis [40]; subsequently our work and Jackson’s work simultaneously found that HadABC also mediated high levels of resistance to ISO when overexpressed [41]. This phenotype was specific as no
Belardinelli & Morbidoni
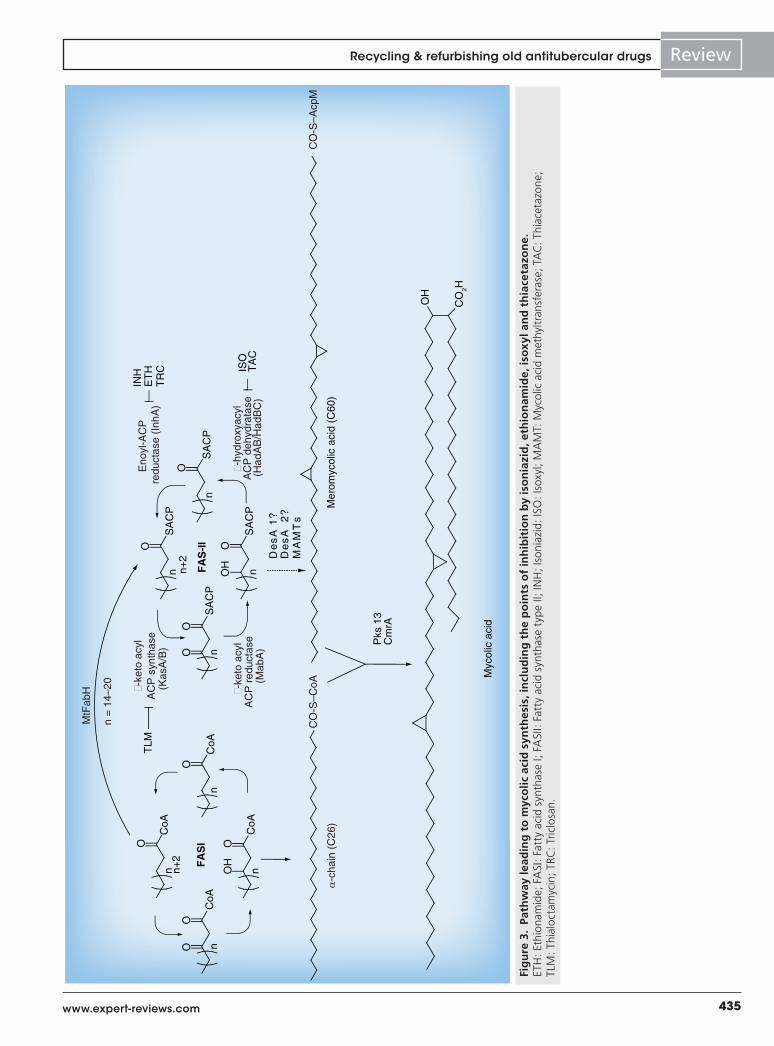
435www.expert-reviews.com
Review
Fig
ure
3.
Path
way
lead
ing
to
myc
olic
aci
d s
ynth
esis
, in
clu
din
g t
he
po
ints
of
inh
ibit
ion
by
iso
nia
zid
, eth
ion
amid
e, is
oxy
l an
d t
hia
ceta
zon
e.ET
H: E
thio
nam
ide;
FA
SI: F
atty
aci
d sy
ntha
se I;
FA
SII:
Fatt
y ac
id s
ynth
ase
type
II; I
NH
; Iso
niaz
id: I
SO: I
soxy
l; M
AM
T: M
ycol
ic a
cid
met
hyltr
ansf
eras
e; T
AC
: Thi
acet
azon
e;
TLM
: Thi
aloc
tam
ycin
; TRC
: Tric
losa
n.
O
n n+2
C
oA
CoA
O
n O
CoA
O
n OH
CoA
O
n
OH
CO
2H
ISO
TA
C
n+2
O
n S
AC
P
INH
ET
HT
RC
FAS-II
FASI
SA
CP
O
n O
SA
CP
O
n OH
SA
CP
O
n
TLM
Pks
13
Cm
rA
Mer
omyc
olic
aci
d (C
60)
Myc
olic
aci
d
De
sA
1?
De
sA
2
?M
AM
Ts
n =
14–
20
MtF
abH
α-c
hain
(C
26)
�-ke
to a
cyl
AC
P s
ynth
ase
(Kas
A/B
)
Eno
yl-A
CP
redu
ctas
e (I
nhA
)
�-ke
to a
cyl
AC
P r
educ
tase
(Mab
A)
�-hy
drox
yacy
lA
CP
deh
ydra
tase
(Had
AB
/Had
BC
)
CO
-S–A
cpM
CO
-S–C
oA
Recycling & refurbishing old antitubercular drugs

Expert Rev. Anti Infect. Ther. 11(4), (2013)436
Review
Key issues
• Tuberculosis is a global health problem, in which resistance to currently available drugs jeopardizes the treatment outcome and dramatically underscores the need for new drugs.
• Mutations rendering the few specifically antitubercular drugs that are used for treatment ineffective are increasingly common, generating multidrug-resistant and extensively drug-resistant strains.
• The synthesis of mycolic acids, essential components of the envelope in all mycobacteria, has been proven a valuable target for drug development.
• The synthesis of mycolic acids is carried out, among other enzymes, by the fatty acid synthase type II (FASII). This synthase already offered targets for antitubercular prodrugs, such as isoniazid and ethionamide.
• The activity of antitubercular prodrugs is hindered by mutations impeding the activation step, thus a reasonable way to overcome that resistance and restore activity is modifying the pharmacophore scaffold to avoid the need of activation.
• In cases in which activity of the prodrug in clinical use is less than optimal or is accompanied by side effects, the improvement of their performance through their combination with ‘booster’ companion molecules increasing the rate of their activation is a feasible and worthy strategy. As a consequence, smaller doses will allow the achievement of therapeutic action with less adverse undesirable side effects.
• The mentioned strategies have produced molecules capable of either replacing prodrugs of known therapeutic value, such as isoniazid, or improve their activity when combined with prodrugs, such as ethionamide.
• Novel FASII inhibitors may be designed using the scaffold of thiacetazone or isoxyl, or improving their structure in order to avoid the activation step. The identification of a new component of the FASII system will also allow undertaking the design of the novel inhibitors affecting this essential step.
other FASII enzyme could protect M. tuberculosis from either TAC or ISO when overexpressed. Moreover, we described spe-cific missense mutations conferring resistance to TAC and ISO that affected hadA or hadC in M. tuberculosis and Mycobacterium kansasii; intriguingly no mutations in hadB were found [40]. Later on, mutations in hadABC were also described by another group, supporting our previous findings [93].
Nonetheless, not only does the determination of essentiality of hadABC need to be confirmed, but also its role as a possible molecular target for TAC and ISO must be assessed. This specific topic is currently underway in our laboratory and will help in confirming whether activated TAC and ISO bind to HadAB and HadBC; thus the three requirements of a protein to be a drug target will be fulfilled: essentiality, high level of resistance by overexpression and binding of the drug or its metabolites.
Thus, taken together, this information suggests that the anti-mycobacterial activity of both prodrugs can be avoided by the bacilli owing to mutations in hadABC, suggesting that both lines of research, the one on these prodrugs and the one on the design of novel drugs targeting HadABC, may be united with the benefit of accelerating the discovery of novel inhibitory molecules.
Expert commentary & five-year viewThe treatment of infections caused by M. tuberculosis has lately been jeopardized by the increase in drug resistance in this micro-organism. Clinical isolates resistant to INH and rifampicin, des-ignated as multidrug-resistant M. tuberculosis gave way to the generation of strains displaying extensive resistance, not only to those first-line antitubercular drugs but also to any fluoro-quinolone, and at least one second-line injectable drug (amikacin, capreomycin or kanamycin). The death toll of patients suffering from infections with these strains (known as extensively drug-resistant tuberculosis) is high, especially in vulnerable groups such as HIV patients. Moreover, strains displaying resistance to all
kinds of therapeutic agents have recently been detected, leaving unconventional drugs, such as neuroleptics, as a last resource for the treatment of those cases on humanitarian grounds. Thus, the discovery of novel antitubercular drugs is an urgent need that has to be met as soon as possible. In this regard, a few molecular targets have been identified, such as the mycobacterial ATP syn-thase and the decaprenyl-phosphoribose 2´ epimerase, leading to the proposal of molecules that will be in the market soon. As a matter of fact, one of such molecules (TMC207, licensed under the name bedaquiline) has just been approved in the USA for clinical use [94,95]. Its also worth mentioning that recently, four different groups have reported several compounds that, instead of acting on the synthesis, inhibit mycolic acid transport across the plasma membrane; analysis of resistant mutants led to the identi-fication of MmpL3 as the transporter of mycolic acids in the form of trehalose monomycolates [96–98]. One of these compounds, designated SQ109, showed promising results and is currently in Phase II clinical trials [99].
The notion that mycolic acids are essential components of the mycobacterial cell envelope, and also players in the interplay with the immune system, has been fundamental in the choice of the synthesis of those uncommon, long-chain fatty acids as a strategi-cal pathway to inhibit. Years of interdisciplinary work from sev-eral groups revealed the mechanism of action of drugs targeting enzymes from the mycolic acid pathway, as well as the ensuing mycobacterial mechanisms of resistance. Combined, both topics shed light on approaches that could help revitalize the use of those drugs or help design pharmacophores on already validated enzymes. A new drug, delamanid (OPC-67683), a nitrodihydro-imidazoxazole derivative, is a new antituberculosis medication that inhibits mycolic acid synthesis and has shown potent in vitro and in vivo activity against drug-resistant strains of M. tubercu-losis, demonstrating the value of targeting the pathway. In this review, we have briefly reviewed the cases of two ‘old’ molecules
Belardinelli & Morbidoni

437www.expert-reviews.com
Review
References1 Young DB. Hunger not of the belly kind.
Tuberculosis (Edinb.) 83(6), 329–330 (2003).
2 Munro AW, McLean KJ, Marshall KR et al. Cytochromes P450: novel drug targets in the war against multidrug-resist-ant Mycobacterium tuberculosis. Biochem. Soc. Trans. 31(Pt 3), 625–630 (2003).
3 Duncan K. Identification and validation of novel drug targets in tuberculosis. Curr. Pharm. Des. 10(26), 3185–3194 (2004).
4 Mitchison DA. Drug resistance in tuberculosis. Eur. Respir. J. 25(2), 376–379 (2005).
5 De Rossi E, Aínsa JA, Riccardi G. Role of mycobacterial efflux transporters in drug resistance: an unresolved question. FEMS Microbiol. Rev. 30(1), 36–52 (2006).
6 Strong M, Goulding CW. Structural proteomics and computational analysis of a deadly pathogen: combating Mycobacterium tuberculosis from multiple fronts. Methods Biochem. Anal. 49, 245–269 (2006).
7 Schreiber M, Res I, Matter A. Protein kinases as antibacterial targets. Curr. Opin. Cell Biol. 21(2), 325–330 (2009).
8 Riccardi G, Pasca MR, Buroni S. Mycobacterium tuberculosis: drug resistance and future perspectives. Future Microbiol. 4(5), 597–614 (2009).
9 Barry CE 3rd, Blanchard JS. The chemical biology of new drugs in the development for tuberculosis. Curr. Opin. Chem. Biol. 14(4), 456–466 (2010).
10 Brennan PJ, Crick DC. The cell-wall core of Mycobacterium tuberculosis in the context
of drug discovery. Curr. Top. Med. Chem. 7(5), 475–488 (2007).
11 Takayama K, Wang C, Besra GS. Pathway to synthesis and processing of mycolic acids in Mycobacterium tuberculosis. Clin. Microbiol. Rev. 18(1), 81–101 (2005).
12 Cole ST, Brosch R, Parkhill J et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393(6685), 537–544 (1998).
13 Barry CE 3rd, Lee RE, Mdluli K et al. Mycolic acids: structure, biosynthesis and physiological functions. Prog. Lipid Res. 37(2–3), 143–179 (1998).
14 Mackaness GB, Smith N. The action of isoniazid (isonicotinic acid hydrazide) on intracellular tubercle bacilli. Am. Rev. Tuberc. 66(2), 125–133 (1952).
15 Middlebrook G. Sterilization of tubercle bacilli by isonicotinic acid hydrazide and the incidence of variants resistant to the drug in vitro. Am. Rev. Tuberc. 65(6), 765–767 (1952).
16 Middlebrook G, Cohn ML. Attenuation of isoniazid-resistant mutants of tubercle bacilli and host hypersensitivity to tuberculin. Proc. Soc. Exp. Biol. Med. 88(4), 568–571 (1955).
17 Middlebrook G, Cohn ML, Schaefer WB. Studies on isoniazid and tubercle bacilli. III. The isolation, drug-susceptibility, and catalase-testing of tubercle bacilli from isoniazid-treated patients. Am. Rev. Tuberc. 70(5), 852–872 (1954).
18 Heym B, Zhang Y, Poulet S, Young D, Cole ST. Characterization of the katG gene encoding a catalase-peroxidase required for
the isoniazid susceptibility of Mycobacte-rium tuberculosis. J. Bacteriol. 175(13), 4255–4259 (1993).
19 Heym B, Saint-Joanis B, Cole ST. The molecular basis of isoniazid resistance in Mycobacterium tuberculosis. Tuber. Lung Dis. 79(4), 267–271 (1999).
20 Musser JM, Kapur V, Williams DL, Kreiswirth BN, van Soolingen D, van Embden JD. Characterization of the catalase-peroxidase gene (katG) and inhA locus in isoniazid-resistant and -susceptible strains of Mycobacterium tuberculosis by automated DNA sequencing: restricted array of mutations associated with drug resistance. J. Infect. Dis. 173(1), 196–202 (1996).
21 Ramaswamy S, Musser JM. Molecular genetic basis of antimicrobial agent resistance in Mycobacterium tuberculosis: 1998 update. Tuber. Lung Dis. 79(1), 3–29 (1998).
22 van Soolingen D, de Haas PE, van Doorn HR, Kuijper E, Rinder H, Borgdorff MW. Mutations at amino acid position 315 of the katG gene are associated with high-level resistance to isoniazid, other drug resistance, and successful transmission of Mycobacterium tuberculosis in The Netherlands. J. Infect. Dis. 182(6), 1788–1790 (2000).
23 Mokrousov I, Narvskaya O, Otten T, Limeschenko E, Steklova L, Vyshnevskiy B. High prevalence of KatG Ser315Thr substitution among isoniazid-resistant Mycobacterium tuberculosis clinical isolates from northwestern Russia, 1996 to 2001. Antimicrob. Agents Chemother. 46(5), 1417–1424 (2002).
hitting the mycolic acid pathway but with flaws that diminished their clinical value: INH, for which new molecules have been proposed, which overcome its weakness; and ETH, for which companion molecules that accelerate its rate of activation have been evaluated. In addition, we discussed the potential of TAC and isoxyl as tools to study HadABC, a critical enzyme of the pathway. A summary of the points of the FASII cycle inhibited by those compounds is shown in Figure 3.
In the near future, in addition to the efforts devoted to anti-tubercular drug discovery through high-throughput screening, we envision a considerable amount of resources will be applied to the improvement or replacement of old drugs with a long history of therapeutical success but with disadvantages stemming from their own chemical structure. It would be of high interest to keep an eye on the clinical evaluation of InhA inhibitors as well as ETH boosters. We believe that recent work on HadABC is also valuable, since it unites two lines of research that were being pursued separately. Thus, inhibitors of HadABC would also be
logical candidates to develop in the near future. In summary, the approach of ‘revamping’ old drugs, or even drugs that were discarded from therapeutical use, may not only prove to be prom-ising but also a real sound way to produce novel antitubercular drugs.
Financial & competing interests disclosureWork in HR Morbidoni’s lab was supported by grants from FONCyT (PICT 38198 and 1063). JM Belardinelli was a fellow of CONICET (Argentina) and is currently a Fellow at the Department of Microbiology, Immunology and Pathology, College of Veterinary Medicine and Biomedical Sciences, Colorado State University, Fort Collins, CO, USA. HR Morbidoni is an independent researcher of CIUNR (Consejode Investigaciones, Universidad Nacional de Rosario, Argentina). The authors have no other relevant affili-ations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Recycling & refurbishing old antitubercular drugs

Expert Rev. Anti Infect. Ther. 11(4), (2013)438
Review
24 Silva MS, Senna SG, Ribeiro MO et al. Mutations in katG, inhA, and ahpC genes of Brazilian isoniazid-resistant isolates of Mycobacterium tuberculosis. J. Clin. Microbiol. 41(9), 4471–4474 (2003).
25 Herrera-León L, Molina T, Saíz P, Sáez-Nieto JA, Jiménez MS. New multiplex PCR for rapid detection of isoniazid-resist-ant Mycobacterium tuberculosis clinical isolates. Antimicrob. Agents Chemother. 49(1), 144–147 (2005).
26 Bergval IL, Schuitema AR, Klatser PR, Anthony RM. Resistant mutants of Mycobacterium tuberculosis selected in vitro do not reflect the in vivo mechanism of isoniazid resistance. J. Antimicrob. Chemother. 64(3), 515–523 (2009).
27 Sherman DR, Mdluli K, Hickey MJ et al. Compensatory ahpC gene expression in isoniazid-resistant Mycobacterium tuberculosis. Science 272(5268), 1641–1643 (1996).
28 Pym AS, Saint-Joanis B, Cole ST. Effect of katG mutations on the virulence of Mycobacterium tuberculosis and the implication for transmission in humans. Infect. Immun. 70(9), 4955–4960 (2002).
29 O’Sullivan DM, McHugh TD, Gillespie SH. The effect of oxidative stress on the mutation rate of Mycobacterium tuberculosis with impaired catalase/peroxidase function. J. Antimicrob. Chemother. 62(4), 709–712 (2008).
30 Bulatovic VM, Wengenack NL, Uhl JR et al. Oxidative stress increases susceptibil-ity of Mycobacterium tuberculosis to isoniazid. Antimicrob. Agents Chemother. 46(9), 2765–2771 (2002).
31 Winder FG, Collins PB, Whelan D. Effects of ethionamide and isoxyl on mycolic acid synthesis in Mycobacterium tuberculosis BCG. J. Gen. Microbiol. 66(3), 379–380 (1971).
32 Quémard A, Lanéelle G, Lacave C. Mycolic acid synthesis: a target for ethionamide in mycobacteria? Antimicrob. Agents Chemother. 36(6), 1316–1321 (1992).
33 Baulard AR, Betts JC, Engohang-Ndong J et al. Activation of the pro-drug ethiona-mide is regulated in mycobacteria. J. Biol. Chem. 275(36), 28326–28331 (2000).
34 DeBarber AE, Mdluli K, Bosman M, Bekker LG, Barry CE 3rd. Ethionamide activation and sensitivity in multidrug-resistant Mycobacterium tuberculosis. Proc. Natl Acad. Sci. USA 97(17), 9677–9682 (2000).
35 Willand N, Dirié B, Carette X et al. Synthetic EthR inhibitors boost antituber-culous activity of ethionamide. Nat. Med. 15(5), 537–544 (2009).
36 Jaju M, Jaju M, Ahuja YR. Combined and individual effects of isoniazid and thiacetazone on human lymphocyte chromosomes in vitro and in vivo. Hum. Toxicol. 3(5), 373–382 (1984).
37 Lambelin G. Pharmacology and toxicology of Isoxyl. Antibiot. Chemother. 16, 84–95 (1970).
38 Harland RD. Stevens–Johnson syndrome with unusual skin features occurring in two patients undergoing treatment for pulmonary tuberculosis with thiacetazone. Tubercle 43, 189–191 (1962).
39 Chintu C, Luo C, Bhat G, Raviglione M, DuPont H, Zumla A. Cutaneous hypersensitivity reactions due to thiaceta-zone in the treatment of tuberculosis in Zambian children infected with HIV-I. Arch. Dis. Child. 68(5), 665–668 (1993).
40 Belardinelli JM, Morbidoni HR. Mutations in the essential FAS II b-hydroxyacyl ACP dehydratase complex confer resistance to thiacetazone in Mycobacterium tuberculosis and Mycobacterium kansasii. Mol. Microbiol. 86(3), 568–579 (2012).
41 Grzegorzewicz AE, Korduláková J, Jones V et al. A common mechanism of inhibition of the Mycobacterium tuberculosis mycolic acid biosynthetic pathway by isoxyl and thiacetazone. J. Biol. Chem. 287(46), 38434–38441 (2012).
42 Banerjee A, Dubnau E, Quemard A et al. inhA, a gene encoding a target for isoniazid and ethionamide in Mycobacterium tuberculosis. Science 263(5144), 227–230 (1994).
43 Banerjee A, Sugantino M, Sacchettini JC, Jacobs WR Jr. The mabA gene from the inhA operon of Mycobacterium tuberculosis encodes a 3-ketoacyl reductase that fails to confer isoniazid resistance. Microbiology 144 (Pt 10), 2697–2704 (1998).
44 Dessen A, Quémard A, Blanchard JS, Jacobs WR Jr, Sacchettini JC. Crystal structure and function of the isoniazid target of Mycobacterium tuberculosis. Science 267(5204), 1638–1641 (1995).
45 Quémard A, Sacchettini JC, Dessen A et al. Enzymatic characterization of the target for isoniazid in Mycobacterium tuberculosis. Biochemistry 34(26), 8235–8241 (1995).
46 Rozwarski DA, Grant GA, Barton DH, Jacobs WR Jr, Sacchettini JC. Modification of the NADH of the isoniazid target
(InhA) from Mycobacterium tuberculosis. Science 279(5347), 98–102 (1998).
47 Mdluli K, Slayden RA, Zhu Y et al. Inhibition of a Mycobacterium tuberculosis beta-ketoacyl ACP synthase by isoniazid. Science 280(5369), 1607–1610 (1998).
48 Vilchèze C, Morbidoni HR, Weisbrod TR et al. Inactivation of the inhA-encoded fatty acid synthase II (FASII) enoyl-acyl carrier protein reductase induces accumula-tion of the FASI end products and cell lysis of Mycobacterium smegmatis. J. Bacteriol. 182(14), 4059–4067 (2000).
49 Vilchèze C, Wang F, Arai M et al. Transfer of a point mutation in Mycobacterium tuberculosis inhA resolves the target of isoniazid. Nat. Med. 12(9), 1027–1029 (2006).
50 Freundlich JS, Yu M, Lucumi E et al. Synthesis and biological activity of diaryl ether inhibitors of malarial enoyl acyl carrier protein reductase. Part 2: 2 -́substi-tuted triclosan derivatives. Bioorg. Med. Chem. Lett. 16(8), 2163–2169 (2006).
51 Heath RJ, Rubin JR, Holland DR, Zhang E, Snow ME, Rock CO. Mechanism of triclosan inhibition of bacterial fatty acid synthesis. J. Biol. Chem. 274(16), 11110–11114 (1999).
52 Bork S, Yokoyama N, Matsuo T, Claveria FG, Fujisaki K, Igarashi I. Growth inhibitory effect of triclosan on equine and bovine Babesia parasites. Am. J. Trop. Med. Hyg. 68(3), 334–340 (2003).
53 Kapoor M, Reddy CC, Krishnasastry MV, Surolia N, Surolia A. Slow-tight-binding inhibition of enoyl-acyl carrier protein reductase from Plasmodium falciparum by triclosan. Biochem. J. 381(Pt 3), 719–724 (2004).
54 Kuo MR, Morbidoni HR, Alland D et al. Targeting tuberculosis and malaria through inhibition of enoyl reductase: compound activity and structural data. J. Biol. Chem. 278(23), 20851–20859 (2003).
55 Parikh SL, Xiao G, Tonge PJ. Inhibition of InhA, the enoyl reductase from Mycobacterium tuberculosis, by triclosan and isoniazid. Biochemistry 39(26), 7645–7650 (2000).
56 Perozzo R, Kuo M, Sidhu Ab et al. Structural elucidation of the specificity of the antibacterial agent triclosan for malarial enoyl acyl carrier protein reductase. J. Biol. Chem. 277(15), 13106–13114 (2002).
57 Rawat R, Whitty A, Tonge PJ. The isoniazid-NAD adduct is a slow, tight-binding inhibitor of InhA, the Mycobacte-rium tuberculosis enoyl reductase: adduct
Belardinelli & Morbidoni

439www.expert-reviews.com
Review
affinity and drug resistance. Proc. Natl Acad. Sci. USA 100(24), 13881–13886 (2003).
58 Bhargava HN, Leonard PA. Triclosan: applications and safety. Am. J. Infect. Control 24(3), 209–218 (1996).
59 Sandborgh-Englund G, Adolfsson-Erici M, Odham G, Ekstrand J. Pharmacokinetics of triclosan following oral ingestion in humans. J. Toxicol. Environ. Health Part A 69(20), 1861–1873 (2006).
60 Taylor TJ, Seitz EP, Fox P, Fischler GE, Fuls JL, Weidner PL. Physicochemical factors affecting the rapid bactericidal efficacy of the phenolic antibacterial triclosan. Int. J. Cosmet. Sci. 26(3), 111–116 (2004).
61 Boyne ME, Sullivan TJ, amEnde CW et al. Targeting fatty acid biosynthesis for the development of novel chemotherapeutics against Mycobacterium tuberculosis: evaluation of A-ring-modified diphenyl ethers as high-affinity InhA inhibitors. Antimicrob. Agents Chemother. 51(10), 3562–3567 (2007).
62 Moir DT. Identification of inhibitors of bacterial enoyl-acyl carrier protein reductase. Curr. Drug Targets. Infect. Disord. 5(3), 297–305 (2005).
63 Betts JC, McLaren A, Lennon MG et al. Signature gene expression profiles discriminate between isoniazid-, thiolacto-mycin-, and triclosan-treated Mycobacte-rium tuberculosis. Antimicrob. Agents Chemother. 47(9), 2903–2913 (2003).
64 Tonge PJ, Kisker C, Slayden RA. Development of modern InhA inhibitors to combat drug resistant strains of Mycobacterium tuberculosis. Curr. Top. Med. Chem. 7(5), 489–498 (2007).
65 Sullivan TJ, Truglio JJ, Boyne ME et al. High affinity InhA inhibitors with activity against drug-resistant strains of Mycobacterium tuberculosis. ACS Chem. Biol. 1(1), 43–53 (2006).
66 am Ende CW, Knudson SE, Liu N et al. Synthesis and in vitro antimycobacterial activity of B-ring modified diaryl ether InhA inhibitors. Bioorg. Med. Chem. Lett. 18(10), 3029–3033 (2008).
67 Freundlich JS, Wang F, Vilchèze C et al. Triclosan derivatives: towards potent inhibitors of drug-sensitive and drug-resist-ant Mycobacterium tuberculosis. ChemMedChem 4(2), 241–248 (2009).
68 Schwartz WS. Comparison of ethionamide with isoniazid in original treatment cases of pulmonary tuberculosis. XIV. A report of the Veterans Administration–Armed Forces
cooperative study. Am. Rev. Respir. Dis. 93(5), 685–692 (1966).
69 Vannelli TA, Dykman A, Ortiz de Montellano PR. The antituberculosis drug ethionamide is activated by a flavoprotein monooxygenase. J. Biol. Chem. 277(15), 12824–12829 (2002).
70 Dover LG, Corsino PE, Daniels IR et al. Crystal structure of the TetR/CamR family repressor Mycobacterium tuberculosis EthR implicated in ethionamide resistance. J. Mol. Biol. 340(5), 1095–1105 (2004).
71 Hanoulle X, Wieruszeski JM, Rousselot-Pailley P, Landrieu I, Baulard AR, Lippens G. Monitoring of the ethionamide pro-drug activation in mycobacteria by 1H high resolution magic angle spinning NMR. Biochem. Biophys. Res. Commun. 331(2), 452–458 (2005).
72 Hanoulle X, Wieruszeski JM, Rousselot-Pailley P et al. Selective intracellular accumulation of the major metabolite issued from the activation of the prodrug ethionamide in mycobacteria. J. Antimicrob. Chemother. 58(4), 768–772 (2006).
73 Nathanson E, Gupta R, Huamani P et al. Adverse events in the treatment of multidrug-resistant tuberculosis: results from the DOTS-Plus initiative. Int. J. Tuberc. Lung Dis. 8(11), 1382–1384 (2004).
74 Frénois F, Engohang-Ndong J, Locht C, Baulard AR, Villeret V. Structure of EthR in a ligand bound conformation reveals therapeutic perspectives against tuberculo-sis. Mol. Cell 16(2), 301–307 (2004).
75 Frénois F, Baulard AR, Villeret V. Insights into mechanisms of induction and ligands recognition in the transcriptional repressor EthR from Mycobacterium tuberculosis. Tuberculosis (Edinb.) 86(2), 110–114 (2006).
76 Flipo M, Desroses M, Lecat-Guillet N et al. Ethionamide boosters: synthesis, biological activity, and structure-activity relationships of a series of 1,2,4-oxadiazole EthR inhibitors. J. Med. Chem. 54(8), 2994–3010 (2011).
77 Flipo M, Willand N, Lecat-Guillet N et al. Discovery of novel N-phenylphenoxyaceta-mide derivatives as EthR inhibitors and ethionamide boosters by combining high-throughput screening and synthesis. J. Med. Chem. 55(14), 6391–6402 (2012).
78 Wang F, Langley R, Gulten G et al. Mechanism of thioamide drug action against tuberculosis and leprosy. J. Exp. Med. 204(1), 73–78 (2007).
79 Dover LG, Alahari A, Gratraud P et al. EthA, a common activator of thiocarba-mide-containing drugs acting on different mycobacterial targets. Antimicrob. Agents Chemother. 51(3), 1055–1063 (2007).
80 Phetsuksiri B, Baulard AR, Cooper AM et al. Antimycobacterial activities of isoxyl and new derivatives through the inhibition of mycolic acid synthesis. Antimicrob. Agents Chemother. 43(5), 1042–1051 (1999).
81 Phetsuksiri B, Jackson M, Scherman H et al. Unique mechanism of action of the thiourea drug isoxyl on Mycobacterium tuberculosis. J. Biol. Chem. 278(52), 53123–53130 (2003).
82 Bhowruth V, Brown AK, Reynolds RC et al. Symmetrical and unsymmetrical analogues of isoxyl; active agents against Mycobacterium tuberculosis. Bioorg. Med. Chem. Lett. 16(18), 4743–4747 (2006).
83 Alahari A, Trivelli X, Guérardel Y et al. Thiacetazone, an antitubercular drug that inhibits cyclopropanation of cell wall mycolic acids in mycobacteria. PLoS ONE 2(12), e1343 (2007).
84 Alahari A, Alibaud L, Trivelli X et al. Mycolic acid methyltransferase, MmaA4, is necessary for thiacetazone susceptibility in Mycobacterium tuberculosis. Mol. Microbiol. 71(5), 1263–1277 (2009).
85 Barkan D, Hedhli D, Yan HG, Huygen K, Glickman MS. Mycobacterium tuberculosis lacking all mycolic acid cyclopropanation is viable but highly attenuated and hyperin-flammatory in mice. Infect. Immun. 80(6), 1958–1968 (2012).
86 Cole ST. Rifamycin resistance in mycobacteria. Res. Microbiol. 147(1–2), 48–52 (1996).
87 Cole ST. Comparative and functional genomics of the Mycobacterium tuberculosis complex. Microbiology 148(Pt 10), 2919–2928 (2002).
88 Sacco E, Covarrubias AS, O’Hare HM et al. The missing piece of the type II fatty acid synthase system from Mycobacterium tuberculosis. Proc. Natl Acad. Sci. USA 104(37), 14628–14633 (2007).
89 Brown AK, Bhatt A, Singh A, Saparia E, Evans AF, Besra GS. Identification of the dehydratase component of the mycobacte-rial mycolic acid-synthesizing fatty acid synthase-II complex. Microbiology 153(Pt 12), 4166–4173 (2007).
90 Sassetti CM, Boyd DH, Rubin EJ. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48(1), 77–84 (2003).
Recycling & refurbishing old antitubercular drugs

Expert Rev. Anti Infect. Ther. 11(4), (2013)440
Review
91 Bhowruth V, Brown AK, Besra GS. Synthe-sis and biological evaluation of NAS-21 and NAS-91 analogues as potential inhibitors of the mycobacterial FAS-II dehydratase enzyme Rv0636. Microbiology 154(Pt 7), 1866–1875 (2008).
92 Brown AK, Papaemmanouil A, Bhowruth V, Bhatt A, Dover LG, Besra GS. Flavonoid inhibitors as novel antimycobacterial agents targeting Rv0636, a putative dehydratase enzyme involved in Mycobacterium tuberculosis fatty acid synthase II. Microbiology 153(Pt 10), 3314–3322 (2007).
93 Gannoun-Zaki L, Alibaud L, Kremer L. Point mutations within the fatty acid synthase type II dehydratase components HadA or HadC contribute to isoxyl resistance in Mycobacterium tuberculosis.
Antimicrob. Agents Chemother. 57(1), 629–632 (2013).
94 Willyard C. Malaria vaccine results present infant immunization quandary. Nat. Med. 18(12), 1723 (2012).
95 Cohen JM, Dlamini S, Novotny JM, Kandula D, Kunene S, Tatem AJ. Rapid case-based mapping of seasonal malaria transmission risk for strategic elimination planning in Swaziland. Malar. J. 12(1), 61 (2013).
96 Grzegorzewicz AE, Pham H, Gundi VA et al. Inhibition of mycolic acid transport across the Mycobacterium tuberculosis plasma membrane. Nat. Chem. Biol. 8(4), 334–341 (2012).
97 Scherman MS, North EJ, Jones V et al. Screening a library of 1600 adamantyl
ureas for anti-Mycobacterium tuberculosis activity in vitro and for better physical chemical properties for bioavailability. Bioorg. Med. Chem. 20(10), 3255–3262 (2012).
98 La Rosa V, Poce G, Canseco JO et al. MmpL3 is the cellular target of the antitubercular pyrrole derivative BM212. Antimicrob. Agents Chemother. 56(1), 324–331 (2012).
99 Tahlan K, Wilson R, Kastrinsky DB et al. SQ109 targets MmpL3, a membrane transporter of trehalose monomycolate involved in mycolic acid donation to the cell wall core of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 56(4), 1797–1809 (2012).
Belardinelli & Morbidoni