enfermedad de gaucher: diagnóstico y avances en el tratamiento€¦ · • de esta forma se dejan...
TRANSCRIPT

Enfermedad de Gaucher: Diagnóstico y
avances en el tratamiento
Dr. Miguel PocovíCatedrático de Bioquímica y Biología Molecular
Universidad de Zaragoza
Barcelona 22 Septiembre 2013
Phillip-Charles-Ernest Gaucher

Enfermedad de Gaucher– una EDL
• La enfermedad lisosomal mas frecuente.
• Autosómica recesiva.
• 1:15 judíos Ashkenazi son portadores y
1: 850 la padecen.
• Otras poblaciones 1:40.000 -1:100.000.
• Dispone de varios tipos de tratamiento.
• Deficiencia de Glucocerobrosidasa lisosomal.

Macrófagos tisulares
Células Kupffer
Matraz de Erlenmeyer
Osteonecrosis
Osteoesclerosis
Osteoclastos
Médula ósea
Anemia, trombocitopenia
Células de Gaucher
Infiltraciones
Fibrosis intersticial
Ocupación de los alveolos
Otros órganos
Corazón
Nódulos linfáticos
Sistema nervioso central
Riñones
Manifestaciones oculares
Manifestaciones cutáneas
Manifestaciones clínicas

Fenotipo Continuo
Hidrops
fetalis
Epilepsia
Mioclonia
Afectación
Bulbar
Movimientos
sacádicos
Asintomaticos Hepatoespleno-
megalia
Sidransky et al 2004
Tipo 1
Tipo 2
Tipo 3

Diagnóstico

¿Qué pretendemos con el
diagnóstico?
• Hacer un diagnóstico precoz, antes que
aparezca patología irreversible en:
• Recién nacidos
• Niños
• Adolescentes
• Familiares
• Población seleccionada, por ejemplo….

Estrategias de búsqueda de
pacientes con EG
• Trombocitopenia de origen no filiado o atribuida a
hiperesplenismo, sin marcadores de hepatopatía crónica de
origen cirrótico o trombocitopenia de origen inmune
• Esplenomegalia de origen no filiado asociada o no a cirrosis
hepática. Pacientes esplenectomizados sin diagnóstico
concreto
• Fractura espontánea de cadera no justificada. Osteomielitis
“aséptica”
• Fosfatasa ácida + esplenomegalia u otros?
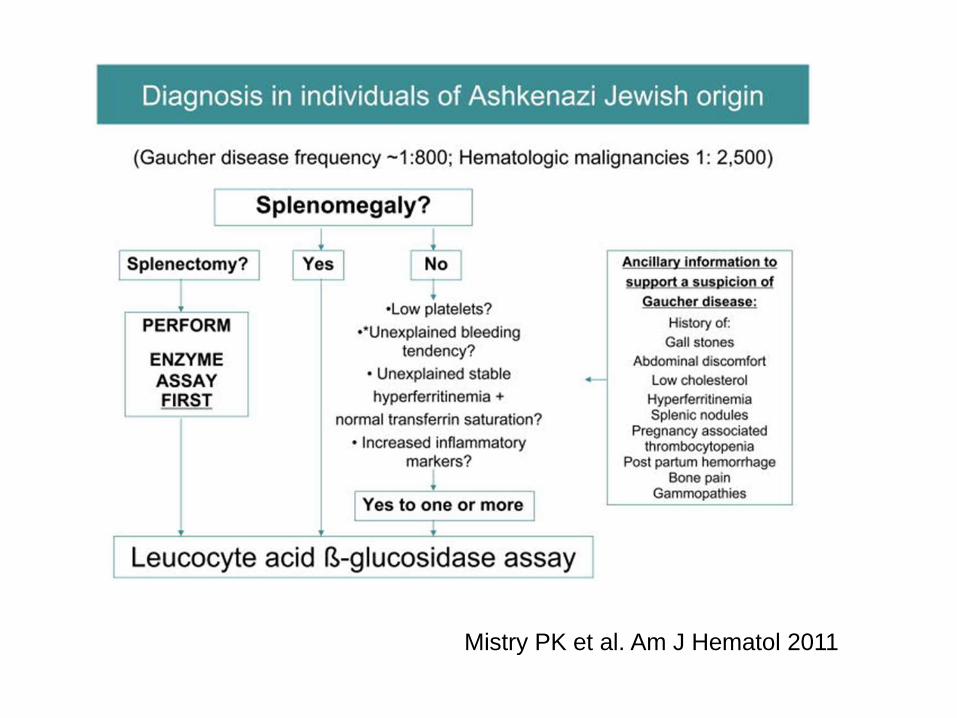
Mistry PK et al. Am J Hematol 2011

Mistry PK et al. Am J Hematol 2011
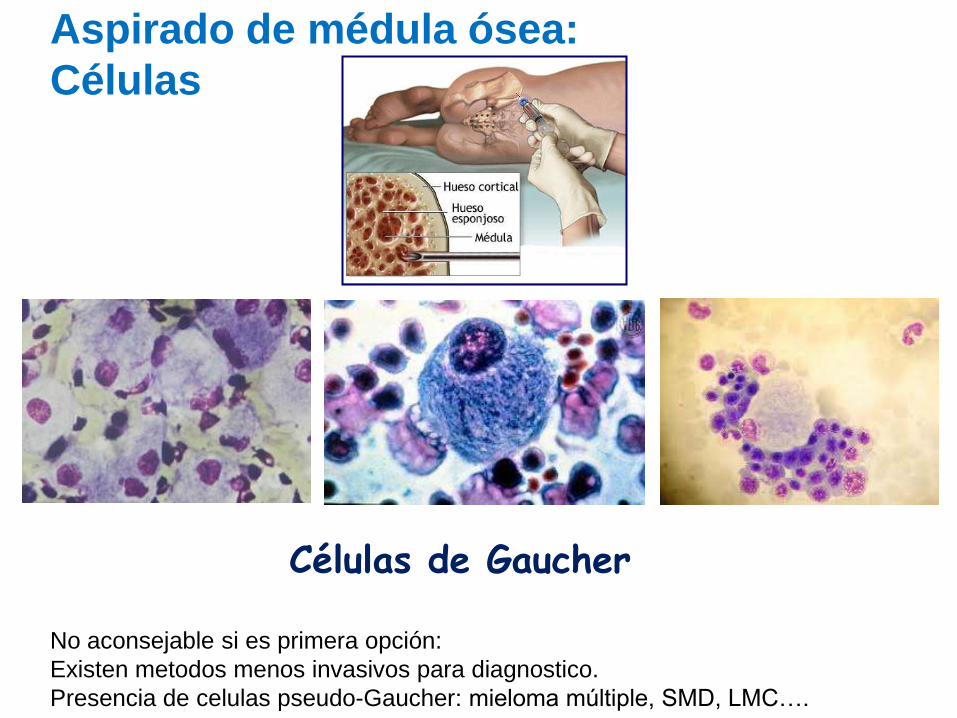
Células de Gaucher
Aspirado de médula ósea:
Células
No aconsejable si es primera opción:
Existen metodos menos invasivos para diagnostico.
Presencia de celulas pseudo-Gaucher: mieloma múltiple, SMD, LMC….

Glucosilceramida Ceramida + Glucosa
glucocerebrosidasa
Leucocitos/fibroblastos
Como se diagnostica la EG? (I)

pH 5
Taurocholato
37ºC, 2h
4-metilumbeliferil-β-D-glucopiranósido(Sigma) β-D-glucopiranosa 4-metilumbeliferona
Como se diagnostica la EG? (II)
4MUλexcitación= 366 nmλemisión = 446 nm

Actividad enzimática (nmol/mgp.h)
0
2
4
6
8
10
12
14
16
18
20
22
24
26
28
30
Pacientes Portadores Sanos
En leucocitos o
fibroblastos
:Costoso
Necesidad de controles. Valores
estándares
Solapamiento
¿Qué nos aporta el análisis enzimático?

DNAActividad
enzimática
-glucocerebrosidasa
Casos especiales: Gota de sangre sobre
papel de filtro (FTA)
Análisis gen
GBA

Importancia de que las gotas de
sangre sean homogéneas
G actividad X 2X 3X
1 gota 2 gotas 3 gotas
Debe depositarse idéntica cantidad que en controles

Diagnóstico genético

Herencia: Autosómica recesiva
Defecto genético: Mutaciones en el gen de GBA (>300)(1)
Gen de GBACromosoma 1
Región q21
7 Kb
11 exones
GBA I II III V VI VII VIII IX X XIIV
Ψ-GBA
Del 55 pb
I II III V VI VII VIII IX X XI
Alu(313 pb)
Alu(626pb)
Alu(320 b)
Alu(277pb)
IV
Enfermedad de Gaucher
(1) www.tau.ac.il/~racheli/genedis/gaucher /gaucher.html
Sujeto sano
Sujeto portador
Sujeto enfermo
Alfonso P. Tesis Doctoral

Missense
V15M
G113E
R120W
M123T
R131C
T134P
P182L
N188S
G195W
R257Q
P266L
E326K
G202R
N = 66
Mutaciones en GBA
encontradas en España
N396T
V398I
D409H
L444P
R463C
R463H
R496H
Stop mut.
W(-4)X
R47X
R163X
R257X
R359X
H311R
W312R
Y313H
G325W
L336P
R359Q
S364N
S364R
N370S
G377S
D380N
P391L
V391I
R395C
Splicing
IVS4-2a>g
IVS5+1g>t
Other mutations
c.(-203)A>G
Recombinations
Rec all gene
Great rec ex5-...10
RecTL
RecNciI
Deletions
large deletion
c.42-65del24
c.225-227delTAC
c.708delC
c.953delT
c.1214-1215delGC
c.1263-1317del55
c.1439-1445del7
c.1451-1452delAC
c.1510-1512delTCT
Insertions
c. 84insG
c.500insT
Giraldo P et al Orphanet J Rare Dis. 2012 Mar 19;7

PacienteDNA
N370SL444PG377SD409Hdel55bp
AnálisisGenético
Familiares
FEETEGRegistro
Solicitante
SiPCR larga 7,3 Kb
PCR anidada
Secuenciación
Re-secuenciaciónNo
Diagrama de flujo para el
diagnóstico genético

Biomarcadores para EG
• Fosfatasa ácida
• Lisozima
-hexosaminidasa (A/B)
• Neopterina
• Adenosin desaminasa (ADA)
• Enzima convertidora de angiotensina (ECA)
• Quitotriosidasa
• CCL18-PARK9

QUITOTRIOSIDASA
Sustrato artificial: 4-metilumbelliferil -D-N, N´, N”-
- Presente en plasma humano:
Determinación de actividad
Función: DesconocidaAntifúngico: Candida, Aspergillus ?Nematodos filiformes: Filarias
Hollak C et al. J Clin Invest. 1994; 93: 1288–1292.

Actividad quitotriosidasa
(nmol/mL.h)
Pacientes Gaucher Portadores No-portadores Niños
n=109 n=121 n=89 n=37
13.991 ± 12.647 71 ±55 45 ±39 37 ±25
300 veces
Giraldo P, et al. Haematologica 2001;86:977

¿Para qué sirve la
quitotriosidasa?
Inicial 12 24 36 48
40.000Paciente # 5001
20.000
QT.nmol/ml.hr
meses
Inicial12 24 36 48 60
10.000
6.000
2.000Paciente # 716
72

Actividad quitotriosidasa en
pacientes con EG vs genotipo
Gen quito. N actividad quito. (nmol/mL.h)
Nor / Nor 74 17.387 ± 12.800
Dup / Nor 41 11.870 ± 10.430*
Dup / Dup 3 0
*p < 0.01
INCONVENIENTE

Genotipo del gen QUITOTRIOSIDASA
75 pb99 pb
ChsPCR
E2E1 E3 E4 E5 E6 E7 E8 E9 E11 E12E10
+/+ +/- -/-
24 pb
Chas
24 pb
Chs
Chas
24 pb
Alelo mayor
Alelo menor ( nulo )

Biomarcador CCL18/PARC
33 ng/mL
(rango 10-72 ng/mL)
948 ng/mL
(rango 237-2285 ng/mL)
Boot RG et al. Blood 2004; 103:33-39
X 29

paciente 1
0,00
200,00
400,00
600,00
800,00
1000,00
0 6 15 24 36
meses de tratamiento
CC
L18
0
2000
4000
6000
8000
10000
12000
Acti
vid
ad
de Q
T
CCL18
QT
paciente 2
0,00
200,00
400,00
600,00
800,00
1000,00
3 13 29 42 52 65 77
meses de tratamiento
CC
L18
0
2000
4000
6000
8000
10000
12000
Acti
vid
ad
de Q
T
CCL18
QT
paciente 3
0,00
200,00
400,00
600,00
800,00
1000,00
0 39 51 60 72 83 99 105
meses de tratamientoC
CL
18
0
2000
4000
6000
8000
10000
12000
Acti
vid
ad
de Q
T
CCL18
QT
Relación entre el descenso de los niveles
plasmáticos de CCL18 y actividad de QT
según nuestra experiencia

Resumen
• Análisis enzimático de Glucocerebrosidasa
– Diagnóstico
• Análisis genético Glucocerebrosidasa
– Estudio familiares (portadores / no portadores)
– Consejo genético
– Pronóstico
• Biomarcadores: quitotriosidasa y/o CCL18/PARC
– Valoración
– Respuesta a tratamientos
– Seguimiento

Posible caso
(Sospecha)
(Hospital)
Lab
(FEETEG)
Courier
Separación
especimenes
leucocitos
Plasma
DNA
-Glucocer
Otras lisoso.
Biomarcadores:
Activ Quitotrios,
CCL18 y MIP1
Diagnostico genético:
Glucocerebrosidasa mutaciones +
Quitotriosidasa genotipo
SMPD1, AGAL NPC1, NPC2 mutaciones
+
FEETEG
Diagnostico
REGISTRO FEETEG
SOLICITANTE
**
*

Análisis
Seguimiento de
pacientes
Muestra
(sangre o plasma
(Hospital)
Courier
Laboratorio*(FEETEG)
Plasma
Biomarcadores:
quitotriosidasa
actividad y
CCL18
Separación
alicuotado
REGISTRO FEETEG
SOLICITANTE
*
*
*

Tratamientos

Palmitoil-CoA + serina
Ceramida
UDP-glucosa
Glucosilceramida(glucocerebrósido)
Gangliósidos
Globósidos
Lactósidos
Ceramida + Glucosa
-Glucocerebrosidasa
Ceramida:glucosil-transferasa
Formación y degradación
de la glucosilceramida

Formación = Degradación
No hay depósito de glucosilceramida
Síntesis + catabolismo Globosidos, Gangliósidos..
Degradación
Formación
GlucocerebrosidasaGLUCOSILCERAMIDA
(SUBSTRATO)
Condiciones normales: Equilibrio

Enfermedad de Gaucher:
Desequilibrio entre entrada y salidaFormación > Degradación
(normal) (disminuida)
Síntesis + catabolismo Globosidos, Gangliósidos..
Degradación
Formación
Glucocerebrosidasa
GLUCOSILCERAMIDA
(SUBSTRATO)
Se deposita glucosilceramida

OPCIONES
TERAPÉUTICAS

Opciones terapéuticas : TES
Síntesis + catabolismo Globos., Gangli..
Degradación
FormaciónImiglucerasa
Velaglucerasa
Taliglucerasa
TES
GlucocerebrosidasaGlucocerebrosidasa
GLUCOSILCERAMIDA
(SUSTRATO)

1978
Descubrimiento de los receptores de manosa 6-
fosfato
Célula
Lisosoma
Lisosoma
Receptor
Manosa-6-P
Lisosoma
Enzima
Enzima
P
P
P
P
P
P
P
P
P
P

1990
Barton, Brady, Grabonski inician los primeros
tratamientos con enzima betaglucosidasa obtenida de placenta
Alglucerasa. Ceredasa. Genzyme Corp

1996Se comercializa la primera
enzima recombinante en CHO
QuikChangeTM Site-Directed Mutagenesis Kit
Strategene
Imiglucerase. Cerezyme. Genzyme-Sanofi

Glicoformas (decoración) típica
que se producen en mamíferos
Asn Asn Asn Asn
Mannosidase I
Asn
ER
Asn
Golgi
Glucose
AsnAsn
N-acetylglucosamine Mannose Galactose Sialic acid Fucose
Asn
Enzymatic
degradation
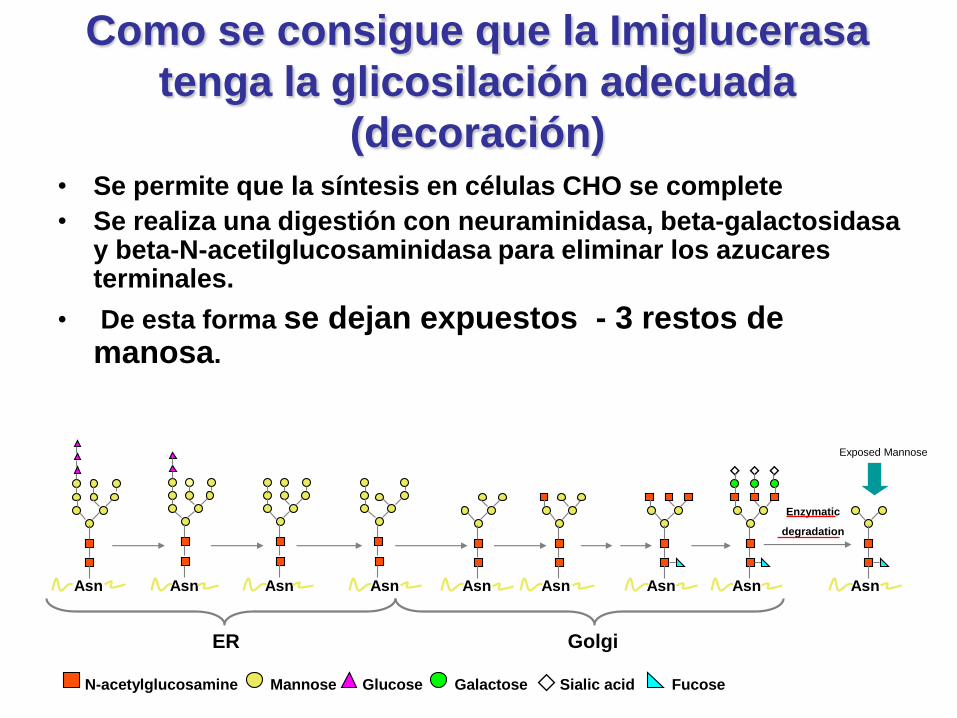
Como se consigue que la Imiglucerasa
tenga la glicosilación adecuada
(decoración)• Se permite que la síntesis en células CHO se complete
• Se realiza una digestión con neuraminidasa, beta-galactosidasay beta-N-acetilglucosaminidasa para eliminar los azucares terminales.
• De esta forma se dejan expuestos - 3 restos de manosa.
Asn Asn Asn Asn
Mannosidase I
Asn
ER
Asn
Golgi
Glucose
AsnAsn
N-acetylglucosamine Mannose Galactose Sialic acid Fucose
Asn
Enzymatic
degradation
Exposed Mannose

Velaglucerasa VPRIV (Shire). Recombinante expresada en células humanas
(comercializada marzo 2011)
Fibroblastos humanos
QuikChangeTM Site-Directed Mutagenesis Kit
Strategene

La glicosilación (decoración) adecuada
de la Velaglucerasa se consigue con
Kifunensine
• Se impide que la cadena de oligosacarido se modifiqueen el Golgi
• Impide que queden enmascaradas las manosas
Asn Asn Asn Asn
Mannosidase I
Asn
ER
Asn
Golgi
Glucose
AsnAsn
N-acetylglucosamine Mannose Galactose Sialic acid Fucose
Asn
Enzymatic
degradation
Exposed Mannose

Taliglucerase. Uplyso (Protalix)/
(Pfizer). Producida en células de zanahoria
(Aprobada por FDA pero no por EMA)

Sintesis de laTaliglucerase
Endoquitinasa
basica
Arabidopsis
thaliana
Quitinasa A
Tabaco
E F D L L V D T M

RE
Golgi
Vacuola
Asn
Asn
Asn
nucleous
Extracellular
space
Distribución de N-Gliproteinasen
células vegetales

Enzima
Señales de
Reconocimiento
N-acetil-glucosamina
Manosa
Glicosilación “Decoración” de las
Enzimas recombinantes
P
P
P
P
P
P
P
P
P

Tekoah Y et al Biosci Rep. 2013 Aug 28. [Epub ahead of print]

Captación de las enzimas
recombiantes
Taliglucerasa
Imiglucerasa
Velaglucerasa
Tekoah Y et al Biosci Rep. 2013 Aug 28. [Epub ahead of print]

Opciones terapéuticas en la EG:
Terapia Reducción Sustrato (TRS)
Síntesis + catabolismo Globos, Gangli..
Degradación
Formación
NB-DNJ, miglustat
(Zavesca®)TRS
Glucocerebrosidasa
GLUCOSILCERAMIDA
(SUSTRATO)
Ceramida:glucosiltransferasa

Palmitoil-CoA + serina
Ceramida
UDP-glucosa
Glucosilceramida(glucocerebrósido)
Gangliósidos
Globósidos
Lactósidos
Ceramida + Glucosa
-Glucocerebrosidasa
Ceramida:glucosil-transferasa

Miglustat(Iminoazucar)
Obtenido
de la corteza de la raíz
de la morera
Morus alba
Bacteria
streptomyces

TRS
N-butil-deoxynojirimicin Miglustat
Zavesca®
Actelion
Genz-112638. Eliglustat (Pendiente
aprobación) Genzyme-Sanofi

Similitud estructural Iminoazucares :
Miglustat vs glucosilceramida
Glucosylceramide
O
HN
OH
C13H27
O
C17H35
O
OH
OHHO
OH
Miglustat
N
OH
OHHO
OH
CH3
Glucose Ceramida

Similitud estructural:
Eliglustat vs Glucosilceramida
ß-Glu-Cer Glucosilceramida
O
HN
OH
C13H27
O
C17H35
O
OH
OHHO
OH
Eliglustat(pendiente aprobación)
Glucosa Ceramida
1-(4´-Hydroxy)phenyl-2-
palmitoylamino-3-pyrrolidino
-1-propanol

Otras opciones terapéuticas:
Chaperonas

5´´ mRNA
Lumen REProteina
Golgi Golgi
Proteina
Normal
Chaperonas
moleculares
¿Qué misión tienen las chaperonas?
LisosomasPH = 5
Degradación
Proteina
mutada
Chaperones
sintéticas

Como funcionan las chaperonas en las Enfermedades de Depósito Lisosomal
Proteína mal plegadaProteina-Chaperona Complejo
Chaperona
farmacológica
Reduce el estres
Celular e inflamación
Reduce el sustrato
acumulado
Sustrato acumulado
Reticuloendoplásmico
Aparato de
Golgi
Lisosoma
Facilita el tráfico

Opciones terapéuticas en EGRecuperar actividad de enzima mutada
del propio paciente
Síntesis + catabolismo Globos., Gangli..
Degradación
FormaciónChaperonas
FarmacológicasChaperonas
GlucocerebrosidasaGlucocerebrosidasa
GLUCOSILCERAMIDA
(SUSTRATO)

Tratamientos aprobados o en desarrollo.
• TES1,2
– Imiglucerasa (Cerezyme®)
– Velaglucerasa (VPRIV™)
– Taliglucerasa (FDA si, EMA no)
• TRS2-4
– Miglustat (Zavesca®)
– Eliglustat/Genz-112638 (pendiente)
• Chaperonas o enzyme enhancement therapy5,6
– Afegostat tartrate/AT2101 (Plicera™; desarrollo
interrumpido)
1. The Pharma Letter Web site. Available at: http://www.thepharmaletter.com/file/96654/protalix-gets-temporary-approval-for-gaucher-drug-taliglucerase-
in-france.html. 2. Sidransky E. Gaucher disease. Medscape Web site. Available at: http://emedicine.medscape.com/article/944157-print. 3. The Medical
News Web site. Available at: http://www.news-medical.net/news/20100212/Genzyme-announces-2-year-data-from-eliglustat-tartrate-Phase-2-trial-for-
Gaucher-disease.aspx. 4. Actelion Web site. http://www1.actelion.com/en/scientists/development-pipeline/phase-4/miglustat-zavesca.page. 5. Cigna
Web site. Available at: http://www.cigna.com/customer_care/healthcare_professional/coverage_positions/pharmacy/ph_1011_coveragepositioncriteria
_zavesca.pdf. 6. Reuters Web site. Available at: http://www.reuters.com/article/idUKN0250346520091002. 7. Pastores GM et al. In: Pagon RA et al.
GeneReviews [Internet]. 1993-2000. Available at: http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=gaucher. 8. Grabowski GA. Lancet.
2008;372:1263-1271.
• Trasplante de médula ósea(TMO)7
– Solo Neuronopáticos
– TMO en algunos casos ha solucionado sintomas
– No aconsejable en mayoríacasos
• Terapia génica8
– Investigación