re: docket no. fda-2012-d-0523; draft guidance for ... · substantive review will help achieve that...
TRANSCRIPT

701 Pennsylvania Avenue, Ste. 800
Washington, DC 20004–2654
Tel: 202 783 8700
Fax: 202 783 8750
www.AdvaMed.org
Bringing Innovation to patient care worldwide
September 27, 2012
Division of Dockets Management (HFA-305)
Food and Drug Administration
5630 Fishers Lane, Room 1061
Rockville, MD 20852
Re: Docket No. FDA-2012-D-0523; Draft Guidance for Industry and Food and Drug
Administration Staff; Refuse to Accept Policy for 510(k)s
Dear Sir/Madam:
The Advanced Medical Technology Association (AdvaMed) provides these comments in
response to a request for comments regarding the Food and Drug Administration (FDA)
Center for Devices and Radiological Health Draft Guidance for Industry and Food and Drug
Administration Staff; Refuse to Accept Policy for 510(k)s. Notice of this draft guidance and
request for comments were published in Fed. Reg. Vol.77, No. 156 (August 13, 2012).
AdvaMed is the world’s largest association representing manufacturers of medical devices,
diagnostic products, and health information systems that are transforming health care through
earlier disease detection, less invasive procedures and more effective treatments. Our
members range from the largest to the smallest medical technology innovators and
companies. More than 70 percent of our members have less than $30 million in domestic
sales annually. We welcome the opportunity to comment on this guidance and look forward
to working with FDA to ensure the revised guidance meets the needs and expectations of
both FDA and industry.
AdvaMed supports FDA’s effort to provide industry with more clarity and transparency on
FDA’s requirements for the content and format of 510(k) submissions. We agree with FDA
that such clarity will enable industry to better meet FDA’s expectations, and has the potential
to improve overall review time. It is essential that FDA resources are used wisely and
effectively. Allowing only well-organized and complete 510(k) submissions to undergo
substantive review will help achieve that goal.
AdvaMed encourages its members to prepare well-organized and complete 510(k)
submissions. The AdvaMed educational division (Medical Technology Learning Institute—
MTLI) conducts two annual workshops on how to prepare good quality 510(k) submissions.

Division of Dockets Management
September 27, 2012
Page 2 of 3
These workshops are open to all industry personnel and benefit from participation by key
CDRH staff who provide information on submission organization and content as well as the
510(k) process itself.
AdvaMed agrees that the purpose of the 510(k) acceptance review, as stated in the
“Background” of the draft guidance, should be “to assess whether a submission is
administratively complete in that it includes all of the information necessary for FDA to
conduct a substantive review and to reach a determination regarding substantial
equivalence…” AdvaMed also agrees with the statement in the Purpose section of the draft
guidance that states “It is critical to distinguish between the completeness of the regulatory
submission, and the quality of the data provided and any studies conducted in support of the
submission. The assessment of the completeness of the 510(k) occurs during the acceptance
review, while the assessment of the quality of the submitted information occurs during the
substantive review.”
However, despite these very clear and concise explanations of the purpose of the acceptance
review stated in the draft guidance, some items in the Acceptance Checklists require
responses that could only be provided if a substantive review has been conducted. For
example, Item G. “Shelf Life” on page 13 of the Traditional 510(k) Checklist asks whether
the storage conditions impact the device safety and effectiveness and if so, does the
submission provide certain information. It would be very difficult for the reviewer
conducting the acceptance assessment to determine if the device could be adversely affected
by the storage conditions. The review should be limited to determining if certain information
is present. Additionally, item 21 in section D “Proposed Labeling” on page 9 requires the
reviewer to determine if there is a device specific guidance, special control or regulation and
that the submitter has followed the applicable guidance, special control or regulation.
Determining if there is a device specific labeling requirement and then assessing if the
submitted labeling meets the requirement goes beyond the bounds of an acceptance review.
The acceptance review should determine that the required elements of a 510(k) are present,
that they are legible and provided in English. It should verify that the submission is
organized in a manner that allows efficient substantive review. In the “Adequacy of
information” section of the draft guidance, it states “…FDA should consider only the
presence or omission of the element or a rationale for the omission of the element…” This
very accurately states the checklist criteria objective.
It is critical that FDA limit its acceptance review to prescreening of 510(k) submissions, that
is, to determining whether an element is present and not whether it is substantively
acceptable. As detailed in Attachment A, the checklists contain some items that could be
interpreted as needing a substantive review. Such substantive review should not occur at this
early phase, when FDA is merely determining whether a 510(k) submission contains all of
the required elements.

Division of Dockets Management
September 27, 2012
Page 3 of 3
Attachment A provides specific comments on the draft guidance and on the three Acceptance
Checklists. These comments reference line numbers applied to the draft guidance text so a
copy of the line-numbered document is provided in Attachment B.
In conclusion and as previously stated, AdvaMed supports the FDA efforts to improve the
clarity and transparency of FDA’s requirements for the content and format of a 510(k)
submission. AdvaMed believes it is important to ensure the most efficient and effective use
of FDA resources and that by ensuring only well-organized and complete 510(k) submissions
are accepted for review, the goal will be achieved. We appreciate the extraordinary efforts
taken by FDA personnel in preparing this draft guidance and support the goals of developing
a guidance that serves the mission of FDA, the needs of industry and the common goal of
benefiting public health.
Sincerely,
Ruey C. Dempsey
Associate Vice President
Technology and Regulatory Affairs

ATTACHMENT A
ADVAMED COMMENTS
Draft Guidance for Industry and FDA Staff
Medical Devices: Refuse to Accept Policy for 510(k)s
Page 4 of 15
Line(s) No. – Line or lines numbers of the guidance
Change – Proposed change to the guidance
Reason – Reason for proposed change
Line(s) No. Change Reason
General
Throughout, modify documents to support intent
as described in lines 47-51; a process to determine
if all the required documents are present, not a
determination if they are present and adequate.
Determination of adequacy of information is not
appropriate.
The guidance is not always consistent. For example, lines 47-
51 explain the difference between completeness and data
quality, and that this guidance is to assess the completeness, not
the quality of the submitted data (quality is assessed during the
substantive review). However, several items in the checklist
require that an assessment be made as to the quality of the data
submitted. Please see Item #21, page 9 of the Acceptance
Checklist for Traditional 510(k)s. Here, during the check in
process, the item requires that a determination be made if the
submission contains data that satisfies any device-specific
guidance, special controls, or regulations, and that the submitter
has followed the recommendations in these documents, or has
otherwise met the applicable statutory or regulatory criteria
through an alternative approach. The underlined portion of this
requirement can only be determined through a review and
understanding of the submission as a whole. This requirement
goes beyond a mere determination if the submission is
complete. Please see item #34, page 15 of the Acceptance
Checklist for Traditional 510(k)s. Here, the checklist requires
that, “All appropriate categories of software verification and
validation documentation provided based on stated level of
concern, as described in Guidance for the Content of Premarket
Submissions for Software Contained in Medical Devices, or the

ATTACHMENT A
ADVAMED COMMENTS
Draft Guidance for Industry and FDA Staff
Medical Devices: Refuse to Accept Policy for 510(k)s
Page 5 of 15
Line(s) No. Change Reason
submitter has provided documentation that it has otherwise met
the applicable statutory or regulatory criteria through an
alternative approach.” Again, a determination if the appropriate
items are provided should be completed during the substantive
review. For the check-in process, one should merely determine
if software verification / validation documents are present.
53
State the requirements for submitting an electronic
copy of the submission both before and after the
issuance of the required guidance. Clarify if the
absence of an electronic copy is a cause for not
accepting a submission (both before and after the
issuance of the electronic copy guidance).
It is our understanding that submission of an electronic copy of
the submission is required after a guidance is issued. Stating
the requirements before and after the guidance is issued will
avoid having to revise this guidance after electronic guidance is
issued. The FDASIA legislation states:
(Sec. 1136(b)):
‘‘(b) DEVICES.—
‘‘(1) IN GENERAL.—Beginning after the issuance of final
guidance implementing this paragraph, presubmissions and
submissions for devices under section 510(k), 513(f)(2)(A),
515(c), 515(d), 515(f), 520(g), 520(m), or 564 of this Act or
section 351 of the Public Health Service Act, and any
supplements to such presubmissions or submissions, shall
include an electronic copy of such presubmissions or
submissions.
‘‘(2) GUIDANCE CONTENTS.—In the guidance under
paragraph (1), the Secretary may—
‘‘(A) provide standards for the electronic copy required
under such paragraph; and
‘‘(B) set forth criteria for waivers of and exemptions
from the requirements of this subsection.’’.

ATTACHMENT A
ADVAMED COMMENTS
Draft Guidance for Industry and FDA Staff
Medical Devices: Refuse to Accept Policy for 510(k)s
Page 6 of 15
Line(s) No. Change Reason
135
Add:
“The start of the FDA review clock for those
submissions accepted for review after the first 15-
calendar-day acceptance review is the date the
FDA received the submission from the sponsor.”
Clarify the initial acceptance review process is part of the FDA
review clock. Refer to lines 237-239.
180-181
Modify the sentence (add underlined wording):
“The acceptance review which occurs prior to the
substantive review will be conducted…”
Ensure that review process and time are clearly stated. Ensure
consistency with line 226.
194 Clarify “in writing”. We suggest that this
communication be done by e-mail.
Email will move the process more quickly.
194
At the end of the sentence in line 194, add the
information provided in the footnote.
Add: “The review clock will not start until the
510(k) submission is accepted for review.
This is important information that should not be buried in the
citation in the footnote.
206
Modify to read: “Upon receipt of the newly
submitted information, FDA staff will ensure the
new information meets the checklist requirements
and complete the screening process within 15
calendar days.”
There is no need to the reviewer to repeat the screening process.
The reviewer needs to determine that the new information
corrects the deficiency found in the original review.
225-227
Revise as follows (modified words are
underlined): “Should FDA fail to complete the
acceptance review within 15 calendar days, the
submission will be considered accepted, the
submitter will be notified in writing, and FDA will
commence with substantive review.”
Include a copy of the Refuse to Accept Checklist.
The use of “should” implies that the reviewer has discretion in
meeting these requirements.
Provide clarity and demonstrate supervisory concurrence.

ATTACHMENT A
ADVAMED COMMENTS
Draft Guidance for Industry and FDA Staff
Medical Devices: Refuse to Accept Policy for 510(k)s
Page 7 of 15
Line(s) No. Change Reason
227-229
Delete sentence beginning “Once a submission has
been accepted. . .
Add: FDA will make every effort to determine
whether all required elements of the 510(k) exist.
On the rare occasion in which FDA
unintentionally overlooks information that should
have been included, FDA may request such
information during the substantive review.
Although we understand that there will be occasions in which
FDA overlooks a missing element during the acceptance
review, this should be a rare occurrence. Otherwise, the
acceptance review will be meaningless, and could result in
increased overall review time.
238-240
Add after line 240:
“The start of the FDA review clock for those
submissions accepted for review after the first 15-
calendar-day acceptance review is the date the
FDA received the submission from the sponsor.”
This section is unclear as to the date the review clock starts for
510(k)s that are accepted without any RTA.
310-319 Move footnote text to body of guidance This is important information and shouldn’t be buried in a
footnote.
341-346
Remove This is sometimes a very subtle question and should not be part
of the acceptance review. A 510(k) may require significant
review to determine whether 510(k) or PMA is the correct
regulatory submission.
449-457
Insert: “This is not a reason to refuse to accept the
submission.” after sentence beginning “It is
likely…
The reviewer should not be assessing the adequacy of the
information during the acceptance review. We believe stronger
text is needed in this section to avoid such practices.

ATTACHMENT A
ADVAMED COMMENTS
Draft Guidance for Industry and FDA Staff
Medical Devices: Refuse to Accept Policy for 510(k)s
Page 8 of 15
Line(s) No. Change Reason
463-468
Replace example.
As discussed with respect to lines 121-126 above, FDA must be
careful not to conduct a substantive review during this
acceptance “pre-screening”. The example provided arguably
requires a substantive review and subjective determination by
the FDA reviewer as it requires an assessment of whether it
complies with applicable guidance. This is problematic in two
ways: (1) it bogs down the prescreening process; and (2) it
effectively turns guidance recommendations into requirements,
without notice and comment rulemaking.
ATTACHMENT A
ADVAMED COMMENTS
Draft Guidance for Industry and FDA Staff
Medical Devices: Refuse to Accept Policy for 510(k)s
Page 9 of 15
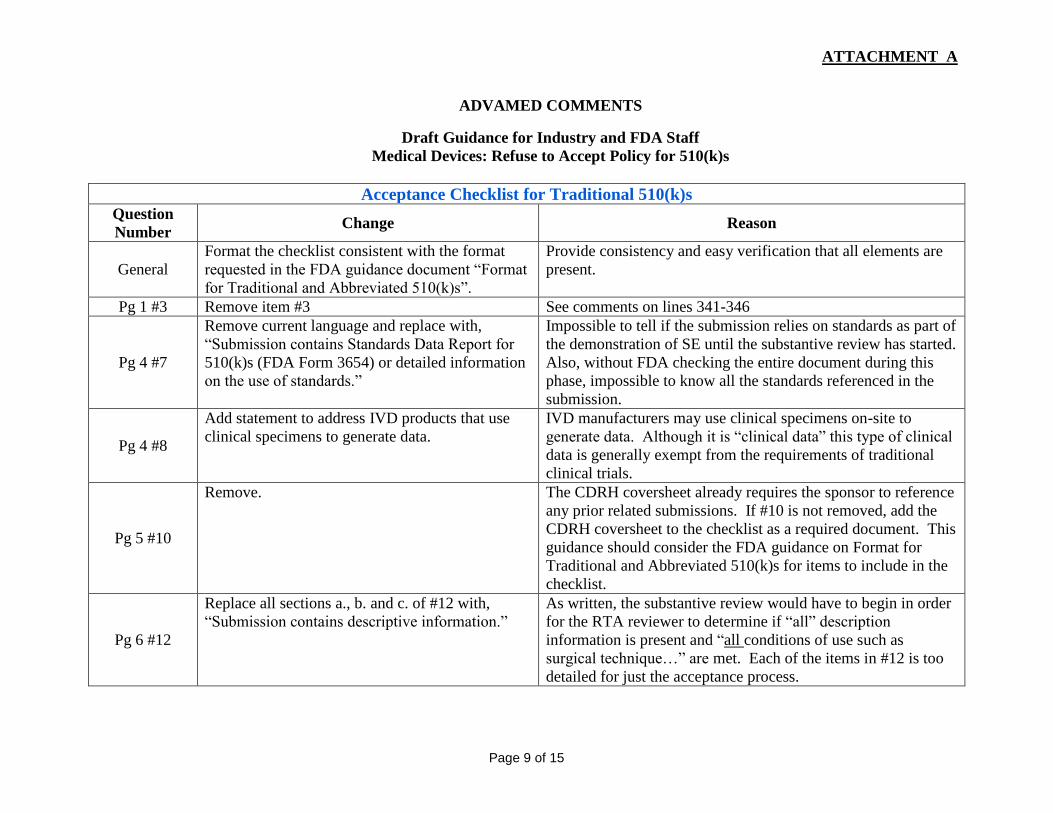
Acceptance Checklist for Traditional 510(k)s Question
Number Change Reason
General
Format the checklist consistent with the format
requested in the FDA guidance document “Format
for Traditional and Abbreviated 510(k)s”.
Provide consistency and easy verification that all elements are
present.
Pg 1 #3 Remove item #3 See comments on lines 341-346
Pg 4 #7
Remove current language and replace with,
“Submission contains Standards Data Report for
510(k)s (FDA Form 3654) or detailed information
on the use of standards.”
Impossible to tell if the submission relies on standards as part of
the demonstration of SE until the substantive review has started.
Also, without FDA checking the entire document during this
phase, impossible to know all the standards referenced in the
submission.
Pg 4 #8
Add statement to address IVD products that use
clinical specimens to generate data.
IVD manufacturers may use clinical specimens on-site to
generate data. Although it is “clinical data” this type of clinical
data is generally exempt from the requirements of traditional
clinical trials.
Pg 5 #10
Remove. The CDRH coversheet already requires the sponsor to reference
any prior related submissions. If #10 is not removed, add the
CDRH coversheet to the checklist as a required document. This
guidance should consider the FDA guidance on Format for
Traditional and Abbreviated 510(k)s for items to include in the
checklist.
Pg 6 #12
Replace all sections a., b. and c. of #12 with,
“Submission contains descriptive information.”
As written, the substantive review would have to begin in order
for the RTA reviewer to determine if “all” description
information is present and “all conditions of use such as
surgical technique…” are met. Each of the items in #12 is too
detailed for just the acceptance process.

ATTACHMENT A
ADVAMED COMMENTS
Draft Guidance for Industry and FDA Staff
Medical Devices: Refuse to Accept Policy for 510(k)s
Page 10 of 15
Acceptance Checklist for Traditional 510(k)s Question
Number Change Reason
Pg 6 #13
Delete:
“Where applicable…dimensions,”
Replace with:
“If the submission contains engineering
drawing(s), schematics, illustrations and/or figures
of the devices, these items are clear and legible.”
As written, the substantive review would have to begin in order
for the RTA reviewer to determine if criteria are met. For
acceptance, merely determine if illustrations are legible. It is
for the substantive reviewer to determine what information is
needed for a determination of SE (e.g., dimensions, illustrations
for each model).
Pg 7 #14a Modify to, “A list of each component or accessory
is provided.”
For components or accessories included in the submission, a
description as detailed as 12a, b, and 13 should not be required.
Only for components or accessories that are part of the system
and under review as part of the 510(k) should require details of
that nature.
This should apply to only those items that are medical devices.
Pg 7 #14b
Modify to, “Each component or accessory that is a
medical device is identified with a 510(k) number,
as 510(k) exempt, or is identified as associated
with the 510(k) under review.”
Pg 8 #17
Remove. This should be part of the substantive review. Additionally, text
is not aligned with statutory language of 513(i) because of
reference to “any differences” language when referring to the
comparison between the device and predicate in determining
NSE.
Pg 8 #18
Delete “advertisements” or add “if available” after
“advertisements”.
The check should merely be for the presence of labeling. The
substantive review should determine what labeling is required
for a determination of SE.
Pg 8 #18b
Remove. This should be part of the substantive review. The check should
merely be to determine if labeling has been included in the
submission.

ATTACHMENT A
ADVAMED COMMENTS
Draft Guidance for Industry and FDA Staff
Medical Devices: Refuse to Accept Policy for 510(k)s
Page 11 of 15
Acceptance Checklist for Traditional 510(k)s Question
Number Change Reason
Pg 9 #21
Remove. This should be part of the substantive review. The wording is
too specific for merely a check-in process; it implies adequate
documentation, which may only be determined during the
substantive review. Here, the reviewer is asked whether the
labeling complies with all of the device-specific guidance,
special controls or regulations, which requires a substantive
review of the content of the labeling. The acceptance review
should be limited to whether the labeling is included in the
510(k) – not whether it complies with FDA’s substantive
expectations. In addition, although this allows for an
“alternative approach”, it risks the reviewer determining
whether that approach is acceptable, and issuing an RTA on that
basis.
Pg 9 #22 Remove. This item would require a substantive review to determine if
labeling complies with 21CFR 809.10.
Pg 10 #23
Modify to read: “Full test report is provided for
each completed test.”
The statement: “..to explain how the data generated from the
test supports a finding of substantial equivalence” implies that
the reviewer will make a qualitative assessment of the
information provided. This would not be appropriate for the
RTA review.
Pg 10 #24
Remove. This should be part of the substantive review. The wording is
too specific for merely a check-in process; it implies adequate
documentation, which may only be determined during the
substantive review.
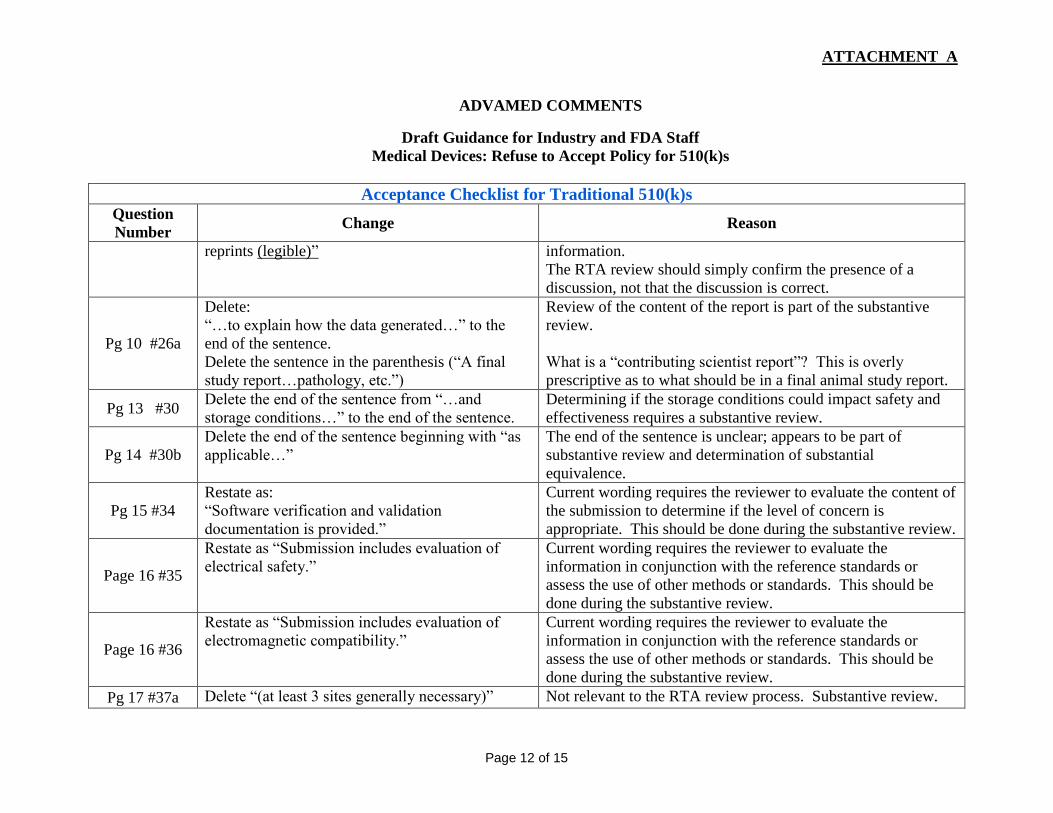
Pg 10 #25 Add the underlined word, “submission includes Ensure the person doing the substantive review has legible
ATTACHMENT A
ADVAMED COMMENTS
Draft Guidance for Industry and FDA Staff
Medical Devices: Refuse to Accept Policy for 510(k)s
Page 12 of 15
Acceptance Checklist for Traditional 510(k)s Question
Number Change Reason
reprints (legible)”
information.
The RTA review should simply confirm the presence of a
discussion, not that the discussion is correct.
Pg 10 #26a
Delete:
“…to explain how the data generated…” to the
end of the sentence.
Delete the sentence in the parenthesis (“A final
study report…pathology, etc.”)
Review of the content of the report is part of the substantive
review.
What is a “contributing scientist report”? This is overly
prescriptive as to what should be in a final animal study report.
Pg 13 #30 Delete the end of the sentence from “…and
storage conditions…” to the end of the sentence.
Determining if the storage conditions could impact safety and
effectiveness requires a substantive review.
Pg 14 #30b
Delete the end of the sentence beginning with “as
applicable…”
The end of the sentence is unclear; appears to be part of
substantive review and determination of substantial
equivalence.
Pg 15 #34
Restate as:
“Software verification and validation
documentation is provided.”
Current wording requires the reviewer to evaluate the content of
the submission to determine if the level of concern is
appropriate. This should be done during the substantive review.
Page 16 #35
Restate as “Submission includes evaluation of
electrical safety.”
Current wording requires the reviewer to evaluate the
information in conjunction with the reference standards or
assess the use of other methods or standards. This should be
done during the substantive review.
Page 16 #36
Restate as “Submission includes evaluation of
electromagnetic compatibility.”
Current wording requires the reviewer to evaluate the
information in conjunction with the reference standards or
assess the use of other methods or standards. This should be
done during the substantive review.
Pg 17 #37a Delete “(at least 3 sites generally necessary)” Not relevant to the RTA review process. Substantive review.

ATTACHMENT A
ADVAMED COMMENTS
Draft Guidance for Industry and FDA Staff
Medical Devices: Refuse to Accept Policy for 510(k)s
Page 13 of 15
Acceptance Checklist for Traditional 510(k)s Question
Number Change Reason
Pg 17 #37b Delete the information within the parenthesis. The information in parenthesis is not relevant to the RTA
review. Should be part of substantive review.
Pg 17 #37c Delete “(detection …LoQ)” The information in parenthesis is not relevant to the RTA
review. Should be part of substantive review.

ATTACHMENT A
ADVAMED COMMENTS
Draft Guidance for Industry and FDA Staff
Medical Devices: Refuse to Accept Policy for 510(k)s
Page 14 of 15
Refuse to Accept Checklist for Abbreviated 510(k)s Question
Number Change Reason
ALL
SEE Traditional comments for items that are
identical (Elements of a Complete Submission
sections).
Where applicable, the Traditional comments apply to the
Abbreviated sections.
Pg 1 #3 Remove item #3 See comments on lines 341-346
Pg 7 #9
Add checklist item for user fee form &
confirmation of payment
Why is the user fee referenced with respect to bundling of
submissions but the user fee form is not a separate entry on the
checklist? It would be more useful to sponsors to have the user
fee item in the checklist as well.
Pg 11 #18 Delete “advertisements” or add “if available” after
“advertisements”.
Advertisements are often developed at the time of 510(k)
submission.
Pg 13 #26a
Delete:
“…to explain how the data generated…” to the
end of the sentence.
Delete the sentence in the parenthesis (“A final
study report…pathology, etc.”)
Review of the content of the report is part of the substantive
review.
What is a “contributing scientist report”? This seems overly
prescriptive as to what should be in a final animal study report.

ATTACHMENT A
ADVAMED COMMENTS
Draft Guidance for Industry and FDA Staff
Medical Devices: Refuse to Accept Policy for 510(k)s
Page 15 of 15
Refuse to Accept Checklist for Special 510(k)s
Question
Number Change Reason
ALL SEE Traditional comments. Where applicable, the Traditional comments apply to the
Special sections, even though there are fewer repeating sections.
Pg 1 #3 Remove. While we recognize this is a requirement for a special 510(k),
this should be determined in the substantive review.
Pg 4 #8
Add checklist item for user fee form &
confirmation of payment.
Why is the user fee referenced with respect to bundling of
submissions but the user fee form is not a separate entry on the
checklist? It would be more useful to sponsors to have the user
fee item in the checklist as well.
Pg 5 #13
Modify to read (add underlined wording):
“Identification of history of changes requiring
letter-to-file made since the previous 510(k)
submission.”
Limit the history to those changes that require letter-to-file
documentation.
Pg 8, #20 Delete “advertisements” or add “if available” after
“advertisements”.
Advertisements are often developed at the time of 510(k)
submission

Draft Guidance for Industry and Food and 1
Drug Administration Staff 2
3
Refuse to Accept Policy for 510(k)s 4
5
This draft guidance, when finalized, will represent the Food and Drug Administration's (FDA's) 6
current thinking on this topic. It does not create or confer any rights for or on any person and 7
does not operate to bind FDA or the public. You can use an alternative approach if the approach 8
satisfies the requirements of the applicable statutes and regulations. If you want to discuss an 9
alternative approach, contact the FDA staff responsible for implementing this guidance. If you 10
cannot identify the appropriate FDA staff, call the appropriate number listed on the title page of 11
this guidance. 12
13
Purpose 14
The purpose of this document is to explain the procedures and criteria FDA intends to use in 15
assessing whether a 510(k) submission meets a minimum threshold of acceptability and should be 16
accepted for substantive review. 17
18
This guidance document updates two existing guidance documents entitled “Center for Devices and 19
Radiological Health’s Premarket Notification (510(k)) Refuse to Accept Policy” issued on June 30, 20
1993 and “510(k) Refuse to Accept Procedures, 510(k) Memorandum K94-1” issued on May 20, 21
1994. Upon issuance as a final guidance document, this guidance will replace those documents. 22
23
Focusing FDA’s review resources on complete submissions will provide a more efficient approach to 24
ensuring that safe and effective medical devices reach patients as quickly as possible. Moreover, with 25
the enactment of the Medical Device User Fee and Modernization Act of 2002 (MDUFMA), the 26
Medical Device User Fee Amendments of 2007 (MDUFA II) and the Medical Device User Fee 27
Amendments of 2012 (MDUFA III),1 FDA agreed to performance goals based on the timeliness of 28
reviews. Acceptance review therefore takes on additional importance in both encouraging quality 29
submissions from sponsors of 510(k) notifications and allowing FDA to appropriately concentrate 30
resources on complete submissions. 31
32
Therefore, we have modified our 510(k) Refuse to Accept (RTA) policy to include an early review 33
against specific acceptance criteria and to inform the submitter within the first 15 34
35
36
1 See Title II of the Food and Drug Administration Safety and Innovation Act (FDASIA) (P.L. 112-144), amending 37 sections 737, 738, and 738A of the Federal Food, Drug, and Cosmetic Act (FD&C Act). 38

calendar days after receipt of the submission if the submission is administratively complete, or if not, 39
to identify the missing element(s). In order to enhance the consistency of our acceptance decisions 40
and to help submitters better understand the types of information FDA needs to conduct a substantive 41
review, this guidance, including the checklists included in the appendices, clarify the necessary 42
elements and contents of a complete 510(k) submission. The process we outline is applicable to all 43
devices reviewed through the 510(k) notification process and has been compiled into checklists for 44
use by FDA review staff. 45
46
It is critical to distinguish between the completeness of the regulatory submission, and the quality of 47
the data provided and any studies conducted in support of the submission. The assessment of the 48
completeness of the 510(k) occurs during the acceptance review, while the assessment of the quality 49
of the submitted information occurs during the substantive review. FDA will base acceptance on the 50
objective criteria outlined in the associated Acceptance Checklist and not on the quality of the data. 51
52
FDA encourages all submitters to provide an electronic copy (eCopy) in place of one of the two hard 53
copies of the 510(k) submission.2 For additional information regarding formatting eCopies for 54
submissions sent to the Center for Devices and Radiological Health (CDRH), please refer to our 55
website for guidelines for submitting: 56
· General information 57
(http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/HowtoMarketYourDev58
ice/PremarketSubmissions/ucm134508.htm) and 59
· Clinical data 60
(http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/HowtoMarketYourDev61
ice/PremarketSubmissions/ucm136377.htm). 62
· 63
For additional information regarding formatting eCopies and submitting hard copies for submissions 64
sent to CBER, please refer to: 65
· “Draft Guidance for Industry: Providing Regulatory Submissions in Electronic Format-66
General Considerations” 67
(http://www.fda.gov/RegulatoryInformation/Guidances/ucm124737.htm) and 68
· “CBER SOPP8110: Submission of Paper Regulatory Applications to CBER” 69
(http://www.fda.gov/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/P70
roceduresSOPPs/ucm079467.htm). 71
· 72
FDA's guidance documents, including this guidance, do not establish legally enforceable 73
responsibilities. Instead, guidance documents describe the Agency's current thinking on a topic and 74
should be viewed only as recommendations, unless specific regulatory or statutory requirements are 75
cited. The use of the word should in Agency guidance documents means that something is suggested 76
or recommended, but not required. 77
78
79
80
81
82
83
2 FDA has issued draft guidance (“eCopy Program for Medical Device Submissions,” available at 84 http://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/UCM313794.85 pdf) to implement section 1136 of the FDASIA, which added Section 745A(b) of the FD&C Act, and provides 86 statutory authority to require eCopy. When final, this guidance will represent the Agency’s thinking on this topic. 87

Background 88
89
The purpose of the 510(k) acceptance review is to assess whether a submission is administratively 90
complete, in that it includes all of the information necessary for FDA to conduct a substantive review 91
and to reach a determination regarding substantial equivalence under section 513(i) of the FD&C 92
Act, 21 U.S.C. § 360c(i). To find a device substantially equivalent under section 513(i) of the FD&C 93
Act, FDA must find that it has the same intended use as the predicate device, and either (1) has the 94
same technological characteristics as the predicate device, or (2) has different technological 95
characteristics, as defined at section 513(i)(1)(B), and the submission contains information, including 96
appropriate clinical or scientific data if necessary, that demonstrates the device is as safe and 97
effective as the predicate and does not raise different questions of safety and effectiveness than the 98
predicate. 99
100
The 510(k) regulations at 21 CFR 807.87 to 807.100 provide greater detail regarding the specific 101
information that each premarket notification submission must contain. For example, the submission 102
must include proposed labeling (807.87(e)), a statement regarding the similarities and differences 103
between the device and others of comparable type (807.87(f)), supporting data (807.87(f) and 104
807.100(b)(2)(ii)(B)), and FDA may request any additional information necessary to determine 105
whether the device is substantially equivalent when the information provided is insufficient to enable 106
such a determination (807.87(l)). Please also refer to our guidance document entitled, “Format for 107
Traditional and Abbreviated 510(k)s” 108
(http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm0843109
65.htm). 110
111
The previous guidances relating to 510(k) RTA policy and the checklist currently used for 112
acceptance review have focused on defining broad issues or principles. Additionally, the previous 113
checklist deals largely with administrative elements but it does not address specific content that is 114
essential for 510(k) review. As a result, FDA accepts many inadequate submissions for review and 115
FDA staff invests significant time in constructing extensive letters requesting all of the additional 116
information needed to conduct a substantive review. This approach is an inefficient use of resources 117
and frequently lengthens review times. For additional information see CDRH’s “Analysis Of 118
Premarket Review Times Under The 119
510(k)Program”(http://www.fda.gov/downloads/AboutFDA/CentersOffices/OfficeofMedicalProduct120
sandTobacco/CDRH/CDRHReports/UCM263386.pdf). The goal of this guidance document is to 121
clarify the content needed in traditional, special, and abbreviated 510(k) submissions to allow FDA 122
to conduct a substantive review, thereby enhancing the quality of received 510(k) submissions and 123
improving overall review time. The review process presented in this document is captured in the 124
Acceptance Checklists for traditional, special, and abbreviated 510(k) submissions, which FDA staff 125
will use during the acceptance review process. 126

Scope 127
128
The information presented in this document is intended to provide FDA staff with a clear, consistent 129
approach for acceptance review for traditional, special, and abbreviated 510(k) notifications and to 130
outline the RTA policy on 510(k)s. 131
132
The acceptance policy does not alter the substantial equivalence decision-making process once the 133
submission has been accepted for review; however, it does alter the start of the FDA review clock for 134
purposes of MDUFA performance goals for those submissions that are not accepted for review. 135
136
This document does not address the monetary aspects or the MDUFA goals associated with 510(k)s. 137
Information pertaining to the fees and payment procedures for submission of a 510(k) notification 138
can be found online; see “Premarket Notification [510(k)] Review Fees” 139
(http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/HowtoMarketYourDevice/Pre140
marketSubmissions/PremarketNotification510k/ucm134566.htm). 141
142
Pre-submission Interaction 143
144
Prior to interacting with review staff, submitters should consult CDRH’s Division of Small 145
Manufacturers, International and Consumer Assistance (DSMICA) or CBER’s Manufacturers 146
Assistance and Technical Training Branch for general information regarding the 510(k) regulations. 147
Before submitting a 510(k) notification, we encourage submitters, especially those who are less 148
familiar with the 510(k) review program or who have novel issues to address, to interact with FDA 149
review staff. Such pre-submission interaction is an important way of improving the quality and 150
completeness of a 510(k). For additional information regarding the Pre-Submission process, please 151
refer to the Draft Guidance “Medical Devices: The Pre-Submission Program and Meetings with FDA 152
Staff.” 3 153
(http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm3103154
75.htm). 155
156
In addition, other FDA guidance documents and resources provide valuable information for 157
preparing 510(k)s, including: 158
· “Guidance for Industry and FDA Staff: Format for Traditional and Abbreviated 510(k)s” 159
(http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/u160
cm084365.htm?utm_campaign=Google2&utm_source=fdaSearch&utm), 161
162
· Other applicable CDRH device-specific and cross-cutting guidance documents, 163
(http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/d164
efault.htm), and 165
· CDRH’s Device Advice 166
(http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/default.htm) 167
168
169
170
171
172
3 Once finalized, this guidance will represent the Agency’s current thinking on this topic. 173

510(k) Refuse to Accept Policies and Procedures 174
175
FDA staff will conduct an acceptance review of all traditional, special, or abbreviated 510(k)s based 176
on objective criteria using the applicable Acceptance Checklist (see Appendices A - C) to ensure that 177
the 510(k) is administratively complete. In order for the submission to be accepted, all administrative 178
elements identified as RTA items should be present or a rationale should be provided for those 179
elements determined by the submitter to be not applicable. The acceptance review should be 180
conducted and completed within 15 calendar days of FDA receiving the 510(k) notification. 181
182
The staff will select the applicable checklist based on the 510(k) type (i.e., traditional, special, or 183
abbreviated). The acceptance review will be conducted on original 510(k) submissions and responses 184
to RTA letters, but not supplements or amendments submitted in response to requests for additional 185
information after a submission has been accepted. The staff should assess whether the submission 186
should be accepted by first answering the preliminary questions below, and then verifying that the 187
submission contains all of the information identified as RTA items in the checklist. The submission 188
should not be accepted and should receive an RTA designation if one or more of the items noted as 189
RTA items in the checklist are not present and no explanation is provided for the omission(s). 190
191
If one or more items noted as RTA items on the Acceptance Checklist are not present, staff 192
conducting the acceptance review should obtain management concurrence and notify the designated 193
510(k) contact person in writing that the submission has not been accepted.4 FDA staff should also 194
provide the submitter with a copy of the completed checklist indicating which item(s) are the basis 195
for the RTA designation. 196
197
The 510(k) submitter may respond to the RTA notification by providing the missing information 198
identified in the checklist. The submitter should submit this information to be included in the file 199
under the originally assigned 510(k) number. A new submission and new user fee are not necessary. 200
Nor should the submitter re-send the entire 510(k) submission, unless FDA notes otherwise (e.g., 201
because the majority of the submission is not in English, or the submission is missing the majority of 202
the items on the checklist). It is sufficient to submit and address only the information requested per 203
the Acceptance Checklist. 204
205
Upon receipt of the newly submitted information, FDA staff should conduct the acceptance screening 206
again following the same procedure within 15 calendar days of receipt of the new information. If the 207
submission is still found to be incomplete, FDA staff should notify the contact person and provide 208
the new checklist indicating the missing item(s). 209
210
211
212
213
214
215
216
4 As outlined in the commitment letter for MDUFA III [FDA, "MDUFA Performance Goals and Procedures" (April 217 18, 2012), available at 218 http://www.fda.gov/downloads/MedicalDevices/NewsEvents/WorkshopsConferences/UCM295454.pdf) (attachment 219 to letter dated July 16, 2012 from Secretary of Health and Human Services Kathleen Sebelius to The Honorable Fred 220 Upton, Chairman, U.S. House of Representatives Committee on Energy & Commerce)], the review clock will not 221 start until the 510(k) submission is accepted for review. 222

When a submission is found acceptable, FDA staff should notify the submission contact person in 223
writing that the 510(k) has been accepted and begin a substantive review of the submission to 224
determine substantial equivalence. Should FDA fail to complete the acceptance review within 15 225
calendar days, the submission should be considered accepted, the submitter should be notified in 226
writing, and FDA should commence with substantive review.5 Once a submission has been accepted, 227
FDA may ask for any information during the substantive review that may have been unintentionally 228
overlooked during the acceptance review. 229
230
FDA Review Clock 231
232
As explained in the commitment letter for MDUFA III referenced in Title II of FDASIA, Public Law 233
112-114, “FDA days begin on the date of receipt of the submission or of the amendment to the 234
submission that enables the submission to be accepted (510(k)) or filed (PMA).”6 Thus, the FDA 235
review clock does not start when a submission is designated RTA. The FDA review clock also would 236
not start if we receive unsolicited amendments during the acceptance review period. Once an 237
application is “Accepted,” the FDA review clock begins as of the date of receipt of the most recent 238
submission or amendment that made the 510(k) complete (even if FDA later requests information 239
that should have been requested during acceptance review). 240
241
242
Refuse to Accept Principles 243
244
In order to use this guidance appropriately, FDA staff should review the following basic principles 245
regarding FDA’s review policies and procedures. 246
247
Acceptance should not be based on a substantive review of the information provided in the 248
510(k) notification. 249
250
It is important to make the distinction between the acceptance review and the substantive review. The 251
acceptance review is conducted to assess whether the submission contains all of the appropriate 252
elements, as identified in the applicable checklist, in order to begin a substantive review. In assessing 253
whether a 510(k) notification should be accepted, submitted information is not evaluated for 254
adequacy to support a finding of substantial equivalence. The checklist is a tool to ensure that the 255
submission contains the necessary information in order to conduct a substantive review (i.e., FDA 256
should not refuse to accept a submission if information is present but inadequate to support a finding 257
of substantial equivalence). The evaluation of the quality of the content and the substantial 258
equivalence decision making process occur within the substantive review once the file has been 259
accepted. 260
261
262
5 In the case of extenuating circumstances such as a government closure during the 15-day review period, the 263 review period may be extended by a comparable number of business days that the FDA buildings are closed. If the 264 submitter receives an automated notice that the acceptance review was not completed because the screening period 265 has exceeded 15 days, FDA may send a correction notice to the submitter. 266 6 FDA, "MDUFA Performance Goals and Procedures" (April 18, 2012), available at 267 http://www.fda.gov/downloads/MedicalDevices/NewsEvents/WorkshopsConferences/UCM295454.pdf) (attachment 268 to letter dated July 16, 2012 from Secretary of Health and Human Services Kathleen Sebelius to The Honorable Fred 269 Upton, Chairman, U.S. House of Representatives Committee on Energy & Commerce)],). 270

Staff should consider the submitter’s justifications for any alternative approaches 271
272
The submitter may provide a rationale for why any criteria in the checklist are not applicable to the 273
device. Likewise, the submitter may provide a rationale for any deviation from a device-specific or 274
cross-cutting guidance document or FDA-recognized standard. It is FDA’s expectation that each item 275
in the checklist will be addressed either by including the requested information or providing a 276
rationale for why is it not applicable or why there is a deviation.7 FDA will not consider a given 277
criterion in the checklist to be “Present” if the submission fails to include either the information 278
requested or a rationale for omission or deviation. See Acceptance Review section below for 279
examples and further explanation. 280
281
The submitter should review device-specific and cross-cutting guidance documents, applicable 282
recognized standards, and applicable regulations. 283
284
Before submitting a 510(k), the submitter should consider the currently available guidance 285
documents and standards, as well as applicable regulations for the proposed device in the preparation 286
of the submission. Staff and industry are encouraged to refer to the product classification database 287
(http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm) to assist in identifying 288
any applicable recognized consensus standards and product specific guidance document(s). 289
290
The Checklist – Preliminary Questions 291
292
Within 15 calendar days of receipt of the 510(k), FDA staff should answer the preliminary questions 293
below, which are included on the first page of the Acceptance Checklists. Depending upon the 294
answers to these preliminary questions, the remainder of the acceptance review may or may not be 295
necessary. If the responses to the preliminary questions and subsequent consultation with the Center 296
personnel identified below indicate that the 510(k) acceptance review should not continue,8 FDA 297
staff should promptly notify the submitter using proper administrative procedures. 298
The preliminary questions are: 299
300
301
302
303
304
305
306
307
308
309
7 The presence of a justification is particularly relevant in the acceptance review stage while the adequacy of such 310 justifications to justify the omission of certain information falls within the scope of the substantive review phase. 311 8 FDA will not process a 510(k) unless it meets the following requirements: i) the submission must be sent with the 312 user fee required by section 738 of the FD&C Act, and ii) the firm must submit the correct number of copies per 21 313 CFR 807.90(c). FDA has issued draft guidance to implement section 1136 of FDASIA, which added Section 314 745A(b) of the FDA&C Act (“eCopy Program for Medical Device Submissions,” available at 315 http://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/UCM313794.316 pdf). Once this guidance is final, at least one copy of the submission will be required to be an eCopy. Since any 317 510(k) not meeting these two requirements will not be processed by the CDRH Document Mail Center or the CBER 318 RPM, they are not included in the checklist. 319

1. Is the product a device (per section 201(h) of the FD&C Act) or a combination product (per 320
21 CFR 3.2(e)) with a device constituent part? 321
322
If the product does not appear to meet the definition of a device under section 201(h) of the FD&C 323
Act, or does not appear to be a combination product with a device constituent part, then the 510(k) 324
lead reviewer should consult with the CDRH Jurisdictional Officer or the CBER Office Jurisdiction 325
Liaison to determine the appropriate action, and inform division management. If FDA staff 326
determines that the product is not a device and is not a combination product with a device constituent 327
part, the 510(k) review team should stop the review and notify the submitter. 328
329
2. Is the application with the appropriate Center? 330
331
If the application is for a single-entity device and appears to be subject to review in a Center different 332
from the one to which it was submitted, or if it is for a combination product with a device constituent 333
part and it appears that a Center different from the one to which it was submitted has the lead, the 334
510(k) team leader should consult with the CDRH Jurisdictional Officer or the CBER Office 335
Jurisdiction Liaison to determine the appropriate action and inform division management. If the 336
510(k) is submitted to CDRH and CDRH staff determines that the application is not subject to 337
CDRH review, or the 510(k) is submitted to CBER and CBER staff determines that the application is 338
not subject to CBER review, the 510(k) review team should stop the review and notify the applicant. 339
340
3. Is a 510(k) the appropriate regulatory submission? 341
342
Staff should determine whether a 510(k) is the appropriate regulatory submission. If a 510(k) is not 343
appropriate (e.g., Class III type and PMA required, or Class I or II type and 510(k)-exempt), staff 344
should make this determination during the acceptance review and notify the submitter of the 345
determination. 346
347
4. Is there a pending PMA for the same device with the same indications for use? 348
349
If there is a pending PMA for the same device, the submitter should withdraw either the 510(k) or the 350
PMA. The review team should consult division management and other Center resources to determine 351
the appropriate action. 352
353
5. If clinical studies have been submitted, is the submitter the subject of the Application 354
Integrity Policy (AIP)?9 355
356
The lead reviewer should refer to the AIP list 357
(http://www.fda.gov/ICECI/EnforcementActions/ApplicationIntegrityPolicy/ucm134453.htm 358
359
360
361
362
363
9 When data in a pending application have been called into question by certain wrongful acts (fraud, untrue 364 statements of material facts, bribery, or illegal gratuities), FDA intends to defer substantive scientific review of such 365 data until completion of a validity assessment and questions regarding reliability of the data are resolved. (See FDA 366 Guide 7150.09 Compliance Policy Guide, Chapter 50 – General Policy – Subject: Fraud, Untrue Statements of 367 Material Facts, Bribery, and Illegal Gratuities, 56 FR 46191.) 368

). If the applicant is on the list, the reviewer should consult the CDRH Office of 369
Compliance/Division of Bioresearch Monitoring (OC/DBM - BIMO) or CBER Office of Compliance 370
and Biologics Quality/Division of Inspections and Surveillance/Bioresearch Monitoring Branch 371
(OCBQ/DIS/BMB) to determine the appropriate action. 372
373
The Checklist – Acceptance Review 374
375
Organizational Elements 376
Although missing one or more of the items in the table of Organizational Elements in the Acceptance 377
Checklists, such as a Table of Contents or page numbers, generally will not lead to an RTA decision, 378
we strongly encourage submitters to incorporate these elements in their submissions to streamline 379
FDA review and decision-making. If, however, the submission is so disorganized that FDA cannot 380
locate the information needed to assess substantial equivalence, or if the submission is so poorly 381
written (e.g., in broken English) that the information submitted to support substantial equivalence 382
cannot be understood, the submission should receive an RTA decision. 383
384
Elements of a Complete Submission (RTA Items) 385
The objective criteria in this checklist outline those elements that are explicitly required by regulation 386
or that are essential to FDA’s substantive review of the submission and determination of substantial 387
equivalence under section 513(i) of the FD&C Act. For example, proposed labels, labeling, and 388
instructions are required by 21 CFR 807.87(e)), while a description of the materials, design, and 389
other features of the device is essential to determining whether its technological characteristics are 390
the same as those of the predicate and whether any differences raise new questions of safety and 391
effectiveness under section 513(i) of the FD&C Act. 392
393
We have also identified several categories and subcategories of data and information that, when 394
applicable, are critical to supporting a statement indicating the device is similar to and/or different 395
from other products of comparable type under 21 CFR 807.87(f) and the substantial equivalence 396
determination. For example, if the new device has direct or indirect patient-contacting components, a 397
biocompatibility assessment will be essential to evaluating whether the new device is as safe as the 398
predicate with respect to the risk of toxicity it poses to the patient. While testing and data would 399
usually be necessary to such an assessment, this is not always the case (for example if the device 400
under review and the predicate are identical in all relevant respects), and acceptance should be based 401
only on the presence of an item or an explanation why the item is not applicable, not the adequacy of 402
such explanation. If the device has no direct or indirect patient-contacting components, no 403
biocompatibility assessment would be necessary and the biocompatibility items on the checklist 404
would be inapplicable. 405
406
Because the applicability of these categories is also critical to the substantial equivalence 407
determination, in order to be accepted, all submissions should include a statement indicating whether 408
these categories apply, as outlined in the Acceptance Checklist (e.g., materials, presence of software, 409
whether the device is intended to be used sterile). When performance data are 410

provided, the submission of full test reports describing how the testing was conducted is crucial to 411
FDA’s assessment of whether the data support a finding of substantial equivalence. 412
413
Where a device-specific guidance document exists for the subject device, the sponsor should follow 414
the recommendations included in that document, or the sponsor should provide a rationale for 415
addressing the scientific issues discussed in the guidance document using an alternative approach in 416
order to meet the applicable statutory and regulatory criteria. In the absence of the recommended 417
information and without a supporting rationale for an alternative approach, the submission should be 418
considered incomplete and not accepted. If special controls have been identified for the device, those 419
controls should be addressed in order for the submission to be accepted. Note, however, that the 420
special controls must be followed in order for the device to be considered in Class II and therefore to 421
support a finding of substantial equivalence. 422
423
Applying the Checklist of RTA Items 424
Using the Acceptance Checklist appropriate to the submission type (Traditional, Abbreviated, or 425
Special), within 15 calendar days of receipt of the 510(k), FDA staff should answer each question for 426
the elements identified as RTA items. For those items that have an option of “yes,” “no,” or “not 427
applicable (N/A)” as an answer, the item should receive an answer of “yes” or “N/A” for the 510(k) 428
submission to be accepted for substantive review. 429
For the first question in each section related to the need for certain performance data (such as 430
biocompatibility, sterilization, software, etc.), staff should indicate whether the submission has 431
addressed one of the options for the 510(k) submission to be accepted for substantive review. For 432
example, the submission should state that either there are or are not direct or indirect (e.g., through 433
fluid infusion) patient-contacting components in order for the submission to be considered complete 434
and accepted for substantive review. 435
436
Elements marked “Not applicable” 437
In developing the checklists, the Agency has considered the general categories and respective 438
subcategories of information that are necessary to conduct a substantive review for the wide range of 439
medical devices that are appropriate for review under 510(k) premarket notification. All such criteria 440
may not be pertinent to a particular device. Staff should select “N/A” for those elements that do not 441
apply to the subject device. For example, the requirements for financial certification and disclosure 442
statements (21 CFR 807.87(i)) only apply to submissions with clinical data. If the submission 443
contains no clinical data, staff should select “N/A.” 444
445
Adequacy of information 446
In order to make the checklist criteria objective, for each RTA item, FDA should consider only the 447
presence or omission of the element or a rationale for the omission of the element or use of an 448
alternative approach during acceptance review. It is likely that FDA staff will encounter scenarios 449
where information is provided, but is incomplete or inadequate. In such instances, FDA staff should 450
answer the question for the respective item as “Yes,” but may communicate the inadequacy or 451
request additional information in the course of the substantive review. For example, the submitter 452
may have provided full test reports for all performance testing; however, FDA may determine that 453
the results of a particular test are not sufficient to support a finding of substantial equivalence and 454
additional justification would be needed. The performance testing 455

criterion would be marked “Yes” in the checklist and the need for additional justification should be 456
communicated to the submitter during the substantive review. 457
458
Elements marked “No” 459
For any acceptance criterion designated as “No,” FDA intends to provide an explanation to describe 460
the missing element(s), if needed. This explanation is particularly important for a criterion in which it 461
may not be immediately apparent to the submitter what necessary information, specifically, is not 462
present. For example, the Device Description section includes an element that states “submission 463
contains all descriptive information recommended in the device-specific guidance document” and a 464
notation of “No” alone may not be sufficient to inform the submitter of what specific piece(s) of 465
information is missing. FDA staff should include a list or statement of the additional information that 466
is necessary to meet the acceptance criteria. This list or statement can be communicated in the 467
comment section on the checklist beside each specific criterion. 468
469
Conversion of Special 510(k) to Traditional 510(k) 470
FDA has developed separate checklists to address the differences in content for special and 471
traditional 510(k) submissions. FDA staff will utilize the appropriate checklist based on the file type 472
as designated by the submitter. In the event that the submitter has submitted a special 510(k), but 473
FDA determines that the file should be converted to a traditional 510(k), FDA will notify the contact 474
person designated in the 510(k) submission of the conversion and the rationale for the conversion. 475
Additionally, FDA staff should provide the completed Acceptance Checklist for traditional 476
submissions indicating which elements are missing. The submitter may respond by providing the 477
identified information and the acceptance review will proceed with the traditional checklist. 478
479
If a 510(k) designated as a special 510(k) qualifies as a special 510(k), but the submission includes 480
performance data, FDA should offer the submitter two options: (1) the data can be removed from the 481
510(k) and staff will proceed with the special 510(k) checklist, or (2) the 510(k) can be converted to 482
a traditional 510(k) and the submitter will provide any other missing information needed for a 483
traditional 510(k) in order to be accepted for substantive review. 484