quimica organica - clubdelquímico upagu
TRANSCRIPT

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
1
UNIVERSIDAD PRIVADA ANTONIO
GUILLERMO URRELO DE CAJAMARCA
FACULTAD DE FARMACIA Y BIOQUIMICA
ESPECIALIDAD DE FARMACIA Y BIOQUÍMICA
IV

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
2
www.clubdelquimico.blogspot.com
APUNTES Y PROBLEMAS DE QUÍMICA ORGÁNICA
INDICE
Capítulo 1: TEORÍA ENLACE Y RESONANCIA Capítulo 2: ÁCIDOS Y BASES EN QUÍMICA ORGÁNICA Capítulo 3: HIDROCARBUROS (Nomenclatura) Capítulo 4: GRUPOS FUNCIONALES (Nomenclatura) Capítulo 5: ALCANOS Y CICLOALCANOS (Análisis Conformacional) Capítulo 6: ESTEREOQUÍMICA Capítulo 7: REACCIONES ORGÁNICAS Capítulo 8: REACCIONES DE LOS ALCANOS Capítulo 9: HALOGENUROS DE ALQUILO Capítulo 10: REACCIONES DE LOS HALOGENUROS DE ALQUILO (Sustituciones
Nucleofílicas) Capítulo 11: HALOGENUROS DE ALQUILO (Reacciones de Eliminación) Capítulo 12: ALCOHOLES Y TIOLES Capítulo 13: ÉTERES, EPÓXIDOS Y SULFUROS Capítulo 14: ALQUENOS Capítulo 15: ALQUINOS Capítulo 16: REACTIVOS ORGANOMETÁLICOS Capítulo 17: ALDEHÍDOS Y CETONAS REACCIONES DE ADICIÓN NUCLEOFÍLICA Capítulo 18: ALDEHÍDOS Y CETONAS (Reacciones de Sustitución Alfa al carbonilo) -
ALDEHÍDOS Y CETONAS a,b-INSATURADOS (Síntesis y Reacciones de sistemas a,b-insaturados)
Capítulo 19: ÁCIDOS CARBOXÍLICOS Capítulo 20: DERIVADOS DE LOS ÁCIDOS CARBOXÍLICOS Capítulo 21: COMPUESTOS DIFUNCIONALES Capítulo 22: SISTEMAS CONJUGADOS Capítulo 23: SUSTITUCIÓN ELECTROFÍLICA AROMÁTICA Capítulo 24: QUÍMICA DE LOS BENCENOS SUSTITUIDOS (Fenoles y Arilaminas) Capítulo 25: HIDROCARBUROS AROMÁTICOS POLICÍCLICOS Capítulo 26: SÍNTESIS Y REACCIONES DE LAS AMINAS Capítulo 27: HETEROCICLOS

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
3
TEORÍA ENLACE Y RESONANCIA
La unión entre dos átomos en una molécula es lo que denominamos
comúnmente enlace. En este capítulo abordaremos en forma sencilla el porqué existen
los enlaces.
Las primeras explicaciones de la naturaleza de los enlaces químicos fueron
dadas por W. Kössel y G.N. Lewis en 1916. Ellos propusieron que existían dos tipos
principales de enlaces químicos:
1.- El Enlace Iónico o Electrovalente (formado por la transferencia de uno o más
electrones de un átomo a otro).
2.- El Enlace Covalente (un enlace que se forma cuando los átomos comparten
electrones).
Los conceptos y explicaciones que se derivan de las proposiciones originales de
Lewis y Kössel son satisfactorios para explicar muchos de los problemas con que
nos enfrentamos en la química orgánica actual.
¿ Por qué existen los enlaces ?
Dos átomos forman un enlace sólo si su interacción es "favorable". Es decir si
desprenden o liberan energía [calor] , (Por el contrario, la ruptura de enlace requiere
energía [calor]).
¿Cuáles son las causas de dicho desprendimiento de energía?
Son dos y se basan en algunas leyes fundamentales de la Física:
(1) Las cargas opuestas se atraen
(2) Los electrones están distribuidos en el espacio
ENLACES IÓNICOS O ELECTROVALENTES
Un enlace iónico se forma cuando dos átomos de electronegatividad muy
distinta se unen entre sí. La pérdida de un electrón del átomo menos electronegativo da
lugar a la formación de un catión; y la ganancia de un electrón por el átomo más
electronegativo da lugar a la formación de un anión.¿Por qué se forman estos iones?
Según la teoría de Lewis-Kössel ambos iones adquieren la estructura electrónica
de un gas noble. Más aún, a partir de los iones individuales se forma una molécula

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
4
cristalina y, en su estado cristalino, los iones tienen energías mucho menores que los de
los átomos a partir de los cuales se forman. Así, se estabilizan al reaccionar, formando
una molécula cristalina.
Ejemplo:
Li = 1s22s1 F = 1s22s22px22py22pz1
Adquieren configuración tipo gas
noble Helio y Neón.
Las sustancias iónicas, por sus grandes fuerzas electroestáticas internas, tienden
a ser sólidas con un punto de fusión muy alto, a menudo superior a 1000 ºC. En
disolventes polares, como el agua, por lo general los iones solvatados conducen la
corriente eléctrica.
ENLACE COVALENTE
Cuando reaccionan dos o más átomos de electronegatividad igual o parecida no
se lleva a cabo una transferencia completa de electrones. En estos casos los átomos
adquieren la estructura del gas noble compartiendo electrones y se forman enlaces
covalentes. Las moléculas covalentes pueden representarse mediante fórmulas en que
los electrones son puntos, pero es más conveniente utilizar fórmulas con guiones en
donde cada guión representa un par de electrones que enlaza a los átomos.
Ejemplo:
Los iones mismos pueden contener enlaces covalentes:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
5
Cuando dos átomos de diferente electronegatividad forman un enlace covalente
los electrones no se comparten equitativamente entre ellos. El átomo de mayor
electronegatividad atrae el par de electrones, acercándolo más a él, y se forma un
ENLACE COVALENTE POLAR. Un ejemplo de un enlace de este tipo es el que se
forma en el cloruro de hidrógeno. El átomo de cloro, que es más electronegativo, atrae
los electrones enlazantes hacia él. Esto hace que el átomo de hidrógeno tenga una
dificiencia de electrones y adquiera una carga positiva parcial ((+)). El átomo de cloro
se hace rico en electrones y aparece en él una carga negativa parcial ((-)).
En términos generales hablamos de enlace polarizado.
Menos Electronegativo : Más Electronegativo
= qd
Una molécula A:B de este tipo es un dipolo, caracterizado por una carga
positiva q en un extremo que se compensa con una negativa q en el otro, separadas por
una distancia d. El producto de dichas cantidades, la carga q por la distancia d, es el
MOMENTO DIPOLAR, .
EL MODELO DE ENLACE DE LEWIS [1]

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
6
ÁCIDOS Y BASES EN QUÍMICA ORGÁNICA
ÁCIDOS Y BASES FUERTES
De acuerdo con la teoría de Bronsted-Lowry, se define un ácido como una
sustancia que puede donar un protón y una base como una sustancia que puede aceptar
un protón.
ácido base base conjugada ácido conjugado del ácido de la base Los iones hidronio, H3O, y los iones hidróxido, -OH, son respectivamente, los
ácidos y bases más fuertes que existen en soluciones acuosas.
NaOH (s) + H2O Na (ac) + -OH (ac)
ACIDOS Y BASES DEBILES
En contraste con los ácidos fuertes, como el HCl y el H2SO4, el ácido acético es
un ácido mucho más débil. Cuando el ácido acético se disuelve en agua, la reacción que
se muestra a continuación no se lleva a cabo en forma completa.
Los experimentos muestran que en una solución 0.1 M a 25 ºC, sólo el 1 % de
las moléculas del ácido acético transfieren sus protones al agua. Como la reacción de
disolución acuosa del ácido acético es un equilibrio, puede describírsela mediante una
expresión para la constante de equilibrio

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
7
Para soluciones acuosas diluidas la concentración de agua es esencialmente
constante (55 moles/lt), de manera que la expresión de la constante de equilibrio puede
rescribirse en términos de una nueva constante, Ka, llamada constante de acidez.
A 25ºC, la constante de acidez del ácido acético es 1.8 10-5. Para cualquier ácido disuelto en agua, pueden escribirse expresiones similares.
Utilizando un ácido hipotético generalizado, HA, la reacción en agua es:
Por esta razón, un valor alto de Ka significa que el ácido es un ácido fuerte y un
valor pequeño de Ka significa que el ácido es débil. Si la constante de acidez es mayor
de 10, a efectos prácticos, el ácido estará totalmente disociado en agua.
La teoría de ácidos y bases fue ampliada enormemente por G.N. Lewis en 1923.
Destruyendo lo que él llamo "el culto del protón", Lewis propuso que los ácidos podían
definirse como receptores de pares de electrones y las bases como donantes de pares de
electrones. En la teoría de Lewis, el protón no es el único ácido, sino también muchas
otras partículas. Por ejemplo, el cloruro de aluminio y el triflururo de boro reaccionan
con aminas en la misma forma que lo hace un protón.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
8
En los ejemplos anteriores, el cloruro de aluminio y el triflururo de boro aceptan
el par de electrones de la amina lo mismo que sucedería con un protón. Esto se debe a
que los átomos centrales de aluminio y boro sólo tienen un sexteto de electrones y por
lo tanto son deficientes de electrones.
Las bases son bastantes similares tanto en la teoría de Lewis como en la teoría
de Bronsted-Lowry.
ÁCIDOS Y BASES EN SOLUCIONES NO ACUOSAS
Muchas de las reacciones ácido-bases orgánicos que comentaremos en este
capítulo no se llevan a cabo en solución acuosa. Muchos de los ácidos que se
encontraran aquí, son mucho más débiles que el agua. Las bases conjugadas de estos
ácidos orgánicos se preparan en amoniaco líquido. La base más potente que puede
emplearse en el amoniaco líquido es el ion amida, -NH2 la base conjugada del amoniaco.
El ion amida, al ser la base conjugada de un ácido sumamente débil, es una base muy
potente.
Recuérdese que: pKa = - log Ka por lo tanto, a medida que el pKa disminuye aumenta
la acidez de los ácidos, y a medida que el pKa aumenta, disminuye la acidez de los
ácidos, aumentando su basicidad, obsérvese la tabla 2.1.
Tabla 2.1.-
Valores de pKa de sustancias orgánicas comunes
ÁCIDO pKa BASE
ciclohexano 45 C6H11-

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
9
NH3 36 NH2-
HCCH 25 HCC-
CH3OH 16 CH3O-
H2O 15,7 HO-
R-NH3+ 10 R-NH2
R-COOH 5 R-COO-
CH3OH2+ -2 CH3OH
PhOH2+ -6,7 PhOH
HPF6 -20 PF6-

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
10
OTROS CRITERIOS A CONSIDERAR:
ELECTRONEGATIVIDAD DEL ÁCIDO
Cuanto más electronegativo sea el elemento que porta el H más polarizado
estará el enlace y más ácido será la familia de compuestos relacionados. A(-)H(+), a
mayor E.N. de A, mayor será la acidez del compuesto, ejemplo:
pKa Ácido Base
43
35
10
18
-2
La basicidad va a disminuir en este orden C- N- O-, cuanto más E.N. es el
elemento, el ácido es más fuerte y la base conjugada del mismo más débil. El nitrógeno
es menos E.N. que el oxígeno y por lo tanto los compuestos nitrogenados son mejores
bases, siendo así el amoniaco una mejor base que el agua.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
11
EFECTO DE RESONANCIA
El efecto de la resonancia estabiliza más a una base que a un ácido, ya que se
produce una deslocalización del par de electrones de la base y por lo tanto estos están
menos disponibles a captar un protón, con lo cual disminuye el pKa. Analicemos el
siguiente ejemplo:
a) CH3COOH pKa = 5 Esta claro que al ver sus valores de pKa, que el ácido acético es mucho más
b) CH3CH2OH pKa = 17 ácido que el etanol
¿Por qué?
Si la carga negativa del acetato se puede deslocalizar por resonancia, el par de
electrones estará menos disponible para actuar, por lo que la base estará estabilizada y
será débil, siendo por lo tanto fuerte el ácido del que deriva.. El etanol será un ácido más
débil porque su base no se encuentra estabilizada por resonancia, su b ase conjugada será
más fuerte ya que los electrones están más disponibles a captar un protón. Se puede
concluir que “cuanto más estable es su base conjugada más fuerte será el ácido” .
Diferencia de acidez entre tipos de compuestos:
Alcoholes y Ácidos Carboxílicos
R-COOH R-COO- pKa 5
R-CH2OH R-CH2O- pKa 17
Hay una fuerte estabilización por resonancia en los iones carboxilato
provenientes de los ácidos carboxílicos, haciendo que sus bases conjugadas sean más
débiles y sus ácidos más fuertes.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
12
Alcoholes y Fenoles
Ph-OH Ph-O- pKa 10
R-CH2OH R-CH2O- pKa 17
El ion PhO- puede estabilizarse por resonancia con participación del anillo
aromático, haciendo que su base conjugada sea lo bastante débil, con respecto a la base
conjugada del alcohol alifático.
Aminas y Amidas
R-CO-NH2 R-CO-NH- pKa = 16
R-CH2-NH2 R-CH2-NH- pKa = 35
La forma básica del amiduro esta estabilizada por resonancia:
La forma ácida también tiene formas resonantes, pero implica separación de cargas y
por lo tanto es menos estable.
En la amina, la forma básica no puede estabilizarse por resonancia por lo tanto es una
especie bastante más básica que la amida, y por ende como ácidos son mucho más
débiles.
Cetonas e Hidrocarburos
R-CO-CH3 R-CO-CH2 pKa = 20
R-CH2CH3 R-CH2CH2 pKa = 43
Nuevamente vemos que la base conjugada de la cetona es una especie que
puede estabilizarse por resonancia disminuyendo su basicidad y aumentando su acidez,
en cambio, el carbanión generado en el hidrocarburo no puede estabilizarse por
resonancia siendo mucho más básico que su homólogo. Por lo tanto los hidrocarburos
son menós ácidos que las cetonas.
Basicidad de una Amina alifática y una Amina aromática
Amina alifática:
ácido base ácido conjugado base conjugada Amina aromática:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
13
En el primer caso la metilamina es una base fuerte dado que no puede
estabilizarse por resonancia, lo que implica que su par de electrones estará siempre
disponible a captar un protón de un ácido, en el segundo caso, en la anilina (amina
aromática) el par de electrones se deslocaliza al interior del anillo, estabilizando a la
base por resonancia, y disminuyendo su basicidad.
Acidez de una amida
Una amida está estabilizada por resonancia, es decir su par de electrones está
deslocalizado y poco disponible a aceptar un protón de ácido, ahí que ellos como
compuestos son relativamente malas bases y buenos ácidos frente a compuestos
similares (aminas por ejemplo).

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
14
Analicemos el siguiente caso:
Tanto la forma ácida como la
forma básica se pueden
estabilizar por resonancia, pero
la forma básica implica
deslocalización de carga, por
lo que es más estable y a su
vez como compuesto es
menos básico. La forma ácida, implica
creación de carga por lo que es
menos estable como estructura
resonante, lo que comprueba
su carácter ácido como
compuesto. Si se tienen el mismo número
de estructuras resonante el
criterio a emplear es el que no
implique creación de carga.
Comparación de la acidez de una cetona, acetil-cetona, acetato de etilo, y malonato de
etilo
pKa = 9 pKa = 10.2 pKa = 14 pKa = 20
Teniendo en cuenta que a menor pKa, mayor acidez:
1. La acetil-acetona: es la más ácida debido a que su base conjugada está muy
estabilizada por resonancia, posee hidrógenos muy ácidos, en a dos grupos
carbonilos.
2. El aceto-acetato de etilo: es el siguiente en acidez debido a que aunque su base
conjugada está muy estabilizada por resonancia, el grupo éster estabiliza menos la
carga negativa que el grupo cetona, debido a que el éster tiene su propia resonancia
interna.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
15
3. El malonato de dietilo: es el que les sigue debido a que las formas resonantes de la
base conjugada estabilizan menos la carga negativa, por la presencia de los dos
grupos éster.
4. La cetona: es la menos ácida debido a que su base conjugada tiene menos
estabilidad por resonancia, posee hidrógenos en a un solo grupo carbonilo, a
diferencia de los casos anteriores.
GRUPOS ATRACTORES DE ELECTRONES POR RESONANCIA
El grupo NITRO: Ayuda a estabilizar la carga negativa en la base conjugada, además el
grupo nitro por si solo ya es estable.
Por ejemplo: el nitrometano es relativamente ácido, tiene un pKa = 10

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
16
El Grupo CIANO: Contribuye un poco menos que el grupo nitro a la estabilidad de la
base conjugada.
El orden de acidez para estos grupos y otros suele ser:
-NO2 -CN -SO2- 3C-
Ejemplos:
Para la base conjugada del
trifenil metano hay 10 posibles
estructuras resonantes, por lo que
deberíamos esperar que fuese un
ácido bastante fuerte, sin
embargo su pKa = 30, lo que
implica que es un ácido bastante
débil lo cual se puede explicar
por efecto estérico de la base y la
perdida de la aromaticidad del
compuestos al deslocalizarse la
carga al interior del anillo.
En este caso a pesar del efecto
estérico, el movimiento de las
cargas sobre el grupo ciano esta
favorecida, debida a la diferencia
de electronegativiodad de los
átomos. Por lo que la forma
básica esta bastante estabilizada y
el ácido del cual proviene es
bastante fuerte, pKa 0
Efectos de resonancia de diversos grupos:
Electrón-dadores -F, -Cl, -Br, -I, -O-, -OR, -OH, -OCOR, -NR2, -SR, -SH, -CH3
Electrón-atrayentes -CN, -CO-, -SO2-, -NO2

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
17
HIBRIDACIÓN DE LOS ÁTOMOS
A medida que aumenta el % de carácter p en los orbitales aumenta el tamaño de
los enlaces y aumenta la basicidad, disminuyendo la acidez de la molécula. Esto se debe
a que los electrones están más disponibles para aceptar protones.
Ejemplos:
Molécula Hibridación del átomo pKa Carácter básico/ácido
hibridación sp3 del "N"
9.8
base fuerte
hibridación sp2 del "N"
5.2
base moderada
hibridación sp del "N" 0 base débil
hibridación sp3 del "C" 40 ácido débil
hibridación sp del "C" 24 ácido fuerte

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
18
EFECTOS INDUCTIVOS Y ELECTROESTÁTICOS
Los grupos donores de electrones por efecto inductivo contribuyen a disminuir
la acidez y aumentan la basicidad de la misma. En cambio los átomos electronegativos
contribuyen a que aumente la acidez y disminuya la basicidad. La polarización de un
enlace por influencia de un átomo o grupo adyacente polar se conoce como efecto
inductivo,
En cambio los halógenos son muy electronegativos y son grupos atractores de
electrones, con lo que polarizan más rápido el enlace a romper, aumentando
considerablemente la acidez de las moléculas, en la Tabla 2.2 se analizan algunos
ejemplos.
Efectos inductivos de diversos grupos:
Electrón-dadores -O-, -CH3, -CO2-
Electrón-atrayentes -F, -Cl, -Br, -I, -OR, -OH, -NR2, -SR, -SH, -CO2H,
-CO2R, -CO-, -CN, -NO2, -SO2-, -C=C-, -CC-, -NR3, -
SR2

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
19
Tabla 2.2.-
Ejemplos de Ácidos Carboxilicos, su pKa y el efecto del sustituyente
Molécula pKa Efecto
3.77
"H" ni da ni quita carga por efecto inductivo.
4.76 efecto inductivo del grupo metilo, aumenta la
basicidad.
2.66 "F" átomo más electronegativo (E.N = 4.0), van
aumentando la acidez con respecto a los otros
halógenos.
2.86 E.N. = 3.0, la acidez disminuye por que al ser
menos electronegativo habrá menos dispersión de
la carga.
2.90 E.N. = 2.8
3.16 E.N. = 2.5
1.29
La presencia de dos átomos de cloro, hace que
aumente la acidez con respecto al primero.
0.05
En este caso hay tres átomos de cloro, aumenta
bastante la acidez con respecto al anterior
4.76 Efecto inductivo más próximo al grupo ácido.
4.88 Efecto inductivo va disminuyendo al crecer la
cadena carbonada.
4.96
Mayor acumulación de carga negativa sobre un
mismo carbono, menor dispersión de carga,
implica mayor basicidad.
5.65
Ídem, al anterior
1.68
El grupo nitro dispersa mejor la carga, aumenta la
acidez.
1.86
La presencia de los metilo aumenta la basicidad,
con respecto al anterior.
2.47 Grupo ciano dispersa carga por efecto de
resonancia.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
20
3.35 El éster dispersa carga por efecto de resonancia,
pero menos que el grupo ciano
3.58 El carbonilo de cetona, dispersa menos carga que
un éster.
3.53 Ahora tenemos un éter, el cual es mas bien un
grupo dador de electrones.
3.83 El hidróxido es dador de electrones, aumenta la
basicidad.
3.77 El hidrógeno, no influye prácticamente en nada.
1.23 El diácido es más fuerte, debido al efecto
inductivo que produce el otro ácido.
4.76 Metilo dador de carga por efecto inductivo,
aumenta la basicidad.
2.83 Este efecto se mantiene cuando hay un átomo de
carbono entre ellos, pero se debilita un poco al
aumentar la distancia entre los dos grupos ácidos.
EFECTO DE LOS PUENTES DE HIDRÓGENO
Los puentes de hidrógenos estabilizan a las bases conjugadas. Por existir una
mayor dispersión de la carga, haciendo que disminuya la basicidad y aumente la acidez
de los mismos. A continuación se ejemplifica este hecho con moléculas que se
diferencian principalmente en su geometría y/o estructura. En primer lugar veamos el
caso de un diácido cis con respecto a su isómero trans.
Se puede observar que la base conjugada esta estabilizada por la formación de
un puente de hidrógeno (pKa = 1.92) en el isómero cis.
En cambio en el isómero trans, este puente de hidrógeno no se puede formar
dado que la geometría de la molécula no se lo permite, lo que hace que aumente su
basicidad al no haber dispersión de carga, (pKa = 3.02).
En el segundo ejemplo analizamos el ácido orto y para hidroxibenzoico. Se
puede concluir que el ácido orto hidroxibenzoico es más ácido que el para, dado que la
base conjugada se puede estabilizar por la formación de un puente de hidrógeno por la
cercanía en el espacio del grupo hidróxido con el carbixilato, cosa que en la posición
para no puede ocurrir.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
21
ácido más fuerte base conjugada estabilizada por puente de hidrógeno
ácido más débil La disposición espacial no permite la formación del puente de hidrógeno
EFECTOS ESTÉRICOS
Recuérdese que los efectos estéricos son los que producen las interacciones
espaciales entre los átomos y/o grupos. Este fenómeno puede influir en la acidez y
basicidad de algunas moléculas. Por ejemplo:
RELACIONES ENERGÉTICAS:
Cuando una reacción química alcanza el equilibrio, la posición del mismo refleja
las energías relativas de reactivos y productos. La constante de equilibrio K se relaciona
con la diferencia de energía libre estándar Gº entre los productos y los reactivos, a
través de la ecuación:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
22
Gº = - RT ln K
en donde R = 1.99 10 -3 kcal/mol-K (8.31 10-3 kJ/mol-K)
T = temperatura
Si los reactivos son de menor energía que los productos (Gº 0; K 1), y la
reacción se dice endergónica. En una reacción exergónica, los productos son de menor
energía que los reactivos (Gº 0; K 1) .
Dos importantes parámetros termodinámicos contribuyen a la diferencia de
energía libre y ,por tanto, a la posición del equilibrio. La entalpia, Hº, o calor de
reacción, que está relacionada con las diferencias en las energías de enlaces entre los
productos y los reactivos. La entropía, Sº, es una medida de las diferencias de libertad
de movimiento entre las moléculas de los productos y los reactivos. Un valor más
positivo de la entropía indica un aumento del grado de libertad al transformarse los
reactivos en productos. La expresión que relaciona entré sí dichos parámetros es:
Gº = Hº - T Sº
El valor de la entalpía, en la mayoría de reacciones orgánicas, es en general
mucho mayor que el del término TSº y, por tanto, su contribución a la diferencia de
energía libre es más importante. En consecuencia, es frecuente hablar de reacciones
refiriéndose a Hº, es decir, como exotérmicas (Hº 0) o endotérmicas (Hº 0). Cuando
Hº TSº, situación que ocurre con frecuencia en sistemas biológicos, casi siempre se
utilizan los términos " exergónico" y "endergónico".
A continuación se muestran las Tablas 2.3 y 2.4, el la primera se muestra la
relación entre la concentración relativa y la energía libre, para equilibrios simples a 25
ºC, la cual resulta ser útil para saber cuanto esta desplazado un equilibrio. En la Tabla
2.4 se muestran los valores de pKa para las sustancias orgánicas más comunes.
Tabla 2.3.-
Relación entre la concentración relativa y la energía libre en el caso de equilibrios
simples, (A B) a 25 ºC.
Población en el equilibrio Gº
Reactivo A, % Reactivo B, % Kc kcal/mol (kj/mol)
99 1 0,01 2,72 11,37
90 10 0,11 1,30 5,53
75 25 0,65 0,65 2,72
50 50 1,00 0 0
25 75 3,00 -0,65 -2,72

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
23
10 90 9.00 -1,30 -5,43
1 99 99.00 -2,72 -11,37
0,1 99,9 999,00 -4,09 -17,10
0,01 99,99 9999,00 -5,46 -22,82
Kc = Constante de equilibrio, la cuál se puede determinar por la relación entre el pKa
del ácido (AH) y el pKa del ácido conjugada de la base (BH).
pKc = pKa(AH) - pKa(BH), por lo que Kc es igual al antilogaritmo de la diferencia
obtenida.
Escala de Acidez
Ácido conjugado pKa Base conjugada Ácido conjugado pKa base conjugada
Ciclohexano
45
C6H11-
CH3-CH3
42
CH3-CH2
-
CH4 40 CH3- Benceno 37 C6H5-
Etileno 36 CH2=CH- NH3 36 NH2
-
-CH3 35 -CH2- CH2=CH-CH3 35 CH2=CH-CH2
-
3-CH 32 -C- -NH2 27 -NH
-
HCCH 25 HCC- 2-NH 23 2-N
-
CH3COCH3 20 CH3COCH2- t-BuOH 19 t-BuO
-
18.5
C2H5OH (ROH)
17
C2H5O
-
RCONHR' 16 RCONR'- CH3OH 16 CH3O
-
H2O 15.7 HO-
15
15
(ROOC)2CH2
13.5
(ROOC)2CH
-
13.4
13.4
(NC)2CH2 11.2 (NC)2CH- CH3COCH2COOR 10.2 CH3COCH-COOR

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
24
RNH3 R2NH2 R3NH
10
RNH2 R2NH R3N
CH3NO2
10.2
-CH2NO2
HCO3- 10.2 CO3
= -OH 10 -O-
9.6
9.3
NH4 9.2 NH3 HCN 9.1 CN-
9.0
8.0
7.2
H2CO3
6.5
HCO3
-
O2NCH2COOCH3 5.8 O2NCH-COOCH3
5.2
-NH(CH3)2 5.1 -N(CH3)2 -NH3 4.6 -NH2
RCOOH 4.50.5 RCOO- 2,4-Dinitrofenol
4.0 (NO2)2-O-
HCOOH 3.7 HCOO- CH2(NO2)2
3.6 -CH(NO2)2
ClCH2COOH
3.8
ClCH2COO
-
2.5
2.4
Cl2CHCOOH
1.3
Cl2CHCOO
-
1.0
2-NH2
1.0
2-NH
Cl3CCOOH 0.9 Cl3CCOO- 2,4,6-Trinitrofenol
0.4
(NO2)3O-
CF3COOH 0 CF3COO- CH3CONH3 0.3
CH3CONH2

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
25
HNO3 -1.4 NO3- -CONH3 -2.0
-CONH2
CH3OH2
-2.0
CH3OH
-2.1
(CH3)2OH -3.8 (CH3)2O t-BuOH2
-4 t-BuOH
-4.0
-4.5
(CH3)2SH
-5.2
(CH3)2S
-5-5
-6.2
-OH2
-6.7
-OH (o -OR)
-6.8
-CH=OH -7.1 -CHO
-9.4
R-CNH
-10
R-CN
-11 H2SO4 ? HSO4-
HBF4 ? BF4- FSO3H ?
FSO3-
HClO4 -20 ClO4- HPF6 -20
PF6-
SbF5.FSO3H -20 SbF5
.FSO3
-

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
26
HIDROCARBUROS
(NOMENCLATURA)
Los hidrocarburos y sus compuestos derivados se pueden clasificar en general en
tres grandes categorías:
1. Hidrocarburos alifáticos, formados por cadenas de átomos de carbono en las que no
hay estructuras cíclicas. Se les denominan en general, hidrocarburos de cadena
abierta o acíclicos.
CH3-CH2-CH3
propano
CH3-CH2-CH2-CH2-CH3
pentano
CH3-CH=CH-CH3
2-buteno
2. Hidrocarburos alicíclicos, o simplemente cíclicos, compuestos por átomos de
carbono encadenados formando uno o varios anillos.
ciclobutano
ciclopentano
biciclo 4,4,0 decano
3. Hidrocarburos aromáticos, que constituyen un grupo especial de compuestos
cíclicos que contienen en general anillos de seis eslabones en los cuales alternan
enlaces sencillos y dobles. Se clasifican, independientemente de los hidrocarburos
alifáticos y alicíclicos, por sus propiedades físicas y químicas muy características.
benceno
naftaleno
fenantreno
FUENTES Y APLICACIONES
El petróleo y los gases naturales a él asociados constituyen en la actualidad la
principal fuente de hidrocarburos. A medida que las reservas de petróleo se van
agotando, sin embargo, cobra creciente interés la posibilidad de convertir parte de las
abundantes reservas mundiales de carbón en hidrocarburos utilizables.
El gas natural se halla compuesto principalmente por metano (CH4). El etano
(C2H6) y el propano (C3H8) representan entre un 5 y un 10 por 100 del total, junto con
trazas de hidrocarburos de C4 y C5 carbonos.
El petróleo líquido es una mezcla compleja, en la que predominan los
hidrocarburos saturados. La industria del petróleo emplea procedimientos de extracción
y de destilación a gran escala para separar el “crudo” en fracciones de utilidad práctica.
Las fracciones líquidas más volátiles son el éter de petróleo (p. eb. 30 - 60 ºC) y la

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
27
ligroína (p. eb. 60 - 90 ºC), compuestos principalmente por hidrocarburos de C 5 hasta C7
átomos de carbonos. La gasolina cubre un amplio rango de compuestos entre C5 y C10
(p. eb. 40 -200 ºC), las demás fracciones importantes del petróleo son el queroseno (p.eb.
175 -325 ºC, de C8 a C14), el gasóleo (p. eb. superior a 275 ºC, de C12 a C18), los aceites
y grasas lubricantes (por encima de C18), el asfalto o coque de petróleo.
NOMENCLATURA
El problema de la nomenclatura de las moléculas orgánicas ha acompañado a la
química orgánica desde sus mismos orígenes y ha empeorado con la multitud de nuevos
compuestos y clases de compuestos que se descubren cada año. Hay que sumar a ésto el
problema de las distintas lenguas (alemán, francés, ingles, danés, español, etc..).
Algunos compuestos han recibido el nombre de sus descubridores. Los nombres de
lugares también han sido utilizados. Bastante nombres de moléculas reflejan la forma de
la misma. Todos estos nombres de moléculas tienen primordialmente interés histórico
aunque muchos se usan todavía comúnmente (nombres comunes), especialmente
cuando el nombre sistemático que describe su estructura es complejo.
La nomenclatura sistemática se introdujo por vez primera en un congreso
químico en Ginebra (Suiza) en 1892 y ha sufrido revisiones continuas desde entonces,
especialmente por la Unión Internacional de Química Pura y Aplicada (IUPAC). Las
reglas de nomenclatura se denominan “reglas de la IUPAC”.
NOMENCLATURA IUPAC PARA LOS HIDROCARBUROS:
Tal como comentamos al principio, los hidrocarburos se clasifican en tres
grandes grupos, en primer lugar veremos las reglas para los hidrocarburos alifáticos, si
el estudiante es capaz de comprenderlas para ellos, el resto es simplemente una
ampliación de las mismas.
Hidrocarburos Alifáticos:
Si bien es cierto que son cadenas lineales de átomos de carbonos, ellos pueden
subdividirse en tres grandes grupos: ALCANOS, ALQUENOS y ALQUINOS.
Alcanos:
Los nombres de los alcanos lineales se dan en la Tabla 3.1. Las raíces
son mayoritariamente latinas o griegas e indican el número de átomos de carbono en la
cadena. Por ejemplo, el nombre heptadecano está formado por la voz griega hepta, siete
y la palabra latina decano, diez. Los cuatro primeros alcanos tienen nombres especiales
que han sido aceptados en el sistema de la IUPAC pero mantienen la terminación “ano”.
TABLA 3.1.-
Todos los alcanos acíclicos obedecen a la fórmula general CnH2n+2.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
28
Fórmula estructural Nombre Fórmula condensada
CH4 metano CH4
CH3-CH3 etano C2H6
CH3-CH2-CH3 propano C3H8
CH3-CH2-CH2-CH3 butano C4H10
CH3-(CH2)3-CH3 pentano C5H12
CH3-(CH2)4-CH3 hexano C6H14
CH3-(CH2)5-CH3 heptano C7H16
CH3-(CH2)6-CH3 octano C8H18
CH3-(CH2)7-CH3 nonano C9H20
CH3(CH2)8-CH3 decano C10H22
La Tabla 3.1., debe ser estudiada con atención ya que sirve de base para la
nomenclatura de un gran número de moléculas orgánicas. Los nombres de los términos
superiores de la serie son los siguientes:
10 decano 20 eicosano 30 triacontano
11 undecano 21 heneicosano 31 hentriacontano
12 dodecano 22 docosano 32 dotricontano
13 tridecano 23 tricosano 33 tritriacontano
14 tetradecano 24 tetracosano 34 tetratriacontano
15 pentadecano 25 pentacosano 35 pentatriacontano
16 hexadecano 26 hexacosano 36 hexatriacontano
17 heptadecano 27 heptacosano 37 heptatriacontano
18 octadecano 28 octacosano 38 actatriacontano
19 nonadecano 29 nonacosano 39 nonatriacontano
40 tatracontano 50 pentacontano 60 hexacontano
41 hentetetracontano 51 henpentacontano 61 henhexacontano
42 dodetetracontano 52 dopentacontano 62 dohexacontano
43 tritetracontano 53 tripentacontano 63 trihexacontano
44 tetratetracontano 54 tetrapentacontano 64 tetrahexacontano
etcétera etcétera etcétera
70 heptacontano 80 octacontano 90 nonacontano
100 hectano 200 dihectano 300 trihectano
Los sustituyentes de tipo alcano se nombran reemplazando la terminación -ano
por -ilo. Algunos homólogos inferiores de los alcanos ramificados tienen nombres
comunes muy usados que utilizan los prefijos -iso y -neo, como es isobutano,
isopentano, neohexano.
CH3- metilo CH3-CH2- etilo CH3-CH2-CH2- propilo

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
29
isopropilo
Es un isoalcano
(ej. n = 1, isopentano)
Es un neoalcano
(ej. n = 1, neohexano)
Los alcanos lineales suelen corresponder a la fórmula general CnH2n+2 , y
sólamente en el caso de ser absolutamente lineal (no ramificado) reciben el nombre de
la Tabla 3.1. Los alcanos ramificados derivan de los sistemas de cadena lineal por
eliminación de un átomo de hidrógeno de un grupo metileno y sustitución por un grupo
alquilo. Tienen la misma fórmula empírica que los alcanos lineales, CnH2n+2 . El
ejemplo más sencillo es el 2-metilpropano, C4H10, con la misma fórmula molecular que
el butano. Por lo tanto ambos compuestos son isómeros.
2-metilpropano
butano
Para homólogos superiores (n4) de los alcanos son posibles más de dos
isómeros, hay tres C5H12, hay cinco C6H14, nueve C7H16. En la Tabla 3.2, se observa el
número de posibles alcanos isómeros CnH2n+2 , el número de posibilidades para conectar
n átomos de carbono entre si y con los 2n+2 átomos de hidrógenos que los envuelven
aumenta dramáticamente con el valor de n.
TABLA 3.2
Número de posibles alcanos isómeros CnH2n+2.
n isómeros
1 1
2 1
3 1
4 2
5 3
6 5
7 9
8 18
9 35
10 75
15 4347
20 366319
Reglas para nombrar a los alcanos remificados:
Para nombrar un hidrocarburo ramificado tal como el siguiente:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
30
se considera que hay un grupo o radical metilo (CH3-) unido una cadena de heptano. El
compuesto es así un metilheptano. (Se prescinde de la “o” final de metilo) Sin embargo,
hay que indicar dónde se encuentra el metilo, ya que los compuestos,
son también metilheptanos. Para determinar la posición del metilo se numera la cadena
más larga y se escribe y menciona dicho número, llamado localizador, delante del
nombre, Así, los tres compuestos anteriores se llaman:
2-metilheptano
3-metilheptano
4-metilheptano
Regla Nº 1:
La cadena más larga se numera de un extremo a otro, de tal forma que se asigne
los números más bajos a los carbonos con cadenas laterales, independientemente
de la naturaleza de los sustituyentes.
Veamos las siguientes moléculas:
Para la primera, la cadena más larga es
de siete carbonos, con dos sustituyentes
metilo en los carbonos C3 y C5, por lo
tanto su nombre correcto es:
3,5-dimetilheptano.
En el segundo ejemplo la cadena más
larga es de cinco carbonos, con tres
sustituyentes metilo en los carbonos C2 y
C4, por lo que su nombre correcto es:
2,2,4-trimetilpentano
Regla Nº 2:
Si en la cadena más larga un sustituyente se repite más de una vez, éstos se
nombran con los prefijos de cantidad di, tri, tetra, penta, hexa, (solamente son
validos para sustituyentes sencillos).
Como consecuencia del segundo ejemplo, nace la siguiente regla.
Regla Nº 3:
Si en un mismo carbono existe más de una vez el mismo sustituyente, el
numero localizador se repite tantas veces como sustituyentes soporte.
Analicemos los siguientes casos:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
31
En este caso la cadena más larga es de
11 carbonos, por lo que el compuesto base
es el undecano, los sustituyentes son el
grupo etilo y el grupo propilo, la
numeración correcta ha de venir por el
lado del etilo dado que asigna valores más
bajos (C4 y C5), si se numerase desde el
lado del propilo tendríamos (C8 y C9), lo
que no esta de acuerdo con la regla Nº 1.
Por lo que el nombre correcto es:
4-etil-5-propilundecano
La cadena base sigue siendo el
undecano y los sustituyentes el grupo etilo
y el grupo propilo, la numeración viene
dado ahora por el grupo propilo dado que
asigna valores más bajos. La única
diferencia en el nombre va a radicar en la
posición de los sustituyentes.
Su nombre correcto sería:
5-etil-4-propilundecano
Regla Nº 4:
Los sustituyentes en un compuesto ramificado se nombran por orden
alfabético, independientemente de la numeración de los mismos, en el caso de
repetirse uno de ellos más de una vez los prefijos de cantidad no se han de
considerar para el orden alfabético.
Hay algunos compuestos que tienen un nombre común aceptado. Estos son
isobutano, isopentano, neopentano e isohexano. Dichos compuestos pueden nombrarse,
pues, de dos formas distintas:
metilpropano o isobutano
2-metilpentano o isohexano
2-metilbutano o isopentano
dimetilpropano o neopentano
Vamos a considerar ahora otros radicales más complejos, radicales que podemos
suponer que proceden de alcanos ramificados. El modo de nombrarlos es tal como se
indica en los siguientes ejemplos:
3-metilbutilo
2-metilbutilo

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
32
1-metilbutilo
2,4-dimetilpentilo
1-etil-1-metilbutilo
4,4-dimetil-2-propilhexilo
Hay unos cuantos radicales que tienen un “nombre propio” admitido por la
IUPAC, estos deben usarse cuando corresponda:
Isopropilo
Isopentilo
Isobutilo
terc-Pentilo
sec-Butilo
Neopentilo
terc-Butilo
Isohexilo
Ejemplo:
4-etil-5-isopropil-3-metil-7-propilundecano
(secuencia: etil, isopropil, metil, propil)
5-(1,2-dimetilpropil)-4-etil-3-metilnonano
(secuencia: dimetilpropil, etil, metil)
Regla Nº 5:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
33
Si en un compuesto hay dos radicales simples y uno complejo, este último se
nombra primero, y a continuación los simples en orden alfabético. En el caso de
que existan dos radicales complejos éstos se nombran en orden alfabético entre sí,
y si éstos tuviesen las mismas palabras se citará en primer lugar aquel que tenga el
número localizador más bajo.
Procedamos a nombrar los siguientes compuestos:
8-(2-metil-1-propilpentil)-6-(1,2,2,3-tetrametilbutil)-tetradecano
6,6,9-trietilpentadecano
(con tres sustituyentes etilo)
6,6,9-tris-(1,1,2-trimetilbutil)-pentadecano
(con tres sustituyentes complejos (1,1,2-trimetilbutil))
Regla Nº 6:
Cuando hay dos o más radicales complejos iguales, para evitar confusiones
con los prefijos sencillos di, tri, tetra, etc., se usan para éstos entonces los prefijos
bis, tris, tetraquis, pentaquis, etc.
Finalmente la regla Nº 7 contempla el caso de que haya varias cadenas de igual
longitud. ¿Cómo se elige entonces la cadena principal? Se toma como principal:
Regla Nº 7:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
34
a) La cadena que tenga el mayor número de cadenas laterales
b) La cadena cuyas cadenas laterales tengan los localizadores más bajos
c) La cadena que tenga el máximo número de átomos de carbonos en las
cadenas laterales más pequeñas.
d) La cadena que tenga cadenas laterales lo menos ramificadas posible.
Alquenos:
Los hidrocarburos que poseen un doble enlace, se nombran cambiando la
terminación -ano (del alcano de igual número de átomos de carbono) por la
terminación -eno.
CH2=CH2
eteno o etileno
CH3-CH=CH2
propeno
1-buteno
2-buteno
Los alquenos suelen responder a la fórmula general CnH2n, siempre y cuando no
sean cíclicos.
La posición del doble enlace o insaturación se indica mediante el
correspondiente localizador. se procura asignar al doble enlace un localizador tan bajo
como sea posible. Si hay ramificaciones se toma como cadena principal la cadena más
larga de las que contenga el doble enlace; es decir el doble enlace tiene primacía sobre
las cadenas laterales en el momento de numerar y elegir la cadena principal.
Ejemplos:
4,5-dimetil-1-hepteno
3-etil-6-metil-2-hepteno
5-etil-3-hepteno
5,6-dimetil-3-hepteno
ISOMERIA CIS - TRANS ó (Z - E) EN LOS ALQUENOS
Cuando dos átomos de carbonos se enlazan mediante un doble enlace, la
estructura resultante es rígida y se generan dos planos diferentes, en el espacio. Así
cuando uno escribe 2-buteno, hay que ser cauteloso dado que existen dos compuestos
distintos, aunque isómeros entre sí, estructuralmente son:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
35
cis-2-buteno
trans-2-buteno
Los dos hidrógenos al mismo lado del
espacio.
Los dos hidrógenos a lados distintos en el
espacio
Analicemos los siguientes ejemplos:
A)
B)
C)
D)
A y B no cabe duda que son el cis y el trans-3,4-dimetil-3-hepteno
respectivamente, obsérvese que en el cis, los dos grupos metilos están a un mismo lado
y en el trans los dos grupos metilos a lados distintos.
C y D sin embargo presentan un problema ¿Cuál es el cis? y ¿Cuál es el trans?
no hay grupos iguales en torno al doble enlace. Los casos como éste han obligado a
introducir una nomenclatura más general ( Z - E ) , que sirva para todos los alquenos.
Este sistema de nomenclatura se basa en comparar la posición relativa de los “grupos
preferentes” a uno y otro lado del doble enlace. La preferencia se establece por el
número atómico: los átomos que están unidos a un carbono determinado del doble
enlace se comparan entre sí; si dichos átomos son iguales se comparan los siguientes
que están unidos y así respectivamente, hasta encontrar la diferencia.
E-1-bromo-1-cloro-propeno Z-1-bromo-1-cloro-propeno
Los átomos más pesados a lados
contrarios del doble enlace
Los átomos más pesados a un mismo
lado del doble enlace
Por lo tanto en los ejemplos anteriores los compuestos C y D se han de nombrar:
(C) Z-3-etil-2,4-dimetil-3-hepteno (D) E-3-etil-2,4-dimetil-3-hepteno

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
36
DIENOS Y POLIENOS:
Cuando un hidrocarburo contiene más de un doble enlace se emplea para
nombrarlo la terminación -adieno, -atrieno, -atetraeno, etc., en lugar de la terminación
-eno. Proceden al nombre los números localizadores de dichos doble enlaces. Los hay
de distintos tipos de polienos:
Clasificación Ejemplo 1 Ejemplo 2
Con dobles enlaces
acumulados
CH2=C=CH2
Aleno
CH2=C=CH-CH2CH3
1,2-pentadieno
Con dobles enlaces
conjugados
CH2=CH-CH=CH2
1,3-butadieno
CH2=CH-CH=CH-CH3
1,3-pentadieno
Con dobles enlaces no
conjugados
1,4-ciclohexadieno
CH2=CH-CH2-CH=CH2
1,4-pentadieno
Alquinos:
Son hidrocarburos con un enlace carbono-carbono triple, obedecen a la
fórmula general CnH2n-2, los triples enlaces son lineal en su disposición espacial. Esta
disposición lineal impide, que pueda obtenerse en la práctica un triple enlace sobre un
anillo pequeño.
H-CC-H etino o acetileno
CH3-CC-H propino
CH3-CH2-CC-H 1-butino
CH3-CC-CH3 2-butino
En el caso de que en un compuesto existan dos o más enlaces triples, estos se
nombran con la terminación -diino, -triino, etc.
3-propil-1,5-heptadiino
10-isopropil-9,9-dimetil-1,4,7,11-dodecatetraino

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
37
Con estos ejemplos queda claro que la numeración de la cadena principal la
mandan los triples enlaces, la que a su vez es aquella que contiene a la mayor cantidad
de triples enlaces.
HIDROCARBUROS CON DOBLES Y TRIPLES ENLACES
Para nombrarlos hay que enunciar tanto el número de dobles enlaces como el de
triples: si hay dos enlaces dobles y uno triple, será un dieno-ino; si hay tres enlaces
dobles y dos triples será un trieno-diino; etc.
Para numerar la cadena principal se procura que recaigan los números más bajos
en las insaturaciones (enlaces dobles y triples), prescindiendo de considerar si son
dobles o triples:
HCC-CH2-CH2-CH=CH-CC-H
3-octeno-1,7-diino
Si se empieza a numerar por la izquierda,
las insaturaciones están localizadas en
1,5,7; si por la derecha, en 1,3,7 (esta
última posición es la preferida).
CH3-CC-CH2-CH=CH-CH=CH-CH2-CH3
5,7-decadien-2-ino
Si se empieza a numerar por la izquierda,
las insaturaciones están en 2,5,7, si por la
derecha, en 3,5,8. La primera posición es la
que debe emplearse.
El problema se plantea cuando, tanto si se empieza a numerar por la izquierda
como por la derecha, los localizadores de las insaturaciones coinciden. En este caso se
da preferencia a los dobles enlaces sobre los triples, en el sentido de que se asigna a
los dobles enlaces los localizadores más bajos.
CH2=CH-CCH 1-buten-3-ino (no 3-buten-1-ino)
8-etil-1,3,8-nonatrien-6-ino
(no 2-etil-1,6,8-nonatrien-3-ino)
4,9,9-trimetil-3-deceno-5,7-diino
(no 2,27-trimetil-7-deceno-3,5-
diino)
RADICALES ALQUENILO Y ALQUINILO
Consideremos el siguiente compuesto:
Junto a dos sustituyentes o radicales alquilo, hay un grupo (-CH=CH-CH3) que
desempeña una función análoga pero que contiene una insaturación. Los grupos o

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
38
radicales univalentes de este tipo adoptan las terminaciones -enilo o -inilo (en o in por
el doble o triple enlace, ilo por tratarse de un radical).
Obsérvese el nombre de los siguientes radicales:
CH2=CH- vinilo (etenilo) CHC- etinilo
CH3CH=CH- 1-propenilo CH3-CC- 1-propinilo
CH2=CH-CH2- alilo (2-propenilo) CHC-CH2- 2-propinilo
HIDROCARBUROS ALICÍCLICOS O CÍCLICOS:
Los hidrocarburos cíclicos se nombran añadiendo el prefijo ciclo- al nombre del
alcano equivalente de cadena abierta.
ciclopropano C3H6
ciclobutano C4H8
ciclopentano C5H10
ciclohexano C6H12
Por ejemplo:
A B C
Resulta más sencillo nombrarlo como derivados de un cicloalcano que no como
derivados de un compuesto de cadena abierta:
A) 1-butil-1-terc-butil-4,4-dimetilciclohexano
B) 1,1,2-trimetilciclopentano
C) 1-etil-1,2,2-trimetilciclopropano

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
39
En cambio, los compuestos siguientes:
1,4-diciclohexil-2-metilbutano 3-ciclopentil-2-ciclopropil-6-metilheptano
Es mejor nombrarlos como derivados de un alcano de cadena abierta.
Los anillos son estructuras que generan planos, por lo tanto dan lugar a la
formación de isómeros espaciales (cis - tras), así por ejemplo, el 1,2-
dimetilciclopentano existen dos compuestos diferentes:
Hay que indicar la diferencia existente entre uno y otro. El de la izquierda tiene
los dos grupos metilos por encima del plano del anillo; el de la derecha, un metilo por
encima y otro por debajo (se acostumbra en señalar con trozo grueso los grupos que
están por encima del plano teórico del ciclo, y en líneas de puntos o rayas los que están
por debajo). Al primer isómero se le añade el prefijo cis, y al segundo el prefijo trans.
Así, los compuestos se llaman, respectivamente:
cis-1,2-dimetilciclopentano
trans-1,2-dimetilciclopentano
Algunos compuestos cíclicos particulares:
1,1’-biciclopropano (biciclopropano)
1,1’-biciclohexano (biciclohexano)
1,2’-dimetil-1,1’-biciclopropano
(1,2’-dimetilbiciclopropano)
HIDROCARBUROS CÍCLICOS CON CARBONOS PUENTES:
Los siguientes alcanos son ejemplos de hidrocarburos con carbonos puente:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
40
El primero es un pentano (tiene 5 átomos de carbono), el segundo un heptano
(tiene 7 átomos de carbono) y el tercero un octano (tiene 8 átomos de carbono). Para
nombrar estos hidrocarburos se añade el prefijo biciclo antes del nombre del alcano, y
se indica entre corchetes el número de átomos de carbono de mayor a menor que hay
ente los cabeza de puente.
biciclo2,1,0pentano
biciclo2,2,1heptano
biciclo2,2,2octano
La forma de representarlos comúnmente es:
Así por ejemplo:
biciclo3,2,1octano biciclo4,3,0nonano biciclo3,3,1nonano
En los sistemas biciclos con sustituyentes o grupos funcionales hay que indicar
la posición de éstos. Para numerar el sistema se empieza por una cabeza de puente y se
continúa por la cadena más larga hacia el otro carbono cabeza de puente, se vuelve
luego por la cadena intermedia y se acaba numerando la más corta. Ejemplo:
cadena larga: 2,3,4
cadena intermedia: 6,7
cadena corta: 8
De acuerdo con lo anterior, los nombres sistemáticos de los compuestos son los
siguientes:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
41
1,8,8-trimetilbiciclo3,2,1octano
5-etil-2,2-dimetilbiciclo2,2,2octano
2,2,6,6-tetrametilbiciclo3,1,1heptano
Obsérvese que, como siempre, se procura que los localizadores, dentro de las
limitaciones impuestas por la regla anterior, sean lo más pequeños posible.
ESPÍRANOS:
Existen unos compuestos bicíclicos en los que un único átomo de carbono forma
parte al mismo tiempo de los dos ciclos. Estos compuestos se conocen con el nombre de
hidrocarburos espiránicos. Para nombrarlos se indica el número de átomos de carbono a
uno y otro lado del carbono espiránico se indican entre corchetes, de menor a mayor.
Ejemplos:
espiro3,4octano espiro2,5octano
Los compuestos cíclicos simples pueden presentar dobles enlaces al interior de
la molécula, en tal caso se nombran con la terminación -eno, y la numeración en tal caso
la manda el doble enlace, en el caso de haber más de uno entonces se nombran con la
terminación -dieno (2 dobles enlaces); -trieno (3 dobles enlaces); etc. En todo caso
tratando de darle los valores más bajos a los localizadores. Analicemos los siguientes
ejemplos:
3-metilciclohexeno
(no es necesario indicar la posición 1 del doble enlace)
5-etil-2-metil-1,3-ciclohexadieno
5-etil-1,7-dimetil-1,3,6-cicloctatrieno
HIDROCARBUROS AROMÁTICOS:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
42
El benceno es un hidrocarburo peculiar ya que, a pesar de que parece un polieno,
su reactividad es menor que la de los alquenos. Se cree que la particular disposición de
los dobles enlaces alternados o conjugados unos con otros y en un ciclo, es la causa de
esta estabilidad inesperada del benceno y de sus derivados.
Benceno (C6H6)
Como muchos de los primitivos derivados del benceno aislados de las plantas
tenían fuerte aroma, se utilizaba la expresión “hidrocarburos aromáticos” para
distinguirlos. Cuando los químicos se dieron cuenta de lo que definía mejor a los
derivados y análogos del benceno era esa estabilidad de la que estamos hablando, se
siguió empleando el término aromaticidad, pero en un doble sentido. Hoy en día, sin
embargo, cuando en Química se dice “un compuesto es aromático”, uno se está
refiriendo a que ese compuesto es más estable de lo esperado, y la expresión no tiene
nada que ver con su buen o mal olor.
Nomenclatura:
Los sustituyentes que pueda haber sobre un anillo bencénico se mencionan
como radicales anteponiéndolos a la palabra benceno.
etilbenceno sec-butilbenceno vinilbenceno
Cuando hay dos sustituyentes, su posición relativa puede indicarse mediante los
números localizadores 1,2-; 1,3- o 1,4-, o mediante los prefijos o- (orto), m- (meta),
o p- (para):
1-etil-2-metilbenceno
o-etilmetilbenceno
1-etil-3-propilbenceno
m-etilpropilbenceno
1,4-dimetilbenceno
p-dimetilbenceno
Si hay tres o más sustituyentes, se procura que reciban los números más bajos
posibles, y en caso de que existan varias opciones la decisión se basará, como norma
general, en el orden de preferencia de los distintos radicales.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
43
2-etil-1-metil-4-propilbenceno 5-alil-1-isopropil-2,3-dimetilbenceno
1-etil-3-(2-metil-3-butenil)benceno 1-terc-butil-4-(1-butinil)-2-etilbenceno
1-butil-5-ciclopentil-2-etil-4-(1-propenil)benceno

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
44
GRUPOS FUNCIONALES
(NOMENCLATURA)
RESUMEN DE LAS PRICIPALES FUNCIONES ORGÁNICAS
Función Grupo Ejemplo Terminación Sustituyente
Alcanos -C-C- CH3-CH3
propano
-ano ....il
Alquenos -C=C- CH2=CH2
propeno
-eno ....enil
Alquinos -CC- CHCH
propino
-ino ....inil
Hidrocarburos
aromáticos benceno
nombre no
sistemático
nombre no
sistemático
acabados en ...il
Derivados
halogenados
R-X CH3CH2CH2Cl
1-cloropropano (cloruro de propilo)
haluro de ...ilo fluoro-
cloro-
bromo-
iodo-
Alcoholes R-OH CH3CH2-OH
etanol
....ol hidroxi-
Fenoles -OH
fenol
nombre no
sistemático
acabados en -
ol
-
Éteres R-O-R CH3-O-CH3
dimetileter
éter ....iloxi- (alcoxi)
Aldehídos R-CHO CH3CH2CHO
propanal
-al formil (-CHO)
Cetonas R-CO-R CH3COCH3
propanona
-ona ....oxo
Ácidos R-COOH CH3CH2COOH -oico carboxi-

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
45
carboxílicos ácido propanoico
Ésteres R-COOR CH3COOCH3
etanoato de metilo
-ato de ....ilo ......iloxicarbonil (-
COOR)
....oiloxi (-OCOR)
Anhídridos R-CO-O-CO-
R
(CH3CO)2O
anhídrido etanoico
anh......oico -
Haluros de
ácido
R-COX CH3COCl cloruro de etanoilo
haluro
de ...oílo
haloformil
(-COX)
Aminas R-NR2 CH3CH2NH2
etanoamina
-amina amino-
Nitrilos o
cianuros
R-CN CH3CH2CN
propanonitrilo
ó
cianuro de etilo
-nitrilo ciano-
Amidas R-CO-NR2 CH3CONH2
etanoamida
-amida amido
DERIVADOS HALOGENADOS:
Se incluye en este apartado todos aquellos hidrocarburos que contienen en su
molécula un átomo de halógeno.
El método que se utiliza con mayor frecuencia para nombrarlos consiste en citar
el nombre del halógeno (fluoro, cloro, bromo, yodo) precediendo al de la molécula
carbonada, he indicando el número localizador. También es aceptable citar el
compuesto como un “haluro de alquilo” (Nomenclatura Función-Radical). Así:
CH3-CH2-CH2-Cl 1-cloropropano
cloruro de propilo
CH3-CHCl-CHCl-CH3 2,3-diclorobutano
-
(CH3)3C-Cl 2-cloro-2-metilpropano
cloruro de terc-butilo
o-diclorobenceno
CH2Br-CH2Br 1,2-dibromoetano
dibromuro de etileno
CH3-CH=CH-CHCl-CH3 4-cloro-2-penteno

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
46
ALCOHOLES, FENOLES y ÉTERES:
Son compuestos de carbono, hidrógeno y oxígeno en los que el oxígeno está
unido por enlaces sencillos a la cadena carbonada.
Un alcohol puede relacionarse con una molécula de agua en la que en lugar de
un H hay un radical alquilo, alquenilo o alquinilo (R); si en lugar de un H hay un
radical fenilo (en general, un radical arilo), se trata de un fenol; si en lugar de los dos H
hay dos radicales, tendremos un éter:
H-O-H Agua
R-O-H Alcohol
Ar-O-H Fenol
R-O-R’
R-O-Ar éter
Ar-O-Ar’
ALCOHOLES:
Para nombrar los alcoholes tenemos dos alternativas principales. En la primera,
llamada Nomenclatura sustitutiva, se considera que se ha sustituido un H de un
hidrocarburo por un OH. Al alcohol se le nombra entonces añadiendo la terminación -
ol al hidrocarburo de referencia.
CH3-CH2-CH3 propano ; CH3-CH2-CH2-OH propanol
El segundo sistema de nomenclatura, igualmente válido, consiste en citar
primero la función y luego el radical como si fuera un adjetivo.
CH3-CH2-CH2- radical propilo ; CH3-CH2-CH2-OH alcohol propílico
Ejemplos:
CH3OH metanol alcohol metílico
CH3-CH2OH etanol alcohol etílico
CH3CH2-CH3 1-propanol alcohol propílico
CH3CHOH-CH3 2-propanol alcohol isopropílico
CH3-CH2-CH2-CH2-OH 1-butanol alcohol butílico
CH3CH2CH(OH)CH3 2-butanol alcohol sec-butílico
Hay casos en que, por la complicación del compuesto, el primer sistema de
nomenclatura resulta más idóneo, por lo que es el único empleado. Veamos los
siguientes ejemplos:
CH3-CH2-CH=CH-CH2CH2-OH 3-hexen-1-ol
CH3-CH=CH-CH2-CH(OH)-CH3 4-hexen-2-ol

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
47
3-hexen-5-in-1-ol
4-metil-2,4-hexadien-1-ol
4-metilciclohexanol
3-ciclopenten-1-ol
1,3-propanodiol
1,2-propanodiol
Habrá observado que la función alcohol tiene “preferencia” frente a las
insaturaciones y radicales: al numerar la cadena se asigna al carbono unido al -OH el
número más bajo posible; por otro lado, el sufijo -ol, por corresponder al grupo
principal, es el último en citarse.
Cuando en un compuesto hay varios tipos de funciones, se plantea el problema
de elegir cuál es la principal. La IUPAC, lo ha solucionado estableciendo una lista de
las diversas funciones, ordenadas según un criterio convencional de preferencia (ver
TABLA 4.2 de las preferencias).
Comentemos algunos ejemplos:
CH3-CH2-CH2-CHOH-CH2-COOH ; ácido 3-hidroxihexanoico
CH3-CH2CHOH-CHO ; 2-hidroxibutanal
En resumen:
Cuando el grupo -OH actúa como función principal sufijo -ol
Cuando el grupo -OH interviene como sustituyente prefijo hidroxi
FENOLES:
Para nombrar los fenoles se utiliza generalmente, como en los alcoholes, la
terminación -ol. En la mayoría de los casos esta terminación se añade al nombre del
hidrocarburo aromático:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
48
fenol
resorcinol
hidroquinona
p-cresol
2-naftol
ÉTERES:
Hay dos sistemas fundamentales para nombrar los éteres. Ambos se especifican
a continuación:
Nomenclatura Sustitutiva Nomenclatura Radicofuncional Ejemplo
metoxietano
etil metil éter
CH3-O-CH2CH3
etoxietileno
etil vinil éter
CH2=CH-O-CH2CH3
metoxibenceno
fenil metil éter
C6H5-O-CH3
Los éteres cíclicos se nombran como oxaciclo............, ejemplos:
oxaciclopropano oxaciclobutano oxaciclopentano oxaciclohexano
ALDEHÍDOS y CETONAS:
Aldehído y cetonas se caracterizan por tener un doble enlace carbono-oxígeno
(grupo carbonilo) en su estructura. La diferencia entre aldehído y cetonas reside en que
en los primeros ese grupo carbonilo se encuentra en el extremo de la cadena carbonada:
Fórmula general de los aldehídos R-CHO
Fórmula general de las cetonas R-CO-R’
ALDEHÍDOS:
Para nombrarlos se emplea la terminación -al. en los ejemplos siguientes se
indica, junto al nombre sistemático, el nombre trivial aceptado en algunos aldehídos.
metanal
formaldehído
etanal
acetaldehído

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
49
propanal propionaldehído
butanal butiraldehído
propenal acrilaldehído (acroleína)
3-fenilpropenal
cinamaldehído
2,3-dihidroxipropanal
gliceraldehído
4-pentanal
2-metil-3,5-hexadienal
3-octen-6-inal
Obsérvese que en todos los ejemplos anteriores se empieza a numerar por el
extremo en que se encuentra el carbonilo ya que el grupo -CHO tiene preferencia sobre
radicales dobles y triples enlaces, y grupos -OH. Sólo cuando en los dos extremos de la
cadena hay grupos aldehído se tienen en cuenta los otros grupos para decidir por dónde
se empieza a numerar.
Cuando en un compuesto hay otras funciones que tienen prioridad sobre la
función aldehído para ser citadas como grupo principal se utiliza el prefijo formil- para
designar al grupo -CHO, al que se le considera entonces como un sustituyente. Por
ejemplo:
ácido 3-formilpentanodioico
El prefijo formil- también se emplea cuando hay tres o más funciones aldehído
sobre el mismo compuesto. En esos casos se puede asimismo utilizar otro sistema de
nomenclatura que consiste en dar el nombre de carbaldehído a los grupos -CHO (los
carbonos de esos -CHO no se numeran; se consideran que no forman parte de la cadena
principal). Este último sistema es el idóneo para compuestos con -CHO unidos
directamente a ciclos. Veamos unos cuantos ejemplos:
3-formilpentanodial o
1,2,3-propanotricarbaldehído
3,6-diformiloctanodial

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
50
ciclopentanocarbaldehído
2,3-naftalenodicarbaldehído
benzaldehído (bencenocarbaldehído)
CETONAS:
Para nombrar las cetonas, o compuestos carbonílicos no terminales, puede
utilizarse la nomenclatura sustitutiva o la radicofuncional, tal como veremos en los
siguientes ejemplos:
Ejemplo Nom. sustitutiva Nom. radicofuncional
propanona
dimetil cetona (acetona)
butanona
etil metil cetona
2-pentanona
metil propil cetona
3-pentanona
dietil cetona
3-buten-2-ona
metil vinil cetona
5-heptin-3-ona
2-butinil etil cetona
ciclohexanona
2-ciclopentenona
ciclohexil ciclopentil
cetona

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
51
difenil cetona
2,4-pentanodiona
1,3-ciclohexanodiona
Se observa claramente en los ejemplos que la numeración la manda el grupo
carbonilo sobre radicales, dobles y triples enlaces, como así mismo sobre el grupo -OH,
en cambio frente a los aldehídos no tienen prioridad, se han de considerar como
sustituyentes, en tal caso se utiliza prefijo oxo-.
ÁCIDOS CARBOXÍLICOS Y DERIVADOS:
Los grupos funcionales que trataremos en este apartado son los ácidos
carboxílicos, los ésteres, los anhídridos de ácido y los haluros de ácido.
ácidos carboxílicos: RCOOH o RCO2H
ésteres: RCOOR’ o RCO2R’
anhídridos: RCO-O-COR o (RCO)2O
haluros de ácido: RCOX
ÁCIDOS CARBOXÍLICOS:
Los ácidos carboxílicos se nombran con la terminación -oico que se une al
nombre del hidrocarburo de referencia. Así:
CH3-CH2-CH3 propano CH3-CH2-COOH ácido propanoico
También puede utilizarse otro sistema, aunque su aplicación se limita
preferentemente a poliácidos y ciclos, que consiste en suponer desglosada la molécula
en un grupo -COOH (grupo carboxilo) y un resto carbonado. Así:
CH3-CH3 etano CH3-CH2-COOH ácido etanocarboxílico
Otros ejemplos de ácidos carboxílicos:
Ejemplo Nombre sistemático Nombre trivial
H-COOH
ác. metanoico
ác. fórmico
CH3-COOH
ác. etanoico
ác. acético
CH3-CH2-COOH
ác. propanoico
ác. propiónico

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
52
CH2=CH-COOH
ác. propenoico
ác. acrílico
CHC-COOH
ác. propinoico
ác. propiólico
CH3-CH=CH-COOH
ác. 2-butenoico
ác. crotónico
CH3-CH=CH-CH2-CH2-COOH
ác. 4-hexenoico
CH3-CC-CH2-CH2-COOH
ác. 4-hexinoico
Veamos a continuación unos cuantos ejemplos de compuestos con dos grupos COOH:
Ejemplo nombre sistemático nombre trivial aceptado
HOOC-COOH
ác. etanodioico ác. oxálico
HOOC-CH2-COOH ác. propanodioico ác. malónico
HOOC-(CH2)2-COOH ác. butanodioico ác. succínico
HOOC-(CH2)3-COOH ác. pentanodioico ác. glutárico
ác.cis-2-butenodioico
o cis-etenodicarboxílico
ác. maleico
ác. trans-2-butenodioico
o trans-etenodicarboxílico
ác. fumárico
ác. o-bencenodicarboxílico
ác- ftálico
Veamos un ejemplo de una molécula que contiene tres o más grupos COOH, existen
dos formas de nombrarla:
ác. 3-carboxi-2-metilhexanodioico ác. 1,3,4-pentanotricarboxílico
SALES:
Los aniones de los ácidos carboxílicos se nombran reemplazando la
terminación -ico del ácido por la terminación -ato.
Ejemplos:
ácido anión sal
CH3-COOH (AcOH)
ác. acético
CH3-COO- (AcO
-)
ión acetato
CH3-COONa (AcONa)
acetato de sodio

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
53
(CH3)3CH-CH2-COOH
ác. 3-metilbutanoico
(CH3)3CH-CH2-COO-
ión 3-metilbutanoico
(CH3)3CH-CH2-COONa
3-metilbutanoato de potasio
ác. 1,3-ciclohexano-
dicarboxílico
ión 1,3-ciclohexano-
dicarboxilato
1,3-ciclohexano-
dicarboxilato de diamonilo
También se puede emplear, preferentemente en los casos complejos, el giro “sal
de sodio, o de calcio, etc...del ácido tal”.
ÉSTERES:
Los ésteres se nombran de forma análoga a las sales ya que hay cierta semejanza
entre ellos: en la sal, un átomo metálica reemplaza al H del ácido; en el éster, es una
cadena carbonada la que reemplaza al H. La diferencia entre una sal y un éster reside en
que el enlace -ONa es predominantemente iónico, y el enlace -O-CH3 es
predominantemente covalente.
ácido carboxílico
sal de ácido carboxílico
éster
Ejemplos de ésteres:
HCOOCH3 metanoato de metilo
o formiato de metilo
CH3-COO-CH2CH3 etanoato de etilo
o acetato de etilo
3-cloropentanoato de de fenilo
3-butenoato de isopropilo

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
54
propanoato de fenilo
Hay casos más complicados. Por ejemplo, aquellos compuestos en los que el
grupo éster no es el prioritario, o cuando hay más de dos grupos éster (si hay solo dos
grupos éster y están sobre la cadena principal no hay ningún problema: se nombra el
compuesto como si se tratara de una sal de un ácido dibásico). Para citar al grupo éster
en los casos complejos caben dos opciones, según que la función principal esté sobre la
porción R ó R’ de la molécula de R-COO-R’. Si “manda” R, el sustituyente -COO-R’ se
nombra como alcoxicarbonil- o ariloxicarbonil- . Si “manda” R’, el sustituyente R-
COO- se nombra como aciloxi- .
Ejemplos:
El grupo principal, de acuerdo con la Tabla de prioridades, es el -
COOH. Se trata, pues, de un ácido el 4-fenilbutanoico, que tiene un
sustituyente -COO-CH2CH3 sobre el fenilo. Dicho sustituyente se
nombra como etoxicarbonil (CO = carbonil; OCH2CH3 = etoxi).
Por lo que el nombre correcto será:
ácido 4-(2-etoxicarbonil)fenil)butanoico
Nos queda por ver un caso particular de esteres, las lactonas; las cuales son
ésteres internos. Se pueden obtener a partir de ciertos hidroxiácidos por pérdida de agua:
-metil--lactona
-lactona
ANHÍDRIDOS DE ÁCIDO:
Anhídrido de ácido equivale a decir “ácido sin agua”. Los anhídridos, en efecto,
provienen de los ácidos por pérdida de una molécula de agua entre 2 grupos carboxilo.
En general, se nombran igual que los ácidos de procedencia:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
55
2 moléculas de anhídrido etanoico
ác. acético (anhídrido acético)
2 moléculas de anhídrido propanoico
ác. propanoico
anhídrido mixto anhídrido etanoico propanoico
HALUROS DE ÁCIDO:
En los haluros de ácido un halógeno está reemplazando al -OH de ácido
carboxilico. El nombre genérico de estos compuestos es haluro de acilo. Ejemplos:
Cloruro de etanoilo (cloruro de acilo)
Fluoruro de butanoilo
Bromuro de benzoilo
Yoduro de ciclohexanocarboxioilo
COMPUESTOS NITROGENADOS:
En este apartado sólo veremos detalles de algunos de los compuestos
nitrogenados más usuales, tales como aminas, nitrilos, nitroderivados y amidas.
AMINAS Y SALES DE AMONIO:
Las aminas y sus correspondientes sales derivan del amoniaco:
amoniaco
cloruro de amonio

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
56
metilamina
cloruro de metilamonio
dimetilamina
clroruro de dimetilamonio
trimetilamina
cloruro de trimetilamonio
Aminas primarias Aminas secundarias Aminas terciarias
fenilamina o anilina
difenilamina
trifenilamina
propilamina
(propanoamina)
N-metilpropilamina
(N-metilpropanoamina)
N-etil-N-metilpropilamina
(N-etil-N-metilpropanoamina)
1,3-propanodiamina
N,N’-dimetilpropanodiamina
N,N-dietil-N’,N’-dimetil-1,3-
propanodiamina
En los casos en que hay varios grupos amina, la forma de nombrar el compuesto
depende de si los átomos de nitrógeno forman parte o no de la cadena principal. Si el
nitrógeno no forma parte de la cadena principal se citan médiente prefijos tales como
amino-, metilamino-, aminometil-, etc.
1,3,5-pentanotriamina
3-aminometil-4-metilamino-1,7-heptanodiamina
Los mismos criterios de nomenclatura que acabamos de ver se utilizan cuando
en el compuesto hay otro grupo que tiene preferencia sobre el grupo amina. Por ejemplo:
ácido 2-aminopropanoico
ácido 2-amino-3-metilbutanoico

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
57
ácido o-aminobenzoico
NITRILOS O CIANUROS:
Son compuestos orgánicos análogos al H-CN (cianuro de hidrógeno o ácido
cianhídrico) se les da el nombre genérico de nitrilos o cianuros. Hay varios sistemas de
nomenclatura para esos compuestos, los más comunes son: A) Añadir el sufijo -nitrilo
al nombre del hidrocarburo de igual número de carbonos; B) Considerarlo como un
dericado del HCN (cianuro de ......). Por ejemplo:
etanonitrilo cianuro de metilo
propanonitrilo cianuro de etilo
4-metilpentanonitrilo
cianuro de isopentilo
benzonitrilo
cianuro de fenilo
Otro sistema de nomenclatura, idónea para casos como los que se indican a
continuación, consiste en emplear el sufijo -carbonitrilo para designar el grupo -CN.
ciclohexanocarbonitrilo
(cianuro de ciclohexilo)
1,1,2,4-butanotetracarbonitrilo
Cuando hay otras funciones que tienen prioridad sobre el grupo -CN se cita éste
mediante el prefijo ciano-.
NITRODERIVADOS:
Los compuestos que contienen u grupo -NO2 se designan mediante el prefijo
nitro- (nunca se considera a dicha función como grupo principal; en otras palabras,
siempre se le nombra como derivado).
Ejemplos de nitroderivados:
nitrometano

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
58
nitropropano
nitrobenceno
1,3-dinitrobenceno
AMIDAS:
Las amidas son una clase de compuestos que podemos considerar que proceden
de sustituir el -OH de un ácido por un -NH2 (o NR2, en general):
Se nombran añadiendo el sufijo -amida al nombre del hidrocarburo de igual número de
átomos de carbono. Por ejemplo:
etanoamida (acetoamida)
butanoamida
benzamida
N-metiletanoamida (N-metilacetoamida)
N,N-dimetiletanoamida
(N,N-dimetilacetoamida)
A veces debe utilizarse la expresión -carboxamida para indicar un grupo -
CONH2. Esto sucede en los compuestos cíclicos principalmente.
ciclopentanocarboamida
N,3-dimetilciclohexanocarboamida
Finalmente, si en un compuesto hay otro grupo funcional que tiene prioridad
sobre la función amida, el grupo -CONH2 se le designa mediante el prefijo carbamoil-;
mientras que un grupo como el -NHCOCH3 recibe el nombre de acetamido-.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
59
COMPUESTOS ORGÁNICOS CON AZUFRE:
El azufre se encuentra en el mismo grupo de la Tabla Periódica que el oxígeno.
Por ello no es de extrañar que existan compuestos orgánicos con azufre análogos a los
alcoholes, aldehídos, cetonas, ácidos carboxílicos, etc. Para nombrar tales compuestos
orgánicos se utiliza el vocablo tio (en griego, azufre), prefijo que también se utiliza en Q.
inorgánica, y cuyo significado práctico viene a ser: “hay un azufre en lugar de un
oxígeno”.
Ejemplo (como grupo principal)
Ejemplo
(como radical)
Tioles
R-SH
CH3-CH2-SH
etanotiol
HS-CH2CH2-COOH
ác. 3-mercaptopropanoico
Sulfuros R1-S-R2 CH3-S-CH3
sulfuro de dimetilo
CH3S-CH2CH2OH
2-(metiltio)etanol
Sales de sulfonio (R)3S+X
- (CH3)3S
+Cl-
cloruro de trimetilsulfonio
Tioaldehídos R-CHS CH3CH2CHS
propanotial
SHS-CH2-
COOCH2CH3
tioformilacetato de
etilo
Tiocetonas R-CS-R CH3-CS-CH2CH3
butanotiona
CH3-CS-CH2-
COOCH3
3-tioxobutanoato de
metilo
TABLA 4.2: ORDEN DE PREFERENCIA PARA LA ELECCION DEL GRUPO
PRINCIPAL
1. Cationes
2. Acidos, en el siguiente orden: R-COOH; R-COOOH, tioácidos.
3. Derivados de ácidos, en el siguiente orden: Anhídridos, Esteres, Haluro de
ácidos, Amidas.
4. Nitrilos, luego isocianuros.
5. Aldehídos, luego tioaldehídos.
6. Cetonas, luego tiocetonas.
7. Alcoholes, en el siguiente orden: fenoles, tioalcoholes, tiofenoles.
8. Hidroperóxidos.
9. Aminas.
10. Eteres, luego tioéters (sulfuros).
11. Peróxidos.
12. Alqueno
13. Alquinos

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
60
14. Alcanos
15. halógenos
16. nitrocompuestos

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
61
ALCANOS Y CICLOALCANOS
(ANÁLISIS CONFORMACIONAL)
FUENTE DE LOS ALCANOS: PETRÓLEO
Muchos alcanos existen de manera natural en plantas y animales. Por ejemplo, la
cubierta cerosa de las hojas de la col contiene nonacosanos (n-C29H60), y el aceite de
algunos pinos contiene heptano. La cera de abejas contiene, entre otras cosas,
eneitriacontano (n-C31H64).
Las principales fuentes de alcanos son, los depósitos mundiales de gas natural y
petróleo, derivados de la descomposición de materia orgánica marina sepultada hace
años. El gas natural consiste principalmente en metano, aunque también contiene etano,
propano, butano e isobutano. Estos hidrocarburos simples se usan en grandes cantidades
para calentar casas habitación, para cocinar alimentos y como combustible en las
industrias. El petróleo es una mezcla compleja de hidrocarburos, la cual es necesario
refinar para obtener diferentes fracciones antes de que pueda usarse.
Productos de la refinación del petróleo
PROPIEDADES DE LOS ALCANOS:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
62
Los alcanos también se llaman a veces parafinas, un nombre derivado del latín
parumaffinis (que significa “de poca afinidad”). este término define muy bien su
comportamiento, ya que los alcanos tienen poca afinidad química hacia otras moléculas,
y son químicamente inertes frente a la mayoría de los reactivos de uso común en
química orgánica. Sin embargo, en condiciones apropiadas loa alcanos reaccionan con
oxígeno, cloro y algunas otras sustancias.
Los alcanos presentan un incremento regular en el punto de ebullición como en
el de fusión conforme aumenta el peso molecular, una regularidad que también se
refleja en otras propiedades. Los parámetros promedio del enlace carbono-carbono son
casi los mismos en todos los alcanos, con longitudes de enlace de 1.54 0.01 Aº y
energías de enlace de 85 3 Kcal/mol. En la Tabla 5.1 puede observarse las
propiedades físicas de algunos alcanos:
TABLA 5.1.-
Propiedades físicas de algunos alcanos
Número de
carbonos
Alcano Punto de fusión
(ºC)
Punto de ebullición
(ºC)
Densidad
(g/mL)
1 Metano -182.5 -164.0 0.5547
2 Etano -183.3 -88.6 0.509 3 Propano -189.7 -42.1 0.5005
4 Butano -138.3 -0.5 0.5788
5 Pentano -129.7 36.1 0.6262 6 Hexano -95.0 68.9 0.6603
7 Heptano -90.6 98.4 0.6837 8 Octano -56.8 125.7 0.7025
9 Nonano -51.0 150.8 0.7176
10 Decano -29.7 174.1 0.7300 20 Eicosano -36.8 343.0 0.7886
30 Triacontano -65.8 450.0 0.8097 4 Isobutano -159.4 -11.7 0.579
5 Isopentano -159.9 27.85 0.6201 5 Neopentano -16.5 9.5 0.6135
8 Isooctano -107.4 99.3 0.6919
La Tabla 5.1 revela asimismo que un incremento en la ramificación tiene el
efecto de reducir el punto de ebullición de un alcano. Así, el pentano hierve a 36.1 ºC, el
isopentano (2-metilbutano) tiene una ramificación y hierve a 27.85 ºC, y el neopentano
(2,2-dimetilpropano) tiene dos ramificaciones y hierve a 9.5 ºC. De modo similar, el
octano hierve a 125.7 ºC, mientras que el iso-octano (2,2,4-trimetilpentano) hierve a
99.3 ºC. Este efecto puede comprenderse si se observa lo que ocurre durante la
ebullición.
Las moléculas no polares, como las de los alcanos, se atraen débilmente entre sí
por efecto de fuerzas intermoleculares de van der Waals. Estas fuerzas, que actúan
sólo a distancias muy cortas, resultan de la polarización inducida de las nubes
electrónicas en las moléculas. Aunque la distribución electrónica en una molécula es
uniforme en promedio durante un periodo, la distribución en un instante dado cualquiera
no es uniforme. Un lado de la molécula puede por casualidad, tener un ligero exceso de

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
63
electrones en relación con el lado opuesto. Cuando esto ocurre, la molécula posee un
momento dipolar temporal y puede provocar que otra molécula cercana adopte
temporalmente un dipolo opuesto, lo cual como resultado induce una ligera atracción
entre las dos moléculas.
Como podría esperarse, las fuerzas de van der Waals aumentan con el tamaño de
la molécula. si bien participan otros factores, al menos parte del incremento en el punto
de ebullición observado al aumentar el número de carbonos en la serie de los alcanos se
debe al incremento en las fuerzas de van der Waals.
El efecto de la ramificación en los puntos de ebullición también puede explicarse
recurriendo a las fuerzas de van der Waals. Los alcanos ramificados tienen una forma
más esférica que los alcanos de cadena lineal; como resultado, tienen menor área
superficial, menos fuerzas de van der Waals, y consecuentemente puntos de ebullición
más bajos.
ISÓMEROS CONSTITUCIONALES DE LOS ALCANOS:
Hay dos maneras que puedan formarse moléculas con la fórmula C4H10: los
cuatro carbonos pueden combinarse en una sola fila (butano), o esa fila puede
ramificarse (isobutano), tal como se indica a continuación:
butano
isobutano
Compuestos como el butano, cuyos carbonos están dispuestos en una sola fila,
se denominan alcanos de cadena lineal, o alcanos normales, mientras que los
compuestos cuya cadena se ramifica, como el 2-metilpropano (isobutano), reciben el
nombre de alcanos de cadena ramificada. Compuestos como las dos moléculas de
C4H10, que tienen la misma fórmula pero difieren estructuralmente, se denominan
isómeros, del griego isos + meros (“hechos de las mismas partes”).
Los isómeros son compuestos que tienen la misma cantidad de átomos de la
misma especie pero que difieren en la forma en que están dispuestos esos átomos.
Compuestos como el butano y el isobutano, cuyos átomos están conectados de manera
distinta, se llaman isómeros constitucionales (o de constitución).

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
64
La isomería constitucional no se limita a los alcanos; se presenta en toda la
química orgánica. Los isómeros de constitución pueden variar en los esqueletos de
carbono (como isobutano y butano), en los grupos funcionales (como alcohol etílico y
éter dimétílico) o en la ubicación de un grupo funcional en la cadena (como
isopropilamina y propilamina). Sin importar la causa de la isomería, los isómeros
constitucionales siempre tienen la misma fórmula pero distinto orden de enlace de los
átomos:
Diferente esqueleto de carbono
C4H10
butano
isobutano
Diferente grupos funcionales
C2H6O
alcohol etílico
éter dimetílico
Diferente posición de los grupos funcionales
C3H9N
isopropilamina
propilamina
CICLANOS:
Si bien hasta aquí sólo se han considerado alcanos de cadena abierta, desde hace
más de 100 años se sabe que también existen compuestos con anillos de átomos de
carbono. Tales compuestos se denominan cicloalcanos o compuestos alicíclicos. Puesto
que los cicloalcanos consisten en anillos de unidades de -CH2-, tienen la fórmula
general (CH2)n o CnH2n, y se representan como polígonos es las estructuras de esqueleto:
ciclopropano ciclobutano ciclopentano ciclohexano cicloheptano
ISOMERÍA CIS - TRANS EN LOS CICLOALCANOS:
En contraste con la libre rotación posible alrededor de los enlaces sencillos en
los alcanos de cadena abierta, en los cicloalcanos existe mucho menor libertad, porque
están geométricamente restringidos. Por ejemplo, el ciclopropano está geométricamente
restringido a ser una molécula planar con estructura rígida, debido a que tres puntos
definen un plano. No es posible rotación alrededor de un enlace carbono-carbono en el
ciclo propano sin romper el anillo.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
65
Los cicloalcanos superiores tienen cada vez mayor libertad, adquiriendo
movimiento y por ende generando diferentes confórmeros, además de isómeros.
La consecuencia más importante de su estructura cíclica es que los cicloalcanos
tienen dos caras o lados, “arriba” y “abajo”, lo cual abre la posibilidad de la isomería
en los cicloalcanos sustituidos. Por ejemplo, existen dos isómeros 1,2-
dimetilciclopropano distintos, uno con los dos metilos del mismo lado del anillo y otro
con un metilo de cada lado. Ambos isómeros son compuestos estables, y no es posible
interconvertirlos sin romper y volver a formar enlaces químicos. Los 1,2-
dimetilciclopropanos son una clase especial de estereoisómeros llamados isómeros cis -
trans. Para distinguirlos se usan los prefijos cis- y trans-; la isomería cis - trans es
común a todos los anillos sustituidos.
Ejemplos:
cis-1,2-dimetilciclopropano trans-1,2-dimetilciclopropano
cis-1,3-dimetilciclobutano trans-1,3-dimetilciclobutano
cis-1-bromo-3-metilciclopentano trans-1-bromo-3-metilcilopentano
ESTEREOQUÍMICA DE ALCANOS Y CICLOALCANOS
La estereoquímica es la rama de la química relacionada con los aspectos
tridimensionales de las moléculas. En este punto pondremos especial atención en el
aspecto espacial de las moléculas dado que estas no son planas sino que tienen y ocupan
un volumen en el espacio.
CONFORMACIONES DE LOS ALCANOS:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
66
Las diferentes configuraciones de los átomos debidas a la rotación alrededor de
un enlace sencillo se denominan conformaciones, y una conformación específica recibe
el nombre de confórmero (isómero conformacional). Sin embargo, a diferencia de los
isómeros constitucionales, los distintos confórmeros no suelen poder aislarse debido a
que se interconvierten con rapidez.
Los químicos representan los isómeros conformacionales de dos maneras, como
se muestra en la figura para el etano. En las representaciones de caballete, el enlace
carbono-carbono se dispone en un ángulo oblicuo y la orientación espacial se indica
representando todos los enlaces C-H. En las proyecciones de Newman[1]
, el enlace
carbono-carbono se representa de frente y los dos átomos de carbono se indican con un
círculo, y los enlaces del carbono de atrás se indican con líneas que salen de la periferia
del círculo. Las ventajas de las proyecciones de Newman son que resultan fáciles de
trazar y permiten visualizar sin dificultad las relaciones entre sustituyentes en los
distintos átomos de carbono.
La rápida rotación alrededor del enlace carbono-carbono interconvierte
las distintas formas.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
67
A pesar de lo dicho acerca de la simetría del enlace sigma (), en realidad no se
observa una rotación perfectamente libre en el etano. En experimentos se ha demostrado
que existe una ligera barrera (2.9 kcal/mol; 12 kJ/mol) a la rotación, y que algunas
conformaciones son más estables (tienen menos energía) que otras. La conformación
más estable (de más baja energía) es aquélla en la cual los seis enlaces carbono-
hidrógeno están lo más alejados posible entre sí (alternados). La conformación menos
estable (de más alta energía) es aquélla en la cual los seis enlaces carbono-hidrógeno
están lo más cercano posible (eclipsados). Entre estas dos conformaciones extremas
existe por supuesto un número infinito de posibilidades.
Gráfica de energía potencial contra rotación del enlace en el etano. Los conformeros
alternados tienen 2.9 kcal/mol menos en energía que los eclipsados.
CONFORMACIONES DEL PROPANO:
El propano es el siguiente en la serie de los alcanos, y en él nuevamente se
encuentra una barrera torsional que se traduce en un ligero impedimento a la rotación
alrededor de los enlaces carbono-carbono. La barrera es ligeramente mayor en el
propano que en etano; 3.4 kcal/mol frente a 2.9 kcal/mol. En el confórmero eclipsado
del propano existen dos interacciones hidrógeno-hidrógeno del tipo de las observadas en
el etano, y una nueva interacción entre un hidrógeno-metilo. Puesto que cada
interacción hidrógeno-hidrógeno en la conformación eclipsada tiene un costo en energía

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
68
aproximadamente de 1.0 kcal/mol, es posible asignar un valor de 3.4 - (2*1.0kcal/mol)
= 1.4 kcal/mol a la nueva interacción hidrógeno-metilo en el propano.
Proyecciones de Newman del propano en las conformaciones alternadas y eclipsadas.
El confórmero alternado es más bajo en energía por 3.4 kcal/mol .

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
69
CONFORMACIONES DEL BUTANO:
La situación conformacional resulta más compleja para los alcanos superiores.
Por ejemplo el caso de la rotación del enlace C 2-C3 del butano.
No todas las conformaciones alternadas del butano tienen la misma energía, y no
todas las conformaciones eclipsadas son iguales. El arreglo de más baja energía,
llamado confórmero anti, es aquel en el cual los dos grupos grandes (metilo) están lo
más separados posible; esto es, a 180º. A medida que ocurre la rotación alrededor del
enlace C2-C3, se llega a una conformación eclipsada en la que existen dos interacciones
metilo-hidrógeno y una interacción hidrógeno-hidrógeno. Si se asignan los valores de
energía de las interacciones eclipsadas que se dedujeron para el etano y propano, es
posible predecir que esta conformación eclipsada está sujeta a mayor tensión que la
conformación anti en 2*1.4 kcal/mol (dos interacciones Me-H) más 1.0 kcal/mol (una
interacción H-H), o sea un total de 3.8 kcal/mol. Esto es exactamente lo que se observa.
Cuando la rotación del enlace continúa se alcanza un mínimo de energía en la
conformación alternada en la cual los grupos metilo están separados 60º, esta última,
que se denomina conformación sesgada o gauche (francés, “torcida”), tiene 0.9
Kcal/mol más energía que la conformación anti, pese a que no presenta interacciones
eclipsadas. Esta diferencia de energía se debe al hecho de que los voluminosos grupos
metilo están más cercanos entre sí en la conformación sesgada, lo que da por resultado
una tensión estérica. La tensión estérica es la interacción repulsiva que ocurre cuando
dos grupos son forzados a acercarse más de lo que permiten sus radios atómicos.
Básicamente, es el resultado del intento de forzar dos átomos a ocupar el mismo espacio.
Cuando el ángulo diedro entre los grupos metilo se aproxima a 0º, se alcanza el
máximo de energía. Puesto que los grupos metilo son forzados a permanecer más cerca
entre sí que en la conformación sesgada, se produce una gran tensión, tanto torcional
como estérica. Se ha estimado una energía de tensión total de 4.5 kcal/mol para esta
conformación, lo que permite calcular un valor de 2.5 kcal/mol para lo interacción
eclipsada metilo-metilo.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
70
Los mismos principios que se aplicaron para el butano son válidos para el
pentano, el hexano, y todos los alcanos superiores. La conformación más favorecida en
cualquier alcano es aquella en la cual todos los enlaces carbono-carbono tienen arreglo
alternados y en la cual todos los sustituyentes grandes guardan una relación anti entre sí.
Un último punto: “Es importante recordar que cuando se dice que un
confórmero particular es “más estable” que otro, ello no significa que esa molécula
adopte y conserve sólo la conformación más estable. Sin embargo, en un instante dado
un porcentaje mayor de moléculas se encuentran en una conformación más estable que
en una menos estable.
CONFORMACIONES Y ESTABILIDAD DE LOS CICLANOS: (Teoría de la
Tensión de Baeyer)
Los químicos de finales del siglo XIX habían aceptado la idea de la existencia de
moléculas cíclicas, pero los límites de las dimensiones factibles de los anillos eran
inciertos. Se conocían numerosos compuestos con anillos de cinco y seis carbonos, pero
no se habían producido anillos mayores o menores. Por ejemplo, no se conocían
ciclopropanos o ciclobutanos, a pesar de los innumerables esfuerzos p or producirlos.
En 1885, Adolf von Baeyer[2]
propuso una interpretación teórica para esta
observación. Baeyer sugirió que, si los carbonos “prefieren” una configuración
tetraédrica con ángulos de enlace de 109º, podría ser que los anillos con menos de cinco

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
71
o más de seis carbonos estuvieran sometidos a demasiada tensión para existir. Baeyer
basó su hipótesis en la noción geométrica simple de que un anillo de tres carbonos
(ciclopropano) debe ser un triángulo equilátero con ángulos de enlace de 60º, un anillo
de cuatro carbonos (ciclobutano) debe ser un cuadrado con ángulos de enlace de 90º, un
anillo de cinco miembros (ciclopentano) debe ser un pentágono regular con ángulos de
enlace de 108º. etc.
Si bien algo de razón en las afirmaciones de Baeyer acerca de la tensión angular
en los anillos pequeños, estaba equivocado al pensar que los anillos pequeños y grandes
estaban demasiado tensionados para existir. Ahora se sabe que es posible producir
anillos de todos los tamaños, desde 3 hasta 30 y más carbonos. No obstante, es muy útil
el concepto de tensión angular - la resistencia de un ángulo de enlace al aumento o
disminución del ángulo tetraédrico ideal - .
TABLA 5.2
Calores de combustión de los cicloalcanos
Cicloalcano
(CH2)n
Tamaño
del anillo
Calor de
combustión
(kcal/mol)
Calor de
combustión
por CH2
(kcal/mol)
Energía de
tensión total
(kcal/mol)
Ciclopropano 3 499.8 166.6 27.6
Ciclobutano 4 655.9 164.0 26.4
Ciclopentano 5 793.5 158.7 6.5
Ciclohexano 6 944.5 157.4 0
Cicloheptano 7 1108 158.3 6.3
Ciclooctano 8 1269 158.6 9.6
Ciclononano 9 1429 158.8 12.6
Ciclodecano 10 1586 158.6 12.0
Cicloundecano 11 1981 158.4 11.0
Ciclododecano 12 1981 157.6 2.4
Ciclotridecano 13 2051 157.8 5.2
Ciclotetradecano 14 2204 157.4 0
Alcano (referencia) - - 157.4 0
Los datos de la Tabla 5.2 muestran claramente que la teoría de Baeyer no es
del todo correcta. Ciclopropano y ciclobutano están bastante tensionados, como lo
predijo Baeyer, pero el ciclopentano lo está más de lo predicho, y el ciclohexano está
totalmente libre de tensión. Para anillos de mayor tamaño no existe un incremento

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
72
regular en la tensión, y los anillos con más de 14 carbonos están nuevamente libres de
tensión. ¿Qué está mal en la teoría de Baeyer ?
Baeyer estaba equivocado por una razón muy simple: Supuso que todos los
anillos eran planos. En realidad, la mayoría de los cicloalcanos no lo son, sino que
adoptan conformaciones tridimensionales plegadas las cuales permiten que los ángulos
de enlace sean tetraédricos. Aun así, el concepto de tensión angular es valioso porque
explica la reactividad de los anillos de tres y cuatro carbonos. En resumen, la teoría de
Baeyer es insuficiente para explicar las energías de tensión observadas y las
configuraciones de los cicloalcanos. Estos adoptan la conformación de energía mínima
por una combinación de tres factores:
1. Tensión angular, debida a aumento o disminución de los ángulos de enlace.
2. Tensión torsional, debida a la presencia de enlaces vecinos eclipsados.
3. Tensión estérica, debida a interacciones repulsivas entre átomos que se acercan
mucho entre sí.
CICLOPROPANO:
Puesto que tres puntos (los tres átomos de carbono) definen un plano, el
ciclopropano debe ser planar y, suponiendo que es una molécula simétrica, debe tener
ángulos de enlace C-C-C de 60º. ¿Cómo puede la teoría del orbital molecular explicar
esta gran distorsión de los enlaces respecto al ángulo tetraédrico normal de 109 º?
La respuesta es que el ciclopropano se describe mejor si se considera que
contiene enlaces curvos. En un alcano no tensionado, la máxima eficacia de enlace se
logra cuando los dos átomos están localizados de modo que sus orbitales superpuestos
apunten directamente uno hacia otro. Sin embargo, en el ciclopropano esto no puede
ocurrir; más bien, deben superponerse en un ángulo pequeño. El resultado de esta
superposición deficiente es que los enlaces del ciclopropano son más débiles y más
reactivos que los enlaces de los alcanos normales.
CICLOBUTANO:
El ciclobutano tiene aproximadamente la misma tensión total (26.4 kcal/mol)
que el ciclopropano (27.6 kcal/mol). Debido a su mayor cantidad de hidrógenos en el
anillo, el ciclobutano tiene mayor tensión torsional que el ciclopropano, si bien tiene
menor tensión angular. Mediciones espectroscópicas indican que el ciclobutano no es
del todo planar, sino que está ligeramente flexionado, de modo que uno de los átomos
de carbono se encuentra 25º arriba del plano formado por los otros tres. El efecto de esta
ligera flexión es un aumento en la tensión angular pero un descenso en la tensión
torsional, hasta que se alcanza un equilibrio de energía mínima entre los dos efectos
opuestos.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
73
CICLOPENTANO:
Baeyer predijo que el ciclopentano debería estar casi libre de tensión, pero los
resultados de mediciones de calores de combustión indican que eso no es verdad.
Aunque el ciclopentano prácticamente no tiene tensión angular, los diez átomos de
hidrógeno introducen considerable tensión torsional en la conformación plana, por tanto,
el ciclopentano adopta una conformación plegada fuera del plano, la cual establece un
equilibrio entre aumento en la tensión angular y decremento en la tensión torsional.
Cuatro de los átomos de carbono del ciclopentano están aproximadamente en el mismo
plano, y el quinto está flexionado fuera del plano. La mayoría de los hidrógenos están
aproximadamente alternados con respecto a sus vecinos. (Conformación sobre)

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
74
CICLOHEXANO:
Los ciclohexanos sustituidos son los más importantes de todos los cicloalcanos,
debido a su amplia distribución en la naturaleza. Un gran número de compuestos,
incluyendo muchos agentes farmacéuticos importantes, contienen anillos de
ciclohexano. Datos experimentales de calores de combustión demuestran que los anillos
de ciclohexano están libres de tensión, tanto angular como torsional. ¿A qué se debe
esto?
La respuesta fue sugerida originalmente por H. Sachse en 1890 y ampliada
después por Ernest Mohr[3]
. Los anillos de ciclohexano no son planos como supusiera
Baeyer, sino que están plegados en una conformación tridimensional que alivia todas las
tensiones. Los ángulos C-C-C del ciclohexano pueden alcanzar el valor tetraédrico libre
de tensión si el anillo adopta la conformación de silla que se muestra a continuación.
Cómo trazar la conformación silla del ciclohexano:
1.- Se trazan dos líneas paralelas, descendentes
hacia la derecha y ligeramente adelantada la inferior
respecto a la superior. Esto significa que cuatro
átomos de carbono del ciclohexano se encuentran en
un mismo plano.
2.- Se localiza el átomo de carbono más alto, arriba
y a la derecha del plano de los otros cuatro, y se
conectan los enlaces.
3.- Se localiza el átomo de carbono más bajo, por
abajo y a la izquierda del plano de los cuatro
carbonos centrales, y se conectan los enlaces.
Obsérvese que los enlaces del átomo de carbono
más alto son paralelos a los enlaces del carbono más
bajo.
Llamada silla, por su semejanza con una silla de alberca. Más aún se observa a
lo largo de cualquiera de los enlaces carbono-carbono en una proyección de Newman,
se halla que en la conformación de silla el ciclohexano no presenta esfuerzo torsional;
todos los enlaces C-H vecinos están perfectamente alternados. La conformación de silla
del ciclohexano es de tanta importancia que todos los químicos orgánicos deben
aprender a dibujarla correctamente.
Enlaces Axiales y Ecuatoriales en el Ciclohexano:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
75
La conformación de silla del ciclohexano tiene muchas consecuencias. Por
ejemplo, que el comportamiento químico de los ciclohexanos sustituidos es regido
directamente por su conformación. Otra consecuencia de la conformación de silla del
ciclohexano es que existen dos tipos de átomos de hidrógenos en el anillo: hidrógenos
axiales e hidrógenos ecuatoriales. El ciclohexano silla tiene seis hidrógenos axiales
que están perpendiculares al anillo (paralelo al eje del anillo) y seis hidrógenos
ecuatoriales que están más o menos en el plano principal del anillo (alrededor del
ecuador del anillo).
MOVILIDAD CONFORMACIONAL DEL CICLOHEXANO:
Puesto que el ciclohexano silla tiene dos clases de posiciones, axiales y
ecuatoriales, podría esperarse que un ciclohexano monosustituido presentara dos formas
isoméricas. Sin embargo, en la realidad esto no se cumple. A temperatura ambiente
existe sólo un metilciclohexano, un bromociclohexano, un ciclohexanol, etc., en virtud
de que el anillo de ciclohexano es conformacionalmente móvil. Las dos conformaciones
de silla pueden interconvertirse con facilidad, con el resultado de que se intercambian
las posiciones axiales y ecuatoriales. Las interconversiones de las conformaciones de
silla, usualmente denominadas interconversión del anillo, se ilustran a continuación.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
76
Interconversión del anillo
La interconversión del anillo del ciclohexano silla
intercambia las posiciones axiales y ecuatoriales
La interconversión del anillo transforma el metilciclohexano axial en
metilciclohexano ecuatorial, tal como se observa a continuación.
El metilciclohexano axial se transforma en metilciclohexano ecuatorial
después de la interconversión del anillo. Mediante determinaciones espectroscópicas se
ha encontrado que la barrera de energía para la interconversión silla-silla es de unas
10.8 kcal/mol (45 kJ/mol), un valor suficientemente bajo para que el proceso ocurra con
gran rapidez a temperatura ambiente. Por ello, sólo se ve una estructura promedio en
rápida interconversión, en vez de isómeros distintos axial y ecuatorial.
CICLOHEXANO EN LA CONFORMACIÓN DE BOTE:
Además de la conformación de silla, el ciclohexano puede adoptar una
conformación de bote, igualmente libre de tensión angular. Sin embargo, no le hemos
puesto mucha atención hasta el momento, porque el ciclohexano en conformación de
bote (ciclohexano bote) es mucho menos estable que el ciclo hexano silla.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
77
Existen dos tipos de átomos de carbono en el ciclohexano bote. Los carbonos 2,
3, 5 y 6 se encuentran en el mismo plano, mientras que los carbonos 1 y 4 están por
encima del plano. Los átomos de hidrógeno interiores en los carbonos 1 y 4 se acercan
entre sí lo suficiente para causar una considerable tensión estérica, y los cuatro pares de
hidrógenos en los carbonos 2, 3, 5 y 6 están eclipsados. Así, el ciclohexano bote tiene
tensión estérica y torsional. Por mediciones experimentales se ha determinado que el
ciclohexano bote es aproximadamente 7.0 kcal/mol menos estable que el ciclohexano
silla, si bien este valor se reduce a unas 5.5 kcal/mol por efecto de una ligera torsión,
que alivia algo de tensión torsional. Aun así, la conformación de bote torcido está
mucho más esforzada que la conformación de silla, y las moléculas adoptan esta
configuración geométrica sólo en circunstancias especiales.
Conformación de bote del ciclohexano. No existe tensión angular en esta
conformación, pero si hay tensiones estérica y torsional.
Conformación de bote y de bote torcido del ciclohexano. La conformación de bote
torcido es de más baja energía que la de bote por 1.5 kcal/mol.
El ciclohexano experimenta un proceso continuo de equilibrio que interconvierte
rápidamente estos átomos axiales y ecuatoriales, tal como se representa en el diagrama
de energía, para el ciclohexano.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
78
CONFORMACIONES DE CICLOHEXANOS MONOSUSTITUIDOS:
Volvamos al caso del metilciclohexano, la diferencia de energía entre los
confórmeros axial y ecuatorial se debe a la tensión estérica causada por las llamadas
interacciones 1,3-diaxiales. Esto es, el grupo metilo axial en C1 está demasiado cerca
de los hidrógenos axiales que se encuentran a tres carbonos de distancia en C3 y C5, lo
que da por resultado una tensión estérica de 1.8 kcal/mol.
La tensión estérica 1,3-diaxial ya debe ser familiar para el estudiante;
anteriormente se le consideró como la tensión estérica entre grupos metilo en el butano
sesgado. Recuérdese que el butano sesgado (gauche) es menos estable que el butano
anti en 0.9 kcal/mol, debido a la interferencia estérica entre átomos de hidrógenos en los
dos grupos metilo. Si un fragmento de cuatro carbonos del metilciclohexano axial se ve
como un butano gauche, es evidente que la interacción estérica es la misma en ambos
casos. Sin embargo, dado que el metilciclohexano tiene dos de tales interacciones, la
tensión estérica es de 2 * 0.9 = 1.8 kcal/mol.
Interconversión del metilciclohexano axial y ecuatorial. La conformación ecuatorial
es más estable en 1.8 kcal/mol.
Lo que es válido para el metilciclohexano también lo es para todos los
ciclohexanos monosustituidos: Un sustituyente casi siempre es más estable en una
posición ecuatorial que un una axial. La cantidad exacta de tensión estérica 1,3-diaxial
en un compuesto específico depende, por supuesto, de la naturaleza y el tamaño del

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
79
grupo axial. Como es de esperar, la cantidad de tensión estérica se incrementa a través
de la serie CH3- CH3CH2- (CH3)2CH- (CH3)3C- simultáneamente con el
volumen de los grupos alquilos cada vez más grandes. Obsérvese que los valores de
la Tabla 5.3 se refieren a interacciones 1,3-diaxiales de los grupos indicados, con un
solo átomo de hidrógeno. Por lo tanto, para obtener el valor de la tensión presente en un
ciclohexano monosustituido estos valores deben duplicarse.
Tensión estérica debida a interacciones 1,3-diaxiales.
Y (kcal/mol) (kJ/mol)
-F 0.12 0.5
-Cl 0.25 1.4
-Br 0.25 1.4
-OH 0.5 2.1
-CH3 0.9 3.8
-CH2CH3 0.95 4.0
-CH(CH3)2 1.1 4.6
-C(CH3)3 2.7 11.3
-C6H5 1.5 6.3
-COOH 0.7 2.9
-CN 0.1 0.4
-H 0 0
ANÁLISIS CONFORMACIONAL DE CICLOHEXANOS DISUSTITUIDOS:
Los ciclohexanos monosustituidos suelen tener el sustituyente en posición
ecuatorial. Sin embargo, en los ciclohexanos disustituidos la situación es más compleja
en virtud de que deben considerarse los efectos estéricos de ambos sustituyentes. Es
necesario analizar todas las interacciones estéricas en las posibles conformaciones antes
de decidir cuál conformación es favorecida.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
80
Analicemos primero el 1,2-dimetilciclohexano ¿Cuál? Existen dos isómeros, cis
y trans, que deben considerarse por separado.
Isómero cis. Ambos grupos metilos están del mismo lado del anillo, y el compuesto
puede existir en cualquiera de las dos conformaciones de silla que se muestran a
continuación:
Conformaciones del cis-1,2-dimetilciclohexano. Las dos conformaciones tienen la
misma energía, puesto que cada una tiene un grupo metilo axial y un grupo metilo
ecuatorial.
Isómero trans. Los dos grupos metilos están en caras opuestas del anillo, y el
compuesto puede existir en cualquiera de las dos conformaciones de silla, pero la
situación en este caso es muy distinta de la del isómero cis. La conformación trans que
aparece a la izquierda de la figura, tiene ambos grupos metilos en posición ecuatorial, y
por tanto solo una interacción gauche entre los metilos (0.9 kcal/mol), y ninguna
interacción 1,3-diaxial. La conformación de la derecha, sin embargo, tiene ambos
grupos metilos axiales, cada uno de ellos presenta dos interacciones 1,3-diaxiales con
los átomos de hidrógenos, lo que hace un total de cuatro interacciones 1,3-diaxiales (0.9
* 4 = 3.6 kcal/mol). Puede predecirse por tanto que el trans-1,2-dimetilciclohexano
existirá casi exclusivamente en la conformación diecuatorial.
Conformaciones del trans-1,2-dimetilciclohexano. La conformación con dos grupos
metilos ecuatoriales es favorecida por 2.7 kcal/mol.
El mismo tipo de análisis conformacional recién realizado para el 1,2-
dimetilciclohexano puede aplicarse a cualquier ciclohexano disustituido o trisustituido.
Por ejemplo, consideremos el cis-1-terc-butil-4-clorociclohexano. Resulta que la gran

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
81
tensión estérica causada por un grupo terc-butilo mantiene eficazmente el anillo de
ciclohexano en una sola conformación.
CONFORMACIONES DE MOLÉCULAS POLICÍCLICAS:
El último punto por considerar acerca de la estereoquímica de los cicloalcanos es
lo que sucede cuando dos o más anillos de cicloalcanos se fusionan para constituir
moléculas policíclicas, como la decalina. La decalina consta de dos anillos de
ciclohexano que se unen para compartir dos átomos de carbono (los carbonos cabeza de
puente) y un enlace, la decalina puede existir en dos formas isoméricas, dependiendo de
si los anillos tienen fusión cis o trans.
Representaciones de la cis- y trans-decalina

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
82
ESTEREOQUIMICA
QUIRALIDAD Y ENANTIÓMEROS:
Los dos objetos representados en la figura 6.1 parecen idénticos a todos los
efectos. A cada arista, cara o ángulo en uno de ellos, le corresponde su arista, cara o
ángulo en el otro. Con todo, los dos objetos no son superponibles entre sí y, por tanto,
son objetos diferentes; se relacionan entre sí como un objeto cualquiera se relaciona con
su imagen en un espejo.
Figura 6.1
Otro par de objetos familiares que se relacionan de esta forma son la mano
izquierda y la mano derecha. En una primera aproximación, parecen idénticas bajo casi
todos los puntos de vista; no obstante, la mano derecha se ajusta perfectamente a
un guante para la mano derecha pero no a un guante para la mano izquierda, por ende
las manos no son idénticas. La propiedad general que define esa relación se denomina
Quiralidad. Cualquier objeto que no es superponible con su imagen especular es quiral.
Por otra parte, si un objeto y su imagen especular pueden hacerse coincidir en el espacio,
se dice que son aquirales.
CAUSA DEL CARÁCTER DERECHO O IZQUIERDO DE LAS MOLÉCULAS: QUIRALIDAD.
Se dice que las moléculas que no son idénticas a sus imágenes especulares, y por
tanto existen en dos formas enantioméricas, son quirales (griego Kheir, mano). No es
posible superponer una molécula quiral y su imagen especular (su enantiómero) de
modo que todos los átomos coincidan.
¿Cómo puede predecirse si cierta molécula es quiral o no? Una molécula no
puede ser quiral si tiene un plano de simetría. Un plano se simetría es un plano
imaginario que corta un objeto en dos mitades que son imágenes especulares exactas
una de la otra. Una molécula que tiene un plano de simetría en cualquiera de sus
posibles conformaciones debe ser superponible con su imagen especular, y por tanto
debe ser no quiral o aquiral.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
83
La causa más común de quiralidad en moléculas orgánicas es la presencia de un
átomo de carbono unido a cuatro grupos distintos. Tales carbonos se denominan
centros quirales o centros estereogénicos o centros asimétricos. Las diferencias no
necesariamente se observan en el átomo inmediatamente contiguo al centro
estereogénico. Por ejemplo, el 5-bromodecano es una molécula quiral porque cuatro
grupos distintos están unidos a C5:
Sustituyentes en el carbono 5
-H
-Br
-CH2CH2CH2CH3 (butilo)
-CH2CH2CH2CH2CH3 (pentilo)
PROPIEDADES FÍSICAS DE LOS ENANTIÓMEROS: ACTIVIDAD ÓPTICA.
La mayoría de las propiedades físicas de los 2-yodobutanos enantioméricos son
idénticas. Tienen idénticos punto de fusión, puntos de ebullición, solubilidades en
disolventes corrientes, densidades, índice de refracción y espectros. Sin embargo,
difieren en un importante aspecto: en la forma en que interaccionan con la luz
polarizada.
La luz puede ser tratada como un movimiento ondulatorio de campos eléctricos
y magnéticos oscilantes que forman ángulos rectos entre sí. Cuando un electrón
interacciona con la luz, oscila a la frecuencia de la luz en la dirección del campo
eléctrico y en fase con él. En la luz normal, los vectores de campo eléctrico de todas las
ondas luminosas se orientan en todos los planos posibles. La luz polarizada plana es
aquella en que los vectores de campo eléctrico de todas las ondas luminosas vibran en el
mismo plano, el plano de polarización.
La luz polarizada plana puede ser producida al pasar luz normal a través de un
polarizador, como una lente polaroid o un dispositivo conocido como prisma de Nicol.
Jean Baptiste Biot[1]
, realizó la notable observación de que cuando un haz de luz
polarizada atraviesa una solución de ciertas moléculas orgánicas, como azúcar o

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
84
alcanfor, el plano de polarización experimenta una rotación. No todas las moléculas
orgánicas tienen esta propiedad, pero se dice que aquellas que hacen girar el plano de la
luz polarizada son ópticamente activas.
Representación esquemática de un polarímetro. La luz polarizada en un plano pasa
por una solución de moléculas ópticamente activas, lo cual hace girar el plano de
polarización.
La magnitud de la rotación puede medirse con un instrumento conocido como
polarímetro, que se representa esquemáticamente en figura de arriba. Primero se coloca
una solución de moléculas orgánicas ópticamente activas en un tubo de muestra;
después se hace pasar por el tubo la luz polarizada en un plano, tras lo cual ocurre la
rotación del plano. la luz se hace pasar luego por un segundo polarizador denominado
analizador. Haciendo girar el analizador hasta que la luz lo atraviesa es posible
encontrar el nuevo plano de polarización, de este modo, la magnitud de la rotación. Este
valor se denota por la letra griega (alfa), y se expresa en grados.
Además de la magnitud de la rotación, también es posible encontrar su sentido.
Respecto a un observador que ve directamente por el extremo del instrumento en el lado
del analizador, algunas moléculas ópticamente activas hacen girar la luz polarizada
hacia la izquierda (en sentido contrario al de las manecillas del reloj) y se dice que son
levógiras o levorrotatorias, mientras que otras la hacen girar hacia la derecha (en el
sentido de las manecillas del reloj) y se dice que son dextrógiras o dextrorrotatorias.
Por convenio, a la rotación hacia la izquierda se le asigna un signo negativo (-), y a la
rotación hacia la derecha se le asigna un signo positivo (+).
Rotación específica:
Para expresar los datos de manera significativa a fin de que puedan hacerse
comparaciones, es necesario establecer condiciones estándar. Por convenio, la rotación
específica, D, de un compuesto se define como la rotación observada cuando se
utiliza luz con longitud de onda de 589 nanómetros (1 mm = 10-9 m) y un trayecto
óptico l de 1 decímetro (1 dm = 10 cm), con concentración de muestra C de 1 g/mL.
(La luz de 589 nm, llamada línea D del Sodio, es la luz emitida por las lámparas de
vapor de sodio.)
Cuando los datos de rotación óptica se expresan de esta manera estándar, la
rotación específica, D, es una constante física característica de cada compuesto con
actividad óptica.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
85
REGLAS DE SECUENCIA PARA ESPECIFICAR LA CONFIGURACIÓN:
Aunque los dibujos constituyan una representación de la estereoquímica, es
difícil traducirlos en palabras. Así, también es necesario un método verbal para explicar
el arreglo tridimensional de los átomos (la configuración) en un centro estereogénico.
En el método acostumbrado se emplean las mismas reglas de secuencia (Z o E).
Revisemos brevemente las reglas de Cahn[2]
-Ingold[3]
-Prelog[4]
a fin de ver cómo
pueden servir para especificar la configuración de un centro estereogénico (carbono
asimétrico).
1. Se observan los cuatro átomos unidos directamente al centro estereogénico y se
asignan prioridades en orden decreciente de número atómico. El grupo con mayor
número atómico tiene la máxima prioridad; el grupo con el menor número atómico
tiene la mínima prioridad.
2. Si no es posible decidir la prioridad aplicando la regla 1, se comparan los números
atómicos de los átomos unidos en segundo lugar en cada sustituyente; si es
necesario se continúa hasta los terceros, cuartos o más átomos, hasta que se
encuentre una diferencia.
3. Los átomos con enlaces múltiples se consideran como un número equivalente de
átomos con enlaces simples. Por ejemplo:
Una vez asignadas las prioridades de los cuatro grupos unidos a un carbono
quiral, se describe la configuración estereoquímica alrededor del carbono quiral
orientando mentalmente la molécula de modo que el grupo de menor prioridad (cuatro)
apunte directamente hacia atrás, lo más alejado posible del observador. Después se
analizan los tres sustituyentes restantes, que se dirigen en forma radial hacia el
observador. Si una flecha curva dibujada desde el sustituyente con mayor prioridad
hacia el de segunda prioridad y de ahí hacia el de tercera (1 2 3) tiene el sentido del
reloj, se dice que el centro estereogénico tiene la configuración R (latín rectus,
“derecho”). Si por el contrario definen una curva en el sentido contrario de un reloj, se
dice que el centro esterogénico tiene la configuración S (latín sinister, “izquierdo”).
Consideremos el ácido láctico como un ejemplo de la forma en que se asigna la
configuración.
1. El primer paso es asignar prioridades.
2. Orientar la molécula, de tal manera que el grupo de menor prioridad esté dirigido
hacia atrás.
Ácido láctico.
Prioridades:
4 -H (baja)
3 -CH3
2 -COOH
1 -OH (alta)

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
86
DIASTERÓMEROS:
Moléculas como las del ácido láctico, alanina y gliceraldehído son relativamente
fáciles de estudiar, puesto que cada una tiene sólo un centro estereogénico y sólo puede
existir en dos configuraciones enantioméricas. Sin embargo, la situación se torna más
compleja en el caso de moléculas que tienen más de un centro estereogénico.
Consideremos por ejemplo el aminoácido esencial treonina (ácido 2-amino-3-
hidroxibutanoico). Dado que la treonina tiene dos centros estereogénicos C2 y C3,
existen cuatro estereoisómeros posibles, como analizaremos a continuación: (Se sugiere
al alumno que verifique por sí mismo que la configuración R,S es correcta.
2R , 3R 2S , 3S 2R , 3S 2S , 3R
Enantiómeros Enantiómeros
Relaciones entre las cuatro treoninas estereoisoméricas
Estereoisómero Es anantiómero de Es diastereómero de
2R , 3R 2S , 3S 2R , 3S y 2S , 3R
2S , 3S 2R , 3S 2R , 3S y 2S , 3R
2R , 3S 2S , 3R 2R , 3R y 2S , 3S
2S , 3R 2R , 3S 2R , 3R y 2S , 3S
¿ Cuál es la relación entre cualquiera dos configuraciones que no sean imágenes
especulares entre sí ? Por ejemplo, ¿cuál es la relación entre el compuesto 2R,3R y el
2R,3S ? Los dos compuestos son estereoisómeros, aunque no son superponibles ni son
enantiómeros. Por lo tanto se dice que son Diastereómeros, los cuales son
estereoisómeros que no son imágenes especulares entre sí.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
87
De los cuatro estereoisómeros posibles de la treonina sólo uno, el 2S,3R,
con D = 29.3º , existe en forma natural en plantas y animales. La mayoría de las
moléculas orgánicas importantes son quirales, y usualmente sólo un estereoisómero se
encuentra en la naturaleza.
COMPUESTOS MESO:
Consideremos un ejemplo más de compuestos con dos centros estereogénicos: el
ácido tartárico. Tracemos su cuatro posibles estereoisómeros:
2R, 3R 2S, 3S 2R, 3S 2S, 3R
Las imágenes especulares 2R,3R y 2S,3S no son superponibles, y son por tanto
un par de enantiómeros. Sin embargo, una observación cuidadosa revela que las
estructuras 2R,3S y 3S,3R son idénticas, lo cual puede corroborarse haciendo girar 180º
una de ellas:
Son Idénticas
La identidad de las estructuras resulta del hecho de que la molécula tiene un
plano de simetría. Dicho plano pasa por el enlace C2-C3, haciendo que una mitad de la
molécula sea la imagen especular de la otra mitad.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
88
MOLÉCULAS CON MÁS DE DOS CENTROS ESTEREOGÉNICOS:
Cuando hay más de un centro quiral en una molécula, dan por resultado un
máximo de 2n estereoisómeros. Por ejemplo, el colesterol contiene ocho centros
estereogénicos, lo que hace posible 28 = 256 estereoisómeros (128 pares de
enantiómeros), aunque muchos están demasiado tensionados para existir. En la
naturaleza existe predominantemente uno de ellos.
Colesterol
(ocho centros quirales)
Una mezcla de 50:50 de dos enantiómeros quirales se denomina mezcla
racémica o racemato, y se denota por el símbolo () o por el prefijo d,l, para indicar
una mezcla de formas dextrogira y levógira. Las mezclas racémicas deben presentar
rotación óptica cero (nula).
PROYECCIONES DE FISCHER:
En 1891, Emil Fischer[5]
sugirió un método basado en la proyección de un átomo
de carbono tatraédrico sobre una superficie plana. Estas proyecciones de Fischer fueron
adoptadas rápidamente, y ahora son un medio estándar para representar la
estereoquímica de centros estereogénicos. En una proyección de Fischer, un átomo de
carbono tetraédrico se representa por dos líneas cruzadas. Por convención, las líneas
horizontales representan enlaces que salen de la página hacia el lector, y las líneas
verticales representan enlaces que se proyectan hacia atrás de la página.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
89
Por ejemplo, el ácido (R)-láctico puede representarse como sigue:
Puesto que una molécula dada puede representarse de muchas maneras, a
menudo es necesario comparar dos proyecciones distintas para ver si representan el
mismo o diferentes enantiómeros. Para comprobar la identidad, es posible hacer girar
sobre el papel las proyecciones de Fischer, pero teniendo cuidado de no cambiar
inadvertidamente el significado de la proyección, Sólo están permitidos dos tipos de
movimientos:
1. Una proyección de Fischer puede hacerse girar 180º sobre la página, pero no 90º ni
270º. La razón de esto es simplemente que una rotación de 180º cumple la
convención de Fischer de que los mismos dos sustituyentes en los enlaces
horizontales siempre salgan del plano hacia el lector y los mismos dos sustituyentes
en los enlaces verticales siempre se proyecten hacia atrás del plano. Y una rotación
de 90º contraría la conversión de Fischer, pues los sustituyentes que estaban en los
enlaces horizontales en una forma quedan verticales después de una rotación de 90º,
invirtiendo su estereoquímica y cambiando el significado de la proyección. Por
ejemplo:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
90
pero:
2. En una proyección de Fischer, un grupo puede mantenerse fijo mientras los otros
tres giran en el sentido del reloj o en el sentido contrario. Por ejemplo:
Éstos son los únicos tipos de movimientos permitidos. Mover una proyección de
Fischer de cualquier otro modo invierte su significado. De este modo, si una proyección
del ácido (R)-láctico se hace girar 90º, resulta una proyección de Fischer del ácido (S)-
láctico.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
91
La rotación de 90º de una proyección de Fischer invierte su significado
ASIGNACIÓN DE CONFIGURACIONES R,S A PROYECCIONES DE
FISCHER:
Es posible asignar las configuraciones estereoquímicas R,S a las proyecciones de
Fischer siguiendo los tres pasos que se indican a continuación:
1. Se asignan prioridades a los cuatro sustituyentes de modo usual.
2. Se realiza uno de los dos movimientos permitidos para colocar el grupo de menor
prioridad (cuarta) en la parte de arriba de la proyección de Fischer.
3. Se determina el sentido de la rotación al pasar de la prioridad 1 a la 2 y a la 3, y
se asigna la configuración R o S como determina la regla. (R en el sentido del
reloj y S en el sentido contrario del reloj).
COMPUESTOS TREO Y ERITRO:
Los prefijos “eritro” y “treo” se utilizan a menudo para designar las
configuraciones relativas en compuestos que poseen dos átomos de carbono quirales
adyacentes que soportan grupos diferentes, pero similares. Estos prefijos derivan de los
carbohidratos eritrosa y treosa.
Eritro corresponde al distereómero que cuando se observa a lo largo del enlace
que conecta los átomos de carbono quirales, posee un rotámero en el que todos los
grupos similares están eclipsados. El diastereómero Treo no posee un rotámero en el
que todos los grupos similares estén eclipsados. Cuando los grupos a átomos unidos a
ambos carbonos quirales son idénticos y se encuentran eclipsados entre si, recuérdese
que es un compuesto Meso.


www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
93
ESTEREOQUÍMICA Y QUIRALIDAD EN CICLOALCANOS:
Este apartado sólo lo comentaremos mediante ejemplos:
1.- El cis-1,2-dibromociclobutano y trans-1,2-dibromociclobutano
No puede existir con su enantiómero o en otras palabras su imagen especular resulta
idéntica
MOLÉCULAS QUIRALES: Son aquellas que pueden existir como
enantiómeros, ejemplo el trans-1,2-dibromociclohexano.
MOLÉCULAS AQUIRALES: Son aquellas que no pueden existir como enantiómeros,
ejemplo el cis-1,2 -dibromociclohexano.
2.- El cis y el trans 1,2-dimetilciclohexano

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
94
Existen como estereoisómeros pero no como
enantiómeros por presentar plano de simetría
3.- El cis y el trans-1,3-dimetilciclohexano
Es el cis-1,3-dimetilciclohexano meso
Son el (+) y (-) trans-1,3-dimetilciclohexano

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
95
[1]
Jean Baptiste Biot (1774-1862), Científico francés, nació en París, Físico del College de france. [2]
R. S. Cahn, Profesor de University College, Londres. [3]
Sir Christopher Ingold, (1883-1979), n. Ilford, Profesor de University College, Londres. [4]
Vlademir Prelog, (1906- ); n. Sarajevo, Profesor de la Universidad de Zagreb, Premio nobel en 1975. [5]
Emil Fischer; (1852 - 1919); Profesor en la Universidad de Berlín; Premio Nobel en 1902.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
96
REACCIONES ORGANICAS
TIPOS DE REACCIONES ORGÁNICAS
Atendiendo a la variación del esqueleto carbonado las reacciones orgánicas
suelen clasificarse en:
Adiciones.- Consisten en procesos en los que el esqueleto carbonado experimenta un
incremento en el número de átomos a través de la incorporación de los átomos del
reactivo y sin ninguna pérdida de los que poseía inicialmente. Ejemplos:
Eliminaciones.- Consisten en procesos en los que el esqueleto carbonado experimenta
una disminución en el número de átomos originales al perderse un fragmento pequeño
(habitualmente no carbonado) por la acción de un reactivo, en cierto sentido pueden ser
consideradas como las reacciones inversas de las adiciones. Ejemplo:
Sustituciones.- Consisten en procesos en los que un átomo o grupo de átomos del
compuesto de partida es reemplazado por un átomo o grupo de átomos procedente del
reactivo. Ejemplos:
Transposiciones.- Consisten en procesos en los que suele modificarse el número de
átomos de carbono presentes en la molécula de partida, variando única y
exclusivamente la disposición relativa de los átomos entre si, resulta frecuente que las
transposiciones puedan ir acompañadas de pérdida de una molécula pequeña no
carbonada, tales como agua, amoniaco, hidrácidos, etc. Ejemplo:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
97
Condensaciones.- Consisten en procesos en los en los que el esqueleto carbonado
experimenta un incremento en el número de átomos a través de la unión de dos
moléculas relativamente pequeñas, con la consiguiente formación de enlaces C-C. No es
infrecuente que la condensación vaya acompañada de la pérdida de una molécula
pequeña tal como agua, amoniaco, etc. Ejemplo:
Degradaciones.- Consisten en procesos en los que el esqueleto carbonado experimenta
una disminución en el número de átomos de carbono, a través de la pérdida de un
fragmento carbonado habitualmente no muy voluminoso. En estas reacciones se rompen
enlaces C-C. Ejemplo:
Inserciones.- Consisten en procesos en los que un resto carbonado o un heteroátomo
portador de un par electrónico enlazante se inserta, es decir, se intercala entre dos
átomos unidos entre sí por un enlace o En estas reacciones se generan dos nuevos
enlaces a partir del par inicial o y el aportado por el reactivo. Ejemplos:
Extrusiones.- Consisten en procesos en los que se expulsa un átomo o grupo de átomos
de una molécula que se encontraba unido a otros dos por un enlace o dos enlaces ,
quedando unido estos dos átomos entre sí por un nuevo enlace o un enlace . Este tipo
de reacción se puede considerar como la inversa a la inserción. Ejemplos:
Oxidación - Reducción (Redox).- Estas reacciones implican transferencia de electrones
o cambio en el número de oxidación. Una disminución en el número de átomos de H
enlazados al carbono y un aumento en el número de enlaces a otros átomos como C,
O, N, Cl, Br, F y S indican que hay oxidación. Ejemplos:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
98
Los nueve tipos de reacciones consideradas aquí son los tipos generales de
reacciones que se conocen en química orgánica, resulta apropiado investigar qué es lo
que se encuentra entre el sustrato y el producto en una reacción típica. Esto es, cuáles
son los intermedios de la reacción.
Intermedios:
Una descripción detallada, paso a paso, de lo que ocurre entre los materiales
iniciales y el producto proporciona la explicación de lo que se llama mecanismo de
reacción.
Un mecanismo implica uno o varios intermedios. A primera vista, la lista de
posibles intermedios es tan larga que descorazonaría a cualquiera. Sin embargo si se
examina mas de cerca los detalles de cualquier reacción orgánica, se llega a una de las
más fascinantes generalizaciones de la disciplina “casi todas las reacciones orgánicas
se desarrollan a través de cuatro tipos de intermedios”. Basándose en la abundancia o
deficiencia de electrones alrededor del átomo de carbono reactivo, los cuatro tipos de
intermedios son:
Los que tienen un átomo de carbono positivamente cargado,
Los que tienen un átomo de carbono negativamente cargado,
Un radical libre neutro con vacante para un electrón y
Un carbono neutro con vacantes para dos electrones.
1.- INTERMEDIOS CON UN ÁTOMO DE CARBONO POSITIVAMENTE
CARGADO.
El carbono puede tener una carga positiva formal debido a una deficiencia de
electrones. Puesto que el intermediario es un catión del carbono, se llama carbocatión.
Carbocatión
Una reacción SN1 típica, como la hidrólisis del cloruro de t-butilo que se muestra
a continuación, se desarrolla a través de un carbocatión intermedio.
Cloruro de t-butilo Carbocatión Alcohol t-butilico
Sin embargo, cuando la carga del carbono no está totalmente establecida, es un
complejo activado (en el estado de transición de una reacción), no se le asigna una carga
formal definida. Se utilizan en su lugar cargas parciales para describir el complejo,
como se indica a continuación:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
99
Las reacciones SN2 se desarrollan típicamente en un sólo paso a través de un
estado de transición. En el ejemplo específico SN2 que sigue, el ion yoduro es
desplazado por el ion hidróxido.
No se trata de igualar aquí un complejo activado con un intermedio. Lo esencial
es que el carbono sigue siendo positivo, no importa cual sea el valor alcanzado por la
carga.
2.- INTERMEDIO CON UN ÁTOMO DE CARBONO CARGADO
NEGATIVAMENTE.
El carbono puede tener un octeto lleno y un exceso de electrones en relación con
el número total de protones en el núcleo. Por consiguiente, puede llevar una carga
formal negativa. Este anión del carbono se conoce apropiadamente como carbanión.
Carbanión
La condensación de Claisen, que se muestra a continuación, constituye una
reacción típica que se desarrolla mediante un carbanión intermedio. Una vez formado, el
carbanión intermedio reacciona con el acetato de etilo que no ha reaccionado todavía,
para desplazar el ion alcoholato (EtO-), con la formación concomitante del acetoacetato
de etilo.
Acetato de etil Carbanión Acetoacetato de etilo

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
100
3.- UN ÁTOMO DE CARBONO SIN CARGA, CON VACANTE PARA UN
ELECTRÓN (RADICAL LIBRE).
En este radical libre, no hay carga en el átomo de carbono, pero dicho
intermedio sigue siendo deficiente en electrones en relación con su capacidad de
electrones totales.
Radical libre
La ruptura homolítica de un enlace covalente puede producir un radical libre
como intermedio. por ejemplo, la bromación del 2-metilpropano, se inicia con la
disociación homolítica del bromo y continua con la formación de un radical libre que
contiene carbono. El producto (bromuro de isopropilo) se forma cuando el radical
isopropilo reacciona con el bromo molecular.
Etapa I
Etapa II
Radical libre isopropilo
Etapa III
4.- UN ÁTOMO DE CARBONO SIN CARGA CON VACANTE PARA DOS
ELECTRONES (CARBENO).
Tampoco aquí hay carga sobre el átomo de carbono, pero éste es deficiente en
electrones y divalente. El intermedio de este tipo se conoce como carbeno. En el
ejemplo que se muestra a continuación, se genera diclorocarbeno a partir de cloroformo
y de una base fuerte, luego se añade el ciclopenteno para formar el producto bicíclico
observado.
diclorocarbeno 6,6-diclorobiciclo (3.1.0) hexano

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
101
La selección de un intermedio es de suma importancia en la definición de un
mecanismo. Una vez que se ha hecho la selección, todas las etapas del mecanismo
propuesto deberán apoyarse en los conceptos fundamentales que les corresponda. Estos
conceptos, son entre otros, polarización, los efectos inductivos, los cálculos de carga
formal, los efectos de resonancia y los de la teoría ácido-base.
Conceptos fundamentales:
Polarización.-Los enlaces covalentes están polarizados con excepción de aquellos entre
átomos idénticos. La dirección de la polarización sigue la escala de electronegatividad
de Pauling. Los elementos más electronegativos que el carbono producirán una mayor
polarización en dichos enlaces. Así por ejemplo, un enlace C-Cl está polarizado con una
ligera carga positiva en el carbono y una ligera carga negativa en el cloro. La
polarización opuesta, esto es, una carga positiva en el cloro y una carga negativa en el
carbono no es consistente con la escala de la electronegatividad. La habilidad para
identificar la correcta polarización de los enlaces covalentes le permite a uno predecir
los sitios de reactividad, o determinar la estabilidad que tienen las moléculas orgánicas o
los intermediarios.
Efecto inductivo.-La polarización puede inducir un dipolo en un enlace próximo al
sustituyente o en uno más alejado. Esto se conoce como el efecto inductivo. Los efectos
inductivos son transmitidos directamente a través de una cadena de átomos dentro de
una molécula pero no a través del espacio vacío, ni por la acción de las moléculas del
disolvente.
Los efectos inductivos pueden ocasionar la liberación de electrones o la captura
de los mismos. Un efecto inductivo en que se liberan electrones se relaciona con la
tendencia de una sección de una molécula o de un sustituyente a despojarse de
electrones, aun cuando esto no implica la donación formal de una unidad completa de
carga. Los grupos alquilo son donadores de electrones (relativos al hidrógeno). Por
ejemplo, al efecto inductivo que interviene en la liberación de electrones del grupo
metilo, se le debe la estabilidad de los carbocationes en la secuencia terciario
secundario primario metilo.
Este hecho se puede explicar cualitativamente por el supuesto de que el grupo
metilo tiene más electrones disponibles para donar que el átomo de hidrógeno.
Un efecto inductivo consistente en la captura de electrones se relaciona con la
tendencia de una sección de una molécula a aceptar electrones. Los grupos o átomos
que presentan los efectos de captura de electrones son más numerosos que aquellos en
que el efecto inductivo se manifiesta en la donación de electrones. En un ejemplo típico,
el bromo en el ácido bromoacético atrae electrones, haciendo que el protón ácido se
pierda con más facilidad que el protón correspondiente al ácido acético.
pKa = 2.86 en Br-CH2CO2H
pKa = 4.76 en CH3-CO2H

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
102
El flujo de electrones se dirige al bromo (alejándose del protón ácido) de la
manera que se indica a continuación:
El anión en el bromoacetato es más estable que el anión en el acetato por una
razón semejante. La carga en el bromoacetato es estabilizada inductivamente por el
bromo receptor de electrones.
ion bromoacetato ion acetato
Los conceptos de carga formal, efectos de resonancia y teoría ácido-base han
sido detallados en otros capítulos (ver ácido-base y teoría de enlace y resonancia).
ALGUNAS PAUTAS PARA ESCRIBIR LOS MECANISMOS DE LAS
REACCIONES:
1. Un mecanismo debe explicar la formación del producto.
2. Típicamente, los compuestos orgánicos aislables (que no sean intermedios)
contienen cuatro enlaces. El carbono, en su estado básico tiene cuatro enlaces (y
sólo cuatro).
3. Las flechas curvas indican la dirección del flujo de electrones dentro de una
estructura.
4. La base más fuerte que puede existir en el agua es el ion hidróxido; el ácido más
fuerte que puede existir en el agua es el ion hidronio, H3O+.
5. En una solución ácida, el ion hidroxilo de un alcohol nunca se pierde, sólo se separa
para formar una molécula de agua.
6. El orden de estabilidad de un carbacatión es terciario secundario primario CH3+.
7. El orden de estabilidad de un radical libre es terciario secundario primario CH3
..
8. El orden de estabilidad de un carbanión es CH 3- primariosecundario terciario.
9. Los átomos de carbono del grupo carbonilo, C=O, son electropositivos; loa átomos
de oxígeno del grupo carbonilo son electronegativo.
10. En general, los efectos de resonancia son más importantes que los efectos inductivos
en la estabilidad de los intermedios.
11. Otros conceptos generales, los cuales comentaremos a continuación:
ENERGÍAS DE DISOCIACIÓN DE ENLACE.-
La energía de disociación del enlace (dada como H en kcal/mol o kJ/mol) es la
energía que se necesita para la homólisis endotérmica de un enlace covalente A:B
A + B ; H = (+). La formación de enlace, lo contrario de esta reacción, es
exotérmica y los valores de H son negativos. Ejemplo:
Rupturas endotérmicas Formación exotérmicas
La reacción en general es exotérmica con un H = (-) 24 kcal/mol

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
103
Velocidades de las reacciones.-
La velocidad de la reacción dA + eB fC + gD, está dada por: velocidad (v) =
kAxB
y donde k es la constante de velocidad a temperatura T. Los valores numéricos de
los exponentes x e y se determinan experimentalmente; no necesitan ser los mismos que
d y e, los coeficientes de la reacción química. La suma de los valores de los exponentes
se define como el orden de la reacción.
Dadas ciertas condiciones, los factores que determinan la velocidad de una
reacción son:
1. El número de colisiones por unidad de tiempo.
2. La entalpía de activación (Energía de activación, E. acti.)
TEORÍA DEL ESTADO DE TRANSICIÓN Y DIAGRAMAS DE ENTALPÍA.
Cuando los reactivos se han encontrado en colisión con suficiente entalpía (H)
de activación y debida orientación, pasan a través de un Estado de Transición (ET)
hipotético en el cual algunos enlaces se rompen y otros se pueden formar.
La relación entre el estado de transición (ET), los reaccionantes (R) y productos
(P) se muestra mediante el diagrama de la figura, para una reacción exotérmica
A + B C + D de un solo paso.
La formación de moléculas con baja entalpía se favorece en el estado de
equilibrio, o sea, C + D. Sin embargo esto se aplica únicamente cuando H de una
reacción predomina sobre TS en la determinación del estado de equilibrio.
TERMODINÁMICA DE LAS REACCIONES.-
La Termodinámica y la velocidad de una reacción determinan si esta reacción
puede ocurrir. La termodinámica de un sistema se describe en términos de varias
funciones importantes.
E, el cambio en la energía, es igual a qv, el calor transferido a/o desde un sistema
a volumen constante: E = qv.
H, el cambio en la entalpía, es igual a qp, el calor transferido a/o desde un sistema
a presión constante: H = qp. Puesto que la mayoría de las reacciones orgánicas se
efectúan a presión atmosférica en recipientes abiertos H se utiliza más a menudo

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
104
que E. El H de una reacción química es la diferencia en las entalpías de los
productos, H(P), y los reaccionantes, H(R): H = HP - HR.
S, es el cambio en la entropía. La entropía es la medida de la distribución al azar.
Mientras mayor sea este azar, más grande será S; mientras más grande sea el orden,
más pequeño será S. Para una reacción: S = SP - SR.
G, es el cambio en la energía libre. A presión constante, G = H - TS, (T =
temperatura absoluta).
REACTIVOS ELECTROFÍLICOS Y NUCLEOFÍLICOS.-
Las reacciones generalmente ocurren en los sitios reactivos de las moléculas y
los iones. Estos sitios se agrupan principalmente dentro de dos categorías. La primera
tiene una alta densidad electrónica porque el sitio es negativo. Tales sitios ricos en
electrones son llamados Nucleofílicos y las especies que poseen tales sitios se
denominan Nucleófilos o donantes de electrones; pueden ser de tres tipos los
nucleófilos:
a) Especies con un par de electrones no compartidos.
b) Especies con el extremo (-) de un enlace polar.
c) Especies con electrones .
La segunda categoría son especies capaces de adquirir electrones. Estos sitios
deficientes de electrones son Electrofílicos y las especies que los poseen se denominan
Electrofilos o receptores de electrones; pueden ser de dos tipos los electrofilos:
a) Especies neutras capaz de adquirir más electrones.
b) Especies con el extremo (+) de un enlace polar.
Pueden ocurrir muchas reacciones por la formación de un enlace entre un sitio
nucleofílico y uno electrofilico.
REPRESENTACIÓN GRÁFICA DE UN MECANISMO DE REACCIÓN:
a) Reacción en un solo paso, con formación de un estado de transición. La reacción del
ion hidróxido con el cloruro de metilo es un ejemplo de una reacción muy general,
conocida como SN2, que implica probablemente un perfil como este.
CH3Cl + OH- CH3OH + Cl
-

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
105
b) Reacción en dos pasos, con formación de un intermedio de reacción. La reacción del
cloruro de t-butilo con el ion hidróxido podría estar representada por este tipo de perfil
de reacción. La cual es una reacción SN1 típica.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
106
[1]
Melvin S. Newman (1908) Nueva York; Ph. D. (1932) Yale Universite; Profesor en la Ohio State
University. [2]
Adolf von Baeyer (1835-1917); Berlín; Ph. D. Berlín (1858); Premio Nobel (1905). [3]
Ernst Mohr (1873-1926); Dresde; Ph.D Kiel (1897), Profesor de la Universidad de Heidelberg.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
107
REACCIONES DE LOS ALCANOS
PREPARACIÓN:
Cada uno de los alcanos menores, desde el metano hasta el n-pentano y
el isopentano, puede obtenerse en forma pura por destilación fraccionada del petróleo y
del gas natural. Más allá de los pentanos, el número de isómeros de cada homólogo se
hace tan grande y las diferencias en sus puntos de ebullición tan pequeños, que resulta
imposible aislar compuestos individuales puros; estos alcanos deben ser sintetizados por
alguno de los métodos que expondremos a continuación.
PREPARACIÓN DE LOS ALCANOS
1.- Hidrogenación de alquenos.
Ejemplo:
2.- Reducción de halogenuros de alquilo.
(a) Hidrólisis de reactivos de Grignard.
Ejemplo:
(b) Reducción con metal y ácido.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
108
Ejemplo:
3.- Acoplamiento de halogenuros de alquilo con compuestos organometálicos.
Ejemplos:
La hidrogenación de alquenos es, por mucho, la más importante de las
reacciones de obtención de alcanos. La única limitación del procedimiento es la
asequibilidad del alqueno apropiado, la cual no es muy seria, pues los alquenos se
pueden preparar fácilmente de alcoholes, que a su vez pueden sintetizarse sin dificultad
en gran variedad de tamaños y formas.
La reducción de un halogenuro de alquilo, ya por medio de un reactivo de
Grignard, ya directamente con metal y ácido, implica sólo el reemplazo de un átomo de
halógeno por uno de hidrógeno; el esqueleto carbonado permanece intacto. Este método
tiene más o menos la misma amplitud que el anterior, puesto que los halogenuros de
alquilo, al igual que los alquenos, generalmente se preparan de alcoholes. El
acoplamiento de los halogenuros de alquilo con compuestos organometálicos es el único
de estos métodos que forma enlaces carbono-carbono, generando un esqueleto
carbonado nuevo y de mayor tamaño.
REACCIONES:
A veces nos referimos a los alcanos empleando el nombre anticuado de
parafinas. Este nombre se les dio para describir lo que parecía una reactividad baja para
estos hidrocarburos.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
109
REACCIONES DE LOS ALCANOS
1.- Halogenación.
Ejemplo:
2.- Combustión.
Ejemplo:
3.- Pirólisis.
HALOGENACIÓN DE LOS ALCANOS:
Cuando una mezcla de metano y cloro se calienta a 120 ºC aproximadamente o
se irradia con la luz de longitud de onda apropiada, tiene lugar una reacción exotérmica.
Esta reacción constituye un proceso industrial importante para la preparación de
cloruro de metilo. Su utilidad como preparación de laboratorio está limitada, ya que la

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
110
reacción no termina con la introducción de un solo átomo de cloro. A medida que la
concentración de cloruro de metilo aumenta, experimenta la reacción de cloración en
competencia con el metano.
El producto real de la cloración del metano con el cloro es una mezcla de cloruro
de metilo (p. eb. -24.2 ºC), cloruro de metileno (CH2Cl2, p. eb. 40.2 ºC), cloroformo
(CHCl3, p. eb. 61.2 ºC), y tetracloruro de carbono (Cl4C), p. eb. 76.8 ºC). La
composición de la mezcla depende de las cantidades relativas de los materiales de
partida utilizadas y de las condiciones de reacción. En este caso resulta fácil separar los
productos por destilación fraccionada debido a la diferencia entre los diferentes punto
de ebullición.
Para la cloración del metano, gran parte de las pruebas experimentales coinciden
con el mecanismo siguiente: la reacción comienza con la homólisis (ruptura homolítica)
de una molécula de cloro en dos átomos de cloro. Desde el momento en que los átomos
de cloro estén presentes en pequeñas cantidades, comienza una reacción en cadena. Un
átomo de cloro reacciona con una molécula de metano para dar lugar a un radical metilo
y una molécula de HCl. El radical metilo reacciona entonces con una molécula de cloro
para dar cloruro de metilo y un átomo de cloro. El átomo de cloro producido puede
reaccionar con otra molécula de metano para continuar la cadena.
Así, el primer paso de la reacción se denomina iniciación, y los dos siguientes
pasos se denominan propagación de la cadena. Cuando dos especies radicales chocan
entre sí, el proceso se denomina terminación.
I.- Iniciación:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
111
II.- Propagación:
III.- Terminación:
En principio, solamente una molécula de cloro ha de experimentar la ruptura
homolítica para transformar muchos moles de metano y cloro en cloruro de metileno y
HCl. En la práctica, el proceso en cadena recorre una media de 10.000 ciclos antes de
finalizar. La terminación del proceso se produce cuando dos radicales chocan entre sí.
La variación total de entalpía Hº corresponde a la cloración, y puede ser
obtenida mediante la suma de las etapas de propagación solamente.
ANALICEMOS LOS DIAGRAMAS DE ENERGÍA PARA LAS ETAPAS DE
PROPAGACIÓN:
Los Hº de cada una de las etapas se calcula en función de las energías de
disociación de enlace, las cuales ya se encuentran calculadas y representadas en tablas.
La ruptura de enlace implica aplicar energía por lo tanto se toma como un valor positivo
(+) para indicar que es energía aplicada al sistema. La formación de enlace implica
liberación de energía, por lo tanto se toma como un valor negativo (-), para indicar que
es una energía liberada por el sistema. La suma total de los enlaces rotos mas los
enlaces formados nos indica el Hº total de la reacción.
Veamos la primera etapa de la propagación:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
112
Es estado de transición estaría representado por:
Veamos la segunda etapa de la propagación:
El estado de transición estaría representado por:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
113
La cloración de los alcanos superiores es similar a la cloración del metano
exceptuando que las mezclas de productos obtenidos son más complejas. El etano no
sólo produce cloruro de etilo, sino también 1,1-dicloroetano y 1,2-dicloroetano.
En el caso del propano, pueden formarse dos productos monoclorados, esto es,
el cloruro de n-propilo y el cloruro de isopropilo aunque no se producen cantidades
iguales.
Cloruro de n-propilo Cloruro de
isopropilo
A 25 ºC, los dos isómeros se producen en la proporción de 43:57
respectivamente. Un tiempo de reacción prolongado da lugar a la mezcla de los cuatro
dicloropropanos posibles.
Examinemos con mayor detalle la monocloración del propano. Recuérdese que
el calor de disociación del hidrógeno secundario es 3 kcal/mol inferior al calor de
disociación correspondiente al hidrógeno primario (ver Tabla 8.1). Se puede anticipar,
entonces, que el hidrógeno secundario sería eliminado más fácilmente por el átomo de
cloro que el hidrógeno primario. Sin embargo existe 6 hidrógenos primarios frente
solamente a dos hidrógenos secundarios que pueden ser reemplazados por el cloro. La
reactividad relativa por hidrógeno es entonces:
Una tendencia similar se observa en la monocloración del 2-metilpropano que
proporciona 36 por 100 de cloruro de t-butilo y 64 por 100 de cloruro de isobutilo.
La reactividad relativa de los hidrógenos terciarios y primarios, por hidrógeno,
resulta ser:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
114
Por esta razón, las velocidades relativas de reacción de los diferentes tipos de
hidrógenos con el cloro radicalario son tal y como de deduce de los valores de los
calores de disociación de enlace para los distintos tipos de hidrógenos.
terciario secundario primario ( 5.1 : 4 : 1 )
TABLA 8.1.-
ENERGÍA DE DISOCIACIÓN DE ENLACES SIMPLES EN kcal/mol
A:B A· + B·
Compuesto D Compuesto D
H-H 104 (CH3)2CH-H 94.5
D-D 106 (CH3)2CH-F 105
F-F 38 (CH3)2CH-Cl 81
Cl-Cl 58 (CH3)2CH-Br 68
Br-Br 46 (CH3)2CH-I 53
I-I 36 (CH3)2CH-OH 80.5
H-F 136 (CH3)2CH-OCH3 80.5
H-Cl 103
H-Br 87.5 (CH3)3C-H 91
H-I 71 (CH3)3C-Cl 78.5
CH3-H 104 (CH3)3C-Br 63
CH3-F 108 (CH3)3C-I 49.5
CH3-Cl 83.5 (CH3)3C-OH 90.5
CH3-Br 70 (CH3)3C-OCH3 78
CH3-I 56 C6H5CH2-H 85
CH3-OH 91.5 CH2=CHCH2-H 85
CH3-OCH3 80 CH2=CH-H 103
CH3CH2-H 98 C6H5-H 103
CH3CH2-F 106 HCC-H 125
CH3CH2-Cl 81.5 CH3-CH3 88
CH3CH2-Br 69 CH3CH2-CH3 85
CH3CH2-I 53.5 CH3CH2CH2-CH3 85
CH3CH2-OH 91.5 CH3CH2-CH2CH3 82
CH3CH2-OCH3 80 (CH3)2CH-CH3 84
(CH3)3C-CH3 80
CH3CH2CH2-H 98 HO-H 119
CH3CH2CH2-F 106 HOO-H 90
CH3CH2CH2-Cl 81.5 HO-OH 51
CH3CH2CH2-Br 69 CH3CH2O-OCH3 44
CH3CH2CH2-I 53.5
CH3CH2CH2-OH 91.5 CH2=CH2 2CH2: 145
CH3CH2CH2-OCH3 80
Reproducida desde: S. W. Benson, “Bond Energies”, J. Chem. F.d., 42, 502 (1965)

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
115
Con alcanos más complejos, las mezclas de cloración son irremediablemente
más complejas. Por ello la cloración de alcanos no es una reacción general adecuada
para preparar cloruros de alquilo. Existe un tipo de compuestos para el que la cloración
tiene utilidad práctica en las preparaciones de laboratorio. Cuando todos los hidrógenos
son equivalentes, existe sólo un producto monoclorado posible. En tales casos, el
producto deseado puede ser separado del hidrocarburo y las especies cloradas mediante
la destilación fraccionada de la mezcla. Dos ejemplos son la cloración del ciclohexano y
la del 2,2-dimetilpropano.
El mecanismo de cloración puede ser aplicado también a los otros halógenos,
pero las reacciones muestran importantes diferencias. En la Tabla 8.2 se recogen las
entalpías totales de la halogenación del metano con diferentes halógenos.
TABLA 8.2
CH4 + X2 = CH3X + HX
X (halógeno) Hº = kcal/mol
F
Cl
Br
I
La reacción con el flúor es tan fuertemente exotérmica que resulta difícil llevar a
cabo una fluoración controlada. La yodación está en el extremo opuesto. La reacción del
metano con el yodo es endotérmica.
La bromación del metano es menos exotérmica que la cloración. De los dos
pasos de propagación de la cadena, solamente uno es relativamente exotérmico:
Por consiguiente, la bromación es mucho más lenta que la cloración. Desde el
punto de vista mecanístico el bromo es mucho más selectivo que el cloro en sus
reacciones con otros alcanos. Por ejemplo, la bromación del propano a 330 ºC en fase
de vapor arroja un 92 por 100 de bromuro de isopropilo y solamente un 8 por 100 del
bromuro de n-propilo.
Los pasos de abstracción de hidrógenos para la formación de los dos isómeros son:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
116
La selectividad del bromo en comparación con la del cloro se hace más evidente
cuando el alcano tiene hidrógenos terciarios. Por ejemplo, cuando el 2,2,3-
trimetilbutano se somete a bromación, se obtiene más de un 96 por 100 del bromuro de
terciario, a pesar de que el alcano tiene solamente un hidrógeno terciario frente a quince
hidrógenos primarios.
(96 por
100)
Así pues, la bromación es un procedimiento un poco más útil que la cloración
para propósitos preparativos. Sin embargo, cuando existe solamente un hidrógeno
terciario y muchos secundarios en una molécula, se seguirán produciendo mezclas muy
complejas.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
117
HALOGENUROS DE ALQUILO
Los halogenuros de alquilo son compuestos que contienen halógeno unido a un
átomo de carbono saturado con hibridación sp3. El enlace C-X es polar, y por tanto los
halogenuros de alquilo pueden comportarse como electrófilos.
Los halogenuros de alquilo pueden obtenerse mediante halogenación por
radicales de alcanos, pero este método es de poca utilidad general dado que siempre
resultan mezclas de productos. El orden de reactividad de los alcanos hacia la cloración
es idéntico al orden de estabilidad de los radicales: terciario secundario primario.
Conforme al postulado de Hammond[1]
, el radical intermedio más estable se forma más
rápido, debido a que el estado de transición que conduce a él es más estable.
Los halogenuros de alquilo también pueden formarse a partir de alquenos. Estos
últimos se unen a HX, y reaccionan con NBS para formar el producto de bromación
alílica. La bromación de alquenos con NBS es un proceso complejo por radicales que
ocurre a través de un radical alilo. Los radicales alilos son estabilizados por resonancia
y pueden representarse de dos maneras, ninguna de las cuales es correcta por sí misma.
La verdadera estructura del radical alilo se describe mejor como una mezcla o híbrido
de resonancia de las dos formas resonantes individuales.
Los alcoholes reaccionan con HX para formar halogenuros de alquilo, pero este
método sólo funciona bien para alcoholes terciarios, R3C-OH. Los halogenuros de
alquilo primarios y secundarios normalmente se obtienen a partir de alcoholes usando
SOCl2 o PBr3. Los halogenuros de alquilo reaccionan con magnesio en solución de éter
para formar halogenuros de alquil-magnesio, o reactivos de Grignard[2]
, RMgX.
Algunos reactivos de Grignard son tanto nucleófilos como básicos, y reaccionan con
ácidos de Bronsted para formar hidrocarburos. El resultado global de la formación del
reactivo de Grignard y su protonación es la transformación de un halogenuro de alquilo
en un alcano (R-X RMgX R-H).
Los halogenuros de alquilo también reaccionan con litio metálico para formar
compuestos de alquil-litio, RLi, que en presencia de CuI forman diorganocupratos o
reactivos de Gilman[3]
, R2CuLi. Estos reaccionan con halogenuros de alquilo para
formar hidrocarburos de acoplamiento como productos.
ESTRUCTURA DE LOS HALOGENUROS DE ALQUILO:
El enlace carbono-halógeno en los halogenuros de alquilo resulta de la
superposición de un orbital de carbono con hibridación sp3 y un orbital de halógeno. Así,
los átomos de carbono de los halogenuros de alquilo tienen configuración geométrica
aproximadamente tetraédrica, con ángulos de enlace H-C-X cercanos a 109º. Los
halógenos aumentan de tamaño al descender en la tabla periódica, incremento que se
refleja en las longitudes de enlaces en la serie de los halometanos.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
118
Como los halógenos son más electronegativos que el carbono, el enlace
resultante entre ambos resulta polarizado, el átomo de carbono tiene una ligera carga
positiva (+), mientras que el átomo de halógeno tiene una ligera carga negativa (
-).
Dado que el átomo de carbono de los halogenuros de alquilo está polarizado
positivamente, estos compuestos son buenos electrófilos. Se verá que gran parte de la
química de los halogenuros de alquilo está determinada por su carácter electrófilo.
OBTENCIÓN DE LOS HALOGENUROS DE ALQUILO
1.- A partir de alquenos por bromación alílica.
Ejemplo:
2.- A partir de alquenos por adición de HBr y HCl.
Ejemplo:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
119
3.- A partir de alcoholes.
(a) Reacción con HX, donde X = Cl, Br o I; Orden de reactividad: 3º 2º 1º.
Ejemplo:
(b) Reacción de alcoholes primarios y secundarios con SOCl2 en piridina.
Ejemplo:
(c) Reacción de alcoholes primarios y secundarios con PBr3, en éter.
Ejemplo:
1.- BROMACIÓN ALÍLICA DE ALQUENOS.
En 1942, Karl Ziegler[4]
informó que al repetir algunos trabajos previos de
Wohl[5]
, observó que los alquenos reaccionan con N-bromosuccinimida (que se abrevia
NBS) en presencia de luz, para formar productos que resultan de la sustitución de
hidrógeno por bromo en la posición adyacente al doble enlace (la posición alílica). Por
ejemplo, el ciclohexeno se convierte en 3-bromocioclohexeno, con un 85% de
rendimiento:
Cicloheheno 3-bromociclohexeno (85%)

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
120
Esta bromación alílica con NBS es muy parecida a la reacción de halogenación
de alcanos: En ambos casos se rompe un enlace C-H de un carbono saturado, y el átomo
de hidrógeno es reemplazado por un halógeno. La analogía es adecuada, pues se ha
demostrado que las bromaciones con NBS efectivamente ocurren a través de un
mecanismo por radicales. Si bien el mecanismo exacto de la reacción es complejo, el
paso crucial que determina el producto implica la sustracción de un átomo de hidrógeno
alílico y la formación del radical correspondiente. El radical reacciona luego con Br2
para formar el producto y un radical bromo, que regresa a la reacción para continuar la
cadena. El Br2 necesario para la reacción con el radical alilo es producido por una
reacción concurrente de HBr con NBS.
Mecanismo:
¿Por qué la bromación ocurre exclusivamente en la posición vecina al doble enlace y no
en cualquiera de las otras posiciones? Una vez más, la respuesta se encuentra
examinando las energías de disociación de enlace para ver las estabilidades de los
diversos tipos de radicales.
Además de influir en la estabilidad, la deslocalización de los electrones no
pareados en el radical alilo tiene consecuencias químicas. Dado que el electrón no
pareado está distribuido en ambos extremos de la del sistema de orbitales pi, la
bromación alílica de un alqueno asimétrico con frecuencia produce una mezcla de
productos. Por ejemplo, la bromación del 1-octeno produce una mezcla de 3-bromo-1-
octeno y 1-bromo-2-octeno. Debido a que el radical alilo intermediario es en este caso
asimétrico, la reacción no es igualmente probable en ambos extremos, y los dos
productos no se forman en cantidades iguales.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
121
Explicación mecanistica:
Dado que en la bromación de alquenos asimétricos se forman mezclas de
productos, la bromación alílica transcurre mejor en alquenos simétricos, como el
ciclohexeno. Los productos de esas reacciones son particularmente útiles para su
conversión en dienos por deshidrohalogenación con base.
2.- OBTENCIÓN DE HALOGENUROS DE ALQUILO A PARTIR DE
ALCOHOLES.
El mejor método para producir halogenuros de alquilo es la síntesis a partir de
alcoholes. El método más sencillo (aunque generalmente el menos útil) para la
conversión de un alcohol en un halogenuro de alquilo consiste en el tratamiento del
alcohol con HCl, HBr o HI:
El método funciona bien cuando es aplicado a alcoholes terciarios, R3C-OH. Los
alcoholes secundarios y primarios también reaccionar pero a velocidades menores y a
temperaturas de reacción considerablemente altas. La reacción de HX con un alcohol
terciario es tan rápida que a menudo se efectúa simplemente burbujeando el HX gaseoso
puro en una solución fría de alcohol en éter. La reacción suele realizarse en unos
cuantos minutos.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
122
La mejor forma de transformar los alcoholes primarios y secundarios en
halogenuros de alquilo es tratándolos con reactivos como el cloruro de tionilo (SOCl2) o
tribromuro de fósforo (PBr3). Estas reacciones normalmente ocurren en condiciones
suaves, menos ácidas, y es menos probable que ocasionen transposiciones catalizadas
por ácido que el método con HX.
Como indican los ejemplos anteriores, el rendimiento de las reacciones con PBr3,
y el SOCl2 generalmente es alto. No suelen interferir otros grupos funcionales como
éteres, carbonilos o anillos aromáticos. Los mecanismo los veremos con más detalles
en capitulas posteriores.
REACCIONES DE LOS HALOGENUROS DE ALQUILO
1.- Formación de reactivos de Grignard.
donde X = Br, Cl, o I
R = Alquilo 1º, 2º, 3º, arilo o vinilo
Ejemplo:
2.- Formación de diorganocupratos (reactivos de Gilman).
R = alquilo 1º, 2º , 3º, arilo o vinilo
Ejemplo:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
123
Ejemplo:
3.- Acoplamiento de compuestos organometalicos.
Ejemplo:
4.- Conversión de halogenuros de alquilo en alcanos.
Ejemplo:
Por el momento solo dejaremos anunciadas estas reacciones para los
halogenuros de alquilo, hay otras pero se clasifican de una manera distinta. En capítulos
posteriores se verán muchos usos más de los reactivos de Grignard y otros
organometálicos.
[1]
Geroge S. Hammond; (1921- ); Director for Chemical Dynamics Allied Chemical
Corporation, Ney Yersey [2]
Francois Auguste Victor Grignard (1871-1935); Profesor de la Universidad de Nancy, Lyon, Francia,
Premio Nobel en 1912. [3]
Henry Gilman (1893-1986), Profesor de la Universidad de Iowa State University [4]
Karl Ziegler, (1898-1976), Instituto Max Planck, Alemania, Premio Nobel en 1963. [5]
Alfred Wohl, (1883-1933), n. Graudentz, Alemania, Profesor de la Universidad de Danzig.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
124
REACCIONES DE LOS HALOGENUROS DE ALQUILO
(SUSTITUCIONES NUCLEOFÍLICAS)
SUSTITUCIÓN NUCLEOFÍLICA
Las sustituciones nucleofílicas son de dos tipos: reacción SN2 y reacción SN1.
En la primera, el nucleófilo entrante ataca al halogenuro desde una posición a 180º
respecto al grupo saliente, lo que da por resultado una inversión de la configuración
cuando en carbono en cuestión es quiral (análoga a la inversión de un paraguas por el
viento) en el átomo de carbono. La reacción tiene cinética de segundo orden, y es
fuertemente inhibida conforme aumenta el volumen estérico de los reactivos. Por tanto,
las reacciones SN2 se ven favorecidas sólo para sustratos primarios y secundarios.
La reacción SN1 ocurre cuando el sustrato se disocia de manera espontánea para
formar un carbocatión en un paso limitante de la velocidad lento, seguido por un ataque
rápido del nucleófilo. En consecuencia, las reacciones SN1 tienen cinética de primer
orden, y ocurren con racemización de la configuración en el átomo de carbono cuando
éste es quiral. Estas reacciones se ven favorecidas para los sustratos terciarios.
REACCIONES SN2
1.- Estas reacciones ocurren con inversión completa de la estereoquímica del
carbono estereogénico.
2.- Estas reacciones presentan cinética de segundo orden y obedecen a la siguiente
ley de velocidad: Velocidad = k · RX · Nu.
3.- Su mecanismo es en un sólo paso, formándose un estado de transición.
Ejemplos.
El mecanismo es concertado, en un solo paso de reacción.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
125
¿Qué mecanismo explica la estereoquímica observada y la cinética de segundo
orden para estas reacciones? En 1937, Hughes e Ingold propusieron la primera
explicación satisfactoria al formular el mecanismo de lo que ellos llamaron reacción
SN2, abreviatura de sustitución nucleofílica bimolecular. (Bimolecular porque, en el
paso cuya cinética se mide, participan dos moléculas: el nucleófilo y el halogenuro de
alquilo).
La característica esencial del mecanismo de reacción SN2 formulado por
Hughes e Ingold es que la reacción transcurre en un solo paso, sin intermedio. Cuando
el nucleófilo entrante ataca el sustrato desde una posición a 180º del grupo saliente, se
forma un estado de transición, en el cual comienza a formarse el enlace nucleófilo
carbono y simultáneamente comienza a romperse el enlace grupo saliente carbono,
invirtiéndose la configuración estereoquímica de la molécula. Este proceso se muestra
para la reacción de (S)-2-bromobutano con ion hidróxido, lo que da por resultado (R)-2-
butanol.
El nucleófilo
-OH utiliza los electrones de su
par no compartido para atacar el carbono del
halogenuro de alquilo en una dirección a 180º del
halógeno saliente. Esto da por resultado un estado
de transición en el cual el enlace C-OH está
parcialmente formado y el enlace C-Br está
parcialmente roto.
La estereoquímica del carbono se invierte a
medida que se forma el enlace C-OH y el ion
bromuro se desprende con el par de electrones del
enlace C-Br original.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
126
CARACTERÍSTICAS DE LA REACCIÓN SN2:
El estudiante ya tendrá una buena idea de cómo ocurren las reacciones SN2, pero
también es necesario saber cómo pueden usarse estas sustituciones y qué variables las
afectan. Algunas reacciones SN2 son rápidas y otras son lentas; algunas tienen altos
rendimiento, y otras rendimientos bajos, etc. La velocidad de una reacción química es
determinada por la energía de activación (G), la diferencia de energía entre los
reactivos y el estado de transición. Otros factores son.
EFECTO ESTÉRICO EN LA REACCIÓN SN2.
La primera variable por observar de una reacción SN2 es el volumen estérico del
halogenuro de alquilo. Como la reacción implica la formación de un estado de
transición parece razonable esperar que un sustrato voluminoso, impedido, dificultará el
acercamiento del nucleófilo, y de este modo será más difícil alcanzar el estado de
transición. En otras palabras los sustratos estéricamente voluminosos, en los cuales el
átomo de carbono está protegido contra el ataque del nucleófilo, reaccionan con mayor
lentitud que los sustratos menos impedidos. Como se muestra para los siguientes casos,
la dificultad para el ataque nucleofílico se eleva a medida que aumenta el tamaño de los
tres sustituyentes unidos al átomo de carbono.
Impedimento estérico a la reacción SN2. Como se indican en estos modelos, el átomo
de carbono en (a), bromometano, es fácilmente accesible, lo que da por resultado una
reacción SN2 rápida. Los átomos de carbono en (b), bromoetano (primario), en (c), 2-
bromopropano (secundario) y en (d), 2-bromo-2-metilpropano (terciario), son
sucesivamente menos accesibles, lo que da por resultado reacciones SN2 cada vez más
lentas.
Las reactividades relativas de algunos sustratos son como sigue:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
127
CH3-Br CH3-CH2-Br (CH3)2-CH-Br (CH3)3-C-Br
metilo primario secundario terc
iario
Ejemplo: Para la reacción R-Br + Nu- R-Nu + Br
- (SN2)
Grupo R Velocidad relativa
CH3- 30
CH3-CH2- 1
R’-CH2CH2- 0.4
(CH3)2-CH- 0.002
(CH3)3-C- 0.001
(CH3)3-C-CH2- 0.00001
NUCLEÓFILO ATACANTE.
La naturaleza del nucleófilo atacante es una segunda variable con notable efecto
en las reacciones SN2. Cualquier especie que sea neutra o cargada negativamente, puede
actuar como nucleófilo, siempre y cuando tenga un par de electrones no compartidos (es
decir, que sea una base de Lewis). Si el nucleófilo tiene carga negativa, el producto es
neutro, pero si el nucleófilo es neutro el producto queda con carga positiva.
Las especies nucleofílicas cargadas suelen ser mejores nucleófilos que las
especies neutras, entre especies de igual naturaleza el mejor criterio a emplear es el del
número atómico, del tal forma que aquellos que tengan el mayor número atómico serán
mejores especies nucleofílicas que las de menor número atómico.
La nucleofília exacta de una especie en una reacción dada depende de muchos
factores, como la naturaleza del sustrato, la identidad del solvente, e incluso la
concentración de los reactivos. Aunque no hay explicaciones precisas para las
nucleofílias observadas pueden detectarse algunas tendencias en los datos:
1. La nucleofília es aproximadamente paralela a la basicidad cuando se comparan
nucleófilos que tienen el mismo átomo atacante. El ion hidróxido por ejemplo, es
más básico y más nucleófilo que el agua. (Tabla 10.1).
2. La nucleofília suele aumentar al descender por una columna en la tabla periódica.
Así, por ejemplo el HS- es más nucleófilo que el OH
-.
3. La correlación de la nucleofília con la basicidad no es útil para comparar átomos de
una misma familia. Al aumentar el número atómico, aumenta la nucleofilia y
disminuye la basicidad. (Tabla 10.2).
Tabla 10.1.-
Nucleofília
decreciente
Nucleófilos con
átomos de
Nitrógeno
Nucleófilos con
átomos de
Oxígeno
Nucleófilos con
Nitrógeno
frente a
Oxígeno
Basicidad
Decreciente
H2N- C2H5O
- H2N
-

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
128
C2H5NH2 HO- HO
-
H3N C6H5O- H3N
C6H5NH2 CH3CO2- H2O
p-NO2C6H4NH2 H2O -
Tabla 10.2.-
Nucleofília
decreciente
Nucleófilos del
Grupo (V)
Nucleófilos del
Grupo (VI)
Nucleófilos del
Grupo (VII)
Basicidad
creciente
R3P RS- I
-
R3N RO- Br
-
- - Cl-
- - F-
GRUPO SALIENTE.
Otra variante que puede influir en gran medida en la reacción SN2 es la
naturaleza del grupo saliente. Dado que éste es expulsado con carga negativa en la
mayoría de las reacciones SN2, es de esperar que los mejores grupos salientes sean
aquellos que estabilicen mejor la carga negativa. Más aún puesto que la estabilidad de
un anión se relaciona con la basicidad, puede decirse que los mejores grupos salientes
serán las bases más débiles (a menor pKa del ácido conjugado que genere, (ver Tabla
10.3). Tan importante como saber cuales son buenos grupos salientes es saber cuales no
lo son, los datos anteriores indican con claridad que F-, OH
-, OR
- y NH2
- no son
desplazados por nucleófilos. En otras palabras fluoruros de alquilo, alcoholes, éteres y
aminas no experimentan reacciones SN2 en circunstancias normales.
Tabla 10.3.-
Relación entre la capacidad como grupo saliente y la acidez del ácido conjugado
Grupo Saliente pKa del ác. conjugado Habilidad como G. Saliente
p-CH3C6H4SO3- (OTs)
I-
Br-
H2O
(CH3)2S
Cl-
0
Muy Buenos Grupos
Salientes
CF3CO2- 0.2 “
H2PO4- 2 “
CH3CO2- 4.8 “
CN- 9.1
NH3 9.2
C6H5O- 10 Buenos Grupos Salientes
RNH2, R3N 10
C2H5S- 10.6
HO- 15.7 Malos Grupos Salientes
CH3O- 15.0 “

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
129
NH2- 36 Muy Malos G. Salientes
CH3- 49 “
DISOLVENTE.
Las velocidades de muchas reacciones SN2 se ven afectadas por el disolvente
que se use. Los disolventes próticos, que contienen grupos -OH, suelen ser los peores
para las reacciones SN2, mientras que los disolventes apróticos polares, que tienen
dipolos fuertes pero carecen de -OH o
-NH, son los mejores.
Los disolventes próticos, como metanol y etanol, retardan las reacciones SN2
porque afectan el nivel de energía del reactivo nucleófilo y no el nivel de energía del
estado de transición. Las moléculas de disolventes próticos son capaces de formar
enlaces de hidrógeno con los nucleófilos cargados negativamente, creando una “jaula”
alrededor del nucleófilo. Esta solvatación estabiliza fuertemente al nucleófilo,
reduciendo su reactividad hacia los electrófilos en la reacción S N2.
Un nucleófilo solvatado
su nucleofília reducida por el incremento en la estabilidad del estado fundamental
En contraste con lo que sucede en el caso de los disolventes próticos, los
disolventes polares apróticos favorecen las reacciones SN2. Son particularmente
valiosos el acetonitrilo, CH3CN, la dimetilformamida, (CH3)2NCHO (DMF), el
dimetilsulfóxido, (CH3)2SO (DMSO); y la hexametilfosforamida (CH3)2N3PO
(HMPA). Estos disolventes son capaces de disolver muchas sales debido a su alta
polaridad, y tienden a rodear a los cationes metálicos en vez de hacerlo con los aniones
nucleófilos, de este modo los nucleófilos no están solvatados y la nucleofilia es mucho
más efectiva.
La polaridad del disolvente es medida generalmente por su constante dieléctrica,
(D), que es una indicación sobre la facilidad del medio de reacción para acomodar
especies cargadas. (ver Tabla 10.4) Las reacciones que transcurren con un mecanismo
SN2, son más favorables en disolventes polares apróticos, y las que transcurren con un
mecanismo SN1 son más favorables en disolventes polares en general, ya que el paso
que controla la velocidad es la formación de un carbocatión.
Tabla 10.4.-
POLAR Disolventes Próticos Cte. Dieléctrica a 25ºC Disolventes apróticos.
H2O 81
HCO2H 59
45 (CH3)2SO (DMSO)
39 CH3CN

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
130
37 HCON(CH3)2 (DMF)
CH3OH 33
30 HMPA
CH3CH2OH 24
23 (CH3)2CO (Acetona)
(CH3)2CHOH 18
(CH3)3COH 11
7 THF
CH3CO2H 6
4 (C2H5)2O (éter)
2 n-C5H12; C6H6; Ccl4
NO POLAR (APOLAR)
Obsérvese que a mayor constante dieléctrica, mayor es la polaridad, es decir un
mayor poder de ionización del disolvente, según el poder ionizante del disolvente y el
tipo de reacción que se esté tratando los efectos del medio de reacción sobre las
velocidades de las reacciones de sustitución nucleófila son:
Mecanismo
Reacción
Efecto de incrementar el
poder ionizante del
disolvente (Polaridad)
SN1 R-L R+ + L- Gran aceleración
SN2 Nu- + R-L R-Nu + L- Pequeña deceleración
SN2 Nu + R-L R-Nu+ + L
- Gran aceleración
SN2 Nu- + R-L
+ R-Nu + L Gran deceleración
REACCIONES SN1
Es notable el hecho de que el panorama es del todo distinto cuando se utilizan
diferentes condiciones de reacción. Cuando se tratan en disolventes próticos con
nucleófilos no básicos en condiciones de neutralidad o acidez, los sustratos terciarios a
menudos reaccionan varios miles de veces más rápido que los primarios o secundarios.
Por ejemplo la reacción de los alcoholes con HX para obtener halogenuros de alquilo
tiene la rapidez máxima en el caso de alcoholes terciarios y la mínima en el caso del
metanol:
R-OH + HX R-X + H2O
R3COH R2CHOH RCH2OH CH3OH
3º 2º 1º Metanol
Más reactivo Menos reactivo
La misma tendencia se observa en muchas reacciones de sustitución en las
cuales los sustratos se calientan con nucleófilos no básicos en solventes próticos: Los
sustratos terciarios reaccionan mucho más rápido que los primarios o secundarios. Por
ejemplo, a continuación se presentan las velocidades relativas de reacción con agua de
algunos halogenuros de alquilo. Se observa que el 2-bromo-2-metilpropano es más de
un millón de veces más reactivo que el bromoetano.
R-Br + H2O R-OH + H-Br

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
131
(CH3)3CBr (CH3)2CHBr CH3CH2Br CH3Br
Reactividad relativa 1,200 0,012 0,001 0,0001
¿Qué sucede? Es claro que se trata de reacciones de sustitución nucleofilica,
aunque el orden de reactividad parece invertido. Estas reacciones no pueden estar
ocurriendo por el mecanismo que hemos venido considerando, y por tanto se concluye
que existe un mecanismo de sustitución alternativo. Este mecanismo alternativo se
llama reacción SN1 (que indica sustitución nucleofilica unimolecular).
CINÉTICA DE LA REACCIÓN SN1.
Se encuentra que la velocidad de la reacción depende sólo de la concentración
del halogenuro de alquilo y es independiente de la concentración del nucleófilo. En
otras palabras, esta reacción es un proceso de primer orden. Sólo una molécula participa
en el paso cuya cinética se determina. La expresión para la velocidad puede escribirse
como sigue:
Velocidad de reacción = Rapidez de desaparición del halogenuro de alquilo = k · RX
La rapidez de esta reacción SN1 es igual a un coeficiente de velocidad k por la
concentración del halogenuro de alquilo. La concentración del nucleófilo no aparece en
la expresión de velocidad. Muchas reacciones orgánicas son muy complejas y
transcurren en pasos sucesivos. Uno de estos pasos suele ser más lento que los otros, y
se le llama paso limitante de la velocidad. Ninguna reacción puede transcurrir más
rápido que su paso limitante de velocidad, el cual actúa como un cuello de botel la.
Diagrama de energía para una reacción SN1. El paso limitante de la velocidad es la
disociación del halogenuro de alquilo.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
132
ESTEREOQUÍMICA DE LA REACCIÓN SN1.
Si es correcto el postulado de que las reacciones SN1 ocurren a través de
carbocationes intermediarios, las consecuencias estereoquímicas deben ser distintas de
las propias de las reacciones SN2. Los carbocationes son especies planares con
hibridación sp2, y por tanto son aquirales. De este modo, si se efectúa una reacción SN1
con un enantiómero de un reactivo quiral y se pasa por un intermediario simétrico
aquiral, el producto debe ser ópticamente inactivo. El carbocatión intermediario
simétrico puede ser atacado por un nucleófilo indistintamente por el lado derecho o por
el lado izquierdo, lo que produce una mezcla 50 : 50 de enantiómeros; es decir, una
mezcla racémica.
CARACTERÍSTICAS DE LA REACCIÓN SN1.
Así como la reacción SN2 es fuertemente influida por variables como disolvente,
grupo saliente, estructura del sustrato y naturaleza del nucleófilo atacante, también lo es
la reacción SN1. Los factores que reducen la energía de activación , ya sea estabilizando
el estado de transición que conduce a la formación del carbocatión intermediario o
elevando el nivel de energía de los reactivos, favorecen las reacciones SN1 haciéndolas
más rápidas. A la inversa, los factores que incrementan la energía de activación, ya sea
desestabilizando el estado de transición que conduce al carbocatión intermedio o
reduciendo el nivel de energía de los reactivos, retardando la reacción S N1.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
133
SUSTRATO.
Según el postulado de Hammond, cualquier factor que estabilice un
intermediario de alta energía estabilizará también el estado de transición que conduce a
dicho intermediario. Dado que el paso limitante de la velocidad en la reacción SN1 es la
disociación unimolecular espontánea del sustrato, es de esperar que la reacción se vea
favorecida siempre que se formen carbocationes intermediarios estabilizados. Esto es
exactamente lo que se observa: cuanto más estable es el carbocatión intermediario, tanto
más rápida es la reacción SN1.
Como resultado de la estabilización por resonancia en los carbocationes alílicos
y bencílicos, los carbocationes tienen el siguiente orden de estabilidad. Nótese que un
carbocatión alílico o bencílico primario es casi tan estable como un carbocatión alquilo
secundario. De manera similar, un carbocatión alílico o bencílico secundario es tan
estable como un carbocatión alquilo terciario.
El orden de estabilidad de los carbocationes es exactamente igual al orden de
reactividad en las reacciones SN1 de los halogenuros de alquilo y los tosilatos. Haciendo
un paréntesis, debe mencionarse que los sustratos alílicos y bencílicos son
particularmente reactivos en las reacciones SN2 así como en las SN1. Los enlaces C-X
alílicos y bencílicos son aproximadamente 10 kcal/mol más débiles que los
correspondientes enlaces saturados, y por tanto se rompen con mayor facilidad.
GRUPO SALIENTE.
Durante el estudio de la reactividad SN2, se dedujo que los mejores grupos
salientes deberían ser los más estables -es decir, las bases conjugadas de ácidos fuertes-.
Para la reacción SN1 se encuentra un orden de reactividad idéntico, debido a que el
grupo saliente participa de manera decisiva en el paso limitante de la velocidad. Así, se
encuentra que el orden de reactividad SN1 es:
TosO- I
- Br
- Cl
- H2O
Más reactivo Menos reactivo
Nótese que en la reacción SN1, la cual a menudo se realiza en condiciones ácidas,
el agua (neutra) puede actuar como grupo saliente. Este es el caso, por ejemplo, cuando
se elabora un halogenuro de alquilo a partir de un alcohol terciario, por reacción con
HBr o HCl. El alcohol se protona primero y después pierde agua para generar un

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
134
carbocatión. La reacción posterior del carbocatión con el ion halogenuro produce el
halogenuro de alquilo.
Mecanismo de la reacción SN1 de un alcohol terciario con HBr para formar un
halogenuro de alquilo. El grupo saliente es agua (neutra).
NUCLEÓFILO.
La naturaleza del nucleófilo atacante es decisiva en la reacción SN2. En la
reacción SN1 no lo es. Por su propia naturaleza las reacciones SN1 proceden a través de
un paso determinante de la velocidad en el cual el nucleófilo agregado no interviene en
la cinética. El nucleófilo no participa en la reacción hasta que ha ocurrido la disociación
limitante de la velocidad. No importa su naturaleza nucleofilica.
DISOLVENTE.
El efecto del disolvente tiene una gran importancia en las reacciones SN1, pero
de manera muy distinta a las reacciones SN2. El postulado de Hammond establece que
cualquier factor que estabilice el carbocatión intermediario incrementará la velocidad de
reacción. La solvatación -estabilización del carbocatión por interacción con
moléculas del solvente- puede tener exactamente tal efecto. Si bien no es fácil definir la
naturaleza precisa de la estabilización del carbocatión por el solvente, podemos
representar las moléculas de este último orientadas alrededor del catión, de manera que
los extremos ricos en electrones de los dipolos del solvente queden frente a la carga
positiva.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
135
Solvatación de un carbocatión en agua. Los átomos de oxígeno (ricos en electrones)
del solvente rodean al catión (con carga positiva) para estabilizarlo.
Las reacciones SN1 ocurren con mucho mayor rapidez en solventes altamente
polares que en solventes no polares. Por ejemplo, en la reacción del 2-cloro-2-metil-
propano se observa un factor de incremento de 100.000 al pasar al del etanol, un
solvente polar, al agua, aún más polar.
(CH3)3C-Cl + ROH (CH3)3C-OR + HCl
Agua Etanol acuoso al 80 % Etanol acuoso al 40 % Etanol puro
100 14 0.1 0.001
Más reactivo REACTIVIDAD Menos reactivo
Debe insistirse en el hecho de que tanto en la reacción SN1 como en la SN2 el
solvente ejerce un gran efecto, aunque por razones distintas. Las reacciones SN2 son
desfavorecidas por los solventes próticos debido a que el nivel de energía del estado
fundamental del nucleófilo atacante disminuye por solvatación. Las reacciones SN1, en
contraste, son favorecidas por los solventes próticos debido a que el nivel de energía del
estado de transición que conduce al carbocatión intermediario disminuye por la
solvatación.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
136
RESUMEN
Características SN1 SN2
Mecanismo
En dos pasos, uno lento y
otro rápido
I)lento R-L R+ + L
-
II)Rápido R+ + Nu
- RNu
En un solo paso
R-L + Nu- R-Nu + L
-
Cinética
De primer orden
De segundo orden
Nucleofilia del reactivo
No afecta a la velocidad
Controla la velocidad,
conjuntamente con el
sustrato.
Estructura del átomo de
carbono saturado
(sustrato)
1) Estabilización por
resonancia
2) FAVORABLE 3º 2º 1º
1) Impedimento estérico
desvaforable
2) CH3 1º 2º 3º
Efectos del disolvente
Favorecida por disolventes
ionizantes (polares)
Favorecidas en disolventes
apróticos.
Estereoquímica
Racemización
Inversión
Condiciones de reacción
Generalmente ácidas
Generalmente básicas.
Reacciones de
competitivas
Eliminación (E1) y
Transposición
Eliminación (E2)

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
137
HALOGENUROS DE ALQUILO (REACCIONES DE ELIMINACIÓN)
REACCIONES DE ELIMINACIÓN:
Cuando un nucleófilo o una base de Lewis atacan a un halogenuro de alquilo,
son posibles dos tipos de reacciones. El reactivo puede atacar al átomo de carbono y
sustituir al halogenuro, o puede atacar a un hidrógeno y causar la eliminación de HX
para formar un alqueno.
Las reacciones de eliminación son más complejas que las de sustitución, por
varias razones. Existe el problema de la regioquímica: ¿Qué productos resultarán de la
deshidrohalogenación de halogenuros asimétricos? En efecto, las reacciones de
eliminación casi siempre producen mezclas de alqueno y usualmente lo más que puede
hacerse es predecir cuál será el producto principal.
Conforme a una regla formulada por el químico ruso Alexander Zaitsev[1]
, las
reacciones de eliminación inducidas por base generalmente tienen como producto el
alqueno más sustituido; es decir, el alqueno con más sustituyentes alquilo en los
carbonos del doble enlace, conocida como regla de Zaitsev. En los siguientes dos
ejemplos el alqueno predominante es el alqueno más sustituido cuando se usa como
base etóxido de sodio en etanol.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
138
La eliminación de HX a partir de halogenuros de alquilo es una reacción
general que constituye un excelente método para elaborar alquenos. Sin embargo, este
asunto es complejo, y las reacciones de eliminación pueden proceder en una variedad de
rutas mecanísticas. En este apartado consideraremos dos de ellas en primera instancia,
las reacciones E1 y E2.
REACCIÓN E2:
La reacción E2 significa eliminación bimolecular y se realiza cuando un
halogenuro de alquilo se trata con una base fuerte como el ion hidróxido (-OH) o ion
alcóxido (-OR). Es la vía de eliminación más común. Al igual que la reacción SN2, la
reacción E2 es un proceso en un paso, sin intermedios pero con un estado de transición.
Cuando la base atacante empieza a sustraer un protón del carbono vecino al grupo
saliente, el enlace C-H comienza a romperse, empieza a formarse un nuevo doble enlace
C-C, y el grupo saliente empieza a separarse, llevándose consigo el par de electrones del
enlace C-X.
Una evidencia que apoya este mecanismo es la cinética de reacción. Dado que
tanto la base como el halogenuro de alquilo participan en el paso limitante de la
velocidad, las reacciones E2 presentan cinética de segundo orden:
Velocidad = k · RX · Base
Una segunda evidencia y más sólida la constituye la estereoquímica de las
eliminaciones E2. Como indican una gran cantidad de datos experimentales, las
reacciones E2 son estereoesfecíficas. La eliminación siempre procede a partir de una
configuración geométrica periplanar, lo cual significa que los cuatro átomos
reaccionantes -el hidrógeno, los dos carbonos y el grupo saliente- se encuentran en un
mismo plano. Existen dos de tales configuraciones: la periplanar sin, (eclipsada, mayor
energía), y la periplanar anti, (alternada de menor energía). De las dos, la
configuración anti es energéticamente la preferida de la molécula.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
139
La base (B:) ataca un hidrógeno vecino
y comienza a sustraer el hidrógeno al
mismo tiempo que empieza a formarse el
doble enlace del alqueno y empieza a
separarse el grupo X.
El alqueno neutro se produce cuando
está totalmente roto el enlace C-H y el
grupo X se ha separado llevándose el par
de electrones del enlace C-X.
La configuración periplanar anti de las eliminaciones E2 tiene consecuencias
estereoquímicas bien definidas que constituyen una sólida evidencia en favor del
mecanismo propuesto. Considérese por ejemplo el meso-1,2-dibromo-1,2-difeniletano,
que experimenta una eliminación E2 cuando se trata con base para formar sólo el
alqueno E (con los dos grupos fenilo en cis) puro no se forma nada del alqueno
isomérico Z, debido a que el estado de transición que conduce al alqueno Z tendría que
tener configuración periplanar sin.
REACCIONES DE ELIMINACIÓN Y CONFORMACIÓN DEL
CICLOHEXANO.
La configuración periplanar anti de las reacciones E2 es de particular
importancia en los anillos de ciclohexano, en los cuales la estructura rígida tipo silla
fuerza una relación específica entre los sustituyentes de átomos de carbono vecinos.
Derek Barton[2]
señaló en 1960 en un artículo decisivo que gran parte de la reactividad

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
140
química de los cicloalcanos sustituidos es controlada por efectos conformacionales. A
continuación se considera la deshidrohalogenación E2 de los clorociclohexanos para ver
el efecto de la conformación sobre la reactividad.
El requisito periplanar anti para las reacciones E2 puede satisfacerse en los
ciclohexanos sólamente si el hidrógeno y el halógeno son diaxiales trans. Si el
halógeno o el hidrógeno son ecuatoriales, la eliminación E2 no puede ocurrir.
Cloro axial : H y Cl son periplanar anti
Cloro ecuatorial : H y Cl no son periplanar anti
Requisitos geométricos para la reacción E2 en los ciclohexanos. Tanto el grupo
saliente como el hidrógeno deben ser axiales para que ocurra la eliminación
periplanar anti.
EFECTO ISOTÓPICO DEL DEUTERIO.
Una evidencia final en apoyo del mecanismo E2 la constituye el fenómeno
conocido como efecto isotópico de deuterio. Se sabe que un enlace carbono-hidrógeno
es más débil en una pequeña cantidad (alrededor de 1.2 kcal/mol) que un enlace
carbono-deuterio correspondiente. Así, un enlace C-H se rompe con mayor facilidad
que un enlace equivalente C-D, y por lo tanto la rapidez de ruptura del enlace C-H es
mayor.
La eliminación de HBr inducida por base del 1-bromo-2-feniletano procede 7.11
veces más rápido que la eliminación correspondiente de DBr a partir de 1-bromo-2,2-
deuterio-2-feniletano:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
141
Este resultado indica que el enlace C-H (o C-D) se rompe en el paso limitante de
la velocidad, y en consecuencia es del todo consistente con el mecanismo propuesto, es
decir en un solo paso.
REACCIÓN E1:
Las eliminaciones E1 se inician con la disociación espontánea de un halogenuro,
parecida a una reacción SN1, pero en este caso a la disociación sigue la pérdida de un
protón del carbocatión intermedio. En efecto el mecanismo E1 normalmente ocurre en
competencia con la reacción SN1 cuando un halogenuro de alquilo se trata con un
nucleófilo no básico en un solvente prótico. Así, los mejores sustratos son aquellos que
también se someten a la reacción SN1, y casi siempre se obtienen mezclas de productos.
Por ejemplo, cuando el 2-cloro-2-metilpropano se calienta a 65ºC en etanol
acuoso al 80%, se obtiene una mezcla de 2-metil-2-propanol (SN1) y 2-metilpropeno
(E1):
Se han obtenido muchas evidencias en apoyo del mecanismo E1. Como es de
esperar, las reacciones E1 presentan cinética de primer orden consistente con un proceso
de disociación espontáneo:
Velocidad = k · R-X
Una segunda evidencia es la estereoquímica de las eliminaciones. A diferencia
de la reacción E2, en las que se requiere una configuración periplanar, en la reacción E1
no existe un requisito geométrico; el carbocatión intermedio puede perder cualquier
protón disponible de una posición vecina. Por tanto, es de esperar que se obtenga el
producto más estable (regla de Zaitsev) en una reacción E1, lo cual es justamente lo que
se observa. La cinética es de primer orden pero su mecanismo es en dos etapas:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
142
DESHIDRATACIÓN DE ALCOHOLES A ALQUENOS:
Para que un sustrato experimente una reacción E1 es necesario que posea un
buen grupo saliente, cuya marcha origine un carbocatión estable. Los alcoholes no
sufren eliminación en condiciones neutras o alcalinas, debido a que el -OH no es un
buen grupo saliente. No obstante, en un medio ácido los alcoholes se protonan en el
oxígeno para dar iones oxonio, capaces de perder agua (H2O), un excelente grupo
saliente.
Así, la reacción de los alcoholes 2ario
y 3ario
con un exceso de un ácido fuerte
constituyen un método establecido para la síntesis de alquenos. Suelen emplearse los
“ácidos sulfúricos y fosfóricos”. La temperatura elevada acelera la reacción y favorece
la eliminación, además, permite que el alqueno destile a medida que se forma, lo que
evita las reacciones secundarias.
Ejemplos:
1.-
2.-
3.-

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
143
En los ejemplos 2 y 3, los resultados están de acuerdo con lo esperado, es decir
la obtención del alqueno más sustituido. Los productos predominantes en el ejemplo 3
se han formado a través de una transposición, lo que demuestra que en la reacción han
intervenido carbocationes como intermedios.
Transposiciones a través de carbocationes: Una propiedad común de los
carbocationes es experimentar transposiciones, siempre y cuando sean capaces de
generar un carbocatión mucho más estable que el precursor.
Transposición de Hidrógeno
Transposición de Metilo
COMPARACIÓN ENTRE LAS REACCIONES E1 y E2.
Características E1 E2
Mecanismo Dos pasos, Intermedio tipo
carbocatión
Un paso, eliminación
concertada, con estado de
transición
Cinética Primer orden Segundo orden

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
144
Transposiciones En ocasiones Nunca
Sustratos Haluros, Tosilatos, alcoholes
en medio ácido
Haluros, tosilatos, hidróxidos
de tetraalquilamonio
Características estructurales
favorables
Halogenuros 3º, y empleando
disolventes polares
Elevada concentración de
base.
Competencia Reacción SN1 Reacción SN2
Correlación entre estructura y reactividad para reacciones de sustitución y de
eliminación.
Tipo de
halogenuro
SN1 SN2 E1 E2
R-CH2-X
(primario)
No ocurre
Altamente
favorecida
No ocurre
Ocurre cuando se
emplean bases
fuertes
R2-CH-X
(secundario)
Puede ocurrir con
halogenuros
bencílicos y
alílicos
Ocurre en
competencia con
la reacción E2
Puede ocurrir con
halogenuros
bencílicos y
alílicos
Es favorecida
cuando se usan
bases fuertes
R3-C-X
(terciario)
Es favorecida en
solventes
hidroxílicos
No ocurre
Ocurre en
competencia con
la reacción SN1
Es favorecida
cuando se
emplean bases
OTRAS REACCIONES DE ELIMINACIÓN:
En las reacciones de eliminación E1 y E2, el producto obtenido
mayoritariamente resulta ser el alqueno más sustituido, pero en muchas ocasiones por
interés sintético es necesario obtener un alqueno menos sustituido en forma mayoritaria.
En tal caso, los métodos anteriores no resultan útiles, y hay que recurrir a otros métodos
descritos en la literatura científica, de los cuales resumiremos algunos.
A.- ELIMINACIÓN DE HOFMANN
La pérdida de una trialquilamina y un átomo de hidrógeno en respecto del
nitrógeno a partir de una sal de amonio cuaternario conduce a un alqueno mediante la
reacción conocida generalmente como eliminación de Hofmann[3]
.
La reacción transcurre normalmente por mecanismo E2 y sigue la preferencia
estereoelectrónica para la eliminación anti. Cuando existen dos tipos diferentes de
hidrógenos en respecto al nitrógeno cuaternario, la eliminación conduce generalmente
al predominio del alqueno menos sustituido. De hecho, Hofmann propuso su regla
sobre la orientación de la eliminación en 1851 como consecuencia de sus estudios sobre
eliminaciones a partir de sales de amonio cuaternario.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
145
La eliminación puede conseguirse tratando una sal de amonio cuaternario con
una base fuerte, si bien es más normal utilizar el hidróxido de amonio cuaternario como
reactivo, con lo que se requiere base externa. El rendimiento en alqueno es
generalmente alto. La reacción posee una utilidad limitada para la síntesis de alquenos,
ya que a menudo es más fácil emplear productos de partida más asequibles. (el
mecanismo será estudiado en el capítulo dedicado a las aminas).
B.- ELIMINACIÓN PIROLÍTICA
Ciertos precursores de alquenos no requieren un reactivo externo que promueva
la eliminación. Los ésteres y los óxidos de amina constituyen dos ejemplos de tales
sustratos. En esos casos, la calefacción aporta suficiente energía para que el grupo
saliente actúe como una base intramolecular. La ruptura térmica de una molécula se
conoce como pirólisis.
Cuando se calienta un éster (300 - 600º), generalmente un acetato que contiene
un átomo de hidrógeno en en su porción alcohólica, se forman un alqueno y un ácido
carboxílico. Se cree que la reacción transcurre de forma concertada a través de un
estado de transición cíclico de seis miembros. Tales reacciones de eliminación pirolítica
se designan a menudo como Ei-eliminación, interna. La reacción es estereoespecifica,
el mecanismo cíclico intramolecular requiere coplanaridad sin de los grupos que se
pierden.
Mecanismo: de la pirólisis de ésteres.
La pirólisis de óxidos de amina (La Reacción de Cope) es un método de
síntesis de alquenos muy relacionado con la pirólisis de ésteres. El átomo de oxígeno de
un óxido de amina posee una carga negativa formal y puede actuar como una base
interna.
Otros ejemplos:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
146
Acetato de treo-2-deuterio E-Estilbeno (trans)
-1,2-difeniletilo
Acetato de Eritro-2-deuterio E-1-Deuterioestilbeno (trans)
-1,2-difeniletilo
El mecanismo de la reacción de Cope será estudiado en capítulo dedicado a las amina.
C.- DESHALOGENACIÓN
En las reacciones de 1,2-eliminación más comunes, el protón es uno de los dos
grupos que se pierde. Cuando en un sustrato están presentes dos átomos de halógeno, la
reacción con base puede conducir a una doble deshidrohalogenación. El ciclohexadieno
se prepara de esta manera a partir del 1,2-dibromociclohexano.
No obstante, si se trata el mismo 1,2-dihalogenuro con yoduro potásico, un
reactivo relativamente no básico, se obtiene ciclohexeno, una monoolefina. La
eliminación de dos átomos de halógeno produce una deshalogenación.
La deshalogenación mediante ion yoduro es un proceso estereoespecífico que
transcurre con un curso estereoquímico anti. El yoduro actúa de la misma manera que la
base en la deshidrohalogenación. En esta reacción E2, la pérdida de los dos átomos de
halógeno está sincronizada con la formación del doble enlace.
El ion yoduro y el cinc metálico son los reactivos más comúnmente utilizados
para la deshalogenación.
D.- DESHIDROGENACIÓN CATALÍTICA.
La eliminación de hidrógeno a partir de un hidrocarburo es un importante
proceso industrial. La deshidrogenación se lleva a cabo a temperaturas elevadas sobre
catalizadores compuestos normalmente de óxidos metálicos. La tecnología de este tipo
de procesos catalíticos continuos es muy sofisticada. Los factores críticos para controlar
los rendimientos y los productos secundarios son la temperatura, la composición del

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
147
catalizador y la velocidad de flujo a través del reactor. El vapor de agua y el nitrógeno
son dos gases portadores utilizados para arrastrar los productos de partida y los
productos de reacción (entre ellos hidrógeno) a través del catalizador. La naturaleza y
concentración de estos gases “inertes” son a menudo variables importantes de la
reacción.
[1]
Alexander M. Zaitsev (o Saytzeff) (1841-1910); n. Kazán, Rusia. [2]
Derek H. R. Barton (1918- ); n. Gravesend, Inglaterra; Premio Nobel (1960). [3]
August W. von Hofmann, (1818-1892), Profesor de la Universidad de Berlín, Alemania.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
148
ALCOHOLES Y TIOLES
Los alcoholes son compuestos que tienen grupos hidroxilo unidos a átomos de
carbono saturado, con hibridación sp3. Esta definición excluye deliberadamente a los
fenoles (grupo hidroxilo unidos a anillos aromáticos) y a los enoles (grupo hidroxilo
unidos a un carbono vinílico), debido a que la química de estos tres tipos de compuestos
es muy diferente. Los alcoholes pueden considerarse los derivados orgánicos del agua,
donde uno de los hidrógenos es sustituido por un grupo orgánico: H-O-H pasa a ser R-
O-H.
Los alcoholes están ampliamente distribuidos en la naturaleza y tienen muchas
aplicaciones industriales y farmacéuticas. El etanol, por ejemplo, es una de las
sustancias orgánicas más simples y mejor conocidas, y se usa como aditivo de
combustibles, como disolvente industrial y en las bebidas; el mentol, un alcohol aislado
de la menta, se usa mucho como saborizante y en perfumería; y el colesterol, un alcohol
esteroide cuya fórmula tiene aspecto complicado, se considera un agente que causa
enfermedades del corazón.
PROPIEDADES DE LOS ALCOHOLES: PUENTE DE HIDRÓGENO
Como ya se comentó, los alcoholes se pueden considerar derivados del agua, en
el sentido de que uno de los hidrógenos ha sido reemplazado por un grupo orgánico. De
este modo los alcoholes tienen casi la misma configuración geométrica del agua. El
ángulo de enlace R-O-H tiene aproximadamente el valor tetraédrico (109º en el metanol,
por ejemplo), y el átomo de oxígeno presenta hibridación sp3.
Los alcoholes son muy distintos de los hidrocarburos y de los halogenuros de
alquilo. No sólo su química es mucho más rica, sino que sus propiedades físicas son
también diferentes. En la Tabla 12.1, se comparan los puntos de ebullición de algunos
alcoholes, alcanos y cloroalcanos simples, se ve que los alcoholes tienen puntos de
ebullición mucho mayores
TABLA 12.1.-
Puntos de ebullición de alcanos, cloroalcanos y alcoholes (ºC)
Grupo alquilo, R Alcano, R-H Cloroalcano, R-Cl Alcohol. R-OH
CH3-
-162
-24
64.5
CH3CH2- -88.5 12.5 78.3
CH3CH2CH2- -42 46.6 97

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
149
(CH3)2CH- -42 36.5 82.5
CH3CH2CH2CH2- -0.5 83.5 117
(CH3)3C- -12 51 83
La causa de sus puntos de ebullición altos es que los alcoholes, al igual que el
agua, están muy asociados en solución debido a la formación de los llamados puentes
de hidrógeno. El átomo de hidrógeno de un grupo -OH en una molécula está polarizado
positivamente, y forma un enlace débil (“puente de hidrógeno”) con el átomo de
oxígeno polarizado negativamente de otra molécula. Aunque los puentes de hidrógeno
tienen una energía de sólo 5 kcal/mol, en contraste con las 103 kcal/mol de un enlace O-
H típico, la presencia de muchos puentes de hidrógeno significa que se debe agregar
energía extra para romperlos durante la ebullición.
Puentes de hidrógeno en los Alcoholes.
PROPIEDAD DE LOS ALCOHOLES: ACIDEZ Y BASICIDAD
Como el agua los alcoholes son débilmente ácidos y débilmente básicos. Como
bases de Lewis débiles, los alcoholes son protonados reversiblemente por los ácidos
para formar iones oxonio, R-OH2+. Como podría esperarse, los alcoholes protonados
son mucho más reactivos que los alcoholes neutros hacia los nucleófilos, debido a que
portan carga positiva.
Como ácido débiles, los alcoholes actúan como donadores de protones. En
solución acuosa diluida, se disocian ligeramente donando un protón al agua.
Los efectos inductivos también son importantes para determinar la acidez de los
alcoholes. Por ejemplo, los sustituyentes halógeno, atrayentes de electrones, estabilizan
un anión alcóxido ayudando a dispersar la carga en una área grande, y haciendo así más
ácido al alcohol. Este efecto inductivo puede observarse comparando la acidez del
etanol (pKa = 16) con la del 2,2,2-trifluoroetanol (pKa = 12.43), o la del terc-butílico
(pKa = 18) con la del perfluoro-2-metil-2-propanol (pKa = 5.4). (véase Tabla 12.2)

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
150
TABLA 12.2.-
Constantes de acidez de algunos alcoholes
Alcohol pKa
(CH3)3COH
CH3CH2OH
HOH (agua)
CH3OH
CF3CH2OH
(CF3)3COH
HCl
18.00 Ácido débil
16.00
15.74
15.54
12.43
5.4
-7.00 Ácido fuerte
Los grupos atrayentes de electrones estabilizan el
alcóxido y reducen el pKa.
pKa = 5.4 pKa = 18
Puesto que los alcoholes son mucho más débiles que los ácidos carboxílicos o
los ácidos minerales, no reaccionan con bases débiles como aminas, ion bicarbonato o
hidróxido metálico. Sin embargo, reaccionan con metales alcalinos y con bases fuertes
como hidruro de sodio (NaH), aminas de sodio (NaNH2), reactivos de alquil-litio (RLi)
y reactivos de Grignard (RMgX). Las sales metálicas de los alcoholes son por sí mismas
bases fuertes, por lo que a menudo se usan como reactivos en química orgánica.
Ejemplos:
RESUMEN DE LAS REACCIONES DE LOS ALCOHOLES:
I.- SÍNTESIS DE ALCOHOLES
REACCIÓN EJEMPLO
a.- Reducción de compuestos carbonílicos
(1) Aldehídos

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
151
Un alcohol primario
(2) Cetonas
Un alcohol secundario
(3) Ésteres
Un alcohol primario
(4) Ácidos carboxílicos
Un alcohol primario
b.- Adición de reactivos de Grignard a compuestos
carbonílicos.
(1) Formaldehí do
Un alcohol primario
(2) Aldehídos
Un alcohol secundario
(3) Cetonas

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
152
Un alcohol terciario
(4) Ésteres
Un alcohol terciario
II.- REACCIONES DE LOS ALCOHOLES
a.- Acidez
ROH + NaH RO-Na
+ + H2
ROH + Na RO-Na
+ + H2
Alcóxidos
b.- Deshidratación
(1) Alcoholes terciarios
(2) Alcoholes secundarios y terciarios
c.- Oxidación
(1) Alcohol primario
Aldehído
Un ácido carboxílico
(2) Alcohol secundario

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
153
Una cetona
III.- SÍNTESIS DE LOS TIOLES
IV.- REACCIONES DE LOS TIOLES
a.- Oxidación de tioles a disulfuros
b.- Otras homologas a la de los alcoholes
OBTENCIÓN DE ALCOHOLES POR REDUCCIÓN DE GRUPOS CARBONILOS.
El método más generalizado para producir alcoholes es la reducción de
compuestos carbonilicos:
Donde H es una agente reductor generalizado
Una reducción orgánica es una reacción en la cual se incrementa el contenido
de hidrógeno o disminuye el de oxígeno, nitrógeno o halógeno de una molécula. A la
inversa, una oxidación orgánica es una reacción en la cual disminuye el contenido de
hidrógeno o se incrementa el de oxígeno, nitrógeno o halógeno de una molécula.
Se dispone de muchos reactivos para reducir cetonas y aldehídos a alcoholes,
pero usualmente se elige el borohidruro de sodio, NaBH4, debido a la seguridad y la
facilidad de su manejo. Este compuesto es un sólido cristalino blanco que se puede
manipular y pesar sin peligro en atmósfera abierta y usar en agua o en solución
alcohólica. Suelen obtenerse altos rendimientos de alcohol. El hidruro de aluminio y
litio, LiAlH4, es un polvo blanco soluble en éter y tetrahidrofurano, es otro agente
reductor que se usa algunas veces para reducción de cetonas y Aldehídos. Si bien es más
potente y reactivo que el NaBH4, el LiAlH4 es también peligroso, y debe ser manejado
por personas experimentadas. Reacciona violentamente con el agua, se descompone
cuando se calienta a más de 125ºC. No obstante estos inconvenientes, el LiAlH4 es un
reactivo en extremo valioso y usa adiario en cientos de laboratorios.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
154
Los ésteres y los ácidos carboxílicos pueden reducirse para formar alcoholes
primarios. Estas reacciones son más difíciles que las reducciones correspondientes de
Aldehídos y cetonas. Por ejemplo, el borohidruro de sodio reduce lentamente los ésteres
y no reduce los ácidos. Así, las reducciones de los ésteres y los ácidos carboxílicos
usualmente se realizan con hidruro de litio y aluminio. Todos los grupos carbonilos,
incluyendo ésteres, ácidos, cetonas y aldehidos, se reducen por medio de LiAlH4 con
alto rendimiento, como lo indican los siguientes ejemplos:
Mecanismo de la reducción con NaBH4 :
Mecanismo de la reducción con LiAlH4:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
155
OBTENCIÓN DE ALCOHOLES POR ADICIÓN DE REACTIVOS DE GRIGNARD
A GRUPOS CARBONILOS.
Los reactivos de Grignard, RMgX, reaccionan con los compuestos carbonílicos
para producir alcoholes de manera semejante a como lo hacen con los hidruros. El
resultado es un método muy útil y general de síntesis de alcoholes.
La reacción consiste en un ataque nucleofílico del
R al carbono electrofílico del grupo carbonilo.
El carácter nucleofílico del grupo alquilo en los reactivos organometálicos de
litio y magnesio puede utilizarse para la síntesis de alcoholes por adición a compuestos
carbonílicos y apertura nucleófila de anillos de oxaciclopropano.
Mecanismo:
Por reacciones de Grignard es posible obtener un gran número de alcoholes,
dependiendo de los reactivos usados, obsérvese los siguientes ejemplos:
Bromuro de ciclohexil Formaldehído Ciclohexilmetanol magnesio (65%)
3-Metilbutanal Bromuro de 3-Metil-1-fenil-1-
butanol fenilmagnesio (73%)
Ciclohexanona 1-Etilciclohexanol
(89%)
Pentanoato de etilo Bromuro de 2-Metil-2-hexanol
(85%) metilmagnesio
REACCIONES DE LOS ALCOHOLES.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
156
Las reacciones de los alcoholes pueden dividirse en dos grupos. Las que ocurren
en el enlace C-O y las que ocurren en el enlace O-H.
En el resumen de las reacciones están representadas casi todas ellas, a
continuación realizaremos algún comentario de las más significativas.
DESHIDRATACIÓN DE ALCOHOLES PARA PRODUCIR ALQUENOS.
Una de las reacciones más importantes del enlace C-O de los alcoholes es la
deshidratación para formar alquenos. Se rompe el enlace carbono-oxígeno, se rompe el
enlace C-H vecino y se forma un enlace pi () de alqueno. (véase capítulo de
eliminación).
Debido a la importancia de la reacción, se ha desarrollado un gran número de
formas alternativas de realizar las deshidrataciones. Uno de los métodos más comunes y
que funciona particularmente bien para los alcoholes terciarios es el método catalizado
por ácido. Las deshidrataciones catalizadas por ácido normalmente siguen la regla de
Zaitsev . En la práctica normal de laboratorio, sólo los alcoholes terciarios se
deshidratan con ácido. Puede hacerse que los alcoholes secundarios reaccionen, pero las
condiciones son más severas (H2SO4 al 75%, 100ºC) y las moléculas sensibles no las
resisten. Los alcoholes primarios son menos reactivos aún que los secundarios, y se
requieren condiciones todavía más severas para la deshidratación (H2SO4 al 95%,
150ºC) .
Dos electrones del átomo de oxígeno se
unen al H+, produciendo un alcohol
protonado intermediario, el cual a su vez
es un buen grupo saliente.
Se rompe el enlace carbono-oxígeno y los
dos electrones del enlace quedan en el
oxígeno, dejando el carbocatión
intermediario.
Dos electrones de un enlace carbono-
hidrógeno vecino forman el enlace pi del
alqueno, y se elimina un H+
(un protón).

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
157
Una vez que se ha reconocido que la deshidratación catalizada por ácido es una
reacción E1, se esclarece la causa por la cual los alcoholes terciarios reaccionan más
rápido. Los sustratos terciarios siempre reaccionan con más rapidez en las reacciones E1
debido a que conducen a carbocationes intermedios muy estabilizados.
Para evitar la necesidad de ácidos fuertes y poder deshidratar alcoholes
secundarios de un modo más suave, se han desarrollado otros reactivos que son eficaces
en condiciones básicas suaves. Uno de tales reactivos, el oxicloruro de fósforo (POCl3),
a menudo es capaz de provocar la deshidratación de alcoholes secundarios y terciarios a
0ºC en piridina (una amina básica) como disolvente.
1-Metilciclohexanol 1-Metilciclohexeno (96%)
Ciclohexanol Ciclohexeno (97%)

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
158
Mecanismo:
El grupo hidroxilo del alcohol reacciona con POCl3 para formar un
diclorofosfato intermedio. La eliminación E2 procede por el mecanismo usual de un
paso, la piridina, una amina básica, sustrae un protón del carbono vecino al mismo
tiempo que el grupo diclorofoafato está saliendo.
CONVERSIÓN DE ALCOHOLES EN HALOGENUROS DE ALQUILO.
Una segunda reacción del enlace C-O de los alcoholes es su conversión en
halogenuros de alquilo. Los alcoholes terciarios se convierten con facilidad en
halogenuros de alquilo por tratamiento con HCl o HBr a 0ºC. Sin embargo, los
alcoholes primarios y secundarios son mucho más resistentes a los ácidos, y se
transforman mejor en halogenuros por tratamiento ya sea con SOCl 2 o con PBr3.
Obtención de cloruros de alquilo con SOCl2.
Mecanismo:
Obtención de bromuros de alquilo con PBr3:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
159
Mecanismo:
A continuación, el HOPBr2 reaccionará con dos o más moléculas del alcohol por
mecanismos análogos al de la primera etapa.
CONVERSIÓN DE ALCOHOLES EN TOSILATOS CON CLORURO DE P-
TOLUENSULFONILO, (P-TOSCL):
Los alcoholes reaccionan con cloruro de p-toluensulfonilo en solución de
piridina para formar tosilatos de alquilo, R-OTs. En esta reacción sólo se rompe el
enlace O-H del alcohol; el enlace C-O queda intacto, y no ocurre cambio en la
configuración si el alcohol es quiral. El tosilato de alquilo resultante tiene
comportamiento químico muy similar al de los halogenuros de alquilo, y como estos
sufre reacciones de sustitución SN1 y SN2 con facilidad.
Ejemplo:
OXIDACIÓN DE ALCOHOLES:
La reacción más importante de los alcoholes es su oxidación para producir
compuestos carbonílicos. Los alcoholes primarios forman aldehídos o ácidos
carboxílicos; los alcoholes secundarios generan cetonas, y los alcoholes terciarios no
reaccionan con la mayoría de los agentes oxidantes, excepto en condiciones más
vigorosas.
La oxidación de los alcoholes primarios y secundarios puede efectuarse con un
gran número de reactivos, como KMnO4, CrO3 y Na2Cr2O7 ó K2Cr2O7. Los alcoholes
primarios se oxidan a aldehídos o ácidos carboxílicos, dependiendo de los reactivos
seleccionados y de las condiciones de reacción. Probablemente el mejor método para
elaborar aldehídos a partir de alcoholes primarios es por medio del clorocromato de
piridinio (PCC), en diclorometano como disolvente.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
160
Todas estas oxidaciones ocurren por rutas estrechamente relacionadas con la
reacción E2. El primer paso implica la reacción entre el alcohol y un reactivo de Cromo
VI para formar un cromato intermedio. Una eliminación bimolecular posterior forma el
producto carbonilo.
Ejemplos:
1-Decanol Ácido decanoico
(93%)
(85%)
4-tercButilciclohexanol 4-tercButilciclohexanona (91%)
Citronelol Citronelal
(82%)
4-metilpentanol 4-metilpentanal

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
161
TIOLES
Los tioles, R-SH, son análogos azufrados de los alcoholes. Los tioles se
nombran por medio del sistema que se usa para los alcoholes, con el sufijo -tiol en lugar
de -ol. El grupo -SH mismo se conoce como grupo mercapto.
La característica física más notable de los tioles es su olor en extremo
desagradable. Por ejemplo, el olor del zorrillo o mofeta se debe principalmente a los
tioles simples 3-Metil-1-butanotiol y 2-Buteno-1-tiol. Al gas natural se le agregan
pequeñas cantidades de tioles con bajo peso molecular que sirven como advertencia
fácil de detectar en caso de fugas.
Los tioles suelen elaborarse a partir de los correspondientes halogenuros de
alquilo por desplazamiento SN2 con un nucleófilo de azufre, como el anión sulfhidrilo, -
SH.
1-Bromooctano 1-
Octanotiol
Los tioles pueden ser oxidados por reactivos suaves, como bromo o yodo, para
formar disulfuros, R-SS-R. La reacción es fácilmente reversible; los disulfuros se
pueden reducir a tioles por tratamiento con cinc y ácido.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
162
ÉTERES, EPÓXIDOS Y SULFUROS
Un éter es una sustancia que tiene dos residuos orgánicos unidos al mismo átomo de
oxígeno, R-O-R’. Los residuos orgánicos pueden ser alquílicos, arílicos o vinílicos, y el
átomo de oxígeno puede formar parte ya sea de una cadena abierta o de un anillo. Tal
vez el éter mejor conocido es el éter dietílico, una sustancia familiar que se ha usado en
medicina como anestésico y se emplea mucho en la industria como disolvente.
En el laboratorio, los éteres normalmente se elaboran por Síntesis de
Williamson o por una secuencia de alcoximercuración-desmercuración. Los éteres
son inertes a la mayoría de los reactivos comunes, pero son atacados por ácidos fuertes
para formar los productos de ruptura. Los epóxidos son éteres cíclicos con anillos de
tres miembros que contienen oxígeno, difieren de los otros éteres cíclicos en su facilidad
de ruptura.
Los sulfuros, R-S-R’, son análogos azufrados de los éteres. Se producen por
una reacción SN2 de tipo Williamson entre un anión tiolato y un halogenuro de alquilo
primario o secundario.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
163
ÉTERES.
Los éteres pueden considerarse derivados orgánicos del agua, en los cuales los
átomos de hidrógeno han sido reemplazados por fragmentos orgánicos; es decir, H-O-H
por R-O-R’. De este modo, los éteres tienen casi la misma configuración geométrica
que el agua. Los enlaces R-O-R` tienen ángulos de enlace aproximadamente tetraédrico
(112º en el éter dimetilíco), y el átomo de oxígeno tiene hibridación sp3.
Hibridación sp
3
La presencia del átomo de oxígeno, electronegativo, hace que los éteres tengan
un ligero momento dipolar y, por tanto, sus puntos de ebullición son un poco más altos
que los alcanos correspondientes. En la Tabla 13.1 se comparan los puntos de ebullición
de algunos éteres comunes con los de los hidrocarburos correspondientes en los que el
átomo de oxígeno se ha sustituido por un -CH2-.
TABLA 13.1
Comparación de los puntos de ebullición de éteres e hidrocarburos .
Éter Hidrocarburo Punto de ebullición (ºC)
Las reacciones de los éteres serán presentadas en forma de tabla, y al final de la
presentación se realizara un comentario de las más significativas.
RESUMEN DE LAS REACCIONES DE LOS ÉTERES.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
164
1.- Formación de éteres.
(a) Síntesis de Williamson.
El halogenuro de alquilo debe ser primario
Recuérdese que los alcóxidos se preparan
desde los alcoholes por tratamiento básico
y/o métales alcalinos.
(b) Alcoximercuración-desmercuración.
Se observa orientación Markovnikov. (Adición Sin)
(c) Epoxidación de alquenos con peroxiácidos.
2.- Reacciones de los éteres.
(a) Ruptura con HX.
Donde HX = HBr o HI
(b) Apertura de anillos de epóxidos catalizada
por base.
La reacción ocurre en el sitio de menor impedimento
estérico (Adición Anti).

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
165
La reacción ocurre en el sitio de menor impedimento
estérico (Adición Anti).
(c) Hidrólisis de epóxidos catalizada por ácido.
La reacción ocurre en el sitio más impedido; se
producen 1,2-dioles trans a partir de epóxidos cíclicos.
(d) Apertura de anillos de epóxido inducida por
ácido.
La reacción ocurre en el sitio más impedido (Adición
Anti).
3.- Formación de sulfuros.
4.- Reacción de oxidación de sulfuros.
(a) Formación de sulfóxidos.
(b) Formación de sulfonas.
SÍNTESIS DE ÉTERES DE WILLIAMSON.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
166
Los alcóxidos metálicos reaccionan con los halogenuros de alquilo primarios y
con los tosilatos a través de una ruta SN2 para producir éteres, un proceso conocido
como síntesis de éteres de Williamson[1]
. Aunque se descubrió en 1850, la síntesis de
Williamson es todavía el mejor método para elaborar éteres, tanto simétricos como
asimétricos.
Ciclopentóxido de potasio Yodometano Éter ciclopentil metil (74%)
Los iones alcóxido necesarios de la síntesis de Williamson normalmente se
obtienen de la reacción de un alcohol con una base fuerte como el hidruro de sodio,
NaH. Una reacción ácido-base entre el alcohol y el hidruro de sodio genera la sal de
sodio del alcohol: ROH + NaH RO
-Na
+ + H2
Alcóxido
Otra forma de obtenerlos es empleando metales alcalinos:
ROH + Metal Alcalino (Na, K) RO
-Na
+ + H2
Alcóxido
ALCOXIMERCURACIÓN - DESMERCURACIÓN DE ALQUENOS.
Una reacción de Alcoximercuración ocurre cuando se trata un alqueno con un
alcohol en presencia de acetato de mercurio. (El trifluoroacetato de mercurio,
Hg(OOCCH3)2, funciona aún mejor). La posterior desmercuración es inducida con
borohidruro de sodio, produciendo un éter. Como lo muestran los siguientes ejemplos,
el resultado neto es la adición Markovnikov del alcohol al alqueno:
Estireno 1-Metoxi-1-feniletano (97%).
Ciclohexeno Éter ciclohexil etílico (100%).
El mecanismo de la reacción de alcoximercuración-desmercuración se inicia con
una adición electrofílica del ion mercúrico al alqueno, seguida de la reacción del catión
intermedio con el alqueno. El desplazamiento de mercurio con el borohidruro de sodio
completa el proceso.
Mecanismo:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
167
Estireno ion mercúrico intermedio
1-Metoxi-1-feniletano
Es posible emplear una amplia variedad de alcoholes y de alquenos en la
reacción de oximercuración.
REACCIONES DE LOS ÉTERES: RUPTURA ÁCIDA.
Los éteres son en su mayoría inertes a la totalidad de los reactivos utilizados en
química orgánica, una propiedad que explica su amplio uso como disolventes inertes.
Halógenos, ácidos suaves, bases y nucleófilos carecen de efecto sobre la mayoría de los
éteres. De hecho, los éteres experimentan sólo una reacción de uso general: se rompen
con ácidos fuertes. El HI acuoso es aún el reactivo preferido para la ruptura de éteres
simples, aunque también puede usarse HBr.
Ejemplos:
Ácido 2-etoxipropanoico
Éter etil isopropílico
Éter etil fenilíco
Éter terc-butil ciclohexílico
Las rupturas ácidas de éteres son reacciones de sustitución nucleofílica típicas
que transcurren por una ruta SN1 o por una ruta SN2, dependiendo de la estructura del
éter. Los éteres alquílicos primarios y secundarios reaccionan por una ruta SN2 en la
cual el ion yoduro o bromuro ataca al éter protonado en el sitio menos sustituido. Los
éteres terciarios, bencílicos y alílicos tienden a romperse a través de un mecanismo S N1.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
168
ÉTERES CÍCLICOS: EPÓXIDOS.
En gran medida, los éteres cíclicos se comportan como los éteres acíclicos. La
química de un grupo funcional éter es la misma, tanto si dicho grupo está en una cadena
abierta como si se encuentra en un anillo. Por ejemplo, los éteres cíclicos comunes
como el tetrahidrofurano y el dioxano a menudo se usan como disolventes debido a que
son inertes, aunque pueden romperse con ácidos fuertes.
El único grupo de éteres cíclicos que se comportan de manera diferente de como
lo hacen los éteres de cadena abierta es el formado por los anillos de tres miembros que
contienen oxígeno, llamados epóxidos u oxiranos. La tensión en el anillo de éter de tres
miembros ocasiona que los epóxidos sean reactivos y les confiere una reactividad
química única.
En el laboratorio, los epóxidos normalmente se elaboran por tratamiento de un
alqueno con un peroxiácido, RCO3H:
Pueden emplearse muchos peroxiácidos para la epoxidación. A escala de
laboratorio, el ácido m-cloroperbenzoico (MCPBA) es el reactivo preferido. Los
peroxiácidos transfieren oxígeno a través de un mecanismo complejo en un solo paso,
sin intermedios. Sin embargo existen pruebas que demuestran que el oxígeno más
alejado del grupo carbonilo es el que se transfiere.
Mecanismo:
Ciclohepteno Peroxiácido 1,2-Epoxicicloheptano Ácido
En otro método para la síntesis de epóxidos se emplean halohidrinas, producidas
por adición electrofílica de HO-X a alquenos. Cuando las halohidrinas se tratan con
base, se elimina HX y se produce un epóxido. Este método en realidad es una síntesis de
éteres de Williamson intramolecular. El átomo de oxígeno (nucleófilo) y el átomo de
carbono (electrófilo) están en la misma molécula.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
169
Mecanismo:
Bromohidrina Sustitución intramolecular Epóxido
REACCIONES DE APERTURA DEL ANILLO DE LOS EPÓXIDOS.
Los anillos de epóxidos pueden romperse por tratamiento con ácido, de modo
muy parecido a como ocurre con los éteres. La principal diferencia es que los epóxidos
reaccionan en condiciones mucho más suaves debido a la tensión en el anillo. Un ácido
mineral acuoso diluido a temperatura ambiente basta para provocar la hidrólisis de los
epóxidos y formar los 1,2-dioles, también llamados glicoles vecinales. La apertura del
anillo de epóxido catalizada por ácido procede por ataque SN2 de un nucleófilo sobre el
epóxido protonado.
Ejemplo:
1,2-Epoxiciclohexano trans-1,2-Ciclohexanodiol (86%)
Los epóxidos protonados también pueden ser abiertos por otros nucleófilos,
además del agua. Por ejemplo, si se emplea HX anhidro, los epóxidos pueden
convertirse en halohidrinas trans.
Los epóxidos, a diferencia de los éteres, también pueden romperse con bases, la
reactividad del anillo de tres miembros es suficiente para que los epóxidos reaccionen
con ion hidróxido a temperaturas elevadas.
Una ruptura nucleofílica de anillo similar se observa cuando los epóxidos se
tratan con reactivos de Grignard. El óxido de etileno se utiliza con frecuencia, y permite

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
170
convertir un reactivo de Grignard en un alcohol primario con dos carbonos más que el
halogenuro de alquilo de partida.
Bromuro de butilmagnesio 1-Hexanol (62%).
REGIOQUÍMICA DE LA APERTURA DEL ANILLO DE EPÓXIDO.
LA DIRECCIÓN EN LA CUAL SE ABREN LOS ANILLOS DE EPÓXIDOS
ASIMÉTRICOS DEPENDE DE LAS CONDICIONES DE REACCIÓN. SI SE EMPLEA UN
NUCLEÓFILO BÁSICO EN UNA REACCIÓN SN2 TÍPICA, EL ATAQUE OCURRE EN EL
CARBONO MENOS IMPEDIDO DEL EPÓXIDO. SIN EMBARGO SI SE EMPLEAN
CONDICIONES ÁCIDAS LA REACCIÓN SIGUE UN CURSO DISTINTO Y EL ATAQUE DEL
NUCLEÓFILO OCURRE PRINCIPALMENTE EN EL ÁTOMO DE CARBONO MÁS
SUSTITUIDO.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
171
ÉTERES CORONA.
Los éteres corona, descubiertos a principios de la década de 1960 por Charles
Pedersen[2]
en la compañía Du Pont, constituyen una adición relativamente reciente a la
familia de los éteres.
Éter 18-corona-6
La importancia de los éteres corona deriva de su extraordinaria capacidad de
solvatar cationes metálicos, secuestrando el metal en el centro de la cavidad del poliéter.
Diferentes éteres corona solvatan diferentes cationes metálicos, dependiendo de la
compatibilidad entre el tamaño del ion y el tamaño de la cavidad. Por ejemplo, el 18-
corona-6 es capaz de formar complejos muy estables con el ion potasio.
KMnO4, solvatado por 18-corona-6,
(este solvato es soluble en benceno)
Los complejos entre éteres coronas y sales inorgánicas son solubles en
disolventes orgánicos no polares, lo que permite efectuar muchas reacciones en
condiciones apróticas que de otra manera tendrían que llevarse a cabo en soluciones
acuosas. Muchas sales inorgánicas, como KF, KCN, y NaN3, pueden hacerse solubles
en disolventes orgánicos empleando éteres coronas. El efecto de su uso para disolver
una sal en un disolvente aprótico polar, como DMSO, DMF o HMPA. En ambos casos,
el catión metálico es fuertemente solvatado, dejando al anión desnudo. Así, la
reactividad SN2 de un anión se incrementa en presencia de un éter corona.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
172
SULFUROS.
Los sulfuros, R-S-R’, son análogos azufrados de los éteres. Se nombran
siguiendo las mismas reglas que para los éteres, usando el término sulfuro en vez de éter.
Sulfuro dimetílico Sulfuro metil fenílico 3-(Metiltio)ciclohexeno
Los sulfuros se elaboran por tratamiento de un halogenuro de alquilo primario o
secundario con un anión tiolato, RS-. La reacción transcurre por un mecanismo SN2, Los
aniones tiolatos son de los mejores nucleófilos que se conocen y los rendimientos que se
obtienen suelen ser altos en estas reacciones de sustitución.
Bencenotiolato de sodio Sulfuro metil fenílico (96%)
Puesto que los electrones de valencia del azufre están más lejos del núcleo y son
retenidos menos fuertemente que los del oxígeno existen algunas diferencias
importantes entre la química de los éteres y la de los sulfuros. Por ejemplo, el azufre es
más polarizable que el oxígeno, y por tanto los compuestos de azufre son más
nucleófilos que sus análogos de oxígeno. A diferencia de los éteres dialquílicos, los
sulfuros dialquílicos son nucleófilos tan eficaces que reaccionan rápidamente con
halogenuros de alquilo primarios a través de un mecanismo SN2. Los productos de tales
reacciones son las sales de trialquilsulfonio, R3S+.
Sulfuro dimetílico Yodometano Yoduro de trimetilsulfonio
Una segunda diferencia entre los sulfuros y los éteres es que los primeros se
oxidan fácilmente. El tratamiento de un sulfuro con peróxido de hidrógeno, H2O2, a
temperatura ambiente, produce el correspondiente sulfóxido (R2SO), y la oxidación
posterior del sulfóxido con un peroxiácido produce una sulfona (R2SO2).
Sulfuro metil fenilíco Metil fenil sulfóxido Metil fenil sulfona
[1]
Alexander W. Williamson (1824 - 1904); n. Wandsworth, Inglaterra; Ph. D. Giessen (1846). [2]
Charles John Pedersen (1904-1989); n. Pusan, Corea, Premio Nobel (1987).

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
173
ALQUENOS Los alquenos son hidrocarburos con un doble enlace carbono-carbono. El doble enlace
es un enlace más fuerte que el enlace sencillo, sin embargo, paradójicamente el doble
enlace carbono-carbono es mucho más reactivo. A diferencia de los alcanos, que
generalmente muestran reacciones más bien no específicas, el doble enlace es un grupo
funcional en el que tienen lugar muchas reacciones con marcado carácter específico.
Históricamente, los hidrocarburos con un doble enlace se conocían con el
nombre de olefinas. Este nombre, más bien raro, proviene del latín oleum, aceite, y
ficare, hacer, producir, y surgió porque los derivados de tales compuestos tenían, a
menudo, apariencia oleaginosa.
En el presente capítulo abordaremos un pequeño resumen de sus reacciones y
métodos sintéticos, resaltando los mecanismos más importantes en cada caso, se finaliza
con los problemas propuestos.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
174
ALQUENOS:
La estructura geométrica del etileno, el alqueno más sencillo, es ampliamente
conocida gracias a los experimentos espectroscópicos y de difracción. La molécula es
plana y de acuerdo con la estructura de Lewis del etileno, el doble enlace de caracteriza
por ser una región con dos pares de electrones.
Según la descripción orbitálistica de las moléculas, se necesita un orbital por
cada par de electrones. Es decir, se necesitan dos orbitales entre ambos carbonos para
alojar el número total de electrones de un doble enlace. El modelo orbitálico del etileno
comprende dos híbridos sp2 de cada carbono que se usa para formar un sencillo Csp
2-
Csp2. Hasta ahora, se ha utilizado el orbital s y dos de los orbitales p de cada carbono,
pero cada carbono aún posee un orbital p. Estos orbitales p son perpendiculares al plano
de los seis átomos, son paralelos entre sí y tienen zonas de solapamiento por encima y
por debajo del plano molecular. Este tipo de unión en la que hay dos zonas de enlace,
por encima y por debajo del plano nodal, se llama enlace . Esta notación se usa para
distinguirla de la del tipo que corresponde al solapamiento de dos orbitales sp2. Dicho
enlace carece de nodos y se denomina enlace .
PROPIEDADES FÍSICAS.
Las propiedades físicas de los alquenos son similares a las de los alcanos
correspondientes. Los alquenos más pequeños son gases a temperatura ambiente.
Comenzando por los compuestos C5, los alquenos son líquidos volátiles. Los alquenos
isómeros tienen puntos de ebullición parecidos y las mezclas sólo pueden ser separadas
mediante una destilación fraccionada realizada con mucho cuidado y con columnas de
gran eficacia.
Los momentos dipolares son pequeños en el caso de los hidrocarburos, pero
permite una distinción entre los isómeros cis y trans. Por ejemplo, el cis-2-buteno tiene
un momento dipolar pequeño mientras que el trans-2-buteno tiene un momento dipolar
nulo debido a su simetría.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
175
ESTABILIDAD DE LOS ALQUENOS.
Aunque la interconversión cis-trans de los isómeros de alquenos no ocurre de
manera espontánea, puede inducirse en condiciones experimentales apropiadas, como
tratamiento con un catalizador ácido fuerte. Si se interconvierte el cis-2-buteno con el
trans-2-buteno y se permite que alcancen el equilibrio, se halla que no tienen la misma
estabilidad. En el equilibrio, la relación de isómeros es de 76% trans y 24% cis. Los
alquenos cis son menos estables que sus isómeros trans debido a la tensión
estérica entre los dos sustituyentes voluminosos en el mismo lado en el doble enlace.
Trans (76%) Cis (24%)
No hay tensión Hay tensión
estérica estérica
Un incremento en el grado de sustitución eleva más la estabilidad. Como regla
general, los alquenos siguen el orden de estabilidad que se pre senta enseguida:
Tetrasustituido > Trisustituido > Disustituido > Monosustituido
Se han propuesto dos explicaciones para el orden de estabilidad observado. La mayoría
de los químicos consideran que el orden se debe principalmente a la hiperconjugación,
un efecto estabilizador que resulta de la superposición entre el orbital pi carbono-
carbono desocupado y un orbital sigma carbono-hidrógeno lleno en un sustituyente
vecino. A mayor número de sustituyentes, más oportunidades existen para la
hiperconjugación y más estable es el alqueno.
Además del efecto de hiperconjugación, también puede utilizarse un argumento
sencillo de energía de enlace para explicar el orden de estabilidad observado en los
alquenos. Un enlace entre carbono sp2 y un carbono sp
3 es algo más fuerte que un
enlace entre dos carbonos sp3. Así, al comparar 1-buteno y 2-buteno, el isómero
monosustiutido tiene dos enlaces sp3-sp
3 y otro sp
3-sp
2, mientras que el isómero
disustituido tiene dos enlaces sp3-sp
2. Los alquenos altamente sustituidos siempre tienen
mayor proporción de enlaces sp3-sp
2 sobre enlaces sp
3-sp
3 que los alquenos menos
sustituidos, y por tanto son más estables.
Además de los Hº de hidrogenación de alguna manera confirman los dos
hechos anteriores, dado que a medida que se va aumentando el grado de sustitución del
alqueno el Hº de hidrogenación va disminuyendo, lo que confirma de alguna manera el
grado de estabilidad, es decir la estabilidad de los alquenos aumenta con el grado de
sustitución.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
176
RESUMEN DE REACCIONES:
REACCIONES EJEMPLOS
1.- Síntesis de alquenos. (véase el capítulo 11, reacciones de
eliminación)
2.- Reacciones de los alquenos.
(a) Hidrogenación ( Adición cis)
(b) Hidrohalogenación
Donde HX, puede ser HCl, HBr o HI, se observa
regioquímica Markovnikov: El H se une
al carbono menos sustituido y el X lo
hace al carbono más sustituido.
Se observa adición anti-Markovnikov, funciona
bien con HBr.
(c) Halogenación (Adición Anti)
Donde X2 = Cl2 o Br2, mecanismo vía ion halonio.
(d) Formación de Halohidrinas (Adición
Anti)
Se observa regioquímica Markovnikov y
estereoquímica anti.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
177
(e) Formación de Haloéteres vecinales
(Adición Anti)
Se observa regioquímica Markovnikov y
estereoquímica anti.
(f) Hidratación
Se observa regioquímica Markovnikov y
estereoquímica anti.
(g) Oximercuración-Desmercuración
Se observa regioquímica Markovnikov; el grupo
OH se une al carbono más sustituido, Adición
sin.
(h) Hidroboración-Oxidación
Se observa adición anti-Markovnikov sin.
(i) Epoxidación
Adición concertada.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
178
(j) Dihidroxilación sin vecinal (cis)
Se observa adición sin.
(k) Dihidroxilación anti vecinal (trans)
Se observa adición anti.
(l) Ozonolisis
Dependiendo del tipo de alqueno, los grupos
carbonilos pueden ser cetonas, aldehídos o
ambos.
(m) Ozonolisis reductiva
Dependiendo del tipo de alqueno, los alcoholes
pueden ser 1º o 2º.
(n) Formación de ciclopropanos
Adición vía carbeno.
Conocida como reacción de Simmons-Smith.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
179
(o) Ruptura oxidativa de alquenos
Se diferencia de la dihidroxilación anti en que el
KMnO4 se usa concentrado y en caliente.
HIDROGENACIÓN DE ALQUENOS.
Los alquenos reaccionan con hidrógeno en presencia de un catalizador adecuado
para formar productos de adición. Los alcanos saturados correspondientes. Platino y
paladio son los dos catalizadores utilizados para la mayoría de las hidrogenaciones de
alquenos. El paladio suele emplearse finamente dividido y con un mineral inerte, como
carbón, a manera de soporte para maximizar el área superficial (Pd/C). El platino se
emplea generalmente como PtO2, reactivo llamado catalizador de Adams[1]
.
La reacción de hidrogenación ha resultado difícil de estudiar mecanísticamente.
Sin embargo, la experiencia ha demostrado que la hidrogenación suele ocurrir con
estereoquímica sin; ambos hidrógenos se unen al doble enlace desde la misma cara. El
primer paso de la reacción es la adsorción del hidrógeno en la superficie del catalizador.
Después se forma un complejo entre el catalizador y el alqueno mediante la
superposición de orbitales vacantes del metal con el orbital pi lleno del alqueno, a
continuación el producto saturado se separa del catizador.
HIDROHALOGENACIÓN.
El protón de ácido fuerte puede adicionarse a un doble enlace para dar una
carbocatión, mediante un ataque electrofílico y posterior captura por el nucleófilo (X-).
Para la reacción se requieren temperaturas bajas, para evitar transposiciones. Si los
carbonos del alqueno no están igualmente sustituidos, el protón del haluro ataca al
carbono menos sustituido, dejando de esa manera un carbocatión más estable y en
consecuencia el halógeno tiende hacia el carbono más sustituido. Este hecho se
denomina regla de markovnikov[2]
.
Mecanismo de adición electrofílico de HX a alquenos.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
180
Ejemplos:
Adiciones radicalarias sobre los alquenos: formación del producto anti-Markovnikov.
El bromuro de hidrógeno puede adicionarse de forma anti-Markovnikov a los
alquenos: en un nuevo mecanismo, el mecanismo de esta reacción de adición no sigue
una secuencia iónica, sino una más rápida en cadena radicalaria. La condición es tratar
al alqueno en HBr y presencia de un peróxido.
Mecanismo de hidrobromación radicalario:
El cloruro de hidrógeno (HCl) y el yoduro de hidrógeno (HI) no dan productos
de adición anti-Markovnikov con los alquenos debido a una cinética desfavorable.
HALOGENACIÓN DE ALQUENOS.
Los reactivos que no contienen átomos de electrófilos pueden atacar
electrofílicamente a los dobles enlaces. Un ejemplo es la halogenación de los alquenos,
que tiene lugar con adición al doble enlace de dos átomos de halógeno para dar un
dihaluro vecinal. Esta reacción va bien con el cloro y el bromo. El flúor reacciona
demasiado violentamente y la formación de un diyoduro es neutra desde el punto de
vista termodinámico.
La estereoquímica de la adición es anti (trans). ¿Cuál es el mecanismo que
explica esta estereoquímica? ¿Cómo ataca el halógeno al doble enlace rico en electrones,
si parece que no contiene ningún centro electrófilo?. La nube electrónica del alqueno
es nucleófila y ataca a un extremo de la molécula de halógeno con desplazamiento
simultáneo del segundo halógeno como anión haluro. El intermedio que resulta es un

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
181
ion halonio cíclico. Un posterior ataque nucleófilo del X- vía una SN2 por la cara
contraria al ion halonio explica claramente una adición anti. Cuando los iones halonio
son simétricos, el ataque es igualmente probable en cualquiera de los dos átomos de
carbono, dando por lo tanto el producto racémico.
Mecanismo de halogenación.
ion halonio
Cuando el halógeno es bromo el intermedio es un ion bromonio cíclico. La
principal característica de esta especie es que el bromo se une a los átomos de carbono
del doble enlace inicial para formar un anillo de tres miembros. Una importante
característica de este ion bromonio cíclico es que explica la estereoquímica de la
bromación. La estructura de este ion es rígida y únicamente puede ser atacada por el
lado opuesto al bromo.
Ejemplos:
1-Cloro.2.metilciclohexeno 1,1,2,Tricloro-2-
metilciclohexano
1-Hexeno 1,2-Dibromohexeno
Ciclohexeno trans-1,2-Dibromociclohexano
racémico
trans-2-Buteno meso-2,3-
Dibromobutano
¿El ion halonio puede reaccionar con otros nucleófilos?. En presencia de otros de
otros nucleófilos, el ion haluro competirá con ellos para atrapar al ion halonio
cíclico. Por ejemplo la bromación del ciclopenteno en presencia de un exceso de ion
cloruro (añadido como sal) da una mezcla de trans-1,2-dibromociclopentano y trans-1-
bromo-2-ciclopentano.
De este mismo modo se pueden explicar la formación de las halohidrinas y
haloéteres vecinales.
Ejemplos:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
182
Ciclopenteno trans-2-Bromociclopentanol
Ciclohexeno Trans-1-bromo-2-metoxiciclohexano

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
183
HIDRATACIÓN ELECTRÓFILA.
Cuando un alqueno se trata con una disolución acuosa de un ácido que contenga
un contraión que sea un mal nucleófilo, como el ácido sulfúrico, el agua juega el papel
de nucleófilo e intercepta al carbocatión que se ha formado después de la protonación
inicial. En la reacción global, los elementos del agua se han adicionado al doble enlace,
o sea, ha tenido lugar una hidratación. Este proceso es el inverso de la deshidratación de
alcoholes inducida en medio ácido. El mecanismo es el mismo pero a la inversa.
Mecanismo de hidratación.
Ejemplo:
Otro método de obtener alcoholes o éteres desde alquenos es la oximercuración-
desmercuración, ya comentada en el capítulo 13. Estos métodos en general conducen al
producto más sustituido, adición Markovnikov.
HIDROBORACIÓN-OXIDACIÓN.
El resultado global de una hidroboración-oxidación, es la adición de agua a un
doble enlace. Al contrario de las hidrataciones descritas anteriormente, esta es anti-
Markovnikov.
El borano, BH3, se adiciona al doble enlace sin activación catalítica. Esta
reacción se denomina hidroboración. La hidroboración, además de ser
estereoespecífica (adición sin), es también regioselectiva. Al contrario de las adiciones
electrófilas descritas con anterioridad, sólo controlan la regioselectividad los factores
estéricos (y no los electrónicos): el boro se une al carbono de menos impedido (menos
sustituido).
La posterior oxidación del producto de hidroboración con peróxido de hidrógeno
en medio básico acuoso, da lugar a la oxidación del alquilborano, produciendo la
formación del alcohol.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
184
Mecanismo propuesto:
Hidroboración
Oxidación
Ejemplos:
HIDROBORACIÓN-HALOGENACIÓN : HIDROHALOGENACIÓN ANTI-
MARKOVNIKOV.
Los alquilboranos también pueden ser precursores de haloalcanos. El resultado
global de esta hidroboración-halogenación es la hidrohalogenación de un alqueno de
forma regioselectiva y estereoespecífica. El halógeno, al contrario de las adiciones
Markovnikov, se coloca en el carbono menos sustituido.
Ejemplos.
EPOXIDACIÓN DE ALQUENOS.
El grupo -OH de los ácidos peroxicarboxílicos, RCO3H, contienen un oxígeno
electrófilo. Estos compuestos reaccionan con los alquenos, adicionando este oxígeno al
doble enlace con formación de oxaciclopropanos. El otro producto de la reacción es el
ácido carboxílico, que se elimina mediante extracciones con base acuosa. (véase el
mecanismo en el capítulo 13, formación de epóxidos).
DIHIDROXILACIÓN ANTI VECINAL (FORMACIÓN DE DIOLES TRANS).
Al tratar un oxaciclopropano con agua, en presencia de cantidades catalíticas de
un ácido o una base, tiene lugar la apertura del ciclo con formación del correspondiente
diol vecinal. En esta reacción el agua ataca nucleofílicamente al anillo de tres miembros
por la parte opuesta a donde se encuentra el oxígeno.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
185
DIHIDROXILACIÓN SIN VECINAL (FORMACIÓN DE DIOLES CIS).
El permanganato potásico reacciona, en solución fría, con los alquenos para dar
los correspondientes dioles vecinales sin. El otro producto de esta reacción es el dióxido
de manganeso, que es un oxidante débil e insoluble en el medio de reacción. En estas
condiciones neutras el MnO2 no reacciona ni con el alqueno ni con el diol.
¿Cuál es el mecanismo de esta transformación? La reacción del enlace con
permanganato constituye una adición concertada con movimiento simultáneo de tres
pares de electrones para dar un éster cíclico que contiene Mn(V). Por razones estéricas,
la única manera de formarse el producto es cuando los dos átomos de oxígeno se
introducen por el mismo lado del doble enlace: sin. Este intermedio es reactivo y en
presencia de agua se hidroliza para dar el diol libre.
La dihidroxilación sin puede conseguirse con mejores rendimientos usando
tetróxido de osmio OsO4, muy similar al permanganato en su modo de acción. Si se
utilizan en cantidades estequiométricas se pueden aislar los ésteres cíclicos, pero
normalmente estos se reducen con H2S o bisulfito, NaHSO3. Sin embargo, al ser el
OsO4 caro y muy tóxico se utilizan cantidades catalíticas en presencia de cantidades
estequiométricas de peróxido de hidrógeno.
Mecanismo de oxidación de los alquenos con permanganato:
Dihidroxilación vecinal sin con tetróxido de osmio.
REACCIÓN DE OZONOLISIS DE ALQUENOS.
En todas las reacciones de adición de alquenos que se han considerado hasta
aquí, el esqueleto carbonado del material de partida ha permanecido intacto. El doble
enlace carbono-carbono se ha transformado en nuevos grupos funcionales, por adición
de diferentes reactivos, pero no han roto o transpuesto enlaces del esqueleto de carbono.
Sin embargo existen reactivos fuertemente oxidantes que rompen los enlaces carbono-
carbono para producir dos fragmentos.
El ozono (O3) es el reactivo más útil para la ruptura de dobles enlaces. Este
compuesto que se elabora convenientemente en el laboratorio haciendo pasar oxígeno a
través de una descarga eléctrica de alto voltaje, se une rápidamente a los alquenos a
bajas temperaturas para formar intermedios cíclicos llamados molozónidos. Una vez

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
186
formados, los molozónidos se transponen con rapidez para formar ozónidos. Los
ozónidos son explosivos, y por tanto nunca se aíslan, sino que suelen tratarse con un
agente reductor, como zinc metálico en ácido acético, para transformarse en compuestos
carbonilicos.
Reacción de ozonólisis.
Alqueno Ozónido Productos carbonílicos
Mecanismo de la ozonolisis: ETAPA 1 Formación y ruptura del molozónido
Molozónido Oxido de carbonilo
ETAPA 2 Formación y reducción del ozónido
Ejemplos.
(Z)-3-Metil-2-penteno 2-
Butanona Etanal
1-Metilciclohexeno 6-Oxoheptanal
Isopropilidenciclohexano Ciclohexanona Aceton
a
OZONOLISIS A ALCOHOLES (OZONOLISS REDUCTIVA).
Al tratar el ozónido con borohidruro sódico se obtienen alcoholes. De esta forma,
un doble enlace puede romperse oxidativamente para producir alcoholes, estamos
hablando de una ozonolisis reductiva.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
187
4-Octeno 1-Butanol (95%)
Otros agentes oxidantes además del ozono también causan ruptura del doble
enlace. Por ejemplo, el permanganato de potasio en solución neutra o ácida provoca la
ruptura del alqueno, formando productos carbonílicos con rendimiento bajo o moderado.
Si en el doble enlace están presentes hidrógenos, se producen ácidos carboxílicos, si hay
dos hidrógenos en un mismo carbono, se forma CO 2.
Ejemplo:
3,7-Dimetil-1-octeno Ácido-2,6-dimetilheptanoico (45%).
ADICIÓN DE CARBENOS A ALQUENOS: SÍNTESIS DE CICLOPROPANOS.
La última reacción de adición a alquenos que se considerará en este capítulo es
la adición de un carbeno a un alqueno para producir un ciclopropano. Un carbeno
R2C: , es una molécula neutra que contiene un carbono divalente con sólo seis
electrones en su capa de valencia. Por ello es altamente reactivo, y sólo puede generarse
sólo como un intermediario de reacción, no como una sustancia aislable.
Uno de los mejores métodos para generar un carbeno sustituido es el tratamiento
de cloroformo, CHCl3, con una base fuerte como hidróxido de potasio. La pérdida de un
protón del CHCl3 genera el anión triclorometano, -:CCl3, el cual libera un ion cloruro
para convertirse en diclorocarbeno, :CCl2. Si se genera diclorocarbeno en presencia de
un doble enlace, ocurre la adición del carbeno, electrófilo, al doble enlace, y se forma
un diclorociclopropano. La adición es estereoespecífica, lo cual significa que se forma
un solo estereoisómero como producto.
Ejemplo:
Ciclohexeno (60%)
cis-2-penteno (65%)
El mejor método para producir ciclopropanos no halogenados es la reacción de
Simmons3 -Smith
4 .
Ejemplo:
Ciclohexeno Biciclo (4.1.0) heptano (92%)

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
188
[1]
Roger Adams, (1889-1971), Profesor de la Universidad de Illinois. [2]
Profesor Vladimir V. Markovnikov, (1838-1904); formuló su regla en el 1869 en la Universidad de
Moscú. 3 Howard E. Simmons, (1929- ), Químico de la Industria E.I. du Pont de Nemours y Cia.
4 Ronald D. Smith, (1930- ), Químico de la Industria E.I. du Pont de Nemours y Cia.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
189
ALQUINOS Los alquinos son hidrocarburos que se caracterizan por poseer un grupo funcional del
tipo triple enlace carbono-carbono. La fórmula general de los alquinos es CnH2n-2. El
acetileno, H-CC-H, que es el alquino más simple, , fue ampliamente usado en la
industria como materia prima para la elaboración de acetaldehído, ácido acético, cloruro
de vinilo y otros productos químicos, pero ahora son más comunes otros procesos más
eficientes en los que se usa etileno como materia prima.
Los átomos de carbono de los alquinos tienen hibridación sp, y el triple enlace
está formado por un enlace sigma () sp-sp y dos pi p-p. Existen relativamente
pocos métodos generales para la síntesis de alquinos. En éste capítulo comentaremos
algo acerca de su estructura, preparación y reacciones de los mismos.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
190
ALQUINOS.
Un triple enlace carbono-carbono resulta de la superposición de dos átomos de
carbono con hidridación sp. Recuérdese que los orbitales híbridos sp del carbono
adoptan un ángulo de 180º entre sí, a lo largo de un eje perpendicular a los ejes de los
dos orbitales no híbridos 2py y 2pz. Cuando dos carbonos con hibridación sp se
aproximan uno a otro para enlazarse, la configuración geométrica es adecuada para que
se forme un enlace sp-sp y dos enlaces p-p; es decir, un triple enlace. De este modo
el acetileno, C2H2, es una molécula lineal con ángulos de enlace H-C-C de 180º.
Formación de un triple enlace carbono-carbono por superposición de dos carbonos
con hibridación sp.
Los alquinos tienen puntos de ebullición muy similares a los de los
correspondientes alcanos y alquenos. El etino (acetileno) es singular por el hecho de no
tener punto de ebullición a presión atmosférica: sublima a -84 ºC. El propino (p.e. -23.2
ºC) y el 1-butino (p.e. 8.1 ºC) son gases, mientras que el 2-butino es prácticamente
líquido (p.e. 27 ºC) a temperatura ambiente. Los alquinos de tamaño medio son líquidos
destilables.
Por analogía con los alquenos, el relativo carácter s de los orbitales híbridos del
carbono de los alquinos sustituidos se traduce en la presencia de momentos dipolares,
excepto en el caso en que los sustituyentes se encuentren distribuidos de forma
totalmente simétrica.
= 0.74 D
= 0.80 D
= 0 D
Por las mismas razones, los alquinos terminales son más ácidos que los alcanos
o alquenos homólogos. El pKa del etino, por ejemplo, es de 25, marcadamente bajo
comparado con los del eteno (pKa = 44) y del etano (pKa = 50).
Hidridación: sp sp
2 sp
3
pKa: 25 44 50.
RESUMEN DE LAS REACCIONES DE LOS ALQUINOS
1.- Formación de alquinos

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
191
(a) Deshidrohalogenación de dihalogenuros vecinales
Ejemplos:
2,3-dibromo-pentano 2-
pentino
2-cloro-2-buteno 2-
butino
(b) Alquilación del ion acetiluro
Acetileno Un alquino
terminal
Un Alquino terminal Un
alquino interno
Ejemplos:
Acetileno 1-butino
2.- Reacciones de los alquinos
(a) Adición de HX, donde X = Br o Cl
Ejemplos:
1-butino 2-bromo-2-buteno 1-butino 1-bromo-1-
buteno

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
192
(b) Adición de X2, donde X = Br o Cl
Ejemplos:
1-butino 1,2-dibromo-1-buteno 1-butino 1,1,2,2,-
tertrabromopentano
(c) Hidratación catalizada por sulfato mercúrico
Una metil cetona
Ejemplos:
1-butino butanona 2-pentino 2-pentanona 3-pentanona
(d) Hidrobaración-oxidación
Ejemplos:
1-
butino butanal
1-ciclopentiletino 2-
ciclopentiletanal
(e) Reducción
1.- Por hidrogenación catalítica
Un alqueno cis
Ejemplos:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
193
1-ciclopentilpropino propilciclopentano 1-ciclopentilpropino cis-1-ciclopentilpropeno 2.- Por litio en amoniaco
Un alqueno trans
Ejemplo:
1-ciclopentilpropino trans-1-ciclopentilpropeno
(f) Acidez: conversión en aniones acetiluro
Ejemplos:
(g) Alquilación de aniones acetiluro
Acetileno Un alquino
terminal
Un Alquino terminal Un
alquino interno
Ejemplos: (véase alquilación del ion acetiluro)
h) Ruptura oxidativa
Ejemplos:
2-butino ácido acético 2-pentino ácido acético ácido
propanoico

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
194
FORMACIÓN DE ALQUINOS: REACCIONES DE ELIMINACIÓN DE
DIHALOGENUROS.
Los alquinos pueden obtenerse por eliminación de HX a partir de halogenuros
de alquilo de manera muy parecida a como se obtenían los alquenos. Sin embargo, dado
que un alquino está doblemente insaturado, es necesario eliminar dos moléculas de HX.
El tratamiento de un 1,2-dihalogenuro (un dihalogenuro vecinal) con un exceso de base
fuerte, como KOH o NaOH, da como resultado una doble eliminación, mediante un
mecanismo E2.
Los dihalogenuros vecinales requeridos se obtienen fácilmente por adición de
bromo o cloro a los alquenos. De este modo, la secuencia global de halogenació-
deshidrohalogenación constituye un muy buen método par transformar un alqueno en un
alquino. Si bien es posible utilizar distintas bases para la deshidrohalogenación,
normalmente se prefiere el amiduro sódico, NaNH2, porque suele tener mayores
rendimientos. La doble deshidrohalogenación ocurre en pasos bien diferenciados a
través de un halogenuro vinílico intermedio, lo cual sugiere que los halogenuros
vinílicos mismos se conviertan en alquinos cuando se tratan con bases fuertes.
Ejemplo:
REACCIONES DE ALQUINOS: ADICIÓN DE HX Y X2.
Como regla general, los reactivos electrófilos se unen a los alquinos del mismo
modo que a los alquenos. Por ejemplo, con HX, los alquinos forman los productos de
adición esperados. Aunque usualmente las reacciones pueden detenerse después de la
adición de un equivalente de HX, un exceso de ácido forma un dihalogenuro. Por
ejemplo, la reacción de 1-hexino con dos equivalentes de HBr produce el 2,2-
dibromohexano. La regioquímica de la adición sigue la regla de Markovnikov. El
halógeno se une al carbono más sustituido del triple enlace, y el hidrógeno lo hace al
menos sustituido. La mayoría de las veces se encuentra en el producto la estereoquímica
trans de H y X. Esto indica un mecanismo similar a los de los alquenos es decir vía
carbocatión. En este caso el intermedio formado es un carbocatión vinílico.
Un alquino Un carbocatión vinílico Un bromuro vinílico
Ejemplo:
1-Pentino 2-Bromo-1-pentino 2,2-
Dibromopentano

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
195
Bromo y cloro también se unen a los alquinos para formar productos de
adición, y también en este caso resulta una estereoquímica trans. La adición es similar a
la de los alquenos.
Ejemplo:
1-Butino (E)-1,2-Dibromo-1-buteno 1,1,2,2-
Tetrabromobutano
HIDRATACIÓN DE ALQUINOS.
Los alquinos pueden adicionar agua en analogía con la hidratación de alquenos.
La adición va en el sentido Markovnikov para dar enoles que tautomerizan a cetonas. La
reacción esta catalizada por los iones mercurio. Los alquinos simétricos internos dan
solamente un compuesto carbonílico; los no simétricos conducen a mezclas de
productos.
Ejemplos:
3-Hexino Único producto (80%)
2-Pentino (50%) (50%)
(91%)
1-Pentino 2-Pentanona (78%)
Los tautómeros son tipos especiales de isómeros que se interconvierten con
rapidez por medio de un equilibrio, llamado ceto-enolico.
Tautómero enol Tautómero ceto
(meno favorecido) (más favorecido).
Mecanismo de la hidratación de un alquino catalizada por ion mercúrico para formar
una cetona. La reacción produce un enol intermediario que rápidamente se
tautomeriza para convertirse en una cetona.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
196
Una cetona Un enol
HIDROBORACIÓN DE ALQUINOS.
Los alquinos terminales pueden reaccionar con borano, aunque en la práctica es
difícil detener la reacción en el paso del borano vinílico. En circunstancias normales,
experimentan una segunda adición al borano vinílico intermedio. Para evitar esta doble
adición puede usarse un borano voluminoso estéricamente impedido, como el bis-(1,2-
dimetilpropil)borano (que se conoce comúnmente como disiamilborano) en lugar del
borano normal (BH3). Cuando un alquino terminal reacciona con disiamilborano, ocurre
la adición de B-H al triple enlace C-C con la regioquímica anti Markovnikov esperada.
Sin embargo, una segunda adición es bastante lenta, puesto que el volumen estérico del
reactivo dialqilborano dificulta el acercamiento al doble enlace. Por lo tanto, la
oxidación del borano vinílico forma primero un enol y finalmente un aldehído.
Ejemplo:
Obsérvese que la secuencia de hidroboración-oxidación es complementaria de la
reacción de hidratación directa de alquinos, ya que resultan productos diferentes. La
hidratación directa de un alquino terminal con ácido acuoso y sulfato mercúrico produce
una metil cetona, mientras que la hidroboración-oxidación del mismo alquino forma un
aldehído:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
197
REDUCCIÓN DE ALQUINOS.
Los alquinos pueden hidrogenarse en las mismas condiciones empleadas para
hidrogenar alquenos. Por lo general, el platino o el paladio sobre carbón se colocan en
suspensión en una solución que contiene al alquino, y la mezcla se pone en contacto con
una atmósfera de hidrógeno. En estas condiciones se produce la completa saturación del
triple enlace.
Ejemplos
5-Decino Decano (96%)
4-Metoxi-1-butino 1-Metoxibutano (100%)
La hidrogenación es un proceso por pasos, lo cual permite en ciertos casos
detener la reacción a nivel del alqueno intermedio. Otra alternativa es evitar la
hidrogenación del alqueno a base de emplear catalizadores modificados. Uno de tales
sistemas es paladio precipitado sobre carbonato cálcico y tratado con acetato de plomo y
quinoleína, y se conoce como catalizar Lindlar[1] . La superficie del metal en el
catalizador de Lindlar adopta una configuración menos activa que la del paladio sobre
carbón, de forma que solamente el primer enlace del alquino, más reactivo, es
hidrogenado. Al tratarse de una adición sin, constituye un método de síntesis
estereoselectiva de alqueno cis.
Ejemplo:
1-Decino cis-5-Deceno (96%)
Un segundo método para la conversión de alquinos en alquenos es emplear
sodio o litio metálico en amoniaco líquido como disolvente. Este método es
complementario al de la reducción de Lindlar, puesto que con él se producen alquenos
trans en vez de alquenos cis. El mecanismo en sí implica la donación de un electrón al
triple enlace para producir un anión radical intermedio (tiene número impar de
electrones) como un anión (tiene carga negativa). Este intermediario acepta un protón
del amoniaco para formar un radical vinílico. La adición de un segundo electrón al
radical vinílico produce un anión vinílico, el cual toma un segundo protón del amoniaco
para formar el alqueno trans como producto.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
198
Ejemplo:
5-Decino trans-5-Deceno (78%)
ACIDEZ DE LOS ALQUINOS: FORMACIÓN DE ANIONES ACETILURO.
La diferencia más notable entre la química de los alquenos y la de los alquinos
es que los alquinos terminales son débilmente ácidos. Cuando un alquino terminal se
trata con una base fuerte, como el amiduro sódico, NaNH2, el hidrógeno terminal es
eliminado y se forma un anión acetiluro:
Anión Acetiluro
ALQUILACIÓN DE ANIONES ACETILURO.
La presencia de un par de electrones no compartidos hace que los aniones
acetiluro sean fuertemente nucleófilos. Como resultado, cuando están disueltos en
tetrahidrofurano estos aniones reaccionan con halogenuros de alquilo, para sustituir el
halógeno y formar un nuevo alquino como producto.
La alquilación de alquinos no se limita al ion acetiluro. Cualquier alquino
terminal puede convertirse en un anión acetiluro correspondiente y alquilarse por
tratamiento con un halogenuro de alquilo para formar un alquino interno. Debido a su
generalidad, la alquilación de acetiluros es el mejor método para formar alquinos
sustituidos a partir de precursores más simples. La alquilación de iones acetiluro se
limita al uso de bromuros y yoduros de alquilo primarios, RCH2X, por razones estéricas,
dado que son sustituciones nucleófilicas del tipo SN2. Además de su reactividad como
nucleófilos, los iones acetiluro son suficientemente fuertes para causar
deshidrohalogenación en vez de sustitución cuando reaccionan con halogenuros de
alquilo secundarios o terciarios.
Ejemplos:
1-Hexino 5-Decino (75%)
Propino 2-Butino (75%)

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
199
1-Pentinilciclohexano
(85%)
RUPTURA OXIDATIVA DE ALQUINOS.
Los alquinos, como los alquenos, pueden romperse por reacción con agentes
oxidantes fuertes, como el permanganato de potasio u ozono. Sin embargo, un triple
enlace suele ser menos reactivo que un doble enlace, y los rendimientos de los
productos de ruptura algunas veces son bajos. La reacción es demasiado compleja
mecanísticamente para ser tratada con detalles. Los productos que se obtienen son
ácidos carboxílicos. Si se oxida un alquino terminal se forma CO2 como uno de los
productos.
Ejemplos:
3-Hexino Ácido propanoico (2 moles)
Butino Ácido propanoico
[1] Dr. Herbert H.M. Lindlar, n. 1909; Hoffman La Roche and Co. A. G., Basilea.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
200
REACTIVOS ORGANOMETÁLICOS
Los reactivos organometálicos son una fuente de carbono nucleofílico, y por lo tanto de
gran utilidad en la química orgánica sobre todo en síntesis. Recuérdese que hasta ahora
siempre hemos visto al carbono en sus compuestos estudiados como un centro
electrófilo.
En este capítulo veremos los métodos de preparación de reactivos
organometálicos, RM. Ellos pueden obtenerse tratando haloalcanos con metales,
particularmente litio y magnesio, en los que un átomo de carbono de un grupo orgánico
se encuentra unido a un metal. Estas especies son bases y nucleófilos fuertes y son
extremadamente útiles en síntesis orgánica.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
201
REACTIVOS ORGANOMETÁLICOS.
Puesto que los metales son elementos electropositivos, los enlaces metal-
carbono pueden tener un alto grado de carácter iónico. Es decir, las estructuras
resonantes dipolares contribuyen a menudo de forma importante a las estructuras de
estos compuestos.
R-M R-M
+ =
R-M
+
El grado de covalencia de este género de enlaces metal-carbono depende
considerablemente del metal y se relaciona con la electronegatividad del mismo. Los
compuestos organometálicos de los metales alcalinos pueden ser considerados
simplemente como sales iónicas del catión metálico alcalino y el correspondiente anión
orgánico denominado carbanión.
Muchos de los compuestos organometálicos se descomponen con el agua; no
obstante, son solubles en varios disolventes orgánicos apróticos. Los disolventes típicos
son los éteres y los alcanos. Hay un elevado número de compuestos organometálicos
usados en síntesis orgánica, debido a su solubilidad en disolventes adecuados y a su
reactividad extrema, no se purifican normalmente y se hace uso de sus disoluciones sin
aislamiento previo.
REACTIVOS DE GRIGNARD.
Los halogenuros orgánicos de muy variadas estructuras, -de alquilo, de arilo y
vinílicos- reaccionan con magnesio metálico en éter o tetrahidrofurano (THF) como
disolvente para producir halogenuros de alquil-magnesio, RMgX. Estos productos,
llamados reactivos de Grignard en honor a su descubridor, Víctor Grignard[1]
, son
ejemplos de compuestos organometálicos, ya que contienen un enlace carbono-metal.
Donde R = alquilo 1º, 2º o 3º, arilo o alquenilo; X = Cl, Br o I.
Ejemplo.
Bromobenceno Bromuro de sec-butilmagnesio
2-Clorobutano Cloruro de sec-
butilmagnesio
Muchas clases de halogenuros orgánicos forman reactivos de Grignard. El
impedimento estérico en los halogenuros no parecen ser un factor determinante en la
formación de los reactivos de Grignard, dado que reaccionan fácilmente halogenuros de
todo tipo, sean primarios, secundarios o terciarios. Los halogenuros de arilo y de
alquenilo también reaccionan, aunque en estos casos es mejor usar tetrahidrofurano
como solvente. El halogenuro puede ser Cl, Br o I, aunque los cloruros son algo menos
reactivos que los bromuros y los yoduros. Los fluoruros orgánicos rara vez reaccionan
con magnesio.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
202
Como podría esperarse en base en lo expuesto sobre a la electronegatividad y la
polaridad de los enlaces, el enlace carbono-magnesio está altamente polarizado, lo cual
hace al átomo de carbono nucleófilo y básico:
En sentido formal, puede considerarse que un reactivo de Grignard es un anión de
carbono o carboanión; es decir, la sal de magnesio de un hidrocarburo ácido. Sin
embargo es más exacto considerar que los reactivos de Grignard contienen un enlace C-
Mg covalente altamente polar, y no un enlace iónico entre C- y
+MgX.
Debido a su carácter nucleófilo/básico, los reactivos de Grignard reaccionan
tanto con ácidos como con una amplia variedad de electrófilos, tal como hemos
comentado a lo largo de los capítulos anteriores. Por ejemplo, reaccionan con dadores
de protones (ácidos de Bronsted) como H2O, ROH, RCOOH o RNH2, para formar
hidrocarburos. La secuencia total es un método sintético útil para transformar un
halogenuro de alquilo en un alcano. R-X R-H, (véase capítulo 8). También
reaccionan con compuestos carbonílicos para dar alcoholes, R-CO-R’ R-CH(OH)-R’,
(véase capítulo 12 y 17).
Ejemplos:
1-Bromodecano Decano (85%) Pr opanona ter-Butanol
REACTIVOS DE ALQUIL-LITIO Y OTROS.
Es posible obtener otros reactivos de organometálicos de manera semejante a
como se hace para los reactivos de Grignard. Por ejemplo, los compuestos de alquil-litio
pueden producirse por reacción de un halogenuro de alquilo con litio metálico:
1-Bromobutano Butil-litio
Los compuestos de alquil-litio son tanto nucleófilos como bases, y su
comportamiento químico es semejante en muchos aspectos al de los halogenuros de
alquil-magnesio. Como los compuestos organolíticos son más difíciles de manejar, se
prefiere el uso de los reactivos de Grignard en el caso de adiciones sencillas a
compuestos carbonílicos. Sin embargo, la mayor reactividad de los reactivos
organolíticos permite su adición a grupos carbonilo bastante impedidos, con buenos
rendimientos.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
203
Otros compuestos organometálicos como los organosódicos y organopotásicos;
son tan reactivos que rara vez se utilizan para adiciones a grupos carbonilos. Deben ser
manejados con sumo cuidado, pues reaccionan con éteres y pueden inflamarse
espontáneamente en contacto con el aire. En cambio, los compuestos organocíncicos
(organocinc) son no demasiado reactivos, por lo que dan lugar a reacciones muy
selectivas. Así, mientras que los reactivos de Grignard reaccionan fácilmente con
ésteres, los derivados de cinc son prácticamente inertes frentes a ellos, aunque, sin
embargo, se adicionan al grupo carbonilo de aldehídos y cetonas. Esta selectividad se
utiliza en la reacción de Reformatsky. Se prepara el derivado organocíncico de un -
halogenoéster y se adiciona a continuación sobre un compuesto carbonílico. La reacción
constituye un método de preparación de -hidroxiésteres.
Bromoacetato de etilo Bromocincacetato de etilo 3-Fenil-
3-hidroxi-
propanoato de etilo
(64%).
Transmetalación.
Los compuestos organometálicos con metales que no sean litio o
magnesio pueden prepararse por una reacción nucleófila del alquil-litio o del reactivo de
Grignard con haluros metálicos. Esta transformación se denomina transmetalación ya
que convierte un organometálico en otro.
Transmetalación general
R-M + M’-X R-M’ + M-X
Transmetalaciones:
4 RMgX + SiO4 SiR4 + 4 MgX2
Tetraalquisilano
2 RMgX + CdX2 CdR2 + 2 MgX2
Dialquilcadmio
3 RLi + AlX3 R3Al + 4 LiX
Trialquilaluminio
RLi + CuI RCu + LiI
Alquilcobre
RCu + RLi R2CuLi
Dialquilcuprato de litio
REACCIONES DE LOS COMPUESTOS ORGANOMETÁLICOS:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
204
1.- Hidrólisis (obtención de alcanos) (véase capítulos 8 y 9).
2.- Acoplamiento de halogenuros de alquilo con compuestos organometálicos
(véase capítulo 9).
3.- Adiciones nucleofílicas. (véase capítulo 10).
4.- Adición de compuestos organometálicos a aldehídos y cetonas (obtención de
alcoholes) (véase capítulo 12).
5.- Adición de compuestos organometálicos a ésteres (obtención de alcoholes)
(véase capítulo 12).
6.- Apertura nucleofílica del oxaciclopropano por compuestos organometálicos
(véase capítulo 13).
[1]
Francois Auguste Victor Grignard (1871-1932); Francia. Nobel en (1912).

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
205
ALDEHÍDOS Y CETONAS REACCIONES DE
ADICIÓN NUCLEOFÍLICA Los aldehídos y cetonas se encuentran entre los compuestos de mayor importancia,
tanto en bioquímica como en la industria química. En el laboratorio, los aldehídos
normalmente se elaboran por ruptura oxidativa de alquenos, por oxidación de alcoholes
primarios o por reducción parcial de cloruros de ácidos o ésteres. Las cetonas se
producen de manera similar por ruptura oxidativa de alquenos, por oxidación de
alcoholes secundarios, o por adición de diorganocupratos a cloruros de ácido.
La reacción de adición nucleofílica es la reacción más importante de los
aldehídos y las cetonas, siendo posible elaborar una gran cantidad de productos por
adición nucleofílica. Las reacciones son aplicables a cetonas y aldehídos, pero en
general estos últimos son más reactivos por razones tanto estéricas como electrónicas.
En el presente capítulo abordaremos la forma de obtención de cetonas y
aldehídos y a continuación las reacciones más significativas de ellos, mostrándose en
forma de resumen.
ALDEHÍDOS Y CETONAS:
Los aldehídos y cetonas se encuentran entre los compuestos de más importancia
tanto en la naturaleza como en la industria química. En la naturaleza, muchas de las
sustancias necesarias para los sistemas vivos son aldehídos y cetonas. En la industria
química se sintetizan grandes cantidades de tales compuestos, que se usan como
solventes o como materias primas para una multitud de otros productos.
Propiedades de Aldehídos y Cetonas.-
El doble enlace carbono-oxígeno de los grupos carbonilos es similar en muchos
aspectos al doble enlace carbono-carbono de los alquenos. El átomo de carbono
carbonílico tiene hibridación sp2 y tres enlaces sigma. El cuarto electrón de valencia
permanece en un orbital p del carbono, y por superposición con un orbital p del oxígeno
forma con él un enlace pi. El átomo de oxígeno tiene otros dos pares de electrones no
compartidos, que ocupan los dos orbitales restantes.
Como los alquenos, los compuestos carbonílicos son planares respecto al doble
enlace, y tienen ángulos de enlace de 120º aproximadamente. Como podría esperarse, el

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
206
doble enlace carbono-oxígeno es más corto (1.22 Aº contra 1.43 Aº) y más fuerte (175
kJ/mol contra 92 kJ/mol) que un enlace sencillo carbono-oxígeno. Los dobles enlaces
carbono-oxígeno se encuentran polarizados debido a la elevada electronegatividad del
oxígeno respecto a la del carbono.
La consecuencia más importante de la polarización del grupo carbonilo es la
reactividad química del doble enlace carbono-oxígeno. En vista de que el carbono
carbonílico tiene carga parcial positiva, éste es un sitio electrófilo y es atacado por
nucleófilos. A la inversa, el oxígeno carboxílico tiene carga parcial negativa y es un
sitio nucleófilo (básico).
Oxígeno nucleófilo; reacciona con ácidos y electrófilos
Carbono electrófilo; reacciona con bases y nucleófilos

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
207
Otra consecuencia de la polaridad del enlace carbonílico es que las moléculas
de aldehídos y cetonas se asocian débilmente, lo cual hace que tengan puntos de
ebullición mayores que los alcanos con peso molecular semejante. Sin embargo, debido
a que no pueden formar puentes de hidrógeno, los aldehídos y las cetonas tienen puntos
de ebullición menores que los alcoholes correspondientes. El formaldehído, el aldehído
más simple, es gaseoso a temperatura ambiente, pero todos los otros aldehídos y cetonas
son líquidos.
Elaboración de Aldehídos y Cetonas.-
Uno de los mejores métodos para la síntesis de aldehídos es la oxidación de
alcoholes primarios y ruptura oxidativa de alquenos. Para la síntesis de cetonas los
métodos son análogos a los de aldehídos (véase capítulo de alcoholes y alquenos), en
todo caso los resumimos en forma de tabla:
SÍNTESIS DE ALDEHÍDOS Y CETONAS
EJEMPLOS
1.- Formación de aldehídos
(a).- Oxidación de alcoholes primarios
(b).- Ozonólisis de alquenos
(c).- Reducción parcial de ésteres
2.- Formación de cetonas
(a).- Oxidación de alcoholes
secundarios
(b).- Ozonólisis de alquenos

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
208
(c).- Acilación de Friedel-Crafts
(d).- Hidratación de alquinos
(e).- Reacción de diorganocupratos
con
cloruros de ácidos
Oxidación de Aldehídos y Cetonas.-
Los aldehídos son rápidamente oxidados para producir ácidos carboxílicos, pero
las cetonas no son reactivas hacia la oxidación excepto en condiciones muy vigorosas.
Esta diferencia de comportamiento es consecuencia de las diferencias estructurales entre
los dos grupos funcionales: Los aldehídos tienen un protón -CHO que puede ser
extraído con facilidad durante la oxidación, pero no así las cetonas.
Muchos agentes oxidantes convierten aldehídos en ácidos carboxílicos, entre
ellos el ácido nítrico caliente y el permanganato de potasio, pero el reactivo de Jones,
CrO3 en ácido sulfúrico acuoso, es una elección más común a la pequeña escala del
laboratorio. Las oxidaciones de Jones ocurren con rapidez a temperatura ambiente y
tienen buenos rendimientos de productos.
Heptanal Ácido heptanoico (85%)
Un inconveniente de la oxidación de Jones es que las condiciones en que se
realiza son ácidas, y las moléculas sensibles pueden experimentar descomposición
catalizada por ácido. Cuando existe esta posibilidad de descomposición, con frecuencia
las oxidaciones de aldehídos se realizan usando una solución diluida de óxido de plata,
Ag2O, en amoniaco acuoso (reactivo de Tollens[1]
).

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
209
Benzaldehído Ácido benzoico
Las cetonas son inertes a la mayoría de los agentes oxidantes comunes, pero
experimentan con lentitud una reacción de ruptura cuando se tratan con KMnO 4 alcalino
caliente. El enlace carbono-carbono próximo al grupo carbonilo se rompe, y se
producen fragmentos de ácidos carboxílicos. La reacción sólo es útil para cetonas
simétricas como ciclohexanona, puesto que apartir de cetonas no simétricas se forman
mezclas de productos.
Ciclohexanona Ácido hexanodioico (79%)
Reacciones de Adición Nucleofílica de Aldehídos y Cetonas.-
La reacción más importante de aldehídos y cetonas es la reacción de adición
nucleofílica. Un nucleófilo ataca al átomo de carbono del grupo carbonilo, electrófilo,
en una dirección aproximadamente perpendicular al plano de los orbitales sp2 del
carbonilo. El carbono carbonílico experimenta una hibridación de sp2
a sp3, y se produce
un ion alcóxido intermediario con configuración geométrica tetraédrica. El nucleófilo
atacante puede tener carga negativa (Nu:-) o ser neutro (:Nu-H). Sin embargo, en este
último caso suele tener un átomo de hidrógeno que puede ser eliminado posteriormente.
HO- ion hidróxido
H- ion hidruro
algunos nucleófilos con carga negativa R3C- un carbanión
RO- un ion alcóxido
NC- ion cianuro
Algunos nucleófilos neutros
H2O
ROH
H3N
RNH2
agua
un alcohol
amoniaco
una amina
La adición nucleofílica a cetonas y aldehídos suele tener dos variantes. (1) El
intermedio tetraédrico puede ser protonado para formar un alcohol, o (2) el átomo de
oxígeno del carbonilo puede eliminarse (como HO- o como H2O) para formar un nuevo
doble enlace carbono-nucleófilo.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
210
(1) (2)
Dos posibles vías de reacción posterior a la adición de un nucleófilo a una cetona o un aldehído
Las principales se ilustran a continuación en la roseta:
Algunas reacciones de adición nucleófilica de cetonas y aldehídos

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
211
Reactividad Relativa de Aldehídos y Cetonas.-
Los aldehídos son en general más reactivos que las cetonas hacia la sustitución
nucleofílica por razones tanto estéricas como electrónicas. Por razones estéricas, porque
la presencia de dos sustituyentes relativamente grandes en las cetonas, contra un solo
sustituyente grande en los aldehídos, hace que los neutrófilos atacantes puedan
aproximarse con mayor facilidad a los aldehídos. Electrónicamente, el mayor grado de
polaridad del grupo carbonilo de los aldehídos los hace más reactivos que las cetonas.
RESUMEN DE REACCIONES DE ADICIÓN NUCLEOFÍLICA
REACCIONES EJEMPLOS
1.- Adición de hidruro: Reducción
2.- Adición de reactivo de Grignard
3.- Adición de HCN: Cianohidrinas
4.- Adición de aminas primarias: Iminas
Iminas
Por ejemplo:
(a) Con hidroxilamina : NH2-OH
se obtiene ; Oximas, R2C=NOH
(b) Con hidrazina, NH2-NHCONH2
se obtiene; semicarbazonas,
R2C=N-NHCONH2
(c) 2,4-dinitrofenilhidrazina, NH2NH-C6H4(NO2)2
se obtiene la 2,4-dinitrofenilhidrazonas,
R2C=NNH-C6H4(NO2)2
Butanona Imina de Butanona
Ciclohexanona Oxima de Ciclohexanona
Benzaldehído Semicarbazona de benzaldehído

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
212
5.-
Adición de aminas secundarias:
Enaminas
6.- Reacción de Wolff-Kishner
7.- Adición de alcoholes: Acetales
8.- Adición de tioles: Tioacetales
9.- Desulfuración de tioacetales con
niquel Raney
10.- Adición de iluros de fósforo:
Reacción de Wittig
Adición Nucleofílica de H2O: Hidratación
Los aldehídos y cetonas reaccionan con agua para producir 1,1-dioles o dioles
geminales (llamados comúnmente gem-dioles). La reacción de hidratación es reversible;
los gem-dioles pueden eliminar agua para regenerar cetonas o aldehídos. La posición
exacta del equilibrio entre gem-dioles y cetonas o aldehídos depende de la estructura del
compuesto carbonílico. Aunque en la mayoría de los casos el equilibrio favorece
fuertemente al compuesto carbonílico.
La adición nucleofílica de agua a cetonas y aldehídos es un proceso lento en
agua pura, pero se cataliza tanto con ácido como con base. Si bien como en todas las

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
213
reacciones catalizadas en ácido o en base no cambian la posición del equilibrio, influyen
fuertemente en la velocidad de la reacción de hidratación.
El ion hidróxido como
nucleófilo se une al
grupo carbonilo de la
cetona o del aldehído
para producir un ion
alcóxido como
intermediario.
El ion alcóxido (básico)
como intermediario
extrae un protón (H+)
del agua para formar un
gem-diol como
producto y regenerar el
ion hidróxido que
catalizó la reacción.
El catalizador ácido
protona al átomo del
carbonilo, básico,
haciendo de la cetona o
aldehído un mucho
mejor aceptor de
nucleófilo.
La adición nucleofílica
de agua, neutra,
produce un gem-diol
protonado.
La pérdida del protón
regenera el catalizador
ácido y forma como
producto un gem-diol
neutro.
Mecanismo de la hidratación catalizada por Base Mecanismo de la hidratación catalizada por
Ácido
Adición Nucleofílica de HCN: Cianohidrinas.
Los aldehídos y las cetonas no impedidas reaccionan con HCN para producir
cianohidrinas, RCH(OH)CN, por tratamiento con HCN, el benzaldehído forma la
cianohidrina llamada mandelonitrilo, con un rendimiento del 88%.
Benzaldehído Mandelonitrilo (88%).
La reacción ocurre muy lentamente con HCN puro, pero es rápida cuando se
agrega una cantidad mínima de base o ion cianuro. Es posible entender este resultado
recordando que el HCN, es un ácido débil con pKa = 9.1, no se disocia ni es nucleófilo.
Sin embargo, el ion cianuro es fuertemente nucleófilo, la adición a cetonas y aldehídos
ocurre por la vía típica de adición nucleofílica. La protonación del intermediario
tetraédrico aniónico produce la cianohidrina y regenera el ion cianuro:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
214
La formación de cianohidrinas es útil debido a las reacciones posteriores que
puede experimentar. Por ejemplo, los nitrilos (RCN) pueden reducirse con LiAlH4 para
producir aminas primarias (RCH2NH2), y también hidrolizarse con ácido acuoso para
formar ácidos carboxilicos. Por tanto, la formación de cianohidrinas constituye un
método para transformar aldehídos o cetonas en otros grupos funcionales al mismo
tiempo que alarga la cadena en un átomo de carbono.
2-Amino-1-
feniletanol Ácido mandélico (90%)
Adición Nucleofílica de Reactivos de Grignard: Formación de Alcoholes.
La síntesis de alcoholes por reacción de reactivos de Grignard con cetonas y
aldehídos es simplemaente una adición nucleofílica. Las adiciones de Grignard suelen
ser irreversibles.
Carbonilo Alcohol
Para más detalles véase capítulo de alcoholes (cap. 12) y reactivos
organometálicos (cap. 16).
Adición Nucleofílica de Hidruro: Reducción.
La reducción de aldehídos y cetonas para producir alcoholes es otra reacción de
adición nucleofílica cuyo mecanismo ha sido estudiado en capítulo 12. para mayor
información remítase a dicho tema.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
215
Adición Nucleofílica de Aminas: Formación de Iminas y Enaminas.
Las aminas primarias, RNH2, experimentan adición a los aldehídos y las cetonas
para producir iminas, R2C=NR. Las aminas secundarias hacen lo mismo para producir
enaminas (-eno + -amina; amina insaturada).
Una imina Una enamina
El ataque nucleofílico a
una cetona o un aldehído
por el par electrónico de
una amina forma un
intermediario tetraédrico
dipolar. Luego se trnasfiere un
protón del nitrógeno al
oxígeno, lo que produce
una carbinolamina
neutra. El catalizador ácido
protona el oxígeno del
hidroxilo el electónico no
compartido del nitrógeno
libera agua, formando un
ion iminio La pérdida de H+ del
nitrógeno genera la
imina (en el caso de ser
amina) como pruducto
final
El grupo “G” puede
ser:
G = H, (NH2-H)
Amoniaco
G = -OH, (NH2-OH)
Hidroxilamina
G = -NH2, (NH2-NH2)
Hidracina
G = R, (NH2-R)
Amina primaria
G = -NHC6H5
(NH2-NHC6H5)
Fenilhidracina
G = -NHCONH2
(NH2-NHCONH2)
Semicarbazida
El producto final
reulta ser:
Imina
Oxima
Hidrazona
Imina
Fenilhidrazona
Semicarbazona
Mecanismo de la formación de imina (o derivado) por la reacción de una cetona o un aldehído con
compuestos derivados del amoniaco.
Las enaminas se forman cuando una cetona o un aldehído reaccionan con una
amina secundaria, R2NH. Hasta la fase del ion iminio, el proceso es idéntico al de
formación de iminas, pero en este punto no existe un protón en el nitrógeno que pueda

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
216
ser perdido para producir un producto neutro. En cambio, se pierde un protón del átomo
de carbono alfa y se obtiene una enamina.
La adición nucleofílica de una amina
secundaria a una cetona o un aldehído,
seguida de la transferencia de un
protón del nitrógeno al oxígeno,
produce una carbinolamina
intermediaria en la vía normal.
La protonación del hidroxilo por el
catalizador ácido convierte aquél en un
mejor grupo saliente.
La eliminación de agua por el par
electrónico del nitrógeno genera un ion
iminio intermediario.
La pérdida de un protón del átomo de
carbono alfa forma la enamina como
producto y regenera el catalizador
ácido.
Ejemplo:
Ciclohexanona Enamina de la
ciclohexanona

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
217
Adición Nucleofílica de Hidrazina: Reacción de Wolff - Kishner.
Una variante importante de la formación de iminas anteriormente descritas es el
tratamiento de una cetona o aldehído con hidrazina (H2N-NH2), en presencia de KOH.
Esta reacción, descubierta independientemente en 1911 por Ludwig Wolff[2]
en
Alemania y por N. M. Kishner[3]
en Rusia, es un método sintético en extremo valioso
para la conversión de cetonas o aldehídos en alcanos, R 2C=O R2CH2.
La reacción de Wolff - Kishner frecuentemente se realiza a 240ºC en
dietilenglicol a ebullición como solvente, pero una modificación en la cual se usa
dimetilsulfóxido como solvente permite que el proceso ocurra a temperatura cercana a
la ambiental.
La reacción de la cetona o el aldehído con la
hidrazina produce una hidrazona en la forma
normal.
La base extrae uno de los protones débilmente
ácidos del -NH2, produciendo un anión
hidrazona. Este anión tiene una forma de
resonancia “alílica” que coloca la carga negativa
en el carbono y el doble enlace entre los
nitrógenos.
La protonación del anión hidrazona se realiza en
el carbono y produce un intermediario neutro.
Entonces la pérdida del nitrógeno inducida por
la base produce un carbanión, que se protona
para formar un alcano neutro como producto.
Ejemplo:
Ciclopropano Metilciclopropano
carbaldehído (72%)

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
218
Reducción de Clemmensen:
Además de la reacción de Wolff-Kishner, existe un segundo proceso, llamado
reducción de Clemmensen[4]
, que también realiza la conversión de cetonas o aldehídos a
los alcanos correspondientes. La reducción de Clemmensen, cuyo mecanismo es
complejo y no se comprende del todo, implica el tratamiento del compuesto carbonílico
con amalgama de cinc, Zn(Hg) y HCl acuoso concentrado. Esta reacción se usa
principalmente cuando la cetona o el aldehído iniciales son sensibles a las condiciones
fuertemente básicas que se requieren para la reducción de Wolff-Kisner.
Propiofenona Propilbenceno
(86%)
Adición Nucleofílica de Alcoholes: Formación de Acetales.
Cetonas y aldehídos reaccionan en forma reversible con alcoholes en presencia
de un catalizador ácido para producir acetales, R2C(OR’)2, antiguamente también
llamados cetales.
Cetona/aldehído Un
Acetal
La adición nucleofílica inicial del alcohol al grupo carbonilo produce un
hidroxiéter llamado hemiacetal, análogo al gem-diol formado por la adición de agua.
Los hemiacetales se forman de manera reversible, normalmente con el equilibrio en
favor del compuesto carbonílico. Sin embargo, en presencia de ácido puede ocurrir una
reacción posterior. La protonación del grupo hidroxilo, seguida por la pérdida de agua a
través de un mecanismo tipo E1, forma un catión (un ion oxonio, R3O+) que une
entonces un segundo equivalente de alcohol para producir el acetal.
Una cetona Un hemiacetal Un acetal

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
219
Mecanismo de la formación de un acetal catalizada por ácido mediante la reacción de cetona o
aldehído con un alcohol.
La protonación del oxígeno carbonílico
polariza fuertemente al grupo carbonilo, y..
..activa a éste para el ataque nucleofílico
por el par no compartido del oxígeno del
alcohol.
La pérdida de un protón produce un
hemiacetal neutro como intermediario
tetraédrico.
La protonación del hidroxilo del
hemiacetal lo convierte en un buen grupo
saliente.
La deshidratación produce un ion oxonio
como intermediario.
La adición de un segundo equivalente de
alcohol origina un acetal protonado.
La pérdida de un protón forma el acetal
neutro como producto.
Los acetales son compuestos extremadamente útiles, debido a que pueden actuar
como grupos protectores de cetonas y aldehídos. Algunas veces sucede que el grupo
funcional interfiere en las reacciones químicas que se intentan en otro lugar de una
molécula compleja. Por ejemplo, si se deseara reducir solamente el grupo éster
del 4-oxopentanoato de etilo, el grupo ceto interferiría. El tratamiento del
cetoéster inicial con LiALH4 reduciría el grupo ceto como el grupo éster, para formar un
diol como producto:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
220
4-Oxopentanoato de etilo 5-Hidroxi-
2-pentanona
El problema anterior, puede evitarse protegiendo al grupo ceto como un acetal.
El la práctica, es conveniente usar etilenglicol (HOCH2CH2OH) como el alcohol
y así formar un acetal cíclico. El mecanismo de formación de un acetal cíclico usando
un equivalente de etilenglicol es exactamente el mismo que cuando se usan dos
equivalentes de metanol u otro monoalcohol. La única diferencia es que ahora los dos
grupos alcohol están en la misma molécula y no en dos moléculas.
Adición Nucleofílica de Tioles: Formación de Tioacetales.
Los tioles, RSH, experimentan adición a aldehídos y cetonas por una ruta
reversible catalizada por ácido para producir tioacetales, R’2C(SR)2. Como es de esperar,
el mecanismo de formación de un tioacetal es idéntico en todos los aspectos al de
formación de un acetal, con la excepción de que se usa un tiol en vez de un alcohol. A
menudo se emplea etanoditiol; el tioacetal se forma rápidamente y con alto rendimiento.
3-Metilciclohexanona Un tioacetal (96%)
Los tioacetales son útiles debido a que experimentan desulfuración cuando se
tratan con níquel pulverizado de preparación especial llamado níquel Raney[5]
(Ni
Raney). Dado que la desulfuración extrae azufre de una molécula reemplazándolo
por hidrógeno, la formación de un tioacetal seguida de la desulfuración con níquel
Raney es un excelente método para la reducción de cetonas o aldehídos a alcanos.
Cetona/Aldehído Tioacetal Alcano
Por ejemplo:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
221
Adición Nucleofílica de Iluros de Fósforo: Reacción de Wittig.
Cetonas y aldehídos se convierten en alquenos por medio de la reacción de
Wittig[6]
. En este proceso, un iluro de fósforo, R2C=(C6H5)3, se une a una cetona o un
aldehído para producir un intermediario dipolar llamado betaina. La betaína
intermadiaria en la reacción de Wittig no se aisla; se descompone para producir un
alqueno y óxido de trifenilfosfina. El resultado neto es el reemplazo del oxígeno del
carbono por el fragmento orgánico que esencialmente estaba unido al fósforo.
Los iluros de fósforo necesarios para la reacción de Wittig se producen
fácilmente por una reacción SN2 de halogenuros de alquilo 1º y algunos 2º (pero no
terciarios) con trifenilfosfina, R3P, son excelentes nucleófilos en reacciones SN2, con
altos rendimientos de sales cristalinas de tetraorganofosfonio. El protón en el carbono
próximo al fósforo con carga positiva es débilmente ácido y puede extraerse con bases
como hidruro de sodio o butil-litio (BuLi) para generar el iluro neutro. (reactivo de
Wittig), Por ejemplo:
Trifenilfosfina Metile
ntrifenilfosforano
La reacción de Wittig es extremadamente útil, y a partir de combinaciones
apropiadas de fosforanos y cetonas o aldehídos es posible elaborar una gran cantidad de
alquenos mono, di- y trisustituídos. Sin embargo, no pueden elaborarse alquenos
tetrasustituídos debido presumiblemente al impedimento estérico de la reacción.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
222
Mecanismo de la reacción de Wittig entre una cetona o aldehído y un iluro de fósforo.
Reacción de Cannizzaro:
Cuando un aldehído sin hidrógenos en el carbono adyacente al grupo -CHO
(como el benzaldehído) se calienta en presencia de ion hidróxido, ocurre una reacción
de dismutación en la cual se producen un equivalente de ácido carboxílico y un
equivalente de alcohol. La reacción fue descubierta en 1853 por S. Cannizzaro[7]
, a lo
cual se debe su nombre reacción de Cannizzaro.
Benzaldehído Ácido benzoico Alcohol
bencílico
La reacción de Cannizzaro ocurre por adición nucleofílica de ion hidróxido al
aldehído para formar un intermediario tetraédrico, que libera ion hidruro como grupo
saliente. Un segundo equivalente de aldehído acepte entonces el ion hidruro en otro
paso de adición nucleofílica. El resultado neto es que una molécula de aldehído
experimenta sustitución acílica de hidruro por hidróxido, de modo que se oxida para
convertirse en un ácido, mientras que una segunda molécula de aldehído experimenta
una adición de hidruro y, por tanto, se reduce para convertirse en un alcohol.
Intermediario tetraédrico
La reacción de Cannizzaro tiene pocas aplicaciones prácticas y se limita al
formaldehído y los benzaldehídos sustituidos.
Reacción de Reformatsky: Preparación de -hidroxiésteres.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
223
Si se trata un éster bromado con cinc metálico en presencia de un aldehído o
cetona se obtiene un -hidroxiéster. Esta reacción, conocida como reacción de
Reformastky, es la forma más importante de preparar -hidroxiácidos y sus derivados.
El -bromo éster y el cinc reaccionan en éter para generar un compuesto
organometálico de cinc, que se adiciona al carbonilo del aldehído o la cetona.
Ejemplos:
Aldehídos y Cetonas con Iluros de azufre como reactivos de transferencia de
metilenos:
Los iluros de azufre son nucleófilos y al igual que en la primera etapa de la
reacción de Wittig, atacan al carbonilo de aldehído o cetona para dar betaínas azufradas.
Sin embargo, a diferencia de las betaínas fosforadas en dichos aductos el sulfuro de
dimetilo, que actúa como grupo saliente es desplazado por ataque nucleofílico del ion
alcóxido con formación de un oxaciclopropano. De esta forma, el iluro de azufre
actúa como agente de transferencia de metileno.
El iluro de azufre se prepara a partir de dimetilsulfuro y yoduro de metilo y
posterior tratamiento básico con BuLi en THF, en un proceso análogo al reactivo de
Wittig.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
224
Ejemplos:
Ciclohexanona Betaína
azufrada (82%)
Oxidación de Baeyer - Villiger.
El carbono carbonílico de los aldehídos y cetonas puede ser atacado por el grupo
hidroxilo de un ácido peroxicarboxílico como si éste procediera de un alcohol. El
resultado es un análogo de un hemiacetal con un grupo peróxido. Estos productos no
son estables y se descomponen a través de un estado de transición de ocho electrones.
En el aducto obtenido a partir de un aldehído, es el hidrógeno el que migra para dar dos
ácidos carboxílicos. Uno se forma por oxidación del aldehído, siendo el otro el
correspondiente al ácido peroxicarboxílico original. En el aducto obtenido a partir de las
cetonas, un grupo alquilo migra de manera análoga para dar un éster. Esta
transformación de denomina oxidación de Baeyer-Villiger[8]
.
Ejemplos:
Ciclohexanona -lactona (90%)
Butanona Acetato de etilo (72%)
Butanal Ácido butanoico
Con cetonas asimétricas es en principio posible obtener dos ésteres diferentes. A
partir de una serie de experimentos se ha establecido una lista de “aptitudes migratorias”,
la cual indica la facilidad relativa de migración de varios sustituyentes. Este orden
depende de la estabilidad del carbocatión en el carbono que migra.
Aptitudes migratorias en la reacción de Baeyer-Villiger
H > terciario > ciclohexilo > secundario fenilo > primario > metilo

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
225
ALDEHÍDOS Y CETONAS
(Reacciones de Sustitución Alfa al carbonilo)
Y
ALDEHÍDOS Y CETONAS-INSATURADOS
(Síntesis y Reacciones de sis temas -insaturados)
En éste capítulo discutiremos la reactividad de aldehídos y cetonas en la posición alfa
(), es decir el carbono contiguo al grupo funcional. El carácter polar del grupo
carbonilo tiene un efecto acidificante sobre los hidrógenos y permite la formación de
alcoholes -insaturados (enoles) y sus aniones correspondientes (iones enolatos).
Los pKa de aldehídos y cetonas van de 19 a 21, valores considerablemente
inferiores a los pKa del eteno (44) o etino (25), aunque mayores que los de los alcoholes
(15-19) ¿Cuál es la razón de la relativa acidez de los aldehídos y cetonas? Existen dos
razones fundamentales: una es el efecto inductivo atrayente de electrones del carbono
carbonílico, polarizado positivamente y otra de mayor importancia, es que los iones
enolatos formados se hallan estabilizados por resonancia.
A continuación veremos las reacciones del carbono , fruto de la enolización
como también los métodos sintéticos y reacciones de sistemas -insaturados de
cetonas y aldehídos.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
226
REACCIONES DE SUSTITUCIÓN ALFA DE GRUPOS CARBONILOS DE
CETONAS Y ALDEHÍDOS:
Las reacciones de sustitución alfa ocurren en la posición contigua al grupo
carbonilo -la posición alfa ()- e implican la sustitución de un átomo de hidrógeno alfa
por algún otro grupo. Se verifican a través de la formación de un enol o ion enolato
como intermedios. Su estudio se iniciará aprendiendo un poco más de estas dos especies.
TAUTOMERÍA CETO-ENOL.
Los compuestos carbonílicos que tienen átomos de hidrógenos en sus carbonos
alfa se interconvierten en forma rápida con sus correspondientes enoles (eno + ol,
alcohol insaturado). Esta rápida interconversión entre dos especies químicamente
distintas es una clase especial de isomería conocida como tautomería. A los isómeros
individuales se les llama tautómeros.
En el equilibrio, la mayoría de los compuestos carbonílicos existen casi
exclusivamente en la forma ceto, y suele ser difícil aislar el enol en forma pura. Por
ejemplo, a temperatura ambiente la ciclohexanona contiene sólo alrededor de 0.0001%
de su tautómero enol, y la acetona sólo alrededor de 0.0000001% de su enol. El
porcentaje del tautómero enol es aún menor en los ácidos carboxílicos y en sus
derivados de acilo, como ésteres y amidas. Si bien los enoles son difíciles de aislar y
en el equilibrio están presentes sólo en pequeña cantidad, son extremadamente
importantes e intervienen en gran parte de la química de los compuestos
carbonílicos.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
227
Ciclohexanona
99.999 9% 0.000 1%
Acetona
99. 999 999 % 0.000 001%
La tautomería ceto-enol de los compuestos carbonílicos eatá catalizada tanto por
ácidos como por bases. La catálisis ácida implica la protonación del átomo de oxígeno
del carbonilo (una base de Lewis), para formar un catión intermediario que puede perder
un protón del carbono y producir enol neutro.
La formación de un enol catalizada por base ocurre vía una reacción ácido-base
entre el catalizador y el compuesto carbonílico. Este último actúa como un ácido prótico
débil y dona a la base uno de sus hidrógenos . Entonces el anión resultante -un ion
enolato- vuelve a protonarse para producir un compuesto neutro.
Mecanismo de la formación del enol catalizada por
Ácido Mecanismo de la formación del enol catalizada por
Base
PREPARACIÓN DE ENOLATOS:
Cuando cetonas y aldehídos se tratan con bases fuertes, éstas provocan la
desprotonación del carbono ; debido al carácter ácido de estos hidrógenos. Los pKa de
aldehídos y cetonas van de 19 a 21, valores considerablemente inferiores a los pKa de
alquenos y alquinos, aunque mayores a los de los alcoholes. Los aniones formados por
desprotonación, los iones enolato, se hallan estabilizados por resonancia.
Producir cantidades estequiométricas de un enolato a partir de un aldehído es
difícil debido a las reacciones secundarias (condensación aldólica). Las cetonas, sin
embargo, pueden desprotonarse con diisopropilamiduro de litio o hidruro potásico.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
228
RESUMEN DE LAS REACCIONES
I.- Síntesis y reacciones de enolatos y enoles
Ejemplos
1.- Preparación de enolatos
2.- Alquilación de enolatos
3.- Alquilación vía enaminas

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
229
4.- Equilibrio ceto-enol
En medio Ácido
En medio Básico
5.- Halogenación
(vía enolato)
(vía enol)
6.- Condensaciones aldólicas
Condensación aldólica mixta (un aldehído no
enolizable)
Condensación entre cetonas

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
230
Condensación aldólica intramolecular:
Se forman anillos sin tensión, preferentemente
(cinco y seis átomos).
II.- Síntesis aldehídos y cetonas -
insaturadas
Ejemplos
6.- Condensaciones aldólicas.
Ver ejemplos anteriores
7.- Bromación-deshidrobromación de
aldehídos y cetonas.
8.- Reacción de Wittig con iluros
estabilizados.
9.- Oxidación de alcoholes alílicos.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
231
10.- Isomerización de aldahídos -insaturados.
III.- Reacciones de aldehídos y cetonas
-insaturados.
Ejemplos
11.- Hidrogenación. (Reducciones)
12.- Adición de halogenos.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
232
13.- Adición de cianuro de hidrógeno.
14.- Adición de agua, alcoholes y aminas.
15.- Adición de reactivos organometálicos.
16.- Condensación con derivados de aminas.
G = -OH, -NH2, -NHR, etc.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
233
17.- Reacción de Michael.
18.-Anelación de Robinson.
Consiste en una : Michael y a continuación
una
Aldólica - intramolecular.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
234
ALQUILACIÓN SENCILLA Y DOBLE DE ENOLATOS.
La mayoría de alquilaciones tiene lugar sobre el carbono , la reacción
constituye un procedimiento general de introducción de un sustituyente alquilo en la
posición contigua a un carbonilo. Uno de los problemas de este tipo de reacción es
controlar la dialquilación. En las condiciones de reacción, la cetona monoalquilada
puede ser desprotonada por el enolato de partida y ser objeto de alquilaciones sucesivas.
Otra complicación es la que surge al alquilar cetonas asimétricas, ambas posiciones
pueden sufrir el ataque del electrófilo.
Ejemplos:
27% 38%
53% 47%
Mecanismo de la alquilación de la ciclohexanona:
ALQUILACIÓN VÍA ENAMINAS.
Las enaminas son ricas en electrones debido a la presencia del sustituyente
nitrógeno. La resonancia entre el par de electrones solitario de dicho nitrógeno y el
doble enlace posibilita el que éste pueda ser atacado por electrófilos. En efecto, las
enaminas en presencia de haloalcanos se alquilan en el carbono dando sales de iminio.
El tratamiento acuoso de éstas las hidroliza por un mecanismo inverso al formulado
para su formación. El resultado final es una cetona alquilada y la amina secundaria
original.
Ejemplos:
Enamina de ciclohexanona 2-Butilcicloxexanona (44%)
2,2-Dimetilbutanal (70%)

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
235
Mecanismo de la alquilación vía enamina para la 3-Pentanona:
2-Metilpentanona
HALOGENACIÓN DE ALDEHÍDOS Y CETONAS A TRAVÉS DE
INTERMEDIOS DE TIPO ENOLATO O ENOL.
Los aldehídos y cetonas reaccionan con halógeno mediante el carbono al
grupo carbonilo. En presencia de ácido, por ejemplo, la halogenación suele detenerse
tras la incorporación del primer halógeno. La velocidad de halogenación catalizada por
ácidos es independiente de la concentración de halógeno, lo cual sugiere un primer paso
determinante de la velocidad que involucra al sustrato carbonílico. Dicho paso es la
enolización. El halógeno ataca a continuación al doble enlace para dar un
halocarbocatión intermedio, estabilizado por el oxígeno. La desprotonación
subsiguiente de dicha especie proporciona el producto.
Ejemplos:
Propanona Bromopropanona
2-Metilciclohexanona 2-Cloro-2-metilciclohexanona

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
236
Mecanismo de la bromación catalizada por ácido de la propanona (acetona):
ETAPA 1.- Enolización
ETAPA 2.- Ataque del halógeno
ETAPA 3.- Desprotonación
La entrada de un segundo halógeno es prácticamente nula. Para poder repetir la
enolización, el compuesto halocarbonílico debe enolizarse de nuevo por el
mecanismo habitual catalizado por ácidos. Sin embargo, la capacidad atrayente de
electrones del halógeno hace que la protonación, el primer paso de la enolización, sea
más difícil que en el compuesto carbonílico original. Los rendimientos de la
monohalogenación suelen ser altos.
La halogenación catalizada por bases es totalmente diferente. En este caso lo
difícil es detener la reacción, por lo que no suele tener utilidad. Con metilcetonas, sin
embargo, el sustituyente trihalometil resultante actúa como grupo saliente en
condiciones básicas y el producto final es en muchos casos un ácido carboxílico y el
trihalometano (Reacción del haloformo).
3-Metil-2-butanona 1,1,1-Tribromo-3-Metil-2-butanona Ácido 2-
Metilpropanoico Bromformo

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
237
Mecanismo de bromación catalizada por base de una metil cetona:
ETAPA 1.- Formación del enolato
ETAPA 2.- Ataque nucleófilo sobre el bromo
ETAPA 3.- Bromación completa
ETAPA 4.- Formación de ácido carboxílico
CONDENSACIÓN ALDÓLICA.- ATAQUE DE ENOLATOS SOBRE LA
FUNCIÓN CARBONILO.
Los enolatos pueden atacar al carbono carbonílico para dar compuestos hidroxi-
carbonílicos. La eliminación subsiguiente de agua conduce a aldehídos y cetonas -
insaturados. La secuencia global de ambos pasos constituye una reacción de
condensación.
La condensación aldólica es general para aldehídos, aunque también puede
realizarse con éxito sobre cetonas. El mecanismo de esta reacción es un ejemplo
característico de la química de enolatos. En condiciones básicas empleadas, existe un

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
238
equilibrio entre aldehídos y su enolato correspondiente. Éste, que se halla rodeado de
aldehído en exceso, utiliza su carbono nucleófilo para atacar al carbonilo de una
molécula de aldehído. La protonación del alcóxido resultante proporciona el aducto
aldólico inicial, el 3-hidroxibutanal, al cual se le ha dado el nombre corriente de aldol.
Mecanismo de formación del aldol:
ETAPA 1.- Formación del enolato.
ETAPA 2.- Ataque nucleófilo.
ETAPA 3.- Protonación.
A temperaturas elevadas el aldol se convierte en su enolato, que sufre una
eliminación de ion hidróxido para dar lugar al producto final. El resultado neto es la
deshidratación del aldol catalizada por hidróxido. Las condensaciones aldólicas suelen
ser de diferentes tipos, entre las que están las intramoleculares que conducen a la
formación de anillos.
Mecanismo de la deshidratación:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
239
Ejemplos de condensaciones aldólicas:
2-Metilpropanal 3-Hidroxi-2,2,4-
trimetilpentanal
PREPARACIÓN Y QUÍMICA DE LOS ALDEHÍDOS Y CETONAS -
INSATURADOS.-
Los aldehídos y cetonas -insaturados contienen dos grupos funcionales. Al
igual que otros compuestos difuncionales, su química puede ser simplemente una
composición de las reactividades de los dos tipos de doble enlace o bien, puede
involucrar a la función enona de forma global. En este apartado revisaremos los
métodos de preparación de dichas moléculas.
Los aldehídos y cetonas -insaturados pueden prepararse por reacciones ya
conocidas por el estudiante que han sido comentadas con anterioridad en este capítulo,
conjuntamente con otras ya estudiadas, como por ejemplo las eliminaciones. Éste es el
caso de la halogenación en el carbono y posterior eliminación en medio básico.
Ciclopentanona 2-Ciclopentenona (73%)
En este caso el doble enlace
carbono-carbono puede formarse en
la posición contigua al carbonilo
mediante la cloración en medio
ácido seguida de una
deshidrocloración catalizada por base,
con buenos rendimientos.
Otro método eficaz de formar cetonas y aldehídos -insaturados es por la
reacción de Wittig. Por ejemplo, el 2-cloroetanal puede convertirse en la sal de fosfonio
y posteriormente ser desprotonado al iluro correspondiente; que reacciona un
aldehído para dar la formación de un aldehído -insaturado.
Ejemplo:
Iluro estabilizado
Heptanal 2-Nonanal (81%)
Los iluros formados de esta
manera no reaccionan con
cetonas. Son posibles otras
reacciones análogas con otros
iluros de alcanoílo.
Otro de los métodos empleados comúnmente para la preparación de los
aldehídos y cetonas -insaturados es la isomerización en medio ácido o básico de
sistemas -insaturados. Así por ejemplo, los compuestos carbonílicos -insaturados
se transponen fácilmente a sus isómeros conjugados. Se dice que el doble enlace
carbono-carbono “entra en conjugación” con el grupo carbonilo.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
240
Ejemplos:
3-Butenal 2-Butenal
3-Ciclohexanona 2-Ciclohexanona
La ruta catalizada por ácidos transcurre a través del dienol conjugado. La
protonación en el extremo más alejado del grupo hidroxilo genera un carbocatión
estabilizado por resonancia que se desprotona en el oxígeno para dar el producto. En la
reacción catalizada por bases, el intermedio es el ion dienolato conjugado, que sufre
reprotonación en el carbono extremo. Ambos mecanismos se ilustran a continuación:
Mecanismo de la isomerización catalizada por ácidos de compuestos carbonílicos
- insaturados:
Mecanismo de la isomerización catalizada por bases de compuestos carbonílicos
-insaturados:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
241
REACCIONES QUE SUFREN LOS ALDEHÍDOS Y CETONAS
INSATURADOS.
Los aldehídos y cetonas -insaturados dan lugar a muchas reacciones
perfectamente predecibles a partir de la química ya conocida de los dobles enlaces
carbono-carbono y carbono-oxígeno. Por ejemplo la hidrogenación con paladio sobre
carbono da el compuesto carbonílico saturado. Ciertos catalizadores especiales
provocan la reducción selectiva del grupo carbonílico sin afectar al doble enlace del
alqueno.
El hidrógeno empleado con
Paladio/Carbono, produce solamente la
hidrogenación del enlace carbono-
carbono, de manera casi cuantitativa
(95%) Cuando se emplea hidrógeno en presencia
de óxido de plata, sulfato de hierro y
acetato de cinc, y además a bajas
presiones de obtiene la hidrogenación del
grupo carbonilo sin dañar el enlace
carbono-carbono.
La halogenación es otra de las reacciones que sufre solamente el doble enlace
carbono-carbono. Por ejemplo la bromación proporciona un compuesto
dibromocarbonílico.
3-Penten-2-ona 3,4-
Dibromo-2-pentanona
ADICIONES 1,4 A ALDEHÍDOS Y CETONAS -INSATURADOS.
Las reacciones anteriormente comentadas pueden clasificarse como adiciones
1,2 a alguno de los enlaces del sistema, ya sea el carbono-carbono o el carbono-
oxígeno. Sin embargo, algunos reactivos se adicionan de forma 1,4 al sistema
conjugado, lo que se denomina adición conjugada. En tales transformaciones la parte
nucleófila del reactivo se une al carbono y la electrófila (normalmente un protón) se
une al oxígeno carbonílico . El producto inicial es un enol, que sufre posteriormente un
reordenamiento a la forma ceto.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
242
Adición 1,2 de un reactivo polar A-B a una
enona conjugada.
Adición 1,4 de un reactivo polar A-B a una
enona conjugada.
Las adiciones 1,4 son realizadas tanto en medio ácido como medio básico.
Ejemplos:
(a) Adición de cianuro de hidrógeno.
Mecanismo de adición:
(b) Adiciones 1,4 de Agua, Alcoholes y Aminas.
Adición de Agua:
Adición de Aminas:
Adición de Alcoholes:
Mecanismo de adición:
(c) Los reactivos organometálicos dan adiciones del tipo 1,2 ó 1,4.
Los reactivos organometálicos pueden adicionarse a la función
carbonilo -insaturada de forma 1,2 ó 1,4. Los reactivos organolíticos, por
ejemplo, reaccionan preferentemente mediante ataque nucleofílico sobre el carbono
carbonílico. En cambio las adiciones 1,4 suelen ocurrir con reactivos del tipo R2CuLi,
mediante un mecanismo bastante complejo para discutir en este capítulo.
Adición 1,2: Adición 1,4:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
243
REACCIÓN DE MICHAEL Y ANELACIÓN DE ROBINSON.
Los iones enolatos dan adiciones conjugadas a aldehídos y
cetonas -insaturados, una reacción que se conoce como reacción de
Michael[1]
. Dicha transformación funciona mejor con enolatos derivados de compuestos
-dicarbonílicos, si bien también tiene lugar con sistemas más sencillos. El mecanismo
de la reacción de Michael incluye el ataque nucleófilo del ion enolato sobre el carbono
del compuesto carbonílico insaturado, seguido de protonación.
Ejemplos de reacción de Michael.
Mecanismo de la reacción de Michael:
Los productos obtenidos por la reacción de Michael en ciertos casos pueden dar
lugar a una posterior condensación aldólica intramolecular, con la formación de un
anillo. La secuencia sintética, que comprende una adición de Michael seguida por una
condensación aldólica intramolecular, se llama también anelación de Robinson[2]
. La
anelación de Robinson ha sido empleada extensamente en la síntesis de anillos.
Ejemplos de Anelación de Robinson:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
244
Producto final de la reacción de
Robinson (86%)
[1]
Dr. Arthur Michael, (1853-1942), Universidad de Harvard. [2]
Sir Robert Robinson, (1886-1975), Universidad de Oxford, Premio Nobel en 1947.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
245
ÁCIDOS CARBOXÍLICOS Los ácidos carboxílicos ocupan un lugar importante dentro de la química orgánica, dado
que sirven para la construcción de derivados relacionados, como ésteres y amidas.
También son importantes en la síntesis orgánica de muchas otras moléculas.
Algunos ejemplos importantes son el ácido cólico, uno de los principales
componentes de la bilis humana, y los ácidos alifáticos de cadena larga como el ácido
oleico y el ácido linoleico, precursores biológicos de grasas y otros lípidos. También se
encuentran en la naturaleza muchos ácidos carboxílicos saturados simples. Por ejemplo,
el ácido acético, CH3CO2H, es el principal componente orgánico del vinagre; el ácido
butanoico, CH3CH2CH2CO2H, es el que da el olor a la mantequilla rancia, y el ácido
hexanoico (ácido caproico), CH3(CH2)4CO2H, es la causa del inconfundible olor de las
cabras y de los calcetines deportivos después de hacer ejercicios.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
246
ÁCIDOS CARBOXÍLICOS: ESTRUCTURA Y PROPIEDADES FÍSICAS.
Cuando se une un grupo hidroxilo a un grupo carbonilo se forma un nuevo
grupo funcional, el grupo carboxilo, dando lugar a los ácidos carboxílicos. Este nuevo
grupo suele representarse como -COOH o -CO2H. Dado que el grupo funcional ácido
carboxílico esta relacionado estructuralmente con las cetonas y con los alcoholes, podría
esperarse ver algunas propiedades familiares a ellos. En efecto, los ácidos carboxílicos
son similares a las cetonas y a los alcoholes en algunos aspectos, aunque también tienen
grandes diferencias. Como en las cetonas, el carbono carboxílico tiene hibridación sp2,
con ángulos de enlace de 120º aproximadamente. Al igual que los alcoholes, los ácidos
carboxílicos están fuertemente asociados por puentes de hidrógeno entre las moléculas.
Este hecho hace que los ácidos carboxílicos tengan un punto de ebullición bastante alto
con respecto a los alcoholes de peso molecular comparable.
Propiedades químicas de los Ácidos:
Formación de dímeros (aumenta el p.eb.)
Dos puentes de hidrógeno
ACIDEZ Y BASICIDAD DE LOS ÁCIDOS CARBOXÍLICOS:
Los ácidos carboxílicos tienen un pKa bajo (pH menor que 7.0) es decir son
compuestos ácidos. Por lo tanto la desprotonación para dar carboxilato es relativamente
fácil, mientras que la protonación es más difícil. Suelen reaccionar con bases como el
hidróxido de sodio y bicarbonato de sodio para formar las sales correspondientes.
Un ácido carboxílico Una sal de ácido carboxílico
(insoluble en agua) (soluble en agua)
Los ácidos carboxílicos suelen disociarse ligeramente en soluciones acuosas
para formar H3O+ y el anión carboxilato, RCOO
-. Como en todo ácido, es posible
definir una constante de acidez, Ka.
Casi en la mayoría de los ácidos carboxílicos, la constante de acidez Ka es del
orden de 10-5
, que corresponde a un pKa de 4.72.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
247
PREPARACIÓN DE LOS ÁCIDOS CARBOXÍLICOS:
De entre los métodos de síntesis de ácidos carboxílicos los más frecuentes son
oxidación, hidrólisis de nitrilos y reacción de reactivos de Grignard con CO2. A
continuación los ilustramos en forma de esquema con un respectivo ejemplo.
Formación de Ácidos Carboxílicos
Método sintético Ejemplo
a) Hidrólisis de otros derivados de ácidos
carboxílicos
Donde X = -Cl, -OR’, -OCOR’, -NHR’.
b) Oxidación de alquilbencenos
c) Ruptura oxidativa de alquenos y
alquinos
d) Oxidación de alcoholes primarios
o aldehídos

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
248
e) Hidrólisis de nitrilos
f) Transposición del ácido bencílico
g) Síntesis malónica
h) Reacción de Kolbe
i) Carboxilación de reactivos organometálicos
j) Oxidación de cetonas
Como puede verse, hay varios métodos para la síntesis de ácidos carboxílicos
por oxidación, carbonatación e hidrólisis de precursores adecuados.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
249
A continuación veremos las reacciones que dan los ácidos carboxílicos, las
cuales pueden agruparse en cinco categorías: Sustitución alfa; Descarboxilación;
Desprotonación; Reducción y Sustitución nucleofílica en el acilo.
REACCIONES DE LOS ÁCIDOS CARBOXÍLICOS
Método sintético Ejemplo
a) Desprotonación
b) Reducción a alcoholes primarios
c) Descarboxilación : Reacción de Hunsdiecker
d) Obtención de Haluros de ácidos
e) Obtención de Anhídridos carboxílicos
Anhídridos cíclicos:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
250
Favorecido en el caso de anillos de cinco o seis miembros.
f) Obtención de Esteres
Esteres cíclicos: (lactonas)
g) Obtención de Amidas
Imidas:
Amidas cíclicas: (lactamas)

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
251
h) Reacción con compuestos organometálicos
i) Formación y reacción de dianiones de
ácidos carboxílicos
Utilidad de la reacción:
j) Bromación: Reacción de Hell-Volhard-Zelinsky
k) Reacciones de desplazamiento de
ácidos 2-bromo carboxílicos.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
252
Los ácidos carboxílicos se pueden identificar fácilmente por métodos
espectroscópicos convencionales. Así, exhiben absorciones características en el
infrarrojo en la zona de 2500 y 3300 cm-1
(debido al OH) y a entre 1710 y 1760 cm-1
(debido al grupo C=O). En la RMN de 13
C, los ácidos carboxílicos también presentan
absorciones en el intervalo de 165 a 185 , y en la RMN de 1H absorben cerca de los 12
.
A continuación se comentarán los mecanismos más significativos de las
reacciones anteriormente mencionadas en las tablas.
La mayoría de los métodos para la preparación de los ácidos carboxílicos, han
sido mencionados con anterioridad en los capítulos precedentes, ejemplo oxidación de
alquenos y alquinos (cap. 14 y 15); oxidación de alcoholes (cap. 12); organometálicos
(cap. 16), etc. Por ésto que haremos un breve comentario de los nuevos métodos aquí
comentados.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
253
HIDRÓLISIS DE NITRILOS:
El grupo funcional nitrilo R-CN, puede ser hidrolizado con ácidos o bases
fuertes en solución acuosa para dar un ácido carboxílico. El método es bueno para
producir ácidos carboxílicos a partir de halogenuros de alquilo (R-X R-CN R-
COOH). La síntesis funciona mejor con halogenuros primarios, ya que puede competir
con reacciones de eliminación E2 con halogenuros secundarios o terciarios.
Mecanismo de la hidrólisis: vía una formación de amida
En medio ácido:
En medio básico:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
254
TRANSPOSICIÓN DEL ÁCIDO BENCÍLICO:
Consiste en un reagrupamiento de una dicetona en medio básico, tal como se
muestra en el siguiente mecanismo.
1) Adición de un ion hidróxido:
2) Transposición
3) Protonación
Ejemplo:
SÍNTESIS MALÓNICA:
Consiste en la obtención de un ácido carboxílico a partir de un malonato de etilo
o metilo, en presencia de una base y un halogenuro de alquilo, con posterior tratamiento
en medio ácido y calor para dar una descarboxilación. Su mecanismo se ilustra a
continuación en forma resumida.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
255
REACCIÓN DE KOLBE:
Es un caso particular en la obtención de ácidos carboxílicos, dado que conduce
solamente a ácidos benzoicos orto sustituidos con hidroxilo. En la reacción de Kolbe[1]
,
el fenóxido ataca al dióxido de carbono para dar la sal del ácido 2-
hidroxibencenocarboxílico. (ácido o-hidroxibenzoico). Su mecanismo se discutirá en el
capítulo de aromáticos.
REACTIVIDAD DEL GRUPO CARBOXILO: MECANISMO DE ADICIÓN-
ELIMINACIÓN.
Aparte de sus propiedades ácido-base, los ácidos carboxílicos muestran una
reactividad en su grupo carbonilo muy similar a la de los carbonilo de aldehído y
cetonas. El grupo carbonílico es susceptible al ataque nucleófilo, el oxígeno puede
reaccionar con electrófilos y los hidrógenos vecinos son ácidos y enolizables. El grupo
hidroxilo por si mismo o modificado puede actuar como grupo saliente dando lugar a
otros derivados carbonílicos. En cualquier caso los ataques nucleofílicos han de seguir
uno de los dos siguientes mecanismos, según las condiciones de la reacción.
A) ADICIÓN-ELIMINACIÓN CATALIZADA POR BASES:
B) ADICIÓN-ELIMINACIÓN CATALIZADA POR ÁCIDOS:
DESCARBOXILACIÓN DE ÁCIDOS CARBOXÍLICOS: REACCIÓN DE
HUNSDIECKER[2]
:
La reacción consiste básicamente en una oxidación. Una sal de plata del ácido
carboxílico se trata con un halógeno, normalmente bromo. El bromuro de plata precipita,
se desprende dióxido de carbono y se forma un bromoalcano en el que el halógeno

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
256
ocupa la posición del grupo carboxilo. Esta reacción permite la conversión de la sal de
un ácido carboxílico en un bromoalcano que contiene un átomo de carbono menos. El
mecanismo se ilustra a continuación:
ETAPA 1) Formación del hipobromito
ETAPA 2) Formación del radical RCOO·
ETAPA 3) Descomposición del radical RCOO·
ETAPA 4) Formación del haloalcano y RCOO·
Ejemplo:
OBTENCIÓN DE HALUROS DE ÁCIDOS A PARTIR DE ÁCIDOS
CARBOXÍLICOS:
Estas reacciones ocurren por un mecanismo similar al de los alcoholes al
reaccionar con los reactivos de PBr3, PCl5 o SOCl2. Inicialmente se forma un derivado
inorgánico del ácido, (formándose así un buen grupo saliente) susceptible de ataque
nucleofílico por parte del halogenuro.
Mecanismo de la formación del Cloruro de ácido con
cloruro de tionilo (SOCl2) Mecanismo de la formación del Bromuro de ácido
con tribromuro de fósforo (PBr3)
La obtención de los demás derivados de ácidos carboxílicos, Ésteres,
Anhídridos y Amidas, suelen seguir uno de los dos mecanismo de adición-eliminación
mencionados con anterioridad, dependiendo de las condiciones de reacción. Los
ésteres por ejemplo se preparan en medio ácido, usando un ácido mineral como
catalizador (sulfúrico o clorhídrico). Las amidas suelen prepararse con aminas y
calentamiento.
REACCIÓN DE LOS ÁCIDOS CARBOXÍLICOS CON COMPUESTOS
ORGANOMETÁLICOS ALQUILLITIO:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
257
Los ácidos carboxílicos con reactivos organometálicos, del tipo alquilitio dan
cetonas, vía la formación del carboxilato correspondiente. A pesar de que el ataque
nucleofílico sobre una especie cargada negativamente es difícil, los reactivos
organolíticos pueden dar la adición nucleofílica sobre el grupo carbonilo del ion
carboxilato. La reacción pasa por un intermedio del tipo dianión del diol geminal el cual
es tratado con agua y la deshidratación de este da la correspondiente cetona.
Mecanismo:
Ejemplos:
REACCIONES DE LOS CARBONOS , VÍA LA FORMACIÓN DE UN
DIANIÓN:
Los ácidos carboxílicos, al igual que otros compuestos carbonílicos, pueden dar
iones enolatos, que participen en reacciones de sustitución nucleófila (vía ion
carboxilato). Los ácidos carboxílicos en presencia de una base fuerte, como lo es el
diisopropilamiduro de litio, (LDA) y un codisolvente muy polar y aprótico, como la
hexametilfosfotriamida, (HMPA) pueden perder un segundo protón para dar el dianión
del ácido carboxílico, que es un potente nucleófilo y que puede dar lugar a alquilaciones,
apertura de oxaciclopropanos y adiciones aldólicas, entre otras reacciones.
Ejemplo: Reacciones en el carbono del ácido 4-metilpentanoico, en presencia de una
base y posteriormente un epóxido (óxido de ciclohexeno), un haluro de
alquilo (Bromuro de etilo), y un cetona (3-pentanona).

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
258
BROMACIÓN DE ÁCIDOS CARBOXÍLICOS EN EL CARBONO : REACCIÓN
DE HELL-VOLHARD-ZELINSKY[3]
:
Los ácidos carboxílicos pueden bromarse en el carbono , pero a diferencia de
aldehídos y cetonas precisan de un catalizador especial, el fósforo. En lás condiciones
de reacción el fósforo reaccionan rápidamente con el bromuro presente para dar el
iniciador, PBr3 (alternativamente puede emplearse el PBr3 directamente). El mecanismo
de esta reacción transcurre a través del bromuro de alcanoílo, formado por reacción del
PBr3 con el ácido carboxílico, tal como se ilustra a continuación:
ETAPA 1.- Formación del bromuro de alcanoílo
ETAPA 2.- Enolización

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
259
ETAPA 3.- Bromación
ETAPA 4.- Intercambio
Ejemplos:
La importancia de la reacción de Hell-Volhard-Zelinsky radica en que el bromo-
derivado obtenido puede dar lugar a otros derivados funcionalizados, por medio de una
sustitución nucleófila.
Ejemplos: Sustitución del bromo, por -OH, -NH2 y -CN, del compuesto
obtenido previamente.
a) Sustitución por -OH.
b) Sustitución por -NH2.
c) Sustitución por -CN.
A continuación se dan problemas propuestos relativos al tema. Éstos han sido
tomados de libros de química orgánica como lo son el: Vollhardt, Morrison y Boyd,
McMurry, otros son aportaciones personales.
[1]
Profesor Herman Kolbe, (1818-1884), Universidad de Leipzig, Alemania. [2]
Dr. Heinz Hunsdiecker, (1904- ); Alemania. [3]
Profesor Carl M. Hell, (1849-1926), U. de Stuttgart, Alemania; Profesor Jacob Volhard, (1834-1910),
U. de Halle, Alemania; Profesor Nicolai Zelinsky, (1861-1953), U. de Moscú.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
260
DERIVADOS DE LOS ÁCIDOS CARBOXÍLICOS Los ácidos carboxílicos y los derivados de ácidos carboxílicos son una clase de
compuestos que se denominan en general Derivados de Acilo, R-CO-Y, donde el grupo
acilo está unido a un sustituyente electronegativo -Y, que puede actuar como grupo
saliente en diversas reacciones de sustitución.
Ácido carboxílico
Halogenuro de ácido
( X = F,Cl, Br, I)
Anhídrido de ácido
Éster
Amida
Nitrilo
La reacciones químicas de los distintos derivados de ácidos carboxílicos están
representadas por un tipo de reacción general: la reacción de sustitución nucleofílica en
el acilo. Mecanísticamente, estas reacciones se realizan por medio de la adición de un
nucleófilo al grupo carbonilo polar del derivado de ácido, seguida de la expulsión de un
grupo saliente del intermediario tetraédrico:
donde Y = Cl, Br, I (halogenuro de ácido); OR’ (éster); OCOR’ (anhídrido) o NHR’
(amida).
La reactividad de un derivado de ácido hacia la sustitución nucleofílica depende
tanto del entorno estérico alrededor del grupo carbonilo como de la naturaleza
electrónica del sustituyente, Y. De este modo, se he encontrado el siguiente orden de
reactividad:
Halogenuro de ácido > Anhídrido > Éster > Amida.
Todos ellos pueden ser obtenidos de los respectivos ácidos carboxílicos, los
cloruros de ácidos a su vez se pueden transformar en cualquier otro derivado de ácido y
así con cada uno de ellos tal como se muestra a continuación en forma de esquema
incluidos los nitrilos. Sus respectivas reacciones se mostraran en forma de resumen con

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
261
un ejemplo, siguiendo la metodología expuesta para los otros grupos funcionales ya
estudiados.
ALGUNAS DE LAS REACCIONES DE SUSTITUCIÓN NUCLEOFÍLICA EN EL ACILO DE LOS
CLORUROS DE ÁCIDO
ALGUNAS DE LAS REACCIONES DE LOS ANHÍDRIDOS DE ÁCIDO

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
262
ALGUNAS REACCIONES DE LOS ÉSTERES
ALGUNAS REACCIONES DE LAS AMIDAS
ALGUNAS REACCIONES DE LOS NITRILOS
RESUMEN GENERAL DE LAS REACCIONES

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
263
Reacciones de los haluros de ácido
Ejemplo
1.-Hidrólisis
2.- Sales carboxílicas
3.- Alcohólisis
4.- Aminólisis
5.- Reacción de Grignard
6.- Reacción con diorganocupratos

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
264
7.- Reducción parcial
Reducción total
Reacciones de anhídrido de ácido
Ejemplo
1.- Hidrólisis
2.- Alcohólisis
3.- Aminólisis
4.- Reducción a alcoholes 1º
Reacciones de los ésteres
Ejemplo
1.- Hidrólisis
2.- Aminólisis
3.- Alcohólisis (Transesterificación)

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
265
4.- Pirolisis de ésteres
5.- Reactivos organometálicos
6.- Reducción parcial hacia aldehído
7.- Reducción total hacia alcoholes 1º
8.- Formación de enolatos con posterior tratamiento con un electrófilo

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
266
9.- Condensación de Claisen
Reacciones de las amidas
Ejemplo
1.- Hidrólisis
2.- Reducción parcial hacia aldehído
3.- Reducción total hacia aminas 1º
4.- Transposición de Hofmann

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
267
Reacciones de los nitrilos
Ejemplo
1.- Hidrólisis
2.- Con reactivos organometálicos
3.- Reducción parcial hacia aldehídos
4.- Reducción totall hacia aminas 1º
5.- Hidrogenación catalitica
6.- Desprotonación y alquilación en el C.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
268
La mayoría de las reacciones de sustitución nucleofílica en los derivados de los
ácidos carboxílicos aquí mencionadas ocurren en dos pasos: adición del nucleófilo y
eliminación del grupo saliente. Aunque algunas veces ambos pasos pueden afectar la
velocidad global de la reacción, suele ser el primero paso el limitante de la velocidad.
Ocurre la adición de un nucleófilo
al grupo carbonilo, lo que
produce un intermedio tetraédrico
Un par de electrones del
oxígeno desplaza al grupo
saliente Y, lo cual genera un
compuesto carbonílico como
producto
El nucleófilo puede ser del
tipo :Nu- o bien :Nu-H. Y es un grupo saliente: -OR,
-NR2, _-Cl, -OCOR’.
El orden de reactividad es Halogenuro de ácido > Anhídrido > Éster > Amida. Una
consecuencia importante de las diferencias de reactividad observadas es que suele ser
posible transformar un derivado más reactivo en uno menos reactivo.
Casi la mayoría de las reacciones de los halogenuros de ácido ocurren por
sustitución nucleofílica en el carbonilo. Por lo que no daremos grandes detalles de cada
una de ellas, veamos el caso de la hidrólisis:
Un haluro Un ácido
de ácido carboxílico
Dado que durante la hidrólisis se genera HCl, a menudo la reacción se realiza en
presencia de piridina o de NaOH como base para secuestrar el HCl e impedir que éste
cause reacciones laterles. La alcohólisis presenta un mecanismo casi idéntico con la
diferencia que el grupo atacante es el alcohol e igual cosa la aminólisis.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
269
La reducción de los halogenuros de ácido, termina convirtiéndolos el alcoholes
primarios cuando se emplean reductores fuertes (LiAlH4) y en aldehídos cuando se
utilizan reductores suaves como por ejemplo el DIDAL , la reducción ocurre por un
mecanismo típico de sustitución nucleofílica en el carbonilo en el cual el ion
hidruro (H-) ataca al grupo carbonilo produciendo un intermediario tetraédrico que
expulsa al ion cloruro, para formar un aldehído, el cual es rápidamente reducido por el
LiAlH4 en un segundo paso para generar el alcohol primario.
Los halogenuros de ácido con reactivos de Grignard suelen producir alcoholes
terciarios en los cuales los dos sustituyentes son idénticos, el mecanismo es igual a la
reducción con LiAlH4, pasándose por una cetona intermedia, la cual reacciona
rápidamente con el segundo equivalente de organometálico, para producir el alcohol,
ejemplo:
Mecanismo:
La obtención de cetonas apartir de halogenuros de ácido, es posible empleando
como agente organometálico un reactivo del tipo diorganocuprato, ejemplo:
La reacciones químicas de los anhídridos de ácido son muy similares a las de
los halogenuros de ácido, con la única diferencia que sus reacciones son más lentas. Los
ésteres sin embargo, son talvez los derivados de ácidos más importantes. Los ésteres
pueden ser obtenidos partiendo de ácidos o anhídridos, aunque el método más amplio es
partiendo de los cloruros de ácido.
Los ésteres presentan un comportamiento químico similar al descrito para los
halogenuros de ácido y anhídridos, pero son menos reactivos hacia los nucleófilos que

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
270
los anteriores; de ahí que las reacciones requieran de un catalizador que suele ser ácido
o básico.
La hidrólisis de ésteres por ejemplo es realizada en medio ácido o básico para
producir un ácido carboxílico más alcohol. La hidrólisis de ésteres en solución básica se
llama saponificación (del latín sapo, saponis, jabón).
Analicemos un ejemplo: Mecanismo de hidrólisis mediante base
Al contrario de lo que ocurre con la hidrólisis catalizada por ácidos, esta
reacción en sus últimas etapas no es de equilibrio, la etapa en la que el ácido se
convierte en la sal del carboxilato, es esencialmente irreversible. Por lo tanto, se
necesita al menos una cantidad estequiométrica de hidróxido (aunque frecuentemente se
emplea un exceso).
Reacción de los ésteres con alcoholes: transesterificación
Esta reacción permite la conversión de un éster en otro sin necesidad de obtener
el ácido libre. La transesterificación es una reacción de equilibrio. Para desplazar el
equilibrio se utiliza normalmente un gran exceso de alcohol, muchas veces como
disolvente. La reacción tiene utilidad dado que las lactonas (ésteres cíclicos) se abren
mediante transesterificación a hidroxiésteres.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
271
Ejemplo:
Amidas a partir de ésteres:
Los ésteres reaccionan con las aminas por un método típico de sustitución
nucleofílica en el acilo para producir amidas. La reacción en si no necesita catalizador
dado que las aminas, son buenos nucleófilos. Sin embargo, la reacción no tiene mucho
uso debido a que se obtienen mayores rendimientos por la aminolisis de los cloruros de
ácido.
Ejemplos:
Benzoato de metilo Benzamida
3-Metilbutanoato de metilo 3-Metil-N-etilbutanoamida
Reactivos de Grignard con ésteres producen alcoholes terciarios:
La reacción de los ésteres con dos equivalentes de un reactivo de Grignard, los
transforma en alcoholes terciarios, mientras que los ésteres del ácido metanoico
(fórmico) conducen a alcoholes secundarios. La reacción en si es una adición
nucleofílica del organometálico a la función carbonílica para dar la sal de magnesio de
un hemiacetal y formación de una cetona intermedia, a continuación tiene lugar otra
adición de un segundo equivalente de reactivo de Grignard sobre el grupo carbonílico
formado. Por ultimo, se obtiene el alcohol al añadir agua a la mezcla de reacción.
Ejemplo:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
272
Mecanismo:
Reducción de ésteres a alcoholes:
Los ésteres se reducen fácilmente por tratamiento con hidruro de litio y aluminio
(LiAlH4) para producir alcoholes primarios, el mecanismo consiste en la unión de un
hidruro al grupo carbonilo, a lo cual sigue la eliminación del ion alcóxido para producir
un aldehído como intermedio. La posterior adición de un hidruro al aldehído forma el
alcohol primario.
Mecanismo:
Éster Aldehído Alcohol
primario
Si en vez de LiAlH4 se usa DIBAL (hidruro de diisobutilaluminio) como agente
reductor, es posible aislar el aldehído. Debe tenerse gran cuidado (usar un equivalente
exacto de hidruro, y efectuar la reacción a -78ºC).
Ejemplo:
Formación de enolatos con posterior tratamiento con un electrófilo:
La acidez de los hidrógenos en de los ésteres es suficiente para que se formen
los enolatos de éster, cuando un éster reacciona con una base fuerte a baja temperatura.
Los enolatos de éster reaccionan como enolatos de cetonas, dando lugar a alquilaciones,
apertura de oxaciclopropanos y reacciones aldólicas.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
273
Ejemplo:
Condensación de Claisen
[1]:
Cuando los enolatos de éster atacan a otra molécula de éster se forma un
producto de condensación de Claisen, dando lugar a un producto conocido como -
ceto-éster. Esto se debe a que los enolatos no deben estar presentes en cantidades
estequiométricas, sino que pueden existir en concentraciones de equilibrio, como ee la
reacción aldólica, Tanto el alcóxido (base) como el éster empleado deben ser derivados
del mismo alcohol para evitar la una transesterificación.
Ejemplo:
Mecanismo:
1) Formación del enolato del éster
2) Adición nucleófila
3) Eliminación

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
274
La condensación de Claisen es esencialmente análoga a la aldólica, pero con un
éster en lugar de un aldehído. En de estar presente ésteres de distinta naturaleza química
se produce una condensación de Claisen cruzada, dando lugar a mezclas de productos, y
en el caso de que una misma molécula posea dos grupos ésteres da lugar a una
condensación de Claisen intramolecular, conocida con el nombre de condensación de
Dieckmann[2]
.
Ejemplos:
a) Condensación de Claisen mixta
b) Condensación de Claisen intramolecular (Dieckmann)

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
275
REACCIONES DE LAS AMIDAS:
Las amidas son mucho menos reactivas que cloruros de ácido, anhídridos de
ácido o ésteres. Por tanto, el enlace amida sirve como la unidad básica a partir de la cual
se forman todas las proteínas.
Las amidas sufren hidrólisis para formar ácidos carbixílicos más aminas cuando
se calientan con ácidos o bases en solución acuosa. Las condiciones que se requieren
para la hidrólisis de amidas son más drasticas que para la hidrólisis de cloruros de ácido
o ésteres, pero los mecamismos son similares.
Ejemplos:
(a) Hidrólisis ácida
(b) Hidólisis básica
Al igual que otros derivados de ácidos carboxílicos, las amidas pueden reducirse
con LiAlH4 o bien LiAl(OR)3H, para obtener alcoholes o aldehídos respectivamente. El
efecto neto de la reducción de una amida es la conversión del grupo carbonilo de la
amida en un grupo metileno (C=O CH2). Este tipo de reacción es específico de las
amidas y no ocurre con otros derivados de ácidos carboxílicos.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
276
Ejemplos:
La reducción con hidruro de litio y aluminio es igualmente eficaz tanto para las
amidas acíclicas como para las cíclicas (lactamas). La reducción de lactamas produce
aminas cíclicas con buenos rendimientos, y constituye un método de síntesis valioso.
Ejemplo:
Una lactama Una amina cíclica
Las amidas se pueden transformar en aminas con un carbono menos respecto a
las amida original mediante la transposición de Hofmann, la que será estudiada en el
capítulo 27, dedicado a las aminas.
REACCIONES DE LOS NITRILOS:
La química de los nitrilos es similar en muchos aspectos a la química de los
compuestos carbonílicos. Al igual que éstos, los nitrilos están fuertemente polarizados,
lo que hace que el átomo de carbono sea electrófilo. Por tanto los nitrilos son atacados
por nucleófilos para producir aniones imina con hibridación sp2
como intermedio, en
una reacción análoga a la formación del ion alcóxido intermedio por adición nicleofílica
a un grupo carbonilo.
Las dos reacciones más importantes de los nitrilos son la hidrólisis y la
reducción. Además, los nitrilos pueden ser parcialmente reducidos e hidrolizados para
producir aldehídos, y se pueden tratar con reactivos de Grignard para producir cetonas.
Los nitrilos pueden hidrolizarse en medio acuoso ácido o básico para dar los
correspondientes ácidos carboxílicos. El mecanismo de estas reacciones procede a
través de una amida intermedia e incluye etapas de adición-eliminación.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
277
Mecanismo de la hidrólisis de nitrilos catalizada por ácidos:
Mecanismo de la hidrólisis de nitrilos catalizada por bases:
Las condiciones para la hidrólisis de nitrilos son normalmente drásticas,
requiriendo altas concentraciones de ácido o base a elevadas temperaturas.
Los reactivos organometálicos reaccionan con los nitrilos para dar cetonas:
Los nucleófilos fuertes, como por ejemplo los reactivos organometálicos, se
adicionan a los nitrilos para dar sales de aniones de imina. El tratamiento coc ácido
acuoso conduce a la imina neutra, que se hidroliza rápidamente a cetona.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
278
Ejemplo:
[1]
Ludwig Claisen, (185-1930), Profesor de la Universidad de Berlin. [2]
Walter Dieckmann, (1869-1925), Profesor de la Universidad de Munich.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
279
COMPUESTOS DIFUNCIONALES Los compuestos difuncionales presentan una gran diversidad química, por lo que
haremos un estudio dedicados sólo a los más significativos, son ejemplos de
compuestos difuncionales Dioles, Hidroxialdehídos, Hidroxicetonas, Hidroxiácidos,
Ácidos dicarboxílicos, Dicetonas, Cetoaldehídos, Cetoácidos, Cetoésteres, etc.
REACCIONES DE LOS DIOLES:
Una reacción importante de los dioles 1,2- y 1,3- es la que dan con aldehídos y
cetonas para formar acetales y cetales cíclicos, (véase el mecanismo en el capítulo 17)
utilizada para proteger al grupo carbonilo. Los dioles con las funciones hidroxi
separadas por más de tres carbonos no presentan, en general, esta reacción, debido a que
el anillo resultante sería de siete o más eslabones.
Ejemplos:
La deshidratación de los 1,2-dioles en presencia de catálisis ácida va
acompañada con frecuencia de una transposición de esqueleto. Por ejemplo, el pinacol
(2,3-dimetilbutano-2,3-diol), reacciona con ácido sulfúrico produciendo la t-
butilmetilcetona, denominada vulgarmente “pinacolona” (Transposición pinacolínica).
Ejemplos:
Pinacol Pinacolona
Mecanismo de la transposición pinacolínica:
La deshidratación de 1,4- y 1,5-dioles produce con frecuencia éteres cíclicos,
sobre todo cuando uno de los grupos hidroxi es terciario.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
280
Ejemplos:
Mecanismo de ciclación:
La reacción se produce por desplazamiento
nucleófilo intramolecular.
Transcurre probablemente a través de un
carbocatión terciario que es atrapado por el
segundo hidroxi secundario.
Los 1,2-dioles se rompen con facilidad por el enlace carbono-carbono entre los
dos carbonos con los grupos hidroxi, cuando se tratan con ácido peryódico HIO4 o
tetracaetato de plomo. Combinada con el proceso de hidroxilación, esta oxidación
constituye un método de ruptura de alquenos, complementario a la ozonólisis. (véase
capítulo 14).
La oxidación peryódica comprende la formación de un diéster cíclico del ácido
peryódico, que por descomposición produce dos fragmentos carbonílicos y ácido yódico.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
281
Ejemplos:
COMPUESTOS -HIDROXICARBONILICOS:
HIDROXIALDEHÍDOS E HIDROXICETONAS
SÍNTESIS:
Los -hidroxialdehídos se obtienen por tratamiento de los ésteres con sodio en
un disolvente inerte tal, como éter o benceno. Estos compuestos se denominan aciloínas
y la reacción recibe el nombre de condensación aciloínica. El producto inicial de la
reacción es la sal disódica de un enodiol, que se hidroliza produciendo la aciloína.
Ejemplo:
La condensación aciloínica es un método útil para la síntesis de compuestos
cíclicos, sobre todo los de tamaño medio (8-13 eslabones). En estos casos, la reacción
debe efectuarse bajo condiciones de alta dilución para evitar reacciones
intermoleculares.
Ejemplos de condensación aciloínica intramolecular:
El mecanismo de esta reacción es similar al del acoplamiento pinacolínico. En
primer lugar, el éster se convierte en anión radicalario por transferencia de un electrón
del metal, quizás directamente al carbono carbonílico polarizado positivamente. A
continuación las especies resultantes se dimerizan dando el dianión del diol
correspondiente.
Mecanismo de la condensación aciloínica:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
282
Los alquenos, los alquinos y los éteres no resultan afectados por estas
condiciones de reacción.
-hidroxicetonas a partir de aldehídos: condensación benzoínicas:
Una segunda aproximación a la preparación de -hidroxicetonas es la
dimerización de aldehídos en presencia del correspondiente catalizador. Por ejemplo, al
tratar el bencenocarboxaldehído (benzaldehído) con una cantidad catalítica de cianuro
sódico en etanol acuoso se obtienen con buen rendimiento la 2-hidroxi-1,2-
difeniletanona.
Ejemplo:
Benzaldehído 2-hidroxi-1,2-
difeniletanona
(Be
nzoína)
El ion cianuro cataliza la reacción únicamente para el caso de aldehídos
aromáticos. Sin embargo, los aldehídos alifáticos sufren el mismo acoplamiento en
presencia de sales de tiazolio.
Ejemplo:
Pentanal 6-Hidroxi-5-decanona
Mecanismo de la condensación benzoínica:
HIDROXIÁCIDOS:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
283
En la naturaleza existen numerosos hidroxiácidos importantes y poseen nombres
vulgares de uso corriente. El ácido láctico es responsable del olor y sabor característicos
de la leche agria. El ácido máleico se encuentra en los jugos de frutas. El ácido tartárico
se conoce desde tiempos inmemoriales como sal monopotásica (crema tartárica) que se
deposita en el poso del vino. El ácido cítrico es un ácido hidroxitricarboxílico que
abunda mucho en la naturaleza.
-hidroxiácidos:
El método más corriente para obtener -hidroxiácidos consiste en la hidrólisis
de -haloácidos, (se obtienen por la reacción de Hell-Volhard-Zelinsky, ver capítulo 20).
Ejemplo:
Ácido butanoico Ác.-2-hidroxi-butanoico
Los -hidroxiácidos se obtienen también por hidrólisis de las cianohidrinas, que
resultan de la reacción de HCN con aldehídos o cetonas (ver capítulo 17). Dado que la
adición de HCN a un grupo carbonilo se invierte en las condiciones fuertemente básicas
necesarias para hidrolizar un nitrilo a un ácido, dicha hidrólisis se efectúa en
condiciones ácidas.
Ejemplo:
2-Hexanona Ácido-2-hidroxi-2-metil-
hexanoico
Transposición del ácido bencílico:
Es posible obtener -hidroxiácidos empleando compuestos -dicarbonílicos,
como -cetoaldehídos y las -cetocetonas, por hidratación en medio básico. Bajo estas
condiciones, la difeniletanodiona (bencilo) se convierte en el ácido
difenilhidroxietanoico (bencílico).
Difeniletanodiona (Bencilo) Sal pótasica del
ácido bencílico
Cuando la reacción ocurre en las -dicetonas cíclicas, la transformación implica
contracción del anillo.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
284
Ejemplo:
Otros hidroxiácidos se obtienen por hidrólisis de lactonas, que se obtienen
mediante la oxidación de Baeyer-Villiger de las cetonas.
Ejemplo:
2-Metil-ciclohexanona Ácido-6-hidroxi-
heptanoico
(-
hidroxiácido)
Si se emplea la cetona cíclica adecuada, se puede obtener el hidroxiácido
deseado, el problema radica que los hidroxiácidos mayores están el equilibrio con su
forma cíclica (lactona). La lactonización, al igual que la esterificación normal, es un
proceso en equilibrio; sólo cuando la lactona tiene un anillo de cinco o seis eslabones
existe una cantidad importante de lactona en el equilibrio.
Ejemplos:
ácido--hidroxibutanoico -butirolactona (27%) (73%)
ácido--hidroxipentanoico -valerolactona (90%) (10%)
PREPARACIÓN DE COMPUESTOS -DICARBONÍLICOS:
Existen los 1,2-compuestos, que aunque son bastante raros, sí poseen ciertas
propiedades químicas interesantes. El grupo más importante de compuestos
dicarbonílicos es el de los 1,3-isómeros debido a su importancia en la síntesis.
Compuestos -dicarbonílicos por condensación de Claisen.
Los compuestos 1,3-dicarbonílicos se obtienen siempre utilizando una reacción
de condensación tipo Claisen (véase el mecanismo en el capítulo 21, páginas 414-416).
Las -dicetonas y los -cetoaldehídos se obtienen por una condensación de Claisen

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
285
cruzada, utilizando una cetona y un éster. En la condensación de Claisen cruzada de
cetonas y ésteres se obtienen buenos resultados, debido a que las cetonas son
notablemente más ácidas que los ésteres, por lo tanto, en el medio básico la cetona se
desprotona en mayor grado que el éster.
Ejemplos:
Etanoato de metilo Acetoacetato de metilo (90%)
(85%)
Los -cetoésteres pueden hidrolizarse para dar lugar a los -cetoácidos los
cuales sufren una DESCARBOXILACIÓN térmicamente de la misma manera que los
1,3-diácidos.
Los ácidos dicarboxílicos experimentan diversas reacciones térmicas, una de
ellas es la descarboxilación cuando se calientan, por ejemplo el ácido málonico se
descarboxila suavemente cuando se calienta a 150 ºC y produce ácido acético.
Ácido málonico Ácido
acético

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
286
Descarboxilación:
El mecanismo de descarboxilación de -cetoácidos es idéntico al descrito para
diácidos, transcurre a través de un estado de transición de seis centros, concertado. El
producto inicial en el caso de un -cetoácido es la forma enólica de la cetona la cual por
equilibrio cetoenolico da el compuesto carnonílico sencillo.
Acidez de los compuestos 1,3-dicarbonílicos:
Los compuestos 1,3-dicarbonílicos que poseen un hidrógeno unido al carbono
que se encuentra entre los dos grupos carbonilo, son ácidos mucho más fuertes que los
aldehídos, cetonas o ésteres normales, debido a que la carga en el ion enolato resultante
puede deslocalizarse en ambos grupos carbonilo.
ion enolato estabilizado por resonancia

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
287
En la Tabla 22.1 se muestran algunos valores de pKa típicos para estos tipos de
sistemas. La acidez de los compuestos 1,3-dicarbonílicos es tan grande que se
convierten en sus bases conjugadas de forma cuantitativa por acción del ion hidróxido
en agua o del ion alcóxido en un disolvente alcohólico.
TABLA 22.1.- Valores de pKa para sistemas -dicarbonílicos y análogos.
Nombre Estructura pKa
2,4-Pentanodiona
(Acetilacetona)
9
2-Cianoetanoato de metilo
(2-Cianoacetato de metilo)
9
3-Oxobutanoato de etilo
(Acetoacetato de etilo)
11
Propanodinitrilo
(Malonodinitrilo)
13
Propanodioato de dietilo
(Malonato de dietilo)
13
Los aniones-dicarbonílicos son nucleófilos:
La elevada acidez de los compuestos -cetocarbonílicos hace que se puedan
utilizar en síntesis, ya que los iones enolato que se obtienen por desprotonación pueden
alquilarse para dar derivados sustituidos.
Ejemplos:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
288
SÍNTESIS ACETOACÉTICA:
Si se emplea acetoacetato de etilo como sustancia de partida, la combinación de
la alquilación, hidrólisis y la descarboxilación proporciona una síntesis de
alquilmetilcetonas.
Acetoacetato de
etilo Una metilcetona
Ejemplos:
SÍNTESIS MALONICA:
Si se emplea el malonato de dietilo en una alquilación con posterior hidrólisis
y descarboxilación el producto final resulta ser un ácido carboxílico simple.
Ejemplos:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
289
La principal limitación de estas síntesis es la eliminación bimolecular que puede
experimentar el derivado halogenado durante el proceso de alquilación, que se produce
por un mecanismo SN2. La importancia de dicha reacción secundaria de eliminación
depende de la estructura del R-X. Los haloalcanos terciarios que reaccionan con aniones
-dicarbonílicos dan preferentemente productos de eliminación. Sin embargo, los
aniones pueden ser atacados por haluros de alcanoilo, -bromoésteres, -bromocetonas
y oxaciclopropanos.
CONDENSACIÓN DE KNOEVENAGEL:
En la condensación de Knoevenagel[1]
, el compuesto -dicarbonílico se trata con
una cantidad catalítica de una base débil, tal como la N-etiletanamina (dietilamina), en
presencia de un aldehído o cetona para dar el producto de condensación aldólica.
Ejemplo:
LA ADICIÓN DE MICHAEL:
Al tratar los aniones -dicarbonílicos con compuestos carbonílicos -
insaturados se producen adiciones 1,4 (adición de Michael). La reacción va bién con
cetonas -insaturadas, aldehídos, nitrilos, y derivados de ácidos carboxílicos,
requiriendo únicamente cantidades catalíticas de base.
Ejemplos:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
290
[1]
Emil Knoevenagel, (1865-1921), Profesor de la Universidad de Heidelberg.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
291
SISTEMAS CONJUGADOS
SISTEMAS ALÍLICOS:
Cuando se trata el 2-buten-1-ol con bromuro de hidrógeno a 0 ºC se produce una
mezcla de 1-bromo-2-buteno y 3-bromo-1-buteno. Una mezcla semejante se obtiene al
tratar 3-buten-2-ol con HBr en las mismas condiciones:
Este hecho experimental se explica mediante la formación de un intermedio
carbocatiónico en el que la carga positiva se deslocaliza entre dos carbonos.
Los intermedios carbocatiónicos de las reacciones precedentes se describen
como híbridos de resonancia de dos estructuras importantes. El más sencillo de tales
cationes es el 2-propen-1-ilo o catión alilo.
catión
alilo híbrido
También se conocen los radicales alilo y los aniones alilo, siendo éstos también
estables por resonancia:
Radical
alilo híbrido
Anión
alilo híbrido
La estabilización por resonancia del sistema (alilo) puede también describirse
en términos de orbitales moleculares, una aproximación mecánico-cuántica más
sofisticada. El esqueleto molecular consta de tres carbonos, cada uno de ellos con
hibridación sp2 y con un orbital p perpendicular al plano molecular, Figura 22.1. Puede

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
292
considerarse como un enlace doble al que se le ha añadido un carbono sp2
adicional. La
molécula sin embargo es simétrica, con las mismas longitudes de enlace C-C.
FIGURA 23.1.- Catión alilo, descripción orbitálica .
Ignorando el esqueleto , podemos combinar matemáticamente los tres orbitales
p para dar tres orbitales . Este procedimiento es análogo a la mezcla de dos orbitales
atómicos para dar los dos orbitales moleculares que describen el enlace , excepto por
la presencia de un tercer orbital. De los tres orbitales moleculares, uno es enlazante y no
tiene ningún nodo (1), uno es no enlazante (tiene la misma energía que los orbitales p
sin interaccionar) y tiene un nodo (2) y uno es antienlazante, con dos nodos (3),
Figura 23.2. Una vez se han derivado los orbitales moleculares podemos llenarlos con el
número adecuado de electrones, Figura 23.3.
FIGURA 23.2.- Orbitales moleculares
del 2-propenilo (obsérvese que el
tamaño de los distintos lóbulos no el mismo).
FIGURA 23.3.- Llenado de los orbitales
moleculares del catión, radical y anión
2-propenilo (alilo).
El nodo que pasa por el carbono central en 2 tiene una consecuencia importante:
cualquier exceso (o defecto) de densidad electrónica se manifestará primordialmente en
los dos carbonos terminales, tal como esperaríamos a partir de las estructuras de
resonancia. Por tanto, en promedio, hay aproximadamente la mitad de una carga

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
293
positiva localizada sobre estos carbonos en el catión 2-propenilo (alilo) y la mitad de
una carga negativa en el anión. La posición central permanece neutra, con su octeto
completo. En el radical neutro cada carbono tiene un electrón y las posiciones
terminales muestran un 50% de carácter radicalario.
CONSECUENCIA DE LA DESLOCALIZACIÓN: QUÍMICA DEL SISTEMA 2-
PROPENILO (ALILO)
CATIÓN ALÍLICO:
Cuando un catión alílico reacciona con un agente nucleofílico, puede reaccionar
en cualquiera de los centros positivos y generalmente se produce una mezcla de
productos. Las reacciones que evolucionan vía cationes alílicos parecen dar con
frecuencia como resultado productos “reordenados”. Dichas reacciones reciben el
nombre de reordenamiento alílicos. Además de formar carbocationes relativamente fácil,
los haluros y alcoholes alílicos también experimentan la sustitución mediante el
mecanismo SN2 más rápidamente que los sistemas análogos saturados.
CONTROL CINÉTICO Y CONTROL TERMODINÁMICO:
Para la discusión del control cinético o termodinámico, analicemos la hidrólisis
del 1-cloro-2-buteno como la del 3-cloro-1-buteno, dado que dan la misma mezcla de
alcoholes. La razón es la presencia del mismo catión alilo intermedio.
Resulta curioso que el producto mayoritario de esta hidrólisis sea el 3-buten-2-
ol, a pesar de que se forme un alqueno terminal, y por ende menos favorecido
termodinámicamente. Debe existir un efecto cinético, es decir, el isómero menos estable
debe formarse más rápidamente, la pregunta es ¿Por qué?. La diferencia reside en la
distribución electrónica en el catión alílico intermedio. Esta molécula es asimétrica, por
lo que deberíamos esperar una distribución desigual de carga entre los carbonos C 1 y C3.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
294
Dicho de otra forma el producto mayoritario procede de un carbocatión secundario
alílico mucho más estable.
Que el producto de la hidrólisis está controlado cinéticamente puede demostrarse
por equilibración de los productos. Calentando la mezcla de butenoles se obtiene la
proporción termodinámica, con predominio del 2-buten-1-ol. En condiciones de baja
temperatura y tiempos cortos de reacción se dice que son de control cinético y las altas
temperaturas y tiempos largos de reacción se dice que son condiciones de control
termodinámico. La situación puede ilustrarse en un diagrama de energía potencial,
Figura 23.4. El alcohol cinético se forma primero pero su formación es reversible lo que
permite que al final se forme lentamente el producto termodinámico.
FIGURA 23.4.- Control cinético frente al control termodinámico para el catión alílico
RADICAL ALÍLICO:
Los radicales alílicos también están estabilizados por resonancia, de una forma
similar a como lo están los cationes alílicos, por lo que en moléculas asimétricas se verá
favorecida la formación de la forma resonante más estable de acuerdo a la estabilidad de
los radicales libres, es decir 3ario
> 2ario
>1ario
.
Una de las reacciones importantes de los radicales alílicos, es justamente la
bromación alílica, mediante el uso del reactivo N-bromosuccinimida. La reacción se
lleva normalmente a cabo en tetracloruro de carbono en el que tanto el reactivo N-
bromosuccinimida como la succinimida producto de la reacción son insolubles. La
reacción tiene lugar en parte sobre la superficie de la N-bromosuccinimida, aunque el
reactivo realmente activo parece ser el bromo formado en disolución diluida a partir de
la reacción de trazas de ácido y humedad con la bromoimida.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
295
El bromo participa luego en la bromación radicalaria en cadena del hidrógeno
alílico. Bajo estas condiciones de alta dilución no tiene lugar la adición del bromo al
doble enlace. Para el mecanismo véase el capítulo 9, páginas 175-177. Otra alternativa
de halogenación alílica es emplear halógeno en bajas concentraciones y temperaturas
elevadas, cuyo mecanismo se comenta en el capítulo 14, página 258.
Ejemplo:
DIENOS CONJUGADOS:
Los dienos conjugados son significativamente más estables de lo que cabría
esperar para un compuesto con los dobles enlaces completamente independientes. Esta
relativamente pequeña pero significativa diferencia se atribuye a dos factores. Primero,
las longitudes de los dobles enlaces son normales esencialmente, pero el enlace sencillo
que los separa es más corto que la distancia de 1.54 Å asociada a los enlaces sencillos
carbono-carbono, véase por ejemplo el 1,3-butadieno.
Esta disminución de la longitud de enlace en parte es consecuencia de la
disminución del carácter s de los orbitales de los carbonos que forman este enlace; el
enlace sencillo entre los dobles enlaces puede ser descrito como Csp2-Csp
2
aproximadamente. Este enlace más corto, es algo más fuerte que los enlaces carbono-
carbono que poseen menos carácter s. Segundo, los orbitales pz sobre C2 y C3 pueden
también solaparse para dar algo de carácter de doble enlace al enlace sencillo C2-C3 (en
el butadieno). Este factor contribuye también algo a la estabilidad extra del sistema de
dobles enlaces conjugados. Figura 23.4.
La prueba de la estabilidad extra de los dienos conjugados proviene de
mediciones de los calores de hidrogenación. Así, el 1,4-pentadieno tiene un Hºhidrog
= 60.8 kcal/mol, mientras que el 1,3-butadieno tiene un Hºhidrog = 57.1 kcal/mol. El
1,3-butadieno es aproximadamente 3,7 kcal/mol más estable de lo predicho
teóricamente.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
296
FIGURA 23.5.
La estructura electrónica del 1,3-butadieno puede describirse mediante cuatro
orbitales moleculares construidos a partir de los cuatro orbitales atómicos p. Los
orbitales moleculares de energía más baja,, no tiene ningún nodo y presenta
interacciones enlazantes entre los distintos lóbulos. El orbital 2 tiene sólo un nodo y
por tanto una interacción antienlazante. Este orbital todavía es enlazante ya que la
interacción antienlazante de los lóbulos centrales se compensa con las interacciones
enlazantes de los extremos. Tanto 1 como 2 tienen una energía menor que los
orbitales p aislados. El orbital 3 tiene dos nodos y, en conjunto es antienlazante,
mientras que 4 es completamente antienlazante, con tres nodos, Figura 23.6. Los cuatro
electrones ocupan los dos orbitales moleculares enlazantes y así se explica la
estabilidad neta del sistema respecto a los cuatros orbitales p independientes.
FIGURA 23.6.- Descripción mediante orbitales moleculares del 1,3-butadieno.
LOS DIENOS CONJUGADOS SON ATACADOS POR ELECTRÓFILOS Y
RADICALES LIBRES.
Los dienos conjugados son centros de elevada densidad electrónica debido a la
presencia de los electrones . De hecho, a pesar de ser termodinámicamente más
estables que los dienos con dobles enlaces independientes, los dienos conjugados son

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
297
más reactivos en presencia de electrófilos y otros reactivos. Así, frente a HX y X2 dan
los producto de adición 1,2- y 1,4-. La generación del primer producto puede
comprenderse fácilmente con la química de alquenos ordinaria (capítulo 14), es el
resultado de una adición Markovnikov a uno de los dobles enlaces. ¿Cómo se explica el
segundo producto?. Veamos dos ejemplos:
(1) Adición de bromo al 1,3-butadieno
¿Cómo es posible explicar la formación de los productos de adición 1,4?. La
respuesta es que en las reacciones participan carbocationes alílicos como intermedios.
(2) Adición de cloruro de hidrógeno al 1,3-butadieno

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
298
CONTROL CINÉTICO CONTRA CONTROL TERMODINÁMICO DE LAS
REACCIONES:
La adición de electrófilos a dienos conjugados a temperatura ambiente o
menores suele formar una mezcla de productos, en la cual el aducto 1,2 predomina
sobre el 1,4. Sin embargo, cuando la misma reacción ocurre a temperaturas mayores, a
menudo cambia la relación de productos y el aducto 1,4 es el que predomina. Por
ejemplo, la adición de HBr a 1,3-butadieno a 0 ºC produce una mezcla 71:29 de aductos
1,2 y 1,4, pero la misma reacción a 40 ºC genera una mezcla 15:85. Además, cuando la
mezcla de productos formada a 0 ºC se calienta a 40 ºC en presencia de más HBr, la
relación de aductos cambia lentamente de 71:21 a 15:85. ¿Cómo pueden explicarse
estas observaciones?
En condiciones suaves a baja temperatura (0 ºC), el HBr se agrega al 1,3-
butadieno bajo control cinético, para formar una mezcla 71:29 de productos con
predominación del aducto 1,2. Puesto que estas condiciones suaves no permite que los
productos alcancen el equilibrio, predomina el producto que se forma primero
(proveniente del carbocatión alílico secundario, formado en primer lugar). Sin embargo,
en condiciones más vigorosas a altas temperaturas (40 ºC), la reacción ocurre
reversiblemente bajo control termodinámico para formar una mezcla 15:85 de productos,
en la que predomina el aducto 1,4, más estable (alqueno disustituido). La mayor
temperatura proporciona más energía para que las moléculas de producto asciendan la
barrera de alta energía que conduce al carbocatión alílico primario, menos estable, y por
tanto resulta una mezcla de productos de equilibrio. Figura 23.7.
FIGURA 23.7.-
REACCIÓN DE CICLOADICIÓN DE DIELS-ALDER:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
299
Una segunda reacción importante de los dienos conjugados es que experimentan
una reacción de adición con alquenos aislados para formar ciclohexenos sustituidos
como productos. Este proceso, llamado reacción de cicloadición de Diels-Alder en
honor a sus descubridores[1],[2]
, es una reacción muy útil en química orgánica debido a
que forma dos enlaces carbono-carbono en un solo paso, y es uno de los pocos métodos
de que se dispone para la formación de moléculas cíclicas.
Ejemplos:
1,3-Butadieno + Eteno Ciclohexeno (20%) 1,3-Butadieno + 3-Buten-2-ona (96%)
El mecanismo de la cicloadición de Diels-Alder difiere del de las otras
reacciones que se han comentado en este capítulo. No es una reacción polar ni por
radicales, sino un proceso pericíclico. Las reacciones pericíclicas no serán estudiadas en
este libro por lo que si el estudiante tiene interés en saber más sobre ellas puede
remitirse a la bibliografía citada al principio del libro de apuntes. La reacción en sí
ocurre en un solo paso, sin intermedios, e implica una redistribución cíclica de los
electrones de enlace. Los dos reactivos simplemente se unen a través de un estado de
transición cíclico en el cual los dos nuevos enlaces carbono-carbono se forman al
mismo tiempo.
La naturaleza concertada de esta reacción puede apreciarse en el estado de
transición deslocalizado en el que los seis electrones se indican por un circulo
punteado o utilizando flechas.
Dieno Dienófilo Producto de la cicloadición [4+2]
En la reacción de Diels-Alder se retiene la estereoquímica del dienófilo:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
300
Ejemplos:
Se obtiene el cis-2-Butendiato de dimetilo,
dado que el reactivo es cis.
Se obtiene el trans-2-Butendiato de dimetilo,
dado que el reactivo es trans.
En la reacción de Diels-Alder se mantiene la estereoquímica del dieno:
Ejemplos:
trans,trans-2,4-Hexadieno Se obtiene producto cis
cis,trans-2,4-Hexadieno Se obtiene producto trans
Otra característica estereoquímica importante de la reacción de Diels-Alder es
que los compuestos dieno y dienófilo se disponen de manera que se forma el endo-
producto en vez que el exo-producto alternativo. Los prefijos endo- y exo- se utilizan
para indicar la estereoquímica relativa al hacer referencia a estructuras bicíclicas, como
norbonanos sustituidos. Un sustituyente en un puente se considera exo si es sin (cis)
respecto al menor de los otros dos puentes, y se considera endo si es anti (trans). Por
analogía, se dice que una reacción de Diels-Alder tiene estereoquímica endo si el
sustituyente que atrae electrones en el dienófilo es sin respecto al puente insaturado de
dos carbonos en el producto.
Cicloadiciones exo:
El producto exo no se forma El producto exo no se forma

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
301
Cicloadiciones endo:
El producto endo se forma en un 100%
La reacción de Diels-Alder normalmente transcurre con selectividad endo; es
decir, sólo se forma el producto endo. Este resultado se conoce como la regla endo.
Cuando los dienófilos son alquinos se obtienen 1,4-ciclohexadienos:
Los alquinos también pueden actuar como dienófilos en cicloadiciones [4+2].
Pueden reaccionar tanto uno solo como los dos enlaces del alquino. La adición simple
lleva a derivados del 1,4-ciclohexadienos.
Ejemplos:
En la tabla 23.1. se presentan algunos de los dienos y dienófilos en la reacción
de Diels-Alder, si uno tiene en cuenta las posibles combinaciones, los productos pueden
resultar bastante interesantes.
TABLA 23.1.- Algunos dienos y dienófilos que participan en reacciones
Diels-Alder
DIENOS NOMBRE DIENÓFILOS NOMBRE
1,3-butadieno
Tetracianoeteno
2,3-Dimetil-1,3-Butadieno
cis-1,2-Dicianoeteno
trans-trans-2,4-Hexadieno
cis-2-Butendiato de dimetilo

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
302
1,3-Ciclopentadieno
trans-2-Butendiato de dimetilo
1,3-ciclohexadieno
Anhídrido 2-Butendioico
5-metilen-1,3-
ciclopentadieno
Butindiato de dimetilo
1,2-Dimetilenciclohexano
Propenal
[1]
Otto Diels (1876-1954); n. Hamburgo; Profesor de la Universidad de Berlín y Kiel; Premio Nobel [2]
Kurt Alder (1902-1958); n. Königshütte; Profesor de la Universidad de Colonia; Premio Nobel en 1950
conjuntamente con Otto Diels, otorgado en reconocimiento de su hallazgo.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
303
SUSTITUCIÓN ELECTROFÍLICA
AROMÁTICA
La sustitución electrofílica aromática es la reacción más importante de los compuestos aromáticos. Es posible introducir al anillo muchos sustituyentes distintos por este proceso. Si se elige el reactivo apropiado pueden efectuarse reacciones de bromación, cloración, nitración, sulfonación, alquilación y acilación, estas seis son reacciones directas, y a partir de ellas se pueden introducir otros grupos.
En este capítulo estudiaremos estas seis reacciones, más las que se puedan obtener a partir de ellas, como así mismo la introducción de un segundo sustituyente.
La reacción más importante de los compuestos aromáticos es la sustitución electrofílica aromática. Es decir la introducción de un electrófilo (E +).
Seleccionando las condiciones y los reactivos apropiados, el anillo aromático se puede halogenar, nitrar, sulfonar, acilar y alquilar. Todas estas reacciones y muchas otras proceden a través de un mecanismo similar.
HALOGENACIÓN DEL BENCENO

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
304
El benceno es normalmente inerte en presencia de halógenos, debido a que los
halógenos no son lo suficientemente electrófilos para destruir su aromaticidad. Sin
embargo, los halógenos pueden activarse mediante ácidos de Lewis como los haluros de
hierro, FeX3 o de aluminio, AlX3, para dar electrófilos más potentes.
MECANISMO DE LA BROMACIÓN:
1) Activación del bromo por un ácido de Lewis (FeBr 3)
Br Br FeBr3 Br Br FeBr3 Br Br FeBr3
2) Ataque electrófilo sobre el benceno por bromo activado
H+ Br Br FeBr3
H
Br + FeBr4
3) El FeBr4
- formado en la etapa anterior actúa ahora como base abstrayendo el protón
del catión hexadienilo.
H
Br + FeBr4Br
+ HBr + FeBr3
En resumen, la halogenación del benceno se hace más exotérmica al pasar del I2
(endotérmica) a F2 (explosiva). Las cloraciones y bromaciones se consiguen utilizando
ácidos de Lewis como catalizadores que polarizan el enlace X-X y activan el halógeno
aumentando su poder electrófilo.
La dificultad termodinámica de la yodación puede evitarse añadiendo una sal de
plata a la mezcla de nitración, que activa y elimina el producto el producto (yoduro) de
la reacción por precipitación.
H
+ I2
I
+ AgI + HNO3AgNO3
Para la fluoración del benceno puede emplearse la reacción de Schiemann.
N2+BF4
-
F
+ N2 + BF3
La sal de diazonio para la reacción de Schiemann a su vez se prepara a partir de
anilina:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
305
NH2 NaNO2
HCl
N2+Cl-
HBF4N2
+BF4-
NITRACIÓN DEL BENCENO:
Los anillos aromáticos se pueden nitrar por reacción con una mezcla de ácido
nítrico y ácido sulfúrico concentrado. Se piensa que en ésta reacción el electrófilo es el
ion nitronio, NO2+
, que se genera a partir del ácido nítrico por protonación y pérdida de
agua.
MECANISMO DE LA NITRACIÓN:
1) Activación del ácido nítrico por el ácido sulfúrico (formación del ion nitronio)
+ H OSO3HHO NOO
H2O NOO
OSO3H+
H2O(-)
NO2
ion nitronio 2) Ataque electrófilico sobre el ion nitronio
NO2H
+
HNO2
3) Abstracción del protón por parte de la base conjugada del ácido sulfúrico.
HNO2 OSO3H+
NO2
+ H2SO4
La nitración de anillos aromáticos es una reacción de particular importancia,
debido a que los nitroarenos que se producen pueden reducirse con reactivos como
hierro o cloruro estannoso para formar aninoarenos (anilina).
SULFONACIÓN DEL BENCENO:
El ácido sulfúrico concentrado no reacciona con el benceno a temperatura
ambiente excepto por protonación. Sin embargo, una forma más reactiva, llamada
“ácido sulfúrico fumante” da lugar a un ataque electrófilico por SO3. El ácido sulfúrico
fumante comercial se prepara por adición de aproximadamente un 8% de trióxido de
azufre (SO3), a ácido sulfúrico concentrado. El electrófilo reactivo es HSO3+
o SO3
neutro, dependiendo de las condiciones de reacción.
MECANISMO DE LA SULFONACIÓN:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
306
H+
OSO
O H
SO3
SO3H
ácido bencenosulfónico (95%)
La sulfonación es reversible, tiene utilidad sintética, debido a que el sustituyente
ácido sulfónico puede utilizarse como grupo protector para dirigir la sustitución.
SO3H
H2O, H2SO4
100 ºC
ALQUILACIÓN DEL BENCENO: REACCIÓN DE FRIEDEL-CRAFTS
En 1877, Charles Friedel1[1]
y James Crafts2[2]
descubrieron que los haloalcanos
reaccionan con benceno en presencia de cloruro de aluminio, para dar el producto de
alquilación. La reacción de Friedel-Crafts es una sustitución electrofílica aromática en la
cual el anillo aromático ataca a un carbocatión electófilico. El carbocatión se genera
cuando el catalizador AlCl3 ayuda al halogenuro de alquilo a ionizarse, de forma muy
similar a como el FeBr3 cataliza las bromaciones aromáticas polarizando al bromo.
MECANISMO DE LA ALQUILACIÓN:
1) Activación del haloalcano
+ AlCl3R CH2 X R CH2 X AlCl3
2) Ataque electrófilo
HR CH2 X AlCl3
+
H
CH2 R+ AlCl3X
3) Pérdida del protón
H
CH2 R+ AlCl3X
CH2R+ HX + AlCl3
Las alquilaciones de Friedel-Crafts intramoleculares pueden utilizarse para la
construcción de un nuevo anillo fusionado con el núcleo bencénico.
ClAlCl3, CS2
25 ºC, 72 h
Las alquilaciones de Friedel-Crafts pueden llevarse a cabo con cualquier
producto de partida, como un alcohol o un alqueno que pueda generar carbocationes:
+ HF0ºC
1[1] Charles Friedel (1832-1899); n. Estrasburgo; Francia. Estudío en la Universidad de Sorbona
2[2] James M. Crafts (1839-1917); n. Boston; USA, Estudío en Harvard (1898).

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
307
Si bien las alquilaciones de Friedel-Crafts son útiles en términos generales para
la síntesis de alquilbencenos, están sujetas a ciertas limitaciones. Una de ellas es que
sólo se pueden usar halogenuros de alquilo, la reacción no funciona con halogenuros de
arilo y halogenuros vinílicos. Una segunda limitación es que las reacciones de Friedel-
crafts no proceden con anillos aromáticos que estén sustituidos con grupos fuertemente
desactivadores. Una tercera limitación es que estas reacciones son difícil de detener en
el producto monosustituido, suelen dar el producto disustituido en para como producto
principal.
ACILACIÓN DE FRIEDEL-CRAFTS
La reacción consiste en la introducción del grupo acilo -COR, al anillo
aromático. El mecanismo de la acilación de Friedel-Crafts es similar al de la alquilación.
El electrófilo reactivo es un catión de acilo estabilizado por resonancia, el cual se genera
por reacción entre el cloruro de acilo y el AlCl3. A diferencia de las alquilaciones, las
acilaciones nunca proceden más de una vez en un anillo , debido a que el acilbenceno
producido es siempre menos reactivo que el material de partida no acilado.
MECANISMO DE LA ACILACIÓN:
R C ClO
+ AlCl3 R C ClO
AlCl3 R C O R C O
H+ R C O
H
CO
R AlCl4C
R
O
+ HCl + AlCl3
+ AlCl4Catión acílico
Las reacciones anteriormente comentadas se pueden resumir dela siguiente forma:
H XXR R
ELECTRÓFILO (X
) REACTIVO REACCIÓN
R+
RBr + AlCl 3
ROH + H+
Alqueno + H+
Alquilación Friedel-Crafts
RCO+
RCOCl + AlCl3 Acilación Friedel-Crafts
NO2+
HNO3 + H2SO4 Nitración
Cl+
Cl2 + FeCl3 Cloración
Br+
Br2 + Fe Bromación
HOSO2+
H2SO4 Sulfonación
ClSO2+
ClSO2OH Clorosulfonación

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
308
Muchos compuestos aromáticos se preparan por desplazamiento nucleofílico a partir de
sales de diazonio. A su vez las sales de diazonio se pueden preparar de la siguiente
manera:
NO2 NH2 N2
H2, Pd/Co biénSn, HCl
HONOo biénNaNO2/HCl
Cl
Sal de Diazonio
ArNH2 ArN2 ArZHONO Z
Z REACTIVO
HO H2O
RO ROH
CN CuCN
Cl CuCl
Br CuBr
I KI
Ar ArH
H H3PO2 o EtOH/H+
F HBF4/
Para la preparación de otros derivados monosustituidos, distintos a los ya
mencionados se procede a partir de alguno de ellos, empleando las reacciones químicas
clásicas, como oxidación, reducción, etc. Las cuales se resumen a continuación en una
especie de tabla.
YR
XR
Y X REACTIVO
REDUCCIÓN:
-NO2
-NH2
H2, Pd/C (o)
Sn, HCl, conc.
-COR -CH(OH)R NaBH4
-COR -CH2R Zn/Hg, HCl, conc.
H2, Pd, Etanol
OXIDACIÓN:
-CH2Cl
-CHO
hexamina
-CH2R -CO2H KMnO4
-CH3
-COR -OCOR R’CO3H
SUSTITUCIÓN:
-CH3
-CH3
-CCl3
-CH2Br
Cl2, PCl5
NBS, CCl4
-CCl3 -CF3 SbF3

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
309
-CN -CO2H HO-, H2O
-Br -NH2 NH2- Na
+, NH3
Otras reacciones importante del anillo benceno son:
REDUCCIÓN DE BIRCH:
Li, NH3 (o Na)
Etanol
HIDROGENACIÓN CATALÍTICA DE ANILLOS AROMÁTICOS:
H2 / Rh / C
Etanol
SUSTITUCIÓN AROMÁTICA A TRAVÉS DE INTERMEDIOS BENCINO:
Cl
NaNH2, NH3 Líq.- NaCl
NH2NH3
SÍNTESIS DE BENCENOS DISUSTITUIDOS:
Una de las formas más seguras de adquirir dominio de la química orgánica es
resolver problemas de síntesis. La habilidad para planear una síntesis satisfactoria en
varios pasos de una molécula compleja requiere un conocimiento práctico de los usos y
las limitaciones de varios cientos de reacciones orgánicas. Es necesario saber no sólo
que reacciones emplear, sino también cuándo utilizarlas. El orden en que se realizan las
reacciones con frecuencia es crítico para el éxito del método total.
Ejemplo: Sintetizar el ácido p-bromobenzoico a partir de benceno.
Es necesario preguntarse “¿Cuál es un precursor inmediato del ácido p-
bromobenzoico?”
? Br COOH
El análisis sintético hacia atrás (retrosintético) revela dos rutas válidas que van
del benceno al ácido p-bromobenzoico.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
310
CH3
Br
CO2H
Br
Br
CH3
KMnO4
Br2FeBr3
CH3Cl
AlCl3
Br2FeBr3
CH3ClAlCl3
Benceno
Un segundo ejemplo de interés es la síntesis del 4-cloro-1-nitro-2-propilbenceno
a partir de benceno, en principio hay tres posibles precursores disustiutidos, pero sólo
uno de ellos es el adecuado.
Cl
NO2
Cl
NO2
Cl
NO2
HNO3H2SO4
Este anillo se encuentra desactivado y no experimenta la alquilaciónde Friedel-Crafts.
Esta molécula no forma elisómero deseado por lareacción de cloración.
p-Cloronitrobenceno m-Cloropropilbenceno o-Nitropropilbenceno
La síntesis final es una ruta de cuatro pasos a partir del benceno:
OCl
O
ClCl
NO2
CH3CH2CClO
AlCl3
Cl2FeCl3
H2, Pd/CEtanol
HNO3
H2SO4
A continuación se realizara un pequeño estudio, para ver como se produce la
entrada de un segundo grupo (E), dada que esta queda sujeta a presencia del grupo ya
presente en anillo bencénico (G). Los sustituyentes en el anillo de benceno afectan tanto
la reactividad del anillo hacia posteriores sustituciones como la orientación de estas
últimas. Los grupos pueden clasificarse en tres categorías: activadores orientadores
orto-para, desactivadores orientadores orto-para, y desactivadores orientadores meta.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
311
GHE
G
HE
G
H E
GHE
HE
G
G
H E
GHE
HE
G
G
H E
G
G
G
+ E
+ E
+ E
G
E
GE
G
E
orto
meta
para La entrada y dirección del electrófilo (E) en la sustitución electrofilica aromática
en bencenos monosustituidos (G) queda gobernada por la naturaleza química del grupo
G, los cuales a su vez se clasifican en activantes fuertes (orientadores orto-para),
activantes moderados (orientadores orto-para), activantes débiles (orientadores orto-
para), desactivantes débiles (orientadores orto-para), desactivantes fuertes
(orientadores meta), a continuación se muestran algunos de ellos en orden decreciente,
de acuerdo a su poder activante o desactivante.
DIRECCIÓN GRUPO ACTIVACIÓN
orto-para -NH2 , -NHR , -NR2
-OH , OR
Activantes fuertes
orto-para -NHCOR
-OCOR
Alquenos
Activantes moderados
orto-para -R (alquilo)
-fenilo
Activantes débiles
orto-para -F , -Cl , -Br , -I Desactivante débiles
Meta -CX3 (X = F, Cl, etc.)
-COOH, -COOR, -COR , -
COH
-SO3H; -NO2; -NR+
3 , -CN
Desactivantes fuertes
La bromación electrófilica, del metilbenceno (tolueno) da sustitución orto y para.
CH3
Br
CH3
Br2 / FeBr3CCl4
CH3
BrCH3
Br
+ +
39% < 1% 60% La bromación no es un caso particular; la nitración y la sulfonación dan los
mismos resultados cualitativos, sustitución principalmente en las posiciones orto y
para , en la molécula del benceno. Explicación mecanistica para el caso del bromo:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
312
CH3
Br
CH3
Br
CH3
Br
orto
meta
para
CH3HBr
CH3
HBr
CH3
H Br
CH3
HBr
HBr
CH3
CH3
H Br
CH3HBr
HBr
CH3
CH3
H Br
CH3
CH3
CH3
+ Br2FeBr3
+ Br2FeBr3
+ Br2FeBr3
Carbocatión 3º, muyestable
Carbocatión 3º, muyestable
Sólo el ataque en las posiciones orto y para da lugar a un catión hexadienilo en
el que una estructura resonante tenga la carga positiva adyacente al sustituyente alquilo,
dejando un carbocatión terciario, en cambio en meta ninguna de las formas resonantes
deja un carbocatión terciario, por tal motivo predominan los productos orto y para dado
que el intermedio es más estable, a su vez el mayoritario es el para por efectos estéricos.
Los sustituyentes desactivantes por efecto inductivo orientan en meta, como por
ejemplo la nitración del trifluorometilbenceno, en donde el único producto es el meta.
CF3
+HNO3
H2SO4
CF3
NO2
Unico producto
explicación mecanistica:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
313
CH3
NO2
CH3
NO2
CH3
NO2
orto
meta
para
CF3HNO2
CF3
HNO2
CF3
H NO2
CF3
HNO2
HNO2
CF3
CF3
H NO2
CF3HNO2
HNO2
CF3
CF3
H NO2
CF3
CF3
CF3
Carbocatión 3º, muyinestable
Carbocatión 3º, muyinestable
HNO3
H2SO4
HNO3
H2SO4
HNO3
H2SO4
Ahora el ataque en orto y para están más desfavorecidos, dado que hay
intermedios inestables, el ataque se produce en meta, de esta manera se evita la
formación de intermedios inestables.
Los halógenos retiran densidad electrónica por efecto inductivo mientras que son
donadores por resonancia, globalmente el efecto inductivo prevalece y los haloarenos
están desactivados, sin embargo la sustitución electrófila tiene lugar principalmente en
las posiciones orto y para.
REGLAS PARA PREDECIR LA ORIENTACIÓN EN LOS BENCENOS
DISUSTITUIDOS
1.- Si todos los sustituyentes se refuerzan entre si, la entrada del tercer grupo no
genera ningún tipo de problemas.
CH3
CH3 CH3
NO2
Ejemplos:
CH3
CH3
Br2FeBr3 CH3
CH3
Br
CH3
NO2
CH3COCl
AlCl3
CH3
NO2
CH3
O

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
314
2.- Si un orientador orto-para y uno meta no se están reforzando, el orientador orto-
para controla la orientación del tercer grupo. (El grupo entrante se dirige principal mente
orto hacia el orientador meta.)
Cl
NO2
CH3
NO2
Principal Principal
Ejemplos:
Cl
NO2
Cl2 / FeCl3
Cl
NO2
Cl
CH3
NO2
SO3/H2SO4
CH3
NO2
SO3H
3.- Un grupo fuertemente activador, que compite con un grupo débilmente
activador,controla la orientación.
OHCH3
(o,p-fuerte)OCH3
CH3
(o,p-moderado)
(o,p-débil)
(o,p-débil)
Principal Ejemplos:
OCH3
CH3
HNO3 / H2SO4
OCH3
CH3
NO2
OHCH3 Br2
FeBr3
OHCH3
Br
4.- Cuando compiten dos grupos débilmente activadores o desactivadores o dos
grupos fuertemente activadores o desactivadores, se obtienen cantidades considerables
de ambos isómeros, hay muy poca preferencia.
Cl
CH3
(o,p-débil)
(o,p-débil)
Cl
CH3
(o,p-débil)
(o,p-débil)
+
Ejemplo:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
315
+
Cl
CH3
Br2FeBr3
Cl
CH3
BrCl
CH3
Br
5.- En la posición de impedimento estérico, entre los sustituyentes meta hay muy
poca sustitución.
CH3
CH3
(Poca sustitución)
Otras reacciones de interés:
Ar CH3
Cl2, Calor
CrO3,(CH3CO)2O
Ar CHCl2
Ar CH(OOCCH3)2Ar CHO
Ar XMgéter
Ar Mg X CO2 Ar COOMgX Ar COOHH
Ar CN H3O Ar COOH
Aldehído
Ácido carboxilico
H2O / H+
¿CUIDADO CON LOS FENOLES? Son activantes fuertes, si se emplean directamente
provocan la polisustitución, si se desea obtener un compuesto disustituido a partir de
ellos se debe proteger el grupo alcohol como acetato, de esa manera conserva su poder
orientador pero disminuye su poder activador.
OHBr
Br
BrBr2FeBr3
OH
Para obtener el para-bromobenceno a partir de fenol se debe proceder de la siguiente
manera:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
316
OH
Ac2O / Py
OAc
Br2FeBr3
OAc
Br
OAcBr
+
NaOH / H2O
OH
Br
OHBr
+
Mayoritario
El mismo cuidado hay que tener con el Anilina, dado que el grupo amino es un
activante fuerte, problema que puede evitarse protegiéndolo como amida.
NH2
Br
Br
BrBr2FeBr3
NH2
NH2
Ac2O / Py
NHCOCH3
Br2FeBr3
NHCOCH3
Br
NHCOCH3
Br+
NaOH / H2O
NH2
Br
NH2
Br+
Mayoritario

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
317
QUÍMICA DE LOS BENCENOS SUSTITUIDOS
(FENOLES y ARILAMINAS)
RESUMEN DE LAS NUEVAS REACCIONES
1. HALOGENACIÓN RADICALARIA
EJEMPLOS: Bromación del tolueno:
Un exceso de halógeno puede conducir a sustituciones múltiples,
tal como se ilustra para la cloración del tolueno:
2. SOLVOLISIS
EJEMPLO: Etanólisis del 4-metilbencenosulfonato de fenilmetilo, preparado por la tosilación del fenol.
4-metilbencenosulfonato de fenilmetilo Bencil, etil, éter
3. REACCIONES DE SN2 DE LOS HALOMETILBENCENOS

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
318
EJEMPLO: Preparación del feniletanonitrilo a partir del bromometilbenceno
4. OZONOLISIS DEL BENCENO
EJEMPLO: Ozonolisis del orto-dimetilbenceno
5. REDUCCIÓN DE BIRCH
Regioselectividad observada para la reducción de Birch:
EJEMPLOS:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
319
6. OXIDACIÓN EN LA CADENA LATERAL
OBTENCIÓN DE ÁCIDOS CARBOXÍLICOS
OTROS POSIBLES OXIDANTES:
OBTENCIÓN DE CETONAS

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
320
EJEMPLOS:
(80%)
FENOLES
A) REACCIONES DE OBTENCIÓN DE FENOLES
7. PREPARACIÓN POR SUSTITUCIÓN AROMÁTICA NUCLEOFILA
Hidroxilación aromática nucleófila directa
Sustitución nucleófila en halobencenos
8. HIDRÓLISIS DE SALES DE ARENODIAZONIO
9. RUPTURA DE ÉTERES
EJEMPLOS:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
321
B) REACCIONES DE FENOLES Y ALCOXIBENCENOS
10. SÍNTESIS DE ÉTERES DE WILLIAMSON
EJEMPLOS:
11. FORMACIÓN DE ÉSTERES (ESTERIFICACIÓN)
EJEMPLOS:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
322
12. REACCIÓN DE KOLBE (CARBOXILACIÓN DE KOLBE[1]
-SCHITT[2]
)
EJEMPLO:
13. TRANSPOSICIÓN DE CLAISEN
EJEMPLO:
14. OXIDACIÓN A QUINONAS
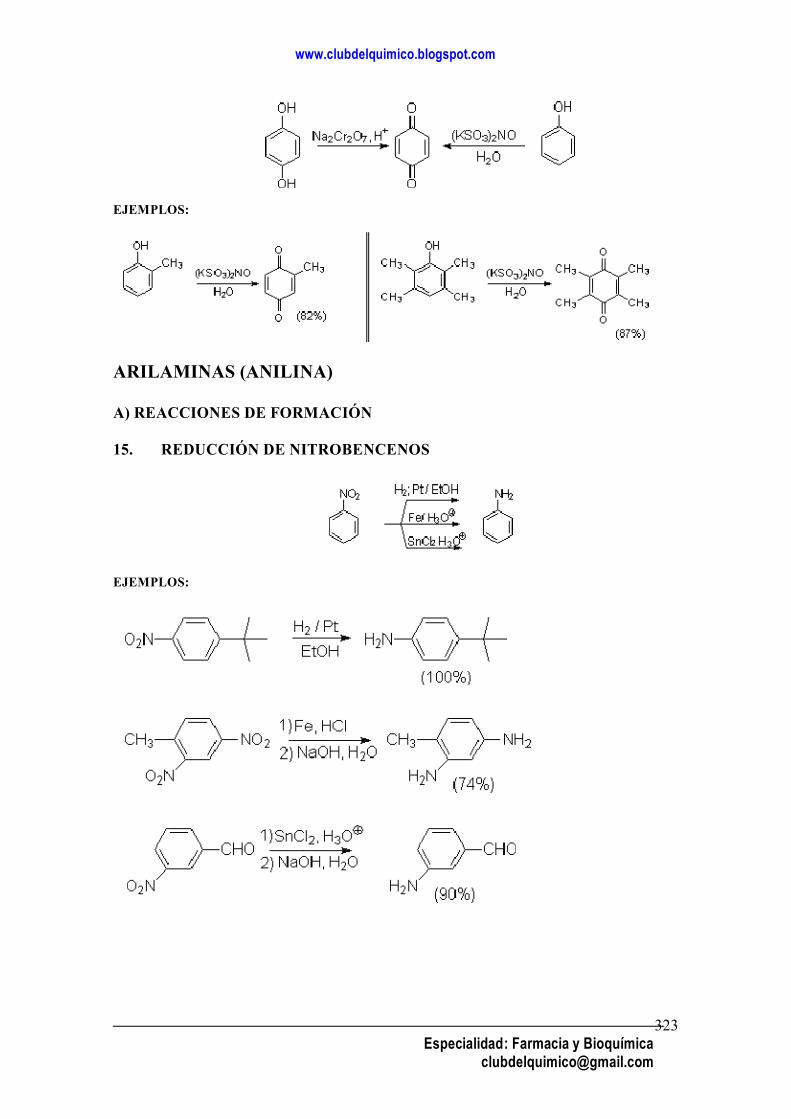
www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
323
EJEMPLOS:
ARILAMINAS (ANILINA)
A) REACCIONES DE FORMACIÓN
15. REDUCCIÓN DE NITROBENCENOS
EJEMPLOS:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
324
B) REACCIONES DE ARILAMINAS
17. FORMACIÓN DE SALES DE ARENODIAZONIO
18. REACCIONES TIPO SANDMEYER
19. ACOPLAMIENTO DE SALES DE DIAZONIO
20. OXIDACIÓN A QUINONAS
EJEMPLOS:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
325
[1]
Herman Kolbe (1818-1884); n. Alemania; Ph.D. Gotinga; Profesor en las Universidades de Marburgo y
Leipzig. [2]
Rudolf Schmitt (1830-1898); n. Wippershain; Alemania; Ph.D. Marburgo; Profesor en la Universidad
de Dresde

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
326
HIDROCARBUROS AROMÁTICOS
POLICÍCLICOS
COMPUESTOS AROMÁTICOS POLICÍCLICOS
Se dice que dos anillos aromáticos que comparten un par de átomos de carbono
que están fusionados. En este capítulo estudiaremos la química del hidrocarburo de
anillos fusionados más sencillo e importante, el naftaleno, C10H8, y trataremos
concisamente la de otros de grado superior.
NAFTALENO
Al contrario de lo que ocurre con el benceno, que es líquido, el naftaleno es un
material cristalino e incoloro con un punto de fusión de 80 ºC. Se le conoce como
repelente e insecticida contra la polilla. El naftaleno se clasifica como aromático porque
sus propiedades se parecen a las del benceno. Su fórmula C10H8, permite suponer un
alto grado de no saturación. Desde el punto de vista químico da las típicas reacciones de
sustitución electrofílica aromáticas, en las que desplaza hidrógeno en forma de ion,
conservándose su sistema anular.
De acuerdo con la teoría de la resonancia, el naftaleno puede considerarse como
un híbrido de resonancia de las tres estructuras I, II y III; su energía de resonancia es de
61 kcal/mol, como lo indica el calor de combustión.
I II III

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
327
De acuerdo con estas representaciones la estructura del naftaleno debería ser
simétrica, con los anillos de benceno planos, casi hexagonales, y con dos planos
especulares perpendiculares biseccionando la molécula. Las medidas cristalográficas de
rayos X confirman esta predicción. Las distancias de los enlaces C-C se desvían sólo
muy ligeramente de las del benceno (1.39 Å) y son claramente muy diferentes de las de
los enlaces sencillos (1.54 Å) y los dobles (1.33 Å). Por conveniencia, representaremos
al naftaleno por medio de la estructura IV, en las que los círculos representan sextetos
aromáticos parcialmente traslapados.
IV
Representación orbitálica del solapamiento del Naftaleno
PREPARACIÓN DEL NAFTALENO:
El método en si consiste en una alcanoilación de Friedel-Crafts con anhídrido
butanodioico (succinico), reducción de Clemmensen de la cetona resultante,
subsiguiente alcanoilación intramolecular de Friedel-Crafts para formar el segundo
anillo, reducción de la cetona bicíclica con borohidruro sódico, deshidratación y
deshidrogenación.
REACCIONES DEL NAFTALENO:
1.- OXIDACIÓN


www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
329
2.- REDUCCIÓN
3.- SUSTITUCIÓN AROMÁTICA ELECTRÓFILICA DEL NAFTALENO
(A) NITRACIÓN
(B) HALOGENACIÓN
(C) SULFONACIÓN

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
330
(D) ACILACIÓN DE FRIEDEL-CRAFTS
¿Por qué la nitración y la bromación se produce preferentemente en el C-1 y
no en el C-2?. La respuesta se puede encontrar inspeccionando las estructuras de
resonancia.
REACTIVADA ELECTRÓFILA DEL NAFTALENO: ATAQUE EN C-1
REACTIVADA ELECTRÓFILA DEL NAFTALENO: ATAQUE EN C-2
Aunque este resultado podría a primera vista indicar que el ataque es
energéticamente similar en cualquier posición, existe una diferencia importante entre los
dos modos: el ataque en C-1 permite dos estructuras en resonancia que mantienen
intacto el un anillo benceno con todos los beneficios de la deslocalización. En cambio
el ataque en C-2 permite únicamente una de tales estructura. Esta es la razón
fundamental de que predomina la sustitución en C-1 en la nitración, bromación y en
la alcanoilación de Friedel-Crafts.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
331
ATAQUE ELECTRÓFILO SOBRE LOS NAFTALENOS MONOSUSTITUIDOS:
Las reglas de orientación en el ataque en bencenos monosustituidos (Capítulo 24)
pueden hacerse fácilmente extensivas al núcleo de naftaleno. El anillo sustituido es el
más afectado por los sustituyentes ya presentes: un grupo activante normalmente dirige
al electrófilo entrante al mismo anillo, mientras que un grupo desactivante lo dirige al
otro.
HIDROCARBUROS BENCÉNICOS TRICÍCLICOS: ANTRACENO Y
FENANTRENO
Debido a sus propiedades, también el antraceno y el fenantreno se clasifican
como aromáticos. La descripción de los orbitales atómicos sigue el mismo esquema
para el naftaleno y conduce al mismo tipo de resultados: estructuras planas con nubes p
que se traslapan parcialmente por encima y por debajo del plano de la molécula.
Desde el punto de vista de enlaces de valencia, se considera que el antraceno es
un híbrido de las estructuras I - IV.
I II III IV
y el fenantreno, un híbrido de las estructuras V - IX. Los calores de combustión indican
que el antraceno tiene una energía de resonancia de 84 kcal/mol, mientras que el
fenantreno tiene una energía de resonancia de 92 kcal/mol.
Por comodidad el antraceno y el fenantreno se puede representar por círculos,
los que representan sextetos aromáticos que se traslapan parcialmente.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
332
PREPARACIÓN DEL ANTRACENO Y FENANTRENO
ANTRACENO:
FENANTRENO:
El método en es muy similar a la obtención del Naftaleno, consisten en una
reacción de Friedel-Crafts del naftaleno con anhídrido butanodioico (succinico) en
nitrobenceno que conduce a la sustitución en C-1 y C-2, los dos productos así obtenidos
se reducen en las condiciones de Clemmensen para a continuación ciclarse y dar el
esqueleto del fenantreno, por reducción, eliminación y deshidrogenación.
REACTIVADA DEL ANTRACENO Y FENANTRENO
REDUCCIÓN:
HALOGENACIÓN:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
333
CICLOADICIONES:
LOS HIDROCARBUROS AROMÁTICOS POLICÍCLICOS Y EL CÁNCER:
Muchos hidrocarburos bencénicos policíclicos son carcinogénicos. La primera
observación de un cáncer por tales compuestos la realizó en 1775 Sir Percival Scott,
cirujano del hospital St. Bartholomew de Londres. Observó que los deshollinadores de
chimeneas eran propensos a cánceres escrotales. Desde entonces, se ha dedicado un
intenso esfuerzo a investigar qué hidrocarburos bencénicos policíclicos tienen
propiedades fisiológicas y cómo su estructura se correlaciona con la actividad. Una
molécula muy bien estudiada es la Benzo[a]pireno, un contaminante ambiental muy
conocido, se produce normalmente en la combustión de materia orgánica, tal como en
los motores de los automóviles y en las calefacciones, en las incineradoras de basuras,
en los incendios forestales, en el humo de los cigarrillos e incluso en la carne asada. La
liberación anual en la atmósfera, sólo en los Estados Unidos, es de 1300 toneladas.


www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
335
SÍNTESIS Y REACCIONES DE LAS AMINAS
PROPIEDADES FÍSICAS
Las aminas son compuestos altamente polares. Las aminas primarias y
secundarias pueden formar puentes de hidrógeno intramoleculares en el estado líquido y
por tanto tienen puntos de ebullición mayores que los alcanos de peso molecular
equivalente. Una característica de las aminas de bajo peso molecular es su olor a
pescado, que en cierta medida es distintivo.
METILAMINA, FORMANDO PUENTES DE
HIDRÓGENO EN EL ESTADO LÍQUIDO
BASICIDAD DE LAS AMINAS:
La basicidad y la nucleofilicidad de las aminas está dominada por el par de
electrones no compartido del nitrógeno, debido a este par, las aminas son compuestos
que se comportan como bases y nucleófilos, reaccionan con ácido para formar sales
ácido/base, y reaccionan con electrófilos en muchas de las reacciones polares que se han
estudiado anteriormente (Ej. Sustituciones Nucleófilicas).
Amina Un ácido Una sal (ion amonio)
BASICIDAD DE ALGUNAS ALQUILAMINAS
NOMBRE ESTRUCTURA pK DEL ION
AMONIO
- AMONIACO :NH3 10.64
AMINAS PRIMARIAS
- METILAMINA
- ETILAMINA
CH3-NH2
CH3CH2-NH2
16.64
10.75
AMINAS SECUNDARIAS
- DIMETILAMINA
- DIETILAMINA
- PIRROLIDINA
(CH3)2NH
(CH3CH2)2NH
10.73
10.94
11.27

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
336
AMINAS TERCIARIAS
- TRIMETILAMINAS
- TRIETILAMINA
(CH3)3N:
(CH3CH2)3N:
9.79
10.75
Sales de aminas: (sales de amonio)
La basicidad de las aminas permite su protonación o alquilación, lo que da lugar
a las sales de amonio. Dependiendo del número de sustituyentes que tenga el nitrógeno,
las sales pueden ser primarias, secundarias o cuaternarias.
La diferencia en el comportamiento de las aminas y sus sales, en cuanto a
solubilidad, puede utilizarse tanto para detectar aminas, como para separarlas de
sustancias no básicas. Un compuesto orgánico insoluble en agua que se disuelve en
ácido clorhídrico acuoso, diluido, debe ser apreciablemente básico, lo que significa que,
de seguro, se trata de una amina. Esta puede separarse de compuestos no básicos por su
solubilidad en ácidos; una vez separada, puede regenerarse alcalinizando la solución
acuosa.
Esteroquímica del Nitrógeno:
El nitrógeno utiliza orbitales sp3, que se dirigen hacia los vértices de un tetraedro.
En consecuencia, las aminas son piramidales como el amoniaco y tienen casi los
mismos ángulos de valencia (180º en la trimetilamina, por ejemplo).
La distribución tetraédrica alrededor del nitrógeno de una amina sugiere que
debería ser quiral si los tres sustituyentes fueran diferentes, puesto que el par solitario
actuaría como el cuarto. Un comportamiento así y su imagen especular, por analogía
con los centros quirales basados en el carbono no son superponibles. Esto se puede
ilustrar con la N-metiletilamina, que es una alcanoamina quiral.
Imágenes especulares de la N-metiletilamina

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
337
Sin embargo, dichos enantiómeros no han podido aislarse. Estudios
espectroscópicos demostraron que la barrera energética entre los dos arreglos
piramidales en torno al nitrógeno, por lo común es tan baja que ambos se
interconvierten rápidamente. La molécula se puede visualizar como un equilibrio entre
las dos formas.
PREPARACIÓN DE LAS AMINAS (SÍNTESIS):
Los diferentes métodos utilizados en el laboratorio para la preparación de
aminas se ilustraran en la siguiente tabla, siguiendo los estilos anteriores (reacción -
ejemplo).
SÍNTESIS DE AMINAS EJEMPLO
1.- Alquilación SN2 con halogenuros de alquilo
(a) Amoniaco
(b) Primaria
(c) Secundaria
(d) Terciaria
Por desgracia, estas reacciones no se obtienen limpiamente después de
que ha ocurrido una sola alquilación. Puesto que las aminas primarias,
secundarias y aun las terciarias tienen reactividad similar, los productos
monoalquilados que se forman inicialmente experimentan una reacción posterior para formar una mezcla de productos, como se ilustra para el 1 -
bromooctano con un exceso al doble de amoniaco, se puede tener mayor
rendimiento del primero empleando un gran exceso de amina inicial, por lo
que este método no es del todo bueno sintéticamente hablando .
2.- Aminas primarias a partir de nitrilos
3.- Aminas primarias a partir de azidas
4.- Aminas primarias a partir de
nitrocompuestos
(a) Nitroalcanos:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
338
(b) Nitroarenos (Nitrobenceno)
5.- Síntesis de Gabriel[1]
, para aminas primarias
6.- Aminas por reducción de amidas
(a) Primarias
(b) Secundarias
(c) Terciarias
7- Aminación reductiva de cetonas/aldehídos
(a) Primarias
(b) Secundarias
(c) Terciarias
(d) Vía oxima
Oxima
8.- Transposición de Hofmann[2]

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
339
9.- Transposición de Curtius
[3]
10.- Transposición de Schmidt[4]
A continuación se realizaran algunos comentarios relacionados con los
mecanismos de las reacciones específicas de aminas como lo son la Síntesis de Gabriel
y las Transposiciones de Hofmann, entre otras.
SÍNTESIS DE GABRIEL:
En la síntesis de aminas de Gabriel, informada en 1887, se hace uso de la
alquilación de imidas, lo que constituye un medio alternativo para producir aminas a
partir de halogenuros de alquilo. En la síntesis de Gabriel se emplea el anión de la imida
cíclica del ácido 1,2-bencenodicarboxílico (ftalimida) como nucleólfilo, analicemos el
proceso completo para la síntesis de la bencilamina:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
340
AMINACIÓN REDUCTIVA DE CETONAS/ALDEHÍDOS:
La aminación reductiva ocurre vía la formación de una imina (Capítulo 17,
página 303-304) como intermediario por una reacción de adición nucleófilica, y a
continuación se reduce la imina. En la reacción de aminación reductiva es posible usar
amoniaco, una amina primaria o una amina secundaria, para producir aminas primarias,
secundarias o terciarias, respectivamente.
Cetona/aldehído Imina Amina
La reacción es posible a la selectividad de los reductores: hidrógeno activado
catalíticamente o cianoborohidruro de sodio, NaBH3CN. Ambos reaccionan más
rápidamente que el doble enlace de imina que con el del grupo carbonilo. El
procedimiento habitual consiste en dejar equilibrar el compuesto carbonílico y la amina
con la imina y el agua en presencia del reductor.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
341
Ciclopentanona Imina de
ciclopentanona Ciclopentanoamina
(NO SE AÍSLA)
TRANSPOSICIÓN DE HOFMANN, CURTIUS Y SCHMIDT:
Los ácidos carboxílicos y los derivados de los ácidos carboxílicos pueden
convertirse en aminas primarias con la pérdida de un átomo de carbono, tanto en la
Transposición de Hofmann y en la Curtius, participa un derivado de ácido, amida y
cloruro de ácido respectivamente, en la de Schmidt, participa un ácido carboxílico, pero
en general proceden por mecanismos similares: Analicemos el de Hofmann
Mecanismo de la transposición de Curtius, vía una azida de acilo:
Cloruro de ácido Azida de acilo Isocianato Amina
La transposición de Schmidt tiene lugar a través de la misma secuencia, pero se
inicia con un ácido carboxílico y conduce directamente a la amina. La adición-
eliminación inicial que produce la azida de alcanoilo está catalizada por ácido sulfúrico,
que se neutraliza una vez terminada la reacción.
Reacciones de las Aminas (Reactividad):
Las reacciones de aminas están controladas por el potencial nucleofílico del
átomo de nitrógeno, la tendencia del nitrógeno a compartir el par de electrones libre,
hace que estás sean buenos nucleófilos, buenas bases y una reactividad excepcional de
los anillos aromáticos con grupos o amino sustituidos (véase Arilaminas, Capítulo 25).

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
342
REACCIONES DE LAS AMINAS EJEMPLO
1.- Alquilación de halogenuros de alquilo Véase la reacción Nº 1 de las Síntesis de aminas
2.- Eliminación de Hofmann
Se favorese la producción del alqueno menos
sustituido.
Propeno (70%)
3.- Sustitución nucleofílica en cloruro de ácidos
(Obtención de amidas)
(a) Primarias
(b) Secundarias
(c) Terciarias
4.- Sulfoamidas
(a) Formación de una Sulfoamida N-sustituida
(b) Formación de una Sulfoamida N,N-disustituida
5.- Reacción de Mannich
6.- Síntesis de óxidos de aminas

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
343
7.- Eliminación de Cope
ELIMINACIÓN DE HOFMANN:
La eliminación de Hofmann consiste en la completa metilación de una amina
con un exceso de yodometano para producir un yoduro de amonio cuaternario, el cual
por calentamiento con óxido de plata sufre una reacción de eliminación para formar un
alqueno. En esta reacción se favorece la formación del alqueno menos sustituido, las
razones de esta selectividad no se comprenden del todo, pero es probable que sean de
origen estérico. Debido al gran tamaño de la trialquilamina como grupo saliente, el ion
hidróxido debe sustraer un hidrógeno de la posición menos impedida, o sea la posición
estéricamente más accesible.
Ejemplo:
Posibles intermedios de reacción:
El intermedio que se forma para
dar lugar al 1-penteno, es
energéticamente más estable,
tiene menor impedimento
estérico, y a la base le es fácil
sustraer el hidrógeno, dando
mayoritariamente éste producto.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
344
En el caso de la formación del
2-penteno, el intermedio es
energéticamente menos estable,
con un mayor impedimento
estérico, el hidrógeno a sustraer
esta más bloqueado
estéricamente por la presencia
del grupo metilo.
La eliminación de Hofmann es útil para la apertura de aminas cíclicas, dando
lugar a dienos, como ejemplo veamos la apertura de la piperidina:
REACCIÓN DE MANNICH:
La reacción de Mannich es una condensación del metanal (formaldehído) con
amoniaco, aminas primarias o secundarias, para formar iones iminio, los cuales son
suficientemente electrófilos para reaccionar con aldehídos y cetonas enolizables
mediante un proceso similar al de la reacción aldólica, con la formación de
compuestos -N-alquilaminocarbonílicos.
Ejemplo de la reacción de Mannich:
Mecanismo:
1) Formación del ion iminio

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
345
2) Enolización
3) Formación del enlace carbono-carbono
ÓXIDOS DE AMINAS Y ELIMINACIÓN DE COPE[5]
:
Las aminas son sensibles a los oxidantes; en presencia de estos, las aminas
primarias y secundarias dan lugar a mezclas complejas. Las aminas terciarias, sin
embargo, se oxidan mediante peróxido de hidrógeno acuoso o ácidos peroxicarboxílicos
a los correspondientes óxidos de aminas, con muy buenos rendimientos. Si un óxido de
amina, posee en el carbono hidrógenos, puede ocurrir una -eliminación por
calentamiento sobre 100ºC, dando lugar a alquenos y N,N-dialquilhidroxilamina.
Ejemplos de formación de óxidos de aminas:
Ejemplo de eliminación de Cope:
Mecanismo:
El mecanismo es similar a la pirólisis de los ésteres, pero tiene lugar a
temperaturas inferiores y es más útil sintéticamente para la obtención de alquenos. Su

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
346
mecanismo es una eliminación sin que transcurre a través de un estado de transición
cíclico de cinco miembros.
Aminas de Origen Natural (Alcaloides):
Las aminas de origen natural se conocen con el nombre de Alcaloides. El
estudio de los alcaloides dio gran auge a la química orgánica en el siglo XIX, y todavía
es un área de investigación fascinante. Entre los alcaloides de origen natural se
encuentran la Morfina (analgésico), La Heroína (no natural, pero se sintetiza desde la
Morfina), al igual que la Codeína. Siendo estos compuestos agentes farmacéuticos
claves.
Morfína
Codeína
Heroína
A nivel industrial tiene gran importancia la 1,6-hexanodiamina, la cual es
necesaria en la manufacturación del nylon.
[1]
Siegmund Gabriel (1851-1924); n. Berlin; Ph.D. Universidad de Berlín (1874), Profesor en la
Universidad de Berlín, fue ayudante del profesor W. von Hofmann. [2]
August W. von Hofmann, (1818-1892), Profesor de la Universidad de Berlín [3]
Theodor Curtius, (1857-1928); Profesor de la Universidad de Heidelberg, Alemania [4]
Karl F. Schmidt, (1887-1971), Knoll AG, Ludwigshafen, Alemania [5]
Arthur C. Cope, (1909-1966), Profesor del Massachusetts Institute of Technology

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
347
HETEROCICLOS
Un compuesto heterocíclico es una sustancia que contiene un anillo formado por más
de un tipo de átomo. Existen compuestos monocíclicos, bicíclicos y mayores, todos
ellos con gran interés para los químicos y bioquímicos.
Pirrol Furano Tiofeno Imidazol Oxazol Tiazol
Pirazol 3-Pirrolina Pirrolidina Piridina Pirimidina Purina
Muchos compuestos naturales presentan en su estructura anillos tipo
heterocíclicos, como los que mostramos a continuación:
Cocaína, estimulante,
anestésico local; se
encuentra en las hojas de la
coca.
Dietilamina del ácido
lisérgico (LSD) psicomimético
Nicotina, se
encuentra en las
hojas secas del
tabaco entre el 2%
y el 8%.
Penicilina G, antibiótico
Como puede verse los compuestos heterocíclicos aparecen en varias moléculas
de interés biológico: son heterociclos los carbohidratos, como también la clorofila y
hemina, que dan el color verde a las hojas y rojo a la sangre, dándole vida a las plantas
y animales. Los sitios reactivos de muchas enzimas y coenzimas son heterociclos. La
herencia tiene su asiento, por último, en la secuencia de unión específica de media
docena de anillos heterocíclicos a largas cadenas de ácidos nucleicos.
NOMENCLATURA DE LOS HETEROCICLOS:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
348
Muchos de los heterociclos tienen nombres comunes. Además, hay varias
formas alternativas de nombrar a los heterociclos que requieren memorización, no son
de aplicación universal y, a veces se prestan a confusión. Para compuestos monocíclicos
sencillos se utiliza prefijos para indicar la presencia y la identidad del heteroátomo: aza-
para nitrógeno, oxa- para oxígeno, tio- para azufre, fosfa- para fósforo y así
sucesivamente.
Oxaciclobutano Azaciclopentano Oxaciclohexano Tiociclohexano 3-metiloxaciclohexano
A continuación resumiremos las formas de preparación de los heterociclos, en
forma de tabla con un respectivo ejemplo, al final de la tabla realizaremos un breve
comentario de las reacciones más significativas, dado que muchas de las utilizadas ya
son conocidas por el estudiante en capítulos anteriores.
PREPARACIÓN DE HETEROCICLOS
REACCIÓN EJEMPLO
1.- SÍNTESIS DE AZACICLOPROPANOS
2.- SÍNTESIS DE OXACICLOPROPANOS
(REACCIÓN DE EPOXIDACIÓN)
3.- SÍNTESIS DE TIOCICLOPROPANOS
4.- SÍNTESIS DE
HETEROCICLOBUTANOS VÍA
UNA SN2 INTRAMOLECULAR.
5.- SÍNTESIS DE
HETEROCICLOPENTANOS VÍA
UNA SN2 INTRAMOLECULAR

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
349
6.- SÍNTESIS DE
HETEROCICLOPENTANOS POR
HIDROGENACIÓN CATALITICA

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
350
7.- SÍNTESIS DE PAAL-KNORR DE 1-
HETERO-
2,4-CICLOPENTADIENOS
8.- SÍNTESIS DE HANTZSCH DE
PIRIDINAS
9.- SÍNTESIS DE FISCHER DE INDOLES
10.- SÍNTESIS DE FRIEDLÄNDER DE
QUINOLINAS
11.- SÍNTESIS DE BISCHLER-
NAPIERALSKI DE
ISOQUINOLINAS
RUTAS PARA LA PREPARACIÓN DE HETEROCICLOPROPANOS:
Los azaciclopropanos pueden prepararse por adición directa de nitrenos,
análogos nitrogenados de los carbenos, a alquenos. Por ejemplo la irradiación o
calefacción del azidocarboxilato de etilo se genera un nitreno que es atrapado por un
alqueno para dar el azaciclopropano correspondiente con rendimientos moderados.
Un nitreno

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
351
Para el caso de la preparación de oxaciclopropanos, el método más generalizado
es la epoxidación de alquenos, ya comentada con anterioridad en este libro (capítulo 13
y 14). Los tiociclopropanos se sintetizan a partir de los oxaciclopropanos
correspondientes, que son fácilmente obtenibles. Un reactivo que permite la conversión
directa de uno a otro es el tiocianato potásico. La transformación es estereoespecífica y
transcurre con inversión en los dos carbonos que reaccionan. El mecanismo de esta
reacción no será comentado en este capítulo.
Para la obtención de anillos de cuatro y cinco eslabones, el método más simple
entre otros es mediante una sustitución nucleofílica intramolecular (síntesis de
Williamson) o por hidrogenación de los correspondientes sistemas insaturados.
SÍNTESIS DE PAAL[1]
-KNORR[2]
DE 1-HETERO-2,4-CICLOPENTADIENOS:
Un método general para éste tipo de anillos es la síntesis de Paal-Knorr. La
molécula deseada se obtiene a partir de un compuesto -carbonílico enolizable por
tratamiento con una amina (para pirroles), o P2O5 (para furanos) o P2S5 (para tiofenos).
Formalmente el proceso puede considerarse como una deshidratación de un doble enol
intermedio (o su equivalente con nitrógeno o azufre) para dar el heterociclo.
Ejemplo:
SÍNTESIS DE HANTZSCH[3]
DE PIRIDINAS:
Las piridinas pueden obtenerse por condensación de productos de partida
acíclicos tales como compuestos carbonílicos con amoniaco. El método más general es
la síntesis de piridinas de Hantzsch. En esta reacción se combinan en varias etapas dos
moléculas de un compuesto -dicarbonílico, un aldehído y amoniaco para dar una
dihidropiridina sustituida, que se oxida fácilmente con ácido nítrico para dar el sistema
aromático.
Mecanismo de la síntesis de Hantzsch de piridinas:
(1) Condensación de Knoevenagel del aldehído con el 3-cetoéster
Enolización del carbono , del
acetoacetato de etilo (abstracción de
H), y condensación con el aldehído,
con posterior deshidratación para la
formación del sistema
-insaturado.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
352
(2) Formación de la enamina del 3-cetoéster con amoniaco
Ataque nucleofílico del amoniaco, al
carbonilo del grupo ceto, con
formación de un -hidroxi-éster,
quien se deshidrata para dar la
formación del sistema -
insaturado.
(3) Adición de Michael.
Consiste en una adición de
Michael vía enamina, para dar
el producto de adición 1,4
sobre el producto de
condensación de Knoevenagel,
con posterior desplazamiento
de un protón para dar como
producto final una
cetoenamina.
(4) Condensación intramolecular de la cetoenamina y tautomerización.
Es una ciclación
intramolecular inducida
por el par de electrones de
la enamina, con posterior
tautomerización hacia el
sistema 1.4-dihidro que es
mucho más estable.
(5) Oxidación y descarboxilación
El compuesto obtenido en la
etapa anterior se oxida
fácilmente con ácido nítrico para
dar el sistema aromático. El
sistema resultante es un éster
3,5-piridindicarboxílico. La
hidrólisis seguida de pirólisis de
la sal de calcio del ácido provoca
la descarboxilación.
SÍNTESIS DE FISCHER[4]
DE INDOLES:
De entre los anillos de benceno condensado con otros anillos, el indol es,
probablemente, el más importante ya que forma parte de muchos productos naturales. E l
indol está relacionado con el pirrol de la misma forma que el naftaleno está relacionado
con el benceno. La forma más general de obtener indoles es la síntesis de indoles de
Fischer. En esta síntesis, la arilhidrazona de un aldehído o cetona se trata con ácido
polifosfórico (PPA) o un ácido mineral, o un ácido de Lewis; ello causa la extrusión de
amoniaco con cierre simultáneo del anillo para dar el heterociclo deseado.
Ejemplo:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
353
El mecanismo de la síntesis de indoles de
Fischer se cree que empieza con la
tautomerización catalizada por ácido de la
arilhidrazona, desde la forma imina a la
forma enamina, tal como se ilustra a
continuación.
SÍNTESIS DE BISCHLER[5]
-NAPIERALSKI[6]
DE ISOQUINOLINAS:
Un método importante para sintetizar derivados de isoquinolina es la síntesis de
Bischler-Napieralski, la cual consiste en la ciclación de derivados acilados de -
feniletilamina por tratamiento con ácidos (a menudo P2O5), lo que genera
dihidroisoquinolinas que pueden aromatizarse. La síntesis en si es una ciclación
intramolecular de tipo Friedel-Crafts.
Ejemplo:
Mecanismo de la síntesis de Bischler-Napieralski:
Si produce el ataque del
carbonilo al catalizador (POCl3),
para luego liberar HCl,
formándose una especie tipo
imina.
Protonación del nitrógeno, lo
cual provoca una reacción tipo
Friedel-Crafts, por parte del anillo,
con posterior regeneración de la
aromáticidad.
Expulsión del catalizador, en
forma de ácido, y posterior
deshidrogenación catalizada por
paladio y calor.
REACCIONES DE LOS HETEROCICLOS
REACCIÓN EJEMPLO
1.- APERTURA NUCLEOFÍLICA DE
HETEROCICLOPROPANOS
En medio ácido, el ataque es en el carbono más sustituido.
Y en medio básico en el menos sustituido.
2.- APERTURA DE ANILLOS DE

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
354
HETEROCICLOBUTANOS
En medio ácido, el ataque es en el carbono más sustituido.
Y en medio básico en el menos sustituido.
3.- SUSTITUCIÓN ELECTROFÍLICA AROMÁTICA
DE LOS 1-HETERO-2,4-CICLOPENTADIENOS
PRODUCTO PRINCIPAL
LAS REACCIONES MÁS FRECUENTES SON: BROMACIÓN CLORACIÓN NITRACIÓN ACILACIÓN DE FRIEDEL-CRAFTS
4.- SUSTITUCIÓN ELECTROFÍLICA AROMÁTICA
EN EL ANILLO DE PIRIDINA
PRODUCTO PRINCIPAL LAS REACCIONES MÁS FRECUENTES SON: NITRACIÓN SULFONACIÓN BROMACIÓN ALQUILACIÓN Y ACILACIÓN NO OCURREN CON
LA PIRIDINA. 5.- SUSTITUCIÓN NUCLEOFÍLICA AROMÁTICA
EN EL ANILLO DE PIRIDINA
(A) REACCIÓN DE CHICHIBABIN
(B) CON REACTIVOS ORGANOMETÁLICOS (R-
LI)

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
355
(C) SUSTITUCIÓN NUCLEOFÍLICA DE
HALOPIRIDINAS
6.- REACCIONES DE LA QUINOLINA E
ISOQUINOLINA (A) SUSTITUCIÓN ELECTROFÍLICA
AROMÁTICA
(B) SUSTITUCIÓN NUCLEOFÍLICA
AROMÁTICA
REACCIÓN DE NITRACIÓN
REACCIÓN DE CHICHIBABIN
APERTURA DE ANILLOS TIPO HETEROCÍCLICOS DE TRES O MÁS
ESLABONES:
Los heterociclopropanos son relativamente reactivos ya que la tensión anular se
libera en las aperturas nucleófilas del anillo. Recuérdese que en condiciones básicas la
reacción tiene lugar sobre el centro menos sustituido, y en condiciones ácidas la
reacción tiene lugar en el centro más sustituido (ver apertura de epóxidos, capítulo 13) .
La reactividad de los heterocicloalcanos con anillos de cuatro o cinco eslabones
está de acuerdo con lo esperado en base a la tensión del anillo: sólo los anillos
tensionados son reactivos, y sus reacciones dan lugar a la apertura del anillo. En
cualquier caso la reacción puede considerarse una sustitución nucleofílica.

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
356
SUSTITUCIÓN ELECTROFÍLICA AROMÁTICA DE LOS 1-HETERO-2,4-
CICLOPENTADIENOS.
El pirrol, furano y tiofeno forman productos de sustitución electrofílica porque
son aromáticos. Cada uno tiene seis electrones en un sistema conjugado cíclico. La
química de estos compuestos es similar a la de los anillos aromáticos bencenoides
activados. Al igual que el benceno, los heterociclos aromáticos de cinco miembros
experimentan reacciones de sustitución electrofílica en lugar de reacciones de adición.
Si se eligen las condiciones de reacción apropiadas, pueden efectuarse halogenaciones,
nitraciones, sulfonaciones y acilaciones de friedel-Cratfs
CONSECUENCIAS DEL ATAQUE ELECTRÓFILO EN C2 Y C3 DE
HETEROCICLOPENTADIENOS AROMÁTICOS:
En donde X = O, N, S
La sustitución electrofilica de estos anillos aromáticos suele ocurrir
preferentemente en el carbono-2, la posición siguiente al heteroátomo, debido a que es
la más rica en electrones (más nucleófila) del anillo. En otras palabras, nótese que el
ataque electrofílico en C2 forma un intermedio catiónico que es más estable (con más
formas resonantes) que el ataque en C3, que forma un catión con sólo dos formas
resonantes. Normalmente se encuentra que orden de reactividad es furano > pirrol >
tiofeno >> benceno.
Ejemplos:
2-NITROPIRROL (83%)
2-CLOROFURANO (65%)
2-ETANOIL-5-
METILTIOFENO (70%)
SUSTITUCIÓN ELECTROFÍLICA AROMÁTICA EN LA PIRIDINA:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
357
La piridina experimenta la sustitución electrófilica muy lentamente. Reacciona
forma muy similar a un anillo bencénico desactivado. Además, la sustitución esperada
para la sustitución electrófila sobre el anillo de la piridina. La halogenación y la
sulfonación pueden efectuarse en condiciones drásticas, pero la nitración tiene muy bajo
rendimiento, y las reacciones de Friedel-Crafts son fallidas; por lo que regular se forma
el producto en la posición 3.
La baja reactividad de la piridina hacia las sustituciones electrofílicas
aromáticas se debe a una combinación de factores. El más importante es el hecho de que
la densidad electrónica del anillo se encuentra disminuida, por el efecto inductivo
atrayente de electrones del átomo de nitrógeno, electronegativo. Un segundo factor que
reduce la reactividad del anillo de piridina hacia el ataque electrofílico es el hecho de
que la formación de complejos ácido-base entre el átomo de nitrógeno del anillo (básico)
y el electófilo atacante coloca una carga positiva en el anillo y por tanto lo desactiva aún
más.
Sustitución electrofílica en posición-2 (La tercera estructura es muy inestable,
contribuye poco a la estabilidad) Sustitución electrofílica en posición-3 Sustitución electrofílica en posición-4 (La segunda estructura es muy inestable,
idems a la posición 2)
La desactivación con respecto al benceno puede atribuirse parcialmente al efecto
inductivo atrayente de electrones del átomo de nitrógeno que desestabiliza el catión
intermedio. De las tres estructuras resonantes que pueden dibujarse para cada posición
del ataque electrófilo, tanto la sustitución en la posición 2 como en la posición 4
implican una forma con contribución muy escasa. (las cuales en donde la carga positiva
se encuentra sobre el átomo de nitrógeno, sin que éste tenga su octeto electrónico
completo). La posición 3, aunque desactivada, es por consiguiente más reactiva frente a
la sustitución electrófila que las posiciones 2 y 4.
Ejemplos:
3-Nitropiridina (22%) Acido-3-piridinasulfónico (71%)
SUSTITUCIÓN NUCLEOFÍLICA SOBRE LA PIRIDINA:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
358
Sustitución nucleofílica en
posición 2 (La tercera estructura es muy
estable, construye bastante a la
estabilidad del híbrido de
resonancia). Sustitución nucleofílica en
posición 3 Sustitución nucleofílica en
posición 4 (La segunda estructura es muy
estable, idems a la posición 2).
La sustitución nucleofílica sobre el anillo de piridina procede con mayor
facilidad, que la sustitución electrofílica, en particular en las posiciones 2 y 4.
El ataque nucleofílico en la posición 2 y 4 genera un carbanión sumamente
estable, dado que la carga negativa puede llegar al átomo de nitrógeno, lo cual es
especialmente estable. El ataque en la posición 3 da una carbanión menos estable que
los anteriores.
Un ejemplo ilustrativo de este hecho el la reacción de Chichibabin[7]
, la cual
consiste en la sustitución de un hidruro por el anión amiduro. Transformaciones
relacionadas con la reacción de Chichibabin tienen lugar cuando se tratan piridinas con
reactivos de Grignard u organolíticos.
Ejemplo:
Los átomos de hidrógenos en de una cadena lateral alquílica en las posiciones
2 o 4 de la piridina tienen una acidez similar a los de una cetona. La reacción de un
grupo carbonilo con el anión alquilo formado mediante una base constituye un método
para la extensión de la cadena lateral de la piridina.
Ejemplo:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
359
Como otras aminas, también la piridina tiene propiedades nucleófilas: reacciona
con halogenuros de alquilo, formando sales de amonio cuaternarias:
Ejemplo:
Como cualquiera otra amina terciaria, puede convertirse a la piridina en su N-
óxido, mediante ácido peroxibenzoico o peróxido de hidrógeno. En contraposición a la
piridina misma, su óxido sufre la nitración principalmente en la posición 4. Lo cuál se
puede explicar por las estructuras resonantes del N-óxido.
Ejemplos:
El N-óxido puede utilizarse a menudo como una forma “activada” de la piridina.
Por tratamiento del N-óxido sustituido con PCl3 se elimina el oxígeno.
Ejemplos:
(90%)

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
360
REACCIONES DE LA QUINOLINA E ISOQUINOLINA:
La quinolina y la isoquinolina son mucho más reactivas que la piridina en las
reacciones de sustitución electrófila. Cuando las reacciones se realizan en disolución
ácida, la reacción se produce sobre la forma protonada y la sustitución tiene lugar en las
posiciones C-% y C-8 del anillo bancénico.
Ejemplos:
Al igual que en el N-óxido de piridina, el N-óxido de quinolina experimenta con
facilidad la nitración y la reacción se produce en el C-4.
Ejemplo:
Tanto la quinolina como la isoquinolina experimentan con facilidad reacciones
de sustitución nucleófila del tipo de la Chichibabin,(véase los ejemplos del cuadro
resumen).
Al igual que las 2- y 4-alquilpiridinas, las 2- y 4-alquilquinolinas y las 1-
alquilisoquinolinas tienen hidrógenos en a que son bastante ácidos y participan en
reacciones catalizadas por bases.
Ejemplo:

www.clubdelquimico.blogspot.com
Especialidad: Farmacia y Bioquímica [email protected]
361
[1]
Karl Paal, (1860-1935), Profesor de la Universidad de Erlangen, Alemania. [2]
Ludwig Knorr, (1859-1921), Profesor de la Universidad de Jena, Alemania. [3]
Arthur Hantzsch, (1857-1935), Profesor de la Universidad de Leipzig, Alemania. [4]
Emil Fischer, es el mismo que propuso la proyección del carbono tetraédrico sobre un plano, (véase
capítulo 6). [5]
A. Bischler, Profesor de la Universidad de Zürich, Alemania. [6]
B. Napieralski, Profesor de la Universidad de Zürich, Alemania. [7]
Alexei E. Chichibabin, (1871-1945), Profesor de la Universidad de Moscú.