quantum chemistry in organic reactions: investigations of
TRANSCRIPT

UCLAUCLA Electronic Theses and Dissertations
TitleQuantum Chemistry in Organic Reactions: Investigations of Mechanism and Stereoselectivity in C-H Bond Functionalizations and Cycloaddition Reactions
Permalinkhttps://escholarship.org/uc/item/3pj1n318
AuthorZOU, LUFENG
Publication Date2013-01-01 Peer reviewed|Thesis/dissertation
eScholarship.org Powered by the California Digital LibraryUniversity of California

UNIVERSITY OF CALIFORNIA
Los Angeles
Quantum Chemistry in Organic Reactions: Investigations of Mechanism and Stereoselectivity in
C-H Bond Functionalizations and Cycloaddition Reactions
A dissertation submitted in partial satisfaction of the
requirements for the degree Doctor of Philosophy
in Chemistry
by
Lufeng Zou
2013

© Copyright by
Lufeng Zou
2013

ii
ABSTRACT OF THE DISSERTATION
Quantum Chemistry in Organic Reactions: Investigations of Mechanism and Stereoselectivity in
C-H Bond Functionalizations and Cycloaddition Reactions
by
Lufeng Zou
Doctor of Philosophy in Chemistry
University of California, Los Angeles, 2013
Professor Kendall N. Houk, Chair
C−H bond functionalization is a powerful method in organic synthesis, since it avoids
prefunctionalization of substrates to introduce desired functionality. However, differentiating
between numerous C−H bonds and effecting site specific and stereoselective chemical
modifications is an ongoing challenge. In the first part of thesis, I will focus on the applications
of quantum mechanical calculations to study C−H activations by organic and metallic reagents.
Dimethyldioxirane (DMDO) is a non-metal C−H oxidation reagent. In Chapter 1, site
selectivities and reactivities in C−H activations by DMDO were studied in substituted
cyclohexanes and trans-decalins, which are models of natural steroids. It is found that the release
of 1,3−diaxial strain in the transition state contributes to the site selectivities and enhanced

iii
equatorial C−H bond reactivities for tertiary C−H bonds. In Chapter 2, C−H activation reactions
that occur with the Grubbs metathesis catalyst were found to be controlled by closed−shell
repulsions between the groups that chelate the ruthenium metal.
In the second part of the thesis, quantum mechanical computations with density functional
theory (DFT) are employed to investigate the stereoselectivities and reactivities in the reactions.
In Chapter 3, the origins of asymmetric conjugative addition of of arylboronic acid to
-substituted enone. In Chapter 4, I have demonstrated the hyperconjugative aromaticity of
5-substituted cyclopentadienes and its effect on the -facial selectivity in Diels-Alder
cycloadditions.

iv
The dissertation of Lufeng Zou is approved.
Miguel A. García-Garibay
Peter M. Felker
Albert M Thomas
Kendall N. Houk, Committee Chair
University of California, Los Angeles
2013

v
To my family and friends.

vi
TABLE OF CONTENTS
ABSTRACT OF THE DISSERTATION ....................................................................... ii
TABLE OF CONTENTS .................................................................................................vi
LIST OF SCHEMES ......................................................................................................... x
LIST OF FIGURES ..........................................................................................................xi
LIST OF TABLES ......................................................................................................... xiii
ACKNOWLEDGEMENTS ...........................................................................................xiv
VITA.................................................................................................................................. xv
Chapter 1. Enhanced Reactivity in Dioxirane C-H Oxidations via Strain Release: A
Computational and Experimental Study ......................................................................... 1
1.1 Abstract ................................................................................................................. 1
1.2 Introduction ........................................................................................................... 1
1.3 Computational Methods ........................................................................................ 4
1.4 Results and Discussions ........................................................................................ 5
1.4.1 Modeling the Dioxirane Oxidation of C-H Bonds .................................... 5
1.4.2 Oxidation of Equatorial and Axial C-H Bonds of Cyclohexane ............. 10
1.4.3 Oxidation of Methylcyclohexane ............................................................ 14
1.4.4 Influence of axial methyl substitution on reactivity ................................ 18
1.4.5 Strain Release and Enhanced Reactivity towards Oxidations in Steroidal Systems
20
1.4.6 Selective Oxidations of Sclareolide ........................................................ 22
1.5 Conclusions ......................................................................................................... 28
1.6 Experimental Section .......................................................................................... 29

vii
1.6.1 General Procedures.................................................................................. 29
1.6.2 General Procedure for Oxidation using TFDO Generated in situ: .......... 29
1.7 References ........................................................................................................... 30
Chapter 2. Formation of Chelated Z-selective Ruthenium Metathesis Catalysts:
Computational Study on C–H Activation Mechanism, Reactivity, and Selectivity ... 35
2.1 Abstract ............................................................................................................... 35
2.2 Introduction ......................................................................................................... 35
2.3 Computational Details ........................................................................................ 38
2.4 Results and Discussions ...................................................................................... 38
2.4.1 Transition state geometries for the C-H activation ................................. 38
2.4.2 The free energy profile for the formation of the chelated ruthenium catalyst 40
2.4.3 Regio- and stereoselectivity in CH activations ....................................... 41
2.4.4 Effects of chelating group and N-substituents on reactivity ................... 43
2.5 Conclusions ......................................................................................................... 48
2.6 References ........................................................................................................... 48
Chapter 3. Mechanism and Enantioselectivity in Palladium-Catalyzed Conjugate Addition
of Arylboronic Acids to -Substituted Cyclic Enones: Insights from Computation and
Experiment ....................................................................................................................... 50
3.1 Abstract ............................................................................................................... 50
3.2 Introduction ......................................................................................................... 51
3.3 Results ................................................................................................................. 53
3.3.1 Effects of Water on Catalyst Turnover.................................................... 53
3.3.2 Effects of Salt Additives on Reaction Rate ............................................. 55
3.3.3 Non-Linear Effect Correlation of Catalyst and Product Enantioenrichment 59

viii
3.3.4 Computational Investigations of the Reaction Mechanism ..................... 61
3.3.5 Experimental and Computational Investigations of the Enantioselectivities 65
3.3.6 Experimental Investigation of Arylpalladium(II) Intermediates and Formation of
the Key C–C Bond ........................................................................................... 74
3.4 Summary and Discussion .................................................................................... 77
3.5 Conclusions ......................................................................................................... 81
3.6 Materials and Methods ........................................................................................ 82
3.7 References ........................................................................................................... 84
Chapter 4. Stereoselectivities of Diels-Alder Reactions of 5-Substituted Cyclopentadienes:
Effects of Hyperconjugative Aromaticity on Reactivity and Stereoselectivity .......... 90
4.1 Abstract ............................................................................................................... 90
4.2 Introduction ......................................................................................................... 90
4.3 Background ......................................................................................................... 92
4.4 Computational Details ........................................................................................ 94
4.5 Results and Discussions ...................................................................................... 95
4.5.1 Comparison of various theoretical levels ................................................ 95
4.5.2 π-Facial stereoselectivity and geometrical distortions of reactants and transition
states, characterized by the C5-X out-of-plane angle ..................................... 100
4.5.3 Distortion/Interaction energies in π-facial stereoselectivity .................. 106
4.5.4 Reaction energies and the reaction pathways ........................................ 109
4.5.5 The origin of substituent-induced bending in the reactants – second order orbital
interaction ....................................................................................................... 114
4.5.6 Diels-Alder reaction energies and hydrogenation energies ................... 115
4.5.7 Hammett plots on the substitution effects- induction and resonance effects 116
4.5.8. Comparison with Torquoselectivity ..................................................... 118
4.6 Conclusions ....................................................................................................... 119

ix
4.7 References ......................................................................................................... 120

x
LIST OF SCHEMES
Scheme 1.1. Selective C-H oxidation. Only one of the five tertiary C-H bonds is
oxidized……………………………………………………………………………………………3
Scheme 1.2. Reaction products in a competition reaction……………………………………….20
Scheme 2.1. Generation of the Z-selective catalyst 3 through C-H activation………………….36
Scheme 2.2. Generation of the Z-selective catalyst 6 through C-H activation………………….37
Scheme 2.3. Three possible sites for C-H activation of 1a, leading to three possible chelated
catalysts 1g……………………………………………………………………………………….41
Scheme 3.1. Asymmetric conjugate addition with (S)-t-BuPyOx ligand…………….………….52
Scheme 3.2. (a) Examination of reaction mass balance. (b) Absence of water prohibits scale-up.
(c) Addition of water facilitates larger scale reactions……………………………………………54
Scheme 3.3. Enantioselectivity-determining step in asymmetric conjugate addition of arylboronic
acids to cyclic enones…………………………………………………………………………….62
Scheme 3.4. (a) Direct formation of C–C bond from arylpalladium(II) cation. (b)
Triphenylphosphine trapping experiments demonstrates configurational stability of arylpalladium
cation………………………………………………………………………………………….….77
Scheme 4.1. The π- facial stereoselectivity………………………………………………………91
Scheme 4.2. Utilization of the π-facial stereoselectivity in total synthesis by David Gin……….91
Scheme 4.3. Benchmarking reactions in anti/syn fashion………………………………………..96
Scheme 4.4. The C5-X out-of-plane angle, θ…………………………………………………..100
Scheme 4.5. Staggered conformation in the transition state for the cyclopentadiene-ethene
reaction...………………………………………………………………………………………..101
Scheme 4.6. Substituent effects on the reactant geometries leading to π- facial
stereoselectivity…………………………………………………………………………………109
Scheme 4.7. Comparison of 5-substituted cyclopentadiene Diels-Alder reactions and
hydrogenations………………………………………………………………………………….115

xi
LIST OF FIGURES
Figure 1.1. Transition state geometries for the reaction of DMDO and isobutene …………...….6
Figure 1.2. Reaction pathway in DMDO oxidations. ……………………………………...…….9
Figure 1.3. Optimized TSs for the reactions of DMDO with equatorial and axial C-H bonds.…10
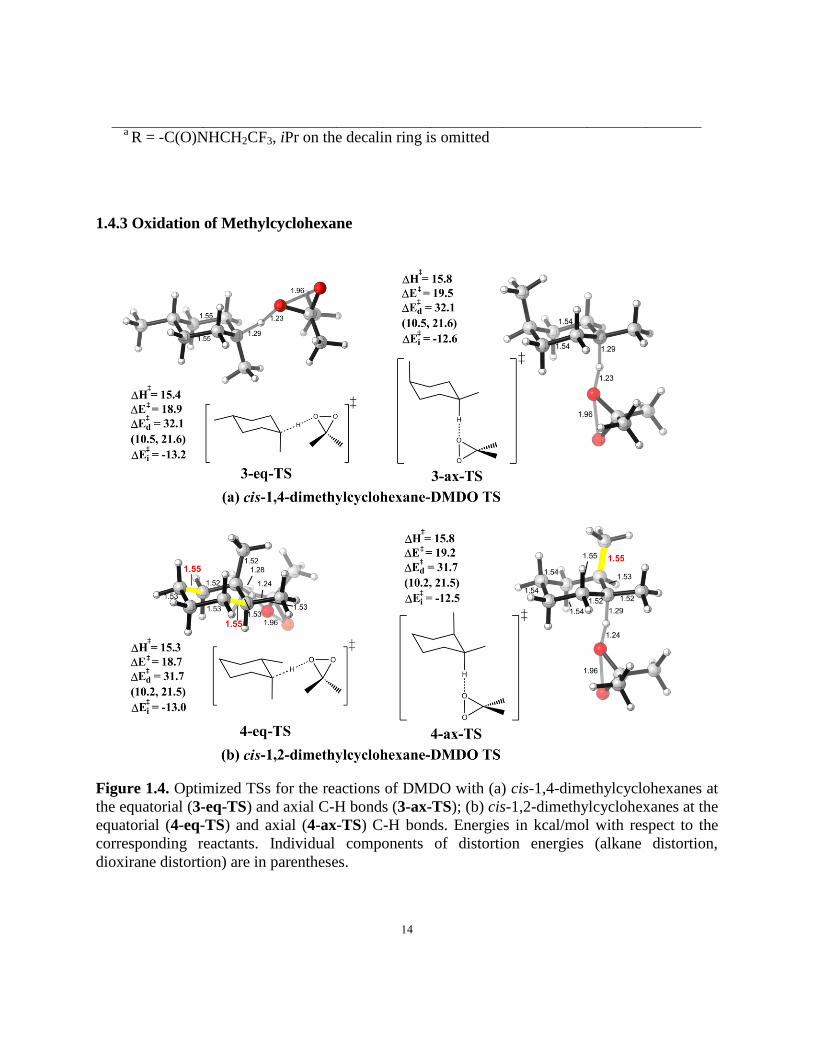
Figure 1.4. Optimized TSs for the reactions of DMDO with (a) cis-1,4-dimethylcyclohexanes at
the equatorial and axial C-H bonds; (b) cis-1,2-dimethylcyclohexanes at the equatorial and axial
C-H bonds…………………………………………………………………………..……………14
Figure 1.5. Breakdown of distortion/interaction energies along the internal reaction coordinates
(IRC) for the reactions of DMDO with (a) 1-methylcyclohexanes and (b) cyclohexanes………16
Figure 1.6. Optimized reactant and transition state geometries for the reaction of DMDO with
substituted cyclohexanes…………………………………………………………………………18
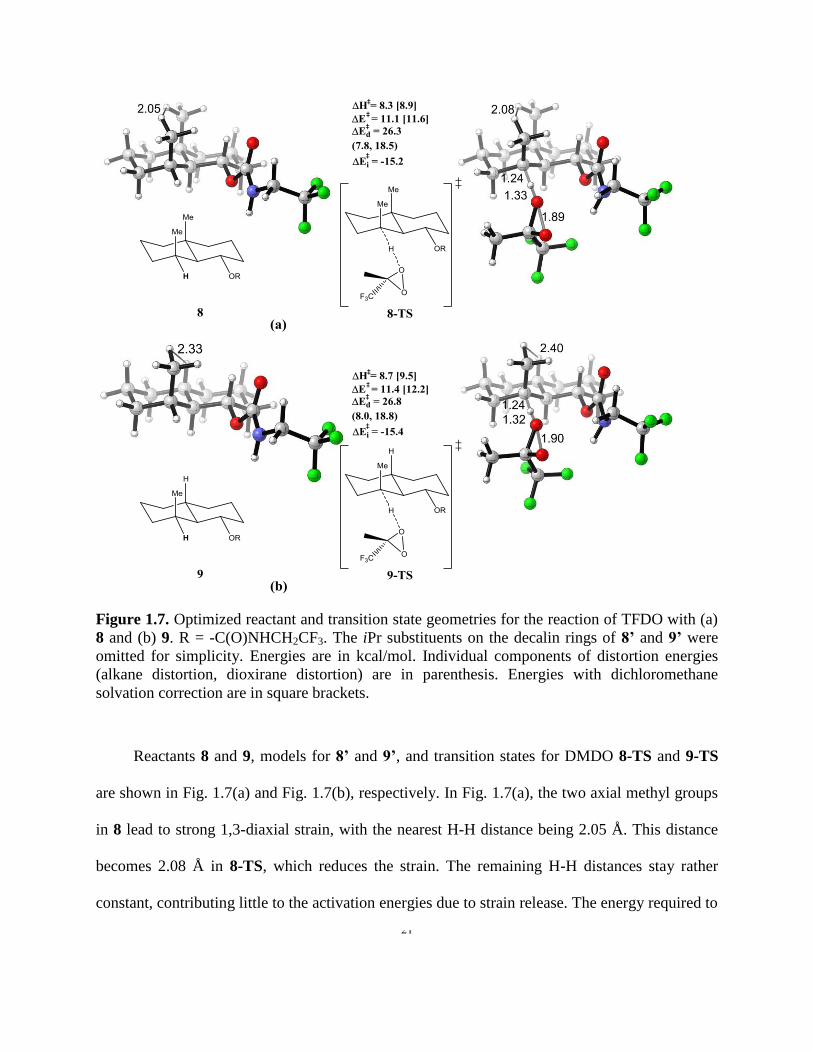
Figure 1.7. Optimized reactant and transition state geometries for the reaction of TFDO with (a)
8 and (b) 9. R = -C(O)NHCH2CF3…………………………………………………….…………21
Figure 1.8. Optimized reactant and transition state geometries for the reaction of TFDO with
sclareolide 10…………………………………………………………………………………….25
Figure 1.9. Optimized reactant and transition state geometries for the reaction of tBu-TFDO with
sclareolide 10. …………………………………………………………………………..……….27
Figure 2.1. Four possible transition state geometries for C-H activation of 4…………………..39
Figure 2.2. The free energy profile of the C-H activation of 4(1a) to form the C-H activated
catalyst (1g)………………………………………………………………………………………40
Figure 2.3. Substrate and TS geometries for C-H activation on three different sites of 1c……..42
Figure 2.4. Steric effects on stereoselectivity for the cis- and trans-TS for C-H activation of
1c…………………………………………………………………………………………………43
Figure 2.5. Substrate and TS geometries for C-H activation on three different sites of 5………46
Figure 3.1. (a) deuterium incorporation using PhB(OH)2. (b) deuterium incorporation using
(PhBO)3. (c) 1H NMR data measuring deuterium incorporation by integral comparison of
-protons relative to H5, control: treatment of ketone 3 to deuterium incorporation conditions..55
Figure 3.2. Determination of linearity between catalyst ee and product ee…………………….60
Figure 3.3. Computed potential energy surface of the catalytic cycle, the alternative direct
nucleophilic addition pathway, and the isomeric carbopalladation pathway …………………...63

xii
Figure 3.4. The optimized geometries of transition states in the enantioselectivity-determining
alkene insertion step of the reaction of 3-methyl-2-cyclohexenone and phenyl boronic acid with
(S)–t-BuPyOx ligand……………………………………………………………………………..67
Figure 3.5. The optimized geometries of transition states in the enantioselectivity-determining
alkene insertion step of the reaction of (a) 3-methyl-2-cyclohexenone and phenyl boronic acid
with (S)-i-PrPyOx ligand; (b) 3-methyl--2-pentenolide and phenyl boronic acid with
(S)-t-BuPyOx ligand…………………………………………………………..…………………70
Figure 3.6. Proposed catalytic cycle for Pd/PyOx-catalyzed conjugate addition of arylboronic
acids to cyclic enones…………………………………………………………………………….80
Figure 4.1. Calculated activation free energies (ΔGX‡) in 35 reactions (kcal/mol)……………..97
Figure 4.2. Comparison of various theories against G4 in predicting π-facial stereoselectivities
ΔΔGX‡ (anti-syn, kcal/mol)………………………………………………………………………99
Figure 4.3. The Diels-Alder transition state geometries for dienophile ethene with
cyclopentadiene, anti/syn to 5-methoxycyclopentadiene, and 5-methborylcyclopentadiene…..101
Figure 4.4. The geometries of reactant diene. Cyclopentadiene, 5-methoxycyclopentadiene. and
5-methborylcyclopentadiene……………………………………………………………………103
Figure 4.5. The correlation between activation energies and distortion energies……………...107
Figure 4.6. The correlation between π- facial stereoselectivities (ΔEa‡(a) - ΔE a
‡(s)) with
differences in anti and syn distortion energies………………………………………………….107
Figure 4.7. The correlation between distortion energies and the reactant C5-X out-of-plane
angle…………………………………………………………………………………………….108
Figure 4.8. The correlation between activation energies (Y axis) and reaction energies……...112
Figure 4.9. The reaction pathways for ethene with cyclopentadiene, methoxycyclopentadiene
and methborylcyclopentadiene ………………………………………………………………...113
Figure 4.10. Orbital interactions on model systems (X= H, –OMe, –BHMe)…………………115
Figure 4.11. Comparison of 5-substituted cyclopentadiene Diels-Alder reaction energies (ΔErxn)
and hydrogenation energies (ΔEHY)…………………………………………………………….116
Figure 4.12. Hammett plot of the selectivity dependence on the inductive effect……………..117
Figure 4.13. Hammett plot of the selectivity dependence on the resonance effect…………….118
Figure 4.14. π-facial stereoselectivity Ea‡(a-s) vs. torquoselectivity Ea
‡(i-o)………………….119

xiii
LIST OF TABLES
Table 1.1. Natural orbital occupancies of frontier orbitals, and activation energies in the
transition states ……………………………………………………………………………………6
Table 1.2. Summary of activation energies and distortion/interaction energies (kcal/mol)…….12
Table 1.3 Experimental and calculated ratios for C2/C3 products from 10……………………..23
Table 1.4 Analysis of site selectivities on 10 for TFDO vs tBu-TFDO…………………………23
Table 2.1. Activation energies for C-H activation of various N-alkyl groups…………………..43
Table 2.2. Activation energies for C-H activation with different N-aryl groups………………..44
Table 2.3. Activation energies for C-H activation of 5- and 6-membered NHC rings………….46
Table 2.4. Activation energies for C-H activation with different carboxylate ligands………….47
Table 3.1. Effect of salt additives on reaction rate………………………………………………56
Table 3.2. Effect of water and NH4PF6 on reaction rate………………………………………...57
Table 3.3. Increased reaction yields with different arylboronic acid substrates under new reaction
conditions………………………………………………………………………………………...59
Table 3.4. Activation energies and enantioselectivities of alkene insertion with (S)-t-BuPyOX,
(S)-i-PrPyOX, (S)-i-BuPyOX, and (S)-PhPyOx ligands…………………………………………68
Table 3.5. Remote ligand substituent effects on activation energies and enantioselectivities of
alkene insertion…………………………………………………………………………………..69
Table 3.6. Activation energies and enantioselectivities of alkene insertion with substrates varying
at the '-position…………………………………………………………………………………71
Table 3.7. Activation energies and enantioselectivities of alkene insertion with various boronic
acids……………………………………………………………………………………………...72
Table 3.8. Enantioselectivity trends in pyridinooxazoline and related ligand frameworks…….72
Table 3.9. Arylpalladium(II) catalyzed conjugate addition……………………………………..75
Table 4.1. Distortion/Interaction energies for various substituents and out-of-plane angle θ…104
Table 4.2. Reaction energies and C5-X out-of-plane angles (θrxn) for various substituents
(kcal/mol), and the relative (anti - syn) reaction energies (ΔΔErxn) and activation energies
(ΔΔEa‡)………………………………………………………………………………………….110

xiv
ACKNOWLEDGEMENTS
Chapter 1
This chapter is a version of:
Zou, L.; Paton, R. S.; Eschenmoser, A.; Newhouse, T. R.; Baran, P. S.; Houk, K. N. J. Org.
Chem. 2013, 78, 4037.
Permission was given for use of this work in this thesis.
Kendall N. Houk was the PI for this work.
Chapter 3
This chapter is a version of:
Holder, J. C.; Zou, L.; Marziale, A. N.; Liu, P.; Lan, Y.; Gatti, M.; Kikushima, K.; Houk, K. N.;
Stoltz, B. M. J. Am. Chem. Soc. 2013, accepted.
Permission was given for use of this work in this thesis.
Kendall N. Houk and Brian M. Stoltz were the PIs for this work.
Sections 3.3.1, 3.3.2, 3.3.3 and 3.3.6 were primarily performed by Brian M. Stoltz and his group.
Sections 3.3.4 and 3.3.5 were primarily performed by Kendall N. Houk and his group. Brian M.
Stoltz and his group wrote the introduction, section 3.2, and the experimentals, section 3.6. Both
groups contribute to the writing of Sections 3.1, 3.4 and 3.5, and various discussions and
proof-reading of the whole manuscript.
Funding
This work was supported by the National Institute of General Medical Sciences, National
Institutes of Health (GM 36700), National Science Foundation (CHE 0614591). Computer time
was provided in part by the UCLA Institute for Digital Research and Education (IDRE)
Hoffman2 cluster, and the Extreme Science and Engineering Discovery Environment (XSEDE).

xv
VITA
LUFENG ZOU
EDUCATION
March 2009 M. S. Chemistry: University of California, Los Angeles
July 2007 B. S. Chemistry: University of Science and Technology of China
HONORS & AWARDS
November 2008 Excellence in First Year Academics and Research, UCLA
PRESENTATIONS
March 2012 ACS Meeting, San Diego
“-Facial Stereoselectivities with 5-Substituted Cyclopentadienes”
PUBLICATIONS
Holder, J. C.; Zou, L.; Marziale, A. N.; Liu, P.; Lan, Y.; Gatti, M.; Kikushima, K.; Houk, K. N.;
Stoltz, B. M. “Mechanism and Enantioselectivity in Palladium-Catalyzed Conjugate Addition of
Arylboronic Acids to -Substituted Cyclic Enones: Insights from Computation and Experiment”
J. Am. Chem. Soc. 2013, accepted
Zou, L.; Paton, R. S.; Eschenmoser, A.; Newhouse, T. R.; Baran, P. S.; Houk, K. N. “Enhanced
reactivity in dioxirane C-H oxidations via strain release: a computational and experimental
study” J. Org. Chem. 2013, 78: 4037.
Bigi, M. A.; Liu, P.; Zou, L.; Houk, K. N.; White, M. C. “Cafestol to Tricalysiolide B and
Oxidized Analogues: Biosynthetic and Derivatization Studies Using Non-heme Iron Catalyst

xvi
Fe(PDP)” Synlett 2012, 2768.
Lan, Y.; Zou, L.; Cao, Y.; Houk, K. N. “Computational Methods To Calculate Accurate
Activation and Reaction Energies of 1,3-Dipolar Cycloadditions of 24 1,3-Dipoles” J. Phys.
Chem. A 2011, 115, 13906.
.

1
Chapter 1. Enhanced Reactivity in Dioxirane C-H Oxidations via Strain Release: A
Computational and Experimental Study
1.1 Abstract
The site- and stereo-selectivities of C-H oxidations of substituted cyclohexanes and
trans-decalins by dimethyldioxirane (DMDO) were investigated computationally with quantum
mechanical density functional theory (DFT). The multi-configuration CASPT2 method was
employed on model systems to establish the preferred mechanism and transition state geometry.
The reaction pathway involving a rebound step is established to account for the retention of
stereochemistry. The oxidation of sclareolide with dioxirane reagents is reported, including the
oxidation by the in situ generated tBu-TFDO, a new dioxirane that better discriminates between
C-H bonds based on steric effects. The release of 1,3-diaxial strain in the transition state
contributes to the site selectivity and enhanced equatorial C-H bond reactivity for tertiary C-H
bonds, a result of the lowering of distortion energy. In addition to this strain release factor, steric
and inductive effects contribute to the rates of C-H oxidation by dioxiranes.
1.2 Introduction
The functionalization of unactivated sp3 C-H bonds is of considerable interest in
contemporary synthetic organic and organometallic chemistry.1-3
Compared to traditional
functional group manipulations and interconversions employed in synthesis, C-H bond
functionalization offers a direct route avoiding pre-functionalization of substrates. However,
differentiating between numerous C-H bonds and effecting site-specific and stereoselective

2
chemical modifications is an ongoing challenge.4 A range of reagents are known for C-H to
C-OH conversion, such as Fe, Cu, Ru, Cr and Pd based systems.5 The pioneering work of
Murray6 and Curci
7 pointed to the use of dimethyldioxirane (DMDO) and
methyl(trifluoromethyl)dioxirane (TFDO) for sp3 C-H oxidation.
8 The dioxiranes are potent
oxidants that lead to conservation of stereochemistry but no intermediate was characterized.9 The
mechanism is still under an ongoing debate and radical products are observed in some
experiments.10-12
Due to their high reactivity, dioxiranes are sometimes generated in situ during
synthesis.13
Recently, a two-phase strategy for terpene total synthesis was invented to holistically
mimic the way such molecules are constructed in nature. In the first phase, the so-called "cyclase
phase", simple terpenes at low oxidation states are stitched together as a prelude to the "oxidase
phase" wherein these molecules are systematically oxidized. In the case of the eudesmane
terpene class this was done using both innate and guided C-H functionalization logic.4 The
purpose of this strategy for retrosynthetic analysis is not to necessarily recapitulate biosynthesis
but rather to uncover new basic reactivity principles and invent new reaction methodology.
The factors that contribute to site selectivity in C-H oxidation reactions has been a question
of interest for some time and continues today.14
Terpenoids contain a host of cyclohexane
derivatives, and the rationalization of reactivities in such systems traces back to Barton’s
fundamental work in the fifties.15,16
Reactivity differences, such as observed for the oxidation of
axial and equatorial secondary alcohols with chromic acid, were explained by steric effects.
Eschenmoser interpreted the enhanced oxidation rates of axial alcohols as compared to their
equatorial epimers as resulting from the release of 1,3-diaxial strain in the transition state of the

3
oxidation of the axial epimers.17
Experimental measurements of the rates and regioselectivites of
C-H oxidation of substituted cyclohexanes by dioxiranes were performed by Curci,18,19
and a
preference for equatorial attack was observed. Furthermore, Curci8 and others
20 delineated a
variety of factors that contribute to site selectivity in oxidations by dioxiranes. Similar factors
have also been reported by M. C. White in C-H oxidation reactions of complex molecules by a
non-heme, iron-based system.21,22
This field has a storied history and has been reviewed
extensively recently.23
In the context of Eschenmoser’s hypothesis based on strain release, a recent example of
site-selective C-H oxidation of a substituted decalin from Baran’s laboratory has been
rationalized.24
Of five unactivated tertiary C-H bonds in the substrate, only equatorial C-H1 is
oxidized to an alcohol by the dioxirane reagent. (Scheme 1.1) Strain release in the transition state
was proposed to account for the preference.25
Based on computational studies from the Houk26
and Bach27
labs, a flattening at the carbon undergoing oxidation is expected in the transition
structure, which alleviates the 1,3-diaxial strain between two axial methyl groups in the
transition state for H1 abstraction and lowers the activation energy for C-H
1 oxidation.
Scheme 1.1. Selective C-H oxidation. Only one of the five tertiary C-H bonds is oxidized24

4
Given the intensive efforts underway to develop substrate controlled selective C-H
activation processes, we undertook calculations at the density functional and
multi-configurational ab initio levels of theory to gain quantitative insights into the exquisite
selectivities reported by Baran. Here we describe computational studies of the transition
structures for C-H oxidation of Baran’s eudesmane decalins and a variety of other simpler
cyclohexanes by dioxirane reagents. We also analyze various substituted cyclohexyl substrates.
For several such cases, we report transition structures and present explanations for the enhanced
reactivity of equatorial C-H bonds in these strained systems. Additionally, we include a new
synthesis of trifluoromethyl, tert-butyl ketone and its use as a reagent in C-H oxidation by in situ
formation of the corresponding dioxirane, tBu-TFDO. A quantitative understanding of the
factors contributing to selective C-H activation is provided, in the context of the
distortion/interaction theory that has been successfully applied to other types of reactions.28-31
1.3 Computational Methods
All density functional theory (DFT) calculations were performed with Gaussian 09.32
Mimina and transition structures were optimized using the unrestricted DFT method, UB3LYP,
with the 6-31G(d) basis set.32
Frequency analyses were carried out on stationary points to verify
that they are minima or saddle points (transition structures - TSs). Energies were recalculated
with UB3LYP/6-311++G(d,p) on these optimized geometries, unless otherwise stated. To ensure
that the correct unrestricted wavefunctions were obtained, a stability test was carried out with
Gaussian keyword stable=opt. Solvation corrections were calculated with the conductor-like

5
polarizable continuum model (CPCM) using the UAHF atomic radii. Multi-determinant
wavefunction theory, Complete Active Space with Second-order Perturbation Theory (CASPT2)
calculations were conducted with MOLCAS 7.4.33
A 10-electron, 10-orbital active space was
employed for CASPT2 calculations with a large cc-pVTZ basis set on the optimized geometries.
1.4 Results and Discussions
1.4.1 Modeling the Dioxirane Oxidation of C-H Bonds
Modeling of dioxirane oxidation has led to different conclusions about the TS geometries.
Goddard34,35
and Bach36
investigated the electronic structure of dioxirane. The O-O bond lengths
in both DMDO and TFDO are 1.51 Å, readily broken due to the ring strain. Early calculations
show that a spiro geometry is preferred over a planar geometry for epoxidations.26,27,37
Houk
explored the geometry and selectivities of dioxirane and cyanodioxirane, a model for TFDO,
with restricted DFT calculations.26
Due to the complexity of the potential energy surface (PES),
and possible intersystem crossing of the singlet and triplet PES, later studies proposed several
different first-order saddle points, associated with three different transition structures.38,39
(Fig.
1.1) We have explored these in more detail in order to determine which is likely to be the most
important transition structure for the reaction.

6
Figure 1.1. Transition state geometries for the reaction of DMDO and isobutane
Table 1.1. Natural orbital occupancies of frontier orbitals, and activation energies in the
transition states a
HOMO LUMO CASPT2 UB3LYPb
TS1-A 1.42 0.58 21.5 19.9c
TS1-B 1.24 0.76 22.5 15.8
TS1-C 1.64 0.37 19.5 20.0
a CASPT2(10,10)/cc-pVTZ, energies in kcal/mol
b UB3LYP/6-311++G(d,p)
c 24.2 kcal/mol with RB3LYP/6-311++G(d,p)
The first transition state, TS1-A, is a spiro transition state with all electrons paired, located
by restricted DFT calculations.26,40
This corresponds to a concerted process in which the t-butyl
alcohol and acetone are produced without the formation of an intermediate. A wavefunction
stability test, however, reveals that this wavefunction is unstable with respect to an unrestricted
wavefunction. That is, an open-shell wavefunction is lower in energy. The closed-shell treatment

7
was further invalidated with the multi-determinant CASPT2, as strong diradical character is
observed in the transition state, indicated by the HOMO/LUMO occupation number of 1.52/0.49.
(Table 1.1)
The second TS, TS1-B, has the O-O bond perpendicular to the breaking C-H bond. This is
found to be a radical hydrogen abstraction process beginning from the 1,3-dioxy radical. The
resulting radical intermediates could then recombine to transfer the OH group to form the
products. For this mechanism, the rate limiting step is the dissociation of the DMDO O-O bond
from the singlet diradical, which requires an activation enthalpy of 23.1 kcal/mol according to
Cremer.41
The bond-dissociation rate determining step has a higher barrier than all three C-H
activation barriers in Table 1.1, so it is disfavored under kinetic control.
The third TS, TS1-C, has the O-O bond aligned with the breaking C-H bond. We use
stable=opt to obtain the correct wavefunction at the initial geometry, then perform each
geometry optimization for transition state using the optimized wavefunction as an initial guess
with the Gaussian keyword guess=read. To make sure the optimized geometries have the correct
wavefunction, the same procedure was repeated on the optimized geometries. Both the UB3LYP
and CASPT2 indicated slight diradical character for the transition state, with <S2> value =
0.5312 with B3LYP, and HOMO/LUMO occupation numbers 1.64/0.37 with CASPT2.
Therefore TS1-C is the more reliable transition state. As CASPT2 becomes formidably
expensive with larger systems, UB3LYP with the stable open-shell wavefunction was employed
for further studies.

8
The mechanism involving TS1-C is analogous to the “oxygen rebound” mechanism
common in iron-oxo oxidations.42,43
The complete reaction pathway was then explored and is
summarized in Figure 1.2. The reactant complex goes through C-H activation transition state
TS1 and forms a weakly-bound radical pair intermediate, which rebounds and forms the final
product. The reaction can be tracked with the forming C-O bond length. As the two reactants
approaches each other, the C-O bond distance decreases until it reaches a minimum at 2.50 Å in
TS1, then increases back to 3.15 Å after hydrogen abstraction, leaving two radical centers in the
intermediate. The C-O bond distance then decreases again, (“rebounds”) through 2.51 Å in TS2
to eventually 1.43 Å in the product. As the second transition state has essentially no barrier, the
radical pair intermediate is expected to have too short a lifetime to escape from the solvent cage,
leading to the retention of stereochemistry as observed in experiments.
The same conclusions hold for the more reactive oxidant, TFDO. The C-H activation free
energy is around 5 kcal/mol lower with TFDO than DMDO, but the preferred transition state is
again obtained with unrestricted and optimized wavefunctions. The overall reaction pathway is
similar and the rate-limiting step is again the C-H activation step with diradical character (not
shown). Based on the similarity of the DMDO and TFDO reaction pathways, the in situ
generated tBu-TFDO is expected to follow the same mechanism. The computational results are
in agreement with Curci’s finding that TFDO is more reactive than DMDO without diminished
selectivity, as an exception to the reactivity-selectivity rule.9

9
Figure 1.2. Reaction pathway in DMDO oxidations. Energies in kcal/mol.

10
1.4.2 Oxidation of Equatorial and Axial C-H Bonds of Cyclohexane
Figure 1.3. Optimized TSs for the reactions of DMDO with equatorial (2-eq-TS) and axial
(2-ax-TS) C-H bonds. Energies in kcal/mol with respect to the corresponding reactants.
Individual components of distortion energies (alkane distortion, dioxirane distortion) are given in
parentheses.
The TSs for axial and equatorial C-H oxidation by DMDO are shown in Figure 1.3. No
significant axial/equatorial preference is found. The activation energy for oxidizing the
cyclohexane equatorial hydrogen is 21.6 kcal/mol, while for the axial hydrogen it is 21.4
kcal/mol.

11
To evaluate the factors governing relative reactivities of C-H bonds quantitatively, a
distortion/interaction energy analysis was conducted. The distortion energy (ΔEd‡) is the energy
required to distort reactants, hydrocarbon plus the oxidant, dimethyldioxirane (DMDO), into the
transition state (activated) geometry. The interaction energy (ΔEi‡) is the energy lowering due to
the interaction of the two distorted reactants. Distortion energies favor attack on the axial
hydrogen by 1.1 kcal/mol, while interaction energies favor equatorial attack by 0.9 kcal/mol.
These two components largely cancel out, leaving essentially no preference (0.2 kcal/mol). The
nearly identical activation energies for 2-eq-TS and 2-ax-TS indicate very weak axial/equatorial
selectivity, if any, as observed experimentally for C-H activations of different types.25
The distortion of the DMDO moiety is about the same for both transition states, and the
difference in the distortion energies mainly comes from the cyclohexane distortion.
Hyperconjugation is a likely factor to the 0.9 kcal/mol difference in interaction energies. (Fig.
1.3) In the 2-eq-TS, there are two C-C bonds anti to the breaking C-H bond, while there are two
antiperiplanar C-H bonds in the 2-ax-TS. The hyperconjugative stabilization of the partial
positive charge in the polarized transition states is larger in 2-eq-TS.20
A slight lengthening of
the antiperiplanar bonds is observed in the optimized transition structures, indicative of this
hyperconjugative interaction.
The distortion energy difference can be attributed to torsional interactions. The Newman
projections of these two transition states are given in Fig. 1.3. The internal C-C-C-C dihedral
angle is 56° in the 2-eq-TS, slightly more distorted than the one in cyclohexane (55°). This angle
is 48° in 2-ax-TS, closer to the more relaxed angle in the cyclohexyl radical (44°). The
relaxation is reflected as a 1.0 kcal/mol lowering in the distortion energy. Hydrogen-hydrogen

12
repulsion also plays a part in the relative stability. As hydrogen is being abstracted from a
tetrahedral sp3 carbon, the carbon flattens and becomes sp
2 in character. As shown in the
Newman projection for 2-eq-TS, this lead to slightly greater eclipsing of the vicinal C-H bonds,
as indicated in the 45° dihedral angle, while that in 2-ax-TS is 50°. Therefore, distortion energy
for 2-eq-TS is higher. This torsional effect favoring axial attack was observed in the case of
nucleophilic attack on cyclohexanone (Felkin’s rule)44,45
and reactions of cyclohexyl radicals,46
in which the axial TS has a more staggered conformation than the equatorial TS. In the reactions
studied here, the cancellation of distortion and interaction energy preferences leads to no intrinsic
selectivity for the secondary C-H bonds of cyclohexane.
This type of analysis has been performed on a variety of cyclohexanes and decalins. The
results are tabulated in Table 1.2.
Table 1.2. Summary of activation energies and distortion/interaction energies (kcal/mol)
Compound
C-H bond for
oxidation ΔH‡ ΔE
‡
ΔEd‡
(alkane)
ΔEd‡
(DMDO)
ΔEd‡
(total)
ΔEi‡
1 tBu-H 16.2 20.0 11.1 21.7 32.7 -12.8
2-eq 17.7 21.6 13.1 22.1 35.2 -13.6
2-ax 17.5 21.4 12.1 22.0 34.1 -12.7
3-eq 15.4 18.9 10.5 21.6 32.1 -13.2
3-ax 15.8 19.5 10.5 21.6 32.1 -12.6

13
3’-ax 16.1 19.6 10.5 21.6 32.2 -12.6
4-eq 15.3 18.7 10.2 21.5 31.7 -13.0
4-ax 15.8 19.2 10.2 21.5 31.7 -12.5
4’-ax 16.4 19.8 10.5 21.7 32.2 -12.4
5-eq 15.4 18.9 10.5 21.6 32.1 -13.2
6 14.7 18.2 9.9 21.3 31.2 -13.0
7 14.2 17.7 9.4 21.1 30.4 -12.8
8a 13.3 16.4 10.1 20.2 30.3 -13.9
9a 13.5 16.8 10.3 20.6 30.9 -14.1
10-eq-C2 18.3 22.2 12.8 22.1 34.9 -12.7
10-eq-C3 18.8 22.7 13.8 23.8 36.0 -13.3
10-ax-C3 18.5 22.2 12.5 22.1 34.5 -12.3
14
a R = -C(O)NHCH2CF3, iPr on the decalin ring is omitted
1.4.3 Oxidation of Methylcyclohexane
Figure 1.4. Optimized TSs for the reactions of DMDO with (a) cis-1,4-dimethylcyclohexanes at
the equatorial (3-eq-TS) and axial C-H bonds (3-ax-TS); (b) cis-1,2-dimethylcyclohexanes at the
equatorial (4-eq-TS) and axial (4-ax-TS) C-H bonds. Energies in kcal/mol with respect to the
corresponding reactants. Individual components of distortion energies (alkane distortion,
dioxirane distortion) are in parentheses.

15
The TSs for tertiary axial/equatorial C-H oxidation of cis-1,4-dimethylcyclohexane by
DMDO are shown in Fig. 1.4(a), as a eudesmane model system with both axial and equatorial
tertiary C-H bonds. The activation energy for the reaction of the equatorial C-H bond in 3-eq-TS
is 0.6 kcal/mol lower than for the axial C-H bond in 3-ax-TS. While interaction energies again
favor equatorial hydrogen abstraction, the distortion energies are essentially the same. The
axial/equatorial selectivity comes from the interaction energies, which are -13.2 kcal/mol for
3-eq-TS and -12.6 kcal/mol for 3-ax-TS. This preference, coming from interaction energies, is
found again in 4-eq-TS and 4-ax-TS, where both axial and equatorial C-H bonds are present in
the same molecule. (Fig. 1.4(b))
The distortion energies are identical for the two transition states, while the interaction
energies are -13.0 kcal/mol and -12.5 kcal/mol, respectively. Just as the case of secondary
hydrocarbons, the lengthening of antiperiplanar C-C bonds is shown in the equatorial TS, leading
to better hyper- conjugation. A more detailed study of how distortion energies change along the
reaction coordinate is summarized in Figure 1.5. Distortion energies are the same since the
tertiary C-H oxidation transition states are earlier than the transition states for secondary C-H
oxidation. With the same distortion energy and favored interaction energy for abstraction of
equatorial hydrogens, the latter is slightly more susceptible to oxidation.

16
Figure 1.5. Breakdown of distortion/interaction energies along the internal reaction coordinates
(IRC) for the reactions of DMDO with (a) 1-methylcyclohexanes and (b) cyclohexanes. The C-H
bonds are functionalized at the equatorial position (for 1-axial-1-methyl-cyclohexane) or axial
position (for 1-equatorial-1-methyl-cyclohexane). Energies in kcal/mol with respect to there
corresponding reactants. The x axis is the “internal reaction coordinate” from calculation, and the
origin is the transition state.
The stereochemistry of the remote methyl group has no effect on the reactivity, only the
stereochemistry of the carbon on which hydrogen is abstracted matters. As in Table 1.2, axial
C-H oxidations give almost identical distortion and interaction energies for
cis-1,4-dimethylcyclohexane (3-ax) and trans-1,4-dimethylcyclohexane (3’-ax), leading to the
same activation energy.

17
The stereochemistry of the methyl group to the reacting center has little impact on the
interaction energy, which is intrinsic whether an axial or equatorial hydrogen is abstracted. As in
Table 1.2, oxidation of cis-1,2-dimethylcyclohexane (4-ax) and trans-1,2-dimethylcyclohexane
(4’-ax) give virtually identical interaction energies. The distortion energy for 4’-ax is raised by
0.5 kcal/mol due to developing torsional strain in the transition state as the reacting site becomes
more sp2 in character. Overall the activation energy for 4’-ax is 0.6 kcal/mol higher than that of
4-ax.
Compared with tertiary C-H bonds, activations of secondary C-H bonds require higher
activation energies, which are around 21.5 kcal/mol in Fig. 1.3, due to the electrophilic nature of
the oxidizing reagent, compared with those activation energies for tertiary C-H bonds, which are
below 20.0 kcal/mol. This agrees with Curci’s experiments that tertiary C-H bonds are generally
more reactive than secondary C-H bonds, and the usual greater stability of tertiary radicals.17
The
differences in activation energies between secondary C-H oxidation (~18 kcal/mol) and tertiary
C-H oxidation (15-16 kcal/mol) is comparable to, or slightly higher, than the difference between
secondary and tertiary C-H bond dissociation enthalpies (BDEs) (98 and 96 kcal/mol,
respectively47
) because there is polarization and partial positive charge build up at the oxidized
carbon in the transition states.

18
1.4.4 Influence of axial methyl substitution on reactivity
Figure 1.6. Optimized reactant and transition state geometries for the reaction of DMDO with
substituted cyclohexanes. Energies are in kcal/mol. Individual components of distortion energies
(alkane distortion, dioxirane distortion) are in parenthesis.
To systematically investigate the effects of 1,3-diaxial methyl-methyl interactions,
reactivities of the tertiary equatorial C-H bonds reacting on axial methylcyclohexane (5),
diaxial 1,3-dimethylcyclohexane (6), and triaxial 1,3,5-trimethylcyclohexane (7) were studied.
The reactants and transition states are shown in Fig. 1.6. The methyl groups in 5, 6, 7 were axial

19
rather than in the more stable equatorial position, to serve as models for rigid terpenes where
the methyl groups are fixed to be axial, as in Baran’s selective oxidation of 1.24
The relative rate
for oxidation of cis-1,3-dimethylcyclohexane (6) is slightly higher than the
trans-1,3-dimethylcyclohexane, as strain release operates in the former isomer, in agreement
with a ratio of 1.4-1.5:1 by TFDO oxidation generated in situ.
As more axial methyl substituents were added (5 to 6 to 7), the nearest H-H distance gets
shorter, and more strain is imposed as the H-H closed shell repulsion increases. In the reactants,
there is no significant H-H repulsion in 5, there is one H-H interaction with a 2.12 Å distance in
6, and there are three pairs of CH-CH interactions of 2.11 Å in 7. In the transition state for
abstraction of equatorial hydrogens, the methyl bends away from the ring, releasing 1,3-diaxial
strain. In 5, the H-H distances are above 2.4 Å, and no strain is released in the TS. In 6, the
closest H-H bond distance increases from 2.12 Å in the reactant to 2.19 Å in TS, releasing strain.
In 7, the two closest H-H distances increase from 2.11 Å to 2.20 Å and 2.21 Å, and further strain
release is expected.
The trend towards lower activation energy was reflected in the decreasing distortion
energies, from 32.1 (5-TS) to 31.2 (6-TS) to 30.4 kcal/mol (7-TS). The interaction energies are
nearly the same, increasing by 0.2 kcal/mol (less negative) with each additional axial methyl
group, but this change is smaller than the increase in distortion energies, which dominates the
trend in reactivity. Most of the difference in distortion energy results from the hydrocarbon
reactant. The steric acceleration proposed to result from strain release by Eschenmoser,17
is
manifested in lower distortion energy in the TS. In other words, the reactants are pre-distorted
towards the TS geometry, and the activation energy is lowered. We have referred to this

20
phenomenon as distortion-acceleration.48
The higher reactivity along the series is also paralleled
by slightly earlier transition states along the series, and so the distortion energy of the dioxirane
decreases in the series.
1.4.5 Strain Release and Enhanced Reactivity towards Oxidations in Steroidal Systems
The strain release (or distortion-acceleration) effect has been taken advantage of by
Baran24
to generate selectivity in C-H oxidations, as in Scheme 1.2. A 1:1 mixture of 8’ and 9’
gave the corresponding oxidation products in a ratio of 3:1. The strain release effect was
proposed to account for the enhancement in reactivity of 8’ as compared with 9’. A similar trend
in reactivity was observed in oxidation by ozone, which produced 8’ and 9’ in a 4:1 ratio.
We modeled the reaction with methyl(trifluoromethyl)dioxirane (TFDO) and
dichloromethane as the solvent. The results are shown in Figure 1.7. A model study with DMDO
and these substrates in the gas phase is summarized in Table 1.2.
Scheme 1.2. Reaction products in a competition reaction
21
Figure 1.7. Optimized reactant and transition state geometries for the reaction of TFDO with (a)
8 and (b) 9. R = -C(O)NHCH2CF3. The iPr substituents on the decalin rings of 8’ and 9’ were
omitted for simplicity. Energies are in kcal/mol. Individual components of distortion energies
(alkane distortion, dioxirane distortion) are in parenthesis. Energies with dichloromethane
solvation correction are in square brackets.
Reactants 8 and 9, models for 8’ and 9’, and transition states for DMDO 8-TS and 9-TS
are shown in Fig. 1.7(a) and Fig. 1.7(b), respectively. In Fig. 1.7(a), the two axial methyl groups
in 8 lead to strong 1,3-diaxial strain, with the nearest H-H distance being 2.05 Å. This distance
becomes 2.08 Å in 8-TS, which reduces the strain. The remaining H-H distances stay rather
constant, contributing little to the activation energies due to strain release. The energy required to

22
distort 8 into 8-TS is 26.3 kcal/mol, mainly from the dioxirane distortion, due to the stiffness of
the ring.
The aforementioned methyl-methyl interaction in 8 is alleviated when the bridge methyl is
replaced by a hydrogen atom for 9 in Fig. 1.7(b). Compared with the case of 8, there is less strain
release. In other words, more energy was required to distort the reactant 9 into its transition states
9-TS, with distortion energy increased to 26.8 kcal/mol; the activation energy is 11.4 kcal/mol.
The 0.5 kcal/mol difference in distortion energy, although rather small, dominates over the
interaction energies, and lead to 0.3 kcal/mol lower activation energy for 8-TS than 9-TS. With
solvent correction, there is 0.6 kcal/mol lower in activation enthalpy for 8-TS, in agreement with
observed 3:1 experimental ratio. For model study with DMDO, predicted activation energy for
8-TS is 0.4 kcal/mol lower than 9-TS (16.4 kcal/mol and 16.8 kcal/mol, respectively). The axial
methyl group gives only 0.4 kcal/mol rate enhancement for the substituted decalin, compared to
0.7 kcal/mol with substituted cyclohexane, which is likely due to the rigidity of the ring, as
reflected in the distortion energies of the ring systems.
1.4.6 Selective Oxidations of Sclareolide
Sclareolide, 10, has five methylenes and ten distinct secondary hydrogens. Experimentally,
10 is oxidized at both the C2 and C3 positions (steroid numbering),49
and ratios of products are
given in Table 1.3.
Electronically, oxidation of C3 is favored because it is remote from the
electron-withdrawing lactone that deactivates the methylenes at C1, C5 and C6 and steric effects

23
favor oxidation at C2. For reagents that behave like small reagents, such as dioxiranes and
Fe(PDP) there is marginal selectivity between oxidation at the C2 and C3 positions. The use of a
manganese porphyrin results in considerably higher site selectivity.50
In the case of the
Rh-mediated amination employing a reagent that behaves like a large reagent, excellent C3
selectivity is observed.5n,24
Table 1.3 Experimental and calculated ratios for C2/C3 products from 10. Energies in kcal/mol.
oxidant C2:C3 ref Calc ΔE‡(C2) ΔE
‡(C3axial) ΔE
‡(C3equatorial)
DMDO 1.5:1a
49 ~1:1
b 22.2
b 22.2
b 22.7
b
TFDO 2.1:1~1:2.0c
49 2.7:1
d 10.2
d 12.1
d 10.9
d
tBu-TFDO 2.4:1e
49 8.3:1
d 11.9
d 14.3
d 13.1
d
Rh2(esp)2f Only C2
49
Mn(TPP)Clg
Fe(PDP)
7:1
1.4:1
50
21
a 1:1 acetone:DCM, <5% conversion
b UB3LYP/6-311++G(d,p), gas phase
c Ratios with a variety of solvents gave from 2.1:1 with hexafluoroisopropanol to 1:2.0 with
acetonitrile.
d UB3LYP/6-311++G(d,p), calculated ratio from activation energies in 298 K with
CPCM/UAHF in acetonitrile
e In acetonitrile solvent
f Amination products
g A ratio of 3:1 was observed for fluorination with Mn(TMP)Cl.
Table 1.4 Analysis of site selectivities on 10 for TFDO vs tBu-TFDO. Energies in kcal/mol.
oxidant TS ΔH‡(solv)
a ΔH
‡ ΔE
‡ ΔEd
‡(10) ΔEd
‡(TFDO) ΔEd
‡(10 ΔEi
‡

24
+TFDO)
TFDO C2 6.7 12.2 15.7 8.7 20.2 28.9 -13.2
TFDO C3-ax 8.8 12.7 15.9 8.5 20.1 28.6 -12.7
TFDO C3-eq 7.5 12.8 16.1 9.2 20.3 29.5 -13.4
tBu-TFDO C2 8.3 14.2 17.8 9.9 20.8 30.7 -12.9
tBu-TFDO C3-ax 10.7 15.1 18.7 10.3 20.8 31.1 -12.4
tBu-TFDO C3-eq 9.5 15.0 18.7 11.1 20.9 32.1 -13.4
a Calculated solvent with CPCM/UAHF in acetonitrile
Table 1.4 shows details of the computed activation barriers with different oxidants. The
bulky tBu group of tBu-TFDO increases the activation energies by about 2 kcal/mol, reflected in
the distortion energies especially from the substrate 10. The interaction energies are essentially
the same, since the methyl group in TFDO and tBu in tBu-TFDO have similar electronic effects.
The reactants and transition states geometries are shown in Figure 1.8 for TFDO and Figure 1.9
for tBu-TFDO. With solvent correction (acetonitrile), the axial TS becomes disfavored probably
due to a higher energy cost for the rearrangement of solvent molecules. Also a higher C2:C3
selectivity is expected. A thorough study of solvent effects is underway.
Figure 1.8 gives the geometries of sclareolide 10 and three transition states for C-H
oxidations on the C2/C3 C-H bonds with TFDO. The methyl-hydrogen strain is released, as the
bond lengths for nearest H-H pairs, which are 2.10 Å and 2.12 Å in 10, increased to 2.11 Å and
2.18 Å in 10-eq-C2-TS, but stays the same on 10-ax-C3-TS and 10-eq-C3-TS. Not surprisingly,
the strain release effect is not as pronounced as is the case with the relief of a methyl-methyl 1,3
diaxial interaction. Accordingly, a mixture of products results.
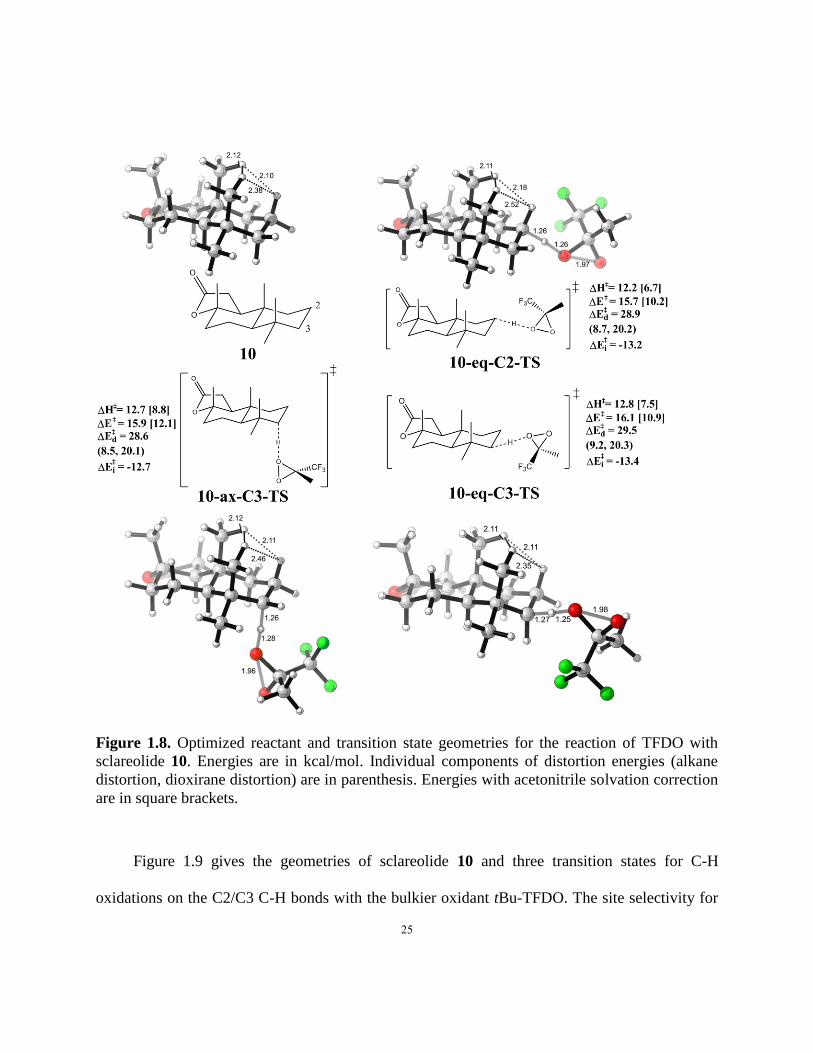
25
Figure 1.8. Optimized reactant and transition state geometries for the reaction of TFDO with
sclareolide 10. Energies are in kcal/mol. Individual components of distortion energies (alkane
distortion, dioxirane distortion) are in parenthesis. Energies with acetonitrile solvation correction
are in square brackets.
Figure 1.9 gives the geometries of sclareolide 10 and three transition states for C-H
oxidations on the C2/C3 C-H bonds with the bulkier oxidant tBu-TFDO. The site selectivity for

26
oxidation on the C2 versus C3 position is predicted to be 0.9 kcal/mol. Experimentally a higher
C2/C3 ratio was observed for tBu-TFDO than for TFDO. The steric effect of the tert-butyl group
raises the activation barriers for tBu-TFDO oxidations compared with TFDO by about 2
kcal/mol. This energy difference is also consistent with the low yield and low levels of reactivity
observed with tBu-TFDO compared with TFDO. The trace conversion affected by tBu-TFDO
may also reflect the low concentration of this reagent in the reaction mixture, as it is formed in situ.
The increased steric effects also render the equatorial hydrogen on C3 as susceptible to oxidation
as the axial hydrogen at the same position.
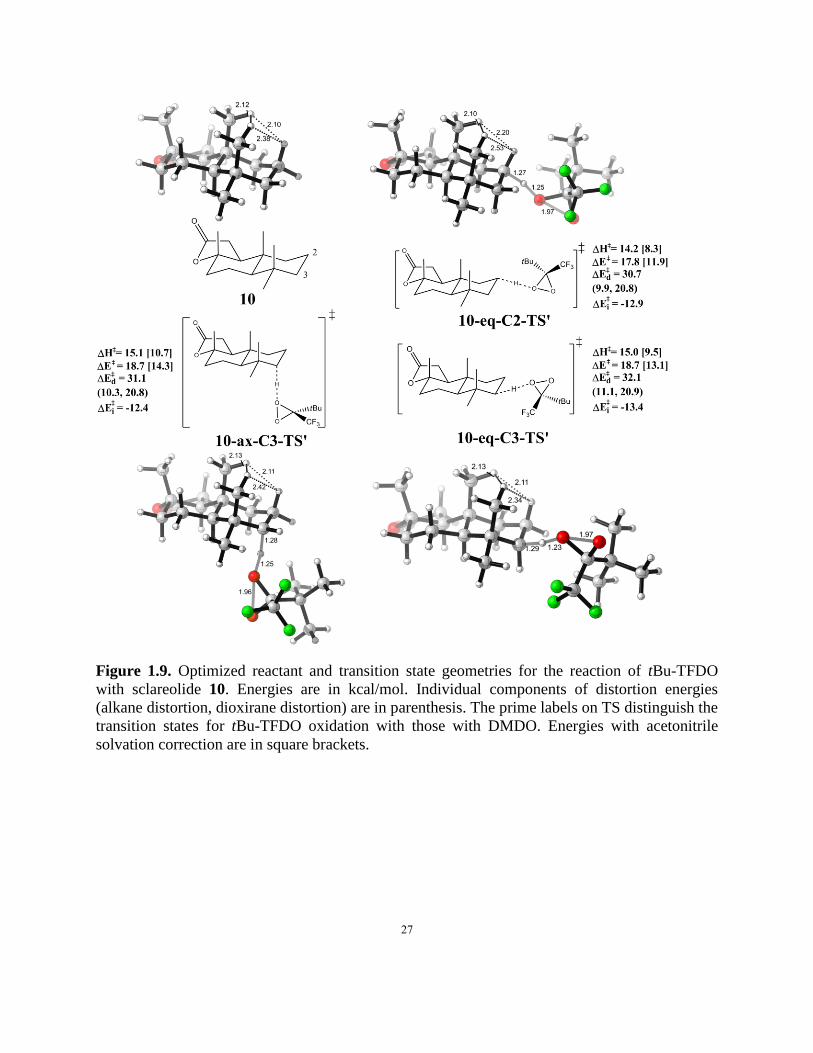
27
Figure 1.9. Optimized reactant and transition state geometries for the reaction of tBu-TFDO
with sclareolide 10. Energies are in kcal/mol. Individual components of distortion energies
(alkane distortion, dioxirane distortion) are in parenthesis. The prime labels on TS distinguish the
transition states for tBu-TFDO oxidation with those with DMDO. Energies with acetonitrile
solvation correction are in square brackets.

28
1.5 Conclusions
The site selectivities and stereoselectivities of sp3 C-H activations in cyclohexane
derivatives by dioxiranes were investigated computationally. CASPT2 calculations established
the appropriate transition state for C-H oxidations by dioxiranes, which corresponds to that
located using unrestricted DFT. The full pathway for dioxirane oxidation was established. The
distortion/interaction model was used to understand the origins of selectivity in each case. In the
absence of axial substitution of cyclohexanes there is no innate preference for either axial or
equatorial C-H activation: eclipsing interactions slightly favor axial activation, but this is
counterbalanced by the greater hyperconjugative stabilization in equatorial activation. The axial
and equatorial C-H bonds of cyclohexane have similar reactivities.
However, with increasing axial substitution, computed activation barriers decrease, in
accord with experiment. The enhancement in equatorial reactivity by geminal and diaxial
substitution arises as a consequence of strain release in going to the transitions state.25
The
distortion/interaction model allows for a quantification of the acceleration that arises as a result
of strain release. The strain release effect is around 0.7 kcal/mol when the strain involving two
1,3-diaxial methyl groups is released. The eudesmane 8 is more reactive than 9 due to the strain
release effect, but the C2/C3 site selectivity for sclareolide is dominated by steric effects and can
be controlled by the bulkiness of the reagents.

29
1.6 Experimental Section
1.6.1 General Procedures
All reactions were carried out under an inert nitrogen atmosphere with dry solvents under
anhydrous conditions unless otherwise stated. Dry acetonitrile (MeCN) was obtained by passing
the previously degassed solvents through activated alumina columns. Product ratios were
determined by analysis of the crude 1H NMR spectra. Yields refer to chromatographically and
spectroscopically (1H-NMR) homogeneous materials, unless otherwise stated. Reagents were
purchased at the highest commercial quality and used without further purification, unless
otherwise stated. Reactions were monitored by thin-layer chromatography (TLC) carried out on
0.25 mm E. Merck silica gel plates (60F-254 or RP-18 F254s) using UV light as the visualizing
agent and an acidic solution of p-anisaldehyde, phosphomolybdic acid, cerric ammonium
molybdate, Seebach’s stain (PMA and CAM), ninhydrin, or potassium permanganate and heat as
developing agents. E. Merck silica gel (60, particle size 0.043-0.063 mm) was used for flash
column chromatography.
1.6.2 General Procedure for Oxidation using TFDO Generated in situ.
The substrate (0.2 mmol) was dissolved in MeCN (6.0 mL, 0.03M) by stirring and
sonicating. To this solution was added aqueous sodium phosphate buffer (6.0 mL, pH = 7.5, 25
mM) and aqueous Na2EDTA (1.3 mL, 40 mM). The solution was then cooled to 4 C (ambient
temperature) and cool (4 C) 1,1,1-trifluoroacetone (0.143 mL, 1.60 mmol, 8.0 equiv) was
added. Oxone
(KHSO5 0.5KHSO4 0.5K2SO4, 738 mg, 2.4 mmol, 12 equiv) was then added

30
in a single portion and the suspension was rapidly stirred for 36 hours. Then, the mixture was
warmed to room temperature and extracted with Et2O (3 15 mL). The combined organic layers
were washed with brine (50 mL), dried over MgSO4, filtered and concentrated.
Similar conditions for in situ dioxirane formation for C-H oxidation have been
developed.51
References 21, 52, and 53 include spectral data for the sclareolide oxidation
products. Reference 54 includes data for the products from oxidation of
1,3-dimethylcyclohexanes. tert-butyl trifluoromethylketone could be synthesized according to
ref. 55. Alternatively, CF3TMS could be added dropwise to a mixture of methyl pivalate and
excess CsF at 0 °C.
1.7 References
(1) Yamaguchi, J.; Yamaguchi, A. D.; Itami, K. Angew. Chem. Int. Ed. 2012, 51, 8960.
(2) Doyle, M. P.; Goldberg, K. I. Acc. Chem. Res. 2012, 45, 777.
(3) Crabtree, R. H. Chem. Rev. 2010, 110, 575.
(4) Brueckl, T.; Baxter, R. D.; Ishihara, Y.; Baran, P. S. Acc. Chem. Res. 2012, 45, 826.
(5) For recent reviews, see: a) Davies, H. M. L.; Beckwith, R. E. J.; Chem. Rev. 2003, 103, 2861;
b) Punniyamurthy, T.; Velusamy, S.;Iqbal, J. Chem. Rev. 2005 105, 2329; c) Godula, K.; Sames,
D. Science 2006, 312, 67; d) Dick, A. R.; Sanford, M. S. Tetrahedron 2006, 62, 2439; e) Davies,
H. M. L.; Manning, J. R. Nature 2008, 451, 417 f) Giri, R.; Shi, B.-F.; Engle, K. M.; Maugel, N.;
Yu, J.-Q. Chem. Soc. Rev. 2009, 38, 3242; g) Daugulis, O.; Do, H.-Q.; Shabashov, D. Acc. Chem.
Res. 2009, 42, 1074. h) Mkhalid, I.A.I.; Barnard, J.H.; Marder, T. B.; Murphy, J. M.; Hartwig, J.
F. Chem. Rev. 2010, 110, 890; i) Shul’pin, G. B. Org. Biomol. Chem. 2010, 8, 4217; j) Jazzar, R;
Hitce, J.; Renaudat, A.; Sofack-Kreutzer, J.; Baudoin, O. Chem. Eur. J. 2010, 16, 2654; k) Lyons,
T. W.; Sanford, M. S. Chem. Rev. 2010, 110, 1147; l) “C-H Activation”: Topics in Current
Chemistry (Eds.: J.-Q. Yu, Z. Shi), Springer, Berlin, 2010, 292. m) Doyle, M. P.; Duffy, R.;

31
Ratnikov, M.; Zhou, L. Chem. Rev. 2010, 110, 704; n) Roizen, J. L.; Harvey, M. E.; Du Bois, J.
Acc. Chem. Res. 2012, 45, 911.
(6) Murray, R. W.; Jeyaraman, R. J. Org. Chem. 1985, 50, 2847.
(7) Mello, R.; Fiorentino, M.; Sciacovelli, O.; Curci, R. J. Org. Chem. 1988, 53, 3890.
(8) Curci, R.; D'Accolti, L.; Fusco, C. Acc. Chem. Res. 2006, 39, 1.
(9) Curci, R.; Dinoi, A.; Rubino, M. F. Pure Appl. Chem. 1995, 67, 811.
(10) Bravo, A.; Bjorsvik, H. R.; Fontana, F.; Minisci, F.; Serri, A. J. Org. Chem. 1996, 61, 9409.
(11) Curci, R.; Dinoi, A.; Fusco, C.; Lillo, M. A. Tetrahedron Lett. 1996, 37, 249.
(12) Bravo, A.; Fontana, F.; Fronza, G.; Minisci, F.; Zhao, L. H. J. Org. Chem. 1998, 63, 254.
(13) Romney, D. K.; Miller, S. J. Org. Lett. 2012, 14, 1138.
(14) Newhouse, T.; Baran, P. S. Angew. Chem. Int. Ed. 2011, 50, 3362.
(15) Barton, D. H. R. J. Chem. Soc. 1953, 1027.
(16) Barton, D. H. R.; Beviere, S. D.; Chavasiri, W.; Csuhai, E.; Doller, D.; Liu, W. G. J. Am.
Chem. Soc. 1992, 114, 2147.
(17) Schreiber, J.; Eschenmoser, A. Helv. Chim. Acta. 1955, 38, 1529.
(18) D'Accolti, L.; Fiorentino, M.; Fusco, C.; Rosa, A. M.; Curci, R. Tetrahedron Lett. 1999, 40,
8023.
(19) Mello, R.; Fiorentino, M.; Fusco, C.; Curci, R. J. Am. Chem. Soc. 1989, 111, 6749.
(20) a) Gonzalez-Nunez, M. E.; Castellano, G.; Andreu, C.; Royo, J.; Baguena, M.; Mello, R.;
Asensio, G. J. Am. Chem. Soc. 2001, 123, 7487; b) Gonzalez-Nunez, M. E.; Royo, J.; Mello, R.;
Baguena, M.; Ferrer, J. M.; de Arellano, C. R.; Asensio, G.; Prakash, G. K. S. J. Org. Chem.

32
2005, 70, 7919.
(21) Chen, M. S.; White, M. C. Science 2010, 327, 566.
(22) Chen, M. S.; White, M. C. Science 2007, 318, 783.
(23) Ishihara, Y.; Baran, P. S. Synlett 2010, 1733.
(24) Chen, K.; Baran, P. S. Nature 2009, 459, 824.
(25) Chen, K.; Eschenmoser, A.; Baran, P. S. Angew. Chem. Int. Ed. 2009, 48, 9705.
(26) Du, X. H.; Houk, K. N. J. Org. Chem. 1998, 63, 6480.
(27) Glukhovtsev, M. N.; Canepa, C.; Bach, R. D. J. Am. Chem. Soc. 1998, 120, 10528.
(28) Paton, R. S.; Kim, S.; Ross, A. G.; Danishefsky, S. J.; Houk, K. N. Angew. Chem. Int. Ed.
2011, 50, 10366.
(29) Lan, Y.; Houk, K. N. J. Am. Chem. Soc. 2010, 132, 17921.
(30) Ess, D. H.; Houk, K. N. J. Am. Chem. Soc. 2008, 130, 10187.
(31) Ess, D. H.; Houk, K. N. J. Am. Chem. Soc. 2007, 129, 10646.
(32) Gaussian 09, Revision B.01, Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.;
Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.;
Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H. P.; Izmaylov, A. F.; Bloino, J.; Zheng, G.;
Sonnenberg, J. L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.;
Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, J. A., Jr.; Peralta, J. E.;
Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin, K. N.; Staroverov, V. N.; Kobayashi,
R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi,
M.; Rega, N.; Millam, N. J.; Klene, M.; Knox, J. E.; Cross, J. B.; Bakken, V.; Adamo, C.;
Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.;
Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Zakrzewski, V. G.; Voth, G. A.; Salvador, P.;
Dannenberg, J. J.; Dapprich, S.; Daniels, A. D.; Farkas, Ö.; Foresman, J. B.; Ortiz, J. V.;

33
Cioslowski, J.; Fox, D. J. Gaussian, Inc., Wallingford CT, 2009.
(33) Aquilante, F.; De Vico, L.; Ferre, N.; Ghigo, G.; Malmqvist, P. A.; Neogrady, P.; Pedersen, T.
B.; Pitonak, M.; Reiher, M.; Roos, B. O.; Serrano-Andres, L.; Urban, M.; Veryazov, V.; Lindh, R.
J. Comput. Chem. 2010, 31, 224.
(34) Harding, L. B.; Goddard, W. A. J. Am. Chem. Soc. 1978, 100, 7180.
(35) Wadt, W. R.; Goddard, W. A. J. Am. Chem. Soc. 1975, 97, 3004.
(36) Bach, R. D.; Andres, J. L.; Owensby, A. L.; Schlegel, H. B.; Mcdouall, J. J. W. J. Am. Chem.
Soc. 1992, 114, 7207.
(37) Shustov, G. V.; Rauk, A. J. Org. Chem. 1998, 63, 5413.
(38) Freccero, M.; Gandolfi, R.; Sarzi-Amade, M.; Rastelli, A. Tetrahedron Lett. 2001, 42, 2739.
(39) Freccero, M.; Gandolfi, R.; Sarzi-Amade, M.; Rastelli, A. J. Org. Chem. 2003, 68, 811.
(40) Fokin, A. A.; Tkachenko, B. A.; Korshunov, O. I.; Gunchenko, P. A.; Schreiner, P. R. J. Am.
Chem. Soc. 2001, 123, 11248.
(41) Cremer, D.; Kraka, E.; Szalay, P. G. Chem. Phys. Lett. 1998, 292, 97.
(42) Ensing, B.; Buda, F.; Gribnau, M. C. M.; Baerends, E. J. J. Am. Chem. Soc. 2004, 126, 4355.
(43) Kumar, D.; de Visser, S. P.; Shaik, S. J. Am. Chem. Soc. 2003, 125, 13024.
(44) Wu, Y. D.; Houk, K. N. J. Am. Chem. Soc. 1987, 109, 908.
(45) Wu, Y. D.; Houk, K. N.; Trost, B. M. J. Am. Chem. Soc. 1987, 109, 5560.
(46) Damm, W.; Giese, B.; Hartung, J.; Hasskerl, T.; Houk, K. N.; Zipse, H. J. Am. Chem. Soc.
1992, 114, 4067.
(47) Luo, Y. R. CRC Handbook of Chemistry and Physics, 91st ed. (CRC Press, Boca Raton, FL,
2010)
(48) Gordon, C. G.; Mackey, J. L.; Jewett, J. C.; Sletten, E. M.; Houk, K. N.; Bertozzi, C. R. J.

34
Am. Chem. Soc. 2012, 134, 9199.
(49) Newhouse, T., PhD thesis with Baran, P. S.
(50) (a) Liu, W.; Groves, J. T. J. Am. Chem. Soc. 2010, 132, 12847; (b) Liu, W.; Huang, X.;
Cheng, M.-J.; Nielsen, R.J.; Goddard, W.A., III; Groves, J.T.; Science 2012, 337, 1322.
(51) Annese, C.; D’Accolti, L.; Fusco, C.; Curci, R. Org. Lett. 2011, 13, 2142.
(52) Cambie, R. C.; Joblin, K. N.; McCallum, N. K. Aust. J. Chem. 1970, 23, 1439.
(53) Choudhary, M. I.; Musharraf, S. G.; Sami, A.; Rahman, A.-u. Helv. Chim. Acta. 2004, 87,
2685.
(54) Senda, Y.; Ishiyama, J.; Imaizumi, S. Tetrahedron 1975, 31, 1601.
(55) Wiedemann, J.; Heiner, T.; Mloston, G.; Prakash, G. K. S.; Olah, G. A. Angew. Chem. Int.
Ed. 1998, 37, 820.

35
Chapter 2. Formation of Chelated Z-selective Ruthenium Metathesis Catalysts:
Computational Study on C–H Activation Mechanism, Reactivity, and Selectivity
2.1 Abstract
The mechanism and origins of formation of chelated Z-selective Ru catalysts were explored
using density functional theory. The key step involves an intramolecular C-H bond activation of
the NHC ligand by carboxylates introduced by anion exchange. The C-H activation follows a
concerted metalation-deprotonation mechanism, where carboxylate abstracts the hydrogen from
the “bottom” position, forming a six-membered ring with the metal. Torsional strains from
closed−shell repulsions between the chelating groups dominate over electronic effects for
determining the barriers for the C-H activation process.
2.2 Introduction
Olefin metathesis is a powerful methodology for the construction of new carbon-carbon
double bonds. Since the discovery of metathesis, nearly 400 ruthenium olefin metathesis
catalysts were reported by the end of 2009.1 Along with improved reactivity and selectivity, the
thermal stability, oxygen- and moisture-tolerance of the catalyst have been greatly improved
over the years. New ligands, especially the N-heterocyclic carbenes and benzylidene ether ligand
had special value.

36
For most olefin metathesis catalysts, E olefins were preferentially obtained due to their
greater thermodynamical stability. A highly Z-selective olefin metathesis catalyst was reported in
2011,2 followed by continuous improvements.
3-5 Z-selective catalysts have already been applied
in several synthetic routes.6 The Z-selective breakthrough was achieved by an unexpected C-H
activation of the previously known E-selective catalyst, 1.2,3
(Scheme 2.1) Intermediate 2 was
obtained by replacing the chloride ligands of 1 with pivalate, and 2 unexpectedly underwent
rapid C-H activation to give a C-chelated, Z-selective catalyst, 3. The Ru-carbon bond to the
mesityl group was formed via intramolecular C-H activation.
Scheme 2.1. Generation of the Z-selective catalyst 3 through C-H activation
An even more efficient Z-selective catalyst, 6, was obtained following the same
procedure, without the detection of anion-exchange intermediates.2-4
The Ru-adamantyl bond
was originally formed by ligand exchange promoted by silver pivalate, but the reaction also
occurs even with sodium pivalate (Scheme 2.2).5

37
Scheme 2.2. Generation of the Z-selective catalyst 6 through C-H activation
Computational studies on ruthenium C-H activation have been reported, but mostly on
(aromatic) sp2 C-H bonds. Cavallo studied computationally the C-H activation of a benzene sp
2
C-H bond as part of a study of the decomposition pathway of the second generation Grubbs
catalyst.7 Berlinguette studied C-H activation on the benzene ligand of bis-tridentate ruthenium
complexes.8 Ruthenium (II) catalyzed sp
2 C-H activation was reviewed by Dixneuf.
9 Few cases
of ruthenium catalyzed intramolecular sp3 C-H activation with carboxylic acid ligands have been
reported.10
Murai,11
Sames,12
and Maes employed the Ru3(CO)12 catalyst for the activation of sp3
C-H bonds adjacent to a nitrogen atom.13,14
Computational studies of both sp2 and sp
3 C-H
activation by various transition metals were recently reviewed by Eisenstein,15
and various
mechanisms in carboxylate-assisted transition-metal-catalyzed C-H bond functionalization is
reviewed by Ackermann.16
We have explored the intramolecular C-H activation that forms Z-selective olefin
metathesis catalyst, using density functional theory. We have found that the mechanism follows
a concerted metallation-deprotonation (CMD) process, which is similar to the palladium
catalyzed catalyzed intramolecular sp3 C-H activation with carboxylic acids studied earlier by

38
Fagnou’s group,17,18
and our group.19
We have investigated transition state (TS) geometries,
explored the origins of selectivities with different types of C-H bonds, and have investigated the
influence of both anionic and NHC ligands on the rates of intramolecular C-H activations.
2.3 Computational Details
All calculations were performed with Gaussian 09.20
Geometry optimizations were
performed with B3LYP and the LANL2D2 basis set for Ru and the 6-31G(d) basis set for other
atoms. Single point energies were calculated at the M06/SDD–6-311+G(d,p) level. Solvation
corrections for single point energies were calculated with the SMD solvation model using THF
as the solvent. The reported free energies and enthalpies include zero-point energies and thermal
corrections calculated at 298 K with B3LYP/SDD-6-31G(d).
2.4 Results and Discussions
2.4.1 Transition state geometries for the C-H activation
C-H activations on the metal centers were proposed to follow several classes of
mechanism, such as oxidative addition, sigma-bond metathesis, and electrophilic activation.21
Among these mechanism, carboxylate-assisted C-H bond functionalizations were mostly
proposed to follow the mechanism of base-assisted deprotonation,16
which was named as
concerted metallation-deprotonation (CMD) by Fagnou.17

39
Depending on the side of attack and the coordination atom of the carboxylate, there are
four different ways the dipivalate intermediate 5 can undergo C-H activation. The four transition
state geometries are shown in Figure 2.1. The carboxylic acid attack from the bottom (TS-A and
TS-C), breaking the Ru-O(benzylidene) bond, or the acid attack from the side (TS-B and TS-D).
In TS-A and TS-B, one of the oxygen of the pivalate carboxylic is coordinated to Ru, while the
other oxygen deprotonates the adamantyl C-H bond, thus forming a six-membered transition
state. In TS-C and TS-D, only one oxygen is involved, forming a four-membered transition state.
The most favored transition state, TS-A, has the forming and breaking bonds in a six-membered
ring with the pivalate oxygen deprotonating from the bottom.
Figure 2.1. Four possible transition state geometries for C-H activation of 4. The Ru-O bond
lengths and TS bond lengths are labeled in Å.

40
2.4.2 The free energy profile for the formation of the chelated ruthenium catalyst
Experimentally, the dicarboxylate intermediate 2 was observed as an intermediate in the
formation of catalyst 3 from 1, but intermediate 5 was not characterized in the formation of
catalyst 6 from 4. No chloride-containing C-H activated catalyst has been observed. We explored
whether a monochloro monopivalate might also give C-H activation. The computed free energy
diagram for C-H activation of 4 (1a) by a dipivalate or a monochlorate, mono pivalate is shown
in Figure 2.2.
Figure 2.2. The free energy profile of the C-H activation of 4(1a) to form the C-H activated
catalyst (1g).
The lower energy pathway (colored in blue) involves an anion exchange step, forming a
stable complex 1c with both Cls replaced with pivalates, which coordinates better with
ruthenium. Isomerization of 1c to a less stable complex 1d gives the geometry of CMD (see the

41
previous section). The rate- and stereo-determining C-H activation step (1e-ts) requires an
overall barrier of 23.5 kcal/mol (1d -> 1e-ts). The complex 1f is formed after the C-H activation
and finally the active catalyst 1g is obtained.
An alternative pathway of C-H activation with only one carboxylate and one chloride ligand
was also studied (colored in pink in Figure 2.2).The free energy barrier (1h-ts) is 28.4 kcal/mol,
around 5 kcal/mol less favored than the dicarboxylate pathway, as the other pivalate is bidentate,
which might give additional stabilization in the transition state.
2.4.3 Regio- and stereoselectivity in CH activations
There are three possible hydrogens which could be subject to replacement by Ru in C-H
activation of the admantyl/mesityl NHC. These are shown in Scheme 2.3 with their
corresponding transition states are shown in Figure 2.3.
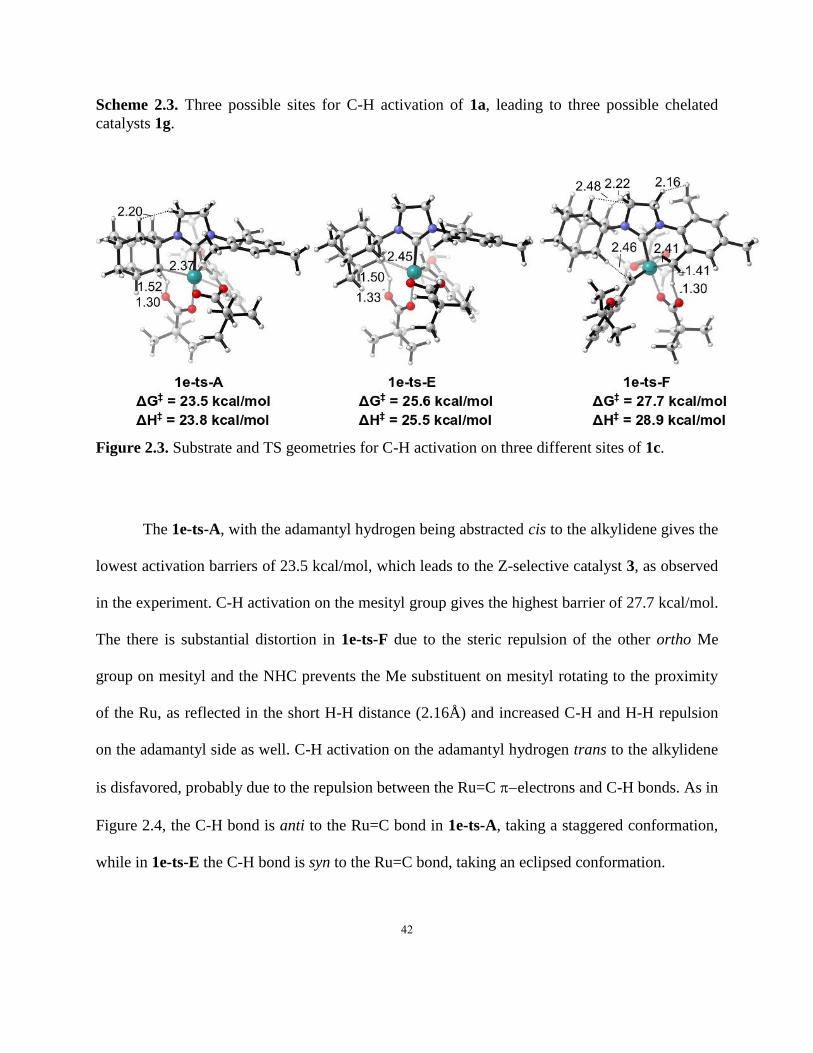
42
Scheme 2.3. Three possible sites for C-H activation of 1a, leading to three possible chelated
catalysts 1g.
Figure 2.3. Substrate and TS geometries for C-H activation on three different sites of 1c.
The 1e-ts-A, with the adamantyl hydrogen being abstracted cis to the alkylidene gives the
lowest activation barriers of 23.5 kcal/mol, which leads to the Z-selective catalyst 3, as observed
in the experiment. C-H activation on the mesityl group gives the highest barrier of 27.7 kcal/mol.
The there is substantial distortion in 1e-ts-F due to the steric repulsion of the other ortho Me
group on mesityl and the NHC prevents the Me substituent on mesityl rotating to the proximity
of the Ru, as reflected in the short H-H distance (2.16Å) and increased C-H and H-H repulsion
on the adamantyl side as well. C-H activation on the adamantyl hydrogen trans to the alkylidene
is disfavored, probably due to the repulsion between the Ru=C electrons and C-H bonds. As in
Figure 2.4, the C-H bond is anti to the Ru=C bond in 1e-ts-A, taking a staggered conformation,
while in 1e-ts-E the C-H bond is syn to the Ru=C bond, taking an eclipsed conformation.

43
Figure 2.4. Steric effects on stereoselectivity for the cis- and trans-TS for C-H activation of 1c.
Atoms not directly not directed linked to the reactive center are omitted for clarity.
2.4.4 Effects of chelating group and N-substituents on reactivity
Table 2.1. Activation energies for C-H activation of various N-alkyl groups
Entry Alkyl-H G‡
H‡ BDE
1
23.5 23.8 98.0

44
2
20.0 20.5 96.4
3
16.2 16.8 98.1
4
15.2 16.4 100.7
5
25.6 26.0 87.4
The reactivity toward C-H activation of several different alkyl groups was studied and is
summarized in Table 2.1. The activation is facilitated with less steric demanding groups. The
activation of the secondary hydrogen on the cyclohexane ring (TS2) requires 20.0 kcal/mol in
free energy, 3.5 kcal/mol lower than TS1. The activation barrier for the primary hydrogen on the
methyl, TS3 and TS4, are significantly lower, which are only 16.2 and 15.2 kcal/mol,
respectively. Replacing the adamantyl group with mesityl increases the activation barrier to 25.6
kcal/mol, probably due to the closed shell repulsion between the methyl group on mesityl and the
NHC rings, as in the case of C-H activation on the mesityl C-H (1e-ts-F).
Table 2.2. Activation energies for C-H activation with different N-aryl groups

45
Entry Aryl group G‡
H‡
1
23.5 23.8
7
23.5 23.1
8
21.3 22.4
9
22.0 23.1
Substituent effects on the N-aryl groups were investigated, and the results are summarized in
Table 2.2. C-H activation of the new catalyst 7 has the same activation free energy as 1.
Enhanced activity towards C-H activation was observed when one or two of the ortho methyl
groups were replaced with fluorine. As in Figure 2.5, there is stabilizing effect of O-H interaction
in 1c, and destabilizing effect from C-H repulsion in 1e-ts-A, both lead to higher barriers. These
effects are absent when the ortho methyl groups are replaced with fluorines.
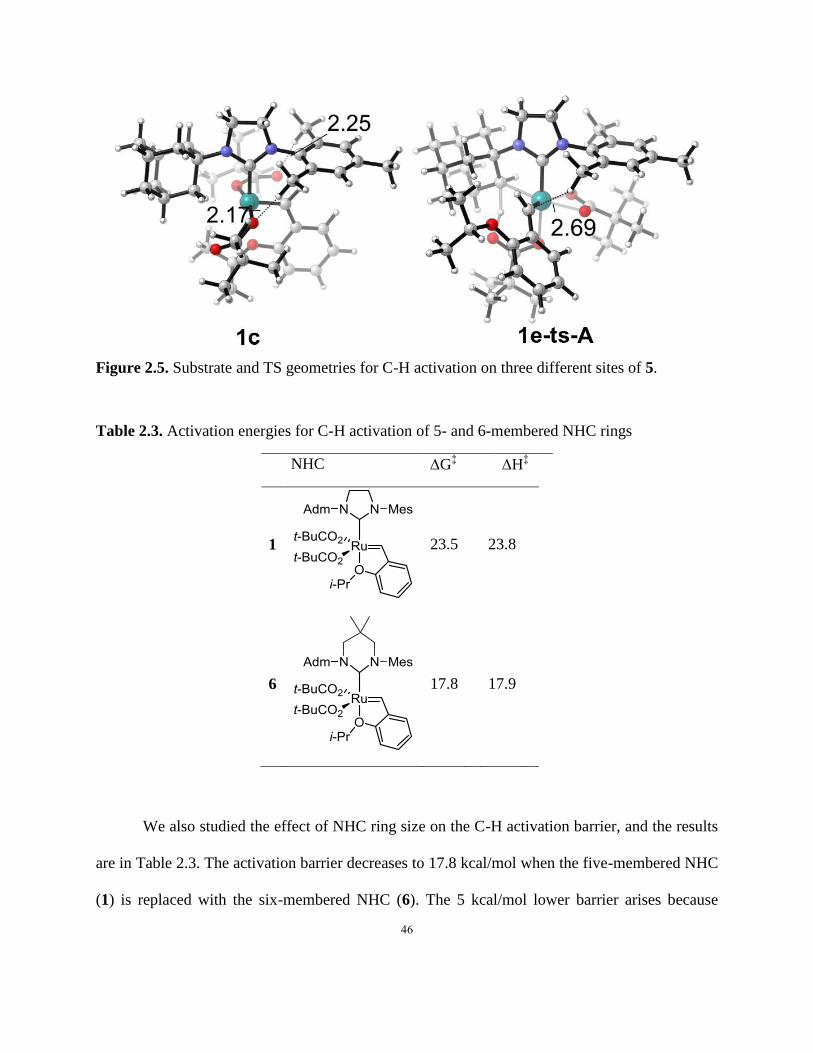
46
Figure 2.5. Substrate and TS geometries for C-H activation on three different sites of 5.
Table 2.3. Activation energies for C-H activation of 5- and 6-membered NHC rings
NHC G‡
H‡
1
23.5 23.8
6
17.8 17.9
We also studied the effect of NHC ring size on the C-H activation barrier, and the results
are in Table 2.3. The activation barrier decreases to 17.8 kcal/mol when the five-membered NHC
(1) is replaced with the six-membered NHC (6). The 5 kcal/mol lower barrier arises because

47
additional carbon in the ring brings the N-adamantyl group closer to the pivalate group, which
facilitates the CH activation. The C-H activation process with 6 is also more exergonic than with
the five-membered complex 1 (reaction energies are -12.6 and -3.0 kcal/mol for 6 and 1,
respectively).
Table 2.4. Activation energies for C-H activation with different carboxylate ligands
Entry Anion RCOO- G
‡ H
‡ pKaH
1 tBuCOO- 23.5 23.8 5.01
10 CH3COO- 24.0 24.4 4.75
11 PhCOO- 26.2 25.5 4.2
The anion effect on the barriers of C-H activation is shown in Table 2.4. Replacing the
pivalate with acetate increases the barrier by 0.5 kcal/mol, while with the benzoate the barrier is
2.7 kcal/mol higher. Notice the strength of the conjugate acid follows the order: benzoic acid
(pKa 4.2) > acetic acid (pKa 4.75) > pivalic acid (pKa 5.01). Since an acid is formed as the
byproduct, formation of a strong acid is less favored, leading to a higher barrier.

48
2.5 Conclusions
The mechanism of C-H activation of the Grubbs II catalyst upon converting halide to
carboxylate ligands has been elucidated. The C-H activation proceeds via a six-membered,
bottom-attack transition state with a high stereoselectivity. Reducing the steric demands of the
N-alkyl group leads to lower activation barriers. The steric effect on the ortho position of N-aryl
group has minor impact on the C-H activation barrier, while substitution of methyl with fluorine
decreases the barrier. Replacing the five membered NHC with six membered NHC also
facilitates the CH activation.
.
2.6 References
(1) Vougioukalakis, G. C.; Grubbs, R. H. Chem. Rev. 2010, 110, 1746.
(2) Endo, K.; Grubbs, R. H. J. Am. Chem. Soc. 2011, 133, 8525.
(3) Keitz, B. K.; Endo, K.; Herbert, M. B.; Grubbs, R. H. J. Am. Chem. Soc. 2011, 133, 9686.
(4) Keitz, B. K.; Endo, K.; Patel, P. R.; Herbert, M. B.; Grubbs, R. H. J. Am. Chem. Soc. 2012,
134, 693.
(5) Rosebrugh, L. E.; Herbert, M. B.; Marx, V. M.; Keitz, B. K.; Grubbs, R. H. J. Am. Chem.
Soc. 2013, 135, 1276.
(6) Herbert, M. B.; Marx, V. M.; Pederson, R. L.; Grubbs, R. H. Angew. Chem. Int. Ed. 2013,
52, 310.
(7) Poater, A.; Cavallo, L. J. Mol. Cat. A. – Chem. 2010, 324, 75.
(8) Muise, S. S. R.; Severin, H. A.; Koivisto, B. D.; Robson, K. C. D.; Schott, E.; Berlinguette,
C. P. Organometallics 2011, 30, 6628.

49
(9) Arockiam, P. B.; Bruneau, C.; Dixneuf, P. H. Chem. Rev. 2012, 112, 5879.
(10) Rousseau, G.; Breit, B. Angew. Chem. Int. Ed. 2011, 50, 2450.
(11) Chatani, N.; Asaumi, T.; Yorimitsu, S.; Ikeda, T.; Kakiuchi, F.; Murai, S. J. Am. Chem. Soc.
2001, 123, 10935.
(12) Pastine, S. J.; Gribkov, D. V.; Sames, D. J. Am. Chem. Soc. 2006, 128, 14220.
(13) Bergman, S. D.; Storr, T. E.; Prokopcova, H.; Aelvoet, K.; Diels, G.; Meerpoel, L.; Maes, B.
U. W. Chem.-Eur. J. 2012, 18, 10393.
(14) Peschiulli, A.; Smout, V.; Storr, T. E.; Mitchell, E. A.; Elias, Z.; Herrebout, W.; Berthelot,
D.; Meerpoel, L.; Maes, B. U. W. Chem.-Eur. J. 2013, 19, 10378.
(15) Balcells, D.; Clot, E.; Eisenstein, O. Chem. Rev. 2010, 110, 749.
(16) Ackermann, L. Chem. Rev. 2011, 111, 1315.
(17) Gorelsky, S. I.; Lapointe, D.; Fagnou, K. J. Am. Chem. Soc. 2008, 130, 10848.
(18) Gorelsky, S. I.; Lapointe, D.; Fagnou, K. J. Org. Chem. 2012, 77, 658.
(19) Giri, R.; Lan, Y.; Liu, P.; Houk, K. N.; Yu, J. Q. J. Am. Chem. Soc. 2012, 134, 14118.
(20) Gaussian 09, Revision B.01, Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.;
Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.;
Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H. P.; Izmaylov, A. F.; Bloino, J.; Zheng, G.;
Sonnenberg, J. L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.;
Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, J. A., Jr.; Peralta, J. E.;
Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin, K. N.; Staroverov, V. N.; Kobayashi,
R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi,
M.; Rega, N.; Millam, N. J.; Klene, M.; Knox, J. E.; Cross, J. B.; Bakken, V.; Adamo, C.;
Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.;
Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Zakrzewski, V. G.; Voth, G. A.; Salvador, P.;
Dannenberg, J. J.; Dapprich, S.; Daniels, A. D.; Farkas, Ö.; Foresman, J. B.; Ortiz, J. V.;
Cioslowski, J.; Fox, D. J. Gaussian, Inc., Wallingford CT, 2009.
(21) Labinger, J. A.; Bercaw, J. E. Nature 2002, 417, 507.

50
Chapter 3. Mechanism and Enantioselectivity in Palladium-Catalyzed Conjugate Addition
of Arylboronic Acids to -Substituted Cyclic Enones: Insights from Computation and
Experiment
3.1 Abstract
Enantioselective conjugate additions of arylboronic acids to -substituted cyclic enones have
been reported previously from our laboratories. Air and moisture tolerant conditions were
achieved with a catalyst derived in situ from palladium(II) trifluoroacetate and the chiral ligand
(S)-t-BuPyOx. We now report a combined experimental and computational investigation on the
mechanism, the nature of the active catalyst, the origins of the enantioselectivity, and the
stereoelectronic effects of the ligand and the substrates of this transformation. Enantioselectivity
is controlled primarily by steric repulsions between the t-Bu group of the chiral ligand and the
-methylene hydrogens of the enone substrate in the enantiodetermining carbopalladation step.
Computations indicate that the reaction occurs via formation of a cationic arylpalladium(II)
species, and subsequent carbopalladation of the enone olefin forms the key carbon-carbon bond.
Studies of non-linear effects and stoichiometric and catalytic reactions of isolated (PyOx)Pd(Ph)I
complexes show that a monomeric arylpalladium-ligand complex is the active species in the
selectivity-determining step. The addition of water and ammonium hexafluorophosphate
synergistically increases the rate of the reaction, corroborating the hypothesis that a cationic

51
palladium species is involved in the reaction pathway. These additives also allow the reaction to
be performed at 40 °C and facilitate an expanded substrate scope.
3.2 Introduction
Asymmetric conjugate addition has become a familiar reaction manifold in the synthetic
chemists’ repertoire.1
Though seminal reports involved highly reactive organometallic
nucleophiles,2
systems were rapidly developed that involved functional-group-tolerant
organoboron nucleophiles. Namely, Hayashi pioneered the use of rhodium/BINAP catalysts for
the asymmetric conjugate addition of a number of boron-derived nucleophiles.3 As an
economical alternative to the rhodium systems, Miyaura pioneered the use of chiral
palladium-phosphine catalysts to address similar transformations4 and Minnaard reported a
palladium-catalyzed asymmetric conjugate addition using a catalyst formed in situ from
palladium trifluoroacetate and commercially available (S,S)-MeDuPhos.5
More recently, asymmetric conjugate addition has become a useful strategy for the challenge
of constructing asymmetric quaternary stereocenters.6 Again, many earlier developed methods
involved highly reactive diorganozinc,7
triorganoaluminum,8
and organomagnesium9
nucleophiles, however, more recently chiral rhodium/diene systems have been shown to
construct asymmetric quaternary stereocenters with functional group-tolerant organoboron
nucleophiles.10
While rhodium systems are highly developed and exhibit a wide substrate scope,
the high cost of the catalyst precursors and oxygen sensitivity of the reactions are undesirable.
Despite progress in palladium-catalyzed conjugate additions for the formation of tertiary

52
stereocenters,11
no conditions were amenable to the synthesis of even racemic quaternary
centers until Lu and coworkers disclosed a dicationic, dimeric palladium/bipyridine catalyst in
2010.12
However, it was not until our recent report that a palladium-derived catalyst was
employed to generate an asymmetric quaternary stereocenter via conjugate addition chemistry.13
Scheme 3.1. Asymmetric conjugate addition with (S)-t-BuPyOx ligand.
We employed a catalyst derived in situ from Pd(OCOCF3)2 and a chiral pyridinooxazoline
(PyOx) ligand,14
(S)-t-BuPyOx (ligand 1, Scheme 3.1). This catalyst facilitates the asymmetric
conjugate addition of arylboronic acids to -substituted enones in high yield and good
enantioselectivity. Importantly, this reaction is highly tolerant of air and moisture, and the chiral
ligand, while not yet commercially available, is easily prepared.15
Initial results with the
Pd/PyOx system were reported rapidly due to concerns over competition in the field. Indeed,
recent publications prove palladium-catalyzed conjugate addition to be a burgeoning field of
research.16
After the initial disclosure, we observed that in addition to catalyzing conjugate
additions to 5-, 6-, and 7-membered enones, the Pd/PyOx catalyst successfully reacted with
chromones and 4-quinolones.17
Intrigued by the broad substrate scope and operational simplicity
of this highly asymmetric process, we conducted a thorough study to optimize the reaction
conditions, including measures to reduce the catalyst loading, lower the reaction temperature,

53
and further generalize the substrate scope. We also performed mechanistic and computational
investigations toward elucidating the catalytic cycle, active catalyst species, and the
stereoelectronic effects on enantioselectivity of this reaction.
3.3 Results
3.3.1 Effects of Water on Catalyst Turnover
In our initial report,13
we were able to demonstrate that the addition of up to 10 equivalents
of water had no deleterious effect. Despite this, water was not considered as an important
additive in the initially reported conditions because the stoichiometric arylboronic acid was
believed to be a sufficient proton source to turn over the catalyst. In considering the overall
reaction scheme, a more precise analysis of the mass balance of the reaction led us to reconsider
the importance of water as a participant in the overall transformation (Scheme 3.2a).
These considerations proved to be essential during the scale up of the reaction. Attempts to
use the original conditions (with no water added) failed to convert enone 2 efficiently, generating
the desired ketone (3) only in moderate yield (Scheme 3.2b). We reasoned that when the reaction
is performed on a small scale under ambient atmosphere the moisture present in the air and on
the glassware could be sufficient to drive the reaction to completion. On a larger scale, however,
where a more significant quantity of water was necessary, this was no longer true. Gratifyingly,
upon the addition of as little as 1.5 equivalents of water to the reaction mixture, both reactivity
and the enantioselectivity were restored (Scheme 3.2c), affording ketone 3 in high yield and ee.

54
Scheme 3.2. (a) Examination of reaction mass balance. (b) Absence of water prohibits scale-up.
(c) Addition of water facilitates larger scale reactions.
We next sought to measure deuterium incorporation at the carbonyl -position as a method
to determine the source of the proton utilized in reaction turnover. Reactions were performed
substituting deuterium oxide for water and observed by 1H and
2H NMR analysis (Figure 3.1).
Using phenylboronic acid, the reaction afforded ketone 3 in similar yield and enantioselectivity
(Figure 3.1a). Likewise, substitution of phenylboroxine ((PhBO)3) for phenylboronic acid and
deuterium oxide for water (Figure 3.1b) resulted in identical yield, albeit with slightly depressed
ee observed in ketone 3. Analysis of ketone 3 by 1H NMR (Figure 3.1c) showed significant
deuterium incorporation at the -position of the carbonyl, even in the presence of phenylboronic
acid.18
As expected, a higher degree of deuterium incorporation was observed in the reaction
where phenyl boroxine was substituted for the boronic acid, however, the similar level of
incorporation in both experiments suggested that the deuterium oxide was the agent assisting
reaction turnover regardless of the use of protic or aprotic boron reagent.
O
P h
O
H
H O B (O H )2P h B (O H )2 H 2 O
OO (S ) - t -B u P y O x (6 m o l % )
P d (O C O C F 3 )2 (5 m o l % )
P h B (O H )2 (2 e q u iv )
C lC H 2 C H 2 C l, 6 0 °C , 1 2 h
5 e q u iv H 2 OP h
9 7 % y ie ld , 9 1 % e e
2 2 m m o l s c a le
OO (S ) - t -B u P y O x (6 m o l % )
P d (O C O C F 3 )2 (5 m o l % )
P h B (O H )2 (2 e q u iv )
C lC H 2 C H 2 C l, 6 0 °C , 1 2 h
N o A d d e d H 2 OP h
In c o m p le te C o n v e rs io n
o n 1 g s c a le (9 m m o l)
2
2
2 3
3
3
(a )
(b )
(c )

55
Figure 3.1. (a) deuterium incorporation using PhB(OH)2. (b) deuterium incorporation using
(PhBO)3. (c) 1H NMR data measuring deuterium incorporation by integral comparison of
-protons relative to H5, control: treatment of ketone 3 to deuterium incorporation conditions.
3.3.2 Effects of Salt Additives on Reaction Rate
Satisfied with our ability to perform the reaction on scale, we turned our attention toward
improving the catalyst activity. We observed that nearly all previous literature reports regarding
palladium-catalyzed conjugate addition utilized cationic precatalysts featuring
weakly-coordinative anions (PF6–, SbF6
–, BF4
– etc.). We reasoned that the substitution of the
trifluoroacetate counterion with a less coordinative species could lead to an increase in reaction
rate. With this goal in mind, we examined a series of salt additives containing weakly
coordinative counterions. We viewed the strategy for the in situ generation of the catalyst as the
more practical and operationally simple alternative to the design, synthesis, and isolation of a
new dicationic palladium precatalyst.
OO
(S ) - t -B u P y O x (6 m o l % )
P d (O C O C F 3 )2 (5 m o l % )
N H 4 P F 6 (3 0 m o l % )
C lC H 2 C H 2 C l, 6 0 °C , 1 2 h
D 2 O
P h
B (O H )2
OO (S ) - t -B u P y O x (6 m o l % )
P d (O C O C F 3 )2 (5 m o l % )
N H 4 P F 6 (3 0 m o l % )
C lC H 2 C H 2 C l, 6 0 °C , 1 2 h
D 2 O
P h
B
OB
O
BO P hP h
P h
(9 9 % y ie ld )
(9 6 % y ie ld )
P h
O
H 1
H 2
H 3
H 4
e n try
b o ro n ic a c id
b o ro x in e
0 .6 7 0 .5 2 0 .8 8 1 .0 0
0 .5 2 0 .4 1 0 .6 6 1 .0 0
H 2 H 3 + H 4 H 5H 1
in te g ra l re la t iv e to H 5
(a )
(b )
(c )
2
2
3
3
D
D
H 5
c o n tro l 0 .9 4 0 .9 0 1 .6 7 1 .0 0

56
We investigated a number of salt additives to test this mechanistic hypothesis (Table 3.1).
Coordinating counterions like chloride (entry 1) shut down reactivity. Pleasingly, as per our
hypothesis, weakly coordinating counterions with sodium cations (entries 2–4) facilitated swift
reaction, albeit with depressed ee. Tetrabutylammonium salts (entries 5–6) encountered slow
reaction times, but good enantioselectivity. Sodium tetraphenylborate (entry 7), however, failed
to deliver appreciable quantities of the quaternary ketone 3, as rapid formation of biphenyl was
observed. Ammonium salts (entires 8–9) provided the desired blend of reaction rate and
enentioselectivity. We concluded that the hexafluorophosphate anion (entry 9) gave the optimal
combination of short reaction time with minimized loss of enantioselectivity.
Table 3.1. Effect of salt additives on reaction rate.a
aConditions: phenylboronic acid (0.5 mmol), 3-methylcyclohexen-2-one (0.25 mmol), water (5
equiv), additive (30 mol %), Pd(OCOCF3)2 (5 mol %) and (S)-t-BuPyOX (6 mol %) in
ClCH2CH2Cl (1 mL) at 40 °C . bGC yield utilizng tridecane standard.
cee was determined by
chiral HPLC. dReaction checked at 83% conversion as determined by GC analysis.
e n trya
t im e (h ) y ie ld (% )b
e e (% )c
1
2
3
2 4 tra c e - - -
2 4 tra c e - - -
6 9 7 8 7
4
5
6
7
2 4 9 8 9 0
5 9 9 8 1
2 4 9 5 8 8
a d d it iv e
n - (B u )4 P F 6
8 8 1d 8 8
N a C l
N a B P h 4
N a P F 6
N a S b F 6
N a B F 4
n - (B u ) 4 B F 4
O O
P h
(S ) - t -B u P y O X , P h B (O H )2
P d (O C O C F 3 )2
a d d it iv e
C lC H 2 C H 2 C l, 4 0 °C
2 3
1 5 9 3 8 98 N H 4 B F 4
1 2 9 6 9 19 N H 4 P F 6

57
Based on our previous observations regarding the beneficial nature of water as an additive,
we next explored the combined effect of water and hexafluorophosphate counterions. We found
addition of both water and ammonium hexafluorophosphate were the most successful for
increasing reactivity (Table 3.2). Water alone is insufficient to alter reactivity (entry 1), though
the use of water with 30 mol % ammonium hexafluorophosphate greatly reduced the reaction
time (entry 2) to only 1.5 hours with minimal effect on yield or ee. Furthermore, this
combination of additives allowed the reaction to proceed at temperatures as low as 25 °C with 5
mol % palladium and 6 mol % ligand, and lowering of catalyst loadings to only 2.5 mol % of
palladium and 3 mol % ligand at 40 °C (entry 3). We determined that optimal conditions for
the reaction with lower catalyst loading to be 5 equivalents of water, 30 mol % ammonium
hexafluorophosphate at 40 °C (entry 4), conditions that reproduce the original result at milder
temperature and lower catalyst loadings. The reaction was extraordinarily tolerant of the amount
of water, with both 10 (entry 5) and 20 (entry 6) equivalents of water having minimal effect on
the yield or ee. Loadings of ammonium hexaflurophosphate can be as low as 5 mol % (entry 7)
or 10 mol % (entry 8) with reactions completed in 24 hours. Stoichiometric additive (entry 9)
gave no additional benefit (entry 4). Thus, we optimized the additive amounts to be 30 mol %
ammonium hexafluorophosphate and 5 equivalents of water.
Table 3.2. Effect of water and NH4PF6 on reaction rate.
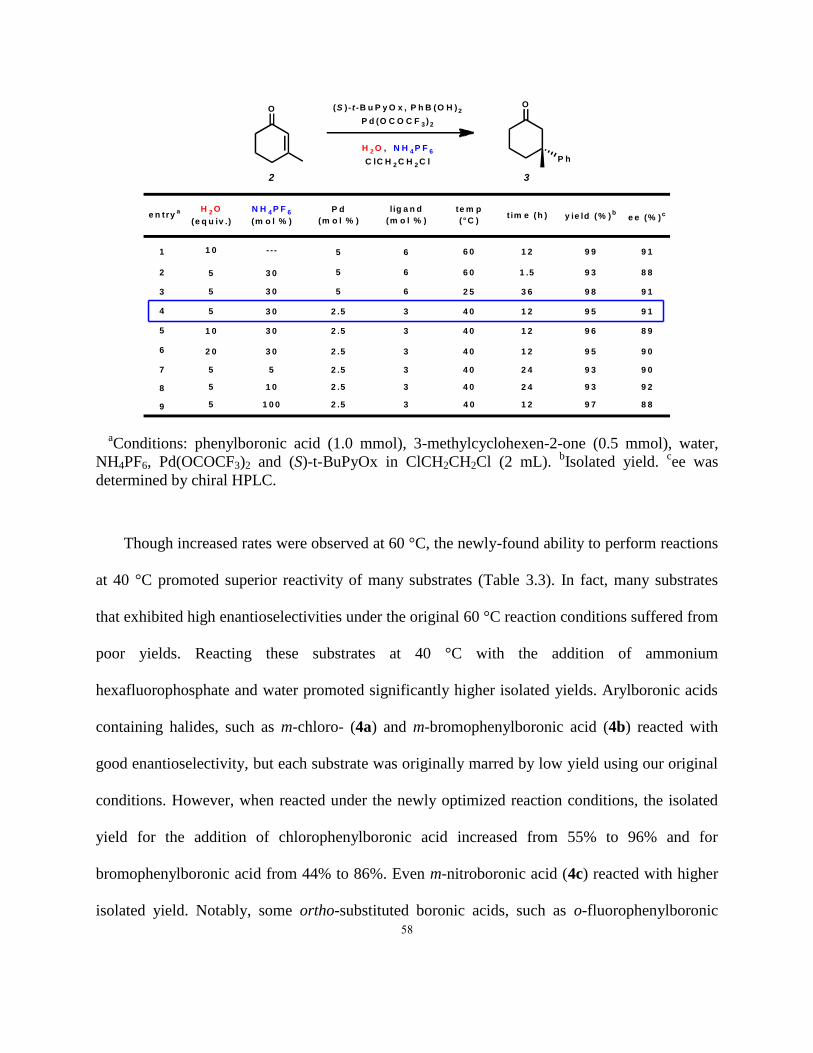
58
aConditions: phenylboronic acid (1.0 mmol), 3-methylcyclohexen-2-one (0.5 mmol), water,
NH4PF6, Pd(OCOCF3)2 and (S)-t-BuPyOx in ClCH2CH2Cl (2 mL). bIsolated yield.
cee was
determined by chiral HPLC.
Though increased rates were observed at 60 °C, the newly-found ability to perform reactions
at 40 °C promoted superior reactivity of many substrates (Table 3.3). In fact, many substrates
that exhibited high enantioselectivities under the original 60 °C reaction conditions suffered from
poor yields. Reacting these substrates at 40 °C with the addition of ammonium
hexafluorophosphate and water promoted significantly higher isolated yields. Arylboronic acids
containing halides, such as m-chloro- (4a) and m-bromophenylboronic acid (4b) reacted with
good enantioselectivity, but each substrate was originally marred by low yield using our original
conditions. However, when reacted under the newly optimized reaction conditions, the isolated
yield for the addition of chlorophenylboronic acid increased from 55% to 96% and for
bromophenylboronic acid from 44% to 86%. Even m-nitroboronic acid (4c) reacted with higher
isolated yield. Notably, some ortho-substituted boronic acids, such as o-fluorophenylboronic
OO
e n trya
t im e (h ) y ie ld (% )b
e e (% )c
1
2
3
1 0
5
1 2 9 9 9 1
te m p
(°C )
6 0
1 .5 9 3 8 86 0
1 2 9 5 9 14 0
P d
(m o l % )
5
4
5
6
7
3 6 9 8 9 12 5
1 2 9 6 8 94 0
1 2 9 5 9 04 0
8
5 2 4 9 3 9 04 0
H 2 O
(e q u iv .)
N H 4 P F 6
(m o l % )
- - -
3 0
5 3 0
5 3 0
1 0 3 0
2 0 3 0
5
9
5 2 4 9 3 9 24 01 0
5 1 2 9 7 8 84 01 0 0
P h
lig a n d
(m o l % )
5
5
6
6
6
2 .5 3
2 .5 3
2 .5 3
2 .5 3
2 .5 3
2 .5 3
(S ) - t -B u P y O x , P h B (O H )2
P d (O C O C F 3 ) 2
H 2 O , N H 4 P F 6
C lC H 2 C H 2 C l
2 3

59
acid (4d), reacted more successfully under the milder reaction conditions, leading to increased
isolated yield of 70%.
Table 3.3. Increased reaction yields with different arylboronic acid substrates under new reaction
conditions.a
aBlue font: reported yield and ee of 5 in the absence of NH4PF6 and water with reactions
performed at 60 °C; red font: yield and ee of 5 with additives. Conditions: boronic acid (1.0
mmol), 3-methylcyclohexen-2-one (0.5 mmol), NH4PF6 (30 mol %), water (5 equiv.),
Pd(OCOCF3)2 (5 mol %) and (S)-t-BuPyOx (6 mol %) in ClCH2CH2Cl (2 mL) at 40 °C . Isolated
yield reported, ee was determined by chiral HPLC.
3.3.3 Non-Linear Effect Correlation of Catalyst and Product Enantioenrichment
Despite optimization of catalytic conditions for this highly enantioselective process, we were
unsure of the nature of the active catalyst. For example, some rhodium conjugate addition
systems have been shown to involve trimeric ligand/metal complexes.19
Furthermore, Lu and
coworkers reported the use of the palladium dimer [(bpy)Pd(OH)]2•2BF4 as a precatalyst for
conjugate addition.12
We aimed to rule out the in situ formation and kinetic relevance of such
dimers in our system. In seeking to support our hypothesized monomeric ligand-metal complex,
4 d4 c
B r N O 2
5 5 % Y ie ld 9 7 % e e
(H O )2 B (H O )2 B (H O )2 BC l (H O )2 B
9 6 % Y ie ld 9 6 % e e
4 4 % Y ie ld 8 6 % e e
8 4 % Y ie ld 8 4 % e e
4 0 % Y ie ld 9 2 % e e
8 1 % Y ie ld 9 1 % e e
3 2 % Y ie ld 7 7 % e e
7 0 % Y ie ld 7 7 % e e
F
4 a 4 b
OO(S ) - t -B u P y O x (6 m o l % )
P d (O C O C F 3 )3 (5 m o l % )
N H 4 P F 6 , H 2 O
C lC H 2 C H 2 C l
4 0 °C , 1 2 hB (O H ) 2
RR
2 4 5

60
we performed a non-linear effect study to determine the relationship between the ee of the ligand
and the ee of the generated product.20
The endeavor was to exclude dimeric (ML)2 complexes
from kinetic relevance, clarifying the monomeric nature of the active catalyst.21
Five reactions
were performed using a catalyst with different level of enantiopurity (racemic, 20%, 40%, 60%
and 80% ee), and the obtained enantioselectivities were plotted against ee of the catalyst mixture
(Figure 3.2). The obtained data clearly demonstrates that a non-linear effect is not present, and
this observation strongly supports the action of a single, monomeric (ML)-type Pd/PyOx catalyst
as the kinetically relevant species.21
While the precise nature of the active catalyst species is
unknown, isolated (PyOx)Pd(OCOCF3)2 serves as an identically useful precatylst, delivering
ketone 3 in 99% yield and 92% ee.
Figure 3.2. Determination of linearity between catalyst ee and product ee.

61
3.3.4 Computational Investigations of the Reaction Mechanism
Despite the results of the non-linear effect study agreeing with the proposed monomeric
Pd/PyOx catalyst, no formal exploration of the mechanism of this transformation has been
reported. Our initial hypothesis concerning the mechanism of the Pd/PyOx-catalyzed asymmetric
conjugate addition were well informed by the seminal work of Miyaura,22
however, the
heterogeneous nature of the reaction medium, undefined nature of the precise catalyst,23
and
complicating equilibrium of organoboron species make kinetic analysis and thorough
mechanistic study extremely challenging.24
Previously, we performed density functional theory (DFT) calculations to investigate the
mechanism of palladium-catalyzed conjugate addition of arylboronic acids to enones, explicitly
studying a catalytic palladium(II)/bipyridine system in MeOH solvent similar to that developed
by Lu.12,25,26
Calculations indicated that the mechanism involves three steps: transmetallation,
carbopalladation (i.e. alkene insertion), and protonation with MeOH. Monomeric cationic
palladium complexes are the active species in the catalytic cycle. The carbopalladation is
calculated to be the rate- and stereoselectivity- determining step (Scheme 3.3). Now, we have
performed DFT investigations on the catalytic cycle of reactions with the Pd/PyOx manifold and
the effects of substituents and ligand on reactivity and enantioselectivity. The calculations were
performed at the theoretical level found satisfactory in our previous study of the Pd/bipyridine
system. Geometries were optimized with BP8627
and a standard 6-31G(d) basis set (SDD basis
set for palladium). Solvent effects were calculated with single point calculations on the gas phase
geometries with the CPCM solvation model in dichloroethane. All calculations were performed
with Gaussian 03.28

62
Scheme 3.3. Enantioselectivity-determining step in asymmetric conjugate addition of
arylboronic acids to cyclic enones.
1O O
B (O H )2
N N
O
t -B u
P d (O C O C F 3 )2
C lC H 2 C H 2 C l, 6 0 °C
2
E n a n t io s e le c t iv ity -D e te rm in in g S te p
3
N
P d
N
O O
H
QuickTim e?and a
PNG decompressor
are needed t o see t his pict ure.

63
Figure 3.3. Computed potential energy surface of the catalytic cycle (shown in black), the
alternative direct nucleophilic addition pathway (via 9-ts, shown in blue), and the isomeric
carbopalladation pathway (via 16-ts, shown in green).
The computed potential energy surface for the catalytic cycle is shown in Figure 3.3. To
simplify the computations of the mechanisms, a model ligand, in which the t-Bu group on the
t-BuPyOx ligand was replaced by H, was used in the calculations of the mechanisms and the full
ligand was used in the calculations of enantioselectivities which will be discussed below.
Calculations on the reaction mechanism with the full ligand scaffold, however, generated a
similar reaction diagram, and the rate- and stereo- determining steps were unchanged (not
shown). The first step involves transmetallation of cationic Pd(II)-phenylborate complex 6 to
generate a phenyl palladium complex. Transmetallation requires a relatively low free energy
barrier of 15.6 kcal/mol (7-ts) with respect to complex 6 and leads to a phenyl palladium

64
complex (8). Complex 8 undergoes ligand exchange to form a more stable phenyl
palladium-enone complex 12, in which the palladium binds to the enone oxygen atom. Complex
12 isomerizes to a less stable complex 13 and then undergoes carbopalladation of the enone
(14-ts) to form the new carbon–carbon bond. The carbopalladation step requires an activation
free energy of 21.3 kcal/mol (12 14-ts), and is the stereoselectivity-determining step. The
regioisomeric carbopalladation transition state 16-ts requires 5.6 kcal/mol higher activation free
energy than 14-ts, indicating the formation of the -addition compound 17 is unlikely to occur.
Coordination of one water molecule to 15 leads to a water-palladium enolate complex 18, and
finally facile hydrolysis of 18 via 19-ts affords product complex 20. Liberation of the product 3
from 20 and coordination with another molecule of phenyl boronic acid regenerates complex 6 to
complete the catalytic cycle. The computed catalytic cycle demonstrates some similarities with
the Pd/bipyridine system in our previous computational investigation, which also involves
monomeric cationic palladium as the active species and a catalytic cycle of transmetallation,
carbopalladation, and protonation (with MeOH instead of H2O).
We also considered an alternative pathway involving direct nucleophilic attack of the phenyl
boronic acid at the enone while the Pd catalyst is acting as a Lewis acid to activate the enone and
directs the attack of the nucleophile (9-ts, Figure 3.3). This alternative pathway requires an
activation free energy of 58.9 kcal/mol, 43.3 kcal/mol higher than the transmetallation transition
state 7-ts. Thus, this alternative pathway was excluded by calculations.

65
3.3.5 Experimental and Computational Investigations of the Enantioselectivities
With the aforementioned optimized reaction conditions and computational elucidation of the
mechanism and stereoselectivity-determining transition states, we explored the effects of ligand
and substrate on enantioselectivities by both experiment and computations. The
enantioselectivity-determining alkene insertion step involves a four-membered cyclic transition
state, which adopts a square-planar geometry. When a chiral bidentate ligand, such as
(S)-t-BuPyOx, is employed, there are four possible isomeric alkene insertion transition states.
The 3D structures of the alkene insertion transition states in the reaction of
3-methyl-2-cyclohexenone with (S)–t-BuPyOx ligand are shown in Figure 3.4. In 1-TS-A and
1-TS-B, the phenyl group is trans to the chiral oxazoline on the ligand, and in 1-TS-C and
1-TS-D, the phenyl group is cis to the oxazoline. 1-TS-A, which leads to the predorminant
(R)-product, is the most stable as the t-Bu group is pointing away from other bulky groups.
1-TS-C leads to the same enantiomer, but with an activation enthalpy 2.6 kcal/mol higher than
1-TS-A. The difference is likely to result from steric effects between the t-Bu on the ligand and
the phenyl group, as indicated by the C-H and C-C distances labeled in Figure 3.4. 1-TS-B and
1-TS-D lead to the minor (S)-product, which are ~3 kcal/mol less stable than 1-TS-A as a result
of the repulsions between the t-Bu on the ligand and the phenyl group. In 1-TS-B, the
cyclohexenone ring is syn to the t-Bu group on the ligand. The shortest H–H distance between
the ligand and the enone is 2.30 Å, suggesting some steric repulsions. In contrast, no
ligand-substrate steric repulsions are observed in 1-TS-A, in which the cyclohexenone is anti to
the t-Bu. 1-TS-A is also stabilized by a weak hydrogen bond between the carbonyl oxygen and
the hydrogen geminal to the t-Bu group on the oxazoline. The O–H distance is 2.16Å. Therefore,

66
the enthalpy of 1-TS-B is 2.3 kcal/mol higher than that of 1-TS-A. This corresponds to an ee of
94%, which is very similar to the experimental observation (93%). Enantioselectivities were
computed from relative enthalpies of the transition states. The selectivities computed from Gibbs
free energies are very similar and are given in the SI.
We then investigated the effects of substituents on the ligand, in particular, at the 4 position
of the oxazoline. The activation enthalpies of four alkene insertion pathways and the computed
and experimental ee for the reaction of 3-methyl-2-cyclohexenone and phenyl boronic acid are
summarized in Table 3.4. The t-Bu substituted PyOx ligand is found to be the optimum ligand
experimentally (Table 3.4, entry 1). Replacing t-Bu with smaller groups, such as i-Pr, i-Bu, or
Ph, dramatically reduces the ee.
The bulky t-Bu substituent on the ligand is essential not only to discriminate the
diastereomeric transition states 1-TS-A and 1-TS-B, but also fix the orientation of the ligand to
point the chiral center cis to the cyclohexenone. The energy difference between 1-TS-C and
1-TS-D, in which the chiral center on the ligand is trans to the cyclohexenone, is diminished.

67
Figure 3.4. The optimized geometries of transition states in the enantioselectivity-determining
alkene insertion step of the reaction of 3-methyl-2-cyclohexenone and phenyl boronic acid with
(S)–t-BuPyOx ligand. Selected H–H, C–H, and O–H distances are labeled in Å.
When the (S)-i-PrPyOx ligand is used, the alkene insertion transition states with phenyl
trans to the oxazoline (2-TS-A and 2-TS-B) are also preferred. Thus, the enantioselectivity is
determined by the energy difference between 2-TS-A and 2-TS-B. The (R)-product (via 2-TS-A)
is favored with a computed ee of 67%, slightly higher than the experimental ee (40%). The
optimized geometries of 2-TS-A and 2-TS-B are shown in Figure 3.5 and the activation energies
of all four transition states are shown in Table 3.4, entry 2. The i-Pr substituted ligand manifests
via similar steric effects to (S)-t-BuPyOx, with, as expected, slightly weaker steric control. The
lower enantioselectivity is attributed to the weaker steric repulsions between the i-Pr and the
cyclohexenone in 2-TS-B than those with the t-Bu in 1-TS-B. The shortest distance between the

68
hydrogen atoms on the ligand and the cyclohexenone is 2.35 Å in 2-TS-B, slightly longer than
the H–H distance in 1-TS-B (2.30 Å). Less steric repulsions with the (S)-i-PrPyOx lead to 2.8
kcal/mol lower activation barriers for 2-TS-B compared to 1-TS-B. The ligand steric effects on
the activation energies of the major pathway TS-A are smaller; the i-Pr substituted 2-TS-A is
only 0.6 kcal/mol more stable than the t-Bu substituted 1-TS-A.
Similarly, when the (S)-i-BuPyOx or (S)-PhPyOx ligands are used, the enantioselectivity is
further decreased to 0.8 kcal/mol (52% ee) for (S)-i-BuPyOx and 1.0 kcal/mol (65% ee) for
(S)-PhPyOx. (Table 3.4, entries 3 and 4). These results agree well with the experimental trend.
Table 3.4. Activation energies and enantioselectivities of alkene insertion with (S)-t-BuPyOX,
(S)-i-PrPyOX, (S)-i-BuPyOX, and (S)-PhPyOx ligands.
TS R1 H
‡ a ee
b
TS-A TS-B TS-C TS-D
1 t-Bu 19.2 21.5 21.8 22.2 94%
[93%]
2 i-Pr 18.6 19.7 20.1 20.0 67%
[40%]
3 i-Bu 18.5 19.3 20.4 20.4 52%
[24%]
4 Ph 18.0 19.0 19.6 20.2 65%
[52%] aThe values are activation enthalpies in kcal/mol calculated at the BP86/6-31G(d)–SDD level
and the CPCM solvation model in dichloroethane. bExperimental ee were obtained under
standard conditions and are given in square brackets.
N
P d
N
OO
R 1
H N
P d
N
O
R 1
H
O
N
P d
N
O
O
R 1
HN
P d
N
O
R 1
H
O
n -T S -A n -T S -B n -T S -C n -T S -D

69
Electronically differentiated PyOx ligands were also studied, and the results are summarized
in Table 3.5. Electron-withdrawing or donating groups at the 4-position of the PyOx ligand
showed minimal effects on the activation barriers, and were calculated to have minimal effect on
product ee. With the electron-withdrawing CF3 and the electron-donating OCH3 on the
4-position of the ligand, the activation enthalpies of alkene insertion increase by only 0.3
kcal/mol and 0.1 kcal/mol, respectively. The calculated ee are essentially identical among these
three ligands. Experimentally, depressed ee was observed with both the 4-CF3 and the 4-OCH3
substituted ligands (entries 5 and 6). This confirms that the enantioselectivity is mainly attributed
to the ligand/substrate steric repulsions.
Table 3.5. Remote ligand substituent effects on activation energies and enantioselectivities of
alkene insertion.
TS R
2 H
‡ a ee
b
TS-A TS-B TS-C TS-D
1 H 19.2 21.5 21.8 22.2 94%
[93%]
5 CF3 19.5 21.7 21.8 22.3 93%
[81%]
6 OCH3 19.3 21.5 21.5 22.2 93%
[78%] aThe values are activation enthalpies in kcal/mol calculated at the BP86/6-31G(d)-SDD level
and the CPCM solvation model in dichloroethane. bExperimental ee are given in square brackets.
N
P d
N
OO
H N
P d
N
O
H
O
N
P d
N
O
OHN
P d
N
O
H
O
n -T S -A n -T S -B n -T S -C n -T S -D
R 2R 2
R 2R 2

70
2 - T S - A
H‡
= 1 8 . 6 k c a l/ m o l
2 - T S - B
H‡
= 1 9 . 7 k c a l/ m o l
7 - T S - A
H‡
= 1 6 . 7 k c a l/ m o l
7 - T S - B
H‡
= 1 8 . 6 k c a l/ m o l
(a )
(b )
Figure 3.5. The optimized geometries of transition states in the enantioselectivity-determining
alkene insertion step of the reaction of (a) 3-methyl-2-cyclohexenone and phenyl boronic acid
with (S)-i-PrPyOx ligand; (b) 3-methyl--2-pentenolide and phenyl boronic acid with
(S)-t-BuPyOx ligand. Selected H–H, C–H, and O–H distances are labeled in Å.
The transition state structures shown in Figure 3.5 indicate that the steric control mainly
arises from the repulsion of the C' hydrogens on the cyclohexenone with the ligand. We then
investigated the effects of substitution at the ' position and replacement of the CH2 group with
O. The reactivity and enantioselectivity of the reactions of lactone (Table 3.6, entry 7) and
','-dimethylcyclohexenone (Table 3.6, entry 8) with the (S)-t-BuPyOx ligand were computed.
The enantioselectivity of lactone is predicted to be lower than cyclohexenone. The enthalpy of
7-TS-B is 1.8 kcal/mol higher than that of 7-TS-A, corresponding to an ee of 88%.
Experimentally, the ee of the lactone product is 59%, also significantly lower than that with
cyclohexenone. The optimized geometries of 7-TS-A and 7-TS-B are shown in Figure 3.5b.

71
Replacing the CH2 group with O decreases the ligand–substrate steric repulsion in 7-TS-B is
smaller than that in 1-TS-B. This results in decreased enantioselectivity.
Methyl substitution at the ' position of cyclohexenone increases the steric repulsion with
the t-Bu group on the ligand. Computations predicted increased enenatioselectivity with
','-dimethylcyclohexenone (99% ee, Table 3.6, entry 8).29
However, experimentally, the ee is
comparable with the reaction of 3-methylcyclohexenone.
Table 3.6. Activation energies and enantioselectivities of alkene insertion with substrates varying
at the '-position.
TS X H‡ a
ee b
TS-A TS-B TS-C TS-D
1 CH2 19.2 21.5 21.8 22.2 94%
[93%]
7 O 16.7 18.6 19.4 19.6 88%
[57%]
8 C(CH3)2 17.9 22.0 20.0 20.8 99%
[90%] aThe values are activation enthalpies in kcal/mol calculated at the BP86/6-31G(d)-SDD level
and the CPCM solvation model in dichloroethane. bExperimental ee are given in square brackets.
Experimental isolated yields: TS-1: 99%, TS-7: 49%, TS-8: 9%.
We also considered the electronic effects of arylboronic acids on enantioselectivity (Table
3.7). Computations predicted that para-electron-withdrawing substituents lead to increases in the
activation barrier in alkene insertion, probably due to the electrophilicity of the -carbon of the
enone, and thus are predicted to afford slightly decreased enantioselectivities. Both
N
P d
N
OX O
H N
P d
N
O
H
X
O
N
P d
N
O
XOHN
P d
N
O
H
X
O
n -T S -A n -T S -B n -T S -C n -T S -D

72
para-acetylphenylboronic acid (9-TS-A) and para-trifluoromethylphenylboronic acid (10-TS-A)
are predicted to react with 92% ee. However, both excellent enantioselectivities (96% ee) and
excellent yields (99% isolated yield) are observed experimentally. Thus, the electronic effects
of phenyl substituents on enantioselectivities are minimal, though slightly increased
enantioselectivities are observed experimentally with the use of electron-withdrawing
substituents.
Table 3.7. Activation energies and enantioselectivities of alkene insertion with various boronic
acids.
TS R3 H
‡ a ee
b
TS-A TS-B TS-C TS-D
1 H 19.2 21.5 21.8 22.2 94%
[93%]
9 CH3CO 21.6 23.7 24.5 24.8 92%
[96%]
10 CF3 21.3 23.4 23.6 24.2 92%
[96%] aThe values are activation enthalpies in kcal/mol calculated at the BP86/6-31G(d)-SDD level
and the CPCM solvation model in dichloroethane. bExperimental ee are given in square brackets.
Experimental isolated yields: TS-1: 99%, TS-9: 1%, TS-10: 99%, TS-11: 99%.
Table 3.8. Enantioselectivity trends in pyridinooxazoline and related ligand frameworks.a
N
P d
N
OO
H N
P d
N
O
H
O
N
P d
N
O
OHN
P d
N
O
H
O
n -T S -A n -T S -B n -T S -C n -T S -D
R 3R 3
R 3
R 3

73
aConditions: 3-methylcyclohexen-2-one (0.25 mmol), phenylboronic acid (0.5 mmol),
ClCH2CH2Cl (1 mL). Yields are isolated yields, ee determined by chiral HPLC.
Permutations of the pyridinooxazoline ligand framework corroborate the calculated data and
suggest that a number of factors affect enantioselectivity. First, the steric demand of the chiral
group on the oxazoline greatly impacts the observed enantioselectivity in the reaction (Table
3.8). Only t-BuPyOx (1) yields synthetically tractable levels of enantioselectivity, while the less
sterically demanding i-PrPyOx (21), PhPyOx (22), and i-BuPyOx (23) all exhibit greatly
diminished selectivity. Oxazoline substitution patterns also affect enantioselectivity.
Substitution at the 4-position appears to be required for high selectivity, as substitution at the
5-position yields practically no enantioselectivity (ligand 24). Electronic variation in the PyOx
framework was observed to have a large effect on the rate of the reaction but, disappointingly,
N
N
O
i -P r
N
N
O
P h
N
N
O
P h
2 1
9 9 % y ie ld
4 0 % e e
2 2
9 9 % y ie ld
5 2 % e e
2 4
9 9 % y ie ld
2 % e e
N
N
O
t -B u
N
N
O
i-P r2 7
1 3 % y ie ld
2 0 % e e
2 8
1 4 % y ie ld
0 % e e
O
B (O H )2
O
lig a n d (6 m o l % )
P d (O C O C F 3 ) 2 (5 m o l % )
C lC H 2 C H 2 C l, 6 0 °C , 1 2 h
+
N
N
O
i-B u
2 3
9 5 % y ie ld
2 4 % e e
N
N
O
t -B u
1
9 9 % y ie ld
9 3 % e e
N
N
O
t -B u
2 5
9 9 % y ie ld
8 1 % e e
N
N
O
t -B u
2 6
9 6 % y ie ld
7 8 % e e
H C F 3 O M e
P h
2 3

74
led to depressed stereoselectivity. CF3-t-BuPyOx (25) afforded the conjugate addition product in
99% yield and 81% ee. Surprisingly, MeO-t-BuPyOx (26) afforded the product in similar yield
and only 78% ee. Finally, substitution at the 6-position of the pyridine (ligands 27 and 28)
greatly diminished both reactivity and selectivity, perhaps due to hindered ligand chelation to
palladium.
Thus, we have concluded that enantioselectivity is controlled by the steric repulsion between
the substituent on the chiral pyridinooxazoline ligand and the cyclohexyl ring. The bulkier t-Bu
substituent on the (S)-t-BuPyOx ligand leads to greater enantioselectivity than the reactions with
(S)-i-PrPyOx or (S)-PhPyOx. Similarly, substrates with less steric demand adjacent the carbonyl
exhibit lower enantioselectivities; for example the reaction of a lactone substrate (Table 3.6,
TS-7) yields lower enantioselectivity due to smaller repulsions between the lactone oxygen and
the t-Bu group.
3.3.6 Experimental Investigation of Arylpalladium(II) Intermediates and Formation of the
Key C–C Bond
Experiments aiming to corroborate the calculated mechanism have been performed. We
sought to observe the formation of the key C–C bond between an arylpalladium(II) species and
the enone substrate in the absence of exogenous phenylboronic acid. Complexes 29 were
synthesized as an intractable mixture of isomers, and were treated with AgPF6 in situ to generate
the [(PyOx)Pd(Ph)]+
cation. Gratifyingly, complexes 29 serve as a competent precatalyst at 5 mol
% loading, and affords ketone 3 in 96% yield and 90% ee (Table 3.9, entry 1). Varying the
amount of complex utilized in proportion with phenylboronic acid, however, leads to significant

75
production of biphenyl above 5 mol % (Table 3.9, entries 2–5). Utilizing even 25 mol % (entry
2) resulted in significant increase in biphenyl production (16% yield), and reduction in yield of
the desired ketone 3 to 79%. Increasing complex loadings to 45 and 65 mol % (entries 3 and 4)
leads to negligible production of ketone 3, and nearly quantitative formation of biphenyl relative
to catalyst loading. Furthermore, attempts to use the complex as a stoichiometric reagent in the
place of PhB(OH)2 lead to no observed product (entry 5), and exclusive formation of biphenyl.
We hypothesize that quantitative generation of the reactive arylpalladium cation intermediate in
high relative concentration leads quickly to disproportionation and formation of biphenyl and
palladium(0). Omission of the AgPF6 in favor of 30 mol % NH4PF6 leads to isolation of only
22% yield of the conjugate addition product (entry 6). Finally, a control experiment demonstrates
that AgPF6 is incapable of catalyzing the reaction itself (entry 7).30
This control further supports
the computational results, which indicate a transmetallation-based mechanism as opposed to a
Lewis acid-catalyzed pathway.
Table 3.9. Arylpalladium(II) catalyzed conjugate addition.a
OO
C o m p le x 2 9 , A g P F 6
H 2 O (5 e q u iv .)
C lC H 2 C H 2 C l, 4 0 °C , 1 2 h P h
B (O H ) 2
e n try c o m p le x 2 9 (m o l % ) A g P F 6 (m o l % ) P h B (O H )2 (m o l % )
y ie ld (% )
3 b ip h e n y l
1
2
3
4
5
5
2 5
4 5
6 5
1 0 5
1 0
5 0
9 0
1 3 0
2 1 0
7 0 2 0 2 0 0
0
4 0
6 0
8 0
1 0 0 9 6
7 9
1 1
5
0
3
1 6
5 3
5 8
8 7
0 0
2 3
6b
5 0 2 2 02 0 0

76
a Conditions: 3-methylcyclohexen-2-one (0.25 mmol), pheylboronic acid (equiv as stated),
ClCH2CH2Cl (1 mL). Yields are isolated yields, ee determined by chiral HPLC. bReaction
performed with 30 mol % NH4PF6
Concerned by our inability to observe consistent product formation at 40 °C, we sought
alternative experimental verification that a putative cationic arylpalladium(II) species is capable
of reacting to form conjugate addition products. Thus, we performed the stoichiometric reaction
of the isomeric phenyl palladium iodide complexes (29) with AgPF6 and 3-methylcyclohexenone
at cryogenic temperatures, allowing the mixture to warm slowly to room temperature before
quenching with trifluoroethanol.31
We observed both conjugate addition product and biphenyl,
with the desired adduct (3) isolated in 30% yield (Scheme 3.4a). Curiously, the conjugate
addition adduct was isolated in only 55% ee. We considered that the isomeric mixture of phenyl
palladium iodide isomers formed configurationally stable cationic species at cryogenic
temperature.32
Repeating the experiment, we substituted triphenylphosphine for
methylcyclohexenone and observed the reaction at low temperature utilizing 1H and
31P NMR
(Scheme 3.4b). Indeed, two 31
P signals corresponding to phosphine-bound palladium(II) species
(30) were observed at 28 and 34 ppm. No isomerization was observed upon warming the
isomeric mixture to room temperature.33
Indeed, we were able to isolate and characterize the
mixture of arylpalladiumphosphine cations. With evidence for the configurationally stable
arylpalladium cation, we rationalized the observed 55% ee for the direct reaction of mixture 29
with methylcyclohexenone corresponds directly to the isomeric ratio of the complex: a ratio of
1.3:1 represents a 56:44 ratio of isomers. Assuming that the major isomer reacts with 92% ee,
and the minor isomer reacts with no stereoselectivity to give racemic products,34
a net
stereoselectivity of 53.7% ee would be predicted for the product. Presuming configurational
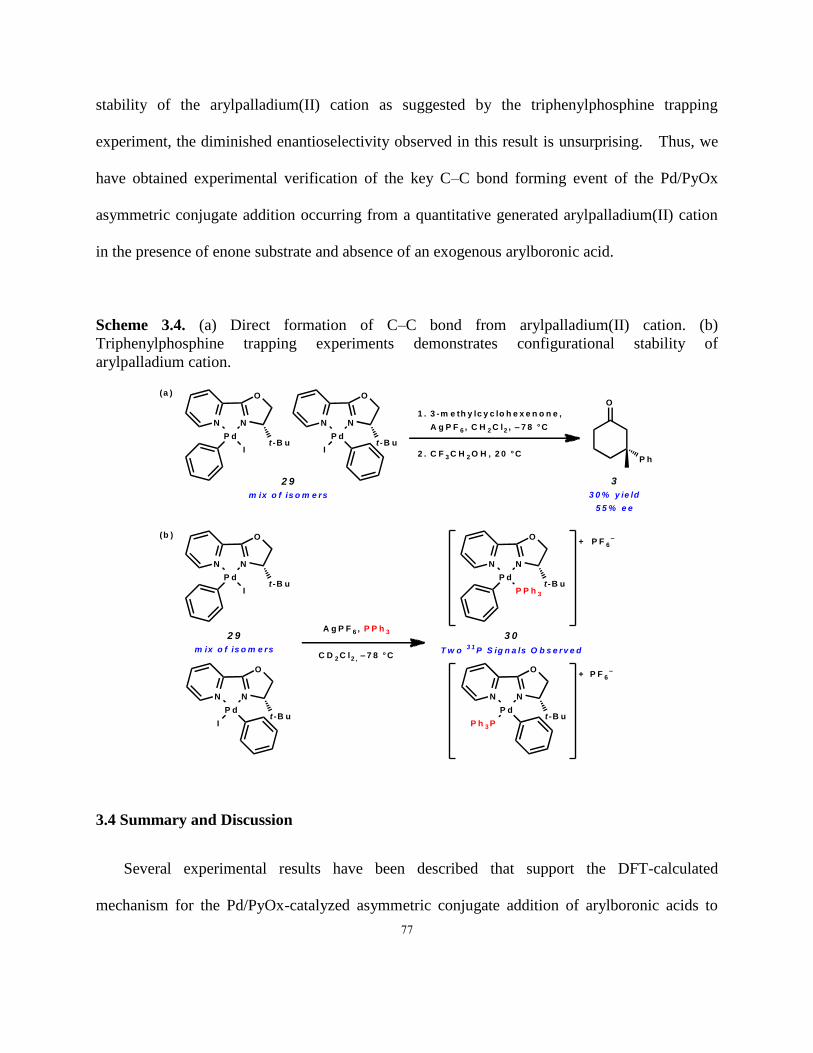
77
stability of the arylpalladium(II) cation as suggested by the triphenylphosphine trapping
experiment, the diminished enantioselectivity observed in this result is unsurprising. Thus, we
have obtained experimental verification of the key C–C bond forming event of the Pd/PyOx
asymmetric conjugate addition occurring from a quantitative generated arylpalladium(II) cation
in the presence of enone substrate and absence of an exogenous arylboronic acid.
Scheme 3.4. (a) Direct formation of C–C bond from arylpalladium(II) cation. (b)
Triphenylphosphine trapping experiments demonstrates configurational stability of
arylpalladium cation.
3.4 Summary and Discussion
Several experimental results have been described that support the DFT-calculated
mechanism for the Pd/PyOx-catalyzed asymmetric conjugate addition of arylboronic acids to
N
P d
I
N
O
t -B u
1 . 3 -m e th y lc y c lo h e x e n o n e ,
A g P F 6 , C H 2 C l2 , – 7 8 °C
2 . C F 3 C H 2 O H , 2 0 °C
O
P h
3
3 0 % y ie ld
5 5 % e e
N
P d
I
N
O
t -B u
N
P d
P P h 3
N
O
t -B u
A g P F 6 , P P h 3
C D 2 C l2 , – 7 8 °C
+ P F 6–
(a )
(b )
2 9
m ix o f is o m e rs
3 0
T w o 3 1
P S ig n a ls O b s e rv e d
N
I
P d
N
O
t -B u
2 9
m ix o f is o m e rs
N
I
P d
N
O
t -B u
N
P h 3 P
P d
N
O
t -B u
+
P F 6–

78
cyclic enone (Figure 3.4); specifically, the role of the palladium catalyst has been addressed.
Calculations and previous experimental work by Miyaura on palladium-catalyzed conjugate
addition suggest that a transmetallation event occurs to transfer the aryl moiety from the boronic
acid species to the palladium catalyst.4
Our calculations indicated that the Pd/PyOx system
operates under a similar manifold, and demonstrated a significant energy difference
(transmetallation is favored by over 40 kcal/mol, Figure 3.4) between the potential roles of the
palladium catalyst, suggesting that the role of the palladium species is not simply that of a Lewis
acid. Furthermore, it is difficult to rationalize the high degree of enantioselectivity imparted by
the chiral palladium catalyst if it is assumed that the metal acts only as a Lewis acid and is not
directly mediating the key C–C bond-forming step.35
Finally, a number of Lewis- and acidic
metal salts were substituted for palladium with no product observed, further indicating that
palladium-catalyzed conjugate addition is likely not a Lewis acid-catalyzed process.36
While highly Lewis-acidic, the role of cationic palladium(II) is to provide a vacant
coordination site for the enone substrate to approach the catalyst. The presence of a cationic
intermediate is further supported by the observed rate acceleration of non-coordinating anionic
additives such as PF6– and BF4
– salts (Table 3.1). Conversely, the addition of coordinating
anions, such as chloride, inhibited the reaction (Table 3.1, entry 1). This counterion effect was
evident even from choice of palladium(II) precatalysts.13
For instance, Pd(MeCN)2Cl2 was not
a suitable precatalyst, nor were any palladium(II) halides. Additionally, Pd(OAc)2 only afforded
modest yield of conjugate adducts, whereas the less coordinating counterion present in
Pd(OCOCF3)2 afforded complete conversion.
Satisfied that the palladium catalyst was not acting as a Lewis acid, we next sought to
demonstrate the viability of the hypothesized arylpalladium(II) species as a catalyst (Table 3.9,

79
entries 1–5). While serving as a suitable precatalyst under reaction conditions, arylpalladium(II)
mixture 29 failed to facilitate the conjugate addition reaction when used in stoichiometric
quantities in the presence of AgPF6 (entry 5). We rationalize this outcome to be the result of the
highly reactive nature of the quantitatively generated arylpalladium(II) cation. Significant
biphenyl formation- occuring even under dilute conditions representative of the catalytic reaction
itself- suggest that alternative reaction pathways, such as disproportionation, readily out compete
the desired insertion reaction. This reactive nature of the arylpalladium(II) cation led us to
propose performing the stoichiometric reactions at low temperatures (Scheme 3.4). Successfully
demonstrating that the key C–C bond could be generated, albeit in modest yield, from
(PyOx)Pd(Ph)(I) (29) corroborates both the precise role of the arylpalladium(II) cation in the
calculated mechanism as well as the transition state put forth in the calculations. However, the
modest yield of this process and requisite cryogenic temperatures prompted, again, consideration
of the role of the arylboronic acid.37
Calculations suggest that the presence of boronic acids as
Lewis basic entities may serve to stabilize these reactive intermediates under the reaction
conditions (Figure 3.3, cationic arylpalladium 8).38
This suggestion is consistent with the
successful use of arylpalladium(II) mixture 29 as a precatalyst in the presence of arylboronic
acids (Table 3.9, entry 1).
Lastly, water (or another proton source) is required for efficient turnover of the reaction.
Considerations of reaction scale (Scheme 3.2) and deuterium incorporation experiments (Figure
3.1) suggest that water is the likely protonation agent, despite numerous other protic sources in
the heterogenous reaction mixture. Attempts to use other, miscible proton sources (MeOH,
phenol, t-BuOH, etc.) typically resulted in 10–15% less enantioselectivity observed.39
2,2,2-Trifluoroethanol can be successfully substituted for water with minimal loss of

80
enantioselectivity, however it affords no supplementary benefit and water is the preferred
additive for all reactions.24
Water in combination with NH4PF6 serves to facilitate milder
reaction conditions (Table 3.2), which in turn greatly increases the synthetic scope with respect
to challenging arylboronic acid nucleophiles (Table 3.3). Synthetic yields were observed to
double in many cases, greatly increasing the utility of these transformations. The functional
group tolerance of the Pd/PyOx system is unprecedented for asymmetric conjugate addition; it
encompasses a wide array of halides, carbonyl functional groups, protected phenols, acetamides,
free hydroxyl groups, and even nitro groups. Many of these groups are incompatible with
traditional rhodium- and copper-catalyzed conjugate additions due to the reactivity with the
nucleophiles used or the strong coordination of the functional group to the metal catalyst.
Figure 3.6. Proposed catalytic cycle for Pd/PyOx-catalyzed conjugate addition of arylboronic
acids to cyclic enones.
L
P d N
N
P d N
N
O
P hP h
OOP d
P d
L
NN
N
L
N
O
O
P hX L
P d
NN B (O H )2
t ra n s m e ta lla t io n
in s e r t io n
p ro to n a t io n
c o o rd in a t io n
+
+
+
H 2 O
3 1
3 2
3 3
3 4 3 5
3
2
+
+

81
The combined results described herein have allowed us to put forth the following catalytic
cycle (Figure 3.6). The cationic catalyst, represented as (PyOx)Pd(X)(L) (31), undergoes
transmetallation with an arylboronic acid to yield cationic (PyOx)Pd(Ar)(L) (32). Substrate
coordination forms cationic arylpalladium(II) 33, which undergoes rate and
enantioselectivity-determining insertion of the aryl moiet -system to afford
C-bound palladium enolate 34. Tautomerization to the O-bound palladium enolate (35), or direct
protonolysis of the C-bound enolate (34), liberates the conjugate addition product (3) and
regenerates a cationic palladium(II) species for reentry into the catalytic cycle.
3.5 Conclusions
In conclusion, we have reported experimental and computational results that corroborate a
single PyOx ligand/metal complex as the active catalytic species in the palladium-catalyzed
asymmetric conjugate addition of arylboronic acids to enones for the construction of quaternary
stereocenters. We have used computational models to rule out a suggested alternative mechanism
in which the palladium catalyst is acting as a Lewis acid to activate the enone. The preferred
mechanism involves formal transmetallation from boron to palladium, rate- and
enantioselectivity-determining carbopalladation of the enone olefin by a cationic palladium
species, and protonolysis of the resulting palladium-enolate. We have taken advantage of these
mechanistic insights to develop a modified reaction system whereby the addition of water and
ammonium hexafluorophophate increase reaction rates, and can facilitate lower catalyst loadings.
The modified conditions have opened the door to new substrate classes that were inaccessible by
the initially published reaction conditions. Furthermore, we have demonstrated that this

82
operationally simple reaction is tolerant of ambient atmosphere and capable of producing
enantioenriched, -quaternary ketones on multi-gram scale. The steric and electronic effects of
the boronic acid and enone substrates and the ligand on enantioselectivities were elucidated by a
combined experimental and computational investigation. The enantioselectivity is mainly
controlled by the steric repulsion of the t-Bu substituent of the oxazoline on the ligand and the
C' position hydrogens of the cyclohexenone substrate in the alkene insertion transition state.
Further investigations of both the scope of this transformation and its application toward
natural product synthesis are current underway in our laboratories.
3.6 Materials and Methods
Unless otherwise stated, reactions were performed with no extra precautions taken to
exclude air or moisture. Air and moisture sensitive reactions were carried out under an
atmosphere of argon using Schlenk and glovebox techniques. Diethyl ether, hexanes, benzene
and toluene were dried and deoxygenated by passing through columns packed with activated
alumina and Q5, respectively. Deionized water was purified with a Barnstead NANOpure
Infinity UV/UF system. Dichloroethane was dried by stirring over CaH2 and subsequent
distillation, followed by deoxygenation by three freeze–pump–thaw cycles. Deuterated solvents
were dried by distillation from CaH2 (CD2Cl2) and deoxygenated by three freeze-pump-thaw
cycles. Commercially available reagents were used as received from Sigma Aldrich unless
otherwise stated. Enone substrates were purchased from Sigma Aldrich (3-methylcyclohexenone,
2-cyclohexene-1-one, chromone) or prepared according to literature procedure.40
Pyridinooxazoline ligands were synthesized according to literature procedures.41
Iodobenzene

83
was obtained from Sigma Aldrich and purified by distillation and deoxygenated by three freeze–
pump–thaw cycles. AgPF6 was obtained from ABCR and used without further purification.
Reaction temperatures were controlled by an IKAmag temperature modulator. Thin-layer
chromatography (TLC) was performed using E. Merck silica gel 60 F254 precoated plates (250
nm) and visualized by UV fluorescence quenching, potassium permanganate, or p-anisaldehyde
staining. Silicycle SiliaFlash P60 Academic Silica gel (particle size 40-63 nm) was used for flash
chromatography. Analytical chiral HPLC was performed with an Agilent 1100 Series HPLC
utilizing a Chiralcel OJ column (4.6 mm x 25 cm) obtained from Daicel Chemical Industries, Ltd
with visualization at 254 nm and flow rate of 1 mL/min, unless otherwise stated. Analytical
chiral SFC was performed with a JASCO 2000 series instrument utilizing Chiralpak (AD-H or
AS-H) or Chiralcel (OD-H, OJ-H, or OB-H) columns (4.6 mm x 25 cm), or a Chiralpak IC
column (4.6 mm x 10 cm) obtained from Daicel Chemical Industries, Ltd with visualization at
210 or 254 nm. 1H and
13C NMR spectra were recorded on a Varian Inova 500 (500 MHz and
125 MHz, respectively) and a Varian Mercury 300 spectrometer (300 MHz and 75 MHz,
respectively). Data for 1H NMR spectra are reported as follows: chemical shift (δ ppm)
(multiplicity, coupling constant (Hz), integration). Data for 1H NMR spectra are referenced to the
centerline of CHCl3 (δ 7.26), CD2Cl2 (δ 5.32), or C2D4Cl2 (δ 3.72) as the internal standard and
were reported in terms of chemical shift relative to Me4Si (δ 0.00). Data for 13
C NMR spectra
were referenced to the centerline of CDCl3 (δ 77.0), CD2Cl2 (δ 53.8), or C2D4Cl2 (δ 43.6) and
were reported in terms of chemical shift relative to Me4Si (δ 0.00). Infrared spectra were
recorded on a Perkin Elmer Paragon 1000 Spectrometer and are reported in frequency of
absorption (cm-1
). High resolution mass spectra (HRMS) were obtained on an Agilent 6200
Series TOF with an Agilent G1978A MultiMode source in electrospray ionization (ESI),

84
atmospheric pressure chemical ionization (APCI) or mixed (MultiMode ESI/APCI) ionization
mode. Optical rotations were measured on a Jasco P-2000 polarimeter using a 100 mm
path-length cell at 589 nm.
3.7 References
1 (a) P. Perlmutter, in Conjugate Addition Reactions in Organic Synthesis, Tetrahedron Organic
Chemistry Series 9; Pergamon, Oxford, 1992; (b) K. Tomioka, Y. Nagaoka, in Comprehensive
Asymmetric Catalysis, Vol. 3 (Eds: E. N. Jacobsen, A. Pfaltz, H. Yamamoto, Springer-Verlag,
New York, 1999; Chapter 31; (c) Gini, F.; Hessen, B.; Feringa, B. L.; Minnaard, A. J. Chem.
Commun. 2007, 710–712.
2 For reviews, see: (a) Harutyunyan, S. R.; den Hartog, T.; Geurts, K.; Minnaard, A. J.; Feringa,
B. L. Chem. Rev. 2008, 108, 2824–2882. (b) Alexakis, A.; Backvall, J. E.; Krause, N.; Pamies,
O.; Dieguez, M. Chem. Rev. 2008, 108, 2796–2893. (c) Lopez, F.; Minnarard, A. J.; Feringa, B.
L. Acc. Chem. Res. 2007, 40, 179–188. (d) Hayashi, T.; Yamasaki, K. Chem. Rev. 2003, 103,
2829–2844. (e) Feringa, B. L. Acc. Chem. Res. 2000, 33, 346–353. (f) Rossiter, B. E.; Swingle,
N. M. Chem. Rev. 1992, 92, 771–806.
3 (a) Shintani, R.; Duan, W.-L.; Hayashi, T. J. Am. Chem. Soc. 2006, 128, 5628–5629. (b)
Hayashi, T.; Takahashi, M.; Takaya, Y.; Ogasawara, M.; J. Am. Chem. Soc. 2002, 124, 5052–
5058. (c) Takaya, Y.; Ogasawara, M.; Hayashi, T. J. Am. Chem. Soc. 1998, 120, 5579–5580.
4 (a) Nishikata, T.; Yamamoto, Y.; Miyaura, N. Angew. Chem., Int. Ed. 2003, 42, 2768–2770.
(b) Nishitaka, T.; Yamamoto, Y.; Miyaura, N. Chem. Lett. 2007, 36, 1442–1443. (c) Nishitaka,
T.; Kiyomura, S.; Yamamoto, Y.; Miyaura, N. Synlett 2008, 2487–2490.
5 Gini, F.; Hessen, B.; Minnaard, A. J. Org. Lett. 2005, 7, 5309–5312.
6 For reviews on the synthesis of quaternary stereocenters, see: (a) Denissova, I.; Barriault, L.
Tetrahedron 2003, 59, 10105–10146. (b) Douglas, C. J.; Overman, L. E. Proc. Natl. Acad. Sci.
U.S.A. 2004, 101, 5363–5267. (c) Christoffers, J.; Baro, A. Adv. Synth. Catal. 2005, 347, 1473–
1482. (d) Trost, B. M.; Jiang, C. Synthesis 2006, 369–396. (e) Mohr, J. T.; Stoltz, B. M. Chem.
Asian J. 2007, 21, 1476–1491. (f) Cozzi, P. G.; Hilgraf, R.; Zimmermann, N. Eur. J. Org. Chem.
2007, 5969–5994.

85
6 For an excellent comprehensive review, see: Hawner, C.; Alexakis, A. Chem. Commun. 2010,
46, 7295–7306.
7 (a) Wu, J.; Mampreian D. M.; Hoveyda, A. H. J. Am. Chem. Soc. 2005, 127, 4584–4585. (b)
Hird, A. W.; Hoveyda, A. H. J. Am. Chem. Soc. 2005, 127, 14988–14989. (c) Lee, K.-S.; Brown,
M. K.; Hird, A. W.; Hoveyda, A. H. J. Am. Chem. Soc. 2006, 128, 7182–7184. (d) Brown, M.
K.; May, T. L.; Baxter, C. A.; Hoveyda, A. H. Angew. Chem., Int. Ed. 2007, 46, 1097–1100. (e)
Fillion, E.; Wilsily, A. J. Am. Chem. Soc. 2006, 128, 2774–2775. (f) Wilsily, A.; Fillion, E. J.
Org. Chem. 2009, 74, 8583–8594. (g) Dumas, A. M.; Fillion, E. Acc. Chem. Res. 2010, 43, 440–
454. (h) Wilsily, A.; Fillion, E. Org. Lett. 2008, 10, 2801–2804.
8 (a) d’Augustin, M.; Palais, L.; Alexakis, A. Angew. Chem., Int. Ed. 2005, 44, 1376–1378. (b)
Vuagnoux-d’Augustin, M.; Alexakis, A. Chem.–Eur. J. 2007, 13, 9647–9662. (c) Palais, L.;
Alexakis, A. Chem.–Eur. J. 2009, 15, 10473–10485. (d) Fuchs, N.; d’Augustin, M.; Humam, M.;
Alexakis, A.; Taras, R.; Gladiali, S. Tetrahedron: Asymmetry 2005, 16, 3143–3146. (e)
Vuagnoux-d’Augustin, M.; Kehrli, S.; Alexakis, A. Synlett, 2007, 2057–2060. (f) May, T. L.;
Brown, M. K.; Hoveyda, A. H. Angew. Chem., Int. Ed. 2008, 47, 7358–7362. (g) Ladjel, C.;
Fuchs, N.; Zhao, J.; Bernardinelli, G. Alexakis, A. Eur. J. Org. Chem. 2009, 4949–4955. (h)
Palais, L.; Mikhel, I. S.; Bournaud, C.; Micouin, L.; Falciola, C. A.; Vuagnoux-d’Augustin, M.;
Rosset, S.; Bernardinelli, G.; Alexakis, A. Angew. Chem., Int. Ed. 2007, 46, 7462–7465. (i)
Hawner, C.; Li, K.; Cirriez, V.; Alexakis, A. Angew. Chem., Int. Ed. 2008, 47, 8211–8214. (j)
Müller, D.; Hawner, C.; Tissot, M.; Palais, L.; Alexakis, A. Synlett 2010, 1694–1698. (k)
Hawner, C.; Müller, D.; Gremaud, L.; Felouat, A.; Woodward, S.; Alexakis, A. Angew. Chem.,
Int. Ed. 2010, 49, 7769–7772.
9 (a) Martin, D.; Kehrli, S.; d’Augustin, M.; Clavier, H.; Mauduit, M.; Alexakis, A. J. Am.
Chem. Soc. 2006, 128, 8416–8417. (b) Kehrli, S.; Martin, D.; Rix, D.; Mauduit, M.; Alexakis,
A. Chem.–Eur. J. 2010, 16, 9890–9904. (c) Hénon, H.; Mauduit, M.; Alexakis, A. Angew.
Chem., Int. Ed. 2008, 47, 9122–9124. (d) Matsumoto, Y.; Yamada, K.-I.; Tomioka, K. J. Org.
Chem. 2008, 73, 4578–4581.
10 (a) Shintani, R.; Tsutsumi, Y.; Nagaosa, M.; Nishimura, T.; Hayashi, T. J. Am. Chem. Soc.
2009, 131, 13588–13589. (b) Shintani, R.; Takeda, M.; Nishimura, T.; Hayashi, T. Angew.
Chem., Int. Ed. 2010, 49, 3969–3971.
11 For excellent review articles, see: (a) Gutnov, A. Eur. J. Org. Chem. 2008, 4547–4554. (b)
Christoffers, J.; Koripelly, G.; Rosiak, A.; Rössle, M. Synthesis 2007, 1279–1300.
12 Lin, S.; Lu, X. Org. Lett. 2010, 12, 2536–2539.
13 Kikushima, K.; Holder, J. C.; Gatti, M.; Stoltz, B. M. J. Am. Chem. Soc. 2011, 133, 6902–
6905.

86
14
PyOx ligand scaffolds are increasingly common in asymmetric catalysis, see: (a) Podhajsky,
S. M.; Iwai, Y.; Cook-Sneathen, A.; Sigman, M. S. Tetrahedron 2011, 67, 4435–4441. (b)
Aranda, C.; Cornejo, A.; Fraile, J. M.; García-Verdugo, E.; Gil, M. J.; Luis, S. V.; Mayoral, J.
A.; Martínez-Merino, V.; Ochoa, Z. Green Chem. 2011, 13, 983–990. (d) Pathak, T. P.;
Gligorich, K. M.; Welm, B. E.; Sigman, M. S. J. Am. Chem. Soc. 2010, 132, 7870–7871. (d)
Jiang, F.; Wu, Z.; Zhang, W. Tetrahedron Lett. 2010, 51, 5124–5126. (e) Jensen, K. H.; Pathak,
T. P.; Zhang, Y.; Sigman, M. S. J. Am. Chem. Soc. 2009, 131, 17074–17075. (f) He, W.; Yip,
K.-T.; Zhu, N.-Y.; Yang, D. Org. Lett. 2009, 11, 5626–5628. (g) Dai, H.; Lu, X. Tetrahedron
Lett. 2009, 50, 3478–3481. (h) Linder, D.; Buron, F.; Constant, S.; Lacour, J. Eur. J. Org. Chem.
2008, 5778–5785. (i) Schiffner, J. A.; Machotta, A. B.; Oestreich, M. Synlett 2008, 2271–2274.
(j) Koskinen, A. M. P.; Oila, M. J.; Tois, J. E. Lett. Org. Chem. 2008, 5, 11–16. (k) Zhang, Y.;
Sigman, M. S. J. Am. Chem. Soc. 2007, 129, 3076–3077. (l) Yoo, K. S.; Park, C. P.; Yoon, C.
H.; Sakaguchi, S.; O’Neill, J.; Jung, K. W. Org. Lett. 2007, 9, 3933–3935. (m) Dhawan, R.;
Dghaym, R. D.; St. Cyr, D. J.; Arndtsen, B. A. Org. Lett. 2006, 8, 3927–3930. (n) Xu, W.; Kong,
A.; Lu, X. J. Org. Chem. 2006, 71, 3854–3858. (o) Malkov, A. V.; Stewart Liddon, A. J. P.;
Ramírez-López, P.; Bendová, L.; Haigh, D.; Kocovsky, P. Angew. Chem., Int. Ed. 2006, 45,
1432–1435. (p) Abrunhosa, I.; Delain-Bioton, L.; Gaumont, A.-C.; Gulea, M.; Masson, S.
Tetrahedron 2004, 60, 9263–9272. (q) Brunner, H.; Kagan, H. B.; Kreutzer, G. Tetrahedron:
Asymmetry 2003, 14, 2177–2187. (r) Cornejo, A.; Fraile, J. M.; García, J. I.; Gil, M. J.;
Herrerías, C. I.; Legarreta, G.; Martínez-Merino, V.; Mayoral, J. A. J. Mol. Catal. A: Chem.
2003, 196, 101–108. (s) Zhang, Q.; Lu, X.; Han, X. J. Org. Chem. 2001, 66, 7676–7684. (t)
Zhang, Q.; Lu, X. J. Am. Chem. Soc. 2000, 122, 7604–7605. (u) Perch, N. S.; Pei, T.;
Widenhoefer, R. A. J. Org. Chem. 2000, 65, 3836–3845. (v) Bremberg, U.; Rahm, F.; Moberg,
C. Tetrahedron: Asymmetry 1998, 9, 3437–3443. (w) Brunner, H.; Obermann, U.; Wimmer, P.
Organometallics 1989, 8, 821–826.
15 The ligand was prepared as described in the literature, see: Brunner, H.; Obermann, U. Chem.
Ber. 1989, 122, 499–507.
16 (a) Xu, Q.; Zhang, R.; Zhang, T.; Shi, M. J. Org. Chem. 2010, 75, 3935–3937; (b) Zhang,
T.; Shi, M. Chem.–Eur. J. 2008, 14, 3759–3764; (c) Gottummukkala, A. L.; Matcha, K.; Lutz,
M.; de Vries, J. G. ; Minnaard, A. J. Chem.–Eur. J. 2012, 18, 6907–6914.
17 Holder, J. C., Marziale, A. N., Gatti, M.; Mao, B.; Stoltz, B. M. Chem.–Eur. J. 2013, 19, 74–
77.
18 We believe the small amount of deuterium incorporation at the methylene adjacent the enone
occurs via substrate enolization during the extended reaction times under the mild reaction
conditions.
19 Duan, W.-L.; Iwamura, H.; Shintani, R.; Hayashi, T. J. Am. Chem. Soc. 2007, 129, 2130–
2138.

87
20
Guillaneux, D.; Zhao, S.-H.; Samuel, O.; Rainford, D.; Kagan, H. B. J. Am. Chem. Soc. 1994,
116, 9430–9439. Further evidence for the monomeric nature of the Pd/PyOx catalyst was
provided by comparison of diffusion rates of various PyOx/Pd complexes by diffusion oriented
spectroscopy (DOSY NMR). These studies suggest an upper bound for the molecular weight of
the solution species of the catalyst, demonstrating that dimeric (ML)2 complexes are not present.
21 (a) Inanaga, J.; Furuno, H.; Hayano, T. Chem. Rev. 2002, 102, 2211–2226. (b) Kagan, H. B.
Synlett 2001, 888–899. (c) Girard, C.; Kagan H. B. Angew. Chem., Int. Ed. 1998, 37, 2922–
2959.
22 Nishitaka, T.; Yamamoto, Y.; Miyaura, N. Organometallics 2004, 23, 4317–4324.
23 The catalyst itself is known to be soluble and not zero valent palladium nanoparticles due to
exclusion by a mercury drop test, see ref 17. For examples of the mercury drop test, see: (a) Ines,
B.; SanMartin, R.; Moure, M. J.; Dominguez, E. Adv. Synth. Catal. 2009, 351, 2124–2132. (b)
Ogo, S.; Takebe, Y., Uehara, K., Yamazaki, T., Nakai, H., Watanabe, Y., Fukuzumi, S,
Organometallics 2006, 25, 331–338.
24 Attempts to study the reaction by NMR initially saw no reaction progress due to the inability
to stir the immiscible reaction mixture in an NMR tube. Substitution of 2,2,2-trifluoroethanol
for water as the super stoichiometric proton source facilitated the reaction to proceed in the
absence of stirring with no detriment to the observed enantioselectivity, however the reaction
was necessarily performed at sufficiently elevated temperature that observation of the initial
rate was not tenable and, thus, kinetic study was abandoned.
25 Lan, Y.; Houk, K. N. J. Org. Chem. 2011, 76, 4905–4909.
26 For related computational studies on palladium-catalyzed conjugate additions of arylboronic
acids to enones: (a) Nishikata, T.; Yamamoto, Y.; Gridnev, I. D.; Miyaura, N. Organometallics
2005, 24, 5025–5032. (b) Sieber, J. D.; Liu, S.; Morken, J. P. J. Am. Chem. Soc. 2007, 129,
2214–2215. (c) Dang, L.; Lin, Z.; Marder, T. B. Organometallics 2008, 27, 4443–4454.
27 (a) Becke, A. D. J. Chem. Phys. 1993, 98, 1372–1377. (b) Becke, A. D. J. Chem. Phys. 1993,
98, 5648–5652. (c) Perdew, J. P.; Chevary, J. A.; Vosko, S. H.; Jackson, K. A.; Pederson, M. R.;
Singh, D. J.; Fiolhais, C. Phys. Rev. B 1992, 46, 6671–6687. (d) Perdew, J. P. Phys. Rev. B 1986,
33, 8822–8824.
28 Gaussian 03, Revision C.02, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A.
Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam,
S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A.
Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T.
Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross,
V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R.

88
Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J.
Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick,
A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J.
Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J.
Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B.
Johnson, W. Chen, M. W. Wong, C. Gonzalez, and J. A. Pople, Gaussian, Inc., Wallingford CT,
2004.
29 Here, it is assumed the isomeric trans and cis phenylpalladium complexes cannot undergo
rapid isomerization before alkene insertion and thus the enantioselectivity is determined by the
energy difference between 8-TS-A and 8-TS-B, since the transmetallation leading to the trans
isomer is favored (see SI). If trans/cis isomerization is faster than alkene insertion, the
enantioselectivity will be determined by the energy difference between 8-TS-A and 8-TS-D (98%
ee).
30 Other Lewis acids also proved incapable of catalyzing the reaction, including AlCl3 and
Sn(OTf)2.
31 These experiments were followed by NMR (
1H,
13C ), however no discrete intermediates were
successfully characterized.
32 Attempts to separate the isomeric mixture of phenylpalladium iodide complexes failed by
both conventional silica gel flash chromatography and preparatory HPLC.
33 Optimization of the cis/trans isomerization transition state of the cationic phenyl
palladium(II) complex failed to locate a TS. Scan of the reaction coordinate indicates the barrier
of isomerization is higher than 10 kcal/mol with respect to the cationic phenyl palladium
complex 11. This suggests the cis/trans isomerization via the dissociative mechanism via
isomerization of the tri-coordinated complex 11 cannot occur.
34 Computations indicated that alkene insertion to the minor isomer of phenyl palladium
complex, in which the Ph is cis to the oxazoline, yields very low enantioselectivity. The chiral
oxazoline is now trans to the enone, and thus the stereocontrol is diminished (see 1-TS-C and
1-TS-D in Figure 3.5).
35 A similar argument about imparted enantioselectivity can be made to rule out the catalytic
activity of palladium(0) nanoparticles. Additionally, a mercury(0) poisoning test has ruled out
the activity of palladium(0) nanoparticles in the Pd/PyOx manifold. See ref. 17.
36 Brønsted acid catalysis was also ruled out, as the substitution of trifluoroacetic acid for
Pd(OCOCF3)2 proved unable to catalyze the reaction. Protic acids are not tenable catalysts in the
absence of palladium salts.

89
37
We have computed the effects of coordination with phenylboronic acid to activate the
carbonyl on the enone in the alkene insertion step. No acceleration on alkene insertion was
observed computationally with either Lewis acid or hydrogen bonding coordination. (Result not
included).
38 The suggestion of boronic acid stabilization of arylpalladium cationic intermediates is
consistent with the observation that boron species lacking hydroxyl groups serve as poor
substrates for the reaction. For example, greatly diminished yields (and high degrees of biphenyl
formation) are observed with the use of NaBPh4 or KF3BPh as the phenyl donor species. (Not
included)
39 See Supporting Information of ref. 13 for details on the sensitivity of enantioselectivity of the
Pd/PyOx system to polar, coordinating solvents. The addition of 5 equiv. of alcoholic co-solvent
as a proton source is generally detrimental to the enantioselectivity and, occasionally, to the yield
of the reaction.
40 Shintani, R.; Yamagami, T.; Kimura, T.; Hayashi, T. Org. Lett. 2005, 7, 5317–5319.
41 (a) i-PrPyOx: Frauenlob, R.; McCormack, M. M.; Walsh, C. M.; Bergin, E. Org. Biomol.
Chem. 2011, 9, 6934–6937. (b) PhPyOx: Malkov, A. V.; Stewart-Liddon, A. J. P.; McGeoch, G.
D.; Ramirez-Lopez, P.; Kocovsk, P. Org. Biomol. Chem. 2012, 10, 4864–4877. (c) i-BuPyOx:
Brunner, H.; Obermann, U. Chem. Ber. 1989, 122, 499–507. (d) 4-OMePyOx, Jensen, K. H.;
Webb, J. D.; Sigman, M. S. J. Am. Chem. Soc. 2010, 132, 17471–17482. (e) 4-CF3PyOx: Jensen,
K. H.; Webb, J. D.; Sigman, M. S. J. Am. Chem. Soc. 2010, 132, 17471–17482. (f) 5-PhPyOx:
Brunner, H.; Obermann, U. Chem. Ber. 1989, 122, 499–507. (g) t-BuQuinOx: He, W.; Yip, K-T.;
Zhu, N-Y.; Yang, D. Org. Lett. 2009, 11, 5626–5628. (h) i-PrQuinOx: Zhang, Y.; Sigman, M. S.
J. Am. Chem. Soc. 2007, 129, 3076–3077.

90
Chapter 4. Stereoselectivities of Diels-Alder Reactions of 5-Substituted Cyclopentadienes: Effects
of Hyperconjugative Aromaticity on Reactivity and Stereoselectivity
4.1 Abstract
In the Diels-Alder cycloaddition of 5-substituted cyclopentadienes, the dienophile could add
either anti or syn to the substituents, which is the π-facial stereoselectivity. A computational analysis
based on distortion/interaction theory is conducted with SCS-MP2, which was employed due to its
agreement with the high-level composite theory G4. Activation and reaction energies are affected
substantially by the 5-substituents and characterized by the bending of C5-X out-of-plane angles to the
C1-C4-C5 plane. Due to the bending of C5-X bonds in the reactant dienes, attack of the dienophiles syn
to a donor and anti to an acceptor are favored since less distortion is required to reach the
corresponding transition states. The bending of C5-X out-of-plane angles in each substituted
cyclopentadiene results from the interactions between the π orbitals of diene and the 5-substituent.
4.2 Introduction
The Diels-Alder cycloaddition is one of the most versatile tools in organic synthesis.1-3
The
generation of complexity with high stereoselectivity provides considerable utility in synthesis and its
atom efficiency stands out in the pursuit of green chemistry.4 In addition to the throughly-studied
exo/endo stereoselectivity, π- facial stereoselectivity provides a potent method of asymmetric induction.
One general selectivity of this type involves the direction of approach of a dienophile to a 5-substituted

91
cyclopentadiene. (Scheme 4.1) When a dienophile, YZ, reacts with a 5-substituted cyclopentadiene,
either syn or anti adducts can be obtained.
Scheme 4.1. The π- facial stereoselectivity
Our interest in the π- facial stereoselectivity of Diels-Alder cycloaddition of 5-substituted
cyclopentadienes was rekindled by the late David Gin,5 who visited UCLA shortly before his tragic
death. In Gin’s total synthesis of aconitine alkaloids, the π- facial stereoselectivity in Diels-Alder
cycloaddition was employed as a key factor to obtain the desired product (3a). Scheme 4.2 shows the
reaction of a cyclopropene dienophile, which adds to the methoxy group on the cyclopentadiene
derivative, with a syn:anti ratio of around 6:1.
Scheme 4.2. Utilization of the π-facial stereoselectivity in total synthesis by David Gin
Despite of its broad application, the origin of the π-facial stereoselectivity remains controversial.
Among the effort of various groups, Burnell and his group first identified the importance of
deformation energy, which we call distortion energy, on stereoselectivities of these reactions. This

92
paper constitutes a theoretical update and an elaboration of the elegant theoretical studies based on
Hartree-Fock theory by Burnell. We developed a straightforward explanation and predictive model
based upon distortion/interaction theory.
4.3 Background
In 1955, the surprisingly contrasteric cycloaddition of 5-acetoxycyclopentadiene to ethylene and
other dienophiles was reported by the Winstein and Woodward groups.6 Since then, a great volume of
examples were identified.
The experimental studies are led by the Burnell group, along with extensive efforts from other
groups as well. Fallis7,8
performed a study of oxygen, sulfur and nitrogen 5-substituents and found that
when the C5 substituent involves N or O bonding to C-5, the dienophile tends to add syn to the
substituent, while a SH substitution on C5 gives mixed products and a SMe on C5 gives a 10:1 ratio on
anti/syn products. Burnell carried out extensive studies on π- facial stereoselectivities of various
5-substituents, and found that steric effects dominate in the absence of heteroatoms for 5-alkyl
cyclopentadienes.9,10
Their investigations of 5-methylcyclopentadiene Diels-Alder reactions revealed
that the stereoselectivity is relatively insensitive to the nature of dienophiles.11
They also studied the
inverse electron demand 1,2,3,4,5-pentachloro-5-X-cyclopentadienes and showed that the π- facial
stereoselectivity follows the same pattern as in nonchlorinated cyclopentadienes.12
Despite numerous experimental investigations, the origin of the π- facial stereoselectivity
remains unsettled, even controversial. The first rationale, in 1973, was Anh's proposal that
stereoselectivities are determined by a stabilizing interaction between the heteroatom lone-pair and the
dienophile π*-LUMO.13
This explains the syn preference of the acetoxy group, but the repulsive

93
interaction between the heteroatom n-orbitals and the diene π-HOMO was neglected. Furthermore,
related heteroatoms, such as the stronger donor S in SMe, give an anti preference.
The Burnell group expanded their experimental studies with extensive calculations with the
Hartree-Fock theory. With bulky hydrocarbon substitutions on C5, the steric effect is dominant and
overrides other factors in π-facial stereoselectivity, as established by Houk and Burnell. For smaller and
heteroatom substituents, Burnell identified the importance of deformation energy on the reactivities and
stereoselectivities of reactions of 5-substituted cyclopentadienes. They showed good correlations
between the diene deformation energies and activation energies. They also showed that the differences
between the syn and anti deformation energies correlate with the differences in syn and anti activation
energies. While the distortion energies of anti additions to 5-substituted cyclopentadienes are in a small
range around the cyclopentadiene, the syn distortion energies are in a much wider range. The
dienophile approaches preferentially at the face with a smaller change of C1-C5-X angle. A modified
steric factor in the reactant diene describing the bond size of C5-X has been proposed. In spite of theses
correlations, the origin of the distortion from the reactant to the transition state (TS) is still unknown.
Various theories attempt to rationalize the π-facial stereoselectivity other than distortion.
Inagaki elaborated the Fukui orbital mixing scheme and argued that syn/anti facial selectivity comes
from the tilting of the orbitals of C1 and C4 in the diene π*-LUMO. This tilting is the consequence of
σ-p orbital mixing between the C1/C4 σ orbital and the π* orbital of the cyclopentadiene, induced by
the heteroatom lone pair p orbital. This argument applies as well for electronegative 5-substituents.14,15
Inagaki extended the arguments to both electronegative and less electronegative heteroatom
substituents.16
Cieplak rationalized the syn preference to the better donor ability of C5-X than C5-H.17
Cieplak’s theory always favors bond formation anti to the best donor, which would lead to a wider

94
range in anti activation energies. This seems quite contrary to the experiment results and to Burnell’s
calculations.
Electrostatic effects have been proposed as an alternative explanation. Kahn and Hehre argues
that the more nucleophilic face of diene is subject to attack by the electron-poor dienophile,18
but it
does not apply to inverse electron demand Diels-Alder additions.12
In summary, the origin of π- facial stereoselectivity is still uncertain. “The general applicability
and predictive value of the proposed explanations are not obvious at the present stage.”19
We have revisited this problem with several types of density functional and ab initio wave
function based methods, and have come up with a new general explanation of this phenomenon. We
report a theoretical exploration of these reactions using high-accuracy methods, both DFT and ab initio.
The distortion/interaction theory, combined with the effects of substituents on the structures of
5-substituted cyclopentadienes, provide a simple explanation on the origin of the π- facial
stereoselectivity.
4.4 Computational Details
All the calculations were performed with Gaussian 09.20
The B3LYP density functional21,22
was
employed with the 6-31G(d) basis set for geometry optimizations. Single point energies were obtained
with a larger 6-311++G(d,p) basis set. Various density functional methods (DFTs) as well as some
wave-functional-based methods (WFTs) and multi-determinant and high level composite method were
tested here. B3LYP has been widely used for geometry optimizations and predicting relative energies,
but to calculate absolute energies, it is subject to significant systematic errors, most relevantly to this
work, on the Diels-Alder reaction of cyclopentadiene and ethene.23
In addition to B3LYP, M06-2X24

95
was tested here since it has been shown to provide more accurate thermodynamics for main-group
elements. B2PLYP-D25,26
was also applied, which incorporates the dispersion correction in the
functional. Furthermore, Grimme’s WFT-based spin-component-scaled second-order Møller-Plesset
perturbation theory (SCS-MP2),27
which exhibits a significant improvement over the original MP2,28
was also used here. High-level composite methods, G3B3,29
CBS-QB3,30,31
and G432
were employed to
compare with the DFT results.33,34
The latest development in the high-level composite method series,
G4, is based on a sequence of single point energy calculations and give a total energy effectively at
CCSD(T,full)/G3LargeXP+HFlimt level. It is applicable for compounds containing first-, second-, and
thired-row main atoms, and exhibits the least mean absolute deviation from 454 experimental energies
in the G3/05 test set, as low as 0.83 kcal/mol, which is accurate enough to beyond the experimental
limit. Due to its accuracy, G4 energies are taken here as the reference values.
4.5 Results and Discussions
4.5.1 Comparison of various theoretical levels
We have investigated the substituent effects on the activation free energies in Scheme 4.3 with 17
5-substituted cyclopentadienes reacting with ethene. There are a total of 35 reactions since ethene
attacking in both syn and anti fashions have been considered except the parent reaction, which is
cyclopentadiene reacting with ethene. The substituents range from a potent π-donor like OMe, to a
strong π-acceptor like BH2. Activation energies in each reaction are calculated with the highest level
method G4 as a reference, along with other high-accuracy composite methods including G3B3 and
CBS-QB3, and the density functional theories including B3LYP, M06-2X, SCS-MP2, B3PLYP-D. For
comparison, free energies was also calculated from the HF/6-31G(d) single point and HF.6-31G(d)

96
geometry, as adopted in Burnell’s previous analysis. The predicted activation energies by various
theoretical methods are plotted against the G4 values. (Figure 4.1)
Scheme 4.3. Benchmarking reactions in anti/syn fashion

97
yHF = 0.82x + 29.4
R2 = 0.76
yB2PLYP-D = 0.81x + 14.9
R2 = 0.82
yB3LYP = 0.98x + 7.3
R2 = 0.95
yM06-2X = 1.10x - 3.8
R2 = 0.98
ySCS-MP2 = 0.96x + 1.1
R2 = 0.99
yG3B3 = 0.97x + 0.3
R2 = 1.00
yMP2 = 1.24x - 15.6
R2 = 0.96
yCBS-Q B3 = 0.99x - 1.6
R2 = 0.99
15.0
20.0
25.0
30.0
35.0
40.0
45.0
50.0
55.0
60.0
65.0
25.0 30.0 35.0 40.0
HF
B2PLYP-D
B3LYP
SCS-MP2
G3B3
M06-2X
CBS-QB3
MP2

98
Figure 4.1. Calculated activation free energies (ΔGX‡) in 35 reactions (kcal/mol). The regression
equation is ΔGX‡ (method) = slope * ΔGX
‡ (G4) + interception.
High-level composite theoretical methods G3B3, CBS-QB3 and G4 agree well with each other.
CBS-QB3 consistently underestimates the free energies by 2.1 kcal/mol. SCS-MP2, G3B3 and
M06-2X give excellent agreement with the G4 values, with 0.3 kcal/mol, 0.5 kcal/mol and 0.6 kcal/mol
MAD, respectively, while the computational cost of SCS-MP2 and M06-2X are much lower.
The HF, B2PLYP-D, and B3LYP barriers are overestimated by around 24 kcal/mol, 9 kcal/mol,
and 7 kcal/mol, respectively. The failure of HF absolute barriers is probably due to the neglect of
correlation energy, while MP2 does not provide an accurate prediction of correlation energies and the
barriers are underestimated by 8 kcal/mol. Therefore, these methods are not suitable for predicting
absolute barriers.
The slope is 0.99 for CBS-QB3, closest to 1, followed by B3LYP (0.98), G3B3 (0.97), and
SCS-MP2 (0.96). These methods are acceptable to predict relative barriers. On the other hand, M06-2X
has a higher slope of 1.10, while MP2 is the highest at 1.24. HF and B2PLYP-D give the smallest slope
of 0.82 and 0.81, respectively. The HF values are scattered around the linear regression line, with the
most significant deviation coming from a 2 kcal/mol underestimation at the lower end for F (syn) and
the higher end for BH2 (both syn and anti). B2PLYP fails in these cases as well, with a 1.5 kcal/mol
underestimation. Therefore the overall slope is decreased greatly for these two methods.
To further test the performance of the theories aforementioned on the π-facial
stereoselectivities, relative anti/syn free energies are compared with G4 values (Figure 4.2).
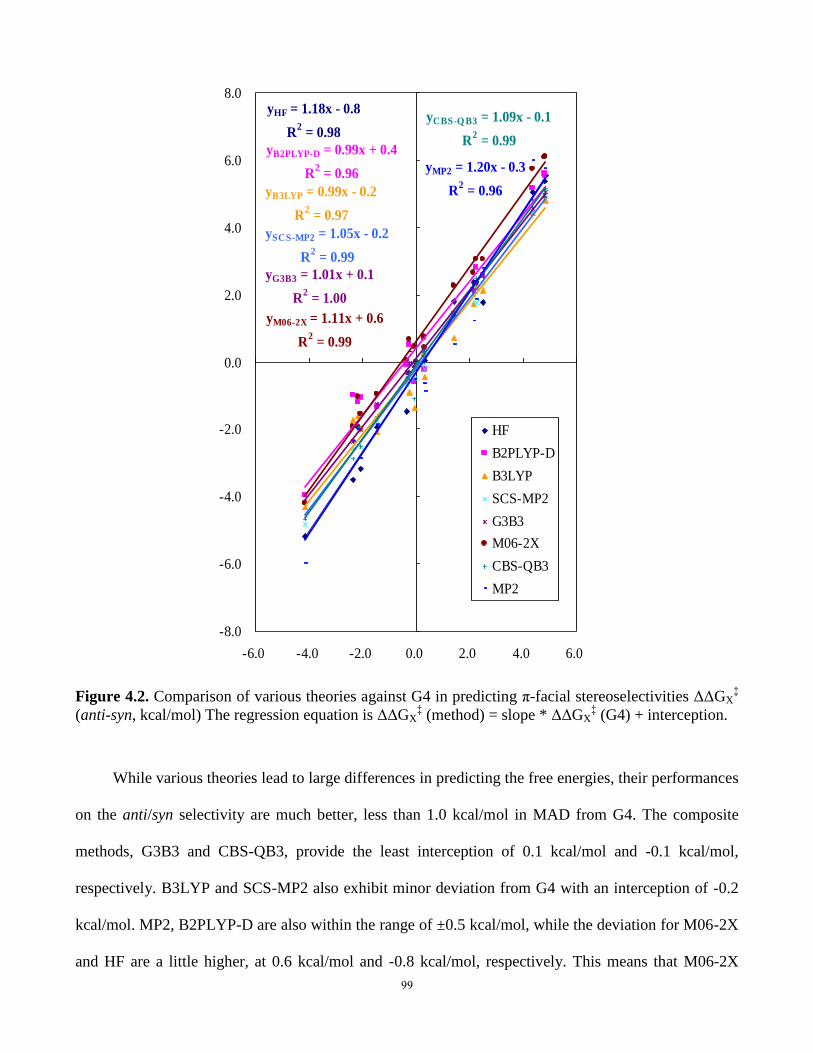
99
Figure 4.2. Comparison of various theories against G4 in predicting π-facial stereoselectivities ΔΔGX‡
(anti-syn, kcal/mol) The regression equation is ΔΔGX‡ (method) = slope * ΔΔGX
‡ (G4) + interception.
While various theories lead to large differences in predicting the free energies, their performances
on the anti/syn selectivity are much better, less than 1.0 kcal/mol in MAD from G4. The composite
methods, G3B3 and CBS-QB3, provide the least interception of 0.1 kcal/mol and -0.1 kcal/mol,
respectively. B3LYP and SCS-MP2 also exhibit minor deviation from G4 with an interception of -0.2
kcal/mol. MP2, B2PLYP-D are also within the range of ±0.5 kcal/mol, while the deviation for M06-2X
and HF are a little higher, at 0.6 kcal/mol and -0.8 kcal/mol, respectively. This means that M06-2X
yHF = 1.18x - 0.8
R2 = 0.98
yB2PLYP-D = 0.99x + 0.4
R2 = 0.96
yG3B3 = 1.01x + 0.1
R2 = 1.00
yB3LYP = 0.99x - 0.2
R2 = 0.97
ySCS-MP2 = 1.05x - 0.2
R2 = 0.99
yM06-2X = 1.11x + 0.6
R2 = 0.99
yCBS-Q B3 = 1.09x - 0.1
R2 = 0.99
yMP2 = 1.20x - 0.3
R2 = 0.96
-8.0
-6.0
-4.0
-2.0
0.0
2.0
4.0
6.0
8.0
-6.0 -4.0 -2.0 0.0 2.0 4.0 6.0
HF
B2PLYP-D
B3LYP
SCS-MP2
G3B3
M06-2X
CBS-QB3
MP2

100
gives systematic overestimation of ΔG‡
a-s favoring syn attack by around 0.6 kcal/mol, while HF
systematically gives 0.8 kcal/mol preference for anti attack. The slopes for G3B3, B3LYP and
B2PLYP-D are closest to 1, being 1.01, 0.99 and 0.99, respectively, followed by a slightly higher slope
of 1.05 with SCS-MP2. Considering both the interceptions and the slopes, G3B3 shows the best
agreement with G4, while B3LYP is the best choice between the non-composite methods, follows
closely by B2PLYP-D and SCS-MP2.
Our analysis with distortion/interaction theories requires an economic and accurate method that is
able to provide both quantitative estimation of absolute energies, and the anti/syn stereoselectivities.
SCS-MP2 provides lowest MAD and SD for absolute barriers, and performance the second best on the
anti/syn stereoselectivities. Due to its excellent performance both on absolute and relative activation
free energies, SCS-MP2 has been employed in the following studies in the present work. All the
energies were calculated by SCS-MP2/6-311++G(d,p)//B3LYP/6-31G(d), unless otherwise specified.
4.5.2 π-Facial stereoselectivity and geometrical distortions of reactants and transition states,
characterized by the C5-X out-of-plane angle
To trace the origin of differences in anti/syn activation barriers, we analyzed the TS geometries
with 5-substituted cyclopentadiene models, including cyclopentadiene (4), 5-methoxycyclopentadiene
(5), and 5-methborylcyclopentadiene (6). The TS structures for ethene with 4 (TS4), with 5 in anti/syn
fashion (TS5a, TS5s), and with 6 in anti/syn fashion (TS5a, TS5s), were plotted in Figure 4.3.
A parameter θ, characterizing the bending of the substituent groups X, is the C5-X out-of plane
angle with respect to the plane defined by C1, C4 and C5. (Scheme 4.4)
Scheme 4.4. The C5-X out-of-plane angle, θ
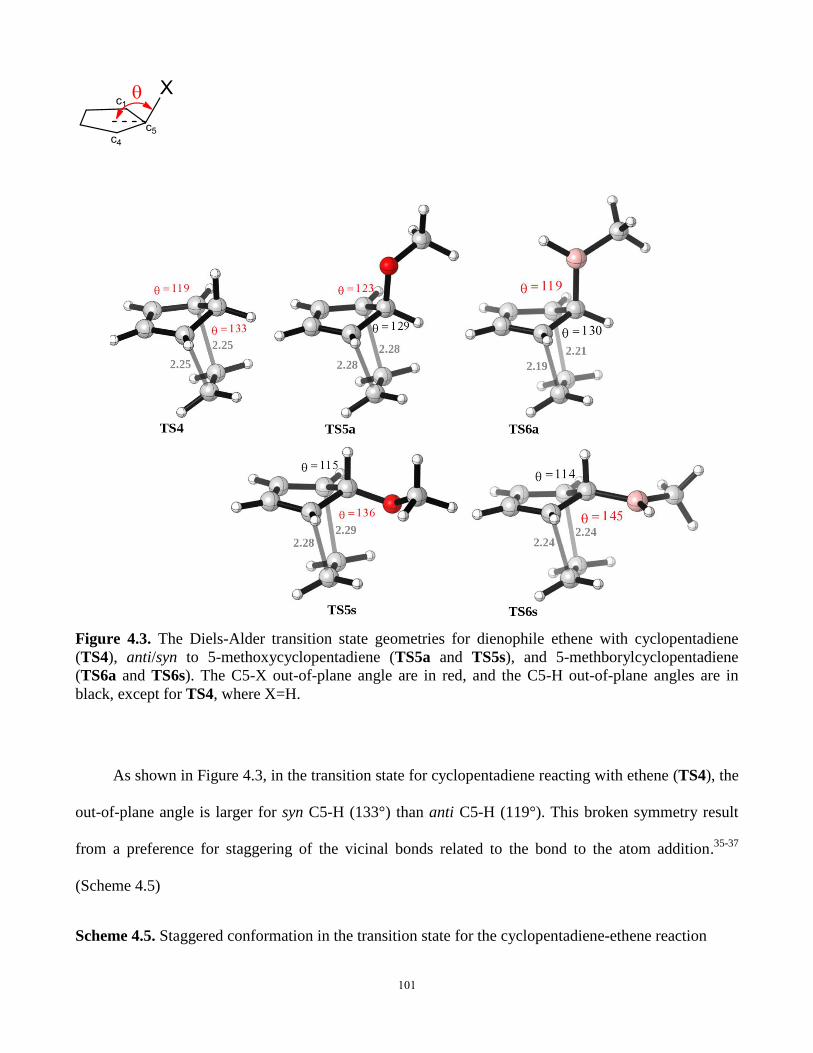
101
Figure 4.3. The Diels-Alder transition state geometries for dienophile ethene with cyclopentadiene
(TS4), anti/syn to 5-methoxycyclopentadiene (TS5a and TS5s), and 5-methborylcyclopentadiene
(TS6a and TS6s). The C5-X out-of-plane angle are in red, and the C5-H out-of-plane angles are in
black, except for TS4, where X=H.
As shown in Figure 4.3, in the transition state for cyclopentadiene reacting with ethene (TS4), the
out-of-plane angle is larger for syn C5-H (133°) than anti C5-H (119°). This broken symmetry result
from a preference for staggering of the vicinal bonds related to the bond to the atom addition.35-37
(Scheme 4.5)
Scheme 4.5. Staggered conformation in the transition state for the cyclopentadiene-ethene reaction

102
When one of the C5-H is substituted by a methoxy or methboryl group, the transition states have
subtle changes in θ on the anti/syn π-faces. Compared with the θ anti to ethene in TS4 (119°), in anti
TSs, it is slightly increased to 123° in TS5a, while it stays the same in TS6a. θ syn to ethene are
slightly decreased from 133° in TS4 to 129° in TS5a and 130 in TS6a. Overall, the changes in θ are
smaller then 5° in anti TSs with the representative 5-substituents, both a strong π-donor like MeO and a
strong π-acceptor like MeBH. On the other hand, the changes in θ with 5-substitution are greater for
syn TSs. The θ anti to ethene in TS4 decreases to 115° in TS5s and 114° in TS6s, while the θ syn to
ethene increases to 136° in TS5s and, most notable, 145° in TS6s.
Other than the change of C5-X and C5-H bending angles, the forming C-C bond distance increases
from 2.25 in TS4 to 2.28 in both TS5a and TS5s with the substitution of MeO, denoting earlier TS, and
decreases to 2.20 in TS6a and almost the same (2.24) in TS6s.
Overall the changes of TS geometries are small, even with the 5-substituents of vastly different
electron demand. The change of out-of-plane angles are within 5° except for the C5-B θ in syn TS
(TS6s), with the changes in anti TSs even smaller than changes in syn TSs. The forming C-C bond
length does change slightly, but it is almost the same for 5 in both anti/syn TS.
Since the activation barrier is the energetic differences between TS and reactant, and the TS
structures are similar, the π-facial stereoselectivity lies in the difference in the reactants. With the same
dienophile ethene, the difference in the diene was analyzed.

103
Figure 4.4. The geometries of reactant diene. Cyclopentadiene (4), 5-methoxycyclopentadiene (5). and
5-methborylcyclopentadiene (6). The C5-X out-of-plane angle are labeled in red. The C5-H
out-of-plane angles are in black, except for 4, where X=H.
Figure 4.4 shows the geometries of the cyclopentadienes 4, 5, and 6. While the C5-X out-of-plane
angles in the transition states are similar, they are quite different in the corresponding reactants. θ in the
symmetrical diene 4 is 127°, while that of 5 is asymmetrical, with the C5-O bond bending away from
the π system, increasing θ to 137°. As we will discuss later, the bending of θ results from a strong
interaction between the 5-substituent and the π system. Due to the bending of C5-O θ, the C5-H θ in 5
also decreases to 119°. On the other hand, C5-B out-of-plane angle θ in 6 has decreased substantially to
109°, while C5-H θ in 6 has increased to 143°.
Comparing with the anti and syn addition transition states in Figure 4.3, the C5-O θ of the reactant
5 (137°) resemble more like that in TS5s (136°) than TS5a (123°). Due to the geometrical similarity,
lower activation barrier for the former is expected. This is supported by our SCS-MP2 computation that
the activation energies are 12.4 kcal/mol (TS5s) and 17.4 kcal/mol (TS5a). On the other hand, the
C5-B θ in reactant 6 is drastically decreased, thus requiring substantial bending out for both anti and
syn TS, however, the difference in θ between 6 (109°) and TS6s (145°) is much greater than TS6a
(119°). Therefore lower barrier for the latter is expected. Indeed our calculation gives 22.4 kcal/mol
and 18.9 kcal/mol for TS6s and TS6a, respectively.
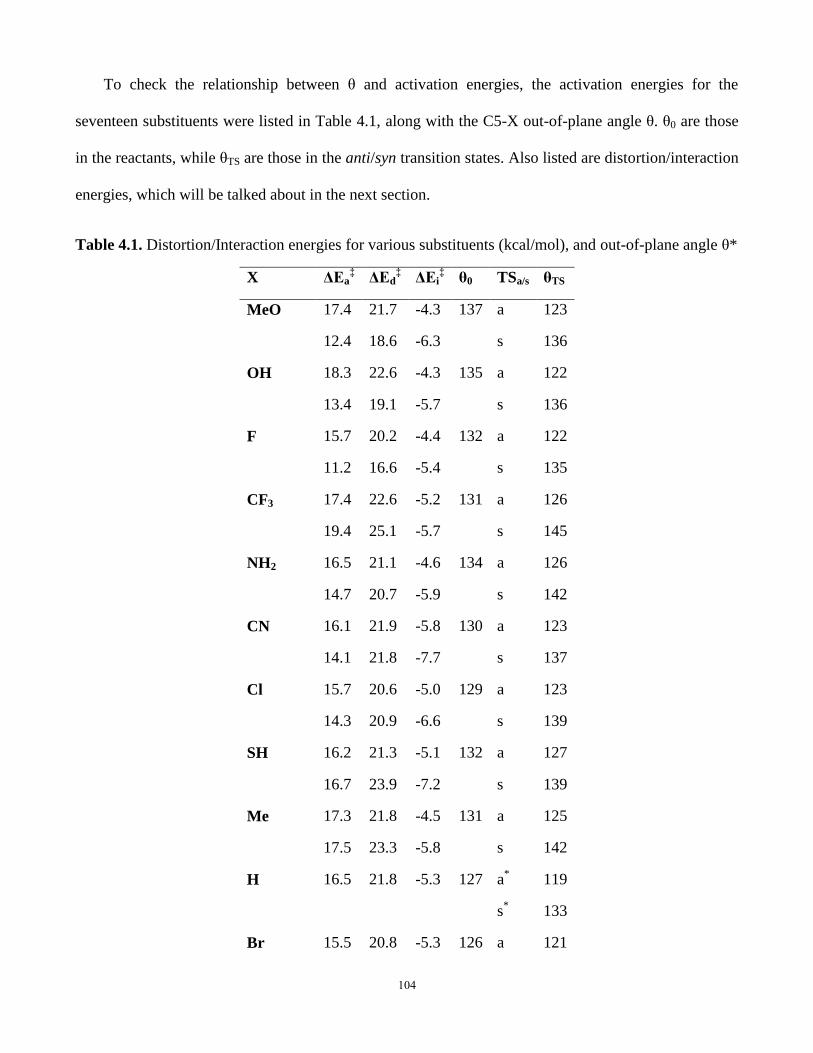
104
To check the relationship between θ and activation energies, the activation energies for the
seventeen substituents were listed in Table 4.1, along with the C5-X out-of-plane angle θ. θ0 are those
in the reactants, while θTS are those in the anti/syn transition states. Also listed are distortion/interaction
energies, which will be talked about in the next section.
Table 4.1. Distortion/Interaction energies for various substituents (kcal/mol), and out-of-plane angle θ*
X ΔEa‡ ΔEd
‡ ΔEi
‡ θ0 TSa/s θTS
MeO 17.4 21.7 -4.3 137 a 123
12.4 18.6 -6.3 s 136
OH 18.3 22.6 -4.3 135 a 122
13.4 19.1 -5.7 s 136
F 15.7 20.2 -4.4 132 a 122
11.2 16.6 -5.4 s 135
CF3 17.4 22.6 -5.2 131 a 126
19.4 25.1 -5.7 s 145
NH2 16.5 21.1 -4.6 134 a 126
14.7 20.7 -5.9 s 142
CN 16.1 21.9 -5.8 130 a 123
14.1 21.8 -7.7 s 137
Cl 15.7 20.6 -5.0 129 a 123
14.3 20.9 -6.6 s 139
SH 16.2 21.3 -5.1 132 a 127
16.7 23.9 -7.2 s 139
Me 17.3 21.8 -4.5 131 a 125
17.5 23.3 -5.8 s 142
H 16.5 21.8 -5.3 127 a* 119
s* 133
Br 15.5 20.8 -5.3 126 a 121

105
16.0 21.8 -5.9 s 138
NO 14.1 19.9 -5.8 116 a 115
14.5 21.2 -6.7 s 132
CH=CH2 16.2 21.3 -5.1 128 a 123
17.1 23.3 -6.2 s 141
CHO 15.3 20.6 -5.3 120 a 120
17.4 24.9 -7.5 s 140
C≡CH 16.9 22.2 -5.3 132 a 124
14.5 21.4 -7.0 s 139
MeBH 18.9 24.6 -5.7 109 a 118
22.4 29.7 -7.3 s 145
BH2 20.0 25.7 -5.7 110 a 116
23.9 30.7 -6.8 s 144
SiH3 17.7 23.4 -5.7 120 a 123
23.0 29.5 -6.5 s 144
* The hydrogen syn or anti to the approaching ethene.
C5-substitutions affect more on the reactant structures than in the TS structures, while the effect is
larger for syn than anti TS. θ0 covers a range of 28°, about twice as large as those of syn and anti θTS,
which are only 13° and 12°, respectively. Substituents involving boron, and the nitrosyl group,
significantly affect the angle and the values lie far from other substituents, which makes the general
trend more difficult to observe. Taking them out make a better statistics of θ0 range from 120°-137°,
while syn and anti θTS range from 135°-145° and 122°-127°, respectively. Therefore, the range of θ0,
θTS (syn) and θTS (anti) are 17:10:5. The anti θTS for the strongest π-donor MeO and strongest acceptor
SiH3 are both 123°, the syn θTS are close (142° and 144°, respectively), while the θ0 with MeO and SiH3
differ by 17° (137° and 120°, respectively).

106
The observation agrees with the activation energies in Table 4.1 that anti activation energies are
similar, while the syn activation energies span a much wider range. The anti activation energies for the
strongest π-donor MeO and strongest acceptor SiH3 are quite close, which are 17.4 and 17.7 kcal/mol,
while the syn activation energies for MeO and SiH3 differ by over 10 kcal/mol, which are 12.4 and 23.0
kcal/mol, respectively. Another example is the halogen series. Anti activation energies for F, Cl, Br are
almost identical, which are 15.7, 15.7 and 15.5 kcal/mol, respectively, while their corresponding syn
activation energies are quite different, which are 11.2, 14.3 and 16.0 kcal/mol. Therefore, the π-facial
stereoselectivity is determined mainly through the reactants rather than transition states.
4.5.3 Distortion/Interaction energies in π-facial stereoselectivity
To provide a quantitative analyze of the activation energy ΔEa‡, we break it into distortion and
interaction energies. The distortion energy ΔEd‡, is the energy required to distort the reactants into the
corresponding TS structure, and the interaction energy ΔEi‡ is the change in energy from the reactant
electron configuration to that of TS. To provide a quantitative account of the geometric and
electrostatic effects in the activation barriers, we applied the distortion/interaction theory to all the 35
reactions and the results are combined with activation energies in Table 4.1.
As demonstrated in Table 4.1, activation energies are dominated by distortion energies, since they
are normally four to five times larger in magnitude than the interaction energies. As illustrated in
Figure 4.5, ΔEd‡ and ΔEa
‡ exhibit excellent linear relationship, while 5-X substituents affects syn TS
more profoundly than the anti TS.
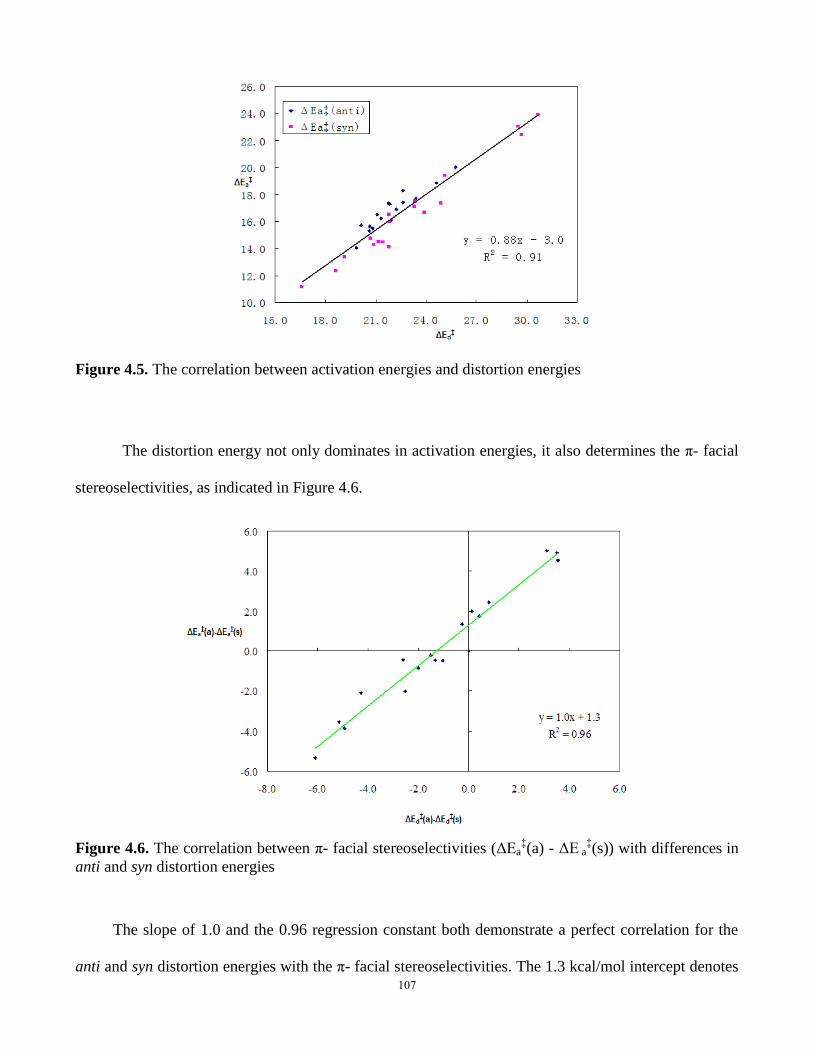
107
Figure 4.5. The correlation between activation energies and distortion energies
The distortion energy not only dominates in activation energies, it also determines the π- facial
stereoselectivities, as indicated in Figure 4.6.
Figure 4.6. The correlation between π- facial stereoselectivities (ΔEa‡(a) - ΔE a
‡(s)) with differences in
anti and syn distortion energies
The slope of 1.0 and the 0.96 regression constant both demonstrate a perfect correlation for the
anti and syn distortion energies with the π- facial stereoselectivities. The 1.3 kcal/mol intercept denotes
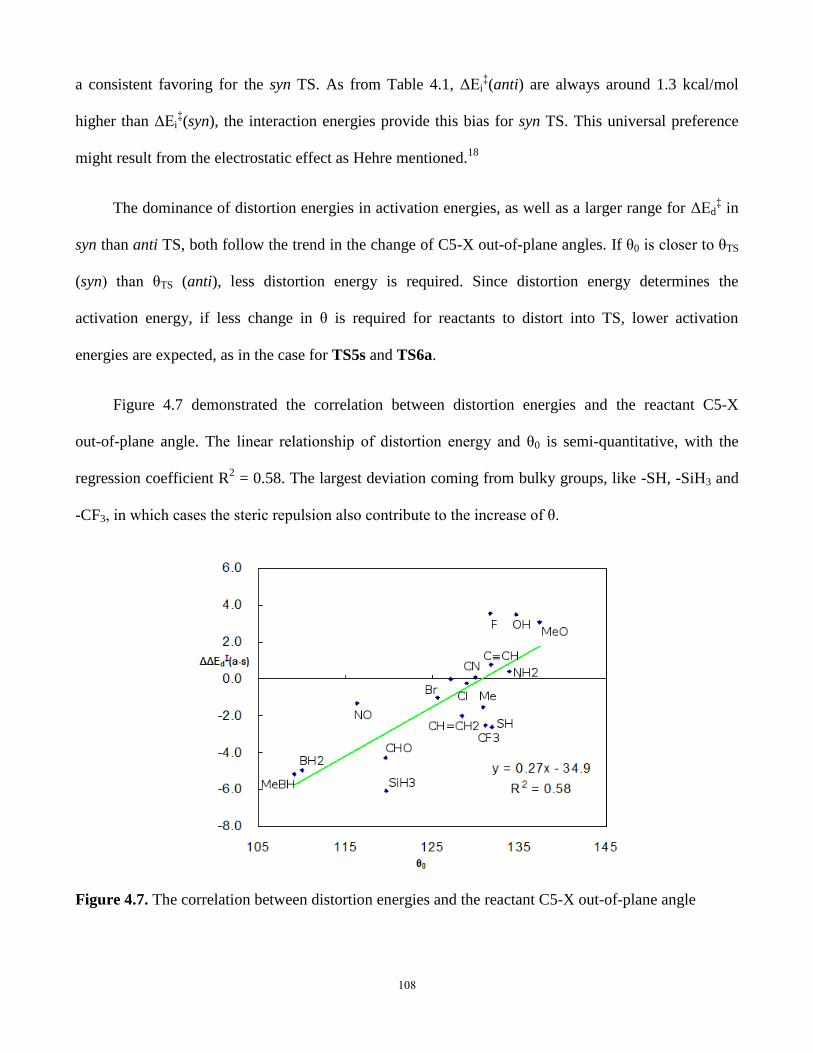
108
a consistent favoring for the syn TS. As from Table 4.1, ΔEi‡(anti) are always around 1.3 kcal/mol
higher than ΔEi‡(syn), the interaction energies provide this bias for syn TS. This universal preference
might result from the electrostatic effect as Hehre mentioned.18
The dominance of distortion energies in activation energies, as well as a larger range for ΔEd‡ in
syn than anti TS, both follow the trend in the change of C5-X out-of-plane angles. If θ0 is closer to θTS
(syn) than θTS (anti), less distortion energy is required. Since distortion energy determines the
activation energy, if less change in θ is required for reactants to distort into TS, lower activation
energies are expected, as in the case for TS5s and TS6a.
Figure 4.7 demonstrated the correlation between distortion energies and the reactant C5-X
out-of-plane angle. The linear relationship of distortion energy and θ0 is semi-quantitative, with the
regression coefficient R2 = 0.58. The largest deviation coming from bulky groups, like -SH, -SiH3 and
-CF3, in which cases the steric repulsion also contribute to the increase of θ.
Figure 4.7. The correlation between distortion energies and the reactant C5-X out-of-plane angle

109
Although the correlation is only semi-quantitative, a simple “127°” criteria works for most cases,
even including some of the bulky groups. The reference θ angle of cyclopentadiene is 127°. For
5-substituted cyclopentadienes, if θ0 of the reactant is above 127°, the reaction proceeds in the syn
fashion, while if θ0 of the reactant is below 127°, the reaction proceeds in the anti fashion. If the θ0 of
the reactant is close to 127°, mixed products is expected. In spite of its simplicity, the “127°” criterion
works for almost all the systems. X = -SH, –CF3 and -CH=CH2 seems to be the only exceptions. The
first two are bulky groups and the cyclopentadiene has to make more distortion in the syn transition
state. In these cases, θ0 gets larger due to the bulky substituent, as reflected in the increasing of θTS in
both syn and anti transition states. X = -CH=CH2 is another exception since the vinyl α-hydrogen is
pointing towards the approaching ethene.
In summary, substituents affect the C5-X out-of-plane angles on the reactants, with those with
increased θ resemble transition states in the syn fashion, and vice versa, leading to the π- facial
stereoselectivity, as in Scheme 4.6.
Scheme 4.6. Substituent effects on the reactant geometries leading to π- facial stereoselectivity
4.5.4 Reaction energies and the reaction pathways
On average, C5-X θTS in syn and anti transition states differs by around 15° (Table 4.1), the
differences in the corresponding products are much smaller. For example, θrxn is only slightly different

110
(1°) on anti/syn faces when X=H, while θTS in different faces differ by 14° when X=H. In addition,
while syn θTS span a much larger range than anti, anti and syn θrxn span similar ranges.
Table 4.2. Reaction energies and C5-X out-of-plane angles (θrxn) for various substituents (kcal/mol),
and the relative (anti - syn) reaction energies (ΔΔErxn) and activation energies (ΔΔEa‡)
X ΔErxn θrxn ΔΔErxn ΔΔEa‡
MeO a -35.8 128 3.5 5.0
s -39.3 126
OH a -37.8 131 0.7 4.9
s -38.5 126
F a -37.9 129 3.3 4.6
s -41.2 125
CF3 a -31.4 131 0.5 -2.0
s -32.0 135
NH2 a -35.6 132 0.8 1.8
s -36.4 126
CN a -35.2 129 1.1 2.0
s -36.3 129
Cl a -36.0 129 2.5 1.4
s -38.5 129
SH a -33.8 133 0.4 -0.4
s -34.2 130
Me a -32.0 131 0.1 -0.2
s -32.1 132
H a -32.2 125 0.0 0.0
s 126
Br a -34.7 128 2.1 -0.5
s -36.8 128

111
NO a -34.7 125 1.5 -0.5
s -36.2 124
CH=CH2 a -32.6 130 0.0 -0.9
s -32.6 131
CHO a -31.4 129 -0.2 -2.1
s -31.2 132
C≡CH a -34.1 130 1.6 2.4
s -35.7 130
MeBH a -24.9 132 -0.6 -3.6
s -24.3 138
BH2 a -23.7 130 -0.6 -3.9
s -23.0 139
SiH3 a -26.3 129 -0.8 -5.4
s -25.5 135
Table 4.2 listed θ values in syn and anti products. The order of the substituents followed the
decreasing syn preference in activation energies by G4, as we did in Table 4.1. For the top five
substituents, which are the strongest π-donors that have N, O or F connecting to C5, anti θrxn is around
4° larger than syn θrxn. On the other hand, the last three substituents in Table 4.2, which are the
strongest π-acceptors, give anti θrxn that are around 6° smaller than the corresponding syn θrxn. θ values
in syn and anti products are almost the same for other substituents.
The syn and anti reaction energies are generally within 1.0 kcal/mol to each other, with the
largest differences occur for MeO and halogen substituents. Although F, Cl and Br lead to very
different π-facial selectivities, as indicated by a range of 5.1 kcal/mol for there ΔΔEa‡(anti - syn), there
relative reaction energies are quite close, with a range of only 1.2 kcal/mol. The rather constant relative
reaction energies, ΔΔEa‡(anti - syn), concur with the trend in θrxn.

112
Figure 4.8. The correlation between activation energies (Y axis) and reaction energies (X axis)
For a specific substituent, the syn/anti reaction energies are closer to each other comparing with
the differences in activation energies, while reaction energies from different substituents span a larger
range then the activation energies, as shown in the different X/Y scales in Figure 4.8. For the syn
transition states, the activation energies correlate better with the reaction energies, while the correlation
for anti transition states is much poorer.
Electron donating groups on C5, such as MeO, increases the activation energy, or destabilize
the diene, while electron-accepting groups like BHMe stabilize the diene. The least reaction energies
for 5-X cyclopentadiene Diels-Alder cycloaddition are around -25 kcal/mol for substituents BHMe,
BH2 and SiH3, and the highest ones are around -40 kcal/mol for F, OMe and OH syn attack.
y = 0.68x + 39.5
R2 = 0.95
y = 0.21x + 23.7
R2 = 0.38
10.0
15.0
20.0
25.0
-45.0 -40.0 -35.0 -30.0 -25.0 -20.0
ΔEa‡(anti)
ΔEa‡(syn)

113
Figure 4.9. The reaction pathways for ethene with cyclopentadiene (4), methoxycyclopentadiene (5)
and methborylcyclopentadiene (6) The reactants has been set as 0 kcal/mol in each pathway.
With the calculated energies, the complete reaction pathways for ethene with the parent
cyclopentadiene (4), and substituted diene with a strong π-donor (5) or π-acceptor (6), were plotted
together in Figure 4.9. Although the electronic demand is very different, the barriers for the two anti
TS, TS5a and TS6a, are close to each other, while the syn activation energies differ by 10 kcal/mol for
TS5s and TS6s. The reaction energy decreases greatly for Prod5s, but less so for Prod5a, while the
reaction energy for Prod6a and Prod6s are almost the same, both around 7.5 kcal/mol the that of
Prod4.

114
4.5.5 The origin of substituent-induced bending in the reactants – second order orbital
interaction
We attribute the bending of out-of-plane angle to a strong second order orbital interaction
between the 5-substituent and the π system. When there are only filled orbitals, like the nonbonding
orbitals on electronegative species like N, O, F, or Cl, the interaction with the π-HOMO of diene raises
the energy, since the interaction of two symmetry matched filled orbitals is unfavored. To minimize the
interaction, C5-X tends to move away from the diene, therefore increasing θ0 of the reactant. In the
case of X=MeO, this interaction is eliminated in the syn product Prod5s but only partially in the anti
product Prod5a, with the the energy of Prod5s even lower than Prod5a. The interaction of the lone
pairs on X with the π*-LUMO of diene could be favored, but their symmetry does not match and thus
not considered.
On the other hand, if there is a vacant orbital close in energy on X and matches the symmetry of
π-HOMO, the interaction aforementioned becomes favorable and C5-X tend to move towards the diene
to maximize the interaction, therefore decreasing θ0 of the reactant. This interaction stabilizes the
reactant and therefore less reaction energies were released in Prod6a and Prod6s.
To test on the proposed rationale, the diene HOMO orbital graphs are plotted in Figure 4.10.
The cyclopentadiene serves as a reference, MeO as a typical π-electron donor since it has nonbonding
lone pair electrons, and MeBH as a typical π-electron acceptor since it has vacant p orbitals.

115
Figure 4.10. Orbital interactions on model systems (X= H, –OMe, –BHMe)
For methoxycyclopentadiene, the interaction between oxygen lone pair and HOMO of diene is
repulsive and C5-X bend away from the diene moiety to minimize orbital mixing. For
5-MeBH-cyclopentadiene, the interaction between boron vacant p orbital and π-HOMO of diene is
favored and C5-X bend towards the diene moiety for enhanced orbital mixing.
4.5.6 Diels-Alder reaction energies and hydrogenation energies
Since hydrogenation of the 5-X cyclopentadienes eliminates the aforementioned secondary
orbital interactions between the substituent and the π system, turning the two conjugated double bond
into one, just the Diels-Alder reaction with ethene, a comparison of the reaction energy of two reactions
is made, as in Scheme 4.7.
Scheme 4.7. Comparison of 5-substituted cyclopentadiene Diels-Alder reactions and hydrogenations
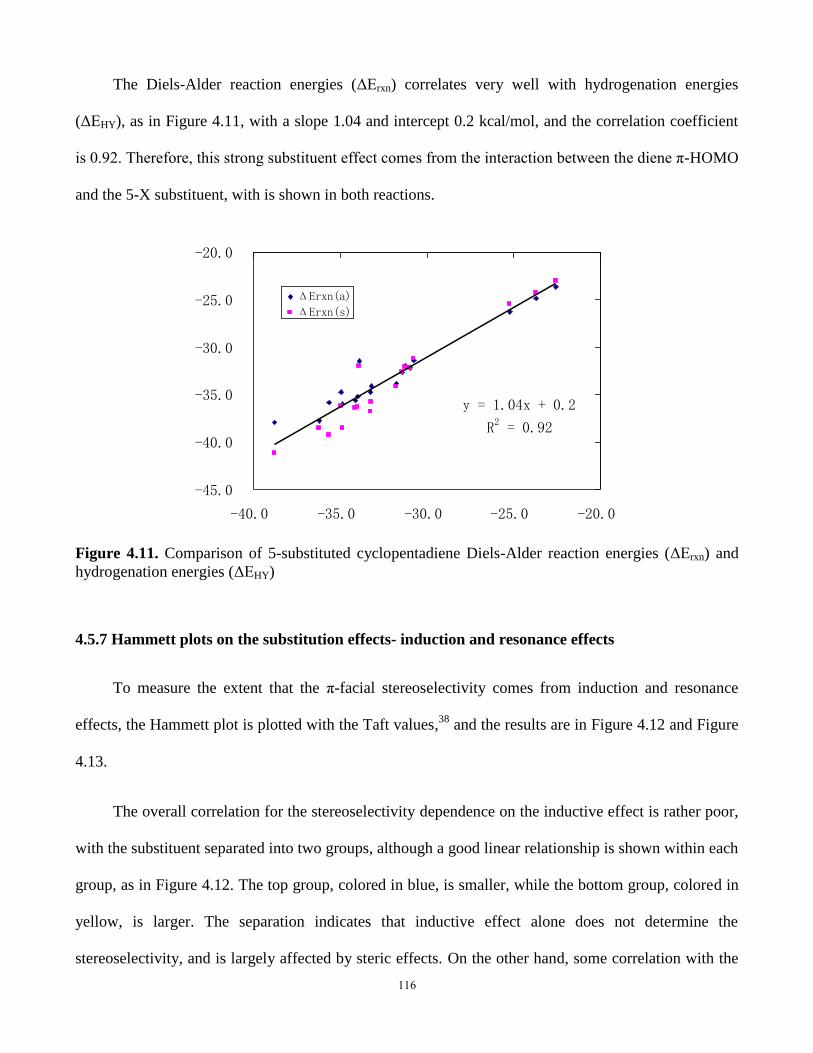
116
The Diels-Alder reaction energies (ΔErxn) correlates very well with hydrogenation energies
(ΔEHY), as in Figure 4.11, with a slope 1.04 and intercept 0.2 kcal/mol, and the correlation coefficient
is 0.92. Therefore, this strong substituent effect comes from the interaction between the diene π-HOMO
and the 5-X substituent, with is shown in both reactions.
Figure 4.11. Comparison of 5-substituted cyclopentadiene Diels-Alder reaction energies (ΔErxn) and
hydrogenation energies (ΔEHY)
4.5.7 Hammett plots on the substitution effects- induction and resonance effects
To measure the extent that the π-facial stereoselectivity comes from induction and resonance
effects, the Hammett plot is plotted with the Taft values,38
and the results are in Figure 4.12 and Figure
4.13.
The overall correlation for the stereoselectivity dependence on the inductive effect is rather poor,
with the substituent separated into two groups, although a good linear relationship is shown within each
group, as in Figure 4.12. The top group, colored in blue, is smaller, while the bottom group, colored in
yellow, is larger. The separation indicates that inductive effect alone does not determine the
stereoselectivity, and is largely affected by steric effects. On the other hand, some correlation with the
y = 1.04x + 0.2
R2 = 0.92
-45.0
-40.0
-35.0
-30.0
-25.0
-20.0
-40.0 -35.0 -30.0 -25.0 -20.0
ΔErxn(a)
ΔErxn(s)
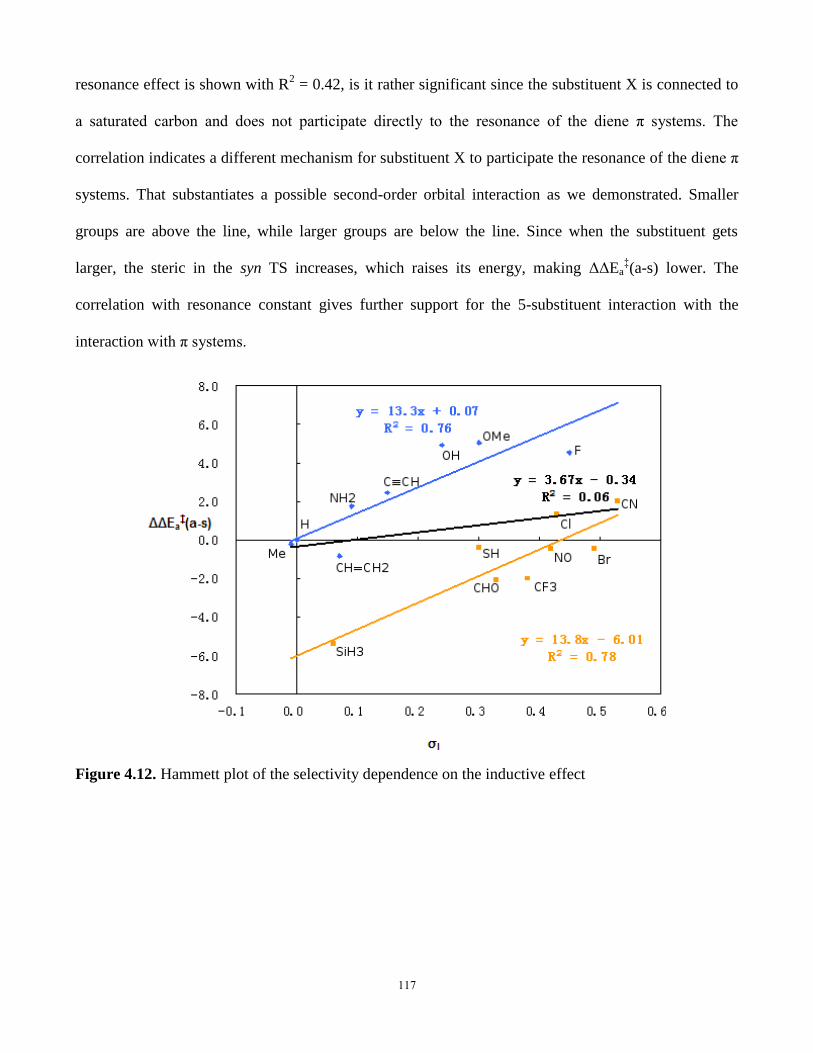
117
resonance effect is shown with R2 = 0.42, is it rather significant since the substituent X is connected to
a saturated carbon and does not participate directly to the resonance of the diene π systems. The
correlation indicates a different mechanism for substituent X to participate the resonance of the diene π
systems. That substantiates a possible second-order orbital interaction as we demonstrated. Smaller
groups are above the line, while larger groups are below the line. Since when the substituent gets
larger, the steric in the syn TS increases, which raises its energy, making ΔΔEa‡(a-s) lower. The
correlation with resonance constant gives further support for the 5-substituent interaction with the
interaction with π systems.
Figure 4.12. Hammett plot of the selectivity dependence on the inductive effect

118
Figure 4.13. Hammett plot of the selectivity dependence on the resonance effect
4.5.8. Comparison with Torquoselectivity
The proposed secondary orbital interaction scheme in the π-facial stereoselectivity could be
compared with the torquoselectivity, which addresses the inward/outward rotation on the ring opening
reaction of 3-X cyclobutene.39,40
With electronegative groups, the secondary orbital interaction favors
syn for π-facial stereoselectivity, while it is repulsive and favors outward rotation for torquoselectivity.
Just as the Diels-Alder cycloaddition of 5-X cyclopentadiene and ethene favors ethene attack
syn to electronegative groups, or π-donor groups, the ring opening of 3-X cyclobutene favors that the
π-donor groups to rotate outward. As in Figure 4.14, a reasonable correlation of the two contrasteric
reactions is given, and torquoselectivity is more sensitive to substituent effects since the substituents
are linked directly to the π systems.
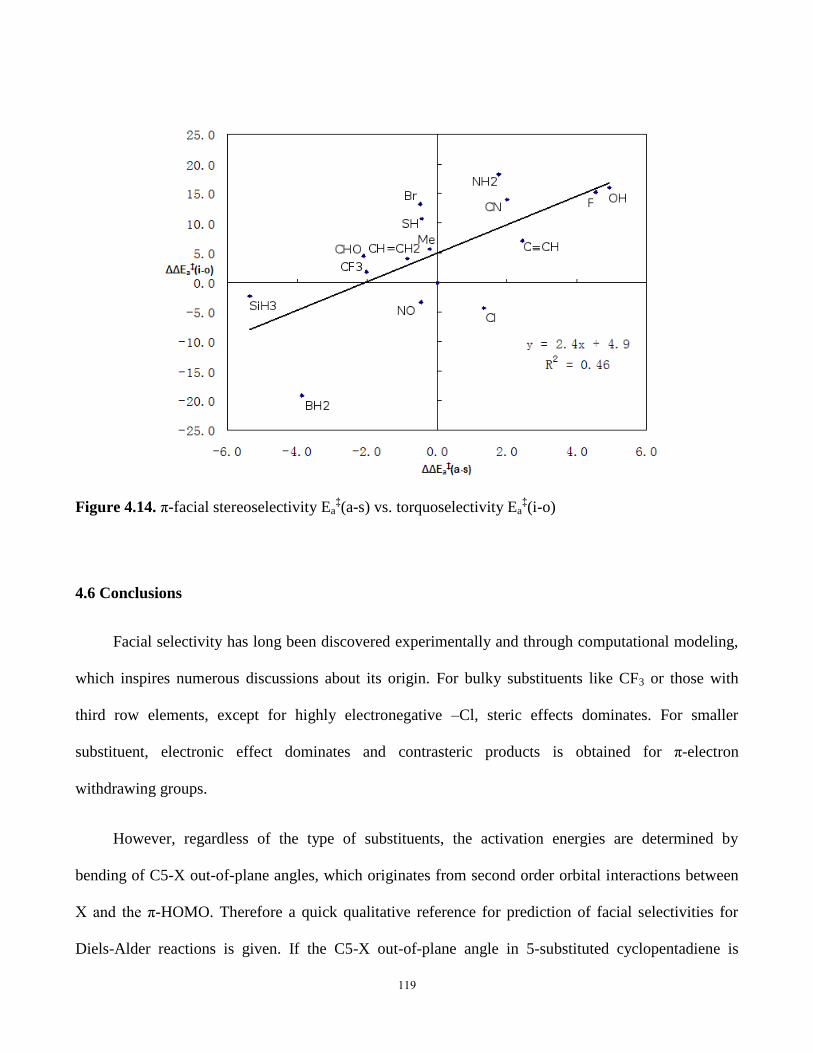
119
Figure 4.14. π-facial stereoselectivity Ea‡(a-s) vs. torquoselectivity Ea
‡(i-o)
4.6 Conclusions
Facial selectivity has long been discovered experimentally and through computational modeling,
which inspires numerous discussions about its origin. For bulky substituents like CF3 or those with
third row elements, except for highly electronegative –Cl, steric effects dominates. For smaller
substituent, electronic effect dominates and contrasteric products is obtained for π-electron
withdrawing groups.
However, regardless of the type of substituents, the activation energies are determined by
bending of C5-X out-of-plane angles, which originates from second order orbital interactions between
X and the π-HOMO. Therefore a quick qualitative reference for prediction of facial selectivities for
Diels-Alder reactions is given. If the C5-X out-of-plane angle in 5-substituted cyclopentadiene is

120
greater than 127°, the angle in cyclopentadiene, syn product is expected, while anti product is expected
when the C5-X out-of-plane angle is less than 127°, except for –SH, –CH=CH2 and –CF3. The
secondary orbital interaction scheme could be expanded to our previous torquoselectivity scheme.
4.7 References
(1) Woodward, R. B.; Katz, T. J. Tetrahedron Lett. 1959, 19.
(2) Woodward, R. B.; Katz, T. J. Tetrahedron 1959, 5, 70.
(3) Woodward, R. B. J. Am. Chem. Soc. 1942, 64, 3058.
(4) Cheong, P. H. Y.; Legault, C. Y.; Um, J. M.; Celebi-Olcum, N.; Houk, K. N. Chem. Rev. 2011,
111, 5042.
(5) Wilmot, J. T. Ph. D. thesis, University of Illinois at Urbana-Champaign, 2010.
(6) Winstein, S.; Shatavsky, M.; Norton, C.; Woodward, R. B. J. Am. Chem. Soc. 1955, 77, 4183.
(7) Macaulay, J. B.; Fallis, A. G. J. Am. Chem. Soc. 1990, 112, 1136.
(8) Macaulay, J. B.; Fallis, A. G. J. Am. Chem. Soc. 1988, 110, 4074.
(9) Xidos, J. D.; Poirier, R. A.; Pye, C. C.; Burnell, D. J. J. Org. Chem. 1998, 63, 105.
(10) Poirier, R. A.; Pye, C. C.; Xidos, J. D.; Burnell, D. J. J. Org. Chem. 1995, 60, 2328.
(11) Burnell, D. J.; Valenta, Z. J. Chem. Soc.-Chem. Commun. 1985, 1247.
(12) Burry, L. C.; Bridson, J. N.; Burnell, D. J. J. Org. Chem. 1995, 60, 5931.
(13) Anh, N. T. Tetrahedron 1973, 29, 3227.
(14) Ishida, M.; Inagaki, S. J. Synth. Org. Chem. Jpn. 1994, 52, 649.
(15) Ishida, M.; Beniya, Y.; Inagaki, S.; Kato, S. J. Am. Chem. Soc. 1990, 112, 8980.
(16) Ishida, M.; Inagaki, S. In Orbitals in Chemistry; Inagaki, S., Ed.; Springer-Verlag Berlin:
Berlin, 2009; Vol. 289, p 183.
(17) Cieplak, A. S. Chem. Rev. 1999, 99, 1265.
(18) Kahn, S. D.; Hehre, W. J. J. Am. Chem. Soc. 1987, 109, 663.
(19) Ohwada, T. Chem. Rev. 1999, 99, 1337.

121
(20) Frisch, M. J. T., G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.;
Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; Nakatsuji, H.; Caricato, M.; Li, X.;
Hratchian, H. P.; Izmaylov, A. F.; Bloino, J.; Zheng, G.; Sonnenberg, J. L.; Hada, M.; Ehara, M.;
Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.;
Vreven, T.; Montgomery, Jr., J. A.; Peralta, J. E.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.;
Kudin, K. N.; Staroverov, V. N.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J.
C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, N. J.; Klene, M.; Knox, J. E.; Cross, J. B.;
Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.;
Cammi, R.; Pomelli, C.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Zakrzewski, V. G.; Voth, G.
A.; Salvador, P.; Dannenberg, J. J.; Dapprich, S.; Daniels, A. D.; Farkas, Ö.; Foresman, J. B.; Ortiz, J.
V.; Cioslowski, J.; Fox, D. J. ; B.01 ed.; Gaussian, Inc.: Wallingford CT, 2009.
(21) Becke, A. D. Phys Rev A 1988, 38, 3098.
(22) Lee, C. T.; Yang, W. T.; Parr, R. G. Phys. Rev. B 1988, 37, 785.
(23) Guner, V.; Khuong, K. S.; Leach, A. G.; Lee, P. S.; Bartberger, M. D.; Houk, K. N. J. Phys.
Chem. A 2003, 107, 11445.
(24) Zhao, Y.; Truhlar, D. G. Theor Chem Acc 2008, 120, 215.
(25) Grimme, S. J. Chem. Phys. 2006, 124.
(26) Schwabe, T.; Grimme, S. Phys Chem Chem Phys 2007, 9, 3397.
(27) Grimme, S. J. Chem. Phys. 2003, 118, 9095.
(28) Moller, C.; Plesset, M. S. Phys Rev 1934, 46, 0618.
(29) Baboul, A. G.; Curtiss, L. A.; Redfern, P. C.; Raghavachari, K. J. Chem. Phys. 1999, 110,
7650.
(30) Montgomery, J. A.; Frisch, M. J.; Ochterski, J. W.; Petersson, G. A. J. Chem. Phys. 2000, 112,
6532.
(31) Montgomery, J. A.; Frisch, M. J.; Ochterski, J. W.; Petersson, G. A. J. Chem. Phys. 1999, 110,
2822.
(32) Curtiss, L. A.; Redfern, P. C.; Raghavachari, K. J. Chem. Phys. 2007, 126.
(33) Zou, L. F.; Shen, K.; Fu, Y.; Guo, Q. X. J Phys Org Chem 2007, 20, 754.
(34) Zou, L. F.; Fu, Y.; Shen, K.; Guo, Q. X. Theochem-J. Mol. Struct. 2007, 807, 87.
(35)Houk, K. N.; Paddon-Row, M. N.; Rondan, N. G.; Wu, Y. D.; Brown, F. K.; Spellmeyer, D. C.;
Metz, J. T.; Li, Y.; Loncharich, R. J. Science 1986, 231, 1108.
(36) Ando, K.; Green, N. S.; Li, Y.; Houk, K. N. J. Am. Chem. Soc. 1999, 121, 5334.
(37) Wu, Y. D.; Houk, K. N.; Paddon-Row, M. N. Angew. Chem. Int. Ed. 1992, 31, 1019.
(38) Hansch, C.; Leo, A.; Taft, R. W. Chem. Rev. 1991, 91, 165.
(39) Rondan, N. G.; Houk, K. N. J. Am. Chem. Soc. 1985, 107, 2099.

122
(40) Kirmse, W.; Rondan, N. G.; Houk, K. N. J. Am. Chem. Soc. 1984, 106, 7989.