qmrnmrnmrnmi stable crcwhexmithiw comkhihbs methods employed in charac-terizing the organolithiuiu...
TRANSCRIPT

Qmrnmrnmrnmi STABLE crcwHExmiTHiw comkhihbs
AHBROVBDt
Gradu»t# Committeei
ofeaeor
Minor Piv®r«s»©r )wi^4
til Gosmiti©©u MeiaWr
Qommitt#© Member : Jy"
W /C C 'V ' t-r Xi' I L' <• • 'L
Director o£/fc^ Department of Ciiemistry
Deais of the Graduate 3c&ool

V/ ON FORMAT IONA.LLY STAPLE CYCLOTlEXYJiLJTH I UK COMTOOHDS
DISSERTATION
Presented to the Graduate Council of the
North Texas State University in Partial
Fulfillment, of tho Requirements
For the Degree of
DOCTOR OF PHILOSOPHY
By
Charles M. Selruarx, B. 3.f M. S.
Denton, Texas
Januar.y , 1Q6&

TABLE OP CONTENTS
Page
LIST OP TABLES iv
LIST OP ILLUSTRATIONS v
Chapter
I. INTRODUCTION 1
II. EXPERIMENTAL PROCEDURES 28
Descriptions of Reagents and Synthesis Procedures
Experimental Methods Employed in Charac-terizing the Organolithiuiu Compounds and Their Derivatives
Experimental Methods Employed in Deriva-tive Porination of the Organolithium Compounds
III. RESULTS AND DISCUSSION 70
BIBLIOGRAPHY 118
1X1

LIST OF TABLES .
Table ' Page
I. Table of Letsinger's Data 3
II. Stereochemistry Investigated Alkenyl
Lithium Compounds . . . . . 11
III. p-Menthane and p-Menthene Retention Times . . . . 45
IV. Characteristic p-Menthene Infrared and Proton Magnetic Resonance Absorptions 46
V. Data Summary for Cyclohexyl Bromide Derivatives 62
VI. Characteristic Rotational Values for
Kenthyllithium Solutions . . . . 75
VII. Spin-Spin Coupling Constants 30
VIII. Calculated Inversion Energies 38
IX. Carbonation Products from Kenthyllithium . . . . 95
X. Bromination Products from Solutions of. Menthyllithium and 4-tert-Butyl-cyclohexyllithium . 103
XI. Product Distribution from Electrophilic Substitution Reactions of the Cyclo-hexyllithium Compounds 110
IV

LIST OP ILLUSTRATIONS
Pigure Page
1. Cyclohexyl Derivatives Studied . . . . . 73
2. c<~Methinyl Proton Resonance Spectra of Ken thy 1 and Neomenthyl Chloride Isomers (a), Bromide Isomers (b)t Iodide Isomers (c), and Alcohol Isomers (d) 81
3. Proton Kagnetic Resonance Spectra of Menthyl Chloride (a) and Ileoiuenthyl Chloride (b) . . . 32
4. Spin-Spin Coupling Diagram of Resonance Patterns 83
5. Proton Magnetic Resonance Spectra of Crystalline l.-entliyllithiuin Dissolved in Toluene (a) and Dimethyl Ether (c). Also Enlargements of o<-Methinyl Proton Spectra in Toluene (af)» Diethyl Ether (b), and Dimethyl Ether (e) 85
6. Inversion Pathways . . . . . . . . . . . 89
7. Infrared Spectra of 3-deutero-p-Menthane in Hydrocarbon Solvent (a) and Dimenthyl Ether Solvent (b) 97
8. <^-Methinyl Proton Resonance of 4-tert-Butylcyclohexyllithiuni Samples of an Original Reaction Solution in Benzene (a) and Crystalline Material Dissolved in Benzene (b), Diethyl Ether (c) Dimethvl Ether (d). . . . . . „ . 101
9. Infrared Spectrum of 4-tert-Butylcyclohexane-1-di in the Carbon-Deuterium Stretching Region . . . . . . « . . . 104

CHAPTER I
INTRODUCTION
Organolitnium compounds have been employed in synthetic
worK for many years. However only during the last decade
has mucn progress been made in establishing the mechanistic
pathways for the reactions of these compounds. To date
only a few detailed mechanism studies are known.
This lack of knowledge may be traced to three major
problems encountered in studying these reactions. The first
of the problems is the inherent sensitivity to moisture and
oxygen displayed by these compounds. A second pernicious
characteristic afforded by these systems with respect to
mechanism studies is their high specific rate constants.
Although both of these are very serious problems they can
be minimized by careful design of the techniques and
apparatus used in conducting such investigations.
The third problem involves elucidation of the conforma-
tional stability of organolithium compounds. The carbon-
lithium bond has been characterized for many systems as
being a fairly labile bond. The necessity of knowing the
conformational stability for such a compound prior to the
initiation of a mechanism investigation is obvious. ?or
this reason configurational stability studies of organolitnium
i

as well as all group IA-IIIA ;netal-orgaiiic compounds have
proven to be a subject of much current interest.
There are a number of important physical methods which
are used in conformational analysis studies. Of these methods
one of the oldest and most easily applied is the measurement
of optical rotation. This technique relates the experimentally
determined rotation to the molecular architecture usually
termed the conformation. Of course absolute configuration
assignments require the application of one or more of the
other techniques, as for example the currently very popular
nuclear magnetic resonance spectroscopy. However the over-
all stereochemical pathways from reactants to products for
many reactions can be followed very easily by rotation
measurements.
Studies ox the stereochemistry of substitution reactions
of organolithium compounds have long been plagued by the
fact that attempts to form optically active compounds, in
which the lithium atom is bound to an asymmetric tetra-
hedrally hybridized carbon resulted in racemic products.
Evidence for compounds possessing this property has been
observed only when the compounds are prepared and consumed
immediately. As a consequence, only the net stereochemistry
from alkyl halides or other starting materials to final
products could be studied; therefore the stereochemical course
of each individual reaction has been long delayed.

The research proposed for this investigation deals with
this problem of synthesizing ana isolating a conformationally
stable al^yllithium compound. Such compounds have not been
previously reported, perhaps because the carbon lithium
bond possesses an adequate decree of ionic character to
cause it to behave as a labile bond when solvated. However
if this is not a true characterization it way be that a
sufficient understanding of the conditions necessary to produce
such a system is still concealed in the unexplored chcmistry
of such compounds. Hints of these conditions can probably
be found in the published literature. Therefore a thorough
knowledge of the literature concerned with this subject is
imperative for the design of experiments which might yield
informative data. Having pointed out the importance assessed
to earlier investigations of this type it is believed that
these studies should be reviewed.
During the early 1930's the entire outlook on organo-
lithium chemistry was changed by the published work of Ziegler
and Colonius (43). Before this time the syntheses of organo-
lithium compounds were very laborious procedures and investi-
gations were directed primarily to understanding the nature
of these compounds. The work of Ziegler and Colonius (43)
outlines a direct synthesis procedure from lithium metal and
alkyl halide. The results of this work yielded an easy
synthetic method for these very reactive compounds. Shortly

' 4
after this method rras reported C-ilnan and co-workers (19, 20)
evaluated the relative reactivities for a number of these
compounds. They observed these reagents to he reactive as,
and in some cases more reactive than the already well in-
vestigated Grignard reagents.
Grignard reagents had already "been characterized as
very useful synthetic reagents. However, as early as 1911
a problem in understanding the stereochemistry of reactions
employing this reagent was realized by Pickard and Kenyon (31).
They generated the Grignard compound in the conversion of
an optically active alkyl iodide to the corresponding alcohol
and obtained only racemic product. Several years later
Porter (32) reported the results from a similar investigation.
In the hope of synthesizing an optically active compound
with one atom of hydrogen and one atom of deuterium attached
to the asymmetric carbon atom, the Grignard reaction was
applied to several optically active organic bromides. Complete
racemization of the bromides was observed in the formation of
the Grignard reagents.
Schwartz and Johnson (33) have also reported the formation
of a racemic anilide product in the reaction between phenyl
isocyanate and Grignard reagents prepared from optically
active 2-bromooctane. This investigation was conducted in
an attempt to gain a better understanding of reactions between
Grignard reagents and phenyl and -6-napthyl isocyanates. These

reactants were being employed at that time as analytical
reagents to detect and determine the concentration of an
organoiaagneaiuia lialide in solution.
In the period from 1900 to 1930 the chemistry of organo-
sodium compounds occupied the interest of several investigators.
With respect to the stereochemistry question Ott (30) reported
that sodium compounds appeared to react with more stereo-
specificity than evidenced for magnesium compounds. He showed
that the action of sodium on optically active «<-phenylethyl
chloride yielded optically active 2,3-diphenylbutane, along
with the meso form of the hydrocarbon, while the recovered
chloride was found to be extensively racemized. Treatment
of the active o(-phenylethyl chloride with magnesium in moist
ether gave only inactive diphylbutane. In this work he
postulated organosodium intermediates, of which Morton (26)
later demonstrated the existence.
V/allis and Adams (39) during this period employed organo-
sodium compounds in the study of a very fundamental stereo-
chemistry problem. This problem involved the question of the
spatial arrangement of groups in compounds containing a
tricovalent carbon atom. Much interest was attached by the
authors to the possibility of the existence of a non-planar
configuration in these compounds. They noted that if such
a spatial arrangement were true, asymmetry would be possible
when the three groups around the central carbon atom were

6
different. Therefore a study of the behavior of such com-
pounds toward polarized light would enable them to determine
the degree of stability of the antirneric configurations.
The authors realized that it was not possible experi-
mentally to resolve such compounds. Therefore they set out
to prepare the corresponding enantiomorphic quadricovalent
compounds, and then to determine whether the rotatory power
of such compounds is retained or lost upon transition into
the tricovalent state and return to the quadricovalent state.
These transitions seemed to be achieved through the formation
of organosodium compounds in the studies of earbanion and
free radical intermediates. The earbanion intermediates were
reported to be derived from a sodium triarylmethyl compound
prepared from an optically active reagent. Evidence for the
existence of a planar configuration was not observed. However,
in a later publication Wittig, Vidal, and Bohnert (41)
demonstrated this observation to be in error. At this time
it was believed that the V / u r t z reaction proceeded by a free
radical mechanism; therefore, this reaction was employed to
generate these intermediates. Only racemic product was produced
in this investigation thereby demonstrating the existence of
a planar figuration for this tricovalent species.
Tarbell and Weiss (36) reported the first investigation
of organolithium stereochemistry. A study of this type had
not been reported earlier because of the lack of a good

7
synthesis method for the lithium compounds. Also, since most
of the stereochemistry previously studied for similar systems
was of magnesium compounds, no encouragement for any success
had been offered. Then the works of Ziegler (4-3) and Ott (30)
were reported and thereby provided a synthetic method and
evidence for conformational stability in organoaliali chemistry.
It is interesting to note that even though the results re-
ported by Y/allis and Adams (39) contradicted Ott's (30)
observations, his wor c was cited as the basis for this study.
These authors found to their disappointment that when
optically active 2-chlorooctane was reacted with lithium,
the acid formed by carbonation was inactive, and the unre-
acted chloride which was recovered had a lower rotation than
the starting material. They pointed out that the reacemi-
zation might have occurred during the carbonation rather than
during the formation of the lithium compound. However, the
latter alternative seemed the more probable, especially in
view of the results reported in earlier magnesium work. The
formation of the active 2,3-diphenylbutane reported by Ott
(30) was then explained as the result of a reaction between
an optically inactive organosodium compound and an optically
active organic chloride,
Letsinger (25) can be credited with providing the
first insignt into the conditions required to demonstrate
conformational stability in organolithium compounds. He

a
was able to prepare optically active 1-methylheptyllithium
from active 2-iodooctane and sec-butyllithiaiii in a 6 per
cent diethyl ether-petroleum ether solvent mixture at -70
degreea centigrade. At these conditions a 20 per cent net
retention of configuration was observed. This work reported
in 1950 actually opened up the area of organolithium stereo-
chemistry.
Characterization of the lithium compound was accomplished
through the carbonation of derivative. The rotation measure-
ments reported were on the isolated acids and not on the
solutions of lithium compound. The carbonation reactions
were initiated two minutes after completion of the tempera-
ture or time variable studies conducted on the lithium
solutions. A summary of these data is listed in Table I.
TABL3 I
TABLE OF LETSIHGER1S DATA
Run Rotation of Temperature tfime at Listed Acia Number Alkyl Iodide (°C) Temperature Rotation
1 -44.36 -70 1 minute -1.13 2 -41.00 -70 60 minutes -0.96 3 -39.85 0 20 minutes -0.00
These data illustrate that time and temperature are certainly
two very real effects on the conformational stability of
organolithium compounds. However, this work does not provide
any evidence as to whether predominant retention or two

9
successive inversion pathways are followed in the conversion
of the organic iodide to the corresponding acid.
Interest in the stereochemistry of lithium alKenyls
became intense shortly after Letsinger (25) published his
work. Investigations by Braude and Coles (7) and Cur tin
and co-workers (11, 12) were reported in 1951. These works
all indicated that a definite increase over the lithium
alkyls in conformational stability was characteristic of
these compounds. This evidence opened up an interesting
and virtually uninvestigated area of research which dominated
the research in organolithium stereochemistry for the next
ten years. Due to the importance of a number of findings
reported during this time period a summary of these works
will be given.
Prior to the 1950's only the alkenyl lithium derivative
of y^-bromostyrene had been synthesized by Wright (42). Charac-
terization by carbonation of the products derived from the
reaction between lithium metal and this halide indicated only
the formation of the trans-alkenyl acid. Since the study of
this system involved the search for a way to obtain stereo-
isomers of l-phenyl-l,3-butadiene the-interest to continue was
subdued.
However, in 1950 Braude and co-workers (6, 8) reported
the synthesis of isobutenyllithium and cyclohexenyllithium
from the corresponding organic bromides and lithium metal.

10
The work was originated because of the desire to employ
©(-alkenyl organometallic derivatives for the synthesis of
various ethylenic systems. The well-known fact that alkenyl
halides containing a halogen atom adjacent to a ethylenic
"bond are very unreactive toward magnesium definitely limited
the selection of soluble metal derivatives to those of
lithium. The outlook for these compounds was somewhat bleax
since the only alicenyl-lithium system known was styryllithium,
which could be syntehsized using magnesium. Furthermore,
Gilman (18) had reported that in an attempt to synthesize
vinyllithium only dehydrohalogenation product was recovered.
Therefore these successful syntheses demonstrated that a limi-
tation to vinyl halides which also yield Grignard reagents did
not exist.
This success then stimulated Baude and Coles (7) to
determine whether the formation of these lithium derivatives
was limited toy?y£-disubstituted and cyclic alkenyl halides
which cannot easily undergo dehydrohalogenation. This led
to the fortunate choice of cis-propenyllithium, which was
found to be conformational^" stable over long periods of
time in refluxing ether. Curtin and co-workers (11, 12) were
also simultaneously investigating similar systems with an
interest in comparing their stereochemical stabilities. Over
the next ten years Curtin proved to be the principal in-
vestigator of the stereochemistry of alkenyl lithium compounds.
Table II includes a list of the systems studied.

11
TAB lull II
STEREOCHEMISTRY INVESTIGATED ALKSNYL LITHIUM COMPOUNDS
Entry Compound Empirical Formula References A trans-Cg^HC: CLiH (42) B cis- and trans-CH^IIC :CLiH (6, 10) C D 3
cis- and trans-ClC6H5C:CLiC6H5 cis- and trans-C6H5HC:CLiC6H5 cis- and trans-CHjHCiCLiCHj
(11, 13., 14) (12, 14, 28, 29) (5, 16)
These compounds were all prepared by the reaction of
the corresponding alkenyl bromides with either lithium
metal or n-butyllithiuin in ether solvent. A definite dif-
ference in the products formed by the two lithium reagents
was observed for the bromides with structure (&). When
lithium metal is employed as the reactant with these bromides,
where both the R»s may be any combination of phenyl, methyl,
and hydrogen, all the lithium compounds, with the exception
of the R = methyl and R' = phenyl case, are successfully
prepared. However when n-butyllithium is employed the
disubstituted acetylenes are obtained as the major products.
Propenyllithium has proven to be an exception. The case
where R = R* = methyl has not been studied.
Rf II /C = C"
R (&•) "Br
No explanation for the difference in these two lithium
reagents has been proposed by any of the investigators. Curtin
and Koehl (14) did postulate why it was possible to prepare

1 2
the two propenyllithium isomers "by the halogen metal exchange
reaction. Their explanation involves a metallation reaction
of the unreaeted bromides (&•) in which all the alkenyllithiums
containing ^-protons except the less reactive pr ope ny11ithi urn
reagents participate.
In reviewing these stereochemical data reported for
these systems three primary observations are projected. The
first factor which appears to affect their conformational
stabilities is the nature of the organic substituents. For
example an isomerization mechanism appears to be available
for the aryl substituted compounds. Curtin and Koehl (14)
have proposed that this mechanism involves the ionization
of the carbon-lithium bond followed by a shift of an aryl
substituent through a linear transition state (b), which is
diagrammed below. In the 'formation of this intermediate the
P-CIC6H4 li ;c=< •
C6H5 C6H5
p_.Ci-.C6H4
7/ C6H5 v- / 7
(b)
(-) P-CI-C6H4 ^C6H5
;c=c^ C6H5 li
negative charge can be distributed in part on the aryl sub-
stituent, thereby generating a favorable situation for
isomerization to proceed.
The second critical factor demonstrated from these
studies was the temperature. At low temperatures (-40 degrees
to -60 degrees centigrade) all the lithium aIkenyIs were found
to exist as conformationally stable cis— and trans—isomers in

13
diethyl ether. Above 0 degrees centigrade, however, inter-
conversion of the aryl substituted lithium a lice ny Is becomes
prominant. For these compounds the cis isomer showed the
greatest tendency to isomerize. In contrast to this the cis-
propenyllithium appeared to be stable in refluxing ether and
the trans-isomer demonstrated only a slight increase in its
tendency to isomerize.
The last and most significant factor was first reported
by Curtin and Koehl (14, 15). They demonstrated the existence
of a definite solvent effect on the stereochemical stability
not only of the lithium alkenyls but also of lithium alkyls.
It was reported that solutions of cis-o^stilbenyllithium and
of cis- and trans-l-p-chlorophenyl-l,2-diphenylvinyllithium
demonstrate greater stereochemical stability in hydrocarbon
solvents than in diethyl ether. Tetrahydrofuran is still more
effective than diethyl ether in promoting isamerization of
these compounds.
These results suggested that a saturated lithium alicyl
might possess more conformational stability than reported
by Letsinger (25) if it existed in an ether-free hydrocarbon
medium. Under these conditions it was observed that optically
active sec-butyllithium could be prepared and carbonated with
greater than 30 per cent retention of configuration. This
synthesis was achieved by employing the metal-metal exchange
reaction between optically active sec-butylmercury and racemic
2-octyllithium in n-pentane solvent.

14
The works of Letsitiger (25) and Cur tin and co-workers
(10, 14, 15) were important pioneering investigations and
illustrated the complexity of the stereochemistry of organo-
lithiui;! compounds. Port her more, the volume of stereochemical
research on both Groups IA and IIA organometallic systems
published since these works is certainly indicative that
these investigators revealed a very challenging area of
research.
One fact that must be emphasized concerning the works
of these two investigators is that they have provided in-
formation regarding only the net stereochemistry from alkyl
halide to final product. At no time were they able to make
any measurements on the organolithium solutions themselves.
Therefore these stability data are derived from determinations
by chemical means only. The fact that the stereochemical
course of each individual reaction could not be determined
by the employed analytical technique initiated investigations
of other methods. Hesmeyanov and Borisov (28) applied their
method of "odd and even cycles" and Allinger and Hermann (2)
employed infrared spectroscopy techniques. However the
results recorded in both of these studies were highly criti-
cized .
Seyferth and Vaughan (35) were among the first to apply
nuclear magnetic resonance spectroscopy to investigate this
stereochemistry. They were able to show unequivocally that

15
the preparation and carbonation procedures employed for the
propenyllithium system involved retention of the geometric
configurations. The oases for these stereochemical assign-
ments were provided "by the differences observed in the
coupling constant values for the two lithium isomers.
This technique has also been employed to study the
conformational stabilities of alkyllithiura compounds in
diethyl ether- An investigation of this type reports the
temperature at which the ^-methylene protons become magnet-
ically equivalent. Non-equivalency of these protons can
be derived primarily from either an unequal population of
rotamers about the carbon-carbon bond or by some inversion
mechanism whereby the protons can exchange places with one
another. For organolithium compounds such a mechanism in a
polar solvent can be conceived through the formation of a
carbanion and a solvated lithium cation. Positions of the
spectral lines as a function of temperature are used to
determine whether changes in population conformers occurs.
Therefore a distinction between these two causes may be
easily obtained.
Most of the published data have been reported on the
inversion rates of alkyl Grignard reagents and dialkyl
magnesium compounds. However Cram (9) has reviewed a private
communication from Roberts, -hitesides, and Witanowski which
reports such a study on 3,3-dimethylbutyllithium in diethyl

16
ether. The lithium compound was found to undergo rapid
inversion above zero degrees centigrade. Frankel, Adams,
and Williams (17) have also claimed such an investigation,
but a review of the data illustrate that magnesium compounds
were primarily the system being examined.
In the early 1950*s Walborsky became interested in the
stereochemical fate of a pair of nobonding electrons in an
orbital which is part of a three-membered ring (37). The
exo-orbitals of, such, a system have been characterized as
possessing more 11 s" character than is reported for tetra-
hedrally hydridized orbitals. Therefore this type of molecule
has a great deal of double bond character associated with it
which should lead to a higher barrier of inversion for such
a pair of electrons.
By I960 both Walborsky and co-workers (37, 38) and
Applequist and Peterson (4) had anticipated that cyclo-
propyllithium compounds might exhibit conformational stabil-
ities comparable to lithium alkenyls. Walborsky and co-
workers (37, 33) investigated the optical stability of a
l-methyl-2,2-diphenylcyclopropyllithium enantiomer. This
study was conducted by chemical means through protonation
with methanol, carbonation, bromination and iodination.
These reactions were carried out in a variety of solvents
such as diethyl ether, 1,2-dimethoxyethane, benzene, petroleum
ether, and various combinations of these solvents over a

17
•temperature range from 0 degrees to 30 degrees centigrade.
Polarimetry measurements were not made on the cyclopropyllithium
solutions. The results recorded from these investigations
demonstrated that the lithium compound could be prepared and
made to undergo electrophilic substitution with essentially
complete stereospecificity.
Applequist and Peterson (4) investigated the configura-
tional stability of the geometrical isomers of cis- and
trans-2-methylcyclopropyllithium. These compounds were
generated by exchange of the bromide isomers with isopro-
pyllithium under various conditions of ether-hydrocarbon
solvent mixtures and temperatures. The resulting solutions
were analyzed by brominolysis and carbonation. These
carbonation reactions were reported to proceed stereospecif- .
ically with retention. However a.lack of over-all stereo-/
specificity was reported for the brominolysis reaction.
The inescapable conclusion for these investigations is
that the cyclopropyllithium reagents do not interconvert at
a significant rate under the conditions employed, oeyferth
and Cohen (34) confirmed these findings by a nuclear magnetic
resonance investigation of the spectrum of cyclopropyllithium
in tetrahydrofuran.
A disagreement on the stereospecificity of the
brominolysis of these lithium reagents is obvious from
these investigations. Applequist speculated that the reason

,d>
for the non-stereospocificity of the brouinolysis was
derived from the existence of either a free radical or a
carboniuiij ion reaction intermediate. The radical arid
carbonium ion possibilities, it pas thought, could be
differentiated in the 2-norbornyl case. According to the
work of rCooyiuan and Vegfcer (23) this system should give
considerable endo-norbornyl bromide in any reaction of
norbornyl radica] with bromine; but exclusively exo-
product was predicted by "finsxein and co-workers (40) if
a bromine and norbornyl cation reaction was predominant.
In the 2-norbornyllithium vjorm Applequist and Chmurny
(3) investigated the stereochemistry of a number of electro-
philic substitution reactions of this lithium compound. The .
direct synthesis method was employed at 20 degrees and 32
degrees centigrade, the temperature of refluxing n-pentane
solvent, for preparing the lithium reagent, /t 20 degrees
different ratios of.endo- and exo-lithium isomers were reported
from the two different chloride isomers. An intermediate ratio,
which was independent of the chloride isomers, of endo- and
exo-lithium epimers was reported for the refluxing solvent
procedure. Average product yields of only 10 per ccnt required

19
that the lithium isomer distributions be determined by
chemical means. To make these isomer distribution claims
for the lithium compounds these authors made two assumptions.
The first assumption is that car Donation of alkyllithium
compounds proceeds by retention of configuration. The second
and more important of the two assumptions is that each isomer
reacts with the same degree of stereospecificity. Neither of
these assumptions was demonstrated experimentally to be valid.
Having solutions which were believed to contain varied
concentrations of the two lithium epimers these investigators
were then able to study the stereochemical pathways afforded
by these isomers. One observation made from these electro-
philic substitution data was that different reagents can give
different product mixtures. This indicates that not all the
reactions of this type proceed with simple retention of con-
figuration. It was also reported that the stereospecificity
of the brominolysis reaction appeared to proceed with prefer-
ential inversion. This unexpected result definitely eliminated
the cation intermediate postulated. However from the stereo-
chemical data alone it was not clear whether or not a radical
or carbonation intermediate could be postulated. It was con-
cluded that concerted aliphatic electrophilic substitutions,
m contrast with nucleophilic substitutions, have both inversion
and retention pathways available.
The nature of 2-norbornyllithium was also studied bv
Mulvaney and Garlund (27) prior to the work reported by

20
Applequist and Chmurny (3). They were interested in the
stereochemistry of alkyllithiuni addition to nonconjugated
olefins. It was their hope that employment of the highly
reactive norborene would facilitate determination of this
stereochemistry. They were able to execute this addition
reaction and characterize the 2-norbornyllithium product
"by chemical means. Evidence for the exo-derivatives only
was reported and the existence of a conformational^" stable
lithium compound was postulated from this observation.
Y ithin the last two'years Hill (21), Krieghoff and
Cowan (24), and Jensen and Nakamaye (22) have all made sig-
nificant contributions to the understanding of the stereo-
chemistry of norbornylmagnesium compounds. Nuclear magnetic
resonance spectroscopy was the primary tool employed in these .
studies. Application of this technique afforded these in-
vestigators a method of studying the organornetallic compounds
themselves instead of their derivatives. Por example Jensen
and Nakamaye (22) reported that each of the isomers in a
mixture of exo- and endo-norbornyl Grignard reagents can readily
be observed and identified from a proton magnetic resonance
spectrum. They were also able to demonstrate by the slow re-
appearance of the selectively destroyed exo-isomer that a
slow equilibrium between the two isomers exists. Armed with
these data they were then able to demonstrate easily the stereo-
chemistry of any individual electrophilic substitution reaction.

21
To date no investigation of this type has been reported
for alkyllithium compounds. Seyferth and co-workers (34, 35)
have determined the stereochemical stabilities of propenyl-
lithiuju and cyclopropyllithium by nuclear magnetic resonance
spectroscopy. There is no direct evidence that the results
obtained from these investigations can be extrapolated to
predict the stereochemistry of alkyl systems. Therefore it
is still unknown whether the observed stereochemistry for
alkyllithium compounds results from two reactions which
both occur by retention or by inversion of configuration.
It is thus obvious that an investigation of this type is
needed.
Such an investigation will require an alkyllithium com-
pound with structural stability comparable to the previously
reviewed systems. The stabilities of these systems appear to
be derived from their molecular geometries—for example, the
bridged norbornyl and trigonally hybridized alkenyl systems.
High energy barriers to most inversion mechanisms are evidently
associated with these geometries. The degree to which this
effect actually exists is definitely of interest, especially
in associated organolithium systems. "Thile it is desirable
to choose a system whose stability to inversion can be varied,
at the same time it must have approximately the bonding
character of simple alkyllithium systems. For such a system
not only an investigation of the stability effects but also

22
the stereochemistry of electrophilic substitutions of simple
alkyllithium compounds would be attainable.
It has been known for some time that the conformational
stabilities of cyclonexyl derivatives are dependent upon the
substituents attached to the ring. Also short of the bridged
derivatives, these systems approximate the bonding character
of secondary al^yl systems. Therefore an investigation of
substituted cyclohexyllithium compounds in a variety of
solvent and temperature conditions has been proposed for
this investigation. Although problems—such as those experi-
enced by Alexandrou (1)—are expected, it seems that definite
contributions to the stereochemistry of alkyllithium com-
pounds are attainable using this system. Furthermore fulfill-
ment of the quest to isolate a conformational^ stable alkyl- •
lithium compound might be observed from the synthesis of a
substituted 'cyclohexyllithium compound.

CHAPTER BIBLIOGRAPHY
1. Alexandrou, N. 13. , "Reaction of n-Butyllithium with cis- and trans-4-tert-Butylcyclohexyl Bromide and Mercuric Bromide," Journal of Organonetallic Chemistry, V (1966), 301. ,L
2. Allinger, Norman I. and Robert L. Hermann, "Infrared Spectra of cis- and trans-Proyenyllithium Stereo-chemistry of the Reaction of Vinyl Chlorides with Lithium Metal," Journal of Organic Chemistry. XXVI (1961), 1040. 6 >L
3. Applequist, Douglas E. and Gwendolyn Heal Chmurny, "Stereochemistry of Electrophilic Substitutions on 2-Norbornyllithium," Journal of the American Chemical Society, LXXXIX (1967) ,"575".
4. Applequist, Douglas E. and Alan H. Peterson, "The Configurational Stability of cis- and trans-2-methyl-cyclopropyllithium and Some Observations on the Stereochemistry of their Reactions with Bromine and Carbon Dioxide," Journal of the American Chemical Society, LXXXIII (1§6I)", Wo2~
5. Bordwell, P. G. and Phillip S. Landis, "Elimination Reactions. VII. A Comparison of Some Eliminations in Open-Chain and Cyclic Systems," Journal of the American Chemical Society, LXXIX (1957), 1593".
6. Braude, E. A. and J. A. Coles, "Alkenylation by Use of Lithium Alicenyls. Part IV. The Condensation of cycloliexeny 1-lithium with Aldehydes and Ketones, and Some Reactions of the Resulting Carbinols. (Studies of Molecular Rearrangement. Part V.)," Journal of the Chemical Society.(1950), 2014.
« "Alkenylation Employing Lithium Alicenyls. Part V. The Formation and Some Reactions of cis-Propenyllithium," Journal of the Chemical Society (1951), 2078.
23

24
3. Brauae, E. A. and C. J. Timmons, "Alkenylation "by Use of Lithium Alkenyls. Part I. The Condensation of isoButenyl-lithium with Benzaldehyde and Acetophenone, and the Oxotropic Rearrangement of the Resulting Carbinols. (Studies in the Molecular Rearrangement. Part III.)," Journal of the Chemical Society (1950), 2000,
9. Craw, Donald J., Fundamentals of Carbanion Chemistry, New York, Academic Press, Inc., 1965.
10. Curtin, David Y. and John W. Crump, "Configurational Stabilities of Stereoisomeric Vinyllithium Compounds." Journal of the American Chemical Society, LXXX (1953), U&T.
11. Curtin, David Y. and Elbert E. Harris, "The Preparation of Stereoisomeric Alx^enyl Lithium Compounds," Journal of the American Chemical Society, LXXIII (1951), £l±b.
12. , "Preparation of Stereoisomeric Alkenyl-lithium Compounds. II. cis-and trans-l,2-Diphenylvinyllithium and U- and /?-Styryllithium," Journal of the American Chemical Society, LXIII (1951), 45197
13. Curtin, David Y. , Harry Johnson, and Edwin G. Ste.iner, "Stereoisomeric Vinyllithium Compounds. III. Reactions with Aldehydes, Ketones, and Methyl Iodide," Journal of the American Chemical Society, LXXVII (19550, vser.
14. Curtin, David Y. and William J. Xoehl, "Effect of Solvent on the Steric Stability of Lithium Reagents," Journal of the American Chemical Society, LXXXIV (1962), 1967.
15. , "Optically Active sec-ButylIithium," Chemistry and Industry (i960), 262.
16. Dreiding, Andre S. and Richard J. Pratt, "The Carboxyl-ation of cis- and trans-2-Butenyl-2-lithium. A Stereospecific Synthesis of Angelic Acid," Journal of the American Chemical Society, LXXVI (1954), 1902.
17- Praenkel, Gideon, David G. Adams, and James Williams, "Structure of Grignard Reagents and Organolithium Compounds," Tetrahedron Letters (1963), 767*

25
18. Gilnan, Henry and A. U. Kaubein, "Ethynyllithium Com-pounds by Halogen-Ketal InterconversionsJournal of the American Chemical Society, LXVII (1945), 1420.
19. Gilman, Henry and R. H. Kirby, "The Relative Reactivities of Organolithium and Organomagnesiun Compounds," Journal of the American Chemical Society, LV (1933), i r o _
20. Gilman, Henry and Myrl Lichtenwalter, "Relative Reactivities of Organometallic Compounds. X. Lithium and Magnesium." Recueil des Travaux Chemiques des Pays-bas, LV (193b), 561.
21. Hill, Alexander B., "Nuclear Magnetic Spectra of the Bornyl and Norbornyl Grignard Reagents," Journal of Organic Chemistry, XXXI (1966), 20.
22. Jensen., Frederick R. and Kay L. Nakamaye, "Preparation of Geometrically Isomeric Grignard Reagents and the Stereochemical Courses of their Reactions," Journal of the American Chemical Society, LXXXVIII (1966),
23. Kooyman, E. C. and G. C. Vegter, "Bicyclanes-I. The Halogenation of 2:2:l-Bicycloheptane (Norbornane)," Tetrahedron, IV (1958), 382.
24. Krieghoff, Nilde G. and Dwaine 0. Cowan, "Stereochemistry of the Grignard Reagents from exo- and endo-Norbornyl Chlorides," Journal of the American Chemical Society, LXXXVIII (19bb),13227
25. Letsinger, Robert L., "Formation of Optically Active 1-Methylheptyllithium," Journal of the American Chemical Society, LXXII (1950), T$42.
26. Morton, Avery A. and Graham M. Richardson, "Condensation by Sodium. XVI. The Formation of Decane in the Wurtz Reaction," Journal of the American Chemical Society, LXII (1940), 1237"
27. Mulvaney, J. E. and Z. G. Gardlund, "Alkyllithium Compounds and Nonconjugated Olefins. The Nature of a Norbornyllithium Compounds," Journal of Organic Chemistry, XXX (1965), 4321.

26
28. Nesmeyanov, A. N. and A. 3. Borisov, "Stereochemistry ox Jilectr ophilic and Homolytic Substitution at the Olefinic Carbon," Tetrahedron, I (1957), 158.
29. Nesaeyanov, A. W., A. S. Borisov, and N. A. Volkenau, "Synthesis of Stereoisomeric Grganomercury Compounds from Organolithium Compounds," Izvestiva Akademii Hauk Soyuza Sovetskiieh Sutsialisticheskikh Respublik r r y w r w .
** • <
30. Ott, Srwin, "Uber die Additionreaktionen der Athylen-Doppelbindung unter Bildung von Athan-Verbindungen rait zwei, durch den Additionsvorgang entstehenden, asymmetrischen ivchlenstoffatomen und der ijinfluss der Reaktionsgeschwindigkeit auf den Reaktionsverlauf," Berichte der Deutschen Cheraischen G-esellschaft, LXI (1923), 21??.:
31. Pickard, Robert Howson and Joseph Kenyon, "VII -Investigations on the Dependence of Rotatory Power on Chemical Constitution. Part I. The Rotations of the Simplest Secondary Alcohols of the Patty Series," Journal of the Chemical Society, LXXXXIX (1911), 45.
32. Porter, C. W. , "Racemization in the Preparation of the Grignard Reagent," Journal of the American Chemical Society, LVII (1935),1436.
33. Schwartz, A. M. and John R. Johnson, "Characterization of Alkyl Halides and Organoinagnesium Halides," Journal of the American Chemical Society, LIII (1931), 1063.
34. Seyferth, Dietmar and Harvey M. Cohen, "The Stability of Cyclopropyllithiujii in Diethyl Ether and in TetrahydrofuranJournal of Organometallic Chemistry, I (1963), 15. ^
35. Seyferth, Dietmar and Lawrence G-. Vaughan, "The Isomeric 1-Propenyllithium Reagents: Their M R Spectra and the Stereochemistry of Their Formation by Direct Reaction and by the Transmetalation Reaction," Journal of Organometallic Chemistry, I (1963), 20T.
36. Tarbell, D. S. and Marvin Weiss, "The Action of Lithium on an Optically Active Aliphatic Chloride," Journal of the American Chemical Society, LXI (1939), 120"3.

2 7
3 7 . " ' a l b o r s ^ y , T i . M . a n d ? . - J . 7 . : : ; p a n t a i o , " ' " y c l o p r o p a n o n . V I . R e t e n t i o n o f O p t i c a l A c l i v i t y r . t t i C n n f i g u r a t i o n i n - c h o O y c l o ' o r o p y l C a r b a n i o n , " j o u r n a j o f t n e A m e r i c a n • d h o . - a c a l S o c i e t y , I X X X I ( 1 ^ 5 ) , 5 3 5 5 * " . '
3 8 . " r a ] l ) o r s . c y f i l . i f . , 1 ? . J . l u p a a t a t o , a n d A . Y o u n g , " C y c ? o p r c p a n e s . X V . T h e O p t i c a l S t a b i l i t y o f l - l l c t l u y 1 , . ? - a i p i i o n y i - o / c l o p r o p y 1 1 i t h i u m , " J o u r n a l o f t h e A m e r i c a n C h o r i o n 1 S o c i e t y , I i X X X V I ( 1 9 6 4 ; ,
3 9 . ' . ' ' a l l i s , ' v v e r e t I S . a n d F r e d e r i c I I . A d a ^ s , " T h e S p a t i a l C o n i i g u r a t i o n o f t h e V a l e n c e s i n T r i e o v a l e n t C a r b o n C o m p o u n d s J o u r n a l o f t h e A m e r i c a n C h e m i c a l S o c i e t y , I V ( 1 9 3 3 ) , 3 3 5 3 " : "
4 0 . W i n s t e i n , S . , 33. C l i p p i n g e r , R o b e r t H o w e , a n d E l l i o t V o g e I f a n g e r , " T h e H o n c l a s s i c a l h o r b o r n y l C a t i o n , " J o u r n a l o f t h e A m e r i c a n C h e m i c a l S o c i e t y , L X X X V I I
3 / 6 .
4 1 . F i t t i g , G e o r g e , P e d e r i c o V i d a l , a n d E r v / i n B o h n e r t , " f i b e r d i e k o n f i g u r a t i v e B e s t a n d i g ^ e i t m e t a i l o r g a n i s e h e r V e r b i n d u n g e n , " B e r i c l i t e d e r D e u t s c h e n C h e m i s c h e n G e s e l l . s c h a f t , i r m i T T l ^ ) 7 ~ 3 y j ;
4 2 . V / r i g h c , G e o r g e 3 ? . , " O r g a n o m e t a l l i c C o m p o u n d s o f S t y r e n e J o u r n a ] o f O r g a n i c C h e m i s t r y , I ( 1 9 3 6 ) , 4 3 7 .
4 3 . Z i e g l e r , K . a n d H . C o l o n i u s , " U n t e r s u c h u n g e n u b e r a l . c a l i -o r g a n i s c h e V e r b i n d u n g e n . V . ? 5 i n e " b e q u e m e S y n t h e o e e i n f a c h e r L i t h i u m a l ^ y l e J u s t u s L i e b i g s A n n a l e n d e r C h e m i e , C C C C L X X I X ( 1 9 3 0 ) , ± 5 5 1 ~~ "

CHAPTER II
.EXPERIMENTAL PROCEDURES
Descriptions of Reagents and Syntheses Procedures
Liaterials
The quality of materials used is a critical factor in
preparing and studying the chemical properties of oxygen
and moisture sensitive organometallic compounds. This is
exemplified by the fact that these compounds are character-
ized primarily through studies of their derivatives. There-
fore considerable attention has been given to determining
the quality of reagents used in this research investigation.
The lithium sand employed for the preparation of the
menthyl- and 4-tert-butylcyclohexyllithium compounds was
prepared from one-half inch diameter lithium rod containing
2 per cent sodium metal (16, 23). The lithium rod, purchased
from the Lithium Corporation of America, was cut into small
chips and added to a 500 milliliter flask containing 100
milliliters of heavy mineral oil. After heating the flask
to a temperature slightly above the melting point of the
lithium metal it was shaken vigorously and cooled very
rapidly, yielding a finely dispersed sand. The sand was
then transferred onto a coarse fritted funnel and washed
free of the mineral oil with dry n-pentane. The lithium
28

29
was kept suspended in the pentane solvent throughout this
washing period and during its addition to an argon-purged
reaction vessel.
Pure grade n-pentane obtained from the Phillips
Petroleum Company was dried by distilling it from lithium
aluminum hydride and stored under a nitrogen blanket. Other
hydrocarbon solvents used in various applications were also
purchased from Phillips Petroleum Company and treated in the
same manner before use.
Prepurified argon and nitrogen gases were used for all
processes which required atmospheres free from water and
oxygen.
Carbon dioxide, compressed air, dimethyl ether, carbon
tetrachloride, bromine, deuterium oxide, and anhydrous
magnesium sulfate were used as obtained from commercial
suppliers. Ethylene dibromide, methyl iodide, and diethyl
ether were always distilled prior to use.
(-)-Me nthy1 Chloride
The procedure reported by Smith and Wright (21) for
the preparation of highly levorotatory menthyl chloride was
utilized for synthesizing this diastereomer. These inves-
tigators found they could increase the levorotation of the
chloride isomer over other known synthetic methods by
treating (-)-menthol with a solution of zinc chloride in
hydrochloric acid, commonly known as the Lucas reagent. After

30
five hours at 35 degreeo centigrade the menthol is con-
verted in 90 per cent yield to a product with a specific
rotation equalling -45 degrees.
T.he procedure for a typical menthyl chloride preparation
consists of dissolving 226 grams of anhydrous zinc chloride
in 154 milliliters of concentrated hydrochloric acid. This
procedure is carried out in a 500 milliliter three—neck
round-bottom flask fitted with a Teflon stirring gland and
a Ilirschberg stirrer. The flask is immersed in an ice water
bath throughout the preparation of the lucas reagent. To r* 2.*
this solution 78 grams of (-)-menthol [<*1^ =-54.8°( n-pentane,
c. ,2.88)] , purchased from the Aldrich Chemical Company,
is added and the temperature is raised to 35 degrees centi-
grade by replacing the ice water bath with a heating mantle.
The reaction vessel is maintained at this temperature with
stirring for five hours.
The product from this reaction was isolated by distillation.
In order to facilitate this process, prior to distillation the
product was extracted with n-pentane solvent and treated with
concentrated sulfuric acid to remove any unsaturates. This
was accomplished by washing the n-pentane extract with 25
milliliters aliquots of sulfuric acid until no yellow
coloration appeared in the acid layer. The hydrocarbon la-'er
was then washed with water and dried over anhydrous magnesium
sulfate. The magnesium sulfate was removed by filtration and

31
the menthyl chloride was concentrated prior to distillation
oy evaporating the n—pentane solvent with a dry air purge.
Kenthyl chloride distillate was obtained at 98 degrees
centigrade and 10 torr, and the specific rotation of this
distillate, recovered in 89 per cent yield was a £ [o<]o =
-43.5°(n-pentane,c. ,5.02)]. A proton magnetic resonance
spectrum of this product demonstrated the characteristic
axial o<-methinyl resonance at 6.75 "3"-
This distillate was also analyzed on a 10 foot by one
quarter inch aluminum tubing gas liquid chromatography
column pa Cited with 20 per cent XF-1150 cyanosilicone substrate
on 60-80 mesh chromosorb P packing material. A temperature
of 110 degrees centigrade and a helium flow of 300 milliliters
per second were employed to obtain retention times of 18.75
minutes for the neomenthyl isomer and 20.5 minutes for the
menthyl isomer. Less than 5 per cent of the neomenthyl
isomer was detected.
( )-Neomenthyl Chloride
A selective synthesis of the neomenthyl chloride
diastereomer has been reported by Horner, Oediger, and
Hoffman (14)• The conversion of (-)-menthol to neomenthyl
chloride was effected by triphenylphosphine dichloride which
proves to be a very selective reagent. The authors reported
a 93 per cent theoretical yield of product with specific
rotation equalling +42.5 degrees. Although the procedure

*7 2
reported by these investigators was followed as closely
as possible typical yields averaging only 60 per cent were
obtained. A number of variations were tried to improve
the yield for this reaction with only limited success.
The procedure followed for preparing this isomer requires
the saturation of 230 milliliters of carbon tetrachloride
solvent with chlorine gas. The saturation point was determined
to be 0.08 grams of chlorine per milliliter. This solution
is added dropwise to a solution consisting of 65.5 grains of
triphenyl phosphine dissolved in 300 milliliters of carbon
tetrachloride solvent under a nitrogen atmosphere at 0 degrees
centigrade. Upon completion of this addition, thereby form-
ing the triphenyl phosphine dichloride reagent, 39 grams of
(-)-menthol are added and the reaction flask is stirred
vigorously and heated to 75 degrees centigrade for a period
of thirty minutes.. This reaction is carried out in a three-
neck round bottom flask equipped with the same stirring
assembly employed in the menthyl chloride preparation and
a Friedrichs reflux condenser equipped with a nitrogen
inlet adapter. A thermometer occupies the other neck of
the reaction flask. Upon cooling, the triphenylphosphine-
hydroxyl chloride by-product precipitates from solution as
a white solid.
Isolation of the neomenthyl chloride is achieved by
distillation following the removal of the- triphenylphsphine

33
hydroxylchloride by filtration. The carbon tetrachloride is
stripped from the solution under reduced pressure and the
diastereomer is distilled over at 91 degrees centigrade and
15 torr. The product recovered in 58 per cent yield was r
observed to possess a specific rotation of <*<1 =4-53.7 (n-
octane ,c. ,1.27) • The characteristic equatorial c<-methinyl
resonance at 5.5^ was observed from a proton magnetic
resonance spectrum of this distillate. Furthermore less
than 5 per cent of the menthyl isomer was detected using
the gas liquid chromatography analysis described in the
menthyl chloride analysis section.
cis-4~tert-Butylc.yclohex.yl Chloride
The method of Horner, Oediger, and Hoffman (14) was
also applied to the preparation of cis-4-tert-butylcyclohexanol
chloride. The only commercially available 4-tert-butyl-
cyclohexanol consists of an 80 per cent trans: 20 per cent cis
mixture of isomers. For this reason one of the selective
chlorinating reagents utilized in the menthyl chloride
synthesis was employed. The phosphine dichloride reagent
proved to be superior to the Lucas reagent for this particular
system.
In a typical synthesis triphenylphosphine dichloride
was prepared by the same procedure as described for the
neomenthyl chloride synthesis. While this reagent is Itept
at 0 degrees centigrade under a nitrogen atmosphere, 35.5

34
grams of commercial 4-tert-butylcyclohexanol dissolved in
250 milliliters of carbon tetrachloride are added, The
mixture is heated to 75 degrees centigrade and allowed to
reflux for two hours. The chloride product is isolated in
the same manner as the neomenthyl chloride. The reaction
supplies only a 35 per cent yield of product which distills
over at 31 degrees centigrade and 10 torr. Characterization
and purity of this distillate was determined by infrared,
nuclear magnetic resonance, and gas chromatographic techniques.
The infrared spectra of both the cis- and the trans-4-
tert-butylcyclohexyl chlorides have been reported by Greene,
Chu, and Walia (13). The infrared spectrum of the chloride
distillate corresponded well with the spectrum given for the
cis-chloride isomer. The strong carbon-chlorine stretching
absorption was observed at 639 wave numbers. According to
the published findings of Larnaudie (17), this corresponds
to the carbon-chlorine stretching region observed for axial
monochlorocyclohexane derivatives.
Separation of the chloride isomers on a gas liquid
phase chromatographic column was accomplished on a 10 foot
by one-quarter inch aluminum tubing column packed with
20 per cent XF-1150 cyanosilicone substrate on 60-80 mesh
Chromosorb P packing material. A temperature of 118 degrees
centigrade and a helium flow of 300 milliliters per minute
were employed to obtain retention times of 17 minutes for

35
the cis isomer and 19*5 minutes for the trans. In a typical
preparation the isolated distillate usually contained from
7 to 10 per cent of the trans isomer. Unreacted cis-4-tert-
batylc/clohexanol has been identified in the chloride
distillate and although its concentration may vary it never
exceeds 10 per cent.
Nuclear magnetic resonance was also employed as one of
the primary methods for characterizing the chloride isomers.
For these two chloride isomers values of 5.75 and 6.55
were recorded for the cis and trans c<-methinyl resonances
respectively. A detailed discussion of the application of
this analytical method to substituted eyclohexyl systems
will be included in a later section.
Preparation of (4-)-Menthyllithium
Menthyllithium was prepared in dry n-pentane under an
argon atmosphere from menthyl or neomenthyl chloride and
lithium sand according to the method originated by Gillian,
Langham, and Moore (12). This method was employed in
preference to other organolithium preparative methods in an
attempt to retard the competing elimination reaction reported
by Alexandrou (1) for a similar system. Yields ranging from
50 to 60 per cent were usually obtained for the menthyl system
by this method. It was observed tnat preparation of the
lithium sand and distillation of the menthyl chloride isomer

36
immediately prior to conducting the men thy llithium synthesis
facilitated the attainment of higher yields.
A one-quarter mole preparation of menthyllithium is
carried out using a 500~milliliter three-neck round bottom
flask equipped with standard taper 24/40 ground glass joints.
The flask is equipped with a Hirschberg stirrer in a Teflon
Asco stirring gland which seals with Neoprene rubber "o"-rings.
A 150-milliliter cylindrical separatory funnel with pressure
equalizing line is used in one neck for the addition of the
menthyl chloride epimer. An argon gas inlet connected by a
standard taper 24/40 joint to a Friedrichs reflux condenser
fills the third neck of the reaction flask. The reaction
flask is heated by a Glascol mantle controlled by a rheostat.
The first step in the preparation is to wash five grams
of lithium sand into the argon purged reaction flask with
dry n-penta-ne. A 43 gram sample of menthyl or neomenthyl
chloride is diluted to 125 milliliters in dry n-pentane and
this solution is loaded into the addition funnel. After
adapting the addition funnel to the reaction flask it is
purged with argon, and the entire reaction assembly is then
adjusted to remain under a positive argon pressure of ap-
proximately one centimeter of mercury. Stirring is started
and the contents of the flask are brought to a slow reflux.
After the reflux has begun, 10 per cent of the menthyl chloride
solution is added to the reaction flask to initiate the re-
action.

37
The initiation of this reaction is slower and not so
obvious as is demonstrated by many of the organolithium
compounds prepared by this method. A color change from
metallic gray to a purplish gray is observed after approx-
imate1^* thirty to forty-five minutes. Upon addition of the
halide the heat is usually reduced, thereby diminishing the
reflux rate. Upon evidence of the slight color change an
increase in the reflux rate can also be observed. If neither
of these two indications of initiation is observed after
forty-five minutes then evidence may- be seen by stopping the
stirring and dropping the heating mantle. If the reaction
has been initiated some metallic particles will sink in the
pentane solvent.
Upon evidence that the reaction has been initiated the
inenthyl chloride solution is added dropwise at a rate which
will maintain reflux of the solvent. It is believed that
reflux should be maintained at all times to eliminate the
production of hot spots in the flask which can cause decom-
position of the organolithium compound and elimination of
hydrogen chloride from the menthyl chloride reactant. Addition
is generally completed after three or four hours. To insure
that the reaction is complete the contents of the flask are
brought to reflux for one hour and then stirred without reflux
for two additional hours.
The reaction product is worked up in-the dry box. It
is first filtered through a medium fritted funnel to separate

38
the filtrate away from the lithium chloride by-product. A
clear yellowish-colored filtrate is usually obtained. If
the solution is- cloudy it should be refiltered to insure
that all of the lithium chloride has been removed. The
solution is then concentrated by removing the pentane
solvent by evaporation. This process, which cools as well
as concentrates, is carried out by applying vacuum to the
flask containing the solution and catching the volatile
solvent in a dry-ice and acetone trap. As the solution is
concentrated and the temperature drops the menthyllithium
is crystallized from the solution. Agitation of the flask
during the evaporation process appears to facilitate the
crystallization step. After the solution has been concen-
trated from a volume of approximately 250 milliliters to
approximately 60 to 70 milliliters, the solution will take
on the appearance of a milky orange liquid if a good yield
of menthyllithium is present. At this point the sample is
filtered through a coarse fritted funnel as rapidly as
possible.
The crystalline menthyllithium isolated on the funnel
frit usually contains p-menthene impurities. The greater
the impurity content the more muddy the consistency of the
product. Washing this material can be accomplished with
pentane solvent. The menthyllithium is very soluble in
n-pentane and therefore care must be taken not to wash the

3y
lithium compound too freely. The usual procedure is to
-.-/ash dorm the sides of the runnel and the solid material
very slo'.vly with a pipette wi.ii.Le vacuum is using applied
to the system. After washing, the vacuum is used to dry
the material for several minutes.
The procedure described above for isolating the
crystalline menthyllithium is repeated two or three times
depending upon yield of carbon bound lithium obtained.
Usually about one-half of the menthyllithium synthesized
can be isolated in this manner. After this quantity has
been removed the concentration of the menthenes becomes
large enough to keep the remaining product in solution.
Isolation of 10 grams of crystalline material is usually
considered a good yield of product.
Recrystallization of the isolated menthyllithium can
be achieved using n-pentane as the solvent. Due to the
high solubility of menthyllithium in this hydrocarbon the
process can be a difficult one to effect. However, it has
been found that if a high concentration of tnenthyllithium
is used in the solution of redissolved crystalline product
the attainment of recrystallized material is greatly
facilitated. The procedure used in obtaining recrystallized
material is the same as described for isolating crystalline
menthyllithium from the reaction mixture. Typical yields
of the recrystallized product averaged about 10 per cent of
the redissolved crystalline material.

40
tr a n s - 4 -1 e r t -B u t y 1 cy c 1 one xy 11 i t h i urn
Alexandrou (1) reported a 25 per cent yield of 4-tert-
butylcyclohexene from the halogen metal exchange reaction of
the trans-bromide with n-butyllithium. He also tried the
direct reaction with copper activated lithium metal as
described by Curtin and Xoehl (6), and a metal-metal exchange
reaction with isomers of 4-tert-butylcyclohexyl mercuric
bromides and n-butyllithium with no improvement. The lacA
of evidence for the formation of the organolithium compound
was indicative that 4-tert-butylcyclohexyllithium would be
more difficult to synthesize than menthyllithium. Since
elimination appeared to be the major reaction, a change from
bromide to chloride reagent was made in an attempt to retard
this process. Also a change to the same direct method re-
ported by Gilman, Langham and Moore (12) to be employed for
the synthesis of menthyllithium was used for this preparation.
One change in the direct method was made after two synthetic
trials yielded only 10 per cent product. This change was to
increase the volume of solvent from 500 to 1000 milliliters.
A reduction in elimination and coupling by-products was noted
by an increase in organolithium compound yield to 50 per cent
when synthesized in the more dilute system.
Because lower yields of organolithium compound are
obtained for this system and the product obtained is only
sparingly soluble in the n-pentane solvent, changes in the

41
isolation procedure for the ^-tert-butylcyclohexyllithiom
compound are required. Separation of the crystalline solid
from the 4-tert-butylcyclohexene by-product was successful
only when the yield of organolithium reached 30 per cent.
In a typical run 1.35 grams of the white crystalline material
was isolated by adding warm n-pentane to the cold cyclohexene-
pentane filtrate. The filtrate had been concentrated by
removal of almost all of the pentane solvent by evaporation.
At this point very little crystalline material had precipitated
from solution. The solution was filtered anyway to claim the
crystals present. As the warm n-pentane being used to wash
the frit filtered into the solution crystalline material im-
mediately appeared. For this same preparation additional
organolithium compound was obtained by repeated washing of
the lithium metal-lithium chloride mud remaining in the re-
action flask.
Experimental Methods Employed in Characterizing the Organolithium Compounds and
Their Derivatives
Double Titration Analysis
For several years there has been a continuing interest
in the analysis of organolithium solutions. This interest
has been stimulated by the fact that mechanistic and struc-
tural studies on these materials are becoming increasingly
more common in the published literature. Since these compounds

42
are very susceptible to oxygen and moisture it is a necessity
to be able to determine the concentration of carbon bound
lithium in the presence of the other basic impurities. The
most common of these impurities are lithium hydroxide, lithium
alkoxide, and lithium oxide. Brown, Ladd, and Newman (3)
have demonstrated that the impurities derived from oxidation
have considerable solubility in solutions of organolithium
compounds in hydrocarbon solvents.
Any one of several methods (4, 5, 9, 10, 11) can be used
to determine the carbon bound lithium concentration with vary-
ing degrees of accuracy and convenience. The "double titration"
method developed by Grilman and Cartledge (10) was the method
chosen for the analysis in this study. One modification was
made in the procedure by the substitution of dry n-pentane
for ether as the solvent. Comparable accuracy for the samples
diluted with the hydrocarbon solvent is achieved with this
modification and the problem of ether purity reported by
Kamienski and Esmay (15) is eliminated.
The procedure involves withdrawing a 3 milliliter
aliquot of the solution to be analyzed and adding it to 10
milliliters of dry n-pentane. A second 3 milliliter aliquot
is withdrawn and also diluted in 10 milliliters of n-pentane.
To this second solution is added dropwise 1 milliliter of
ethylene dibromide. The solution is swirled gently as the
halide is added and then allowed to stand for two minutes.
Both of these solutions are prepared in the dry box.

43
Upon removal of these samples from the dry box they are
hvdrolvzed with distilled water. The hydrolysis reaction (£t)
vieIds tf
R Li + HOH —» HH •+ LiOH (Ct)
hydrocarbon and lithium hydroxide. At this point all of
the lithium is converted to lithium hydroxide with the
exception of the carbon bound lithium previously reacted
with ethylene dibromide in sample number two. This reaction (b)
KLi 4- BrCH2CH2I3r—* RBr + LiBr + CH2CH2 (b)
converts all of the carbon bound lithium to organic halide
and lithium bromide. The concentration of hydroxide in
each hydrolyzed san^le is then determined by standard acid
and base titration methods using phenolphthalein indicator.
The agitation called for by Gilrnan and Cartledge (10) when
the second sample is near its end point is supplied by a
magnet stirrer and stirring bar. The difference between the
basic normalities of the two samples yields the molarity of
the carbon bound lithium and the normality of the ethylene
dibromide sample is a measure of the normality of oxygen
bound lithium.
Gas Liquid Chromatography
Some of the major by-products generated during the
menthyllithium or 4-tert-butylcyclohexyllithium syntheses
are the olefin derivatives of these compounds. Therefore
the second analytical method developed to determine the

44
parity of solutions derived from these compounds is designed
to determine the olefin concentration. It is also used as a
cliec,-; for the concentration of hydrocarbon bound to lithium
through a carbon lithium bond. This analysis employs gas
liquid chromatography for evaluating the olefin content
in these solutions.
Zubyk and Conner (24) reported the separation of cis-
and trans-p-menthanes, 3-p-menthene, and 1-p-menthene
using a packed Carbowax 4000 chromatograph column. An eight
foot by one-quarter inch aluminum tubing column packed with
10 per cent Carbowax 4000 substrate on 30-60 mesh size
Chromosorb W was prepared for this analysis. Comparable
separations to those reported by Zubyk and Conner (24) were
achieved at a temperature of 90 degrees centigrade and with
a helium carrier gas flow of 120 milliliters per minute.
A Model A-350B Aerograph chromatograph equipped with dual
thermal conductivity detectors was the instrument used for
this analysis.
The retention times of the components of a p-menthane
and p-menthene mixture for this column are listed in Table III.
It is evident from these data that the 2-p-menthene and
3-p-menthene are not separated on this column. Therefore
only the total amount of 2- and 3-p-menthenea can be deter-
mined by this analysis.

45
TABLE III
p-LIENTHANE AND p-MENTHSNB RETENTION TIMES
Compound Retention Time (Minutes)
cis-p-menthane . 3.00 trans-p-menthane . 4.05 trans-8(9)-p-ftienthene 5.25 2-p-menthen e 5.05 3-p-menthen e . . 5.85 1-p-menthene . . 8.70
The p~menthene compounds listed in Table I are charac-
terized by first concentrating each component by fractional
distillation on a Nester/Paust Annular Teflon Spinning Band
distillation column. The fractions, which do not represent
pure p-menthenes due to the inefficiency of the distillation,
are then analyzed on the Carbowax 4000 column, and infrared
and nuclear magnetic resonance spectra recorded for each
fraction. The data recorded from the infrared and proton
magnetic resonance analyses, along with the characteristic
boiling points reported for these compounds, are summarized
in Table 17.
Good agreements between these infrared wave length
values and those reported by Eschinazi and Pines (8) are
observed. Therefore from these data the menthene compositions
OA each distillate faction are established and assignments
of the olefinic proton resonances are made.

46
TABL^ IV
CHARACTERISTIC p-LIENTHENE INFRARED AND PROTON MAGNETIC RESONANCE ABSORPTIONS
Boiling Infrared Proton Compound Point Wave Lengths Resonances Compound
(°C) (0) (Liicrons) (4" Values)
1-p-uenthene 176 8.69 11.0 12.56 4-69 2-p-menthene 169 9.0 12.05 13.79 4.52 3-p-menthene 166 8.81 11.18 12.38 4.69 trans-8(9)-p-menthene 165.5 6.15 11.3 5.38
The p-inenthanes were characterized primarily from the
hydrogenation of a mixture of p-menthenes. The procedure
reported by Sauvage, Baker, and Husaey (20) for the hydro-
genation of cyclohexene over platinum oxide at one atmosphere
of hydrogen in acetic acid was used. A 10 gram sample of
the p-menthene mixture was shaken under these conditions
for approximately thirty hours. The sample was then
extracted in hydrocarbon solvent and analyzed by gas chroma-
tography and infrared spectroscopy. The retention times
reported in Table I for the two p-methanes were observed
and the infrared spectra were compared with those reported
in Sadtler (19) as spectra numbers 1846 and 20221 and by
Eschinazi and Pines (8).
When the hydrocarbon layer of a hydrolyzed sample from
a typica1 menthyllithium solution is analyzed by gas chroma-
tography the concentration of p-menthenes can vary from only-
trace quantities to 10 to 15 per cent. This concentration is

47
dependent upon the age and origin of the sample. The con-
centration of 4-tert—butylcyclohexene is also dependent
upon these two factors. Characterizations of the 4-tert-
butylcyclohexene and the saturated compound were accomplished
by employing the same techniques as those described for the
menthyl compounds.
Polarimetry
The possibility exists that the reaction between
optically active menthyl chlorides and lithium metal can
yield optically active enantiomers of the lithium compounds.
In order to determine whether this phenomenon occurs an
analytical procedure for measuring the rotations of the air
and moisture sensitive solutions was designed. For other
organic compounds standard procedures were employed for ob-
taining rotation data.
Optical rotatory dispersion curves were measured for
solutions of menthyllithium prepared from both menthyl and
neomenthyl chloride. These curves were measured over a
range of 600 to 320 millimicrons using a Gary Model 60
instrument.
Routine optical rotations were taken on a Model 80
Rudolph Visual High Precision Polarimeter equipped with
both sodium and mercury light sources. The instrument can
be read to 0.01 degrees directly. Polarimoter tubes adapted
with tapered ground glass porta and ntoppera and modified

48
with Teflon 'washers sealing the standard 15.5 millimeter
glass cndplates were used. Dry benzene, n-octane, and n-
pentane were the solvents used for these determinations.
All of the samples studied were prepared in the dry box.
The first step in the procedure employed to obtain
these readings is to dissolve the recrystallized rnenthyl-
lithiom in dry hydrocarbon solvent. The solution is then
filtered through a medium fritted funnel in an attempt to
eliminate any insoluble or suspended impurities. Aliquots
are tali.en for titration and gas chromatography analyses to
determine the concentration of carbon bound lithium and
olefin and alkoxide impurities. Another sample of the
solution is loaded into the polarimeter tube and taken out
of the dry box. Ten or more rotation readings are taken
and recorded as rapidly as possible. As soon as this is
completed the tube is rinsed several times and loaded with
solvent; rotation readings are then recorded for this
standard.
These optical rotational data are reported as specific
rotations which can be calculated from the observed rota-
tional readings and the concentration of carbon bound
lithium. The specific rotation can be determined from the
following expression:
t 100 - T T

C<
1'J
t — specific rotation of a substance* at a A specified wave length (A) and temperature (t)
— til® observed rotation measured in decrees at A a specified wave length (A) and temperature (5)
1 = length of liquid path measured in decimeters
c = concentration of solute expressed in grains per 100 milliliters of solution
Infrared Spectroscopy
This analytical technique was used to aid in the charac-
terizations not only of the organolithium compounds, but
also of the organic derivatives derived from these compounds.
The samples were prepared for these investigations using
well-known techniques, such as distillation, isolation from
gas-liquid phase chromatographic columns, and crystallisation.
Exceptions were the air and moisture sensitive compounds which,
were prepared in the dry box. These samples were run either
in tightly sealed cesium bromide liquid cells (0.1 millimeter
path length) or as postassium bromide pellets. These pellets
were prepared in the dry bos using a Mini-Press purchased
from the 7/ilks Scientific Corporation.
Two Perkins-31mer Infrared Spectrometers were used to
record these spectra. The Model 621 instrument was used
primarily with some spectra recorded on the Model 237. Care
was taken to insure that the instruments were calibrated
properly with the recommended standards.

50
Nuclear iJagnetic Resonance Spectroscopy 7*
The principal physical method used in the conformational
analyses of the organolithiuin compounds was nuclear magnetic
resonance spectroscopy . As in most of the reported conforma-
tional applications of nuclear magnetic resonance spectroscopy,
the hydrogen nucleus was utilized. The accessibility of in-
strumentation was the factor which dictated the choice of
a proton magnetic resonance study.
Most of the spectra recorded in this investigation were
obtained from a Yarian Associates A-60 Model High Resolution
Nuclear Magnetic Resonance Spectrometer. This instrument
was equipped with a variable temperature controller. However
the instrument's probe was limited to a temperature range of
-60 to 200 degrees centigrade. Spectra were also recorded on •
an A-60A Spectrometer which was equipped with a variable
temperature probe that could be cooled to -100 degrees centi-
grade. For both spectrometers the sweep was properly cali-
brated in the usual manner using tetramethylsilane and
chloroform absorptions. The variable temperature controller
was calibrated with methanol.
Because of the design of the detection system of the
A-60 and A60A Models a definite phase problem is encountered
at low temperatures. In order to eliminate this problem
spectra recorded over a temperature range from room tempera-
ture to -100 degrees centigrade were obtained from a Model

51
IIR-oO Varian Spectrometer. The Varian HA-100 Model High
Resolution Spectrometer was also employed in this in-
vestigation. This instrument was used primarily in studies
where changes in chemical shifts would aid in the charac-
terization of the compounds under investigation.
The procedure for the preparation of samples for this
analytical method are mostly routine. Only one sample
preparation procedure for the oxygen and moisture sensitive
compounds needs elaboration. This is the procedure employed
when dimethyl ether is used as the solvent.
The requirements of this procedure are to /.eep the
organolithium solution free from oxygen and moisture and at
the same time to Keep the sample below a temperature of
-25 degrees centigrade. This latter requirement is neces-
sitated by the low boiling point of dimethyl ether. For a
typical run these requirements are fulfilled by first sealing
a nuclear magnetic resonance sample tube onto an outer 14/55
standard tapered ground glass joint. A sample of crystalline
organolithium compound is then loaded into the sample tube.
An adapter consisting of a Kontes 4 millimeter hollow plug
vacuum stopcocii adapted with both male and female 14/55
standard tapered ground glass joints is fitted on xhe sample
tube. The stopcoc^ is closed in order to retain the dry box
atmosphere in the sample tube, and the entire assembly is
then removed from the dry box. It is then immediately

52
connected "by the remaining female glass joint to a liigh
vacuum manifold and the sample tube is evacuated.
After the system has been evacuated it is cooled to
the temperature of liquid nitrogen by raising a Dewar flask
filled with the liquid around the sample tube. Dimethyl
ether gas is condensed through a manifold port onto the
crystalline sample. The extremely cold temperature solid-
ifies the ether solvent so that the sample tube can be
evacuated once again. While continuing evacuation the
sample tube is sealed off with a torch directly below the
glass joint which had been adapted to the tube. The sample
is then warmed in a dry ice-acetone bath to -78 degrees
centigrade. ' ,7ith snaking the crystalline solute dissolves
at this temperature in the dimethyl ether solvent. The
temperature-of the solution can then be regulated to any-
desired temperature by adjusting the temperature of the
acetone bath with either dry ice or acetone additions.
Dry toluene and cyclopentane were used as solvents
for low temperature studies requiring hydrocarbon solvents.
n-Pentane can also be used but interference from side bands
in the high magnetic region limits its usefulness. The
samples for these studies are dissolved in hydrocarbon solvent
in the dry box at room temperature and special tight fitting
stoppers purchased from M R Specialties Incorporated are used -
to seal the sample tubes.

53
Experimental Llethods Employed in Derivative Formation of the Organolithium
Compounds
Car conation
Solutions of redissolved crystalline compounds, of the
remaining mother liquors, and original reaction solutions
for both of the organolithium systems studied are carbonated
in two ways. The first method consists of preparing a
mixture of crushed dry ice in n-pentane solvent and then
placing it inside a plastic glove "bag under a nitrogen
purge. The solution to be carbonated is then removed from
the dry box in a tightly stoppered flask and also placed
into the glove bag. The final step is to remove the stopper
and very quickly pour the contents of the flask over the
dry ice-n-pentane slurry. The reaction flask is then removed
from the glove bag, covered with a loosely fitting cover, and
allowed to sit for several hours while the excess carbon
dioxide evolves.
In order to determine whether the stereochemistry of
» this reaction is altered by changes in reaction procedures
a second carbonation procedure was employed for the menthol
system. This method differs from the low temperature pro-
cedure described above first, by the fact that it is conducted
at room temperature, and second, in the use of carbon dioxide
gas as the reactant. In a typical run the solution of organo-
lithium is loaded into a round bottom flask in the dry box.

54
" It is then capped, brought oat of the box, and secured to
a lattice rac;:. A gas dispersion tube purging with carbon
dioxide gas is then quickly inserted into the reaction flask
and the flow of gas is continued until the solution gives
a negative Gilman test.
Solvent effects on the reaction pathway were studied
as well. The method of carbonation using a dry ice-n-pentane
slurry is employed for these investigations. Bolh diethyl
and dimethyl ethers are used as solvents. The procedures
for preparing solutions in these two solvents differs from
that used for hydrocarbon solvents.
The preparation of a solution using dimethyl ether as
the solvent haa been essentially outlined in the procedure
for preparing nuclear magnetic resonances samples in this
solvent. However, due to a number of appartus changes made
to facilitate carrying out this process in larger quantities,
the following description is included.
In the dry box a sample of crystalline organolithium
compound is loaded into a 100 milliliter single-necic round
bottom flask equipped with a 24/40 standard tapered outer
joint. The flask is then sealed with an adapter that consists
of a Lontes 4- millimeter hollow plug vacuum stopcock sealed
to inner 24/40 and outer 14/35 standard tapered joints. Keep-
ing the stopcock closed, the flask and adapter are removed from
the dry box and connected to a high vacuum-manifold. The at-
mosphere above the lithium compound is then evacuated.

55
After the system has been evacuated it is cooled to the
temperature of liquid nitrogen by raising a Dewar flask filled
with the liquid around the f]ask. Dimethyl ether gas is con-
densed through a manifold port onto the crystalline sample.
The extremely cold temperature solidifies the ether solvent
so that the flask can be evacuated once again. At this
point the manifold and flask are pressurized to atmospheric
pressure with argon. Under this argon atmosphere the system
is warmed to the desired temperature. The flask is isolated
from the room atmosphere by dosing the adapter stopcock and
taken into the glove bag. The carbonation procedure is the
same as for other samples. Except for a few alterations this
same procedure is used in carrying out the carbonation reaction
on diethyl ether solutions. The only major change is neces-
sitated by the physical property differences of the two
solvents. A bulb to bulb distillation is required to transfer
the diethyl ether solvent into the reaction flask. In ad-
dition, when diethyl ether is the solvent, only a temperature
of below 0 degrees centigrade is required.
Characterization of the products produced from these
reactions is pursued by two different methods for the menthyl
system. The first method involves isolating the carboxylic
acids formed; the procedure used is the method outlined by
Smith and Wright (21).
The crude acid is obtained in yields as high as 90 per
cent. It .possesses a melting point between 62 and 65 degrees

56
centigrade and a specific rotation of -56.1°(CH3CH20H,
c. 5.2). Sruitn and Wright (21) reported the welting point
and specific rotation values for the 3-p-menthane car "boxy lie
acid isomer to "be equal to 60 to 63 degrees centigrade and
-4-2.6°(CH3CH20H, C. 816). They also reported the 3-p-
heomenthane acid isomer to have a higher melting ^oint and a
positive specific rotation value. A lacx of evidence for the
formation of the highest melting acid placed serious doubt
on the degree of sensitivity offered by this analysis method.
This is the reason the second and most effective method was
incorporated.
The procedure for this method employs the conversion
of the acid diastereoners into their respective esters. This
is accomplished by first carbonating a pentane solution of a
lithium compound over dry ice and then extracting the residual
hydrocarbon solution with dilute hydrochloric acid. The
hydrocarbon layer is then separated from the aqueous layer
and dried over anhydrous magnesium sulfate. This drying agent
is later removed by filtration and the carboxylic acid-n-
pentane solution is added dropwise to a cold ether solution
of diaamethane. This reagent is synthesized using either
the method reported by Moore and Reed (18) or the method of
Arndt (2). The former was found to be the favored method.
After the dropwise addition of the organic acid solution has
been completed the resulting solution is allowed to sit from

57
six to ei^ai hoars. Au thir> ti„.e ilic remaining excess
diazoi.i2 oiir,us is 'Invon oA.' by x'Si ] uxmg line solution v/ioxi
a steam bath. "Disappearance of the yellow coloration in-
dicates unao this atop Las been comp]eted and the reaction
pro duct 3 are concentrator; by 3olvont evaporation an .1 analyzed.
T-lie menthyl est or epimer mixture is easily isolated from
other menthyl derivatives by fractional distillation. This
is accomplished at 93. p to 94 degrees centigrade and J tori-
pressure. A typica"1 product isolated in such a fraction
possesses a 'specific rotation of -39• 7°( 11-C5H12,c. 5.0) and
characteristic ester infrared absorptions. However the
identity of the ester is confirmed by a .100 megacycle proton
magnetic resonance spectrum which shows an axial <-iuetliinyl
proton resonance at a lvalue of 2.33. This resonance is
obscured by other absorption in the 60 megacycle spectrum.
A detailed discussion on conformational analysis of cyclo-
ne xyl systems by nuclear magnetic resonance spectroscopy will
be included in the following chapter.
A gas liquid chromatographic analysis of the esters is
made at 145 degrees centigrade with a 240 milliliter per
minute helium flow on a ten foot by one-quarter inch aluminum
tubing column packed with a 15 per cent polypropylene glycol
substrate on 30-60 mesh Oiiromosorb V7 pacIcing material. The
use of the thermal conductivity detector instrument allows
the characterization of the two ester isomers by isolation

53
of the column effluent into micr o-infrared cells. This
technique was especially useful in characterizing the low
concentration isomer whose concentration ranged from only
5 per cent to 30 per cent, depending upon the solvent
employed in preparing the organolithiuij solution. Again,
both compounds demonstrated characteristic infrared ab-
sorptions of organic esters.
A second chromatographic analysis which gives better
separation of the ester compounds and the menthyl alcohol
impurities is carried oat at 105 degrees centigrade with
2 milliliters per minute carrier gas flow rate on a 150
foot by 0.01 inch Carbowax K-1540 G-olay column. A Perkin-
Slmer Model F-ll gas chromatograph equipped with a hydrogen
flame detector is used in this analysis. Retention times
of 11 and 13.5 minutes on the polypropylene glycol column
and 16 and 18.7 minutes on the Carbowax column are recorded
for the two isomers.
Gtolow and Boyce (22) have reported the physical
properties and the nuclear magnetic resonance spectra for
the cis and trans isomers of methyl 4-tert~butylcyclohexane-
carboxylate. Using these data, along with infrared and
carbon and hydrogen analyses, the compounds separated on
the Carbowax K-1540 and polypropylene glycol chromatography
columns were identified. Retention times recorded on these
columns using the conditions described above were 23.0 and

59
26.0 minutes on the Carbowax Ii-1540 column and 24.8 and
28.5 minutes on the polypropylene glycol column for the
cis and trans isomers.
Deuteration
The reactions between deuterium oxide and the menthyl
ana tcrt—buti;-lcyclohexyl lithium compounds undor investi-
gation were studied. The use of both n-pentane and dimethyl
ether as solvents for these reactions requires, as was
described for carbonation, two different procedures for
carrying out tnese investigations. The sample preparations
are conducted in the same manner as those described for
carbonation in the two different solvents. After these
steps are completed the deuterium oxide is introduced onto
the samples through a rubber septum cap with a syringe.
Therefore the reaction flask used for the dimethyl ether
solution must have a second opening which is sealed off with
the septum cap. In addition, magnetic stirring bars are
placed into the- flasks prior to sample introductions for both
systems. These are used for stirring the solution as the
deuterium oxide is being added.
The hydrocarbon layer containing the product is isolated
from the excess deuterium oxide with a separatory funnel and
dried over anhydrous magnesium sulfate. The drying agent
is later removed by filtration and the sample is concentrated
by evaporating the solvent with a stream of dry air. This

60
sample is then analyzed using the infrared spectrometer,
gas chromatography, and optical rotation instrumentation
previously described. The infrared stretching frequencies
of the caroon—deuterium bond are the most important data
recorded from these analyses. These values occur between
2200 and 2100 wave numbers for the deutero oyclohexane
derivatives studied. Carbon tetrachloride is the solvent
used in recording these spectra.
Bromination
Bromination procedures for both molecular bromine and
ethylene dibromide reagents are employed using both n-pentane
and dimethyl ether solvents and temperatures ranging from
room temperature to -70 degrees centigrade.
The preparation of the organolithium solutions in the
two different solvents is accomplished with the same methods
described for the carbonation reactions. One-hundred-milliliter
two-neck round bottom flasks equipped with 14/35 tapered ground
glass joints were used. One neck of the flask is modified
with a 4- millimeter stopcock inserted between the flask and
the joint. This entry port is used primarily to keep an
atmosphere of argon above the sample throughout the reaction.
Both of the brominating agents are introduced into the
reaction flask by the dropwise addition of n-pentane solutions
of these two reagents. The addition funnels employed for
this process are first loaded with the n-pentane-bromination

61
reagent solutions and then attached to the reaction vessel
through the second nock of tne flask. This is accomplished
while the entire s;/stem is being purged with argon gas.
The syste:;i is then equilibrated with an argon atmosphere
and the reaction is completed by the addition of the brominat-
ing reagent over a period of thirty minutes. The solution
in the reaction flask is stirred with a magnetic stirring
bar and the desired temperature is achieved by immersing the
flask in either a dry ice-acetone or ice water bath.
The reaction products are isolated by evaporating the
n-pentane solvent and, if large enough quantities of re-
actants are initially used, the products can be distilled.
The menthyl products are isolated at 32 and 83 degrees
centigrade and 7 torr; the 4-tert-butylcyclohexyl bromide
isomers are isolated at 99 to 102 degrees centigrade and
13 torr. The isolated products are then characterized using
infrared, nuclear magnetic resonance, and gas chromatographic
techniques. Infrared characterization data for substituted
cyclohexyl bromide systems has been reported by Eliel and
Haber (7). These data along with an analysis of the charac-
teristic nuclear magnetic resonance absorptions observed for
the different ©<-methinyl proton conformations support the
quantitative chromatographic determinations for the dis-
trioution of products. A summary of the findings from nuclear
magnetic resonance and infrared studies is included in
Table V.

62
TABLE V
DATA SIOIARY FOR CYCL0H3XYL BR Oil IDE DERIVATIVES
C ompound Characteristic
Infrared Wave dumber (cm"-'-)*
Characteristic^ Proton Magnetic Resonance Value
tie n thy 1 Bromide 686 6.30
Neomenthyl Bromide 665 5.62
trans-4-tert-Butyl-cyclohexyl Bromide 690 6.07
cis-4-tert-Butyl-cyclohexyl Bromide 663 5.38
*31iel and Haber (7) iiave reported characteristic infrared ranges of 690-710 cm~l and 670-685 cm~l for equa-torial and axial cyclohexyl bromide derivatives.
Separation of the bromide isomers of the menthyl and
4-tert-outylcyclohexyl systems is achieved on a 150 foot by
0.01 inch K-1540 Carbowax Golay chromatographic column. The
conditions employed for these analyses are a temperature of
88 degrees centigrade with a carrier gas flow rate of 2
milliliters per minute. Retention times of 16.2 and 17
minutes for the neomenthyl and menthyl bromide isomers, and
18.6 and 19-0 minutes for the cis- and trans-4—tert-butyl-
cyclohexyl bromide isomers are observed.
Ox idation
The procedure used to carry out the reaction between
molecular oxygen and menthyllithiuiu solutions is identical
to the one used for the room temperature carbonation of

63
mentliyllithiuw-n-po;itane solutions. Samples from tho orig-
inal reaction solution before crystallization and samples
from solutions of redissolved crystalline material in n-pentane
solvent are studied. Dry compressed air is bubbled through
these solutions at room temperature for one hour • Dui in^
-this time the solution is stirrod with a magnetic s lining
bar.
Upon completion of the oxidation the contents of the
flask are hydrolyzed and acidified with dilute hydrochloric
acid. The aqueous layer is then removed and the remaining
hydrocarbon solution is dried over magnesium sulfate. This
drying agent is easily removed by filtration and the filtrate
concentrated by solvent evaporation. The reaction products
are analyzed by gas liquid chromatography and nuclear mag-
netic resonance methods. An 8 foot by one-quarter inch
aluminum tubing column packed with 10 per cent Carbowax 4000
substrate on 30-60 mesh size Chromosorb V/ at 135 degrees
centigrade with a helium carrier gas flow of 105 milliliters
per minute is used for the chromatographic analysis. Re-
tention times of 16.7 and 21 minutes for the neomenthol and
menthol isomers are recorded for this analysis. Character-
istic ®<-methinyl proton resonances for the neomenthyl and
menthyl isomers occur at 5.97 and o.SQ't respectively.

u -
Iorti-LithiLU.I 'Bxchange
This reaction is carried out by adding a "benzene solution
of methyl iodide to a solution of mentliyllithium in n-pentane
solvent. The methyl iodide solution was added by pipette
and allowed to react for one minute. The product is then
quiclcly filtered through a fritted funnel. This entire
procedure is carried out in the dry box.
Upon removal of the filtrate from the dry box it is
hydro!yzed and the hydrocarbon layer is dried over magnesium
sulfate. The menthyl iodide solution is isolated from the
drying agent by filtration and then isolated from other
menthyl derivatives by fractional distillation. The menthyl
iodide fraction is collected at 115-116 degrees centigrade
and 10 torr pressure. This fraction is analyzed by proton
magnetic resonance and polarimetry methods. The character-
istic «=< -methinyl proton resonance patterns are observed from
the neomenthyl and menthyl iodide isomers at 5.24 and 5.89^.
The specific rotation value determined for this fraction
equals -31.5°(CD3C1, c. 13.0).
Decomposition
The decomposition is carried out on samples of crystal-
line methyllithium dissolved in dry n-decane. Both sealed
nuclear magnetic resonance sample tubes and. the same two-neck
flasks employed for bromination reactions are used as re-
action vessels. The organolithium solutions are prepared

65
and loaded into these reaction vessels in the dry box. At
the same ti^e samples for carbon "bound lithium concentration
and gas chromatographic analyses are taken.
The nuclear magnetic resonance sample tubes used for
reaction vessels are adapted with taped glass joints and
stopcock" adapter as described for the sample tubes used in
nuclear magnetic resonance studies of the organolithium
compounds in dimethyl ether solvent. As in these studies,
when the samples are removed from the dry box they are
frozen, evacuated, and then sealed with a glass torch. The
samples in the larger volume reaction flask are immediately
purged and then equilibrated, under an argon atmosphere when
brought out of the dry box.
The variable temperature attachment on the nuclear
magnetic resonance spectrometer is employed in carrying out
the decompositions of the samples sealed in the nuclear
magnetic resonance sample tubes. Spectra of the olefinic
proton absorptions are recorded repeatedly at the desired
elevated temperatures until the reaction is complete. For
the larger volume samples the elevated temperatures are
furnished by heated oil baths. These samples are stirred
with magnetic stirring bars.
The reactions are terminated by the addition of water
and then the hydrocarbon layers are separted and dried in
the same manner as the deuterated compounds described earlier.

66
The anal.-sis of Vut> p-.:ietheties and p-nienthane ooppositions
fron a topical reaction is ;.,ade using gas liquid ciiroi,.a-
tography. Tito inability of the packed Carbowax 4000 column
described earlier to separate the 2- and 3-p-i.ientiienes re-
quired the use of another column. This separation is achieved
on a 200 foot "by 0.01 inch 95 per cent Squalane and 5 per cent
Atpet Golay column at 83 degrees centigrade and with a carrier
gas flow of 2 milliliters per minute. The same infrared and
nuclear Magnetic resonance techniques described earlier are
again applied for identification of the elution order of
these compounds. Retention times of 13.5 and 18.8 minutes
are recorded for the p-3-menthene and p-2-nienthene.

CHAPTER BIBLIOGRAPHY
1. Alexandrou, h. E. , "Reaction of n-But,, llitnium with c is — and tr a ns -4~tsrt-Bat y lc,/clohex;-1 Bromide and Iviercoric Bromide," Journal of Or^anowctallic Chemistry, V (1966), 301.
2. Arndt, P., "DiazomethaneOrganic Syntheses, XV (1935), 3-
3. Brown, Theodore L., J. A. Ladd, and G. K. Newman, "Interaction of Alkyllitnium Compounds with Base. Complex Formation Between Ethyllithiuin and Lithium Ethoxide in Hydrocarbon Solvents," Journal of Organo-r.ietallic Chemistry, III (1965), 1.
4- Clifford, A. L. and R. R. Olsen, "The Analytical Chemistry of Or ganome tallies. Quantitative Determination of Organoalkalies," Analytica 1 Chernistry,, XXXII (19-60), 544.
5. Collins, Peter P., Conrad Xamiens.^i, Donald L. Esmay, and R. B. Ellestad, "Oxidimetric Determination of n-Butyllithi..,:. in Hydrocarbons Using Vanadium .Tcr.toxide," Analytical Chemistry, XXXIl'I (1961), 463.
6. . Cur tin, D. Y. and J. hoenl, "Effect of Solvent on the Steric Stability of Lithium Reagents," Journal of the American Chemical Society, LXXXIV (1962), I967.
7. Sliel, Ernest L. and Ralph G. Haber, "Conformational Analysis. V. The Reaction of cis- and trans-4-t-Butylcyclohexanol and trans-4-Methylcyclohexanol with Phosphorus Pentabromide. Syntheses of Alkylcyclohexyl Bromides," Journal of Organic Chemistry, XXIV (1959) 143.
8. Sschinazi, H. E. and Herman Pines, "Study in the Terpene Series. XXVI. Disproportionation of d-Limonene in the Presence of Palladium Hydroxide-Barium Sulfate Catalyst," Journal of the American Chemical Society, LXXVII (1956)","1176:
9.. Everson, W. L., "Determination of Butyllithium in Hydrocarbons by Thermometric Titration with Butanol," Analytical Chemistry, XXXVI (1964), 054.
67

68
10. Oilman, Henry and j'rank K. Cart ledge, "The Analysis of Organolithium Compounds," Journal of Organometallic Cher, is try, II (1964), 447. '
11. Gilman, Henry and A. U. Haubein, "The Quantitative Analysis of Alkyllithium Compounds," Journal of the American Chemical Society, LXVI (1944), 1515>.
12. Gilman, Henry, 7,rright Langham, and Ared W. Moore, "Some Interconversion Reactions of Organolithium Compounds." Journal of the American Chemical Society, LXII (1940), T5TT*
13* Greene, Frederick 3 . , Chin-Chium Chu, and Jasjit V/alia, "Decomposition of Alkyl Hypochlorites. The 4-t-Butylcyclohexyl Radical," Journal of Organic Chemistry, XXIX (1964), 1285.
14. Horner, von Leopold, Hermann Oediger, ana Hellmut Hoffman, "Reaktionen Mit Triphenyl-Phosphin-Dihalo-geniden," Justus Liebigs Annalen der Chemie, DCXXVI (1959), 26"
15. Kamienski, Conrad and Donald 1. Esmay, "Analysis and Stability of n-3utyllithium Solutions in n-Heptane," Journal of Organic Chemistry, XXV (i960), 115.
16. _ , "Effect of Sodium in the Preparation of Organolithium Compounds," Journal of Organic Chemistry, XXV (i960), 1807.
17. Larnaudie, M. Marcel, "Isome'rie des derives mono-substitues der cyclohexane," Comptes Rendus Hebdomadaires Des Se'ances De PAcad&nie Des Sciences. 0 o m V (1052) ,"1541
18. Moore, James A. and Donald "3. Reed, "Diazomethane," Organic Syntheses, XV (1935), 3.
15* Sadtler Standard Infrared Spectra, Philadelphia. The Sadtler Research Laboratories, Spectra Numbers 1846 and 20221.
20. Sauvage, James Frederick, Robert H. Baker, and Allen S. Hussey, "The Hydrogenation of Cyclohexene over Platinum Oxide," Journal of the American Chemical Society, LXXXII (1$66)',""6( 01

69
21. Smith, J. G. and George ?. //right, "The Diastereomeric Menthyl Chlorides Obtained j?roia (-)Menthol," Journal of Organic Chemistry, XVII (1952), 1116.
22. Stolow, Robert D. and Charles B. Boyce, "Preparation of cis- and tran3-4-t-Butyl-<<,«<-diirtethylcyclohexane-methanol," Journal of Organic Chemistry, XXVI (1961), 7426.
23. Walborsky, II. II. and M. S. Aronoff, "Cycl opropanes XIX. Reaction of Lithium Metal with Optically Active Halides," Journal of Organometallic Chemistry, IV (1965), 415T
24. Zubyk, W. J. and A. Z. Conner, "Analysis of Terpene Hydrocarbons and Related Compounds by Gas Chroma-tography," Analytical Chemistry, XXXII (I960), 912.

CHAPTER III
RESULTS AND DISCUSSION
Derivatives of cyclohexyllithium have been proposed as
the compounds to incorporate in this investigation of the
conformational stability and the stereochemistry of a number
of characteristic reactions of alkyllithium compounds. This
selection evolved from the requirement that the organic
moieties of these lithium compounds possess different degrees
of conformational stability. The barrier to inversion for
the substituted cyclohexyl products has been known for some
time to be dependent upon the nature of the organic sub-
stituent. Therefore a model system for this investigation
should be provided by a series of these compounds.
The first objective of this investigation is to determine
whether a configurationally stable alkyllithium compound
can be synthesized and isolated. From the viewpoint of the
lithium compound there appear to be three main factors which
can enhance this stability. The first factor is the experi-
mental conditions employed in synthesizing and isolating the
compound. It is obvious from the review of earlier investi-
gations of this type that the selections of both the tempera-
ture and the solvent are critical.
70

71
The second factor, which Curtin and Koehl (9) predicted
to be an integral ingredient of the inversion mechanism of
these systems, is the degree of association demonstrated by
these compounds. The extent of association will be primarily
predetermined by the choice of the solvent,. Even so this
phenomenon might still influence the populations of the
different conformers available to the organic constituents,
especially for systems as large as cyclohexyl derivatives.
The last factor concerns the effect of the conforma-
tional stability of the organic moiety on the overall
stability of the alkyllithium compound. This effect would
definitely be a primary contributor to the overall stability
of non-bridged cyclic systems. Therefore with the initial
objective of synthesizing a configurationally stable ,
cyclohexyllithium compound this effect is of primary im-
portance in- selecting the compound to study.
Conformational analysis has been applied most intensively
to cyclohexane and its substitution products. Because of
steric interactions and an increase in the number of gauche
conformers the axial position of a substituent is less
favored than an equatorial position. For the mono-substituted
products these effects become pronounced when an isopropyl
substituent is considered. If a still bulkier substituent
is examined, such as the tert-butyl group, then an axial

7 0
orientation is no longer possible. For di- and. tri-
substituted products these effects are magnified and
preference for the equatorial substituent may be observed.
The first substituted cyclohexyl product to be in-
vestigated as a lithium derivative is the menthyl system,
diagrammed in Figure 1 (a, b). This sytem is capable of
exhibiting not only conformational stability but also
optical activity. Incorporation of the optical property
into the system provides a convenient method for determining
whether one conformation of the compound is preferred over
another. It will also serve, along with proton magnetic
resonance and chemical derivative data, as evidence for
conformational stability.
Some concern might originate because this highly sub-
stituted molecule contains asymmetric centers other than the
carbon atom to which the lithium atom is bound. It is
reasonable to question whether neighboring group and asym-
metric induction effects potentially complicate the stereo-
chemistry studies being undertaken. Gram (8) pointed out in f
his review on the stereochemistry of organomercury compounds
that these same questions arose. In these cases simpler
systems were investigated and the same conclusion regarding
the stereochemistry for both groups was reached. For the
lithium system the same logic will be applied and in this
investigation both menthyl and 4-tert-butyl.cyclohexyl systems
will be studied.

73
Selection of the menthyl derivative also involved the
availability of the starting organic chloride. Smith and
Wright (25) and Horner and co-workers (12) have reported
stereospecific syntheses for menthyl chloride (a^) and
neomenthyl chloride (th) epimers from (-)-menthol (a2).
This alcohol is commercially available in a very pure form.
In contrast only an 80:20 isomer mixture of the trans- (c2)
1 2 3 4 5 6 CI OH D Br I C02H
(c) (d)
Fig. 1—Cyclohexyl derivatives studied
and cis-4-tert-butylcyclohexanols (d2) is available. Further-
more syntheses of the corresponding chlorides, (cj) and (di),
have not been studied so extensively and are therefore less
refined than those listed for the menthyl system.
Carbon bound lithium yields ranging from 50 to 60 per
cent were obtained from the direct reaction of lithium metal
with either menthyl- or neomenthyl chloride. This productivity

74
decreased to only 30 per cent for the cis-4-tert-butyl-
cyclohexyl chloride reaction. From these reaction solutions
crystalline, pyrophoric organolithium compounds were isolated.
The 4-tert-butylcyclohexyllithium product was observed to have
a relatively lower solubility in hydrocarbon solvent than the
inenthyllithium product. This property has a definite effect
on the amount of material which can be crystallized from a
solution. Only about half of the soluble menthyl derivative
can be isolated while almost all of the 4-tert-butylcyclohexyl
compound is obtainable.
Although the syntheses of these two cyclic organolithium
compounds have not been reported, this accomplishment comprises
only half of the first objective of this investigation. Com-
pletion of conformational analysis investigations of these
systems constitutes the second and most demanding half of
this study. This obviously requires installation of other
analysis methods. One of the oldest and most accessible
techniques employed in investigations of this type is a
measure of the rotation of polarized light. Since the menthyl-
lithium products could demonstrate optical activity, polarimetry
studies were pursued first.
Hydrocarbon solutions of the redissolved crystalline
lithium products derived from both the menthyl and neomenthyl
chloride isomers were observed to be optically active. Further-
more, both solutions were observed to be highly dextrorotatory

75
and the near identities of their specific rotations suggest
that the composition of the two solutions is identical. Only
small differences were recorded between the specific rotations
of an original reaction solution and a solution comprised of
dissolved crystalline material. Due to the high dextro-
rotatory specific rotational values reported "by McNiven and
Reed (19) for the p-2-menthene and p-3-menthene compounds
no measurements were recorded for the mother liquor solutions.
Table VI contains characteristic data recorded in these studies.
TABLE VI
CHARACTERISTIC ROTATIONAL VALUES FOR IviENTHYLLITHItM SOLUTIONS
Entry Chloride Isomer
Specific Rotation Menthyllithium Solution Specific Rotation
1 + 50.8° (n-C8H18,c. 5.24) +53.4° (C6H6, c. 3.40)^
2 -39.9 (C6H6,C. 5.00) +42.1 (C5H10, C. 6.28)-1
3 -40.0 (n-CgHgOgjC. 5.00) (**) (n-C5H12, c. 5.41)
-40.0 (n-C0pI]i g,c. 5.00) +56.0 (n-C H-j j* c. 3.65)^
^Solution of dissolved crystalline material.
**The rotational character for this original reaction solution was found to be dextrorotatory. Due to the presence of highly dextrorotatory p-menthenes in the solution a specific rotation value would be meaningless.
The accuracy of these values is only as good as the sampling
technique employed for obtaining the rotational values and
concentration values for the inenthyllithium. solutions.

76
Special attention was given to standardizing these techniques
from one ana3.ysis to another in order to minimize experi-
mental errors. This effort was then extended "by attempting
to determine whether the specific rotation values recorded
were subject to variation with time. Therefore a time
versus specific rotation study was conducted on a raenthyl-
lithiuu-cyclopentane solution. No change in rotation was
observed over a period of one hour.
Optical rotatory dispersion analyses were employed to
investigate the observation that the specific rotations were
effectively equivalent, irrespectibe of the chloride isomer
reagent. It was possible that one of the solutions could
possess a plane curve which starts on the positive side of
the zero rotation axis in the visible and crosses over this
axis in the ultraviolet region. However both solutions gave
almost identical dextrorotatory plane dispersion curves
from 600 to 300 millimicrons.
V/ith respect to the conformational analysis determination
the conclusions derived from these rotational studies were at
their best disappointing. The data indicates that the com-
position of the two solutions is probably identical. 7hether
this composition is comprised of a rapid or slowly equilibrating
equilibrium mixture or of a high concentration of one of the
menthyl- or neomenthyllithium isomers is not known. Further-
more, no predictions with respect to any of these possibilities
can be made from the size of the observed rotation values.

77
Head (21) has reported the rotational contributions of
tlie menthyl and isopropyl substituents for the neomenthyl
alcohol and amine isomers. Even with the assumption that
these contributions remain unchanged for the lithium compounds,
a lack of evidence of rotational contributions of lithium
substituents renders these values ineffective, furthermore
an investigation of the changes in rotational values with
different substituents for almost any system simply clouds
the situation even more. An example may be found in a
review of rotational values reported by V/alborsky and co-
workers (26) for the following conformational^ equivalent
compounds.
V
- CH~
^ / / W / /
X H Br I C02II
*1 -128° +116° +171° -36°
Infrared spectra of crystalline lithium compounds
suspended in a potassium bromide disc, a solution of dis-
solved crystalline material, and an original reaction
solution sample were recorded. In these spectra the charac-
teristic cyclohexyl ring and carbon-lithium absorptions were
observed in their respective frequency regions. Total analyses
of these spectra to establish complete structures were not
attempted. Furthermore, due to the broad shapes of the carbon-
lithium absorption, conformational assignments were not attempted.

78
These spectra were recorded primarily for purposes of com-
parison. No major changes were observed between any of
these three scans. This illustrates that the composition of
the crystalline material constitutes the primary composition
for both the original reaction solution and the solution
prepared from the crystalline material. It is also evident
that these data complement the findings reported in the
optical activity studies. However, the infrared data re-
quires simultaneous crystallization of the two epimeric
lithium compounds if a rapidly attainable equilibrium
exists in solution.
It is obvious at this point that another method of
investigation is needed. From a review of the published
literature a great deal of conformational analysis data for
cyclohexyl systems has been reported from nuclear magnetic
resonance investigations. Furthermore this tool has proven
to be very effective in a number of organometallic stereo-
chemistry studies. For these reasons incorporation of
proton magnetic resonance spectroscopy as a conformational
analysis instrument was one of the primary objectives of
this investigation.
Experimental observations by Lemieux and co-workers (17,
18) and Brownotein and Miller (5) demonstrated the usefulness
of proton magnetic resonance spectroscopy in studying con-
formations of substituted cyclohexyl ring compounds. Lemieux

79
and co-workers (17, IS) demonstrated the existence of a
chemical shift difference between the signals for protons
in axial and equatorial conformations. They observed that
the signal for protons in an axial orientation occurs at
higher fields than those for equatorial hydrogens.
Employment of this property as a conformational analysis
tool is often ineffective due to the existence of small
chemical shift differences and the complexity of the spin
coupling. For similar compounds Brownstein and Miller (5)
noted that conformational determinations could "be made "by
recording the widths of the two proton absorptions. They
reported that the width of the resonance peak of the ring
protons was always considerably greater in the disubstituted
isomers which had diequatorial conformations than in isomers
with axial-equatorial conformations. They further demon-
strated that spin-spin coupling is greater between two protons
that are axial to one another than between axial-equatorial
protons, which in turn is greater than for two equatorial
protons. Since the completion of these early investigations
by Lemieux and co-workers (17, 18) and Brownstein and filler
(5) a number of investigations of these types have been con-
ducted on a variety of similar compounds. Dryer (10) has
reported approximate coupling values for such systems; these
values are listed in Table VII.

80
TABLE VII
SPIN-SPIN COUPLING CONSTANTS
Type (XY) Coupling Constants (eps)
a,a 5-10 /—V a»e 2-4 < > e,e .2-4
K-C— C-H I I x y
These observations are applicable in characterizing
the menthyl conformers. Illustrations of the observed
o<-nenthinyl proton signals for a few menthyl epimers are
shown in Figure 2. Complete spectra of the menthyl and neo-
menthyl chlorides are shown in Figure 3. In agreement with
the previously described observations it can be seen in
Figure 2 that the broadest absorption occurs at a higher
field strength. This absorption therefore corresponds to
the o<_menthinyl proton in the axial position. Likewise the
absorption occurring at the lower field position is a much
sharper signal corresponding to the equatorial proton. The
axial and equatorial absorptions for various menthyl isomers
generally demonstrate signal widths of 30 and 10 cycles per
second respectively.
These width differences are readily explained in a
breakdown of the coupling constants generating each resonance
pattern. The high field absorption is derived from two axial-
axial and one axial-equatorial splitting. However the low
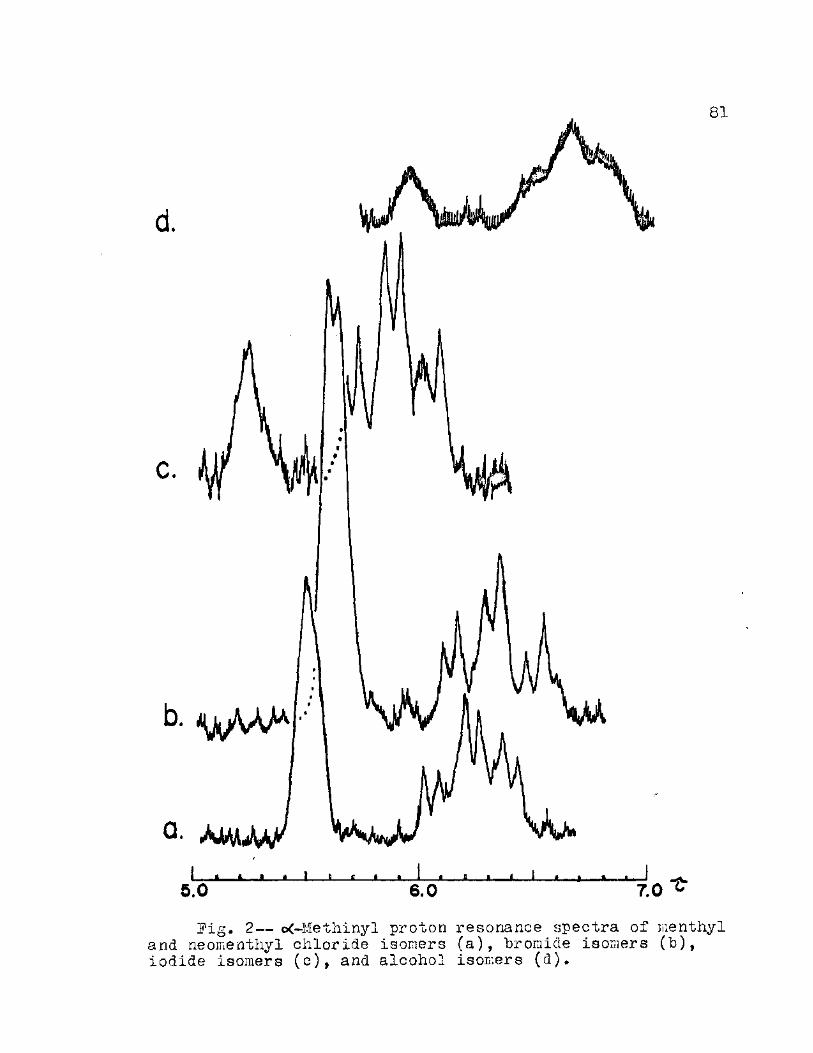
d.
c.
b.
Amu
81
5.0 6.0 7.0 C
Pig. 2 — o(-.Methinyl proton resonance spectra of menthyl and neomenthyl chloride isomers (a), bromide isomers (t>), iodide isomers (c), and alcohol isomers (d).

82
3--Proton magnetic resonance spectra of menthyl chloride (a) and neomenthyl chloride ("b).

83
field signal evolves from two axial-equatorial and one
equatorial-equatorial couplings. Using coupling values from
Table YII and the qualitative diagrams shown in Figure 4,
the two resonance patterns can be duplicated..
O A
axial D.e
0 to 20 L
30 0
quatorial e,e
10 20 JO
,?ig. 4—Spin-spin coupling diagram of resonance patterns.
Both of these characteristic patterns have been computer-
synthesized by Glaze (27). The IA0C00U II program reported
by Castellano and Botimer—By (6) was employed in these
calculations. Employing this procedure, while using vicinal
axial-axial and axial-equatorial coupling constants of 13 and
4 cycles per second and a geminal coupling of -15 cycles per

84
second, a high field pattern that could be almost super-
imposed upon the proton magnetic resonance spectrum was
generated.
A typical 60 megacycle proton magnetic resonance
spectrum of a solution of the crystalline lithium compound
dissolved in hydrocarbon solvent is illustrated in Figure 5a.
The K-methinyl proton signal appears as a multiplet signal
30 cycles per second wide at 10.5^• This high field ab-
sorption is characteristic of protons attached to a caroon
atom bound to lithium. Benzene, toluene, and cyclopentane
solvents were used in recording spectra of this type.
The broad triplet pattern of the o<-methinyl proton
closely resembles the high field patterns recorded for other
menthyl conformer compounds. However the absence of a second .
o<-methinyl resonance complicates such an interpretation. An
observation reported by Krieghoff and Cowan (16) is definitely
related to any speculations concerning where a second ab-
sorption might occur. They found the differences in chemical
shifts between the two norbornyl Grignard c<-proton absorptions
to be consistent with the corresponding differences for a large
number of exo-endo pairs of norbornyl compounds. Prom Figure 2
the chemical shift differences between menthyl epimers appear
to be approximately 45 to 47 cycles per second. Bo evidence
for a second absorption either up or down field from the
10.5 T absorption was observed.
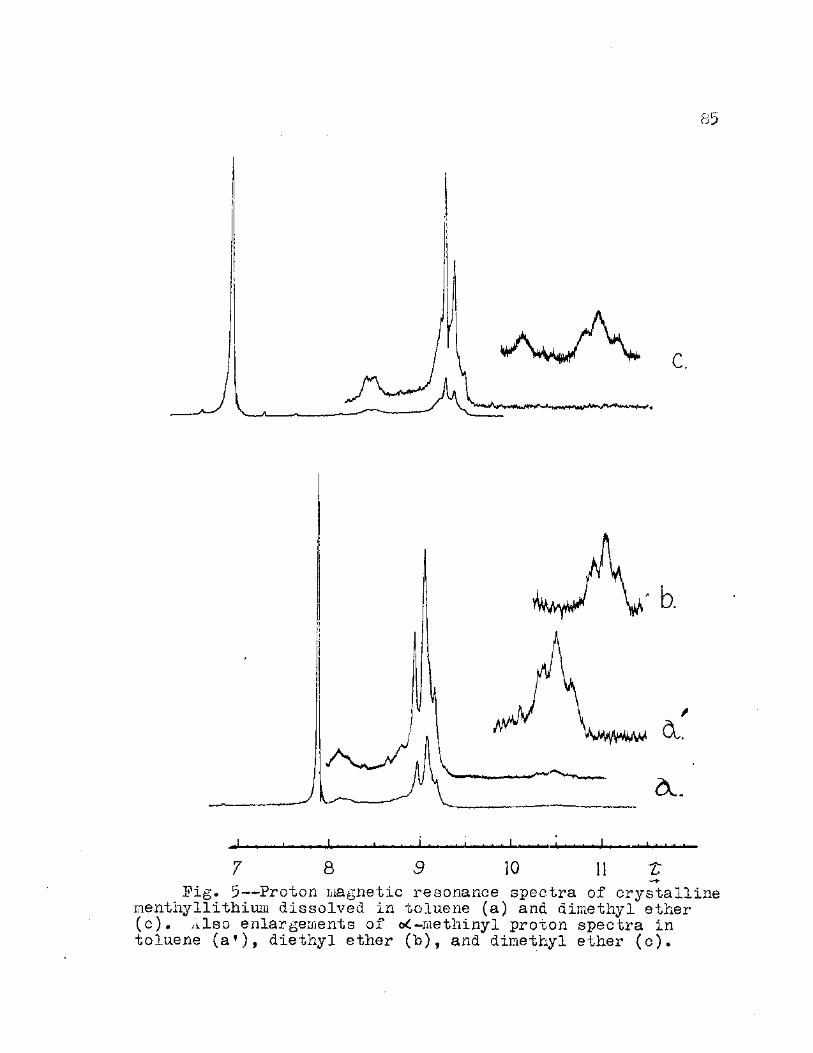
85
MAW/****"1"'**
C.
7 8 10 11 Pig. 5—Proton magnetic resonance spectra of crystalline
menthyllithium dissolved in toluene (a) and dimethyl ether (c). Also enlargements of oi-methinyl proton spectra in toluene (a')» diethyl ether (b), and dimethyl ether (c).

oo
not
The trend reported by Krieghoff and Cowan (16) need
necessarily exist for the menthyl system. The possi-
bility exists that the broad nultiplet absorption might be
comprised of two absorptions overlapping one another. A
100 megacycle spectrum was obtained in benzene solvent to
investigate this possibility. The c<-proton resonance at
this frequency could be superimposed upon that obtained at
60 megacycles. These two spectra at different frequencies
were recorded over a time duration of several hours. During
this time period no visible change of the oO-methinyl resonance
was observed. This indicates that if an equilibrium mixture
of the two isomers exists, it does so in a rapidly equili-
brating manner. Otherwise some changes in the resonance
pattern may be expected as the slowly equilibrating equili-
brium mixture is attained.
An investigation was initiated at this point to determine
whether the observed resonance pattern was effected by changes
in the solution temperature. Such an investigation should be
very informative if the observed resonance is derived from a
coalescence of the two epimer proton resonances. An increase
in the temperature would definitely facilitate any slow in-
version process. Likewise a decrease in the rate of a rapid
inversion process should be obtained by a lowering of the
temperature. Either effect would be easily noted by a definite
broadening of the resonance pattern. Spectra were recorded

87
from 50 decrees centigrade to -35 decrees centigrade with
no observable changes in the resonance width. These spectra
were recorded over a time period of several hours. The fact
that the -methinyl resonance does not alter in width from
50 to -35 degrees centigrade over a period of several hours
is conclusive evidence that a slow interconversion between
the menthyl- and neomenthyllithium compounds does not occur.
However, this observation does not preclude a very rapid
interconversion with a barrier energy of less than 12
kilocalories per mole.
This energy barrier was determined by assuming that the
resonance observed is that of the menthyl epimer and that the
neomenthyl isomer resonance occurs 45 to 47 cycles per second
down field. This frequency difference is approximately the
chemical shift difference observed for a number of equatorial-
axial pairs of menthyl compounds. Recalling that the difference
in chemical shift is directly related to the rate constant for
a first order process allows the activation energy to be
computed using the Arrhenius equation. Table VIII lists the
energy values computed for the expected range of unimolecular
frequency factors.
An assessment of this energy barrier must be made before
determining whether the observed resonance is derived from a
single stable conformer or a rapidly equilibrating mixture
of both conformers. To assess this value.some idea of the

63
TABIiS VIII
CALCULATED INVERSION ENERGIES
Frequency Factor Energy* (sec-1) (kcal/mole)
11 o 1 10XJ" 3. 1012 . 9.0 101 3 9.7 101 4 10.6 101 5 11.6
*Values determined at -35 degrees centigrade and a rate constant equalling 45 sec-1.
process requring such energy must be envisioned. In accordance
with popular concepts of the nature of the carbon lithium bond
this inversion pathway can be envisioned through a carbanion
intermediate. However the fact that organolithium compounds
exist as associated species in hydrocarbon solvent would re-
quire a complex dissociation step prior to the formation of
the intermediate. Curtin and Koehl (9) have speculated that
a complex ion containing a number of alkyl lithium units is
involved in the racemization mechanism of optically active
sec-butyllithium in ether solvents. Both of the proposed
processes are diagrammed in Figure 6.
Miller and co-workers (15) have recently investigated
barriers to intramolecular motion which leads to inversion.
They reported barrier energies for XY3 acyclic carbanions
to be in the range of 10 kilocalories per mole. They also
reported that if carbanions are involved in the inversion of
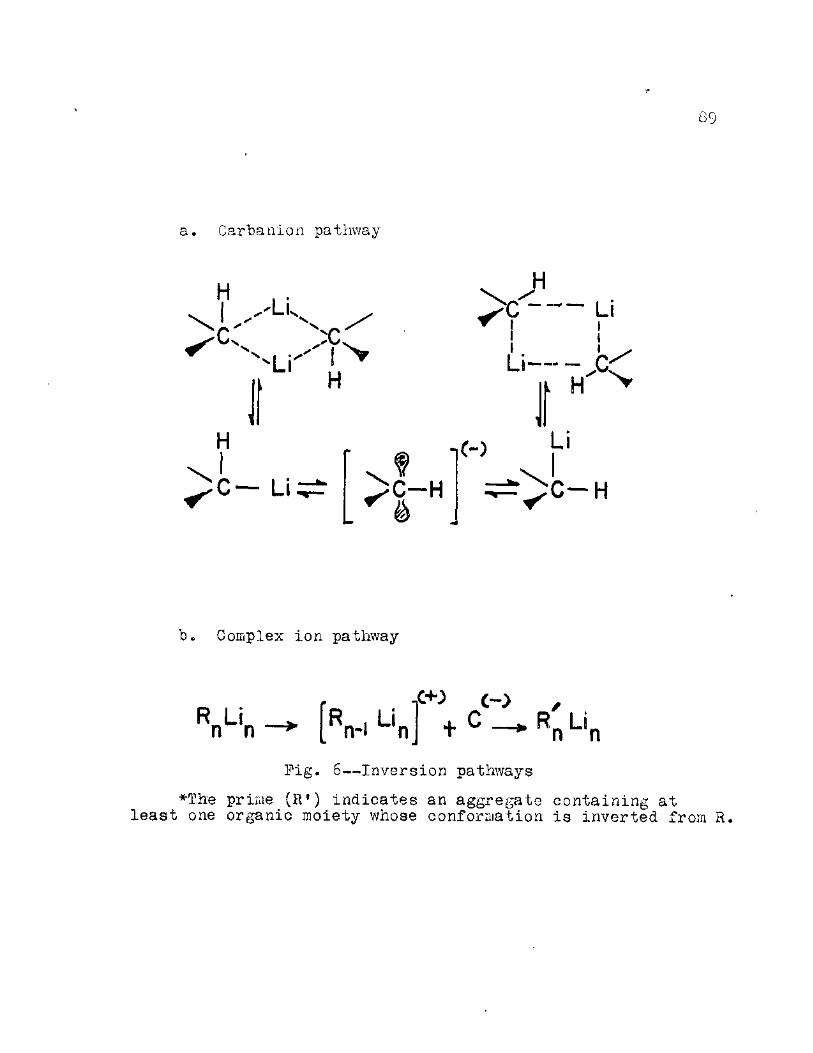
a. Carbanion pathway
89
H I c;
.'Lk
H
H
Li
if-)
^ C - H
H C " 1 Li
Li— H ^ I
Li I C—H
"b. Complex ion pathway
R Li n n
R_ i Li rH n
w c~>
R n L L n n
Pig. 6—Inversion pathways
*The prime (R *) indicates an aggregate containing at least one organic moiety whose conformation is inverted from R.

90
cyclic specics, the "barriers would be higher than those
reported for acyclic analogs. It should be emphasized that
the 12 kilocalories per mole energy value reported earlier
is derived from a different process than the 10 kilocalories
per mole value reported here. This lower energy value in-
cludes only the energy required to invert the carbanion and
not the energy to form this radical.
It is well-known that alkyllithium compounds exist as
associated species in hydrocarbon solution. Therefore the
first step in forming a carbanion intermediate is to break
up these associated systems. Morton and Fetters (20) have
reported an enthalpy of dissociation of 37 kilocalories per
mole for dimeric polyisoprenyllithium. Brown (4) estimates
from nuclear magnetic resonance studies that the dis-
sociation of hexameric ethyllithium to a dimer and a tetramer
requires 16 to 18 kilocalories per mole. Furthermore ad-
ditional energy will be required in forming the carbanion
from an alkyllithium monomer or dimer. One therefore gets
the impression that the inversion barrier would be much
higher than the 12 kilocalories per mole value determined
if carbanion fo^.ation ie involved.
On the other hand the complex ion pathway in hydrocarbon
solvent would definitely represent an even higher energy
process than the carbanion pathway. Prom a mass spectrum
analysis of ethyllithium Brown and co-workers (3) have

91
reported the energy required to dissociate the hexameric
aggregate of this compound. This ionization process (a)
diagrammed below requires 111*5 kilocalories per mole of
Li^Rg + e' • LigR^" •+• R + 2e'
(a)
energy. This example illustrates the approximate energy
requirement necessary to consider the complex ion pathway.
It is evident from this discussion that the observed
resonance is not derived from a rapidly equilibrating mixture
of the two lithium epimers. The correct assignment therefore
appears to be the «<-methinyl proton absorption of a con-
figurationally stable menthyl- or neomenthyllithium compound.
Based solely on the shape and width of the signal one might
predict it to be the menthyl conformer.
Evidence of the absorption frequency and characteristic
shape of the other epimer resonance would certainly enhance
the configurational assignment for both signals. It seemed
that employment of & more basic solvent, such as an ether,
might facilitate some exchange pathway and thereby yield an
inverted product. Such an effect could generate a sufficient
concentration of the second epimer so that a resonance signal
would be observed.
The first solvent tried was diethyl ether. Above zero
degrees centigrade this solvent reacts in a very vigorous
manner with the lithium solute. At lower temperatures this
reaction is retarded and resonance signals were recorded.

92
No change in the characteristic shape of the resonance from
that observed in hydrocarbon solvent was recorded. However
a shift in absorption frequency was observed with the resonance
occurring at 11.00 ^ (5b) instead of 10.5 (5a*) as reported
earlier. This type of shift for these electron deficient
systems in more polar solvent is not uncommon. The facts
that no change in the absorption pattern is observed and
that no other signal appears are certainly in agreement with
a similar investigation reported by Cram (8). He cited from
a private communication of Roberts, Whitesides and Witanowski
that 3,3-dimathylbutyllithiuin appears to be configurationally
stable in diethyl ether below zero degrees centigrade.
Application of dimethyl ether, which io known for its
ability to solvate polar systems, as the solvent produced
the long awaited evidence for a second absorption. The new
absorption occurred at 10.13 "2" (5c) and had the characteristic
shape and width expected for a neomenthyl o<-methinyl proton
absorption. The characteristic broad multiplet, which oc-
curred at 11.00 ^ (5b) in diethyl ether, was also present at
10.95 jh (5c) in this solvent. Integration of these two
signals indicated a 75:25 ratio of the menthyl-neomenthyl
methinyl resonances. These results were recorded at -45
degrees centigrade.
The fact that these two absorptions were separated by
50 cycles per second, and that they demonstrated the expected

93
signal widths and shapes of a raenthyl-neonienthyl epimer
pair was very informative. In order to determine whether
the same results could "be observed for another ether solvent,
tetrahydrofuran, which can also be characterized as a polar
solvent, was employed. As with dimethyl ether both resonances
were observed and the two spectra could be superimposed. The
fact that identical spectra were obtained in two different
ether solvents provides good evidence that the second
resonance is not derived from a lithium derivative of the
solvent. To further demonstrate that these two resonances
are derived from the menthyl- and neomenthyllithium oC-protons
an investigation employing chemical means was undertaken.
Seyferth and co-workers (23, 24) have demonstrated
that carbonation of alkenyl- and cyclopropyllithium compounds
proceeds with retention of configuration. Although this
stereospecific pathway has not been demonstrated for cyclic
lithium compounds it has been demonstrated for norbornyl
Grignard reagents by Jensen and Nakamaye (14). These data
certainly predict a retention pathway for the menthyl com-
pounds under investigation. Therefore the first attempt to
demonstrate the origin of the observed resonances by chemical
means employs this reaction.
Solutions originating from a variety of sources, such as
the original reaction solution, solutions derived from dis-
solving crystalline material in different hydrocarbon and

94
ether solvents, and mother liquor solutions were carbonated.
Two procedures were employed in carrying out these reactions.
The first process involved pouring the solution of lithium
compound over a slurry of dry ice and n-pentane at -70 degrees
centigrade; the other procedure involved "bubbling carbon
dioxide gas through the organolithium solution. The resulting
acid products were either isolated through crystallization or
esterified with diazomethane. The results obtained from
these systems are recorded in Table IX.
It is easily observed from these data that 3-p-methane
carboxylic acid is the primary derivative produced from
carbonation of any of the three solutions containing the
lithium compound in hydrocarbon solvent. This evidence for
only one isomer is definitely complemented by the optical
rotation and infrared data reviewed earlier. Furthermore,
upon replacing this solvent with dimethyl ether, a substantial
increase in the other acid epimer is produced. This ob-
servation together with the fact that the same isomer dis-
tribution i,n dimethyl ether is observed by chemical means
and from proton magnetic resonance spectra is very informative.
It establishes that the resonance assignments made earlier
from proton resonance spectra analyses were correct. However,
the most important conclusion to be drawn from these data is
that the menthyllithiuin exists as a conformational^ stable
compound in hydrocarbon solvent.

9 5
CO
o
Ph
X EH
a
rH
c 0
o •H
n o5 O A1 CD
£h !H •* 0
r H £ >> O
,-q ffi - P H S3 0
*P *"0 O 0 3 Esl
* d t>5 O H ^ cd
PM C2 <cj
S3 O O w
•H - P cd
CD
O T5 rO 0) !h O Cd O
O Jh Ph
• P a 0 >
r H O
CO
a , a o w
*H •P 3
H o
CO
a • H ^ 5
*H u
o
03
0 i H T3 CO - H •H r H - p eg *H W S3
M
<11, T3 rC> nd t : ;
O r H i n KO r H i—I m !>- i—I r-H m r H H~ K \ J>5 * • • * W ** t>5 t>> • • • • • •
r a i n ^ i r \ k n r c j & i i \ & v o o
a d S3 J3 c 0) 0 0 0) 0 0)
f-^ f~HS »-h|
?h ?H %-i ^ £-r rC5 0 d ) r d n d C D <D <13 <d 0 <d 0 0 *H - P P *H *H P - P - H «H - P *H *P *P O C Q C D O O C Q C D O O G O O C O C O
< $ p q i 3 < i } < t | p q | i q < $ « 4 p £ | < $ w p q
M H I H M
c i a a a S3 S3 o o o o o o
P P p P P r Q ^ ^ ^ fn ?H ^ 03 O O Sh
<d * d * d »d f d f d * d ^ t>3 t>>
j x j i x j w w W f r j UJ
d s3 $3 o o o
fn Jh ^ cd cd cd o o o o o o u u u
nd «d * d
M M H
Sh Jh 0) CD ,£} <3 - P •P W W
r H H >5 l ^ r^H ^ - P
£ 5 8 > v H *H
W f l f i
a , o
rQ u 03 i O O ,
< i j < j ; f q o o o < a J < ^ < ^ f q o < i J < t J
c d o 5 c d a 3 a 3 c d r O r Q r j D r Q r Q o 3 a 3 M M M M M f H I — I M H M H H M
a) CO • 03
• H 0 H H i H 3 - P cd T * O
p 0 d CO o nd t>» o O
u o • P4 P^
S n h CD O 0 cd *H JH > t i ) a
CH # h 03 0
o n3 0 0 (D *H r*H J> £1( N - P
* •H CO O 0 ^ CD • H CO
* d 0 Js 0 *H $ rH 0 £3 r H o CD U O 05
1—1 a d > r*H O - P JH O • H X 03 S3
P -H ^ a
O o • H
P -H ^ a • H !H
r H • P 0 o a 03 ^
- P ro o H • P ^ £3 •H 1—J o o CD - p - p JH c H 6 S3 o •% £3 o a) o3 0 O G CD 0} 0 H •H O S3 3 t H
u T i • H H P4f~-l 0 O >> CD 03 CJ 0 ,Cj
rQ u a o p 4 - p M ' H CD S3
O ^ 0 p4 0 • H H3 S
CD r c ! JH 0 <3 * o S3 O S3 cd 0 *H d 03 A
*C5 - P O ^ CI S3 £3
H pq 03 0 •H J>> pq 03 Pi O r-\
O • * f P4«H Jh a
r H O i i*0 cd CD ^ 0 S3 S fn O"1 o *H - H
+D CD *H • H •P JH S3 •f--5 H H P4 CD •P t>3 0
CD SH pi CO r H 0 nd cd
r'3 O 4^ O
03 r«3 O JH 0 W 6H
««
^ - x r H
M tS3 • H JH
^ 0 03 rO 03 O • d - P
**—"*H Jh 0 p»
05 g
w o 03 JH 05
^ 3 o

96
In order "to obtain additional chemical evidence for
these observations the reaction between deuterium oxide and
(+) -menthyllithium was studied. This reaction was selected
because a large amount of conformational analyses data for
the carbon-deuterium stretching frequencies in cyclohexyl
compounds has been reported. Corey, Sneen} Danaher, Young,
and Rutledge (7) have published evidence that the stretching
frequencies for the monodeutero compounds exhibit two bands.
In the case of the axial deuterium these bands are separated
characteristically by 25 wave numbers and in the case of
equatorial deuterium they are separated by 15 wave numbers.
Furthermore, the absorptions due to axial deuterium for the
compounds studied in this investigation occur at nearly the
same place; those due to equatorial deuterium occur at higher
frequencies. The final distinction noted was that the higher
frequency band of the doublet is more intense for axial whereas
the low frequency band is more intense for equatorial deuterium.
Armed with these conformational analysis data and a very
similar system the following results were recorded.
3-deutero-p-Menthane is produced in the reaction between
deuterium oxide and menthyllithium. "The infrared spectra
illustrated in Figure 7 demonstrates that the axial and
equatorial deuterium product distributions are dependent
upon the solvent employed. Deuterolysis in hydrocarbon or
diethyl ether solvent produces a fairly broad single absorption

97
a.
L
2500 cm 2000 2500
b.
2138
2167
cm -i 2000
Pig. 7—Infrared spectra of 3-deutero~p-rienthane in hydrocarbon solvent (a) and dimethyl ether solvent ("b).

9$
at 2167 wave numbers. The width of this absorption evidently
maslcs out the low intensity band, thereby eliminating charac-
terization of the absorption with respect to the position or
the characteristic separation from the unobserved low in-
tensity band. However the observed absorption does occur
in the frequency range characteristic of equatorial deuterium
compounds.
Two absorptions are observed in the spectrum of the
deuterium products isolated from the same deuterolysis re-
action conducted in dimethyl ether solvent. A distinct
shoulder absorption on the 2167 wave number band was recorded
at 2138 wave numbers. The .fact that only two and not all
four absorption bands are present eliminates an inanediate
characterization. However the observation that these two
absorptions are separated by 29 wave numbers does exclude
the possibility that they both belong to a single deuterium
compound. Furthermore the fact that the 2138 wave number
absorption is found in the frequency range characteristic
of axial deuterium cyclohexyl derivatives is in agreement
with this observation. Therefore the assignments of these
two bands as the strong equatorial and axial deuterium
stretching frequencies can be confidently made from these data,
It is noteworthy that the carbonation and deuterolysis
chemical evidence definitely compliments the results reported
from the proton magnetic resonance investigation. Reviewing

99
these data along witli the optical activity and infrared
results, the following conclusions regarding the inenthyl system
are derived:
1. Isomerization of the two chloride reagents occurs
in their reaction with lithium metal in hydrocarbon solvent.
2. This reaction yields a single optically active
menthyllithium isomer as the primary product of the reaction.
IIo more than 5 per cent of the net carbon "bound lithium
resides in an axial conformation.
3. The (+)-menthyllithium isomer prepared demonstrates
configurational stability for indefinite time periods in
hydrocarbon and cold diethyl ether solvents.
4. In contrast to the last observation some isomeri-
zation process exists at temperatures below -20 degrees
centigrade in dimethyl ether and tetrahydrofuran solvents.
This process yields a mixture of the menthyl- and neomenthyl-
lithium epimers. Both of these compounds exhibit configurational
stability on the nuclear magnetic resonance time scale at these
temperature.
5. Both carbonation and deuterolysis reactions of these
lithium isomers proceeds by configurational retention pathways.
A conformational analysis investigation of the 4-tert-
butylcyclohexyllithiuia product was undertaken upon completion
of the menthyllithium characterization. Due to the success
experienced with the menthyl system the same techniques were

100
employed in tlie investigation of the 4-tert-butylcyclohexyl
compound. However a few procedure changes were required be-
cause of the inherent low solubility property demonstrated
by this compound in hydrocarbon solvents. ?or example,
carDonation and deuterolysis reactions were conducted on
the crystalline product suspended by agitation in such
solvent instead of on true solutions of the dissolved
crystalline material.
Proton magnetic resonance spectra of the o<-methinyl
resonances in various solution are recorded in Figure 8.
A spectrum of the original reaction solution is diagrammed
i*1 illustration 8a. Due to the problem of side bands occur —
2 ing in the field region of interest when n—pentane solvent
is employed this solution had been concentrated by removal
of a large portion of this solvent; subsequently benzene
standard was added. The spectrum shows a broad unresolved
multiplet resonance at 10.59^ and two other absorptions 43
and 55 cycles per second down field from this resonance.
Employment of the frequency difference of 48 to 50 cycles
per second recorded in the menthyllithium investigation
provided no evidence for the characterization of either of
these two lower field resonances. It was believed that these
two resonances were derived from by-products of the synthesis.
In an effort to investigate further the possibility that
both of the lithium isomers exist in hydrocarbon solution a

101
b.
iiti , I a_ I • • - • 1 i mJLm _ a _ J i i a.
10.0 11.0
-ig. -8— ©<-Methinyl proton resonance of 4-tert-butyl-cyclohexyllitlviuia samples of an original reaction solution in benzene (a) and crystalline material dissolved in benzene ("b), diethyl ether (c), dimethyl ether (d).

102
spectrum of dissolved crystalline product was required. At
this point the low solubility property of the crystalline
material was realized. Spectrum 8b was recorded on a typical
saturated benzene solution of this material. It was soon
observed however that addition of diethyl ether solvent to
a proton magnetic resonance sample tube containing undissolved
crystalline material in a saturated benzene solution yielded
a recordable spectrum as shown in spectrin 3c. No appreciable
concentration of the two resonances in question from spectrum
8a were observed. Also the broad multiplet is resolved into
the characteristic broad triplet resonance observed in the
menthyl spectra. It is noteworthy that no shift in the ab-
sorption frequency of this resonance occurred upon addition
of diethyl ether.
These data compiled from spectra 8a, 8b, and 8c suggest
that solutions prepared from the crystalline material in
either hydrocarbon or diethyl ether solvents are primarily
composed of the trans-lithium isomer. If either of the
other low field resonances are derived from the cis-isomer
then either its concentration is generated during the synthesis
or a slow equilibrium exists between these two isomers.
Dimethyl ether solvent was employed at this point to
determine whether either of these two low field absorptions
were produced. Spectrum 8d illustrates the resonances re-
corded in this solvent. The characteristic broad triplet

103
resonance occurs at 10.05/" and a second resonance at 10.03^.
Obviously a frequency shift of the resonances occurs in this
solvent. It ras also noted that the 10.03 jh resonance shifts
to 9.S5 Ih with changes in temperature over extended time
periods. The resonance position of this particular absorption
corresponds to the position of the 10.03^ resonance recorded
in spectrum 3a. These two resonances exhibited an approximate
30:20 high field to low field resonance ratio at -50 degrees
centigrade. An inversion of this ratio can be observed as
the temperature for such a solution is warmed from -70 degrees
to -30 degrees centigrade. The observation that this new
resonance could be concentrated yields an ideal system for
characterizing this new absorption by chemical means.
In contrast to the results recorded for the menthyl
system, the applications of carbonation and deuterolysis re-
actions produced only evidence for the existence of the trans-
lithium isomer in all of the previously described solutions.
For example, no observable difference in the carbon-deuterium
stretching frequency spectra of the 4-tert-butylcyclohexane-
1-d] product derived from crystalline lithium compound dissolved
in any of the solvents of interest could be detected. A typical
spectrum of the carbon-deuterium absorption region is illustrated
in Figure Q. The observed absorption doublet may be easily
characterized from the works of Corey and co-workers (7) as
the symmetric and asymmetric stretching modes for the equatorial
deuterium conformation.

104
v — v / V
2184
2167
2500 cm 2000
Fig. 9—Infrared spectrum of 4~tert-butylcyclohexane-l-d- in the carbon-deuterium stretching region.

105
In the case of 'the carbonation reaction the only
product detected after esterification with diazoraethane
was the trans-ester isomer. This result was observed to
he independent of the solvent employed. However a new
component in the product mixture of a dimethyl ether solution
was detected "by gas liquid chromatography. This solution h&d
"been characterized "by a proton magnetic resonance analysis
to be concentrated in the unassigned low field resonahce. A
complete characterization of this component was not obtained
through employment of preparative chromatography combined with
other analysis techniques, but sufficient evidence was recorded
to demonstrate that it was not the cis-ester isomer.
The conclusions to be drawn from these data are that the
4-tert-butylcyclohexyllithium compound prepared from the cis- .
chloride isomer and lithium metal is the trans compound.
Furthermore, in hydrocarbon and diethyl ether solvents this
isomer demonstrates configurational stability and indicates
a definite preference for the lithium atom to reside in the
equatorial position. However in dimethyl ether solvent the
behavior of the lithium compound is not so well understood.
The recorded data suggest the existence of some interaction
between dimethyl ether and the lithium compound. Reactions
of this type, such as metalation and methylene insertion
reactions, have been reported by Ziegler and Gellert (28).
However these reactions occurred only at temperatures above

106
-20 decrees centigrade. The fact that some similar process
could occur at slightly lover temperatures appears reasonable,
but whether the interaction involves the cis- or trans-lithium
isomer is the question of interest. It is unfortunate that
the evidence presented does not indicate whether isomerization
occurred in this solvent.
Combining the conformational analysis data reported from
the investigations of the menthyl and 4-tert-butylcyclohexyl
systems, one main conclusion is evident. This noteworthy ob-
servation is the preference demonstrated by the lithium atom
to reside in an equatorial conformation in substituted cyclo-
hexyllithium compounds. One might find it difficult to explain
this phenomenon from the size of the covalent radius of the
lithium atom. However, recalling the fact that these compounds
exist as associated species in hydrocarbon solvent, makes it
easier to illustrate that this functionality is large enough
to demand a particular conformation. One other observation
common to both systems is that carbonation and deuterolysis
reactions bpth proceed with configurational retention pathways.
Initially there was some concern over complication of the
stereochemistry of the menthyl system reactions by neighboring
group and asymmetric induction effects. These results obviously
indicate that if such effects are present they are definitely
insignificant for these two reactions.

107
It is evident that sufficient data has not been reported
to postulate mechanisms for these two reactions. However a
prediction that these two reactants form similar assisted
intermediates does not appear to "be an unrealistic explanation
for the recorded results. Obviously such a prediction could
not be made for a' reaction which yields configurationally
inverted products. Applequist and Chiuurny (1) have reported
evidence for such a reaction involving bromine and norbornyl-
liohium in hydrocarbon solvent. In order to determine whether
the stereochemistry of such a reaction is complicated by
neighboring group and asymmetric induction effects the
bromination reactions of menthyl- and 4-tert-butylcyclohexyl-
lithium compounds were investigated. The results recorded
from these reactions are listed in Table X. These results
demonstrate that a definite temperature effect on the stereo-
chemistry of this reaction exists. At low temperatures pre-
dominant inversion of configuration is observed for the bromide
derivatives of these two lithium reagents. However at higher
temperatures this product distribution is reversed for the
4—tert-butyIcyclohexyl bromides. Therefore from these data
and the findings reported by Applequist and Chmurny (1) it
is evident that solvent, temperature, and the nature of the
organic moiety of the lithium reagent can all effect the
stereochemistry pathway followed in this electrophilic sub-
stitution reaction.

103
TABLE X
BROMIHATIOF PRODUCTS PR OK SOLUTIONS OP IvIENTIiYLLITHIUM AND 4-TERT-BUTYLGYCLOHEXYLLITHIUM
Lithium Compound Reactants and Conditions
Product Ratio Equatorial Axial
4-tert-butyl-cyclohexyllithium
Original reaction solution, n-pentane solvent, -70°C. 23/77
4-tert-"butyl-cyclohexyllithium Crystalline material,
n-pentane solvent, 70°C. 21/79
Mother liquor solution, n-pentane solvent, -70°C. 21/79
Crystalline material, n-pentane solvent, 0°C. 37/63
Mother liquor solution, n-pentane solvent, 0°C. 58/42
Crystalline material, dimethyl ether solvent. -70°C. 59/41
Menthyllithium Crystalline material, n-pentane solvent, -70°C. 38/62
Menthyllithium Crystalline material, n-pentane solvent, 0°C. 65/35

109
Nov/ the question arises as to whether the nature of *A
the electrophile should be included in this list of variables
when electropirilic substitution reactions of alley H i thiuin
compounds are being considered in general. Data concerning
this question are included in the recorded product dis-
tributions of Table XI. These data were obtained from an
investigation of substitution reactions between the mentliyl-
and 4-tert-butylcyclohexyllithium reagents and a variety of
electrophiles.
One explanation for the observed variations in the
reaction products listed in Table XI has been published by
Applequist and Chmurny (l). These authors have reported
that retention and inversion pathways for such substitution
reactions appear to be inherently equal. However one is
favored over the other by changes in the conditions and re-
agents employed in carrying out the reaction. For example,
they speculated that carbon dioxide way have an affinity for
lithium great enough to proceed by a four-membered transition
state in spite of some inherent preference for inversion.
It is noteworthy that this explanation is based on the
assumption that the lithium reagent did not isomerize in the
presence of the bromide electrophile. The fact that this
assumption is not only questionable but unnecessary is
demonstrated by the conformational stability data and the
observed variations in product distributions data for different

110
TAB HE XI
PRODUCT DISTRIBUTION PROM ELECTROPHILIC SUBSTITUTION REACTIONS OP THE CYCLOHEXYLLITHIUE COMPOUNDS
Electrophile Conditions Products* Product Ratio Equatorial Axial
Carbon dioxide n-C5Hi2,25°C &6, b 6 95/5 Carbon dioxide (CH3CH2) 0,-40°C a6 »b6 96/4 Carbon dioxide (CH3)20,-40°C a6,b6 70/30 Carbon dioxide n-C5Hi2,25°C °6 » 6 95/5 Carbon dioxide (CIl3)20,-40OC C6,d6 95/5
Deuterium oxide 11-0511x2,25°C a3 ,b 3 * *
Deuterium oxide (CH3CH2)20,-40°C a3,b3 *-x Deuterium oxide (CH3)20,-40°C a3,b3 **
Deuterium oxide n-C5ll3 2,25°C C3,d3 * *
Deuterium oxide (CH3CH2)20,25°C °3>d3 Deuterium oxide (CH3)20,-70
oC c3,d3 **
Ethylene dibromide n-C5H12,25°C a4 >^4 97/3 Ethylene dibromide (CH3)20,-70°C a4 ,b4_ 88/12 Ethylene dibromide n-C5Hi2,25°C c4 »^4 95/5
Bromine n-C H-, 2 ,-70°C a4 j^4 38/62 Bromine n-C5Hx2,0°C a 4 >0 4 65/35 Bromine n-C5Hl2,-70°C c 4»d4 21/79 Bromine n-C5Hi2,25
0C C4»d4 37/63 Bromine (CH3)20,-70°C c4»d4 60/40
Oxygen n-C5H12>25°C a2>'b2 80/20
Kethyl iodide n-C5Hi2j25°C a5,b5 80/20
*A description of the products can be found in Figure 1.
**Since these samples were analyzed by infrared spectro-copy only a qualitative estimate of the ratio can be reported, Except for the spectrum of the dimethyl ether solution of menthyllithiura the carbon-deuterium absorption was primarily derived from an eqiaatorial conformation.

Ill
elcctrophiles all included in this investigation. These
data suggest that the character of the electrophilic agent
determines the pathway through an isomerization effect on
the lithium reagent, furthermore this effect is facilitated
to different degrees "by the conditions employed in conducting
the reaction. The proposal of this effect requires that the
stereochemistry pathway followed in such reactions "be one of
retention. Such a process can he easily envisioned for alkyl-
lithium compounds as proceeding by an assisted mechanism.
Although these conclusions are only speculations they seem
more consistent with the prevailing concepts of organometallic
mechanisms.
Pyrolytic elimination of lithium hydride from lithium
alkyls has been a characteristic reaction of interest since
the work of Schorigin (22) was reported in 1910. An in-
vestigation of this reaction has been initiated for the con-
figuration lly rigid (4-)-menthyllithiuLi compound. ]?rom this
study two main goals are sought. The first of these is to
report the stereochemistry of a characteristic elimination
reaction along with all the previously listed substitution
reaction data. The second and most important goal is to
compare the results recorded from the decomposition of this
conformationally rigid system with results reported by Glaze
and co-workers (11) for sec-butyllithiurn. These authors
proposed that sec-butyllithium decomposes by a cis-elimination

112
mechanism. They further speculated that the association
character of this alkyllithium compound had a definite effect
011 the pathway exhibited for this reaction. Whether a similar
pathway will he exhibited for the menthyllithium compound can
be demonstrated from an analysis of the decomposition products,
The following illustration demonstrates how this can be deter-
mined.
H
Li II
pseudo-trans
il
H
2-p-nienthene
CH3
H H
Mixture of 3-p-Menthene 2-p-menthene
Evidence for a cis-elimination mechanism has also been
demonstrated for the thermal decomposition of (-)—menthyl
chloride by Barton and co-workers (2). This mechanism
assignment was based on the 3:1 ratio of p-3-rnenthene and

113
These p-2-monthene products .Corned in the decomposition.
auoiiors concluded that the (-)-menthyl chloride pyrolyzed
by a Juonogeneous unimolecular mechanism. They proposed
the following four centered transition state (a) as being
the product yielding intermediate. They explained the
C^=C i i i « II--CI (a)
preference for a cis- over a trans-elimination process for
this system as being derived from the nature of the cyclo-
hexy.;. ring. This system was believed to be capable of
minimizing the activation energy for the proposed transition
state by allowing this intermediate to lie in a single
plane. On the other hand Huckel and co-workers (13) have
demonstrated that under the conditions of an E2 reaction
with sodium ethoxide a trans-elimination mode of reaction is
preferred. This assignment was also based on the observed
pioduco distribution, which in this case yields only
p-2-menthene.
A 45:55 ratio of p-2-menthene to p-3-menthene products
was recorded in the thermal decomposition of (+)-menthyllithium(
Although this distribution is somewhat different from that
recorded for the chloride derivative it definitely indicates
that the lithium compound decomposes by a cis-elimination
mechanism. Whether the proposed pathway is derived by the

114
effects noted by Barton and co-workers (2) or by Glaze and
co-tvor Iters (11) is not known at this time. .Furthermore
the effect of alkoxide concentration on such a process is
unknown. This concentration of alkoxide might produce the
variation in product distributions between the chloride i
reaction and the lithium decomposition by facilitating
some E2 reaction products. Both of these questions are
interesting but additional data will be required before they
can be answered.

CHAPTER BIBLIOGRAPHY
1. App.Tequist, Douglas E. and Gwendolyn Weal Ghuiurny, "Stereochemistry of Electrophilic Substitution on 2-Norbornyllithiuin," Journal of the American Chemical Society, LXXXIX (1967}, 075. '
2. Barton, D. R.H., A. J. Head, and R. J. Williams, "Stereospeeificity in Thermal Elimination Reactions, Part II. The Pyrolysis of (-)-Menthyl Chloride," Journal of the Chemical Society (1952), 453.
3. Berkowitz, Joseph, D. A. Bafus, and Theodore L. Brown, "The Mass Spectrum of Ethyllithium Vapor," Journal of Physical Chemistry, LXV (I96I), 1380.
4. Brown, Theodore L., "On the Mechanism of Organolithium Reactions in Hydrocarbon Solution," Journal of Organometallic Chemistry, V (1966), 19I.
5. Brownstein, S. and R. Miller, "Conformational Analysis of Disubstituted Cyclohexanes by Proton Resonance Spectroscopy," Journal of Organic Chemistry, XXIV (1959), 1886. b ^
6. Castellano, S. and A. A. Bothner-By, :Analysis of HMR Spectra by Least Squares," Journal of Chemical Physics, XXXXI (1964), 3863.
7. Corey, E. J., R. A. Sneen, M. G. Danaher, R. L. Young, and R. L. Rutledge, "A Method for the Location of Deuterium Atoms Attached to Oyclohexane Rings," Chemistry and Industry (1954), 1294.
8. Cram, Donald J., fundamentals of Carbanion Chemistry, Hew York, Academic Press, Inc., Ifj6^.
9. Curtin, David Y. and 7'illiam J. Koehl, "Effect of Solvent on the Steric Stability of Lithium Reagents," Journal of the American Chemical Society. LXXXIV (1962), T$6T.
10. Dryer, John R., Applications of Absorption Spectroscopy of Organic Compounds, Englewood Cliffs, Prentice-Hall, Inc., 1965.
115

11. Glaze, 'Yilliam H., Jacob Lin, and 3. G. .Pelton. "The yi olysis of Unsolvated Alkyllithium Compounds," Journal of Organic Chemistry, XXXI (I966), 2643.
12. Horner, von Leopold, Hermann Oediger, and Hellmut Hoffman, "Reaktionen Mit Triphenyl-Phosphin-
DCXXVIg( 19^9 j1'"2^ustus L i e b i^ s Annalen der Chemie ,
15* Walte]f> Werner lappe, and Gunter Legutke, Abspalbungreairtionen und Ihr Sterischer Verlauf "
Eiebi^s Annalen der Chemie, DXXXXIII (1940),
14. Jensen, Frederick R. and Kay L. Nakamaye, "Preparation Isomeric Grignard Reagents and the
Stereochemical courses of their Reactions," Journal ' of^t^e American Chemical Society, LXXXVIII (I966)t
15. Koeppl G. i. D. 6. Sagatys, G. S. Kirshnamurthy, and filler, "Inversion Barriers of Pyramidal
(XY3 and Related Planar ( XY) Species," Jo^nafof the American Chemical Society. LXXXIX (I967), 33967
16* ^ild® a M Dwaine 0. Cowan, "Stereochemistry Reage^ts from exo- and endo-Norbornyl
ixx^in (xgroTr&ssf — 4-merlc?-n- Sississi society,
17# Le .ieux, R. U., R. K. Eullnig, H. J. Bernstein, and I' +* "Configurational Effects in the ?ro*°P Magnetic Resonance Spectra of Acetylated carbohydrates," Journal of the Amerinan Society. LXXIX (IgbV), 1UU5Z ^nemical
18.
|?o^nTE|netio'Resoiiantrsp«tra of Sii MeSfcered S o c f e ^ ; P ™ 3 ( i 9 f g f ^ 9 § £ African Cheaical
IS5(ai9l2TrS MineS'" ^£ilft"e 3o£e,ioal

117
20. Morton, M. and L. J. Fetters, "Homogeneous Anionic Polymerization V. Association Phenomena in Organo-lithium Polymerization," Journal of Polymer Science, A2 (1964), 3311. ;
21. Read, John, "Recent Progress in the Menthone Chemistry,,!
Chemical Reviews, ¥11 (1930), 1.
22. Schorigin, P., "Uber "bis Natiumalkyle Hire Reaktion hi it der Atheru," Berichte "Der Deutschen Chemischen Gesellschaft, XXXXIII (l'jl&TT W 5 T .
23. Seyferth, Dietmar and Harvey M. Cohen, "The Stability of Cyclopropyllithium in Diethyl Ether and in Tetra-hydrofuran," Journal of Organometallic Chemistry, I (1963), 15.
24. Seyferth, Dietmar and Lawrence G. Vaughan, "The Isomeric 1-Propenyllithium Reagents: Their iMR Spectra and the Stereochemistry of Their Formation by Direct Reaction and by Their Transmetalation Reaction," Journal of Organometa11ic Chemistry, I (1963), 201.
25. Smith, J. G. and George F. '/right, "The Diastereomeric Menthyl Chlorides Obtained from (-)Menthol," Journal of Organic Chemistry, XVII (1952), 1116.
26. Walborsky, H. L I . , F. J. Impastato, and A. E . Young, "Cyclopropanes. XV. The Optical Stability of 1-Methy1-2,2-diphenyl-cyclopropyllithium," Journal of the American Chemical Society, LXXXVI (1§64), 3283.
27. William H. Glaze, unpublished notes, Department of Chemistry, North Texas State University, Denton, Texas, 1967.
28. Zeigler, von Karl and II. G. Gellert, "XVII. Reaktionen Zwischen Lithium-alky leu und Athern," Justus Liebigs Annalen der Chemie, DLXVII (1950), 1851

BIBLIOGRAPHY
Books
Cram, Donald J., Fundamentals of Carbanion Chemistry, New York, Academic Press, Inc., 1965.
Dryer, John R., Applications of Absorption Spectroscopy of Organic Compounds, Englewood Cliffs, Prentice-Hall, Inc., vjoTI
Sadtler Standard Infrared Spectra, Philadelphia, The Sadtler Research laboratories, Spectra Numbers 184-6 and 20221.
Articles
Alexandrou, N. E., "Reaction of n-Butyllithium with cis-and trans-4-tert-Butylcyclohexyl Bromide and Mercuric Bromide," Journal of Organonieta11ic Chemistry, V (1966), 30TI
Allinger, Norman L. and Robert L. Hermann, "Infrared Spectra of cis- and trans-Propenyllithium Stereochemistry of the Reaction of Vinyl Chlorides with Lithium Metal," Journal of Organic Chemistry, XXVI (1961), 1040.
Applequist, Douglas E. and Gwendolyn Heal Chmurny, "Stereo-chemistry of Electrophilic Substitutions on 2-Norbornyl-lithium," Journal of the American Chemical Society, LXXXIX (1967), 8?5:
Applequist, Douglas E. and Alan H. Peterson, "The Configura-tional Stability of cis- and trans-2-methylcyclopropyl-lithium and Some Observations on the Stereochemistry of their Reactions with Bromine and Carbon Dioxide," Journal of the American Chemical Society, LXXXIII (I96I), 862.
Arndt, J*., "Diazomethane," Organic Syntheses, XV (1935), 3.
Barton, D. R. H., A. J. Head, and R. J. Williams, "Stereo-specificity in Thermal Elimination Reactions. Part II. The Pyrolysis of (-)-Menthyl Chloride," Journal of the Chemical Society (1952), 453.
118

119
Berkov/itz, Joseph, D. A. Bafus, and Theodore L. Br own, "The Ilass Spectrum of Ethyllithium _Vapor," Journal of Physical Chemistry, LXV (I96I), 1380.
Bordwell, P. G. and phillip S. Landis, "Elimination Reactions. VII. A Comparison of Some Eliminations in Open-Chain and Cyclic Systems," Journal of the American Chemical Society, LXXIX (1957), 1593.
Braude, 3. A. and J. A. Coles, "Alkenylation by Use of Lithium Alkenyls. Part IV. The Condensation of cycloHexenyl-lithium with Aldehydes and. Ketones, and Some Reactions of the Resulting Carbinols. (Studies in Molecular Rearrangement. Part V.)," Journal of the Chemical Society (1950), 2078.
, "Alkenylation Employing lithium Alkenyls. Part V. The Formation and Some Reactions of cis-Propenyllithium," Journal of the Chemical Society (1951), 2078.
Braude, E. A. and C. J. Timmons, "Alkenylation by Use of Lithium Alkenyls. Part I. The Condensation of isoButenyl-lithium with Benzaldehyde and Acetophenone, and the Oxotropic Rearrangement of the Resulting Carbinols. (Studies in the Molecular Rearrangement. Part III.)," Journal of the Chemical Society (1950), 2000.
Brown, Theodore L., "On the Mechanism of Organolithium Reactions in Hydrocarbon Solution," Journal of Organometallic Chemistry, V (1966), 1^1.
Brown, Theodore L., J. A. Ladd, and G. II. Newman, "Inter-action of Alkyllithium Compounds with Base. Complex Formation Between Ethyllithium and Lithium Ethoxide in Hydrocarbon Solvents," Journal of Organometallic Chemistry, III (1965), 1. '
Brownstein, S. and R. Miller, "Conformational Analysis of Disubstituted Cyclohexanes by Proton Resonance Spectro-scopy," Journal of Organic Chemistry, XXIV (1959), 1886.
Castellano, S. and A. A. Bothner-by, "Analysis of M R Spectra by Least Squares," Journal of Chemical Physics, XXXXI (1964), 3863.
Clifford, A. L. and R. R. Olsen, "The Analytical Chemistry or Organometallics. Quantitative Determination of Organoalkalies," Analytical Chemistry, XXXII (i960), 544.

120
Collins, Peter ?., onrad Eamienski, Donald L. Esmay, and R. B. Ellestad, "Oxidimetric Determination of n-Butyllithium in Hydrocarbons Using Vanadium PeQt-oxide," Analytical Chemistry, XXXIII (I96I), 468.
Corey, S. J., R. A. Sneen, M. G. Danaher, R. L. Young, and R. ,L. Rutledge, "A tlethod for the Location of Deuterium ..toms Attached to "yclohexane Rings," Chemistry and Industry (1954), 1294.
Curtin, David Y. and John 7. Crump, "Configurational Stabil-ities of Stereoisomeric Vinyllithium Compounds," Journal of the American Chemical Society, LXXX (1953), 1922.
Curtin, David Y. and Elbert E. Harris, "The Preparation of Stereoisomeric Alkenyl Lithium Compounds," Journal of the American Chemical Society, LXXIII (1951), 2716.
, "Preparation of Stereoisomeric Alkenyl-lithium Compounds. II. cis-and trans-1,2-Diphenylvinyllithium and o<- and y^-Styryllithiuin," Journal of the American Chemical Society, LXIII (1951), 45197"
Curtin, David Y., Harry Johnson, and Edwin G. Steiner, "Stereoisomeric Vinyllithium Compounds. III. Re-" actions with Aldehydes, Ketones, and Methyl Iodide," Journal of the American Chemical Society, LXXVII (1955),
Curtin, David Y. and William J. Koehl, "Effect of Solvent on the Steric Stability of Lithium Reagents," Journal of the American Chemical Jociety, LXXXIV (19627" 1967.
.» "Optically Active sec-Butyllithiura," Chemistry and Industry (i960), 262.
Dreiding, Andre S. and Richard J. Pratt, "The Carboxylation of cis- and trans-2-Butenyl-2-lithiun. A Stereospecific Synthesis of Angelic Acid," Journal of the American Chemical Society, LXXVI (1954)," 19027"'
Eliel, Ernest L. and Ralph G. Haber, "Conformational Analysis. V. The Reaction of cis- and trans-4-t-Butylcyclohexanol and trans-4-Methylcyclohexanol with Phosphorus Penta-bromide. Syntheses of Alkylcyclohexyl Bromides," Journal of Organic Chemistry, XXIV (1959)» 143.

121
Eschinazi, E. E. and Herman Pines, "Study in the Terpene Series. XXVI. Lisproportionation of d-Limonene in the Presence of Palladium Hydroxide-Barium Sulfate Catalvst," Journal of the American Chemical Society, IlXXVIII (lg^b;, 11757
Everson, W. L., "Determination of Butyllithium in Hydrocarbons "by Thermometric Titration with Butanol," Analytical Chemistry, XXXVI (1964), 854.
Fraenkel, Gideon, David G. Adams and James Williams, "Structure of Grignard Reagents and Organolithium Compounds," Tetrahedron Letters (1963), 767.
Gilman, Henry and Frank K. Cartledge, "The Analysis of Organolithium Compounds," Journal of Organometallic Chemistry, II (1964), 447. •
Gilman, Henry and A. U. Ilaubein, "Ethynyllithium compounds "by Halogen-Lietal Interconversions." Journal of the American Chemical Society, EXVII (19?5), 142(57
"The Quantitative Analysis of Alkyllithium Compounds," Journal of the American Chemical Society, LXVI (1944), 1515.
Gilman, Henry and R. H. Kirby, "The Relative Reactivities of Organolithium and Organomagnesium Compounds," Journal . of the American Chemical Society, LV (1933), 1265.
Gilman, Henry, Wright Langham, and Fred W. Moore, "Some Interconversion Reactions of Organolithium Compounds." Journal of the American Chemical Society, M i l (1940), 2327.
Gilman, Henry and Ivlyrl Lichtenwalter, "Relative Reactivities of Organometallic Compounds. X. Lithium and Magnesium," Recueil des Travaux Chemiques des Pays-bas, LV (1936), 561.
Glaze, William H., Jacob Lin, and E. G. Helton, "The Pyrol-ysis of Unsolvated Alkyllithium Compounds," Journal of Organic Chemistry, XXXI (1966), 2643.
Greene, Frederick D. s Ohin-Chium Chu, and Jasjit Walia, "Decomposition of Alkyl Hypochlorites. The 4-t-Butylcyclohexyl Radical," Journal of Organic Chemistryt XXIX (I964), 1285.

122
Hill, Alexander E., "Nuclear Magnetic Spectra of the Bornyl and Norbornyl Grignard Reagents," Journal of Organic Chemistry, XXXI (i960), 20.
Horner, von Leopold, Hermann Oediger, and Hellmut Hoffman, "Reactionen Mit Triphenyl-Phosphin-Dihalogeniden," Justus Liebigs Annalen der Chernie, DCXXVI (1959), 26.
Huckel, von Walter, Werner Tappe, and Gunter Legutke, "Abspaltungreaktionen und liar Sterischer Verlauf," Justus Liebigs Annalen der Chemie, DXXXXIII (1940), 191*
Jensen, Frederick R. and Kay L. Nakamaye, "Preparation of Geometrically Isomeric Grignard Reagents and the Stereo-chemical Courses of their Reactions," Journal of the American Chemical Society, LXXXVIII (1966), "345F.
Kamienski, Conrad and Donald L. Esmay, "Analysis and Stability of n-Butyllithiurn Solutions in n-Heptane," Journal of Organic Chemistry, XXV (I960), 115.
, "Effect of Sodium in the Preparation of Organolithium Compounds," Journal of Organic Chemistry, XXV (i960), 1807.
Koeppl, G. W., D. S. Sagatys, G. S. Kirshnamurthy, and Sidney I. Miller, "Inversion Barriers of Pyramidal (XY3) and Related Planar ( XY) Species," Journal of the . American Chemical Society, LXXXIX (I967), 5396.
Krieghoff, Nilde G. and Dwaine 0. Cowan, "Stereochemistry of the Grignard Reagents from exo- arid endo-Norbornyl Chlorides," Journal of the American Chemical Society, IiXXXVIII (196b) ,13227
Larnaudie, M. Marcel, "Isomerie des derive's monosubstitues des cyclohexane,"fComptes Rendus Hebdomadaires Des Seances De Ti'AcademieDes Sciences, CCXXXV (1952) 154-.
Lemieux, R. U., R. K. Kullnig, H. J. Bernstein, and W. G. Schneider, "Configurational Effects in the Proton Magnetic Resonance Spectra of Acetylated Carbohydrates," Journal of the American Chemical Society, LXXIX (1957)« 1005.
, "Configurational Effects on the Proton Magnetic Resonance Spectra of Six-Membered Ring Compounds, "Journal of the American Chemical Society, LXXX (1958), 6098..

123
Let singer, Ro"bert L., "Formation of Optically Active ?-Methylheptyllithium," Journal of the American Chemical Society, LXXII (1950), 45771 '
McNiven, Heal t,. and John Reed, "Researches in the Menthane Series. Part XVII. Configurations of Menthols and l.Ienthylamines," Journal of the Chemical Society (1952), ] 53.
Moore, James A. and Donald E. Reed, "Diazomethane," Organic Syntheses, XV (1953), 3.
Morton, Avery A. and Graham M. Richardson, "Condensation by Sodium. XVI. The Formation of Decane in the Wurtz Reaction," Journal of the American Chemical Society, LXII (1940), 123':
Morton, M. and 1. J. Fetters, "Homogeneous Anionic Polymer-ization V. Association Phenomena in Organolithium Polymerization," Journal of Polymer Science, A2 (1964), 33U.
Mulvaney, J. E. and Z. G. Gardlund, "Alkyllithium Compounds and Uonconjugated Olefins. The Nature of a Norbornyl-lithium Compound," Journal of Organic Chemistry, XXX (1965), 4321.
ITesmeyanov, A. H. and A. E. Borisov, "Stereochemistry of Electrophilic and Homolytic Substitution at the Olefinic Carbon," Tetrahedron, I (1957), 153.
ITesmeyanov, A« N.» A. E. Borisov, and II. A. Volkenau, "Synthesis of Stereoisomeric Organomercury Compounds from Organolithium Compounds," Izvestiya Akademii Nauk Soyuza Sovetskikh Sotsialisticheskikh' Respublik (1954)
Ott, Erv/in, "liber die Additionreaktionen der Athylen-Doppelbindung unter Bildung von Xthen-Verbingdungen mit zv/ei, durch den Additionsvorgong entstehenden, asyiruiietrischen Kohlenstoffatomen und der Einfluss der Reaktionsgeschwindigkeit auf den Reaktionsverlauf," Berichte der Deutschen Chemischen Gesellschaft, TjXI TO2S), 2T2T.
Pickard, Robert Howson and Joseph Kenyon, "VII - Investigations on the Dependence of Rotatory Power on Chemical Consti-tution. Part I. The Rotations of the Simplest Secondary Alcohols of the Fatty Series," Journal of the Chemical Society, LXXXXIX (1911), 45.

124
Porter, 0. W., "Raccmisation in the Preparation of the Grignard Reagent," Journal of the American Chemical Society, LVII (1935), H36.
Read, John, "Recent Progress in the Menthons Chemistry," Chemical Reviews, VII (1930), 1.
Sauvage, James Frederick, Robert H. Baker, and Allan S. Kussey, "The Kydrogenation of Cyclohexane over Platinum Oxide," Journal of the American Chemical Society, ixxxii (19 0), smo~.
Schorigin, P., "Uber bis Hatiumalkyle Ihre Reaktion mit der Atheru," Berichte Der Deutschen Chemischen GeselLschaft» XXXXIII (I9IO), 19517 *
Schwartz, A. II. and John R. Johnson, "Characterization of Alkyl Halides and Organomagnesium Halides," Journal of the American Chemical Society, 1III (1931), 1065.
Seyferth, Dietmar and Harvey M. Cohen, "The Stability of Cyclopropyllithium in Diethyl Ether and in Tetrahydro-furan," Journal of Organometallic Chemistry, I (1963), 15.
Seyferth, Dietmar and Lawrence G. Vaughan, "The Isomeric 1-Propenyllithium Reagents: Their KMR Spectra and the Stereochemistry of Their Formation by Direct Reaction and by the Transmetalation Reaction," Journal of Organometallic Chemistry, I (1963), 20TT
Smith, J. G. and George P. Wright, "The Diastereomeric Menthyl Chlorides Obtained Prom (-)Menthol, Journal of Organic Chemistry, XVII (1952), 1116.
Stolow, Robert D. and Charles B. Boyce, "Preparation of cis- and trans-4~t-Butyl-o(,«<-dimethylcyclohexanementhanol,n
Journal of Organic Chemistry, XXVI (19 61), 7426.
Tarbell, D. 3. and Marvin Weiss, "The Action of Lithium on an Optically Active Aliphatic Chloride," Journal of the American Chemical Society, LXI (1939), 1203.
Walborsky, H. M. and M. S. Aronoff, "Cyclopropanes XIX. Reaction of Lithium Metal with Optically Active Halides," Journal of Organometallic Chemistry. IV (1965), 418.
Walborsky, H. M. and P. J. Impastato, "Cyclopropanes. VI. Retention of Optical Activity and Configuration in the Cyclopropyl Oarbanion," Journal of the American Chemical Society. LXXXI (1959), 5&55~.

125
\7a I b o r S I' , h.. • • , ? • J « I».iptHS"Li3 SO , 3 il(. 1 . E • /OUQjj j^ 11 Gj d o — "oropa i ies . X V . T h e O p t i c a l S t a b i l i t y ox 1— r!s ohy J.— 2 , 2 - d I p i i e n y l - c y c " * o p r o p y i l i t n i c u , " J o u r n a 7 o f t h e A m e r i c a n gi' iei-i ica]. S o c i e t y , LXXXV1 ( I ' joA- ) t~~y2'o2 •
V-fa"11 i s . E v e r e t t 3 . a n d ? r e d e r i c I I . Ada:. iS, " i ' l io S p a t i a l ;on f i g u r a t i o n o f the V a l e n c e s i n S r i o o v a l e n t C a r b o n
OoiJipoundsi" J o u r n a l o f t h e A m e r i c a n G h e r . i c a l S o c i e t y , LV ( 1 9 3 3 ) , 3 6 3 6 .
E i n s t e i n , S . , 75. G l i p p i n g e r , R o b e r t i l o u e , a n d E l l i o t V o g e l - ^ f a n g e r . " T h e H o n c l a s s i o a l I T o r b o r n y l C a t i o n , " J o u r n a l o f t h e A n e r i c a n C h e m i c a l S o c i e t . y , L X X X V I I ( 1 9 6 5 ) 1 3 7 b .
W i t t i g , G e o r g e , F e d e r i c o V i d a l , a n d E r v / i n B o h n e r t , " i f b e r d i e k o n f i g u r a t i v e B c s t a nd i g k e i t rue t a i l o r ga n i s c h e r V e r b i n d u n g e n , " B e r i c h t e d e r D e u t s c h e n G h e m i s c h e n G e s e l l s c n a i t , I i X X X I I I (~1 "J > 0 ) "j 3 5 9 *
W r i g h t , G e o r g e 3?., " O r g a n o u e t a l l i c Compounds o f S t y r e n e , " J o u r n a l o f O r g a n i c C h e m i s t r y , I ( 1 9 3 6 ) , 4 5 7 .
Z i e g l e r , k . a n d I I . C o l o n i u s , " U n t e r s u c h u n g e n u b e r a l ^ a l i -o r g a n i s c h e V e r b i n d u n g e n . V . E i n e bequeme S y n t h e s e e i n f a c h e r L i t h i u m a l k y l e J u s t u s L i e b i g s A n n a l e n d e r G h e m i e , GCGGLXXIX ( 1 9 3 0 ) , T W .
Z i e g l e r , v o n k a r l a n d I I . G . G e l l e r t , " X V I I . R e a k t i o n e n
Z w i s c h e n L i t h i u m - a l k y l e n und A t h e r n , " J u s t u s I d e b i g s A n n a l e n d e r C h e u i i e , D I t X V I I ( 1 9 5 0 ) , 1 8 5 .
U n p u b l i s h e d M a t e r i a l s
V f i l l i a m H . G l a z e , u n p u b l i s h e d n o t e s , D e p a r t m e n t o f C h e m i s t r y , K o r t h T e x a s S t a t e U n i v e r s i t y , B e n t o n , T e x a s , 1 9 6 7 .