public resources for bioinformatics databases : how to find relevant information. analysis tools
TRANSCRIPT

Public Resources for Bioinformatics
•Databases : how to find relevant information.
•Analysis Tools

Public Resources (II) – Analysis tools
Web-based analysis tools – easy to use, but often with less customization options.
Stand-alone analysis tools – requires installation and configuration, but provides more customizatio0n options.
Commercial analysis tools Scripting for bioinformatics projects

web-based tools
• Identification of web-based bioinformatics resources. – Portals, lists, – Google search
• Organization–Book mark.–html page.

web-based tools
Practice –retrieve genomic sequence from Ensemble and perform reverse
complementation with SMS
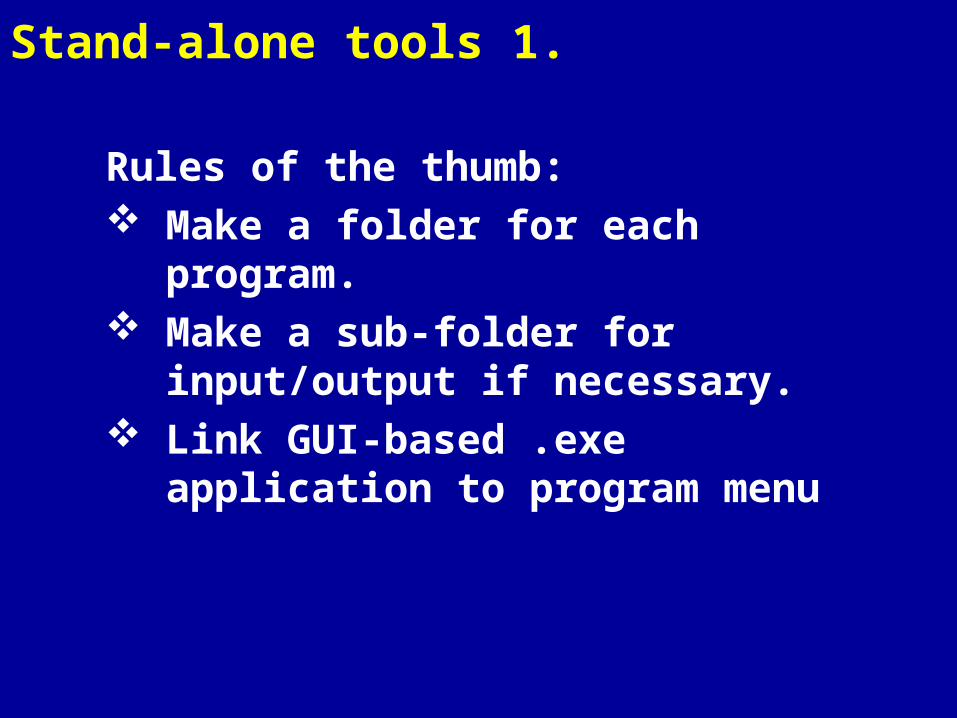
Stand-alone tools 1.
Rules of the thumb: Make a folder for each program. Make a sub-folder for input/output
if necessary. Link GUI-based .exe application to
program menu
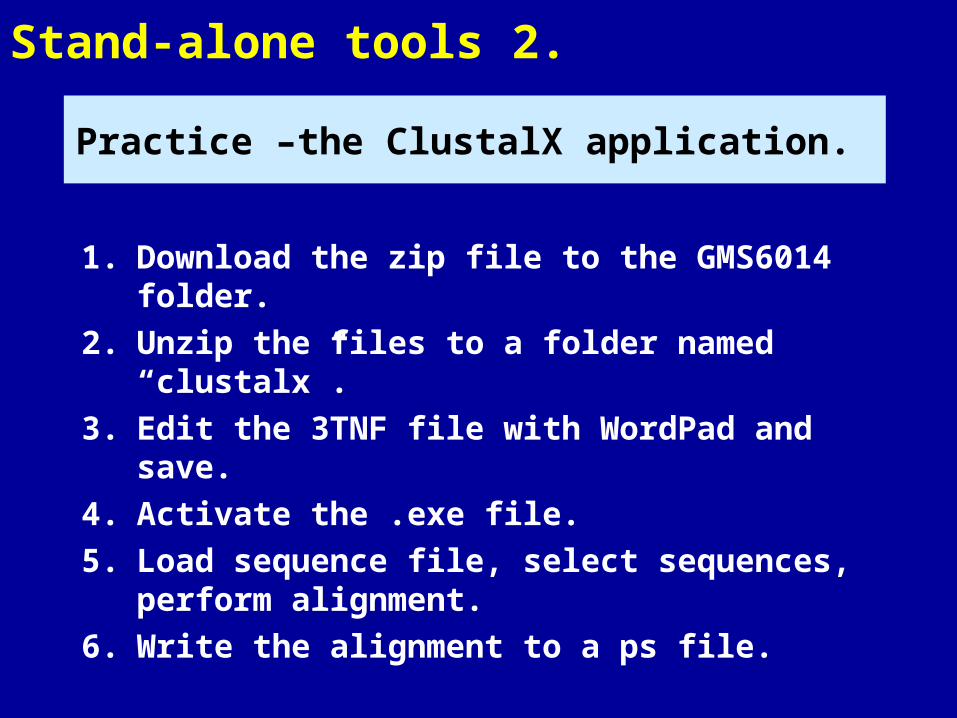
Stand-alone tools 2.
1. Download the zip file to the GMS6014 folder.
2. Unzip the files to a folder named “clustalx”.
3. Edit the 3TNF file with WordPad and save.
4. Activate the .exe file.
5. Load sequence file, select sequences, perform alignment.
6. Write the alignment to a ps file.
Practice –the ClustalX application.
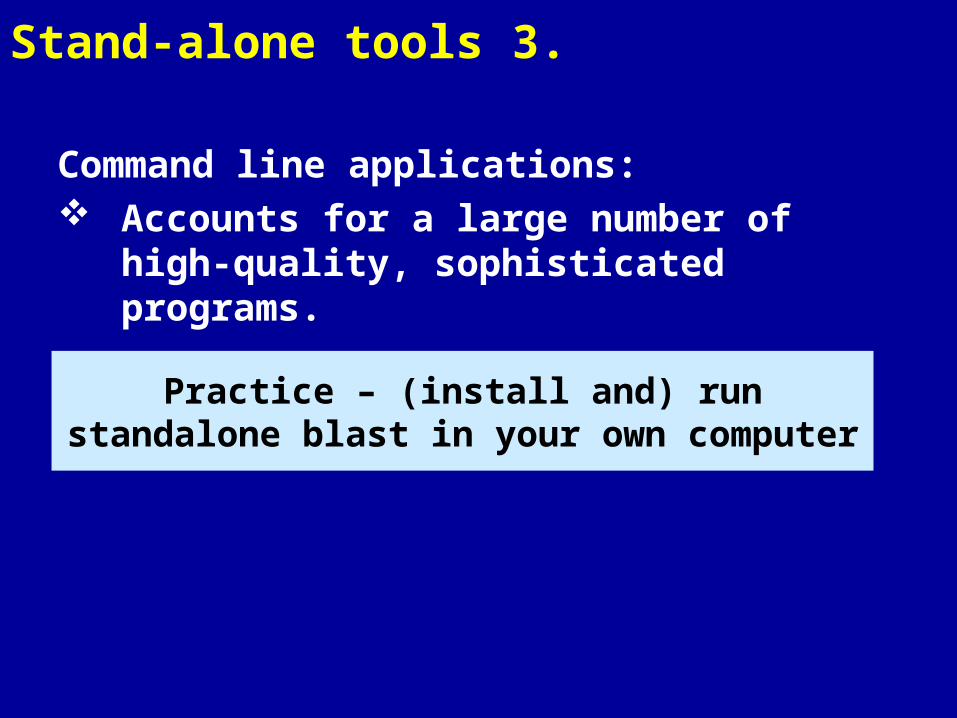
Stand-alone tools 3.
Command line applications: Accounts for a large number of high-quality,
sophisticated programs.
Practice – (install and) run standalone blast in your own computer

Identifying the ortholog of TNF (Tumor necrosis factor) in mosquito genomes
Pet Projects:
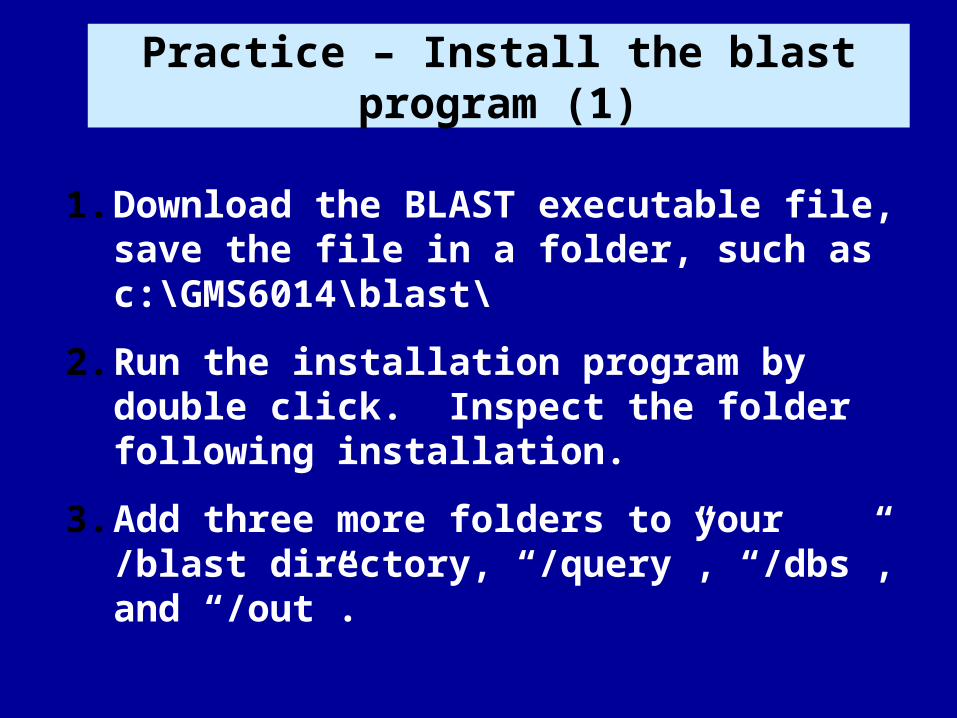
Practice – Install the blast program (1)
1. Download the BLAST executable file, save the file in a folder, such as c:\GMS6014\blast\
2. Run the installation program by double click. Inspect the folder following installation.
3. Add three more folders to your /blast directory, “/query”, “/dbs”, and “/out”.

Practice – Install the blast program (2)
5. Inspect the contents of the doc, data, and bin folder. Move the programs from blast\bin to the blast folder.
6. Bring a command (cmd) window by typing “cmd” in the StartRun box.
7. Go to the blast folder by typing “cd C:\GMS6014\blast”
8. Try to run the program by typing “blastall”, read the output.
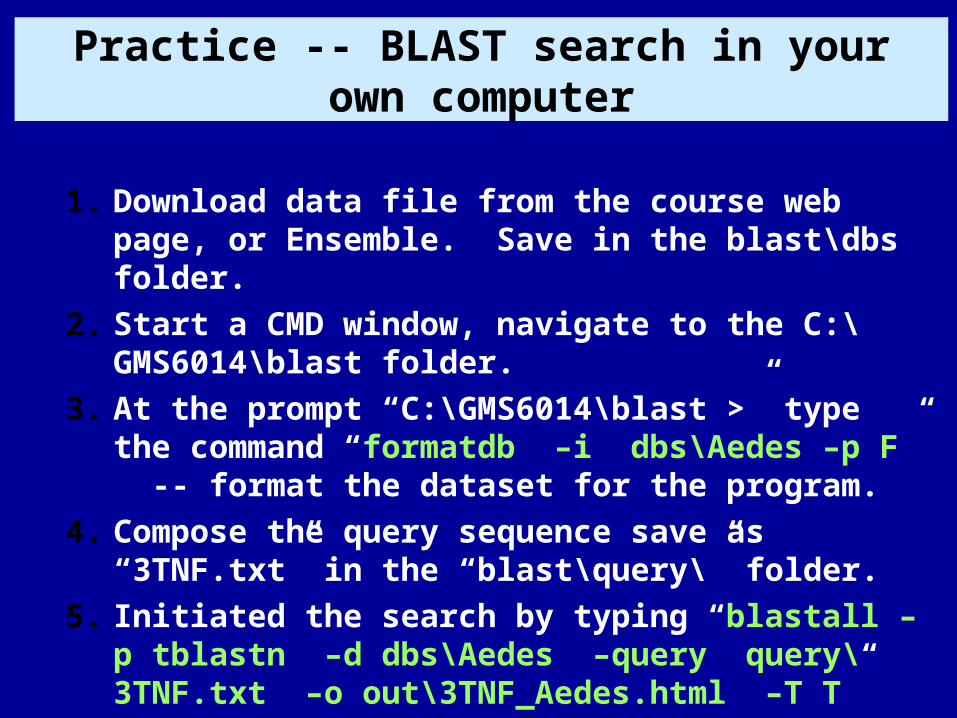
Practice -- BLAST search in your own computer
1. Download data file from the course web page, or Ensemble. Save in the blast\dbs folder.
2. Start a CMD window, navigate to the C:\GMS6014\blast folder.
3. At the prompt “C:\GMS6014\blast >” type the command “formatdb –i dbs\Aedes –p F” -- format the dataset for the program.
4. Compose the query sequence save as “3TNF.txt” in the “blast\query\” folder.
5. Initiated the search by typing “blastall –p tblastn –d dbs\Aedes –query query\3TNF.txt –o out\3TNF_Aedes.html –T T”

What’s in a command?
formatdb –i dbs\Aedes –p F
Program – format database for search.
Feed me the input file name
Tell me is it a protein sequence file?
For more info, refer to the “user manual” file in the blast\doc folder.

Advantages of Running BLAST at Your Own Machine
Do it at any time, no waiting on the line.
Search for multiple sequences at once.
Search a defined data set.
Automate Blast analysis.
Combine Blast with other analysis.
…..

BLAST is a program implemented in C/C++
void BlastTickProc(Int4 sequence_number, BlastThrInfoPtr thr_info)
{
if(thr_info->tick_callback &&
(sequence_number > (thr_info->last_db_seq + thr_info->db_incr))) {
NlmMutexLockEx(&thr_info->callback_mutex);
thr_info->last_db_seq += thr_info->db_incr;
thr_info->tick_callback(sequence_number, thr_info->number_of_pos_hits);
thr_info->last_tick = Nlm_GetSecs();
NlmMutexUnlock(thr_info->callback_mutex);
}
return;
}
/*
Sends out a message every PERIOD (i.e., 60 secs.) for the index.
THis function runs as a separate thread and only runs on a threaded
platform.
Should I care ?

Programming language comparison
/* TRANSLATION: 3 or 6 frame translate cDNA sequences*/
//---------------------------------------------------------------------------#include "translation.hpp"
int main(int argc, char **argv){ int num_seq=0;
char string[MAXLINE]; DSEQ * dseq;
infile.getline (string,MAXLINE);
if (string[0]=='>') strncpy (dbname,string,MAXLINE); while (!infile.eof()) { dseq=Get_Lib_Seq (); if (dseq->reverse==0) Translation (&dseq->name[1], dseq->seq); else Translation (&dseq->name[1], dseq->r_seq); num_seq++; if (num_seq%1000==0) { cout<<num_seq<<endl; cout<<dseq->name<<endl; } delete dseq; }
infile.close(); outfile.close(); cout<<num_seq<<" translated"<<endl; getch();
return 0;}
DSEQ* Get_Lib_Seq(){ int i,n; char str[MAXLINE]; DSEQ* dseq; n = 0; dseq=new DSEQ; strcpy (dseq->name, dbname);
while(infile.getline(str,MAXLINE)) { if (str[0] == '>') { strcpy( dbname, str); break; }
for(i=0;i<strlen(str);i++) { if(n==MAXSEQ) break; dseq->seq[n++] = str[i]; } } dseq->seq[n]='\0';
if(n==MAXSEQ) cout<<"WARNING: sequence"<<dbname<<"too long!"<<endl; dseq->len=n; if (dseq->name[9]=='3') Reverse (dseq); else dseq->reverse=0; return dseq;}
void Reverse (DSEQ* dseq) //Reverse dseq{ int i,j; j=0; for (i=(dseq->len-1);i>0;i--) { if (dseq->seq[i]=='A'||dseq->seq[i]=='a') dseq->r_seq[j++]='T'; if (dseq->seq[i]=='C'||dseq->seq[i]=='c') dseq->r_seq[j++]='G'; if (dseq->seq[i]=='G'||dseq->seq[i]=='g') dseq->r_seq[j++]='C'; if (dseq->seq[i]=='T'||dseq->seq[i]=='t') dseq->r_seq[j++]='A'; if (dseq->seq[i]=='N'||dseq->seq[i]=='n') dseq->r_seq[j++]='N'; } dseq->r_seq[j++]='\0'; dseq->reverse=1;}void Translation (char name[], char seq[]){ char ppseq[MAXSEQ/3];
for (int f=0; f<3; f++) { outfile<<">"<<"F_"<<f<<name<<endl; int j=0; int len=strlen(seq); for( int i=f; i<len; i=i+3) ppseq[j++]=Translate(&seq[i]); ppseq[j++]='\0'; int m=strlen(ppseq)/50; // output 50 aa per line for (int n=0; n<=m; n++) { for (int i=n*50; i<50*(n+1); i++) { outfile<<ppseq[i]; if (ppseq[i]=='\0') break; } outfile<<endl; } }}
char Translate(char s[]){ int c1,c2,c3;
char P, code[3];
//***standard translation table, A(0),C(1), G(2), T(3)*****
char table [4][4][4]= {{{'K','N','K','N'},{'T','T','T','T'},{'R','S','R','S'},{'I','I','M','I'}}, {{'Q','H','Q','H'},{'P','P','P','P'},{'R','R','R','R'},{'L','L','L','L'}}, {{'E','D','E','D'},{'A','A','A','A'},{'G','G','G','G'},{'V','V','V','V'}}, {{'*','Y','*','Y'},{'S','S','S','S'},{'*','C','W','C'},{'L','F','L','F'}}};
//*********** table2 for n at 3rd position********************char table2 [4][4]={{'X','T','X','X'},{'X','P','R','L'}, {'X','A','G','V'},{'X','S','X','X'}}; strncpy (code, s, 3); c1=Convert(code[0]); c2=Convert(code[1]); c3=Convert(code[2]); if (c1>=4 || c2>=4) P='X'; //can be Optimized further here by considering....
else { if (c3>=4) P=table2[c1][c2]; else P=table[c1][c2][c3];
//P=table[Convert(code[0])][Convert(code[1])][Convert(code[2])]; } return (P);}
int Convert (char c){ char s=c;
if (s=='A'||s=='a') return (0); if (s=='C'||s=='c') return (1); if (s=='G'||s=='g') return (2); if (s=='T'||s=='t'||s=='U'||s=='u') return (3); if (s=='N'||s=='n') return (4); else return (5);}
f#Translation -- read from fasta DNA file and translate into three frames
#
import string
from Bio import Fasta
from Bio.Tools import Translate
from Bio.Alphabet import IUPAC
from Bio.Seq import Seq
ifile = "S:\\Seq\\test.fasta"
parser = Fasta.RecordParser()
file =open (ifile)
iterator = Fasta.Iterator (file, parser)
cur_rec = iterator.next()
cur_seq = Seq (cur_rec.sequence,IUPACUnambiguousDNA())
translator = Translate.unambiguous_dna_by_id[1]
translator.translate (cur_seq)
Translation : C Translation : Python

Observe: scripting is not that difficult
Example: Python and bioPython.
1. Simple python scripts.
2. Batch Blast with a Python script.
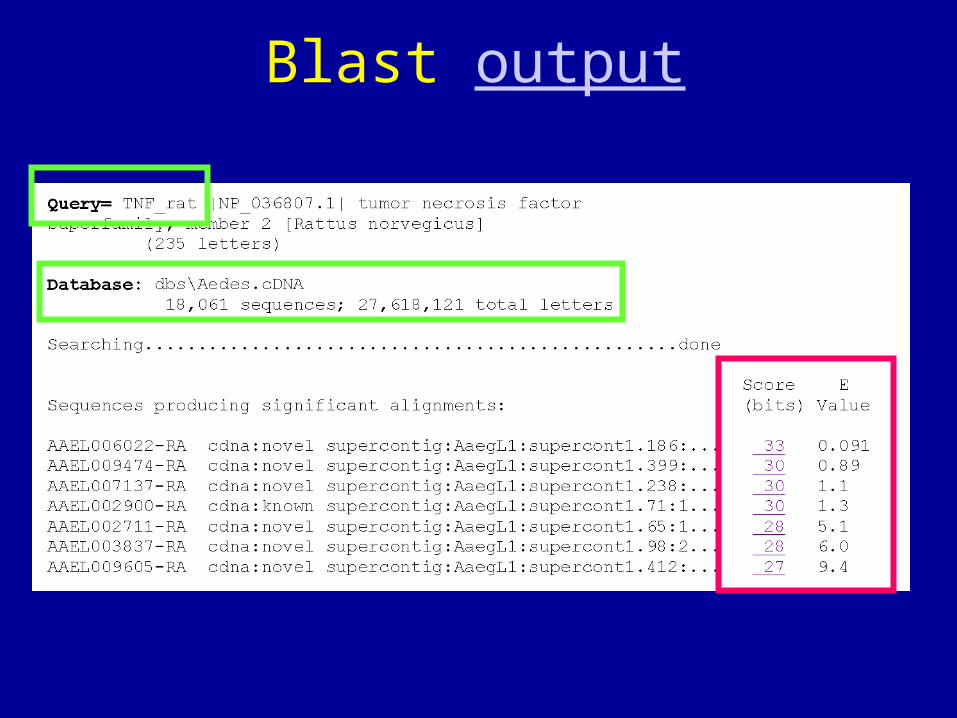
Blast output

Questions after the Blast search?
Questions:• Is this a expressed gene in the Aedes mosquito?
- Gene prediction & gene structure
• Is this the true ortholog of TNF?
- Fundamentals of sequence comparison
• What can we learn from the comparison of sequences?
-- protein dommains/motifs.

If you care:
1.) Data structure and Algorithm
char: name
char: sequenceSEQ
Identify the best alignment for two sequences (p69-73)
Seq1: MA-DSV—WC..
Seq2: MALD-IHWS..
int: seq_length

Programming languages
C/C++
Java - Biojava
Python - Biopython
Perl - Bioperl
Efficiency, Power Simplicity, Fast Dev.

Observe: programming is not that difficult
Example: Python and bioPython.
1. Simple python scripts.
2. Batch Blast with a Python script.
