proteomics software at msi. · proteomics software at msi. • proteomics : emerging technology •...
TRANSCRIPT

Proteomics software at MSI.
Pratik Jagtap Minnesota Supercomputing institute
http://www.mass.msi.umn.edu/

Proteomics software at MSI.
• proteomics : emerging technology • proteomics workflow • search algorithms • de novo analysis : Peaks • Statistical validation of protein identification • Quantitative tools • Targeted proteomics • Data dissemination : Tranche
Proteomics Fifteen Years Ago…
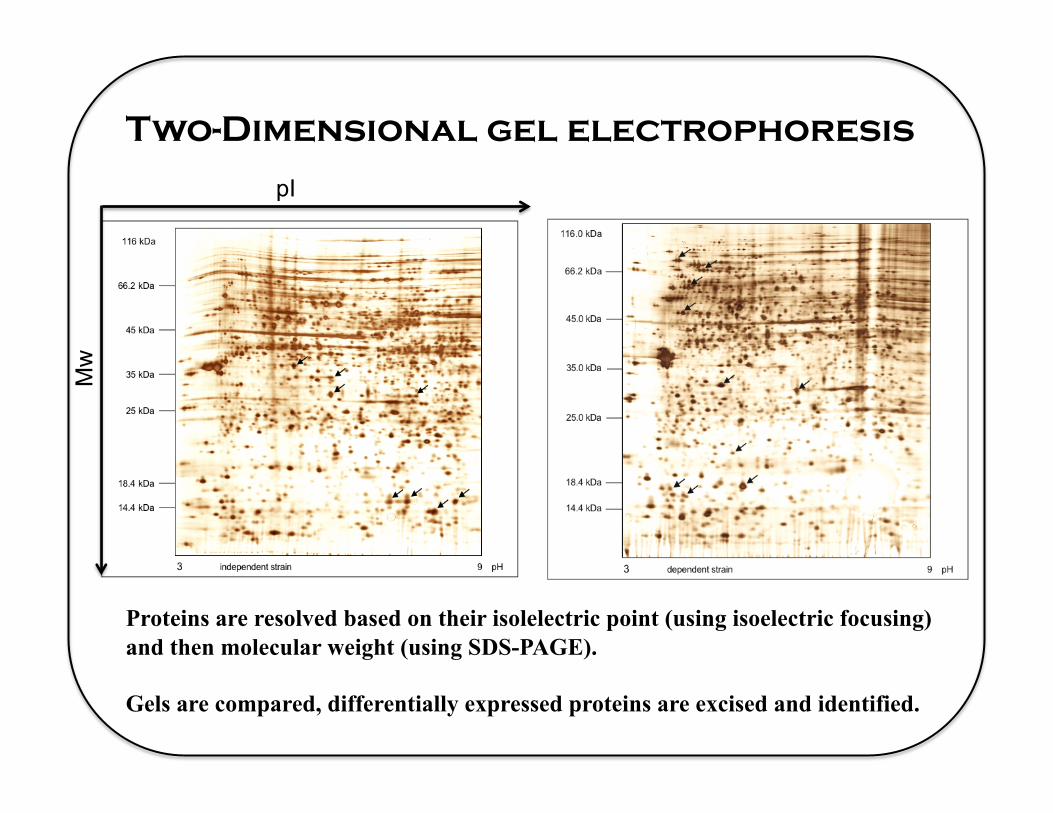
Two-Dimensional gel electrophoresis
pI
Mw
Proteins are resolved based on their isolelectric point (using isoelectric focusing) and then molecular weight (using SDS-PAGE).
Gels are compared, differentially expressed proteins are excised and identified.

Proteomics Fifteen Years Ago…
Search algorithm
Mass Spectrometry
Data Extrac5on. Analysis So9ware that correlates the protein ID to the excised gel spot.

Genomics
Genomics : Number of Databases for search and Shotgun analysis.
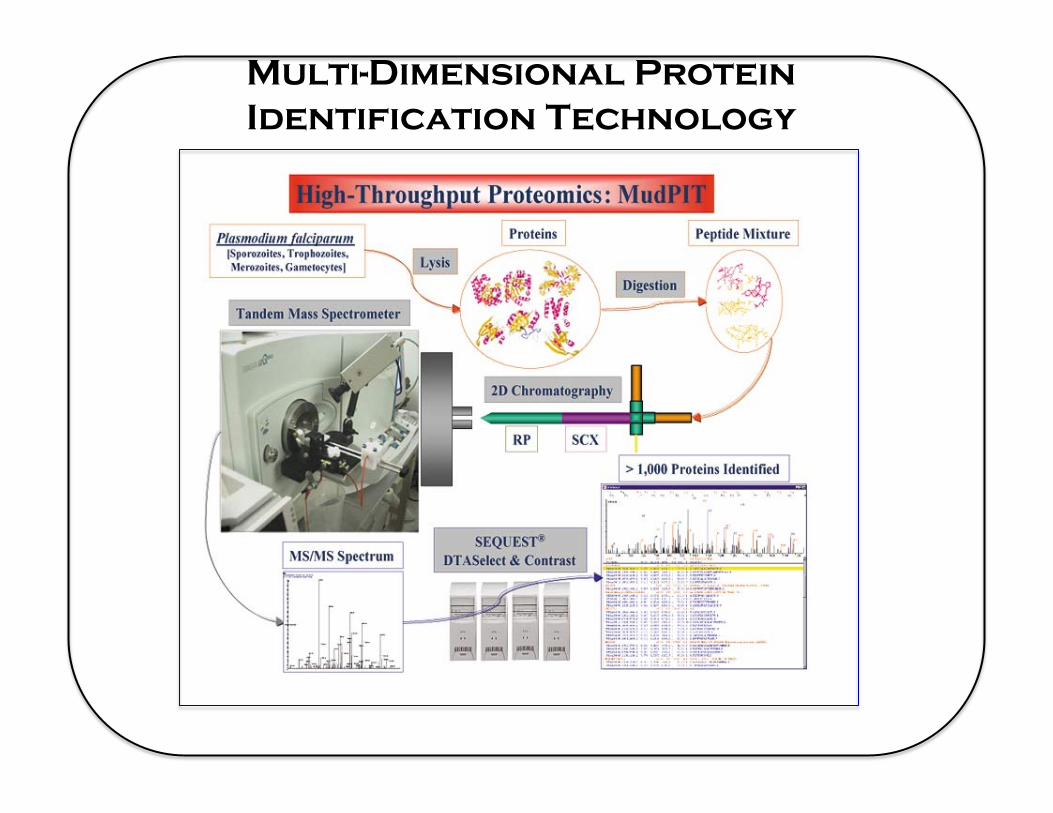
Multi-Dimensional Protein Identification Technology
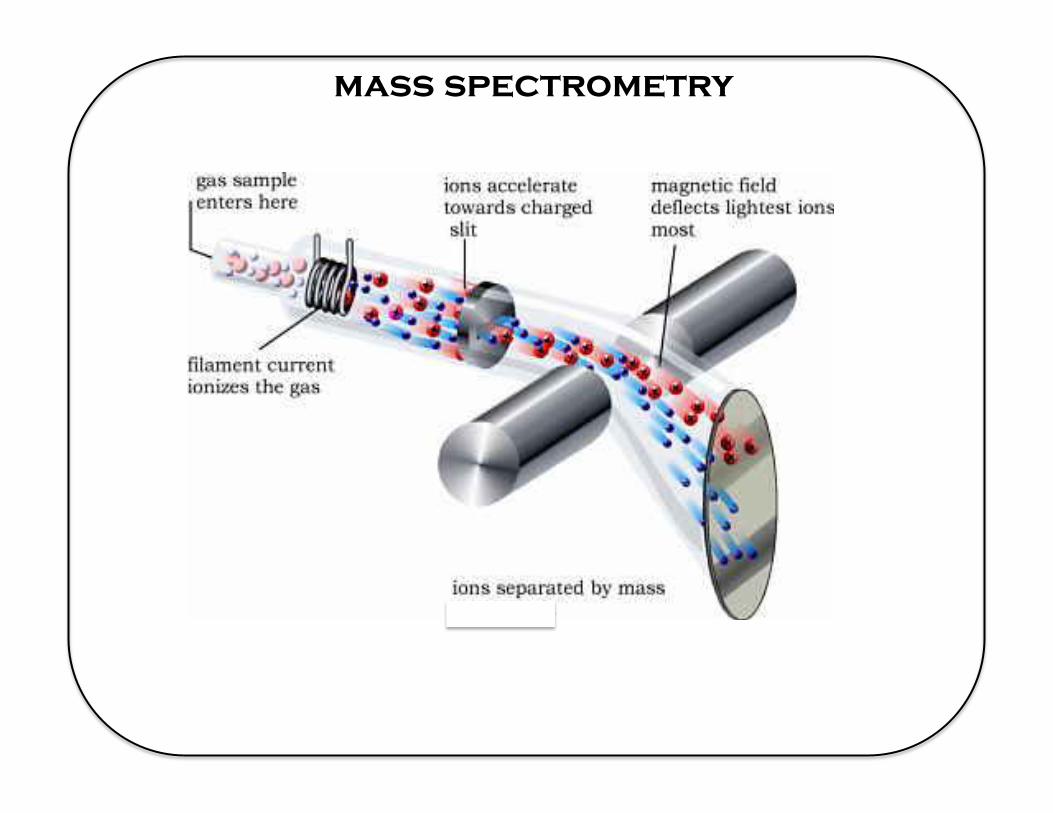
mass spectrometry

Mass Spectrometers & data formats
Thermofinnigan Xcalibur / .raw Life Technologies Analyst / .wiff ; .t2d
Waters Masslynx / .raw Bruker .baf
Mascot
Sequest
X! tandem OMSSA
ProteinPilot

Proteomics workflow
Protein Peptide
Mass spectrum
Fragmentation
Search against database.
mass spectrum In
tens
ity
Mass / charge
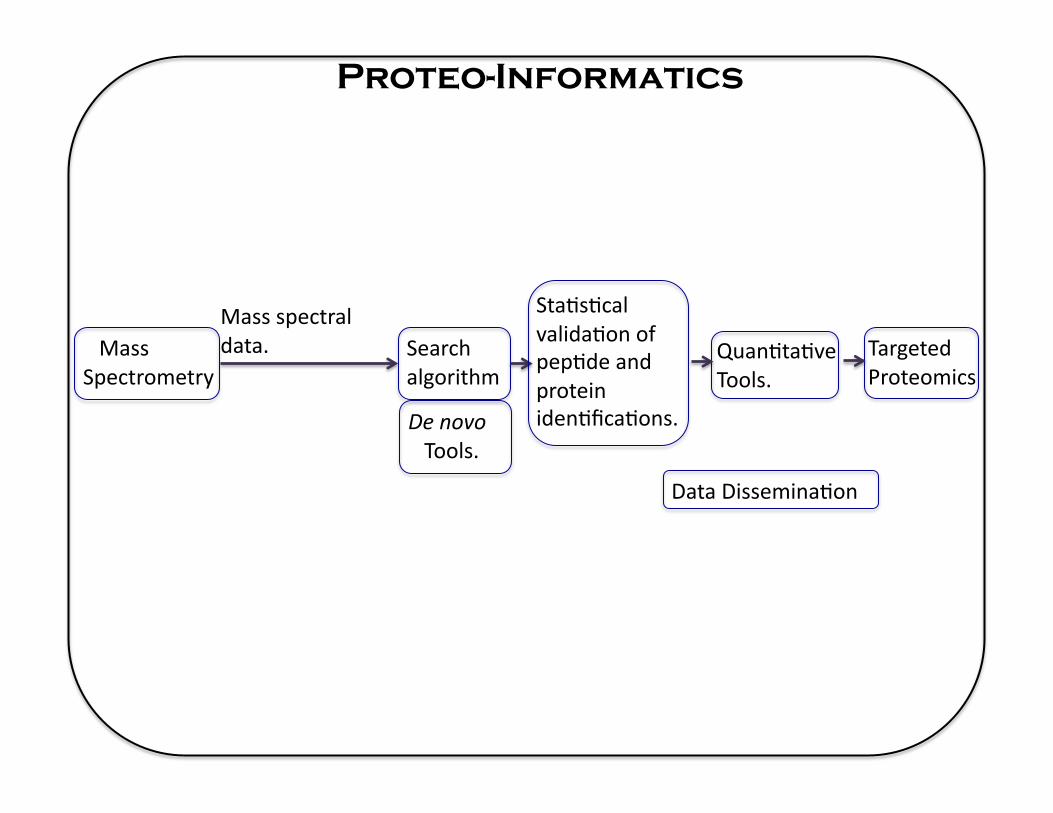
Proteo-Informatics
Mass Spectrometry
Mass spectral data. Search
algorithm
Sta5s5cal valida5on of pep5de and protein iden5fica5ons. De novo
Tools.
Data Dissemina5on
Quan5ta5ve Tools.
Targeted Proteomics
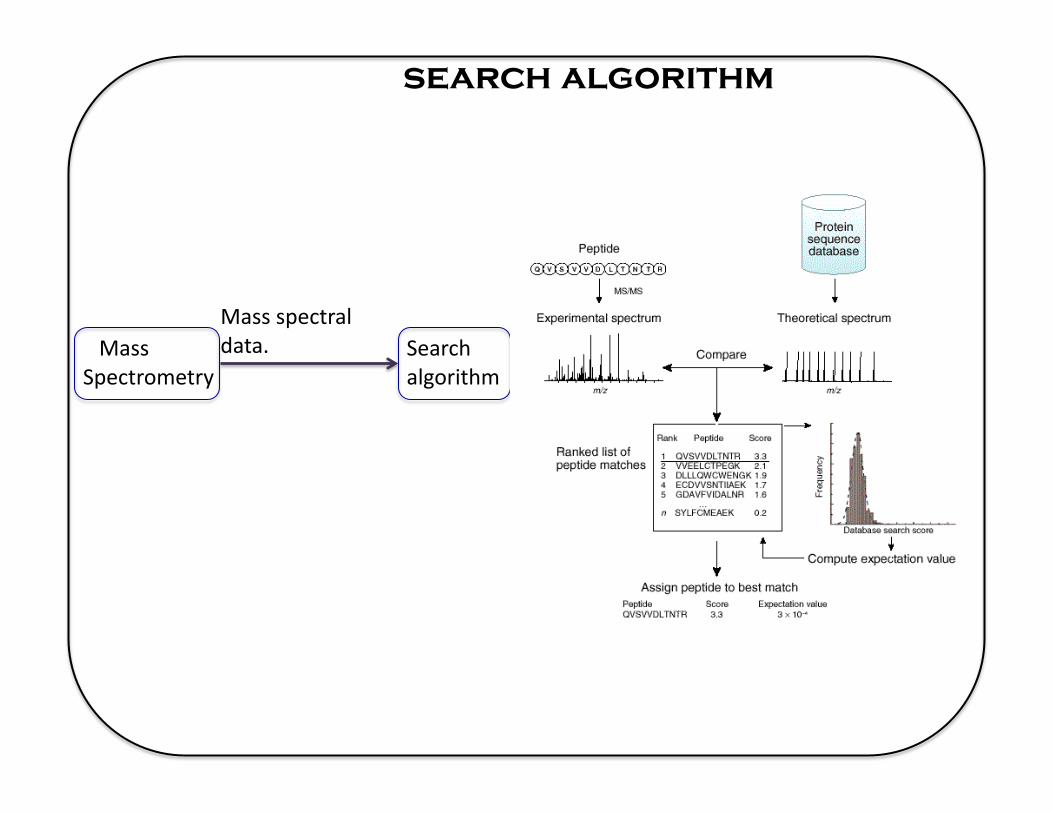
search algorithm
Mass Spectrometry
Mass spectral data. Search
algorithm

search algorithm
Mass Spectrometry
Mass spectral data. Search
algorithm
Sequest
X!tandem
OMSSA
MaxQuant RDC : sdvlapp32
ProteinPilot CPC9 ; CGL 138.
Mascot https://sequest7.msi.umn.edu/mascot

Protip / TINT
Raw Data from
Orbitrap mzxml format
dta format
X!TANDEM search
Scaffold Analysis
Scaffold Viewer
MASCOT search
SEQUEST search
Mgf format
OMSSA search
Powered by
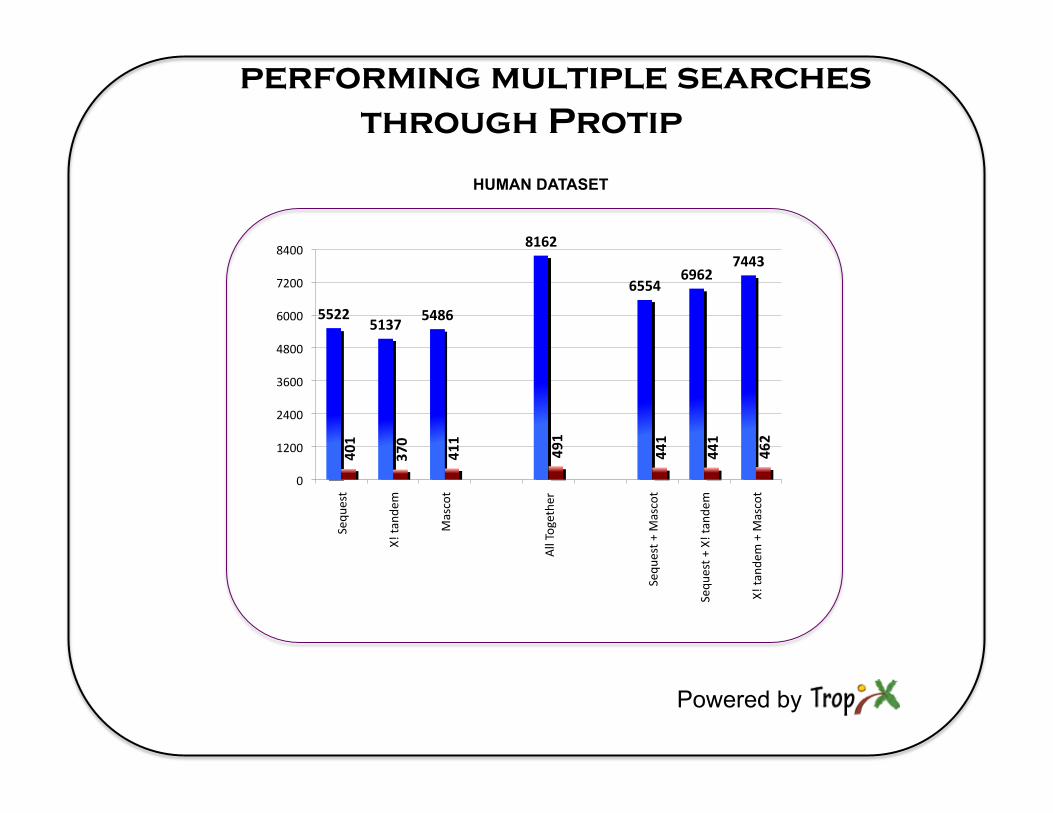
performing multiple searches through Protip
MASCOT search
# of
pro
tein
s
5522 5137
5486
8162
6554 6962
7443
401
370
411
491
441
441
462
0
1200
2400
3600
4800
6000
7200
8400
Sequ
est
X! ta
ndem
Mascot
All Together
Sequ
est +
Mascot
Sequ
est +
X! tande
m
X! ta
ndem
+ M
ascot
HUMAN DATASET
Powered by
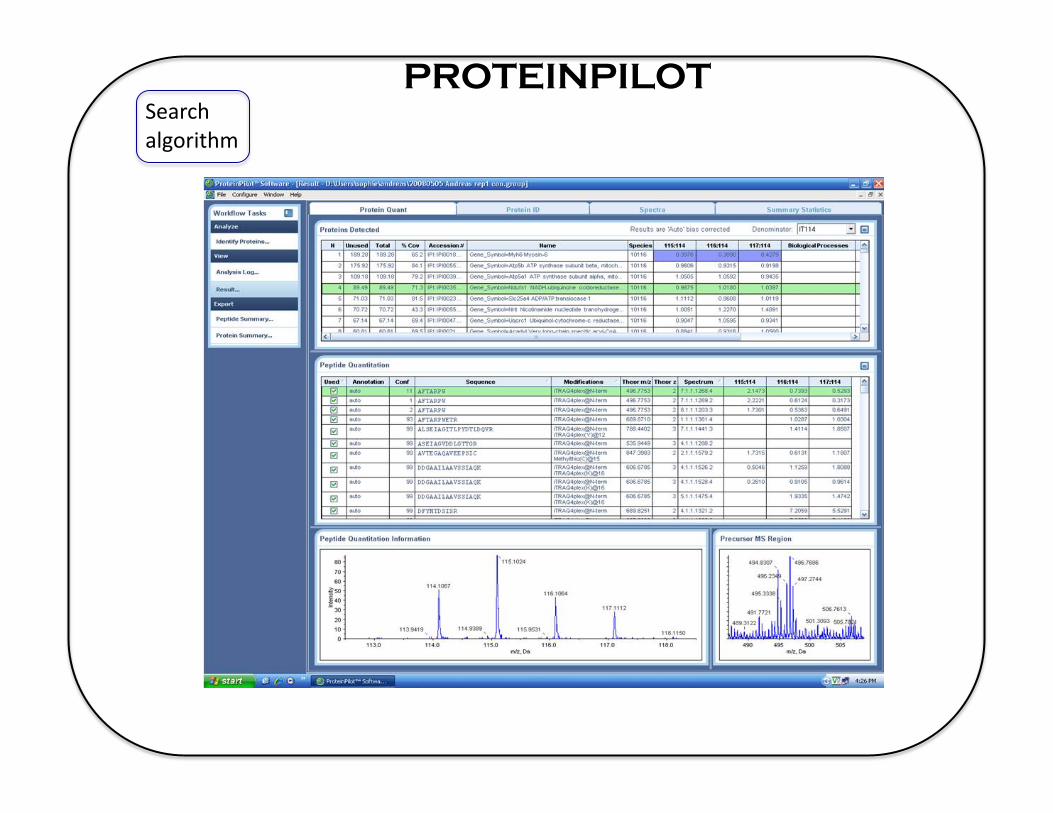
PROTEINPILOT Search algorithm
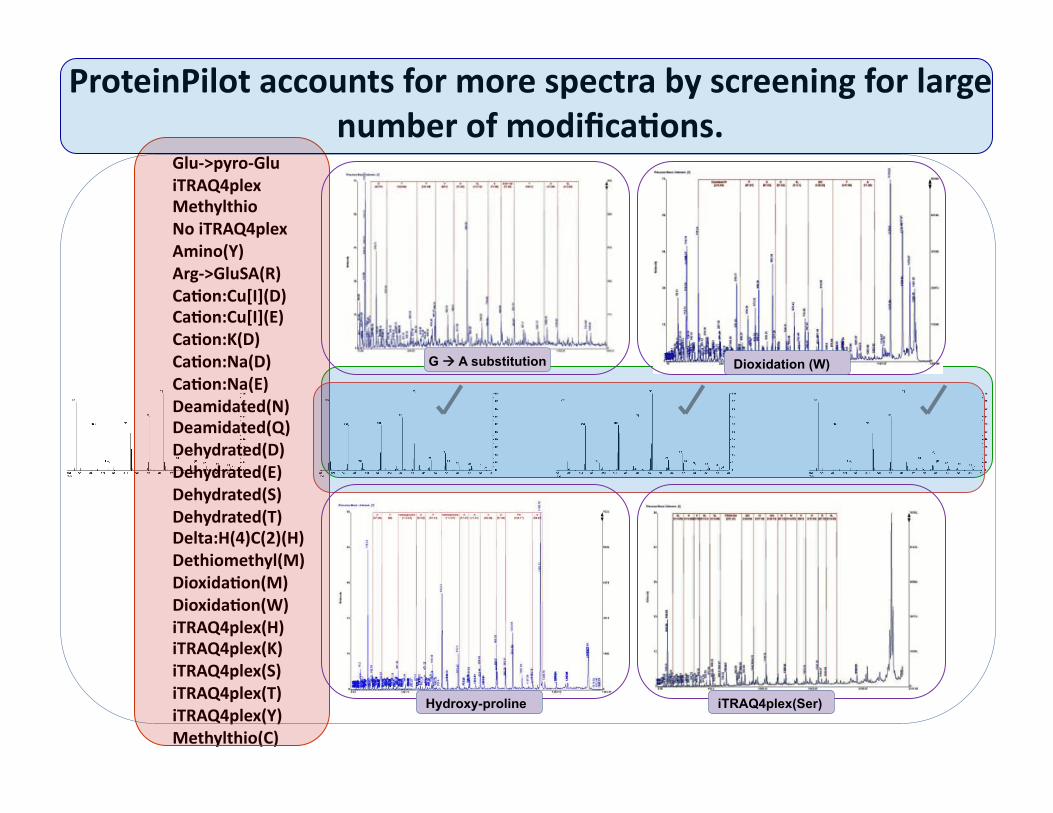
ProteinPilot accounts for more spectra by screening for large number of modifica@ons.
Glu‐>pyro‐Glu iTRAQ4plex Methylthio No iTRAQ4plex Amino(Y) Arg‐>GluSA(R) Ca@on:Cu[I](D) Ca@on:Cu[I](E) Ca@on:K(D) Ca@on:Na(D) Ca@on:Na(E) Deamidated(N) Deamidated(Q) Dehydrated(D) Dehydrated(E) Dehydrated(S) Dehydrated(T) Delta:H(4)C(2)(H) Dethiomethyl(M) Dioxida@on(M) Dioxida@on(W) iTRAQ4plex(H) iTRAQ4plex(K) iTRAQ4plex(S) iTRAQ4plex(T) iTRAQ4plex(Y) Methylthio(C)
Hydroxy-proline
G A substitution
iTRAQ4plex(Ser)
Dioxidation (W)

maxquant Search algorithm
• MaxQuant is an integrated suite of algorithms specifically developed for high-resolution, quantitative MS data.
• MaxQuant detects peaks, isotope clusters and stable amino acid isotope-labeled (SILAC) peptide pairs as three-dimensional objects in m/z, elution time and signal intensity space.
• By integrating multiple mass measurements, mass accuracy in the p.p.b. range is achieved.
• MaxQuant quantifies several hundred thousand peptides per SILAC-proteome experiment.
http://www.maxquant.org/

data in different formats
Protein DB Sequence with ID
Protein ID
Homologous DB
Sequence with putative ID
SPIDER
No DB Sequence but no ID de novo
Multiple software
high confidence Sequence ID
inChorus
quantity Protein ID
PEAKS Q
PTMs Protein ID
PTM Finder
PEAKS Options De novo Tools.

PEAKS resources at MSI • PEAKS Online
– http://sequest5.msi.umn.edu:8080/peaksonline/ – Get a password from Tu. – Set up a search using peaklist. – Monitor your search status. – Links for .anz files that can be used further in PEAKS
Client.
• PEAKS Client v4.5 – PEAKS Client available at CGL : CPC7 and CPC10. – Use remote access to cpc7.msi.umn.edu or
cpc10.msi.umn.edu – Use your .anz file (generated from Online search) for
further analysis.
De novo Tools.
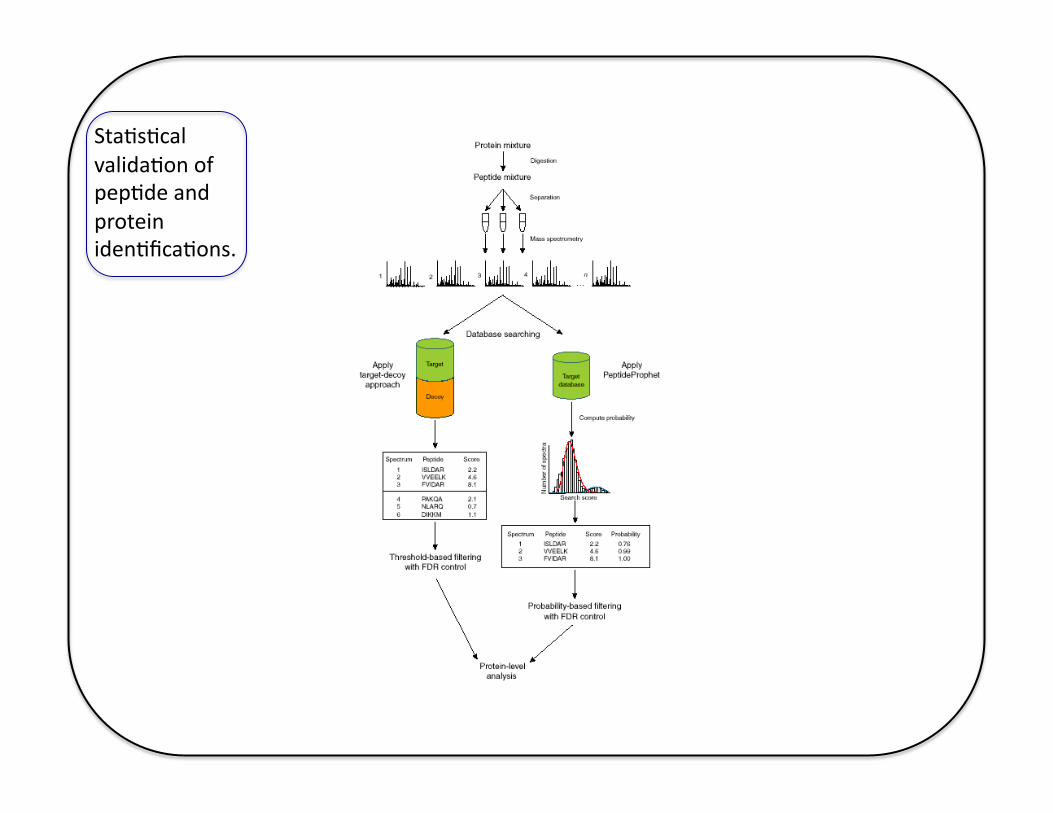
Sta5s5cal valida5on of pep5de and protein iden5fica5ons.

Scaffold
Sta5s5cal valida5on of pep5de and protein iden5fica5ons.
https://www.msi.umn.edu/sw/scaffold-for-pro
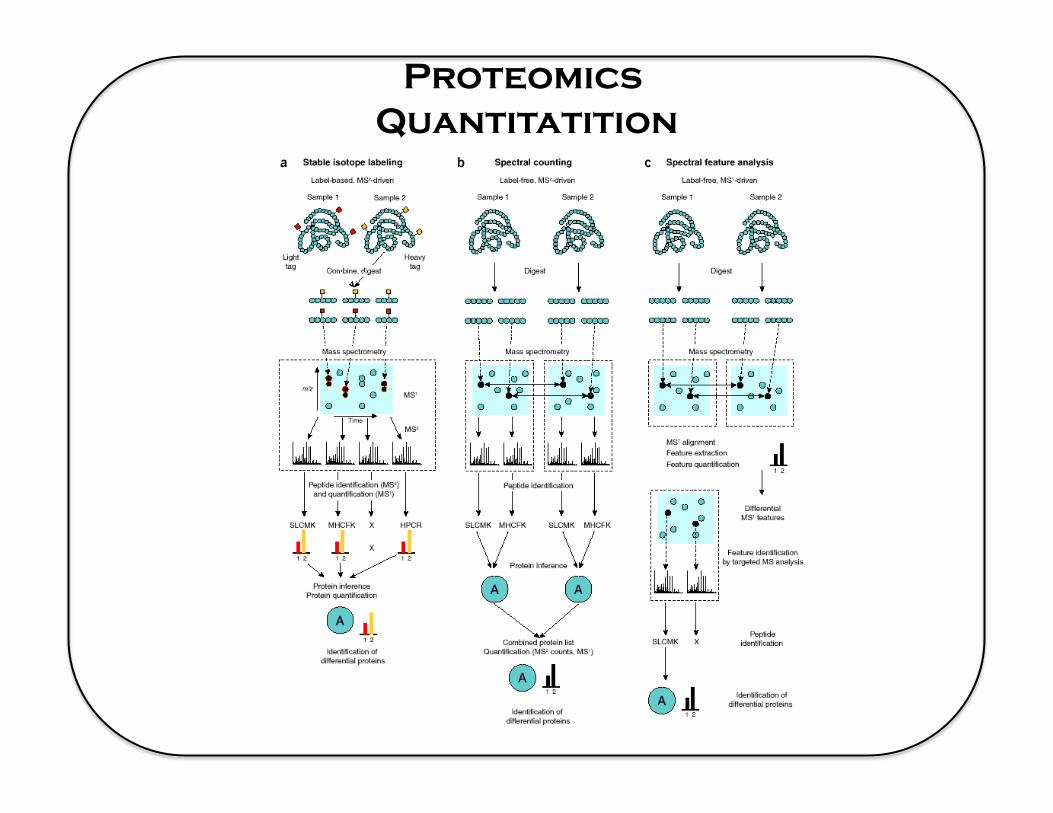
Proteomics Quantitatition
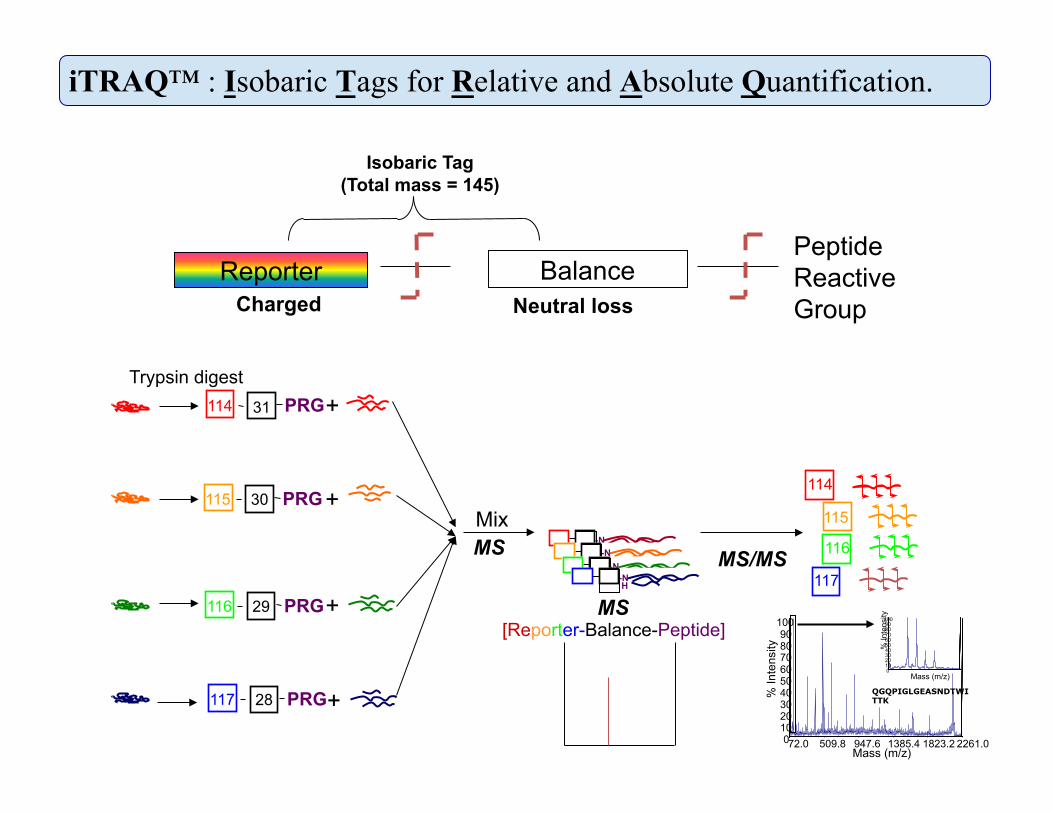
iTRAQ™ : Isobaric Tags for Relative and Absolute Quantification.
31 114 PRG +
+
+
+
30 115 PRG
29 116 PRG
28 117 PRG
Trypsin digest
[Reporter-Balance-Peptide] MS
-N H -N
H -N H -N
H
Mix MS MS/MS
117
116
115
114
Mass (m/z) 0 10 20 30 40 50 60 70 80 90 100
% In
tens
ity
72.0 509.8 947.6 1385.4 1823.2 2261.0 Mass (m/z)
0 10 20 30 40 50 60 70 80 90
100
% In
tens
ity
QGQPIGLGEASNDTWITTK
Charged Neutral loss
Isobaric Tag (Total mass = 145)
Reporter Balance Peptide Reactive Group
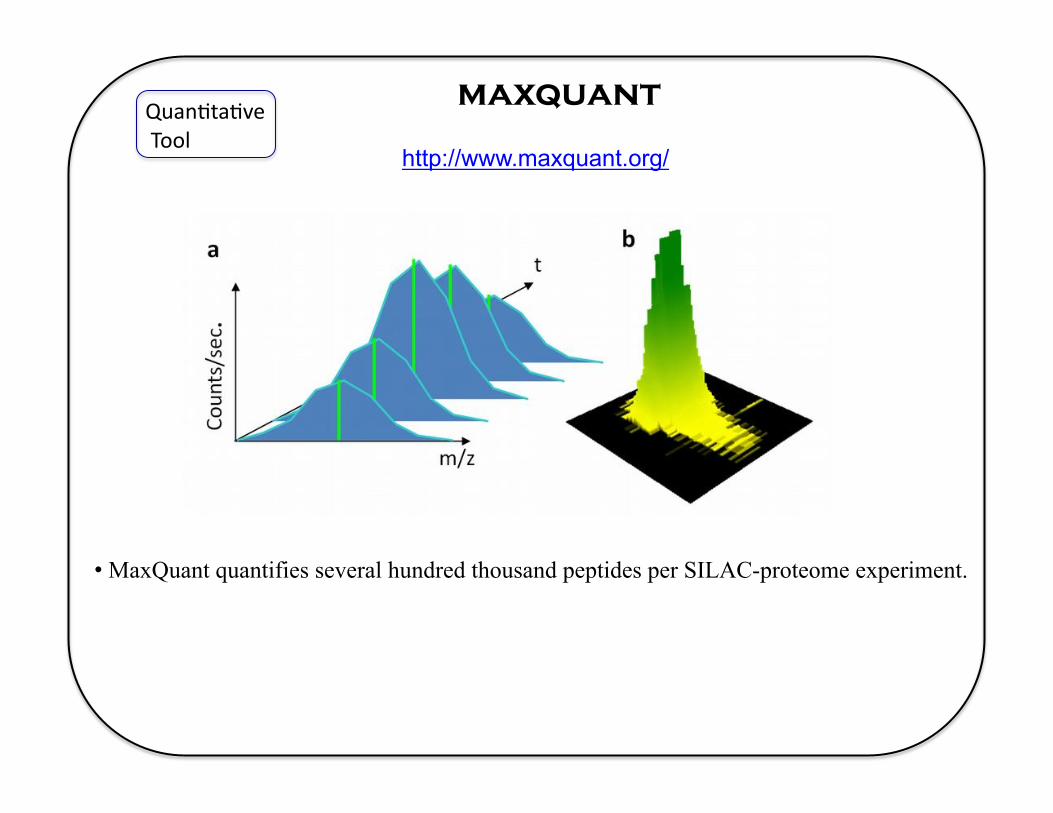
maxquant Quan5ta5ve Tool
http://www.maxquant.org/
• MaxQuant quantifies several hundred thousand peptides per SILAC-proteome experiment.
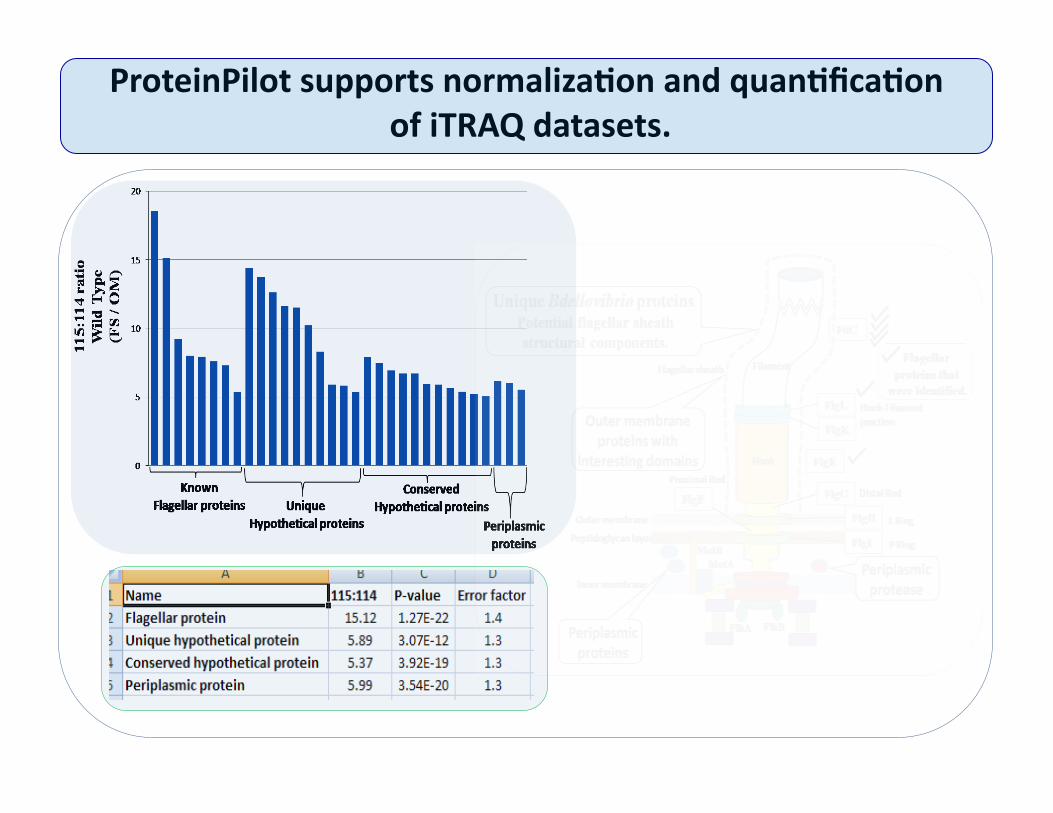
ProteinPilot supports normaliza@on and quan@fica@on of iTRAQ datasets.
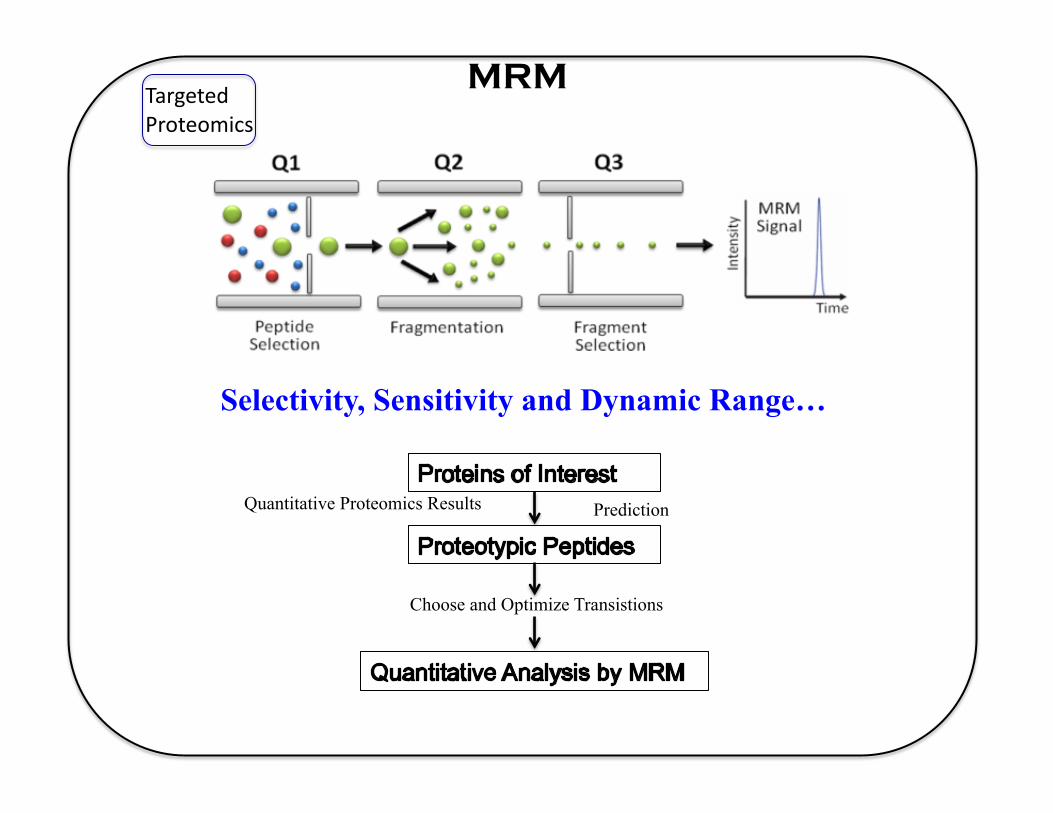
MRM Targeted Proteomics
Quantitative Proteomics Results Prediction
Choose and Optimize Transistions
Selectivity, Sensitivity and Dynamic Range…
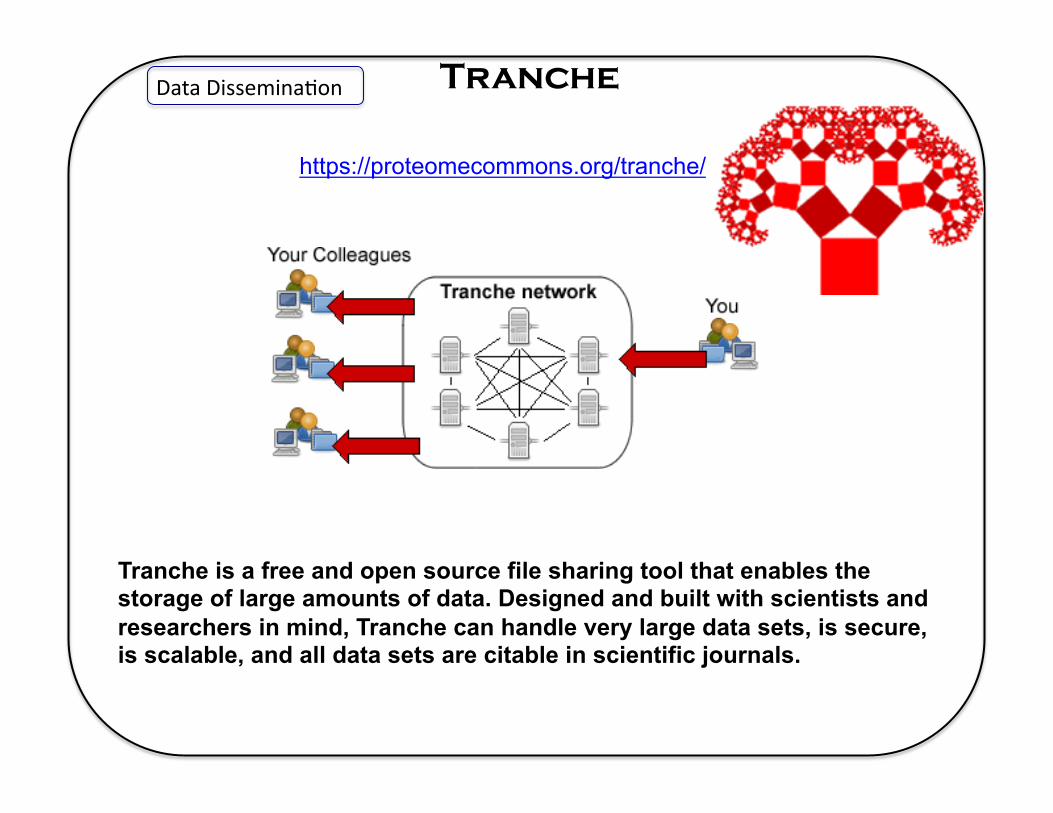
Tranche Data Dissemina5on
https://proteomecommons.org/tranche/
Tranche is a free and open source file sharing tool that enables the storage of large amounts of data. Designed and built with scientists and researchers in mind, Tranche can handle very large data sets, is secure, is scalable, and all data sets are citable in scientific journals.

LAST WORD…
Questions ? Pratik Jagtap
[email protected] http://twitter.com/pratikomics
Sequest X! tandem OMSSA MaxQuant ProteinPilot Mascot Scaffold PEAKS TPP Tranche Trans-proteomic Pipeline Pipeline ProTIP
http://sitemaker.umich.edu/iwsmoi