protein 4 from escherichia coli - journal of bacteriology - american
TRANSCRIPT

Vol. 174, No. 10
Deletion of an Additional Domain Located between SXXKand SXN Active-Site Fingerprints in Penicillin-Binding
Protein 4 from Escherichia coliHARALD MOTTL, PATRICK NIELAND, GIJS DE KORT, JOHN J. WIERENGA, AND WOLFGANG KECK*
Department of Biochemistry, BIOSON Research Institute, University of Groningen,Nijenborgh 4, 9747 AG Groningen, The Netherlands
Received 25 November 1991/Accepted 5 March 1992
It was suggested previously that the primary structure of penicillin-binding protein 4 (PBP4) is new andunique among proteins that interact with penicillin. Our proposal that PBP4 carries an additional domain,located between the active-site fingerprints SXXK and SXN, was investigated by mutational deletion analysis.A clustered set of internal deletions was created in this region by exonuclease treatment of the dacB codingDNA, starting from two internal restriction sites. PBP4 mutants carrying internal deletions were selected byscreening for immunoreactive forms of PBP4 with reduced molecular weight that were still active with respectto penicillin binding. DNA sequencing revealed 24 distinct PBP4 mutants with internal deletions ranging from37 to 113 amino acids. The amino- and carboxy-terminal end points of the deletions were not randomlydistributed but tended to cluster in certain areas. Overproduction of the individual mutated forms of PBP4resulted in accumulation of the major portion of the proteins in the particulate cell fraction. The yield of solubleand active mutated forms of the protein was reduced from below 1% to 79% of the level obtained for the nativeprotein. The deletions that were introduced had minor effects on the deacylation rate of bound benzylpenicillin.Two pairs of cysteine residues (Cys-139-Cys-153 and Cys-197-Cys-214) that are located in the deletable regionmay form disulfide bridges.
In a previous communication we reported the nucleotidesequence of the Escherichia coli dacB gene, which codes forpenicillin-binding protein 4 (PBP4). Homology studies withother proteins that interact with penitillin suggested thatPBP4 and P-lactamases of class A have an evolutionaryrelationship (16). Sequence comparisons suggested thatPBP4 contains a stretch of 188 amino acids located betweenthe SXXK and SXN fingerprints that is not present in classA ,-lactamases (19). This feature is unique among penicillin-interacting proteins. Examination of published three-dimen-sional structures of ,-lactamases revealed that the proposedinsertion point of the extra sequence lies in a very flexibleloop at the periphery of the model structure (7). The size andthe position of the additional sequence suggest that it mightbe folded as a structurally independent domain. This hypo-thetical domain organization of PBP4 might be proven byremoval of the proposed peripheral domain. The classicalstrategy for probing the domain organization of a proteinconsists of limited digestion with proteases and analysis ofthe functional properties of the products. However, thisstrategy cannot be applied to the domain organization ofPBP4, because the domain under study is situated within theprimary structure of the protein between active-site finger-prints and any digestion with proteases would inevitablydestroy the enzyme. Therefore, we decided to use geneticengineering techniques to create internal deletions by exo-nuclease treatment in the proposed region and to screen forPBP4 derivatives with reduced molecular weight but stillactive with respect to penicillin binding. In this paper wedescribe the construction, isolation, and characterization ofsuch mutants.
* Corresponding author.t Present address: Academic Hospital Leiden, 2300 RC Leiden,
The Netherlands.
MATERIALS AND METHODS
DNA manipulation. E. coli MC1061 was used as a host forthe subcloning and expression of protein (12). The host forM13 phages was JM101 (13). Site-directed mutagenesis wasperformed with strain BW313 (9). Enzymes were obtainedfrom Boehringer, Mannheim, except for Bal 31, which was
purchased from Promega. The enzymes were used accordingto the manufacturer's instructions.The mutagenesis and sequencing primers were synthe-
sized by Eurosequence, Groningen. The sequence of themutagenesis primer that was used for introducing the BamHIsite at bp 837 of the dacB gene was CGCTGACGGGATCCCTGCCAC. The mutated base (position 840) is underlined.Phage M13mpl8dacB was used in the site-directed mutagen-esis experiment (14), which was performed exactly as de-scribed by Kunkel et al. (9). Mutated M13 clones wereselected by BamHI restriction endonuclease digestion ofreplicative-form phage DNA. The mutation was transferredto the expression plasmid pROFI2AB by ligating a 450-bpHpaI-NcoI fragment from the mutated M13mpl8dacB intothe HpaI-NcoI sites of plasmid pROFI2AB (14).
Construction of deletion mutants. The CsCl gradient-puri-fied expression plasmid pROFI2AB was linearized in threedifferent experiments with restriction endonuclease NcoI orBamHI or a combination of both enzymes. The linearizedplasmids were then treated with the exonuclease Bal 31 tocreate nested sets of deletion mutants exactly as describedby Sambrook et al. (18). The Bal 31 reaction was stoppedafter 2, 5, 10, 20, and 30 min of incubation; 30 min was
sufficient to remove about 500 bp from each end. Thesamples were then pooled, and the protruding ends producedby Bal 31 were filled in with T4 DNA polymerase and theKlenow fragment of E. coli DNA polymerase I. The plas-mids were religated, cut again with NcoI or BamHI to enrich
3261
JOURNAL OF BACTERIOLOGY, May 1992, p. 3261-32690021-9193/92/103261-09$02.00/0Copyright C 1992, American Society for Microbiology
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
b on
22
Febr
uary
202
2 by
103
.87.
106.
102.

3262 MOTTL ET AL.
for clones carrying a deletion, and transformed into E. coliMC1061.
Detection of mutated forms of PBP4. Kanamycin-resistanttransformants were streaked out on fresh Luria broth (LB)-agar plates supplied with 50 Lg of kanamycin sulfate andgrown overnight at 30°C and for another 2 h at 42°C to inducethe expression of the dacB gene. The colonies were trans-ferred to nitrocellulose membranes and lysed as describedby Stanley with minor modifications (20). PBP4 was thenimmunochemically detected with a polyclonal antiserum (8).PBP4-overproducing clones were grown in 3 ml of LBmedium at 30°C overnight in a shaking water bath. Expres-sion of PBP4 was induced by incubating the cultures for 2 hat 42°C. A 3-ml culture was spun down in a microfuge andresuspended in 50 LI of ice-cold 0.2 M Tris-Cl (pH 8.0). Afterthe addition of 50 [lI of ice-cold 0.2 M Tris-Cl (pH 8.0)-i mMEDTA-1 M sucrose-120 ,ug of lysozyme per ml, 100 [LI ofice-cold water was added (23). The lysis of the cells wascompleted by incubating the reaction vessels for 10 min in asonication bath. After 30 min on ice, 10 pl of 1 M MgCl2 and1 ,ug of DNase were added and sonification was continuedfor another 10 min. Cell debris was spun down for 5 min at20,000 x g at 40C. The penicillin-binding proteins in 20 RI ofthe supernatant were radiolabeled by incubation with 2.5 [L1of [1251]amoxycillin for 30 min at 4°C. Sodium dodecylsulfate-polyacrylamide gel electrophoresis (SDS-PAGE)loading buffer (10 ,ul) was added, and then the mixture wasdenatured (3 min, 100°C) and separated by SDS-PAGE on a10% polyacrylamide gel. Penicillin-binding proteins weredetected by autoradiography (3 days at -80°C) with oneintensifying screen.
Penicillin-binding clones were identified by the appear-ance of extra bands with a molecular mass that was reducedrelative to that of native PBP4. The plasmid DNA wasisolated, and the DNA sequence of the dacB gene wasdetermined by using Sequenase (U.S. Biochemical) as rec-ommended by the manufacturer.
Expression studies. Expression of the mutated proteinswas studied by inoculating 500 ml of LB (supplemented with50 ,ug of kanamycin sulfate per ml, 10 mM MgSO4, and 0.5%glucose) with 9 ml of a fresh overnight culture of a deletionmutant plasmid in host strain MC1061 grown in the abovemedium at 300C in a shaking water bath. The 500-ml cultureswere grown under aeration at 30°C until they reached anoptical density at 600 nm of 0.4. The cultures were thentransferred to a 39 or 42°C water bath. After 90 min, thecultures were cooled on ice and the cells were collected bycentrifugation (15 min, 10,000 x g, 4°C). The cell pellet wasresuspended homogeneously in 7.5 ml of ice-cold 0.2 MTris-Cl (pH 8.0)-10 mM MgCl2-1 ,ug of DNase per ml (theDNase was occasionally substituted by 5 U of Benzonase[Merck]). After the addition of 7.5 ml of ice-cold 0.2 MTris-Cl (pH 8.0)-12 mM EDTA-1 M sucrose-120 ,ug oflysozyme per ml in 15 ml of water, 300 ,ul of 1 M MgCl2 wasadded and the suspension was incubated for 10 min on ice.Finally, phenylmethylsulfonyl fluoride in ethanol was addedto a final concentration of 40 ,ug/ml. Lysis was completed byultrasonication (two times for 1 min, 400-W output, Sonicsand Materials sonicator). The suspension was separated intosoluble and particulate fractions by ultracentrifugation (60min, 150,000 x g, 4°C). A low-speed centrifugation step toseparate cell debris was found to be unnecessary; completelysis was achieved with this protocol. The protein concen-tration was determined by the biuret method (2).
Disulfide bonds. Free sulfide functions in PBP4 weredetermined with Ellman reagent [3 mM solution of 5,5'-
dithiobis(2-nitrobenzoic acid)] exactly as described previ-ously (17). Purified PBP4 (994 ,ug in 64.4 RI) corresponding to20 nmol was diluted in 1 ml of 6.4 M guanidinium chloride in0.1 M sodium phosphate buffer (pH 7.3). After 50 ,ul ofEllman reagent was added, the increase of A412 was mea-sured and corrected for the background A412 of the reactionmixture. Oxygen was removed from the solutions by sparg-ing with helium, and the experiment was performed at 25°C.A molar extinction coefficient (E) of 14,000 M-1 cm-' wasused to calculate free sulfide functions. Disulfide bridges inPBP4 and its mutated derivatives were detected by compar-ing the relative mobilities of reduced and nonreduced pro-teins on SDS-PAGE (1, 5).
Typically, 20 pl1 of protein solution was mixed with 5 ,ul ofSDS-PAGE loading buffer and denatured for 3 min a 100°C.For reducing or nonreducing conditions, loading buffer wasused with or without 1.7 mM mercaptoethanol, respectively.The samples were separated by SDS-PAGE, and the pro-teins were electrotransferred onto nitrocellulose membranes(BA85; Schleicher & Schull) and stained immunochemicallyfor PBP4. Pure PBP4 was used as a marker. In a similarexperiment, Cibacron Navyblue 2GE-modified TSK-Fracto-gel HW65-F was used for the enrichment of mutated PBP4proteins, which were treated directly with reducing andnonreducing loading buffers (15). With this method, theelectrotransfer and the immunochemical staining could beomitted and the PBP4 mutant proteins could be stained withCoomassie brilliant blue after separation by SDS-PAGE.
Screening of mutant proteins for affinity to Navyblue 2GE.Dye-substituted Fractogel (25-pl settled bed volume) wasequilibrated with 50 mM glycine-NaOH (pH 10.5) and incu-bated (10 min, 4°C) with 100 pL1 of the soluble fraction (1mg/ml) from the respective mutant. The supernatant wasremoved, and the resin was washed two times with 1 ml ofequilibration buffer. The bound protein was eluted by theaddition of 50 RI of 50% SDS-PAGE loading buffer and thendenatured for 3 min at 100°C. Analysis of 25 pA of the eluatewas done by SDS-PAGE (15).
Interaction with benzylpenicillin. Active-site titration ofsoluble extracts with [14C]benzylpenicillin was performed asdescribed previously (14). The half-life values of the PBP4-penicillin complexes were measured as follows. Solubleextracts (1,200 plA) were labeled with 12 nmol of [14C]ben-zylpenicillin (26 kBq) for 15 min at 37°C and chased with1,200 nmol of unlabeled benzylpenicillin (10 Rl). The solu-tions were incubated at 37°C, 100-pi samples were removedin duplicate every 40 min and precipitated as describedpreviously (14), and the remaining radioactivity was deter-mined. The half-life of the PBP4-penicillin complex wascalculated from a first-order decay function.SDS-PAGE and fluorography. Discontinuous SDS-PAGE
was performed as described by Laemmli (10). The separatinggel contained 10% T and 2.7% C, and the stacking gelcontained 4% T and 2.7% C. Gels were 100 by 100 by 0.5 mmor 160 by 130 by 1 mm in size. Penicillin-binding proteinswere labeled in 20-pA fractions with various amounts of[35S]benzylpenicillin as indicated in the figure legends. Afterincubation for 10 min at 37°C, the reaction was stopped byboiling the sample for 3 min in SDS sample buffer. SDS-PAGE was performed, and the gel was treated with sodiumsalicylate (3). Labeled penicillin-protein complexes weredetected by fluorography with Kodak X-Omat AR film at-800C.Radiolabeled compounds. [35S]benzylpenicillin (0.41 TBq/
mmol) was obtained from New England Nuclear. Theamounts necessary for labeling the PBPs were dried under
J. BACTERIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
b on
22
Febr
uary
202
2 by
103
.87.
106.
102.

PROBING THE DOMAIN ORGANIZATION OF PBP4 3263
TABLE 1. Sequence data of mutants with deletions in PBP4'
Amt of activeDeletion No. of Position of Class (with PBP4 produced Double bands in Deacylation rate ofclone residues deletion respect to (pmol/mg) at: penicillin-binding bound benzylpenicillindeleted disulfide bonds) assay at 42°C (10-3/min)
39°C 42°C
pORAF2 57 116-172 I <10 10 + NDpORAF3 37 124-160 I 30 10 - NDpORAF4 71 124-194 ISS 350 80 + 4.6pORAF5 96 124-219 I 260 40 + 5.4pORAF6 103 124-226 II 60 30 + NDpORAF7 108 124-231 II <10 <10 + NDpORAF8 58 125-182 ISS 20 120 - NDpORAF9 96 126-220 II 20 20 + NDpORAF10 98 125-222 II 30 60 + NDpORAF11 95 126-220 II 50 40 + NDpORAF12 98 126-223 II 10 30 + NDpORAF13 104 126-229 II 80 340 - NDpORAF14 113 126-238 II <10 60 + NDpORAF15 103 127-229 II 360 20 + 5.5pORAF16 33 129-161 I 100 30 - NDpORAF17 91 129-219 II 270 50 + 5.1pORAF18 103 129-231 II 42 370 - 4.9pORAF19 44 130-173 I 90 60 - NDpORAF20 62 130-191 ISS 790 240 + 5.8pORAF21 90 130-219 II 420 100 + 5.5pORAF22 89 130-218 II 480 210 + 1.7pORAF23 92 130-221 II 140 90 - 2.6pORAF24 89 131-219 II 480 240 + 5.5pORAF25 87 133-219 II 760 270 + 6.0C214-*S 650 210 + 3.2pROFI2AB ND 1,030 + 5.6
a In this table we use the numbering of amino acids of PBP4 published earlier (16). The C214-.S site-directed mutant is described in Materials and Methods.The wild-type expression plasmid pROFI2AB was reported previously (14). Start and end points of the deletions refer to the actually deleted amino acids. Theamino acid sequences of deletion mutants at the fusion points remained unchanged with three exceptions (pORAF3, Ala-123 changed to Ser; pORAF13, His-125changed to Gin; pORAF15, Asp-126 changed to Glu). The classification of the mutants into classes I, ISS, and II refers to the number of pairs of cysteine residueswhich have been deleted in the respective mutant. In case I mutants, Cys-139-Cys-153 is deleted and no more disulfide bridge is formed. In class ISS mutants,the Cys-139-Cys-153 pair is deleted but Cys-197-Cys-214 still forms a disulfide bridge. In class II mutants, both pairs of cysteines are deleted. The occurrenceof double bands in the penicillin-binding assay was judged on the basis of the fluorographs shown in Fig. 4. Exposure times shorter than those used in Fig. 4 werealso used to identify the clones. Only clones that showed double bands with approximately the same intensity were recorded. The amount of benzylpenicillinbound per milligrams of soluble protein was determined in soluble fractions prepared after induction of expression for 90 min at 39 and 42°C as described inMaterials and Methods. The deacylation rate of bound benzylpenicillin was determined as described in detail in Materials and Methods. Due to the low expressionof some of the mutant enzymes, this parameter could not be determined with sufficient precision for all deletion mutants. ND, not determined.
vacuum, and the protein solutions were added to the drysamples. [14Clbenzylpenicillin was obtained from Amershamat a specific activity of 2.16 GBq/mmol. [1251]amoxicillin wasprepared exactly as described previously (11). Briefly, 33 ,ulof 0.1 M sodium phosphate buffer (pH 7.5) containing 20 ,ugof amoxycillin was mixed with 2 RI of 125INa (100 mCi/ml;Amersham) and 10 RI1 of chloramine T (2 mg/ml). After 1 minof incubation at room temperature, 5 ,ul of cold INa (2mg/ml) was added, and the reaction was continued for 40 minat room temperature. The remaining iodine was then re-duced by the addition of 10 ,ul of sodium metabisulfite (2mg/ml). All solutions were made in the above-mentionedphosphate buffer. The reaction mixture was used withoutfurther purification for the labeling of penicillin-bindingproteins.
RESULTSConstruction of deletion mutants. The methodology de-
scribed above proved to be efficient for the preparation andscreening of internal deletion mutants of PBP4. The experi-ment was first performed by creating internal deletionsstarting from the NcoI site at amino acid position 132. Theyield of clones producing immunologically reactive PBP4upon induction of expression was 23 out of 486 kanamycin-
resistant transformants, which is equivalent to 4.7%. Thisfigure remained astonishingly constant when deletions weremade starting either from the BamHI site, which had beencreated at position 214 by site-directed mutagenesis, or fromboth the BamHI and NcoI sites. All together, 98 positiveclones emerged from the colony immunoblotting experi-ments; 44 of these clones showed a PBP4 with reducedmolecular weight but intact penicillin-binding activity. Sincesome clones occurred several times, as determined by DNAsequencing, the actual number of distinct clones was re-duced to 24, all of which maintained penicillin-bindingactivity.
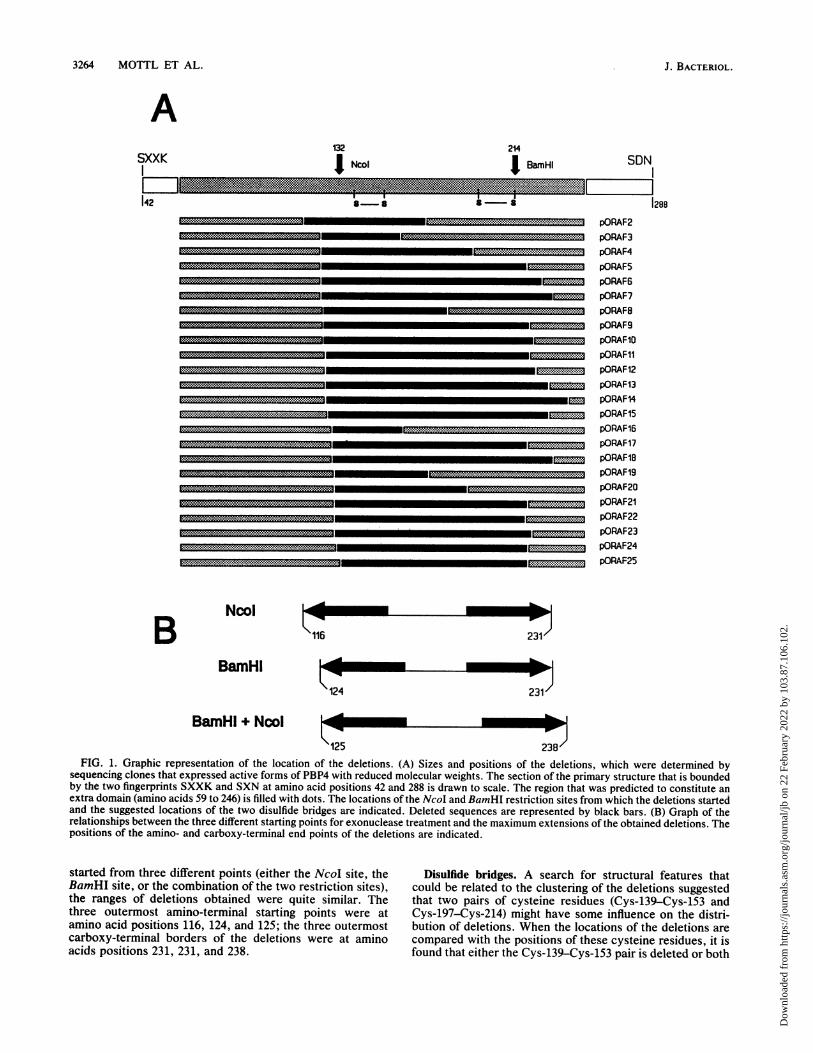
Distribution of deletions within the PBP4 primary sequence.The amino- and carboxy-terminal borders of the deletionsintroduced are summarized in Table 1 and Fig. 1A. It isremarkable that, with only one exception that starts at aminoacid 116, all deletions start within the same region of 9 aminoacids between positions 124 and 133. The carboxy-terminalborders of the deletions show a greater variability, but againin a majority of 16 from 24 deletion clones the end pointstend to cluster in a region of 13 amino acids betweenpositions 218 and 231. The sizes of the deletions introducedare actually independent of the starting point of the exonu-clease treatment (Fig. 1B). Although exonuclease action
VOL. 174, 1992
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
b on
22
Febr
uary
202
2 by
103
.87.
106.
102.
3264 MOTTL ET AL.
A214
I BanHI
...... .........*- - i
ve.4eI I~~~~-~~~~-
=_I =
231
BamHI
BamHI + NCoI125
124 231
238FIG. 1. Graphic representation of the location of the deletions. (A) Sizes and positions of the deletions, which were determined by
sequencing clones that expressed active forms of PBP4 with reduced molecular weights. The section of the primary structure that is boundedby the two fingerprints SXXK and SXN at amino acid positions 42 and 288 is drawn to scale. The region that was predicted to constitute anextra domain (amino acids 59 to 246) is filled with dots. The locations of the NcoI and BamHI restriction sites from which the deletions startedand the suggested locations of the two disulfide bridges are indicated. Deleted sequences are represented by black bars. (B) Graph of therelationships between the three different starting points for exonuclease treatment and the maximum extensions of the obtained deletions. Thepositions of the amino- and carboxy-terminal end points of the deletions are indicated.
started from three different points (either the NcoI site, theBamHI site, or the combination of the two restriction sites),the ranges of deletions obtained were quite similar. Thethree outermost amino-terminal starting points were atamino acid positions 116, 124, and 125; the three outermostcarboxy-terminal borders of the deletions were at aminoacids positions 231, 231, and 238.
Disulfide bridges. A search for structural features thatcould be related to the clustering of the deletions suggestedthat two pairs of cysteine residues (Cys-139-Cys-153 andCys-197-Cys-214) might have some influence on the distri-bution of deletions. When the locations of the deletions arecompared with the positions of these cysteine residues, it isfound that either the Cys-139-Cys-153 pair is deleted or both
2
132
I NcolSXXK
['4
B
SDN
1288pORAF2pORAF3pORAF4pORAF5pORAF6pORAF7pORAFSpORAF9pORAFIOpORAFIIpORAF12pORAF13pORAF14pORAF15pORAF16pORAFi7pORAFIBpORAF19pORAF20pORAF21pORAF22pORAF23pORAF24pORAF25
NCoI116
m
J. BACTERIOL.
I
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
b on
22
Febr
uary
202
2 by
103
.87.
106.
102.
PROBING THE DOMAIN ORGANIZATION OF PBP4 3265
_ _+--4- +
20 8 C
6 5. . . . . .
OC B S 1 15 20 25
B+ -+--I
C 4 20
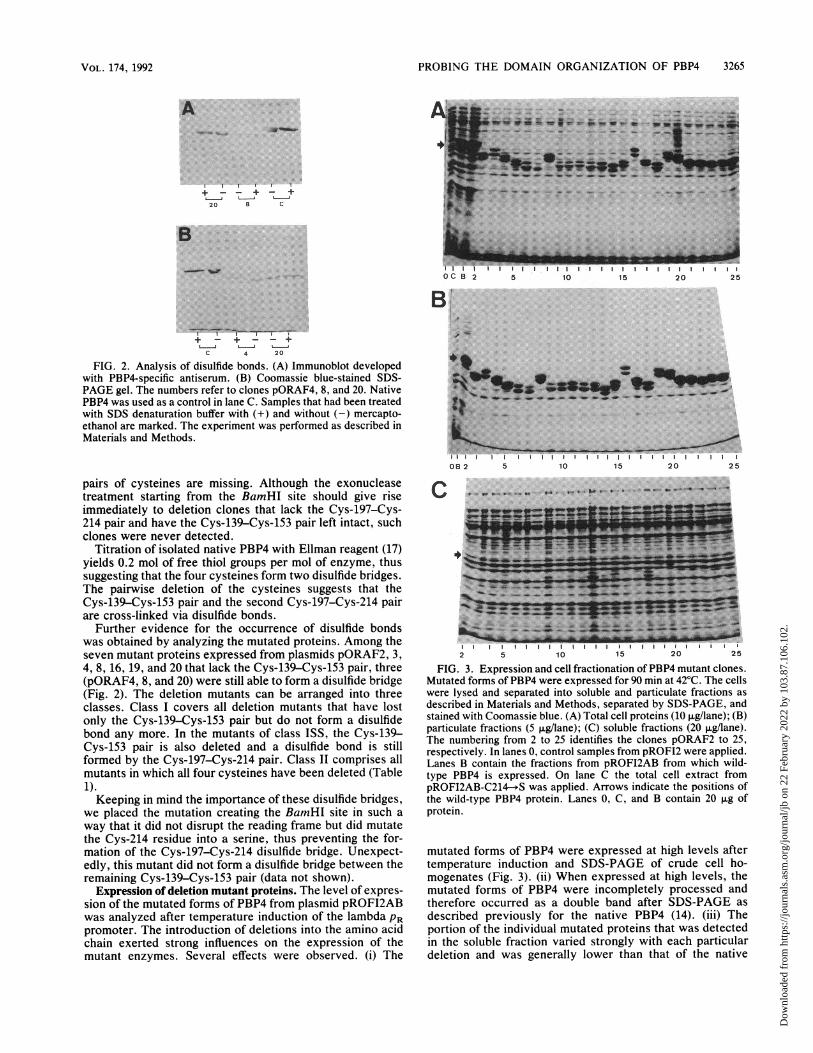
FIG. 2. Analysis of disulfide bonds. (A) Immunoblot developedwith PBP4-specific antiserum. (B) Coomassie blue-stained SDS-PAGE gel. The numbers refer to clones pORAF4, 8, and 20. NativePBP4 was used as a control in lane C. Samples that had been treatedwith SDS denaturation buffer with (+) and without (-) mercapto-ethanol are marked. The experiment was performed as described inMaterials and Methods.
pairs of cysteines are missing. Although the exonucleasetreatment starting from the BamHI site should give riseimmediately to deletion clones that lack the Cys-197-Cys-214 pair and have the Cys-139-Cys-153 pair left intact, suchclones were never detected.
Titration of isolated native PBP4 with Ellman reagent (17)yields 0.2 mol of free thiol groups per mol of enzyme, thussuggesting that the four cysteines form two disulfide bridges.The pairwise deletion of the cysteines suggests that theCys-139-Cys-153 pair and the second Cys-197-Cys-214 pairare cross-linked via disulfide bonds.
Further evidence for the occurrence of disulfide bondswas obtained by analyzing the mutated proteins. Among theseven mutant proteins expressed from plasmids pORAF2, 3,4, 8, 16, 19, and 20 that lack the Cys-139-Cys-153 pair, three(pORAF4, 8, and 20) were still able to form a disulfide bridge(Fig. 2). The deletion mutants can be arranged into threeclasses. Class I covers all deletion mutants that have lostonly the Cys-139-Cys-153 pair but do not form a disulfidebond any more. In the mutants of class ISS, the Cys-139-Cys-153 pair is also deleted and a disulfide bond is stillformed by the Cys-197-Cys-214 pair. Class II comprises allmutants in which all four cysteines have been deleted (Table1).Keeping in mind the importance of these disulfide bridges,
we placed the mutation creating the BamHI site in such away that it did not disrupt the reading frame but did mutatethe Cys-214 residue into a serine, thus preventing the for-mation of the Cys-197-Cys-214 disulfide bridge. Unexpect-edly, this mutant did not form a disulfide bridge between theremaining Cys-139-Cys-153 pair (data not shown).
Expression of deletion mutant proteins. The level of expres-sion of the mutated forms of PBP4 from plasmid pROFI2ABwas analyzed after temperature induction of the lambda PRpromoter. The introduction of deletions into the amino acidchain exerted strong influences on the expression of themutant enzymes. Several effects were observed. (i) The
OB 2 5 10 15 20 25
2 5 10 15 20 25
FIG. 3. Expression and cell fractionation of PBP4 mutant clones.Mutated forms of PBP4 were expressed for 90 min at 420C. The cellswere lysed and separated into soluble and particulate fractions asdescribed in Materials and Methods, separated by SDS-PAGE, andstained with Coomassie blue. (A) Total cell proteins (10 p.g/lane); (B)particulate fractions (5 p.g/lane); (C) soluble fractions (20 ~.g/lane).The numbering from 2 to 25 identifies the clones pORAF2 to 25,respectively. In lanes 0, control samples from pROFL2 were applied.Lanes B contain the fractions from pROFI2AB from which wild-type PBP4 is expressed. On lane C the total cell extract frompROF12AB-C214---*S was applied. Arrows indicate the positions ofthe wild-type PBP4 protein. Lanes 0, C, and B contain 20 p.g ofprotein.
mutated forms of PBP4 were expressed at high levels aftertemperature induction and SDS-PAGE of crude cell ho-mogenates (Fig. 3). (ii) When expressed at high levels, themutated forms of PBP4 were incompletely processed andtherefore occurred as a double band after SDS-PAGE 'asdescribed previously for the native PBP4 (14). (iii) Theportion of the individual mutated proteins that was detectedin the soluble fraction varied strongly with each particulardeletion and was generally lower than that of the native
VOL. 174, 1992
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
b on
22
Febr
uary
202
2 by
103
.87.
106.
102.
3266 MOTTL ET AL.
__ _ * | D N s | | | | a~~~~~~~~~~~~~~~,7
B 2 5 10 15 20 25 C
B_
B 2 5 10 15 20 25 C
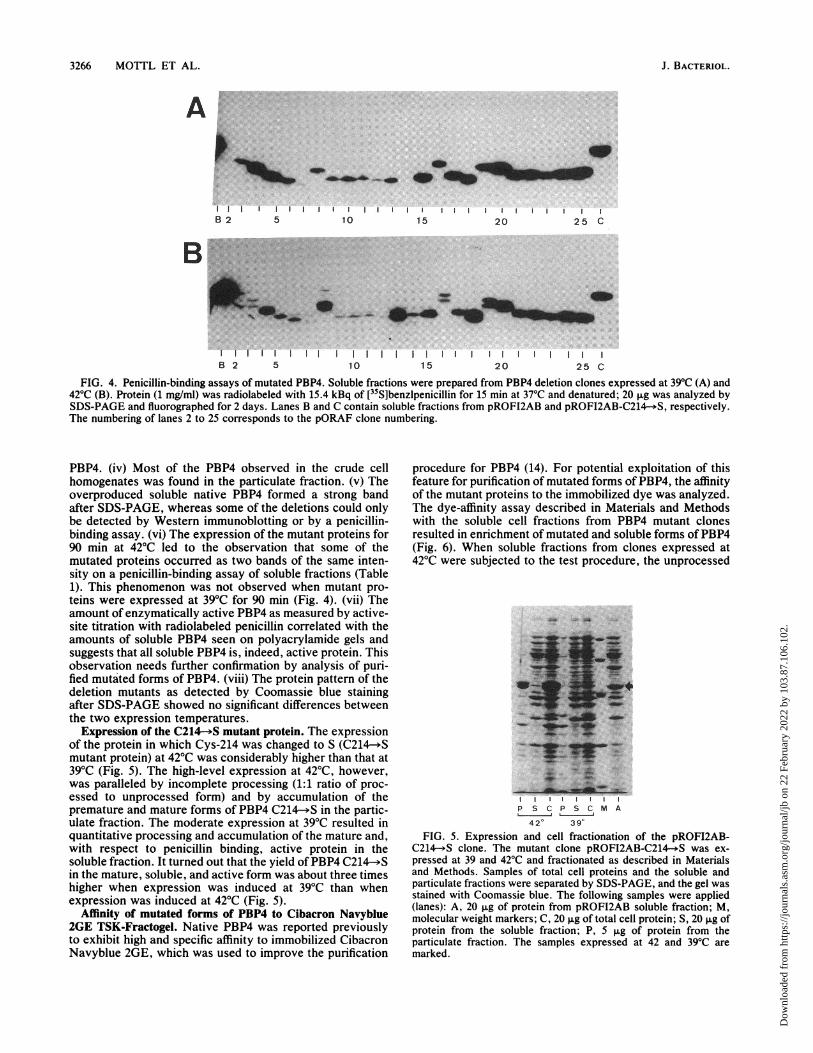
FIG. 4. Penicillin-binding assays of mutated PBP4. Soluble fractions were prepared from PBP4 deletion clones expressed at 39'C (A) and42'C (B). Protein (1 mg/ml) was radiolabeled with 15.4 kBq of [35S]benzlpenicillin for 15 min at 37'C and denatured; 20 ,ug was analyzed bySDS-PAGE and fluorographed for 2 days. Lanes B and C contain soluble fractions from pROFI2AB and pROFI2AB-C214-.S, respectively.The numbering of lanes 2 to 25 corresponds to the pORAF clone numbering.
PBP4. (iv) Most of the PBP4 observed in the crude cellhomogenates was found in the particulate fraction. (v) Theoverproduced soluble native PBP4 formed a strong bandafter SDS-PAGE, whereas some of the deletions could onlybe detected by Western immunoblotting or by a penicillin-binding assay. (vi) The expression of the mutant proteins for90 min at 42°C led to the observation that some of themutated proteins occurred as two bands of the same inten-sity on a penicillin-binding assay of soluble fractions (Table1). This phenomenon was not observed when mutant pro-teins were expressed at 39°C for 90 min (Fig. 4). (vii) Theamount of enzymatically active PBP4 as measured by active-site titration with radiolabeled penicillin correlated with theamounts of soluble PBP4 seen on polyacrylamide gels andsuggests that all soluble PBP4 is, indeed, active protein. Thisobservation needs further confirmation by analysis of puri-fied mutated forms of PBP4. (viii) The protein pattern of thedeletion mutants as detected by Coomassie blue stainingafter SDS-PAGE showed no significant differences betweenthe two expression temperatures.
Expression of the C214-+S mutant protein. The expressionof the protein in which Cys-214 was changed to S (C214--Smutant protein) at 42°C was considerably higher than that at39°C (Fig. 5). The high-level expression at 42°C, however,was paralleled by incomplete processing (1:1 ratio of proc-essed to unprocessed form) and by accumulation of thepremature and mature forms of PBP4 C214-*S in the partic-ulate fraction. The moderate expression at 39°C resulted inquantitative processing and accumulation of the mature and,with respect to penicillin binding, active protein in thesoluble fraction. It turned out that the yield of PBP4 C214->Sin the mature, soluble, and active form was about three timeshigher when expression was induced at 39°C than whenexpression was induced at 42°C (Fig. 5).
Affinity of mutated forms of PBP4 to Cibacron Navyblue2GE TSK-Fractogel. Native PBP4 was reported previouslyto exhibit high and specific affinity to immobilized CibacronNavyblue 2GE, which was used to improve the purification
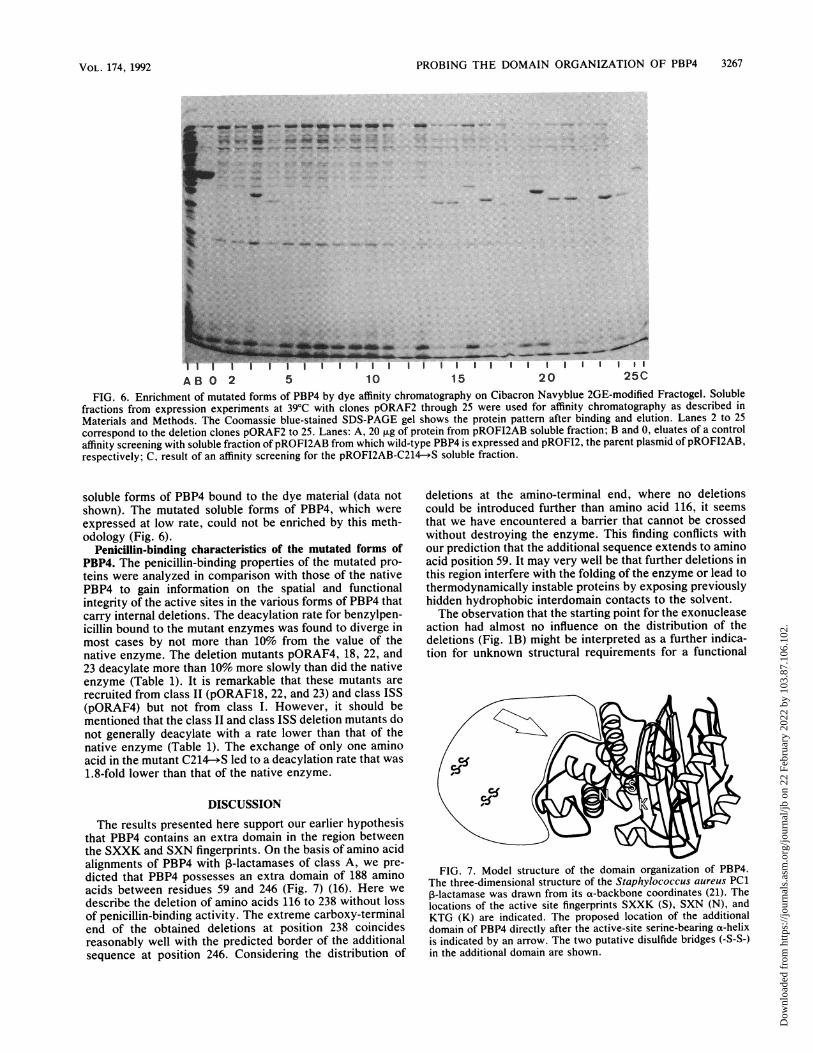
procedure for PBP4 (14). For potential exploitation of thisfeature for purification of mutated forms of PBP4, the affinityof the mutant proteins to the immobilized dye was analyzed.The dye-affinity assay described in Materials and Methodswith the soluble cell fractions from PBP4 mutant clonesresulted in enrichment of mutated and soluble forms ofPBP4(Fig. 6). When soluble fractions from clones expressed at42°C were subjected to the test procedure, the unprocessed
P S C P S C M A
420 390FIG. 5. Expression and cell fractionation of the pROFI2AB-
C214,-S clone. The mutant clone pROFI2AB-C214-*S was ex-pressed at 39 and 42'C and fractionated as described in Materialsand Methods. Samples of total cell proteins and the soluble andparticulate fractions were separated by SDS-PAGE, and the gel wasstained with Coomassie blue. The following samples were applied(lanes): A, 20 ,ug of protein from pROFI2AB soluble fraction; M,molecular weight markers; C, 20 ,ug of total cell protein; S, 20 ,ug ofprotein from the soluble fraction; P, 5 ,ug of protein from theparticulate fraction. The samples expressed at 42 and 39'C aremarked.
J. BACTERIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
b on
22
Febr
uary
202
2 by
103
.87.
106.
102.
PROBING THE DOMAIN ORGANIZATION OF PBP4 3267
AB 0 2 5 10 15 20 25C
FIG. 6. Enrichment of mutated forms of PBP4 by dye affinity chromatography on Cibacron Navyblue 2GE-modified Fractogel. Solublefractions from expression experiments at 39°C with clones pORAF2 through 25 were used for affinity chromatography as described inMaterials and Methods. The Coomassie blue-stained SDS-PAGE gel shows the protein pattern after binding and elution. Lanes 2 to 25correspond to the deletion clones pORAF2 to 25. Lanes: A, 20 ,ug of protein from pROFI2AB soluble fraction; B and 0, eluates of a controlaffinity screening with soluble fraction of pROFI2AB from which wild-type PBP4 is expressed and pROFI2, the parent plasmid of pROFI2AB,respectively; C, result of an affinity screening for the pROFI2AB-C214- S soluble fraction.
soluble forms of PBP4 bound to the dye material (data notshown). The mutated soluble forms of PBP4, which wereexpressed at low rate, could not be enriched by this meth-odology (Fig. 6).
Penicillin-binding characteristics of the mutated forms ofPBP4. The penicillin-binding properties of the mutated pro-teins were analyzed in comparison with those of the nativePBP4 to gain information on the spatial and functionalintegrity of the active sites in the various forms of PBP4 thatcarry internal deletions. The deacylation rate for benzylpen-icillin bound to the mutant enzymes was found to diverge inmost cases by not more than 10% from the value of thenative enzyme. The deletion mutants pORAF4, 18, 22, and23 deacylate more than 10% more slowly than did the nativeenzyme (Table 1). It is remarkable that these mutants arerecruited from class II (pORAF18, 22, and 23) and class ISS(pORAF4) but not from class I. However, it should bementioned that the class II and class ISS deletion mutants donot generally deacylate with a rate lower than that of thenative enzyme (Table 1). The exchange of only one aminoacid in the mutant C214-*S led to a deacylation rate that was1.8-fold lower than that of the native enzyme.
DISCUSSIONThe results presented here support our earlier hypothesis
that PBP4 contains an extra domain in the region betweenthe SXXK and SXN fingerprints. On the basis of amino acidalignments of PBP4 with ,B-lactamases of class A, we pre-dicted that PBP4 possesses an extra domain of 188 aminoacids between residues 59 and 246 (Fig. 7) (16). Here wedescribe the deletion of amino acids 116 to 238 without lossof penicillin-binding activity. The extreme carboxy-terminalend of the obtained deletions at position 238 coincidesreasonably well with the predicted border of the additionalsequence at position 246. Considering the distribution of
deletions at the amino-terminal end, where no deletionscould be introduced further than amino acid 116, it seemsthat we have encountered a barrier that cannot be crossedwithout destroying the enzyme. This finding conflicts withour prediction that the additional sequence extends to aminoacid position 59. It may very well be that further deletions inthis region interfere with the folding of the enzyme or lead tothermodynamically instable proteins by exposing previouslyhidden hydrophobic interdomain contacts to the solvent.The observation that the starting point for the exonuclease
action had almost no influence on the distribution of thedeletions (Fig. 1B) might be interpreted as a further indica-tion for unknown structural requirements for a functional
FIG. 7. Model structure of the domain organization of PBP4.The three-dimensional structure of the Staphylococcus aureus PC1P-lactamase was drawn from its a-backbone coordinates (21). Thelocations of the active site fingerprints SXXK (S), SXN (N), andKTG (K) are indicated. The proposed location of the additionaldomain of PBP4 directly after the active-site serine-bearing a-helixis indicated by an arrow. The two putative disulfide bridges (-S-S-)in the additional domain are shown.
VOL. 174, 1992
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
b on
22
Febr
uary
202
2 by
103
.87.
106.
102.

3268 MOTTL ET AL.
PBP4 enzyme that do not tolerate deletions beyond position116 toward the SXXN fingerprint. The distribution of thedeletions is not affected by structural characteristics of theDNA or by the specificity of the exonuclease used. DNAsequencing of some randomly picked primary clones clearlyindicated that the deletions on the DNA level are randomlydistributed and do not show the restriction that is found inthe expressed mutated proteins.To our knowledge this is the first study that reports on the
successful creation of mutants with such large internaldeletions between two active-site fingerprints of an enzymewithout destroying its activity. In a previous publication wedescribed the temperature dependence for the expressionlevel of native PBP4. Expression at 42°C yielded dispropor-tionaly more active PBP4 than did expression at 40 or 37°C.In addition we reported the occurrence of an unprocessedprecursor of PBP4 in the particulate fraction exclusivelyafter induction at 42°C (14). After induction at 42°C, themutated forms of PBP4 show a diverse expression that doesnot follow obvious rules but illustrates the complex nature ofthe forces that govern protein expression and folding pro-cesses.The contribution of the two disulfide bridges to the struc-
tural integrity of the extra domain of PBP4 is stressed by ourresults. The great impact of the C214--S mutation, which islocated within the additional domain region, on the deacy-lation rate (1.8-fold slower than that of wild-type protein) isdifficult to understand, especially when it is compared withthe impact of mutated forms of the protein that carrydeletions of more than 100 amino acids within this domain. Itseems that the core structure of PBP4 that carries thepenicillin-binding domain is a rather adaptable structure,since it can tolerate such gross changes. The deletableportion of the structure, however, seems to have someinfluence at least on the deacylation rate of the penicillin-protein complex, since a single amino acid exchangeC214-*S in this region led to a 1.8-fold lower deacylationrate.The deletion mutants offer new ways to study the physi-
ological function of PBP4 and especially to investigate thefunction of the deleted part of the primary structure. Wehave demonstrated that the purification method developedfor the wild-type enzyme is also useful for the purification ofmutant enzymes. This will allow a more thorough character-ization of the enzymological properties of the mutated pro-teins. The fact that the mutated forms of PBP4 still interactwith the Cibacron dye furthermore indicates that the bindingsite for the dye is not situated in the deleted portion of theamino acid sequence. It further provides evidence that,besides the penicillin-binding site, a second site is kept intactand suggests that the scaffolding of these mutant proteins iscorrect.Then et al. showed recently that boronic acids, which are
specific competitive inhibitors for P-lactamases, also com-pete with the binding of P-lactams to PBP4 but do notinteract with the other penicillin-binding proteins of E. coli(21). Our proposal that PBP4 is related to the ,-lactamases ofclass A, which was purely based on sequence alignments, issupported by these characteristics. The data of Then et al.also suggest conformational differences between the activesite of PBP4 and those of other E. coli penicillin-bindingproteins.The physiological relevance of the unique domain organi-
zation of PBP4 has not been elucidated. However, when weconsider the proposed location of the extra domain directlyafter the active-site serine-bearing a-helix and in the vicinity
of the SXN fingerprint, we might speculate about a regula-tory function of the extra domain with regard to the autolyticDD-endopeptidase activity ofPBP4 (Fig. 7). This speculationis supported by results from Hakenbeck and Messer, whofound that the endopeptidase activity in synchronized E. colicells increased in a nonlinear manner indicative of a regula-tion phenomenon (6).
It was recently observed that changes in the compositionof peptidoglycan are linked to P-lactamase induction (22).Galleni et al. found that an increased amount of PBP4 leadsto hyperinduction of the chromosomal P-lactamase AmpC(4). Although this observation is preliminary, it suggests thatPBP4, which may be dispensible under laboratory growthconditions, is more than just another DD-carboxypeptidaseof E. coli. With the mutants described here we can analyzewhether the extra domain of PBP4 is, indeed, involved in theregulation of the endopeptidase activity and/or in the induc-tion of the chromosomal ,B-lactamase or whether it has someother, still unknown function.
REFERENCES1. Allore, R. J., and B. H. Barber. 1984. A recommendation for
visualizing disulfide bonding by one-dimensional SDS/PAGE.Anal. Biochem. 137:523-527.
2. Bergmeyer, H. U. 1983. Methods of enzymatical analysis, vol. 2,3rd ed., p. 86-88. Verlag Chemie, Weinheim, Germany.
3. Chamberlain, J. P. 1979. Fluorographic detection of radioactiv-ity in polyacrylamide gels with the water-soluble fluor, sodiumsalicylate. Anal. Biochem. 98:132-135.
4. Galleni, M., E. Bartowski, and S. Lindquist. 1990. Abstr. 90thAnnu. Meet. Am. Soc. Microbiol. 1990, p. 187. AmericanSociety for Microbiology, Washington, D.C.
5. Gurusinghe, A., P. A. Ryan, and P. F. Davis. 1986. Readyidentification of disulfide-bonded polypeptides by one-dimen-sional sodium dodecyl sulfate-polyacrylamide slab gel electro-phoresis. Electrophoresis 7:96-98.
6. Hakenbeck, R., and W. Messer. 1977. Activity of murein hydro-lases in synchronized cultures of Escherichia coli. J. Bacteriol.129:1239-1244.
7. Herzberg, O., and J. Moult. 1987. Bacterial resistance to lactamantibiotics: crystal structure of P-lactamase from Staphylococ-cus aureus PC1 at 0.25 nm resolution. Science 236:694-701.
8. Korat, B., H. Mottl, and W. Keck. 1991. Penicillin-bindingprotein 4 from Escherichia coli: molecular cloning of the dacBgene, controlled overexpression and alterations in murein com-position. Mol. Microbiol. 5:675-684.
9. Kunkel, T. A., J. D. Roberts, and R. A. Zakour. 1987. Rapid andefficient site-specific mutagenesis without phenotypic selection.Methods Enzymol. 154:367-382.
10. Laemmli, U. K. 1970. Cleavage of structural proteins during theassembly of the head of bacteriophage T4. Nature (London)227:680-685.
11. Masson, M. J., and R. Labia. 1983. Synthesis of a 125I-radiola-belled penicillin for penicillin-binding protein studies. Anal.Biochem. 128:164-168.
12. Meissner, P. S., W. P. Sisk, and M. L. Berman. 1987. Bacterio-phage lambda cloning system for the construction of directionalcDNA libraries. Proc. Natl. Acad. Sci. USA 84:4171-4175.
13. Messing, J. 1983. New M13 vectors for cloning. MethodsEnzymol. 101:21-78.
14. Mottl, H., and W. Keck. 1991. Purification of penicillin-bindingprotein 4 of Escherichia coli as a soluble protein by dye-affinitychromatography. Eur. J. Biochem. 200:767-773.
15. Mottl, H., and W. Keck. Submitted for publication.16. Mottl, H., P. Terpstra, and W. Keck. 1991. Penicillin-binding
protein 4 of Escherichia coli shows a novel type of primarystructure among penicillin-interacting proteins. FEMS Micro-biol. Lett. 78:213-220.
17. Riddles, P. W., R. L. Blakeley, and B. Zerner. 1983. Reassess-ment of Ellman's reagent. Methods Enzymol. 91:49-60.
18. Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular
J. BACTERIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
b on
22
Febr
uary
202
2 by
103
.87.
106.
102.

VOL. 174, 1992 PROBING THE DOMAIN ORGANIZATION OF PBP4 3269
cloning: a laboratory manual, 2nd ed. Cold Spring HarborLaboratory, Cold Spring Harbor, N.Y.
19. Spratt, B. G., and K. D. Cromie. 1988. Penicillin-binding pro-teins of gram negative bacteria. Rev. Infect. Dis. 10:699-711.
20. Stanley, K. K. 1983. Solubilization and immune-detection of,-galactosidase hybrid proteins carrying foreign antigenic deter-minants. Nucleic Acids Res. 11:4077-4092.
21. Then, R. L., C. Hubschwerlen, and R. Charnas. 1990. Antago-nism between some novel boronic acids and cephalosporins ininducible strains of Enterobacter cloacae. Poster presentation,
50 years of penicillin application, Berlin.22. Tuomanen, E., S. Lindquist, S. Sande, M. Galleni, K. Light, D.
Gage, and S. Normark. 1991. Coordinate regulation of P-lacta-mase induction and peptidoglycan composition by the ampoperon. Science 251:201-204.
23. Witholt, B., M. Boekhout, M. Brock, J. Kingma, H. van Heer-ikhuizen, and L. de LeU. 1976. An efficient and reproducibleprocedure for the formation of spheroblasts from variouslygrown Escherichia coli. Anal. Biochem. 74:160-170.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
b on
22
Febr
uary
202
2 by
103
.87.
106.
102.