prof. dr. sc. eduard vrdoljak centar za onkologijuneuron.mefst.hr/docs/graduate...
TRANSCRIPT

PROCES RAZVOJA LIJEKA
Klinički razvoj lijeka
Modul 1
Prof. dr. sc. Eduard VrdoljakCentar za onkologiju
KBC Split

Klinički razvoj lijeka
• Obuhvaća oko 60% vremenskog razdoblja razvoja lijeka
• 2003. godine – registriran jedan od 13 lijekova podvrgnutih kliničkom ispitivanju
• Važno je čim ranije u kliničkom ispitivanju definirati djelotvornost i sigurnosni profil lijeka
Clin Pharm & Ther 2008: 84(2):263-266.

Vrijeme potrebno za razvoj lijeka
14,4014,10
11,60
8,10
02468
1012141618
1963-1969 1970-1979 1980-1989 1990-1998
God
ine
Source: Tufts University, News Release, 11/30/2001
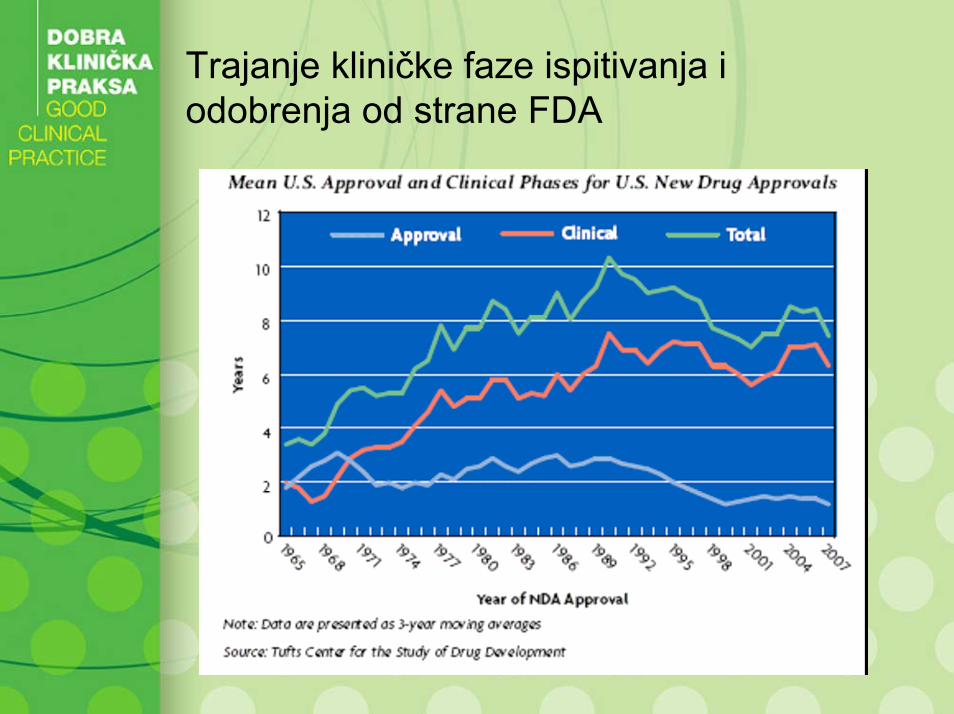
Trajanje kliničke faze ispitivanja i odobrenja od strane FDA

Faze kliničkog razvoja lijeka
Faza Svrha/cilj Ispitanici Broj Trajanje (godine)
IPrva primjena u ljudi, farmakokinetika (PK), farmakodinamika (PD)
Zdravi dobrovoljci,
bolesnici
Promjenjiv, do par stotina
Promjenjivo, 1 do 2 godine
II
PD, odnos doze i učinka, rana
učinkovitost (“dokaz koncepta“)
Bolesnici (+zdravi
dobrovoljci)
Promjenjiv, do više stotina
Promjenjivo, 1 do 2 godine
IIIPotvrda
učinka/sigurnosti Bolesnici Promjenjiv, do više tisuća
Promjenjivo, 3 do 5 godina
IVUčinak/sigurnost
“dokazana u praksi“, optimizacija
Bolesnici, odobrene indikacije
Promjenjiv Promjenjiv
Regulatorno odobrenje Regulatorno odobrenje –– pupušštanje u promettanje u promet

Podjela kliničkih ispitivanja po cilju
Tip kliničkog ispitivanja Cilj
Humana farmakologija • Procjena podnošljivosti• Opis farmakokinetike i farmakodinamike• Ispitivanje metabolizma lijeka i interakcija• Procjena aktivnosti
Terapijsko-istraživački • Ispitivanje primjene za ciljne indikacije• Procjena doze za sljedeća ispitivanja• Dati okvire za dizajn terapijsko dokazujućih
ispitivanja, njihovih ciljeva i metodologije
Terapijsko-dokazujući • Demonstrirati / potvrditi učinkovitost• Dokazati sigurnosni profil• Dati odgovarajući okvir za procjenu odnosa
koristi / rizika u svrhu registracije lijeka • Ustanoviti odnos doze i učinka
Terapijsko-primjenjivi • Poboljšati razumijevanje odnosa koristi / rizika u općoj ili posebnoj populaciji i/ili uvjetima
• Utvrditi rjeđe nuspojave• Poboljšati preporuke za doziranje

Osobitosti kliničkih studija faze I
• Prva primjena novog lijeka u ljudi – zdravi dobrovoljci ili bolesnici (npr. za citostatike, anti-HIV i druge NTI lijekove)
• Farmakokinetika (Cmax, Tmax, area under the curve [AUC]; klirens lijeka…)
• Rana evaluacija podnošljivosti:– Relevantni raspon doze (“dose range”)
– Toksičnost ograničena dozom (“dose-limiting toxicity”)
• Rana evaluacija neškodljivosti
• Prva farmakodinamska svojstva (primarna, sekundarna), ranaocjena učinkovitosti
• Vodi ka novim mogućnostima puta primjene, doza, režima doziranja; PK i PK/PD modeliranje
• Vrijeme i broj ispitanika mogu znatno varirati
• Mora se definirati doza koja će se primijeniti u studijama faze II

Tipične studije u fazi I
• Prva primjena u čovjeka (FIH – “first in human”):– Jednokratne i višestruke doze lijeka uz povećanje doza
(“single and multiple dose escalation”)• Povećanje doze:
– Logaritamska skala– Udvostručenje doze– Fibonaccijeva skala
– Potrebno utvrditi maksimalnu podnošljivu dozu (“maximum tolerated dose”; MTD)
• Utjecaj hrane na farmakokinetiku lijeka• Interakcije lijekova• Posebne populacije:
– Osobe s oštećenjem jetre ili bubrega– Spori metabolizatori
• Utjecaj spola i dobi• Bioraspoloživost različitih formulacija

• Interval doziranja:– Na temelju farmakokinetskih podataka (aktivna
tvar, metabolit)– Na temelju parametara o neškodljivosti – Empirijski
• Maksimalna doza:– Maksimalna podnošljiva doza (“maximum
tolerated dose”; MTD)– Plato odnosa PK/PD– dio NOAEL

Ispitanici:• Zdravi dobrovoljci:
– Obično muškarci 18-45 godina koji….• Ne uzimaju nikakve lijekove i…• Imaju uredan zdravstveni status
• Pacijenti:– Koji imaju blaži oblik bolesti i….– Ne uzimaju drugu terapiju
• Etičke dvojbe:– Trebaju li u fazi I sudjelovati zdravi dobrovoljci ako
postoji povećan rizik za neželjeni događaj ?• npr. slučaj superagonista TGN1412
– Treba li u fazu I uključivati bolesnike ako zbog sudjelovanja u fazi I možda neće moći uzimati lijek kad on dođe na tržište
• Npr. protutijela na monoklonsko protutijelo– Koliko su zdravi dobrovoljci informirani o riziku
sudjelovanja u pokusima faze I ?

Faza I
• Studije faze I su nerijetko otvorene, ali mogu biti i različitog stupnja “skrivenosti” tretmana
• Tipično je kontrola niža doza lijeka ili placebo
• Česti su križni dizajni
• Po statističkoj logici – tipično su studije “nejednakosti”

Izbor prve doze u čovjeka
Prema : – NOAEL – najveća doza kod koje nisu
uočene nuspojave (“No Observed AdverseEffect Level”) u pokusu na životinjama
• Smjernice FDA (2005) o izračunavanju maksimalne preporučene početne doze na temelju NOAEL
– MABEL – minimalna doza koja postiže očekivani biološki učinak (“MinimalAnticipated Biological Effect Level”)
– Mikrodoziranje – nefarmakološka doza– PK/PD modeliranje
• Prva doza obično 1/100-1/25 dio NOAEL

MABEL vs. NOAEL
• MABEL se temelji na zauzetosti receptora i na krivulji doza-odgovor – Relevantni životinjski modeli– Koriste se manje doze iz farmakodinamskih studija– Ne postoji jednostavni algoritam za izračunavanje
• NOAEL se temelji na toksičnim učincima– U ovim dozama mogu biti prisutni minimalni učinci– Zauzetost receptora se ne uzima u obzir– Općenito, doza je veća nego MABEL
• U slučaju nedoumice, uvijek je bolje uzeti manju dozu
• Bolje započeti s najmanjom dozom koja je aktivna, nego s najvećom koja je sigurna

Prva doza lijeka TGN1412TOKSIKOLOGIJA• Utvrditi NOAEL (no
observed adverse effectlevel)
• Pretvoriti NOAEL u HED (human equivalent dose)
– Prilagoditi ekspoziciji čovjeka
– Prilagoditi razlikama između vrsta u pogledu afiniteta/potentnosti
• Primijeniti ≥ 10-struki faktor sigurnosti i razmotriti PAD:
FARMAKOLOGIJA• Utvrditi MABEL
(minimal anticipated biological effect level) u čovjeka
– Prilagoditi ekspoziciji čovjeka
– Uključiti predviđeno trajanje učinka
– Prilagoditi razlikama između vrsta u pogledu afiniteta/potentnosti
Maksimalna preporučena početna doza
50.0 mg/kg
16.0 mg/kg
1.6 mg/kg 0.1 mg/kg 0.001 mg/kg

• Kompletna faza Ib– Novi metodološki koncept u istraživanju
lijekova za maligne bolesti– Individualna faza Ib može uključivati
kombinaciju novoga lijeka s nekim drugim lijekom
– Istovremeno nekoliko individualnih faza Ibobjedinjenih u jednom protokolu
• Npr. ispitivani lijek u kombinaciji sa nekoliko lijekova već prisutnih na tržištu u više skupina

Rani razvoj lijeka treba nam dati sljedeće podatke:
• Režim doziranja (raspon doza, interval doziranja)
• Preliminarno definiranje odnosa doza/odgovor• Parametre o neškodljivosti koje treba pratiti• Smjernice za kriterije uključenja/isključenja

Faza 0
• FDA’s Guidance on Exploratory IND studies (2006.)• Ograničena primjena u čovjeka, prije faze I
– Ne ispituje se terapijski učinak niti maksimalna podnošljiva toksična doza
• Zbog ubrzanog razvoja lijekova, prije svega onkoloških• U ranom stadiju razvoja može se donijeti odluka o tome
zavrjeđuje li lijek daljnja ispitivanja
• Mikrodozne studije:– Jednokratna primjena subterapijskih doza u 10-15
bolesnika prije faze I– Mikrodoza < 1/100 doze koja ima farmakološki učinak
(< 100 µg lijeka; za proteine < 30 nmol)– Daju preliminarni uvid u farmakokinetiku– Ne daju podatke o terapijskom učinku i neškodljivosti

Faza 0
• Farmakodinamske studije– Utvrditi je li mehanizam djelovanja u čovjeka
identičan onome iz pretkliničkih studija• Farmakodinamski biomarkeri (npr. vezanje
monoklonskog protutijela za receptor, smanjenje aktivnosti tirozin kinaze)
– Mogu se davati i ponavljane doze, u trajanju do 7 dana
• Faza 0 kod monoklonskih protutijela– sekvencijalno uključenje ispitanika– jednokratna mikro-doza sa evaluacijom:
• farmakodinamskih biomarkera• ex-vivo farmakoloških mjerenja• farmakokinetike• neškodljivosti i podnošljivosti

Monoklonska protutijela

Osobitosti kliničkih studija faze II
• Bolesnici (ponekad i zdravi dobrovoljci)
• PD svojstva: vrsta i jakost učinka, reakcija na dozu, obično se temelji na “surogatnim“ parametrima, npr. EEG za antiepileptike ili pokazatelji koštane pregradnje za bisfosfonate
• Daljnja PK svojstva (npr. kod zatajenja bubrega ili jetre, kritične interakcije s drugim lijekovima)
• Eksplorativni učinak i sigurnost (“dokaz koncepta“) – na temelju “čvrstih” kliničkih ishoda, npr. smanjenje broja napadaja, smanjenje broja fraktura
• Definiranje doze i režima doziranja za glavna ispitivanja učinka/sigurnosti
• Identifikacija mogućih sigurnosnih problema

• Faza IIa :– Pilot studija koja uključuje mali broj pacijenata (20-40)– Cilj: “dokaz koncepta” i prikupljanje podataka o
djelotvornosti
• Faza IIb:– Uključen veći broj pacijenata (150-500)– Čvršći dokazi o učinkovitosti, odnosu doza/učinak i
neškodljivosti– Potrebno je dobiti podatke neophodne za dizajn,
doziranje, ciljeve i metode koji će se primjenjivati u fazi III

Faza II
• Studije faze II su tipično dvostruko slijepe, ali mogu biti i otvorene i različitog stupnja “skrivenosti” tretmana
• “Kontrola” su i placebo, i druge doze i drugi aktivni tretmani (npr. “zlatni standard”)
• Vrlo raznoliki dizajni
• Po statističkoj logici – tipično su studije “nejednakosti”

• Postoji trend da se objedine pojedini dijelovi studija faze I i II (Ib/IIa)– Promatranje farmakodinamskih markera u zdravih
dobrovoljaca ili– Integracija male skupine pacijenata (IIa) u dio
pokusa faze I• Demonstrirati čim ranije “dokaz koncepta”
(“proof of concept”)• Studije faze I i II sve su više orijentirane prema
donošenju odluke o daljnjem razvoju lijeka(“decision making oriented”)

Osobitosti kliničkih studija faze III
• Bolesnici (ciljane indikacije)
• Bolesnici (indikacije) i doziranje – sukladno ciljanoj uporabi
• Nedvojbeno utvrđivanje učinkovitosti i sigurnosti
• Sveobuhvatna, multicentrična, multinacionalna dugoročna
• Obično je naglasak na učinku ; ponekad sigurnost/učinak
• Mora jasno pokazati učinkovitost na temelju “čvrstih” kliničkih
ishoda, npr. broj napadaja, fraktura kosti, preživljavanja, i sl.
• Mora polučiti dugoročne podatke o sigurnosti
• Mora biti jednostavno i jasno sročena

Faza III
• Studije faze III MORAJU biti dvostruko slijepe (ili, za neke indikacije može PROBE otvoreni dizajn)
• Tipična kontrola je placebo ili drugi aktivni tretman (npr. “zlatni standard”)
• Jednostavni dizajn – tipično paralelne grupe, “svi odjednom” ili sekvencijske studije (posebno kod preživljenja)
• Po statističkoj logici – studije nejednakosti, neinferiornostiili ekvivalencije (prema “zlatnom standardu”)
• Postoje i integrirani pokusi faze IIb i III (IIb/III) – adaptivni dizajn

Primarni ciljevi - primjeri
• Lijekovi za liječenje malignih bolesti:– Smanjenje veličine tumora– Postotak odgovora (“reponse rate”)– Vrijeme do progresije bolesti (“TTP”)– Preživljenje bez progresije (“PFS”)– Ukupno preživljenje
• Antihipertenzivi:– Vrijednosti krvnog tlaka– Kardiovaskularni morbiditet– Kardiovaskularni/ukupni mortalitet

Osobitosti postupka ocjene rizika/koristi u svrhu puštanja u promet
• Proizvodna i farmaceutska kvaliteta• Djelotvornost
Klinički podaci, posebno faza III + sve studije s “čvrstim”kliničkim ishodima
• SigurnostToksikologija i sigurnosna farmakologija poglavito u vezi korištenja lijeka u trudnoći i dojenju i izravne toksičnosti
Svi klinički podaci (posebno dugoročne ekspozicije)
Plan praćenja lijeka u općoj primjeni (eng. pharmacovigilance) i plan upravljanja rizicima

Razvoj GCP standarda i zakonska regulativa kliničkog ispitivanja
1938 – Američka uprava za hranu i lijekove (FDA)
1947 – Nirnberški kodeks
1962 – Kefauver–Harrisov amandman (SAD) [IND]
1964 – 2000 – Deklaracija iz Helsinkija
1981 – FDA propisi o informiranom pristanku i etičkimpovjerenstvima za nadzor kliničkih ispitivanja
1997 – ICH–GCP smjernice
2001 – EU direktiva za klinička ispitivanja
1986 – Državne GCP smjernice
1968 – Zakon o lijekovima UK-a [CTC, CTX]
1977 – FDA CFR: Obveze za sponzore, nadzornike i istražitelje
Eliksir sulfanilamida – 1937
Talidomid – 1960
Nirnberški proces – 1940
Istrage klin.ispit. od strane FDA – 1974

Regulativa u RH
• Zakon o lijekovima (NN 71/2007, NN 45/2009)
• Pravilnik o kliničkim ispitivanjima i dobroj kliničkoj praksi (NN 121/2007)
• Pravilnik o praćenju nuspojava nad lijekovima i medicinskim proizvodima (NN 29/2005)

U pripremi modula sudjelovali:
• Doc.dr.sc. Vladimir TrkuljaZavod za farmakologiju Medicinski fakultet Sveučilišta u Zagrebu
• Prof.dr.sc. Mladen BobanZavod za farmakologijuMedicinski fakultet Sveučilišta u Splitu
• Dr. Suzana Mimica MatanovićKlinička bolnica OsijekMedicinski fakultet Sveučilišta u Osijeku

“Quality means doing it right whenno one is looking. “
Henry Ford