preclinical pharmacological evaluation of a novel …...small molecule therapeutics preclinical...
TRANSCRIPT

Small Molecule Therapeutics
Preclinical Pharmacological Evaluation of a Novel MultipleKinase Inhibitor, ON123300, in Brain Tumor Models
Xiaoping Zhang1, Hua Lv1, Qingyu Zhou3, Rana Elkholi2, Jerry E. Chipuk2, M.V. Ramana Reddy2,E. Premkumar Reddy2, and James M. Gallo1
AbstractON123300 is a low molecular weight multikinase inhibitor identified through a series of screens that
supported further analyses for brain tumor chemotherapy. Biochemical assays indicated that ON123300
was a strong inhibitor of Ark5 and CDK4, as well as growth factor receptor tyrosine kinases such as b-typeplatelet-derived growth factor receptor (PDGFRb). ON123300 inhibited U87 glioma cell proliferation with
an IC50 3.4 � 0.1 mmol/L and reduced phosphorylation of Akt, yet it also unexpectedly induced Erk
activation, both in a dose- and time-dependent manner that subsequently was attributed to relieving Akt-
mediated C-Raf S259 inactivation and activating a p70S6K-initiated PI3K-negative feedback loop. Cotreat-
ment with the EGFR inhibitor gefitinib produced synergistic cytotoxic effects. Pursuant to the in vitro
studies, in vivo pharmacokinetic and pharmacodynamic studies of ON123300 were completed in mice
bearing intracerebral U87 tumors following intravenous doses of 5 and 25 mg/kg alone, and also at the
higher dose concurrently with gefitinib. ON123300 showed high brain and brain tumor accumulation based
on brain partition coefficient values of at least 2.5. Consistentwith the in vitro studies, single agentON123300
caused a dose-dependent suppression of phosphorylation ofAkt aswell as activation of Erk in brain tumors,
whereas addition of gefitinib to the ON123300 regimen significantly enhanced p-Akt inhibition and
prevented Erk activation. In summary, ON123300 demonstrated favorable pharmacokinetic characteristics,
and future development for brain tumor therapy would require use of combinations, such as gefitinib, that
mitigate its Erk activation and enhance its activity. Mol Cancer Ther; 13(5); 1105–16. �2014 AACR.
IntroductionGlioblastoma multiforme represents the most common
primary brain tumor in adults and is among themost lethalof all cancers. Despite multimodality treatment consistingof surgical resection followed by concurrent or sequentialtreatment with radiation and chemotherapy, the prognosisfor patients with glioblastoma multiforme is poor, with amedian survival time of approximately 14 months (1).Standard chemotherapy is based on DNA alkylatingagents, most often temozolomide; however, due to modestlong-term benefits, there is substantial interest to identifymolecularly targeted agents that can be used in combina-tion with temozolomide.
Receptor tyrosine kinase (RTK)/RAS/PI(3)K signalingcascades are among the most frequently altered signalingpathways in glioblastoma multiforme that are likely keyrequirements for disease progression in a majority of glio-blastoma multiforme (2–5). RTK signaling hyperactivationare most commonly caused by EGFRmutation/amplifica-tion or PDGFR amplification/overexpression, largelymediated through the PI3K/Akt/mTOR and Ras/mito-gen-activated protein kinase (MAPK) downstream signal-ing pathways. Pathologic fibroblast growth factor receptor1 (FGFR1) signaling also occurs in glioblastoma multi-formes which exhibit FGFR1 kinase domain gain-of-func-tion mutations (6). These pathway aberrations have stim-ulated an effort to discover novel modulators, albeit notnecessarily directed at glioblastoma multiforme, due inpart to its orphan disease status and the more restrictiverequirements to identify novel compounds with adequateblood brain barrier (BBB) transport (7, 8).
Our own efforts in anticancer drug discovery have ledto a number of agents in clinical trials (9–13), and morerecently we have employed a pharmacokinetic/phar-macodynamic-driven drug development paradigm toidentify agents suitable for brain tumor chemotherapy(14–16). Application of a pharmacokinetic/pharmaco-dynamic-driven approach to the ON123 series, 154 lowmolecular weight moieties, produced ON123300 as thelead compound (Fig. 1A) that possessed favorable
Authors' Affiliations: Departments of 1Pharmacology and Systems Ther-apeutics and 2Oncological Sciences, Icahn School of Medicine at MountSinai, New York, New York; and 3Department of Pharmaceutical Science,University of South Florida, Tampa, Florida
Note: Supplementary data for this article are available at Molecular CancerTherapeutics Online (http://mct.aacrjournals.org/).
Corresponding Author: James M. Gallo, Department of Pharmacologyand System Therapeutics, Icahn School of Medicine at Mount Sinai, OneGustave L. Levy Place, Box 1603, New York, NY 10029. Phone: 212-241-7770; Fax: 212-996-7214; E-mail: [email protected]
doi: 10.1158/1535-7163.MCT-13-0847
�2014 American Association for Cancer Research.
MolecularCancer
Therapeutics
www.aacrjournals.org 1105
on March 22, 2020. © 2014 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst February 25, 2014; DOI: 10.1158/1535-7163.MCT-13-0847

pharmacokinetic properties, including the ability topenetrate the BBB. A biochemical kinase screen indicat-ed ON123300 was a multitargeted kinase inhibitor withprimary targets of Ark5 and CDK4, PDGFRb, FGFR1,proto-oncogene Ret receptor tyrosine kinase, and proto-oncogene Fyn tyrosine-protein kinase. Ark5 is a memberof the AMPK family and found to be directly phosphor-ylated and activated by Akt to prevent cell death. It wasreported that Ark5-mediated mTOR phosphorylationinduced by IGF-1 plays a key role in tumor malignancyand transient RNAi-mediated ARK5 knockdown causedsignificant reductions in cell proliferation and brain inva-sion in a glioma xenograft mouse model (17–20). CDK4has been shown to be responsible for hyperphosphoryla-tion of tumor suppressor protein Rb releasing its inhibi-tion on G1–S cell-cycle progression through activation ofthe transcription factor E2F (21). CDK4-silenced cellsundergo apoptosis and displayed decreased colony for-
mation capacity (22). Small-molecule inhibitors of CDK4andCDK6have shownpreclinical efficacy in glioblastomamultiforme models (23, 24). Given the interesting andpotential importance of ON123300’s targets and its pos-itive pharmacokinetic profile we undertook a detailedpharmacokinetic/pharmacodynamic analysis in a com-mon preclinical brain tumor model (U87MG).
Materials and MethodsON123300 (Fig. 1A), ON1231120, and ON1231320 com-
pounds were supplied by Dr. M.V. Reddy (Departmentof Oncological Sciences, Icahn School of Medicine atMount Sinai; ref. 16). Gefitinib was purchased from LCLaboratories. Temozolomide and b-actin antibody waspurchased from Sigma-Aldrich, and IRDye 800CW-con-jugated and Alexa Fluor 680-conjugated secondary anti-bodies were from Rockland and Invitrogen, respectively.
0
1
2
3
4
JNKERK2ERK1CREBAkt1pan
p38γ
Control
ON123300
Pix
el d
en
sity
Targets
*
**
**
**
**
**
p-Akt S473
β-Actin
p-rpS6
p-Rb S780
p-p70S6
p-Erk
Akt
Erk1/2
p70S6
rpS6
p-Ark5
+ − − − Control A
B
C− + − − ON123300
− − + − ON1231120
− − − + ON1231320
Ark5
CN
ONN
N
N
N
HN
Figure 1. ON123300 regulatedMAPKandAkt pathways. A, chemical structure ofON123300. B,U87 cells cultured on 10-cmplatewere treatedwith 6.3mmol/LON123300 for 1 hour and cell lysates were analyzed by the phospho-MAPK Array Kit (see Supplementary Fig. S1); selected proteins shown above.ON123300 treatment decreased phosphorylated Akt, CREB, and JNK pan expression levels. In contrast, p-Erk and p38g were increased in cells treated withON123300. C, U87 cells cultured on 10-cm plate were treated with 6.3 mmol/L ON123300, 70 mmol/L ON1231120, or 10 mmol/L ON1231320 for 1 hour, andthen cell homogenates were separated by SDS-PAGE gel and expression of p-Akt, phosphorylation levels of its downstream proteins, and p-Erk weredetected by Western blotting. Total proteins and b-actin expression were used as control. ON123300 inhibited phosphorylation of Akt and its downstreamsignaling components, P70S6K, 40S ribosomal protein S6 (rpS6) and Rb S780 (decreased to 40.1%� 5.7%; 31.8%� 2.1%; 60.5%� 1.0%; 54.5%� 6.3%relatively to control), yet increased p-Erk (increased to 120%� 6.9% relative to control). Data, measured mean values and SD of at least three experiments.�, P < 0.05; ��, P < 0.01.
Zhang et al.
Mol Cancer Ther; 13(5) May 2014 Molecular Cancer Therapeutics1106
on March 22, 2020. © 2014 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst February 25, 2014; DOI: 10.1158/1535-7163.MCT-13-0847

All other antibodies were purchased from Cell SignalingTechnology. Ninety-six–well p-Akt and p-Erk whole-celllysate kits were purchased from Meso Scale Discovery.Human phospho-MAPK array kit was purchased fromR&D Systems. All other chemicals and solvents wereobtained from commercial sources.U87MG (U87) and U251 human glioma cells were
purchased from American Type Culture Collection inAugust 2011. U87/EGFRvIII and U87/PTEN cell lineswere a generous gift from Dr. Webster Cavenee (Univer-sity of California-San Diego, La Jolla, CA) and Dr. PaulMischel (University of California Los Angeles, LosAngeles, CA) obtained in December 2009, respectively.Cells were authenticated usingWestern blotting assays inJune 2013. GBM10 and GBM39 cells were obtained fromDr. Jann Sarkaria (MayoClinic, Rochester,MN) inDecem-ber 2011. Cells were cultured in Dulbecco’s modifiedEagle medium supplemented with 10% standard FBS,100 U/mL penicillin, and 100 U/mL streptomycin andmaintained in ahumidified atmosphere of 5%CO2 in air at37�C.NIH SWISS nude mice were purchased from Taconic
and maintained in the American Association for theAccreditation of Laboratory Animal Care accredited Lab-oratory Animal Resources of Icahn School of Medicine atMount Sinai (New York, NY). All study procedures wereapproved by the Institutional Animal Care and UseCommittee.
In vitro cytotoxicity and combination drug studiesThe cytotoxicity of ON123300 was determined using a
colorimetric sulforhodamine B (SRB)-based assay (25).Suspensions of glioma cells (100 mL containing 2 � 103
cells) were seeded in 96-well plates and allowed to attachto the surface by overnight incubation. The cellswere thentreatedwith increasing concentrations ofON123300 for 72hour. At the end of the treatment, cells were fixed with10% (v/v) trichloroacetic acid (TCA) and stained with0.4% SRB. The optical densities were measured with aSpectraMax M2 microplate reader (Molecular Devices) ata wavelength of 570 nm. A Sigmoid Emax model (Win-Nonlin, Pharsight Corporation) was used to calculate IC50
values (mean of three independent studies), which weredefined as the drug concentration that was required toreduce the number of viable cells to 50% compared withcontrol treatment (vehicle alone). For combination stud-ies, cells were treated with ON123300 (from 0.03 to 16mmol/L) and gefitinib (from 0.16 to 80 mmol/L) alone ortogether at a fixed concentration ratio of 0.2, or treatedwith ON123300 (from 0.1 to 25 mmol/L), temozolomide(from 10 to 2,500 mmol/L), alone or together at a fixedconcentration ratio of 0.01 for 72 hours and thenprocessedusing the SRB assay. The resultant cell proliferation datawere used to calculate the combination indexes at desig-nateddegrees [50% (ED50), 75% (ED75), and 90% (ED90)] ofcell toxicity, respectively, using the CompuSyn program(ComboSyn, Inc.; ref. 26). Combination indices of <1indicate synergy and those >1 antagonisms.
Flow cytometric analysisU87 cells seeded on 6-well plates were cultured over-
night and treated with ON123300 and gefitinib alone orconcurrently for 24 hours. The cell mediumwas aspiratedand the remaining cells washed and then collected byscraping in 1mLPBS. Cellswere centrifuged at 500� g for5 minutes at 4�C and then resuspended in 0.5 mL PBS at adensity of 2� 106 cells/mL. After adding 4.5 mL 70% ice-cold ethanol the cellswere kept on ice for 2hours, and thenwashed and incubated in PBS containing 20 mg/mL pro-pidium iodide, 0.2 mg/mL DNase-free RNase and 0.1%(v/v) Triton X-100 for 30 minutes at room temperature.The cell cycle of intact/attached cells was analyzed on aflow cytometer (FACSCalibur, BD Biosciences) and onlylive cells were gated and quantitated by FlowJo software(Tree Star, Inc). For Annexin V studies, U87 cells seededon 12-well plates were cultured overnight and treatedwith ON123300 and gefitinib alone or concurrently for24 hours. Both floating and attached cells were har-vested, stained with AnnexinV-FITC 633 (10 mg/mL) inbinding buffer (10 mmol/L HEPES pH 7.4, 150 mmol/LNaCl, 5 mmol/L KCl, 1 mmol/L MgCl2, 1.8 mmol/LCaCl2), and analyzed by flow cytometry (FACScan, BDBiosciences; 5,000 events/sample).
Live cell microscopyU87 cells were grown in 6-well plates overnight and
treatedwithON123300, gefitinib or the combination for 24hours. Treated cells were then directly visualized with anOlympus IX70 fluorescence live cell microscope (Olym-pus Imaging) controlled by InVivo software (MediaCybernetics).
Protein sample preparationCell lysates from treated cells or brain tumors were
prepared in a cell lysis buffer (27) containing 1 mmol/Lphenylmethylsulfonylfluoride, phosphatase inhibitor I,phosphatase inhibitor II, and protease inhibitor cocktailat a ratio of 600 mL of buffer to 80%–90% confluent cells in10 cmplate or 20mLof buffer to 1mgof brain tumor tissue.After adding cell lysis buffer the cells and tissues werebrief sonicated, incubated on ice for 30 minutes, and thencentrifuged at 21,000 � g for 10 minutes at 4�C. Thesupernatants were collected and stored at �80�C. Proteinconcentrations of both supernatants were measured by aBSA assay according to the manufacturer’s instructions.
Phospho-MAPK arrayHumanphospho-MAPKarrayswereprocessed accord-
ing to the manufacturer’s protocol. Briefly, U87 cellstreatedwith 6.3 mmol/LON123300 for 1 hourwere rinsedwith ice-cold PBS and solubilized with lysis buffer toreach a cell concentration of 1 � 107/mL. Cell super-natants were collected after cell lysates were rocked at4�C for 30 minutes and then centrifuged for 5 minutes.Subsequently, 200 mg of the cell supernatants were adjust-ed to final volume of 1.5 mL with array buffer 1 followedby the addition of 20 mL of a detection antibody cocktail
Preclinical PK/PD Evaluation of ON123300 in Brain Tumor
www.aacrjournals.org Mol Cancer Ther; 13(5) May 2014 1107
on March 22, 2020. © 2014 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst February 25, 2014; DOI: 10.1158/1535-7163.MCT-13-0847

and incubated at room temperature for 1 hour. The pre-pared sample–antibody mixtures were added to mem-branes that had beenpreincubatedwith array buffer 5 andthen incubated overnight at 4�C. Next day, the mem-branes were washed 3 times with washing buffer andincubated with diluted Streptavidin-HRP antibody for 30minutes at room temperature. The membranes were thendeveloped by Chemi Reagent Mix and exposed to X-rayfilm.
Western blot assayEqual amounts of protein samples (20 mg) were electro-
phoresed on SDS-polyacrylamide gels and transferred topolyvinylidene fluoride membranes that were blockedwith 5% nonfat milk at room temperature for 1 hour, andsubsequently probedwith various primary antibodies for1 hour at room temperature or overnight at 4�C. The blotswere washed and then incubated with IRDye 800CW-conjugated or Alexa Fluor 680-conjugated secondaryantibodies at room temperature for 1 hour and thenquantitated for protein using an Odyssey 2.1 (LI-CORBiotechnology) systemand the ImageJ software.AllWest-ern blots were repeated at least three times.
Meso Scale Discovery assayMeso Scale Discovery (MSD) 96-well multispot p-Akt/
total Akt and p-Erk/total Erk assays were carried out asper the manufacturer’s protocol with minor modifica-tions. Briefly, plates were blocked with the MSD blockingsolution for 1 hour at room temperature while shakingand then washed 4 times with Tris wash buffer. For eachwell, 25 mL of protein (20 mg for Akt and 5 mg for Erk) wasadded to the plate in duplicate and incubated overnight at4�C. The plates were washed four times with Tris washbuffer and incubated with 25 mL of detection antibody ineach well at room temperature for 2 hours with shaking.The plates were analyzed on a SECTOR 6000 instrument(Meso Scale Discovery) after washing with Tris washbuffer and addition of 150 mL of read buffer. Both thephosphor- and total signals were corrected for back-ground (BSA spots) and for any effects of the lysis buffer.The percentage of phosphorylation against total proteinwas calculated and the expression of phosphorylation ineach treated group was expressed as percentage relativeto the control group.
Orthotopic glioma model and dosing studiesTheU87 gliomamodel used in this studywas described
previously. Briefly, NIH Swiss nude mice were anesthe-tized with a cocktail (ketamine:acepromazine:xylazine:saline ¼ 3:2:1:24 volume ratio; ketamine: 100 mg/mL;acepromazine: 10 mg/mL; xylazine: 20 mg/mL) at anintraperitoneal dose of 5 mL/kg. After securing in astereotactic apparatus, mice were implanted with a sus-pension of U87 cells (106 cells in 10 mL PBS) into thecaudate putamen at a position 0.7 mm anterior and 2.2mm lateral from the bregma at a depth of 2.5 mm using a10 mLHamilton syringe (Hamilton Co.). Once the animals
recovered from anesthesia (about 20 minutes) they werereturned to the animal care facilities, and provided foodandwater ad libitum. Mice weremonitored daily and thenentered into the pharmacokinetic/pharmacodynamicstudies upon the appearance of clinical symptoms (i.e.,unkempt appearance, arched back, unsteady gait) or abody weight loss of 2 g over 2 consecutive days.
ON123300 was administered to groups of brain tumor–bearing mice as an intravenous bolus at a dose of either 5or 25mg/kg via a tail vein. Blood, normal brain, and braintumorwere collected from3 to 4mice at each timepoint (5,15, 30 minutes and 1, 2, 4, 6, 10 hours postadministration)in which the mice were briefly anesthetized with isoflur-ane and exsanguinated by cardiac puncture. Plasma wasseparated by centrifugation (14,000 rpm, 2 minutes, 4�C)and brain tissues obtained by gross dissection. Normalbrain and brain tumor samples were rinsed thoroughlywith 0.9% saline solution and together with plasma sam-ples stored at �80�C until analyzed by liquid chromatog-raphy-tandem mass spectrometry (LC/MS-MS). For theON12300-GFN treatment study, ON123300 was adminis-tered at an intravenous dose of 25 mg/kg via a tail vein 1hour after an oral dose of 200 mg/kg gefitinib (set timepoint of gefitinib administration as of -1), and had blood,normal brain, and brain tumor samples collected at eachtime point (0, 0.08, 0.25, 0.5, 1, 2, 4, 6, 10, 17 hours post-ON123300 administration) and analyzed as described forON123300 only treatment.
LC/MS-MS analysisAll plasma and tissue samples from the pharmacoki-
netic studies were analyzed with an electrospray ioniza-tion LC/MS-MS system (HPLC; QTrap 5500, AppliedBiosystems) described previously (16). To 10 mL samplesof plasma, normal brain or brain tumor homogenate (20%w/w tissue/water), 40 mL of cold acetonitrile was used toprecipitate proteins followed by centrifugation at 15,000rpm for 5 minutes. For both plasma and brain samples, 10mL aliquots of the resultant supernatant were injected intothe LC/MS-MS system. Data collection and analysis wereperformed using Analyst 1.5.1 software (Applied Biosys-tems MDS Sciex).
Pharmacokinetic and pharmacodynamic data andstatistical analysis
Noncompartmental analyses (NCA) were performedusingWinNonlinPhoenix, version6.3 software [PharsightCorporation] to estimate pharmacokinetic and pharma-codynamic parameters that included for each compoundthe areas between the effect–time curve in brain tumor(ABEC), and the areas under the drug concentration–timecurve in plasma, brain, and brain tumor (AUCp, AUCb,and AUCbt, respectively), half-life (t1/2), systemic clear-ance (CL), and the apparent volume of distribution atsteady state (Vss). The normal brain or brain tumor par-tition coefficient (Pb and Pbt) was calculated as the ratio ofeither theAUCborAUCbt over the correspondingAUCp.All AUC calculations were based on the area to the last
Zhang et al.
Mol Cancer Ther; 13(5) May 2014 Molecular Cancer Therapeutics1108
on March 22, 2020. © 2014 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst February 25, 2014; DOI: 10.1158/1535-7163.MCT-13-0847

observable concentration using the linear-log interpola-tion method. Data were statistically evaluated by a Stu-dent t test or one-way ANOVA with the level of signif-icance chosen as P < 0.05.
ResultsIn vitro studiesOur previous pharmacokinetic/pharmacodynamic-
driven screen of 154 ON123 compounds indicatedON123300 had potential as an anticancer drug for braintumor chemotherapy based on in vitro cytotoxicity and anintegrated panel of in vitro pharmacokinetic assays (i.e.,metabolic stability, BBB permeability, and plasma andbrain tissue binding; ref. 16). Further delineation of its invitro cytotoxicity ofON123300 toU87 cells indicated it hadan IC50 equal to 3.4 (mean)� 0.1 (SD) mmol/L. In addition,ON123300 inhibition of cell proliferation in a panel of 11glioma models, including a patient-derived model(GBM10), was quite comparable with that observed inU87 cells (Supplementary Fig. S1). A previously com-pleted biochemical screen of 288 kinases using recom-binant proteins of ON123300 found the following pri-mary targets; Ark5 (IC50 ¼ 5 nmol/L) and CDK4 (IC50 ¼3.9 nmol/L), as well as appreciable inhibitory actionagainst, PDGFRb (IC50 ¼ 26 nmol/L), FGFR1 (IC50 ¼ 26nmol/L), proto-oncogene Ret receptor tyrosine kinase(RET, IC50 ¼ 9.2 nmol/L) and proto-oncogene Fyntyrosine-protein kinase (FYN, IC50 ¼ 11 nmol/L).To determine how ON123300 target modulation influ-
enced cell signaling, wemeasured the activities of severalcritical molecules in both the PI3K/Akt/mTOR and Ras/MAPK signaling cascades using a phospho-MAPK arraycontaining 26 different capture antibodies (Supplemen-tary Fig. S2). Selected proteins that were significantlydifferent (P < 0.05) in treated and control are shownin Fig. 1B. ON123300 decreased expression of phosphor-ylatedAkt, CREB, and JNKpan decreased,whereas p-Erkand p-p38g increased in cells. On the basis of thesedisparate findings related to Akt and Erk, we furtherevaluated these and related proteins, including Ark5, akey target, by Western blot analyses. ON123300 inhibitedphosphorylation of Akt and its downstream signalingcomponents, P70S6K, 40S ribosomal protein S6 (rpS6),and Rb S780 (see Fig. 1C). In conjunction with p-Ark5 andp-Akt inhibition there was an increase in p-Erk uponexposure toON123300 (see Fig. 1C). TwoadditionalON123series compounds, ON1231120 and ON1231320, had neg-ligible effects on these proteins. As measured by Westernblot and MSD assays the opposite effects of ON12300 onboth p-Akt and p-Erk were concentration-dependent withan IC50 for p-Akt inhibition equal to 5.2 mmol/L (Fig. 2A),which roughly correlated to that for cytotoxicity, and 100%increase in p-Erk at a concentration of about 7 mmol/Lcompared with vehicle. Using the same experimental sys-temthe time-dependent behaviorofp-Akt inhibitionandp-Erk activation is shown in Fig. 2B in which the effects ofON123300 on p-Akt can be seen as early as 10 minutes, anadir at 30minutes and sustained inhibition over 6 hours at
6.3 mmol/L of ON123300. The p-Erk time profile wasoscillatory in nature with a rapid increase at 5 minutesfollowed by a decline and then a second peak at 2 hourswith another modest decline over 6 hours yet still elevatedabout 2-fold relative to pretreatment. Similar dose- andtime-dependent behavior on p-Akt and p-Erk was alsoobserved in two other glioma models; U251 glioma cellsand GBM39 cells, a patient-derived brain tumor model(Supplement Fig. S3). Specifically, in the GBM39 model at4 mmol/LON123300 (close to that used in U87 cells), nadirp-Akt inhibition was 55% and peak p-Erk activation was174% relative to the control.
ON123300 did not directly inhibit Akt based on thebiochemical kinase screen but did show appreciableinhibitory action against upstream tyrosine kinase recep-tors (PDGFRB, FGFR1, andVEGFR3) thatwe inferredwasthe cause for reduced Akt activity. To elucidate theimportance of FGFR1 and PDGFRB signaling in Aktinhibition, we examined their response upon stimulationby their cognate ligands, bFGF (20 ng/mL) and PDGF-BB(50 ng/mL), respectively (28, 29). The activation status ofthe receptor and Akt and Erk were determined by West-ern blot analysis (Fig. 2C) in U87 cells that were serum-starved for 18 hours and then treated for 2 hours with 3mmol/L ON123300 before stimulation with each ligandfor 10minutes. Ligand stimulation with bFGF and PDGF-BB induced high levels of p-Akt and p-Erk in control cellsthat was abolished completely for p-Akt and partially forp-Erk by ON123300.
Previous studies reported that mTOR inhibition acti-vates Erk by release of a concomitant negative feedbackloop in which activation of p70S6K suppresses itsupstream signaling component PI3K and causes inacti-vation of Erk (30, 31). To test whether ON123300-inducedErk activationwasmediated through this PI3K/Ras/Raf/Erk negative feedback loop, PI3K activity was inhibitedeither by pretreating cells with 50 mmol/L LY294002 or 1mmol/L wortmannin for 1 hour or augmented by usingU87/PTEN cells that possess normal PTEN activity. Asshown in Fig. 2D, abrogation of the PI3K feedback bypharmacologic inhibition of PI3K or by overexpression ofPI3K suppressor PTEN reduced Erk activation in cellstreated with either vehicle or ON123300. These data sug-gested that Erk activation may be partially modulated byrelease of feedback inhibition via PI3K activity but mayalso involve other signaling pathways. Under certainconditions, ligand concentration-dependent and time-dependent, Akt directly phosphorylates C-Raf at a highlyconserved serine residue S259 and this phosphorylationresults in recruiting 14-3-3 protein binding to C-Raf caus-ing inactivation leading to Erk inactivation (32, 33). Thus,inhibition of Akt by ON123300 could prevent phosphor-ylation of C-Raf S259 via this cross-talk mechanism andpromoteRaf and thenErk activation.ON123300 treatmentalone, in either U87 or U87PTEN cells, partially decreasedp-C-Raf S259 expression (Fig. 2D) and when combinedwith LY294002 or wortmanin even greater inhibition of p-C-Raf S259 was achieved supporting this pathway
Preclinical PK/PD Evaluation of ON123300 in Brain Tumor
www.aacrjournals.org Mol Cancer Ther; 13(5) May 2014 1109
on March 22, 2020. © 2014 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst February 25, 2014; DOI: 10.1158/1535-7163.MCT-13-0847

connection in Erk activation. In summary, our resultsdemonstrated that ON123300-induced Erk activation iscaused by the release of the p70S6K/PI3K/Ras/Raf/Erknegative feedback loop and by suppression of Akt-induced Raf inhibition. Figure 2E illustrates the proposedmechanisms involved in ON123300 signaling.
In vitro combination studiesOn the basis of the fact that ON123300 activated Erk
through PI3K and Raf mechanisms, we considered thatthe addition of the EGFR inhibitor gefitinib could preventthis negative effect and enhance ON123300 activity. First,the in vitro cytotoxicity assays and associated combination
index values being less than 1 (range 0.25–0.7) for theON123300-gefitinib combination indicated synergy for alldesignated degrees of cell toxicity in all glioma cell lines(Fig. 3A; ref. 26). We completed a combination studywithON123300 and temozolomide and found an antagonisticeffect based on the combination indices being >1 (Sup-plementary Table S1). Although initially perplexing thisantagonism could be explained by the ability of temozo-lomide to induce Erk1/2 in U87 cells (34, 35), yet furtherinvestigations would be indicated to further characterizethis combination. To investigate the molecular mechan-isms underlying the synergistic cytotoxic effect of thecombination of ON123300 and gefitinib Western blot
0 2 4 6 8 10 μmol/L
p-Akt
p-Erk
β-Actin
Erk
Akt
0
50
100
0
50
100A
C D E
B
0 4 8 12 16
p-Akt
Cell growth
ON123300 (μmol/L)
Perc
enta
ge o
f cell
pro
life
ratio
n
Pe
rce
nta
ge
of p
rote
in
exp
ressio
n (
rela
tive
)
50
100
150
200
250
0 2 4 6 8 10Perc
enta
ge o
f p-E
rk(re
lative)
ON123300 (μmol/L)
0 0.08 0.25 0.5 1 2 4 6 h
p-Akt
β-Actin
p-Erk
Akt
Erk
0
50
100
0 1 2 3 4 5 6
Time (h)
Pe
rce
nta
ge
of p
-Akt
(rela
tive)
0
100
200
300
0 1 2 3 4 5 6
Time (h)
Perc
enta
ge o
f p-E
rk(r
ela
tive)
p-PDGFR
p-Erk
p-Akt
− + + − − hFGF− − − + + PDGF-BB − − + − + ON123300
FGFR
p-FGFR
Erk
Akt
PDGFR
β-Actin
− − + + − − − − LY294002
− − − − + + − − Wortmannin− + − + − + − + ON123300
p-Akt
p-Erk
Erk
PTEN
U87 PTENU87
Akt
p-PTEN
p-C-RafS259
C-Raf
β-Actin
FGFR1 PDGFRB
Ras PI3K
Raf Akt
MEK
rpS6
P70S6K
ERK1/2mTOR
Rheb
TSC1/2 Ark5
PTEN
Figure 2. ON1233300 regulated p-Akt and p-Erk activities through PDGFRB and FGFR1 signaling pathways, P70S6K/PI3K/Akt negative feedbackloops and cross-talk between PI3K/Akt and Ras/Erk pathways. U87 cells were incubated with increasing concentrations of ON123300 for 1 h (A) orwith 6.3 mmol/L of ON123300 for indicated times (B). The p-Akt and p-Erk expression levels against total Akt and Erk expressions were visualizedby Western blot analysis or quantified by MSD assays. C, U87 cells were serum starved for 24 hours and then treated for 2 hours with 3 mmol/LON123300 before stimulation with bFGF (20 ng/mL) and PDGF-BB (50 ng/mL) for 10 minutes. The activation status of the receptor, downstreamsignal molecules, Akt and Erk, were determined by Western blot analysis. D, U87 cells or U87 PTEN cells cultured on 6-well plate were treatedwith 6.3 mmol/L ON123300 for 1 hour after pretreating with vehicle, 50 mmol/L LY294002, or 1 mmol/L wortmannin for 1 hour. The p-Akt, p-Erk, p-C-RafS259, and p-PTEN expression levels were visualized by Western blot analysis. Total proteins and b-actin expression were used as control. E, overviewof the biochemical pathways involved in ON123300 targeting. Inhibition reactions are designated by plungers. Data, percentages of the meanvalue and mean � SD of three experiments.
Zhang et al.
Mol Cancer Ther; 13(5) May 2014 Molecular Cancer Therapeutics1110
on March 22, 2020. © 2014 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst February 25, 2014; DOI: 10.1158/1535-7163.MCT-13-0847

analyses of U87 cells were completed that demonstratedgefitinib not only enhanced ON123300 blockade of p-Akt,p-Ark5, andp-mTOR (S2481 and 2448), but also decreasedp-Erk levels relative to control (Fig. 3B). The favorableresponses to gefitinib are attributed to EGFR inhibitionthat is upstream of both the PI3K/Ras/Raf/Erk negativefeedback loop and Akt-Raf activation that led to Erkactivation.To examine the effects of the gefitinib and ON123300
combination on cell dynamics, live cell microscopy andflow cytometry were completed. Consistent with cytotox-icity assays, the live cell microscopy showed a dramaticdecrease of total cell number of U87 cells following singleor combination treatments for 24 hours (Fig. 3C). Flowcytometry of live cells demonstrated significant effects oncell-cycle progression following 24 hours drug exposures(Fig. 3D) with ON123300 alone and in combination withgefitinib causing an approximate 2.5-fold increase in thepercentage of cells in the G2–M phase [38.2% � 01.4%(alone) 35.8% �0.4% (combined) vs. 10.4% � 0.6% con-trol], whereas gefitinib caused cell-cycle redistribution
mainly at the G1 phase (80.9% � 2.5% vs. 66% � 0.9%).ON123300-induced G2–M phase arrest may be throughAkt inhibition as it has been reported that Akt promotesG2–M cell-cycle progression by inducing phosphoryla-tion-dependent 14-3-3 theta binding and cytoplasmiclocalization of WEE1 (36, 37) that switches the balance ofregulator activities and causes the initial activation ofcyclin B-Cdc2 at the meiotic G2–M phase transition.ON123300 could interfere with this action by inhibitionof Akt. In addition, based on Annexin V staining,ON123300 induced cell death by apoptosis in a dose-dependent manner. Gefitinib treatment alone did notcause apoptotic cell death and in combination withON123300 had no effect on ON123300-induced apoptosis(Fig. 3E).
In vivo pharmacokinetic/pharmacodynamic studiesFrom our prior pharmacokinetic screening analyses of
the ON123 series, ON123300 was the lead compound thatpossessed favorable pharmacokinetic characteristics sug-gestive of distribution into brain (16). To determine
0
0.25
0.5
0.75
1
U87 U87 VIII U87 PTEN
ED50 ED75 ED90
Co
mb
ina
tio
nin
de
x
Cell lines
A
C D E
B
p-Erk
p-Akt
Akt
Erk
β-Actin
− + − + ON123300 − − + + GFN
p-mTOR S2448
p-mTORS2481
mTOR
p-Ark5
Ark5
0
25
50
75
100
Ctrl 300 GFN Combp-A
ktexp
ressio
n
level (r
ela
tive)
Treatment
0
50
100
Ctrl 300 GFN Combp-E
rke
xp
ressio
n
level (r
ela
tive)
Treatment
GFN Comb
Control ON123300
0.0
0.2
0.4
0.6
0.8
1.0
1.2
Control ON123300 GFN Comb
Cell-
cycle
dis
trib
ution S G2 G1
Treatment
0
25
50
75
CombGFN6.3 μmol/L
2.1 μmol/L
1 μmol/L
Ctrl
Ap
op
tosis
ce
ll (p
erc
en
tag
e)
Treatment
ON123300
****
** **
Figure 3. Mechanisms of action of concurrent treatment with ON123300 and gefitinib (GFN). A, combination indices for ON123300 and gefitinib combinationtherapy. U87 parental, U87 VIII, U87/PTEN cells were treated with different concentration of ON123300, gefitinib, or with a fixed concentration ratio of0.2 of both for 72 hours. Combination indices were computed using the CompuSyn program for designated degrees of toxicity; ED50, ED75, and ED90. B,immunoblot analysis was performed on Akt, Erk, mTOR, and Ark5 expression after treatment of U87 cells with 6.3 mmol/L ON123300, 13.3 mmol/L gefitinib orboth for 1 hour. p-Akt and p-Erk expression levels were quantitated by ImageJ software. The expression of p-mTOR S2448 and p-mTOR S2481 were86%� 3.1% and 102%� 5.2%when treated with ON123300 alone and reduced to 64%� 2.8% and 84%� 4.5%when treated in combination. C–E, aftertreatment of U87 cells with 6.3 mmol/L ON123300, 13.3 mmol/L gefitinib, or both for 24 hours, cells were analyzed by live cell imaging for morphology (C),and by flow cytometry for cell cycle (D) and apoptosis quantification (E). Scale bars, 20 mm. Data, percentages of the mean value and mean � SD ofthree experiments. ��, P < 0.01.
Preclinical PK/PD Evaluation of ON123300 in Brain Tumor
www.aacrjournals.org Mol Cancer Ther; 13(5) May 2014 1111
on March 22, 2020. © 2014 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst February 25, 2014; DOI: 10.1158/1535-7163.MCT-13-0847
whether these findings translated to an in vivo setting, aseries of pharmacokinetic/pharmacodynamic studies ofON123300 alone and combined with gefitinib were con-ducted in mice bearing intracerebral U87 tumors. Theseefforts were based on the measurement of two biomar-kers, p-Akt andp-Erk in brain tumor samples andplasma,normal brain, and brain tumor concentrations ofON123300 and GFN following doses.
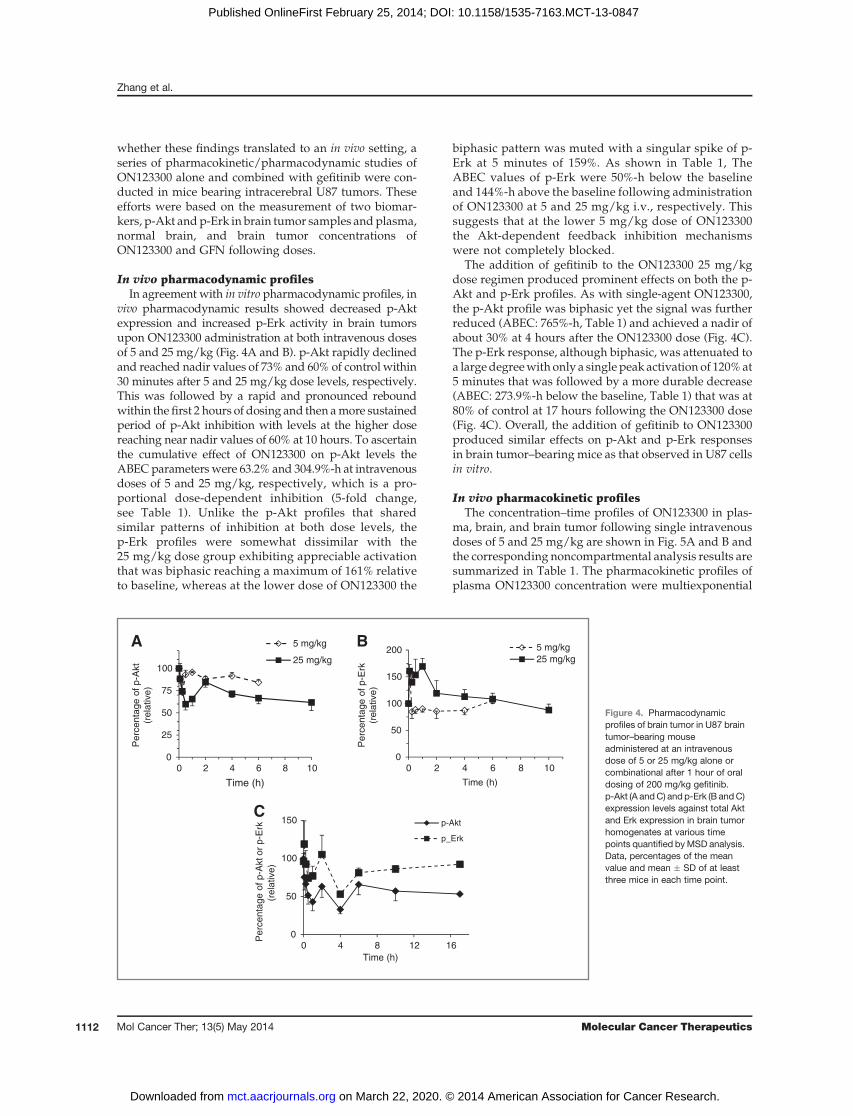
In vivo pharmacodynamic profilesIn agreement with in vitro pharmacodynamic profiles, in
vivo pharmacodynamic results showed decreased p-Aktexpression and increased p-Erk activity in brain tumorsupon ON123300 administration at both intravenous dosesof 5 and 25 mg/kg (Fig. 4A and B). p-Akt rapidly declinedand reached nadir values of 73% and 60% of control within30 minutes after 5 and 25 mg/kg dose levels, respectively.This was followed by a rapid and pronounced reboundwithin the first 2 hours of dosing and then amore sustainedperiod of p-Akt inhibition with levels at the higher dosereaching near nadir values of 60% at 10 hours. To ascertainthe cumulative effect of ON123300 on p-Akt levels theABECparameterswere 63.2% and 304.9%-h at intravenousdoses of 5 and 25 mg/kg, respectively, which is a pro-portional dose-dependent inhibition (5-fold change,see Table 1). Unlike the p-Akt profiles that sharedsimilar patterns of inhibition at both dose levels, thep-Erk profiles were somewhat dissimilar with the25 mg/kg dose group exhibiting appreciable activationthat was biphasic reaching a maximum of 161% relativeto baseline, whereas at the lower dose of ON123300 the
biphasic pattern was muted with a singular spike of p-Erk at 5 minutes of 159%. As shown in Table 1, TheABEC values of p-Erk were 50%-h below the baselineand 144%-h above the baseline following administrationof ON123300 at 5 and 25 mg/kg i.v., respectively. Thissuggests that at the lower 5 mg/kg dose of ON123300the Akt-dependent feedback inhibition mechanismswere not completely blocked.
The addition of gefitinib to the ON123300 25 mg/kgdose regimen produced prominent effects on both the p-Akt and p-Erk profiles. As with single-agent ON123300,the p-Akt profile was biphasic yet the signal was furtherreduced (ABEC: 765%-h, Table 1) and achieved a nadir ofabout 30% at 4 hours after the ON123300 dose (Fig. 4C).The p-Erk response, although biphasic, was attenuated toa largedegreewith only a single peak activation of 120%at5 minutes that was followed by a more durable decrease(ABEC: 273.9%-h below the baseline, Table 1) that was at80% of control at 17 hours following the ON123300 dose(Fig. 4C). Overall, the addition of gefitinib to ON123300produced similar effects on p-Akt and p-Erk responsesin brain tumor–bearing mice as that observed in U87 cellsin vitro.
In vivo pharmacokinetic profilesThe concentration–time profiles of ON123300 in plas-
ma, brain, and brain tumor following single intravenousdoses of 5 and 25 mg/kg are shown in Fig. 5A and B andthe corresponding noncompartmental analysis results aresummarized in Table 1. The pharmacokinetic profiles ofplasma ON123300 concentration were multiexponential
0
25
50
75
100
1086420
5 mg/kgA B25 mg/kg
Time (h)
Pe
rce
nta
ge
of p
-Akt
(re
lative
)
0
50
100
150
200
1086420
5 mg/kg
25 mg/kg
Time (h)
Pe
rce
nta
ge
of p
-Erk
(re
lative
)
0
50
100
150C
1612840
p-Akt
p_Erk
Pe
rce
nta
ge
of p
-Akto
r p
-Erk
(rela
tive)
Time (h)
Figure 4. Pharmacodynamicprofiles of brain tumor in U87 braintumor–bearing mouseadministered at an intravenousdose of 5 or 25 mg/kg alone orcombinational after 1 hour of oraldosing of 200 mg/kg gefitinib.p-Akt (A andC) and p-Erk (B andC)expression levels against total Aktand Erk expression in brain tumorhomogenates at various timepoints quantified byMSD analysis.Data, percentages of the meanvalue and mean � SD of at leastthree mice in each time point.
Zhang et al.
Mol Cancer Ther; 13(5) May 2014 Molecular Cancer Therapeutics1112
on March 22, 2020. © 2014 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst February 25, 2014; DOI: 10.1158/1535-7163.MCT-13-0847

and overall declined fairly rapidly with terminal elimi-nation half-lives of 1.5 hours. The AUCp values did notincrease in a dose-proportional manner, being 1,206 ng-h/mLat 5mg/kgand4,224ng-h/mLat 25mg/kgor a 3.5-fold increase that translated into an increase in CL at thehigher dose that could reflect saturable plasma proteinbinding for a low clearance drug. Previously, we showedthatON123300 is highly bound (99.4%) to plasmaproteinsin mice (16). The apparent volume of distribution ofON123300 was elevated at the higher dose also consistentwith saturable plasma protein binding (Table 1).ON123300 rapidly penetrated into brain with peak con-centrations at 5 minutes of 5735 ng/g and 12432 ng/g atdoses of 5 and 25 mg/kg, respectively. Distribution ofON123300 into normal brain as measured by the brainpartition coefficient (Pb)was equal to 2.6 and 2.8 at the lowand high doses, respectively. As expected in brain tumorswhere the BBB is compromised distribution of ON123300was greater andmore variable with brain tumor partitioncoefficients (PBT) equal to 5.7 and 15 at the 5 and 25mg/kgdose levels, respectively. Whether the elevated partition-ing into brain tumor at the 25 mg/kg dose could partiallybe attributed to saturation of BBB efflux transporters isunknownat this time; however, basedonprevious studiesit is unlikely to be a substrate for theABC transporters (16).
The concentration–time profiles of ON1233300 in micepretreated with a single 200 mg/kg oral dose of gefitnibwere similar in nature to those inmice treatedwith single-agent ON1233300 (See Fig. 5C and Table 1). There was amarked reduction (33%) in the total clearance ofON123300 in the presence of gefitinib. Normal brain andbrain tumor partitioning of ON123300 was analogous tothat when ON123300 was given alone, and althoughdistribution into brain tumor was less in the presence ofgefitinib it is more likely a reflection of interanimal var-iability in BBB integrity because normal brain distributionwas unaltered.
DiscussionDespite the plethora of new anticancer drugs under
study, appreciable improvements in patient survival havenot yet followed. Specifically, with respect to braintumors, successful chemotherapy must not only inhibitkey pharmacodynamic targets, but overcome the BBB thatcan greatly limit drug access to the desired target. Byapplication of a pharmacokinetic/pharmacodynamic-driven drug development strategy ON123300 surfacedas the lead candidate in theON123 series based on toxicityto glioma cells and proficient BBB penetration and accu-mulation in normal brain (16). The current investigation
Table 1. Pharmacokinetic and pharmacodynamic parameters after the administration of 5 and 25 mg/kgON123300 (5mg/kg and 25mg/kg) alone or with oral dosing of 200mg/kg gefitinib [25mg/kgþ gefitinib] onmice bearing U87 tumors
Dose
Tissue Parameter Unit 5 mg/kg 25 mg/kg 25 mg/kg þ GFN
Plasma AUCp h�ng/mL 1,206 4,224 6,351t1/2 H 1.5 1.5 2.3CL L/h/kg 4,146 5,919 3,937Vss L/kg 3,597 10,193 7,078
Brain AUCb h�ng/mL 3,123 11,679 19,794Cmax ng/mL 5,753 12,432.5 16,300Tmax h 0.083 0.083 0.25Pb 2.6 2.8 3.1
Brain tumor AUCbt h�ng/mL 6,886 63,221 49,440Cmax ng/mL 6,493 27,250 20,900Tmax h 0.083 1 0.5Pbt 5.7 15 7.8ABEC (%_h) p-Akt (%h) 63.2 304.9 765.0
p-Erk (%h) 50.0 144.1a 273.9
NOTE: NCA were performed using WinNonlin Phoenix, version 6.3 software (Pharsight Corporation) to estimate pharmacokinetic andpharmacodynamic parameters that included for each compound the areas under the drug concentration–time curve in plasma, brain,and brain tumor (AUCp, AUCb, and AUCbt, respectively), the areas between the effect-time curve in brain tumor (ABEC), half-life (t1/2),systemic clearance (CL), and the apparent volume of distribution at steady state (Vss). The normal brain or brain tumor partitioncoefficient (Pb and Pbt) was calculated as the ratio of either the AUCb or AUCbt over the corresponding AUCp. All AUC calculationswere based on the area to the last observable concentration using the linear-log interpolationmethod. It is assumed that the density oftissues equals 1 g/mL.aABEC above the baseline.
Preclinical PK/PD Evaluation of ON123300 in Brain Tumor
www.aacrjournals.org Mol Cancer Ther; 13(5) May 2014 1113
on March 22, 2020. © 2014 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst February 25, 2014; DOI: 10.1158/1535-7163.MCT-13-0847

was designed to characterize the pharmacokinetics/phar-macodynamics of ON123300 and assess its potential inbrain tumor chemotherapy by focusing on the compo-nents in the Ras/MAPK and PI3K/Akt/mTOR pathwaysthat are often dysregulated in patients with brain tumor(7, 8, 38).
In U87 glioma cells, ON123300 did inhibit p-Akt in adose- and time-dependent manner, yet unexpectedlyalso stimulated p-Erk that would be detrimental to itsanticancer activity. The observation that inhibitorsalong the RTK/PI3K/Akt/mTOR pathway produceundesirable anti-complimentary effects has been notedpreviously (29–33, 39–41). Specifically, the PDGFRinhibitor imatinib elicited Erk activation that was liganddependent, in which imatinib inhibited signaling medi-ated by PDGF-BB, but not by PDGF-AA or stem cellfactor (29, 40). The mTOR1 inhibitor rapamycin and itsanalogues can activate Ras and its downstream counter-parts by relief of the brake supplied by PI3K andtriggered by p70S6K (30, 31). This p70S6K/PI3K/Ras/Raf/MEK feedback loop exists in both cancer patientsand in preclinical animal models that appeared drugschedule-dependent as we observed (30, 31). In addi-tion, Akt antagonizes Raf activity by direct phosphor-ylation of S259, and thus, treatment with a PI3K or Aktinhibitor, such as LY294002, results in Erk hyperactiva-tion by decreasing inhibitory S259 phosphorylation of c-Raf (32, 33). Our data demonstrate that ON123300 treat-ment appears to cause MAPK activation not only byrelief of PI3K feedback inhibition triggered by p70S6K,but also by downregulation of c-Raf S259 phosphoryla-tion. The overall action of ON123300—decreased Akt
activity and activated Erk—was observed in three dif-ferent glioblastoma multiforme models, including apatient-derived model that supported the need to findanother drug that could synergize with ON123300 andprevent Erk activation.
Our previous studies of single-agent gefitinib sug-gested it would be effective in mitigating the rapid Erkactivation caused by ON123300 due to its rapid andappreciable penetration of the BBB and its inhibitoryaction upstream of PI3K and Akt and their points ofinteraction with the MAPK pathway (42–44). Otherstudies employing gefitinib or an EGFR inhibitor alsosuggested tumor growth and Erk suppression whenused in combination with either an mTOR inhibitor(rapamycin) or Akt inhibitor (45). In the latter study(45), the Akt inhibitor activated Erk yet when combinedwith gefitinib (150 mg/kg three times/week) growth ofNCI-H292 tumors in mice were significantly retardedcompared with the Akt inhibitor alone. These positivepreclinical findings of EGFR inhibitors and Akt/mTORpathway inhibitors have not yet translated to the clinicas noted for the combinations of gefitinib and ever-olimus or sirolimus (46–48). Pharmacodynamic analy-ses of intratumoral MAPK, PI3K, and mTOR signalingwere not completed leaving the cause of the lack ofefficacy unresolved (46–48). Other agents, possiblythose directly inhibiting Raf\MEK\Erk signal cascades,such as the MEK inhibitor AZD6244, may also curtailON123300-induced Erk activation (49, 50); however,with the gefitinib–ON123300 combination not onlywas Erk activation suppressed, Akt inhibition was alsoenhanced.
5
50
500
5,000
6543210
5 mg/kgA BP
B
BT
Time (h)
C
ON
12
33
00
co
nce
ntr
atio
n
(ng
/mL
)
5
50
500
5,000
50,000
1086420
25 mg/kg
P
B
BT
Time (h)
ON
12
33
00
co
nce
ntr
atio
n
(ng
/mL)
1
10
100
1,000
10,000
181614121086420
ON
123300 c
oncentr
ation
(ng
/mL
)
Time (h)
25 mg/kg+GFN
P
B
BT
Figure 5. Pharmacokinetic profilesof ON123300 in U87 brain tumor–bearing mouse concurrentlytreated with ON123300 alone orwith GFN. ON123300 wasadministered into U87 braintumor–bearing mouse at anintravenous dose of 5 or 25 mg/kgvia a tail vein alone or after 1 hour oforal dosing of 200 mg/kg gefitiniband plasma (P), normal brain (B),and brain tumor (BT) samples werecollected at various time pointsand analyzed by LC/MS-MS.ON123300 concentration (A–C) inplasma and tissue samples at anintravenous dose of 5 mg/kg (A),25 mg/kg alone (B), or pretreatedwith GFN (C). Data, mean � SD ofat least three mice in each timepoint.
Zhang et al.
Mol Cancer Ther; 13(5) May 2014 Molecular Cancer Therapeutics1114
on March 22, 2020. © 2014 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst February 25, 2014; DOI: 10.1158/1535-7163.MCT-13-0847

In summary, through a series of in vitro and in vivopharmacokinetic/pharmacodynamics investigationsof ON123300 we confirmed it possessed suitable phar-macokinetic properties for brain tumor chemotherapyaccentuated by brain:plasma partitioning of more than1. Of particular interest and significance was this mul-tikinase inhibitor produced opposing effects on thePI3K/Akt/mTOR and Ras/MAPK pathways that wereattributed to release of feedback inhibition mechan-isms that were dose-dependent, and would likely cur-tail future development as a single agent. Because thisfeature may be characteristic of inhibitors of the PI3K/Akt/mTOR pathway, it is worthwhile to consider con-founding pathway activations early in the drug devel-opment scheme and whether rational combinationtherapy can overcome, and in the process, improvedisruption of oncogenic pathways.
Disclosure of Potential Conflicts of InterestM.V. Ramana Reddy is a consultant in Onconova Therapeutics, Inc and
has ownership interest in several patents on kinase inhibitors throughTemple University. E. Premkumar Reddy is a director, a consultant/advisory board member of Onconova Therapeutics Inc, and has receivedcommercial research grant from and has ownership interest (includingpatents) in the same.Nopotential conflicts of interestwere disclosed by theother authors.
DisclaimerThe outcome of this research project could affect the value of these
patents and of Onconova Therapeutics.
Authors' ContributionsConception and design: X. Zhang, M.V. Ramana Reddy, J.M. GalloDevelopment of methodology: X. Zhang, H. Lv, Q. Zhou, E.P. ReddyAcquisition of data (provided animals, acquired and managed patients,provided facilities, etc.): H. Lv, R. Elkholi, J.E. Chipuk, E.P. ReddyAnalysis and interpretation of data (e.g., statistical analysis, biostatis-tics, computational analysis): X. Zhang, R. Elkholi, J.E. Chipuk, E.P.Reddy, J.M. GalloWriting, review, and/or revisionof themanuscript:X.Zhang, J.E.Chipuk,E.P. Reddy, J.M. GalloAdministrative, technical, or material support (i.e., reporting or orga-nizing data, constructing databases): X. ZhangStudy supervision: J.M. GalloOther: Design and synthesis of all ON123300 and related compounds,M.V. Ramana Reddy.
Grant SupportThis work was supported by the NIH grants CA127963 (to J.M. Gallo),
P01CA-130821 (to E.P. Reddy), and CA157740 (to J.E. Chipuk).The costs of publication of this article were defrayed in part by the
payment of page charges. This article must therefore be hereby markedadvertisement in accordance with 18 U.S.C. Section 1734 solely to indicatethis fact.
Received October 8, 2013; revised January 15, 2014; accepted January 28,2014; published OnlineFirst February 25, 2014.
References1. Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med
2008;359:492–507.2. Squatrito M, Holland EC. DNA damage response and growth factor
signaling pathways in gliomagenesis and therapeutic resistance. Can-cer Res 2011;71:5945–9.
3. Network CGAR. Comprehensive genomic characterization defineshuman glioblastoma genes and core pathways. Nature 2008;455:1061–8.
4. Nazarenko I, Hede SM, He X, Hedr�en A, Thompson J, Lindstr€om MS,et al. PDGF and PDGF receptors in glioma. Ups J Med Sci 2012;117:99–112.
5. Uhrbom L, Hesselager G, Nist�er M, Westermark B. Induction of braintumors in mice using a recombinant platelet-derived growth factor B-chain retrovirus. Cancer Res 1998;58:5275–9.
6. Beenken A,Mohammadi M. The FGF family: biology, pathophysiologyand therapy. Nat Rev Drug Discov 2009;8:235–53.
7. Bai RY, Staedtke V, Riggins GJ. Molecular targeting of glioblastoma:drug discovery and therapies. Trends Mol Med 2011;17:301–12.
8. WickW,WellerM,WeilerM,Batchelor T, YungAW,PlattenM.Pathwayinhibition: emergingmolecular targets for treating glioblastoma. NeuroOncol 2011;13:566–79.
9. Jimeno A, Li J, MessersmithWA, Laheru D, RudekMA,Maniar M, et al.Phase I study of ON 01910.Na, a novel modulator of the Polo-likekinase 1 pathway, in adult patients with solid tumors. J Clin Oncol2008;26:5504–10.
10. Olnes MJ, Shenoy A, Weinstein B, Pfannes L, Loeliger K, Tucker Z,et al. Directed therapy for patients with myelodysplastic syndromes(MDS) by suppression of cyclin D1 with ON 01910.Na. Leuk Res2012;36:982–9.
11. Roschewski M, Farooqui M, Aue G, Wilhelm F, Wiestner A. Phase Istudy of ON 01910.Na (Rigosertib), a multikinase PI3K inhibitor inrelapsed/refractory B-cell malignancies. Leukemia 2013;27:1920–3.
12. Seetharam M, Fan AC, Tran M, Xu L, Renschler JP, Felsher DW, et al.Treatment of higher risk myelodysplastic syndrome patients unre-sponsive to hypomethylating agents with ON 01910.Na. Leuk Res2012;36:98–103.
13. Ma WW, Messersmith WA, Dy GK, Weekes CD, Whitworth A, Ren C,et al. Phase I study of Rigosertib, an inhibitor of the phosphatidylino-sitol 3-kinase and Polo-like kinase 1 pathways, combined with gem-citabine in patients with solid tumors and pancreatic cancer. ClinCancer Res 2012;18:2048–55.
14. Nuthalapati S, Zhou Q, Guo P, Lv H, Cosenza S, Reddy MV, et al.Preclinical pharmacokinetic and pharmacodynamic evaluation of novelanticancer agents, ON01910.Na (Rigosertib, Estybon) and ON013105,for brain tumor chemotherapy. Pharm Res 2012;29:2499–511.
15. Lv H, Wang F, Ramana Reddy MV, Zhou Q, Zhang X, Reddy EP, et al.Screening candidate anticancer drugs for brain tumor chemotherapy:pharmacokinetic-driven approach for a series of (E)-N-(substitutedaryl)-3-(substitutedphenyl)propenamide analogues. InvestNewDrugs2012;30:2263–73.
16. Lv H, Zhang X, Sharma J, Reddy MV, Reddy EP, Gallo JM. Integratedpharmacokinetic-driven approach to screen candidate anticancerdrugs for brain tumor chemotherapy. AAPS J 2013;15:250–7.
17. Lu S, Niu N, Guo H, Tang J, GuoW, Liu Z, et al. ARK5 promotes gliomacell invasion, and its elevatedexpression is correlatedwith poor clinicaloutcome. Eur J Cancer 2013;49:752–63.
18. Kusakai G, Suzuki A, Ogura T, Kaminishi M, Esumi H. Strong asso-ciation of ARK5with tumor invasion andmetastasis. J Exp Clin CancerRes 2004;23:263–8.
19. Suzuki A, Lu J, Kusakai G-i, Kishimoto A, Ogura T, Esumi H. ARK5 is atumor invasion-associated factor downstream of Akt signaling. MolCell Biol 2004;24:3526–35.
20. Suzuki A, Kusakai G-i, Kishimoto A, Lu J, Ogura T, Lavin MF, et al.Identification of a novel protein kinase mediating Akt survival signalingto the ATM protein. J Biol Chem 2003;278:48–53.
21. Kato J,MatsushimeH, Hiebert SW, EwenME, Sherr CJ. Direct bindingof cyclin D to the retinoblastoma gene product (pRb) and pRb phos-phorylation by the cyclin D-dependent kinase CDK4. Genes Dev1993;7:331–42.
22. Deng X, Ma L, Wu M, Zhang G, Jin C, Guo Y, et al. miR-124 radio-sensitizes human glioma cells by targeting CDK4. J Neurooncol2013;114:263–74.
Preclinical PK/PD Evaluation of ON123300 in Brain Tumor
www.aacrjournals.org Mol Cancer Ther; 13(5) May 2014 1115
on March 22, 2020. © 2014 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst February 25, 2014; DOI: 10.1158/1535-7163.MCT-13-0847

23. Cen L, Carlson BL, SchroederMA, Ostrem JL, Kitange GJ,Mladek AC,et al. p16-Cdk4-Rb axis controls sensitivity to a cyclin-dependentkinase inhibitor PD0332991 in glioblastoma xenograft cells. NeuroOncol 2012;14:870–81.
24. Michaud K, Solomon DA, Oermann E, Kim JS, ZhongWZ, PradosMD,et al. Pharmacologic inhibition of cyclin-dependent kinases 4 and 6arrests the growth of glioblastoma multiforme intracranial xenografts.Cancer Res 2010;70:3228–38.
25. Fricker SP. The application of sulforhodamine B as a colorimetricendpoint in a cytotoxicity assay. Toxicol In Vitro 1994;8:821–2.
26. Chou TC. Theoretical basis, experimental design, and computerizedsimulation of synergism and antagonism in drug combination studies.Pharmacol Rev 2006;58:621–81.
27. Zhou Q, Guo P, Wang X, Nuthalapati S, Gallo JM. Preclinical phar-macokinetic and pharmacodynamic evaluation of metronomic andconventional temozolomide dosing regimens. J Pharmacol Exp Ther2007;321:265–75.
28. Luo JC, Lin HY, Lu CL, Wang LY, Chang FY, Lin HC, et al. Dexameth-asone inhibits basic fibroblast growth factor-stimulated gastric epi-thelial cell proliferation. Biochem Pharmacol 2008;76:841–9.
29. Servidei T, Riccardi A,SanguinettiM,Dominici C,Riccardi R. Increasedsensitivity to the platelet-derived growth factor (PDGF) receptor inhib-itor STI571 in chemoresistant glioma cells is associatedwith enhancedPDGF-BB-mediated signaling and STI571-induced Akt inactivation.J Cell Physiol 2006;208:220–8.
30. Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A,et al. Inhibition of mTORC1 leads toMAPK pathway activation througha PI3K-dependent feedback loop in human cancer. J Clin Invest2008;118:3065–74.
31. Carracedo A, Baselga J, Pandolfi PP. Deconstructing feedback-sig-naling networks to improve anticancer therapy with mTORC1 inhibi-tors. Cell Cycle 2008;7:3805–9.
32. Zimmermann S, Moelling K. Phosphorylation and regulation of Raf byAkt (protein kinase B). Science 1999;286:1741–4.
33. Moelling K, Schad K, Bosse M, Zimmermann S, Schweneker M.Regulation of Raf-Akt cross-talk. J Biol Chem 2002;277:31099–106.
34. Watt HL, Rachid Z, Jean-Claude BJ. Receptor activation and inhibitionin cellular response to chemotherapeutic combinational mimicries: theconcept of divergent targeting. J Neurooncol 2010;100:345–61.
35. Sun S, Lee D, Lee NP, Pu JK, Wong ST, Lui WM, et al. Hyperoxiaresensitizes chemoresistant human glioblastoma cells to temozolo-mide. J Neurooncol 2012;109:467–75.
36. OkumuraE, Fukuhara T,YoshidaH,HanadaSi S,KozutsumiR,MoriM,et al. Akt inhibits Myt1 in the signalling pathway that leads to meioticG2/M-phase transition. Nat Cell Biol 2002;4:111–6.
37. Katayama K, Fujita N, Tsuruo T. Akt/protein kinase B-dependentphosphorylation and inactivation of WEE1Hu promote cell
cycle progression at G2/M transition. Mol Cell Biol 2005;25:5725–37.
38. Huang S, Armstrong EA, Benavente S, Chinnaiyan P, Harari PM. Dual-agent molecular targeting of the epidermal growth factor receptor(EGFR): combining anti-EGFR antibody with tyrosine kinase inhibitor.Cancer Res 2004;64:5355–62.
39. George D. Targeting PDGF receptors in cancer–rationales and proof ofconcept clinical trials. Adv Exp Med Biol 2003;532:141–51.
40. Dong Y, Jia L,Wang X, Tan X, Xu J, Deng Z, et al. Selective inhibition ofPDGFR by imatinib elicits the sustained activation of ERK and down-stream receptor signaling in malignant glioma cells. Int J Oncol2011;38:555–69.
41. Chandarlapaty S, Sawai A, Scaltriti M, Rodrik-Outmezguine V, Grbo-vic-Huezo O, Serra V, et al. AKT inhibition relieves feedback suppres-sion of receptor tyrosine kinase expression and activity. Cancer Cell2011;19:58–71.
42. Wang S, Guo P, Wang X, Zhou Q, Gallo JM. Preclinical pharmacoki-netic/pharmacodynamic models of gefitinib and the design of equiv-alent dosing regimens in EGFR wild-type and mutant tumor models.Mol Cancer Ther 2008;7:407–17.
43. Wang S, Zhou Q, Gallo JM. Demonstration of the equivalent pharma-cokinetic/pharmacodynamic dosing strategy in a multiple-dose studyof gefitinib. Mol Cancer Ther 2009;8:1438–47.
44. Sharma J, Lv H, Gallo JM. Intratumoral modeling of gefitinib pharma-cokinetics and pharmacodynamics in an orthotopic mouse model ofglioblastoma. Cancer Res 2013;73:5242–52.
45. Rao RD, Mladek AC, Lamont JD, Goble JM, Erlichman C, James CD,et al. Disruption of parallel and converging signaling pathways con-tributes to the synergistic antitumor effects of simultaneousmTORandEGFR inhibition in GBM cells. Neoplasia 2005;7:921–9.
46. Nguyen TD LA, Lis E, Rosen N, Shaffer DR, Scher HI, Deangelis LM,et al. A pilot study to assess the tolerability and efficacy of RAD-001(everolimus) with gefitinib in patientswith recurrent glioblastomamulti-forme (GBM). Journal of Clinical Oncology ASCO Annual MeetingProceedings 2006;24.
47. Reardon DA, Quinn JA, Vredenburgh JJ, Gururangan S, Friedman AH,Desjardins A, et al. Phase 1 trial of gefitinib plus sirolimus in adults withrecurrent malignant glioma. Clin Cancer Res 2006;12:860–8.
48. Kreisl TN, Lassman AB, Mischel PS, Rosen N, Scher HI, Teruya-Feldstein J, et al. A pilot study of everolimus and gefitinib in thetreatment of recurrent glioblastoma (GBM). J Neurooncol 2009;92:99–105.
49. See WL, Tan IL, Mukherjee J, Nicolaides T, Pieper RO. Sensitivity ofglioblastomas to clinically available MEK inhibitors is defined byneurofibromin 1 deficiency. Cancer Res 2012;72:3350–9.
50. Tang Y, Dai Y, Grant S, Dent P. Enhancing CHK1 inhibitor lethality inglioblastoma. Cancer Biol Ther 2012;13:379–88.
Mol Cancer Ther; 13(5) May 2014 Molecular Cancer Therapeutics1116
Zhang et al.
on March 22, 2020. © 2014 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst February 25, 2014; DOI: 10.1158/1535-7163.MCT-13-0847

2014;13:1105-1116. Published OnlineFirst February 25, 2014.Mol Cancer Ther Xiaoping Zhang, Hua Lv, Qingyu Zhou, et al. Inhibitor, ON123300, in Brain Tumor ModelsPreclinical Pharmacological Evaluation of a Novel Multiple Kinase
Updated version
10.1158/1535-7163.MCT-13-0847doi:
Access the most recent version of this article at:
Cited articles
http://mct.aacrjournals.org/content/13/5/1105.full#ref-list-1
This article cites 49 articles, 18 of which you can access for free at:
Citing articles
http://mct.aacrjournals.org/content/13/5/1105.full#related-urls
This article has been cited by 1 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://mct.aacrjournals.org/content/13/5/1105To request permission to re-use all or part of this article, use this link
on March 22, 2020. © 2014 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst February 25, 2014; DOI: 10.1158/1535-7163.MCT-13-0847