praveen balimane addresses slas admet special interest group at slas2015
TRANSCRIPT

1
Dr. Praveen Balimane Senior staff fellow Division of Clinical Pharmacology-1 at OCP/OTS/CDER/FDA 2015 SLAS ADMET SPECIAL INTEREST GROUP MEETING Washington DC Moderator: David M. Stresser, Ph.D. Corning® GentestSM Contract Research Services
“Transporter Evaluation in Drug Development.”

2
ADMET Special Interest group - Mission
•Advance drug discovery and development by promoting the discussion and dissemination of topics and ideas for the integration of higher throughput technologies with methods for determining toxicity, pharmacokinetics and metabolism.
•Accelerate the drug discovery pipeline and shorten the time of the development of new drugs that cure illnesses and improve quality of life.

3
Past Speakers and topics
Year Speaker Topic 2012 Michael Fisher, Alnylam Metabolic Stability assays
2013 Adrian Fretland, Lilly Impact of regulatory guidance on in vitro DDI testing
2014 David Stresser, Corning Time-dependent inhibition of P450
2015 Praveen Balimane, FDA Transporter Evaluation in Drug Development
All slide decks from past talks are available on our Linked-In page:

Transporter Evaluation in Drug Development
ADMET Special Interest Group SLAS Meeting
Washington D.C (Feb 11th, 2015) 4
Praveen Balimane, Ph.D.
Office of Clinical Pharmacology Office of Translational Sciences
CDER, FDA

Disclaimer
The contents of this presentation are my own personal opinions and do not necessarily
reflect the official views and/or policy of the FDA or any government agency.
5

Topics
• Overview- ADMET, Transporters DDI – Decision trees (Pgp and OATP) – Novel transporters- MATE, BSEP’s – Hepatic transporters: safety interplay – Renal transporters: creatinine
• Open forum
6

7
TUFT’s REPORT: Total cost of developing a drug is 2.6 Billion $$
Joseph Dimasi et. al., TUFTS center for study of Drug Development, Nov- 2014
Higher than the GDP of Bhutan, Somalia,
Aruba……..many more
BUMPER approval Rate in 2014 41 novel meds !!! - 17 first-in-class - 17 orphan/rare

Success depends on several moving parts…
8
Discovery Development

9 Compound selection process
Solubility
Transporter
pKa/LogP
In vivo PK
Permeability
MetStab
Toxicity CYP450
Induction
Safety
IDEAL Compound
Dial Out
Liability
SAR SPR
Dial In
Properties
Advance Developable compounds
Efficacy

10
Impact of Transporters
• Global effect on ADMET • Targeted drug accumulation in organs – efficacy & safety
• DDI’s: anticipate and manage • Polymorphism & clinical variability

Transporter-based Drug-Drug Interactions are Clinically Relevant
Transporter Perpetrator drug Victim drug Clinical Effect OATP (hepatic)
Cyclosporin Pravastatin 10-fold ↑ Cyclosporin Rosuvastatin 7-fold ↑ Rifampicin Glyburide 2-fold ↑ Rifampicin Bosentan ~6-fold ↑
OAT (renal)
Probenecid Cephradine 4-fold ↑ Probenecid Acyclovir 1.5-fold ↑ Probenecid Methotrexate ~2-fold ↑
OCT (renal)
Cimetidine Metformin 1.5-fold ↑ Cimetidine Dofetilide 1.5-fold ↑
P-gp (gut) Quinidine Digoxin ~2-fold ↑ Dronedarone Digoxin 2.6-fold ↑
11
OATP’s, Efflux (P-gp, BCRP), OAT’s, OCT’s

Regulatory Guidance/Guideline on Drug Interactions • U.S. Food and Drug Administration (FDA)’s Draft Guidance for
Industry: Drug Interaction Studies—Study Design, Data Analysis, Implications for Dosing, and Labeling Recommendations (2012) (http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM292362.pdf)
–In addition to P-gp, transporter-related drug interaction evaluations and decision trees are included for additional transporters (BCRP, OATP1B1/3, OAT1/3 and OCT2)
• European Medicines Agency (EMA) Guideline on the Investigation of Drug Interactions (2012) (http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf)
• Pharmaceuticals Medical Devices Agency (PMDA) Draft Guideline on Drug Interactions (2013) (http://search.e-gov.go.jp/servlet/Public?CLASSNAME=PCMMSTDETAIL&id=495130206)
12

Since 2007, 40-60% of NME drug labels contain transporter information
(N=183).
0
10
20
30
40
50
60
70
2003 2004 2005 2006 2007 2008 2009 2010 2011
Year of approval
% o
f N
ME
PIs
wit
h t
ran
spo
rter
. in
form
ati
on
Transporter information has been increasingly included in the FDA Approved New Molecular Entities (NMEs) Labeling
(2003-2011)
Agarwal S, et al. Pharm Res. 2013, 30:899-910; Lee S-C, et al, book chapter, 2014; Yu J, et al., DMD, 2014
86%
15% 18%15%
8%1.4%
9.5%1.4%
0%
10%20%
30%40%
50%60%
70%80%
90%
% o
f N
ME
P-gp BCRPOATP
OCT OAT MATEMRP BSEP
Transporters
P-gp is the mostly studied transporter (N=74)
13
In 2012, 79% of NME labels contain transporter information (N=33) and 96% were P-gp; BCRP: 36%; OATP: 48%; OCT: 33%; OAT: 27%; MATE (N=1, 3%); MRP (N=5, 15%);
BSEP (N=2, 6%).
2012-2013 70-80%

14 14
The Challenges to Study Transporter DDI
• The issues presented by transporters are significantly more complex than for metabolizing enzymes – Involved in absorption, distribution and excretion: multiple processes of
concern – Broad tissue distribution: different effects at different sites – Functional redundancy: different transporters and different subfamilies – Uptake and efflux transporters: need to consider both to assess the overall
effect – Applicability of kinetic parameters and their interpretation – Measuring drug exposure in plasma may not reflect impact on a drug’s
disposition (e.g., toxicity)
Tweedie D, et al, Clin Pharm Ther, July 2013

15
NME as a Substrate Does the drug level depend on a given transporter?
• Route of elimination – Hepatic major – Renal major – Rate limiting step
• Physicochemical properties of the drug
– e.g., BCS or BDDCS
• Structure – e.g., OATs for anions and OCTs for cation – Caveat: some cations transported by OATs (cimetidine, sitagliptin) – similarity to known substrates

Evaluation of NME as a Substrate for Transporters
Determine whether
NME is a P-gp and/or BCRP
substrate in vitro
All NMEs
Hepatic or biliary secretion major?
e.g., ≥ 25% total clearance?
Renal active secretion major?
e.g., ≥ 25% total clearance?
Refer to P-gp and BCRP decision tree
for the need to conduct in vivo studies
Determine whether NME is an OATP1B1
or OATP1B3 Substrate in vitro
Determine whether NME is an OAT1, OAT3
or OCT2 substrate in vitro
Refer to OATP1B1/1B3 decision tree for the need to conduct in
vivo studies
Refer to OAT1/3 and OCT2/MATE decision tree
for the need to conduct in vivo studies
Yes or unknown Yes or unknown
(modified from page 31 of 75- FDA 2012 draft guidance) ;
Other trasnporters,
e. g. , MRP, may need to be evaluated.
16
Also consider MATEs
Tweedie D, et al. Clin Pharm Ther, July 2013

NME as an Inhibitor Does the drug affect a given transporter?
• Inhibitors can be substrates or non-substrates for a given transporter.
• The need to study depends on whether drugs are likely co-administered with known substrates of major human transporters.
• Their concentration (free, total, Cmax etc.) in target site dictates their effect
• All drug-related moieties (parent, metabolites, active/inactive) can act as inhibitors
17

18
Transporter Inhibitor Decision Trees
P-gp/BCRP OATP1B1/OATP1B3
OAT1/OAT3/OCT2/MATEs
Goal: Determine whether in vivo studies are needed based on in
vitro assessment. It is not intended to use in vitro
data to determine the magnitude of an in vivo interaction.
FDA 2012 Draft DDI Guidance

19
Examples of Transporter-Related PMR/PMC 2011-2012
Year Drug Name (Brand Name)
Transporter-Related PMR or PMC
2011 VILAZODONE HYDROCHLORIDE (VIIBRYD)
DDI with digoxin (P-gp)
2011 BOCEPREVIR (VICTRELIS)
DDI with digoxin (P-gp)
2011 RILPIVIRINE (EDURANT)
DDI with digoxin (P-gp)
2011 EZOGABINE (POTIGA)
Substrate of renal transporters DDI with digoxin (P-gp)
2011 RIVAROXABAN (XARELTO)
Renal impairment plus P-gp/moderate CYP3A inhibitor
2012 IVACAFTOR (KALYDECO)
DDI with digoxin (P-gp)
2012 EVG/COBI/FTC/TFV (STRIBILD)
In vitro as substrate and/or inhibitor of major transporters as stated in the guidance (plus MRP2, MRP4, BSEP, MATE1 and OCT1).
2012 TERIFLUNOMIDE (AUBAGIO)
DDI with rosuvastastin (BCRP and OATP1B1)
PMR/PMC: Postmarketing requirement/Postmarketing commitment Tweedie D, et al. CPT, July 2013

P-GP DECISION TREE
20

21
P-gp Inhibition Decision Tree
-Initially proposed in Zhang L et. al., Xenobiotica, 38(7–8): 709–724, 2008 -2012 FDA draft Drug Interaction Guidance
[I]1 is total Cmax
Bi-directional transport assay with a probe P-gp substrate (e.g. in Caco-2 or MDR1-overexpressing
polarized epithelial cell lines)
Net flux ratio of a probe substrate decreases with increasing concentrations of the
investigational drug
Net flux ratio of the probe substrate is not affected with increasing concentrations of the
investigational drug.
Poor or non-inhibitor Probably a P-gp inhibitor
Determine Ki or IC50 of the inhibitor
An in vivo drug interaction study with a P-gp
substrate is not needed.
An in vivo drug interaction study with a P-gp substrate
such as digoxin is recommended.
[I]1/IC50 (or Ki) ≥ 0.1 or
[I]2/IC50 (or Ki) ≥ 10
[I]1/IC50 (or Ki) < 0.1 and
[I]2/IC50 (or Ki) < 10
[I]2 (gut concentration)/IC50≥ 10 is New (Not in 2006 draft DDI
Guidance).
[I]2 is Dose/250 mL
Different from ITC Whitepaper (unbound
Cmax)

22
In Vitro and In Vivo Digoxin Data Recent NDA approvals (2003-2010)
Drug name [I]1/IC50 (unbound Cmax)
[I]1/IC50 (total Cmax)
[I]2/IC50 Digoxin Cmax
(% Change)
Digoxin AUC
(% Change) Lapatinib <0.1 1 1355 NA 180
Dronedarone <0.1 0.09 1349 NA 150
Ranolazine <0.1 0.04 2987 46 60
Darunavir <0.1 0.33 146 15 58
Tolvaptan <0.1 0.23 109 30 20
Etravirine <0.1 0.04 76 19 18
Tetrabenazine <0.1 0.01 6 13 2
Maraviroc <0.1 0.01 13 4 0.5
Deferasirix <0.1 0.003 4.3 -8.7 -8
Lacosamide <0.1 0.01 1 4.8 2.4
Sitagliptin <0.1 0.02 2 18 11
-Agarwal S, Zhang L, Huang, S-M, Clin Pharmacol Ther 89(1): February 2011 (poster presentation at the annual ASCPT meeting, Dallas, TX, March 2-5, 2011);
-Agarwal, Arya and Zhang, JCP, 2012.
False Positive
False Negative
~82% predictive !!

23
Igut (Ient) Algorithm Exploration
• Because [I]2 assumes that the entire dose is dissolved in the gut, the use of [I]2/IC50 criteria may lead to false positives, especially for drugs with low solubility.
• A new algorithm, [I]gut/IC50, was explored to determine whether this algorithm could potentially reduce the false positive rate by considering the actual absorption of the drugs into the enterocytes – [I]gut ([I]ent) is defined as Fa×ka×Dose/Qen
Agarwal, Arya and Zhang, JCP, 2012; Agarwal S, et al, Clin Pharmacol Ther : February 2012 (poster presentation at the annual ASCPT meeting, National
Harbor, MD, March 14-17, 2012) (Poster Session III-3, 7-8 am, March 17, 2012)

24
Igut (Ient) Algorithm Exploration Dataset of 24 drugs that have both in vitro and in
vivo P-gp inhibition data (digoxin as the substrate) 12 drugs showed positive interaction with digoxin in
vivo 12 drugs showed negative interaction with digoxin in
vivo 5 False positives and 1 false negative
[I]gut values were determined from inhibitors’ in
vivo PK data. Data Sources:
Zhang et al; Xenobiotica. 2008 Jul;38(7-8):709-24. Agarwal et al; J Clin Pharmacol. 2012 Feb 7. [Epub ahead of print]. Fenner at al; Clin Pharmacol Ther. 2009 Feb;85(2):173-81.

25
Igut (Ient) Algorithm Exploration
1 (8%)
11 (92%)
10 (83%)
2 (17%)
FN TP
TN FP
1 (8%)
11 (92%)
7 (58%)
5 (42%)
[I]gut,/IC50 ≥2 [I]2/IC50≥ 10
Predicted
Observed
• A distinct [I]gut/IC50 cut off value that could eliminate all 5 false positives in our dataset of 24 drugs was not identified. • [I]gut/IC50 cutoff of ≥2 (as “predicted positive”) appears to classify 3 out of 5 FPs (based on [I]2/IC50 ≥ 10) as “true negatives”, reducing false positive rate from 42% to 17% without changing FN rate. • Talinolol remains as a false negative by either algorithm. • False positives and false negatives may be caused by mechanisms that cannot be captured in the in vitro P-gp inhibition assay. • [I]gut/IC50 algorithm needs further validation to confirm its utility as an additional algorithm.
Agarwal S, et al, Clin Pharmacol Ther : February 2012 (poster presentation at the annual ASCPT meeting, National Harbor, MD, March 14-17, 2012) (Poster Session III-3, 7-8 am, March 17, 2012)

EMERGING TRANSPORTERS
26

27
2nd International Transporter Consortium Transporter Workshop (March 2012)
Zamek-Gliszczynski et al. Clin Pharmacol Ther, November 2012
Red: Critical transporter proteins to evaluate prospectively Green: additional one to evaluate prospectively Yellow: retrospective evaluation
27

28
Emerging Transporters -Impact on a Broad Range of Drugs
• Multidrug And Toxin Extrusion Transporters: MATEs
• Drugs and Conjugate Efflux Pumps of the ABCC Family (MRP2, other MRPs)
• Bile Salt Export Pump (ABCB11)
2nd ITC Transporter Workshop (March 2012)

29
MATE (SLC47A) Transporters • Efflux transporters
– Proton-antiproters
• MATE1 – Liver and kindey
• MATE2 and MATE2K – Kidney
Hillgren K, et al, CPT, 94, 52-63, 2013

30
Clinical Importance • Polymorphism of MATE1/2 has been linked to clinical
effects in metformin-treated subjects • Reduced metformin response
• MATEs mediates clinical drug-drug interactions (DDIs) previously attributed to OCT2 – Overlapping substrate between MATEs and OCT2
• MATEs also transport anionic compounds and zwitterions – Some differential specificity of inhibitors
• Inhibition of MATEs may increase tissue concentration of substrate drugs – Renal toxicity consideration if the substrate drug is renal
toxic
Hillgren K, et al, CPT, 94, 52-63, 2013

31
Putative MATE-Mediated Clinical DDIs
Hillgren K, et al, CPT, 94, 52-63, 2013 and references therein
MATE Mediates Clinical Drug Drug Interactions Previously Attributed to OCT2
Perpetrators inhibit BOTH the MATE’s and OCT

32
Recommendation from ITC • MATEs need to be considered for prospective
investigation along with OCT2 and OATs.
NME as a substrate NME as an inhibitor
Hillgren K, et al, CPT, 94, 52-63, 2013
(Change in creatinine clearance may indicate renal transporter DDI)

33
BSEP (ABCB11) • An efflux transporter expressed on the
canilicular membrane of the hepatocytes • Secrete bile acids to bile
– Bile acids are taken up by multiple transporters including NTCP and OATPs.
Hillgren K, et al, CPT, 94, 52-63, 2013

34
Clinical Importance • Mutations in the ABCB11 gene lead to accumulation of bile
salts in the liver and progressive intrahepatic cholestasis. – The clinical spectrum of ABCB11 mutations covering benign
recurrent intrahepatic cholestasis type 2 to progressive familial intrahepatic cholestasis type 2 (PFIC2), also known as BSEP deficiency syndrome
– Other common polymorphism in ABCB11 is c.1331T>C (p.V444A) leads to lower BSEP levels.
• Inhibition of BSEP can lead to increased bile salts in the liver that may lead to cholestasis. – Targeted inactivation of BSEP in mice is known to cause persistent
cholestasis
CPT, 94, 52-63, 2013

Hepatic Transporters
Interplay with hepato-TOX
35

Transporters- DILI
36

37 Role of BSEP Transporters in DILI
Tox. Sciences, 118, 2, 485-500, 2010
200 marketed drugs used to assess the relationship between BSEP inhibition and liver injury
IC50 < 25 uM
Cyclosporin Nefazodone
Rosiglitazone Rifampin Ritanovir
Troglitazone Bosentan
IC50 > 100 uM
Asprin Antipyrine
Caffeine Cimetidine
Desipramine Famotidine Metformin
Nadolol Sulfasalazine
Timolol Verapamil

38
• >600 compounds • When factoring for exposure, 95% of the
annotated compounds with a Css/BSEP IC50 ratio ≥ 0.1 were associated with some form of liver injury.
• Drugs with a Css/BSEP IC50 ratio ≥ 0.1 and a Css/MRP IC50 ratio ≥ 0.1 had almost a 100% correlation with some evidence of liver injury in humans.
• integration of BSEP and MRP2 data is a useful tool for informing the potential for liver injury due to altered bile acid transport.
ToxSci Advance Access published November 5, 2013
Morgan et al., TOXICOLOGICAL SCIENCES, 2013

39
Morgan et al., TOXICOLOGICAL SCIENCES, 2013

40
Recommendation from ITC • Restrospective testing
– At this stage, it is impossible to define a value for a BSEP inhibition constant that will realistically predict significant BSEP-mediated DILI.
– In vitro characterization of BSEP–drug interactions is certainly warranted after the appearance of cholestatic issues in clinical trials or safety studies
Systematic studies required with ALL relevant transporters (BSEP, NTCP, MRP2, OATP,?) to assess
the “causal link”

RENAL TRANSPORTERs
Inter play with creatinine
41

Creatinine-Drug Interactions • Creatinine = biomarker probe to predict the kidney function (GFR) • Creatinine is found to be a substrate of multiple renal transporters including
OCT2, MATE1, MATE2K, and OAT2. • Increase in serum creatinine can be due to :
– renal toxicity or – inhibition of creatinine transport pathways by new molecular entities.
42 Lepist E-I, et al., Kidney Int. 2014, 86(2):350-7.
Huang Y, AAPS Webinar, May 2014

Increase in serum creatinine (without alteration in renal function)?
• Common features by a group of drugs in the literature and in NDA submissions: – ~10-30% increase in sCr in clinical trials accompanied by
decrease in CLcr – No effect on actual GFR (aGFR) as assessed by inulin, sinistrin,
iohexol, iothalamate, or Cr-EDTA – No impact on various renal function biomarkers (e.g., albumin,
blood urea nitrogen (BUN), Cystatin C, β-microglobulin, N-acetyl-β-glucosaminidase (NAG), para-aminohippurate (PAH), etc.)
– The increase in sCr generally has rapid-onset upon drug administration and is reversible, returning to baseline after discontinuation of the drugs.
43 V Arya, X Yang, et. al., ASCPT 2014, Atlanta, GA.

Inhibition of renal transporters may account for the increase in serum creatinine
44
Can increase in creatinine concentration be used as an “indicator” of in vivo renal transporter inhibition by the new molecular entity?
V Arya, X Yang, et. al., ASCPT 2014, Atlanta, GA.
Can in vitro inhibition of renal transporters (MATE’s, OCT) be an early predictor of potential increase in creatinine concentration in clinic
and

45
Summary • Transporters should be considered in the overall drug
development strategy – May be a critical factors contributing to DDI, toxicity and efficacy
• Novel transporters: – MATEs to be prospectively studied for new drugs as their substrates or
inhibitors along with other renal transporters (OCT2 and OATs). – MRP2 and BSEP may play a role in liver toxicity and should be studied if
there is preclinical or clinical signs of liver toxicity to understand the mechanisms.
– Other transporters may be important for drug delivery and drug target and should be studied on a case-by-case basis.
• Emerging science and novel models (KO- cell lines, humanized animal models etc.) will continue to shape the transporter field and Regulatory Guidance's

46
Acknowledgements • Lei Zhang • Shiew-Mei Huang • Sheetal Agarwal • Jaya Vaidyanathan • Ping Zhao • Kellie Reynolds • Vikram Arya • Xinning Yang • Leslie Chinn • Other FDA Transporter Scientific Interest Group Members • ITC members • IQC members • Sabbatical scientists at the FDA

Transporters, like CYPs, are being recognized as proteins that can play a pivotal role in dictating the ADME properties of drugs. A thorough understanding of potential roles of transporters in drug interactions and toxicity is important in drug development. The talk will provide a high level overview of various transporter evaluation initiatives at the agency. Some of the topics which will be discussed: On-going efforts on decision trees within the DDI guidance, novel emerging transporters impacting ADME, inter-play of hepatic transporters and liver-toxicity, and inter-play of renal transporters and renal function etc.
47
Transporter Evaluation in Drug Development
48
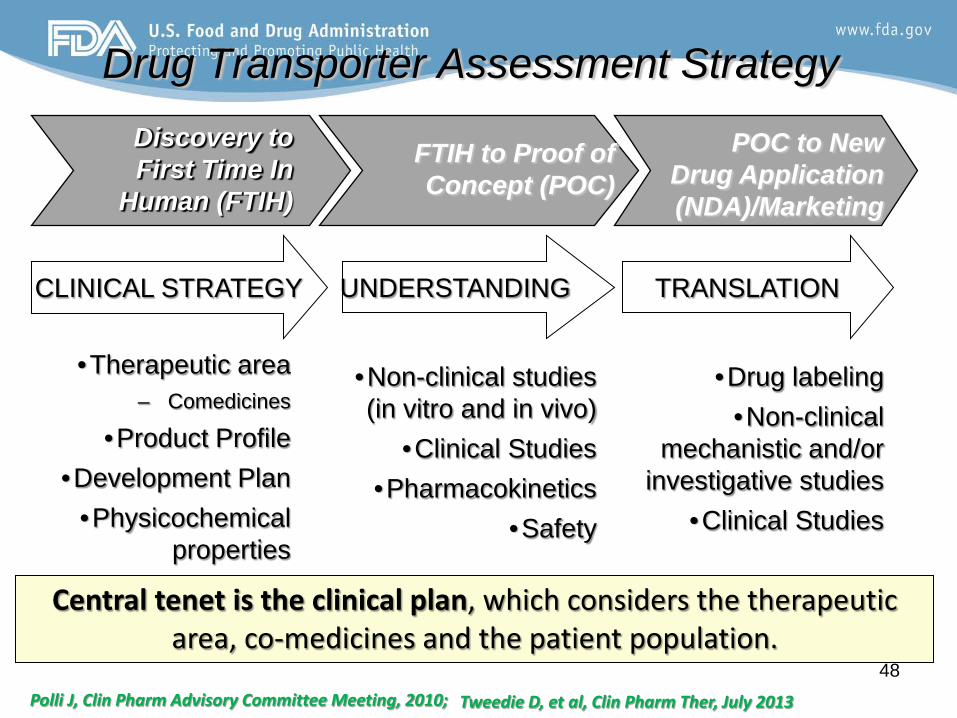
Drug Transporter Assessment Strategy
Central tenet is the clinical plan, which considers the therapeutic area, co-medicines and the patient population.
CLINICAL STRATEGY
•Therapeutic area – Comedicines
•Product Profile •Development Plan
•Physicochemical properties
UNDERSTANDING TRANSLATION
Discovery to First Time In
Human (FTIH)
FTIH to Proof of Concept (POC)
POC to New Drug Application (NDA)/Marketing
•Drug labeling •Non-clinical
mechanistic and/or investigative studies
•Clinical Studies
•Non-clinical studies (in vitro and in vivo)
•Clinical Studies •Pharmacokinetics
•Safety
Polli J, Clin Pharm Advisory Committee Meeting, 2010; Tweedie D, et al, Clin Pharm Ther, July 2013

49
P-gp is the Most Studied Transporter 2003-2006
2007-2011
Total # of approved NMEs 87 95
# (%) of NME labeling that have information on a specific transporter
16 (18%)
41 (43 %)
# of NME labeling that have information on P-gp
12
39
# of NME labeling that have transporter information other than P-gp such as BCRP,
OATP, etc
4
12
Transporter-related PMR* or PMC*
P-gp
20
16
Agarwal, Fan and Zhang (manuscripts in preparation)
*PMR: Post-marketing requirement; PMC: Post-marketing commitment

50
Transporters and Liver Toxicity
• Drug-induced liver injury could be multi-factorial. – BSEP inhibition shows correlation but not all leads to
drug-induced liver injury (DILI). DILI can be caused by other mechanisms.
– Other transporter involvement? • Uptake transporters? • Efflux transporters?
– Factors affecting BSEP expression? – Metabolites?
• A comprehensive panel may need to be evaluated to predict the risk.

51
In Vitro and In Vivo Digoxin Data Recent NDA approvals (2003-2010)
In vivo outcome of 9 /11 NMEs (82%) were accurately predicted. Two false positives: etravirine and maraviroc
The 2 false positives partially may be attributed
to potential P-gp induction effects that may off-set their inhibition effects Etravirine is a CYP3A inducer Maraviroc did not interact with midazolam in vivo
It is a weak P-gp inhibitor (IC50 ~183 uM, I2/IC50 ~12)
-Agarwal S, Zhang L, Huang, S-M, Clin Pharmacol Ther 89(1): February 2011 (poster presentation at the annual ASCPT meeting, Dallas, TX, March 2-5, 2011); -Agarwal, Arya and Zhang, JCP, 2012.

52
P.52-63
Acknowledgements: KM Hillgren* D Keppler* AA Zur KM Giacomini B Stieger CE Cass L Zhang* ITC *Corresponding authors